
Submitted:
17 May 2024
Posted:
20 May 2024
You are already at the latest version
Abstract
The growing interest in benchtop NMR systems is visible in a vast number of applications in science and industry. Low-field nuclear magnetic resonance (LF-NMR) became an instant in technologies that productions are dependent on the reliable structure identification, grade and purity control, chemical process inspection or reaction monitoring. Beyond the benefits of being cost-, space-, maintenance- and user friendly, insufficient sensitivity and low signal resolution are the main drawbacks, due to which the low-field NMR (LF-NMR) in the fields of organic chemistry and synthesis remains limited to solely academic teaching purposes. Herein, we discuss the possibilities of LF-NMR for the needs of organic chemistry and synthesis on a set of compounds 1-4 achieved as the products of student’s laboratory course and organic derivatives 5-8 that are from our research interests. Chemical structure determination of all compounds 1-8 presented herein was performed using the benchtop-type NMR instrument operating at 60 MHz (Pulsar Oxford Ltd.). Structure of novel compounds 5-7 prepared under the aims of our research was characterized by combined 1H/19LF-NMR and compared with an appropriate high-field NMR data from Bruker Ascend Instrument (400 MHz). Finally, possibilities of the low-field NMR as an analytical tool for the research purposes in the fields of organic chemistry and synthesis are documented on a set of 1H and 2D 1H-1H COSY for azomethine 8.
Keywords:
low-field NMR
; benchtop NMR
; high-field NMR
; structure
; synthesis
1. Introduction
Although 1H NMR spectroscopy in combination with 13C-(19F) NMR represents the most powerful tool for the precise structural identification, the availability of the method – in terms of real-time acquisition, the possibility of self-operation and self-availability of spectra, is limited. Conventional high-field NMR (HF-NMR) spectrometers are large and expensive. Operation and maintenance of HF-NMR spectrometers depends on a trained staff and the machinery is kept in a special laboratory requiring cryogenic cooling because of the use of solenoidal superconducting magnets [1]. The release of benchtop-type NMR instruments onto the market [2,3] have enabled the access to NMR spectroscopy for a broad research community, including students. Most benchtop spectrometers that operate at low magnetic fields (1-2 T) are compact and may be used in any type of laboratory [4]; they also possess the additional benefits of being inexpensive and user-friendly in the process of data acquisition. Importantly, cryogenic cooling is unnecessary as permanent compact magnets (NdFeB, SmCo) are used instead of solenoids [5,6]. Low-field NMR (LF-NMR) spectroscopy using commercially available benchtop-type NMR spectrometers from Oxford, Bruker, Magritek, Nanalysis and ThermoScientific operating at 40 – 100 MHz are capable for 1D 1H NMR, 19F NMR, 13C NMR and 2D 1H-1H COSY experiments [7,8]. Although LF-NMR is ubiquitously used in industry and science [9,10,11,12,13], in the fields of organic chemistry and synthesis, the technique is ultimately applied as a pedagogical tool allowing undergraduate students to practice in measurement and analysis of the NMR spectra or for reaction monitoring [14,15,16,17,18]. Nevertheless, identification of the expected products by students making use of benchtop-NMR closely models the analytical approach performed on a scientific level towards accurate structural description. Since the sensitivity at low-fields can be overcome by increasing the concentration of the sample and the resolution is adjustable by acquisition period, herein we have selected three sets of compounds (Figure 1) to explore the usability of LF-NMR in organic chemistry and synthesis for rapid and precise chemical structural characterization. The structure of 1-4 was evaluated by 1H LF-NMR on a PULSAR Oxford compact spectrometer operating at 60 MHz. The fluorine-substituted derivatives 5-7 were characterized by the means of 1H and 19F LF-NMR at 60 MHz and 56.5 MHz, respectively. The comparative HF-NMR data (400 MHz for 1H / 376 MHz for 19F, Bruker Ascend TM) were obtained for 5-7. In the case of azomethine 8 1H and 2D (1H-1H) COSY involving a benchtop NMR were used for profiling highlighting the profitability of LF NMR in a field of organic chemistry and synthesis either as educational tool or science.
2. Results
2.1. LF-NMR Structural Analysis for De-Novo Synthetized Organic Compiounds
Since 2020, when the benchtop NMR operating at 60 MHz (Pulsar Oxford) began to be employed in our instructional organic laboratories, the synthesis of almost all the prescribed experiments has been accompanied by 1H NMR structure characterization. Depending on the educational degree, students either receive knowledge on proton nuclear magnetic resonance under the teacher´s supervision or gain experience through generating the NMR data on their own and proceeding through the entire process towards the 1H NMR solution. Although the instalation of the benchtop-type instrument in the instructional laboratory have enabled the in-time structural analysis subsequently upon reaction completion, undergraduate students as well as gratuates and some young researchers before gaining complete practical independence have to pass out the basic expertise under the supervision of expert in both - data acqusition and data analysis. For successful data analysis the assignment of protons in the structure of elucidated organic compound with the signal (peak) in 1H NMR spectra is neccessary. Such key-step represents a challenge quite often also for skilled chemists. Non-experts are able to solve the problem only by use of available databases [19] and for that reason, students are preparing and analysis compounds with already known structures. Also, prediction of proton spectra using on-line nmr-predictor allows students to quickly identify the correct structure [20,21,22,23].
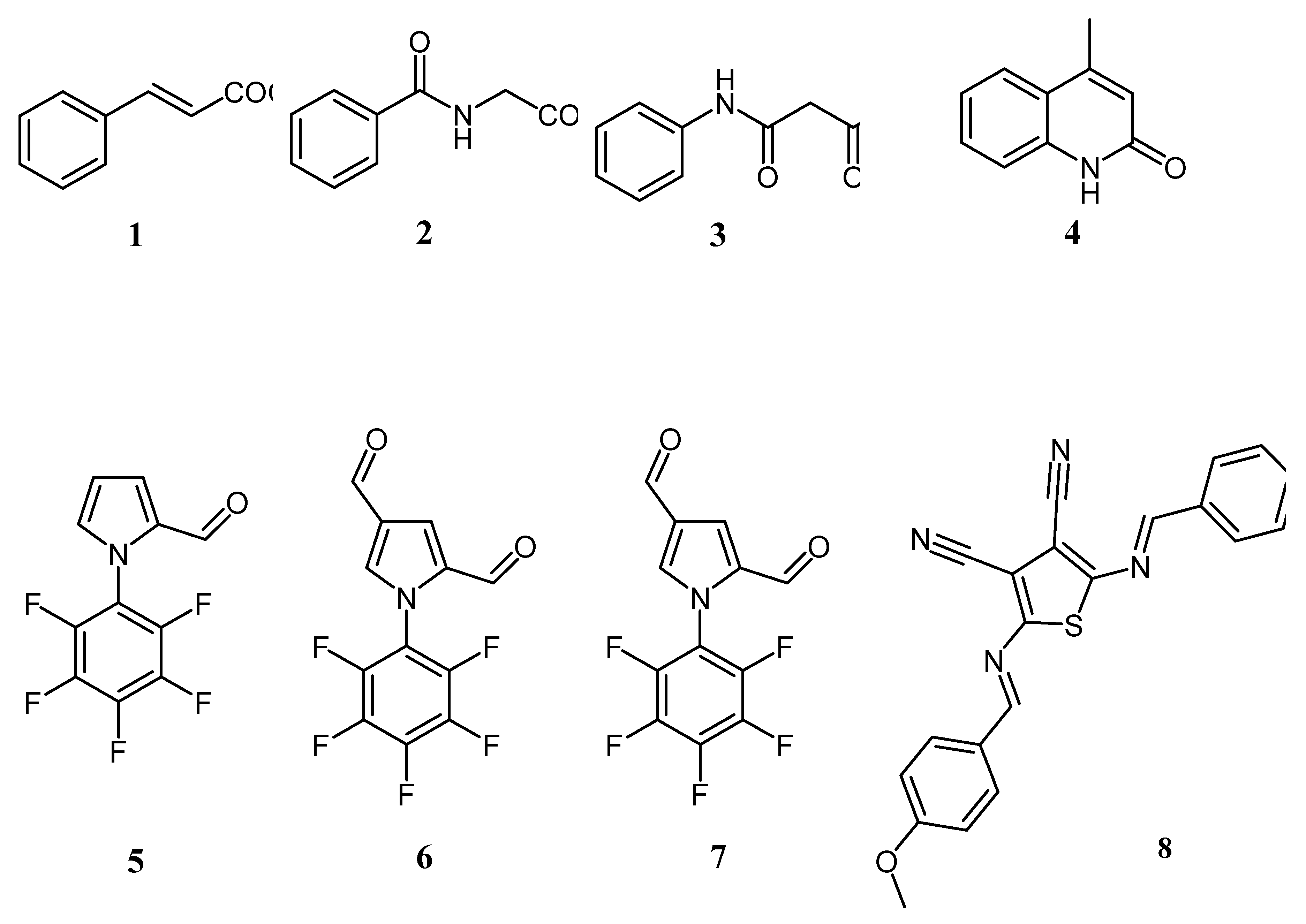
The above detailed algorithm have been applied for structure elucidation of a set of four compounds, namely cinnamic acid (1) (Figure 2), hippuric acid (2) (Figure 3), acetanilide (3) and 4-methylquinoline-2-ol (4) (Figure 4), that have been prepared during the undegraduate laboratory course. At the end, beyond gaining the skill in synthesis, students were able to prepare the sample and performe the NMR acquisition finalizing their experiments with chemical structure identification as a result of correct interpretation the spectra.
