Submitted:
20 May 2024
Posted:
21 May 2024
You are already at the latest version
Abstract
Abstract Incretin mimetics, also known as glucagon-like peptide-1 (GLP-1) receptor agonists, are a class of medications used to treat type 2 diabetes by mimicking the actions of incretin hormones in the body. These hormones are responsible for promoting insulin release from the pancreas in response to nutrient intake, as well as decreasing glucagon secretion, slowing gastric emptying, and promoting satiety. By imitating these actions, incretin mimetics help to regulate blood sugar levels in individuals with diabetes and glucose intolerance. One of the key benefits of incretin mimetics is its ability to lower blood sugar levels without causing hypoglycemia. This is especially important for individuals with diabetes who may experience dangerous drops in blood sugar levels when using other medications. Additionally, incretin mimetics have been shown to promote weight loss in some individuals, making them a valuable option for those struggling with obesity in addition to diabetes. Incretin mimetics are administered by injection, once or twice daily, depending on the specific medication. They are often combined with other diabetes medications, such as metformin or insulin, for optimal blood sugar control. These medications are generally well-tolerated, with common side effects including nausea, vomiting, and diarrhea, which usually diminish over time as the body adjusts to the medication. Studies have shown that incretin mimetics can help improve cardiovascular outcomes in individuals with diabetes, including reducing the risk of heart attacks and strokes. This is particularly significant given the increased risk of cardiovascular disease in individuals with diabetes. Incretin mimetics also have been shown to preserve pancreatic beta-cell function, which is responsible for producing insulin. This can help slow the progression of diabetes and reduce the need for higher insulin doses over time. Despite their many benefits, incretin mimetics are not without limitations. They can be expensive compared to other diabetes medications, which may make them less accessible for some individuals. Additionally, there have been concerns about the potential risk of pancreatitis and pancreatic cancer associated with the use of these medications. However, more research is needed to fully understand these risks. It is important for healthcare providers to carefully assess each individual's unique needs and medical history when considering the use of incretin mimetics. They may not be appropriate for everyone, particularly those with a history of pancreatitis or pancreatic cancer. Patients should also be educated about these medications' proper administration and potential side effects to ensure their safety and efficacy. Overall, incretin mimetics have emerged as an important therapeutic option for individuals with type 2 diabetes, offering a unique mechanism of action that can help improve blood sugar control, promote weight loss, and reduce cardiovascular risk. With ongoing research and development in this field, incretin mimetics continue to show promise as valuable tools in managing diabetes and its associated complications and comorbidities.
Keywords:
Obesity
; Diabetes
; GLP-1
; Incretins
; mimetics
; adverse reactions
Introduction
Diabetes Mellitus type 2 (T2DM) is a metabolic disease where pancreatic beta cells cannot sustain increased demand for insulin; thus, deficiency results progressively. The common feature in Type 2 Diabetes is relative insulin deficiency, so there is an insufficient reduction in the resistance to insulin action. Management of diabetes is aimed at improving hyperglycemia symptoms and minimizing the long-term microvascular and macrovascular complications such as diabetic nephropathy, neuropathy, retinopathy, and cardiovascular effects. Compared to the general population, individuals with type 2 diabetes have a significantly greater risk of both fatal and nonfatal cardiovascular events. The complications often contribute to low quality of life among patients and increased rate of mortality due to various factors [1] but mainly due to cardiovascular events [2]. The treatment modalities range from dietary and lifestyle management to oral antidiabetics and injectable therapies. The risk of macro-vascular problems is increased by the substantial correlation observed between the severity of hyperglycemia, metabolic alterations resulting from type 2 diabetes, and vascular damage. The discovery of medications that regulate hyperglycemia and affect other metabolic risk variables to enhance cardiovascular outcomes is in high demand. Obesity, aging, and genetic vulnerability are factors in the rising prevalence of Type 2 diabetes mellitus. Insulin resistance, the functional failure of pancreatic beta cells, and excessive or incorrect glucagon output are the leading contributors to the development of type 2 diabetes. Currently, T2DM is considered a systemic illness in which hyperglycemia is caused by a malfunction in several organs and tissues [3]. Diabetes is a chronic condition that impacts the patient's day-to-day activities and thus requires a sustained change in lifestyle activities, including dietary choices. Educating the patient is critical to achieving and maintaining a healthy lifestyle and managing the disease. Each patient's management should be individualized, considering comorbidities and personal and cultural beliefs [4]. Diabetes type 2 is a long-term comorbidity brought on by decreased insulin production and actions due to peripheral tissue insulin resistance, mainly in skeletal muscle and adipose tissue. Combined with β-cell malfunction in addition to causing microangiopathy, homeostatic hyperglycemia increases the risk of microangiopathy, leading to cardiovascular diseases. Economic disadvantages, a decline in healthy life expectancy, and a decline in patient quality of life are all caused by complications from these vascular conditions. Furthermore, obese patients with type 2 diabetes need more rigorous glycemic control and weight management because obesity is linked to the pathophysiology of the disease and is a risk factor for macroangiopathy [5,6]. Medication and lifestyle modifications like diet and exercise can help with treatment. However, for people with chronic diabetes, proper medication is crucial.
Obesity is a major factor that contributes to the development and complications of diabetes mellitus. Patients who are obese are at a higher risk of developing type 2 diabetes due to the increased resistance to insulin. Obesity can cause dysfunction in the production and utilization of insulin, leading to elevated blood glucose levels. Furthermore, obesity can exacerbate the complications of diabetes, such as cardiovascular disease, kidney failure, and neuropathy. These complications can significantly impact the quality of life of patients and increase the risk of mortality. Moreover, obesity complicates the management of diabetes as it can make it more difficult to control blood sugar levels through diet and medication alone. Patients who are obese may require higher doses of insulin or other medications to achieve optimal glycemic control. Additionally, obesity can also lead to other comorbidities, such as hypertension and dyslipidemia, which further increase the risk of cardiovascular events in patients with diabetes (Figure 1). Therefore, effective strategies for managing obesity in diabetic patients are essential to prevent and mitigate the complications associated with this chronic condition [7,8].
The majority of Type 2 diabetes treatment drugs that target various pathways have been linked to adverse effects, including edema, obesity, hypoglycemia, and diminished insulin sensitivity. However, recently, treatments that target incretin hormones have come to light as potentially safe options for the management of type 2 diabetes. The incretin pathway regulates the human body's daily blood glucose level. Most of the total insulin secreted by β-cells in response to oral glucose consumption is accounted for by it [9]. Together with lifestyle changes and lowering cardiovascular risk, metformin is still the drug of choice for type 2 diabetes and is advised among those agents for first-line treatment. The American Diabetes Association's guidance encourages the adoption of a patient-centered strategy to direct further therapy, considering factors like cost, side effects, weight fluctuations, hypoglycemia risk, and efficacy with additional medication. Add-on therapy for metformin includes thiazolidinediones, dipeptidyl peptidase-4 inhibitors, insulin, sodium-glucose co-transporter two inhibitors, glucagon-like peptide-1 receptor agonists, luminal glucosidase inhibitors, diet, and sulfonylureas [6,10,11].
Incretin Hormones
Incretin hormones include glucagon-like peptide 1 (GLP-1) and Gastric inhibitory peptide, also known as a glucose-dependent insulinotropic polypeptide (GIP), facilitating insulin hormone release from pancreatic beta cells [12]. The dipeptidyl peptidase-4 enzyme rapidly breaks down the incretin hormones. The incretin effect entails the increased insulin production response when a glucose stimulus is given orally rather than intravenously. The incretin effect is markedly diminished in patients with diabetes [13]. This has led to the development of two major incretin-based therapeutic approaches. This is caused by a decreased GIP capacity to induce insulin release. This may be linked to a general dysfunction of beta cells or particular abnormalities in the GIP signaling pathway. Deterioration of glycemic regulation may be related to a decreased incretin action, which affects post-prandial glycemic excursions. On the other hand, GLP-1's insulinotropic efficacy is significantly less compromised. This indicates that exogenous GLP-1 can increase insulin secretion, decrease glucagon secretion, and lower plasma glucose levels throughout fasting and post-prandial periods (Figure 2). This has prompted the creation of drugs that lower blood sugar based on incretin actions [14].
The incretin hormones released from the gastrointestinal tract's epithelium significantly influence normal glucose tolerance. They prevent an excessive rise in post-prandial glucose levels by promoting insulin secretion in a glucose-dependent manner. The incretin action is dose-dependent, meaning that even as the meal's carbohydrate content increases, similar post-prandial glucose elevations are generated [15]. The insulin responses of oral and intravenous glucose treatments that produce comparable glucose excursions are typically compared to quantify the incretin effect. The incretin effect may be accountable for up to five times spikes in post-prandial glucose clearance, as indicated by contrasts of the amounts of glucose that must be infused intravenously to resemble the concentrations induced by the oral administration [13].
The oral glucose delivery causes the two gut incretin hormones, GIP and GLP-1, to be released more. This, in turn, enhances insulin secretion, leading to an increase in glucose disposal. This explains why the incretin effect relies on the quantity of carbohydrates consumed [15]. The proportion of carbohydrates consumed affects the incretin secretion and the insulin response. The incretin effect accounts for up to 70% of the insulin released in response to glucose intake in normal healthy individuals. Endocrine L-cells, primarily found in the mucosa of the distal section of the small intestine and colon, metabolize proglucagon to produce glucagon-like peptide-1, a 30-amino acid polypeptide. On the other hand, glucose-dependent insulinotropic peptide is produced from the endocrine K-cells located in the mucosa of the duodenum and upper jejunum. It is a 42-amino acid polypeptide. Glucagon-like peptide is rapidly broken down by the ubiquitous enzyme dipeptidyl peptidase 4, with a brief half-life of about 1.5 minutes. In contrast, glucose-dependent insulinotropic peptide typically has a longer half-life of approximately 7 minutes. The hormones have no risk of causing hypoglycemia despite enhancing insulin secretion from the beginning of a meal intake [16] because these hormones have no insulinotropic activity at lower glucose concentrations. Insulin gene expression and synthesis are both improved by glucagon-like peptide-1. Moreover, it has trophic and protective properties on beta cells and, in a glucose-dependent way, potently suppresses pancreatic glucagon release. Glucose-dependent insulinotropic peptide, on the other hand, has been demonstrated to increase glucagon secretion. The pancreatic beta-cells G-protein coupled receptors allow the hormones to exert their insulinotropic action [14].
These two incretin hormones have various other roles besides influencing the endocrine pancreas. Several brain regions have glucagon-like peptide-1 receptors. These receptors are thought to stimulate the perception of satiety, especially when combined with the glucagon-like peptide-1-induced slowing of gastrointestinal motility mediated by the vagus nerve, decrease food consumption, and control body weight. The glucagon-like peptide-1 hormone is responsible for the delay in gastric emptying, which helps reduce post-meal glucose elevations. On the other hand, GIP has no role in gastric emptying delay. Therefore, the effect of delayed gastric emptying is only noted with the GLP-1 analogs. However, a reduction in delayed gastric emptying to the successive doses of the glucagon-like peptide-1 mimetics may be emphasized [16].
