
Submitted:
22 May 2024
Posted:
24 May 2024
You are already at the latest version
Abstract
Introduction: A plethora of biological molecules regulate chondrogenesis in the epiphyseal growth plate. Disruptions of quantity and function of these molecules can manifest clinically as stature abnormalities of various etiologies. Traditionally, the growth hormone/insulin-like growth factor 1 (IGF1) axis represents the etiological center of final stature attainment. Of note, little is known about the molecular events that dominate the growth of the craniofacial complex and its correlation with somatic stature.
Aim: Given the paucity of the relevant data, this review discusses available information regarding potential applications of lateral cephalometric radiography as a potential clinical indicator of genetic short stature in children.
Materials and Methods: A literature search was conducted in the PubMed electronic database using the key words: cephalometric analysis and short stature; cephalometric analysis and achondroplasia; cephalometric analysis and hypochondroplasia; cephalometric analysis and skeletal abnormalities; cephalometr* and SHOX; cephalometr* and CNP; cephalometr* and ACAN; cephalometr* and CNVs; cephalometr* and IHH; cephalometr* and FGFR3; cephalometr* and Noonan syndrome; cephalometr* and “Turner syndrome”; cephalometr* and achondroplasia.
Results: In individuals with genetic syndromes causing short stature, linear growth of the craniofacial complex is confined, following the pattern of somatic short stature regardless of its etiology. The angular cephalometric measurements differ from the average height normal individuals and are suggestive of a posterior placement of the jaws and a vertical growth pattern of the face.
Conclusion: The greater part of the existing literature regarding cephalometric measurements in short statured children with genetic syndromes provides qualitative data. Furthermore, cephalometric data for individuals affected with specific rare genetic conditions causing short stature, should be the focus of future studies. These quantitative data are required to potentially establish cut-off values to refer for genetic testing based on craniofacial phenotypes.
Keywords:
short stature
; genetics
; genetic syndromes
; craniofacial morphology
; cephalometric radiograph
; growth hormone/insulin-like growth factor 1 axis
; epiphyseal growth plate
; cartilage extracellular matrix
1. Introduction
Growth failure is a clinical entity comprised of two constituents, namely short stature (quantitative linear body growth) and/or growth faltering (linear body growth rate)[1]. Short stature in childhood and adolescence represents the most common reason for referral to pediatric endocrinology out-patient clinics[2,3,4]. Short stature is commonly defined to be height that is less than two standard deviations below the average for that specific age, gender, and population, and, on standard growth charts, a drop below the third centile. The American Academy of Pediatrics recommends plotting the data on the World Health Organization charts for children aged less than two years and the Centers for Disease Control and Prevention’s 2000 charts for children aged two years or older[5,6]. For the Greek population the most widely utilized charts are the Chiotis et al. 2003 growth charts[7].
In addition to the above definition, a child may be within the normal height range according to the height charts, and still be considered as having idiopathic short stature if she or he is much shorter than the parents. Finally, restricted growth (dwarfism) is a special form of short stature common among many syndromes and is defined as adult stature less than 147 cm, regardless of gender.
A short statured child can fall into one of three categories, which are useful for clinical purposes, namely primary growth failure, secondary growth failure and idiopathic short stature[8]. Primary growth failure is of genetic origin, as one or more genetic defects cause(s) disruption of the events taking place at the epiphyseal growth plate, directly impeding its function. Secondary growth failure is associated with etiologies extrinsic to the growth plate, including endocrine, paracrine, or nutritional factors, physical mechanisms, extracellular fluid parameters, proinflammatory cytokines and extracellular matrix molecules. Diagnosis of idiopathic short stature (ISS) is set by exclusion of the former two categories, therefore short stature with no recognizable genetic, endocrine, paracrine, nutritional, or other disorder.
Children with ISS have normal birth size and normal body proportions. ISS can either be asymptomatic or symptomatic, when there is one or more symptoms present, as revealed by clinical examination or medical history[6]. Additionally, ISS is further divided into familial and non-familial (sporadic), and each category can be characterized by normal or delayed puberty. In familial ISS, both the child and one of the parents are below 2 standard deviations (SD) and the child’s curve runs close and parallel to the parents’ curve. In sporadic ISS both parents are of normal stature, but the child is below 2SD or under the target height. Treatment of children with ISS with recombinant human growth hormone (rhGH), albeit controversial, improves linear growth and adult height; a height difference of 5.3 cm in males and 4.7 cm in females was noted between a study group and a control group[2].
2. The multifactorial Etiology of Short Stature
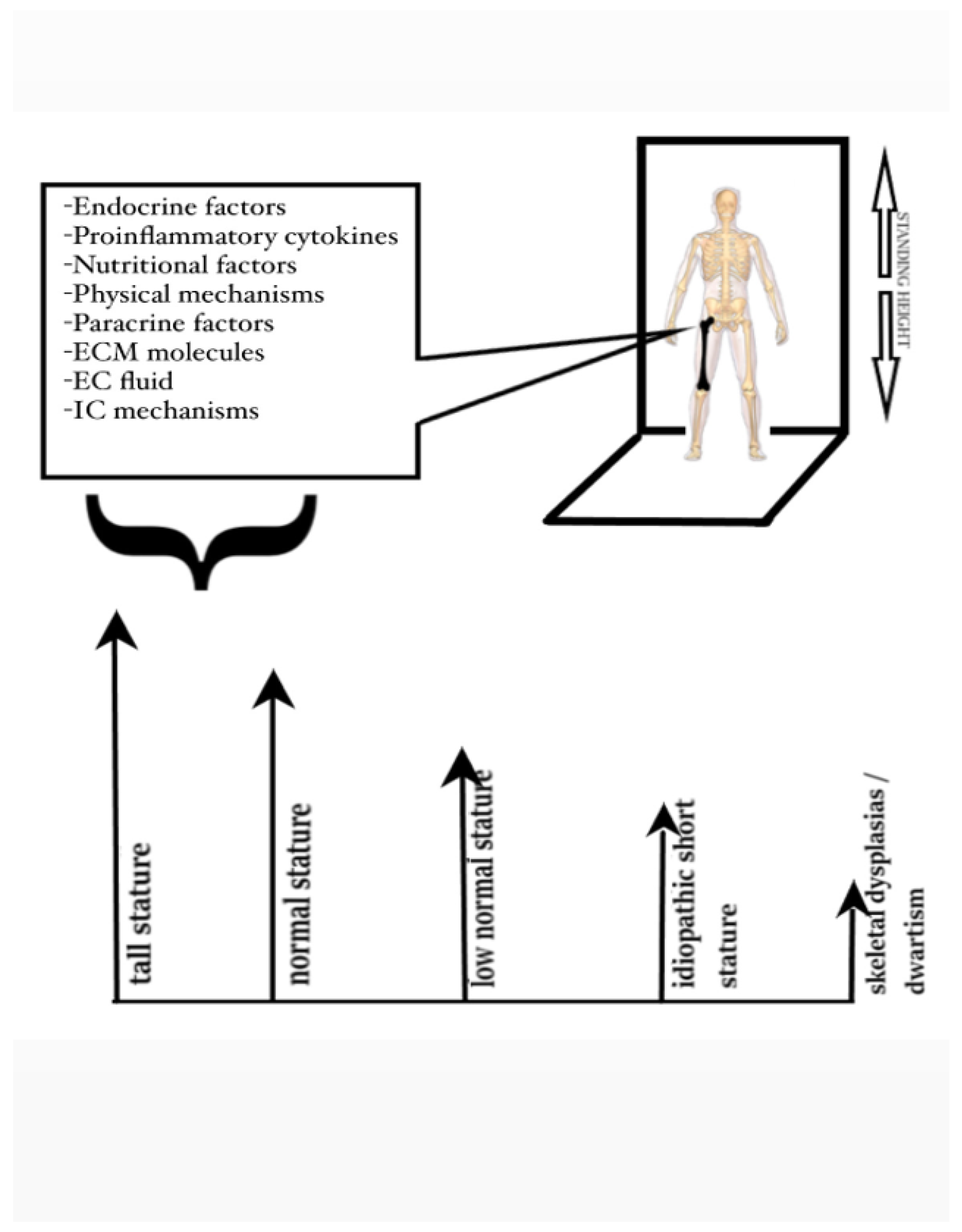
Epiphyseal growth plate (EGP) is the key biological structure responsible for long bones elongation through induction of chondrogenesis. This biological process determines the standing adult height. For many years, it was assumed that the sole mechanism regulating chondrogenesis at the EGP was through mesenchymal cell differentiation to chondrocytes and later chondrocyte maturation and proliferation, a process regulated by the human growth hormone/insulin-like growth factor 1 axis. However, advances in the fields of molecular biology and medical genetics have overturned this tenet and shed light upon the biological factors orchestrating events at the EGP. Chondrogenesis in the EGP is strictly regulated by many intrinsic (endogenous to the growth plate) and extrinsic (exogenous to the growth plate) biological factors, which act either as activators or as inhibitors of chondrocyte metabolism and growth. These factors include endocrine, paracrine, or nutritional factors, physical mechanisms, extracellular fluid parameters, proinflammatory cytokines, extracellular matrix molecules and intracellular mechanisms. Genetic defects affecting one of the above mechanisms can severely disrupt the events taking place at the EGP during chondrogenesis and cause stature abnormalities.
Stature is a multifactorial phenotypic trait influenced by multiple genetic and environmental factors. The genetic component is vast, as about 420 genetic loci contribute to adult stature, by encoding mainly proteins expressed in the EGP[9]. Some single gene defects can result in severe growth disorders, while most mutations or polymorphisms in various genetic loci cause low normal stature or idiopathic short stature. This observation highlights the fact that some of the genes involved play a crucial role in human growth, while some others a minor one. In addition, sequence variants in the same genetic locus can be expressed as different clinical entities. The phenotypic manifestation is the net result of the gene involved and the type of mutation in the gene (gain of function, altered function, impaired function or complete loss of function). Therefore, tall stature and dwarfism in severe skeletal dysplasias may be considered merely the two extreme ends of the same clinical spectrum between normal stature, low normal stature, and idiopathic short stature (Figure 1). This approach to stature abnormalities has dictated the classification of short stature proposed by the European Society for Pediatric Endocrinology based on the causes of short stature into primary (of genetic origin) and secondary (of endocrine or other origin). This classification is less centered around pathophysiology and constitutes a more useful approach for clinical purposes[8,10,11,12,13,14].
Figure 1.
(A) Lateral cephalometric tracing, (B) cephalometric landmarks and the lines connecting them that are referred as cephalometric reference planes. The most utilized cephalometric landmarks and planes are depicted. PO: porion; S: sella; N: nasion; OR: orbitale; PNS: posterior nasal spine; ANS: anterior nasal spine; A: A-point, B: B-point, Go: gonion, Pg: pogonion, Gn: gnathion, Me: menton; SN: Anterior cranial base plane; sBa: Posterior cranial base plane; PoOr: Frankfurt horizontal plane; PNS-ANS: Palatal plane; GoMe: Mandibular plane; Y-axis SPg or SGn; NA plane; NB plane; NPg: Facial plane.
Figure 1.
(A) Lateral cephalometric tracing, (B) cephalometric landmarks and the lines connecting them that are referred as cephalometric reference planes. The most utilized cephalometric landmarks and planes are depicted. PO: porion; S: sella; N: nasion; OR: orbitale; PNS: posterior nasal spine; ANS: anterior nasal spine; A: A-point, B: B-point, Go: gonion, Pg: pogonion, Gn: gnathion, Me: menton; SN: Anterior cranial base plane; sBa: Posterior cranial base plane; PoOr: Frankfurt horizontal plane; PNS-ANS: Palatal plane; GoMe: Mandibular plane; Y-axis SPg or SGn; NA plane; NB plane; NPg: Facial plane.
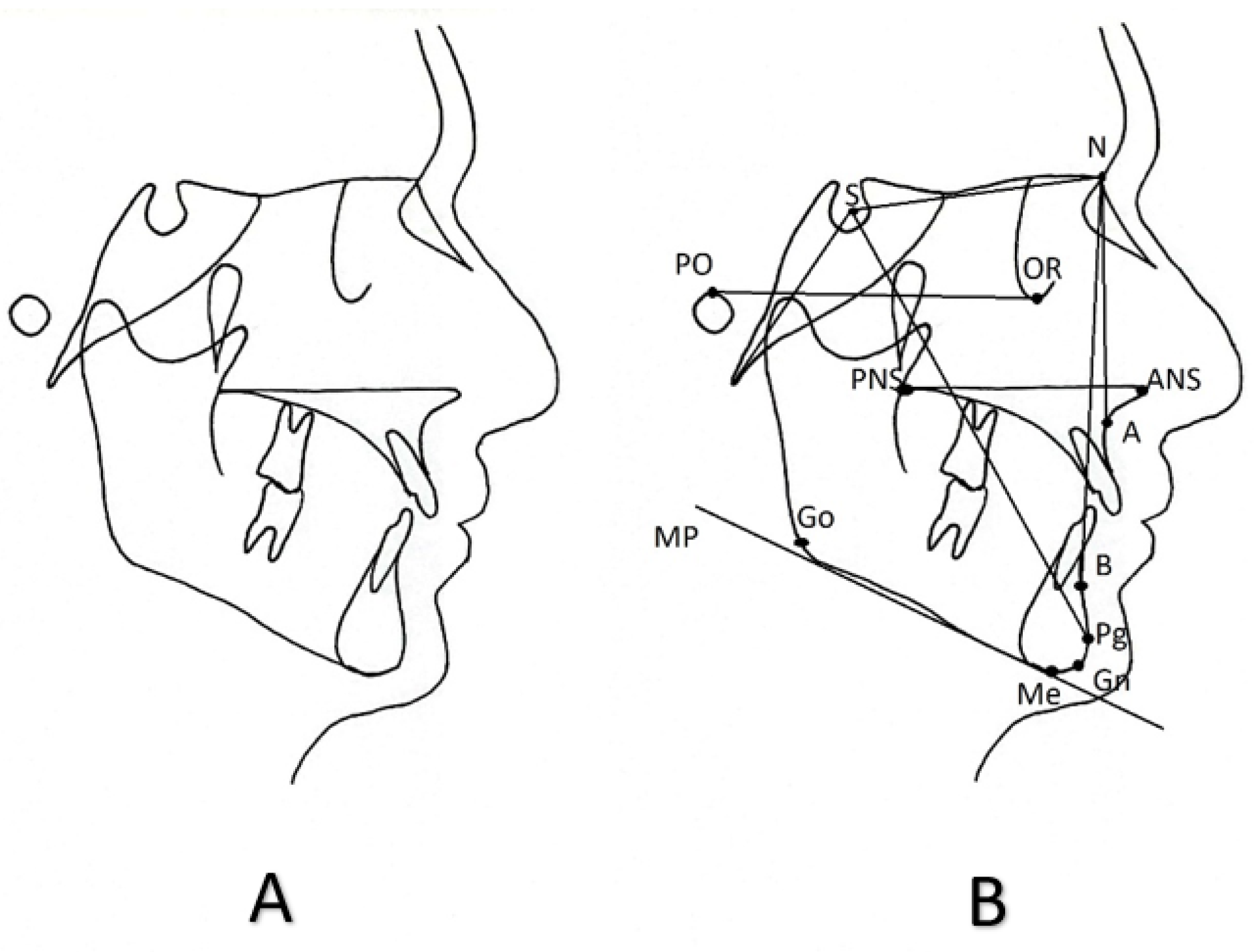
Figure 2.
