
Submitted:
23 May 2024
Posted:
23 May 2024
You are already at the latest version
Abstract
Grey mullets comprise different species which represent the most ubiquitous teleost families in the planet coastal waters. They represent an important proportion of the Mediterranean lagoons production and have been recently considered a cultivated marine fish. This study aimed to explore the intestinal bacterial communities of 34 wild-caught grey mullets in order to study their composition and the variation during different seasons. Thirty four healthy wild-caught fish were sampled and the V3-V4 hypervariable regions of 16S rRNA were sequenced. Sixty prokaryotic phyla were identified and the most abundant ones were: Proteobacteria (mean relative abundance 35.4%±17.9), Actinobacteriota (mean relative abundance 16.4%±9.9) and Firmicutes (mean relative abundance 10.1%±10.9). Bacteria belonging to the phylum Chloroflexi were rele-vant in autumn, Spirochaetota, Verrucomicrobiota, Fusobacteriota and Cyanobacteria were particularly abundant in winter while Bacteroidota characterized summer fish. A total of three hundred thirty-two prokaryotic families were identified and the twenty-six most abundant ones were: Rhodociclaceae (Proteobacteria), Brevinemataceae (Spirocheaetota) and Fusobacteriaceae (Fusobacteriota) and the Staphylococcaceae (Firmicutes). This study sheds light on the ecology and high variation in the complex gut microbial community structure of Mediterranean grey mullets, the potential biotechnological role for fish nutrition and health and for preserving the aquatic habitat, a prerequisite for a sustainable aquaculture.
Keywords:
microbiome
; ecosystem
; Mugilidae
; 16S barcode
; bioremediation
; fish gut
; aquaculture
1. Introduction
The interest on intestinal microbiome of aquatic animals is getting more and more pivotal because its knowledge can contribute to the development of effective strategies for fish rearing in captivity, by manipulating gastrointestinal (GI) microbial communities in order to promote the health and the productivity through novel therapeutics and feed additives [1,2]. Moreover, a flexibility in the gut microbiome may play a role for the biological diversity conservation, enabling fish to colonize new and different aquatic environments [3]. Fish GI tract is a complex ecosystem composed of a dynamic consortium of microor-ganisms which play critical roles in the nutrition, energy source, host health to reduce or inhibit pathogenic microbes and for the safety of the environment [4]. Fish gut microflora can be divided into two groups: the resident (autochthonous) and the transient (alloch-thonous) communities [5]. The resident can adhere to and colonize the mucosal surfaces, or occur within the epithelial tissues, while the transient communities are char-acterized by non-adherent free-living microorganisms although they inhabit microniches especially during periods of stress [6]. Intestinal microbiota of aquatic animals has higher fluidity than terrestrial animals and changes in relation to various factors such as tem-perature, salinity and trophic level [7-10]. The studies on Mediterranean fish species indi-cated that it is influenced by species, sex, age [11], physiology other than epigenetic factors (season, feeding regimen, water temperature and salinity) [12,13]. However, the existence of a core gut microbiota within and between different species independent of diet and ge-ographic location does exist [14]. Thus, the monitoring of bacteria present in healthy wild fish in their natural environment is the first step for understanding their possible use in captivity [15]. Research on intestinal bacteria of a wide range of fish species have mostly reported on the isolation, identification and evaluation of cultivable bacteria; however, only a small part(<2%) of the GI microbiota may be cultured and these types of procedures fail to provide information of the microbial community as a whole. Metabarcoding se-quencing by various next-generation sequencing (NGS) platforms has emerged as a method to discover new groups of micro-organisms with greater accuracy in environ-mental systems in spatial and temporal scales [5,10,16]. Illumina MiSeq (Illumina, USA) has been widely used for 16S rRNA gene sequencing of gastrointestinal tract microbiota of freshwater [17] and marine fish [16,18]. This technology has let to identify groups of bac-teria which, otherwise, would be unknown, especially those that are not culturable on synthetic media.
Fish belonging to the Mugilidae family, commonly known as grey mullets’ group, comprise a great number of species and they are one of the most ubiquitous teleost fami-lies in the planet coastal waters [19]. They have omnivorous and herbivorous feeding hab-its [20] and have been recently considered a cultivated marine fish which can be fed with “alternative” energy as insects and cost-effective materials, plants, production discards etc., contributing to the realization of the goal of sustainability in aquaculture [21]. In this sense, grey mullets represent an interesting resource for aquacultural use since, the poten-tial for fish large scale production faces numerous challenges represented by ecological carrying capacity of existing sites and environmental impact [22]. Moreover, the valoriza-tion of fish species less considered for the market is a way for preserving the more valued ones and the biodiversity of the aquatic environment [23].
Among Mediterranean Mugilidae, M. cephalus (Linnaeus, 1758), C. ramada (Risso, 1827), C. labrosus (Risso, 1827), C. saliens (Risso, 1810) and C. auratus (Risso, 1810) are some of the most representative fish species in Sardinian lagoons [22,24]. Above all, flathead grey mullet, M. cephalus, represents about 50% of world mullets production ([19], possess-es high economic value and is appreciated in the food market for its eggs processed to ob-tain a seafood which is known with different names such as Avgotaracho (Grece), Karasumi (Japan) or Bottarga (Italy), depending on the geographical production area [25, 26]. In Italy, grey mullets represent an important proportion of the production of coastal lagoons [27] and their culture is carried out in extensive systems, based on natural cycles and dynamics [28]. Production is based on wild fry availability and it cannot compete with intensive cage culture at sea, but it aims to combine environmental compatibility with economic sustainability. This is an advanced form of coastal lagoon management and represents one of the most interesting examples of coastal lagoon management in the world [29]. According to official sources in 2015, the Italian organic mullet species (Mugil cephalus, Chelon aurata, Chelon saliens, and Chelon labrosus) production is estimated at 80 tonnes (source EUMOFA).
Earlier studies reported the composition and the predicted functions of the associated gut microbiota on Mugil cephalus of different ages from Northwest Pacific marine envi-ronments [30], from Chinese coastal marine areas [31] and on Mediterranean wild thick–lipped grey mullets (Chelon labrosus, Risso 1827) [23]. However, the intestinal microbiota of wild grey mullets from Mediterranean aquatic environments still deserve more explora-tions and remains an important topics of scientific interest.
In this work, a metabarcoding study on 34 wild mullets from a Mediterranean transi-tional aquatic environment has been carried out in order to analyze, as a first step, the structure of their intestinal bacterial communities (both resident and transient), during different seasons. These results shed light on the variation in the complex gut microbial community structure of Mediterranean grey mullets and its potential biotechnological role for fish and the aquatic environment.
2. Materials and Methods
2.1. Study Area and Fish Samplings
Thirty four healthy wild-caught mullets, destined for the local food market, were captured by professional fisheries on September 27th 2018 (autumn), February 10th 2019 (winter), July 10th 2019 (summer) and May 18th 2021 (spring) from Santa Giusta Lagoon (Central west coast of Sardinia (Italy) (coordinates: Lat 39°52’N, Long 8°35’E).
This aquatic area (8.6 km2 and 1.0 m mean depth) is a research site of the “Marine Ecosystems of Sardinia” of the Italian Long-Term Ecological Research network (www.lteritalia.it; https://deims.org/6f7581f0-e663-4681-bf9d-4668d6c3f2ba), recognized as a Site of Community Importance for European Union (SCI ITB030037) and is designat-ed by the Sardinian Government as a protected area for animals (INFS code: OR0211) ().
2.2. Fish Measurements and Preparation of Gut Samples
Body and gut weight and total length were measured. The intestine (mean weight 14 ± 6 g) was aseptically removed, diluted (10%, w/v) in saline solution (0.90% NaCl) and homogenized in plastic bags by Stomacher® 400 (FermionX Ltd, Worthing, UK) at room temperature. The whole intestines of fish were collected. The samples for microbiological analyses (homogenates) were made up by mixing the guts of two-three individuals of the same species and immediately stored at -80°C until DNA extraction. The pooling of the samples was performed to minimize the individual variations in the fish [34,35].
2.3. Environmental Characterization
To explore the seasonal dynamics in the lagoon along the study period, a total of 49 water samples were collected. Water samplings were carried out monthly in autumn 2018, winter and summer 2019 and in May 2021. Temperature, salinity and dissolved oxygen (DO) were measured in situ using a YSI 6600 v2 (YSI Inc., Yellow Springs, USA) multi-parameter probe.
Samples for nutrient analyses were collected at about 30 cm depth. Nutrients were analyzed within a few hours after sampling. Concentrations of inorganic nutrients such as reactive phosphorus (PO4), ammonium (NH4), nitrate (NO3), nitrite (NO2), and re-active silica (SiO4) were determined on the filtered samples according to Strickland and Parsons, 1972 [36]. Total dissolved inorganic nitrogen (DIN) was calculated as the sum of NH4, NO3, and NO2. Chlorophyll a (Chla) was determined following the SCOR-UNESCO protocol 1997.
