
Submitted:
21 May 2024
Posted:
22 May 2024
You are already at the latest version
Abstract
Despite a large amount of research on synchronous and mutually induced kidney and heart damage, the basis of the disease is still not fully clarified. Healthy mitochondria are essential for normal kidney and heart function. Mitochondrial dysfunction occurs when the clearance or process of generation and fragmentation of mitochondria is disturbed. The kidney is the second organ after the heart in the number of mitochondria. Kidney tubules are rich in mitochondria due to the high energy requirements for absorption processes of large amounts of ultrafiltrate and dissolved substances.
The place of action of oxidative stress is the influence on the balance in the production and breakdown of mitochondrial reactive oxygen species .
A more precise determination of the place and role of key factors that play a role in the onset of the disease is necessary for understanding the nature of the onset of the disease and the creation of therapy in the future.
The narrative review integrates results found in previously performed studies which have evaluated oxidative stress participation in renocardial syndrome type 3.
Keywords:
oxidative stress
; acute heart failure
; cardiorenal syndrome
1. Introduction
Acute kidney injury is a complex clinical syndrome. A general definition of acute kidney injury (AKI) says that is an abrupt decrease in functional reserves of glomeruli resulting in the increase in nitrogenous waste products in persons who had a normal anatomic quantum of healthy renal tissue until then. According to the last nomenclature for kidney function and disease, a definition and classification of K/DIGO guideline from 2012 is still accepted and according to it, acute kidney injury is a subgroup of acute kidney diseases. It is an abrupt decrease in the intensity of glomerular filtration, which is manifested and determined by the increase in serum creatinine and different urine volume or oliguria. The time, during which the renalfunction decreases, is different ,and it is measured in hours and days [1]. The occurrence of acute kidney injury is associated with a prolonged stay during hospital treatment, high mortality and increased cost of treating patients[2,3,4,5]. In addition, the onset, severity, number of episodes, duration and outcome of acute kidney function loss are affected by the underlying disease, comorbidities, altered pharmacodynamic profile of drugs and risk factors[6,7]. Due to the frequent occurrence of multiorgan dysfunction in acute kidney injury, the involvement of the scientific and professional public is especially dedicated to understanding the connection between the kidneys and other organs[8,9,10]. Heart failure is a structural or functional disorder of the heart that prevents normal inflow or unhindered emptying of the ventricles, whereby circulatory failure is not a mandatory phenomenon. Acute decompensated heart failure caused by the sudden development of renal injury is a life-threatening condition whose main clinical feature, in addition to cardiogenic shock and acute pulmonary oedema, may also be severe arrhythmias[11]. Acute cardiorenal and renocardial connection is based on hemodynamic and non-hemodynamic mechanisms and metabolic abnormalities that, together with other mechanisms, accelerate the functional damage of both organs[12,13,14,15].
Acute kidney injury is not only an indicator of the severity of the disease but activates other mechanisms in which it plays a leading role and causes long-term consequences on the function of other organs and systems[16,17,18,19,20]. It became clear that the universal clinical use of biomarkers in different forms of acute kidney injury is not possible because each etiopathogenetic mechanism leads to different molecular, cellular and functional changes [21,22,23,24,25,26].
The fact is that all clinical manifestations of acute kidney injury cannot be explained by one pathophysiological mechanism[27,28,29]. One of the basic pathophysiological mechanisms that in acute kidney damage affects the damage to other organs is oxidative stress.
Oxidative stress has been identified as a pathogenetic mechanism in various acute and chronic diseases, including primary and secondary kidney and heart diseases. The result is an imbalance between the creation of reactive oxygen species (ROS) and the endogenous antioxidant capacity[30,31,32]. It plays a key role in primary heart and kidney disease mechanisms. The capacity of oxidative stress to introduce the affected cells in the body into processes from apoptosis to necrosis is realized with the help of ROS, which, although created only from one source, are capable of initiating a series of new reactions with short-term and long-term consequences[33,34,35,36].
The current research aims to diagnose numerous forms of acute kidney damage more precisely (such as subacute, subclinical, transitory, prolonged or severe) and apply treatment according to the initial lesion and prevent potential damage to other organs.
Bearing in mind that acute kidney injury is not just a naive observer but an actor in acute disease, this narrative review deals with the current topic related to cardiovascular consequences caused by local and systemic oxidative stress during acute kidney injury.