Factors affecting the homeostasis of the epiphyseal growth plate and the phenotypic manifestations of stature abnormalities resulting from complex interactions among these factors.
Figure 2.
Factors affecting the homeostasis of the epiphyseal growth plate and the phenotypic manifestations of stature abnormalities resulting from complex interactions among these factors.
3. Clinical Assessment of Children with Short Stature
Currently, there is no widely accepted diagnostic algorithm when assessing children with short stature referred to pediatric clinics. The clinician usually obtains a thorough medical history, including chief complaint (commonly being the short stature), history of the presenting complaint, past medical history, family history and pedigree, review of systems, and then carries out a thorough clinical examination. Meticulous measurements of height, body weight, head circumference, sitting height, arm span, body proportions (such as sitting height/height ratio, arm span/height ratio and upper arm/lower arm ratio) and pubertal status (rated according to Tanner)[1] are of paramount importance, as there might exist subtle disproportions that go by unnoticed even to the most experienced clinician. In addition, assessment of fontanelles, dentition status and any apparent dysmorphic features should be done. Radiologic imaging of left hand and wrist is necessary for bone age assessment and evaluation of any incidental pathologic morphologic findings. Also, a blood sample is usually collected for preliminary biochemical assessment.
Clues collected from biochemical testing may lead the pediatrician into classifying the possible etiology of short stature into primary (genetic) or secondary. There are no clear guidelines as to when genetic testing must be addressed, but it relies heavily upon the clinical suspicion of the physician based on the findings from the physical, radiologic and laboratory evaluation of the patient. Findings that arise suspicion for short stature of genetic etiology include developmental delay, intellectual disability, behavioral problems, one apparently affected parent (with special attention on body proportions and dysmorphic features), family members with early onset arthritis or discopathy, microcephaly, relative macrocephaly, disproportionate ratios, extreme short stature or apparent dysmorphic features. The growth chart can also provide valuable indications about a possible genetic etiology, especially if there is a growth curve starting with a low or low-normal birth length, decreasing length SD for two to three years, followed by a stable height SD in childhood and a further decrease of height SD during adolescence. The genetic etiology of stature abnormalities may be further elucidated in the future as more genetic tools are added in our diagnostic toolkit. For specific conditions, such as Noonan syndrome, scoring systems have already been proposed[15].
4. Cephalometric Assessment of Children with Short Stature
Cephalometric radiography is an extra-oral radiographic technique used to display the craniofacial complex in standardized repeatable conditions. The cephalometric radiologic system contains a special mechanical part called the cephalostat, which bears a plug inserted into the external ear canal stabilizing the head and ensuring a constantly predetermined head position in a three-dimensional (3D) space. This results in a radiograph captured under standardized conditions that can be repeated with precision when it is required, because differences between radiographs should be real and not the result of different capture conditions. This technique offers the opportunity to conduct comparisons between radiographs of the same individual taken during different times or between radiographs of different individuals.
The cephalometric radiograph is part of daily dental and orofacial practice, especially in oral and maxillofacial surgery and orthodontics. It is used extensively in pre-treatment planning for assessing the quantitative extent of the existing oral and maxillofacial structure abnormalities and for verifying the clinical diagnosis, as well as post-treatment for the evaluation of the therapeutic result. There are two types of cephalometric x-rays, namely the lateral cephalometric x-ray and the frontal or postero-anterior cephalometric x-ray, with regard to the direction through which the x-ray beam is headed towards the face. Discrepancies affecting the structures of the craniofacial complex can be classified into 3 large categories as it regards the skeletal plane to which they are orientated: anomalies in the antero-posterior (sagittal) plane, anomalies in the vertical (frontal) plane, and anomalies in the transverse plane. Most of the existing abnormalities concern the first two planes and the lateral cephalometric x-ray. The use of the frontal cephalometric x-ray is very limited due to the extensive structural noise that the radiologic image contains, but it is the only technique that can assess abnormalities in the transverse plane, such as a posterior cross-bite or a facial asymmetry.
To provide diagnostic information, the cephalometric radiograph undergoes a process called “tracing”. This can be done either by hand or digitally with the use of applications on a computer. Specific bony or soft tissue points on the cephalometric tracing are identified and marked as cephalometric landmarks. The lines connecting the cephalometric landmarks are called “cephalometric reference planes”, despite being two dimensional structures. Finally, various linear, angular, or even radial measurements are conducted between the cephalometric landmarks and cephalometric planes. This process constitutes the cephalometric analysis. Figure 1 depicts a lateral cephalometric tracing conducted by hand on a transparent paper and indicates the most common cephalometric landmarks and reference planes.
5. Cephalometric Data for Genetic Syndromes Associated with Short Stature
There are several studies assessing specific genetic syndromes that present with short stature. To review the literature, a search was conducted in the PubMed electronic database using the following search criteria: cephalometric analysis and short stature; cephalometric analysis and achondroplasia; cephalometric analysis and hypochondroplasia; cephalometric analysis and skeletal abnormalities; cephalometr* and SHOX; cephalometr* and CNP; cephalometr* and ACAN; cephalometr* and CNVs; cephalometr* and IHH; cephalometr* and FGFR3; cephalometr* and Noonan syndrome; cephalometr* and Turner syndrome; cephalometr* and achondroplasia. Extensive cephalometric analysis studies have been conducted for common genetic syndromes, such as achondroplasia, Turner syndrome and Noonan syndrome. Cephalometric data for rarer genetic syndromes are scarse.

Table 1.
Cephalometric data for common genetic syndromes associated with short stature. (NA: not available data).
Table 1.
Cephalometric data for common genetic syndromes associated with short stature. (NA: not available data).
| GENETIC SYNDROMES ASSOCIATED WITH SHORT STATURE | DATA FROM LATERAL CEPHALOMETRIC RADIOGRAPHS | |||
|---|---|---|---|---|
| Skull | Antero-posterior plane | Vertical plane | Teeth | |
| Achondroplasia (1 in 15,000 to 40,000) |
Skull deformities Pneumatized frontal sinuses Significantly reduced posterior cranial base length Acute cranial base angle Significantly reduced length of the cribriform plate of the ethmoidal bone Remarkably increased anterior sphenoidal length |
Posteriorly placed and smaller maxilla and an anteriorly or normally placed mandible Skeletal class III that can result in an anterior cross-bite |
Reduced upper anterior facial height Posterior tilt of the nasal floor (palatal plane) High coronoid process |
Maxillary incisors labially proclined |
| Turner syndrome (1 in 2,000 to 3,000) |
Reduced posterior cranial base length Increased cranial base angle Smaller and thiner calvarium Fused cervical vertebrae Inferiorly and anteriorly placed external acoustic meatus Smaller,more delicate and less pneumatized mastoid procecess Large and excessively pneumatized shenoidal sinuses Smaller sella turcica Premature calcification of the petroclinoid ligament Reduced facial/cerebral skull ratio |
Reduced length of the maxilla Reduced length of the mandible Posteriorly positioned maxilla Posteriorly positioned mandible Posteriorly positioned chin Skeletal class II Transverse plane Facial asymmetry Posterior cross-bite as a result from the transversal dimension reduction of the maxilla |
Hyperdivergent skeletal planes Reduced posterior facial height Increased anterior facial height |
Occlusal plane angle is remarkably tilted Maxillary incisors lingually inclined Short teeth roots Remarkably low tongue position Pharyngeal airway space narrower in all its dimensions |
| Noonan syndrome (1 in 1,000 to 2,500) |
NA | Class I molar relationship and class III cuspid relationship, Class I skeletal relationship Class II skeletal relationship |
Vertical mandibular growth pattern with hyperdivergent planes and increased vertical angles. Both an increased and a decreased vertical overjet have been reported |
Labially inclined maxillary mandibular incisors Palatally inclined maxillary incisors and labially inclined mandibular incisors Lingually inclined mandibular incisors |
| Idiopathic growth hormone deficiency | Reduced anterior cranial base length | Reduced maxillary length and mandibular length |
Reduced anterior facial height and ramus height | NA |
| Prader-Willi syndrome (1 in 10,000 to 20,000) |
Reduced cranial base angle | Skeletal class II with posteriorly placed mandible and reduced mandibular and maxillary length Skeletal class III with anteriorly placed mandible |
Vertical growth direction and increased anterior facial height In cases of skeletal class III, horizontal growth direction |
Soft tissue excess In cases of skeletal class III, lingually inclined mandibular incisors and labially inclined maxillary incisors |
| Muenke syndrome (1 in 30,000) |
Decreased instracranial volume Significantly reduced anterior cranial base and skull length Increased angle between cranial base and Frankfort horizontal plane Hypertelorism Frontal bossing |
Reduced length of the maxilla and midface deficiency Reduced length of the mandible Posteriorly placed maxilla Transverse plane Increased facial width Significant skeletal asymmetry |
Reduced upper and lower anterior facial height Hypedivergent skeletal planes Increased gonial angle Anterior open bite |
NA |
Table 2.
Cephalometric data for less common genetic syndromes associated with short stature. (NA: not available data).
Table 2.
Cephalometric data for less common genetic syndromes associated with short stature. (NA: not available data).
| GENETIC SYNDROMES ASSOCIATED WITH SHORT STATURE | DATA FROM LATERAL CEPHALOMETRIC RADIOGRAPHS | |||
|---|---|---|---|---|
| Skull | Antero-posterior plane | Vertical plane | Teeth | |
| 22q11.2 deletion syndrome | Increased cranial base angle | Posteriorly placed mandible | NA | NA |
| 49, XXXXY syndrome (1 in 85,000 to 100,000) |
NA | Anteriorly placed mandible | NA | Lingually inclined mandibular incisors |
| Catania brachydactylous type of acrofacial dysostosis | NA | No distinctive abnormalities | NA | NA |
| Beckwith-Wiedemann syndrome (1 in 11,000) |
NA | Dental class I | Vertical growth pattern Anterior open bite |
Macroglossia |
| Bloch-Sulzberger syndrome (1,2 in 100,000) |
NA | Reduced maxillary length | Hyperdivergent skeletal planes | Lingually inclined maxillary incisors |
| Chronic acid sphingomyelinase deficiency (1 in 250,000) |
NA | Posteriorly placed maxilla and mandible and skeletal class II |
NA | Increased nasolabial angle Convex profile Retroinclination of maxillary and mandibular incisors |
| Cockayne syndrome (2 to 3 in a million) |
Hypodevelopment | Posteriorly placed and shorter mandible and skeletal class II | NA | NA |
| Syndromic craniosynostosis with fused spheno-occipital synchondrosis (1 in 100,000) |
Moderate and severe upward anterior cranial base inclination |
Severe midface deficiency Higher percentage of severe Class III skeletal pattern |
Severely hyperdivergent skeletal planes Severely forward condyle position |
NA |
| Ectodermal dysplasia 1, hypohidrotic (1 in 10,000 to 100,000) |
NA | Reduced length and posterior placement of the maxilla Anteriorly placed mandible with protruding chin Skeletal class III |
Hyperdivergent skeletal planes Reduced anterior facial height Reduced upper anterior facial height |
First maxillary molars located in higher positions |
| Ectodermal dysplasia anhidrotic or Rapp-Hodgkin syndrome | NA | Mildly to moderately reduced mandibular length with anterior mandibular placement Maxilla placed closer to the anterior cranial base |
NA | NA |
| Ellis-Van Creveld syndrome (1 in 60,000 to 200,000) |
NA | Skeletal class I or class II with posteriorly placed mandible Class III with anteriorly placed mandible, or posteriorly placed maxilla |
Hyperdivergency of the skeletal planes, or normal vertical growth direction, or even horizontal growth pattern | Mandibular and maxillary incisor retroclination Upper lip retrusion Lower lip retrusion Both concave and convex profiles have been reported |
| Hajdu-Cheney syndrome (less than 100 cases described) |
Increased cranial base length Enlarged, elongated, and wide open sella turcica with slender clinoids |
Posteriorly placed maxilla and mandible | NA | NA |
| Hallerman-Streiff syndrome (less than 200 people worldwide) |
NA | Skeletal and dental class II due to shorter and posteriorly placed mandible | Vertical growth pattern with an opening of the gonial angle, a large anterior open bite, and an excessive increase in the lower anterior facial height | NA |
| Kabuki syndrome (1 in 32,000) |
NA | Posteriorly placed maxilla and mandible with a skeletal Class I pattern | Increased lower anterior facial height and anterior open bite | NA |
| Klippel-Feil syndrome (1 in 40,000) |
Fused cervical vertebrae | Skeletal class I | Vertical growth pattern | NA |
| Langer-Giedion syndrome (extremely rare) |
NA |
Posteriorly placed maxilla and mandible | NA | NA |
| Larsen syndrome (1 in 100,000) |
Orbits positioned posteriorly relative to the anterior cranial base | Posteriorly positioned maxilla and mandible with skeletal Class III pattern Transverse plane Hypertelorism Narrow maxillary basal arch Reduced maxillary and mandibular dental arch widths |
Increased vertical angles with a large Gonial angle Growth tendency of the mandible toward the postero-inferior direction |
Mandibular primary incisors lingually inclined |
| Methylmalonic aciduria and homocystinuria (1 in 200,000) |
Head rotated and bent towards the left shoulder, which is located in a lower position than the right one Horizontal planes of both maxillary bones converge towards the right |
NA | NA | NA |
| Moebius syndrome (1 in 50,000 to 500,000) |
NA | Posteriorly placed mandible with reduced length and skeletal class II | Increased maxillary height resulting in a vertical growth pattern | Proclined maxillary and mandibular incisors Protrusion of upper and lower lips Long upper lip |
| Congenital or childhood onset myotonic dystrophy type I (1 in 9,000) |
NA | Increased ANB angle and reduced facial angle | Hyperdivergent skeletal planes with mandibular plane angle and intermaxillary angle increased | NA |
| Rubinstein-Taybi syndrome (1 in 100,000 to 125,000) |
Brachycephaly | Skeletal class II | NA | NA |
| Seckel syndrome (1 in 10,000) |
Small skull with an extremely short anterior cranial base and maxillary length Differences in the morphology of the sella turcica observed between girls and boys |
NA | NA | NA |
| Silver-Russell syndrome (1 in 30,000 to 100,000) |
NA | Skeletal class II with posteriorly placed mandible Class I and III have also been reported |
NA | NA |
| Simpson-Golabi-Behmel syndrome | Increased anterior cranial base length | Increased length of the maxilla and the mandible with a skeletal class III pattern | Increased lower anterior facial height | NA |
| Solitary Median Maxillary Central Incisor syndrome (1 in 50,000 |
Hypoplastic sella turcica Cervical vertebral maturation (CVM) at stage CS2 |
Skeletal class III with an anterior cross bite as a result of reduced maxillary length and anteriorly placed mandible | Vertical growth pattern | Convex profile Airway patency Maxillary and mandibular incisal proclination |
| Treacher-Collins syndrome (1 in 50,000) |
Reduced length of both the anterior and posterior cranial base and a reduced cranial base angle | Posteriorly placed maxilla with reduced length Posteriorly placed mandible with a characteristic reduction of the mandibular length Reduced maximum ramus width |
Hyperdivergent skeletal planes and increased gonial angle Both the anterior and posterior facial heights are decreased |
The maxillary and functional occlusal planes are tipped upwards posteriorly |
| Williams syndrome (1 in 7,500 to 18,000) |
Reduced anterior cranial base length |
Posteriorly placed chin |
Hypedivergent skeletal planes Unusual proportion of upper to lower anterior facial height and posterior to anterior facial height |
NA |
Table 2.