The Dipartimento Specialistico Regionale Idrometeoclimatico (SAR-ARPAS: http://www.sar.sardegna.it/) provided daily data of rainfall (Rain).
Rain data were obtained by summing daily rainfall values to get seasonal accumula-tions.
2.4. DNA Extraction, Amplification and Sequencing
DNA was extracted from ca. 0.750 g of homogenate (n=12) obtained by mixing the guts of two-three individuals of the same species.
All extractions were performed according to the PowerSoil DNA Isolation Kit (Qi-agen®, Hilden, Germany) following the manufacturer’s instructions. DNA quality and quantity were assessed by spectrophotometry (NanoDrop®, Wilmington, DE, USA) and fluorometrically (Qubit® Life technologies, Paisley, UK) to ensure optimal measurement of DNA quantity and purity. The V3-V4 region of the 16S rRNA gene was amplified using universal bacterial primer pair 341F (5’-CCTACGGGNBGCASCAG-3’) and 785R (5’-GACTACNVGGGTATCTAATCC-3’). The PCR mixtures, in a final volume of 25 µl, were as follow: 10 ng of template DNA, 0.5 U of Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific, Waltham, USA), 1X Phusion HF buffer, 0.5 µM of each primer and 200 µM of each dNTP. PCRs (98°C for 4 min; 35 cycles of 98°C for 20 s, 57°C for 30 s, 72°C for 30 s; 72°C for 5 min) were set up in triplicate in order to smooth possible in-tra-sample variance. PCR products were visualized on 1.5% agarose gels, then amplicon triplicates were pooled and purified using 0.8X volumes of AMPure XP beads (Beckman Coulter, Brea, CA, USA).
The pooled PCR products were indexed and subsequently normalized according to the “16S Metagenomic Sequencing Library Preparation” protocol, with minor adjust-ments: 1) 0.5 U of Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific, Waltham, USA) was used for each reaction and 2) PCR amplicons were purified using 0.7X volumes of AMPure XP beads (Beckman Coulter, Brea, CA, USA). Finally, amplicon libraries were equally pooled and sequenced using the Illumina MiSeq system, on the 2 x 300 bp format (Illumina, San Diego, CA, USA). The 16S amplicon sequences generated for this study can be found in the Sequence Reads Archive (SRA) at NCBI under the accession number PRJNA893889.
2.5. Bioinformatic Analyses
Raw reads were initially processed with Cutadapt, v. 2.1 to remove primer sequences and reads shorter than 100 bp [37]. All further analyses were conducted in R, v. 4.1 (R CoreTeam, 2019). At first, using DADA2, v. 1.20 [38], forward reads were trimmed at 270 bases and reverse reads at 170 bases, also truncating reads where bases with quality 2 were found and allowing 0 Ns and maximum expected errors equal to 2. At the end of this process, reads shorter than 20bp were discarded. Resulting reads were then denoised and merged to obtain the amplicon sequence variants (ASVs) in the samples. Taxonomy annotation was performed in DADA2 using the SILVA database v. 138 clustered at 99% identity [39]. Subsequent analyses were performed using the phyloseq R package [40] for data handling and further filtering. All ASVs assigned to Eukarya, mitochondria and chloroplast, or not assigned at phylum level, were removed; only ASVs with total counts above 10 reads across all the samples were retained for further analyses. Rarefaction curves were produced with vegan R package. Alpha diversity indexes were computed with phyloseq dedicated functions; beta diversities principal coordinates analysis plots were computed on Bray-Curtis distances matrixes. In order to describe the different mi-crobial communities, their differences and similarities, relative abundances of ASVs were computed in all samples. A Venn diagram was drawn to reveal the unique and shared families in the different samples. ASVs were grouped at family levels and analyzed with nVennR package.
3. Results
3.1. Fish Biometry
Morphological traits of the wild mullets are represented by the equation showed in Figure 2.
The graphic shows the relationship between total weight and length of the mullets and the determination coefficient (R2) indicates a good correlation (r=0.916) between these bio-metric variables.
3.2. Aquatic Environmental Characterization
Physical and chemical parameters of the aquatic environment along the period of study are reported in Table 1. Seasonal rain accumulates showed the highest mean values (396.2 mm) in autumn 2018 and the lowest ones (40 mm) in summer 2019. Mean temperature data showed seasonal variations with the highest values recorded in summer 2019 (26.83±2.03°C) and the lowest ones in winter 2019 (12.59±2.19°C). Mean salinity data in-dicated an increase along the seasons from autumn 2018 (20.84 psu±7.54) to summer 2019 (35.10 psu±2.99). The lowest DO (5.78 mgL-1±0.67) mean values were registered in summer 2019 while the highest (10.03 mgL-1±1.07) ones in winter. Regarding nutrients, the highest mean values were detected in autumn for all those considered (Table 1). The lowest mean chlorophyll a content (0.55 mg m-3±0.26) was detected in spring while and the highest mean value in autumn (22.42 mg m-3±4.84).
3.3. 16S rDNA Sequencing
3.3.1. Diversity Analysis
16S barcode sequencing yielded a total of 4,995,563 row sequences. After the initial steps of filtering, denoising, merging and chimera removal, a total of 2,618,159 sequences were obtained. From these, 12,045 amplicon sequence variants (ASVs) were identified considering only those that were not assigned to eukaryotic phyla. Considering all samples, ASVs with fewer than 10 sequences were discarded, resulting in a final number of 7817 ASVs. The rarefaction curves showed that the sequencing effort was sufficient to assess the biodiversity of the samples (see Supplementary Material). Table 2 shows the number of total counts relative to the ASVs remaining after the different filtration steps, the number of ASVs identified in the microbiota of each fish intestinal sample in each season, and the microbial α-diversity expressed by Shannon and Simpson indexes. No significant difference was found among the α-diversity indexes of samples.
3.3.2. Composition of Intestinal Microbiota of Grey Mullets
3.3.2.1. Phyla
The 7817 ASVs identified in the dataset were assigned to 60 different phyla (see Supplementary Table S1). Figure 3 shows the relative abundances of the phyla identified in the mullets during the various seasons. Phyla with a median abundance of more than 1% or a variance greater than 85% of the entire dataset were considered in the analyses. In this regard, Acidobacteriota, Actinobacteriota, Bacteriodota, Chloroflexi, Cyanobacteria, Desulfobacterota, Firmicutes, Planctomycetota, Proteobacteria and Verrucomicrobiota had a median abundance of more than 1%, while Spirochaetota and Fusobacteriota showed an overall low abundance but high variability among the samples, as they both accounted for more than 40% of the prokaryotic community in MUG-SG06 sample during winter. All the remaining 48 phyla were included in the “Other” group (Figure 3 and Supplementary Table S1).
The gut microbiota of grey mullets was dominated by Proteobacteria (35.4%±17.9), fol-lowed by Actinobacteriota (16.4%±9.9) and Firmicutes (10.1%±10.9) which together accounted for 61.9% of total population identified, and constituted the “core” microbial group. In this “core” microbiome, the Actinobacteriota resulted significantly higher in fish captured in May (spring) (84.68%) with respect to February (winter) (43.82%), July (summer) (37.21%) and September (autumn) (29.8%) (P<0.01) and the Firmicutes were at the highest number in spring, but they were significantly higher in summer with respect to winter and autumn and also in autumn with respect to winter (P<0.05) (Figure 4). Generally, the highest number of bacterial phyla was observed in summer; however, the few phyla that appeared specific to a given season, always showed low abundances, so they should not have a strong effect on the structure and dynamics of the microbial communities in the fish gut (Supplementary Table S1).
To explore the prokaryotic diversity of single fish intestinal sample, the three phyla with the highest abundance were examined in each of them (Supplementary Table S2). These identified phyla were represented by Actinobacteriota, Bacteriodota, Chloroflexi, Cyanobacteria, Firmicutes, Fusobacteriota, Proteobacteria, Spirochaetota and Verrucomicrobiota. On average, the top three phyla identified in each sample comprised more than 75% of the total abundance and this means that these phyla accounted for most of the microbial community in the gut of each sample. The only exceptions were MUG-SG01, MUG-SG02 and MUG-SG07 in which, however, the most abundant three phyla represented more than 65% of the total abundances.
Considering the gut microbiome during the different periods of study, the ASVs belonging to Chloroflexi phylum were particularly relevant for fish captured in September (autumn) (MUG-SG01 and MUG-SG02), while much lower abundances of this phylum were found in all the other samples captured in the other seasons (see Supplementary Table S1 and Figure 4). Remarkably, the C. labrosus samples caught in February (winter) (MUG-SG05 and MUG-SG06) showed marked differences with respect to each other. In particular, fewer ASVs were identified in the MUG-SG06 sample, with a large abundance of Spirochaetota (45.8%) and Fusobacteriota (40.4%) as already highlighted, which were at very low abundance or even absent in all the other samples (see Supplementary Table S1).
