
Submitted:
18 May 2024
Posted:
21 May 2024
You are already at the latest version
Abstract
Atherosclerotic cardiovascular disease, a leading cause of morbidity and mortality worldwide, is a chronic inflammatory disease, linked to a state oxidative stress, with several well-identified culprits, of which low-density-lipoprotein cholesterol (LDL-C) and lipoprotein(a). No specific treatment is currently available to lower lipoprotein(a). It is, therefore, of paramount importance to identify nutritional factors that could lower lipoprotein(a). Growing evidence shows that although reducing saturated fatty acid (SFA) intake decreases LDL-C, it could increase lipoprotein(a). Optimal dietary recommendations may therefore differ depending on an in-dividual’s baseline lipoprotein(a) and LDL-C levels. Assessing the diet-induced true LDL-C response and net balance of these two atherogenic entities is difficult because LDL-C measurement is confounded by lipoprotein(a) cholesterol content. We have estimated, for the first time, the net atherogenic result of diet-induced variations of both LDL-C and lipoprotein(a), thanks to our new concept of LDLapoB risk equivalent=apoB+lp(a)x5 (in nmol/L). This is illustrated, via the rare case of a physician with very high lipoprotein(a) and hypobetalipoproteinemia. She experiences twice a considerable lipoprotein(a) increase (+32%, + 28mg/dL, + 67nmol/L) and higher LDLapoB risk equivalent (138mg/dL versus 123mg/dL) on a mediterranean diet, low in SFA. A reasonable SFA intake, despite causing marginal LDL-C increase, may be advisable in patients with high lipoprotein(a) who need personalized dietary recommendations.
Keywords:
oxidative stress
; LDL-cholesterol
; lipoprotein(a)
; diet
; saturated fat
; carbohydrate
; atherosclerotic cardiovascular disease
; risk assessment
; risk prevention
1. Introduction
Atherosclerotic cardiovascular disease (ASCVD), which is a leading cause of morbidity and mortality worldwide, is a chronic inflammatory disease of the vascular system. This is linked to a state of oxidative stress, with several well-identified culprits. [1] One of them is Oxidized LDL (low density lipoprotein) particles, which stimulate this inflammatory response within the vessel walls. Lowering LDL cholesterol (LDL-C) by medical therapy or diet plays an important role in reducing cardiovascular risk and preventing chronic disease. This has been well-studied and current nutritional guidelines from around the world recommend reducing saturated fatty acid (SFA) intake to reduce LDL-C. [2,3]
However, we have another offender. Lipoprotein(a) ( lp(a)) is an LDL particle to which a molecule of apo(a) has been disulfide bonded to apo B100, and an established independent risk factor for ASCVD. [4] Mendelian randomizations have estimated that lp(a) atherogenicity is approximately 6-fold higher than that of LDL on a per apolipoprotein B (apoB) particle basis. [5] Lp(a) is the preferential carrier of oxidized phospholipids in the plasma and major contributor to oxidative stress.[4]
Specific, effective Lp(a)-lowering therapies are in phase II/III clinical trials (NCT04023552, NCT05581303), but currently no specific treatment is available. It is, therefore, of paramount importance to identify lifestyle factors that could lower lp(a) levels. Unfortunately, for many decades, lp(a) has been considered a non-modifiable risk-factor because it is predominantly genetically determined. This has limited research in this area and high-level evidence is lacking. Despite this, interest on this topic is rapidly expanding and data is continuously showing that although reducing SFA intake decreases LDL-C levels, it may increase lp(a) levels [6,7,8,9,10,11]. The recent European Atherosclerosis Society guidelines state that low SFA, high carbohydrate diets increase lp(a) by 10-15%. [4] Optimal dietary recommendations may therefore differ depending on an individual’s baseline lp(a) and LDL-C levels. However, LDL-C measurement is confounded by lp(a) cholesterol content which presents significant interindividual heterogeneity. Therefore, assessing the true LDL-C response and net atherogenic balance of these two highly atherogenic entities which take opposite directions in response to SFA intake is very difficult. In the era of precision medicine, it is of utmost importance to identify which patients should benefit from personalized dietary recommendations.