1.1. Oxidative Stress and Mechanisms of Its Occurrence
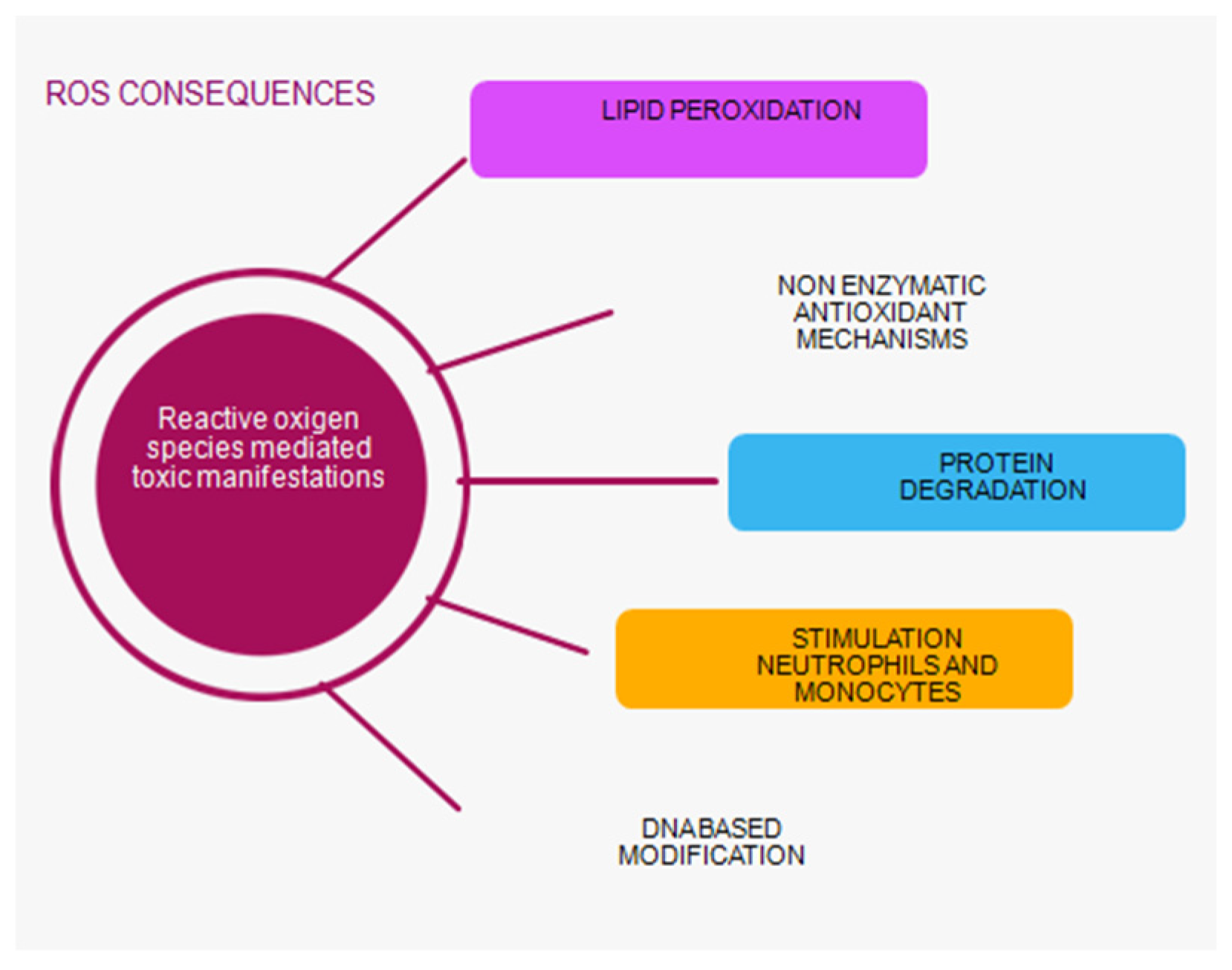
Oxidative stress occurs as a result of a disturbed balance between oxidant activity, i.e. creation of free radicals and the function of the antioxidant system in the body. Damage to the structure of the cell and disruption of its function occurs during a large accumulation of oxidants and antioxidants. This imbalance plays a major role in the oxidative damage of the cell, on the molecules of proteins, lipids, carbohydrates, enzymes and nucleic acids. In the role of oxidases, there are reactive oxygen species (ROS) formed in these reactions, which can be used as biomarkers of oxidative stress. ROS in small concentrations is important for maintaining homeostasis at a given moment. A small increase in ROS and reactive nitrogen species (RNS) levels, which are responsible for normal redox signalling, promotes cell proliferation and differentiation [36,37,38]. Oxidative stress can directly affect DNA and initiate mutagenic lesions, and indirectly by initiating autocatalytic lipid peroxidation, it can influence the formation of DNA adducts. During lipid peroxidation, which is the most significant negative consequence of synthesizing ROS, genotoxic decomposition products are created, of which the indicator of the level of lipid peroxidation is TBARS, thiobarbituric acid, and its end product[39,40]. It has been shown that oxidized proteins, especially products of oxidative modification of protein - AOPP, are created during the activation of oxidative activity at the level of neutrophils and monocytes. Stimulation of these cells causes the so-called monocyte respiratory burst and the secretion of various inflammatory mediators. Their measurement is also a marker of monocyte activation (Scheme 1)[41,42].
The final and most mutagenic product of lipid peroxidation is malonyl dialdehyde (MDA). It is a marker of oxidative stress that indicates the degree of cell membrane damage induced by free radicals. Intense lipid peroxidation of cell membranes leads to loss of turgor of cell membranes, reduction of membrane potential and increase of their permeability to hydrogen ions, leading to cell rupture[43,44,45]. Thiols are the main components of extracellular non-enzymatic antioxidant mechanisms. Thiol (R-SX sulfhydryl group) is an organic sulfur compound that can remove free radicals. A reduced concentration of free thiol groups enables oxidative stress to be maintained. Thiols are a biomarker for systemic reduction status and degree of systemic oxidative stress. Other antioxidant compounds such as glutathione, homocysteine, and cysteine contain low molecular weight (LMB) thiols and are less important for antioxidant capacity. Free thiols have the largest share because a high level of systemic free thiols indicates a more favourable redox status. Plasma proteins contain the largest amount of redox-active thiol groups. Thiols are a biomarker for identifying patients at risk of progression and development of acute kidney injury (AKI). Thiols as a pathophysiological indicator are key transduction elements in redox signalling, which is why they are recognized as indicators of oxidative stress. Research has shown that there is a correlation between low levels of thiols and AKI, which confirmed once again that oxidative stress plays an important role in the pathophysiology of AKI[46,47,48,49].
1.2. Oxidative Stress in Patients with AKI
In the kidneys, the blood flow and glomerular filtration rate are kept constant with the help of mechanisms of autoregulation, myogenic response and tubuloglomerular feedback, which are under the influence of nitrogen oxide (NO) and superoxide anion (O2)[50]. These vasodilator molecules protect renal endothelial and mesangial cells from apoptosis and fibrosis by stimulating antioxidant genes, while NOS inhibition reduces renal blood flow and increases renal vascular resistance even at concentrations that do not affect blood pressure[51]. ROS produced by mitochondria can have a renoprotective effect by moderately increasing the concentration of hypoxia-inducible factors (HIF) in endothelial cells and stimulating the synthesis of the main regulator of the oxidative stress response (NpF2) nuclear factor erythroid 2-related factor 2[52].
The main factor for maintaining physiological tone in the blood vessels of the kidneys, endogenous O2, is induced by NADPH oxidase (NOX) in the blood vessels. NADPH oxidase and xanthine oxidase are enzymes that are involved in the occurrence of oxidative stress. In polymorphonuclear leukocytes, under the action of NADPH oxidase, free radicals are created during the process of converting oxygen molecules into superoxide anions. Further, the superoxide anion is converted into hydrogen peroxide H2O2. Both products of NADPH oxidase activity, superoxide anion and hydrogen peroxide are precursors for the creation of stronger oxidants by creating conditions for other reactions[53]. Nitrosative stress occurs when the superoxide anion reacts with nitrogen oxide, and on this occasion, the toxic product peroxynitrite is created[54].