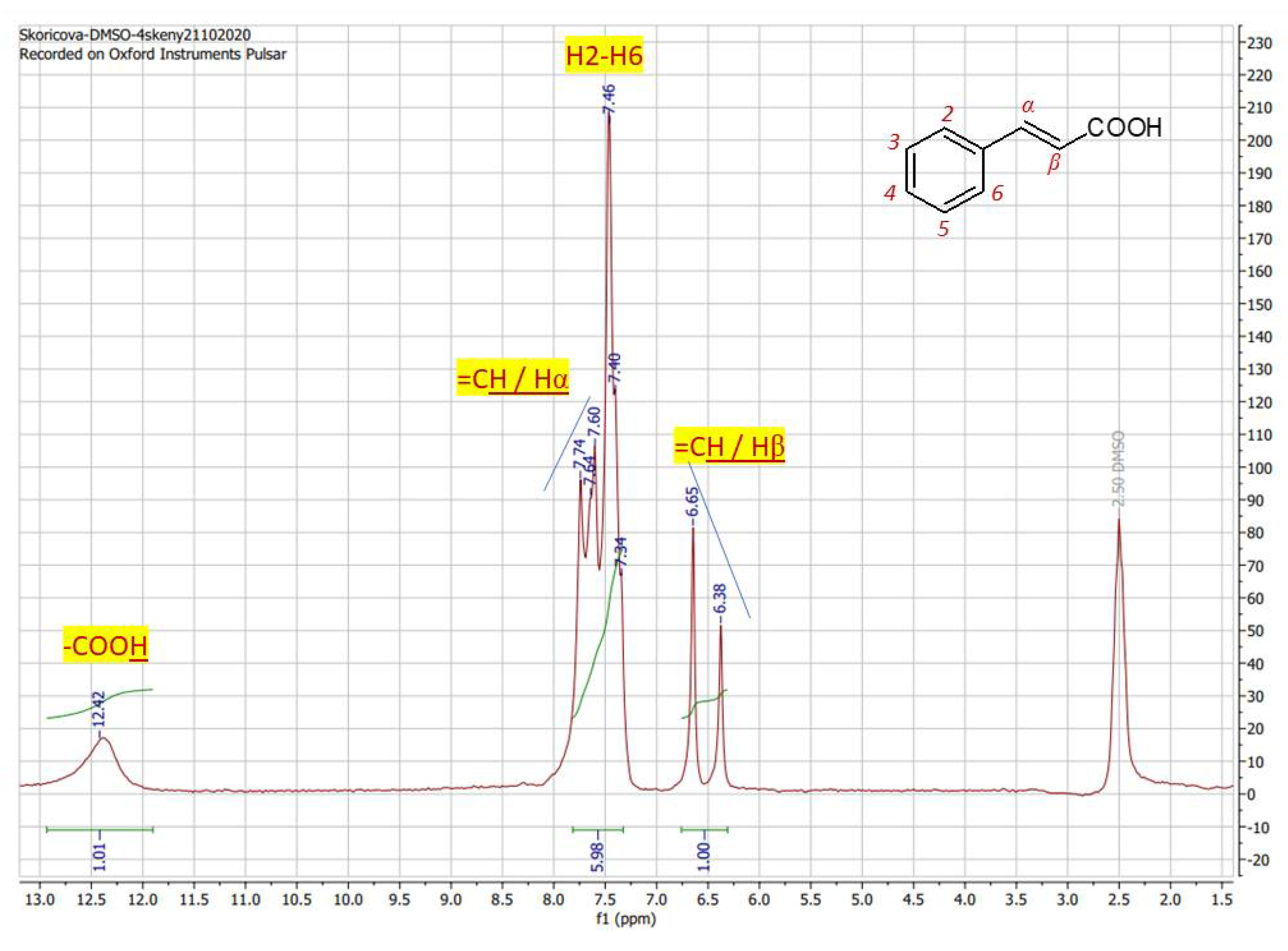
In the proton spectrum of cinnamic acid (1) (Figure 2) obtained from a DMSO-d6 solution at 4 scans (8 mins) the characteristic carboxyl proton is displayed as sharp signal at 12.42 ppm, that is obvious for carboxylic acids. Aromatic peaks of five protons H2-H6 are splitted into a multiplet type of signal in the range of 7.74 – 7.34 ppm. Generally, signal splitting of aromatic hydrogen atoms is obvious also in the spectra taken at high-magnetic fields.The splitting pattern of H2/H6 in the form of doublet is expected at 7.62 ppm but is overlayed with a signal for a double bond proton Hα at 7.67 ppm. Double bond proton Hβ is observed as a clear doublet at 6.52 ppm with interaction constant 3J = 16.2 Hz confirming the E-izomer of cinnamic acid. For such situation, when the double bond proton is overlayed by other signals the „roof“ effect is helpfull to find the appropriate pair, as is marked (blue line) within the spectrum. In spite of overlayed signals as the consequence of supressed sensitivity, spectrum of cinnamic acid obtained at low field (60 MHz) is thoroughly interpretable and represents a valid spectral result comparable to those, obtained on HF-NMR instrument. The interactions 1/6/1 (sum. = 8 protons) corresponds with formula of cinnamic acid C9H8O2.
Figure 2.
1H NMR spectrum of cinnamic acid in DMSO-d6 recorded at 60 MHz with proton assigment according to the structure (4 scans / 8 mins).
Figure 2.
1H NMR spectrum of cinnamic acid in DMSO-d6 recorded at 60 MHz with proton assigment according to the structure (4 scans / 8 mins).
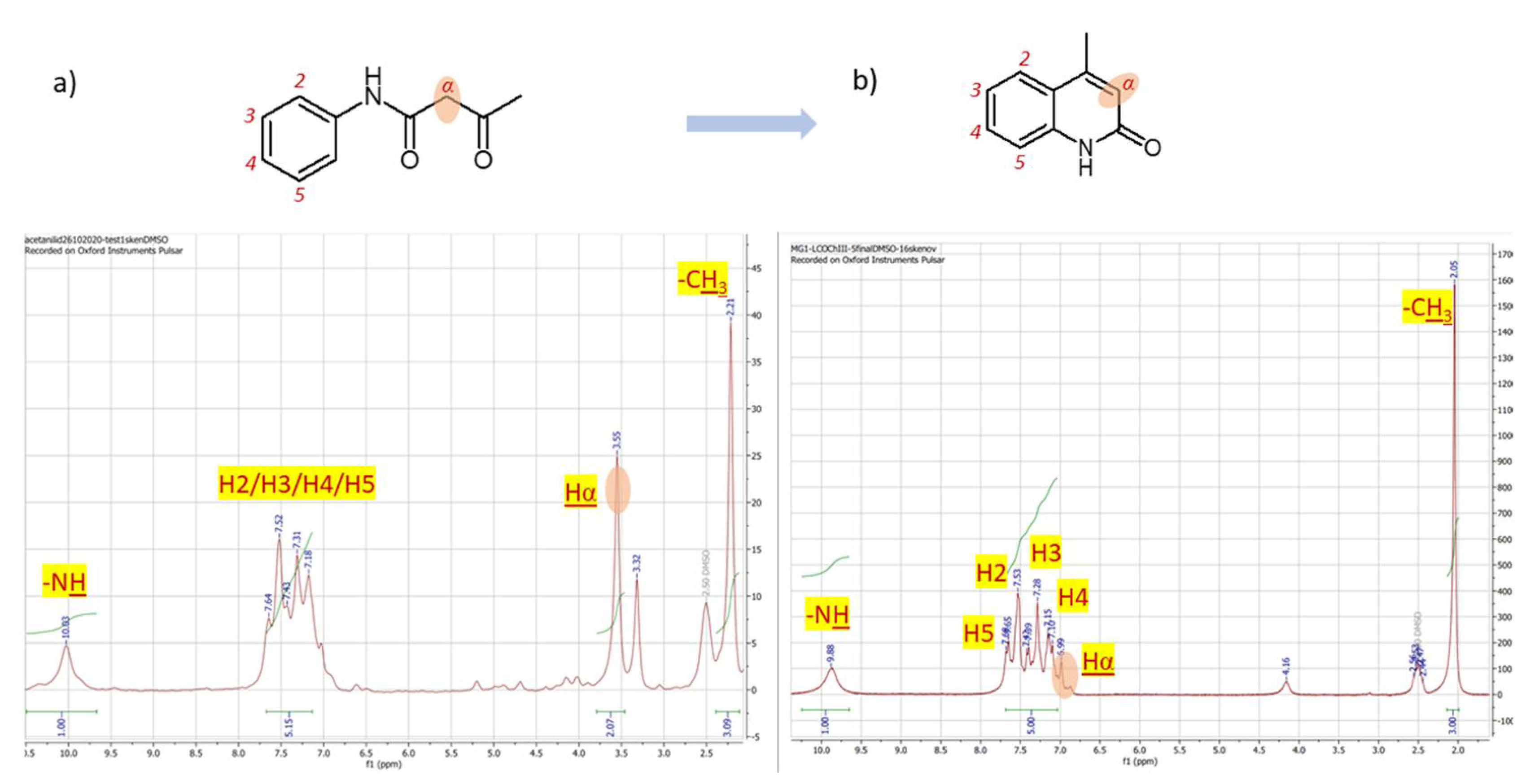
The proton spectrum of hippuric acid (2) (Figure 3) that posses a little bit more complex structure than the cinnamic acid exhibit significantly higher sensitivity and signal resolutiom even in an area of aromatic signals (6.5 – 8.0 ppm). Acid proton from carboxylic group is shifted to 12.53 ppm (broad singlet) as is expected for proton of the functional group of carboxylic acids. The nitrogen proton from NH group is represented by signal at 8.78 ppm. On this signal the non-standardly high resolution occurs, since the splitting with either neighboring protons of CH2 or aromtic protons is transformed to a triplet form of signal instead of broad singlet. Aromatic protons H2/H6 are represented by doublets at 7.84 ppm with 3J = 3.6 Hz. Aromatic proton H4 is viewed at 7.43 ppm as doubked and aromatic part is complemented with doublet at 7.50 ppm with 3J = 3.6 Hz for H3/H5. Integration 1/1/2/3/2 (sum. = 9 protons) corresponds with compound formula C9H9NO3. ince the acetanilide (3) acts as a substrate for subsequent synthesis of 4-methylquinoline-2-ol (4) that is achieved upon treatment with P2O5, by observing the changes of proton spectra profiles of both compounds allows to examine the completion of reaction process. Although the resolution of proton spectra of acetanilide (Figure 4a) is comparably lower, since only 4 scan (8 mins) were used to achieve spectrum the main difference the aliphatic protons of substrate (Fig. 6a, Hα) that are through the reaction converted to an aromatic one (Figure 4b, H Hα) is visible immediately upon comparison of the two spectra. While the group of aliphatic protons in the structure of acetanilide is assigned with signal at 3.55 ppm (Figure 4a) as singlet with integration equal to 2, the aromatic proton in the structure of product 4-methylquinoline-2-ol is observed in the range of aromatic protons at 6.99 ppm (Figure 4b) with integration of one proton. Since the spectrum of 4-methylquinoline-2-ol was taken at 16 scans (32 minutes totall time) the spectrum 6b is with better resolution showing the possibility to distinguish aromatic protons as following: H5 at 7.67 ppm, H2 at 7.53 ppm, H3 at 7.28 ppm and H4 at 7.13 ppm. The structure complemented with the signal of amine group proton at 9.88 observable as broad singlet and three protons of methyl group 2.05 ppm. Methyl protons of substrate on Fig. 6a correspond with chemical shift = 2.21 ppm and NH proton at 10.03ppm. Aromatic protons H2-H5 of acetanilide are splitted in a multiplet-type signal 7.64 – 7.18 ppm. The integration 1/5/2/3 (sum. = 11) correspond with formula of acetanilide C10H11NO2 and 1/5/3 (sum. = 9) is in accordance with formula C10H9NO of 4-methylquinoline-2-ol.
Figure 3.
1H NMR spectrum of hippuric acid in DMSO-d6 recorded at 60 MHz with proton assigment according to the structure (8 scans / 16mins).
Figure 3.
1H NMR spectrum of hippuric acid in DMSO-d6 recorded at 60 MHz with proton assigment according to the structure (8 scans / 16mins).
