
Submitted:
22 May 2024
Posted:
23 May 2024
You are already at the latest version
Abstract
A partial mitochondrial DNA Cytochrome Oxidase subunit I (mtCOI) gene haplotype variant of the coconut rhinoceros beetle (CRB) Oryctes rhinoceros classed as ‘CRB-G (clade I)’ has been the focus of much research since 2007 with reports of invasions into new Pacific Island locations (e.g., Guam, Hawaii, Solomons Islands). For numerous invasive species, inference of invasion biology via whole genome is superior to assessments via the partial mtCOI gene. Here, we explore CRB draft mitochondrial genomes (mitogenomes) from historical and recent collections, with assessment focused on individuals associated within the CRB-G (clade I) classification. We found that all Guam CRB individuals possessed the same mitogenome across all 13 protein coding genes and differed from individuals collected elsewhere, including ‘non-Guam’ individuals designated as CRB-G (clade I) by partial mtCOI assessment. Two alternative ATP6 and COIII partial gene primer sets were developed to enable distinction between CRB (clade I) that invaded Guam and CRB-G (Clade I) individuals collected elsewhere. Phylogenetic analyses based on concatenated ATP6-COIII genes showed that only Guam CRB-G (clade I) individuals clustered together, and therefore Guam was not the source of the CRB that invaded the other locations in the Pacific assessed in this study. The use of the mtCOI and/or mtCOIII genes for initial molecular diagnosis of CRB remained crucial, and assessment of more native CRB populations will further advance our ability to identify the provenance of CRB invasions being reported within the Pacific and elsewhere
Keywords:
DNA barcoding
; comparative mitogenome analysis
; Asiatic rhinoceros beetle
; hitchhiker plant pest
1. Introduction
The mitochondrial DNA (mtDNA) genome (mitogenome) is largely maternally inherited and generally consists of 13 protein coding genes (PCG’s), 22 tRNA genes, 2 rRNA genes, and an AT-rich region that is low in nucleotide complexity (e.g., Crozier 1990). Due to the maternal inheritance nature, the mitogenome in general does not undergo recombination (e.g., Saville et al. 1998; Leducq et al. 2017). Hebert et al. (2003) demonstrated the use of the partial mtDNA cytochrome oxidase subunit I (mtCOI) gene sequence to aid in species diagnostics, and this system helped transform understanding of species diversity (Hebert et al. 2004; Rugman-Jones et al. 2013; Tay et al. 2016). Multiple partial mtCOI databases (e.g., BOLD; GenBank) subsequently provided considerable contribution to disentangling species status (e.g., De Barro et al. 2011; Behere et al. 2007; Tay et al. 2017a). However, the use of partial mtCOI is not without its limitations; association with endosymbionts, effect of selective sweep, and impact of pseudogenes, amongst other factors, can all lead to inaccurate interpretations (Hurst and Jiggins, 2005; Tay et al. 2017b). Analysis of population-wide partial mtCOI gene diversity has also found that the 5’ gene region typically has low nucleotide diversity (i.e., conserved nucleotide sequence) in some arthropod groups such as Hemiptera, Lepidoptera, and Coleoptera (e.g., Tay et al. 2022a; Tay et al. 2022b; Behere et al. 2007). Subsequently, an over reliance on partial mtCOI gene sequences has resulted in some misidentification of species status (e.g., Tay et al. 2017b), and some misunderstandings of population dynamics (e.g., Goergen et al. 2016 vs. Tay et al. 2022a; see also review by Tay et al. 2023). Examples of confounded interpretations include the invasion history of Spodoptera frugiperda (Cock et al. 2017; Nagoshi et al. 2018, Nagoshi et al. 2019) and population expansion patterns of Helicoverpa species (Leite et al. 2014 vs. Arnemann et al. 2018).
As technology advances and costs decrease, it is now easier and cheaper than ever before to obtain greater genetic data from specimens to provide richer information content for analysis and interpretation. Combining sequence data from multiple mtDNA genes, full mitochondrial DNA genomes (mitogenomes) and/or whole genomes is now regularly being shown to provide superior analysis to partial mtCOI genes alone for all applications. Examples include: identifications of species (e.g., Walsh et al. 2019), sub-species (e.g., Anderson et al. 2016; Elfekih et al. 2021; Zhang et al. 2022), hybrids (e.g., Anderson et al. 2016; Elfekih et al. 2021; Valencia-Montoya et al. 2020), populations (Tay et al. 2022a; Rane et al. 2023), patterns like demographic expansion (e.g., Pearce et al. 2017; Lannucci et al. 2021), and pest incursion histories (e.g., Tay 2016; Otim et al. 2018; Benelli et al. 2023; Li et al. 2022).
Here, we examine draft full mitogenomes of the coconut rhinoceros beetle (CRB; Oryctes rhinoceros), a pest that causes economic yield losses to coconut and oil palm (Bedford, 1980; Indriyanti et al. 2019). These mitogenomes were generated through whole genome sequencing (WGS) from multiple geographically distinct locations to develop additional molecular markers for tracking and monitoring genetically distinct populations of this species. Particular attention is given to the CRB-G (clade I) group determined using partial mtCOI gene assessment (Marshall et al. 2017), because of the current biosecurity focus on this mitocnondrial haplotype variant due to its reported resistance to known isolates of the Oryctes rhinoceros Nudivirus (OrNV) biological control agent (Marshall et al. 2017), and new incursions within the Pacific region (Paudel et al. 2023; Marshall et al. 2023). Specifically, we test whether or not the Guam CRB-G (clade I) was the source population for the CRB-G (clade I) in Solomon Islands and in Palau as suggested in some publications (e.g., Datt 2020, Caasi 2023). We do this by identifying two partial mitochondrial gene regions that more confidently differentiated CRB (clade I) individuals that invaded Guam from other CRB individuals, including other individuals classed as CRB-G (clade I) using the partial mtCOI gene, but which were collected on other Pacific islands (e.g., Solomon Islands, Palau). We discuss the benefits of mitogenomes as resources for developing alternative diagnostic markers, and assess efficacies of the partial mtCOI gene as the current preferred standard diagnostic DNA marker to distinguish CRB populations.
2. Material and Methods
2.1. Samples
CRB samples collected between 2019 and 2022 were sourced from Guam, Palau, Indonesia, Malaysia, Hawaii, and Philippines (Table 1). The gut of each specimen was dissected, preserved in 100% ethanol, and stored at -20 °C until needed for DNA extraction. Additionally, a historic specimen collected from Guam (04-Or5; circa 2014) was used as a reference to enable matching of more recently collected CRB individuals from Guam that classed within the CRB-G (clade I) haplotype grouping sensu Marshall et al. (2017).