The reaction known as the Fenton reaction of classic oxidative stress occurs as a result of the reaction of hydrogen peroxide and intracellular iron. The Haber Weiss reaction occurs as a result of the interaction of superoxide anion and hydrogen peroxide, the product of which is the hydroxyl radical[55].
Chlorinated stress occurs when hydrogen peroxide in the presence of chlorine, and under the action of myeloperoxidase (MPO) from polymorphonuclear leukocytes, turns into hypochlorous acid, which reacts with endogenous amines, creating chloramines[56]. Degradation of arachidonic acid in the process of lipid peroxidation by enzymatic and non-enzymatic reactions produces MDA. Once created, it undergoes oxidation under the influence of enzymes from the mitochondria or reacts with proteins on the cell membrane or with DNA, causing mutagenic damage. The concentration of MDA in the urine is directly proportional to the degree of kidney tubule damage, which is why it can be a good indicator of tubule damage in acute renal failure. Processes of stimulated lipid peroxidation increase the risk for ischemia-reperfusion damage. An increase in MDA concentration is not only a sign of activity but also a consequence of the accumulation of oxidative stress molecules that damaged kidneys cannot excrete[57,58,59].
1.3. The Role of Mitochondria in Acute Renocardial Syndrome
Mitochondria play a major role in acute renocardial syndrome because they make up a third of the cell's volume, play a decisive role in interactions with other organelles in the cell, and affect every biochemical process in the cell. The amount of energy produced by mitochondria almost completely meets the body's needs.
The antioxidant capacity of mitochondria reduces H2O2 to water and thus physiologically ensures a low level of ROS. In disturbed conditions, ROS are produced in larger quantities and exceed the antioxidative capacity of the organelle. As a consequence of accumulation, mitochondrial ROS (mtROS) rapidly react with NO, release H2O2 into the cytoplasm and form an environment for nitrification and worsening of oxidative damage outside the mitochondria[60].
The essence of preserving cell vitality and mitochondrial function is the balance between the production and elimination of mtROS.
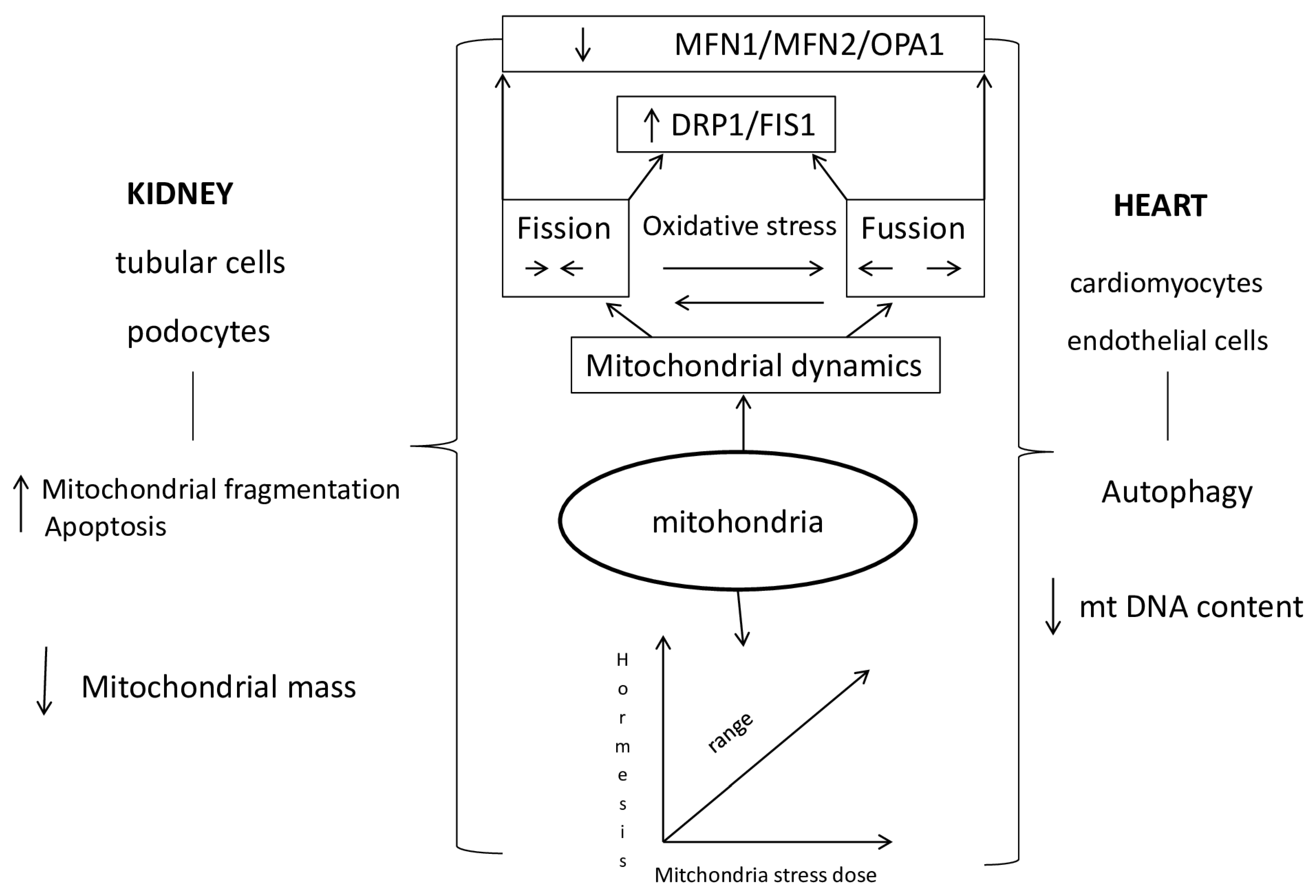
Once disturbed homeostasis in mitochondria caused by oxidative stress further increases the production of ROS and leads to the so-called "ROS induced ROS to release" vicious circle, disruption of mitochondrial function and morphology, impaired mitochondrial wall permeability and cell death. Reactive oxygen species stress increases the tendency for mutations that reduce ATP synthesis, leading to the inactivation of the calcium channel pump and a consequent increase in intracellular calcium, while activating phospholipase promotes the breakdown of phospholipids on the membrane[61,62]. Mitochondrial dysfunction is the basis of kidney and heart diseases. The most important event in kidney damage during AOB is tubule cell apoptosis, which leads to mitochondrial fragmentation, resulting in reduced energy metabolism and increased ROS generation. An increased amount of mt ROS affects the function of kidney and heart cells in acute renocardial syndrome. Mitochondrial morphological changes, fission and fusion, are mediated by guanosine triphosphatases (GTPases). /The fusion process of the outer membrane is regulated by mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2), while the fusion of the inner membrane is controlled by optic atrophy 1 (OPA1). There is a direct connection between the morphology of mitochondria and the energy state of the cell. Mitochondrial fragmentation is dependent on Mfn2 expression. Greater expression of mitofusin 2 stimulates mitochondrial oxidative phosphorylation OXPHOS reactions in mitochondrial metabolism. A decrease in the activity of the effector OPA1 promotes mitochondrial fragmentation and reduces oxygen consumption.// Fission is regulated by protein 1, which is related to dynamin-related protein 1 (Drp1), otherwise, a cytosolic protein that activates the mitochondrial membrane. Activation of Drp1 in damaged mitochondria of the heart during acute kidney injury plays an important role in cardiac dysfunction[63]. Impaired fission fission (FIS1) is directly caused by impaired function of mitochondrial dynamic proteins. The consequence of this disorder is the accumulation of mtDNA mutations and consequently damaged proteins. In addition, the parts of mitochondria produced by fission produce larger amounts of reactive oxygen species that damage cells with oxidative stress. The size of mitochondrial fragments is related to the performance of mitochondrial oxidative phosphorylation (OXPHOS).
At the level of damaged heart cells, as a result of gene ablation, increased protein expression and insufficient function of inhibitory mechanisms, there is an increase in the number of mitochondria, a change in their shape, apoptosis and severe contractile damage of cardiomyocytes[64].