Cephalometric data for less common genetic syndromes associated with short stature(NA: not available data).
Table 2.
Cephalometric data for less common genetic syndromes associated with short stature(NA: not available data).
| GENETIC SYNDROMES ASSOCIATED WITH SHORT STATURE | DATA FROM LATERAL CEPHALOMETRIC RADIOGRAPHS | |||
|---|---|---|---|---|
| Skull | Antero-posterior plane | Vertical plane | Teeth | |
| 22q11.2 deletion syndrome | Increased cranial base angle | Posteriorly placed mandible | NA | NA |
| 49, XXXXY syndrome (1 in 85,000 to 100,000) |
NA | Anteriorly placed mandible | NA | Lingually inclined mandibular incisors |
| Catania brachydactylous type of acrofacial dysostosis | NA | No distinctive abnormalities | NA | NA |
| Beckwith-Wiedemann syndrome (1 in 11,000) |
NA | Dental class I | Vertical growth pattern Anterior open bite |
Macroglossia |
| Bloch-Sulzberger syndrome (1,2 in 100,000) |
NA | Reduced maxillary length | Hyperdivergent skeletal planes | Lingually inclined maxillary incisors |
| Chronic acid sphingomyelinase deficiency (1 in 250,000) |
NA | Posteriorly placed maxilla and mandible and skeletal class II |
NA | Increased nasolabial angle Convex profile Retroinclination of maxillary and mandibular incisors |
| Cockayne syndrome (2 to 3 in a million) |
Hypodevelopment | Posteriorly placed and shorter mandible and skeletal class II | NA | NA |
| Syndromic craniosynostosis with fused spheno-occipital synchondrosis (1 in 100,000) |
Moderate and severe upward anterior cranial base inclination |
Severe midface deficiency Higher percentage of severe Class III skeletal pattern |
Severely hyperdivergent skeletal planes Severely forward condyle position |
NA |
| Ectodermal dysplasia 1, hypohidrotic (1 in 10,000 to 100,000) |
NA | Reduced length and posterior placement of the maxilla Anteriorly placed mandible with protruding chin Skeletal class III |
Hyperdivergent skeletal planes Reduced anterior facial height Reduced upper anterior facial height |
First maxillary molars located in higher positions |
| Ectodermal dysplasia anhidrotic or Rapp-Hodgkin syndrome | NA | Mildly to moderately reduced mandibular length with anterior mandibular placement Maxilla placed closer to the anterior cranial base |
NA | NA |
| Ellis-Van Creveld syndrome (1 in 60,000 to 200,000) |
NA | Skeletal class I or class II with posteriorly placed mandible Class III with anteriorly placed mandible, or posteriorly placed maxilla |
Hyperdivergency of the skeletal planes, or normal vertical growth direction, or even horizontal growth pattern | Mandibular and maxillary incisor retroclination Upper lip retrusion Lower lip retrusion Both concave and convex profiles have been reported |
| Hajdu-Cheney syndrome (less than 100 cases described) |
Increased cranial base length Enlarged, elongated, and wide open sella turcica with slender clinoids |
Posteriorly placed maxilla and mandible | NA | NA |
| Hallerman-Streiff syndrome (less than 200 people worldwide) |
NA | Skeletal and dental class II due to shorter and posteriorly placed mandible | Vertical growth pattern with an opening of the gonial angle, a large anterior open bite, and an excessive increase in the lower anterior facial height | NA |
| Kabuki syndrome (1 in 32,000) |
NA | Posteriorly placed maxilla and mandible with a skeletal Class I pattern | Increased lower anterior facial height and anterior open bite | NA |
| Klippel-Feil syndrome (1 in 40,000) |
Fused cervical vertebrae | Skeletal class I | Vertical growth pattern | NA |
| Langer-Giedion syndrome (extremely rare) |
NA |
Posteriorly placed maxilla and mandible | NA | NA |
| Larsen syndrome (1 in 100,000) |
Orbits positioned posteriorly relative to the anterior cranial base | Posteriorly positioned maxilla and mandible with skeletal Class III pattern Transverse plane Hypertelorism Narrow maxillary basal arch Reduced maxillary and mandibular dental arch widths |
Increased vertical angles with a large Gonial angle Growth tendency of the mandible toward the postero-inferior direction |
Mandibular primary incisors lingually inclined |
| Methylmalonic aciduria and homocystinuria (1 in 200,000) |
Head rotated and bent towards the left shoulder, which is located in a lower position than the right one Horizontal planes of both maxillary bones converge towards the right |
NA | NA | NA |
| Moebius syndrome (1 in 50,000 to 500,000) |
NA | Posteriorly placed mandible with reduced length and skeletal class II | Increased maxillary height resulting in a vertical growth pattern | Proclined maxillary and mandibular incisors Protrusion of upper and lower lips Long upper lip |
| Congenital or childhood onset myotonic dystrophy type I (1 in 9,000) |
NA | Increased ANB angle and reduced facial angle | Hyperdivergent skeletal planes with mandibular plane angle and intermaxillary angle increased | NA |
| Rubinstein-Taybi syndrome (1 in 100,000 to 125,000) |
Brachycephaly | Skeletal class II | NA | NA |
| Seckel syndrome (1 in 10,000) |
Small skull with an extremely short anterior cranial base and maxillary length Differences in the morphology of the sella turcica observed between girls and boys |
NA | NA | NA |
| Silver-Russell syndrome (1 in 30,000 to 100,000) |
NA | Skeletal class II with posteriorly placed mandible Class I and III have also been reported |
NA | NA |
| Simpson-Golabi-Behmel syndrome | Increased anterior cranial base length | Increased length of the maxilla and the mandible with a skeletal class III pattern | Increased lower anterior facial height | NA |
| Solitary Median Maxillary Central Incisor syndrome (1 in 50,000 |
Hypoplastic sella turcica Cervical vertebral maturation (CVM) at stage CS2 |
Skeletal class III with an anterior cross bite as a result of reduced maxillary length and anteriorly placed mandible | Vertical growth pattern | Convex profile Airway patency Maxillary and mandibular incisal proclination |
| Treacher-Collins syndrome (1 in 50,000) |
Reduced length of both the anterior and posterior cranial base and a reduced cranial base angle | Posteriorly placed maxilla with reduced length Posteriorly placed mandible with a characteristic reduction of the mandibular length Reduced maximum ramus width |
Hyperdivergent skeletal planes and increased gonial angle Both the anterior and posterior facial heights are decreased |
The maxillary and functional occlusal planes are tipped upwards posteriorly |
| Williams syndrome (1 in 7,500 to 18,000) |
Reduced anterior cranial base length |
Posteriorly placed chin |
Hypedivergent skeletal planes Unusual proportion of upper to lower anterior facial height and posterior to anterior facial height |
NA |
5.1. Achondroplasia (OMIM 100800)
Achondroplasia is a rare genetic disorder with an estimated birth prevalence of about 4 per 100,000 live births worldwide. Achondroplasia is caused by mutations (G1138A and G1138C) in the fibroblast growth factor receptor 3 (FGFR3) gene located at chromosome 4p16.3. It is an autosomal dominant disorder, but about 80% of cases are sporadic, due to a de novo mutation in offspring of unaffected parents[16]. Lateral cephalometric radiographs of individuals with achondroplasia demonstrate:
a) Skull: enlarged skull calvaria, frontal bossing, high degree of pneumatization of the frontal sinuses, acute occipital prominence, and a short, deformed and depressed nasal bone. Normal length of the anterior cranial base, remarkably reduced length of the posterior cranial base and acute cranial base angle[17,18,19,20]. The length of the cribriform plate of the ethmoidal bone is remarkably reduced, but the anterior sphenoidal length is remarkably increased, possibly compensating for the shortened cribriform plate length, and suggesting that growth in the length of the anterior cranial base takes place primarily by adaptation at one site, the spheno-ethmoidal synchondrosis. The reduced posterior cranial base length possibly results from hypoplasia of the bone that is preformed in cartilage with possible early closure of the spheno-occipital synchondrosis. The exaggerated closure of the cranial base angle in achondroplasia may be related to an increased brain size and possibly earlier than normal closure of the intersphenoidal synchondrosis[18].
b) Antero-posterior plane: Posteriorly placed and smaller maxilla, anteriorly or normally placed mandible of normal size with a normal gonial angle, antero-posterior arrangement of the jaws, expressed as skeletal class III pattern of malocclusion resulting in anterior cross-bite[17,18,19,20].
c) Vertical plane: Reduced upper anterior facial height, posterior tilt of the nasal floor (palatal plane) and high coronoid process[17,18,19].
d) Teeth and soft tissue: Maxillary incisors proclined relative to the Frankfurt horizontal plane and mandibular incisor angulations normal relative to the mandibular plane[19].
Lateral cephalometric radiographs of achondroplastic transgenic mice expressing a constantly active mutant of MEK1 in chondrocytes revealed significantly reduced skull length but significantly increased cranial arch length. The area of the brain case was significantly larger and more circular. The maximum diameter was normal, but the minimum diameter, roughly equivalent to brain case height, was increased[21].