2.2. Whole Genome Sequencing (WGS)
We used the Qiagen Blood and Tissue DNA extraction kit (Duesseldorf, Germany) and the manufacturer’s protocol to extract genomic DNA. All extracted DNA was eluted in 200 µL EB and kept frozen until used for WGS. We assessed the quality of the extracted DNA using Qubit 2.0 prior to sequencing. WGS was carried out by the Australian Genome Resource Facility (AGRF) in Melbourne, Australia, or by AZENTA Life Sciences in China. The WGS data returned an average of 25x coverage, 150 bp paired-end reads/sample, assuming a genome size of approximately 350 Mbp.
2.3. Mitogenome Assembly and Annotation
We assembled all mitogenomes by importing the raw sequence reads into Geneious Prime 2022.2.2 (Build 2022-08-18 14:34) (Biomatters Ltd., Auckland), and used the published mitogenome (MT457815, Filipovic et al. 2021) as the reference sequence. We used Geneious Mapper with ‘Low Sensitivity / Fastest’ option and selecting no fine tuning (i.e., None (fast / read mapping)) during the mitogenome assembling process. Although we received pair-ended reads for all samples, mitogenomes were assembled using forward reads only due to the high genome coverage for each sample. All assembled mitogenomes were initially annotated using the MITOS program and selecting invertebrate mitochondrial genetic code (Bernt et al. 2013). As a final quality assessment, the annotated CRB mitogenomes were visually inspected. The assembled and annotated mtCOI, ATP6, and COIII genes used in this study are available from the CSIRO data repository (Tay et al. 2024).
2.4. Mitogenome Identity Assessment
The non-recombination nature of the mitogenome implies that CRB individuals classified as CRB-G (clade I) based on the partial mtCOI gene assessment method of Marshall et al. (2017) (e.g., Solomon Islands MT457815, Taiwan MW632131) would share mitogenome identity with our reference Guam specimen (i.e., 04-Or5; Table 1), if a single source of invasion entered into Guam and subsequently spread from to other locations. To assess this, randomly selected CRB specimens from Guam (i.e., NZ-20-738; Guam-01_GDoA, Guam-02_GDoA, Guam-09_GDoA, Guam-13_GDoA, Guam-17_GDoA; Table 1) that were collected in more recent times (2020 and 2021) were compared with the representative historical Guam individual (04-Or5) to visually assess and confirm mitogenome identity. This was then followed by comparison with all other CRB individuals including CRB-G type individuals collected from elsewhere (Table 1). Individuals were compared based on the partial mtCOI sequence analysis (described in Marshall et al. 2017) as well as sequence similarity of other mitochondrial genes assessed by this work.
2.5. Alternative CRB Marker Development to Identify the Original CRB Population that Invaded Guam
To identify candidate mitochondrial genes as alternative DNA markers specific to individuals from Guam, all mitogenomes generated from this study, as well as publicly available CRB mitogenomes from GenBank, were downloaded and aligned within GenBank using MAFFT V7.490 (Katoh and Standley 2013; Katoh et al. 2002) with default setting options (i.e., algorithm: FFT-NS-2; Scoring matrix: 200PAM / k = 2; Gap open penalty: 1.53; Offset value: 0.123). We visually identified candidate polymorphic sites unique to individuals from Guam (i.e., 04-Or5, NZ-20-738, Guam-01_GDoA, Guam-02_GDoA, Guam-09_GDoA, Guam-13_GDoA, Guam-17_GDoA), but absent in all other CRB individuals (Table 1). SNPs identified were analysed for potential restriction endonucleases to develop polymerase-chain reaction (PCR) restriction fragment length polymorphism (RFLP) solutions (PCR-RFLP), for a simple and easy-to-use approach to confidently differentiate CRB-G (clade I) that invaded Guam from all other genetically distinct CRB, including CRB from elsewhere classed as CRB-G (clade I) by partial COI assessment. Design and analysis of PCR-primers were through the Primer Analysis Software version 7.60 (Molecular Biology Insights, Inc., Cascade, CO, USA). Primers were optimised for minimal false primer annealing sites, minimal primer dimer and duplex formation, and minimal/no hairpin structure, with a Ta (theoretical annealing temperature) of ≥60 °C (calculated as Ta = 4(G + C) + 2(A + T)), and an optimal amplicon length of between 500-600 bp to facilitate ease of Sanger sequencing. The candidate restriction endonuclease was initially selected for a single cut site with in silico analysis of all different mitochondrial DNA haplotypes in Enzyme X version 3.3 <http://nucleobytes.com/enzymex/>. We reconfirmed primer efficacies and RFLP conditions by randomly selecting and analysing DNA from specimens collected from Guam and elsewhere, as well as by PCR-Sanger sequencing to confirm primer amplification accuracy. We used the restriction digestion conditions as recommended by the manufacturer of the BmpI restriction enzyme (New England BioLabs). Visualisation of the RFLP was on a 1.5% 1x TAE agarose gel.
2.6. Mitogenome Analysis
The mitogenomes from the GenBank database and those generated from this study were aligned to estimate pairwise nucleotide identity and distances (p-dist) between: (i) full mtCOI gene vs. full ATP6 gene, and (ii) full mtCOI vs. full COIII genes. The related O. nasicornis mitogenome (0K484312) was included to provide comparison of inter-specific nucleotide distance with CRB. We also inferred phylogenies of the CRB individuals based on the widely used partial mtCOI gene region (676 bp) versus our proposed alternative mitochondrial ATP6 and COIII partial gene regions (excluding nucleotides at primer annealing sites, see Marshall et al. 2017). The APT6 and COIII partial gene sequences were concatenated before phylogeny inference. We used IQ-Tree (Trifinopoulos et al. 2016) and selected the ‘Auto’ option for estimating substitution models, and 1,000 bootstrap alignments to estimate branch support using the ultrafast bootstrap approximation (UFBoot) (Hoang et al. 2018) algorithm. We used Dendroscope 3 (Huson et al. 2007) for visualisation and post analysis editing for both COI and ATP6+COIII phylogenies.