Stabilization of the structure of mitochondrial cristae, increase in respiratory function of mitochondria, reduction of ROS release, apoptosis in heart cells, as well as improvement of heart function, is achieved by increasing the concentration of intracellular NAD+ and the enzyme responsible for its production (nicotinamide mononucleotide adenosyl transferase)[65,66].
1.4. Mechanisms of Mitochondrial Dysfunction in Cardiorenal Syndrome Type 3
Mitochondria play a major role in cell function by generating ATP through oxidative phosphorylation of fatty acids. They adapt to the needs of the cell and the extracellular space by forming a network of mitochondria that change in number and appearance[67,68,69].
Fusion of mitochondria serves to keep themselves healthy and increase their capacity for oxidative phosphorylation. Deficiency of Mfn2 in the proximal epithelial cells of the kidneys caused an exceptional susceptibility to apoptosis and a significant fragmentation of mitochondria. In adults, the deficiency of Mfn1 and 2 leads to hypertrophy and dilatation of the heart muscle [70,71].
Mitochondria fission removes damaged mitochondria and changes the arrangement of the contents in the new cell created during mitosis. Early renal tubule cell damage is manifested by DRP1 activation in cisplatin-induced nephrotoxicity. In diabetics, the main contribution to the occurrence of oxidative stress is the upregulation of DRP1, while the downregulation of DRP1 improves the morphology of mitochondria and reduces the apoptosis of kidney cells. After unilateral ureteral obstruction, downregulation of DRP1 improves mitochondrial function and reduces the proliferation of fibroblasts by hypoxia-stimulated TGFβ1, a key inducer of fibrosis in the renal proximal tubules. Mitochondrial fission is important for heart muscle function. Increased expression of DRP1 and Mfn1 was found in the heart muscle of patients with heart failure, while overexpression of Mfn2 in an experimental model slowed endothelial dysfunction and the initiation of atherosclerosis. Inhibition of DRP1 in cardiac muscle reduces mitochondrial fragmentation and myocardial infarct volume [73,74,75].
In the human body, the heart, blood vessels and kidneys are the organs with the largest number of mitochondria because they need the largest amount of energy for their function.
During acute kidney damage as well as acute heart damage, the cells of these organs are insufficient for the oxidation of fatty acids, due to which ATP is depleted and lipids accumulate in the cell, and an alternative path in the tubule cells in the kidneys for energy restoration and ATP synthesis is achieved with the help of enzymes glycolysis, while this modification in the heart has not been insufficiently researched for the time beeing[76,77,78].
Reactive oxygen species (mtROS) are removed in mitochondria using enzymatic and non-enzymatic reactions. Superoxide anion (O2-) is converted into H2O2 by means of superoxidase dismutase (SOD). The main roles in mitochondria for reducing the concentration of hydrogen peroxide (H2O2) are played by mitochondrial catalase and glutathione peroxidase [79,80,81].
Preclinical research has shown that acute kidney damage affects the metabolism in heart muscle cells through signaling pathways that are a consequence of mitochondrial function, and cause disruption of the metabolism of pyruvate, glyoxylate, dicarboxylic acid, starch, sucrose and amino acid synthesis. The main regulator of myocardial damage in acute kidney damage is growth factor receptor binding protein 2 (Grb2). Activation of this protein or the use of Grb2 inhibitors promotes inhibition of the Akt/mTOR signaling pathway and thus leads to disruption of mitochondrial function in cardiomyocytes [82,83,84,85].
The idea of hormesis was presented for the first time in the 16th century in connection with the bimodal response of cells to external factors, while the term mitohormesis was introduced in 2006 for the response of mitochondria in subepithelial cells to a dose of stress. The knowledge that the majority of proteins in mitochondria are coded via nuclear DNA pointed to the fact of the importance of retrograde signaling from mitochondria and the adaptive response of the nucleus for mitochondrial recovery [86,87,88,89,90]. In the cells, signal molecules inform the cytosolic system about stress in the mitochondria, so that the accumulation of proteins in the cytosol of the cell activates the transcription mechanisms that are part of the mitohormetic response. The most important signaling pathway of mitochondrial stress in the cytosolic signaling system of the cell is ROS (reactive oxygen species) from mitochondria [91,92,93,94,95]. High concentrations of mitochondrial ROS are harmful, while low concentrations are a stimulus for numerous signaling pathways (Scheme 2.).
