
Submitted:
17 May 2024
Posted:
20 May 2024
You are already at the latest version
Abstract
The marine environment is the largest ecological habitat on Earth, albeit one of the least explored, particularly in terms of its microbial inhabitants. The marine fish gut is host to a diverse microbial community from which diverse bioactive molecules can be sourced. Due to the unique environmental pressures these microbial communities experience, the bioactive molecules they produce often evolve unique adaptations that give them diverse structures and activities, differentiating them from terrestrial homologues. Of particular interest, due to their structural and functional diversity, are the ribosomally-synthesized antimicrobial peptides (bacteriocins). With increasing pressure from emerging antibiotic-resistant disease and industrial demand for novel therapeutics, the marine fish gut microbiome represents a relatively untapped resource of novel bacteriocins that could prove beneficial to human health and aquaculture. This review presents an overview of the marine fish gut microbiome and explores its potential as a source of bacteriocins for human health with considerations for applications and future research in this area.
Keywords:
marine fish
; gut microbiome
; bacteriocins
; bioprospecting
; antimicrobial
1. Introduction
The marine environment is the largest ecological habitat on the planet. The Earth’s oceans cover over 70% of the planet’s surface and reach depths of over 10,000m [1]. Each layer, or zone, of the ocean is its own ecological niche that is distinguishable based on factors such as temperature, salinity and sunlight availability [2]. The top layers (epipelagic and mesopelagic zones) sustain a high abundance of diverse life and are very metabolically active. In contrast, the deep ocean layers (bathypelagic, abyssal and hadal zones) are subject to low temperatures, high hydrostatic pressure and lack of sunlight [3]. From hydrothermal vents to the deep, dark trenches, marine fish inhabit extreme environments and have as a result acquired diverse and unique physiological and molecular adaptations.
The marine environment is also a rich source of diverse natural bioactive compounds with therapeutic and economic potential. People have been aware of the healing properties of the sea and marine natural products for thousands of years; ancient Greeks, for example, used marine invertebrates to treat a wide range of health conditions, particularly disorders of the skin, digestive and genitourinary systems [4].
To date, there are 15 clinically approved drugs derived from marine organisms, ranging from peptides to antibodies and covering a wide variety of biological activities. Many more are in various stages of clinical trials [5]. We are in an age when there is a huge demand for novel antimicrobials and alternative treatments to antibiotics. Antimicrobial resistance (AMR) is an extremely concerning and global threat to human health. It has been estimated that in 2019 alone, AMR was associated with 4.95 million deaths [6]. Traditional antibiotic treatments have become less effective, multidrug-resistant pathogens are more common yet the development of new antibiotic-based treatments has slowed dramatically over the past few decades [7]. From 2014 to 2018 over 250 preclinical antimicrobial compounds from marine organisms were reported [8,9,10]. The majority of these compounds were polyketides (35%), terpenoids (26%), alkaloids (17%) and peptides (14%), as well as lipids, lipopeptides and several other molecule types. Just over half of these molecules were produced by bacteria or fungi, and, significantly, these producers were isolated from higher organisms, particularly from marine sponges.
The mammalian gut microbiome is a well-established source of beneficial bacteria and compounds with therapeutic potential [11,12], and undoubtedly, this scenario is paralleled in the fish gut microbiome. There is growing interest in bioprospecting the fish gut microbiome to find novel bioactive compounds with therapeutic potential and for applications in aquaculture [13]. The gut microbiome of fish has crucial biological functions in digestion, immune system modulation, stress response, and protection from pathogens/infection [14]. These functions are often mediated by microorganisms and microbial metabolites, such as bacteriocins [15]. Bacteriocins are low molecular weight (<10 kDa), ribosomally-synthesized peptides with antimicrobial activity produced by bacteria that are immune to their own bacteriocin. The antagonistic activity of bacteriocins can be bacteriocidal or bacteriostatic, and the range of susceptible target organisms can be described as broad or narrow. The classification system for bacteriocins by Cotter et al. [16] proposed two main classes of bacteriocins, based largely on the extent to which they are modified and the mature peptide structure. In this scheme class I bacteriocins are highly modified (e.g. lantibiotics or lanthipeptides), class II are unmodified bacteriocins, and larger (>10kDa), heat-labile antimicrobial peptides were reclassified as ‘bacteriolysins’ (formerly class III bacteriocins) [16]. The class I lantibiotics are synthesized as prepeptides with unique lanthionine and/or β-methyllanthionine residues that undergo extensive posttranslational modification resulting in the formation of signature (methyl)lanthionine rings. Lanthipeptides can be further divided into subclasses I–V based on the enzyme(s) involved in their biosynthesis, modification, and mode of action [17]. The class II bacteriocins are subdivided into four groups (classes IIa-d): the anti-listerial pediocin-like peptides (IIa), two-component peptides (IIb), cyclic/circular bacteriocins (IIc) and the linear and non-pediocin-like peptides (IId) [18]. Bacteriocins are emerging as promising alternatives to conventional antibiotics due to their spectrum of activity – which can be broad or narrow, thermostability and their capacity for bioengineering and generation of variants with value-added properties [19]. Attention is now turning to fish microbiomes as novel sources of bioactive molecules such as bacteriocins. Conventionally, the diversity of fish microbiomes has been determined using culture-dependent methods and has very likely been underestimated in terms of its abundance and richness. However, ongoing advances in culture-independent methodologies, such as metagenomic sequencing, are enabling more accurate analysis of this microbial niche. Because of the huge expanse of the marine environment, marine microorganisms are among the least explored and least accessible and thus the application of culture-independent metagenomic analysis becomes even more valuable. The marine fish gut microbiome is essentially an untapped reservoir for novel antimicrobials.
In this review, we discuss the diversity of the gut microbiomes of marine fish, with a focus on finfish: Chondrichthyes (cartilaginous fish) and Osteichthyes (bony fish). We also discuss current research on the discovery of bacteriocins from marine fish gut microbiota, their applications, and the current prospects of this microbial niche as a source of novel antimicrobials.
2. The Diversity of the Marine Fish Gut Microbiome
The marine fish gut is host to a diverse microbial community largely consisting of bacteria, with reported counts ranging from 104 – 109 [20], as well as fungi, archaea and viruses. This community begins to form at the fish larval stage, with early colonizers originating from the egg surface, the surrounding water and the first feed [21]. The diversity of the fish gut microbiome is then influenced by biotic and abiotic factors such as host phylogeny, trophic level (diet) and environmental salinity [22], as well as, though to a lesser extent, environmental pH and temperature [20,23]. The fish gut microbiome is an important and unique niche within the marine environment, with reports that the composition of microorganisms found in marine fish gut can differ from that of the surrounding waters and can even contain species that are rarely, if at all, found in the surrounding water [24].
The composition of the marine fish gut microbiome has already been extensively reviewed [20,25,26]. Early studies on fish microbiome diversity were limited to the culturable organisms [27,28], however, with the emergence of next-generation sequencing (NGS) and metagenomic technologies there is now a growing number of culture-independent studies that have been able to provide a more comprehensive description of the fish gut microbiome [29]. Findings from more recent studies which have utilized culture-independent methods are given in Table 1. The most frequently reported phylum is Pseudomonadota (formerly Proteobacteria), followed by Bacillota (Firmicutes), Actinomycetota (Actinobacteria) and Bacteroidota (Bacteroidetes). Other abundant phyla include Mycoplasmatota (Tenericutes), Cyanobacteriota (Cyanobacteria), Fusobacteriota (Fusobacteria) and Chloroflexota (Chloroflexi). At a lower taxonomic level, Vibrionales are frequently detected, particularly species of Vibrio and Photobacterium, as well as members of the families Moraxellaceae, Pseudomonadaceae and Micrococcaceae (Table 2). Host diet is a significant determinant of the predominant taxa detected within the gut microbiome. Results reported by Sullam et al. [22] suggested that Bacteroidales and Clostridiales dominate the gut microbiomes of herbivorous marine fish, whilst Vibrionales and Alteromonadales are dominant in carnivorous and omnivorous marine fish. Similarly, Huang et al. [30] demonstrated that host feeding habits could be differentiated by “indicators”, specific bacterial taxa. It was suggested that these indicators were associated with specific symbiotic functional activities such as the production of bioactive enzymes, thus aiding in digestion within the gut.
The majority of studies of marine fish gut microbiota have focused on 16S rRNA sequence data, profiling the bacterial portion of this community. However, one recent study analyzed whole-metagenome shotgun sequencing data from the intestines of various deep-sea fish of the northern Atlantic Ocean [31]. Overall, the most abundant phyla detected were Pseudomonadota, Bacillota, Actinomycetota and Bacteroidota, in agreement with previous studies. However, cluster analysis showed that the proportions of these phyla varied significantly between some samples. Furthermore, in one cluster a significant proportion of Eukaryota was detected, specifically Ascomycota, Basidiomycota and Euryarchaeota. In terms of the eukaryotic species, there have been suggestions that yeasts are commensals of the fish microbiome with species such as Debaryomyces hansenii, Saccharomyces cerevisiae and red-pigmented Rhodotorula dominating [25,32]. The main archaeal groups reported in studies of marine fish gut microbiota are Crenarchaeota and Euryarchaeota [33,34]. Euryarchaeota have also been identified in deep-sea fish microbiota samples [31]. These are two of the major groups of planktonic archaea found in marine environments and are key contributors to nutrient cycling in pelagic waters and deep-sea sediment [35]. It has been suggested that the digestive tract of fish is an important habitat for archaeal groups within the marine environment, particularly the obligate anaerobic Euryarchaeota [34].
