
Submitted:
22 May 2024
Posted:
23 May 2024
You are already at the latest version
Abstract
Staphylococcus aureus bacteremia continues to be associated with significant morbidity and mortality, despite improvements in diagnostics and management. Persistent infections pose a major challenge to clinicians and have been consistently shown to increase the risk of mortality and other infectious complications. S. aureus, while typically not considered an intracellular pathogen, has been proven to utilize an intracellular niche, through several phenotypes including small colony variants, as a means for survival that has been linked to chronic, persistent, and recurrent infections. This intracellular persistence allows for protection from the host immune system and leads to reduced antibiotic efficacy through a variety of mechanisms. These include antimicrobial resistance, tolerance, and/or persistence in S. aureus that contribute to persistent bacteremia. This review will discuss the challenges associated with treating these complicated infections and the various methods that S. aureus uses to persist within the intracellular space.
Keywords:
antibiotic resistance
; small colony variant
; relapse
; recurrence
; bloodstream
; host-pathogen
Introduction
Antibiotic resistance in Staphylococcus aureus continues to pose significant risk to the healthcare landscape due to limited antimicrobial options [1]. However, persistent S. aureus infections present a major challenge to clinicians during treatment of “susceptible infections” in patients. This is particularly true for persistent S. aureus bacteremia, characterized by repeatedly positive Staphylococcus aureus cultures in the bloodstream despite apparently appropriate antibiotic therapy [2]. This condition is associated with increased morbidity, mortality, and healthcare costs, making its management a priority in clinical practice [2,3]. In this perspective, we overview the problem of persistent bacteremia caused by S. aureus and highlight the intracellular proclivity of the organism as a mechanism for survival in the host and defense against antibiotic treatments.
The Clinical Challenges of Persistent Staphylococcus aureus Bacteremia (SAB)
Through the years, the definition of persistent SAB continues to be refined with the contemporary definition geared toward early switching to alternative regimens [2]. This has aligned with rapid advances in molecular organism identification (i.e., MALDI-TOF and others) and automated antimicrobial susceptibility testing [4,5]. However, this definition is not standardized and therefore allows for a range of practice interpretation of when to consider changes in antibiotic therapy [2]. Traditionally, and informed from historical clinical trials, the definition of persistent bacteremia derived from the average duration of bacteremia among clinical trials of antibiotic therapy – around 7 days [6,7]. Since then, several studies have suggested earlier definitions of persistent SAB (3-5 days duration of positive cultures) due to improved outcomes noted with shorter durations of bacteremia [3,8,9]. For example, we argued persistent or prolonged bacteremia be considered as > 4 days duration of positive cultures based upon the observed increase in mortality and cytokine markers associated with this threshold from regression analysis [8,10]. This time frame corresponds with the final identification and susceptibility profile of S. aureus obtained from blood cultures accounting for time to growth in blood culture vials, identification by molecular methods, and phenotypic susceptibility. However, recently Holland et al proposed considering persistent bacteremia earlier, after one calendar day, for further diagnostic evaluation and consideration of antibiotic failure for implementing alternative treatment regimens [2]. This suggestion is supported by a study by Kuehl et al, which found that patients who had positive blood cultures beyond 24 hours of antibiotic therapy were at a higher risk of mortality and metastatic infections [11]. The overwhelming clinical evidence implies an excessive morbidity and mortality risk of persistent bacteremia for patients and recommends a low threshold for early antibiotic changes for rapid bacteremia clearance.
Treatment Approaches for Persistent SAB.
Several treatment options still exist for S. aureus; however, the treatment of complicated bacteremia is limited to only a handful of approved agents. This includes anti-staphylococcal β-lactams (nafcillin or oxacillin) and targeted cephalosporins (i.e., cefazolin) for MSSA, while vancomycin or daptomycin are typically reserved for MRSA. Recently, ceftobiprole received FDA approval for MSSA and MRSA bacteremia and endocarditis based on a randomized controlled trial demonstrating non-inferiority to daptomycin [12], and ceftaroline has been used similarly without indication [13,14,15]. Following induction with these antibiotics, other agents are commonly used as step down therapy including linezolid (IV/PO), the lipoglycopeptides dalbavancin and oritavancin, and many oral options including β-lactams and trimethoprim-sulfamethoxazole, and clindamycin [16]. Currently an ongoing adaptive clinical trial aims to assess the efficacy of adjunctive clindamycin for susceptible SAB [17]. Clearly, there are a diverse range of options to treat SAB. However, these options are considerably narrowed when confronted with MRSA or in patients with persistent bacteremia [3,16].
Successful management of persistent SAB requires a comprehensive approach that addresses both host and pathogen factors. Initial management involves timely and appropriate antibiotic therapy guided by antimicrobial susceptibility testing [18]. However, given the potential for antibiotic resistance and/or tolerance, combination therapy with agents targeting different bacterial pathways has been used successfully to improve bacterial clearance [3,16]. This is of highest concern in persistent MRSA bacteremia, where treatment options are extremely limited once the standard-of-care agents deem unsuccessful. Any randomized controlled trial of SAB implements a non-inferiority design [12,19,20,21,22]. No randomized controlled trial exists to understand optimal treatment of persistent SAB, and therefore treatment approaches are guided by observational and retrospective case-control studies. From this evidence, treatment paradigms have been proposed to consider how to address persistent MRSA bacteremia and include combination therapy with priority towards daptomycin plus ceftaroline followed by daptomycin plus an antistaphylococcal beta-lactam, then finally vancomycin plus a hydrophilic beta-lactam to reduce the risk of nephrotoxicity (e.g., cefazolin, ceftaroline) [16]. In addition to antibiotic therapy, the removal of potential sources of infection, such as infected intravascular devices or sites of metastatic infection, is essential. This may involve the removal or exchange of indwelling catheters, debridement of infected tissues, or surgical drainage of abscesses. Close monitoring for complications, such as infective endocarditis or metastatic infections, is also imperative during persistent bacteremia [16].
While antibiotic therapy is crucial for a successful outcome in SAB, host factors also play a significant role in the persistence of S. aureus and must be carefully evaluated and managed [23]. Underlying conditions that compromise host immune function, such as diabetes mellitus or immunosuppressive therapy, must be evaluated and managed when possible. Some host characteristics can be either modifiable or non-modifiable. For example, patients with diabetes can have blood glucose managed within normal range, however the underlying immunologic suppression from this conditional is unchanged. Optimal supportive care, including nutritional support and management of comorbidities, is also essential to enhance host defenses and improve outcomes. However, the host-pathogen dynamic in SAB remains poorly understood [24,25,26]. This is evident from the failed vaccination attempts in human trials aimed at preventing S. aureus invasive infections despite promising animal model data [24]. Only recently have we begun to study and better understand the underlying host immunologic pathology occurring at the onset and during SAB. From these studies, it is noted that profound cytokine imbalances exist in patients with SAB [8,24,25,27,28]. The pro-inflammatory cytokine response appears to be important during early initial response and clearance bacteremia (e.g. IL-1β, IL-2, IL-6, TNF-α, and glutamine) [8,28]. In terms of survival outcome, high levels initially and throughout the duration of bacteremia appear to be detrimental either to excessive pro-inflammatory response (e.g. IL-6 and TNF) or host-immune paralysis (e.g., IL-10, IL-6, IL-17, CCR2, T4, adiponectin) [10,24,29,30]. Related to persistent bacteremia, a dampening of the pro-inflammatory cytokines IL-1β and TNF-α has been linked to duration of bacteremia a result of the pathogens’ response to antibiotic exposure and ability to evade host defenses through multiple mechanisms [8,10,28,29].
In summary of its clinical importance and treatment challenges, persistent SAB poses a significant clinical problem, characterized by ongoing bloodstream infection despite appropriate therapy. Successful management requires a multifaceted approach that addresses both pathogen and host factors. Timely and appropriate antibiotic therapy, removal of potential sources of infection, optimization of host defenses, and efforts to prevent recurrent infections are key components of management. Ultimately, persistent SAB represents a complex clinical syndrome that requires a multidisciplinary approach involving infectious disease specialists, clinical microbiologists, pharmacists, nurses, and other healthcare professionals. Collaboration between specialties is essential to ensure timely diagnosis, appropriate treatment, and ongoing management of this challenging condition. However, identification of why S. aureus persists from an organism, host, and mechanistic level remains key to solving this issue.
Small Colony Variant S. aureus Result in Reduced Antibiotic Efficacy and Infection Persistence
Although reduction in the duration of SAB has been a noble goal over the last three decades, the mechanistic understanding of how and why S. aureus persists when susceptible to antibiotic therapy remains elusive. There are several proposed mechanisms contributing to this phenomenon. S. aureus has metabolic adaptations to survive and hide from the immune systems of humans. One of the most studied phenotypes contributing to persistence is the small colony variant (SCV) of S. aureus. This is a unique phenotype characterized by distinct morphological and growth characteristics [31]. SCVs contribute to persistence of many invasive infections including bacteremia, pneumoniae, and osteomyelitis, among others, and SCVs typically exhibit slow growth rates and form small, non-pigmented colonies on blood agar [31,32,33]. SCVs remain difficult to detect using standard laboratory methods for surveillance studies due to lack of dedicated screening methods and noted instability of the phenotype, but they may contribute to up to 30% of invasive S. aureus infections [32].
Small colony variants were first described in 1911 as a slow growing subpopulation [34] and later noted for their difference in “color, texture, and viscidity” [35]. Early research identified SCV occurrence correlated to harsh environmental conditions such as high salt content [36]. The clinical impact of SCVs arose in the early 1990s by linking this phenotype as a cause of persistent and relapsing S. aureus infections [31]. Since this time, the understanding of the clinical relevance and emergence of SCVs appeared through genetic mutations or adaptive responses to various environmental stresses during infection in patients, such as exposure to antibiotics or host immune defenses. These mutations often result in alterations in metabolic pathways, leading to changes in bacterial physiology and virulence [37].These mutations often result in alterations in metabolic pathways, leading to changes in bacterial physiology and virulence [37]. Clinical manifestations of SCV infections vary depending on the site of infection (e.g., endocarditis vs osteomyelitis) and underlying host factors [37,38].
A hallmark feature of SCVs is their reduced susceptibility to antibiotics, particularly aminoglycosides and cell wall-active agents like β-lactams [32,39]. This reduced susceptibility is attributed to decreased metabolic activity and alterations in membrane potential, which impair antibiotic uptake and efficacy through reduction in the production of active targets such as peptidoglycan and penicillin-binding proteins [37,40,41,42]. In addition, resistance to these key antibiotics, particularly beta-lactams for MSSA, SCVs have noted reduced susceptibility to standard of care agents for MRSA, vancomycin through reduced peptidoglycan formation and daptomycin since active membrane potential is required for activity [32,40,43]. Management of SCV infections is complex and often involves prolonged courses of antibiotic therapy tailored to the specific susceptibility profile of the isolate. Antimicrobials with high intracellular uptake, such as fluoroquinolones, may be preferred when susceptible, to treat the intracellular reservoir [40]. Combination therapy may be necessary to enhance bacterial clearance and prevent the emergence of resistance, but the optimal combination for SCV treatment is not established [39,40]. The lipoglycopeptide oritavancin, which has dual inhibition of peptidoglycan and cell membrane synthesis may have specific activity against S. aureus SCVs [44,45]. Natural products also are being explored against SCVs including tomatidine and its derivatives noted to block F0F1ATPase [46]. These molecules also synergize with aminoglycosides and prevent SCV formation [47]
Antimicrobial Resistance, Tolerance, and Persistence in S. aureus Contribute to Persistent Bacteremia
While major attention is rightfully given toward antimicrobial resistance in S. aureus, the occurrence of antimicrobial tolerance and persistence also represents significant issues for antimicrobial therapy [48,49,50,51]. The observations of antimicrobial tolerance and persistence in S. aureus represent a growing area of interest and investigation [52,53]. Antimicrobial tolerance is distinct from antimicrobial resistance in that tolerant strains appear to be susceptible upon MIC testing in liquid culture, yet the bacterial population remains viable and can survive transient antimicrobial pressure (Figure 1). These tolerant populations may enter a dormancy state or reduce cellular processes to survive antimicrobial pressure through reduction in active targets (e.g., reduced cell wall replication) [52]. Antimicrobial tolerance may be acquired through either genetic mutation triggered from environmental conditions (e.g., reduced oxygen or nutrients) or antibiotics [50,52,54]. In contrast to tolerance, persistence is a subset of the bacterial population that can survive high concentrations of antimicrobial exposure. During treatment, the majority of the population may be killed by therapeutic concentrations, leaving the “persister cells” remaining to subvert antimicrobial treatment (Figure 1) [52]. Similar to tolerance, persisters may enter a state of dormancy and altered metabolism during this phase [55,56,57]. Once the antimicrobial exposure is removed, these cells may begin to replicate leading to infection recurrence [55]. While SCVs described previously in detail can also be tolerant and persisters to antibiotics, these phenomena are not restricted to SCVs. Different mechanisms for SCVs and persisters exist, but a commonality between these two phenotypes is decreased ability to make ATP [58].