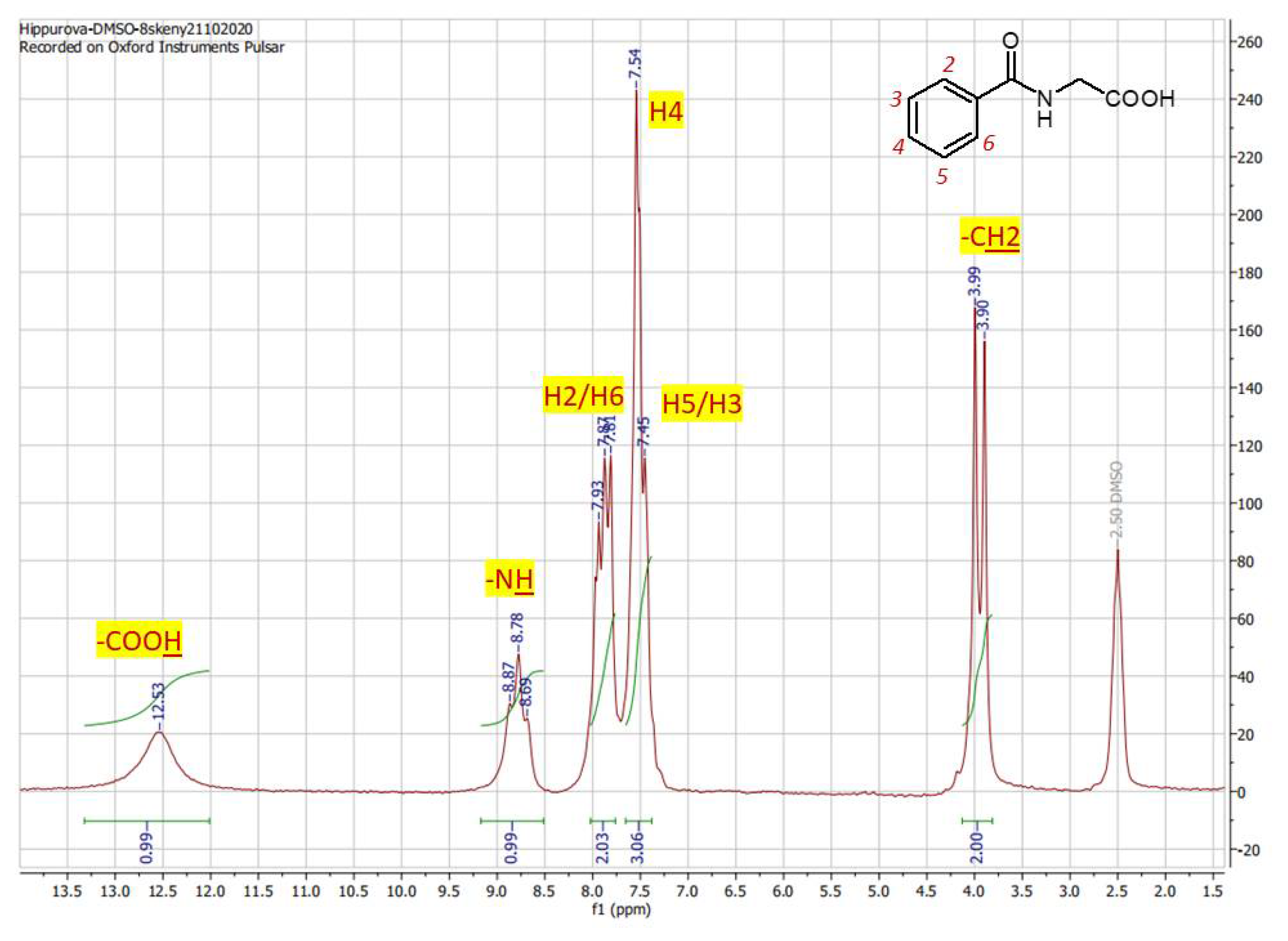
Figure 4.
1H NMR spectrum of a) acetanilide (60 MHz, DMSO-d6, 2 scans, 4 mins) and b) 4-methylquinoline-2-ol (60 MHz, DMSO-d6, 16 scans, 32 mins).
Figure 4.
1H NMR spectrum of a) acetanilide (60 MHz, DMSO-d6, 2 scans, 4 mins) and b) 4-methylquinoline-2-ol (60 MHz, DMSO-d6, 16 scans, 32 mins).
2.2. Synthesis and Combined 1H / 19F NMR for Structure Analysis of Novel Pentafluorophenylpyrroles 5-7
Compounds that possess intermolecular interactions such as hydrogen bonds, charge–transfer (CT) interactions, and Lewis acid–base interactions are most favoured in electronic materials design. Particularly, oligo- and polymers have been identified as promising opto-electronic materials due to their potential mechanical flexibility, structural tunability, solution processability and cost-effectiveness. Polyfluorobenzenes are attractive among the known and utilized organic materials as they exhibit stacking interactions on surfaces leading to long-range order in covalent organic frameworks [24,25]. The synthesis of such compounds is therefore still urgent and the presence of a formyl group is important in subsequent structural modifications since it undergoes a number of reactions. Therefore, 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) was chosen as the substrate for the synthesis of novel formyl and vinyl substituted derivatives - 1-pentafluorophenyl-1H-pyrrole-2,4-dicarbaldehyde (6) and 1-pentafluorophenyl-2-vinyl-1H-pyrrole (7) (Scheme 1). The synthesis of aldehyde 5 followed the Vilsmeier-Haack-Arnold (VHA) protocol and the reaction proceeded at 80 °C for 8 hours [26]. Herein, the slight modification of approach towards the monoformylated compound 5 is presented (Scheme 1) and the reaction proceeded at a lower temperature (25 °C) and extended reaction time (24 hours, Method A, Scheme 1). This manner is more convenient when compared to that originally described [26] in terms of more convenient isolation and with a yield increase of up to 90 %. Trials towards the synthesis of pyrrole-2-carbaldehyde 5 were extended with the approach described for the formylation of pyrrole-type derivatives. The reaction relies on the use of oxalyl chloride (COCl)2 instead of phosporus oxychloride (POCl3) (Method B) according to [27]. Nevertheless, the same product 5 has been isolated in a lower yield with the modified process under the standard VHA conditions (82 % vs. 92 %). As the formyl group possesses a significant deactivation effect on a pyrrole core through subsequent electrophilic formylation under the standard VHA reaction condition (Scheme 1), the Cα/Cβ biscarbonyl 6 was isolable in a significant minority in a mixture with substrate 5.
Under the conditions of the Wittig reaction the formyl group (-CH=O) is transformed into the vinylene-type substituent (-HC=CH-). Nowadays it is known that derivatives with vinylene-linkers are important for opto-electronic materials since the linkage ensures effective π-conjugation. Our target vinylene-type derivative 7 is rather important as a substrate for subsequent polymerization reaction towards polyfluorofenylpyrroles. Its synthesis was performed from a monoformylated substrate 5 under the standard Wittig-type reaction behaviour (Scheme 1). The reaction produced a mixture of desired product 7 and starting material 5 in the ratio of 5 : 7 = 6 : 1. Although the reactions towards 6 and 7 proceeded only with partial conversion, such type of derivatives combines the polarized aromatic backbone (C6F5) with a modifiable pyrrole core.
Considering the close absolute values of the gyromagnetic ratio of isotopes 1H and 19F with γ (1H) = 26.75105 and γ (19F) = 25.18034 rad·s-1T-1, in combination with the fact that γ in combination with the field strength has the main impact on NMR sensitivity, 19FNMR spectroscopy utilizes the substantial resolution and sensitivity. Therefore, the presence of the fluorine atom in any organic molecule opens a convenient route for structural identification by 19F NMR. Given the synthetic importance of pentafluorophenylpyrroles as building blocks for functional oligo- and polymers, the effectiveness of their structural characterization force the process of materials design. Using the LF 19F NMR we were able to identify the structure of previously reported pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) (Figure 5, left). The 19F signals at the chemical shifts δ = -143.40 ppm (dq, JF-F = 22.9, JH-F = 4.8 Hz) for F2/F6, -149.74 ppm and -158.17 ppm (q, JF-F = 22.9, 4.8 Hz) for F3/F5, respectively, coupling to the proton of carbonyl group on the pyrrole (CH=O) and the Cα-proton of the pyrrole core, that are accompanied with the triplet at -149.74 ppm, were explicitly acquired from LF-NMR data (Figure 5, left). In combination with 1H LF-NMR (Figure 6, left) from which the proton signals are readily assigned and identified the data achieved from the benchtop NMR in this particular case were of comparably higher-quality than those which have been achieved by HF-NMR (Figure 5 and 6 – right). Additionally, the challenging aspect of combined the 1H/19F LF-NMR experiment increases the pedagogical value in terms of accessing the slightly underrated fluorine NMR spectroscopy in the interest of undergraduate students when dealing with research into fluorine-containing derivatives. In the case of novel 1-pentafluorophenyl-1H-pyrrole-2,4-dicarbaldehyde (6) and pentafluorophenyl-2-vinyl-1H-pyrrole (7) the capability of LF-NMR regarding both 19F and 1H is rather limited. These compounds were synthesized for the first time, and, to the best of our knowledge, their spectral data were previously unknown by either LF or by HF-NMR. Moreover, the reactions towards both derivatives 6 and 7 have proceeded as an incomplete conversion with desired products in minor abundance in a mixture with the substrate 5. Therefore, the spectral data characterizes an appropriate mixture of 5/6 (Figure 7) or 5/7 and the structure from both LF/HF-NMR can only be deduced. However, with the spectra of pure 5 at hand, the signal estimation for novel compounds relies on the exclusion principle. According to the signal splitting to a doublet with δ = 9.69 and 9.71 ppm with an additional chemical shift at 9.90 ppm in the LF-proton NMR (Figure 7, left) and a signal with δ= 9.59 (2xH) ppm with an additional chemical shift at 9.85 ppm in the HF-proton NMR (Figure 8, right) illustrated for compound 6 (Figure8), it can be assumed that the mixture contains more formylated products. From the most shielded proton at 9.90 ppm (LF-NMR) or 9.85 ppm (HF-NMR) that is a part of bisformylated derivative 6, the ratio of substrate 5 / product 6 was determined to be 8 : 1 from both LF- and HF-proton NMR. More convenient manipulation of the spectral width enables a better signal assignment for pyrrole-core protons at 7.33 ppm and 6.67 ppm for Hβ and Hα, respectively. In the case of novel compounds 6 and 7 the better signal distinguishing between the desired product and the substrate is achieved from the fluorine spectra (Figure 7). While in the 19F HF-NMR signals are distributed significantly from each other (Figure 7, right) with product determination at -148.2 ppm for F2/F6, -153.5 ppm for F4 and -159.8 ppm for F3/F5, the effective adjustment of the spectral width allows the same signal designation from 19F LF-NMR (Figure 7, left) with chemical shifts at -143.5 ppm for F2/F6, -149.0 ppm for F4 and finally at -158.0 ppm for F3/F5. Surprisingly, the signal splitting in LF-NMR for compound 6 is of better quality compared to HF-NMR data, assuming that H-F splitting of F2/4 with the neighbouring protons of the pyrrole core is equal to JH-F = 22.5 Hz. In the case of vinylated derivative, that have been isolated in a reaction mixture in minority, the best resolution is achievable via high-field NMR data for both, 1H and 19F NMR spectra (Figure 9 and Figure 10)
2.3 Synthesis and Throughout Structural Characterization of Thiophene-Centered Azomethine 8.
Among the various small molecules synthesized for organic electronics, azomethines with imine linkage (-C=NH-) known as Schiff bases are recognized as an alternative for typical conjugated materials containing vinylene-type bonding (-HC=CH-) [28,29]. Azomethines with a thiophene core are observed undergoing a reversible electrochemical oxidation process, exhibit a narrow energy band gap, high thermal stability, and generally act as p-type (hole transporting) materials [30]. The general route towards azomethines is comparably more convenient as an approach than their vinylene-type counterparts. Based on previous results, we have found that the phosphorus oxychloride (P2O5) exhibits a notable efficiency in Schiff-type condensation towards thiophene-centred azomethines [31]. From here, we followed a procedure utilizing P2O5 in ethanol (Scheme 2). Hence, the yield of previously described compound 2,5-bis-[(4-methoxy-benzylidene)-amino]-thiophene-3,4-di-carbonitrile (8) increased up to 60% from 56% [30].