The 463 amino acid GLP-1 receptor has eight hydrophobic domains. The N-terminal extracellular hydrophobic domain homologs expressed in several tissues and organs, including the heart, kidney, gut, pancreatic islets, stomach, and lung, are highly retained. After GLP-1R is activated, intracellular calcium and cyclic adenosine monophosphate (AMP) levels rise quickly, and subsequent insulin release is glucose-dependent. With a half-life of only 1-2 minutes, the enzyme dipeptidyl peptidase-4 (DPP-4) quickly inactivates the GLP-1 hormone. Amino acid changes at specific locations in the C-terminus and in the N-terminus also play a direct role in receptor engagement and resistance to DPP-4 inhibition, extending the half-life and impact of the receptor [2]. Only a small percentage of the various forms of GLP-1 in circulation are physiologically active. GLP-1 amide, a significant secretory product, is the active form of GLP-1. After entering the bloodstream, GLP-1 amide has a half-life of less than two minutes because the DPP-4 enzyme quickly cleaves it between positions 8 and 9 to produce the N-terminally truncated metabolite GLP-1 amide, which is physiologically inert and does not bind to the GLP-1 receptor. When delivered therapeutically, metabolites may have advantageous glucose-regulatory and cardio-protective effects, such as lowering oxidative stress in vascular tissues, protecting beta cells, and preventing gluconeogenesis and oxidative stress in hepatocytes. Vasodilation and cardiomyocyte viability are directly impacted by GLP-1 amide and its metabolites, which also improve cardiac function. For instance, while GLP-1[9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36] amide acts through a GLP-1R-independent mechanism, GLP-1[7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36] amide acts through a GLP-1R-dependent pathway to affect the cardiovascular system. Since targeting GLP-1R activation or GLP-1 degradation may have various cardiovascular implications, these metabolites have a wide range of therapeutic potential [13].
The heart contains glucagon-like peptide-1 receptors, and most research indicates that glucagon-like peptide-1 protects the myocardium. Moreover, it has been discovered that GLP-1 lowers human concentrations of free fatty acids and minimizes the post-prandial rise in triglycerides. It also has diuretic and natriuretic effects by modifying renal Na+/H+ exchange, which helps lower blood pressure [14]. Other roles of glucagon-like peptide-1 include the facilitation of increased glucose uptake within the muscles, decreased rate of glucose production from the liver, and neuroprotection. Patients diagnosed with type 2 diabetes have a significantly diminished incretin effect. The characteristic in concern is likely the main cause of these patients' incapacity to release enough insulin to prevent hyperglycemia after oral glucose [2]. The diminished incretin impact in patients with type 2 diabetes appears to be caused by diminished post-prandial production, decreased insulinotropic potency, and diminished insulinotropic effect of glucose-dependent insulinotropic peptide.
Glucagon-like Peptide-1 Receptor Agonists (GLP-1RAs)
Incretin Mimetics are hormone-like agents that can be used in addition to metformin and or sulfonylurea drugs in the management of Type 2 diabetes. In some cases, the GLP-1 analogs have been used to manage obesity. These agents act like incretin hormones such as glucagon-like peptide-1. They stimulate the pancreas to produce more insulin by binding to the GLP-1 receptors. These drugs include liraglutide, albiglutide, lixisenatide, exenatide, semaglutide, and dulaglutide. Although they can be taken in place of or in addition to insulin, incretin mimetics are not meant to be used in place of anti-diabetic medications. GLP-1 receptor agonists should be utilized selectively following metformin failure in patients with known atherosclerotic cardiovascular disease or in individuals without established cardiovascular disease but with high-risk signs, according to the current American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) consensus protocol. A pre-filled pen is used to administer these medications beneath the skin due to poor oral bioavailability [15].
The amount that GLP-1 receptor agonists reduce plasma glucose in fasting and post-prandial states varies according to the formulation. Long-acting GLP-1 agonists such as dulaglutide, Liraglutide, albiglutide, and exenatide extended-release lower blood glucose by increasing insulin secretion and decreasing glucagon. Short-acting GLP-1 mimetics such as exenatide short-acting and lixisenatide primarily lower post-prandial plasma glucose by delaying stomach emptying [17]. The role of incretin mimetics in promoting weight loss and a comparatively reduced risk of hypoglycemia compared to other anti-hyperglycemic medications are the main advantages of glucagon-like peptide-1 receptor agonists. The glucagon-like peptide-1 receptor agonists are structurally similar to the incretin hormone GLP-1. The incretin mimetics have distinctions in potency and chemical makeup; these therapies also vary significantly in how long they take to act. The mimetics are modified to resist breakdown by the dipeptidyl peptidase 4 enzyme. The components are not orally active due to rapid gastrointestinal inactivation and thus have to be administered via subcutaneous injections [18]. The incretin-based mimetics are considered in patients with contraindications or intolerance to the first-line agent, metformin. It can also be used in patients who do not achieve their glycated hemoglobin target within three months of therapy and, particularly, in patients with atherosclerosis, chronic renal disease, and heart failure. Some of the GLP-1 analogs, such as semaglutide and Liraglutide, have been approved for obesity management and in overweight patients with comorbidities. The major barriers to prescribing these drugs are higher costs and tolerability [18].
The incretin mimetic agents have also been demonstrated to reduce total cholesterol and systolic and diastolic blood pressure and to encourage weight loss. The benefit of weight loss is facilitated by directly acting on the hypothalamus to cause an increased satiety, thus reducing caloric intake. Patients with type 2 diabetes mellitus frequently develop atherosclerotic cardiovascular disease, which is also a leading cause of mortality in this population. Research has shown that in individuals with type 2 diabetes, reducing cardiovascular risk factors can help avoid atherosclerotic cardiovascular disease. GLP-1 agonists have significant cardiovascular benefits, including improving the overall cardiac output, reducing infarction size, and decreasing the general risk for cardiovascular events. They additionally enhance left ventricular ejection fraction, myocardial contractility, coronary blood flow, and endothelial function [19].
The primary adverse effect is mild to moderate nausea that typically subsides after a few weeks. Hypoglycemia, a well-known adverse effect of several pre-existing antidiabetic therapy options, is rare and mostly happens when sulfonylurea is used in concert with incretin mimetics. Insulin and incretin mimetics should not be used concurrently [20]. The other primary adverse reactions of Glucagon-like peptide-1 agonists are vomiting and diarrhea, which can cause acute kidney injury through volume reduction. Side effects may also include headaches, infections, moderate tachycardia, dyspepsia, and dizziness.
Patients who use this class of medication should be counseled that eating when feeling full may cause temporary, moderate nausea. If nausea is experienced, the dosage of these drugs should be increased cautiously [18]. Erythema and pruritus at the injection site are other frequent side effects, particularly with the longer-acting drugs in this group of agents. Antibodies may develop against specific GLP-1 analogs and may impair the effectiveness of these agents, especially exenatide. This immunogenicity could result in Anaphylaxis or responses at the injection site [15]. Antibody formation has been reported more frequently with the weekly administration of exenatide than with the twice-daily dosing. Due to statistically negligible glycemic improvement and exacerbated hypoglycemia effects, combination therapy with GLP-1 agonists and dipeptidyl peptidase-4 inhibitors is not currently advocated.
Reactive oxygen species partially oxidize low-density lipoproteins (LDL) cholesterol when it traverses through the intima layer of arterial blood arteries, producing oxidized LDL molecules and may contribute to the formation of atheromas. By producing adhesion molecules such as vascular cell adhesion protein 1 and E-selectin, the interaction between monocytes and macrophages with oxidized LDL and free radicals stimulates additional infiltration of monocytes. When oxidized LDL stimulates monocytes, they develop into macrophages. Pro-inflammatory cytokines macrophages. Through phagocytosis, macrophages absorb lipid particles and inhibit the production of Krüppel-like factor 2 that inhibits endothelial nitric oxide synthase, resulting in decreased Nitric oxide (NO) production and inhibiting vasodilation via NO-mediated vascular smooth muscle relaxation [21]. Macrophages become foam cells in a condition where reactive oxygen species and oxidized LDL predominate. These cells can undergo apoptosis and discharge their lipid content into the lipid core of developing atherosclerotic plaques. The thick fibrous cap covering stable plaques, primarily made of collagen, serves as a barrier against rupture. When atherogenesis advances, the fibrous cap is broken down by matrix metalloproteinase through proteolysis, endothelial cells then undergo apoptosis, and more necrotic regions emerge. Plaque rupture, thrombus development, and bleeding into areas of necrotic plaque are the outcomes of this.
The expression of GLP-1 receptors in vascular smooth muscle cells, monocytes, endothelial cells, and macrophages can have a variety of consequences that may impede the development or rupture of atherosclerotic plaques. First, GLP-1, exenatide, liraglutide, and semaglutide lower the generation of ROS. GLP-1 receptor stimulation, such as that provided by GLP-1, exenatide, dulaglutide, and Liraglutide, effectively reduces the activation of adhesion molecules and monocytes and macrophages caused by oxidized LDL and its subsequent activation [20]. For instance, exenatide reduces the buildup of monocytes in the vascular wall. Endothelial cells result in vascular smooth muscle relaxation and endothelium-derived vasodilation by suppressing endothelin synthesis, producing more nitric oxide, and expressing more nitric oxide synthase. Lixisenatide, Liraglutide, and dulaglutide are examples of drugs that preferentially produce M2 macrophages from monocytes. This leads to an increase in the otherwise repressed development of KLF-2. Following GLP-1 receptor stimulation, there is a decrease in reactive oxygen species exposure, which hinders the process of foam cell formation, reduces caspase-mediated foam cell apoptosis, and limits the development of necrosis in the core of atherosclerotic plaques [19].
Moreover, GLP-1 receptor activation decreases the proliferation of vascular smooth muscle and its potential migration into plaques. Exenatide stabilizes endothelial cell integrity. Semaglutide decreases the amount of plaque hemorrhage. Metalloproteinase expression is downregulated, keeping fibrous caps intact and inhibiting plaque rupture. Plaque stabilization and a slowdown of plaque progression are the overall outcomes [20]. GLP-1 receptor agonists significantly decrease atherosclerotic plaques' creation, extent, and vulnerability. Renal benefits have been reported following the use of incretin mimetics. GLP-1 receptor agonists decrease albumin excretion in the urine, prevent new-onset macroalbuminuria, or delay the rate at which the estimated glomerular filtration rate (eGFR) declines over time. Most of the processes underlying these kidney benefits are unclear [18]. Although significant decreases in renal composite outcomes have been reported, these reports mostly depended on prevailing effects that prevented new-onset chronic macro-albuminuria.
Some of the high-risk variables are the presence of albuminuria, eGFR < 60 ml/min, left ventricular hypertrophy, coronary artery stenosis >50%, and individuals who are ≥ 55 years of age. Additionally, it is possible to avoid hypoglycemia and weight gain by using incretin mimetics. Going one step further, the ESC guidelines suggest using GLP-1 receptor agonists or SGLT-2 inhibitors as first-line treatment for patients at high or very high risk or with existing atherosclerotic cardiovascular disease. Extremely high-risk variables include diabetes for a length of more than ten years without target organ damage, three or more main risk factors, and any additional risk factors [14]. Approximately 30 to 60 percent of individuals with type 2 diabetes would be eligible for a GLP-1 RA based on these guidelines. However, in actual clinical practice, the proportion of patients on incretin mimetics medication is very low. The apparent discrepancy between clinical practice and guidelines has several causes. First, the expense of treating with GLP-1 receptor agonists is similar to that of an intensified insulin treatment regimen, including glucose monitoring expenses, but significantly more than that of most oral glucose-lowering medications. While several cost-effectiveness assessments have indicated that the overall benefits of GLP-1 RA therapy far exceed the direct treatment costs, the cost of the GLP-1 receptor agonists currently on the market continues to be a significant barrier in most nations. In addition, certain patients may be reluctant to begin incretin mimetics due to the requirement for daily or weekly injections. Third, patients who have a history of pancreatitis, diabetic retinopathy, or medullary thyroid cancer may not be able to take GLP-1 RAs due to contraindications [18].
Increased glucagon secretion contributes to the underlying cause of hyperglycemia in type 2 diabetes. The use of incretin as a medication for type 2 diabetes has been actively explored as it both increases insulin secretion and inhibits the production of glucagon. Nonetheless, because type 2 diabetes reduces the responsiveness of GIP in β-cells, research into GLP-1-related medications has resulted in the usage of GLP-1 receptor agonists in managing the condition [20].