An added layer to antimicrobial tolerance and persistence is the ability for S. aureus to produce robust biofilms [59,60]. These are complex communities of bacteria encased within a self-produced extracellular matrix consisting of polysaccharides, proteins, and extracellular DNA [61]. Biofilms provide protection against antibiotics and host immune responses, allowing bacteria within the biofilm to survive and persist despite antibiotic exposure [62,63]. Both tolerant bacterial populations and persister cells occur deep within the biofilm, and treatment is further compromised by lack of antibiotic penetration through the extracellular matrix [64,65]. The low oxygen environment of biofilms combined high bacterial burden may also reduce ATP and membrane potential [66]. In bacteremia, biofilms are known to play an important role in the pathology at the infection sources including endocarditis, prosthetic devices, osteomyelitis. and osteomyelitis [60].
Antimicrobial tolerant and persistent S. aureus poses a significant clinical challenge as it can lead to treatment failure, recurrent infections, and the spread of antibiotic resistance [52,67]. Arguably, antimicrobial tolerance is responsible for persistent SAB (consecutive days of bacteremia) while persister cells are responsible for bacteremia recurrence (positive cultures after day(s) of negative cultures) (Figure 1). Effective management of antimicrobial tolerant/persistent S. aureus infections often requires a multifaceted approach, including the use of combination antibiotic therapy, removal of biofilm-associated infections, and the development of novel antimicrobial strategies to target tolerant/persistent bacterial populations [52]. However, the challenge of identifying these strains is a significant barrier for early recognition and initiation of targeted therapy. Currently, the assays to test for tolerance / persistent S. aureus are experimental, and identifying these strains only occurs through examination of an altered colony phenotype (e.g., SCV) or infection progression despite antibiotic therapy [48]. Understanding potential reservoirs for S. aureus tolerant populations and persistent cells is important for improving the response to antimicrobial therapy.
Staphylococcus aureus Proclivity for Intracellularly Growth and Persistence
Although S. aureus is traditionally considered an extracellular pathogen, there is now substantial evidence supporting the presence of an intracellular niche for this organism [68]. Using in vitro assays, S. aureus has been shown to survive within a wide variety of cell types, including epithelial cells, endothelial cells, fibroblasts, osteoblasts, keratinocytes, macrophages, and neutrophils [55,69,70,71,72,73]. Evidence of intracellular S. aureus persistence as a cause of human disease has been harder to prove, although a few studies have successfully done so. The presence of intracellular S. aureus during human infections have now been demonstrated in patients with recurrent rhinosinusitis, tonsillitis, osteomyelitis, and bacteremia [74,75,76,77,78,79]. Importantly, these studies all examined patients with recurrent or chronic infections. This evidence highlights intracellular survival as an important part of the lifecycle of persistent S. aureus infections.
The evidence for intracellular S. aureus as a contributing mechanism of persistent S. aureus infections is evident by the research and understanding of SCVs in this environment. S. aureus SCVs have a noted preference for increased intracellular uptake because of bacterial surface proteins that bind host receptors, such as epithelial cells and macrophages [58]. This intracellular lifestyle enables SCVs to evade innate host defenses and antibiotics, leading to chronic or recurrent infections that are challenging to eradicate, contributing to persistent or relapsing infections. However, this proclivity for intracellular invasion is not restricted to SCVs, and S. aureus regardless of colony phenotypes become intracellular pathogens.
Intracellular S. aureus Reduce Immune Recognition and Activation
S. aureus that persist intracellularly do so by avoiding the normal immune system activation during acute infection. Much of the typical immune activation is regulated by the accessory gene regulator (agr) system. Agr is a quorum-sensing system that regulates the expression of many toxins and other inflammatory factors [80] including α-toxin, which is known to induce inflammation and cell death when produced in an intracellular environment [81,82,83]. Several studies have tested clinical isolates from persistent S. aureus infections and found that these strains tend to have an agr-deficiency [58,84,85,86]. Similarly, SCVs have been shown to have decreased agr expression [58]. In vitro studies have demonstrated that these agr-deficient strains have a higher rate of cellular uptake but lower induction of inflammatory responses [80,87]. SCVs have demonstrated lower production of IL-1β, IL-6, and IL-12 than wild-type S. aureus after infection of epithelial cells [88]. In fact, a mouse model of chronic osteomyelitis infection has demonstrated that inflammatory markers return to normal levels during the infection, despite the persistent bacteria remaining intracellularly [89]. In addition, a study of patients with chronic rhinosinusitis with nasal polyps found no increase in the number of eosinophils, lymphocytes, and neutrophils in tissue samples, despite the presence of intracellular S. aureus [75]. Overall, this decreased immune activation allows for S. aureus to hide in the intracellular environment and largely avoid the host immune system.
There are multiple methods that S. aureus can utilize to survive in an intracellular environment. For S. aureus to enter host cells, upregulation of alternative sigma factor B (SigB) is required. This regulatory system modulates the S. aureus stress response through transcription of genes that control resistance to heat, oxidative and antibiotic stresses, and contributes to the SCV phenotype [58]. After uptake into host cells, S. aureus are typically taken up into phagosomes. This is consistent for both phagocytic cells and non-professional phagocytes such as epithelial cells, osteoblasts, and fibroblasts [82,90,91,92]. The phagolysosome environment has many antibacterial properties, including possessing an acidic pH of 4.5 [26]. S. aureus are capable of not only tolerating an acidic environment, but also deacidifying the environment through production of ammonia [93]. Importantly, the acidic environment enhances expression of the agr system, and appears to be necessary for the survival of S. aureus intracellularly [94,95,96]. This effect is likely strain specific [97,98]. S. aureus is also capable of protecting itself from defensins and other antimicrobial peptides within the phagosome environment. The GraRS system leads to upregulation of multiple peptide resistance factor (MrpF) which confers protection from host defense peptides by enhancing lysinylation of phosphatidylglycerol to the outer portion of cellular membrane [99]. Similarly, the dlt operon (dltABCD) and oatA lead to alterations in membrane teichoic acids and peptidoglycan, respectively, which lead to protection from the phagosome environment [99,100]. Finally, S. aureus may produce protective enzymes (catalase, superoxide dismutase, etc.) to protect themselves from reactive oxygen species produced within the phagosome [101,102]. An increase in SodM, an enzyme responsible for detoxifying reactive oxygen species, was discovered in S. aureus isolates from patients with persistent cystic fibrosis infections [103]. In vitro assays have determined that this increase is noted specifically after internalization of S. aureus into airway epithelial cells [104].
Despite these adaptations to the harsh phagosome environment, many S. aureus will exit the phagosome as a method to increase survival. This phagosomal escape is an agr mediated process, as agr mutant strains are not capable of escaping to the cytosol. An increase in agr expression has been measured just prior to phagosomal escape [105,106]. Phenol-soluble modulins (PSMs), agr-dependent cytotoxic peptides that are activated during the stringent response, have proven to play an important role in escape into the cytosol [105,107,108,109,110]. The concentration of PSMs has been similarly shown to increase just prior to S. aureus phagosomal escape has proven to be a required component in this process [107,111]. Other S. aureus escape factors, including a non-ribosomal peptide synthetase and a Tet38 efflux pump, have been identified but their mechanisms are less clear [112,113]. In addition to these mechanisms, S. aureus may escape phagosomal destruction during overwhelming infection. In these situations, host cells are not able to tackle the high bacterial burden and become exhausted, creating an intracellular niche for S. aureus replication [114,115,116]. After phagosomal escape, replication within the cytosol leads to host cell lysis and release of S. aureus [98]. These released cells may be re-phagocytosed, leading to a cycle that maintains a portion of intracellular S. aureus [116,117]. This maintenance of intracellular S. aureus can also contribute to spreading of infection to other host sites, when mobile phagocytes are infected [116,118].
The above survival mechanisms appear to contradict the fact that persistent infections are often caused by agr-deficient S. aureus strains, as these strains would be unable to escape the phagosome due to lack of PSMs [58,84,85,86]. Indeed, these agr deficient strains employ different survival methods that do not require PSM-mediated phagosomal escape. Siegmund et al. determined that PSM-deficient S. aureus have a higher overall survival rate within endothelial cells [119]. The surviving bacteria were co-localized with LC3, a marker of autophagy, indicating that these bacteria can survive within vesicles, likely by interfering with lysosomal recruitment and the autophagy process. Unlike the process of phagosome escape and host cell lysis presented above, this PSM-independent process allows for a consistent niche of intracellular growth.
Anaerobic Metabolism of S. aureus Correlates to Intracellular Growth
There is a growing body of evidence to suggest that S. aureus utilize anaerobic metabolism during persistent intracellular infections. Figure 2 presents a working model of key interactions of intracellular S. aureus metabolism and host-factors leading to persistence. S. aureus SCVs have demonstrated a decrease in TCA cycle activity and corresponding increase in glycolytic activity compared to wild-type strains, an effect that is consistent across all auxotrophic types of SCVs [42]. This effect, however, does not seem to be restricted to S. aureus with the SCV phenotype. One study conducted whole genome sequencing of 206 MRSA isolates from patients with persistent bacteremia [120]. They found frequent mutations in genes involved in the TCA cycle (citZ and odhA). Mutations in these genes have been linked to antibiotic tolerance [56,57], which may help explain persistence despite adequate antimicrobial treatment. Similarly, S. aureus isolates from persistent cystic fibrosis infections have downregulation of many proteins involved in amino acid metabolism, fatty acid metabolism, and energy metabolism [103,121]. These studies also found a reduction in expression of proteins in the phosphoenolpyruvate carbohydrate phosphotransferase systems, which are responsible for the transport of glucose into the bacterial cell, a necessary precursor to glycolysis.