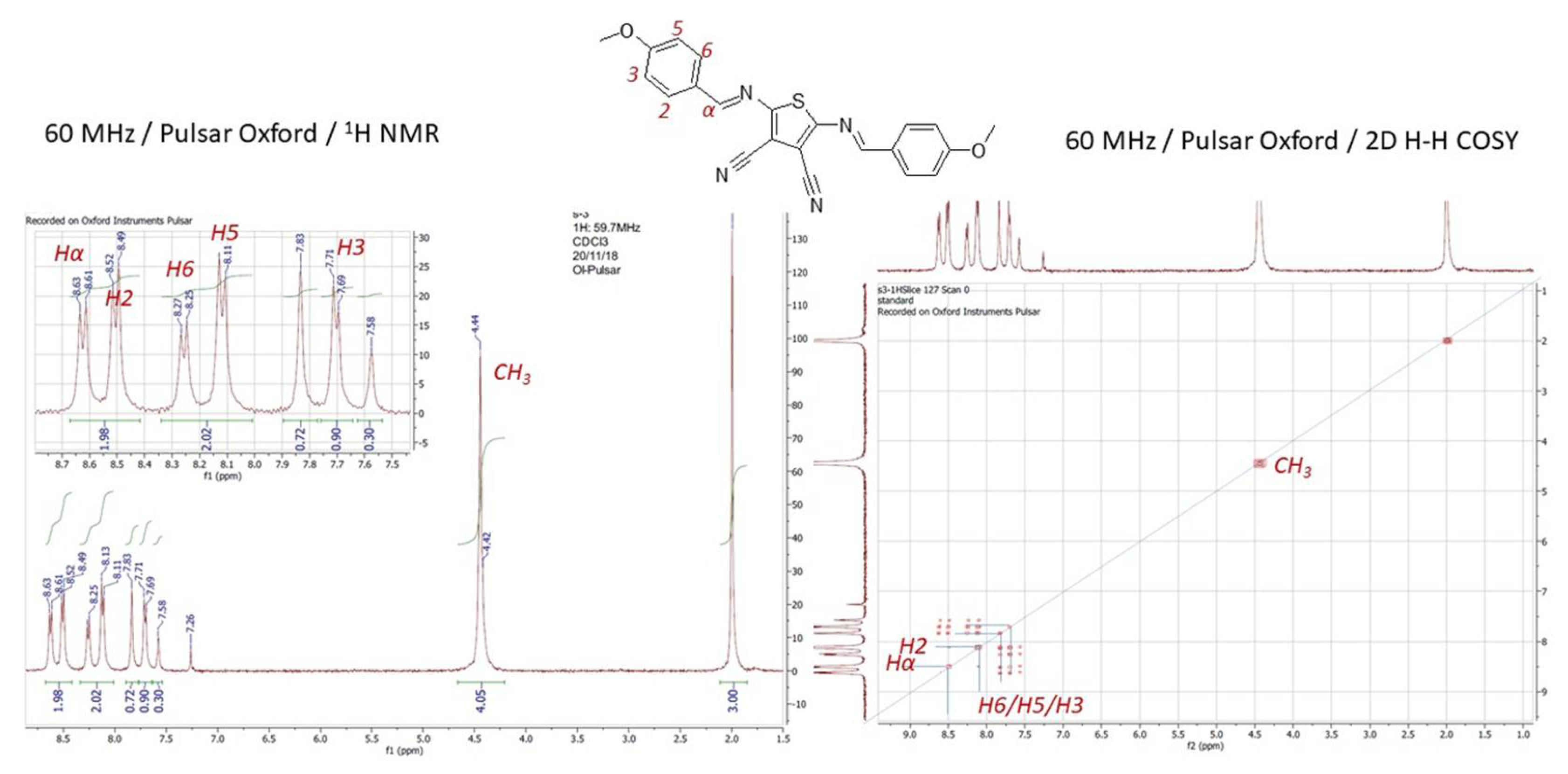
Chemical structure of the 2,5-bis-[(4-methoxy-benzylidene)-amino]-thiophene-3,4-dicarbo-nitrile (8) have been for the first time fully assigned by the combination of routine 1H NMR and 2D – COSY (1H-1H) spectra acquired at LF, as is shown on Figure 11.
Interestingly, one of the structure determining protons assigned to the proton of azometine-linkage (-HC=N-) appear in 1D spectrum as doublet at δ = 8.62 ppm, and a corresponding cross-peak signal was observed in the 2D COSY spectrum. The aromatic protons in the structure of 8a are readily detectable in both 1H and 2D spectrum with the appropriate signal splitting at δ(H2) = 8.52 ppm, δ(H6) = 8.26 ppm and δ(H5) = 8.12 ppm. Interestinlgy, the signal of the proton H3 is splitted into an unexpected signal splitting pattern at δ(H3-phenyl-core 1) = 7.70 ppm as doublet while the same proton on the other phenyl core is separated unconventionally to doublet with δ = 7.83 ppm with integration of 2/3 and δ = 7.58 integrated to 1/3. Such type of equivocal dyadic cleavage in aromatic fragment occurs probably as the consequence of the sample overheating due to the long duration of NMR measurement. It is obvious, also during the acquisition at the high magnetic fields, that the long irradiation affects the sample overheating causing the spin dephasing because of the internal magnetic field (B1) inhomogeneity. As result of such behaviour the signal degradation with a broader bandwidth occurs within the spectra as have occurred by both, 1H and 2D NMR experiments for 8.
3. Discussion
Our results on the proton NMR (Figure 2 – Figure 4), combined proton-fluorine (Figure 5 - Figure 10) and demonstrate that low-field NMR spectroscopy holds great potential to move forward from instructional laboratory to organic and synthetic chemistry research fields with benefit of access the ultimative spectral data promptly.
Although the NMR spectrsoscopy with compact instruments is nowadays extended in academic teaching to the best of our knowledge, there is only one report until now postulating the possible utilization of LF-NMR further in behalf of organic chemistry research and synthesis [32]. Herein, through a set of compounds 1-4 with known chemical structure and novel derivatives 5-8 we have demonstrated the practical benefits of LF-NMR for structural characterization of compounds used as substrates for gaining insights into reaction processes.
Summarizing, the combination of 1H/19F LF-NMR enables the chemical-structural investigation of small polyfluorinated organic compounds as pure compounds or mixtures in almost the same quality as is offered by HF-NMR. At this stage, it can be concluded that for fluoro-substituted organic molecules with low-molecular weights, LF-NMR meets the criteria of quantitative structural elucidation achieved in-time with the benefits of being cost- and user friendly. Neverthelless, characterization of the mixture of vinylene 7 and its substrate 5 was rather unappropriate according to the low-field NMR acquisition and the HF-NMR have been used for qualitative elucidation of the chemical structure. On the other hand, by combined 1H- and 1H-1H 2D (COSY) at 60 MHz the combarably valuable spectral sheet have been achieved as by HF-NMR.
4. Materials and Methods
4.1. General
All commercially available chemicals were used as received without further purification. Deuterated solvents – dimethylsulfoxide-d6 (DMSO-d6) and chloroform (CDCl3) with content of 99.96 % of D and 99.8 % of D, respectively, were used for the sample preparation. Content of standard tetramethylsilane TMS have been adjusted by its dropping directly into a prepared sample. Presented compounds 1-4 were synthesized during the undegraduate laboratory course in a range of years 2018-2023. Each year minimum of 5 / maximum of 15 repeated experiments for each compound were realized.
All spectra were processed using MNova (version 11.0, MestreLab Research, S.L., Santiago de Compostela, Spain) following an identical protocol, which included zero filling to 64 K and apodization with a 0.3 Hz exponential window function prior to Fourier transformation, manual phase and baseline correction, and referencing to the signal of the internal standard tetramethylsilane (δ/ppm = 0). Residual solvent peaks were reffered either to 7.26 ppm for CDCl3 or 2.50 ppm for DMSO-d6 in combination with 3.33 ppm for water.
Phosphorus oxychloride (CAS: 10025-87-3, 99 % reagent grade), N,N-dimethylformamide (CAS: 68-12-2, 99.5 % reagent grade), 1-pentafluorophenyl-1H-pyrrole (CAS: 344452-95-5, reagent grade), dichloroethane (CAS: 107-06-2, 99 % spectrophotometric grade), potassium carbonate (CAS: 584-08-7, 99 % reagent grade), diethyl ether (CAS: 60-29-7, 99 % reagent grade), toluene (CAS: 108-88-3, 99 % ACS grade), dichloromethane (CAS: 75-09-2, 99 % ACS grade), n-hexane (CAS: 110-54-3, 99 % analytical grade), Ethyl acetate (CAS: 141-78-6, 99.7 % technical grade); Methylphenylphosphonium bromide (CAS: 1779-49-3, 98 % reagent grade), and all the solvents were purchased from commercial suppliers (Sigma-Aldrich, Alfa-Aesar, TCl, Lancaster, Merck, Ossila, Ontario Chemicals) and used as received. Starting materials: 1-Pentafluorophenyl-1H-pyrrole-2-carbaldehyde 5; 1-Pentafluorophenyl-1H-pyrrole-2,4-dicarbaldehyde 6; 1-Pentafluorophenyl-2-vinyl-1H-pyrrole 7 was prepared according to the below described manner. All of the performed reactions were monitored by thin layer chromatography (TLC) on plates precoated with silica gel (Merck60F254) and visualized using a UV lamp operating at 254/365 nm wavelenghts. The synthesized compounds were fully characterized by 1H, 19F, 13C NMR (Pulsar Oxford 60 MHz, Varian Unity 400 MHz), column chromatography using Silica gel Kiesegel 60.
4.2. Synthesis
Compounds 1-4 were synthesized according to known procedures as following, cinnamic acid (1) [33], hippuric acid (2) [34], acetanilide (3) [35], 4-methylquinoline-2-ol (4) [36] and azomethine 8 [31].