Incretin Mimetics Classes
The incretin mimetics are broadly categorized into two classes. Exendin-4 (39-amino acid peptide) and human GLP-1 molecules are the two groups. Exendin-4 and GLP-1 have 53% sequence homology. The exendin-4 formulations include exenatide and lixisenatide. The human GLP-1 backbone molecules include dulaglutide, semaglutide, albiglutide, and Liraglutide. This kind of medication for type 2 diabetes has the advantage of delaying stomach emptying and, in the event of elevated blood sugar, blocking pancreatic alpha-cell glucagon production. Moreover, GLP-1 receptor agonists can increase pancreatic beta-cell proliferation while reducing their apoptosis rate [12,22].
Exenatide
Exenatide (Figure 3), commonly marketed as Bydureon and Byetta, is a synthetic exendin-4 that was first extracted from the saliva of a Gila lizard. It has an amino acid substituted at position 2 and shares about 53%. Similar structural homology to native glucagon-like peptide-1. It mimics the effects of glucagon-like peptide-1. It binds and activates the glucagon-like peptide-1 receptors on the beta cells of the pancreas, after which insulin synthesis and secretion is initiated. It is administered subcutaneously and reaches peak plasma concentrations after about 2 hours due to its rapid absorption rate. The half-life is approximately 2 hours. It thus requires twice daily dosing, one hour before morning and evening meals to achieve its full effect on glycemic control [23].
An extended-release formulation that is administered once weekly is also available. Unlike the native glucagon-like peptide-1 hormone, exenatide does not become degraded by the ubiquitous enzyme dipeptidyl peptidase-4. The kidneys clear it through glomerular filtration. When used as an add-on therapy to metformin, it shows massive improvements in glycemic control and a reduction in fasting plasma glucose concentrations. Exenatide extended-release is one of the regularly used medications that control type 2 diabetes effectively. However, it has no cardio-protective properties and has the least effect among analogies on weight and glycosylated hemoglobin [4]. Generally, it is well tolerated, with mild side effects of nausea, vomiting, diarrhea, and headache. It should, however, not be administered to patients with end-stage renal failure because it is primarily excreted in the kidneys.
Liraglutide
Liraglutide (Figure 4) is an acylated mammalian glucagon-like peptide-1 administered once daily. It is commonly marketed as Victoza for T2DM and Saxenda for weight reduction. It has a similar structural homology to native glucagon-like peptide-1 of about 97%. The analog is obtained via recombinant DNA technology [22]. It has a half-life of 13 hours and is therefore administered once daily. It improves Insulin sensitivity, especially in patients who have been seen to have uncontrolled Type 2 diabetes mellitus. The GLP-1 peptide backbone has a fatty-acyl moiety, which confers DPP-4 resistance and non-covalent binding to serum albumin, resulting in an extended half-life. The recommended starting dose is 0.6 mg/day for one week, after which it should be titrated weekly up to a maximum of 1.8 mg/day. Liraglutide has significantly decreased HbA1c levels, post-prandial glycemia, and fasting glycemia. Additionally, it is linked to a decreased risk of weight gain and hypoglycemia [23].
Liraglutide is used as an adjunct to diet and lifestyle management to improve glycemic control in patients with type 2 diabetes. If used concurrently with an insulin secretagogue, consider reducing the latter's dose. Liraglutide is indicated to reduce the occurrence of major cardiovascular events. It improves insulin sensitivity through the upregulation of glucose transporter-4. AMP-activated protein kinase activation within the muscle and liver facilitates insulin sensitivity augmentation and promotes glucose metabolism [15].
Liraglutide also reduces oxidative stress and inflammation, which contribute to the amelioration of insulin resistance. The agent also improves endothelial and cardiac function. Low-grade inflammation is a factor in the pathogenesis of type 2 diabetes mellitus and may be crucial in the onset of diabetes-related problems, such as cardiovascular disease. In Type 2 diabetes patients with a history of cardiovascular disease or who are at high risk, treatment with glucagon-like peptide-1 receptor agonists such as Liraglutide lowers the risk of cardiovascular complications [24]. It has been approved for secondary prevention of cardiovascular disease. Liraglutide contains anti-apoptotic, anti-inflammatory, antioxidant, and neuroprotective properties that may make it useful in treating neurological diseases. It also traverses the blood-brain barrier. The metabolite GLP-1 amide, which is generated when DDP-4 cleaves liraglutide, mediates the anti-inflammatory effects of Liraglutide following intracerebral hemorrhage [25].
Liraglutide minimizes endogenous glucose production and glucagon release [22]. The listed extra pancreatic effects contribute to insulin sensitivity. Liraglutide also slows down gastric emptying and decreases appetite, thus contributing to weight loss. Saxenda is used as an additional therapy in conjunction with a lower-calorie diet and more physical activity for the long-term control of weight in people with at least one weight-related condition, such as hypertension, dyslipidemia, and obesity. Some of the side effects associated with Liraglutide include headache, constipation, heartburn, malaise, and a rash at the site of injection. The medication should be properly stored away from light and heat. Unused administration injection pens should not be frozen but refrigerated at 4-8 0 C. It should not be used in patients with type 1 diabetes and diabetic ketoacidosis [24].
Dulaglutide
Dulaglutide (trade name Trulicity) is a long-acting agent with two stable glucagon-like peptide-1 moieties connected with an immunoglobulin fragment (GLP-1(7-37) covalently linked to an Fc fragment of human IgG4 (Figure 5). It is administered once weekly at any time of the day, with or without food. The initial dose is 1.5 mg subcutaneously but can gradually increase to 3 mg. It can be injected into the abdomen, upper arm, or thigh. The injection site should be rotated after each dose. It should be stored away from light. The two should not be injected into the same area when using insulin. It has a half-life of 5 days. Metabolism is the breakdown into amino acids through protein catabolism pathways. It should be stored at room temperature or refrigerated at 2OC. It should never be frozen. It is commonly marketed as Trulicity. It has similar efficacy to other glucagon-like peptide-1 agonists and has better glycemic control. It lowers glycated hemoglobin, fasting, and post-prandial glucose levels [26]. It also has a role in promoting weight loss. It improves the beta cell function of the pancreas by promoting cellular regeneration, thus improving the cell mass. It also prevents cell apoptosis, leading to slow progression of Type 2 diabetes. This effect on cellular regeneration delays the need for initiation of insulin therapy.
Dulaglutide provides cardio-protective benefits through blood pressure lowering and improvement of lipid levels. It lowers the low-density lipoproteins cholesterol. Dulaglutide has the potential to raise the number of endothelial cells and endothelial progenitor cells in peripheral blood, decrease persistent inflammation, enhance the function of endothelial cells and endothelial progenitor cells, and eventually boost the elasticity of arterial arteries in Type 2 Diabetes Mellitus patients. It is considered second-line therapy in addition to diet and exercise in patients who have problems achieving glycemic control on monotherapy with metformin. Dulaglutide has more benefits in lowering fasting plasma glucose than post-prandial blood glucose. It acts on the principle of glucose-dependent mechanism; therefore, there is no significant risk for profound hypoglycemia. Dulaglutide has mild gastrointestinal adverse effects such as nausea, vomiting and diarrhea. The symptoms worsen within the first two weeks but decrease over time. No dose adjustment is required in patients with renal or hepatic impairment when administering dulaglutide [27,28].
A 2023 study reported that dulaglutide, an antidiabetic agent, can protect mice from sepsis-induced lung injury [29]. Sepsis can potentially trigger the release of inflammatory factors by inducing an overabundance of inflammatory cytokines, reactive oxygen, and nitrogen species. By triggering the cyclic adenosine monophosphate/protein kinase A and cyclic guanosine monophosphate/protein kinase G signaling channels, the generated reactive nitrogen and reactive oxygen species damage the mitochondria and cause mitochondrial apoptosis [4]. Sepsis-related cardiac damage has been linked to oxidative stress, which is primarily brought about by an overabundance of reactive oxygen species. The activation of lipopolysaccharides (LPS) greatly induces oxidative stress in cardiac cells, as evidenced by increased nitric oxide generation and reduced glutathione levels. Following dulaglutide treatment, the oxidative stress state is reduced, implying that dulaglutide protects against LPS-induced oxidative stress injury to cardiac cells. Two key markers of myocardial cell dysfunction frequently employed in evaluating acute myocardial infarction are creatine-kinase myoglobin and troponins. Stimulation with lipopolysaccharide markedly increased the expressions of creatine-kinase myoglobin in myocardial cells, showing that lipopolysaccharide is the cause of the malfunction of myocardial cells. Dulaglutide greatly lowered the dysfunction of cardiac cells caused by lipopolysaccharide, suggesting that the drug may have a protective effect against myocardial dysfunction brought on by sepsis [30]. In sepsis patients, nitric oxide synthase stimulates the formation of nitric oxide. An excessively generated nitric oxide and superoxide radicals react to produce peroxynitrite, which additionally interferes with cardiac function, modifies cardiac load, down-regulates the β-adrenergic receptor, hinders type I calcium channel function, and reduces the functioning of the mitochondrial electron transport chain complex in cardiac cells [30]. Dulaglutide therapy drastically lowers the increased production of nitric oxide and heightened expression of nitric oxide synthase generated by lipopolysaccharide, suggesting that dulaglutide inhibits inflammatory factor-mediated nitric oxide synthase activation [26,31].
Semaglutide
Semaglutide (Figure 6) has the structure of an acylated mammalian glucagon-like peptide with an amino acid substitution at position two. It is commonly marketed as Ozempic and Wegovy (different doses and strengths) as a subcutaneous injection and Rybelsus, an oral formulation [13]. Semaglutide is prescribed as an add-on to diet and exercise in individuals with type 2 diabetes mellitus to improve glycemic control. It is also indicated to lower the probability of serious cardiovascular events in people with a history of cardiovascular disease and type 2 diabetes, such as nonfatal myocardial infarction and nonfatal stroke. No dose adjustment is necessary in patients with renal failure or hepatic impairment when administering semaglutide.
Semaglutide has also been marketed as Wegovy, a weight loss agent and is recommended in conjunction with a lower-calorie diet and exercise to help individuals with obesity (BMI ≥30 kg/m2) or overweight (BMI ≥27 kg/m2) and with at least one weight-related comorbid disease to lose weight. It is also used in individuals with existing cardiovascular disease who are also obese or overweight at increased risk for major adverse cardiovascular events. Owing in part to the rising incidence of obesity, heart failure with preserved ejection fraction is now known as the most common form of heart failure. The onset and progression of heart failure with preserved ejection fraction are significantly influenced by excess obesity and type 2 diabetes. Compared to individuals without type 2 diabetes, patients with heart failure who have unaltered ejection fraction are more likely to have type 2 diabetes, which is linked to worse hemodynamic and clinical outcomes, such as a higher burden of symptoms and reduced functional ability. Few effective treatments are available for this particular population [32].
Semaglutide, like other glucagon-like peptide-1 agonists, increases pancreatic beta-cells glucose-dependent insulin release, inhibits erroneously high glucagon secretion, and slows stomach emptying. As a weight loss agent, it works as a physiological regulator of appetite and limits the intake of calories centrally. It is thought that delayed stomach emptying plays a more significant role in regulating post-prandial hyperglycemia than insulin secretion. Semaglutide has demonstrated excellent hypoglycemia and weight-reducing properties [13]. Semaglutide, in contrast to conventional GLP-1 RAs, translocates directly to the brain stem, hypothalamus, and lateral septal nucleus, where it has effects on central neurons. Semaglutide has shown better effects in concurrent body weight loss assessments than other GLP-1 RAs. Semaglutide has a different anorexigenic mechanism than other GLP-1 RAS, which may account for some of its strong weight reduction effects. Weight loss linked to semaglutide is thought to be unrelated to gastrointestinal adverse events, such as nausea and vomiting, and is thought to occur independently of these reactions [33].