3. Results
3.1. Mitochondrial Genome Analysis
Mitochondrial genomes were assembled and annotated from an average of 1,472 fragments (mean standard deviation 997 fragments) per sample. Across all the mitochondrial COI, ATP6, and COIII gene sequences, nucleotide differences between CRB individuals were low (<2% difference) suggesting that all were the same species (i.e., O. rhinoceros) (Table S1). The assembled and annotated mitogenome from the Guam 04-Or5 specimen (collected in 2014) provided evidence that all Guam individuals examined here shared the same mitogenome (Table 1). Nucleotide differences within the mitochondrial ATP6 and COIII genes were identified in Guam individuals, but these nucleotide polymorphisms were absent from specimens collected elsewhere, including those classed as CRB-G (clade I) by partial COI assessment. For the ATP6_T4430C and COIII_C5390T SNPs identified from the partial ATP6 for COIII genes (see Table 1), there were, on average, 1,876 and 1,740 reads at each of these nucleotide sites to confirm differentiation of CRB-G (clade I) that invaded Guam from other CRB (i.e., equivalent to the diagnostic SNP for ATP6 and COIII being independently confirmed an average of 1,876 and 1,740 times, respectively).
Pairwise nucleotide analysis of the complete mtCOI gene sequence vs. complete ATP6 gene sequence, and also the complete mtCOI gene sequence vs. the complete COIII gene sequence, showed that the seven Guam CRB, one Solomon Islands CRB (MT457815), one Taiwan (MW632131), two Palau CRBs (Palau-03, Palau-04), and four Philippines CRB (Phil-01. -02, -03, -04) specimens analysed shared 100% identity across the complete mtCOI gene sequence. However, when the comparison included the full ATP6 and full COIII gene sequences, only the Guam individuals remained 100% identical to each other. CRB from Solomon Islands (MT457815), Taiwan (MW632131), Palau (Palau-03, Palau-04), and Philippines (Phil-01. -02, -03, -04) all had polymorphisms in these two alternative mitochondrial marker genes (Table 1).
3.2. Alternative Primers to Identify the Original Invasive CRB Population Present in Guam
Two alternative sets of primers were developed (Table 2) to distinguish CRB-G (clade I) that invaded Guam from other CRB, including those collected elsewhere classed as CRB-G (clade 1) by partial COI assessment. One primer amplifies a partial ATP6 gene region of 494 bp length, and the other amplifies a partial COIII gene region of 469 bp length. Optimal PCR annealing temperature for both ATP6 and COIII was 52˚ C with 1.0 µM primer concentration for both ATP6 and COIII primer pairs, and 0.5 mM dNTPs concentration and 1 unit of DNA polymerase in a 50 µL PCR reaction volume.
3.3. Phylogeny
Phylogenetic analysis was carried out using specimens (see Table 1) with available mitogenome DNA sequence data to allow comparison using the mitochondrial gene regions from COI, COIII, and ATP6. Based on the widely used mtCOI partial gene, Figure 1a three clades could be recognised. One clade (red branches) included seven Guam CRB-G (clade I) specimens, four Philippines (green circles) and two individuals from Palau (yellow circles). The other two clades (blue branches) included six individuals from Indonesia, eight from Malaysia, and two from Palau B. Phylogenetic analysis based on concatenation of both partial ATP6 and partial COIII genes returned a different population clustering pattern (Figure 1b). Together, the use of the ATF6 and COIII gene regions showed that Guam CRB-G (clade I) individuals (red branches) clustered by themselves, whereas the Philippines, Malaysian, and Indonesian individuals clustered largely according to their geographical distributions. CRB from Palau (yellow circles) appeared to have multiple origins, clustering with specimens collected from both the Philippines and Indonesia. However, branch node confidence values for Indonesia (54-78) and Philippines (46-48) were low, suggesting longer sequence lengths from both mitochondrial and inclusion of nuclear genes, as well as more samples, are required for confident assessment. Notably, CRB populations in Malaysia appeared to be consisted of two diverse evolutionary lineages based on both COI and the concatenated ATP6-COIII partial genes, with the unique ON764799 and ON764801 individuals originating from both coconut palm and oil palm hosts from the state of Johor (Anggraini et al. 2023).
4. Discussion
In this study, we characterised and reanalysed the draft mitogenomes of CRB individuals from both the native (i.e., Indonesia, Malaysia, Philippines, Taiwan) and exotic (i.e., Guam, Palau, Solomon Islands) ranges. This is also the first time the mitogenome of all recently collected Guam CRB individuals analysed in this study were found to shared sequence identity with specimens historically collected from Guam by possessing the same mitogenome sequence across all 13 protein coding genes (results not presented), and specifically to the ATP6 and COIII genes that exhibited nucleotide differences with CRB from other locations. This resulted in the ATP6 and COIII protein coding genes being used as alternative DNA markers for differentiating Guam-specific CRB-G (clade I) from the other tested CRB individuals. The remaining individuals from elsewhere, however, including those designated as CRB-G (clade I) (based on the partial mtCOI assessment approach), did not share the same maternal lineage as the Guam CRB-G (clade I) individuals. In other words, the multi-gene assessment (albeit with a limited number of specimens), provided strong supporting evidence that the CRB invasion into Guam was distinct from the CRB invasions detected in from Solomon Islands and in Palau, and therefore Guam was not the source of the CRB that invaded these other locations. Our finding based on full mitogenome and especially the ATP6 and COIII genes vs. the widely used partial COI gene in the CRB studies reported to-date (e.g., Marshall et al. 2017; Reil et al. 2018; Etebari et al. 2020) indicated that the reported mitogenome of CRB-G from Solomon Islands identified from the partial COI gene signature (Filipović et al. 2021) represented a mistaken identity. Increasing sampling of CRB from Guam, Palau, and Solomon Islands is needed to further increase confidence of the specificity of the ATP6 and COIII alternative markers to differentiate CRB-G (clade I) from other CRB populations.
For the PCR-RFLP primers focused on the partial ATP6 gene sequence, separation is based on a BpmI restriction site. Individuals classed as Guam CRB-G (clade I) produced two fragments (i.e., 271 bp and 223 bp), whereas all other CRB remained uncut (i.e., 494 bp) (Table 2). A second primer set was developed based on the COIII gene that can also differentiate between Guam CRB-G (clade I) from other CRB; however, this diagnostic method requires sequence analysis (such as through Sanger sequencing) to detect the presence of a ‘C’ or a ‘T’ base at nucleotide position 5,390 (see Table 1). Although these new markers improve the differentiation between CRB that invaded Guam and other CRB populations, assessment of more CRB individuals from native populations (e.g., Malaysia, Singapore, Sri Lanka, India, Bangladesh, Myanmar, Cambodia, Laos, Vietnam, southern China, Indonesia, Philippines, Taiwan, Thailand), will be needed to provide a more robust confirmation of CRB invasion histories. Also, for all work using molecular diagnostics of CRB, use of either the mtCOI or mtCOIII genes is recommended as an initial approach to first confirm that samples are O. rhinoceros. For example, while the T4430C SNP site within ATP6 from Guam specimens was a T, it was also a ‘T’ in O. narsicornis (see Table 1). Therefore, a direct PCR-RFLP without first confirming species status could lead to misidentification of O. rhinoceros among other Oryctes spp.