4.2.1. Synthesis of 1-Pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5)
Method A
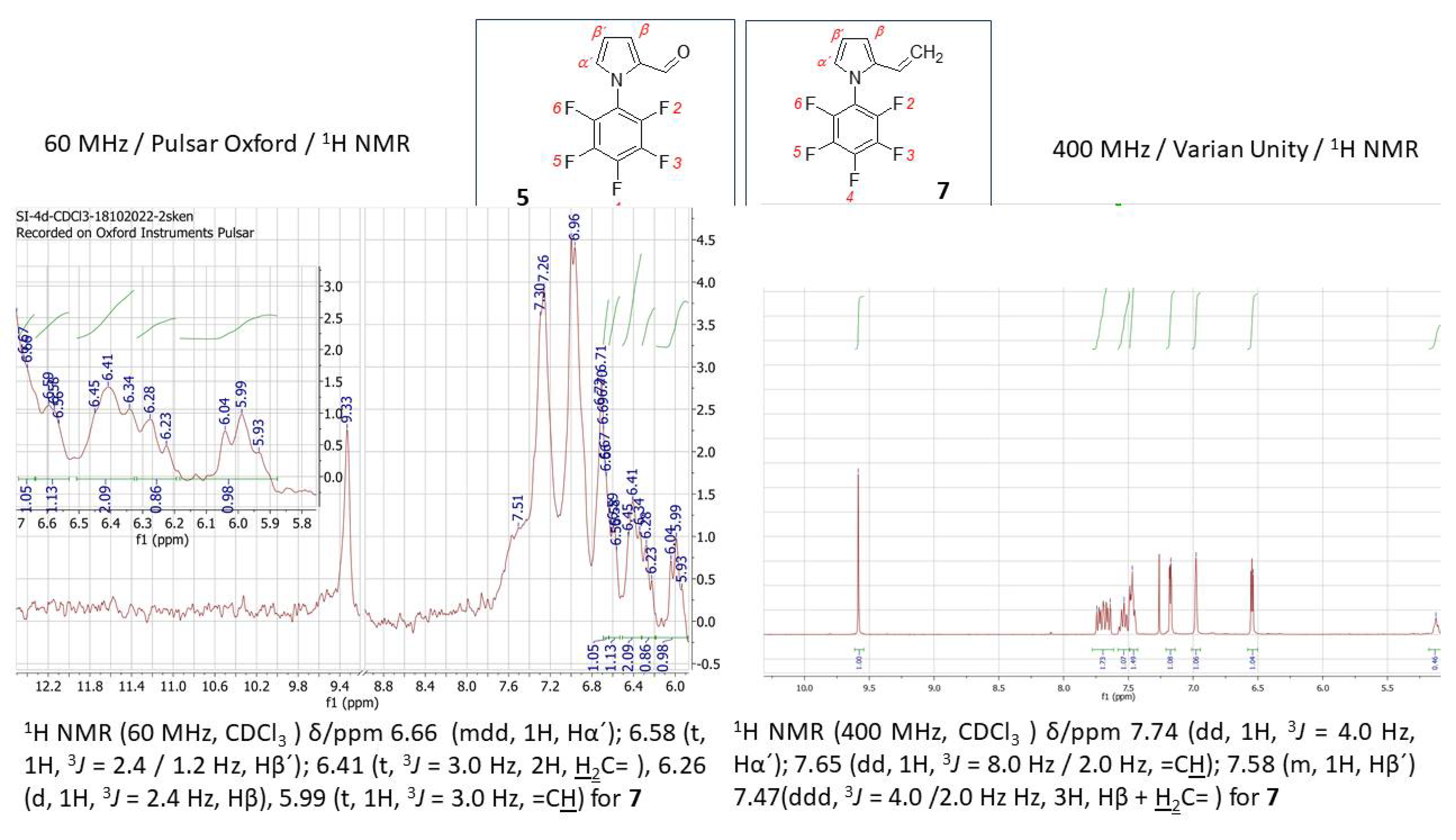
1-Pentafluorophenyl-1H-pyrrole (0.8 g, 3.5 mmol) was dissolved in N,N-dimethylformamide (2.6 mL, 2.5 g, 0.03 mol) and cooled to 0 °C. POCl3 (2.5 mL, 0.027 mol) was carefully added to this mixture so that the temperature did not rise above 10-15 °C. After adding the entire amount of POCl3, the mixture was cooled for another 15 minutes. Subsequently dichloromethane (4 mL) was added and the reaction mixture was stirred at room temperature overnight. It was then cooled to 0 °C and saturated K2CO3 solution was added until the pH was slightly adjusted on pH neutral. The mixture was purified by extraction with diethyl ether (3x30 mL) and column chromatography with n-hexane : EtOAc = 9 : 1 as eluent. Expected product (C11H4F5NO, 261,15 g.mol−1) was obtained as a light-grey solid with a m.p. = 70-73 °C and in 92.30 % yield (0.844 g). 1H NMR (60 MHz, CDCl3) δ/ppm 9.65 (s, 1H, CHO); 7.22 (d,1H, 3J = 1.8 Hz, Hα´); 7.03 (s, 1H, Hβ); 6.62 (d, 1H, 3J = 1.8 Hz, Hβ´). 1H NMR (400 MHz, CDCl3) δ/ppm 9.58 (s, 1H, CHO); 7.18 (d,1H, 3J = 4.0 Hz, Hα´); 6.98 (s, 1H, Hβ); 6.54 (t, 1H, 3J = 4.0 Hz, Hβ´). 19F NMR (59.7 MHz, CDCl3) δ: -143.40 (dd, J = 22.9, 4.8 Hz, 2F, F2,F6), -149.74 (t, J = 22.9 Hz, 1F, F4), -158.17 (dd, J = 19.6, 3.3 Hz, 2F, F3/F5). 19F NMR (376 MHz, CDCl3) δ: -147.13 (dd, J = 19.6, 3.3 Hz, 2F, F2,F6), -153.42 (t, J = 21.5 Hz, 1F, F4), -161.83 (dd, J = 19.6, 3.3 Hz, 2F, F3/F5).
13C NMR (101 MHz, CDCl3) δ: 178,66 (s); 145,01 (m); 142,77 (m); 142,49 (m); 140,29 (m); 138,96 (m); 136,41 (m); 132,72 (s); 131,60 (s); 124,98 (s); 112,33 (s).
Method B
N,N-Dimethylformamide (0.15 mL, 0.14 g, 1.94 mmol) was cooled to 0°C. To this cooled solvent, POCl3 (0.30 mL, 0.49 g, 3.21 mmol) was gradually added, ensuring that the temperature did not exceed 10-15°C. After the complete addition, the mixture was allowed to cool for another 10 minutes. Meanwhile, a mixture of 1-pentafluorophenyl-1H-pyrrole (1 g, 4.29 mol) and dichloroethane (5 mL, 6.25 g, 0.063 mol) was prepared, which was added to the formylation reagent POCl3/DMF. The resulting mixture was stirred for 7 hours at 80 °C. After the reaction was completed, the mixture was cooled to room temperature, and the pH was adjusted to alkaline using a mixture of ice and K2CO3. Subsequently, the mixture was extracted with diethyl ether (5 x 30 mL) and purified by column chromatography with eluent (toluene). Expected product (C11H4F5NO, 261.15 g.mol−1) was obtained as a grey-white solid with a m.p. = 75-78 °C and in 96.47 % yield (0.489 g). 1H NMR (400 MHz, CDCl3) δ/ppm 9.58 (s, 1H, CHO); 7.18 (d,1H, 3J = 4.0 Hz, Hα´); 6.98 (s, 1H, Hβ); 6.54 (t, 1H, 3J = 4.0 Hz, Hβ´).
4.2.3. Synthesis of 1-Pentafluorophenyl-1H-pyrrole-2,4-dicarbaldehyde (6)
1-Pentafluorophenyl-1H-pyrrole-2-carbaldehyde (0.5 g, 1.91 mmol) was dissolved in N,N-dimethylformamide (8 mL, 7.6 g, 0.103 mol) and cooled to 0 °C. Subsequently, it was introduced into the prepared mixture, and POCl3 (8 mL, 13.12 g, 0.086 mol) was meticulously added to ensure that the temperature did not exceed 10-15 °C. After adding the entire amount of POCl3, the mixture was cooled for another 15 minutes and then mixed at room temperature overnight. The solution was subsequently cooled to 0 °C, and a saturated K2CO3 solution was incrementally added until achieving a slightly adjusted neutral pH. The mixture was purified by extraction with diethyl ether (5x30 mL) and column chromatography with n-hexane : EtOAc = 9 : 1 as eluent. Expected product (C12H4F5NO2, 289.16 g.mol−1) was obtained as a mixture of substances 5 and 6 a ratio of 6 : 5 = 8 : 1 in a yellowish solid state with a m.p. = 50-53 °C and in 85.24 % yield (0.47 g). 1H NMR (60 MHz, CDCl3) δ: 9.90 / 9.71 (s, 2H, CHαO + CHβO), 7.33 (s, 1H, Hβ), 6.67 (d, J = 2.9 Hz, 1H, H2´) for 6. 1H NMR (400 MHz, CDCl3) δ: 9.85 / 9.59(s, 2H, CHαO + CHβO), 7.48 (s, 1H, Hβ), 6.85 (d, J = 2.9 Hz, 1H, H2´) for 6. 19F NMR (59.7 MHz, CDCl3) δ: -143.5 (dd, J = 22.5 Hz, 14.3 Hz, 2F, F2/F6), -149.0 (t, J = 22.5 Hz, 1F, F4), -158.0 (t, J = 22.5 Hz, 2F, F3/F5) for 6. 19F NMR (376 MHz, CDCl3) δ: -148.2 (d, 2F, F2/F6), -153.5 (t,1F, F4), -159.8 (d, 2F, F3/F5) for 6. 13C NMR (101 MHz, CDCl3) δ: 184 (s); 177,64 (s); 144,01 (m); 141,50 (m); 137,94 (m); 135,38 (m); 131,69 (s); 130,56 (s); 123,96 (s); 11,31 (s); 1109 (s).
4.2.4. Synthesis of 1-Pentafluorophenyl-2-vinyl-1H-pyrrole (7)
Methylphenylphosphonium bromide (0.7 g, 1.9 mmol) was suspended in 1,4-dioxane (20 mL, 20.6 g, 0.234 mol). To this suspension was added a solution of K2CO3 (4.6 g, 0.033 mol) in water (1 mL) and the mixture was stirred at room temperature. 1-Pentafluorophenyl-1H-pyrrole-2-carbaldehyde (0.5 g, 1.9 mmol) in 1,4-dioxane (2 mL, 2.06 g, 0.023 mol) was added and the mixture was stirred at 110 °C for 24–48 hours. Subsequently, the solvent was evaporated, to which a small amount of water and diethyl ether were added. This mixture was extracted and purified by column chromatography with a THF : n-hexane = 3 : 7 elution mixture. Expected product (C12H6F5N, 259,17 g.mol−1) was obtained as a mixture of 5 and 7 in a ratio 5 : 7 = 6 : 1 oil structure brown color and in 95.53 % yield (0.47 g). 1H NMR (400 MHz, CDCl3) δ/ppm 6.66 (m (d), 1H, Hα´); 6.58 (t, 1H, 3J = 2.4 / 1.2 Hz, Hβ´); 6.41 (t, 3J = 3.0 Hz, 2H, H2C= ), 6.26 (d, 1H, 3J = 2.4 Hz, Hβ), 5.99 (t, 1H, 3J = 3.0 Hz, =CH). 1H NMR (400 MHz, CDCl3) δ/ppm 7.74 (dd, 1H, 3J = 4.0 Hz, Hα´); 7.65 (dd, 1H, 3J = 8.0 Hz / 2.0 Hz, =CH); 7.58 (m, 1H, Hβ´) 7.47(ddd, 3J = 4.0 /2.0 Hz Hz, 3H, Hβ + H2C= ). 19F NMR (59.7 MHz, CDCl3) δ: -147.5 (d, 2F, F3/F5), for F4 and F2/F6 signals not detectable for 7. 19F NMR (376 MHz, CDCl3) δ: -148.8 (d, 2F, F2,F6), -154.5 (d, 1F, F4), -157.3 (d, 2F, F3/F5) for 7. 13C NMR (101 MHz, CDCl3) δ: 144,96 (s); 142,49 (s); 135,22 (s); 133,25 (m); 128,76 (m).
5. Conclusions
Herein, we highlight the LF-NMR spectroscopy as profitable analytical tool for structure determination in organic laboratory immediately upon synthesis. Results are illustrated on a set of 1H NMR spectral data for a set of compounds 1-4 representing the products of undergraduate laboratory course. Combined 1H and 19 F LF-NMR data for pentafluorophenylpyrroles 5-7 as the products of our research efforts were compared with their HF-NMR counterparts. Finally, the set of 1H, 2D 1H-1H COSY and 13C NMR data acquisition on a benchtop-type system for azomethine 8 have been used for its structure evaluation. Moreover, benchtop NMR represents the spectral instrument of daily use with advantages of being environmentally benign and energy & costs & time saving.
Author Contributions
MG – synthesis of pharmaceuticals and 1H LF-NMR measurements, literature database investigation and summary of theorethical knowledge; PT – 19F NMR signal assignement and solution, HF-NMR data retrieving, BP -19F LF-NMR data acquisition; SI – synthesis of fluoro-containing compounds, ZT - synthesis of azomethine and fluoro-containing compounds, manuscript conceptualization, writing of the manuscript.
Funding
This research was funded by the Slovak Research and Development Agency and Ministry of Education, Research and Youth of the Slovak Republic , projects APVV-19-0087 and APVV-20-0564; VEGA 1/0086/21.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Laukien, D. D.; Tschopp, W. H. Superconducting magnet design. Concepts Magn. Reson., 1994, 6, 255-273. [CrossRef]
- Blümich, B. Low-field and benchtop NMR. J. Magn. Reason., 2019, 306, 27-35. [CrossRef]
- Blümich, B. Introduction to compact NMR: A rewiev of methods. TrAC Trends Anal. Chem., 2016, 83, 2–11. [CrossRef]
- Anders, J.; Dreyer, F.; Krüger, D.; Schwartz, I.; Plenio, M. B.; Jelezko, F. Top. Curr. Chem., 2013, 335, 1–22.