For this purpose, we have estimated, for the first time, the net atherogenic result of diet-induced variations of both LDL-C and lp(a), thanks to our new concept of LDLapoB risk equivalent. This is illustrated, via the rare case of a physician with very high lp(a) and hypobetalipoproteinemia.
2. Materials and Methods
A 37-year-old female physician tested her lp(a) because of family history of premature coronary artery disease. No medication, no significant past medical history, unremarkable clinical presentation, body mass index 19.7kg/m². Overnight fasting laboratory results are shown in Table 1.
LDL-C was calculated using the Friedwald equation which includes the cholesterol contained in lp(a). Lp(a) was always measured in the same laboratory, at the same time in the menstrual cycle, in both nmol/L and mg/dL with the Turbidimétrie-Tina-quant Lp(a) Gen.2 COBAS Roche assay. There was no conversion from mg/dL to nmol/L because of the well-established isoform size heterogeneity. [4]
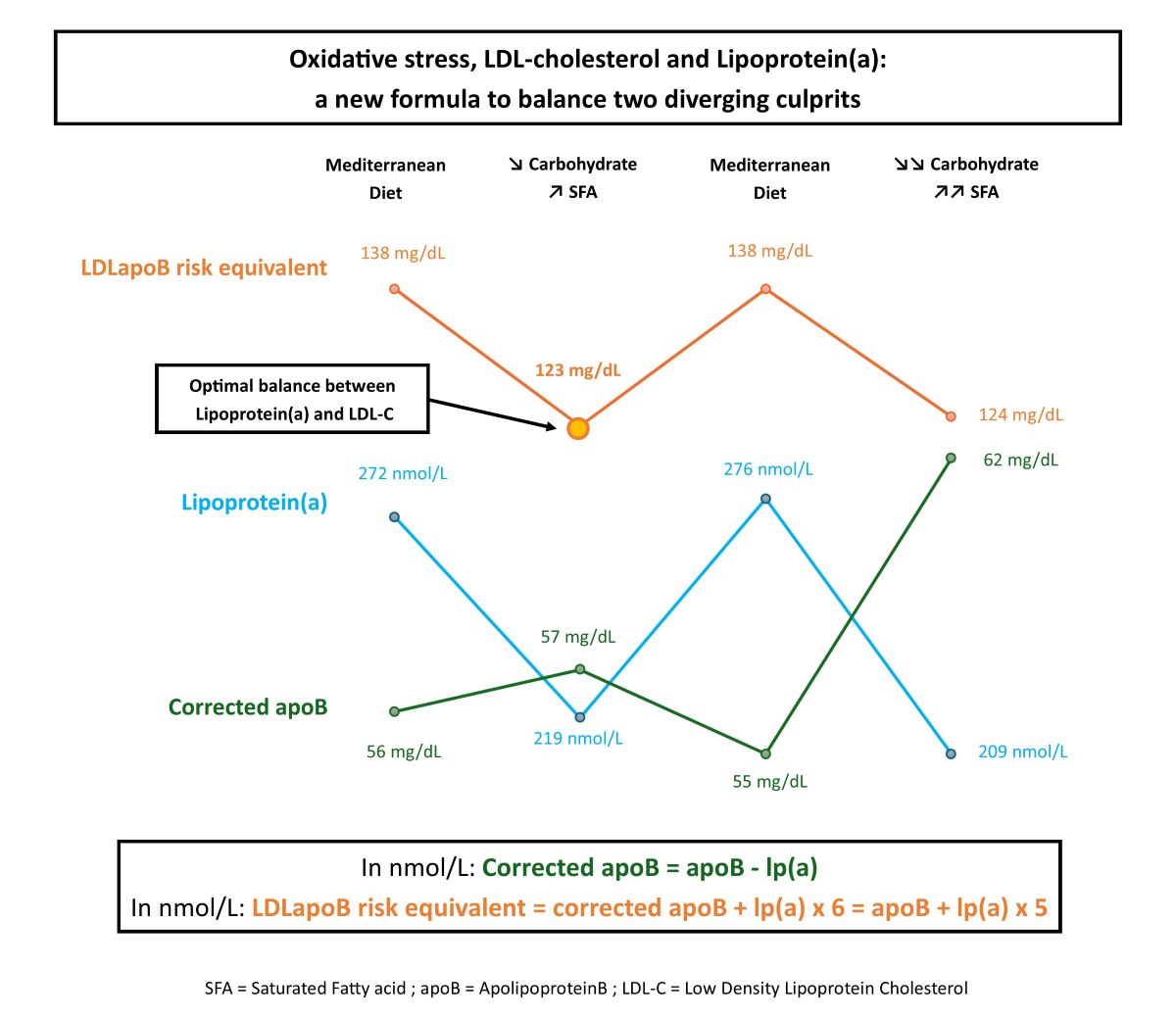
Corrected apoB (ApoB other than lp(a)) was calculated as follows:
In nmol/L: Corrected apoB=apoB-lp(a)
We assumed that, particularly in this case, with very low triglycerides, variations in corrected apoB broadly reflect variations in LDLapoB. Although this does not correspond exactly to the LDL-C response as the cholesterol content of LDLapoB particles can vary particularly with fat intake modulation, LDL-C and LDLapoB are highly correlated and with regards to risk equivalence LDLapoB is more accurate as apoB particle number has been shown to be a superior marker of cardiovascular risk than LDL-C. [12]
Mendelian randomizations estimate that lp(a) atherogenicity is approximately 6-fold higher than that of LDL on a per apoB particle basis [5] Therefore, LDLapoB risk equivalent was calculated as follows:
In nmol/L: LDLapoB risk equivalent=corrected apoB+lp(a)x 6=apoB+lp(a)x 5