Semaglutide helps in lowering the low-density lipoproteins cholesterol. Just like other GLP-1 RAs semaglutide does not, however, increase high-density lipoprotein cholesterol levels. The inhibition of cardiovascular events may be achieved by a combination of hypoglycemia brought on by increased insulin secretion and modifications in lipid metabolism brought on by these incretin mimetics [33] and decreases in the magnitude of chronic systemic inflammation [33]. Semaglutide positively impacts fatty liver since it decreases triglycerides and significantly improves liver function markers. It is metabolized through the proteolytic breakdown of the peptide framework, followed by successive beta-oxidation of the side chain of fatty acids. Primary excretion is through urine and feces [35]. Administration is subcutaneously through the abdomen, upper arm, or thigh. Rybelsus is available as an oral formulation for ease of administration and patient convenience.
If required, the weekly injection day may be adjusted as long as there are at least two days between administrations. Whenever administering an injection in a particular body area every week, it's important to use a different injection site. Examine the medication visually before using it; it must appear clear and colorless. Whenever particulate matter and coloring are apparent, do not administer the medication. While semaglutide and insulin can be injected into the same general body area, the injection sites should not be close to one another to overlap absorption areas, when used with insulin. Always administer the two drugs as separate injections to prevent intramolecular complex formations that may contribute to the inactivation of both agents. If the dose has been missed for less than five days, administer the injection immediately. But suppose the missed period has extended more than five days. In that case, the patients can resume their usual once-weekly administration plan by skipping the missed dose and administering the following one on the day that it is planned [35].
Rybelsus is usually taken orally 30 minutes before the first meal in the morning. The tablets should be swallowed wholly and not crushed or split to increase effectiveness. In case of a missed dose, it should be skipped for that day and resume the schedule the next day. If a subcutaneous dose of Wegovy is missed and the next one is more than two days away, give the skipped dose as soon as feasible. Whenever a dose is missed, and the next one is less than 48 hours away, do not give the missing dose; instead, commence dosing on the next regularly scheduled day of the week. To minimize the likelihood of abdominal discomfort upon starting treatment again, restart dosing as usual if more than two doses are missed. When necessary, reinitiate treatment and adhere to the increased dose schedule [36].
The instructions for oral semaglutide with insulin should be thoroughly explained to the patients. It should be considered to lessen the risk of hypoglycemia. Patients' expectations should be considered while starting oral semaglutide treatment and informed about crucial treatment-related concerns [37]. As a result of the way it is formulated, oral semaglutide is less likely to be absorbed when food or liquids are present in the stomach due to nutrient-drug interactions. As a result, the prescribing physician should advise taking oral semaglutide whole when fasting, for instance, when waking up, with a sip of water, and at least half an hour before consuming any food or liquids or taking any other oral prescriptions. While gastrointestinal adverse effects are frequent with glucagon-like peptide-1 receptor agonists, most individuals receiving oral semaglutide are not affected by them. Counseling patients about the possibility of feeling full, satisfied, or even nauseated is important. Patients should be informed that nausea usually resolves and should adhere to the medication [37]. There have been several reports [38] recently published that are indicative of new and some serious side effects of semaglutide, including hyperemesis gravidarum (HG) [39], psychiatric adverse events [40], biliary disease [41], pancreatitis [42], appendicitis [43], bowel obstruction [44], gastroparesis [45], and elevations in liver enzymes [46].
Albiglutide
Albiglutide (Figure 7), a complex polypeptide, is commonly marketed as Tanzeum, made as a lyophilized for reconstituted agent. It is available as 30mg/pen or 50mg/pen. It is also an incretin mimetic used as an adjunct to diet and exercise to manage diabetes mellitus type 2. It is comprised of two GLP-1 molecules fused with the human serum albumin. It is administered as 30 mg SC once a week, and if the glycemic response is insufficient, it can be increased to 50 mg weekly. It slows gastric emptying and increases glucose-dependent insulin secretion. Compared to other GLP-1 receptor agonists, albiglutide causes fewer nausea episodes, which may be explained by the fact that it is a significant biochemical entity that does not cross the blood-brain barrier [47].
Through attaching to particular receptors on pancreatic beta cells and boosting insulin production, albiglutide mimics the action of GLP-1 and may help individuals with type 2 diabetes achieve better glycemic control. Albiglutide is a polypeptide made from recombinant DNA that is made up of two tandem repeats of a fused 30-amino acid sequence. With only one amino acid alteration, the albiglutide amino acid sequence is very similar to endogenous human GLP-1. The amino acid alteration makes it resilient to dipeptidyl peptidase 4 (DPP4) degradation. The binding of two recombinant molecules to human serum albumin effectively prolongs the plasma half-life of albiglutide [48].
Albiglutide is contraindicated in patients with diabetic ketoacidosis, Type 1 diabetes, Gastroparesis, and patients with medullary thyroid carcinoma. Its peak plasma time takes about 3-5 days. Metabolism follows that of human serum albumin in the vascular endothelium, with a half-life of up to 5 days. It is administered subcutaneously to the abdomen, upper arm, or thigh at any time of the day without regard to meals. If a dose is missed, provide it as soon as the patient remembers, but no later than three days. After that, patients can start taking their medication on the day it is normally administered [13]. Patients should be instructed to wait until their next regularly scheduled weekly administration if the missing dose occurred more than three days ago.
GLP-1 Mimetics such as albiglutide may have a cardio-protective effect via reducing endoplasmic reticulum stress, controlling autophagy, and promoting anti-inflammatory pathways. The main indirect effects of GLP-1 RAs on the ventricles are caused by the beneficial modulation of inflammation, endothelial function, and glucose uptake, as ventricular cardiomyocytes express GLP-1 receptors at relatively low levels. Furthermore, research suggests that GLP-1RAs modulate extracellular matrix alterations to attenuate cardiac remodeling after myocardial infarction. Heart failure is characterized by calcium overload. By inhibiting the phosphorylation of the ryanodine receptor 2 and blocking the activation of calmodulin-dependent protein kinase II, GLP-1 RA therapy modifies cytosolic Ca2+ concentrations. GLP-1 mimetics demonstrate their anti-oxidative stress properties by preventing the generation of intracellular and mitochondrial reactive oxygen species in methylglyoxal-treated cells [49].
Although albiglutide is generally well tolerated, rare adverse events that may occur include pancreatitis and hypersensitivity reactions. The common side effects include the area of injection reactions, diarrhea, nausea, vomiting, dizziness, headache, and lethargy. The extended circulation half-life of the albiglutide-albumin hybrid is caused by its larger molecular size, which also hinders renal clearance [47]. At the GLP-1 molecule's cleavage location, a Gly8Ala mutation inhibited DPP-4 degradation. However, a different approach was taken with albiglutide, where the terminal arginine was connected to albumin, and two copies of DPP-4-resistant GLP-1 analogs were coupled through the N-terminus.
Tirzepatide
Tirzepatide (Figure 8) is commonly marketed as Mounjaro for Type 2 Diabetes Mellitus, and Zepbound brand for weight loss. Introducing GLP-1 activity into the GIP sequence led to the discovery of tirzepatide. A balanced dual agonist, it exhibits equivalent affinity for GIPR compared to native GIP but binds GLP-1R with about five times less affinity than native GLP-1 [50]. This indicates that GIPR activity is favorably over GLP-1R activity, which induces a dose-dependent reduction in HbA1c. It also plays a role in promoting weight loss. It is a once-weekly subcutaneous injectable peptide from the native gastric inhibitory polypeptide (GIP) structure family, and it has agonist activity toward both GLP-1 and GIP receptors. It has an additive value on certain endogenous hormone pathways triggered by nutrients linked to energy regulation. Tirzepatide has a potency ratio for GIPR activity and a C20 unsaturated di-acid acyl chain that influences albumin binding. The molecule's general characteristics allow for once-weekly administration [50]. This pharmacokinetic characteristic may have benefits beyond convenience, such as improved clinical effectiveness at high and sustained doses of selective GLP-1R agonists.
Zepbound is administered in conjunction with a diet low in calories and increased physical exercise to help adults who have at least one weight-related comorbid condition such as hypertension, dyslipidemia, type 2 diabetes mellitus, obstructive sleep apnea, or cardiovascular disease and are obese or overweight to help in weight management. Dual actions on pancreatic beta cells help promote insulin production. GIP-driven factor helps improve the white adipose tissue role and a greater anorexigenic property from the signals of both receptor pathways in the brain. The dual action is meant to improve glycemic control and help in weight reduction [50].
Lixisenatide
Lixisenatide (Figure 9) is commonly marketed as Adlyxin. Lixisenatide is a peptide with 44 amino acids. Similar to exenatide, it is derived from exendin-4 but has six lysine residues added to its C-terminus in place of a proline residue. The amino acid at position 44, the C-terminal, is amidated. Lixisenatide is roughly 55% bound to plasma proteins and binds to the GLP-1 receptors with a high degree of affinity that can reach four times that of human GLP-1. The molecule is rapidly absorbed after subcutaneous injection, and it takes about two hours to reach a peak plasma concentration. Lixisenatide has a half-life of about 3 hours [13].
The glomerular filtration process is the principal method of elimination of lixisenatide. Cytochrome P450 isoenzyme activity is unaffected by lixisenatide. However, because lixisenatide decreases stomach emptying, the absorption rate of oral medications such as warfarin, ethinyl estradiol, acetaminophen and others may be slowed down. Despite this, concurrent usage Lixisenatide has not been linked to any documented clinically relevant side effects. When taking lixisenatide with drugs that have a limited therapeutic range, like warfarin, or those that depend on concentration, such as antibiotics, precaution should be taken. Furthermore, drugs that would not want a delayed impact should be taken at least an hour before lixisenatide. It is recommended to use oral contraceptives one hour before or 11 hours after its injection [53].
It has a longer half-life attributed to its exendin-4 backbone. It is administered once a day, 10 mcg via injection. After two weeks, the dose is titrated to 20 mcg. Slower stomach emptying caused by lixisenatide has a significant impact on post-prandial glycaemia. Reducing HbA1c, fasting, and post-prandial glycemia are all efficient when used as monotherapy. In animal trials, lixisenatide is administered peripherally and crosses the blood-brain barrier. Retinal filtration, tubular reabsorption, and metabolic catabolism are thought to be the routes by which lixisenatide is eliminated. Its metabolites' composition and possible mechanisms of action are unknown. It is the most recent once-daily GLP-1 agonist to be licensed for the treatment of type-2 diabetes. Lixisenatide is linked to modest decreases in body weight, a considerable reduction in glycated hemoglobin (HbA1c) of 0.5–1% and decreases in post-prandial and fasting plasma glucose. Regarding reducing HbA1c, lixisenatide has been demonstrated to be non-inferior to exenatide given twice daily and tends to be well-tolerated [53].
Lixisenatide's most often reported side effects are gastrointestinal symptoms, such as nausea, vomiting, and diarrhea. The first three to six weeks of lixisenatide medication are when these side effects usually decrease in intensity and tend to be dose dependent. There have also been reports of immunogenic effects with this injectable drug. In comparison to patients who do not have anti-lixisenatide antibodies, people who develop these antibodies may experience a slightly greater frequency of allergic responses and injection site reactions. Antibody status is not routinely checked. However, patients should consider switching to a different antidiabetic medication if their glycemic control significantly worsens or if they have substantial injection site reactions [54].