- Yu, H.Y.; Myoung, S.;Ahn, S. Recent applications of benchtop nuclear magnetic resonance spectroscopy. Magnetochem., 2021, 7, 121, 1– 6. [CrossRef]
- Gouilleux, B.; Silva Elipe M.V. Benchtop cryofree NMR. Magn. Reson. Chem., 2024, in press. [CrossRef]
- Castaing-Cordier, T.; Bouillaud, D.; Farjon, J.; Giraudeau, P. Recent advances in benchtop NMR spectroscopy and its applications. Ann. Rep. NMR Spectr., 2021, 103, 191-258. [CrossRef]
- Giberson, J.; Scicluna, J.; Legge, N; Longstaffe, J. Developments in benchtop NMR spectroscopy 2015-2020. Annu. Rep. NMR Spectrosc., 2021, 102, 153 – 246. [CrossRef]
- Rhee, Y.; Shilliday, E. R.; Matviychuj, Y.; Nguyen, T.; Robinson, N.; Holland, D. J.; Connolly, D. J.; Johns, M. L. Detection of honey using benchtop 1H NMR spectroscopy. Analyt. Meth., 2023, 13, 1690-1699. [CrossRef]
- Draper, S. L.; McCarney, E. R.; Benchtop nuclear magnetic resonance in forensic chemistry, Magn. Res. Chem., 2023, 61, 106 – 129. [CrossRef]
- Remy, C.; Danoun, S.; Delample, M.; Morris, C.; Gilard, V.; Balayssac S. Characterization of fatty acid forms using benchtop NMR in omega-3 oil supplements. Magn. Reson. Chem., 2024, 62, 328-336. [CrossRef]
- McCarney, E.R.; Kristoffersen, K.A.; Anderssen K.E. Quantitative at-line monitoring of enzymatic hydrolysis using benchtop diffusion nuclear magnetic resonance spectroscopy. Magn. Reson. Chem., 2024, 62, 452-462. [CrossRef]
- Araneda, J. F.; de Alwis Weerasekera, H.; Leclerc, N. C.; Luk, S. B.; Riegel, S. D. Determination of copolymer compositions in polyhydroxyalkanoates using 1H benchtop nuclear magnetic resonance spectroscopy, Anal. Methods, 2023, 28, 3476 – 3482. [CrossRef]
- Riegel, S. D.; Leskowitz, G. M. Benchtop NMR spectrometers in academic teaching. TrAC Trends Anal. Chem., 2016, 82, 12 – 26.. [CrossRef]
- Jenne, A.; Soong, R.; Downey, K.; Biswas, R.G.; Decker, V.; Busse, F.; Goerling, B.; Haber, A.; Simpson, M.J.; Simpson A.J. Brewing alcohol 101: An undergraduate experiment utilizing benchtop NMR for quantification and process monitoring. Magn. Reson. Chem., 2024, 62, 429-438. [CrossRef]
- Maschmeyer, T. ; Russell, D.J.; Napolitano, J.G. ; Hein J.E. Reaction monitoring via benchtop nuclear magnetic resonance spectroscopy: A practical comparison of on-line stopped-flow and continuous-flow sampling methods. Magn. Reson. Chem., 2024, 62, 310-322. [CrossRef]
- Soong, R.; Downey, K.; Moser, A.; Monje, P.; Jenne, A.; Biswasm R. G.; Bastawrous, M.; Mamujdar, R.; Lysak, D. H ; Adamo, A.; Goerling, B.; Decker, V.; Busse, F.; Dominguez, S.; Sauer, F.; Mikchaylichenko, S.; Luk, V.; Simpson, A. J. A CASE (Computer-Assisted StructureElucidation) for Bench-Top NMR Systems in the Undergraduate Laboratory for De Novo Structure determination: How Well Can We DO? J. Chem. Educ., 2022, 99, 3780 – 3788.
- van Bramer, S. E.; Bastin, L. D. Spectroscopy Data for Undergraduate Teaching. J. Chem. Edu., 2023, 100, 3897-3902. [CrossRef]
- Morrison, R. W.; Zhang, M. Application of benchtop NMR in the organic chemistry instructional laboratory, Magn. Res. Chem., 2020, 58, 1187-1192. [CrossRef]
- de-Sousa, A.; Hemmer, M.; Gasteiger, J. Prediction of 1H NMR Chemical Shifts Using Neural Networks trained for chemical shifts. Anal. Chem., 2007, 74, 80-90. [CrossRef]
- Banfi, D.; Patiny, L. www.nmrdb.org: Resurrecting and processing NMR spectra on-line Chimia., 2008, 62: 280-281.
- Castill, A. M.; Patiny, L.; Wist, J. Fast and Accurate Algorithm for the Simulation of NMR spectra of Large Spin Systems. J. Magn. Res., 2011, 209, 123 – 130. [CrossRef]
- Glögger, S.; Blümich, B.; Appelt, S. NMR Spectroscopy for Chemical Analysis at Low-Magnetic Fields. Topp. Curr. Chem. 2013, 335, 1 – 22. [CrossRef]
- Liu, L.-H.; Yan M., Perfluorphenylazides: new applications in surface functionalization and nanomaterials synthesis. Acc. Chem. Res., 2010, 43, 1434 - 1443.
- Braunecker, W. A.; Hurst, K. E.; Ray, K. G.; Owczarczyk, Z. R.; Martinez, M. B.; Leick, N.; Keuhlen, A.; Sellinger, A.; Johnson, J. C. Phenyl / Perfluorphenyl Stacking Interactions Enhance Structural Order in Two-Dimmensional Covalent Organic Frameworks. Cryst. Growth Des., 2018, 18, 4160 - 4166. [CrossRef]
- Hrnčáriková, K.; Végh, D. Synthesis and Reactivity of New 1-Pentafluorophenyl-1H-pyrrole Derivatives. Molecules, 2003, 8, 536 - 540.
- Martynovskaya, V.; Shchrebakova, V. S.; Ushakov, I. A.; Borodyna, T. N.; Ivanov, V. Expedient Synthesis of new class of organic building blocks: 1-Allenylpyrrole-2-carbaldehydes. Tetrahedron Lett., 2020, 61, 52 – 58.
- Koole, M.; Frisenda, R.; Petrus, M. L.; Perrin, M. L.; van der Zant, H. S. J.; Dingemans, T. J. Charge Transport through Conjugated Azomethine-based Single Molecules for Optoelectronic Applications. Org. Electron., 2016, 34, 38 - 41. [CrossRef]
- Bourgeaux, M.; Skene, W. G. A Highly Conjugated p- and n-Type Polythiophenoazomethine: Synthesis, Spectroscopic, and Electrochemical Investigation. Macromol., 2007, 40, 1792 – 1795. [CrossRef]
- Pająk, A. K.; Kotowicz, S.; Gnida, P.; Małecki, J. G.; Ciemięga, A.; Łuczak, A.; Jung, J.; Schab-Balczerak, E. Synthesis and Characterization of New Conjugated Azomethines End-Capped with Amino-thiophene-3,4-dicarboxylic Acid Diethyl Ester. Int. J. Mol. Sci., 2022, 23, 8160 (1-19). [CrossRef]
- Tokárová, Z.; Maxianová, P.; Váry, T.; Nádaždy, V.; Végh, D.; Tokár, K. Thiophene-centered azomethines: Structure, photophysical and electronic properties. J. Mol. Struct., 2020, 1204, 127492. [CrossRef]
- Blümich, B.; Singh, K. Desktop NMR and Its Applications From Materials Science To Organic Chemistry. Angew. Chem. 2018, 57, 6996 – 7010. [CrossRef]
- Su, F.; Chen, W.-Ch. Synthesis of cinnamic acid from benzaldehyde and acetone. J. Chim. Chem. Soc. 1956, 3, 87-91. [CrossRef]
- Vogel’s Textbook of Practical Organic Chemistry by Brian S. Furniss, Antony J. Hannaford, Peter W. G. Smith & Austin R. Tatchell; Fifth Edition; Page No. 1156 (1989).
- Xu, L,; Wu, X.; Xiao, J. (2011). Asymmetric Direct Reductive Amination of β-Keto Amides. Science of Synthesis: Stereosel. Synth., 2011, 2, 291.
- Jung, J. C.; Jung, Y. J.; Park, O.-S. A convenient one-pot synthesis of 4-hydroxycoumarin, 4-hydroxythiocoumarin, and 4-hydroxyquinolin-2(1H)-one. Synth. Commun., 2001, 31, 1195 – 1200. [CrossRef]
Figure 1.
The structure of investigated compounds by means of LF-NMR: Compounds 1-4 from the category of undergraduate products from laboratory course; the set of small-molecules 5-8 from a category of our current research interests.
Figure 1.
The structure of investigated compounds by means of LF-NMR: Compounds 1-4 from the category of undergraduate products from laboratory course; the set of small-molecules 5-8 from a category of our current research interests.
Scheme 1.
Synthesis organic small-molecules 5-7 from a category of pentafluorophenylpyrroles that are currently of our research focus, herein presented according to the 1H and 19F LF-NMR (PULSAR Oxford 60 MHz, 56.5 MHz) in combination with 1H /19F HF-NMR (Bruker Ascend TM 400 MHz, 376 MHz).
Scheme 1.
Synthesis organic small-molecules 5-7 from a category of pentafluorophenylpyrroles that are currently of our research focus, herein presented according to the 1H and 19F LF-NMR (PULSAR Oxford 60 MHz, 56.5 MHz) in combination with 1H /19F HF-NMR (Bruker Ascend TM 400 MHz, 376 MHz).