Lp(a) was >95th percentile [4] and corrected apoB <5th percentile for age and sex [13]. The rest of the blood tests were unremarkable, including normal thyroid function, liver tests, renal function, fat soluble vitamins. High sensitivity CRP was undetectable. Computed tomographic angiogram and carotid ultrasound showed no atheroma. After consulting a lipid specialist, in view of her family’s lipid profiles, a polygenic origin of hypobetalipoproteinemia was suggested. Statins were not considered beneficial because of naturally low apoB. A heart healthy lifestyle and a mediterranean diet (MD) , low in SFA were recommended.
Having read the EAS guidelines which state that low SFA, high carbohydrate diets increased lp(a) by 10-15% [4], she tested this in 4 steps at 2-month intervals. Weight and physical activity remained unchanged, implying isocaloric diets. Protein and low carbohydrate fruit and vegetable intake did not vary significantly.
- Diet :1 when elevated lp(a) was discovered: rich in SFA and carbohydrate. Low in polyunsaturated fatty acid (PUFA), monounsaturated fatty acid (MUFA).
- Diet 2: MD as recommended (rich in whole grain complex carbohydrates, PUFA and MUFA from extra virgin olive oil and nuts, oily fish, poor in SFA).
- Diet 3: A quarter of the carbohydrate was replaced with SFA. MUFA, PUFA unchanged.
- Diet 4: MD same as diet 2.
- Diet 5: Half of the carbohydrate replaced with SFA. MUFA, PUFA unchanged.
3. Results
On a MD, lp(a) was 32% higher (+ 28mg/dL, + 67nmol/L) than when SFA replaced some carbohydrate. Lp(a) appeared to increase specifically with the carbohydrate/SFA ratio. MUFA and PUFA seemed neutral. LDLapoB risk equivalent was significantly higher (138mg/dL versus 123mg/dL) on a MD, despite similar apoB levels, due to a much higher proportion of lp(a) in total apoB (20% versus 14.5%). The least atherogenic diet, as assessed by LDLapoB risk equivalent, was diet 3: a MD with a moderate increase in the proportion of SFA. On diet 5, corresponding to the highest SFA intake, the risk increase induced by corrected apoB increase (probably associated with LDL-C increase) exceeded the benefit induced by further lp(a) lowering.