5.2. Turner syndrome (OMIM 309585)
Turner syndrome is a common chromosomal aberration caused by a complete or partial loss of one copy of the X chromosome (45X0) or by mosaicism of a 45X0 cell line with a normal 46XX cell line with an incidence of up to 1 per 2000 live-born girls[22]. The 45X0 karyotype is estimated to be present in 3% of all conceptions, but almost 99% of these abnormal fetuses spontaneously abort, usually during the first trimester of the pregnancy, accounting for up to 10% of all spontaneous abortions[22]. The cephalometric data available for individuals with Turner syndrome include:
a) Skull: Posterior cranial base exhibiting reduced length and cranial base angle being increased (increased angle of flexion), causing a flattening of the cranial base[23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39], smaller dimensions of the calvarium of the skull[35,37], reduced thickness of the calvarium[39], fused cervical vertebrae[40], a more inferiorly and anteriorly placed external acoustic meatus[41], smaller and more delicate mastoid processes and of reduced pneumatization, larger and excessively pneumatized sphenoidal sinuses, smaller Sella turcica[42], premature calcification of the petro-clinoid ligament in patients before the age of 20[39], small facial part of the skull compared with the cerebral part[39], retarded development of the cranial skeleton affecting appositional and sutural growth, as well as growth in the condylar cartilage and the spheno-occipital synchondrosis[43]. Most prominent discrepancies from normal controls are observed in 45X0 individuals, while milder phenotype is noted in individuals bearing an isochromosome and even milder differences in mosaic 45X0/46XX individuals[24,25,35].
b) Antero-posterior plane: Full Turner individuals (45X0) exhibit the most remarkable aberrations of craniofacial morphology[24,25,35], including reduced length of the maxilla[24,25,26,29,30,32,33,35,42,44,45,46,47,48], reduced length of the mandible[25,26,27,30,31,32,34,35,36,37,42,45,46,47], posteriorly positioned maxilla[24,25,26,27,28,34,35,37,42,44,45,46,49,50,51], posteriorly positioned mandible in relation to the anterior cranial base[24,25,26,27,28,33,35,37,42,44,45,46,49,50,51], and posteriorly positioned chin[49]. Cases of normal maxillary[34,36] or mandibular length[44] have also been reported. In fact, Rzymski et al. have reported a disproportionately large mandible in females with Turner syndrome, as if it did not fit in with the remaining facial bones, that was squared with pronounced angles, resembling a man's jaw[39]. The above arrangement of the jaw bones leads to a skeletal class II pattern in most individuals with Turner syndrome[32,45,46,47]. However, skeletal class III pattern has also been reported, but with both the maxilla and the mandible being posteriorly positioned[50]. Growth hormone has a beneficial effect on maxillary and mandibular growth, as shown by such treatment of individuals with Turner syndrome who have demonstrated a class I skeletal pattern[52,53,54].
c) Vertical plane: The cephalometric angles assessing the anomalies in the vertical plane (MP-SN, MP-FH, MP-PP) seem to be increased in individuals with Turner syndrome, constituting the skeletal planes as hyperdivergent, and implying a posterior/downward inclination of the mandible with a vertical growth pattern of the face[24,25,26,31,33,34,35,37,40,43,45,49,50,55], a reduction of the posterior facial height, and an increase in the anterior facial height. Other findings include the presence of a deep bite resulting from the increased vertical overjet[37,46], a shortened distance of glenoid fossa and gonion from the Sella[26], as well as an anterior open bite[47]. Individuals with Turner syndrome treated with growth hormone demonstrated normalization of the vertical characteristics after the treatment[52,53,54], while posterior facial height, mandibular ramus height, and anterior facial height were mostly influenced[53].
d) Coronal plane: Individuals with Turner syndrome exhibited facial asymmetry[40,45] and posterior cross-bite as a result from the transversal dimension reduction of the maxilla[32,47].
e) Teeth and soft tissue: The occlusal plane angle is remarkably tilted, the maxillary incisors are lingually inclined, and the upper and lower lips are 1mm and 5mm forward of the E line[50]. In addition, both the maxillary and the mandibular incisors reportedly exhibit normal inclination and position in relation to their skeletal base[44], the teeth roots are short[47], the position of the tongue is remarkably low[56] and the pharyngeal airway space is narrower in all its dimensions[27,46].
5.3. Noonan Syndrome (OMIM 163950)
Noonan syndrome is characterized by genotypic and phenotypic variance, which makes it difficult to identify mildly affected individuals. It is a common genetic abnormality with an incidence of one in 1,000 to 2,500 live births for the severe phenotype, but mild cases may be as common as one in 100 live births. Familial recurrence is consistent with an autosomal dominant mode of inheritance, but de novo mutations are more common, accounting for 60% of cases[57]. The cephalometric data available for individuals with Noonan syndrome include the following:
a) Skull: In subjects with Noonan syndrome, the distance by which the odontoid tip extended past McGregor's line is significantly greater than that of the control subjects, the third and fourth cervical vertebrae are in significantly superior positions and also significantly superior to the hyoid bone in subjects with Noonan syndrome compared to controls. There is no difference in the position of the hyoid bone between the groups[58]. Head circumference of all females with Noonan syndrome is normal, whereas in 50% of males it is below the 3rd percentile for age[59].
b) Antero-posterior plane: Skeletal abnormalities in the antero-posterior plane do not seem to demonstrate a specific pattern in individuals with Noonan syndrome. However, the length of the jaws seems to be reduced. There are reports of class I malocclusion[60,61,62]. Several reports indicate a posteriorly placed mandible[15,63,64,65].
c) Vertical plane: Most reports indicate a downwards mandibular growth pattern with hyperdivergent planes and increased vertical angles[15,62,63,64,66]. Nevertheless, cases of a horizontal growth pattern[60], an increased vertical overjet[62,63] and a decreased vertical overjet[61,65] have been reported.
5.4. Idiopathic Growth Hormone Deficiency (262400 type IA; 612781 type IB; 173100 type II; 307200 type III; 618157 type IV)
All linear measurements were reduced in a study of isolated growth hormone deficiency (IGHD) individuals[67]. Total maxillary length was the most reduced parameter followed by posterior cranial base length, total mandibular length, total posterior facial height, total anterior facial height, mandibular corpus length and anterior cranial base length. Angular measurements were in the normal range, except for increased gonial angle. Posterior facial height/anterior facial height and lower-anterior facial height/anterior facial height ratios were not different from those of the control group[67].
Growth hormone treatment had a reportedly favorable influence on the craniofacial growth pattern of 21 boys with idiopathic short stature (ISS) and 25 boys with growth hormone deficiency (GHD). Before human growth hormone treatment, all the craniofacial structures had reduced length, as well as a retrognathic facial type and a skeletal class II tendency[68]. After treatment, the jaw and the cranial base angles were increased in children with short stature, while the angles between the anterior cranial base and the mandibular plane and between the maxilla and mandible were larger than normal. The proportions between anterior and posterior face heights and between upper and lower anterior face heights were also smaller[68]. During the treatment period, an overall enhancement in growth of the facial skeleton was observed in boys with short stature. The changes induced by growth hormone yielded a more prognathic growth pattern, a more anterior position of the jaws in relation to the cranial base and increased anterior rotation of the mandible.
The mandibular corpus length and anterior face height of the growth hormone-treated boys were greater at the end of the study compared with the boys in the reference group. No differences in growth response were noted either between the GHD and ISS boys. The only change that remained significantly correlated with orthodontic treatment was the alteration in mandibular ramus height, showing a larger change in the boys who had not undergone orthodontic therapy. Another study noted that in individuals with idiopathic growth hormone deficiency that did not receive growth hormone treatment, the anterior cranial base length, anterior facial height, maxillary length, mandibular length and ramus height were smaller, while treated individuals had a significantly larger upper anterior facial height, maxillary length, and ramus height[69].
5.5. Prader-Willi Syndrome (OMIM 176270)
Among 42 individuals with Prader-Willi syndrome (PWS), 12 demonstrated a posteriorly placed mandible with a skeletal class II pattern, vertical growth direction and increased anterior facial height, 10 individuals had reduced cranial base angle, a skeletal class II pattern and an increased lower anterior facial height, while 20 had an anteriorly placed mandible, a skeletal class III pattern with anterior growth direction, lingually inclined mandibular incisors and labially inclined maxillary incisors[70].
In another report of 20 subjects with PWS, the length of the maxilla, the length of the mandible at both the ramus and the mandibular body, and the posterior and anterior facial heights were significantly reduced, while the angular measurements did not differ significantly from normal values. The antero-posterior cephalometric radiographs revealed a significant reduction in maxillary skeletal width, mandibular skeletal width, and interzygomatic distance[71]. Similar findings were noted in most individuals of another cohort of PWS patients. The mandibular and maxillary length, ramus height, posterior facial height, and mid-facial height were all significantly smaller, while the overall small bony structures in contrast with the relatively large soft tissue draping was observed especially in obese adults. The data suggest that a characteristic bony model might be created for PWS, which could be of use both in the diagnosis and treatment of such patients by orthodontists[72]. A positive correlation between cephalometric measurements, gender, crown length of permanent left central incisor and combined mesiodistal diameter of permanent maxillary anteriors with somatic stature was found in 70 individuals with PWS[73].
5.6. Down Syndrome (OMIM 190685)
Individuals with Down syndrome showed reduced anterior cranial base length with an increased cranial base angle, reduced mandibular length and upper anterior and lower anterior facial height with a normal posterior facial height[74]. Maxillary length and facial convexity were reduced when compared with the control group with a class I skeletal pattern, although the anteroposterior position of the maxilla was normal. Individuals with Down syndrome had reduced maxillary length with flatter cranial base and thus differed from typical class III skeletal pattern. Maxillary deficiency was not prominent in the face of individuals with Down syndrome because of the overall reduction in craniofacial dimensions[74]. A report of children with Down syndrome with and without hypodontia indicated that individuals with hypodontia presented over the years a reduced ANB angle and a decrease in vertical angles compared to subjects without hypodontia[75]. In another report of Down syndrome patients, the cranial base angle was normal, and the length of the anterior cranial base was reduced in 54% of the individuals, while the jaws presented normal relations in both the antero-posterior and the vertical planes[76]. An anterior cross-bite was observed in 38% and a reduced interincisal angle in 77%. The lower incisors were anteriorly placed in 84% and were labially inclined in 77%. The upper incisors were anteriorly placed in 77% of the sample, while the lower lip protruded in 84% of them[76]. The most common abnormality observed in Down syndrome patients from Pakistan was fusion between C2 and C3. This anomaly was found in 20% of subjects with skeletal class I, 50% of subjects with skeletal class II and 53.3% with skeletal class III patterns[77]. The highest frequencies of partial cleft at the level of C1 and occipitalization were observed in subjects with skeletal class II and III malocclusions, respectively, however, none of the subjects showed fusion between C1 and C2 or dehiscence. Furthermore, the association of cervical vertebral anomaly was highest with skeletal class III and lowest with skeletal class I malocclusions[77].
5.7. Muenke Syndrome (OMIM 602849)
Muenke craniosynostosis or coronal synostosis syndrome is caused by a point mutation (C749G) in the FGFR3 gene resulting in a Pro250Arg (P250R) substitution[78]. The cephalometric data for these patients include:
a) Skull: Decreased intracranial volume, significantly reduced length of the skull length and significant reduction of the anterior cranial base length[78,79,80]. The angle between the cranial base and the Frankfurt horizontal plane is increased[78]. The anterior part of the skull is characterized by a significant increase of the intercoronal distance. Bilateral interorbital and anterior interorbital distances are increased, confirming a hypertelorism typical for this syndrome. The "frontal bossing" frequently found in brachycephaly is characterized by the increased sagittal extension of the forehead and by the increased height of the frontal prominence. Following surgery (fronto-orbital advancement), the measurements defining the morphology of the forehead are reduced and appear to be constant throughout the follow-up[80].
b) Antero-posterior plane: Reduced length of the maxilla[79,80] and reduced length of the mandible[78,79]. The severely reduced maxillary length is held accountable for the midface deficiency in those individuals[80,81]. The maxilla is posteriorly placed, while the mandible is normally placed[82]. Most patients exhibit class I skeletal relation[82] but class II and III have also been described[82].
5.8. Other Genetic Syndromes
22q11.2 deletion syndrome (OMIM 611867): In 20 patients the cranial base angle was increased, and the mandible was posteriorly placed[84].
49, XXXXY syndrome: In a 9-year-old boy the maxilla was placed in a normal antero-posterior position, the mandible was slightly anteriorly placed, and the mandibular incisors were lingually inclined[85].
Acrofacial dysostosis - Catania brachydactylous type (OMIM 101805): Analysis of cephalometric and hand-wrist radiographs of a mother and daughter showed no distinctive diagnostic abnormalities[86].
Beckwith-Wiedemann syndrome (OMIM 130650.): Most of 25 children (76%) exhibited hyperdivergent skeletal planes with a dental class I relation. About 40% had an anterior open bite and 16% showed also unilateral posterior cross-bite. No statistically significant differences were observed among the different phenotypes of Beckwith-Wiedemann syndrome[87].
Bloch-Sulzberger syndrome (OMIM 308300): In a case of an 8-year-old female patient with cleft lip and palate, the length of the maxilla was reduced, the skeletal planes were hyperdivergent, and the maxillary incisors were lingually inclined[88].
Chronic acid sphingomyelinase deficiency: Four individuals had a posteriorly placed maxilla, and one patient had a normal antero-posterior maxillary positioning. Three patients had a good nasolabial angle and two had an increased nasolabial angle. The mandible was posteriorly placed in four and only one patient had an anteriorly placed mandible. The convex profile was predominant, as well as maxillary and mandibular retrusion and skeletal class II pattern. Three patients showed good positioning and two showed retroinclination of the upper incisors. In the evaluation of the lower incisors, three patients showed good positioning, one showed a lower inclination, and one showed a greater inclination[89].
Cockayne syndrome (OMIM 216400): Cephalometric analysis of 9 patients revealed hypodevelopment of the skull, reduced length of the mandible, posteriorly placed and shorter mandible and skeletal class II pattern[90].
Craniosynostosis, syndromic with fused or open spheno-occipital synchondrosis: Patients with syndromic craniosynostosis usually have more severe symptomatology in comparison to patients with single suture synostosis[91]. The fused group of patients showed a higher percentage of severe midface deficiency than the non fused group (70% versus 10%). The fused group showed relatively higher percentages of severe class III skeletal pattern (100% vs. 70%), severely hyperdivergent skeletal planes (40% vs. 10%), severely forward condyle position (30% vs. 0%), and moderate and severe upward anterior cranial base inclination (90% vs. 50%) than the open group. However, the two groups exhibited the same distribution of moderately and severely retrusive orbital position[92].
Εctodermal dysplasia 1, hypohidrotic, X-linked (OMIM 305100): In 48 patients the maxilla exhibited reduced length and was posteriorly placed. The length of the mandible was normal, but it was anteriorly placed with a protruding chin. The skeletal pattern was class III, and the anterior facial height was reduced. Male patients with numerically more clinical manifestations of ectodermal dysplasia exhibited a larger reduction in the length of the maxilla and a more posteriorly placed maxilla, as well as larger vertical angles and an increased anterior facial height, in comparison with male patients bearing less manifestations of ectodermal dysplasia[93]. In another report, 6 patients exhibited reduced length of the maxilla and an anteriorly placed mandible. Hyperdivergent skeletal planes, reduced anterior facial height, reduced upper anterior facial height were also present. The first maxillary molars were located in higher positions suggesting that the vertical and antero-posterior maxillary growth retardation, rather than the lack of occlusal support due to hypodontia, leads to the reduced anterior facial height in individuals with ectodermal dysplasia[94].
Ectodermal dysplasia, anhidrotic, with cleft lip/palate or Rapp-Hodgkin syndrome (OMIM 129400): In 3 patients of a family with a rare form of ectodermal dysplasia, the length of the maxilla was mildly to moderately reduced. The maxilla was placed closer to the anterior cranial base, but it was positioned within normal limits anterior-posteriorly relative to the forehead (Lande's angle). The presence of an anteriorly placed mandible also contributed to a concave skeletal profile[95].