The CRB is a hitchhiker pest (Hoffmann et al. in press) and is continuing to disperse to new locations, being recently reported in the Marshall Islands (The Marshall Islands Journal 2023) and multiple Hawaiian islands (HDOA 2024). Notably, our results found that Palau CRB appear to have multiple origins (Figure 1b). The node confidence support estimates in Figure 1b displayed a range of values, with some of the individuals (e.g., from Palau, Indonesia) appearing low (less than 60), which limited the power of inference for better understanding the invasion history of this pest across its distributional ranges. It is likely that future detailed genetic assessments of CRB will provide the resolving power required to further elucidate CRB invasion histories.
Increasingly, WGS and multigene approaches have provided greater analytical power than partial genome assessments, and are therefore rapidly becoming more widely adopted for the interrogation of demographic history and evolutionary relationships of some of the world’s most significant transboundary invasive plant pests (Anderson et al. 2016; Anderson et al. 2018; Zhang et al. 2022; Elfekih et al. 2018; Elfekih et al. 2021; Tay et al. 2022a; Rane et al. 2023), including CRB (Reil et al. 2018; Etebari et al. 2021; Filipović et al. 2021). Given that the WGS/multigene approaches can provide more comprehensive evidence than single gene analyses (e.g., partial mtCOI), and we have found exactly this result with this analysis, we suggest that a detailed study using these more detailed genetic assessments is needed to further improve current understanding of CRB invasion biology.
Supplementary Materials
The following are available online at Preprints.org.
Acknowledgements
This project was funded by the Australian Government Department of Foreign Affairs and Trade (DFAT) Administered (aid) Simple Grant Agreement 77092). Indonesian CRB samples were provided under Republic of Indonesia Ministry of Agriculture Agricultural Quarantine Agency Permit numbers 2021.1.2005.0.K13.E.00003 No. 4299301 and 2022.1.2005.0.K12.E.00002 No. 5896762. CRB samples from the Philippines were gathered under the Gratuitous Permit DENR8-GP No. 2022-02 (2022.01.10) provided by the Department of Environment and Natural Resources 8 of the Republic of the Philippines and conducted under the VSU-IP 2021-10 (BIO-CAMP) and VSU-IP 2022-2 (CRB) projects. All other CRB individuals sourced for this study did not require collection/export permits.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anderson, C.J.; Tay, W.T.; McGaughran, A.; et al. Population structure and gene flow in the global pest, Helicoverpa armigera. Molecular Ecology 2016, 25, 5296–5311. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Oakeshott, J.G.; Tay, W.T.; et al. Hybridization and gene flow in the mega-pest lineage of moth, Helicoverpa. Proceedings of the National Academy of Sciences U.S.A. 2018, 115, 5034–5039. [Google Scholar] [CrossRef] [PubMed]
- Anggraini, E.; Vadamalai, G.; Lih, L.K.; et al. Variants in the mitochondrial genome sequence of Oryctes rhinoceros (Coleoptera: Scarabaeidae) infected with Oryctes rhinoceros nudivirus in oil palm and coconut plantations. Scientific Reports 2023, 13, 16850. [Google Scholar] [CrossRef] [PubMed]
- Arnemann, J.A.; Roxburgh, S.; Walsh, T.; et al. Multiple incursion pathways for Helicoverpa armigera in Brazil show its genetic diversity spreading in a connected world. Scientific Reports 2018, 9, 19380. [Google Scholar] [CrossRef] [PubMed]
- Ayivi, S.P.G.; Tong, Y.; Storey, K.B.; et al. The mitochondrial genomes of 18new Pleurosticti (Coleoptera: Scarabaeidae) exhibit a novel trnQ-NCR-trnI-trnM gene rearrangement and clarify phylogenetic relationships of subfamilies within Scarabaeidae. Insects 2021, 12, 1025. [Google Scholar] [CrossRef] [PubMed]
- Bedford, G.O. Biology, Ecology, and control of palm rhinoceros beetles. Annual Review of Entomology 1980, 25, 209–229. [Google Scholar] [CrossRef]
- Behere, B.T.; Tay, W.T.; Russell, D.A.; et al. Mitochondrial DNA analysis of field populations of Helicoverpa armigera (Lepidoptera: Noctuidae) and of its relationship to H. zea. BMC Evolutionary Biology 2007, 7, 117. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; et al. MITOS: Improved de novo Metazoan Mitochondrial Genome Annotation. Molecular Phylogenetics and Evolution 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Benelli, G.; Lucchi, A.; Anfora, G.; et al. European grapevine moth, Lobesia botrana. Part I: Biology and ecology. Entomologia Generalis 2023, 43, 261–280. [Google Scholar] [CrossRef]
- Caasi, J.A.S.; Guerrero, A.L.; Yoon, K.; et al. A mathematical model of invasion and control of coconut rhinoceros beetle Oryctes rhinoceros (L.) in Guam. Journal of Theoretical Biology 2023, 570, 11525. [Google Scholar] [CrossRef]
- Cheng, C.-T.; Jeng, M.-L.; Tsai, J.-F.; et al. Two mitochondrial genomes of Taiwanese rhinoceros beetles, Oryctes rhinoceros and Eophileurus chinensis (Coleoptera: Scarabaeidae). Mitochondrial DNA B Resources 2021, 6, 2260–2262. [Google Scholar] [CrossRef] [PubMed]
- Cock, M.J.W.; Beseh, P.K.; Buddie, A.G.; et al. Molecular methods to detect Spodoptera frugiperda in Ghana, and implications for monitoring the spread of invasive species in developing countries. Scientific Reports 2017, 7, 4103. [Google Scholar] [CrossRef] [PubMed]
- Crozier, R.H. From population genetics to phylogeny: Uses and limits of mitochondrial DNA. Aust. Syst. Biol. 1990, 3, 111–124. [Google Scholar] [CrossRef]
- Datt, N.; Gosai, R.C.; Ravuiwasa, K.; Timote, V. Key transboundary plant pests of Coconut [Cocos nucifera] in the Pacific Island countries – a biosecurity perspective. Plant Pathology & Quarantine 2020, 10, 152–171. [Google Scholar]
- De Barro, P.J.; Liu, S.-S.; Boykin, L.M.; Dinsdale, A.B. Bemisia tabaci: a statement of species status. Annual Review of Entomology 2011, 56, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Etebari, K.; Hereward, J.; Sailo, A.; et al. Examination of population genetics of the coconut rhinoceros beetle (Oryctes rhinoceros) and the incidence of its biocontrol agent (Oryctes rhinoceros nudivirus) in the South Pacific Islands. Current Research in Insect Science 2021, 1, 100015. [Google Scholar] [CrossRef] [PubMed]