4. Discussion
This is a striking example of an individual who may not benefit from current nutritional guidelines for ASCVD prevention. The net benefit seems to be in favor of lp(a) lowering, via preservation of a reasonable SFA intake, despite marginal LDL-C increase. Our results are in line with expanding evidence, showing lp(a) increase with SFA reduction, and specifically with carbohydrate replacement [6,7,8,9,10,11]. Furthermore, in response to SFA increase: high density lipoprotein-cholesterol (HDL-C) increased as expected, and LDL particle size probably increased making them less atherogenic, as suggested by the decrease in triglycerides /HDL-C ratio [14]. MUFA and PUFA seem neutral, which is in agreement with the OMNI trial showing that unsaturated fat is the best strategy for SFA replacement in patients with high lp(a) [11]. The PREDIMED trial also showed that lp(a) increase was greater in the low total fat diet (implying a higher carbohydrate intake) than on the MDs rich in unsaturated fat [15]. Interestingly, despite higher LDL-C and apoB, diet 5 is less atherogenic than the MDs, due to a much lower proportion of lp(a) in total apoB. In patients with high lp(a), the best option might be to recommend a MD maintaining the antioxidants (carotenoids, polyphenols) and healthy unsaturated fatty acids, but to suggest a reasonable increase in the proportion of SFA from dairy products which have been shown not to increase cardiovascular risk [16].
For most of the population with normal lp(a) levels, 30% variation has no clinical relevance. However, the higher the baseline lp(a), the greater the baseline risk and the larger the potential risk increase. Although lp(a) is causally related to ASCVD, uncertainty regarding risk reduction caused by lp(a) reduction remains. The post-hoc analysis of the FOURIER trial (with the limitations inherent to all post-hoc analyses), showed that with evolocumab, in secondary prevention, over 2.2 years, relative risk reduction (RRR) was 15% per 25nmol/L lp(a) decrease. RRR would be 40% for 67nmol/L here [17].
Unexplained lp(a) temporal variations have been reported [18] and random variation cannot be totally excluded in our case. However, similar increase and decrease twice with the same intervention over a short period of time, makes this very unlikely, especially as our results coincide with many other papers. In fact, these unexplained temporal variations could be due to changes in diet which were shown to modify lp(a) levels in only two weeks in a personal experiment by another physician. Under more drastic conditions than ours, he showed even greater absolute and relative changes in lp(a) levels with carbohydrate/ fat intake modulation. Interestingly, the diet-induced lp(a) variation persisted under statins [19].
We suggest that lp(a) be measured a second time in patients with high lp(a) who have adopted a MD or reduced SFA. Specific, effective Lp(a)-lowering therapies are in phase II/III clinical trials (NCT04023552, NCT05581303). If lp(a) increases beyond the inclusion cut-off, patients could become eligible. Additionally, in many countries, social security reimbursement eligibility criteria replicate clinical trial inclusion criteria. Becoming eligible for potentially highly effective treatments could have a considerable impact on an individual’s prognosis.
Beyond the nutritional scope, our formula can be used by clinicians in many other circumstances. It can gauge the residual risk in patients with high lp(a) and low LDL-C (often thanks to lipid lowering therapy). On diet 4, apoB 69 mg/dL seems reassuring. However, the LDLapoB risk equivalent of 138 mg/dL conveys a totally different picture. It can also be used to assess the net benefit of statin therapy, which can increase lp(a) despite lowering LDL-C, by comparing LDLapoB risk equivalent before and after treatment. [20].
We hope our paper will stimulate further research. Although evidence is increasing, more randomized controlled trials need to confirm that lp(a) increases with the carbohydrate/ SFA ratio. Patients with very high lp(a) need to be included in these trials to record large absolute variations, which could be clinically meaningful (as in our case). However, in view of current guidelines, testing the effect of increased SFA intake in patients with very high lp(a), could be prohibitive due to ethical concerns. Nevertheless, as the lp(a) reduction in response to SFA increase appears to persist under statin therapy, the diet-induced LDL-C increase could be counterbalanced by medical therapy. [19]. Nonetheless, foods are complex with possible pleiotropic effects on sometimes unknown risk factors. The final step would be randomized clinical trials analyzing the effect of diet-induced lp(a) reduction on ASCVD outcomes.