The redox regulator Rex inhibits important genes involved in anaerobic metabolism including lactate dehydrogenase and alanine dehydrogenase. Rex plays an important role in activating anaerobic metabolism in response to intracellular uptake or in response to high concentrations of NADH or NO. RsaG is a sRNA that inhibits the expression of Rex [122,123]. RsaG is expressed in response to high levels of glucose-6-phosphate, as frequently found in the host cell cytosol or mucoid secretions. An increase in RsaG has been noted during in vitro internalization assays using both myoblasts and macrophages, as well as after exposure to mucus- secreting epithelial cells. This increase in RsaG therefore triggers a switch to anaerobic metabolism via de-repression of Rex-regulated proteins. Similarly, inhibition of Rex has been observed in response to high levels of NO, a signaling molecule utilized during wound repair that is present at high levels in lung epithelial cells in patients with inflammatory diseases such as asthma or COPD [124,125,126]. Rex is also the major regulator of staphylococcal respiratory response AB (srrAB), a two-component system that regulates virulence factors and genes involved in anaerobic metabolism [127,128,129]. Mutations in srrAB allow for S. aureus SCVs to grow rapidly, yet retain aminoglycoside resistance that was acquired during the SCV phase [130]. S. aureus glycolysis is also necessary for the upregulation of itaconate production, as demonstrated in a mouse pulmonary infection model. High itaconate levels in turn lead to production of extracellular polysaccharide and biofilm formation [126].
Like bacterial cells, host cell metabolism also plays an important role in immune response. Hypoxia-inducible factor 1a (hif1α) is an important gene that controls both immune response and cellular metabolism [131]. HIF1α activation stimulates production of pro-IL-1β, an important part of the inflammasome response and promotes glycolysis. Importantly, HIF1α expression is induced during S. aureus infection of human cells, and the metabolic state of the bacterial cell plays an important role in this expression [131,132]. S. aureus mutants deficient in cellular glycolysis are unable to stimulate host cell HIF1α expression and therefore host cell glycolysis [131]. Contrarily, S. aureus SCV infection stimulate more host glycolysis than wild-type S. aureus infection, an effect that has been demonstrated in multiple host cell types [132]. Bacterial glycolysis appears to also play an important role in establishing infection, an effect that has been demonstrated using an in vivo mouse cutaneous infection model. Mice that were infected with glycolysis-deficient S. aureus demonstrated a significantly lower bacterial burden than those infected with wild-type strains [131]. These findings highlight the complex and important interplay between bacterial and host metabolic processes.
Host cell anaerobic metabolism has an important secondary cellular consequence, the induction of necroptosis pathways [132]. Necroptosis is a type of host cell death that, unlike apoptosis, releases viable bacteria, thus promoting bacterial persistence [133]. Necroptosis is further upregulated by SCVs compared to wild type S. aureus and is not activated when host cells are infected by glycolysis-deficient S. aureus. An in vivo mouse model demonstrated that mice unable to utilize necroptosis had greater S. aureus persistence compared to wild-type mice. This pathway provides an important mechanistic link between bacterial anaerobic metabolism and infection persistence.
Another notable effect of anaerobic metabolism is the inhibition of trained immunity. Trained immunity is the process wherein the innate immune system develops memory of an infection and is therefore strengthened against re-infection with the same organism [134]. Unlike the adaptive immune response, this memory is thought to be accomplished through a series of epigenetic changes and is highly regulated by the amount of intracellular fumarate [135,136]. Fumarate is a TCA cycle substrate that induces both the trained immunity response and glycolysis [137]. Infection with S. aureus SCVs leads to lower amounts of intracellular fumarate than infection with wild-type strains, an effect that is mediated by an increase in fumC activity, an enzyme that breaks down fumarate [132]. Therefore, SCVs do not induce trained immunity to the degree of wild-type S. aureus as validated using an in vivo mouse model. Even in the presence of a mixed SCV and wild-type population, the excess fumC activity from the SCV isolates prevents the trained immunity response. This increases susceptibility of infection upon future S. aureus exposure.
Intracellular S. aureus Are More Resistant to the Effects of Antibiotics through Multifaceted Mechanisms
The ability for S. aureus to survive in the intracellular environment provides protection from many antibiotics. These mechanisms are outlined in Figure 3. First, most antibiotics are either unable to penetrate into the intracellular space or lack sufficient penetration for microbial killing. For example, β-lactams, an important class in the clinical management of S. aureus infections, do not accumulate within phagocytes, despite their ability to diffuse through membranes [138]. This is thought to be due to their weak acidity, which leads to lower accumulation within the already acidic intracellular space [139]. Aminoglycosides are unable to cross the membrane due to their polarity but have been noted to enter the intracellular space by endocytosis [140]. This process requires a longer duration of drug exposure and ultimately leads to localization within lysosomes. However, optimizing aminoglycoside exposures for this effect is limited by their well-known toxicities, namely nephrotoxicity and ototoxicity [141]. Conversely, lincosamides (clindamycin), macrolides, and fluoroquinolones all demonstrate accumulation within the intracellular space [138].
Interestingly, the intracellular accumulation of antibiotics does not always correlate with the antibiotic’s activity within that space. One study evaluating intracellular activity of a variety of antibiotics found that intracellular activity was consistently lower than extracellular, despite the high intracellular accumulation of some of the antibiotics used [142]. Overall, the extent of antibiotic activity was greatly dependent on both the concentration and time of antibiotic exposure for all agents. This disconnect between intracellular concentration and activity points to other important factors that affect antibiotic activity within the intracellular environment.
Intracellular bacteria undergo several changes that lead to increased antibiotic resistance. In SCVs, the pattern of antimicrobial resistance is dependent on the specific auxotrophic type. Hemin or menadione auxotrophic SCVs are typically resistant to aminoglycosides [32]. The inhibition of electron transport within these cells leads to a reduction in the electrochemical gradient across the membrane. This gradient is required for the uptake of aminoglycosides into S. aureus. The other type of SCVs, thymidine auxotrophs, are resistant to sulfa antibiotics (trimethoprim-sulfamethoxazole) as a result of disruption of the tetrahydrofolic acid pathway [143]. Thymidine auxotrophy results in reduced ClpC activity, which is needed to activate aconitase, an enzyme involved in the intraconversion of the tricarboxylic acids citrate, cis-aconitate, and isocitrate in the TCA cycle [144]. Hence the reduced TCA activity results in a SCV similar to menadione and hemin auxotrophs [143,145]. The slow-growing SCV phenotype also leads to decreased susceptibility to cell wall-active antibiotics, such as β-lactams and vancomycin [146]
Because all three SCV auxotrophic classes lead to disruption of the electron transport chain, SCVs are deficient in ATP. This ATP depletion has been linked to the formation of persister cells from exponential phase normal-colony phenotype S. aureus [147]. An in vitro study found that persister cells arise due to a stochastic change from exponential to stationary growth phase, a change that was accompanied by a large decrease in ATP levels. This change to stationary phase was associated with a 100- to 1000-fold increase in survival after antibiotic challenge. Another study found that inactivation of enzymes involved in the TCA cycle (sucA or sucB) increased the formation of persister cells in stationary phase cultures [56]. Interestingly, this study did not find a consistent decrease in ATP levels amongst persister cells formed during the stationary phase, but it did note lower membrane potential. This effect was confirmed pharmacologically using a proton motive force inhibitor, which increased the formation of persister cells.. As noted earlier, tomatidine and related derivatives inhibit bacterial ATP synthase (F0F1ATPase), which specifically kills S. aureus SCVs [46]. This selectivity in S. aureus, and not mammalian cells, points to the use of F0F1ATPase in auxotrophic SCVs to create a membrane gradient despite low ATP [148]. Inhibition of this enzyme is fatal for S. aureus as it collapses the membrane potential but leaves untargeted host cells unharmed [47].
Peyrusson et al. demonstrated that intracellular S. aureus persisters are produced because of antibiotic exposure. These bacteria exist in a non-dividing state and display activation of the stringent response and other stress responses [55]. The decreased metabolic state associated with intracellular persistence leads to downregulation of many cellular processes that are the targets of bactericidal antibiotics, such as fluoroquinolones, aminoglycosides, and β-lactams and causes widespread antibiotic tolerance. Collectively, the intracellular host environment along with the altered S. aureus metabolism (through multiple mechanisms) required to survive in this environment dramatically reduces the effect of common antibiotics used in bacteremia treatment, setting the stage for treatment failure.
Conclusions
Mortality from SAB remains unacceptably high over the course of decades of improvements in diagnostics and therapeutics. In this review, we have summarized evidence on the role of tolerant and persistent populations of S. aureus compounded by their proclivity for anaerobic metabolism and intracellular invasion to escape host recognition and antibiotic treatment. This evidence suggests that intracellular S. aureus are a significant contributor to persistent bacteremia and treatment failure in patients. Current therapeutics either lack activity against tolerant and persistent populations due to their inability to kill quiescent cells and/or lack sufficient host cell penetration to eliminate intracellular S. aureus. Further mechanistic understanding of S. aureus intracellular invasion combined with targeting these bacterial populations for therapeutic treatment will be essential to reducing the prevalence of persistent SAB among patients.