- Elfekih, S.; Tay, W.T.; Gordon, K.; et al. Standardized molecular diagnostic tool for the identification of cryptic species within the Bemisia tabaci complex. Pest Management Science 2018, 74, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Elfekih, S.; Tay, W.T.; Polaszek, A.; et al. On species delimitation, hybridization and population structure of cassava whitefly in Africa. Scientific Reports 2021, 11, 7923. [Google Scholar] [CrossRef] [PubMed]
- Filipović, I.; Hereward, J.; Rašić, G.; et al. The complete mitochondrial genome sequence of Oryctes rhinoceros (Coleoptera: Scarabaeidae) based on long-read on nanopore sequencing. PeerJ 2021, 9, e10552. [Google Scholar] [CrossRef]
- Goergen, G.; Lava-Kumar, P.; Sankung, S.B.; et al. First report of outbreaks of the fall armyworm Spodoptera frugiperda (J E Smith) (Lepidoptera, Noctuidae), a new alien invasive pest in West and Central Africa. PLoS ONE 2016, 11, e0165632. [Google Scholar] [CrossRef]
- HDOA (Hawaii Department of Agriculture) Coconut rhinoceros beetle information. Posted on 15 January 2014. https://hdoa.hawaii.gov/pi/main/crb/ (last accessed on 10 April 2024).
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; et al. Biological identifications through DNA barcodes. Proceedings of the Royal Society of London Series B 2003, 270, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Penton, E.H.; Burns, J.M.; et al. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 14812–14817. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, Q.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: improving the ultrafast bootstrap approximation. Molecular Biology and Evolution 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.D.; Blas, A.; Tay, W.T. Biosecurity interceptions of Coconut Rhinoceros Beetle Oryctes rhinoceros. Management of Biological Invasions. in press.
- Hurst, G.D.D.; Jiggins, F.M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proceedings of the Royal Society Series B 2005, 272, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Richter, D.C.; Rausch, C.; Dezulian, T.; Franz, M.; Rupp, R. Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics 2007, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Indriyanti, D.R.; Utami, Z.T.; Setiati, N.; Soesilowati, E.; Slamet, M. Identification of insect pests that attack the coconut plants in Jepara regency. Journal of Physics: Conference Series 2019, 1321, 032030. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.-I.; et al. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research 2002, 440, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Lannucci, A.; Benazzo, A.; Natali, C.; et al. Population structure, genomic diversity and demographic history of Komodo dragons inferred from whole-genome sequencing. Molecular Ecology 2021, 20, 6309–6324. [Google Scholar] [CrossRef]
- Leducq, J.-B.; Henault, M.; Charron, G.; et al. Mitochondrial recombination and introgression during speciation by hybridization. Molecular Biology and Evolution 2017, 38, 1947–1959. [Google Scholar] [CrossRef]
- Leite, N.A.; Alves-Pereira, A.; Corrêa, A.S.; et al. Demographics and genetic variability of the new world bollworm (Helicoverpa zea) and the Old World bollworm (Helicoverpa armigera) in Brazil. PLoS ONE 2014, 9, e113286. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Fu, K.Y.; Guo, W.C.; Wang, T.Z.; Lu, Y.B. The complete mitochondrial genome of Tuta absoluta (Lepidoptera: Gelechiidae) and genetic variation in two newly invaded populations in China. Journal of Asia-Pacific Entomology 2022, 25, 101988. [Google Scholar] [CrossRef]
- Marshall, S.D.G.; Moore, A.; Vaqalo, M.; et al. A new haplotype of the coconut rhinoceros beetle, Oryctes rhinoceros, has escaped biological control by Oryctes rhinoceros nudivirus and is invading Pacific Islands. Journal of Invertebrate Pathology 2017, 149, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.D.G.; Paudel, S.; Mansfield, S.; et al. Coconut rhinoceros beetle in Solomon Islands: a tale of two invasions. Biological Invasions 2023, 25, 2659–2678. [Google Scholar] [CrossRef]
- Nagoshi, R.N.; Goergen, G.; Tounou, K.A.; et al. Analysis of strain distribution, migratory potential, and invasion history of fall armyworm populations in northern Sub-Saharan Africa. Scientific Reports 2018, 8, 3710. [Google Scholar] [CrossRef]
- Nagoshi, R.N.; Dhanani, I.; Asokan, R.; et al. Genetic characterization of fall armyworm infesting South Africa and India indicate recent introduction from a common source population. PLoS ONE 2019, 14, e0217755. [Google Scholar] [CrossRef]
- Otim, M.H.; Tay, W.T.; Walsh, T.K.; et al. Detection of sister-species in invasive populations of the fall armyworm Spodoptera frugiperda (Lepidoptera: Noctuidae) from Uganda. PLoS ONE 2018, 13, e0194571. [Google Scholar] [CrossRef] [PubMed]
- Paudel, S.; Mansfield, S.; Villamizar, L.F.; et al. Can biological control overcome the threat from newly invasive coconut rhinoceros beetle populations (Coleoptera: Scarabaeidae)? A review. Annals of the Entomological Society of America 2023, 114, 247–256. [Google Scholar] [CrossRef]
- Pearce, S.L.; Clarke, D.F.; East, P.D.; et al. Genomic innovation, transcriptional plasticity and gene loss underlying the evolution and divergence of two highly polyphagous and invasive Helicoverpa pest species. BMC Biology 2017, 15, 63. [Google Scholar] [CrossRef]
- Rane, R.; Walsh, T.K.; Lenancker, P.; et al. Complex multiple introductions drive fall armyworm invasions into Asia and Australia. Scientific Reports 2023, 13, 660. [Google Scholar] [CrossRef] [PubMed]
- Reil, J.B.; Doorenweerd, C.; San Jose, M.; et al. Transpacific coalescent pathways of coconut rhinoceros beetle biotypes: Resistance to biological control catalyses resurgence of an old pest. Molecular Ecology 2018, 27, 4459–4474. [Google Scholar] [CrossRef] [PubMed]
- Rugman-Jones, P.F.; Hoddle, C.D.; Hoddle, M.S.; Stouthamer, R. The Lesser of Two Weevils: Molecular-Genetics of Pest Palm Weevil Populations Confirm Rhynchophorus vulneratus (Panzer 1798) as a Valid Species Distinct from R. ferrugineus (Olivier 1790), and Reveal the Global Extent of Both. PLoS ONE 2013, 8, e78379. [Google Scholar] [CrossRef]
- Saville, B.; Kohli, Y.; Anderson, J.B. mtDNA recombination in a natural population. Proceedings of the National Academy of Sciences of the United States of America 1998, 95, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Tay, W.T. Rapid molecular DNA identification method for the European invasive grapevine moth Lobesia botrana. 2016. Australia: CSIRO. Available online: https://www.wineaustra- lia.com/getmedia/72f4848c-82c7-45b2-ba10-a179add3d6df/ CSE-1301-Final-Report.