Maintaining mitochondrial homeostasis improves kidney function [96]. Mitohormetic pathways can be blocked by the use of antioxsidants, while the use of DRP1 inhibitors blocks mitochondrial fragmentation and apoptosis to protect the kidney from ischemia and acute kidney damage caused by cisplatin [97,98]. Inhibition of mitochondrial fission and stimulation of mitochondrial fusion can be a therapeutic goal in the treatment of atherosclerosis [99,100].
2. Conclusions
The exact mechanism of renocardiac syndrome type 3 is complex and not yet understood. There is a link between acute kidney injury and the pathophysiology of developing cardiovascular disease. We showed a greater understanding of the development of cardiorenal mechanisms. Acute kidney injury is associated with acute and chronic cardiovascular complications. This kind of interaction is possible in different ways and is very dynamic. Further research with area at risk mechanisms and control of oxidative stress will improve the management of acute renocardial syndrome.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable .
Acknowledgments
I thank Tasic Katarini for her administrative support .
Declaration of Conflicting Interests
The author have no potential conflicts of interest to disclose.
References
- K/DIGO AKI Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Intern Suppl. 2012, 2, S3–S106. [Google Scholar]
- Sawhney, S.; Fraser, D.S. Epidemiology of AKI: Utilizing Large Database to determine the Burden of AKI. ACKD 2017, 24, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Rizo-Topete, L.M.; Rosner, M.H.; Ronco, C. Acute Kidney Injury Risk Assessment and the Nephrology Rapid Response Team. Blood Purif. 2016, 43, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Macedo, E.; Mehta, R.L. Preventing Acute Kidney Injury. Crit Care Clin. 2015. [Google Scholar] [CrossRef]
- K/DIGO AKI Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Intern Suppl. 2012, 2, S3–S106. [Google Scholar]
- Youden, W.J. Index for rating diagnostic tests. Cancer 1950, 3, 32–35. [Google Scholar] [CrossRef]
- Kellum, J.A.; Ronco, C.; Bellomo, R. Acute kidney disease and the community. Lancet 2016, 387, 1974–1976. [Google Scholar] [CrossRef] [PubMed]
- Hertzberg, D.; Rydén, L.; Pickering, J.W.; Sartipy, U.; Holzmann, M.J. Acute kidney injury-an overview of diagnostic methods and clinical management. Clin. Kidney J. 2017, 10, 323–331. [Google Scholar] [CrossRef]
- Feltes, C.M.; Van Eyk, J.; Rabb, H. Distant-organ changes after acute kidney injury. Nephron Physiol. 2008, 109, 80–84. [Google Scholar] [CrossRef]
- Tecson, K.M.; Hashemi, H.; Afzal, A.; Gong, T.A.; Kale, P.; McCullough, P.A. Community-Acquired Acute Kidney Injury as a Risk Factor of de novo Heart Failure Hospitalization. Cardiorenal Med. 2019, 9, 252–260. [Google Scholar] [CrossRef]
- ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2021. Eur Heart J 2021, 42, 3599–3726. [CrossRef]
- Pliquett, R.U. Cardiorenal Syndrome: An Updated Classification Based on Clinical Hallmarks. J. Clin. Med. 2022, 11, 2896. [Google Scholar] [CrossRef] [PubMed]
- Virzì, G.M.; Breglia, A.; Castellani, C.; Ankawi, G.; Bolin, C.; de Cal, M.; Cianci, V.; Angelini, A.; Vescovo, G.; Ronco, C. Lipopolysaccharide in systemic circulation induces activation of inflammatory response and oxidative stress in cardiorenal syndrome type 1. J. Nephrol. 2019, 32, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Virzì, G.M.; Breglia, A.; Brocca, A.; de Cal, M.; Bolin, C.; Vescovo, G.; Ronco, C. Levels of Proinflammatory Cytokines, Oxidative Stress, and Tissue Damage Markers in Patients with Acute Heart Failure with and without Cardiorenal Syndrome Type 1. Cardiorenal Med. 2018, 8, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Virzì, G.M.; Clementi, A.; de Cal, M.; Brocca, A.; Day, S.; Pastori, S.; Bolin, C.; Vescovo, G.; Ronco, C. Oxidative Stress: Dual Pathway Induction in Cardiorenal Syndrome Type 1 Pathogenesis. Oxidative Med. Cell. Longev. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Matheny, M.E.; Greevy, R.A.; Eden, S.K.; Perkins, A.M.; Parr, S.K.; Fly, J.; Abdel-Kader, K.; Himmelfarb, J.; Hung, A.M.; et al. Acute Kidney Injury and Risk of Incident Heart Failure Among US Veterans. Am. J. Kidney Dis. 2018, 71, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Gammelager, H.; Christiansen, C.F.; Johansen, M.B.; Tønnesen, E.; Jespersen, B.; Sørensen, H.T. Three-year risk of cardiovascular disease among intensive care patients with acute kidney injury: a population-based cohort study. Crit. Care 2014, 18, 492–492. [Google Scholar] [CrossRef] [PubMed]
- Dieter, B.P.; Daratha, K.B.; McPherson, S.M.; Short, R.; Alicic, R.Z.; Tuttle, K.R. Association of Acute Kidney Injury with Cardiovascular Events and Death in Systolic Blood Pressure Intervention Trial. Am. J. Nephrol. 2019, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Sohel, B.M.; Rumana, N.; Ohsawa, M.; Turin, T.C.; Kelly, M.A.; Al Mamun, M. Renal function trajectory over time and adverse clinical outcomes. Clin. Exp. Nephrol. 2016, 20, 379–393. [Google Scholar] [CrossRef]
- Di Lullo, L.; Reeves, P.B.; Bellasi, A.; Ronco, C. Cardiorenal Syndrome in Acute Kidney Injury. Semin. Nephrol. 2019, 39, 31–40. [Google Scholar] [CrossRef]