Patients with a known sensitivity to lixisenatide or any of its inactive ingredients should not use this medication. Lixisenatide should be avoided in individuals with type-1 diabetes mellitus as it has not been evaluated in this population. Lixisenatide and DPP-4 inhibitors have not been studied and should not be combined. When lixisenatide is taken in combination with other drugs that lower blood glucose directly, the risk of hypoglycemia increases. If lixisenatide is introduced to these regimens, such as basal insulin therapy or sulfonylurea prescription, a decrease may be required due to the possible risk of hypoglycemia. Mild-to-moderate renal impairment does not necessitate adjusting the dosage. However, monitoring is advised for these individuals since they may be more vulnerable to unfavorable outcomes, such as dehydration, which could deteriorate their kidney function. Individuals who suffer from severe renal impairment need to be constantly monitored for worsening renal function and an increased chance of adverse events, especially those related to the gastrointestinal system [53,55].
Patients are advised to adhere to a one-step program for dose escalation when prescribed lixisenatide. Initially, patients should use the green pre-filled beginning pen with preset doses, administering 10 mcg subcutaneously once daily for 14 days. Using the burgundy pre-filled maintenance pen with preset doses, the dose should be adjusted to a once-daily maintenance dose of 20 mcg beginning on day 15. The medication must be administered to patients one hour before their first meal [53]. Patients must activate each pen once. Additionally, like Liraglutide and most insulin pens, lixisenatide pens that are not in use should be refrigerated, but pens that are in use can remain at room temperature.
Lixisenatide is not as difficult to administer as once weekly dulaglutide. However, it is easier than certain once-weekly GLP-1 agonists that come with pen devices meant to mix medication into a solution. Patients who find the dial mechanism of liraglutide pens difficult to use but prefer daily administration may find the preset lixisenatide dose slightly more convenient. When prescribed the Adlyxin beginning pack, patients must be reminded to use the appropriate pen. Since there are no significant, apparent variations in weight reduction, glycemic parameter effects, tolerability, or overt administration-related benefits, the decision on the choice of incretin mimetic is usually based on the cost and patient factors [53,54,55].
Conclusions
In conclusion, the management of diabetes is aimed at improving hyperglycemia symptoms and minimizing long-term microvascular and macrovascular complications such as diabetic nephropathy, neuropathy, retinopathy, and cardiovascular effects. The discovery of medications that regulate hyperglycemia and affect other metabolic risk variables to enhance cardiovascular outcomes is in high demand. Obesity, aging, genetic vulnerability, and diversity are factors in the rising prevalence of Type 2 diabetes mellitus. Insulin resistance, the functional failure of pancreatic beta cells, and excessive or incorrect glucagon output are the main causes of type 2 diabetes. Diabetes type 2 is a long-term comorbidity brought on by decreased insulin production as a result of peripheral tissue insulin resistance and β-cell malfunction. In addition to causing microangiopathy, homeostatic hyperglycemia increases the risk of macroangiopathy, leading to cardiovascular diseases. Economic disadvantages, a decline in healthy life expectancy, and a decline in patient quality of life are all caused by complications from these vascular conditions. Furthermore, obese patients with type 2 diabetes need more rigorous glycemic control and weight management strategies because obesity is linked to the pathophysiology of the disease and is a risk factor for macroangiopathy [7]. Medication and lifestyle modifications like diet and exercise can help with effective treatment for obesity and Type 2 diabetes. In addition, simultaneous administration of a low dose of a luminal α-glucosidase inhibitor agent with the GLP-1 agent with or without insulin may help to reduce the potential side effects of both agents and further improve the clinical efficacy of the therapeutic regimen. Glucagon-like peptide-1 (GLP-1) receptor agonists (GLP-1RAs) are a valuable therapy alternative for people with type 2 diabetes since they target several of the disease's pathophysiology problems. GLP-1 RAs lower glucagon secretion slows the pace of gastric emptying and enhances glucose-dependent insulin secretion. Additionally, they are linked to decreased hunger, which results in weight loss. These drugs have been linked to dose-limiting adverse effects such as nausea, vomiting, and diarrhea. Efforts have been made to increase tolerability and efficacy by looking into dose-escalation regimens that enable larger doses linked to better efficacy while minimizing undesirable effects. Glucagon-like receptor agonists have both central and peripheral effects. These agents have been shown to minimize food intake and reduce appetite. This eventually contributes to weight loss, improved insulin sensitivity, and improvements in beta-pancreatic cell function. Administration of incretin mimetics enhances insulin release, inhibits glucagon production, and consequently slows gastric emptying. It results in better glycemic control, as shown by reduced glycated hemoglobin and decreased fasting glucose and post-prandial glucose levels. The most common adverse effects of GLP-1 agonists are vomiting, diarrhea, and nausea, which can cause acute kidney injury by volume contraction. There may also be headaches, infections, moderate tachycardia, dyspepsia, and dizziness.
Patients who use this class of medication should be cautioned that eating when feeling full may cause temporary, moderate nausea. If nausea is experienced, the dosage of these drugs should be increased gradually. Erythema and pruritus at the injection site are other frequent side effects, particularly with the longer-acting drugs in this class of medications. There is a minimal chance of having a minor hypoglycemic episode, although as of yet, no significant hypoglycemic episodes have been reported in the available research. Antibodies that patients may develop against specific GLP-1 analogs may impair the effectiveness of these drugs, especially exenatide. Anaphylaxis or responses at the injection site could result from this immunogenicity. Pregnancy and hypersensitivity are two conditions that make GLP-1 agonist use contraindicated and prevent the prescription of this family of drugs. For women of reproductive age, the recommended course of action when using GLP-1 agonists following conception is unclear since controlled studies of the safety of incretin use during pregnancy are lacking. Individuals with serious gastrointestinal conditions such as Gastroparesis and inflammatory bowel disease should also avoid GLP-1 analogs. Very recently, it has been reported that monotherapy with GLP-1 is associated with a risk of new-onset nonproliferative diabetic retinopathy, Diabetic retinopathy, and diabetic macular edema [56,57].
In summary, the increasing use of incretin mimetics therapy during T2DM and associated comorbidities, including obesity and overweight and glucose-intolerant conditions, shows a promising future, whether used as mono- or combination therapy [58,59,60], or when combined with nutraceuticals, lifestyle, or dietary factors that may augment the physiologic responses [61].
References
- Tan Q, Akindehin SE, Orsso CE, Waldner RC, DiMarchi RD, Müller TD and Haqq AM (2022) Recent Advances in Incretin-Based Pharmacotherapies for the Treatment of Obesity and Diabetes. Front. Endocrinol. 13:838410. [CrossRef]
- Ahmad, E., Lim, S., Lamptey, R., Webb, D. R., & Davies, M. J. (2022). Type 2 diabetes. The Lancet, 400(10365), 1803-1820.s.
- Collins, L., & Costello, R. A. (2023). Glucagon-Like Peptide-1 Receptor Agonists. In StatPearls. StatPearls Publishing.
- Teague, M., Martinez, A., Walker, E., El-Rifai, M., & Carris, N. W. (2023). Use and Interchange of Incretin Mimetics in the Treatment of Metabolic Diseases: A Narrative Review. Clinical Therapeutics, 45(3), 248-261. [CrossRef]
- Hussain, U., Das, A. K., Ghosh, S., and Sil, P. C. An overview on the role of bioactive α-glucosidase inhibitors in ameliorating diabetic complications. Food and Chemical Toxicology, Volume 145, November 2020, 111738.
- Ruze, R., Liu, T., Zou, X., Song, J., Chen, Y., Xu, R., ... & Xu, Q. (2023). Obesity and type 2 diabetes mellitus: Connections in epidemiology, pathogenesis, and treatments. Frontiers in endocrinology, 14, 1161521.
- Usman, M. S., Davies, M., Hall, M. E., Verma, S., Anker, S. D., Rosenstock, J., & Butler, J. (2023). The cardiovascular effects of novel weight loss therapies. European heart journal, 44(48), 5036–5048. [CrossRef]
- Yashi, K., & Daley, S. F. (2023). Obesity and Type 2 Diabetes. In StatPearls. StatPearls Publishing.
- Collins, L., & Costello, R. A. (2023). Glucagon-Like Peptide-1 Receptor Agonists. In StatPearls. StatPearls Publishing.
- Tulp OL. Effect of delayed luminal carbodhydrate uptake via α-glucosidase inhibition on plasma glycemic parameters and lipid profiles in adult wistar fatty rats. Gastroenterol Hepatol Open Access. 2022;13(5):168‒173. [CrossRef]
- Hossain U, Das AK, Ghosh S, Sil PC. An overview on the role of bioactive α-glucosidase inhibitors in ameliorating diabetic complications. Food Chem Toxicol. 2020;145:111738. [CrossRef]
- Al Musaimi, O (2024). Exploring FDA-Approved Frontiers: Insights into Natural and Engineered Peptide Analogues in the GLP-1, GIP, GHRH, CCK, ACTH, and α-MSH Realms. Biomolecules, 14, 264. [CrossRef]
- Nauck, M. A., Mirna, A. E. A., & Quast, D. R. (2023). Meta-analysis of head-to-head clinical trials comparing incretin-based glucose-lowering medications and basal insulin: An update including recently developed glucagon-like peptide-1 (GLP-1) receptor agonists and the glucose-dependent insulinotropic polypeptide/GLP-1 receptor co-agonist tirzepatide. Diabetes, Obesity and Metabolism, 25(5), 1361-1371.
- Jinnouchi H, Sugiyama S, Yoshida A; et al. Liraglutide, a glucagon-like peptide-1 analog, increased insulin sensitivity assessed by hyperinsulinemic-euglycemic clamp examination in patients with uncontrolled type 2 diabetes mellitus. J Diabetes Res. 2015;2015:706416. [CrossRef]
- Boer, G. A., & Holst, J. J. (2020). Incretin Hormones and Type 2 Diabetes—Mechanistic Insights and Therapeutic Approaches. Biology, 9(12). [CrossRef]
- Jia, S., Wang, Z., Han, R., Zhang, Z., Li, Y., Qin, X., … & Yang, J. (2021). Incretin mimetics and sodium-glucose co-transporter 2 inhibitors as monotherapy or add-on to metformin for treatment of type 2 diabetes: A systematic review and network meta-analysis. Acta Diabetologica, 58, 5-18.
- Sposito, A. C., Berwanger, O., de Carvalho, L. S. F., & Saraiva, J. F. K. (2018). GLP-1RAs in type 2 diabetes: Mechanisms that underlie cardiovascular effects and overview of cardiovascular outcome data. Cardiovascular diabetology, 17(1), 157. [CrossRef]
- Nauck, M. A., Quast, D. R., Wefers, J., & Meier, J. J. (2021). GLP-1 receptor agonists in the treatment of type 2 diabetes–state-of-the-art. Molecular metabolism, 46, 101102.
- Wright, S. C., Motso, A., Koutsilieri, S., Beusch, C. M., Sabatier, P., Berghella, A., ... & Lauschke, V. M. (2023). GLP-1R signaling neighborhoods associate with the susceptibility to adverse drug reactions of incretin mimetics. Nature communications, 14(1), 6243.
- Hansen, K. B., Vilsbøll, T., & Knop, F. K. (2010). Incretin mimetics: A novel therapeutic option for patients with type 2 diabetes – a review. Diabetes, Metabolic Syndrome and Obesity, 3, 155–163. [CrossRef]
- Ruan, Z., Yang, L., Shi, H., Yue, X., Wang, Y., Liang, M., & Hu, H. (2021). The cost-effectiveness of once-weekly semaglutide compared with other GLP-1 receptor agonists in type 2 Diabetes: A systematic literature review. Expert review of pharmacoeconomics & outcomes research, 21(2), 221-233.