- Tay, W.T.; Beckett, S.J.; De Barro, P.J. (2016). Phosphine resistance in Australian Cryptolestes species (Coleoptera: Laemophloeidae): perspectives from mitochondrial DNA cytochrome oxidase I analysis. Pest Management Science 2016, 72, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Tay, W.T.; Walsh, T.K.; Downes, S.; Anderson, C.; et al. Mitochondrial DNA and trade data support multiple origins of Helicoverpa armigera (Lepidoptera, Noctuidae) in Brazil. Scientific Reports 2017, 7, 45302. [Google Scholar] [CrossRef] [PubMed]
- Tay, W.T.; Elfekih, S.; Court, L.N.; et al. The trouble with MEAM2: Implications of pseudogenes on species delimitation in the globally invasive Bemisia tabaci (Hemiptera: Aleyrodidae) cryptic species complex. Genome Biology and Evolution 2017, 9, 2732–2738. [Google Scholar] [CrossRef] [PubMed]
- Tay, W.T.; Rane, R.; Padovan, A.; et al. Global population genomic signature of Spodoptera frugiperda (fall armyworm) supports complex introduction events across the Old World. Communications Biology 2022, 5, 297. [Google Scholar] [CrossRef]
- Tay, W.T.; Court, L.N.; Hoffmann, B.D.; et al. Draft mitogenomes of the invasive ant Lepisiota frauenfeldi (Mayr 1855) (Hymenoptera: Formicidae). Mitochondrial DNA Part B 2022, 7, 1183–1185. [Google Scholar] [CrossRef]
- Tay, W.T.; Meagher, R.L., Jr.; Czepak, C.; Groot, A.T. Spodoptera frugiperda: Ecology, evolution and management options of an invasive species. Annual Review of Entomology 2023, 68, 299–317. [Google Scholar] [CrossRef]
- Tay, W.T.; Popa-Baez, A.; Dulla, G.; et al. Mitochondrial COI, ATP6, and COIII complete sequence database for coconut rhinoceros beetles (Oryctes rhinoceros) from native and introduced ranges. CSIRO Data Access Portal 2024. [Google Scholar] [CrossRef]
- The Marshall Islands Journal Rhino beetle takes root. 19 October 2023. Available online: https://marshallislandsjournal.com/rhino-beetle-takes-root/ (accessed on 17 March 2024).
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–235. [Google Scholar] [CrossRef] [PubMed]
- Valencia-Montoya, W.A.; Elfekih, S.; North, H.L.; Meier, J.I.; et al. Adaptive introgression across semipermeable species boundaries between local Helicoverpa zea and invasive Helicoverpa armigera moths. Molecular Biology and Evolution 2020, 37, 2568–2583. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.K.; Perera, O.; Anderson, A.; et al. Mitochondrial DNA genomes of five major Helicoverpa pest species from the Old and New Worlds (Lepidoptera: Noctuidae). Ecology and Evolution 2019, 9, 2933–2944. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Tay, W.T.; et al. Population genomics provides insights into lineage divergence and local adaptation within the cotton bollworm. Molecular Ecology Resources 2022, 22, 1875–1891. [Google Scholar] [CrossRef]
Figure 1.
Phylogenetic analysis using (a) partial mtCOI gene sequence (676 bp), and (b) concatenated partial APT6 (446 bp) and partial COIII (422 bp) gene sequences. (a) Three clades are evident based on partial mtCOI genes. One clade (red) contains all individuals from Guam and Philippines (green circles), two from Palau (yellow circles), one from Taiwan (aqua blue circle), and one from Solomon Islands (purple circle). Two (major and minor) clades (blue) do not contain any individuals from Guam but include all individuals from Malaysia (i.e., two Malaysian CRB (ON764799, ON764801) in the minor but evolutionary divergent clade). The major blue clade also included all CRB individuals from Indonesia as well as two Palauan CRB individuals (PALAU-01, PALAU-02). (b) The phylogeny from partial ATP6 and COIII concatenated sequences showed different population demographic patterns, with all Guam individuals clustering together (red), whereas Philippines (green), Malaysia (khaki), and Indonesia (pink) largely clustered according to geography. CRB specimens from Palau (yellow circles) appeared to have multiple origins involving at least Philippines and Indonesia, whereas Taiwan (aqua blue circle) and Solomon Islands (purple circle) appeared to have closer affinity with Philippines CRB individuals but with low (<50%) bootstrap node support values. The Oryctes narsicornis sample (OK484312) was included as an outgroup.
Figure 1.
Phylogenetic analysis using (a) partial mtCOI gene sequence (676 bp), and (b) concatenated partial APT6 (446 bp) and partial COIII (422 bp) gene sequences. (a) Three clades are evident based on partial mtCOI genes. One clade (red) contains all individuals from Guam and Philippines (green circles), two from Palau (yellow circles), one from Taiwan (aqua blue circle), and one from Solomon Islands (purple circle). Two (major and minor) clades (blue) do not contain any individuals from Guam but include all individuals from Malaysia (i.e., two Malaysian CRB (ON764799, ON764801) in the minor but evolutionary divergent clade). The major blue clade also included all CRB individuals from Indonesia as well as two Palauan CRB individuals (PALAU-01, PALAU-02). (b) The phylogeny from partial ATP6 and COIII concatenated sequences showed different population demographic patterns, with all Guam individuals clustering together (red), whereas Philippines (green), Malaysia (khaki), and Indonesia (pink) largely clustered according to geography. CRB specimens from Palau (yellow circles) appeared to have multiple origins involving at least Philippines and Indonesia, whereas Taiwan (aqua blue circle) and Solomon Islands (purple circle) appeared to have closer affinity with Philippines CRB individuals but with low (<50%) bootstrap node support values. The Oryctes narsicornis sample (OK484312) was included as an outgroup.