References
- Rodvold, K.A.; McConeghy, K.W. , Methicillin-resistant Staphylococcus aureus therapy: Past, present, and future. Clin Infect Dis 2014, 58 (Suppl. S1), S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Holland, T.L.; Bayer, A.S.; Fowler, V.G. , Persistent Methicilin-Resistant Staphylococcus aureus Bacteremia: Resetting the Clock for Optimal Management. Clin Infect Dis 2022, 75, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Kullar, R.; Sakoulas, G.; Deresinski, S.; van Hal, S.J. , When sepsis persists: A review of MRSA bacteraemia salvage therapy. J Antimicrob Chemother 2016, 71, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.A.; Perez, K.K.; Forrest, G.N.; Goff, D.A. , Review of rapid diagnostic tests used by antimicrobial stewardship programs. Clin Infect Dis 2014, 59 (Suppl. S3), S134–S145. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Fang, F.C. , Diagnostic Stewardship: Opportunity for a Laboratory–Infectious Diseases Partnership. Clinical Infectious Diseases 2018, 67, 799–801. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, N.; Quinn, E.L.; Saravolatz, L.D. , Trimethoprim-sulfamethoxazole compared with vancomycin for the treatment of Staphylococcus aureus infection. Ann Intern Med 1992, 117, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.P.; Fromm, B.S.; Reddy, B.R. , Slow response to vancomycin or vancomycin plus rifampin in methicillin-resistant Staphylococcus aureus endocarditis. Ann Intern Med 1991, 115, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Rose, W.E.; Eickhoff, J.C.; Shukla, S.K.; Pantrangi, M.; Rooijakkers, S.; Cosgrove, S.E.; Nizet, V.; Sakoulas, G. , Elevated serum interleukin-10 at time of hospital admission is predictive of mortality in patients with Staphylococcus aureus bacteremia. J Infect Dis 2012, 206, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Kullar, R.; McKinnell, J.A.; Sakoulas, G. , Avoiding the Perfect Storm: The Biologic and Clinical Case for Reevaluating the 7-Day Expectation for Methicillin-Resistant Staphylococcus aureus Bacteremia Before Switching Therapy. Clin Infect Dis 2014, 59, 1455–1461. [Google Scholar] [CrossRef]
- Rose, W.E.; Shukla, S.K.; Berti, A.D.; Hayney, M.S.; Henriquez, K.M.; Ranzoni, A.; Cooper, M.A.; Proctor, R.A.; Nizet, V.; Sakoulas, G. , Increased Endovascular Staphylococcus aureus Inoculum Is the Link Between Elevated Serum Interleukin 10 Concentrations and Mortality in Patients With Bacteremia. Clin Infect Dis 2017, 64, 1406–1412. [Google Scholar] [CrossRef]
- Kuehl, R.; Morata, L.; Boeing, C.; Subirana, I.; Seifert, H.; Rieg, S.; Kern, W.V.; Kim, H.B.; Kim, E.S.; Liao, C.H.; Tilley, R.; Lopez-Cortes, L.E.; Llewelyn, M.J.; Fowler, V.G.; Thwaites, G.; Cisneros, J.M.; Scarborough, M.; Nsutebu, E.; Gurgui Ferrer, M.; Perez, J.L.; Barlow, G.; Hopkins, S.; Ternavasio-de la Vega, H.G.; Torok, M.E.; Wilson, P.; Kaasch, A.J.; Soriano, A.; International Staphylococcus aureus collaboration study, g. ; the Escmid Study Group for Bloodstream Infections, E.; Sepsis, Defining persistent Staphylococcus aureus bacteraemia: Secondary analysis of a prospective cohort study. Lancet Infect Dis 2020, 20, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Holland, T.L.; Cosgrove, S.E.; Doernberg, S.B.; Jenkins, T.C.; Turner, N.A.; Boucher, H.W.; Pavlov, O.; Titov, I.; Kosulnykov, S.; Atanasov, B.; Poromanski, I.; Makhviladze, M.; Anderzhanova, A.; Stryjewski, M.E.; Assadi Gehr, M.; Engelhardt, M.; Hamed, K.; Ionescu, D.; Jones, M.; Saulay, M.; Smart, J.; Seifert, H.; Fowler, V.G., Jr.; Group, E.S. , Ceftobiprole for Treatment of Complicated Staphylococcus aureus Bacteremia. N Engl J Med 2023, 389, 1390–1401. [Google Scholar] [CrossRef] [PubMed]
- Sakoulas, G.; Moise, P.A.; Casapao, A.M.; Nonejuie, P.; Olson, J.; Okumura, C.Y.; Rybak, M.J.; Kullar, R.; Dhand, A.; Rose, W.E.; Goff, D.A.; Bressler, A.M.; Lee, Y.; Pogliano, J.; Johns, S.; Kaatz, G.W.; Ebright, J.R.; Nizet, V. , Antimicrobial Salvage Therapy for Persistent Staphylococcal Bacteremia Using Daptomycin Plus Ceftaroline. Clin Ther 2014. [Google Scholar] [CrossRef] [PubMed]
- Rose, W.E.; Schulz, L.T.; Andes, D.; Striker, R.; Berti, A.D.; Hutson, P.R.; Shukla, S.K. , Addition of ceftaroline to daptomycin after emergence of daptomycin-nonsusceptible Staphylococcus aureus during therapy improves antibacterial activity. Antimicrob Agents Chemother 2012, 56, 5296–5302. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Aung, G.; Thomas, A.; Jahng, M.; Johns, S.; Fierer, J. , The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: A retrospective case series of 10 patients. J Infect Chemother 2013, 19, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Rose, W.; Fantl, M.; Geriak, M.; Nizet, V.; Sakoulas, G. , Current Paradigms of Combination Therapy in Methicillin-Resistant Staphylococcus aureus (MRSA) Bacteremia: Does it Work, Which Combination, and For Which Patients? Clin Infect Dis 2021, 73, 2353–2360. [Google Scholar] [CrossRef]
- Campbell, A.J.; Dotel, R.; Braddick, M.; Britton, P.N.; Eisen, D.P.; Francis, J.R.; Lynar, S.; McMullan, B.; Meagher, N.; Nelson, J.; O'Sullivan, M.V. N.; Price, D.J.; Robinson, J.O.; Whelan, A.; Tong, S.Y. C.; Bowen, A.C.; Davis, J.S. , Clindamycin adjunctive therapy for severe Staphylococcus aureus treatment evaluation (CASSETTE)-an open-labelled pilot randomized controlled trial. JAC Antimicrob Resist 2022, 4, dlac014. [Google Scholar] [CrossRef]
- van Hal, S.J.; Jensen, S.O.; Vaska, V.L.; Espedido, B.A.; Paterson, D.L.; Gosbell, I.B. , Predictors of mortality in Staphylococcus aureus Bacteremia. Clin Microbiol Rev 2012, 25, 362–386. [Google Scholar] [CrossRef]
- Fowler, V.G., Jr.; Boucher, H.W.; Corey, G.R.; Abrutyn, E.; Karchmer, A.W.; Rupp, M.E.; Levine, D.P.; Chambers, H.F.; Tally, F.P.; Vigliani, G.A.; Cabell, C.H.; Link, A.S.; DeMeyer, I.; Filler, S.G.; Zervos, M.; Cook, P.; Parsonnet, J.; Bernstein, J.M.; Price, C.S.; Forrest, G.N.; Fatkenheuer, G.; Gareca, M.; Rehm, S.J.; Brodt, H.R.; Tice, A.; Cosgrove, S.E.; Endocarditis, S. a.; Bacteremia Study, G. , Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med 2006, 355, 653–665. [Google Scholar] [CrossRef]
- Pujol, M.; Miro, J.M.; Shaw, E.; Aguado, J.M.; San-Juan, R.; Puig-Asensio, M.; Pigrau, C.; Calbo, E.; Montejo, M.; Rodriguez-Alvarez, R.; Garcia-Pais, M.J.; Pintado, V.; Escudero-Sanchez, R.; Lopez-Contreras, J.; Morata, L.; Montero, M.; Andres, M.; Pasquau, J.; Arenas, M.D.; Padilla, B.; Murillas, J.; Jover-Saenz, A.; Lopez-Cortes, L.E.; Garcia-Pardo, G.; Gasch, O.; Videla, S.; Hereu, P.; Tebe, C.; Pallares, N.; Sanllorente, M.; Dominguez, M.A.; Camara, J.; Ferrer, A.; Padulles, A.; Cuervo, G.; Carratala, J.; Investigators, M.B. T. , Daptomycin Plus Fosfomycin Versus Daptomycin Alone for Methicillin-resistant Staphylococcus aureus Bacteremia and Endocarditis: A Randomized Clinical Trial. Clin Infect Dis 2021, 72, 1517–1525. [Google Scholar] [CrossRef]
- Davis, J.S.; Sud, A.; O'Sullivan, M.V.; Robinson, J.O.; Ferguson, P.E.; Foo, H.; van Hal, S.J.; Ralph, A.P.; Howden, B.P.; Binks, P.M.; Kirby, A.; Tong, S.Y.; Combination Antibiotics for, M.R. S. a. s. g.; Australasian Society for Infectious Diseases Clinical Research, N. , Combination of Vancomycin and beta-Lactam Therapy for Methicillin-Resistant Staphylococcus aureus Bacteremia: A Pilot Multicenter Randomized Controlled Trial. Clin Infect Dis 2016, 62, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.C.; Lye, D.C.; Yahav, D.; Sud, A.; Robinson, J.O.; Nelson, J.; Archuleta, S.; Roberts, M.A.; Cass, A.; Paterson, D.L.; Foo, H.; Paul, M.; Guy, S.D.; Tramontana, A.R.; Walls, G.B.; McBride, S.; Bak, N.; Ghosh, N.; Rogers, B.A.; Ralph, A.P.; Davies, J.; Ferguson, P.E.; Dotel, R.; McKew, G.L.; Gray, T.J.; Holmes, N.E.; Smith, S.; Warner, M.S.; Kalimuddin, S.; Young, B.E.; Runnegar, N.; Andresen, D.N.; Anagnostou, N.A.; Johnson, S.A.; Chatfield, M.D.; Cheng, A.C.; Fowler, V.G., Jr; Howden, B.P.; Meagher, N.; Price, D.J.; van Hal, S.J.; O’Sullivan, M.V. N.; Davis, J.S.; Network, f. t. A. S. f. I. D. C. R. , Effect of Vancomycin or Daptomycin With vs Without an Antistaphylococcal β-Lactam on Mortality, Bacteremia, Relapse, or Treatment Failure in Patients With MRSA Bacteremia: A Randomized Clinical Trial. JAMA 2020, 323, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Fowler, V.G., Jr.; Miro, J.M.; Hoen, B.; Cabell, C.H.; Abrutyn, E.; Rubinstein, E.; Corey, G.R.; Spelman, D.; Bradley, S.F.; Barsic, B.; Pappas, P.A.; Anstrom, K.J.; Wray, D.; Fortes, C.Q.; Anguera, I.; Athan, E.; Jones, P.; van der Meer, J.T.; Elliott, T.S.; Levine, D.P.; Bayer, A.S.; Investigators, I.C. E. , Staphylococcus aureus endocarditis: A consequence of medical progress. JAMA 2005, 293, 3012–3021. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.S.; Fowler, V.G.; Shukla, S.K.; Rose, W.E.; Proctor, R.A. , Development of a vaccine against Staphylococcus aureus invasive infections: Evidence based on human immunity, genetics and bacterial evasion mechanisms. FEMS Microbiol Rev 2020, 44, 123–153. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Rose, W.; Schrodi, S.J. , Complex host genetic susceptibility to Staphylococcus aureus infections. Trends Microbiol 2015, 23, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Horn, J.; Stelzner, K.; Rudel, T.; Fraunholz, M. , Inside job: Staphylococcus aureus host-pathogen interactions. Int J Med Microbiol 2018, 308, 607–624. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.; van Kessel, K.P.; van Strijp, J.A. , Staphylococcal innate immune evasion. Trends Microbiol 2005, 13, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Minejima, E.; Bensman, J.; She, R.C.; Mack, W.J.; Tuan Tran, M.; Ny, P.; Lou, M.; Yamaki, J.; Nieberg, P.; Ho, J.; Wong-Beringer, A. , A Dysregulated Balance of Proinflammatory and Anti-Inflammatory Host Cytokine Response Early During Therapy Predicts Persistence and Mortality in Staphylococcus aureus Bacteremia. Crit Care Med 2016, 44, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Volk, C.F.; Burgdorf, S.; Edwardson, G.; Nizet, V.; Sakoulas, G.; Rose, W.E. , Interleukin (IL)-1beta and IL-10 Host Responses in Patients With Staphylococcus aureus Bacteremia Determined by Antimicrobial Therapy. Clin Infect Dis 2020, 70, 2634–2640. [Google Scholar] [CrossRef]
- Wozniak, J.M.; Mills, R.H.; Olson, J.; Caldera, J.R.; Sepich-Poore, G.D.; Carrillo-Terrazas, M.; Tsai, C.M.; Vargas, F.; Knight, R.; Dorrestein, P.C.; Liu, G.Y.; Nizet, V.; Sakoulas, G.; Rose, W.; Gonzalez, D.J. , Mortality Risk Profiling of Staphylococcus aureus Bacteremia by Multi-omic Serum Analysis Reveals Early Predictive and Pathogenic Signatures. Cell 2020, 182, 1311–1327. [Google Scholar] [CrossRef]