- Frandsen, C. S., Dejgaard, T. F., Andersen, H. U., Holst, J. J., Hartmann, B., Thorsteinsson, B., & Madsbad, S. (2017). Liraglutide as adjunct to insulin treatment in type 1 diabetes does not interfere with glycaemic recovery or gastric emptying rate during hypoglycaemia: A randomized, placebo-controlled, double-blind, parallel-group study. Diabetes, Obesity and Metabolism, 19(6), 773-782not interfere with glycaemic recovery or gastric emptying rate during hypoglycaemia: A randomized, placebo-controlled, double-blind, parallel-group study.
- Hvarchanova, N. (2024, January). Incretin-mimetic drugs—Nature, benefits, and risks. In Varna Medical Forum (Vol. 13, No. 1).
- Zobel, E. H., Ripa, R. S., Von Scholten, B. J., Rotbain Curovic, V., Kjaer, A., Hansen, T. W., Rossing, P., & Størling, J. (2021). Effect of Liraglutide on expression of inflammatory genes in type 2 diabetes. Scientific Reports, 11(1), 1-8. [CrossRef]
- Wronka M, Krzemińska J, Młynarska E, Rysz J, Franczyk B. New Insights into the Use of Liraglutide-Impact on Cardiovascular Risk and Microvascular Outcomes. Biomedicines. 2023;11(4):1159. [CrossRef]
- Scott LJ. Dulaglutide: A Review in Type 2 Diabetes. Drugs. 2020;80(2):197-208. [CrossRef]
- Gao X, Di Y, Lv Y; et al. A pharmacokinetic study comparing the biosimilar HEC14028 and Dulaglutide (Trulicity®) in healthy Chinese subjects. Clin Transl Sci. 2024;17(4):e13775. [CrossRef]
- Arslanian SA, Hannon T, Zeitler P; et al. Once-Weekly Dulaglutide for the Treatment of Youths with Type 2 Diabetes. N Engl J Med. 2022;387(5):433-443. [CrossRef]
- Wang Y, Deng F, Zhong X; et al. Dulaglutide provides protection against sepsis-induced lung injury in mice by inhibiting inflammation and apoptosis. Eur J Pharmacol. 2023;949:175730. [CrossRef]
- Wang, R., Wang, N., Han, Y., Xu, J., & Xu, Z. (2021). Dulaglutide alleviates LPS-induced injury in cardiomyocytes. ACS omega, 6(12), 8271-8278.
- Xie D, Li Y, Xu M, Zhao X, Chen M. Effects of dulaglutide on endothelial progenitor cells and arterial elasticity in patients with type 2 diabetes mellitus. Cardiovasc Diabetol. 2022;21(1):200. [CrossRef]
- Kosiborod, M. N., Petrie, M. C., Borlaug, B. A., Butler, J., Davies, M. J., Hovingh, G. K., ... & Shah, S. J. (2024). Semaglutide in patients with obesity-related heart failure and type 2 diabetes. New England Journal of Medicine.
- Okamoto, A., Yokokawa, H., Nagamine, T., Fukuda, H., Hisaoka, T., & Naito, T. (2021). Efficacy and safety of semaglutide in glycemic control, body weight management, lipid profiles and other biomarkers among obese type 2 diabetes patients initiated or switched to semaglutide from other GLP-1 receptor agonists. Journal of Diabetes & Metabolic Disorders, 20, 2121-2128.
- Tulp OL, Einstein GP. Review: Obesity and its associated inflammatory cytokines pose significant risk factors for COVID-19 outcomes. Advances in Obesity, Weight Management and Control. 2022;12(1):14‒20. [CrossRef]
- Ruan Z, Yang L, Shi H; et al. The cost-effectiveness of once-weekly semaglutide compared with other GLP-1 receptor agonists in type 2 Diabetes: A systematic literature review. Expert Rev Pharmacoecon Outcomes Res. 2021;21(2):221-233. [CrossRef]
- Rakhat Y, Wang L, Han W; et al. Oral Semaglutide under Human Protocols and Doses Regulates Food Intake, Body Weight, and Glycemia in Diet-Induced Obese Mice. Nutrients. 2023;15(17):3765. Published 2023 Aug 28. [CrossRef]
- Bucheit, J. D., Pamulapati, L. G., Carter, N., Malloy, K., Dixon, D. L., & Sisson, E. M. (2020). Oral semaglutide: A review of the first oral glucagon-like peptide 1 receptor agonist. Diabetes technology & therapeutics, 22(1), 10-18.
- Sodhi, M., Rezaeianzadeh, R., Kezouh, A., & Etminan, M. (2023). Risk of Gastrointestinal Adverse Events Associated With Glucagon-Like Peptide-1 Receptor Agonists for Weight Loss. JAMA, 330(18), 1795–1797. [CrossRef]
- Okeke, I. G., Camarda, A. R., Okeke, R., & Chaughtai, S. (2024). Semaglutide-induced Hyperemesis Gravidarum. JCEM case reports, 2(2), luad167. [CrossRef]
- Tobaiqy, M., & Elkout, H. (2024). Psychiatric adverse events associated with semaglutide, liraglutide and tirzepatide: A pharmacovigilance analysis of individual case safety reports submitted to the EudraVigilance database. International journal of clinical pharmacy, 46(2), 488–495. [CrossRef]
- Rubino, D. M., Greenway, F. L., Khalid, U., O'Neil, P. M., Rosenstock, J., Sørrig, R., Wadden, T. A., Wizert, A., Garvey, W. T., & STEP 8 Investigators (2022). Effect of Weekly Subcutaneous Semaglutide vs Daily Liraglutide on Body Weight in Adults With Overweight or Obesity Without Diabetes: The STEP 8 Randomized Clinical Trial. JAMA, 327(2), 138–150. [CrossRef]
- Singh, S., Chang, H. Y., Richards, T. M., Weiner, J. P., Clark, J. M., & Segal, J. B. (2013). Glucagonlike peptide 1-based therapies and risk of hospitalization for acute pancreatitis in type 2 diabetes mellitus: A population-based matched case-control study. JAMA internal medicine, 173(7), 534–539.
- Casella, S., & Galli, K. (2024). Appendicitis: A Hidden Danger of GLP-1 Receptor Agonists?. The Journal of pharmacy technology : jPT : Official publication of the Association of Pharmacy Technicians, 40(2), 108–111. [CrossRef]
- Gudin, B., Ladhari, C., Robin, P., Laroche, M. L., Babai, S., Hillaire-Buys, D., & Faillie, J. L. (2020). Incretin-based drugs and intestinal obstruction: A pharmacovigilance study. Therapie, 75(6), 641–647. [CrossRef]
- Kalas, M. A., Galura, G. M., & McCallum, R. W. (2021). Medication-Induced Gastroparesis: A Case Report. Journal of investigative medicine high impact case reports, 9, 23247096211051919. [CrossRef]
- Newsome, P., Francque, S., Harrison, S., Ratziu, V., Gaal, L. V., Calanna, S., Hansen, M., Linder, M., & Sanyal, A. (2019). Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Alimentary Pharmacology & Therapeutics, 50(2), 193-203. [CrossRef]
- Nowell, J., Blunt, E., & Edison, P. (2023). Incretin and insulin signaling as novel therapeutic targets for Alzheimer’s and Parkinson’s disease. Molecular Psychiatry, 28(1), 217-229.
- Tschöp M, Nogueiras R, Ahrén B. Gut hormone-based pharmacology: Novel formulations and future possibilities for metabolic disease therapy. Diabetologia. 2023;66(10):1796-1808. [CrossRef]
- Pandey S, Mangmool S, Parichatikanond W. Multifaceted Roles of GLP-1 and Its Analogs: A Review on Molecular Mechanisms with a Cardiotherapeutic Perspective. Pharmaceuticals (Basel). 2023;16(6):836. [CrossRef]
- Willard, F. S., Douros, J. D., Gabe, B. N., Showalter, A. D., Wainscott, D. B., Suter, T. M., Capozzi, M. E., Stutsman, C., Cardona, G. R., Urva, S., Emmerson, P. J., Holst, J. J., Coghlan, M. P., Rosenkilde, M. M., Campbell, J. E., & Sloop, K. W. (2020). Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight, 5(17). [CrossRef]
- Aronne, L. J., Sattar, N., Horn, D. B., Bays, H. E., Wharton, S., Lin, W. Y., Ahmad, N. N., Zhang, S., Liao, R., Bunck, M. C., Jouravskaya, I., Murphy, M. A., & SURMOUNT-4 Investigators (2024). Continued Treatment With Tirzepatide for Maintenance of Weight Reduction in Adults With Obesity: The SURMOUNT-4 Randomized Clinical Trial. JAMA, 331(1), 38–48. [CrossRef]
- Tobaiqy, M., & Elkout, H. (2024). Psychiatric adverse events associated with semaglutide, liraglutide and tirzepatide: A pharmacovigilance analysis of individual case safety reports submitted to the EudraVigilance database. International journal of clinical pharmacy, 46(2), 488–495. [CrossRef]
- Leon, N., LaCoursiere, R., Yarosh, D., & Patel, R. S. (2017). Lixisenatide (Adlyxin): A Once-Daily Incretin Mimetic Injection for Type-2 Diabetes. Pharmacy and Therapeutics, 42(11), 676. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5642155/.
- Lee J, Kim R, Kim MH; et al. Weight loss and side-effects of liraglutide and lixisenatide in obesity and type 2 diabetes mellitus. Prim Care Diabetes. 2023;17(5):460-465. [CrossRef]
- Niedermier V, Ayers G, Springer S. Lixisenatide (Adlyxin) for Type 2 Diabetes Mellitus. Am Fam Physician. 2017;96(4):257-258.
- Eleftheriadou A, Riley D, Zhao SS; et al. Risk of diabetic retinopathy and diabetic macular oedema with sodium-glucose cotransporter 2 inhibitors and glucagon-like peptide 1 receptor agonists in type 2 diabetes: A real-world data study from a global federated database. Diabetologia. [CrossRef]
- Wai KM, Mishra K, Koo E; et al. Impact of GLP-1 Agonists and SGLT-2 Inhibitors on Diabetic Retinopathy Progression: An Aggregated Electronic Health Record Data Study. Am J Ophthalmol. [CrossRef]
- Liu, C., Zou, Y., & Qian, H. (2020). GLP-1R agonists for treating obesity: A patent review (2015-present). Expert Opinion on Therapeutic Patents, 30(10), 781–794. [CrossRef]
- Gong, B., Yao, Z., Zhou, C., Wang, W., Sun, L., & Han, J. (2024). Glucagon-like peptide-1 analogs: Miracle drugs are blooming? European Journal of Medicinal Chemistry, 269, 116342. [CrossRef]
- Cowart, K. (2019). Oral Semaglutide: First-in-Class Oral GLP-1 Receptor Agonist for the Treatment of Type 2 Diabetes Mellitus. Annals of Pharmacotherapy. [CrossRef]
- Rizvi SAA, Einstein GP, Tulp OL, Sainvil F, Branly R. Introduction to Traditional Medicine and Their Role in Prevention and Treatment of Emerging and Re-Emerging Diseases. Biomolecules. 2022;12(10):1442. Published 2022 Oct 9. [CrossRef]
Figure 1.
Complications of obesity. COPD, chronic obstructive pulmonary disease; IBD, inflammatory bowel disease; PCOS, polycystic ovary syndrome; T2D, type 2 diabetes. Reproduced under the Creative Commons Attribution License terms from, Tan et al., [1].
Figure 1.
Complications of obesity. COPD, chronic obstructive pulmonary disease; IBD, inflammatory bowel disease; PCOS, polycystic ovary syndrome; T2D, type 2 diabetes. Reproduced under the Creative Commons Attribution License terms from, Tan et al., [1].