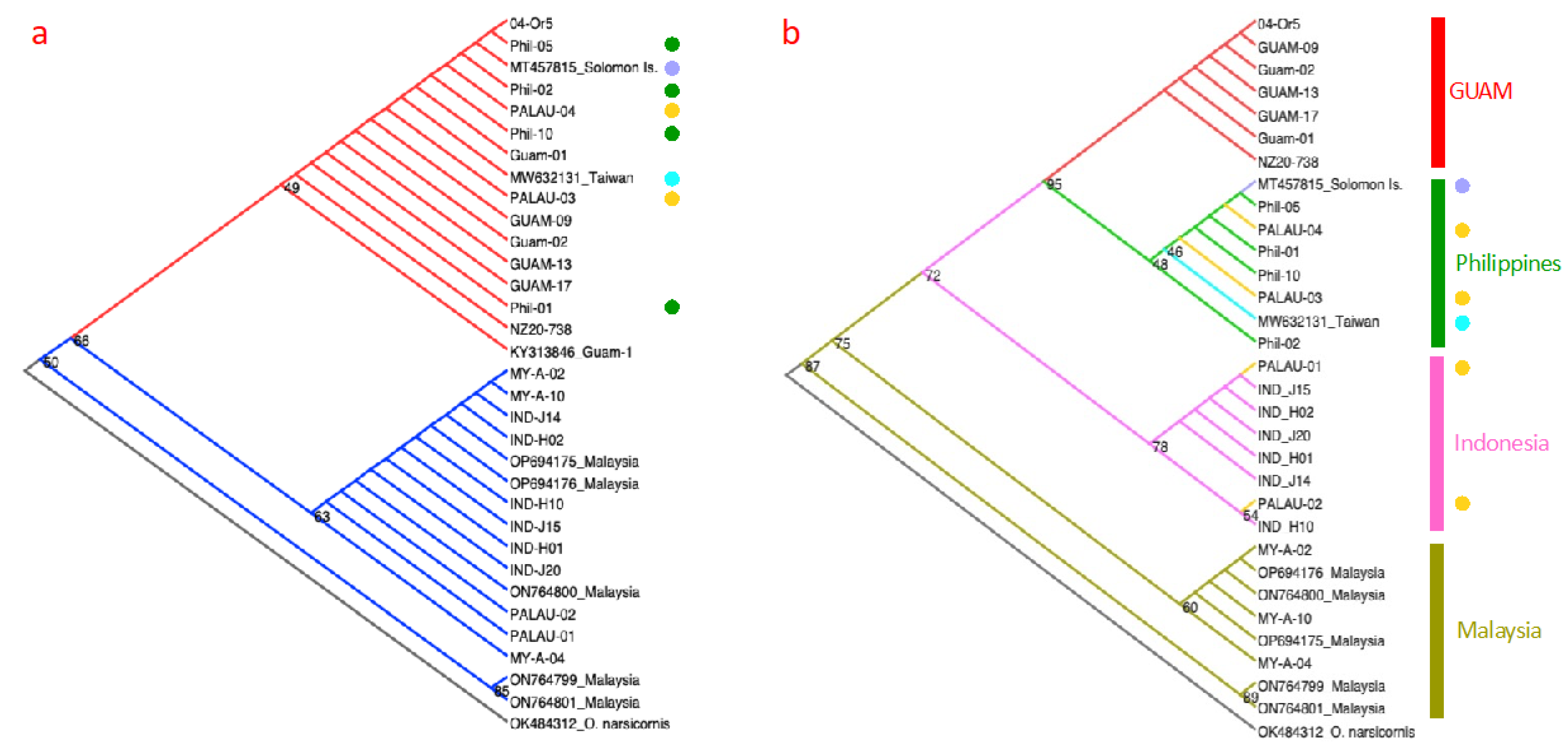

Table 1.
Details of Oryctes rhinoceros (CRB) samples used in this study, including GenBank accession numbers of publicly available assembled and annotated mitogenomes and single nucleotide polymorphisms (SNPs) differentiating CRB-G (clade I) that invaded Guam from other CRB using the mitochondrial cytochrome oxidase subunit I (mtCOI), ATP synthase membrane subunit 6 (ATP6) and cytochrome oxidase subunit III (COIII). A related Oryctes nasicornis mitogenome (GenBank OK484312; Ayivi et al. 2021) was included to provide comparison of inter-specific nucleotide distance with CRB. A historic CRB specimen collected from Guam (sample 04-Or5), classed as CRB-G (clade I) using the partial mtCOI gene was included to generate a Guam-type reference mitogenome for sequence comparison with the other CRB specimens analysed. Nucleotide positions followed annotation of MT457815 (Filipović et al. 2021).
Table 1.
Details of Oryctes rhinoceros (CRB) samples used in this study, including GenBank accession numbers of publicly available assembled and annotated mitogenomes and single nucleotide polymorphisms (SNPs) differentiating CRB-G (clade I) that invaded Guam from other CRB using the mitochondrial cytochrome oxidase subunit I (mtCOI), ATP synthase membrane subunit 6 (ATP6) and cytochrome oxidase subunit III (COIII). A related Oryctes nasicornis mitogenome (GenBank OK484312; Ayivi et al. 2021) was included to provide comparison of inter-specific nucleotide distance with CRB. A historic CRB specimen collected from Guam (sample 04-Or5), classed as CRB-G (clade I) using the partial mtCOI gene was included to generate a Guam-type reference mitogenome for sequence comparison with the other CRB specimens analysed. Nucleotide positions followed annotation of MT457815 (Filipović et al. 2021).