Viruses are the most abundant entities on earth, and it is no different in the marine environment. It has been estimated that there are between 106 and 108 viruses per ml of seawater [36]. A recent study by Geoghegan et al. [37] used a metagenomic-based approach to characterize the viromes of several marine fish species: They identified viral sequences that represented 11 viral families, with Astroviridae, Picornaviridae, Arenviridae, Reoviridae and Hepadraviridae dominating. They suggested that many of the identified viruses were diet or microbiome -associated and that host phylogenetics is a significant determinant of virome diversity.
Efforts have been made to define a “core microbiome” for marine fish, however this has proven a difficult task given the diversity of marine fish and the factors that affect their microbiome diversity. Fish gut microbiota members can also be transient [20]. Instead, it has been suggested that, perhaps, functional diversity is more important than taxonomic/phylogenetic diversity [29,30]. Furthermore, many of these studies have noted that a high percentage of operational taxonomic units (OTUs) could not be taxonomically assigned at the genus level, indicating the novelty and high biodiscovery potential of the marine fish gut microbiomes. For example, Huang et al. [30] reported that over 70% of OTUs from coastal fish gastrointestinal samples were unassigned at genus level, whilst Johny et al. [38] reported over 90% of the deep-sea fish gut OTUs could not be assigned at genus level.
The gut microbiome of marine fish is involved in the regulation of host processes including digestion, stress and immune responses, reproduction, and metabolism [14,22]. Many of these processes are mediated by the production of microbial metabolites, including antimicrobials, and bioactive enzymes.

Table 1.
The predominant phyla reported in recent culture-independent studies of marine fish gut microbiota. (phyla are listed in order of abundance where possible).
Table 1.
The predominant phyla reported in recent culture-independent studies of marine fish gut microbiota. (phyla are listed in order of abundance where possible).
| FISH SPECIES | Sample | Predominant Phyla | Ref. |
|---|---|---|---|
|
Gadus morhua (Atlantic Cod) |
Intestinal contents |
Pseudomonadota, Bacteroidota, Bacillota |
[39] |
|
Siganus fuscescens (Mottled spinefoot rabbitfish) |
Intestinal contents |
Pseudomonadota, Bacillota, Bacteroidota, Fusobacteriota, Mycoplasmatota, Cyanobacteriota |
[40] |
| Various White Sea (arctic) fish |
Posterior intestine |
Pseudomonadota, Bacillota, Actinomycetota, Bacteroidota, Mycoplasmatota, Fusobacteriota |
[41] |
| Various Mediterranean fish | Midgut | Pseudomonadota, Bacillota, Bacteroidota, Actinobacteriota, Patescibacteria, Fusobacteriota, Planctomycetota, and Dependentiae |
[42] |
| Coastal fish of Hong Kong | Gastrointestinal contents | Pseudomonadota, Bacillota, Mycoplasmatota | [30] |
| Various deep-sea fish of Atlantic Ocean | Intestinal contents |
Pseudomonadota, Bacteroidota, Bacillota, Actinomycetota, Ascomycota, Basidiomycota, Euryarchaeota, Spirochaetes |
[31] |
| Centroscyllium fabricii (Black dogfish shark) | Gut contents | Actinomycetota, Pseudomonadota, Acidobacteriota (Acidobacteria), Bacillota, Chloroflexota | [43] |
| Benthobatis moresbyi (Dark Blind Ray) | Gut contents | Actinomycetota, Pseudomonadota, Acidobacteriota, Chloroflexota, Bacillota | [38] |
Ref. = reference.
Table 2.
Recent culture-independent studies of marine fish gut microbiome diversity. The most abundant/predominant taxa reported at family and genus level are given.
Table 2.
Recent culture-independent studies of marine fish gut microbiome diversity. The most abundant/predominant taxa reported at family and genus level are given.
| Fish | Sample | Abundant genera | Abundant Families | Ref. |
|---|---|---|---|---|
| Centroscyllium fabricii(Black dogfish shark) | Gut contents | Acinetobacter, Thalassobacillus, Alteromonas, Leeuwenhoekiella, Corynebacterium, Pseudonocardia, Pseudomonas | NR | [43] |
| Benthobatis moresbyi(Dark Blind Ray) | Gut contents | Acinetobacter | Moraxellaceae, Koribacteraceae, Nitrospiraceae | [38] |
| White Sea (arctic) fish | Posterior intestine | Streptococcus, Sphingomonas, Micrococcus, Chthoniobacter, Pseudomonas, Corynebacterium, Staphylococcus, Acinetobacter, Propionibacterium, Vibrio, Photobacterium, Bacillus | Moraxellaceae, Vibrionaceae, Pseudomonadaceae, Propionibacteriaceae, Corynebacteriaceae, Micrococcaceae | [41] |
| Various Mediterranean fish | Midgut | Pseudoalteromonas, Bradyrhizobium, Diaphorobacter, Mycoplasma, Clostridium, Thaumasiovibrio, Microbulbifer | Xanthobacteraceae, Comamonadaceae, Pseudoalteromonadaceae, Clostridiaceae, Vibrionaceae, Propionibacteriaceae, Staphylococcaceae, Mycoplasmataceae, Flavobacteriaceae, and Peptostreptococcaceae | [42] |
| Various Antarctic fish | Rhodococcus, Thermus, Acinetobacter, Propionibacterium, Streptococcus, and Mycoplasma | NR | [33] | |
| Coastal fish of Hong Kong | Gastrointestinal contents | Clostridium, Photobacterium, Ralstonia, Acinetobacter, Thermus, Ralstonia, | NR | [30] |
NR = not reported, Ref. = reference.
3. Bacteriocins from Marine Fish Gut Microbiota
Bioactive molecules representing several different bacteriocin classes and bacteriocin-like peptides have been identified in isolates of marine fish gut microbiomes, as outlined in Table 3. These peptides exhibit activity against a wide range of gram-positive and gram-negative bacteria, and even against fungi in some cases. Though in many of these studies the chemical structure of these bacteriocins/peptides is yet to be determined, it again highlights the biodiscovery potential and novelty of this microbial niche. Notably, these bacteriocins are all from bacilli or lactic acid bacteria (LAB) strains, some of which are novel species or strains.
3.1. Bacteriocins from LAB
Lactic acid bacteria (LAB) are important members of the fish gut microbiome with cultivable species from marine fish including Carnobacterium spp., Lactobacillus spp., Lactococcus spp., Enterococcus spp. and Pediococcus spp. [44]. Studies have shown that beneficial LAB in the fish gut may have a significant role in promoting host fish health by improving resistance to infection through immune system modulation and the production of antimicrobial molecules, such as bacteriocins [44].
Pilet at al. [45] screened for antimicrobial-producing strains from the intestines of salmon and trout using Listeria species as target organisms. They identified two antimicrobial-producing species, Carnobacterium piscicola V1 and Carnobacterium divergens V41. C. piscicola V1 was found to produce two class IIa bacteriocins, piscicocins V1a and V1b. The purified peptides were active against the same spectrum of gram-positive indicator strains, which included C. divergens, E. faecalis and L. monocytogenes. Furthermore, piscocin V1a was significantly more potent than V1b and was deemed to be a novel bacteriocin based on amino acid sequence analysis [46]. In a subsequent study, C. divergens V41 was found to produce divercin V41, also a novel class IIa bacteriocin. The amino acid sequence of divercin V41 was shown to be most similar to those of pediocin PA-1 and enterocin A [47]. Furthermore, piscicocins V1a and V1b and divercin V41 were optimally produced at low temperatures (≤20°C) and in the presence of NaCl (up to 4%) [48], likely an adaptation to the marine environment.
Enterococcus mundtii Tw56 was isolated from the intestine of an Odontesthes platensis specimen from the Patagonian region of Argentina, an area subject to relatively low temperatures. Cell-free supernatant (CFS) from this strain was able to inhibit gram-positive and gram-negative strains including Enterococcus spp., Listeria spp., Pseudomonas aeruginosa and Shewanella putrefaciens. It was deduced through thermal and chemical stability assays and PCR that the CFS contained mundticin KS, a class IIa bacteriocin [49].