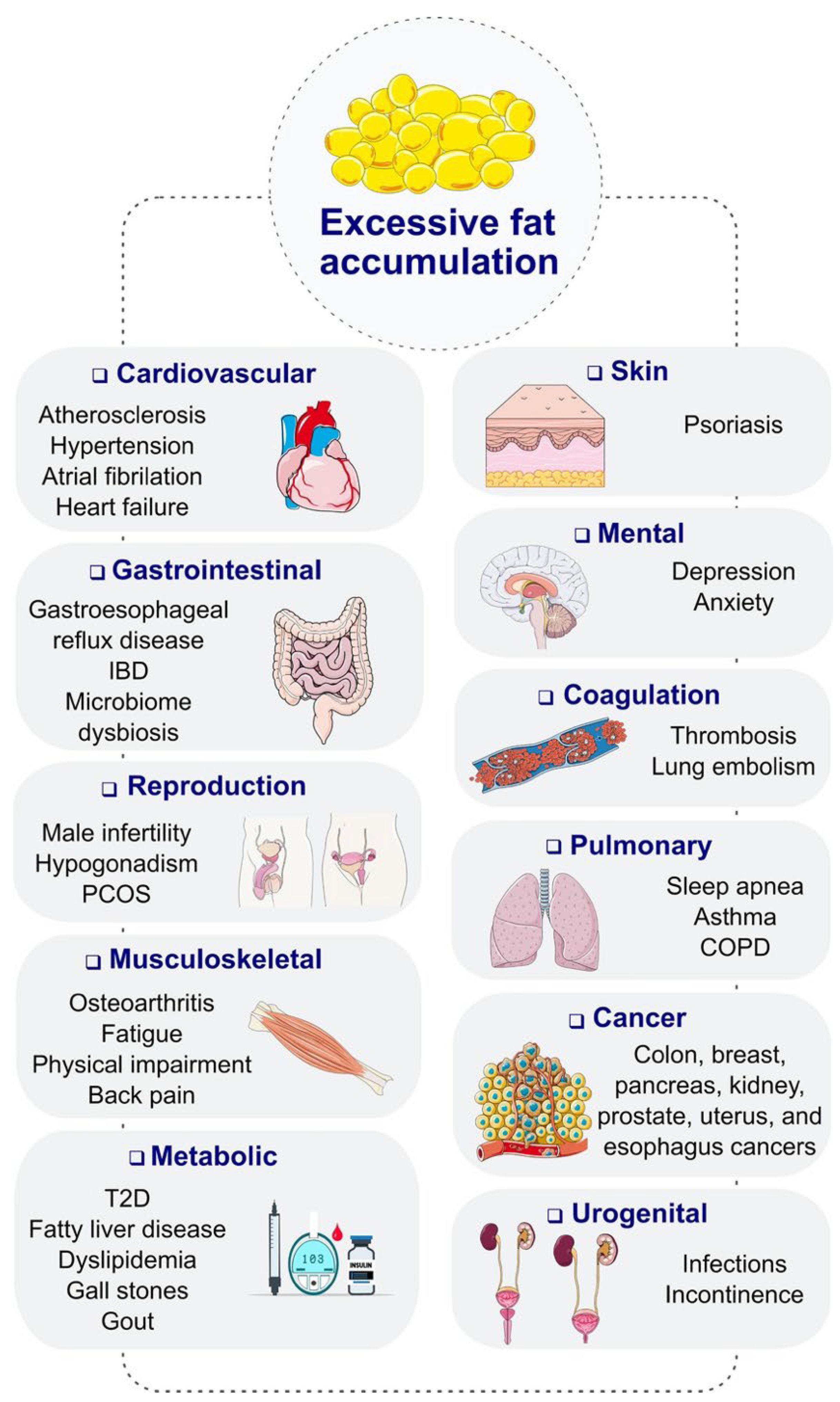
Figure 2.
Mechanism of the insulinotropic effects of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
Figure 2.
Mechanism of the insulinotropic effects of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
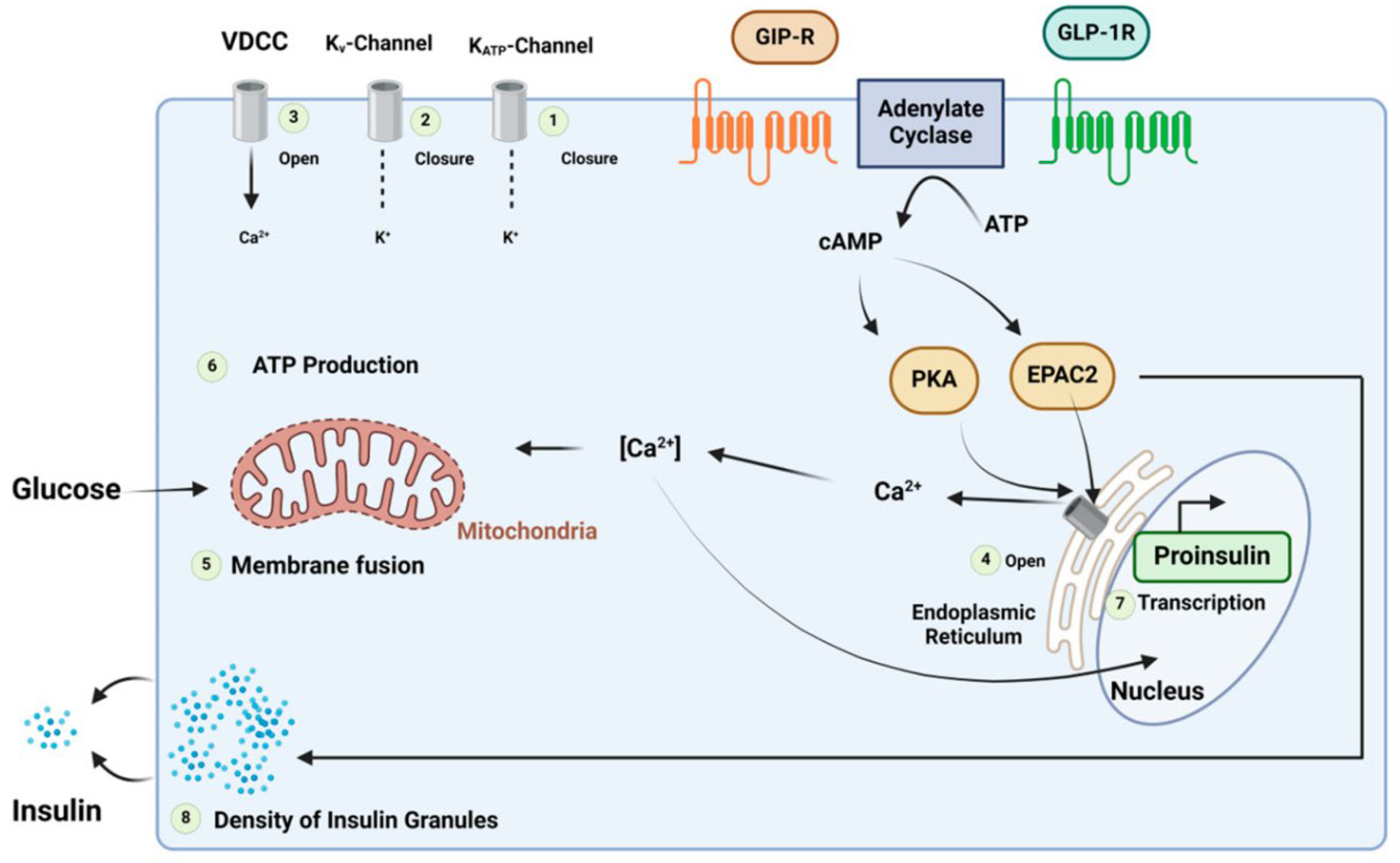
Figure 3.
Chemical structure of Exenatide. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
Figure 3.
Chemical structure of Exenatide. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
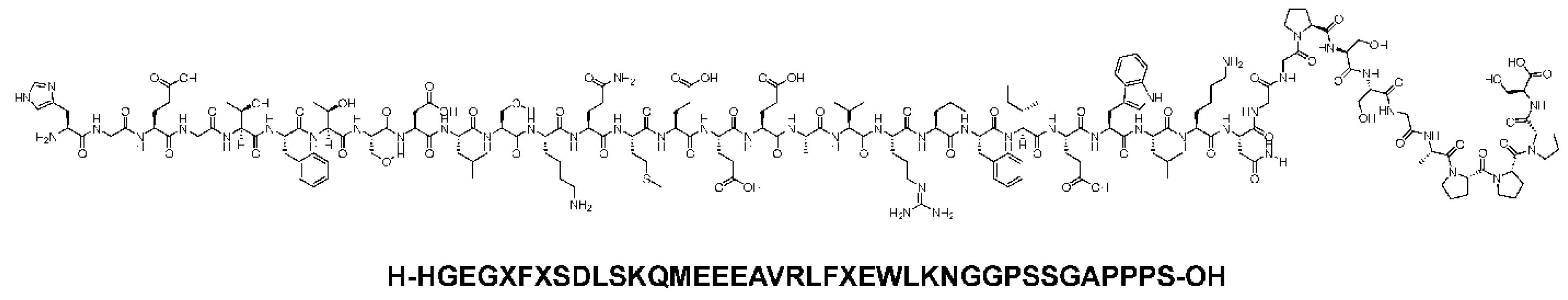
Figure 4.
Chemical structure of liraglutide. Black: peptide backbone; red: hexadecanoyl; pink: Glu. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
Figure 4.
Chemical structure of liraglutide. Black: peptide backbone; red: hexadecanoyl; pink: Glu. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
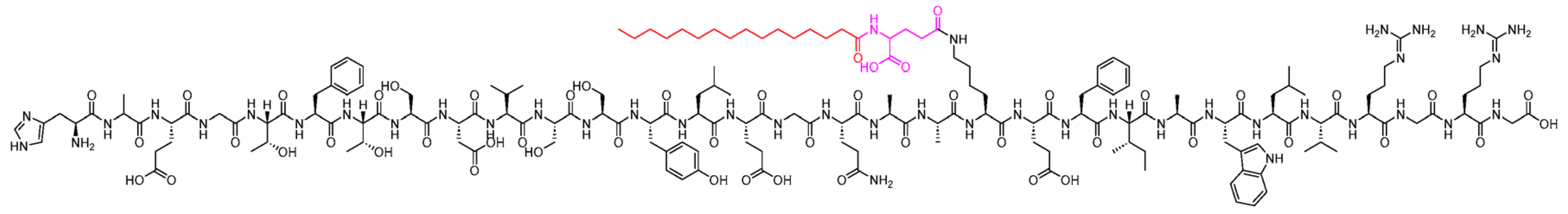
Figure 5.
Chemical structure of Dulaglutide. Black: peptide backbone; red: hexadecanoyl; pink: Glu. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
Figure 5.
Chemical structure of Dulaglutide. Black: peptide backbone; red: hexadecanoyl; pink: Glu. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
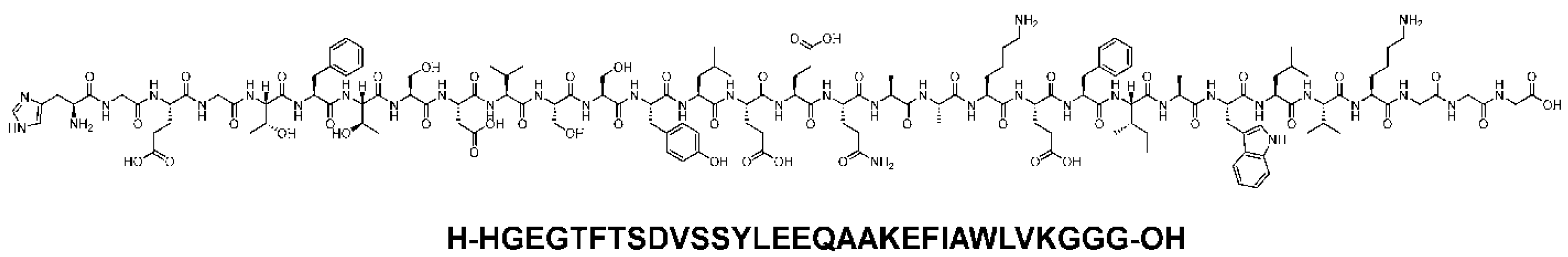
Figure 6.