3.3.2.2. Families
Looking at deeper taxonomy levels, a total of 332 families were identified. Not all the identified ASVs were assigned to known families and were therefore removed from the analysis. In this regard, 70.1% of the ASVs, comprising 84% of the reads, remained. However, most of the known families had total abundances in all samples below 1%, meaning that they had low impact on the overall microbial communities. On the other hand, 88 families overpass this threshold (see Supplementary Table S3). In order to have a broad picture of the identified families and their impact on the microbial communities of the mullets' gut, their abundances were further investigated. Figure 5 shows the relative abundances of all the identified families, highlighting those having median abundance in the samples higher than 1% or variance higher than 75% of the entire dataset. All remaining families have been enclosed in the “Other” group, which reaches a large fraction of the overall abundance in some samples. However, some families were found to have a large impact in terms of abundance on the communities, as discussed below.
Sixty microbial families were shared among all fish and represent different amounts of the overall microbial communities ranging from 37.8% to 81.8% with a median value of 54.6% (see Supplementary Table S3). Eleven known families were found to be ubiquitous in all fish and seasons. These families were Staphylococcaceae, Comamonadaceae, Bei-jerinckiaceae, Propionibacteriaceae, Rhodobacteraceae, Rubritaleaceae, Ilumatobacterace-ae, DEV007, Halieaceae, Pirellulaceae, and Desulfocapsaceae (see Supplementary Table S3). These families had average abundances of less than 5% in all gut microbial communities analyzed. Nevertheless, some trends could be highlighted in certain seasons, such as: Staphylococcaceae in May (spring), which reached 35.8% abundance; Rhodobacteraceae and Rubritaleaceae in February (winter), exceeding 20% abundance; Comamonadaceae in July (summer) had an abundance higher than 14%; and Beijerinckiaceae in September (autumn), which had around 10% of the counts (see Supplementary Table S3 and Figure 5). In most other cases abundances were lower than 1.9%. A total of twenty-six most abun-dant bacterial families was found. Looking at the three most abundant families within each intestinal microbial community of the different mullet samples, on average, these three families represented 38.4% of the total abundance, even if they showed very high variability (st. dev: 21.9) (Figure 6). This is due to the fact that in half of the samples the three mostly abundant families do not reach 30% of the total abundance while in some others they largely exceed 50% (see Supplementary Table S4).
The seasonal distribution of intestinal bacterial families shows that Rhodociclaceae (Proteobacteria) (47.7%) was the most abundant group in September (autumn), Brevinemataceae (Spirocheaetota) (45.8%) and Fusobacteriaceae (Fusobacteriota) (40.4%) were dominant in February (winter), and Staphylococcaceae (Firmicutes) (35.8%) prevails in May (spring). On the other hand, the relative abundance of the first three families in the fish caught in July (summer) did not appear particularly high, as only Comamonadaceae and Xanthomonadaceae (both Proteobacteria, respectively 14.92% and 11.39%) exceeded 10% abundance in MUG-SG09 (see Supplementary Table S4). The other less abundant families which characterized each season were: Chromatiaceae (Proteobacteria), Caldilineaceae (Chloroflexi), Beijerinckiaceae (Proteobacteria), Anaerolineaceae (Chloroflexi) and Desul-fobaccaceae (Desulfobacterota) in September (autumn); Rubritaleaceae (Verrucomicrobio-ta), Rhodobacteraceae (Proteobacteria), Nostocaceae (Cyanobacteria), Shewanellaceae (Proteobacteria), Halieaceae (Proteobacteria), Thermoanaerobaculaceae (Acidobacteriota) in February (winter); Xanthomonadaceae (Proteobacteria), Clostridiaceae (Firmicutes), Chitinophagaceae (Bacteroidota), Spirosomaceae (Bacteroidota) identified in July (summer), and Pasteurellaceae (Proteobacteria), Brevinemataceae (Spirochaetota), Micrococcaceae (Actinobacteriota), Mycoplasmataceae (Firmicutes), Vibrionaceae (Proteobacteria), Carnobacteriaceae (Firmicutes), Pirellulaceae (Planctomycetota) observed in May (spring) (see Supplementary Table S3).
4. Discussion
In this study next generation sequencing technologies (NGS) and bioinformatics analysis allowed us to gain a greater knowledge of the microbial communities (both the resident and the transient) associated with the gut of Mediterranean wild grey mullets in response to a variety of environmental aquatic factors. The study revealed the structure of a complex ecosystem highly influenced by the aquatic environment during different seasons. The taxonomic information gathered has been important to define the healthy gut microbiota and the identification of indicator species that could be used in the future to monitor the gut health status of fish at its developmental stage. The fish specimens analyzed represent the typical edible wild fauna from Sardinian coastal lagoons and other Mediterranean transitional aquatic environments [27]. Research attention has been focused on the aquaculture of M. cephalus, which represents a traditionally harvested and consumed fish in various European countries as Italy, Spain, France, especially appreciated in Tunisia, Egypt and Taiwan and a suitable species for feeding populations in developing countries [20,26]. Moreover, the culture of grey mullets Mugilidae species is considered a priority within the current strategies of sustainable European aquaculture [21]. To the authors’ knowledge, various studies were made on the gut microbiota of cultured [41] and wild mullets [23,31] and the present study has provided more information on the biodiversity of this ecosystem and their biotechnological potential. Previous studies on Mediterranean grey mullets had showed some interesting biotechnologically traits of intestinal cultivable bacteria as a source of bioactive compounds with immunological and bioremediation functions [42].
The outlined microbiome confirms that the acquisition and maintenance of the gut microbiota is a complex process, which is dictated by both environmental factors and host physiological pressures. Indeed, each captured grey mullet of this study is characterized by a specific breeding period which determines the migratory behavior that depends on spawning, endocrine mechanisms, photoperiod, temperature and feeding activity [43]. This work have revealed the presence in the mullets of dominant main phyla: Gram negative (Proteobacteria) and Gram positive (Actinobacte-riota and Firmicutes) which accounted for 61.9% of total prokaryotic population identified across all the intestinal samples and constitute the “core” microbial community in the mullets’ gut. This suggests the potential role of these core taxonomic groups for vital functions in the nutrition and/or the immunity of the fish. From an ecological point of view, it was interesting to observe a seasonal influence on the gut microbial composition with a dominance, in spring, of Actinobacteriota and Firmicutes which were also detected in the intestinal microflora of other marine and freshwater fish [14,44-46].
The present study detected the occurrence of the phylum Chloroflexi, which was par-ticularly abundant in the C. ramada individuals caught in autumn (both samples were above 29.5%). Other papers reported the presence of Chloroflexi in the microbial commu-nities of M. cephalus gut [30] and in a wide range of aerobic and anaerobic habitats in-cluding sediments, hot spring, and methanogenic reactors, where these bacteria are sup-posed to have a role in sludge stabilization and breakdown [47]. These authors reported that bacteria of Chloroflexi phylum, commonly isolated from sludge matrices, have a role in bioremediation processes, being able to degrade complex polymeric organic com-pounds to low molecular weight substrates (sludge granulation). Moreover, Liang et al. (2016) [48] described Chloroflexi as component of bacterial communities from petrole-um reservoir and its involvement in toluene degradation. In particular, members of Chloroflexi phylum belonging to the Anaerolineaceae family, found at an abundance of more than 6% in the mullets collected in autumn, are described in literature as methano-genic bacteria (hydrocarbon degrading), frequently encountered in presence of petroleum. The presence of bacteria able of degrading toxic substances as aromatic compounds (sty-rene and fluorobenzoate) on Chelon labrosus was also pointed out by means of PICRUSt functional analysis [23], although their role is still unclear and most of this group of mi-croorganisms remains uncultured and understudied [49].
In this study, the most abundant bacterial family was represented by Rhodocyclaceae (47.7%) (beta-Proteobacteria) as observed by Le and Wang (2020) [30] in the gut of M. cephalus from the Taiwan Strait. Interestingly, the Rhodocyclaceae species were described by different authors for producing bioactive metabolites and, in particular, to be able to transform perchlorate into harmless chloride [50-52]. In this regard, Guarino 2020 [52] re-ported about the genera Azospira and Dechloromonas of Rhodocyclaceae family, able to transform perchlorate into harmless chloride, which are widely distributed in different environments such as soil and groundwater. Nowadays, perchlorate (ClO4-) is an ubiqui-tous ion released into the environment by anthropogenic activity although significant quantities of perchlorate are naturally formed in the atmosphere, especially during thun-derstorms [51,53,54]. The main effect on human beings is its action on the thyroid gland by inhibiting iodide uptake and synthesis of thyroid-stimulating hormone, with serious impairments of growth, metabolism, and reproduction. Another dominant bacterial fam-ily identified in the mullets is the Brevinemataceae (Spirocheaetota). This microbial group was found to be very abundant in winter (45.8%) as also reported by Le (2020) [30] for the gut of Mugil cephalus and by García-Márquez (2022) [23] for C. labrosus individuals. Members of Brevinemataceae family are described by other authors for producing butyrate [55] which may have an intestinal barrier function and support the mucosal immunity [56]. Throughout this study, another family represented by Staphylococcaceae (Firmicutes) was detected significantly in the gut of fish sampled in spring (35.8%). Different papers described several biotechnological activities of Staphylococcaceae [57] and its capacity of degrading hydrocarbon [48]. Generally, the Firmicutes phylum is regarded as beneficial bacteria to the host since it comprises the group of lactic acid bacteria which are highly studied for their probiotic properties [58,59]. In this regard, the Lactobacillales order was identified with an abundance of more than 1% in all fish species caught in spring and summer, seasons with environmental conditions more suitable for this type of bacteria.