Ellis-Van Creveld syndrome (OMIM 225500): Among 7 individuals, 2 demonstrated a class I skeletal pattern, 4 individuals a class III (due to anteriorly placed mandible in 2 patients and to posteriorly placed maxilla in 2 patients), and one individual a class II due to the posteriorly placed mandible. Regarding the vertical plane, 4 patients showed a hyperdivergency of the skeletal planes, 2 patients were in the normal range, and one patient showed a horizontal growth pattern. Incisor retroclination was prominent in the mandibular and maxillary arches in 4 patients, while soft tissue analysis indicated upper lip retrusion in 4 patients and lower lip retrusion in one patient. One patient presented with a convex profile, whereas 2 patients presented with concave profiles[96]. A high degree of agreement was observed between two methods of determining the skeletal age (degree of cervical vertebral maturation in lateral cephalometric radiography and hand-wrist method) in 178 patients with short stature, especially in girls in the familial short-stature group, whereas boys had higher agreement in the nonfamilial short-stature group[97].
Hajdu-Cheney syndrome (OMIM 102500): In a case of a 22-year-old female, cephalometric analysis revealed anteroposterior and vertical hypoplasia of the midface. The cranial base was increased. The maxilla and the mandible were placed posteriorly, although their relation was within normal limits. The Sella turcica was enlarged, elongated, and wide open with slender clinoids[98].
Hallermann-Streiff syndrome (OMIM 234100): In a 10-year-old girl the mandible had reduced length and was posteriorly placed, resulting in a skeletal and dental class II. In the vertical plane, the patient presented a vertical growth pattern with an opening of the gonial angle, a large anterior open bite, and an excessive increase in the lower anterior facial heigh[99]t.
Hemifacial microsomia (OMIM 141400): In 183 patients, the ratios of the affected side to the unaffected side of the lateral distance of the mandibular condyle, the mandibular ramus height, and the length of the body of the mandible were significantly reduced. The inclination of the body of the mandible was significantly larger in the side with hemifacial microsomia than in the unaffected side, and the extent of the mandibular ramus was significantly lower than in the unaffected side. The affected/unaffected ratios of the extent of the angle of the mandible and the inclination of the body of the mandible were increased. Moreover, the length and the inclination of the body of the mandible had significant correlations with the distance of the shift of the menton[100].
Kabuki syndrome (OMIM 147920.): A male patient had a posteriorly placed maxilla and mandible with a skeletal class I pattern, increased lower anterior facial height and a severe anterior open bite. During the follow-up, the antero-posterior growth of the maxilla and mandible showed improvement. The skeletal pattern changed to skeletal class III from skeletal class I during the follow-up period. Both the upper and the lower anterior facial height increased[101].
Klippel-Feil syndrome (OMIM 616549.): Cephalometric analysis of a case of an 8-year-old female revealed a class I skeletal pattern with a vertical growth pattern and fused cervical vertebrae[102]. A case of a 12-year-old female with Turner syndrome combined with Klippel-Feil syndrome demonstrated a skeletal class III pattern[40].
Langer-Giedion syndrome (OMIM 190350): A 10-year-old child had both a posteriorly placed maxilla and mandible[103].
Larsen syndrome (OMIM 150250): In a case of a 5-year-old girl both the maxilla and the mandible were posteriorly positioned forming a skeletal class III pattern. The vertical angles were increased with a large gonial angle, and growth tendency of the mandible toward the postero-inferior direction. Inclination of the maxillary primary incisors was within the normal range, while the mandibular primary incisor inclined more lingually. The orbits were also positioned posteriorly relative to the anterior cranial base. The antero-posterior cephalometric radiograph revealed hypertelorism and a narrow maxillary basal arch and maxillary and mandibular dental arch widths[104].
Methylmalonic aciduria and homocystinuria (OMIM 277410): A 11-year-old patient, who developed that disorder during the neonatal period, exhibited a postural alteration in which the head was rotated and bent towards the left shoulder, which was lower than the right one. That alteration was responsible for breaking muscular and skeletal balance in the frontal plane, thus causing the horizontal planes of both maxillary bones to converge towards the right, as highlighted by the postero-anterior cephalogram[105].
Moebius syndrome (OMIM 157900.): Among 24 patients with classic and incomplete types, 66% presented either micrognathia or retrognathia, 95% showed mandibular hypoplasia, and 75% had a skeletal class II pattern. Maxillary height was increased resulting in a vertical growth pattern. Upper and lower incisors tended towards proclination, and upper and lower lips protruded over cephalometric markings, and a long upper lip was evidenced in 41% of the patients. No statistically significant differences were noted when comparing classic and incomplete Moebius syndrome[106].
Myotonic dystrophy type I (OMIM 160900) congenital or childhood onset: Larger ANB angle values and smaller facial angle values were observed in 15 patients. In the vertical plane, the skeletal planes were hyperdivergent with mandibular plane angle and the intermaxillary angle increased. During a 5-year follow-up period, the intermaxillary angle remained the same in individuals with myotonic dystrophy type I, whereas this angle decreased in healthy individuals[107].
Orofaciodigital syndrome type I, also called Papillon-Léage-Psaume syndrome (OMIM 311200): Two sisters had an apparent dolichocephaly and a leptoprosopic face, a skeletal open bite, and a bimaxillary retrognathism[108].
Rubinstein-Taybi syndrome (OMIM 180849): Most of 8 individuals demonstrated a skeletal class II pattern and brachycephaly[109].
Seckel syndrome (OMIM 210600): 4 siblings (2 girls and 2 boys) had remarkably small skulls with an extremely short anterior cranial base and maxillary length. Differences in the morphology of the Sella turcica were observed between girls and boys[110].
Silver-Russell syndrome (OMIM 180860): Skeletal class II pattern was observed in 18 individuals, class I in 3 patients, and class III in one individual, while almost all patients (n = 21/22) displayed a molar dental class II. All the patients with a skeletal class II pattern had a posteriorly placed mandible with a reduced ramus length and short horizontal branch. Among the patients with class I pattern, 2 presented a reduced length of the maxilla and a posteriorly positioned mandible, and 1 patient presented anterior placement of both the maxilla and the mandible. The sole patient with skeletal class III pattern showed a shorter and posteriorly placed mandible associated with a shorter maxilla. The hyoid bone was in a normal position in 15 cases, accessioned (from 2 to 4 mm up) in 4 cases, and lowered (from 1 to 9 mm down) in 3 cases[111].
Simpson-Golabi-Behmel syndrome (OMIM 312870): A 6-year-old boy and an 8-year-old boy exhibited increased length of the anterior cranial base, and increased length of the maxilla and the mandible[112,113]. The 6-year-old had a class I skeletal pattern and hyperdivergent skeletal planes with increased lower anterior facial height and increased gonial angle[112]. The 8-year-old boy had a skeletal class III pattern, and an increased lower anterior facial height[113].
Solitary median maxillary central incisor syndrome (OMIM 147250): In a case of an 11-year-old girl with panhypopituitarism, a hypoplastic Sella turcica was observed in addition to a skeletal class III pattern with a convex profile, as a result of reduced maxillary length and anteriorly placed mandible. Other findings included hyperdivergency of the skeletal planes, cervical vertebral maturation at cervical stage 2, airway patency, anterior crossbite, upper and lower incisal proclination. No family history for skeletal class III pattern was reported[114].
Treacher-Collins syndrome (OMIM 154500.): Reduced length of both the anterior and posterior cranial base and reduced cranial base angle was observed in 20 patients. The maxilla was posteriorly placed and exhibited reduced length. The mandible was also posteriorly placed with a characteristic reduction of the mandibular length and decreased maximum ramus width. The skeletal planes were hyperdivergent and the gonial angle was increased. Both the anterior and posterior facial heights were decreased, although the lower anterior facial height was increased. The maxillary and functional occlusal planes were tipped upwards posteriorly[115].
Williams-Beuren syndrome, also called Williams syndrome (OMIM 194050): 8 patients had reduced length of the anterior cranial base, although the cranial base angle was normal. The chin was posteriorly placed, although the mandible had a normal position in the antero-posterior plane. The skeletal planes were hyperdivergent, although total anterior facial height was normal. Despite the normal total anterior facial height, there was an unusual proportion of upper to lower anterior facial height and posterior to anterior facial height[116]. In 17 individuals the most affected parameters were those regarding the mandibular incisors[117].
6. Cephalometric Data of Gene Variants Associated with Short Stature
There are several studies assessing specific pathogenic or likely pathogenic genetic variants associated with short stature. To review the literature, a search was conducted in the PubMed electronic database using the following search criteria: cephalometric analysis and short stature; cephalometric analysis and gene variant; cephalometr* and SHOX; cephalometr* and GHR. No studies were found providing cephalometric data in individuals with copy number variations (CNVs) or some variants in genes known to be associated with growth disorders[118]. For example, no studies were found providing cephalometric data in individuals with mutations in the Aggrecan (ACAN) gene located on 15q26, which cause spondyloepimetaphyseal dysplasia aggrecan type (OMIM 612813), spondyloepiphyseal dysplasia type Kimberley (OMIM 608361) and short stature with advanced bone age (OMIM 165800).
6.1. Growth Hormone Receptor Gene (GHR Gene)
GHR gene (5p13.1–p12) codes for the growth hormone receptor protein. Mutations in this gene have been associated with Laron syndrome (OMIM 262500), also known as the growth hormone insensitivity syndrome (GHIS), a disorder characterized by proportional dwarfism. Individuals carrying the CA genotype of the rs6184 single nucleotide polymorphism in GHR gene showed both significantly decreased values for ANB angle (tendency for class III skeletal pattern) and increased mandibular length. No statistically significant differences amongst genotype groups of rs6180 single nucleotide polymorphism were observed. Moreover, the CA genotype of rs6184 SNP and the AA haplotype were highly associated with class III skeletal pattern[119].
6.2. Short Stature Homebox Containing Gene (SHOX Gene)
SHOX gene is located in pseudoautosomal region 1 on the distal end of the short arm of chromosomes Xp22.33 and Yp11.32. SHOX haploinsufficiency can phenotypically express as idiopathic short stature, Leri-Weill dyschondrosteosis (OMIM 127300) or Langer mesomelic dysplasia (OMIM 249700). In a case of a 24-year-old male with Leri-Weill dyschondrosteosis, the lateral cephalogram showed a posteriorly placed maxilla, a posteriorly placed mandible with reduced ramus and body length, increased mandibular angle, and a class III skeletal relation with hyperdivergent skeletal planes[120]. A 17-year-old female with Leri-Weill dyschondrosteosis had an abnormal craniofacial morphology due to craniosynostosis through premature fusion of the squamosal sutures and partial closure of the coronal sutures. The skull calvarium was bulging through the partially synostosed coronal and totally synostosed squamosal sutures[121]. A 10-year-old male with Leri-Weill dyschondrosteosis and congenital conductive loss of hearing had frontal bossing, low nasal bridge, maxilla deficient in both the antero-posterior and vertical plane and a slightly anteriorly placed mandible, with a posteriorly placed chin, resulting in a mesio-relation of the jaws and a negative horizontal overjet of the incisors. The lower anterior facial height was normal[122].
6.3. Natriuretic Peptide Precursor C Gene (NPPC Gene)
NPPC gene resides on chromosome 2q24-qter. Mutations in the gene express as acromesomelic dysplasia Maroteaux type (OMIM 602875) or disproportionate short stature. Cephalometric data exist only in mice with NPPC gene mutations[123]. The craniofacial complex morphology in knockout mice not expressing the NPPC gene and transgenic mice overexpressing the NPPC gene were compared to that of wild type mice. Three dimensional reconstructions of microCT skull images demonstrated reduced skull lengths in the knockout mice. Nose length, nasal bone length and maxilla length were significantly reduced. The mandible was also affected, exhibiting shorter length, but the phenotypic aberration was milder. Transgenic mice exhibited increased cranial length with increased nose length, nasal bone length and maxilla length, but their mandibular length did not differ from wild type mice. Skull width and interorbital distances were normal. The nasal, premaxilla, maxilla, and frontal bones were more markedly affected than the neurocranium, resulting in hypoplasia in knockout mice skulls or hyperplasia in transgenic mice skulls in the sagittal plane. Knockout mice showed mandibular hypoplasia at the center of the condylar process and angular process in the sagittal direction. No difference was noted among the mandibles of wild type and transgenic mice. The skull base was shorter in knockout mice and longer in transgenic mice compared with wild type mice. Both the occipital and sphenoid bones that comprise the skull base were significantly shorter in knockout mice and longer in transgenic mice than those of wild type mice[123].
6.4. Indian Hedgehog Gene (IHH Gene)
Mutations in IHH gene (2q35-36) are responsible for brachydactyly type A1 (OMIM 112500), and acrocapitofemoral dysplasia (OMIM 607778). When 50 female mice were fitted with orthodontic functional appliances placing their mandibles in a more anterior position, the mandibular advancement triggered IHH gene expression in condylar cartilage and increased the replicating mesenchymal cell population[124]. These findings suggest that IHH gene acts as a mediator of mechanotransduction that converts mechanical signals resulting from anterior mandibular displacement to stimulate cellular proliferation in condylar cartilage.
6.5. Fibroblast Growth Factor Receptor 3 Gene (FGFR3 Gene)
The FGFR3 gene (4p16.3) encodes the most significant receptor of fibroblast growth factor, a paracrine signal molecule acting directly upon the growth plate chondrocytes, by binding to its receptors on the chondrocyte cell membrane. FGFR3 bears an extracellular part responsible for binding the ligand and an intracellular part responsible for mediating the intracellular signal transduction through activating Ras/MAPK pathway, which leads to chondrocyte cease of multiplication. The biological significance of this interlude is to give time to the newly differentiating osteoblasts to form new bone and for the biological processes of chondrogenesis and osseogenesis to take place in conjuction.