- Proctor, R.A.; Balwit, J.M.; Vesga, O. , Variant subpopulations of Staphylococcus aureus as cause of persistent and recurrent infections. Infect Agents Dis 1994, 3, 302–312. [Google Scholar]
- von Eiff, C. , Staphylococcus aureus small colony variants: A challenge to microbiologists and clinicians. Int J Antimicrob Agents 2008, 31, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Kahl, B.C.; Belling, G.; Becker, P.; Chatterjee, I.; Wardecki, K.; Hilgert, K.; Cheung, A.L.; Peters, G.; Herrmann, M. , Thymidine-dependent Staphylococcus aureus small-colony variants are associated with extensive alterations in regulator and virulence gene expression profiles. Infect Immun 2005, 73, 4119–4126. [Google Scholar] [CrossRef] [PubMed]
- Kolle, W.; Hetsch, H. , Die experimentelle Bakteriologie und die Infektionskrankheiten mit besonderer Berücksichtigung der Immunitätslehre. Urban und Schwarzenberg, Berlin, Germany 1911, 1. [Google Scholar]
- Bigger, J.W.; Boland, C.R.; O'meara, R.A. Q. , Variant colonies of Staphylococcus aureus. The Journal of Pathology and Bacteriology 1927, 30, 261–269. [Google Scholar] [CrossRef]
- Youmans, G.P. , Production of Small-Colony Variants of Staphylococcus aureus. Proceedings of the Society for Experimental Biology and Medicine 1937, 36, 94–96. [Google Scholar] [CrossRef]
- Proctor, R.A.; von Eiff, C.; Kahl, B.C.; Becker, K.; McNamara, P.; Herrmann, M.; Peters, G. , Small colony variants: A pathogenic form of bacteria that facilitates persistent and recurrent infections. Nature Reviews Microbiology 2006, 4, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Kahl, B.C.; Becker, K.; Löffler, B. , Clinical Significance and Pathogenesis of Staphylococcal Small Colony Variants in Persistent Infections. Clin Microbiol Rev 2016, 29, 401–427. [Google Scholar] [CrossRef] [PubMed]
- Maduka-Ezeh, A.N.; Greenwood-Quaintance, K.E.; Karau, M.J.; Berbari, E.F.; Osmon, D.R.; Hanssen, A.D.; Steckelberg, J.M.; Patel, R. , Antimicrobial susceptibility and biofilm formation of Staphylococcus epidermidis small colony variants associated with prosthetic joint infection. Diagn Microbiol Infect Dis 2012, 74, 224–229. [Google Scholar] [CrossRef]
- Garcia, L.G.; Lemaire, S.; Kahl, B.C.; Becker, K.; Proctor, R.A.; Denis, O.; Tulkens, P.M.; Van Bambeke, F. , Antibiotic activity against small-colony variants of Staphylococcus aureus: Review of in vitro, animal and clinical data. Journal of Antimicrobial Chemotherapy 2013, 68, 1455–1464. [Google Scholar] [CrossRef]
- Baltch, A.L.; Ritz, W.J.; Bopp, L.H.; Michelsen, P.; Smith, R.P. , Activities of daptomycin and comparative antimicrobials, singly and in combination, against extracellular and intracellular Staphylococcus aureus and its stable small-colony variant in human monocyte-derived macrophages and in broth. Antimicrob Agents Chemother 2008, 52, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Kriegeskorte, A.; Grubmuller, S.; Huber, C.; Kahl, B.C.; von Eiff, C.; Proctor, R.A.; Peters, G.; Eisenreich, W.; Becker, K. , Staphylococcus aureus small colony variants show common metabolic features in central metabolism irrespective of the underlying auxotrophism. Front Cell Infect Microbiol 2014, 4, 141. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.N.; Bayer, A.S.; Weidenmaier, C.; Grau, T.; Wanner, S.; Stefani, S.; Cafiso, V.; Bertuccio, T.; Yeaman, M.R.; Nast, C.C.; Yang, S.J. , Phenotypic and genotypic characterization of daptomycin-resistant methicillin-resistant Staphylococcus aureus strains: Relative roles of mprF and dlt operons. PLoS ONE 2014, 9, e107426. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Schweizer, F.; Karlowsky, J.A. , Oritavancin: Mechanism of action. Clin Infect Dis 2012, 54 (Suppl. S3), S214–S219. [Google Scholar] [CrossRef] [PubMed]
- Bouza, E.; Burillo, A. , Oritavancin: A novel lipoglycopeptide active against Gram-positive pathogens including multiresistant strains. Int J Antimicrob Agents 2010, 36, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne Boulet, M.; Isabelle, C.; Guay, I.; Brouillette, E.; Langlois, J.P.; Jacques, P.E.; Rodrigue, S.; Brzezinski, R.; Beauregard, P.B.; Bouarab, K.; Boyapelly, K.; Boudreault, P.L.; Marsault, E.; Malouin, F. , Tomatidine Is a Lead Antibiotic Molecule That Targets Staphylococcus aureus ATP Synthase Subunit C. Antimicrob Agents Chemother 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Langlois, J.P.; Millette, G.; Guay, I.; Dube-Duquette, A.; Chamberland, S.; Jacques, P.E.; Rodrigue, S.; Bouarab, K.; Marsault, E.; Malouin, F. , Bactericidal Activity of the Bacterial ATP Synthase Inhibitor Tomatidine and the Combination of Tomatidine and Aminoglycoside Against Persistent and Virulent Forms of Staphylococcus aureus. Front Microbiol 2020, 11, 805. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gefen, O.; Ronin, I.; Bar-Meir, M.; Balaban, N.Q. , Effect of tolerance on the evolution of antibiotic resistance under drug combinations. Science 2020, 367, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Levin-Reisman, I.; Ronin, I.; Gefen, O.; Braniss, I.; Shoresh, N.; Balaban, N.Q. , Antibiotic tolerance facilitates the evolution of resistance. Science 2017, 355, 826–830. [Google Scholar] [CrossRef]
- Miller, C.R.; Monk, J.M.; Szubin, R.; Berti, A.D. , Rapid resistance development to three antistaphylococcal therapies in antibiotic-tolerant staphylococcus aureus bacteremia. PLoS ONE 2021, 16, e0258592. [Google Scholar] [CrossRef]
- Berti, A.D.; Shukla, N.; Rottier, A.D.; McCrone, J.S.; Turner, H.M.; Monk, I.R.; Baines, S.L.; Howden, B.P.; Proctor, R.A.; Rose, W.E. , Daptomycin selects for genetic and phenotypic adaptations leading to antibiotic tolerance in MRSA. J Antimicrob Chemother 2018, 73, 2030–2033. [Google Scholar] [CrossRef] [PubMed]
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. , Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nature Reviews Microbiology 2016, 14, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Berti, A.D.; Hirsch, E.B. , Tolerance to antibiotics affects response. Science 2020, 367, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Meredith, E.M.; Harven, L.T.; Berti, A.D. , Antimicrobial Efficacy against Antibiotic-Tolerant Staphylococcus aureus Depends on the Mechanism of Antibiotic Tolerance. Antibiotics (Basel) 2022, 11. [Google Scholar]
- Peyrusson, F.; Varet, H.; Nguyen, T.K.; Legendre, R.; Sismeiro, O.; Coppee, J.Y.; Wolz, C.; Tenson, T.; Van Bambeke, F. , Intracellular Staphylococcus aureus persisters upon antibiotic exposure. Nat Commun 2020, 11, 2200. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bojer, M.S.; George, S.E.; Wang, Z.; Jensen, P.R.; Wolz, C.; Ingmer, H. , Inactivation of TCA cycle enhances Staphylococcus aureus persister cell formation in stationary phase. Sci Rep 2018, 8, 10849. [Google Scholar] [CrossRef] [PubMed]
- Zalis, E.A.; Nuxoll, A.S.; Manuse, S.; Clair, G.; Radlinski, L.C.; Conlon, B.P.; Adkins, J.; Lewis, K. , Stochastic Variation in Expression of the Tricarboxylic Acid Cycle Produces Persister Cells. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Tuchscherr, L.; Loffler, B.; Proctor, R.A. , Persistence of Staphylococcus aureus: Multiple Metabolic Pathways Impact the Expression of Virulence Factors in Small-Colony Variants (SCVs). Front Microbiol 2020, 11, 1028. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. , Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef]
- Donlan, R.M.; Costerton, J.W. , Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev 2002, 15, 167–193. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J. , The biofilm matrix. Nature Reviews Microbiology 2010, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Raad, I.; Hanna, H.; Jiang, Y.; Dvorak, T.; Reitzel, R.; Chaiban, G.; Sherertz, R.; Hachem, R. , Comparative activities of daptomycin, linezolid, and tigecycline against catheter-related methicillin-resistant Staphylococcus bacteremic isolates embedded in biofilm. Antimicrob Agents Chemother 2007, 51, 1656–1660. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Hook, M. , Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat Rev Microbiol 2014, 12, 49–62. [Google Scholar] [CrossRef]
- Aslam, S. , Effect of antibacterials on biofilms. Am J Infect Control 2008, 36, S175–e9. [Google Scholar] [CrossRef] [PubMed]
- Mah, T.F.; O'Toole, G.A. , Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 2001, 9, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Theis, T.J.; Daubert, T.A.; Kluthe, K.E.; Brodd, K.L.; Nuxoll, A.S. , Staphylococcus aureus persisters are associated with reduced clearance in a catheter-associated biofilm infection. Front Cell Infect Microbiol 2023, 13, 1178526. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Rolston, K.V. , Vancomycin tolerance, a potential mechanism for refractory gram-positive bacteremia observational study in patients with cancer. Cancer 2006, 106, 1815–1820. [Google Scholar] [CrossRef] [PubMed]
- Garzoni, C.; Kelley, W.L. , Staphylococcus aureus: New evidence for intracellular persistence. Trends Microbiol 2009, 17, 59–65. [Google Scholar] [CrossRef]
- Xu, D.; Hu, G.; Luo, J.; Cheng, J.; Wu, D.; Cheng, L.; Huang, X.; Fu, S.; Liu, J. , Staphylococcus aureus induces mitophagy to promote its survival within bovine mammary epithelial cells. Vet Microbiol 2023, 280, 109697. [Google Scholar] [CrossRef]
- Rollin, G.; Tan, X.; Tros, F.; Dupuis, M.; Nassif, X.; Charbit, A.; Coureuil, M. , Intracellular Survival of Staphylococcus aureus in Endothelial Cells: A Matter of Growth or Persistence. Front Microbiol 2017, 8, 1354. [Google Scholar] [CrossRef]
- Perez, K.; Patel, R. , Survival of Staphylococcus epidermidis in Fibroblasts and Osteoblasts. Infect Immun 2018, 86. [Google Scholar] [CrossRef] [PubMed]
- Abu-Humaidan, A.H.; Elven, M.; Sonesson, A.; Garred, P.; Sorensen, O.E. , Persistent Intracellular Staphylococcus aureus in Keratinocytes Lead to Activation of the Complement System with Subsequent Reduction in the Intracellular Bacterial Load. Front Immunol 2018, 9, 396. [Google Scholar] [CrossRef] [PubMed]