- Mehta, S.; Chauhan, K.; Patel, A.; Patel, S.; Pinotti, R.; Nadkarni, G.N.; Parikh, C.R.; Coca, S.G. The prognostic importance of duration of AKI: a systematic review and meta-analysis. BMC Nephrol. 2018, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G. Ptolemy and Copernicus Revisited. Clin. J. Am. Soc. Nephrol. 2018, 13, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.J.; Hsu, C.; Parikh, R.V.; Leong, T.K.; Tan, T.C.; Walia, S.; et al. Non-recovery from dialzsis-requiring acute kidneyinjurz and short-term mortality and cardiovascular risk: a cohort study. BMC Nephrol. 2018, 19, 134. [Google Scholar] [CrossRef]
- Bhatraju, P.K.; Zelnick, L.R.; Chinchilli, V.M.; Moledina, D.G.; Coca, S.G.; Parikh, C.R.; Garg, A.X.; Hsu, C.-Y.; Go, A.S.; Liu, K.D.; et al. Association Between Early Recovery of Kidney Function After Acute Kidney Injury and Long-term Clinical Outcomes. JAMA Netw. Open 2020, 3, e202682–e202682. [Google Scholar] [CrossRef]
- Mehta, R.L. Renal Recovery After Acute Kidney Injury and Long-term Outcomes Is Time of the Essence? JAMA Netw. Open 2020, 3, e202676–e202676. [Google Scholar] [CrossRef]
- Vaara, S.T.; Bhatraju, P.K.; Stanski, N.L.; McMahon, B.A.; Liu, K.; Joannidis, M.; Bagshaw, S.M. Subphenotypes in acute kidney injury: a narrative review. Crit. Care 2022, 26, 1–10. [Google Scholar] [CrossRef]
- Perinel, S.; Vincent, F.; Lautrette, A.; Dellamonica, J.; Mariat, C.; Zeni, F.; et al. Transient and Persistent Acute Kidney Injury and the Risk of Hospital Mortality in Critically Ill Patients: Results of a Multicenter Cohort Study. Crit Care Med. 2015, 43, e269–e275. [Google Scholar] [CrossRef] [PubMed]
- Bhatraju, P.K.; Mukherjee, P.; Robinson-Cohen, C.; O’keefe, G.E.; Frank, A.J.; Christie, J.D.; Meyer, N.J.; Liu, K.D.; Matthay, M.A.; Calfee, C.S.; et al. Acute kidney injury subphenotypes based on creatinine trajectory identifies patients at increased risk of death. Crit. Care 2016, 20, 1–10. [Google Scholar] [CrossRef]
- Flannery, A.H.; Bosler, K.; Ortiz-Soriano, V.M.; Gianella, F.; Prado, V.; Lambert, J.; Toto, R.D.; Moe, O.W.; Neyra, J.A. Kidney Biomarkers and Major Adverse Kidney Events in Critically Ill Patients. Kidney360 2021, 2, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.C.; Zager, R.A. Catalytic iron mediated renal stress responses during experimental cardiorenal syndrome 1 (“CRS-1”). Transl. Res. 2021, 237, 53–62. [Google Scholar] [CrossRef]
- Johnson, A.C.; Delrow, J.J.; Zager, R.A. Tin protoporphyrin activates the oxidant-dependent NRF2-cytoprotective pathway and mitigates acute kidney injury. Transl. Res. 2017, 186, 1–18. [Google Scholar] [CrossRef]
- Virzì, G.M.; Clementi, A.; Brocca, A.; de Cal, M.; Ronco, C. Molecular and Genetic Mechanisms Involved in the Pathogenesis of Cardiorenal Cross Talk. Pathobiology 2016, 83, 201–210. [Google Scholar] [CrossRef]
- Himmelfarb, J.; McMonagle, E.; Freedman, S.; Klenzak, J.; McMenamin, E.; Le, P.; Pupim, L.B.; Ikizler, T.A. The PICARD Group Oxidative Stress Is Increased in Critically Ill Patients with Acute Renal Failure. J. Am. Soc. Nephrol. 2004, 15, 2449–2456. [Google Scholar] [CrossRef]
- Kasuno, K.; Shirakawa, K.; Yoshida, H.; Mori, K.; Kimura, H.; Takahashi, N.; Nobukawa, Y.; Shigemi, K.; Tanabe, S.; Yamada, N.; et al. Renal redox dysregulation in AKI: application for oxidative stress marker of AKI. Am. J. Physiol. Physiol. 2014, 307, F1342–F1351. [Google Scholar] [CrossRef]
- Just, A. Nitric Oxide and Renal Autoregulation. Kidney Blood Press. Res. 1997, 20, 201–204. [Google Scholar] [CrossRef]
- Tomsa, A.M.; Alexa, A.L.; Junie, M.L.; Rachisan, A.L.; Ciumarnean, L. Oxidative stress as a potential target in acute kidney injury. PeerJ 2019, 7, e8046. [Google Scholar] [CrossRef]
- Dennis, J.M.; Witting, P.K. Protective role for antioxidants in acute kidney disease. Nutrients 2017, 9, 718. [Google Scholar] [CrossRef]
- Araujo, M.; Welch, W.J. Oxidative stress and nitric oxide in kidney function. Curr. Opin. Nephrol. Hypertens. 2006, 15, 72–77. [Google Scholar] [CrossRef]
- Nilakantan, V.; Hilton, G.; Maenpaa, C.; Van Why, S.K.; Pieper, G.M.; Johnson, C.P.; Shames, B.D. Favorable balance of anti-oxidant/pro-oxidant systems and ablated oxidative stress in Brown Norway rats in renal ischemia-reperfusion injury. Mol. Cell. Biochem. 2007, 304, 1–11. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Anderstam, B.; Ann-Christin, B.-H.; Valli, A.; Stenvinkel, P.; Lindholm, B.; Suliman, M.E. Modification of the oxidative stress biomarker AOPP assay: Application in uremic samples. Clin. Chim. Acta 2008, 393, 114–118. [Google Scholar] [CrossRef]
- Selmeci, L.; Seres, L.; Antal, M.; Lukács, J.; Regöly-Mérei, A.; Acsády, G. Advanced oxidation protein products (AOPP) for monitoring oxidative stress in critically ill patients: a simple, fast and inexpensive automated technique. cclm 2005, 43, 294–7. [Google Scholar] [CrossRef]
- Mahzari, S.; Hosseinian, S.; Hadjzadeh, M.-A.; Mohebbati, R.; Noshahr, Z.S.; Rad, A.K. Kidney dysfunction and oxidative stress in doxorubicin-induced nephrotic rat: Protective role of sesame oil. Saudi Journal of Kidney Diseases and Transplantation 2021, 32, 1243–1252. [Google Scholar] [CrossRef]