5. Conclusions
The findings of the present work provides interesting insights on the diversity and biotechnological potential of the symbiotic intestinal communities hosted by Mediterranean grey mullets and corroborate the concept that their gut is a peculiar and unique eco-logical niche whose microbiome is complex and highly variable, colonized by beneficial bacteria which play a role in the nutrition and the health of these fish and the aquatic en-vironment. The present studies highlight the peculiar role of “cleaner” and “low impact” fish, generally attributed to the grey mullets and commonly used for purifiing aquacultural plants[60]. The presence in the mullets gut of microbial groups with a potential role in bioremediation, detected by this work, adds new important insights on their intestinal ecology for a rational use in aquaculture, in accordance with the EU policy for innovative technology and environmentally and commercially sustainable aquaculture [22]. On the base of these results, future research could be driven to the mullet for preserving their intestinal microbiome which is an added value to protect fish and to preserve the aquatic habitat, a prerequisite of a sustainable aquaculture.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: the abundances for each identified phyla; Table S2: the relative abundances of the three most abundant phyla in each sample; Table S3: the abundances of all known families identified; Table S4: the relative abundances of the top three families identified in each sample; Figure S1: rarefaction curve.
Author Contributions
“Conceptualization, R.F. and A.V.; Methodology, R.F., G.S., C.T.S., G.B and F.D.; Software, F.D.; Validation, F.D. and A.V.; Formal analysis, F.D. and C.T.S.; Investigation, R.F., G.S., C.T.S.; Resources, N.F.; Data Curation, R.F., F.D. and A.V.; Writing—original draft preparation, R.F and A.V.; Writing—Review and Editing, R.F., G.S., F.D. and A.V.; Visualization, all the authors; Supervision, R.F.; Project administration, R.F., A.V. and N.F.; Funding acquisition, N.F. All authors have read and agreed to the published version of the manuscript. All authors have read and approved the final manuscript.
Funding
“This research was funded by Sardinian Region contribution: L.R. n. 6/2012 art-3, comma34. Interventi regionali per l’attuazione della strategia comunitaria in agricoltura.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.
Acknowledgments
The authors would like to thank dr. Riccardo Diciotti and dr. Jacopo Culurgioni for collecting grey mullets samples and sharing their expertise for the identification of fish; the Santa Giusta Cooperative for providing fish.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Hai, N.V. The use of probiotics in aquaculture. J. Appl. Microbiol. 2015, 119, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.K; Rasmussen, B. B.; Castex, M.; Gram, L.; Bentzon-Tilia, M. The aquaculture microbiome at the centre of business creation. Microb. Biotechnol. 2017, 10, 1279–1282. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; DiBattista, J.D.; Stat, M.; Bunce, M.; Boyce, M.C.; Fairclough, D.V.; Travers, M.J; Huggett, M.J. The Microbiome of the gastrointestinal tract of a range-shifting marine herbivorous fish. Front. Microbiol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, A. R.; Chao, R.; Ringø, E.; Zhou Z., G. Progress in fish gastrointestinal microbiota research. Rev. Aquac. 2018, 10, 626–640. [Google Scholar] [CrossRef]
- Legrand, T.P.R.A.; Wynne, J.W.; Weyrich, L.S.; Oxley, A.P.A. A microbial sea of possibilities: current knowledge and prospects for an improved understanding of the fish microbiome. Rev. Aquac. 2020, 12, 1101–1134. [Google Scholar] [CrossRef]
- Banerjee, G.; Ray, A.K. Bacterial symbiosis in the fish gut and its role in health and metabolism. Symbiosis 2017, 72, 1–11. [Google Scholar] [CrossRef]
- Denev, S.; Staykov, Y.; Moutafchieva, R.; Beev, G. . Microbial ecology of the gastrointestinal tract of fish and the poten-tial application of probiotics and prebiotics in finfish aquaculture. Int. Aquat. Res. 2009, 1, 1–29. [Google Scholar]
- Guerreiro, I.; Serra, C. R.; Enes, P.; Couto, A.; Salvador, A.; Costas, B.; Oliva-Teles, A. Effect of short chain fructooligo-saccharides (scFOS) on immunological status and gut microbiota of gilthead sea bream (Sparus aurata) reared at two temperatures. Fish and Shellfish Immunol. 2016, 201649, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Ringø, E.; Zhou, Z.; Vecino, J. L.; Wadsworth, S.; Romero, J.; Krogdahl, Å.; Olsen, R.E.; Dimitroglou, A.; Foey, A.; Da-vies, F.; Owen, M.; Lauzon, H.L.; Martinsen, L.L.; De Schryver, P.; Bossier, P.; Sperstad, S.; Merrifield, D.L. Effect of die-tary components on the gut microbiota of aquatic animals. A never-ending story? Aquacul. Nutr. 2016, 22, 219–282. [Google Scholar] [CrossRef]
- Sullam, K.E.; Essinger, S.D.; Lozupone, C.A.; O'Connor, M.P.; Rosen, G.L.; Knight, R.; Kilham, S.S.; Russell, J. A. Envi-ronmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol. Ecol. 2012, 21, 363–3378. [Google Scholar] [CrossRef]
- Piazzon, M. C.; Naya-Català, F.; Simó-Mirabet, P.; Picard-Sánchez, A.; Roig, F. J.; Calduch-Giner, J. A.; Sitjà-Bobadilla, A.; Pérez-Sánchez, J. Sex, Age, and Bacteria: How the Intestinal Microbiota Is Modulated in a Protandrous Hermaphro-dite Fish. Front. Microbiol. 2019. [CrossRef]
- Floris, R.; Manca, S.; Fois, N. Microbial ecology of intestinal tract of gilthead sea bream (Sparus aurata Linnaeus, 1758) from two coastal lagoons of Sardinia (Italy). Trans. Water Bullet. 2013, 7, 4–12. [Google Scholar] [CrossRef]
- Floris, R.; Sanna, G.; Satta, C.; Piga, C.; Sanna, F.; Lugliè, A.; Fois, N. Intestinal Microbial Ecology and Fillet Metal Chemistry of Wild Grey Mullets Reflect the Variability of the Aquatic Environment in a Western Mediterranean Coastal Lagoon (Santa Giusta, Sardinia, Italy). Water 2021, 13, 879. [Google Scholar] [CrossRef]
- Nikouli, E.; Meziti, A.; Antonopoulou, E.; Mente, E.; Kormas, K.A. Gut Bacterial Communities in Geographically Dis-tant Populations of Farmed Sea Bream (Sparus aurata) and Sea Bass (Dicentrarchus labrax). Microorganisms 2018, 6, 92. [Google Scholar] [CrossRef]
- Tarnecki, A.M.; Patterson, W.F.; Arias, C.R. Microbiota of wild-caught Red Snapper Lutjanus campechanus BMC. Microbi-ology 2016, 16, 245. [Google Scholar] [CrossRef]
- Yukgehnaish, K.; Kumar, P.; Sivachandran, P.; Marimuthu, K.; Arshad, A.; Paray, B. A.; Arockiaraj, J. Gut microbiota metagenomics in aquaculture: factors influencing gut microbiome and its physiological role in fish. Rev. Aquac. 2020, 12, 1903–1927. [Google Scholar] [CrossRef]
- Kashinskaya, E.N.; Belkova, N.L.; Izvekova, G.I.; Simonov, E.P.; Andree, K.B.; Glupov, V.V.; Baturina, O.A; Kabilov, M.R.; Solovyev, M.M. A comparative study on microbiota from the intestine of Prussian carp (Carassius gibelio) and their aquatic environmental compartments, using different molecular. J. Appl. Microbiol. 2015, 119, 948–961, The Society for Applied Microbiology methods. [Google Scholar] [CrossRef]
- Dehler, C. E.; Secombes, C. J.; Martin Samuel, A.M. Environmental and physiological factors shape the gut microbiota of Atlantic salmon parr (Salmo salar L.). Aquaculture 2017, 467, 149–157. [Google Scholar] [CrossRef]
- Crosetti, D. Biology, Ecology and Culture of Grey Mullet (Mugilidae). In first ed.; CRC Press Taylor & Francis Group, Boca Raton, FL, USA, 2016; pp. 42–127.