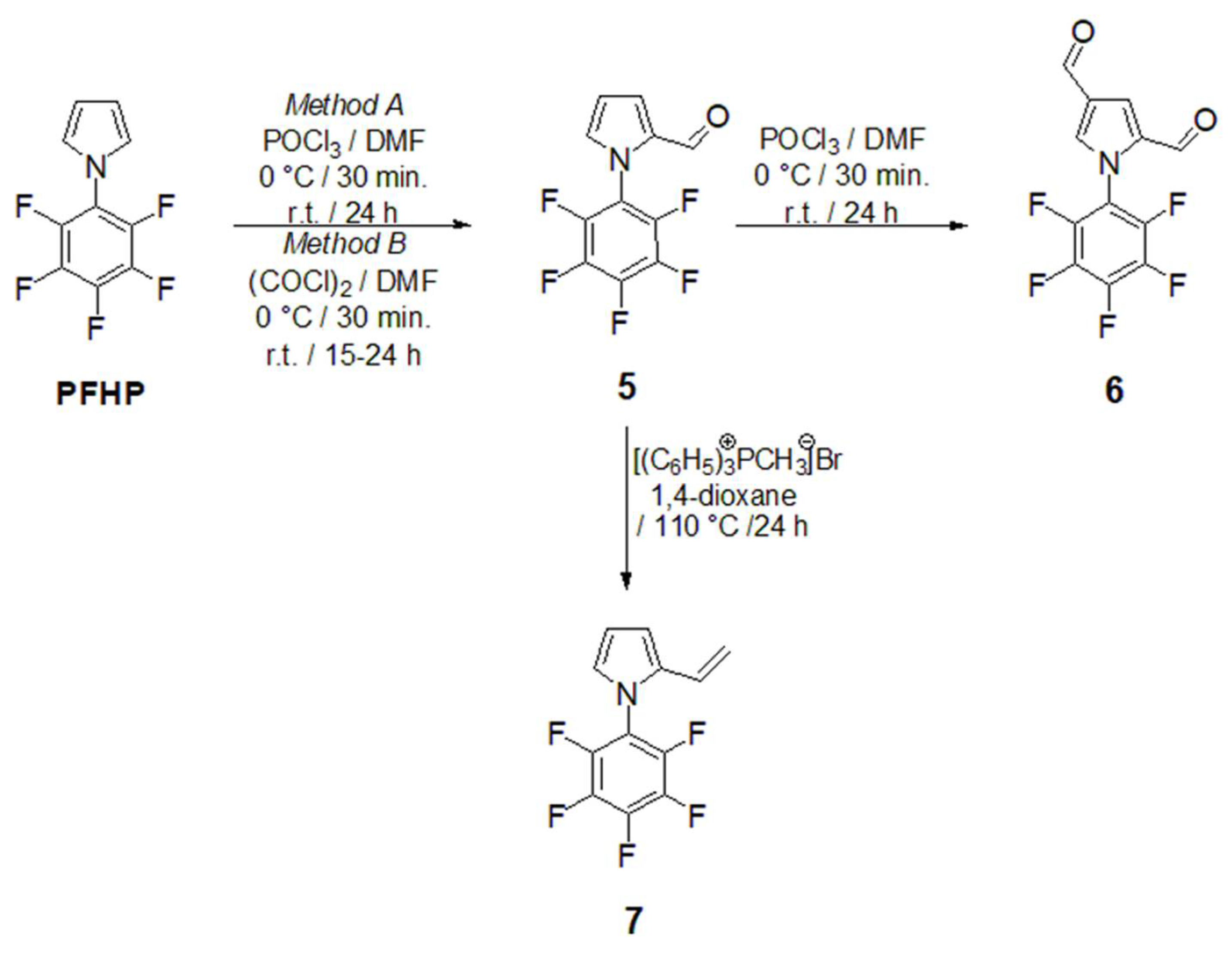
Figure 5.
19F NMR spectrum 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) obtained from a 60 MHz Pulsar Oxford benchtop NMR at 56.5 MHz (for 19F isotope, left) and from a 400 MHz Bruker Ascend TM at 376 MHz (for 19F isotope, right).
Figure 5.
19F NMR spectrum 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) obtained from a 60 MHz Pulsar Oxford benchtop NMR at 56.5 MHz (for 19F isotope, left) and from a 400 MHz Bruker Ascend TM at 376 MHz (for 19F isotope, right).
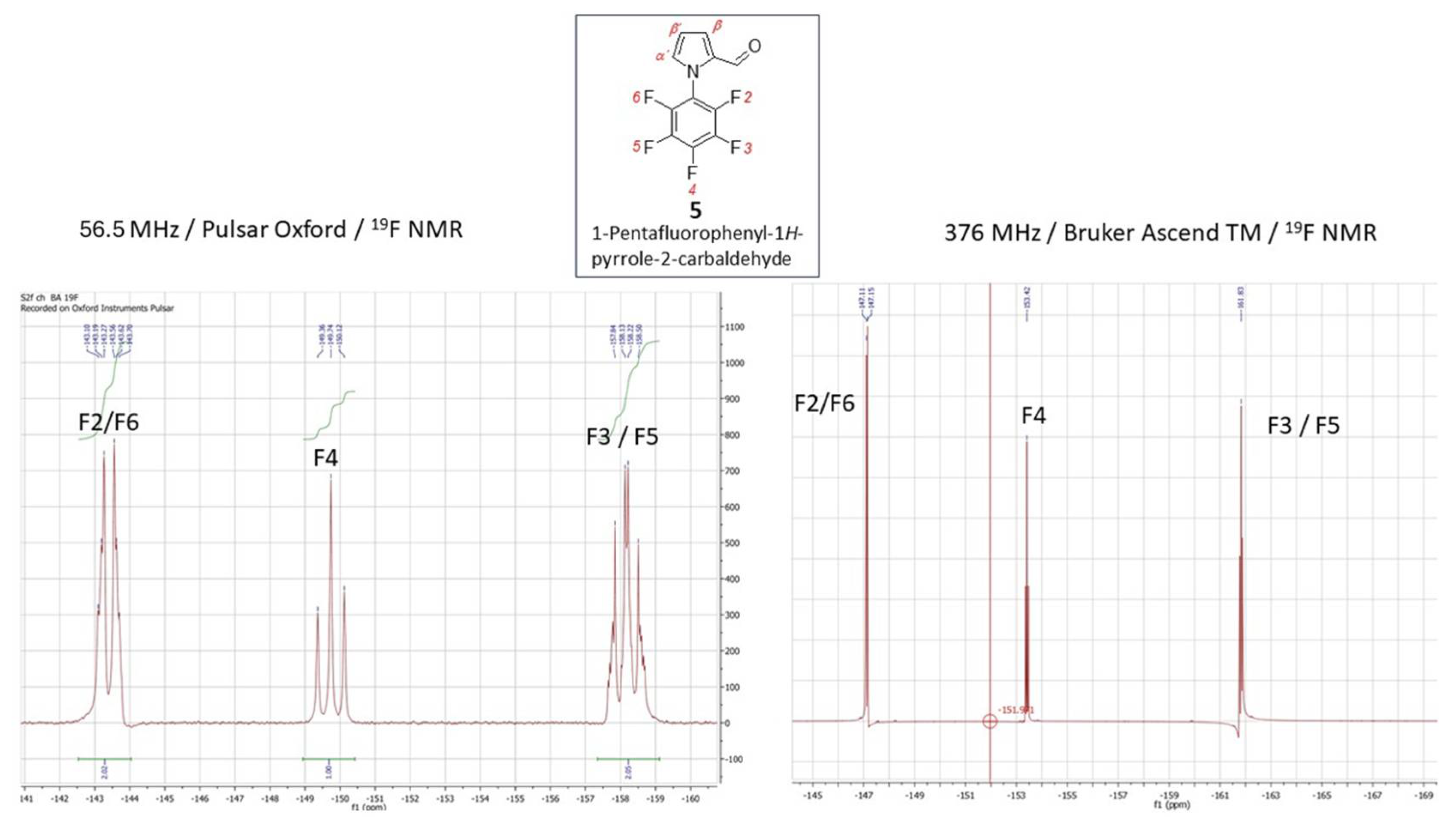
Figure 6.
1H NMR spectrum 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) obtained from a 60 MHz Pulsar Oxford benchtop NMR at 400 MHz (fleft) and from a 400 MHz Bruker Ascend TM (right).
Figure 6.
1H NMR spectrum 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) obtained from a 60 MHz Pulsar Oxford benchtop NMR at 400 MHz (fleft) and from a 400 MHz Bruker Ascend TM (right).
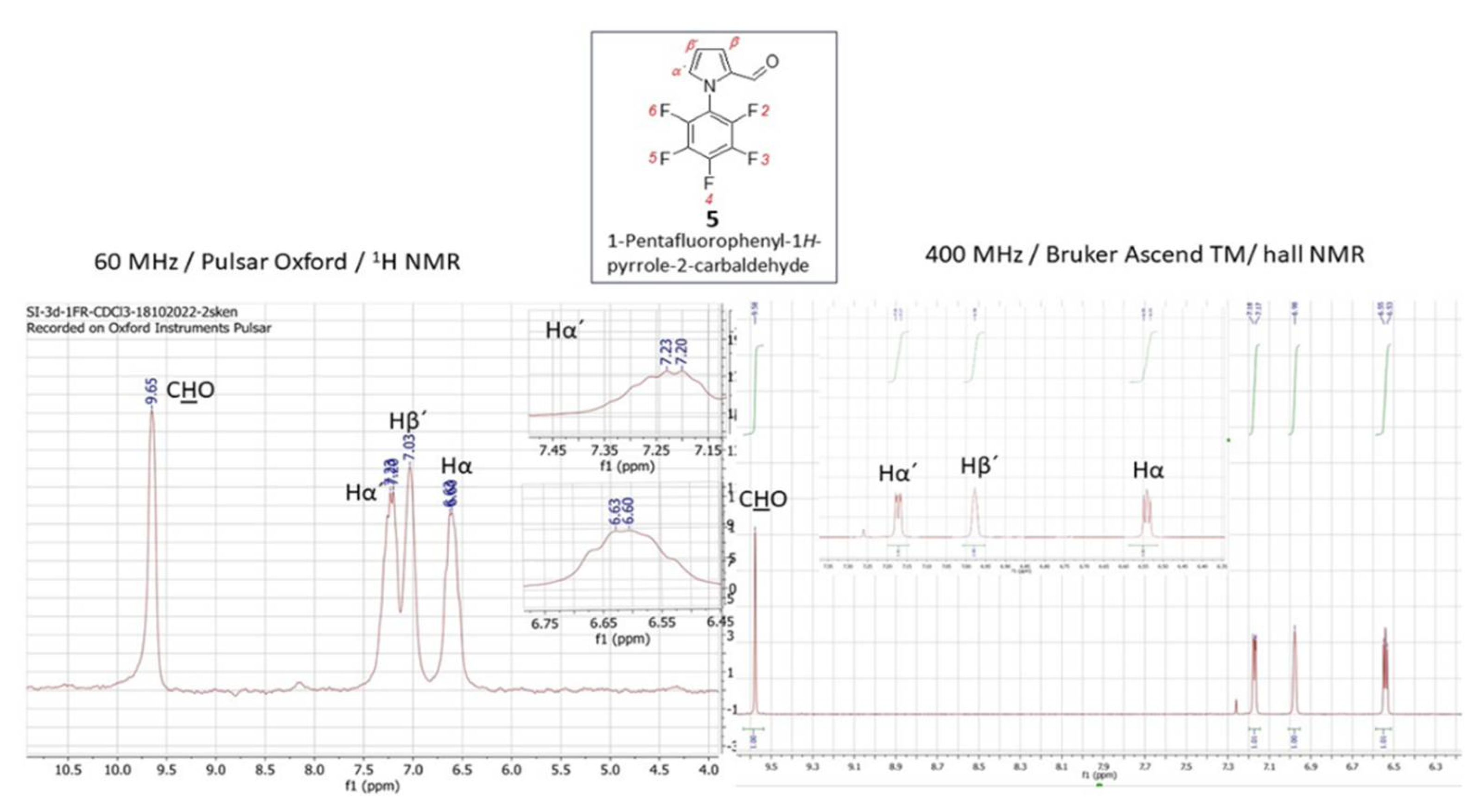
Figure 7.
19F NMR spectrum for a mixture of 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) / 1-pentafluorophenyl-1H-pyrrole-2,4-dicarbaldehyde obtained from a 60 MHz Pulsar Oxford benchtop NMR at 56.5 MHz (for 19F isotope, left) and from a 400 MHz Bruker Ascend TM at 376 MHz (for 19F isotope, right).
Figure 7.
19F NMR spectrum for a mixture of 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) / 1-pentafluorophenyl-1H-pyrrole-2,4-dicarbaldehyde obtained from a 60 MHz Pulsar Oxford benchtop NMR at 56.5 MHz (for 19F isotope, left) and from a 400 MHz Bruker Ascend TM at 376 MHz (for 19F isotope, right).

Figure 8.
1H NMR spectrum for a mixture of 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) / 1-pentafluorophenyl-1H-pyrrole-2,4-dicarbaldehyde obtained from a 60 MHz Pulsar Oxford benchtop NMR at 60 MHz and from a 400 MHz Bruker Ascend TM.
Figure 8.