Two separate studies found nisin Z -producing Lactococcus lactis strains from marine fish intestines with activity against the fish pathogens, Streptococcus iniae and Lactococcus garvieae, respectively [50,51]. Nisin Z is a natural variant of the class I bacteriocin (lantibiotic) nisin A, differing only by a single amino acid (His27Asn) [52] and contains the characteristic lanthionine and methyllanthionine rings of this bacteriocin class (Figure 1). As with other nisin peptides, nisin Z acts by binding to and forming pores in the membrane of target cells leading to cell death [53]. Sequeiros et al. [50] demonstrated this in their study when they exposed L. garvieae to supernatant from the nisin Z-producing fish isolate (L. lactis TW34) and reported a drop in viable cell counts by 6 logs (colony forming units [CFU]/mL) within one hour but without cell lysis, thus indicating bactericidal activity. Heo et al. [51] prepared partially purified nisin Z in a solution containing varying concentrations of NaCl from 0-4% w/v. They showed that nisin Z prepared in 3.5% NaCl, a concentration similar to that of seawater, was the most effective against S. iniae. This demonstrates the increased effectiveness of the marine-derived nisin Z under native conditions.
More recently, Li et al. [54] isolated a novel class IId bacteriocin, CAMT6 from the marine fish isolate Enterococcus durans YQ-6. CFS from this strain possessed a broad spectrum of activity, targeting both gram-positive and gram-negative bacteria, and also inhibited fungi. Measurement of CFS conductivity and cell morphology imaging of L. monocytogenes after exposure to CAMT6 demonstrated that the mode of action of CAMT6 involved disruption of the cell membrane, causing leakage of cell contents. Furthermore, the purified CAMT6 peptide exhibited anti-Listeria activity in a chicken breast model and anti-biofilm activity, demonstrating a potential application in food preservation. The amino acid sequence of CAMT6 is unusual in that, at 12 amino acids in length, it is much shorter than previously reported bacteriocins from enterococci and did not share homology with bacterial-derived proteins, but, rather, was most similar to antimicrobial peptides from humans and sheep.
3.2. Bacteriocins from Bacilli
Bacilli isolated from marine fish have also been found to produce highly stable, and often novel, bacteriocins that can target a broad range of target strains:
Bindiya et al. (2015) screened isolates from the gut contents of a deep-sea shark (Centroscyllium fabricii) against various indicator strains. Amongst the antimicrobial producers, they identified Bacillus amyloliquefaciens BTSS-3, a strain with activity against pathogenic bacteria including Salmonella enterica Typhimurium, Clostridium perfringens, S. aureus, Proteus vulgaris, and several Bacillus species [55]. The purified antimicrobial molecule designated BaCf3, was subsequently shown to possess characteristics of being a bacteriocin: it had a mass of ~3000 Da, was pH tolerant (pH 2.0–13.0) and highly thermostable (4-100°C) and retained activity even after autoclaving [56]. Sequence compositional analysis found Bacf3 to be rich in hydrophobic amino acids, such as cysteine and glycine, and was predicted to contain at least one disulfide bridge. The predicted tertiary structure consisted of three anti-parallel β-sheets and resembled that of laterosporulin, a defensin-like bacteriocin from Brevibacillus sp. strain GI-9 [58]. Based on its hydrophobic nature and structural similarity to defensin-like bacteriocins, it was predicted that Bacf3 targeted the cell membrane. This was confirmed by microscopic analysis of Bacillus circulans target cells after exposure to Bacf3, whereby leakage of intracellular material and disruption to the cell membrane was observed [57]. Furthermore, Bacf3 was shown to also have anti-biofilm and anti-cancer activities [56,57]. This is one of the first studies that has purified a novel bacteriocin from a deep-sea fish gut isolate, and, significantly, it retained antimicrobial activity under low temperatures (4°C) and after being subjected to high pressure and heat (autoclaving).
Formicin, a novel two-component lanthipeptide (class I bacteriocin), was discovered by screening bacterial isolates from the intestine of Atlantic mackerel (Scomber scombrus) for antimicrobial activity. The peptide, produced by a Bacillus paralicheniformis isolate, was found to inhibit a range of gram-positive indicator organisms including clinically relevant pathogens such as Listeria monocytogenes, Staphylococcus aureus, Streptococcus mutans, several species of Enterococcus and Clostridia, including Clostridioides difficile [59]. Formicin differs from other two-component lanthipeptides in that the α-peptide contains fewer hydrophobic amino acids and has an overall positive charge of +2, and the β-peptide uniquely contains a negatively charged C-terminus due to the presence of an aspartate residue in the penultimate position (Figure 2a,b). The discovery of formicin and its contribution to scientific discovery in Ireland was commemorated by the Irish national postal service (An Post) together with Science Foundation Ireland by the release of a special issue stamp in 2018 (Figure 2c).
Novel bacteriocin-like molecules derived from bacilli of the marine fish gut microbiome have also been reported, such as CAMT2 [60] and BpSl14 [61], produced by Bacillus amyloliquefaciens ZJHD-06 and Bacillus safensis, respectively. Both molecules exhibited a broad spectrum of activity, targeting both gram-negative and gram-positive strains and even fungi. Elucidation of their chemical structures found that they were more similar to eukaryotic AMPs than to bacterial peptides.
Recent studies combining in vitro and genomic screening have also highlighted the antimicrobial potential of novel Bacilli isolated from fish [62,63]. For example, Bacillus sp. GFP-2, a novel strain of Bacillus velezensis, was isolated from intestinal contents of whitespotted bamboo sharks (Chiloscyllium plagiosum) and found to inhibit the growth of Bacillus subtilis and Escherichia coli. Analysis of the genome sequence of strain GFP-2 found that it encoded genes for various AMPs, such as LCI and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) -related peptides, various antimicrobial secondary metabolites including difficidin, bacillysin, macrolactin and fengycin, as well as lanthipeptides [64].
3.3. Bacteriocins from Actinobacteria
Actinobacteria are abundant in the marine environment, and those associated with higher organisms such as sponges, molluscs and fish are known to produce a wide variety of bioactive compounds with diverse biological activities [65]. One of the most significant antimicrobials from a marine-derived actinomycete is anthracimycin – an antibiotic with potent activity against Bacillus anthracis, the pathogen responsible for causing anthrax. Anthracimycin was discovered by screening Streptomyces isolates from marine sediment [66]. Indeed, Streptomyces are among the most frequently identified producers of bioactive compounds from marine environments, particularly those found in association with higher organisms [65]. Several recent studies have screened Actinobacteria from marine fish microbiomes for antimicrobial activity: Vignesh et al. [67] isolated Actinobacteria from marine fish gut samples that had antimicrobial activity against S. enterica, S. aureus and E. coli. Furthermore, one of the isolates, a Streptomyces strain, also demonstrated anti-fungal and quorum-sensing inhibitory activity. More recently, Vadivel et al. [68] isolated antimicrobial and anti-quorum sensing Streptomyces from marine fish gut samples. Several studies have highlighted that the key to identifying novel bioactive and antimicrobial compounds may now be in the “rare” Actinobacteria – those that are not as easily cultivated. These include genera such as Salinispora, Actinomadura, Microbacterium and Micrococcus [65,69]. Sanchez et al. [70] identified various Actinobacteria, including Rhodococcus, Microbacterium and Micromonospora species (which would be considered part of the “rare” Actinobacteria group) from marine fish digestive tract samples and detected antimicrobial activity against pathogenic Vibrio spp., B. subtilis, S. aureus and E. faecium. Though these studies didn’t identify specific antimicrobial molecules, they highlight the biodiscovery and antimicrobial potential of marine fish gut-derived Actinobacteria.
4. ‘Extreme’ Marine Environment Impact on Microbial Products
Another interesting aspect of the marine environment is the spectrum of ‘extremes’ to which it is subjected, for example, fluctuating temperatures (from hydrothermal vents to near-freezing deep waters), sunlight exposure (or complete lack thereof), salinity and increasing hydrostatic pressure with depth (approx. one atmosphere every 10m). Such conditions have effects on the molecular structure and functionality of proteins found in these niches. Cold-adapted enzymes, for example, may require more flexible intramolecular bonding (such as electrostatic and hydrophobic interactions) as a result of the effect of low temperature on entropy. Such ‘flexible’ interactions can operate at a relatively reduced energy cost. By comparison, mesophilic enzymes require more thermostable intramolecular bonding [71].
However, the challenges of protein production in the deep sea are different. In short, these effects include compression of the protein and consequently a reduction in protein volume. Secondly, high pressure can lead to protein unfolding as a result of the penetration of water into the protein structure [71]. High rigidity and stability are therefore required to negate the denaturing effects of high pressure. Adaptations include the production of piezolytes, cold-shock proteins, certain transcription factors and chaperones, as well as changes within protein structures themselves [72,73]. However, there are reports that deep-sea proteins are highly active under, if not at least tolerant to, high pressure. Lactate dehydrogenase (LDH) from the deep-sea fish, Coryphaenoides armatus, for example, was shown to be more tolerant to high temperature and high pressure than a homologous LDH from a shallow-water fish species. Sequence analysis of the LDH proteins found differences in over 20 amino acid residues in the deep-sea LDH that were suggested to enhance protein stability and enzyme activity [74]. Studies of various other proteins from bathypelagic and abyssal fish species have reported that this tolerance to high pressure is primarily achieved through changes in protein primary structure, mainly amino acid substitutions which result in less flexibility sites that are consequently more resistant to compression (i.e. changes in volume) and can therefore maintain protein functionality [75,76]. Lemaire et al. [77] analyzed hydrophobic ligand binding proteins from an abyssal fish species and after aligning the protein sequences with their non-deep-sea homologs they suggested that post-translational modification, particularly glycosylation of ligand binding domains, is also important for high-pressure tolerance in deep-sea proteins. There is also strong evidence that osmolytes, in particular Trimethylamine N-oxide (TMAO), are crucial to maintaining protein integrity at great depths [78,79]. Martin et al. [73] found a Photobacterium species that produced a novel class of osmolytes, termed “piezolytes”, the intracellular concentrations of which responded to changes in hydrostatic and osmotic pressures.