- Koven, W.; Gisbert, E.; Meiri-Ashkenazi, I.; Nixon, O.; Israeli, D.; Tandler, A.; Soria, H.N.; Solovyev, M; , Rosenfeld, H. The effect of weaning diet type on grey mullet (Mugil cephalus) juvenile performance during the trophic shift from car-nivory to omnivory. Aquaculture 2020, 518, 734848. [Google Scholar] [CrossRef]
- Boyd, C.E.; D’Abramo, L.R.; Glencross, B.D.; Huyben, D.C.; Juarez, L.M.; Lockwood, G.S.; McNevin, A.A.; Tacon, A.G.J.; Teletchea, F.; Tomasso, J.R.; Tucker C., S.; Valenti W., C. Achieving sustainable aquaculture: Historical and current per-spectives and future needs and challenges. J. World Aquacult. Soc. 2020, 51, 578–633. [Google Scholar] [CrossRef]
- Alexander, K.; Potts, T.; Freeman, S.; Israel, D.; Johansen, J.; Kletou, D.; Meland, M.; Pecorino, D.; Rebours, C.; Shorten, M.; Angel, D.L. The implications of aquaculture policy and regulation for the development of integrated multi-trophic aquaculture in Europe. Aquaculture 2015, 443, 16–23. [Google Scholar] [CrossRef]
- García-Márquez, J.; Cerezo, I. M.; Figueroa, L. F.; Abdala-Díaz, R. T.; Arijo, S. First Evaluation of Associated Gut Micro-biota in Wild Thick-Lipped Grey Mullets (Chelon labrosus, Risso 1827). Fishes 2022, 7, 209. [Google Scholar] [CrossRef]
- Cataudella, S.; Crosetti, D.; Massa, F. (eds). Mediterranean coastal lagoons: sustainable management and interactions among aq-uaculture, capture fisheries and the environment. Studies and Reviews; General Fisheries Commission for the Mediterranean. No 95. Rome, FAO, 2015; pp. 278.
- Caredda, M.; Addis, M.; Pes, M.; Fois, N.; Sanna, G.; Piredda, G.; Sanna, G. Physico-chemical, colorimetric, rheological parameters and chemometric discrimination of the origin of Mugil cephalus’ roes during the manufacturing process of Bottarga. Food Res. Int. 2018, 108, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Khemis, I.B.; Hamza, N.; Sadok, S. Nutritional quality of the fresh and processed grey mullets (Mugilidae) products: a short review including data concerning fish from freshwater. Aquat. Living Resour. 2019, 32, 2. [Google Scholar] [CrossRef]
- Vallainc, D.; Concu, D.; Gimenez, G.; Papiol, G.G.; Loi, B.; Leggieri, F.; Brundu, G.; Chindris, A.; Sanna, G.; Fois, N.; An-tognarelli, F.; Rossini, P. Producing flat-head grey mullet Mugil cephalus (Linnaeus, 1758) fries in captivity from sexual-ly mature adults collected in Sardinian lagoons. Aquac. Rep. 2021, 21, 100844–10. [Google Scholar] [CrossRef]
- Pellizzato, M. Classic and modern valliculture. In The State of Italian Marine Fisheries and Aquaculture; Ministero delle Politiche Agricole, Alimentari e Forestali (MiPAAF); Cataudella, S., Spagnolo, M. (Eds.); Roma, Italy, 2011; pp. 237–238.
- Cataudella, S.; Tancioni, L.; Cannas, A. L’acquacoltura estensiva. In Acquacoltura Responsabile verso le Produzioni Acquat-iche del terzo Millennio; Cataudella, S., Bronzi, P. (Eds.); Unimar-Uniprom; Rome, Italy, 2001; pp. 283–306.
- Le, M.H.; Wang, D. Structure and membership of gut microbial communities in multiple fish cryptic species under po-tential migratory effects. Sci. Rep. 2020, 10, 7547. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Yi, H.; Lai, H.; Li, H.; Liu, X.; Chen, Q.; Chen, J.; Zhang, Z.; Wei, X.; Huang, C.; Lin, L.; Xin, G.; Li, G. Intestinal mi-crobiota of the four omnivorous fishes revealed by 16S rRNA metabarcoding from the habitats of oyster reefs. Ecol. In-dic. 2023, 154, 110895. [Google Scholar] [CrossRef]
- Cottiglia, M. Guide per il riconoscimento delle specie animali delle acque lagunari e costiere italiane - Pesci lagunari. Consiglio Nazionale delle Ricerche (Roma), AQ/1/90, vol.1, 1980; pp.
- Trape, S.; Harrison, I. J.; Diouf, P. S.; Durand, J. D. Redescription of Liza bandialensis (Teleostei: Mugilidae) with an iden-tification key to mullet species of Eastern Central Atlantic. C. R. Biol. 2012, 335(2), 120–128. [Google Scholar] [CrossRef]
- Hovda, M. B.; Lunestad, B. T.; Fontanillas, R.; Rosnes, J. T. Molecular characterisation of the intestinal microbiota of farmed Atlantic salmon (Salmo salar L.). Aquaculture 2007, 272, Issues 1–4, Pages 581-588, ISSN 0044-8486. [Google Scholar] [CrossRef]
- Skrodenyte-Arbaciauskiene, V.; Sruoga, A.; Butkauskas, D.; Skrupskeli, K. Phylogenetic analysis of intestinal bacteria of freshwater salmon Salmo salar and sea trout Salmo trutta trutta and diet. Fish Sci. 2008, 74, 1307–1314. [Google Scholar] [CrossRef]
- Strickland, J.D.H.; Parsons, T.R. A Practical Handbook of Seawater Analysis, 2nd ed. Fisheries Research Board of Canada, Ottawa, ON, Canada, 1972; p. 167.
- Martin, M. Cutadapt removes adapter sequences form high-throughput sequencing reads. EMBnet.j. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Ofek, T.; Lalzar, M.; Laviad-Shitrit, S.; Izhaki, I.; Halpern, M. Comparative Study of Intestinal Microbiota Composition of Six Edible Fish Species. Front. Microbiol. 2021, 12, 760266. [Google Scholar] [CrossRef] [PubMed]
- Floris, R.; Sanna, G.; Mura, L.; Fiori, M.; Culurgioni, J.; Diciotti, R.; Rizzo, C.; Lo Giudice, A.; Laganà, P.; Fois, N. Isola-tion and Identification of Bacteria with Surface and Antibacterial Activity from the Gut of Mediterranean Grey Mullets. Microorganisms 2021, 9, 2555. [Google Scholar] [CrossRef] [PubMed]
- Katselis, G.; Koukou, K.; Dimitriou, E.; Koutsikopoulos, C. Short-term seaward fish migration in the Messo-longhie-Etolikolagoons (Western Greek coast) in relation to climatic variables and the lunar cycle. Estuar. Coast. Shelf Sci. 2007, 73, 571–582. [Google Scholar] [CrossRef]
- Ghanbari, M.; Wolfgang, K.; Konrad, J.; Domig, A. New view of the fish gut microbiome: Advances from next-generation sequencing. Aquaculture 2015, 448, 464–475, ISSN 0044-8486,. [Google Scholar] [CrossRef]
- Egerton, S.; Culloty, S.; Whooley, J.; Stanton, C.; Ross, RP. The Gut Microbiota of Marine Fish. Front. Microbiol. 2018, 9, 873. [Google Scholar] [CrossRef]
- Gadoin, E.; Durand, L.; Guillou, A.; Crochemore, S.; Bouvier, T.; Roque, E.; Dagorn, L.; Auguet, J.C.; Adingra, A.; Des-nues, C.; Bettarel, Y. Does the Composition of the Gut Bacteriome Change during the Growth of Tuna? Microorganisms 2021, 27, 9, 1157. [Google Scholar] [CrossRef]
- Speirs, L. B. M.; Rice, D. T. F.; Petrovski, S.; Seviour, R. J. The Phylogeny, Biodiversity, and Ecology of the Chloroflexi in Activated Sludge. Front. Microbiol. 2019. [CrossRef]
- Liang, B.; Li-Ying, W.; Zhou, Z.; Mbadinga, S. M.; Zhou, L.; Liu, J. F.; Yang, S.Z.; Gu, J. D.; Mu, B. Z. High Frequency of Thermodesulfovibrio spp. and Anaerolineaceae in Association with Methanoculleus spp. in a Long-Term Incubation of n-Alkanes-Degrading Methanogenic Enrichment Culture. Front. Microbiol. [CrossRef]
- Bovio, P.; Cabezas, A.; Etchebehere, C. Preliminary analysis of Chloroflexi populations in full-scale UASB methano-genic reactors. J. Appl. Microbiol. 2019. [CrossRef] [PubMed]
- Achenbach, L.A.; Michaelidou, U.; Bruce, R.A.; Fryman, J.; Coates, J.D. Dechloromonas agitata gen. nov., sp. nov. and Dechlorosoma suillum gen. nov., sp. nov., two novel environmentally dominant (per)chlorate-reducing bacteria and their phylogenetic position. Int. J. Syst. Evol. Microbiol. 2001, 51, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.; Salamone, A.; Nerenberg, R. Kinetics of a chlorate-accumulating, perchlorate-reducing bacterium. Water Res. 2008, 42, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Guarino, F.; Motta, O.; Turano, M.; Proto, A.; Vigliotta, G. . Preferential Use of the Perchlorate over the Nitrate in the Respiratory Processes Mediated by the Bacterium Azospira sp. OGA 24. Water 2020, 12, 2220. [Google Scholar] [CrossRef]
- Dasgupta, P.K.; Martinelango, P.K.; Jackson, W.A.; Anderson, T.A.; Tian, K.; Tock, R.W.; Rajagopalan, S. The origin of naturally occurring perchlorate: the role of atmospheric processes. Environ. Sci. Technol. 2005, 39, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Kumarathilaka, P.; Oze, C.; Indraratne, S.P.; Vinthanage, M. Perchlorate as an emerging contaminant in soil, water and food. Chemosphere 2016, 150, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Lokesh, J.; Abdelhafiz, Y.; Siriyappagouder, P.; Pierre, R.; Sørensen, M.; Fernandes, J.M.O.; Kiron, V. Macroalga-Derived Alginate Oligosaccharide Alters Intestinal Bacteria of Atlantic Salmon. Front. Microbiol. 2019, 10, 2037. [Google Scholar] [CrossRef]
- Liu, H.; Wang, J.; He, T.; Becker, S.; Zhang, G.; Li, D.; Ma, X. Butyrate: A Double-Edged Sword for Health? Adv. Nutr. 2018, 9, 21–29. [Google Scholar] [CrossRef]
- Li, X.; Yan, Q.; Ringø, E.; Wu, X.; He, Y.; Yang, D. The influence of weight and gender on intestinal bacterial community of wild largemouth bronze gudgeon (Coreius guichenoti, 1874). BMC Microbiol. 2016, 16, 191. [Google Scholar] [CrossRef]
- Kumar, R.S.; Kanmani, P.; Yuvaraj, N.; Paari, K.; Pattukumar, V.; Arul, V. Purification and characterization of enterocin MC13 produced by a potential aquaculture probiont Enterococcus faecium MC13 isolated from the gut of Mugil cephalus. Can. J. Microbiol. 2011, 57, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Carnevali, O.; Maradonna, F.; Gioacchini, G. Integrated control of fish metabolism, wellbeing and reproduction: The role of probiotic. Aquaculture 2017, 472, 144–155. [Google Scholar] [CrossRef]
- Bronzi, P.; Rampacci, M.; Ugolini, R. L’acquacoltura intensiva. In Cataudella, S., Bronzi P. (Eds.), Aquacoltura responsabile verso le Produzioni Acquatiche del terzo Millennio. Unimar-Uniprom, Rome, Italy, 2001; pp. 348–349.