- Prajsnar, T.K.; Serba, J.J.; Dekker, B.M.; Gibson, J.F.; Masud, S.; Fleming, A.; Johnston, S.A.; Renshaw, S.A.; Meijer, A.H. , The autophagic response to Staphylococcus aureus provides an intracellular niche in neutrophils. Autophagy 2021, 17, 888–902. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.; Vaudaux, P.; Francois, P.; Schrenzel, J.; Huggler, E.; Kampf, S.; Chaponnier, C.; Lew, D.; Lacroix, J.S. , Evidence of an intracellular reservoir in the nasal mucosa of patients with recurrent Staphylococcus aureus rhinosinusitis. J Infect Dis 2005, 192, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Bassiouni, A.; Drilling, A.; Psaltis, A.J.; Vreugde, S.; Wormald, P.J. , The persistence of intracellular Staphylococcus aureus in the sinuses: A longitudinal study. Rhinology 2017, 55, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Zautner, A.E.; Krause, M.; Stropahl, G.; Holtfreter, S.; Frickmann, H.; Maletzki, C.; Kreikemeyer, B.; Pau, H.W.; Podbielski, A. , Intracellular persisting Staphylococcus aureus is the major pathogen in recurrent tonsillitis. PLoS ONE 2010, 5, e9452. [Google Scholar] [CrossRef] [PubMed]
- Ellington, J.K.; Harris, M.; Webb, L.; Smith, B.; Smith, T.; Tan, K.; Hudson, M. , Intracellular Staphylococcus aureus. A mechanism for the indolence of osteomyelitis. J Bone Joint Surg Br 2003, 85, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wijenayaka, A.R.; Solomon, L.B.; Pederson, S.M.; Findlay, D.M.; Kidd, S.P.; Atkins, G.J. , Novel Insights into Staphylococcus aureus Deep Bone Infections: The Involvement of Osteocytes. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Torlakovic, E.; Hibbs, J.R.; Miller, J.S.; Litz, C.E. , Intracellular bacteria in blood smears in patients with central venous catheters. Arch Intern Med 1995, 155, 1547–1550. [Google Scholar] [CrossRef]
- Grundmeier, M.; Tuchscherr, L.; Bruck, M.; Viemann, D.; Roth, J.; Willscher, E.; Becker, K.; Peters, G.; Loffler, B. , Staphylococcal strains vary greatly in their ability to induce an inflammatory response in endothelial cells. J Infect Dis 2010, 201, 871–880. [Google Scholar] [CrossRef]
- Menzies, B.E.; Kourteva, I. , Staphylococcus aureus alpha-toxin induces apoptosis in endothelial cells. FEMS Immunol Med Microbiol 2000, 29, 39–45. [Google Scholar] [PubMed]
- Schnaith, A.; Kashkar, H.; Leggio, S.A.; Addicks, K.; Kronke, M.; Krut, O. , Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem 2007, 282, 2695–2706. [Google Scholar] [CrossRef] [PubMed]
- Soong, G.; Chun, J.; Parker, D.; Prince, A. , Staphylococcus aureus activation of caspase 1/calpain signaling mediates invasion through human keratinocytes. J Infect Dis 2012, 205, 1571–1579. [Google Scholar] [CrossRef]
- Tuchscherr, L.; Medina, E.; Hussain, M.; Volker, W.; Heitmann, V.; Niemann, S.; Holzinger, D.; Roth, J.; Proctor, R.A.; Becker, K.; Peters, G.; Loffler, B. , Staphylococcus aureus phenotype switching: An effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol Med 2011, 3, 129–141. [Google Scholar] [CrossRef]
- Loffler, B.; Tuchscherr, L.; Niemann, S.; Peters, G. , Staphylococcus aureus persistence in non-professional phagocytes. Int J Med Microbiol 2014, 304, 170–176. [Google Scholar] [CrossRef]
- Tuchscherr, L.; Loffler, B. , Staphylococcus aureus dynamically adapts global regulators and virulence factor expression in the course from acute to chronic infection. Curr Genet 2016, 62, 15–17. [Google Scholar] [CrossRef]
- Haslinger-Loffler, B.; Kahl, B.C.; Grundmeier, M.; Strangfeld, K.; Wagner, B.; Fischer, U.; Cheung, A.L.; Peters, G.; Schulze-Osthoff, K.; Sinha, B. , Multiple virulence factors are required for Staphylococcus aureus-induced apoptosis in endothelial cells. Cell Microbiol 2005, 7, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.J.; Drilling, A.J.; Cooksley, C.; Bassiouni, A.; Kidd, S.P.; Psaltis, A.J.; Wormald, P.J.; Vreugde, S. , Reduced Innate Immune Response to a Staphylococcus aureus Small Colony Variant Compared to Its Wild-Type Parent Strain. Front Cell Infect Microbiol 2016, 6, 187. [Google Scholar] [CrossRef]
- Horst, S.A.; Hoerr, V.; Beineke, A.; Kreis, C.; Tuchscherr, L.; Kalinka, J.; Lehne, S.; Schleicher, I.; Kohler, G.; Fuchs, T.; Raschke, M.J.; Rohde, M.; Peters, G.; Faber, C.; Loffler, B.; Medina, E. , A novel mouse model of Staphylococcus aureus chronic osteomyelitis that closely mimics the human infection: An integrated view of disease pathogenesis. Am J Pathol 2012, 181, 1206–1214. [Google Scholar] [CrossRef]
- Seidl, K.; Solis, N.V.; Bayer, A.S.; Hady, W.A.; Ellison, S.; Klashman, M.C.; Xiong, Y.Q.; Filler, S.G. , Divergent responses of different endothelial cell types to infection with Candida albicans and Staphylococcus aureus. PLoS ONE 2012, 7, e39633. [Google Scholar] [CrossRef]
- Jarry, T.M.; Cheung, A.L. , Staphylococcus aureus escapes more efficiently from the phagosome of a cystic fibrosis bronchial epithelial cell line than from its normal counterpart. Infect Immun 2006, 74, 2568–2577. [Google Scholar] [CrossRef] [PubMed]
- Hudson, M.C.; Ramp, W.K.; Nicholson, N.C.; Williams, A.S.; Nousiainen, M.T. , Internalization of Staphylococcus aureus by cultured osteoblasts. Microb Pathog 1995, 19, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Weinrick, B.; Dunman, P.M.; McAleese, F.; Murphy, E.; Projan, S.J.; Fang, Y.; Novick, R.P. , Effect of mild acid on gene expression in Staphylococcus aureus. J Bacteriol 2004, 186, 8407–8423. [Google Scholar] [CrossRef] [PubMed]
- Kubica, M.; Guzik, K.; Koziel, J.; Zarebski, M.; Richter, W.; Gajkowska, B.; Golda, A.; Maciag-Gudowska, A.; Brix, K.; Shaw, L.; Foster, T.; Potempa, J. , A potential new pathway for Staphylococcus aureus dissemination: The silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS ONE 2008, 3, e1409. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.Y.; Schwartz, J.; Thoendel, M.; Ackermann, L.W.; Horswill, A.R.; Nauseef, W.M. , agr-Dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J Innate Immun 2010, 2, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Tranchemontagne, Z.R.; Camire, R.B.; O'Donnell, V.J.; Baugh, J.; Burkholder, K.M. , Staphylococcus aureus Strain USA300 Perturbs Acquisition of Lysosomal Enzymes and Requires Phagosomal Acidification for Survival inside Macrophages. Infect Immun 2016, 84, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Watkins, K.E.; Unnikrishnan, M. , Evasion of host defenses by intracellular Staphylococcus aureus. Adv Appl Microbiol 2020, 112, 105–141. [Google Scholar] [PubMed]
- Lacoma, A.; Cano, V.; Moranta, D.; Regueiro, V.; Dominguez-Villanueva, D.; Laabei, M.; Gonzalez-Nicolau, M.; Ausina, V.; Prat, C.; Bengoechea, J.A. , Investigating intracellular persistence of Staphylococcus aureus within a murine alveolar macrophage cell line. Virulence 2017, 8, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Jack, R.W.; Otto, M.; Collins, L.V.; Staubitz, P.; Nicholson, G.; Kalbacher, H.; Nieuwenhuizen, W.F.; Jung, G.; Tarkowski, A.; van Kessel, K.P.; van Strijp, J.A. , Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med 2001, 193, 1067–1076. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Kuiack, R.C.; McGavin, M.J.; Heinrichs, D.E. , Staphylococcus aureus Uses the GraXRS Regulatory System To Sense and Adapt to the Acidified Phagolysosome in Macrophages. mBio 2018, 9. [Google Scholar] [CrossRef]
- Karavolos, M.H.; Horsburgh, M.J.; Ingham, E.; Foster, S.J. , Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology (Reading) 2003, 149 Pt 10, 2749–2758. [Google Scholar] [CrossRef]
- Cosgrove, K.; Coutts, G.; Jonsson, I.M.; Tarkowski, A.; Kokai-Kun, J.F.; Mond, J.J.; Foster, S.J. , Catalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J Bacteriol 2007, 189, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Treffon, J.; Block, D.; Moche, M.; Reiss, S.; Fuchs, S.; Engelmann, S.; Becher, D.; Langhanki, L.; Mellmann, A.; Peters, G.; Kahl, B.C. , Adaptation of Staphylococcus aureus to Airway Environments in Patients With Cystic Fibrosis by Upregulation of Superoxide Dismutase M and Iron-Scavenging Proteins. J Infect Dis 2018, 217, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Treffon, J.; Chaves-Moreno, D.; Niemann, S.; Pieper, D.H.; Vogl, T.; Roth, J.; Kahl, B.C. , Importance of superoxide dismutases A and M for protection of Staphylococcus aureus in the oxidative stressful environment of cystic fibrosis airways. Cell Microbiol 2020, 22, e13158. [Google Scholar] [CrossRef] [PubMed]
- Munzenmayer, L.; Geiger, T.; Daiber, E.; Schulte, B.; Autenrieth, S.E.; Fraunholz, M.; Wolz, C. , Influence of Sae-regulated and Agr-regulated factors on the escape of Staphylococcus aureus from human macrophages. Cell Microbiol 2016, 18, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Jarry, T.M.; Memmi, G.; Cheung, A.L. , The expression of alpha-haemolysin is required for Staphylococcus aureus phagosomal escape after internalization in CFT-1 cells. Cell Microbiol 2008, 10, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- Grosz, M.; Kolter, J.; Paprotka, K.; Winkler, A.C.; Schafer, D.; Chatterjee, S.S.; Geiger, T.; Wolz, C.; Ohlsen, K.; Otto, M.; Rudel, T.; Sinha, B.; Fraunholz, M. , Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin alpha. Cell Microbiol 2014, 16, 451–465. [Google Scholar] [CrossRef]
- Wang, R.; Braughton, K.R.; Kretschmer, D.; Bach, T.H.; Queck, S.Y.; Li, M.; Kennedy, A.D.; Dorward, D.W.; Klebanoff, S.J.; Peschel, A.; DeLeo, F.R.; Otto, M. , Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 2007, 13, 1510–1514. [Google Scholar] [CrossRef]
- Queck, S.Y.; Jameson-Lee, M.; Villaruz, A.E.; Bach, T.H.; Khan, B.A.; Sturdevant, D.E.; Ricklefs, S.M.; Li, M.; Otto, M. , RNAIII-independent target gene control by the agr quorum-sensing system: Insight into the evolution of virulence regulation in Staphylococcus aureus. Mol Cell 2008, 32, 150–158. [Google Scholar] [CrossRef]
- Geiger, T.; Francois, P.; Liebeke, M.; Fraunholz, M.; Goerke, C.; Krismer, B.; Schrenzel, J.; Lalk, M.; Wolz, C. , The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog 2012, 8, e1003016. [Google Scholar] [CrossRef]
- Chatterjee, S.S.; Joo, H.S.; Duong, A.C.; Dieringer, T.D.; Tan, V.Y.; Song, Y.; Fischer, E.R.; Cheung, G.Y.; Li, M.; Otto, M. , Essential Staphylococcus aureus toxin export system. Nat Med 2013, 19, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Blattner, S.; Das, S.; Paprotka, K.; Eilers, U.; Krischke, M.; Kretschmer, D.; Remmele, C.W.; Dittrich, M.; Muller, T.; Schuelein-Voelk, C.; Hertlein, T.; Mueller, M.J.; Huettel, B.; Reinhardt, R.; Ohlsen, K.; Rudel, T.; Fraunholz, M.J. , Staphylococcus aureus Exploits a Non-ribosomal Cyclic Dipeptide to Modulate Survival within Epithelial Cells and Phagocytes. PLoS Pathog 2016, 12, e1005857. [Google Scholar] [CrossRef] [PubMed]