In the context of bacteriocins from deep-sea bacteria, they may possess unique adaptations in their transcription, structure, and functionality as a consequence of the deep-sea environmental conditions, such as resistance to high hydrostatic pressure and low temperatures.
5. Applications of Marine Fish -Derived Bacteriocins
One of the applications for marine-derived bacteriocins is as an alternative to antibiotics in the treatment of fish diseases within the aquaculture industry [80,81]. Fish and fish products are significant, globally traded commodities, with millions of tonnes consumed each year, either directly as food, as fishmeal or for fish oil. Outbreaks of disease in commercial fish significantly impact global supply, economics and human health [82].
Many studies have also explored the application of bacteriocins from marine fish microbiomes, or the producing strains, in biopreservation of food products, such as seafood products [48,49] and chicken [54]. Schelegueda et al. [49], for example, assessed the potential use of mundticin KS from E. mundtii Tw56 in food biopreservation. The bacteriocins exhibited antimicrobial activity against Listeria innocua over a range of pH (2.0-10.0), and after heat treatment (up to 121°C x 15 min). Furthermore, CFS from the producing strain retained full activity after storage at -30°C for 1 year, indicating its potential for use in frozen foods. Duffes [48], studied the anti-Listeria activity of the bacteriocinogenic strains, C. pisccola V1 and C. divergens V41, in vacuum-packed cold smoked salmon over a period of up to 4 weeks. In co-culture assays at 8°C, both strains reduced the viability of L. monocytogenes by 5-7-fold (log CFU/g) compared to L. monocytogenes alone. The inhibitory effects were even greater at 4°C, with C. piscola V1, for example, reducing the counts of L. monocytogenes to less than 10 CFU/g after 4 weeks. Crude bacteriocin extracts from the Carnobacterium strains also demonstrated effective anti-Listeria activity, particularly at 4°C whereby L. monocytogenes was below detectable levels after 1 week. Significantly, the crude extracts were more effective at inhibiting Listeria than nisin under these conditions.
Bacteriocinogenic isolates from marine fish are generating great interest in the aquaculture industry for use as probiotics. For example, Shastry et al. [83] demonstrated that Enterococcus lactis RS5 (Table 3) exhibited not only bacteriocin production but also resistance to bile salts, low pH (53% viability at pH 1.5) and protease digestion - key attributes of probiotic strains. Nguyen et al. [84] explored the probiotic potential of the nisin Z-producing L. lactis WFLU12, originally isolated from the gut of olive flounder [51] (also Table 3). They reported that fish on a diet supplemented with the antimicrobial strain, WFLU12 (approx. 109 CFU/g), exhibited increased growth and were more resistant to infection by Streptococcus parauberis, compared to the control group.
There is also the potential for use as treatments for infection in humans; indeed, many of the bacteriocins listed in Table 3 are capable of targeting human pathogens. Formicin, for example, exhibited effective activity against several clinically relevant species including C. difficile [59]. This is particularly important in cases of antimicrobial resistance whereby treatment with conventional antibiotics has become less effective and novel alternatives are sought.
6. Challenges, Metagenomics and Future Prospects
One of the (first) challenges in searching for novel bacteriocins, and other useful metabolites, from novel marine sources is the cultivation of the producing microorganisms. Without the correct conditions, potentially novel marine microorganisms, and thus their metabolites, may be lost when attempting to culture in vitro [85]. Environmental pressure, temperature and dissolved oxygen concentration are just a few factors to be taken into account as they can all affect the diversity of isolated microorganisms and the metabolites they produce [86,87,88,89]. Often, modified or selective media is required and long incubation times of days, or even weeks, for antimicrobial production to occur. Sanchez et al. [70], for example, implemented the use of selective media for the isolation of bioactive marine Actinobacteria from fish. Vadivel et al. [68] also explored optimization of antimicrobial production in Streptomyces maritimus SQA4 (from squid) using the “one factor at a time” method, whereby one factor or variable in the cultivation step is modified at a time. They observed that substituting carbon, nitrogen, and salt sources, as well as altering pH, had varying effects on antimicrobial activity. Furthermore, production of the bioactive metabolite was detected when the strain was cultivated on solid medium after two days but was not detected when in broth culture until after 10 days of incubation. The growth of some marine bacteria may also be dependent on the presence of specific signaling molecules found in their natural environment [90].
Advances in genome-mining methods for bacteriocins and other antimicrobial molecules have already been extensively reviewed [91]. Genomic DNA screening has been used to great effect by several groups in the identification of bacteriocin genes. Genome sequence analysis, using BAGEL3 bacteriocin mining software, allowed for the identification of the formicin biosynthetic operon, from which the masses of the core bacteriocin peptides could be predicted. This was a critical step in the purification and characterization of formicin, as the peptide masses found using colony mass spectrometry did not match any previously characterized bacteriocins [59].
As mentioned above, culture-independent and metagenomic methods are being used to great effect in characterizing the taxonomic diversity of the marine fish gut microbiome. Several studies have also employed the use of metagenomic sequencing to characterize functionality, the presence of antimicrobial resistance genes and bioactive metabolite genes in marine fish microbiota [92,93,94], including those from rarer deep-sea fish species (Figure 3). The culture-independent nature of metagenomics allows for a more comprehensive understanding of these gut communities and removes the bias of sampling only culturable microbiota. Few studies, however, have used metagenomics approaches for screening marine fish microbiomes for bacteriocins specifically. Yi et al. [95] used metagenomic sequencing to study functional dynamics in the gut microbiota of several aquatic animals, including marine and freshwater fish. By comparative gene sequence analysis against multiple protein databases, they identified numerous bacteriocin-associated genes, including secretion systems, and immunity-related membrane transporter systems. However, they identified few core peptide biosynthetic genes, suggesting a role in antimicrobial resistance rather than production by the microbiota.
Whilst in silico genomics screening can identify bacteriocin biosynthetic gene clusters, there is still a need for peptide purification but expression in the native host can present another challenge. Another emerging technology is bacteriocin “reincarnation”. This method involves cloning of the biosynthetic operon of an “inactive” antimicrobial into a heterologous host, thereby enabling controlled expression of the antimicrobial gene. Collins et al. [96] utilized this method to reincarnate pediocin-like bacteriocin structural genes that had been identified during in silico analysis of the Lactobacillus pangenome, yet these strains did not exhibit in vitro antimicrobial activity. Ten pediocin-like bacteriocin genes were cloned with a (pediocin) signal sequence and heterologously expressed in E. coli and Lacticaseibacillus paracasei and displayed antimicrobial activity against L. innocua. This study demonstrates the potential for such methods in the expression of such “inactive” bacteriocins, that may be identified in genomic sequences of marine-fish-derived bacteria, and allow for their heterologous expression under more familiar, terrestrial, conditions.
6. Conclusions
With the increasing demand for novel antimicrobial therapeutics to combat antimicrobial resistance, there is a pressing need to source novel bioactive compounds. The marine environment is vast, with a multitude of factors affecting the diversity of marine life and their biomolecules. The marine fish gut microbiome is no exception, and it is emerging as a productive source of antimicrobial molecules, such as bacteriocins. Key aspects of marine life, such as high salinity, hydrostatic pressure and a range of environmental temperatures all have an impact on the structural and functional diversity of marine antimicrobial molecules, including bacteriocins, often distinguishing them from terrestrial homologs. Characterizing the true diversity of microorganisms in marine fish gut microbiomes and the bioactive molecules they produce still presents challenges but may be key in determining the true potential of this niche. Recent applications of culture-independent technologies, such as metagenomics and next-generation sequencing, have begun to bridge this knowledge gap and are already being used to great effect to resolve the taxonomic diversity of the fish gut microbiome. Bacteriocins from the marine fish gut can be used to combat disease, either directly or indirectly as products of probiotic strains, and for the biopreservation of food products. The marine fish gut microbiome is a potential treasure trove of novel bacteriocins and other antimicrobials but limitations in accessibility and technology have meant its true bioprospective potential has yet to be truly explored.
Author Contributions
S.U.L., conceptualization, writing - original draft, and visualization; C.S., C.H., supervision, writing - review, and editing; R.P.R., funding acquisition, resources, supervision, writing - review, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was co-funded by a Teagasc Walsh Fellowship (grant number 2017218), Science Foundation Ireland (SFI) under Grant Number SFI/12/RC/2273_P2 and the European Union (ERC, BACtheWINNER, 101054719). Views and opinions expressed are, however, those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them.