1H NMR spectrum for a mixture of 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) / 1-pentafluorophenyl-1H-pyrrole-2,4-dicarbaldehyde obtained from a 60 MHz Pulsar Oxford benchtop NMR at 60 MHz and from a 400 MHz Bruker Ascend TM.
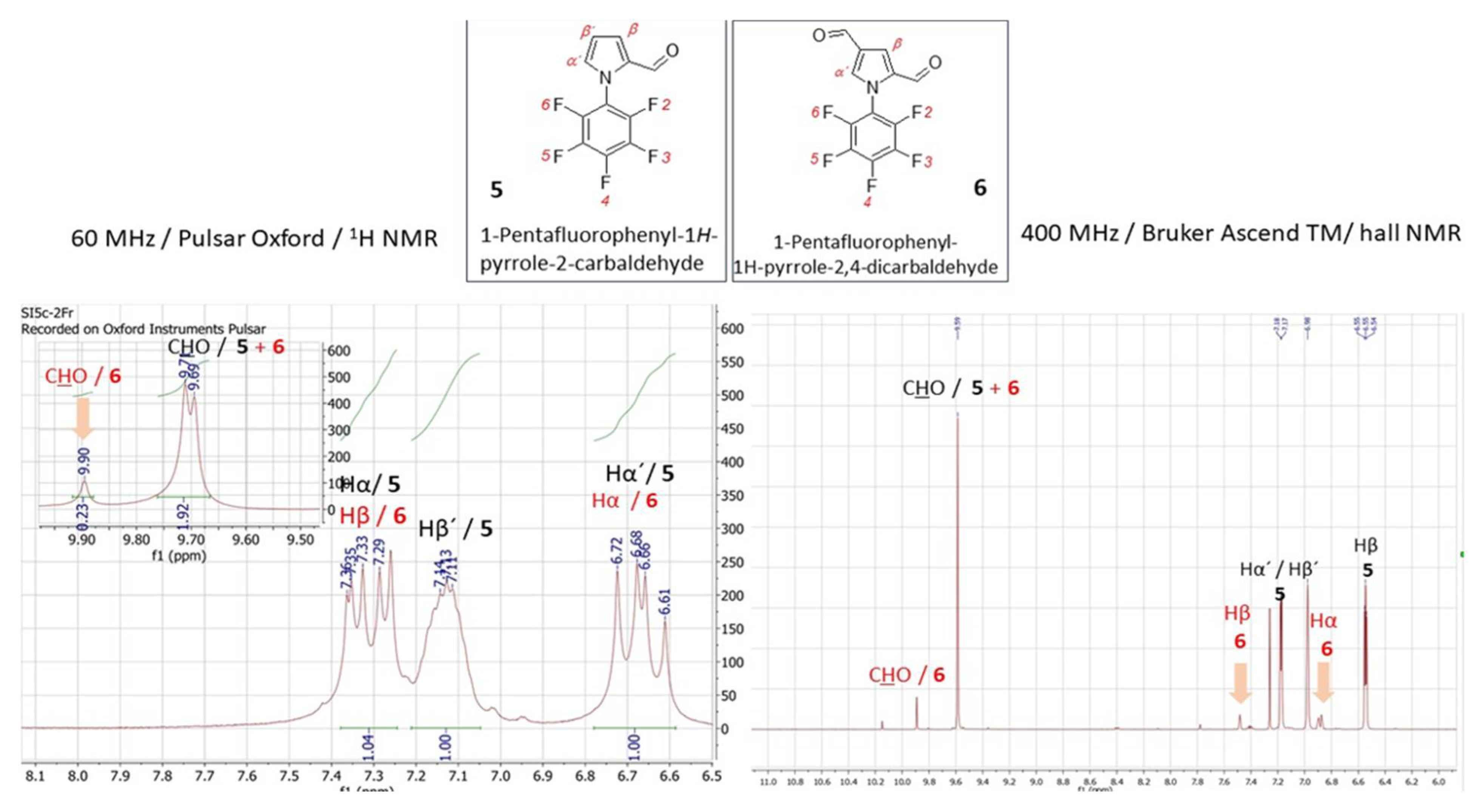
Figure 9.
19F NMR spectrum for a mixture of 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) / 1-pentafluorophenyl-2-vinyl-1H-pyrrole (7) obtained from a 60 MHz Pulsar Oxford benchtop NMR at 56.5 MHz (for 19F isotope, left) and from a 400 MHz Bruker Ascend TM at 376 MHz (for 19F isotope, right.
Figure 9.
19F NMR spectrum for a mixture of 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) / 1-pentafluorophenyl-2-vinyl-1H-pyrrole (7) obtained from a 60 MHz Pulsar Oxford benchtop NMR at 56.5 MHz (for 19F isotope, left) and from a 400 MHz Bruker Ascend TM at 376 MHz (for 19F isotope, right.
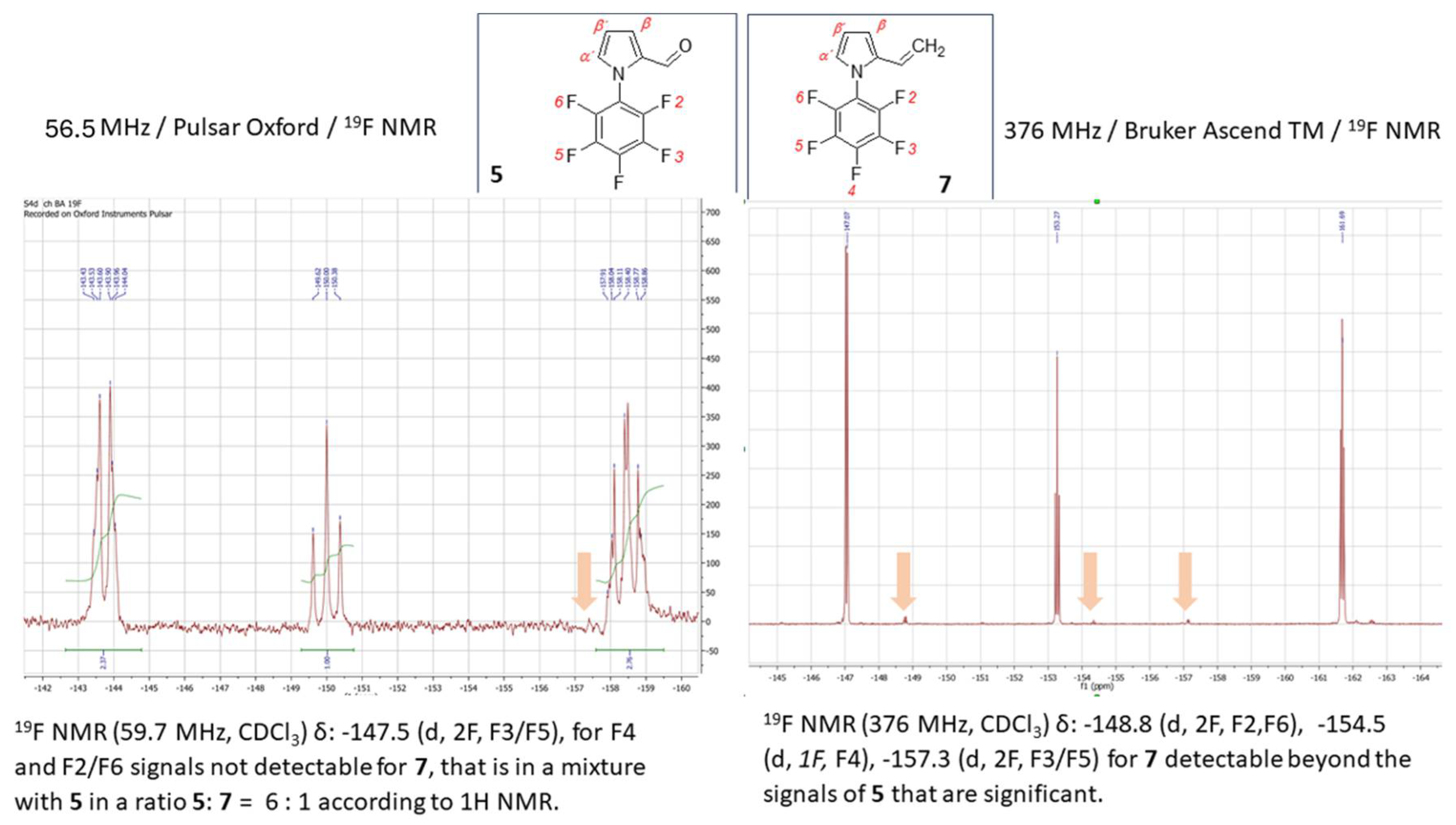
Figure 10.
1H NMR spectrum for a mixture of 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) / 1-pentafluorophenyl-2-vinyl-1H-pyrrole (7) obtained from a 60 MHz Pulsar Oxford benchtop NMR (left) and from a 400 MHz Bruker Ascend TM (right).
Figure 10.
1H NMR spectrum for a mixture of 1-pentafluorophenyl-1H-pyrrole-2-carbaldehyde (5) / 1-pentafluorophenyl-2-vinyl-1H-pyrrole (7) obtained from a 60 MHz Pulsar Oxford benchtop NMR (left) and from a 400 MHz Bruker Ascend TM (right).
Scheme 2.
Synthesis of thiophene-centered azomethine 8.
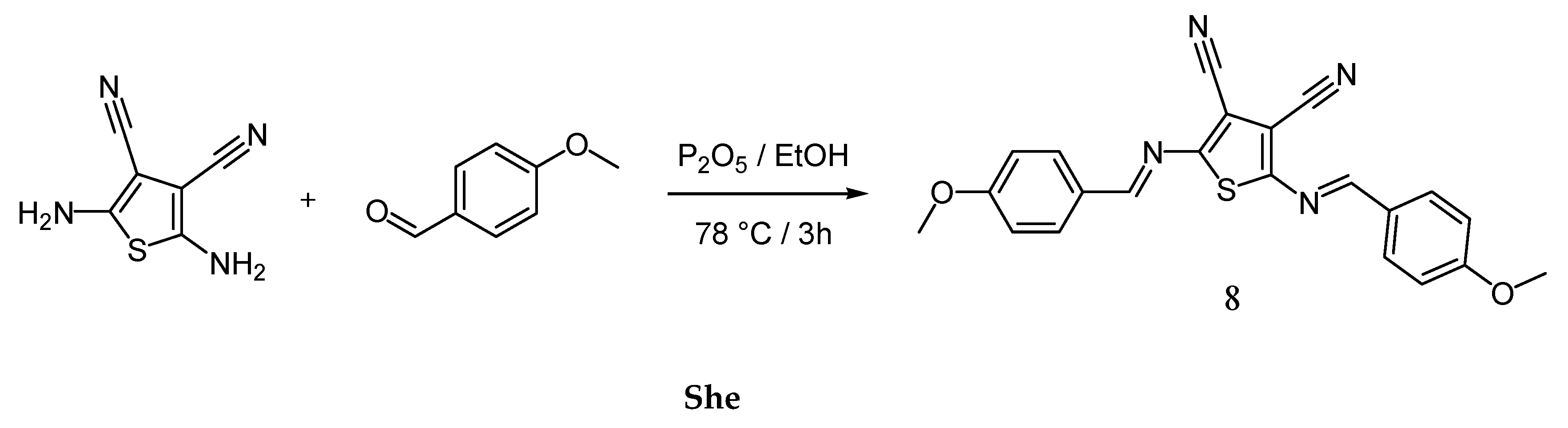
Figure 11.
Low-field 1H NMR and 2D 1H-1H (COSY) NMR for azomethine 8.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