5. Conclusions
In the era of precision medicine, it is of utmost importance to identify which patients should benefit from personalized dietary recommendations, particularly in situations like high lp(a) where no specific treatment is available. We have estimated, for the first time, the net atherogenic result of diet-induced variations of both LDL-C and lp(a), thanks to our new concept of LDLapoB risk equivalent. In individuals with high lp(a), maintaining a reasonable SFA intake may be advisable.
Author Contributions
Conceptualization, M.B.R.; methodology, M.B.R. and B.V.T.; validation, M.B.R. and B.V.T.; formal analysis, M.B.R. and B.V.T.; resources, M.B.R. and B.V.T.; writing—original draft preparation, M.B.R. and B.V.T.; writing—review and editing, M.B.R. and B.V.T. Both authors have read and agreed to the published version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
This case report illustrating our formula was specifically discussed with the physician and informed consent to publish was obtained.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Batty M, Bennett MR, Yu E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022;11:3843. [CrossRef]
- Fogacci F, Borghi C, Cicero AFG. Diets, Foods and Food Components’ Effect on Dyslipidemia. Nutrients 2021;13:741. [CrossRef]
- Fernandez ML, Raheem D, Ramos F, Carrascosa C, Saraiva A, Raposo A. Highlights of Current Dietary Guidelines in Five Continents. Int J Environ Res Public Health 2021;18:2814. [CrossRef]
- Kronenberg F, Mora S, Stroes ESG, Ference BA, Arsenault BJ, Berglund L, et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J 2022;43:3925–46. [CrossRef]
- Björnson E, Adiels M, Taskinen M-R, Burgess S, Chapman MJ, Packard CJ, et al. Lipoprotein(a) Is Markedly More Atherogenic Than LDL: An Apolipoprotein B-Based Genetic Analysis. J Am Coll Cardiol 2024;83:385–95. [CrossRef]
- Enkhmaa B, Petersen KS, Kris-Etherton PM, Berglund L. Diet and Lp(a): Does Dietary Change Modify Residual Cardiovascular Risk Conferred by Lp(a)? Nutrients 2020;12:2024. [CrossRef]
- Fogacci F, Di Micoli V, Sabouret P, Giovannini M, Cicero AFG. Lifestyle and Lipoprotein(a) Levels: Does a Specific Counseling Make Sense? J Clin Med 2024;13:751. [CrossRef]
- Law HG, Meyers FJ, Berglund L, Enkhmaa B. Lipoprotein(a) and diet-a challenge for a role of saturated fat in cardiovascular disease risk reduction? Am J Clin Nutr 2023;118:23–6. [CrossRef]
- Enkhmaa B, Berglund L. Non-genetic influences on lipoprotein(a) concentrations. Atherosclerosis 2022;349:53–62. [CrossRef]
- Law HG, Khan MA, Zhang W, Bang H, Rood J, Most M, et al. Reducing saturated fat intake lowers LDL-C but increases Lp(a) levels in African Americans: the GET-READI feeding trial. J Lipid Res 2023;64:100420. [CrossRef]
- Haring B, von Ballmoos MCW, Appel LJ, Sacks FM. Healthy dietary interventions and lipoprotein (a) plasma levels: results from the Omni Heart Trial. PloS One 2014;9:e114859. [CrossRef]
- Glavinovic T, Thanassoulis G, de Graaf J, Couture P, Hegele RA, Sniderman AD. Physiological Bases for the Superiority of Apolipoprotein B Over Low-Density Lipoprotein Cholesterol and Non-High-Density Lipoprotein Cholesterol as a Marker of Cardiovascular Risk. J Am Heart Assoc. 2022;11(20):e025858. [CrossRef]
- Jungner I, Marcovina SM, Walldius G, Holme I, Kolar W, Steiner E. Apolipoprotein B and A-I values in 147576 Swedish males and females, standardized according to the World Health Organization-International Federation of Clinical Chemistry First International Reference Materials. Clin Chem 1998;44:1641–9.