Institutional Review Board Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Greenaway SF, Sullivan KD, Umfress SH, Beittel AB, Wagner KD. Revised depth of the Challenger Deep from submersible transects; including a general method for precise, pressure-derived depths in the ocean. Deep Sea Research Part I: Oceanographic Research Papers 2021;178:103644. [CrossRef]
- National Oceanic and Atmospheric Administration USDoC. Layers of the Ocean. https://www.noaa.gov/jetstream/ocean/layers-of-ocean [accessed March 28, 2023.
- Somero GN. Biochemical ecology of deep-sea animals. Experientia, journal article 1992;48(6):537-543. [CrossRef]
- Voultsiadou E. Therapeutic properties and uses of marine invertebrates in the ancient Greek world and early Byzantium. Journal of Ethnopharmacology 2010;130(2):237-247. [CrossRef]
- 2023. Approved Marine Drugs. https://www.marinepharmacology.org/approved [accessed August 14, 2023.
- Collaborators AR. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. The Lancet; 2022. p. 629-655.
- Frieri M, Kumar K, Boutin A. Antibiotic resistance. Journal of Infection and Public Health 2017;10(4):369-378.
- Mayer AMS, Guerrero AJ, Rodríguez AD, Taglialatela-Scafati O, Nakamura F et al. Marine Pharmacology in 2014–2015: Marine Compounds with Antibacterial, Antidiabetic, Antifungal, Anti-Inflammatory, Antiprotozoal, Antituberculosis, Antiviral, and Anthelmintic Activities; Affecting the Immune and Nervous Systems, and Other Miscellaneous Mechanisms of Action. Marine Drugs 2020;18(1):5. [CrossRef]
- Mayer AMS, Guerrero AJ, Rodríguez AD, Taglialatela-Scafati O, Nakamura F et al. Marine Pharmacology in 2016–2017: Marine Compounds with Antibacterial, Antidiabetic, Antifungal, Anti-Inflammatory, Antiprotozoal, Antituberculosis and Antiviral Activities; Affecting the Immune and Nervous Systems, and Other Miscellaneous Mechanisms of Action. Marine Drugs 2021;19(2):49. [CrossRef]
- Mayer AMS, Pierce ML, Howe K, Rodríguez AD, Taglialatela-Scafati O et al. Marine pharmacology in 2018: Marine compounds with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis and antiviral activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Pharmacological Research 2022;183:106391. [CrossRef]
- Halloran K, Underwood MA. Probiotic mechanisms of action. Early Human Development 2019;135:58-65.
- Wang L, Ravichandran V, Yin Y, Yin J, Zhang Y. Natural Products from Mammalian Gut Microbiota. Trends in Biotechnology 2019;37(5):492-504. [CrossRef]
- Wanka KM, Damerau T, Costas B, Krueger A, Schulz C et al. Isolation and characterization of native probiotics for fish farming. BMC microbiology 2018;18(1):119-119. [CrossRef]
- Butt RL, Volkoff H. Gut Microbiota and Energy Homeostasis in Fish. Frontiers in endocrinology 2019;10:9-9. [CrossRef]
- FAO/WHO. Evaluation of Health and Nutritional Properties of Powder Milk and Live Lactic Acid Bacteria. 2001.
- Cotter PD, Hill C, Ross RP. Bacteriocins: developing innate immunity for food. Nature Reviews Microbiology 2005;3(10):777-788.
- Montalbán-López M, Scott TA, Ramesh S, Rahman IR, van Heel AJ et al. New developments in RiPP discovery, enzymology and engineering. Natural Product Reports, 10.1039/D0NP00027B 2021;38(1):130-239.
- Nissen-Meyer J, Rogne P, Oppegard C, Haugen SH, Kristiansen EP. Structure-Function Relationships of the Non-Lanthionine-Containing Peptide (class II) Bacteriocins Produced by Gram-Positive Bacteria. Current Pharmaceutical Biotechnology 2009;10(1):19-37.
- Cotter PD, Ross RP, Hill C. Bacteriocins — a viable alternative to antibiotics? Nature Reviews Microbiology 2013;11(2):95-105.
- Egerton S, Culloty S, Whooley J, Stanton C, Ross RP. The Gut Microbiota of Marine Fish. Frontiers in microbiology 2018;9:873-873.
- Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM et al. Evidence for a core gut microbiota in the zebrafish. The ISME journal 2011;5(10):1595-1608.
- Sullam KE, Essinger SD, Lozupone CA, O'Connor MP, Rosen GL et al. Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Molecular ecology 2012;21(13):3363-3378.
- Ward NL, Steven B, Penn K, Methé BA, Detrich WH. Characterization of the intestinal microbiota of two Antarctic notothenioid fish species. Extremophiles, journal article 2009;13(4):679-685.
- Troussellier M, Escalas A, Bouvier T, Mouillot D. Sustaining Rare Marine Microorganisms: Macroorganisms As Repositories and Dispersal Agents of Microbial Diversity. Frontiers in microbiology 2017;8:947-947.
- Romero J, Ringø E, Merrifield D. The Gut Microbiota of Fish. 2014.
- Ou W, Yu G, Zhang Y, Mai K. Recent progress in the understanding of the gut microbiota of marine fishes. Marine Life Science & Technology 2021;3(4):434-448.
- Yano Y, Nakayama A, Yoshida K. Population Sizes and Growth Pressure Responses of Intestinal Microfloras of Deep-Sea Fish Retrieved from the Abyssal Zone. Applied and Environmental Microbiology 1995;61(12):4480-4483.
- Ohwada K, Tabor PS, Colwell RR. Species composition and barotolerance of gut microflora of deep-sea benthic macrofauna collected at various depths in the atlantic ocean. Appl Environ Microbiol 1980;40(4):746-755.
- Ghanbari M, Kneifel W, Domig KJ. A new view of the fish gut microbiome: Advances from next-generation sequencing. Aquaculture 2015;448:464-475.
- Huang Q, Sham RC, Deng Y, Mao Y, Wang C et al. Diversity of gut microbiomes in marine fishes is shaped by host-related factors. Molecular ecology 2020;29(24):5019-5034.
- Collins FWJ. An investigation into antimicrobial production in the Lactobacillus genus and the fish microbiome. PhD Thesis Thesis, University College Cork; 2019.
- Andlid T, Juárez R-V, Gustafsson L. Yeast colonizing the intestine of rainbow trout (Salmo gairdneri) and turbot (Scophtalmus maximus). Microbial Ecology, journal article 1995;30(3):321-334.
- Song W, Li L, Huang H, Jiang K, Zhang F et al. The Gut Microbial Community of Antarctic Fish Detected by 16S rRNA Gene Sequence Analysis. BioMed research international 2016;2016:3241529-3241529.
- van der Maarel MJEC, Sprenger W, Haanstra R, Forney LJ. Detection of methanogenic archaea in seawater particles and the digestive tract of a marine fish species. FEMS Microbiology Letters 1999;173(1):189-194.
- Vuillemin A, Wankel SD, Coskun ÖK, Magritsch T, Vargas S et al. Archaea dominate oxic subseafloor communities over multimillion-year time scales. Science advances 2019;5(6):eaaw4108-eaaw4108.
- Suttle CA. Viruses in the sea. Nature 2005;437(7057):356-361.
- Geoghegan JL, Di Giallonardo F, Wille M, Ortiz-Baez AS, Costa VA et al. Virome composition in marine fish revealed by meta-transcriptomics. Virus Evolution 2021;7(1):veab005.
- Johny TK, Saidumohamed BE, Sasidharan RS, Bhat SG. Inferences of gut bacterial diversity from next-generation sequencing of 16S rDNA in deep sea blind ray - Benthobatis moresbyi. Ecological Genetics and Genomics 2018;9:1-6.
- Star B, Haverkamp TH, Jentoft S, Jakobsen KS. Next generation sequencing shows high variation of the intestinal microbial species composition in Atlantic cod caught at a single location. BMC Microbiol 2013;13:248.
- Jones J, DiBattista JD, Stat M, Bunce M, Boyce MC et al. The Microbiome of the Gastrointestinal Tract of a Range-Shifting Marine Herbivorous Fish. Frontiers in Microbiology, Original Research 2018;9.
- Burtseva O, Kublanovskaya A, Fedorenko T, Lobakova E, Chekanov K. Gut microbiome of the White Sea fish revealed by 16S rRNA metabarcoding. Aquaculture 2021;533:736175. [CrossRef]
- Kormas K, Nikouli E, Kousteni V, Damalas D. Midgut bacterial microbiota of 12 fish species from a marine protected area in the Aegean Sea (Greece). Microbial Ecology 2023;86(2):1405-1415. [CrossRef]
- Johny TK, Saidumohamed BE, Sasidharan RS, Bhat SG. Metabarcoding data of bacterial diversity of the deep sea shark, Centroscyllium fabricii. Data in Brief 2018;21:1029-1032. [CrossRef]
- Ringø E, Hoseinifar SH, Ghosh K, Doan HV, Beck BR et al. Lactic acid bacteria in finfish—An update. Frontiers in microbiology 2018;9:1818.