| Sample code | Country | Specimen collection date | Haplotype designation based on partial mtCOI (Marshall et al. 2017) | mtCOI_G1779A | Designation based on partial ATP6 and COIII (this study) | ATP6_T4430C | COIII_C5390T |
|---|---|---|---|---|---|---|---|
| 04-Or5 | Guam | 2014 | CRB-G (clade I) | G | Guam | T | C |
| NZ-20-738 | Guam | 2020 | CRB-G (clade I) | G | Guam | T | C |
| Guam-01_GDoA | Guam | 2022 | CRB-G (clade I) | G | Guam | T | C |
| Guam-02_GDoA | Guam | 2022 | CRB-G (clade I) | G | Guam | T | C |
| Guam-09_GDoA | Guam | 2022 | CRB-G (clade I) | G | Guam | T | C |
| Guam-13_GDoA | Guam | 2022 | CRB-G (clade I) | G | Guam | T | C |
| Guam-17_GDoA | Guam | 2022 | CRB-G (clade I) | G | Guam | T | C |
| MT457815 | Solomon Is. | 2019 | CRB-G (clade I) | G | not Guam | C | T |
| MW632131 | Taiwan | 2002 | CRB-G (clade I) | G | not Guam | C | T |
| MY-A-02 | Malaysia | 2022 | CRB-S (clade IV) | A | not Guam | C | T |
| MY-A-04 | Malaysia | 2022 | CRB-S (clade IV) | A | not Guam | C | T |
| MY-A-10 | Malaysia | 2022 | CRB-S (clade III) | A | not Guam | C | T |
| ON764800 | Malaysia | 2021 | CRB-S (clade III) | A | not Guam | C | T |
| OP694176 | Malaysia | 2021 | CRB-S (clade III) | A | not Guam | C | T |
| OP694175 | Malaysia | 2021 | CRB-S (clade IV) | A | not Guam | C | T |
| ON764799 | Malaysia | 2020 | CRB-S (clade II) | A | not Guam | C | T |
| ON764801 | Malaysia | 2021 | CRB-S (clade II) | A | not Guam | C | T |
| PALAU-01 | Palau | 2022 | CRB-S (clade IV) | A | not Guam | C | T |
| PALAU-02 | Palau | 2022 | CRB-S (clade IV) | A | not Guam | C | T |
| PALAU-03 | Palau | 2022 | CRB-G (clade I) | G | not Guam | C | T |
| PALAU-04 | Palau | 2022 | CRB-G (clade I) | G | not Guam | C | T |
| Phil-01 | Philippines | 2022 | CRB-G (clade I) | G | not Guam | C | T |
| Phil-02 | Philippines | 2022 | CRB-G (clade I) | G | not Guam | C | T |
| Phil-05 | Philippines | 2022 | CRB-G (clade I) | G | not Guam | C | T |
| Phil-10 | Philippines | 2022 | CRB-G (clade I) | G | not Guam | C | T |
| IND-H01 | Indonesia | 2021 | CRB-S (clade III) | A | not Guam | C | T |
| IND-H02 | Indonesia | 2021 | CRB-S (clade IV) | A | not Guam | C | T |
| IND-H10 | Indonesia | 2021 | CRB-S (clade III) | A | not Guam | C | T |
| IND-J14 | Indonesia | 2022 | CRB-S (clade IV) | A | not Guam | C | T |
| IND-J15 | Indonesia | 2022 | CRB-S (clade IV) | A | not Guam | C | T |
| IND-J20 | Indonesia | 2022 | CRB-S (clade IV) | A | not Guam | C | T |
| OK484312 | unspecified | unspecified | Not applicable | T | Not applicable | T | T |
Note: Annotation of the mtCOI, ATP6 and COIII genes in the samples used in this study was based on the published mitochondrial genome (MT457815) from a Solomon Islands individual (Filipović et al. 2021) associated within the CRB-G (clade I) haplotype grouping (based on the mtCOI partial gene characterisation). Additional GenBank accessions included are: MW632131 (Cheng et al. 2021), ON764800, OP694176, OP694175, ON764799, ON764801 (Anggraini et al. 2023), and OK484312 (Ayivi et al. 2021; O. nasicornis).
Table 2.
PCR primer sets for ATP6 (for PCR-RFLP) and COIII were developed to differentiate CRB that invaded Guam from other CRB (including CRB classed as CRB-G (clade I) using the partial mtCOI gene in other locations; sensu Marshall et al. 2017). The restriction enzyme BpmI, which has a restriction digest site of CTCCAG, cuts the partial ATP6 gene associated with the Guam CRB-G (clade I) population once to produce two fragments of 271 and 223 bp due to the presence of a ‘T’ at nucleotide position (nt) 4430, whereas for ‘non-Guam’ individuals this partial gene amplicon of 494 bp remains undigested (see Table 1). The PCR-Sanger sequencing primers for COIII targets a partial 469 bp gene region from nucleotide position (nt) 5017 to 5485. COIII identification of Guam CRB-G (clade I) is through the detection of the ‘C’ base at nt5390, which is a ‘T’ nucleotide in all other CRB (see Table 1). Primer locations followed the nucleotide position of MT457815 (Filipović et al. 2021).
Table 2.
PCR primer sets for ATP6 (for PCR-RFLP) and COIII were developed to differentiate CRB that invaded Guam from other CRB (including CRB classed as CRB-G (clade I) using the partial mtCOI gene in other locations; sensu Marshall et al. 2017). The restriction enzyme BpmI, which has a restriction digest site of CTCCAG, cuts the partial ATP6 gene associated with the Guam CRB-G (clade I) population once to produce two fragments of 271 and 223 bp due to the presence of a ‘T’ at nucleotide position (nt) 4430, whereas for ‘non-Guam’ individuals this partial gene amplicon of 494 bp remains undigested (see Table 1). The PCR-Sanger sequencing primers for COIII targets a partial 469 bp gene region from nucleotide position (nt) 5017 to 5485. COIII identification of Guam CRB-G (clade I) is through the detection of the ‘C’ base at nt5390, which is a ‘T’ nucleotide in all other CRB (see Table 1). Primer locations followed the nucleotide position of MT457815 (Filipović et al. 2021).
| Nucleotide position | Primer name: primer sequence (5’-3’) | Restriction enzyme | CRB-G (clade I) (Marshall et al. 2017) | Other CRB |
| nt4192-4216 | CRB-ATP6-F: ATGAATTCAAACTTTTAATTGGACC | BpmI (CTCCAG) | T | C |
| nt4685-4663 | CRB-ATP6-R: GGAGTAAAGAGTTCTAAGGATAG | 271+223 bp | 494 bp | |
| nt5017-5039 | CRB-COIII-F: CTTAGCTCCTACAATCGAATTAG | Uncut | C | T |
| nt5485-5462 | CRB-COIII-R: TCTACCTCATCAGTAAATGGAAAT | 469 bp | 469 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