Mutations in the FGFR3 gene manifest a perfect example of how different mutations in the same gene are expressed as different clinical entities, all belonging to the same clinical spectrum. Activating mutations in FGFR3 gene cause permanent activation of the FGFR3 regardless of the presence of its ligand, resulting in ceasing the chondrogenesis in the growth plate. The exact extent of this growth faltering will be determined by the exact nature of the activating mutation. Different activating mutations in the FGFR3 gene may cause achondroplasia (OMIM 100800), thanatophoric dysplasia type I (OMIM 187600)- which is a short-limb dwarfism syndrome usually lethal in the perinatal period-, as well as hypochondroplasia (OMIM 146000) with clinical features including short-limbed dwarfism, lumbar lordosis, short and broad bones and caudal narrowing of interpediculate distance of lumbar spine. Lastly, a less significant activating mutation in the FGFR3 gene causes moderately short height with relative macrocephaly for height, a clinical entity that may be considered as idiopathic short stature.
One individual with posteriorly placed maxillae, diagnosed through a lateral cephalometric radiograph, had two mutations (A213G and A223G ) in exon 3 of the FGFR3 gene in compound heterozygosity[125]. 6 patients with non-syndromic isolated sagittal synostosis had a common FGFR3 polymorphism 294C>T (Asn294Asn). The intracranial volumes were assessed with 3D CT but no statistical difference was found between them and those of healthy individuals[126]. Among 8 patients with synostotic frontal plagiocephaly (unilateral coronal synostosis), 3 patients had FGFR3 Pro250Arg mutation, 2 patients had mutations in FGFR2 gene and 3 patients in Twist gene. Two features were strongly associated with a detectable mutation in all patients with synostotic frontal plagiocephaly: asymmetrical brachycephaly (retrusion of both supraorbital rims) and orbital hypertelorism. Other abnormalities in the craniofacial region and extremities were clues to a particular mutation in FGFR2, FGFR3, or Twist genes. Neither macrocephaly nor degree of nasal angulation, nor relative vertical position of the lateral canthi correlated with mutational detection[127].
6.6. Parathyroid Hormone-Like Hormone (PTHLH) or Parathyroid Hormone-Related Protein (PTHrP)
Mutations in the PTHLH gene (12p11.22) cause brachydactyly type E (OMIM 613382), an autosomal dominant syndrome characterized by short stature, brachydactyly and learning difficulties. In an experimental animal model, it was observed that higher levels of PTHrP expression coincided with the slowing of chondrocyte hypertrophy[128]. Mandibular advancement in rats increased the number of differentiated chondroblasts and triggered PTHrP expression, which retarded their further maturation to allow for more growth and subsequent increase of the cartilage volume.
Discussion
Disruptions of the biological factors regulating the molecular events taking place at the epiphyseal growth plate during the developmental time period can result in stature abnormalities[129]. Short stature is characterized as either primary or secondary when a recognizable genetic or an endocrine disease is responsible for its occurrence[1,130]. Sometimes, despite the meticulously carried out diagnostic work-up no underlying cause can be unveiled, and, thus, short stature is characterized as idiopathic (ISS)[130]. It is possible that ISS constitutes a manifestation of a milder phenotypic expression of some known genetic or endocrine disease presenting with very few or even without any clinical findings and, thus, remains undiagnosed[129]. It would be of great clinical importance if there was a diagnostic tool capable of recognizing the underlying cause in such borderline cases where the physical findings are normal or close to normal.
Little is known about the correlation of the standing body height with the quantitative extend of growth of the craniofacial complex, and even less is known about the changes that are induced in the craniofacial complex in individuals with genetic conditions that cause short stature. If such a relation is established, then specific causes of short stature could be attributed to specific craniofacial abnormalities. A useful, inexpensive, widely available, and daily utilized diagnostic tool for assessing the craniofacial complex is the cephalometric radiograph, which may quantify the degree of oral and maxillofacial growth retardation. The assessment of the cephalometric radiograph requires an experienced general dentist, a specialized orthodontist or an oral and maxillofacial surgeon and this constitutes a disadvantage of the method. If each genetic cause of short stature presents with a specific pattern of cephalometric measurements aberrations, then the cephalometric radiograph could be used in guiding the further genetic testing or even in setting the diagnosis on its own.
Cephalometric evaluation of children with reduced stature due to genetic causes demonstrates that these individuals exhibit abnormal growth of the craniofacial complex[131]. Craniofacial growth seems to follow the linear somatic retardation, with the individuals exhibiting reduced linear measurements and abnormal angular measurements when compared to the reference values of normal individuals. Both the maxilla and the mandible are shorter and posteriorly placed and their antero-posterior arrangement results in skeletal class II pattern and a retrognathic profile of the face. The angular measurements in the vertical plane constitute the skeletal planes as hyperdivergent and imply a posterior/downward inclination of the mandible with a vertical growth pattern of the face, reduced posterior facial height and increased anterior facial height.
Some syndromes present with characteristic aberrations in the cephalometric measurements, whereas other genetic disorders present with similar and non-distinguishable pathologic craniofacial characteristics. For instance, in individuals with Turner syndrome, the most commonly affected craniofacial structure is the cranial base, with the posterior cranial base exhibiting reduced length and the cranial base angle being increased (increased angle of flexion) causing a flattening of the cranial base, while in individuals with achondroplasia, the most prominent manifestation is the significantly reduced posterior cranial base length and the acute cranial base angle, while milder phenotypes fall between these two conditions[24,25,35]. Thus, if studies could propose a cut-off value for the cranial base angle above which the diagnosis of Turner syndrome is probable and below which the diagnosis of achondroplasia is probable, enabling clinicians to further expand these measurements in the diagnosis of other genetic disorders solely based on their craniofacial phenotype.
Interestingly, the greater part of the existing literature regarding cephalometric measurements in short statured children with genetic syndromes provide qualitative data, whereas quantitative data are required to potentially establish the above-mentioned cut-off values. Moreover, the literature is scarce offering a few scientific papers providing cephalometric data for individuals affected with rare genetic conditions causing short stature. Consequently, more studies providing quantitative cephalometric data in large populations of affected individuals need to be conducted.
Funding Statement
This research did not receive any funding.
Author Contributions
G.P. conceived the study, researched the literature and authored the original draft of the manuscript. N.Z. researched the literature, designed the figures and authored the original draft of the manuscript. G.P.C. thoroughly reviewed and revised the manuscript. C.Y. designed and supervised the study, as well as authored the final draft of the manuscript. All authors have reviewed and consented to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article. Please direct any additional inquiries to the corresponding author.
Conflicts of Interest
It is declared by the authors that they have no conflicts of interest.
References
- Wit, J.M.; Oostdijk, W.; Losekoot, M.; Van Duyvenvoorde, H.A.; Ruivenkamp, C.A.L.; Kant, S.G. MECHANISMS IN ENDOCRINOLOGY: Novel Genetic Causes of Short Stature. Eur J Endocrinol 2016, 174, R145–R173. [Google Scholar] [CrossRef] [PubMed]
- Paltoglou, G.; Dimitropoulos, I.; Kourlaba, G.; Charmandari, E. The Effect of Treatment with Recombinant Human Growth Hormone (RhGH) on Linear Growth and Adult Height in Children with Idiopathic Short Stature (ISS): A Systematic Review and Meta-Analysis. Journal of Pediatric Endocrinology and Metabolism 2020, 33, 1577–1588. [Google Scholar] [CrossRef]
- Rogol, A.D.; Hayden, G.F. Etiologies and Early Diagnosis of Short Stature and Growth Failure in Children and Adolescents. J Pediatr 2014, 164. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.G.; Dattani, M.T.; Clayton, P.E. Controversies in the Diagnosis and Management of Growth Hormone Deficiency in Childhood and Adolescence. Arch Dis Child 2016, 101, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Grummer-Strawn, L.M.; Reinold, C.; Krebs, N.F. Centers for Disease Control and Prevention (CDC) Use of World Health Organization and CDC Growth Charts for Children Aged 0-59 Months in the United States. MMWR Recomm Rep 2010, 59, 1–15. [Google Scholar] [PubMed]
- Rogol, A.D. Children with Asymptomatic Short Stature: What Is an Appropriate Evaluation? J Pediatr 2013, 163, 937–938. [Google Scholar] [CrossRef] [PubMed]
- Height and Weight of Children of Hellenic Origin Aged 0-18 Years (2000-2001): Comparison with Data Collected during the Period 1978-1979 | Request PDF. Available online: https://www.researchgate.net/publication/284813940_Height_and_weight_of_children_of_Hellenic_origin_aged_0-18_years_2000-2001_Comparison_with_data_collected_during_the_period_1978-1979 (accessed on 30 April 2024).
- Wit, J.M.; Charmian Quigley, by A.; Ranke, M.B. International Classification of Pediatric Endocrine Diagnoses. Horm Res Paediatr 2016, 86, 212–214. [CrossRef]
- Wood, A.R.; Esko, T.; Yang, J.; Vedantam, S.; Pers, T.H.; Gustafsson, S.; Chu, A.Y.; Estrada, K.; Luan, J.; Kutalik, Z.; et al. Defining the Role of Common Variation in the Genomic and Biological Architecture of Adult Human Height. Nat Genet 2014, 46, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Powls, A.; Botting, N.; Cooke, R.W.; Pilling, D.; Marlow, N. Growth Impairment in Very Low Birthweight Children at 12 Years: Correlation with Perinatal and Outcome Variables. Arch Dis Child Fetal Neonatal Ed 1996, 75, F152–F157. [Google Scholar] [CrossRef]
- Saunders, C.L.; Lejarraga, H.; Del Pino, M. Assessment of Head Size Adjusted for Height: An Anthropometric Tool for Clinical Use Based on Argentinian Data. Ann Hum Biol 2006, 33, 415–423. [Google Scholar] [CrossRef]
- Ranke, M.B. Towards a Consensus on the Definition of Idiopathic Short Stature. Horm Res 1996, 45, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B. Short Stature and Altered Body Proportions. Paediatric Orthopaedic Diagnosis 2015, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Del Pino, M.; Orden, A.B.; Arenas, M.A.; Fano, V. Argentine References for the Assessment of Body Proportions from Birth to 17 Years of Age. Arch Argent Pediatr 2017, 115, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Duncan, W.J.; Fowler, R.S.; Farkas, L.G.; Ross, R.B.; Wright, A.W.; Bloom, K.R.; Huot, D.J.; Sondheimer, H.M.; Rowe, R.D.; Opitz, J.M. A Comprehensive Scoring System for Evaluating Noonan Syndrome. Am J Med Genet 1981, 10, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Coi, A.; Santoro, M.; Garne, E.; Pierini, A.; Addor, M.C.; Alessandri, J.L.; Bergman, J.E.H.; Bianchi, F.; Boban, L.; Braz, P.; et al. Epidemiology of Achondroplasia: A Population-Based Study in Europe. Am J Med Genet A 2019, 179, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Pineau, M.; Farrow, E.; Nicot, R.; Ferri, J. Achondroplasia: Orocraniofacial Features and Orthodontic-Surgical Management Guidelines Proposal. Journal of Craniofacial Surgery 2018, 29, 2186–2191. [Google Scholar] [CrossRef] [PubMed]
- Mm, C.; Gf, W.; Phillips, C. A Morphometric Analysis of the Craniofacial Configuration in Achondroplasia. J Craniofac Genet Dev Biol Suppl 1985. [Google Scholar]
- Celenk, P.; Arici, S.; Celenk, C. Oral Findings in a Typical Case of Achondroplasia. 2003; 31, 236–238. [Google Scholar] [CrossRef]
- Shinohara, M.; Funakoshi, Y.; Takaishi, Y.; Hieda, T. [A Case Report on the Achondroplasia and Its Dental Findings]. Shoni Shikagaku Zasshi 1991, 29, 159–166. [Google Scholar] [PubMed]