- Pilet M-F, Dousset X, Barré R, Novel G, Desmazeaud M et al. Evidence for Two Bacteriocins Produced by Carnobacterium piscicola and Carnobacterium divergens Isolated from Fish and Active Against Listeria monocytogenes. Journal of Food Protection 1995;58(3):256-262. [CrossRef]
- Bhugaloo-Vial P, Dousset X, Metivier A, Sorokine O, Anglade P et al. Purification and amino acid sequences of piscicocins V1a and V1b, two class IIa bacteriocins secreted by Carnobacterium piscicola V1 that display significantly different levels of specific inhibitory activity. Appl Environ Microbiol 1996;62(12):4410-4416. [CrossRef]
- Metivier A, Pilet M-F, Dousset X, Sorokine O, Anglade P et al. Divercin V41, a new bacteriocin with two disulphide bonds produced by Carnobacterium divergens V41: primary structure and genomic organization. Microbiology 1998;144(10):2837-2844. [CrossRef]
- Duffes F, Leroi F, Boyaval P, Dousset X. Inhibition of Listeria monocytogenes by Carnobacterium spp. strains in a simulated cold smoked fish system stored at 4°C. International Journal of Food Microbiology 1999;47(1):33-42.
- Schelegueda LI, Vallejo M, Gliemmo MF, Marguet ER, Campos CA. Synergistic antimicrobial action and potential application for fish preservation of a bacteriocin produced by Enterococcus mundtii isolated from Odontesthes platensis. LWT - Food Science and Technology 2015;64(2):794-801.
- Sequeiros C, Garcés ME, Vallejo M, Marguet ER, Olivera NL. Potential aquaculture probiont Lactococcus lactis TW34 produces nisin Z and inhibits the fish pathogen Lactococcus garvieae. Archives of Microbiology 2015;197(3):449-458.
- Heo W-S, Kim E-Y, Kim Y-R, Hossain MT, Kong I-S. Salt effect of nisin Z isolated from a marine fish on the growth inhibition of Streptococcus iniae, a pathogen of streptococcosis. Biotechnology Letters 2012;34(2):315-320.
- Mulders JW, Boerrigter IJ, ROLLEMA HS, SIEZEN RJ, de VOS WM. Identification and characterization of the lantibiotic nisin Z, a natural nisin variant. European Journal of Biochemistry 1991;201(3):581-584.
- Breukink E, van Kraaij C, Demel RA, Siezen RJ, Kuipers OP et al. The C-Terminal Region of Nisin Is Responsible for the Initial Interaction of Nisin with the Target Membrane. Biochemistry 1997;36(23):6968-6976.
- Li Q, Chen Q, Wu Y, Chen Z, Liu Y et al. Purification, characterization and structural identification of a novel bacteriocin produced by marine original Enterococcus durans YQ-6, and its inhibition of Listeria monocytogenes. LWT 2023;173:114329.
- Bindiya ES, Tina KJ, Raghul SS, Bhat SG. Characterization of Deep Sea Fish Gut Bacteria with Antagonistic Potential, from Centroscyllium fabricii (Deep Sea Shark). Probiotics Antimicrob Proteins 2015;7(2):157-163.
- Bindiya ES, Tina KJ, Sasidharan RS, Bhat SG. BaCf3: highly thermostable bacteriocin from Bacillus amyloliquefaciens BTSS3 antagonistic on food-borne pathogens. 3 Biotech 2019;9(4):136. [CrossRef]
- Saidumohamed BE, Johny TK, Raveendran AT, Sheela UB, Sreeranganathan M et al. 3D Structure Elucidation and Appraisal of Mode of Action of a Bacteriocin BaCf3 with Anticancer Potential Produced by Marine Bacillus amyloliquefaciens BTSS3. Re:GEN Open 2022;2(1):45-56. [CrossRef]
- Singh PK, Chittpurna, Ashish, Sharma V, Patil PB et al. Identification, purification and characterization of laterosporulin, a novel bacteriocin produced by Brevibacillus sp. strain GI-9. PLoS One 2012;7(3):e31498. [CrossRef]
- Collins FWJ, O'Connor PM, O'Sullivan O, Rea MC, Hill C et al. Formicin - a novel broad-spectrum two-component lantibiotic produced by Bacillus paralicheniformis APC 1576. Microbiology 2016;162(9):1662-1671.
- An J, Zhu W, Liu Y, Zhang X, Sun L et al. Purification and characterization of a novel bacteriocin CAMT2 produced by Bacillus amyloliquefaciens isolated from marine fish Epinephelus areolatus. Food Control 2015;51:278-282.
- Saidumohamed BE, Baburaj AP, Johny TK, Sheela UB, Sreeranganathan M et al. A magainin-2 like bacteriocin BpSl14 with anticancer action from fish gut Bacillus safensis SDG14. Analytical Biochemistry 2021;627:114261.
- Emam AM, Dunlap CA. Genomic and phenotypic characterization of Bacillus velezensis AMB-y1; a potential probiotic to control pathogens in aquaculture. Antonie van Leeuwenhoek 2020;113(12):2041-2052.
- Yi Y, Zhang Z, Zhao F, Liu H, Yu L et al. Probiotic potential of Bacillus velezensis JW: Antimicrobial activity against fish pathogenic bacteria and immune enhancement effects on Carassius auratus. Fish Shellfish Immunol 2018;78:322-330.
- Wu J, Xu G, Jin Y, Sun C, Zhou L et al. Isolation and characterization of Bacillus sp. GFP-2, a novel Bacillus strain with antimicrobial activities, from Whitespotted bamboo shark intestine. AMB Express 2018;8(1):84.
- Valliappan K, Sun W, Li Z. Marine actinobacteria associated with marine organisms and their potentials in producing pharmaceutical natural products. Applied Microbiology and Biotechnology 2014;98(17):7365-7377. [CrossRef]
- Jang KH, Nam S-J, Locke JB, Kauffman CA, Beatty DS et al. Anthracimycin, a Potent Anthrax Antibiotic from a Marine-Derived Actinomycete. Angewandte Chemie International Edition 2013;52(30):7822-7824. [CrossRef]
- Vignesh A, Ayswarya S, Gopikrishnan V, Radhakrishnan M. Bioactive potential of actinobacteria isolated from the gut of marine fishes. 2019.
- Vadivel M, Venugopal G, Angamuthu V, Manikkam R, Joseph J et al., editors. Exploration of Fish Gut Associated Actinobacteria for its Anti-Microbial and Anti-Quorum Sensing Properties. International Seminar on Promoting Local Resources for Sustainable Agriculture and Development (ISPLRSAD 2020); 2021: Atlantis Press.
- Subramani R, Sipkema D. Marine Rare Actinomycetes: A Promising Source of Structurally Diverse and Unique Novel Natural Products. Marine Drugs 2019;17(5):249.
- Sanchez LM, Wong WR, Riener RM, Schulze CJ, Linington RG. Examining the Fish Microbiome: Vertebrate-Derived Bacteria as an Environmental Niche for the Discovery of Unique Marine Natural Products. PLOS ONE 2012;7(5):e35398.
- Gerday C, Aittaleb M, Arpigny JL, Baise E, Chessa J-P et al. Psychrophilic enzymes: a thermodynamic challenge. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology 1997;1342(2):119-131.
- Mykytczuk NCS, Wilhelm RC, Whyte LG. Planococcus halocryophilus sp. nov., an extreme sub-zero species from high Arctic permafrost. Int J Syst Evol Microbiol 2012;62(Pt 8):1937-1944. [CrossRef]
- Martin D, Bartlett DH, Roberts MF. Solute accumulation in the deep-sea bacterium Photobacterium profundum. Extremophiles, journal article 2002;6(6):507-514. [CrossRef]
- Brindley AA, Pickersgill RW, Partridge JC, Dunstan DJ, Hunt DM et al. Enzyme sequence and its relationship to hyperbaric stability of artificial and natural fish lactate dehydrogenases. PloS one 2008;3(4):e2042-e2042. [CrossRef]
- Porter ML, Roberts NW, Partridge JC. Evolution under pressure and the adaptation of visual pigment compressibility in deep-sea environments. Molecular Phylogenetics and Evolution 2016;105:160-165.
- Morita T. Structure-based analysis of high pressure adaptation of α-actin. Journal of Biological Chemistry, Article 2003;278(30):28060-28066.
- Lemaire B, Karchner SI, Goldstone JV, Lamb DC, Drazen JC et al. Molecular adaptation to high pressure in cytochrome P450 1A and aryl hydrocarbon receptor systems of the deep-sea fish Coryphaenoides armatus. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2018;1866(1):155-165.
- Yancey PH, Blake WR, Conley J. Unusual organic osmolytes in deep-sea animals: adaptations to hydrostatic pressure and other perturbants. Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology 2002;133(3):667-676.
- Somero GN. Protein adaptations to temperature and pressure: complementary roles of adaptive changes in amino acid sequence and internal milieu. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology 2003;136(4):577-591.
- Desriac F, Defer D, Bourgougnon N, Brillet B, Le Chevalier P et al. Bacteriocin as Weapons in the Marine Animal-Associated Bacteria Warfare: Inventory and Potential Applications as an Aquaculture Probiotic. Marine Drugs 2010;8(4):1153-1177.