- Irigaray, P.; Caccamo, D.; Belpomme, D. Oxidative stress in electrohypersensitivity self-reporting patients: Results of a prospective in vivo investigation with comprehensive molecular analysis. Int. J. Mol. Med. 2018, 42, 1885–1898. [Google Scholar] [CrossRef]
- Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef]
- Pfaff, A.R.; Beltz, J.; King, E.; Ercal, N. Medicinal Thiols: Current Status and New Perspectives. Mini-Reviews Med. Chem. 2020, 20, 513–529. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxidants Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef]
- Gillis, B.S.; Arbieva, Z.; Gavin, I.M. Analysis of lead toxicity in human cells. BMC Genom. 2012, 13, 344–344. [Google Scholar] [CrossRef]
- Baylis, C.; Qiu, C. Importance of nitric oxide in the control of renal hemodynamics. Kidney Int. 1996, 49, 1727–1731. [Google Scholar] [CrossRef]
- Pavlakou, P.; Liakopoulos, V.; Eleftheriadis, T.; Mitsis, M.; Dounousi, E. Oxidative Stress and Acute Kidney Injury in Critical Illness: Pathophysiologic Mechanisms—Biomarkers—Interventions, and Future Perspectives. Oxidative Med. Cell. Longev. 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Lushchak, V.I. Classification of oxidative stress based on its intensity. EXCLI J. 2014, 13, 922–937. [Google Scholar]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive Oxygen Species and Neutrophil Function. Annu. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free Radicals, Antioxidants in Disease and Health. Free Radic. Antioxid. 2008, 4, 89. [Google Scholar] [CrossRef]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 1–12. [Google Scholar] [CrossRef]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and physiological role of reactive oxygen species - the good, the bad and the ugly. Acta Physiol. 2015, 214, 329–348. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Verhaar, M.C.; Westerweel, P.E.; van Zonneveld, A.J.; Rabelink, T.J. Free radical production by dysfunctional eNOS. Heart 2004, 90, 494–495. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Wagner, R. Mitochondrial Outer Membrane Channels: Emerging Diversity in Transport Processes. BioEssays 2018, 40, e1800013. [Google Scholar] [CrossRef] [PubMed]
- Krüger, V.; Becker, T.; Becker, L.; Montilla-Martinez, M.; Ellenrieder, L.; Vögtle, F.-N.; Meyer, H.E.; Ryan, M.T.; Wiedemann, N.; Warscheid, B.; et al. Identification of new channels by systematic analysis of the mitochondrial outer membrane. J. Cell Biol. 2017, 216, 3485–3495. [Google Scholar] [CrossRef]
- Baines, C.P.; Gutiérrez-Aguilar, M. The still uncertain identity of the channel-forming unit(s) of the mitochondrial permeability transition pore. Cell Calcium 2018, 73, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef]
- Takemura, K.; Nishi, H.; Inagi, R. Mitochondrial Dysfunction in Kidney Disease and Uremic Sarcopenia. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, E.; Kazachenko, A.; Vyssokikh, M.; Vasileva, A.; Tcvirkun, D.; Isaev, N.; Kirpatovsky, V.; Zorov, D. The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int. 2007, 72, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Zhang, B.; Li, Y.; Xu, X.; Lv, J.; Jia, Q.; Chai, R.; Xue, W.; Li, Y.; Wang, Y.; et al. Mithochondrial Dysfunction:An Emerging Link in the pathophysiology of Cardiorenal Syndrome. Front Cardiovasc. Med. 2022, 9, 837270. [Google Scholar] [CrossRef] [PubMed]
- Ravarotto, V.; Bertoldi, G.; Innico, G.; Gobbi, L.; Calò, L.A. The Pivotal Role of Oxidative Stress in the Pathophysiology of Cardiovascular-Renal Remodeling in Kidney Disease. Antioxidants 2021, 10, 1041. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Boncompagni, C.; Ramaccini, D.; Pedriali, G.; Bouhamida, E.; Tremoli, E.; Giorgi, C.; Pinton, P. Comprehensive Analysis of Mitochondrial Dynamics Alterations in Heart Diseases. Int. J. Mol. Sci. 2023, 24, 3414. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Zhang, J.; Gomez, H.; Kellum, J.A.; Peng, Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 2023, 19, 401–414. [Google Scholar] [CrossRef]
- Mallick, R.; Duttaroy, A.K. Modulation of endothelium function by fatty acids. Mol. Cell. Biochem. 2021, 477, 15–38. [Google Scholar] [CrossRef]
- Palm, C.L.; Nijholt, K.T.; Bakker, B.M.; Westenbrink, B.D. Short-Chain Fatty Acids in the Metabolism of Heart Failure – Rethinking the Fat Stigma. Front. Cardiovasc. Med. 2022, 9, 915102. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Lopez-Diaz, A.M.; Guerrero-Mauvecin, J.; Miguel, V.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Tubular Mitochondrial Dysfunction, Oxidative Stress, and Progression of Chronic Kidney Disease. Antioxidants 2022, 11, 1356. [Google Scholar] [CrossRef]
- Tang, C.; Cai, J.; Yin, X.-M.; Weinberg, J.M.; Venkatachalam, M.A.; Dong, Z. Mitochondrial quality control in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, M.; Xiong, L.; Fan, J.; Zhou, Y.; Li, H.; Peng, X.; Zhong, Z.; Wang, Y.; Huang, F.; et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Li, S.; Lin, Q.; Shao, X.; Zhu, X.; Wu, J.; Wu, B.; Zhang, M.; Zhou, W.; Zhou, Y.; Jin, H.; et al. Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free. Radic. Biol. Med. 2020, 152, 632–649. [Google Scholar] [CrossRef] [PubMed]