- Bloom, M.W.; Murakami, S.; Cody, D.; Montufar-Solis, D.; Duke, P.J. Aspects of Achondroplasia in the Skulls of Dwarf Transgenic Mice: A Cephalometric Study. Anat Rec A Discov Mol Cell Evol Biol 2006, 288, 316–322. [Google Scholar] [CrossRef]
- Doswell, B.H.; Visootsak, J.; Brady, A.N.; Graham, J.M. Turner Syndrome: An Update and Review for the Primary Pediatrician. Clin Pediatr (Phila) 2006, 45, 301–313. [Google Scholar] [CrossRef]
- Tecuta-Busoi, A.-I.; Matei, M.; Florescu, L.M.; Gheonea, I.A. Developmental Abnormalities of the Skull Base in Patients with Turner Syndrome. Curr Health Sci J 46, 329–335. [CrossRef] [PubMed]
- Rizell, S. Dentofacial Morphology in Turner Syndrome Karyotypes. Swed Dent J Suppl 2012, 7–98. [Google Scholar]
- Rizell, S.; Barrenas, M.-L.; Andlin-Sobocki, A.; Stecksen-Blicks, C.; Kjellberg, H. 45,X/46,XX Karyotype Mitigates the Aberrant Craniofacial Morphology in Turner Syndrome. The European Journal of Orthodontics 2013, 35, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Perkiomaki, M.R. The Relationship of Distinct Craniofacial Features between Turner Syndrome Females and Their Parents. The European Journal of Orthodontics 2005, 27, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Eklund, M.; Kotilainen, J.; Evälahti, M.; Waltimo-Sirén, J. Cephalometric Analysis of Pharyngeal Airway Space Dimensions in Turner Syndrome. Eur J Orthod 2012, 34, 219–225. [Google Scholar] [CrossRef]
- Dumancic, J.; Kaic, Z.; Varga, M.L.; Lauc, T.; Dumic, M.; Milosevic, S.A.; Brkic, H. Characteristics of the Craniofacial Complex in Turner Syndrome. Arch Oral Biol 2010, 55, 81–88. [Google Scholar] [CrossRef]
- Andersen, E.; Sonnesen, L.; Kjaer, M.S.; Fischer Hansen, B.; Kjær, I. The Prenatal Cranial Base Complex and Hand in Turner Syndrome. Eur J Orthod 2000, 22, 185–194. [Google Scholar] [CrossRef]
- Simmons, K.E. Growth Hormone and Craniofacial Changes: Preliminary Data from Studies in Turner’s Syndrome. Pediatrics 1999, 104, 1021–1024. [Google Scholar] [CrossRef]
- Hass, A.D.; Simmons, K.E.; Davenport, M.L.; Proffit, W.R. The Effect of Growth Hormone on Craniofacial Growth and Dental Maturation in Turner Syndrome. Angle Orthod 2001, 71, 50–59. [Google Scholar] [CrossRef]
- Corvo, G.; Tartaro, G.P.; Stoppoloni, F.; Balzano, G. [Cephalometric Evaluation of Patients with Turner Syndrome. Authors’ Experience]. Minerva Stomatol 1998, 47, 127–133. [Google Scholar]
- Rongen-Westerlaken, C.; vd Born, E.; Prahl-Andersen, B.; Rikken, B.; Teunenbroek, V.; Kamminga, N.; vd Tweel, I.; Otten, B.J.; Delamarre vd Waal, H.A.; Drayer, N.M. Shape of the Craniofacial Complex in Children with Turner Syndrome. J Biol Buccale 1992, 20, 185–190. [Google Scholar] [PubMed]
- Rongen-Westerlaken, C.; Born, E. vd; Prahl-Andersen, B.; Teunenbroek, A. v; Manesse, P.; Otten, B.; Tweel, I. vd; Kuijpers-Jagtman, A.; Delemarre vd Waal, H.; Drayer, N.; et al. Effect of Growth Hormone Treatment on Craniofacial Growth in Turner’s Syndrome. Acta Paediatr 1993, 82, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Midtbo, M.; Wisth, P.J.; Halse, A. Craniofacial Morphology in Young Patients With Turner Syndrome. The European Journal of Orthodontics 1996, 18, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Peltomäki, T.; Alvesalo, L.; Isotupa, K. Shape of the Craniofacial Complex in 45,X Females: Cephalometric Study. J Craniofac Genet Dev Biol 1989, 9, 331–338. [Google Scholar]
- Jensen, B.L. Craniofacial Morphology in Turner Syndrome. J Craniofac Genet Dev Biol 1985, 5, 327–340. [Google Scholar]
- Rzymski, K.; Kosowicz, J. Abnormal Basal Angle of the Skull in Sex Chromosome Aberrations. Acta Radiol Diagn (Stockh) 1976, 17, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Rzymski, K.; Kosowicz, J. The Skull in Gonadal Dysgenesis a Roentgenometric Study. Clin Radiol 1975, 26, 379–384. [Google Scholar] [CrossRef]
- Park, J.H.; Tai, K.; Sato, Y. Management of Klippel-Feil Syndrome Combined with Turner Syndrome: A Case Report. Int J Orthod Milwaukee 2013, 24, 37–42. [Google Scholar] [PubMed]
- Horowitz, S.L.; Morishima, A.; Vinkka, H. The Position of the External Ear in Turner’s Syndrome. Clin Genet 1976, 9, 333–340. [Google Scholar] [CrossRef]
- Weiss, E.; Loevy, H.; Saunders, A.; Pruzansky, S.; Rosenthal, I.M.; Opitz, J.M. Monozygotic Twins Discordant for Ullrich-Turner Syndrome. Am J Med Genet 1982, 13, 389–399. [Google Scholar] [CrossRef]
- FILIPSSON, R.; LINDSTEN, J.; ALMQVIST, S. Time of Eruption of The Permanent Teeth, Cephalometric and Tooth Measurement and Sulphation Factor Activity in 45 Patients with Turner’s Syndrome with Different Types of X Chromosome Aberrations. Acta Endocrinol (Copenh) 1965, 48, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Ahiko, N.; Baba, Y.; Tsuji, M.; Horikawa, R.; Moriyama, K. Investigation of Maxillofacial Morphology and Oral Characteristics with Turner Syndrome and Early Mixed Dentition. Congenit Anom (Kyoto) 2019, 59, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Cazzolla, A.P.; Lo Muzio, L.; Di Fede, O.; Lacarbonara, V.; Colaprico, A.; Testa, N.F.; Giuseppe, T.; Zhurakivska, K.; Marzo, G.; Lacaita, M.G. Orthopedic-orthodontic Treatment of the Patient with Turner’s Syndrome: Review of the Literature and Case Report. Special Care in Dentistry 2018, 38, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Aristizábal, J.F.; Smit, R.M. Two-Phase Orthodontic Treatment in a Patient with Turner Syndrome: An Unusual Case of Deep Bite. The Cleft Palate-Craniofacial Journal 2015, 52, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Russell, K.A. Orthodontic Treatment for Patients with Turner Syndrome. American Journal of Orthodontics and Dentofacial Orthopedics 2001, 120, 314–322. [Google Scholar] [CrossRef] [PubMed]
- OGIUCHI, H.; TAKANO, K.; TANAKA, M.; HIZUKA, N.; TAKAGI, S.; SANGU, Y.; SHIZUME, K.; KAWANISHI, I. Oro-Maxillofacial Development in Patients with Turner’s Syndrome. Endocrinol Jpn 1985, 32, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Svanberg, C.; Norevall, L.-I.; Ekman, B.; Wahlberg, J.; Bågesund, M. Cephalometric Analysis of Adults with Turner Syndrome. Swed Dent J 2016, 40, 33–41. [Google Scholar]
- Takeyama, H.; Honzawa, O.; Hozaki, T.; Kiyomura, H. A Case of Open Bite with Turner’s Syndrome. American Journal of Orthodontics and Dentofacial Orthopedics 1990, 97, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J.; Redman, R.S.; Shapiro, B.L. Effect of X-Chromosome Aneuploidy on Jaw Growth. J Dent Res 1965, 44, 269–282. [Google Scholar] [CrossRef]
- Bagattoni, A.; Lardani, L.; Vanni, A.; Costi, T. Craniofacial and Occlusal Features of Individuals with Turner Syndrome: A Cephalometric Study. J Biol Regul Homeost Agents 2021. [Google Scholar] [CrossRef]
- Juloski, J.; Dumančić, J.; Šćepan, I.; Lauc, T.; Milašin, J.; Kaić, Z.; Dumić, M.; Babić, M. Growth Hormone Positive Effects on Craniofacial Complex in Turner Syndrome. Arch Oral Biol 2016, 71, 10–15. [Google Scholar] [CrossRef]
- Juloski, J.; Glisic, B.; Scepan, I.; Milasin, J.; Mitrovic, K.; Babic, M. Ontogenetic Changes of Craniofacial Complex in Turner Syndrome Patients Treated with Growth Hormone. Clin Oral Investig 2013, 17, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Babic, M.; Glisic, B.; Scepan, I. Mandibular Growth Pattern in Turner’s Syndrome. The European Journal of Orthodontics 1997, 19, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Perkiomaki, M.R.; Alvesalo, L. Palatine Ridges and Tongue Position in Turner Syndrome Subjects. The European Journal of Orthodontics 2008, 30, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Bhambhani, V.; Muenke, M. Noonan Syndrome. Am Fam Physician 2014, 89, 37–43. [Google Scholar] [PubMed]
- Miyamoto, J.J.; Yabunaka, T.; Moriyama, K. Cervical Characteristics of Noonan Syndrome. The European Journal of Orthodontics 2014, 36, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Ranke, M.B.; Heidemann, P.; Knupfer, C.; Enders, H.; Schmaltz, A.A.; Bierich, J.R. Noonan Syndrome: Growth and Clinical Manifestations in 144 Cases. Eur J Pediatr 1988, 148, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Chatzistavrou, E.; Andreadis, G. Noonan Syndrome in 12-Year-Old Male: Case Report and Orthodontic Management of the Occlusion. Balkan Journal of Dental Medicine 2020, 24, 118–126. [Google Scholar] [CrossRef]
- Kawakami, M.; Yamamoto, K.; Shimomura, T.; Kirita, T. Surgical Orthodontic Treatment for Open Bite in Noonan Syndrome Patient: A Case Report. The Cleft Palate-Craniofacial Journal 2016, 53, 253–258. [Google Scholar] [CrossRef]
- Emral, M.E.; Akcam, M.O. Noonan Syndrome: A Case Report. J Oral Sci 2009, 51, 301–306. [Google Scholar] [CrossRef]
- Bagattoni, S.; Costi, T.; D’Alessandro, G.; Toni, S.; Gatto, M.R.; Piana, G. Craniofacial and Occlusal Features of Children with Noonan Syndrome. Am J Med Genet A 2021, 185, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Cardiel Ríos, S.A. Correction of a Severe Class II Malocclusion in a Patient with Noonan Syndrome. American Journal of Orthodontics and Dentofacial Orthopedics 2016, 150, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Al Qabbani Pedodontist, H.; Al Habib hospital, S. Noonan Syndrome: A Case Report. International Journal of Oral Health Dentistry 2017, 3, 238–239. [Google Scholar] [CrossRef]
- Okada, M.; Sasaki, N.; Kaihara, Y.; Okada, R.; Amano, H.; Miura, K.; Kozai, K. Oral Findings in Noonan Syndrome: Report of a Case. J Oral Sci 2003, 45, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Neto, L.A.; Melo, M.F.B.; Franco, A.A.; Oliveira, A.H.A.; Souza, A.H.O.; Valença, E.H.O.; Britto, I.M.P.A.; Salvatori, R.; Aguiar-Oliveira, M.H. Cephalometric Features in Isolated Growth Hormone Deficiency. Angle Orthod 2011, 81, 578–583. [Google Scholar] [CrossRef]
- Kjellberg, H.; Wikland, K.A. A Longitudinal Study of Craniofacial Growth in Idiopathic Short Stature and Growth Hormone-Deficient Boys Treated with Growth Hormone. The European Journal of Orthodontics 2007, 29, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Funatsu, M.; Sato, K.; Mitani, H. Effects of Growth Hormone on Craniofacial Growth. Angle Orthod 2006, 76, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, G.; Stenehjem, J.S.; Axelsson, S.; Saeves, R. Craniofacial and Dentoalveolar Morphology in Individuals with Prader–Willi Syndrome: A Case-Control Study. Orphanet J Rare Dis 2022, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Giuca, M.R.; Inglese, R.; Caruso, S.; Gatto, R.; Marzo, G.; Pasini, M. Craniofacial Morphology in Pediatric Patients with Prader–Willi Syndrome: A Retrospective Study. Orthod Craniofac Res 2016, 19, 216–221. [Google Scholar] [CrossRef]
- Schaedel, R.; Poole, A.E.; Cassidy, S.B. Cephalometric Analysis of the Prader-Willi Syndrome. Am J Med Genet 1990, 36, 484–487. [Google Scholar] [CrossRef]
- Gopinath, T.; Ganesh, S.; Subramani, V. Role of Facial Index and Odontometric Parameters in the Establishment of Stature and Gender of Individuals. J Pharm Bioallied Sci 2021, 13, 1068. [Google Scholar] [CrossRef] [PubMed]
- Jesuino, F.A.S.; Valladares-Neto, J. Craniofacial Morphological Differences between Down Syndrome and Maxillary Deficiency Children. The European Journal of Orthodontics 2013, 35, 124–130. [Google Scholar] [CrossRef] [PubMed]
- van Marrewijk, D.J.F.; van Stiphout, M.A.E.; Reuland-Bosma, W.; Bronkhorst, E.M.; Ongkosuwito, E.M. The Relationship between Craniofacial Development and Hypodontia in Patients with Down Syndrome. The European Journal of Orthodontics 2016, 38, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, J.; Biedma, B.; Rodríguez, M.; Mora, M.; Cunqueiro, M.; Pazos, M. Cephalometrics in Children with Down’s Syndrome. Pediatr Radiol 2002, 32, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Faruqui, S.; Fida, M.; Shaikh, A. Cervical Vertebral Anomalies in Skeletal Malocclusions: A Cross-Sectional Study on Orthodontic Patients at the Aga Khan University Hospital, Pakistan. Indian Journal of Dental Research 2014, 25, 480. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, E.B.; Wu, J.K.; Sullivan, S.R.; Vasudavan, S.; Padwa, B.L.; Rogers, G.F.; Mulliken, J.B. Craniofacial Growth in Patients With FGFR3Pro250Arg Mutation After Fronto-Orbital Advancement in Infancy. Journal of Craniofacial Surgery 2011, 22, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Kidwai, F.K.; Mui, B.W.H.; Almpani, K.; Jani, P.; Keyvanfar, C.; Iqbal, K.; Paravastu, S.S.; Arora, D.; Orzechowski, P.; Merling, R.K.; et al. Quantitative Craniofacial Analysis and Generation of Human Induced Pluripotent Stem Cells for Muenke Syndrome: A Case Report. J Dev Biol 2021, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, E.; Eulert, S.; Bill, J.; Würzler, K.; Phan The, L.; Reuther, J. Typische Merkmale Des Kraniofazialen Wachstums Beim FGFR3-Assoziierten Koronarnahtsynostosesyndrom (so Genannte Muenke-Kraniosynostose). Mund-, Kiefer- und Gesichtschirurgie 2003, 7, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Muenke, M.; Gripp, K.W.; McDonald-McGinn, D.M.; Gaudenz, K.; Whitaker, L.A.; Bartlett, S.P.; Markowitz, R.I.; Robin, N.H.; Nwokoro, N.; Mulvihill, J.J.; et al. A Unique Point Mutation in the Fibroblast Growth Factor Receptor 3 Gene (FGFR3) Defines a New Craniosynostosis Syndrome. Am J Hum Genet 1997, 60, 555–564. [Google Scholar]