- Sahoo TK, Jena PK, Patel AK, Seshadri S. Bacteriocins and their applications for the treatment of bacterial diseases in aquaculture: a review. Aquaculture Research 2016;47(4):1013-1027.
- FAO. The State of World Fisheries and Aquaculture 2020 . Sustainability in action. Rome, Italy2020. Report No.: 978-92-5-132692-3 Contract No.: 63.
- Shastry RP, Arunrenganathan RR, Rai VR. Characterization of probiotic Enterococcus lactis RS5 and purification of antibiofilm enterocin. Biocatalysis and Agricultural Biotechnology 2021;31:101897. [CrossRef]
- Nguyen TL, Park C-I, Kim D-H. Improved growth rate and disease resistance in olive flounder, Paralichthys olivaceus, by probiotic Lactococcus lactis WFLU12 isolated from wild marine fish. Aquaculture 2017;471:113-120. [CrossRef]
- Deming JW, Baross JA. Survival, Dormancy, and Nonculturable Cells in Extreme Deep-Sea Environments. In: Colwell RR, Grimes DJ (editors). Nonculturable Microorganisms in the Environment. Boston, MA: Springer US; 2000. pp. 147-197.
- Choi Eun J, Nam S-J, Paul L, Beatty D, Kauffman Christopher A et al. Previously Uncultured Marine Bacteria Linked to Novel Alkaloid Production. Chemistry & Biology 2015;22(9):1270-1279.
- López R, Monteón V, Chan E, Montejo R, Chan M. Oxygen limitation favors the production of protein with antimicrobial activity in Pseudoalteromonas sp. Brazilian journal of microbiology : [publication of the Brazilian Society for Microbiology] 2012;43(3):1206-1212.
- Nakayama A, Yano Y, Yoshida K. New Method for Isolating Barophiles from Intestinal Contents of Deep-Sea Fishes Retrieved from the Abyssal Zone. Applied and Environmental Microbiology 1994;60(11):4210-4212. [CrossRef]
- Zeng X, Xiao X, Wang P, Wang FP. Screening and characterization of psychrotrophic, lipolytic bacteria from deep-sea sediments. Journal of Microbiology and Biotechnology 2004;14:952-958.
- Nichols D, Lewis K, Orjala J, Mo S, Ortenberg R et al. Short Peptide Induces an “Uncultivable” Microorganism To Grow In Vitro. Applied and Environmental Microbiology 2008;74(15):4889.
- Zhong Z, He B, Li J, Li Y-X. Challenges and advances in genome mining of ribosomally synthesized and post-translationally modified peptides (RiPPs). Synthetic and Systems Biotechnology 2020;5(3):155-172. [CrossRef]
- Johny TK, Puthusseri RM, Bhat SG. Metagenomic landscape of taxonomy, metabolic potential and resistome of Sardinella longiceps gut microbiome. Archives of Microbiology 2021;204(1):87. [CrossRef]
- Collins FWJ, Walsh CJ, Gomez-Sala B, Guijarro-García E, Stokes D et al. The microbiome of deep-sea fish reveals new microbial species and a sparsity of antibiotic resistance genes. Gut Microbes 2021;13(1):1-13. [CrossRef]
- Stevenson SJ, Lee KC, Handley KM, Angert ER, White WL et al. Substrate degradation pathways, conserved functions and community composition of the hindgut microbiota in the herbivorous marine fish Kyphosus sydneyanus. Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology 2022;272:111283. [CrossRef]
- Yi Y, Liang L, Wang Z, Ai P, You X et al. A Comparative Metagenomics Study on Gastrointestinal Microbiota in Amphibious Mudskippers and Other Vertebrate Animals. Animals 2019;9(9):660. [CrossRef]
- Collins FWJ, Mesa-Pereira B, O'Connor PM, Rea MC, Hill C et al. Reincarnation of Bacteriocins From the Lactobacillus Pangenomic Graveyard. Front Microbiol 2018;9:1298. [CrossRef]
Figure 1.
The structure of the class I bacteriocin (lantibiotic) nisin Z, a natural nisin A variant, which differs by one amino acid (position 27 where Asn replaces Ala, highlighted in yellow). Nisin Z -producing Lactococcus lactis strains have been found in marine fish intestinal samples by two separate studies, Sequeiros et al. [50] and Heo et al. [51]. Figure adapted from Mulders et al. [52].
Figure 1.
The structure of the class I bacteriocin (lantibiotic) nisin Z, a natural nisin A variant, which differs by one amino acid (position 27 where Asn replaces Ala, highlighted in yellow). Nisin Z -producing Lactococcus lactis strains have been found in marine fish intestinal samples by two separate studies, Sequeiros et al. [50] and Heo et al. [51]. Figure adapted from Mulders et al. [52].
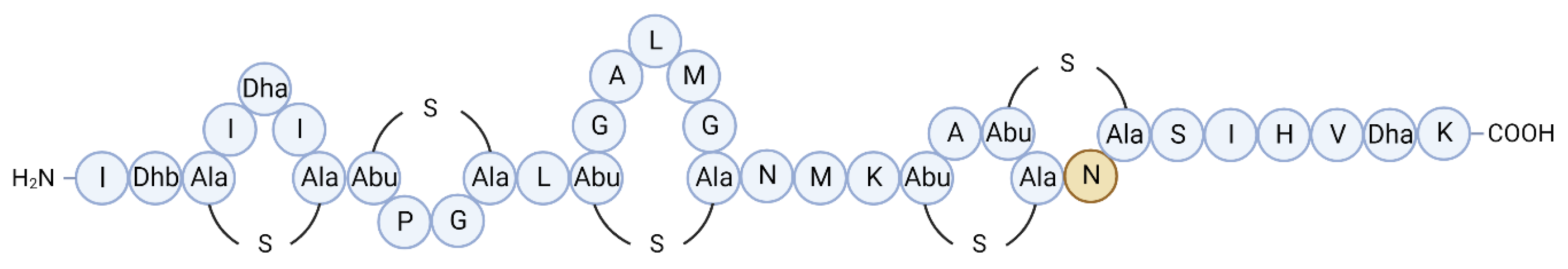
Figure 2.
Predicted structures and core peptide sequences of the α (Frcα) and β (Frcβ) peptides of Formicin, a novel two-component lanthipeptide produced by Bacillus paralicheniformis APC 1576, a mackerel gut isolate. Formicin’s secondary structure differs from that of other similar lanthipeptides, yet it still retains a broad spectrum of activity against gram-positive pathogens. Figure adapted from Collins et al. [57]. The discovery of formicin and its contribution to scientific research was commemorated by the release of a postal stamp released by An Post and Science Foundation Ireland, celebrating scientific discoveries in Ireland (c).
Figure 2.
Predicted structures and core peptide sequences of the α (Frcα) and β (Frcβ) peptides of Formicin, a novel two-component lanthipeptide produced by Bacillus paralicheniformis APC 1576, a mackerel gut isolate. Formicin’s secondary structure differs from that of other similar lanthipeptides, yet it still retains a broad spectrum of activity against gram-positive pathogens. Figure adapted from Collins et al. [57]. The discovery of formicin and its contribution to scientific research was commemorated by the release of a postal stamp released by An Post and Science Foundation Ireland, celebrating scientific discoveries in Ireland (c).
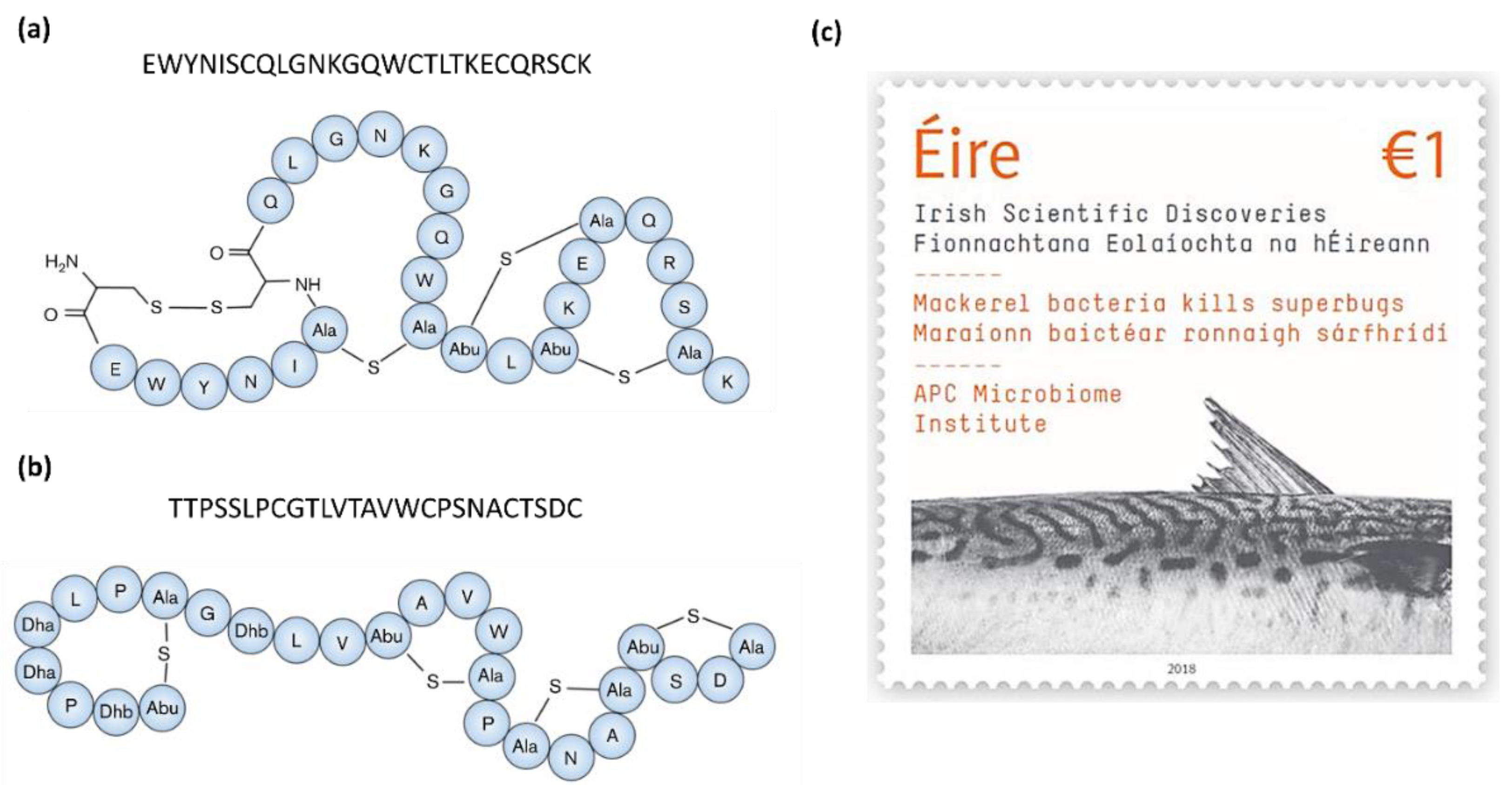
Figure 3.