- Yokoyama K, Tani S, Matsuo R, Matsumoto N. Increased triglyceride/high-density lipoprotein cholesterol ratio may be associated with reduction in the low-density lipoprotein particle size: assessment of atherosclerotic cardiovascular disease risk. Heart Vessels 2019;34:227–36. [CrossRef]
- Fitó M, Estruch R, Salas-Salvadó J, Martínez-Gonzalez MA, Arós F, Vila J, et al. Effect of the Mediterranean diet on heart failure biomarkers: a randomized sample from the PREDIMED trial. Eur J Heart Fail 2014;16:543–50. [CrossRef]
- Yuan M, Singer MR, Pickering RT, Moore LL. Saturated fat from dairy sources is associated with lower cardiometabolic risk in the Framingham Offspring Study. Am J Clin Nutr 2022;116:1682–92. [CrossRef]
- O’Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I, et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019;139:1483–92. [CrossRef]
- Marcovina SM, Viney NJ, Hughes SG, Xia S, Witztum JL, Tsimikas S. Temporal variability in lipoprotein(a) levels in patients enrolled in the placebo arms of IONIS-APO(a)Rx and IONIS-APO(a)-LRx antisense oligonucleotide clinical trials. J Clin Lipidol 2018;12:122-129.e2. [CrossRef]
- Scholl, JG. Does a ketogenic diet lower a very high Lp(a)? A striking experiment in a male physician. BMJ Nutr Prev Health 2020;3:413–5. [CrossRef]
- Tsimikas S, Gordts PLSM, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J. 2020;41(24):2275-2284. [CrossRef]

Table 1.
Diet-induced changes in fasting lipid panels.
| Diet 1 | Diet 2 MD |
Diet 3 | Diet 4 MD |
Diet 5 | |
|---|---|---|---|---|---|
| Total cholesterol | 156 mg/dL | 149 mg/dL | 156 mg/dL | 153 mg/dL | 179 mg/dL |
| HDL cholesterol | 63 mg/dL | 55 mg/dL | 67 mg/dL | 57 mg/dL | 74 mg/dL |
| Triglycerides | 44 mg/dL | 37 mg/dL | 39 mg/dL | 41 mg/dL | 42 mg/dL |
| LDL cholesterol | 84 mg/dL | 87 mg/dL | 81 mg/dL | 88 mg/dL | 97 mg/dL |
| Lp(a) | 100 mg/dL 239 nmol/L |
113 mg/dL 272 nmol/L |
91 mg/dL 219 nmol/L |
115 mg/dL 276 nmol/L |
87 mg/dL 209 nmol/L |
| Apolipoprotein B | / | 70 mg/dL 1400 nmol/L |
68 mg/dL 1359 nmol/L |
69 mg/dL 1380 nmol/L |
72 mg/dL 1440 nmol/L |
| Corrected apoB | / | 56 mg/dL 1128 nmol/L |
57 mg/dL 1140 nmol/L |
55 mg/dL 1104 nmol/L |
62 mg/dL 1231 nmol/L |
| LDLapoB risk equivalent | / |
138 mg/dL 2760 nmol/L |
123 mg/dL 2454 nmol/L |
138 mg/dL 2760 nmol/L |
124 mg/dL 2485 nmol/L |
| lp(a)/ apoB ratio | / | 19.4 % | 16.1 % | 20 % | 14.5 % |
| TG/HDL-C ratio | 0.70 | 0.67 | 0.58 | 0.74 | 0.57 |
| Hemoglobin A1c | 5.3 % | 5.2 % | 5.2 % | 5.3 % | 5.2 % |
* Abbreviations: HDL-C=high-density lipoprotein cholesterol, LDL-C=low-density lipoprotein cholesterol, apoB=apolipoprotein B, TG=triglycerides, MD= mediterranean diet.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