- Giese, B.; Glowinski, F.; Paprotka, K.; Dittmann, S.; Steiner, T.; Sinha, B.; Fraunholz, M.J. , Expression of delta-toxin by Staphylococcus aureus mediates escape from phago-endosomes of human epithelial and endothelial cells in the presence of beta-toxin. Cell Microbiol 2011, 13, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Clauditz, A.; Resch, A.; Wieland, K.P.; Peschel, A.; Gotz, F. , Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Infect Immun 2006, 74, 4950–4953. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Heit, B.; Heinrichs, D.E. , Antimicrobial Mechanisms of Macrophages and the Immune Evasion Strategies of Staphylococcus aureus. Pathogens 2015, 4, 826–868. [Google Scholar] [CrossRef] [PubMed]
- Jubrail, J.; Morris, P.; Bewley, M.A.; Stoneham, S.; Johnston, S.A.; Foster, S.J.; Peden, A.A.; Read, R.C.; Marriott, H.M.; Dockrell, D.H. , Inability to sustain intraphagolysosomal killing of Staphylococcus aureus predisposes to bacterial persistence in macrophages. Cell Microbiol 2016, 18, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Heit, B.; Heinrichs, D.E. , Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol 2016, 18, 514–535. [Google Scholar] [CrossRef] [PubMed]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; Kim, J.; Park, S.; Darwish, M.; Lee, B.C.; Hernandez, H.; Loyet, K.M.; Lupardus, P.; Fong, R.; Yan, D.; Chalouni, C.; Luis, E.; Khalfin, Y.; Plise, E.; Cheong, J.; Lyssikatos, J.P.; Strandh, M.; Koefoed, K.; Andersen, P.S.; Flygare, J.A.; Wah Tan, M.; Brown, E.J.; Mariathasan, S. , Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Siegmund, A.; Afzal, M.A.; Tetzlaff, F.; Keinhorster, D.; Gratani, F.; Paprotka, K.; Westermann, M.; Nietzsche, S.; Wolz, C.; Fraunholz, M.; Hubner, C.A.; Loffler, B.; Tuchscherr, L. , Intracellular persistence of Staphylococcus aureus in endothelial cells is promoted by the absence of phenol-soluble modulins. Virulence 2021, 12, 1186–1198. [Google Scholar] [CrossRef]
- Elgrail, M.M.; Chen, E.; Shaffer, M.G.; Srinivasa, V.; Griffith, M.P.; Mustapha, M.M.; Shields, R.K.; Van Tyne, D.; Culyba, M.J. , Convergent Evolution of Antibiotic Tolerance in Patients with Persistent Methicillin-Resistant Staphylococcus aureus Bacteremia. Infect Immun 2022, 90, e0000122. [Google Scholar] [CrossRef]
- Sriramulu, D.D.; Nimtz, M.; Romling, U. , Proteome analysis reveals adaptation of Pseudomonas aeruginosa to the cystic fibrosis lung environment. Proteomics 2005, 5, 3712–3721. [Google Scholar] [CrossRef] [PubMed]
- Pagels, M.; Fuchs, S.; Pane-Farre, J.; Kohler, C.; Menschner, L.; Hecker, M.; McNamarra, P.J.; Bauer, M.C.; von Wachenfeldt, C.; Liebeke, M.; Lalk, M.; Sander, G.; von Eiff, C.; Proctor, R.A.; Engelmann, S. , Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol Microbiol 2010, 76, 1142–1161. [Google Scholar] [CrossRef] [PubMed]
- Desgranges, E.; Barrientos, L.; Herrgott, L.; Marzi, S.; Toledo-Arana, A.; Moreau, K.; Vandenesch, F.; Romby, P.; Caldelari, I. , The 3'UTR-derived sRNA RsaG coordinates redox homeostasis and metabolism adaptation in response to glucose-6-phosphate uptake in Staphylococcus aureus. Mol Microbiol 2022, 117, 193–214. [Google Scholar] [CrossRef]
- Luo, J.D.; Chen, A.F. , Nitric oxide: A newly discovered function on wound healing. Acta Pharmacol Sin 2005, 26, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.; Knight, D.; Burgess, S.; Franklin, P.; Horak, F.; Legg, J.; Moeller, A.; Stick, S. , Epithelial inducible nitric oxide synthase activity is the major determinant of nitric oxide concentration in exhaled breath. Thorax 2004, 59, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Palma Medina, L.M.; Becker, A.K.; Michalik, S.; Surmann, K.; Hildebrandt, P.; Gesell Salazar, M.; Mekonnen, S.A.; Kaderali, L.; Volker, U.; van Dijl, J.M. , Interaction of Staphylococcus aureus and Host Cells upon Infection of Bronchial Epithelium during Different Stages of Regeneration. ACS Infect Dis 2020, 6, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.D.; Snyder, D.J.; Putnam, N.E.; Valentino, M.D.; Hammer, N.D.; Lonergan, Z.R.; Hinger, S.A.; Aysanoa, E.E.; Blanchard, C.; Dunman, P.M.; Wasserman, G.A.; Chen, J.; Shopsin, B.; Gilmore, M.S.; Skaar, E.P.; Cassat, J.E. , Bacterial Hypoxic Responses Revealed as Critical Determinants of the Host-Pathogen Outcome by TnSeq Analysis of Staphylococcus aureus Invasive Infection. PLoS Pathog 2015, 11, e1005341. [Google Scholar] [CrossRef] [PubMed]
- Throup, J.P.; Zappacosta, F.; Lunsford, R.D.; Annan, R.S.; Carr, S.A.; Lonsdale, J.T.; Bryant, A.P.; McDevitt, D.; Rosenberg, M.; Burnham, M.K. , The srhSR gene pair from Staphylococcus aureus: Genomic and proteomic approaches to the identification and characterization of gene function. Biochemistry 2001, 40, 10392–10401. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wu, Y.; Zhu, T.; Han, H.; Liu, H.; Xu, T.; Francois, P.; Fischer, A.; Bai, L.; Gotz, F.; Qu, D. , Staphylococcus epidermidis SrrAB regulates bacterial growth and biofilm formation differently under oxic and microaerobic conditions. J Bacteriol 2015, 197, 459–476. [Google Scholar] [CrossRef]
- Cao, S.; Huseby, D.L.; Brandis, G.; Hughes, D. , Alternative Evolutionary Pathways for Drug-Resistant Small Colony Variant Mutants in Staphylococcus aureus. mBio 2017, 8. [Google Scholar] [CrossRef]
- Wickersham, M.; Wachtel, S.; Wong Fok Lung, T.; Soong, G.; Jacquet, R.; Richardson, A.; Parker, D.; Prince, A. , Metabolic Stress Drives Keratinocyte Defenses against Staphylococcus aureus Infection. Cell Rep 2017, 18, 2742–2751. [Google Scholar] [CrossRef] [PubMed]
- Wong Fok Lung, T.; Monk, I.R.; Acker, K.P.; Mu, A.; Wang, N.; Riquelme, S.A.; Pires, S.; Noguera, L.P.; Dach, F.; Gabryszewski, S.J.; Howden, B.P.; Prince, A. , Staphylococcus aureus small colony variants impair host immunity by activating host cell glycolysis and inducing necroptosis. Nat Microbiol 2020, 5, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Kitur, K.; Wachtel, S.; Brown, A.; Wickersham, M.; Paulino, F.; Penaloza, H.F.; Soong, G.; Bueno, S.; Parker, D.; Prince, A. , Necroptosis Promotes Staphylococcus aureus Clearance by Inhibiting Excessive Inflammatory Signaling. Cell Rep 2016, 16, 2219–2230. [Google Scholar] [CrossRef]
- Kumar, R.; Kanev, L.; Woods, S.D.; Brenner, M.; Smith, B. , Managing hyperkalemia in high-risk patients in long-term care. Am J Manag Care 2017, 23, S27–S36. [Google Scholar]
- Saeed, S.; Quintin, J.; Kerstens, H.H.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; Sharifi, N.; Janssen-Megens, E.M.; Ter Huurne, M.; Mandoli, A.; van Schaik, T.; Ng, A.; Burden, F.; Downes, K.; Frontini, M.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Ouwehand, W.H.; van der Meer, J.W.; Joosten, L.A.; Wijmenga, C.; Martens, J.H.; Xavier, R.J.; Logie, C.; Netea, M.G.; Stunnenberg, H.G. , Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef]
- Arts, R.J.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.C.; Wang, S.Y.; Habibi, E.; Goncalves, L.G.; Mesquita, I.; Cunha, C.; van Laarhoven, A.; van de Veerdonk, F.L.; Williams, D.L.; van der Meer, J.W.; Logie, C.; O'Neill, L.A.; Dinarello, C.A.; Riksen, N.P.; van Crevel, R.; Clish, C.; Notebaart, R.A.; Joosten, L.A.; Stunnenberg, H.G.; Xavier, R.J.; Netea, M.G. , Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab 2016, 24, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. , Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Tulkens, P.M. , Intracellular distribution and activity of antibiotics. Eur J Clin Microbiol Infect Dis 1991, 10, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Renard, C.; Vanderhaeghe, H.J.; Claes, P.J.; Zenebergh, A.; Tulkens, P.M. , Influence of conversion of penicillin G into a basic derivative on its accumulation and subcellular localization in cultured macrophages. Antimicrob Agents Chemother 1987, 31, 410–416. [Google Scholar] [CrossRef]
- Aubert-Tulkens, G.; Van Hoof, F.; Tulkens, P. , Gentamicin-induced lysosomal phospholipidosis in cultured rat fibroblasts. Quantitative ultrastructural and biochemical study. Lab Invest 1979, 40, 481–491. [Google Scholar]
- Cosgrove, S.E.; Vigliani, G.A.; Fowler, V.G., Jr.; Abrutyn, E.; Corey, G.R.; Levine, D.P.; Rupp, M.E.; Chambers, H.F.; Karchmer, A.W.; Boucher, H.W. , Initial low-dose gentamicin for Staphylococcus aureus bacteremia and endocarditis is nephrotoxic. Clin Infect Dis 2009, 48, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Barcia-Macay, M.; Seral, C.; Mingeot-Leclercq, M.P.; Tulkens, P.M.; Van Bambeke, F. , Pharmacodynamic evaluation of the intracellular activities of antibiotics against Staphylococcus aureus in a model of THP-1 macrophages. Antimicrob Agents Chemother 2006, 50, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Proctor, R.A.; Kahl, B.; von Eiff, C.; Vaudaux, P.E.; Lew, D.P.; Peters, G. , Staphylococcal small colony variants have novel mechanisms for antibiotic resistance. Clin Infect Dis 1998, 27 (Suppl. S1), S68–S74. [Google Scholar] [CrossRef]
- Somerville, G.A.; Chaussee, M.S.; Morgan, C.I.; Fitzgerald, J.R.; Dorward, D.W.; Reitzer, L.J.; Musser, J.M. , Staphylococcus aureus aconitase inactivation unexpectedly inhibits post-exponential-phase growth and enhances stationary-phase survival. Infect Immun 2002, 70, 6373–6382. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, I.; Kriegeskorte, A.; Fischer, A.; Deiwick, S.; Theimann, N.; Proctor, R.A.; Peters, G.; Herrmann, M.; Kahl, B.C. , In vivo mutations of thymidylate synthase (encoded by thyA) are responsible for thymidine dependency in clinical small-colony variants of Staphylococcus aureus. J Bacteriol 2008, 190, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F.; Miller, M.H. , Emergence of resistance to cephalothin and gentamicin during combination therapy for methicillin-resistant Staphylococcus aureus endocarditis in rabbits. J Infect Dis 1987, 155, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Conlon, B.P.; Rowe, S.E.; Gandt, A.B.; Nuxoll, A.S.; Donegan, N.P.; Zalis, E.A.; Clair, G.; Adkins, J.N.; Cheung, A.L.; Lewis, K. , Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat Microbiol 2016, 1. [Google Scholar] [CrossRef]
- Mitchell, G.; Gattuso, M.; Grondin, G.; Marsault, E.; Bouarab, K.; Malouin, F. , Tomatidine inhibits replication of Staphylococcus aureus small-colony variants in cystic fibrosis airway epithelial cells. Antimicrob Agents Chemother 2011, 55, 1937–1945. [Google Scholar] [CrossRef]
Figure 1.
Concept of antimicrobial resistance, tolerance, and persistence in S. aureus and potential impact on treatment outcomes.
Figure 1.
Concept of antimicrobial resistance, tolerance, and persistence in S. aureus and potential impact on treatment outcomes.