Chemical structure of Semaglutide. Black: peptide backbone; red: 17-carboxyheptadecanoyl (C18 diacid); pink: Glu; blue: 8-amino-3,6-dioxaoctanoic acid (ADO). Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
Figure 6.
Chemical structure of Semaglutide. Black: peptide backbone; red: 17-carboxyheptadecanoyl (C18 diacid); pink: Glu; blue: 8-amino-3,6-dioxaoctanoic acid (ADO). Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
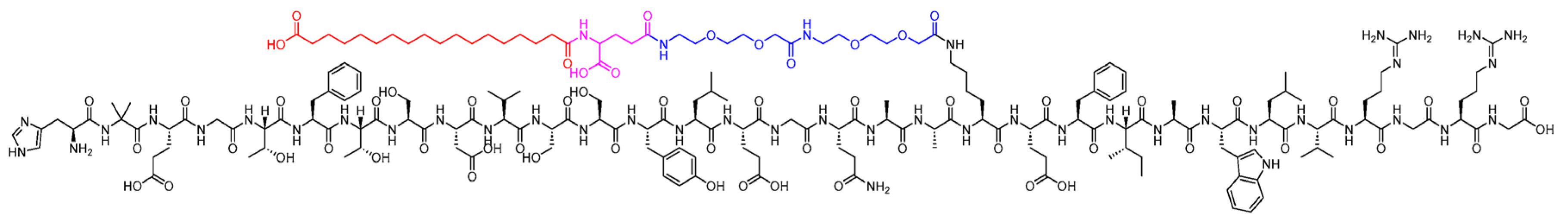
Figure 7.
Chemical structure of Albiglutide. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
Figure 7.
Chemical structure of Albiglutide. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
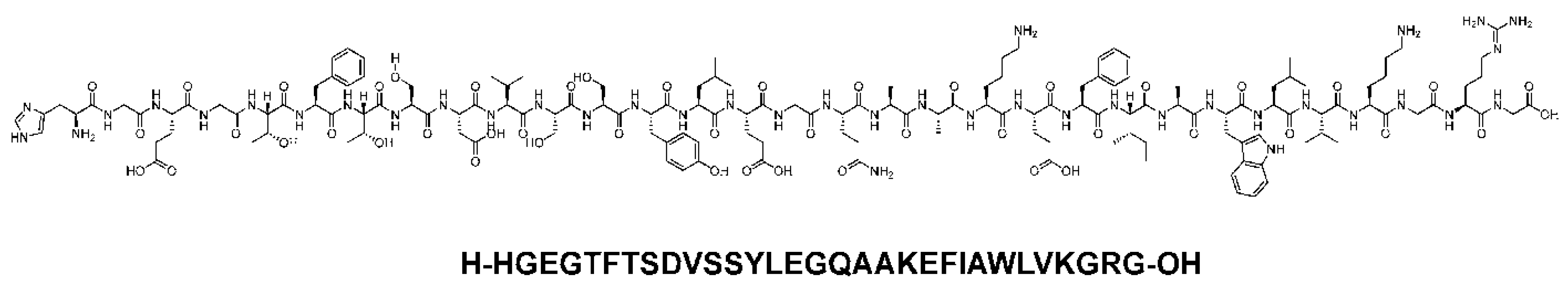
Figure 8.
Chemical structure of tirzepatide. Pink: C20 fatty acid diacidic moiety. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
Figure 8.
Chemical structure of tirzepatide. Pink: C20 fatty acid diacidic moiety. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
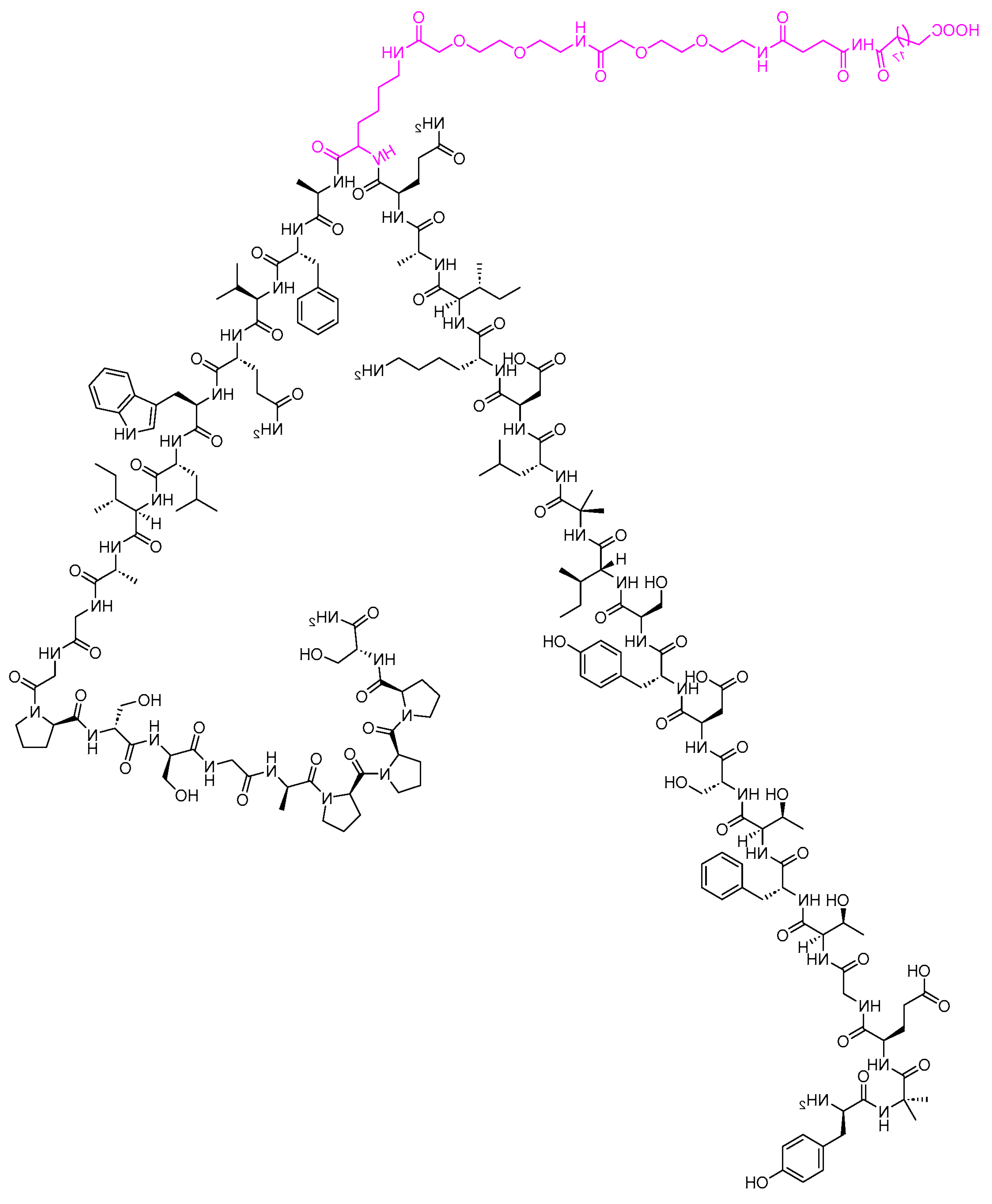
Figure 9.
Chemical structure of lixisenatide. Blue: difference from exenatide. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
Figure 9.
Chemical structure of lixisenatide. Blue: difference from exenatide. Reproduced under the Creative Commons Attribution License terms from, Al Musaimi [12].
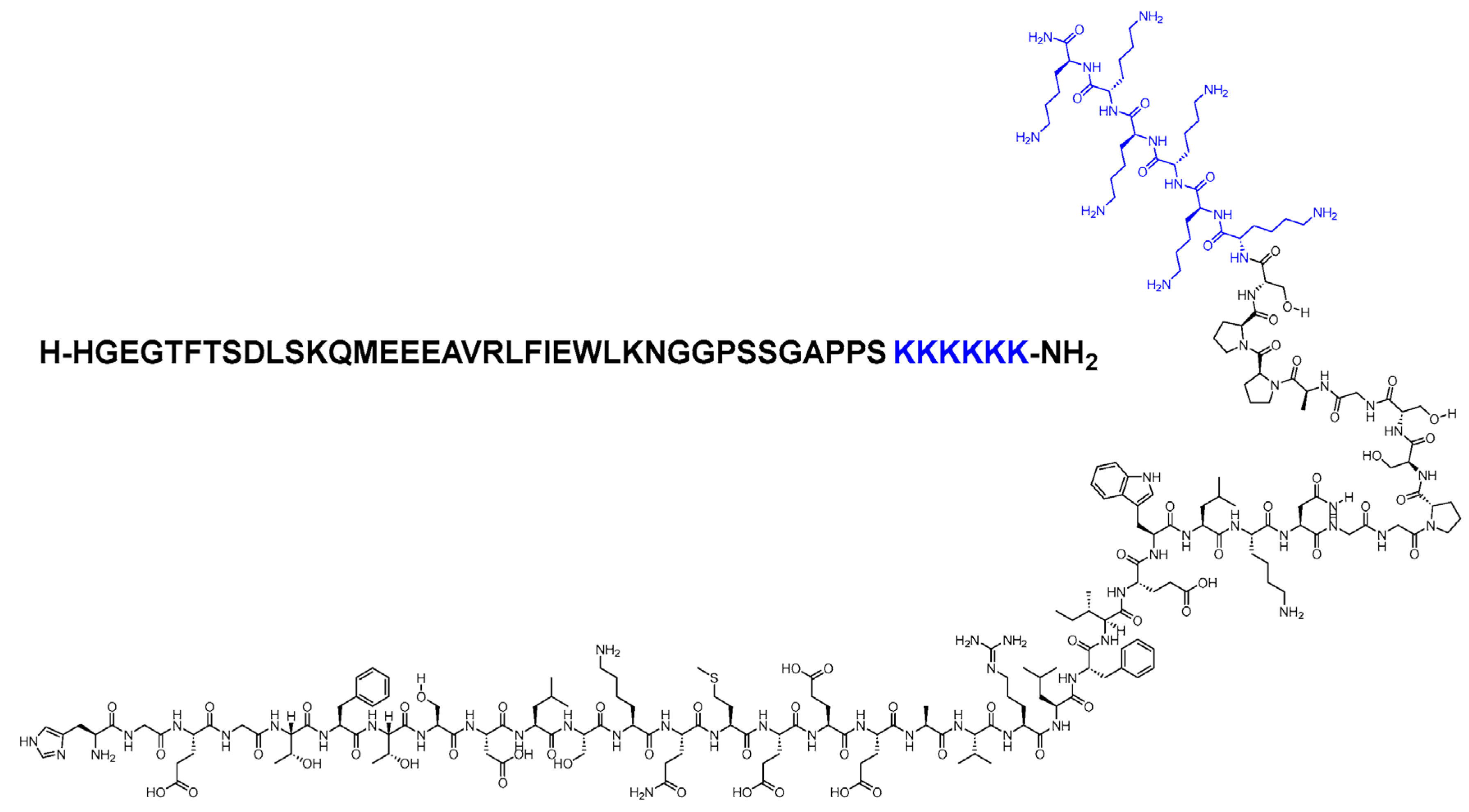
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