- Samra, F.; Bauder, A.R.; Swanson, J.W.; Whitaker, L.A.; Bartlett, S.P.; Taylor, J.A. Assessing the Midface in Muenke Syndrome: A Cephalometric Analysis and Review of the Literature. Journal of Plastic, Reconstructive & Aesthetic Surgery 2016, 69, 1285–1290. [Google Scholar] [CrossRef]
- Arnaud, E.; Meneses, P.; Lajeunie, E.; Thorne, J.A.; Marchac, D.; Renier, D. Postoperative Mental and Morphological Outcome for Nonsyndromic Brachycephaly. Plast Reconstr Surg 2002, 110, 6–12. [Google Scholar] [CrossRef]
- Lewyllie, A.; Roosenboom, J.; Indencleef, K.; Claes, P.; Swillen, A.; Devriendt, K.; Carels, C.; Cadenas De Llano-Pérula, M.; Willems, G.; Hens, G.; et al. A Comprehensive Craniofacial Study of 22q11.2 Deletion Syndrome. J Dent Res 2017, 96, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Lia, E.N.; Otero, S.A.M.; Ferraz, M.; Gonçalves, L.P.V. Oral Aspects of 49, XXXXY Syndrome: A Case Report. J Dent Child (Chic) 2007, 74, 136–139. [Google Scholar]
- Wulfsberg, E.A.; Campbell, A.B.; Lurie, I.W.; Eanet, K.R. Confirmation of the Catania Brachydactylous Type of Acrofacial Dysostosis: Report of a Second Family. Am J Med Genet 1996, 63, 554–557. [Google Scholar] [CrossRef]
- Defabianis, P.; Mussa, A.; Ninivaggi, R.; Carli, D.; Romano, F. Maxillo-Facial Morphology in Beckwith-Wiedemann Syndrome: A Preliminary Study on (Epi)Genotype-Phenotype Association in Caucasians. Int J Environ Res Public Health 2022, 19. [Google Scholar] [CrossRef]
- Nojima, K.; Onoda, M.; Nishii, Y.; Sueishi, K. Orthodontic Treatment for Bloch-Sulzberger Syndrome in Patient with Cleft Lip and Palate. Bull Tokyo Dent Coll 2017, 58, 259–267. [Google Scholar] [CrossRef]
- Bender, C. V.; da Silveira, H.L.D.; dos Santos, N.S.; Cavagni, J.; Rados, P. V.; John, A.B.; De Souza, C.F.M.; Giugliani, R.; Visioli, F. Oral, Dental, and Craniofacial Features in Chronic Acid Sphingomyelinase Deficiency. Am J Med Genet A 2020, 182, 2891–2901. [Google Scholar] [CrossRef] [PubMed]
- Bloch-Zupan, A.; Rousseaux, M.; Lauge, V.; Schmittbuhl, M.; Mathis, R.; Desforges, E.; Koob, M.; Zaloszyc, A.; Dollfus, H.; Laugel, V. A Possible Cranio-Oro-Facial Phenotype in Cockayne Syndrome. Orphanet J Rare Dis 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Yapijakis, C.; Pachis, N.; Sotiriadou, T.; Vaila, C.; Michopoulou, V.; Vassiliou, S. Molecular Mechanisms Involved in Craniosynostosis. In Vivo 2023, 37, 36–46. [Google Scholar] [CrossRef]
- Yang, I.-H.; Chung, J.H.; Cho, I.-S.; Kim, S.; Baek, S.-H. Effect of Early Spheno-Occipital Synchondrosis Fusion in Preadolescent Patients With Syndromic Craniosynostosis on Craniofacial Skeletal Patterns: A Preliminary Study Using Cephalometric Analysis. J Craniofac Surg, 33, 179–182. [CrossRef]
- Wang, H.; Hung, K.; Zhao, K.; Wang, Y.; Wang, F.; Wu, Y. Anatomical Analysis of Zygomatic Bone in Ectodermal Dysplasia Patients with Oligodontia. Clin Implant Dent Relat Res 2019, 21, 310. [Google Scholar] [CrossRef]
- Nakayama, Y.; Baba, Y.; Tsuji, M.; Fukuoka, H.; Ogawa, T.; Ohkuma, M.; Moriyama, K. Dentomaxillofacial Characteristics of Ectodermal Dysplasia. Congenit Anom (Kyoto) 2015, 55, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.C.; Kyrkanides, S. Cephalometric Analysis of Rapp-Hodgkin Syndrome. J Med Genet 1994, 31, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Tuna, E.B.; Koruyucu, M.; Kürklü, E.; Çifter, M.; Gençay, K.; Seymen, F.; Tüysüz, B. Oral and Craniofacial Manifestations of Ellis–van Creveld Syndrome: Case Series. Journal of Cranio-Maxillofacial Surgery 2016, 44, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Danaei, S.M.; Karamifar, A.; Sardarian, A.; Shahidi, S.; Karamifar, H.; Alipour, A.; Ghodsi Boushehri, S. Measuring Agreement between Cervical Vertebrae and Hand-Wrist Maturation in Determining Skeletal Age: Reassessing the Theory in Patients with Short Stature. American Journal of Orthodontics and Dentofacial Orthopedics 2014, 146, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Bazopoulou-Kyrkanidou, E.; Vrahopoulos, T.P.; Eliades, G.; Vastardis, H.; Tosios, K.; Vrotsos, I.A. Periodontitis Associated With Hajdu-Cheney Syndrome. J Periodontol 2007, 78, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Bahar Tuna, E.; Sulun, T.; Rosti, O.; El Abdallah, F.; Kayserili, H.; Aktoren, O. Craniodentofacial Manifestations In Hallermann-Streiff Syndrome. CRANIO® 2009, 27, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Tokura, T.; Miyazaki, A.; Igarashi, T.; Dehari, H.; Kobayashi, J.; Miki, Y.; Ogi, K.; Sonoda, T.; Yotsuyanagi, T.; Hiratsuka, H. Quantitative Evaluation of Cephalometric Radiographs of Patients With Hemifacial Microsomia. The Cleft Palate-Craniofacial Journal 2019, 56, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Tuna, E.; Marşan, G.; Gençay, K.; Seymen, F. Craniofacial and Dental Characteristics of Kabuki Syndrome: Nine Years Cephalometric Follow-Up. Journal of Clinical Pediatric Dentistry 2012, 36, 393–400. [Google Scholar] [CrossRef]
- Ozdiler, E.; Akcam, M.O.; Sayin, M.O. Craniofacial Characteristics of Klippel-Feil Syndrome in an Eight Year Old Female. J Clin Pediatr Dent 2000, 24, 249–254. [Google Scholar]
- Katge, F.A.; Rusawat, B.D.; Shivasharan, P.R.; Patil, D.P. Langer-Giedion Syndrome: A Rare Case Report. J Dent (Shiraz) 2016, 17, 238–241. [Google Scholar]
- Yasunaga, M.; Ishikawa, H.; Yanagita, K.; Tamaoki, S. An Orthodontic Perspective on Larsen Syndrome. BMC Oral Health 2021, 21, 1–8. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Tagariello, T.; Piana, G. Oral and Craniofacial Findings in a Patient with Methylmalonic Aciduria and Homocystinuria: Review and a Case Report. Minerva Stomatol 2010, 59, 129–137. [Google Scholar]
- Telich-Tarriba, J.E.; Amador-Lara, A.; Quiroz-Barrios, J.; Cardenas-Mejia, A. Cephalometric Analysis of the Craniofacial Morphology in Patients With Moebius Syndrome. Journal of Craniofacial Surgery 2021, 32, 2446–2448. [Google Scholar] [CrossRef]
- Fontinha, C.; Engvall, M.; Sjögreen, L.; Kiliaridis, S. Craniofacial Morphology and Growth in Young Patients with Congenital or Childhood Onset Myotonic Dystrophy. Eur J Orthod 2018, 40, 544–548. [Google Scholar] [CrossRef]
- Romero, M.; Franco, B.; del Pozo, J.S.; Romance, A. Buccal Anomalies, Cephalometric Analysis and Genetic Study of Two Sisters with Orofaciodigital Syndrome Type I. The Cleft Palate-Craniofacial Journal 2007, 44, 660–666. [Google Scholar] [CrossRef]
- Martins, F.; Roussen, A.C.; Rezende, N.; Hiraoka, C.; Zamunaro, M.; Gallottini, M. Oral and Cephalometric Study in Brazilian Rubinstein–Taybi Syndrome Patients. Special Care in Dentistry 2022, 42, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Kjær, I.; Hansen, N.; Becktor, K.B.; Birkebæk, N.; Balslev, T. Craniofacial Morphology, Dentition, and Skeletal Maturity in Four Siblings with Seckel Syndrome. The Cleft Palate-Craniofacial Journal 2001, 38, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Vo Quang, S.; Galliani, E.; Eche, S.; Tomat, C.; Fauroux, B.; Picard, A.; Kadlub, N. Contribution of a Better Maxillofacial Phenotype in Silver–Russell Syndrome to Define a Better Orthodontics and Surgical Management. J Stomatol Oral Maxillofac Surg 2019, 120, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, T.; Kitai, N.; Iguchi, Y.; Murakami, S.; Yanagi, M.; Takada, K. Craniofacial Morphology in a Patient with Simpson-Golabi-Behmel Syndrome. The Cleft Palate-Craniofacial Journal 2003, 40, 550–555. [Google Scholar] [CrossRef]
- Bayram, M.; Yildirim, M.; Seymen, F. Clinical and Oral Findings of a Patient with Simpson–Golabi–Behmel Syndrome. European Archives of Paediatric Dentistry 2015, 16, 63–66. [Google Scholar] [CrossRef]
- Nota, A.; Ehsani, S.; Pittari, L.; Gastaldi, G.; Tecco, S. Rare Case of Skeletal Third Class in a Subject Suffering from Solitary Median Maxillary Central Incisor Syndrome (SMMCI) Associated to Panhypopituitarism. Head Face Med 2021, 17, 49. [Google Scholar] [CrossRef]
- Kapadia, H.; Shetye, P.R.; Grayson, B.H.; McCarthy, J.G. Cephalometric Assessment of Craniofacial Morphology in Patients With Treacher Collins Syndrome. Journal of Craniofacial Surgery 2013, 24, 1141–1145. [Google Scholar] [CrossRef]
- Mass, E.; Belostoky, L. Craniofacial Morphology of Children with Williams Syndrome. Cleft Palate Craniofac J 1993, 30, 343–349. [Google Scholar] [CrossRef]
- Danneels, F.; Verdonck, A.; Indencleef, K.; Declerck, D.; Willems, G.; Cadenas De Llano-Pérula, M. Determination of Craniofacial and Dental Characteristics of Individuals with Williams-Beuren Syndrome by Using 3D Facial Scans and Radiographs. Orthod Craniofac Res 2022, 25, 359–367. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, Y.; Shao, Q.; Zhang, C.; Kou, S.; Yang, W.; Zhang, M.; Ban, B. Clinical and Genetic Evaluation of Children with Short Stature of Unknown Origin. BMC Med Genomics 2023, 16, 1–13. [Google Scholar] [CrossRef]
- Tobón-Arroyave, S.I.; Jiménez-Arbeláez, G.A.; Alvarado-Gómez, V.A.; Isaza-Guzmán, D.M.; Flórez-Moreno, G.A.; Pérez-Cano, M.I. Association Analysis between Rs6184 and Rs6180 Polymorphisms of Growth Hormone Receptor Gene Regarding Skeletal-Facial Profile in a Colombian Population. Eur J Orthod 2018, 40, 378–386. [Google Scholar] [CrossRef]
- Depeyre, A.; Schlund, M.; Nicot, R.; Ferri, J. Dental and Maxillofacial Signs in Leri-Weill Dyschondrosteosis. J Oral Maxillofac Surg 2019, 77, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Al Kaissi, A.; Shboul, M.; Kenis, V.; Grill, F.; Ganger, R.; Kircher, S.G. Leri-Weill Dyschondrosteosis Syndrome: Analysis via 3DCT Scan. Medicines (Basel) 2019, 6, 60. [Google Scholar] [CrossRef]
- De Leenheer, E.M.R.; Kuijpers-Jagtman, A.M.; Sengers, R.C.A.; Oudesluijs, G.G.; Rappold, G.A.; Cremers, C.W.R.J. Congenital Conductive Hearing Loss in Dyschondrosteosis. Ann Otol Rhinol Laryngol 2003, 112, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Nakao, K.; Okubo, Y.; Yasoda, A.; Koyama, N.; Osawa, K.; Isobe, Y.; Kondo, E.; Fujii, T.; Miura, M.; Nakao, K.; et al. The Effects of C-Type Natriuretic Peptide on Craniofacial Skeletogenesis. J Dent Res 2013, 92, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.H.; Rabie, A.B.M.; Hägg, U. Indian Hedgehog: A Mechanotransduction Mediator in Condylar Cartilage. J Dent Res 2004, 83, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanya, R.; Prasad, S.; Prasad, R.; Mogra, S.; Shetty, V.; Rao, V. Mutations in FGFR3 Gene Associated with Maxillary Retrognathism. Indian Journal of Dental Research 2019, 30, 185. [Google Scholar] [CrossRef]
- Anderson, P.J.; Netherway, D.J.; McGlaughlin, K.; David, D.J. Intracranial Volume Measurement of Sagittal Craniosynostosis. Journal of Clinical Neuroscience 2007, 14, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Mulliken, J.B.; Gripp, K.W.; Stolle, C.A.; Steinberger, D.; Müller, U. Molecular Analysis of Patients with Synostotic Frontal Plagiocephaly (Unilateral Coronal Synostosis). Plast Reconstr Surg 2004, 113, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Rabie, A.B.M.; Tang, G.H.; Xiong, H.; Hägg, U. PTHrP Regulates Chondrocyte Maturation in Condylar Cartilage. J Dent Res 2003, 82, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.; Sävendahl, L.; De Luca, F.; Dauber, A.; Phillip, M.; Wit, J.M.; Nilsson, O. Short and Tall Stature: A New Paradigm Emerges. Nat Rev Endocrinol 2015, 11, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Wit, J.M.; Kamp, G.A.; Oostdijk, W. Towards a Rational and Efficient Diagnostic Approach in Children Referred for Growth Failure to the General Paediatrician. Horm Res Paediatr 2019, 91, 223–240. [Google Scholar] [CrossRef]
- Davidopoulou, S.; Chatzigianni, A. Craniofacial Morphology and Dental Maturity in Children with Reduced Somatic Growth of Different Aetiology and the Effect of Growth Hormone Treatment. Prog Orthod 2017, 18. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