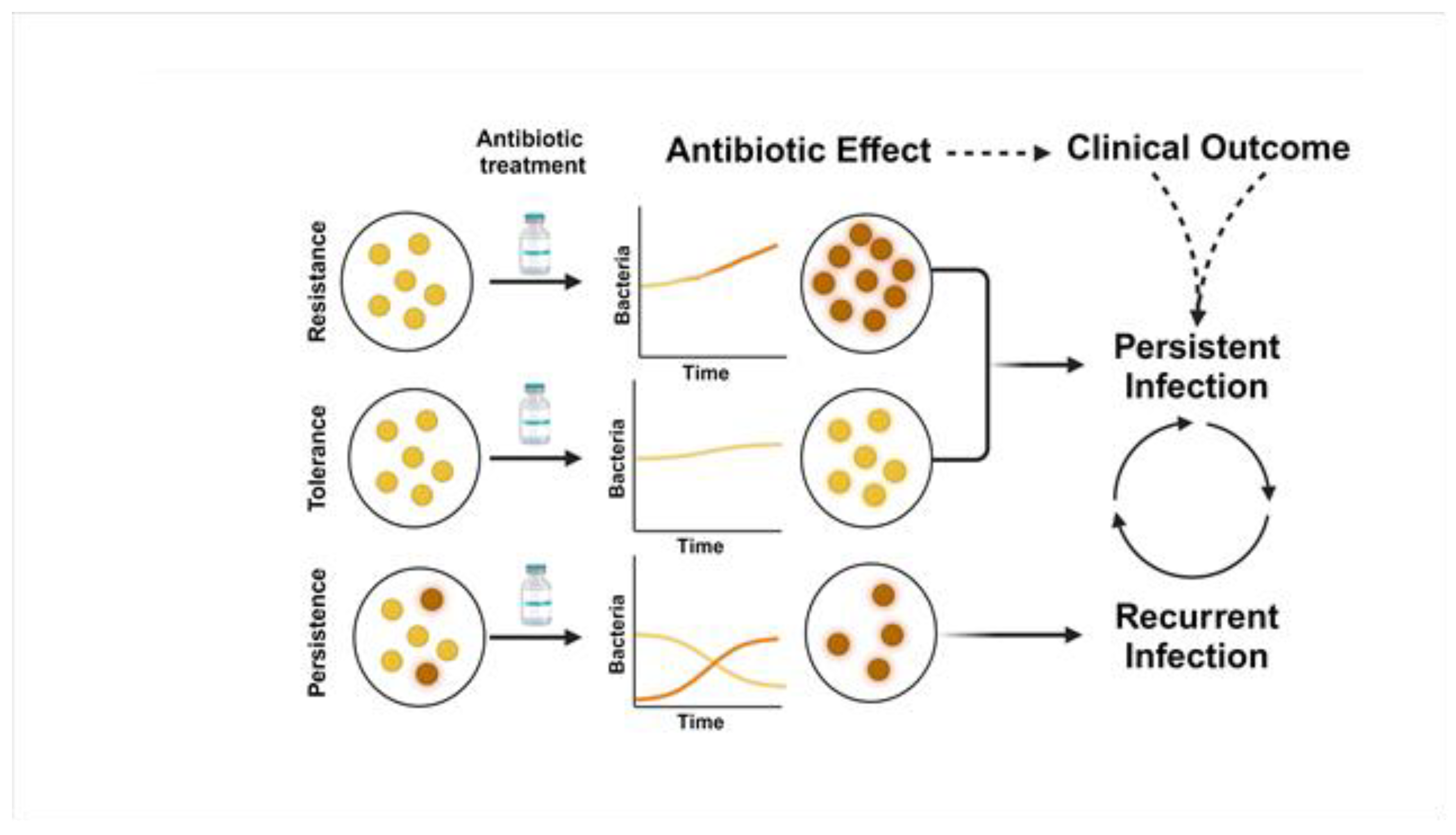
Figure 2.
Model of intracellular lifecycle and anaerobic metabolism of S. aureus contributing to persistent infection.
Figure 2.
Model of intracellular lifecycle and anaerobic metabolism of S. aureus contributing to persistent infection.
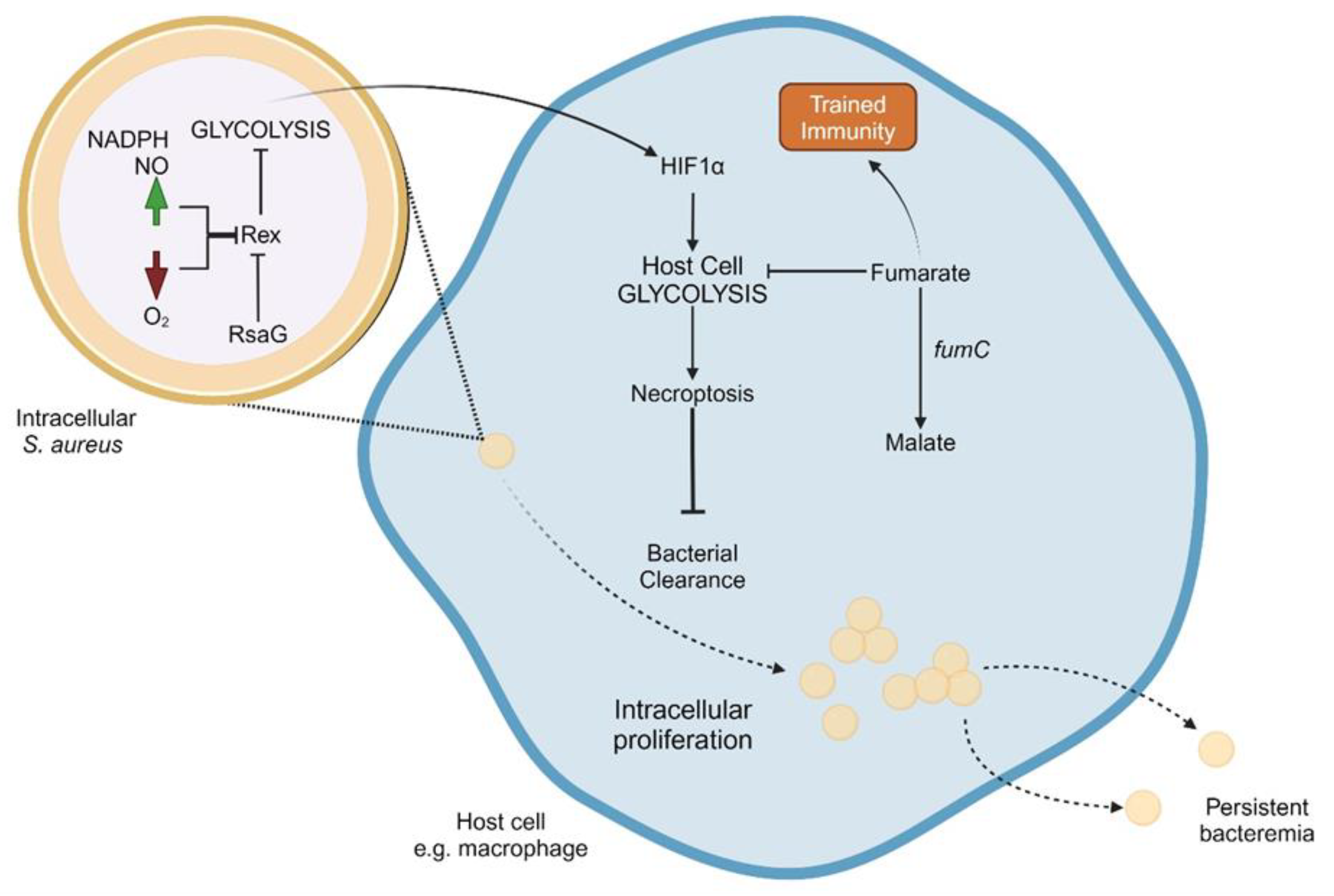
Figure 3.
Mechanisms of reduced antibiotic activity against intracellular S. aureus.
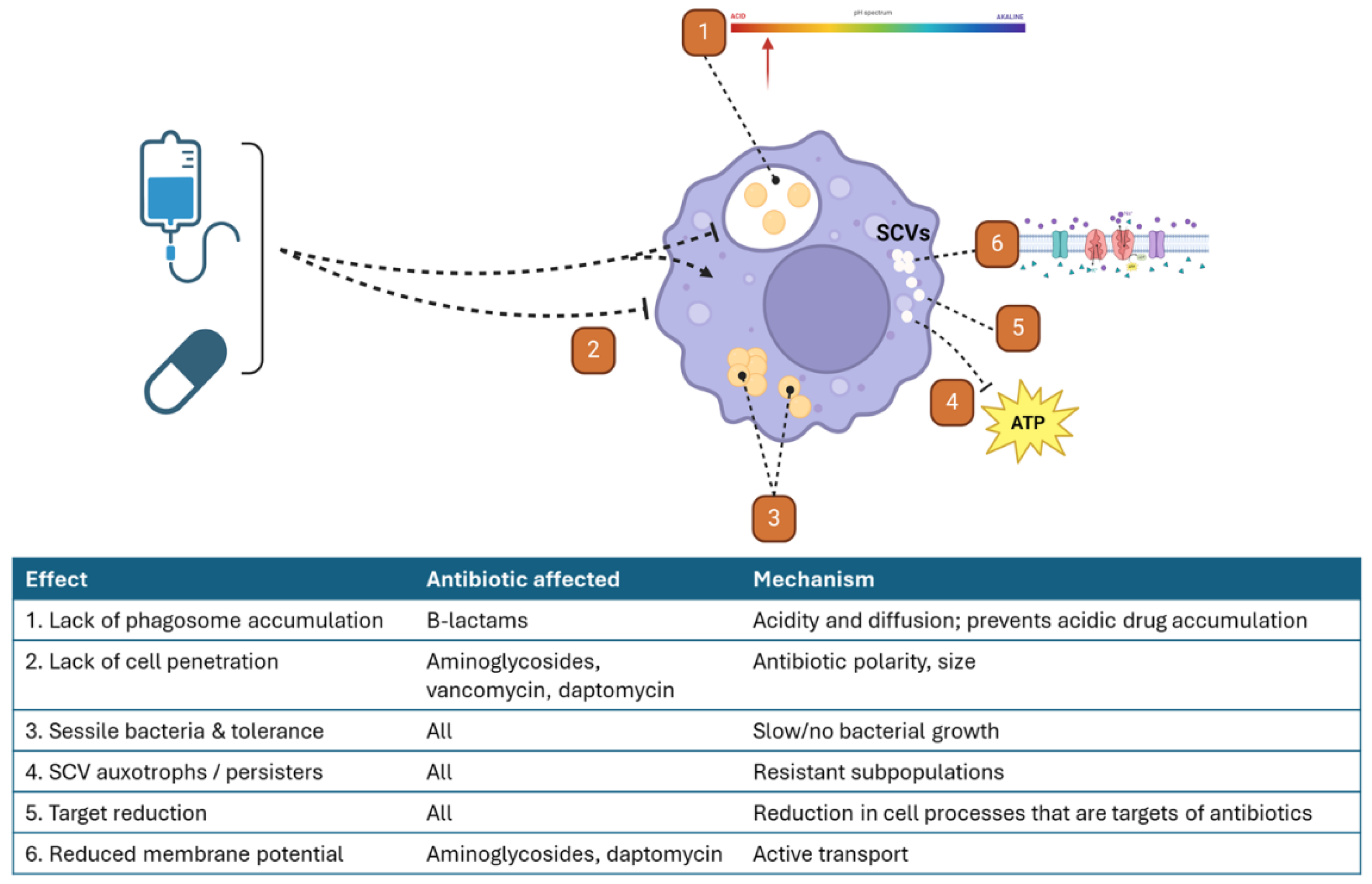
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