Figure 1.
Study area.
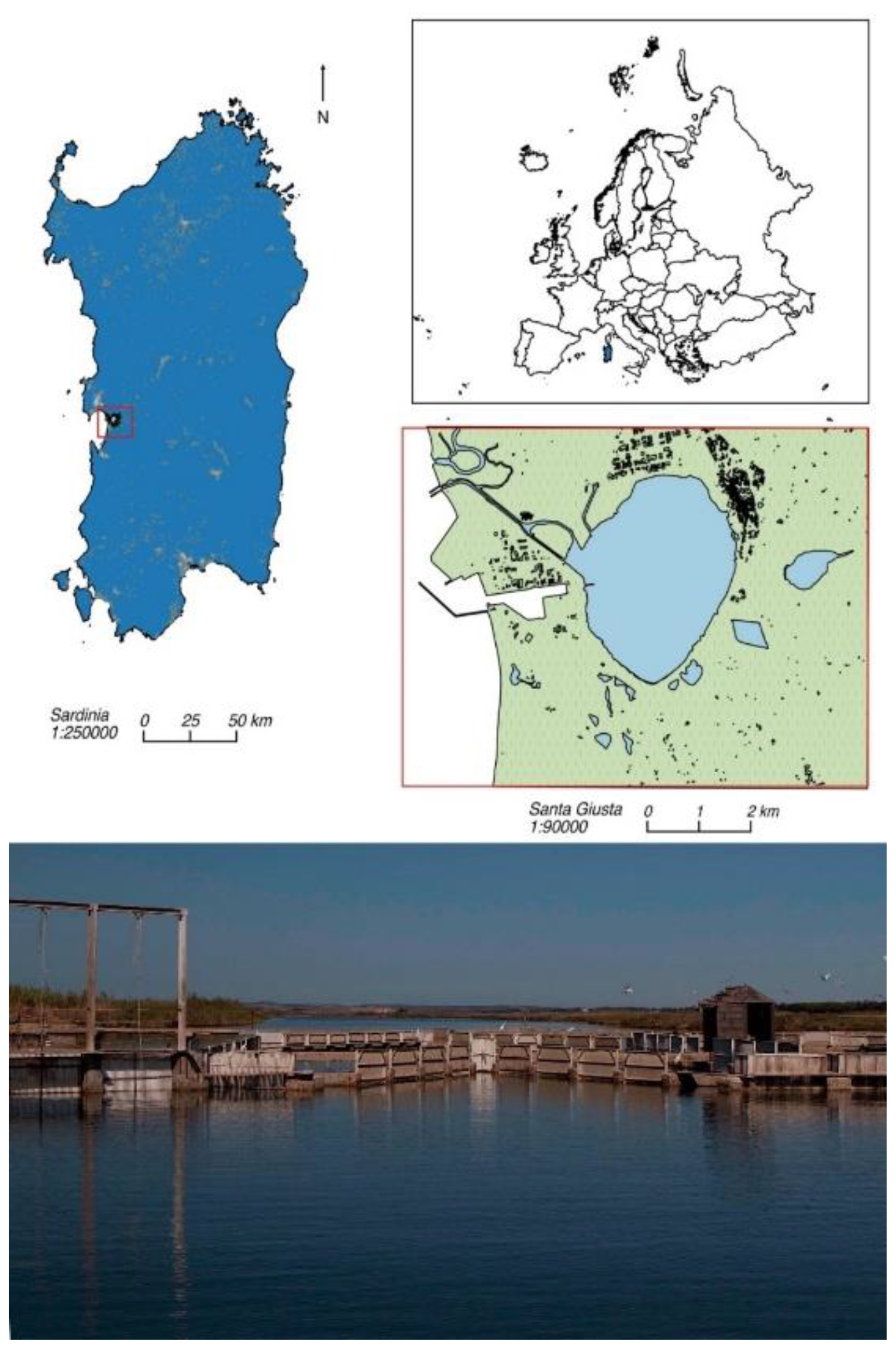
Figure 2.
Fish length (cm) and weight (g) of the mullets analyzed in the study.
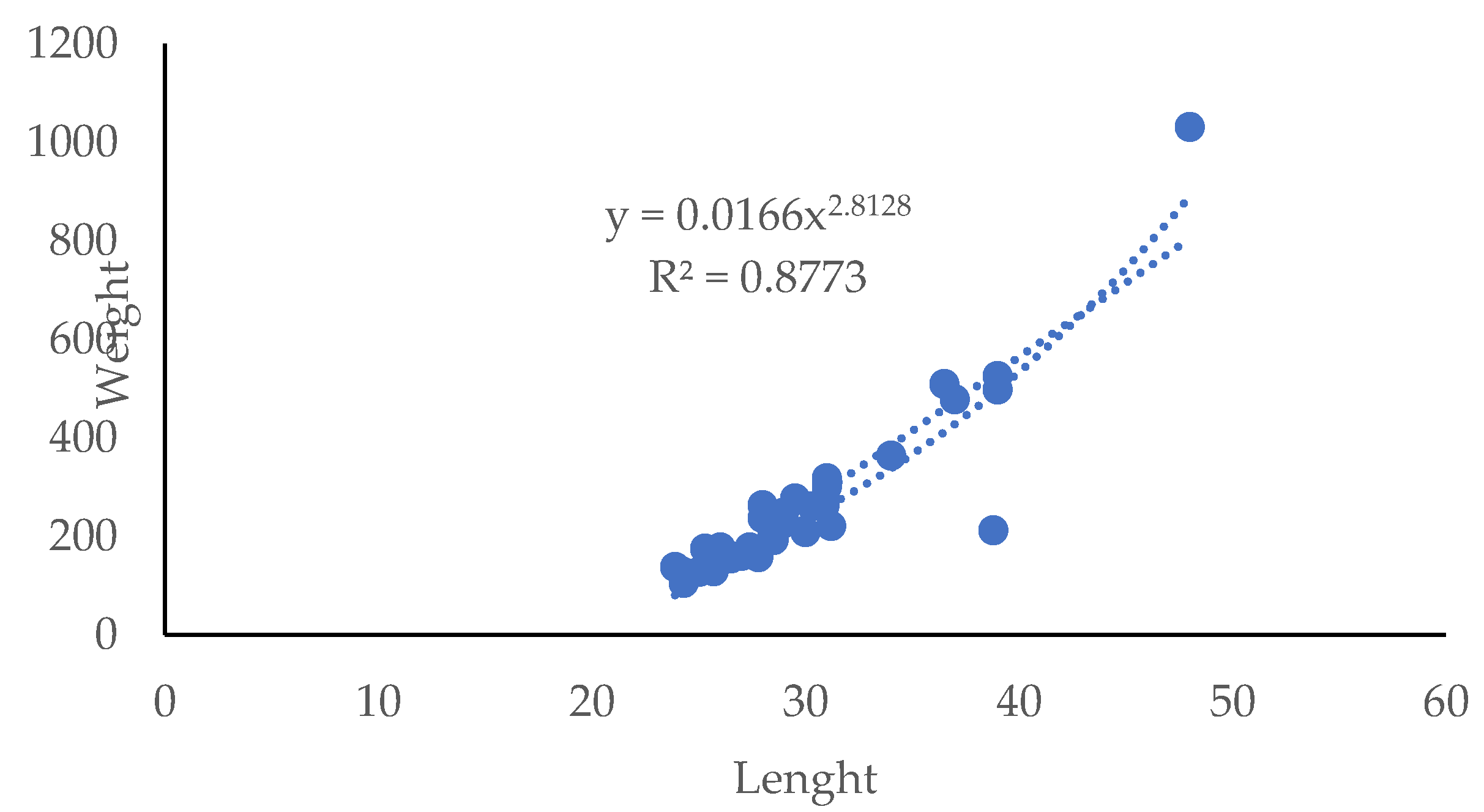
Figure 3.