Culture-independent, metagenomics-based studies have enhanced our analysis of the microbiomes of marine fish, including rarer/unusual species such as Anoplogaster cornuta (a) Bathysaurus ferox (b) Centroscyllium fabricii (c) and Cottunculus thomsonii (d). Figure adapted from Collins et al. [93].
Figure 3.
Culture-independent, metagenomics-based studies have enhanced our analysis of the microbiomes of marine fish, including rarer/unusual species such as Anoplogaster cornuta (a) Bathysaurus ferox (b) Centroscyllium fabricii (c) and Cottunculus thomsonii (d). Figure adapted from Collins et al. [93].
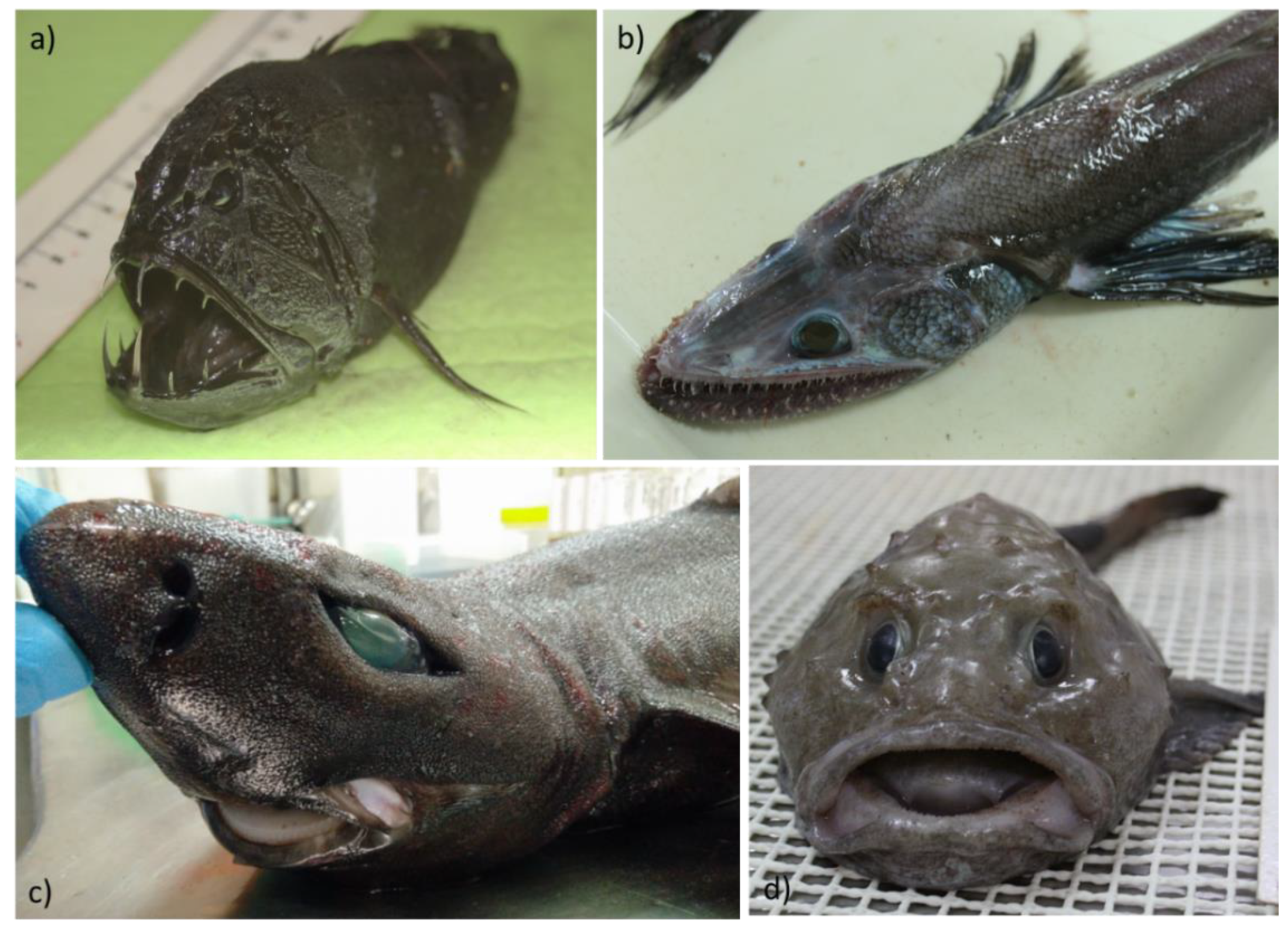
Table 3.
Bacteriocins, bacteriocin-like inhibitory substances and other AMPs identified from marine fish gut microbiome isolates. Many class IIa bacteriocins have been isolated from this microbial niche, covering a wide spectrum of sensitive organisms.
Table 3.
Bacteriocins, bacteriocin-like inhibitory substances and other AMPs identified from marine fish gut microbiome isolates. Many class IIa bacteriocins have been isolated from this microbial niche, covering a wide spectrum of sensitive organisms.
| Molecule | Producer | Host, source | Susceptible organism(s) | Ref. |
|---|---|---|---|---|
| Class I bacteriocins (lanthipeptide subclass II) | ||||
| Formicin | Bacillus paralicheniformis APC 1576 | Scomber scombrus, intestine | Clostridia spp., Bacillus spp., Listeria spp., Enterococcus spp., Streptococcus mutans, M. luteus | [52] |
| Class IIa bacteriocins | ||||
| Divercin V41 | Carnobacterium divergens V41 | salmon or trout, intestine | Carnobacterium piscicola, Listeria spp. | [42] |
| Mundticin KS | Enterococcus mundtii Tw56 | Odontesthes platensis, intestine | Enterococcus spp., Listeria spp., M. luteus, Pseudomonas aeruginosa, Shewanella putrefaciens | [44] |
| Enterocin R5 |
Enterococcus lactis RS5 |
Sillago indica, gut | E. coli, S. enterica Typhimurium, S. aureus, P. aeruginosa. B. subtilis, B. cereus, Proteus vulgaris | [76] |
| Nisin Z | Lactococcus lactis subsp. lactis | Paralichthys olivaceus, intestine | Streptococcus iniae | [46] |
| Nisin Z | Lactococcus lactis TW34 | Odontesthes platensis, intestine | Lactococcus garvieae | [45] |
| Piscidins Vla, Vlb | Carnobacterium piscola V1 | salmon/trout, intestine | Listeria spp. | [41] |
| Class IId bacteriocins | ||||
| CAMT6 |
Enterococcus durans YQ-6 |
Larimichthys polyactis, NR |
S. aureus, Bacillus spp., S. haemolyticus, P. acnes, Salmonella paratyphi, V. parahaemolyticus, P. foulis, E. aerogenes, Fusarium sylvaticum, Aspergillus fumigatus |
[49] |
| Other AMPs /bacteriocin-like inhibitory substances | ||||
| BaCf3 | Bacillus amyloliquefaciens BTSS3 | Centroscyllium fabricii, intestine |
Bacillus spp., Clostridium perfringens, Salmonella Typhimurium, Proteus vulgaris |
[50,51] |
AMP = antimicrobial peptide; NR = not reported, Ref = reference.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