- Tagaya, M.; Kume, S.; Yasuda-Yamahara, M.; Kuwagata, S.; Yamahara, K.; Takeda, N.; Tanaka, Y.; Chin-Kanasaki, M.; Nakae, Y.; Yokoi, H.; et al. Inhibition of mitochondrial fission protects podocytes from albumin-induced cell damage in diabetic kidney disease. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2022, 1868, 166368. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Schirone, L.; Ameri, P.; Basso, C.; Catalucci, D.; Modica, J.; Chimenti, C.; Crotti, L.; Frati, G.; Rubattu, S.; et al. The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. 2020, 178, 2060–2076. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, X.; An, P.; Luo, J.; Luo, Y. Mitochondrial Homeostasis in VSMCs as a Central Hub in Vascular Remodeling. Int. J. Mol. Sci. 2023, 24, 3483. [Google Scholar] [CrossRef]
- Cooper, H.A.; Cicalese, S.; Preston, K.J.; Kawai, T.; Okuno, K.; Choi, E.T.; Kasahara, S.; Uchida, H.A.; Otaka, N.; Scalia, R.; et al. Targeting mitochondrial fission as a potential therapeutic for abdominal aortic aneurysm. Cardiovasc. Res. 2020, 117, 971–982. [Google Scholar] [CrossRef]
- Pabla, N.; Bajwa, A. Role of Mitochondrial Therapy for Ischemic-Reperfusion Injury and Acute Kidney Injury. Nephron 2021, 146, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef]
- Buchke, S.; Sharma, M.; Bora, A.; Relekar, M.; Bhanu, P.; Kumar, J. Mitochondria-Targeted, Nanoparticle-Based Drug-Delivery Systems: Therapeutics for Mitochondrial Disorders. Life 2022, 12, 657. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.-J.; Wang, Z.-Y.; Xu, L.; Chen, X.-H.; Li, X.-X.; Liao, W.-T.; Ma, H.-K.; Jiang, M.-D.; Xu, T.-T.; Xu, J.; et al. HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020, 36, 101671. [Google Scholar] [CrossRef]
- Yao, M.; Liu, Y.; Sun, M.; Qin, S.; Xin, W.; Guan, X.; Zhang, B.; He, T.; Huang, Y. The molecular mechanisms and intervention strategies of mitophagy in cardiorenal syndrome. Front. Physiol. 2022, 13, 1008517. [Google Scholar] [CrossRef] [PubMed]
- Santovito, D.; Egea, V.; Bidzhekov, K.; Natarelli, L.; Mourão, A.; Blanchet, X.; Wichapong, K.; Aslani, M.; Brunßen, C.; Horckmans, M.; et al. Noncanonical inhibition of caspase-3 by a nuclear microRNA confers endothelial protection by autophagy in atherosclerosis. Sci. Transl. Med. 2020, 12, eaaz2294. [Google Scholar] [CrossRef]
- Bugga, P.; Alam, J.; Kumar, R.; Pal, S.; Chattopadyay, N.; Banerjee, S.K. Sirt3 ameliorates mitochondrial dysfunction and oxidative stress through regulating mitochondrial biogenesis and dynamics in cardiomyoblast. Cell. Signal. 2022, 94, 110309. [Google Scholar] [CrossRef]
- Yu, R.; Lendahl, U.; Nistér, M.; Zhao, J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front. Endocrinol. 2020, 11, 374. [Google Scholar] [CrossRef]
- Merry, T.L.; Chan, A.; Woodhead, J.S.T.; Reynolds, J.C.; Kumagai, H.; Kim, S.-J.; Lee, C. Mitochondrial-derived peptides in energy metabolism. Am. J. Physiol. Metab. 2020, 319, E659–E666. [Google Scholar] [CrossRef]
- Gyuraszova, M.; Gurecka, R.; Babickova, J.; Tothova, L. Oxidative Stress in the Pathophysiology of Kidney Disease: Implications for Noninvasive Monitoring and Identification of Biomarkers. Oxidative Med. Cell. Longev. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Agborbesong, E.; Li, X. The role of mitochondria in acute kidney injury and chronic kidney disease and its therapeutic potential. Int J Mol Sci 2021, 22, 20. [Google Scholar] [CrossRef] [PubMed]
- Takemura, K.; Nishi, H.; Inagi, R. Mitochondrial Dysfunction in Kidney Disease and Uremic Sarcopenia. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Xia, F.; Li, L.; Peng, X.; Liu, W.; Zhang, Y.; Fang, H.; Zeng, Z.; Chen, Z. Novel Insights into the Molecular Features and Regulatory Mechanisms of Mitochondrial Dynamic Disorder in the Pathogenesis of Cardiovascular Disease. Oxidative Med. Cell. Longev. 2021, 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Graciani, K.M.; Chapa-Dubocq, X.R.; MacMillan-Crow, L.A.; Javadov, S. Association Between L-OPA1 Cleavage and Cardiac Dysfunction During Ischemia-Reperfusion Injury in Rats. Cell. Physiol. Biochem. 2020, 54, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Takemura, K.; Nishi, H.; Inagi, R. Mitochondrial Dysfunction in Kidney Disease and Uremic Sarcopenia. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Toan, S.; Li, R.; Zhou, H. Melatonin fine-tunes intracellular calcium signals and eliminates myocardial damage through the IP3R/MCU pathways in cardiorenal syndrome type 3. Biochem. Pharmacol. 2020, 174, 113832. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, X.; Wang, X.; Cui, S.; Liu, R.; Liu, J.; Fu, B.; Gong, M.; Wang, C.; Shi, Y.; et al. Grb2 Induces Cardiorenal Syndrome Type 3: Roles of IL-6, Cardiomyocyte Bioenergetics, and Akt/mTOR Pathway (Publication with Expression of Concern. See vol. 10, 2022). Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
Scheme 1.
Role ROS in the kidney and heart relationchip.
Scheme 2.
Mithochondrial Dysfunction in Cradiorenal Syndrome type 3.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