Abundances of the identified phyla. Phyla with median abundance lower than 1% across the samples are grouped in the “Other” category showed at the bottom of each stacked bar. Abundances in y axis are in percentage.
Figure 3.
Abundances of the identified phyla. Phyla with median abundance lower than 1% across the samples are grouped in the “Other” category showed at the bottom of each stacked bar. Abundances in y axis are in percentage.

Figure 4.
Relative abundances of the most abundant phyla identified in each intestinal microbial community of the mullets during the various seasons. The relative abundances of these nine top phyla are shown across all samples except when zero. Abundances in x axis are in logarithmic scale.
Figure 4.
Relative abundances of the most abundant phyla identified in each intestinal microbial community of the mullets during the various seasons. The relative abundances of these nine top phyla are shown across all samples except when zero. Abundances in x axis are in logarithmic scale.
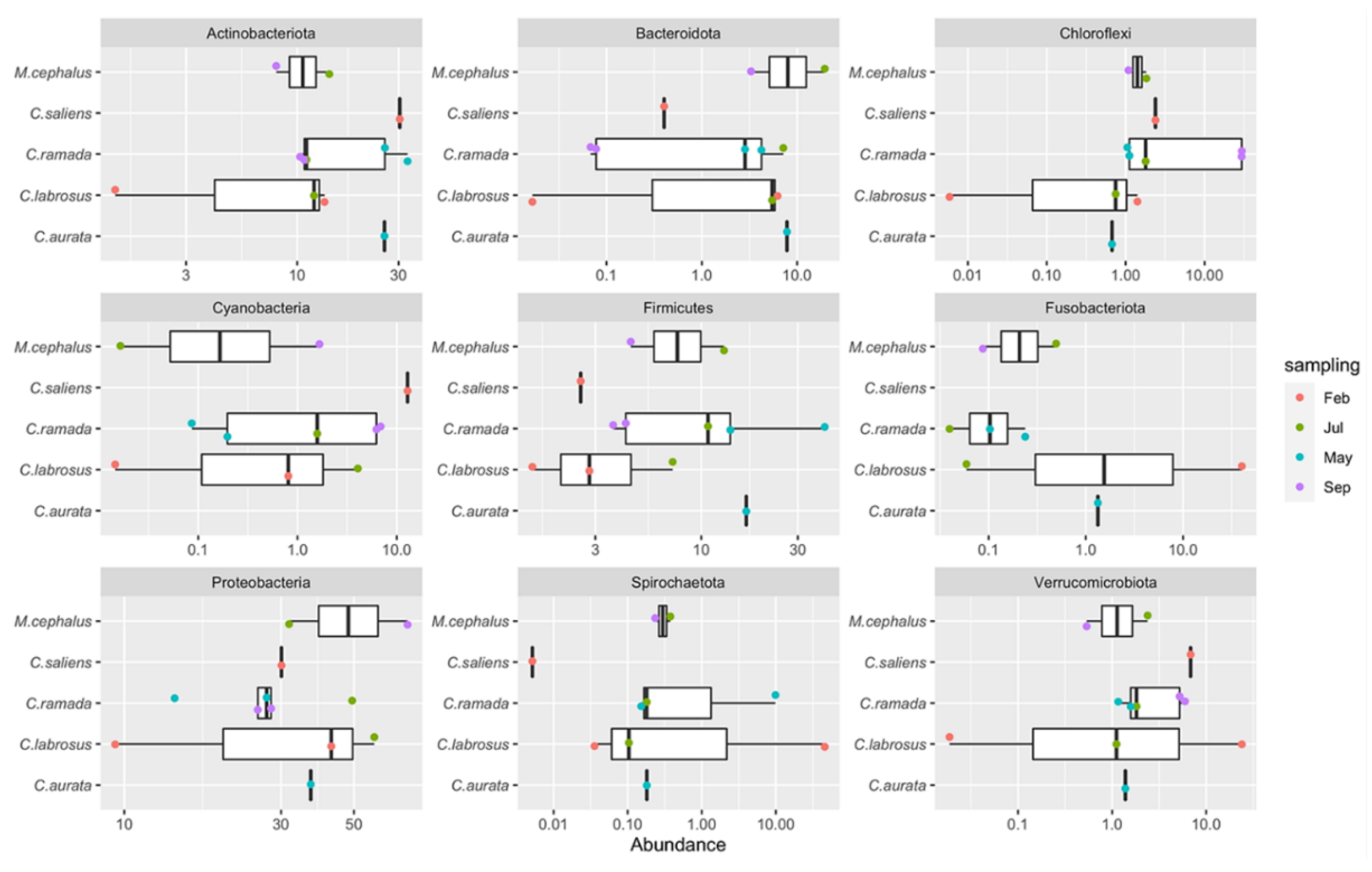
Figure 5.
Known families identified in the samples. Families with median abundance below 1% across all samples or showing a variance lower than 75% of the entire dataset are grouped in “Other”.
Figure 5.
Known families identified in the samples. Families with median abundance below 1% across all samples or showing a variance lower than 75% of the entire dataset are grouped in “Other”.
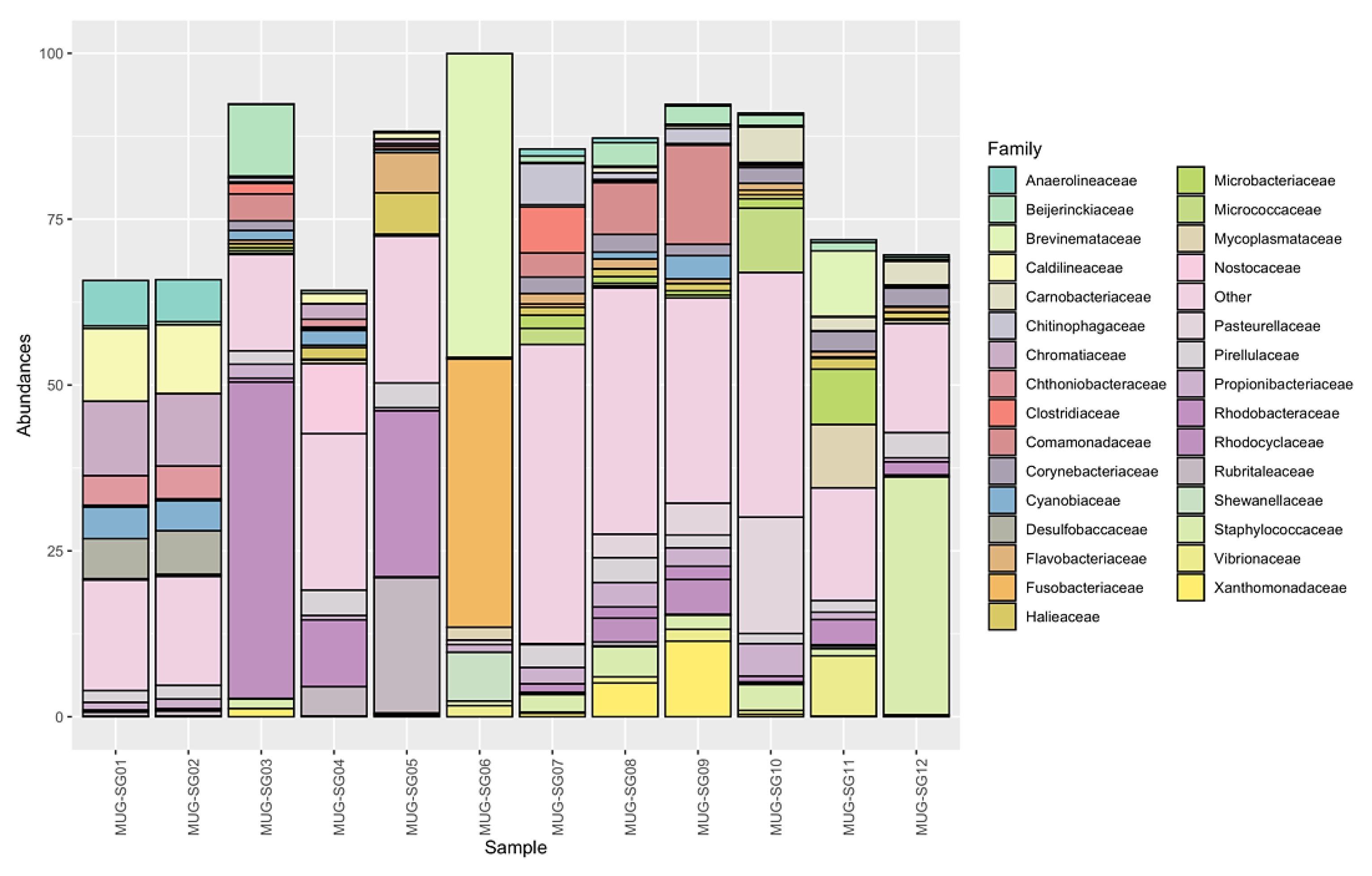
Figure 6.
The most abundant families and their relative abundances across the different gray mullets. Zero values are not shown. Abundances in x axis are in logarithmic scale.
Figure 6.
The most abundant families and their relative abundances across the different gray mullets. Zero values are not shown. Abundances in x axis are in logarithmic scale.
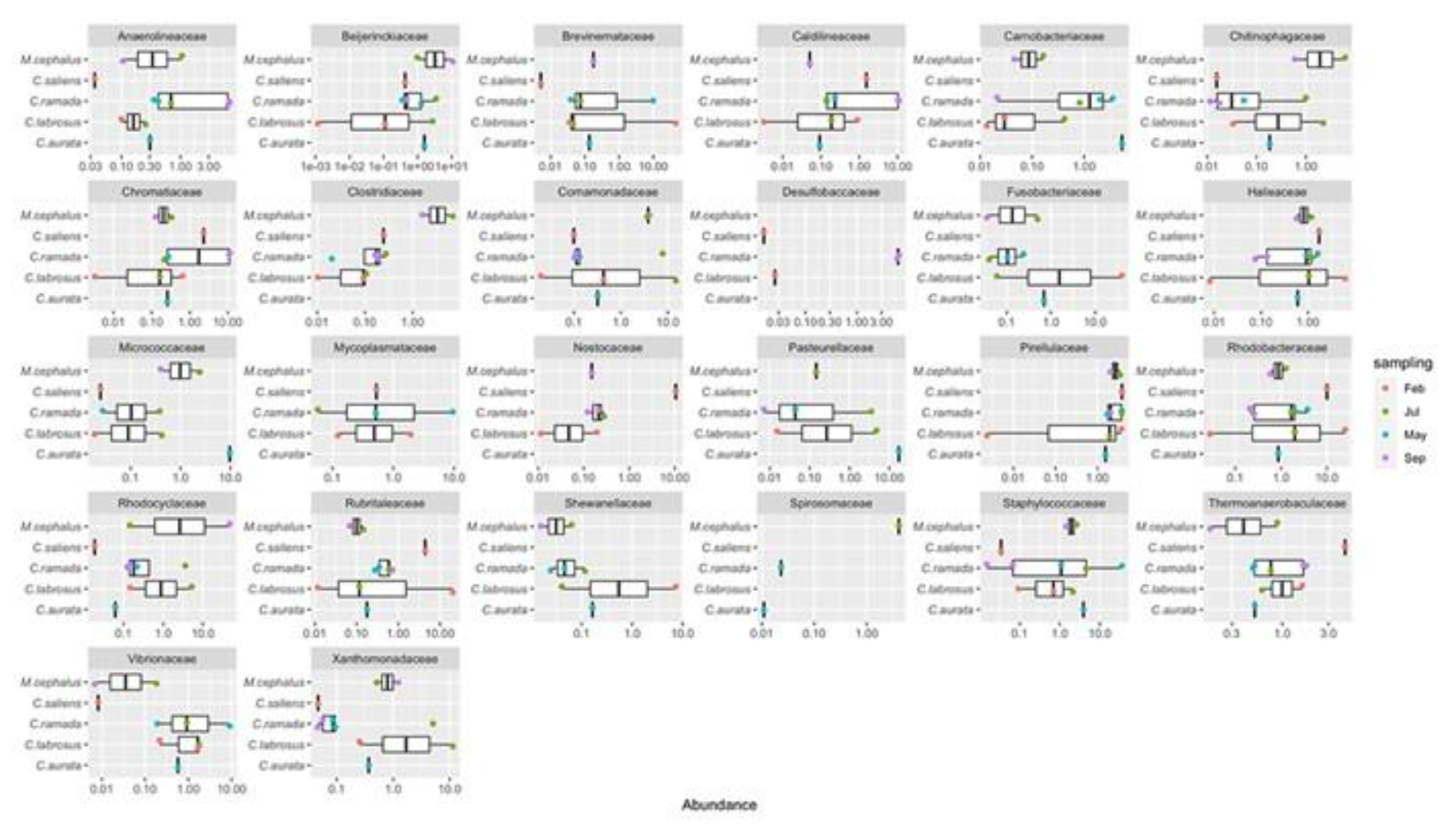

Table 1.
Mean values±sd of the environmental parameters in Santa Giusta lagoon during sampling seasons (DO: dissolved oxygen; DIN: dissolved inorganic nitrogen; Chla: chlorophyll a).
Table 1.
Mean values±sd of the environmental parameters in Santa Giusta lagoon during sampling seasons (DO: dissolved oxygen; DIN: dissolved inorganic nitrogen; Chla: chlorophyll a).
| Autumn | Winter | Summer | Spring | |
|---|---|---|---|---|
| Total rains (mm) | 396.2a | 106.2a | 40.0b | 69.4b |
| Temperature (°C) | 14.53±4.22a | 12.59±2.19a | 26.83±2.03b | 19.65±1.38c |
| Salinity (psu) | 20.84±7.54a | 22.83±3.85a | 35.10±2.99b | 27.33±2.38ab |
| DO (mgL-1) | 9.57±1.85a | 10.03±1.07a | 5.78±0.67b | 7.01±0.73c |
| DIN (µM) | 4.91±0.75a | 2.60±1.32b | 3.48±1.71ab | 3.42±1.57ab |
| PO4 (µM) | 1.96±1.02a | 1.20±0.49b | 0.45±0.26c | 0.77±0.13bc |
| SiO4 (µM) | 296.73±38.53a | 63.61±71.38b | 96.01±52.21b | 32.01±8.06b |
| Chla (mg m-3) | 22.42±4.84a | 3.70±2.82b | 3.43±2.11b | 0.55±0.26c |
Table 2.
Number of amplicon sequence variants (ASVs) and microbial α-diversity indexes of mullets’ intestinal microbiome. .
Table 2.
Number of amplicon sequence variants (ASVs) and microbial α-diversity indexes of mullets’ intestinal microbiome. .
| Sample code | Season | Host species | Total counts | Observed ASVs | Shannon | Simpson |
|---|---|---|---|---|---|---|
| (N. fish) | ||||||
| MUG-SG01 | Autumn | C. ramada (3) | 26,486 | 457 | 5.02 | 0.98 |
| MUG-SG02 | Autumn | C. ramada (3) | 28,282 | 487 | 5.13 | 0.98 |
| MUG-SG03 | Autumn | M. cephalus (3) | 292,010 | 705 | 3.36 | 0.76 |
| MUG-SG04 | Winter | C. saliens (2) | 96,743 | 1,211 | 5.21 | 0.96 |
| MUG-SG05 | Winter | C. labrosus (3) | 78,504 | 984 | 5.45 | 0.98 |
| MUG-SG06 | Winter | C. labrosus (3) | 342,004 | 150 | 1.95 | 0.76 |
| MUG-SG07 | Summer | M. cephalus (3) | 345,355 | 1,081 | 5.99 | 0.99 |
| MUG-SG08 | Summer | C. ramada (3) | 291,064 | 1,343 | 5.93 | 0.99 |
| MUG-SG09 | Summer | C. labrosus (2) | 127,093 | 747 | 5 | 0.96 |
| MUG-SG10 | Spring | C. auratus (3) | 110,687 | 514 | 4.75 | 0.96 |
| MUG-SG11 | Spring | C. ramada (3) | 311,797 | 717 | 4.68 | 0.95 |
| MUG-SG12 | Spring | C. ramada (3) | 242,564 | 1,810 | 4.45 | 0.9 |
| Mean ±sd | 191,049 ± 124,210 | 850 ± 458 | 4.74 ± 1.12 | ± 0.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



