Submitted:
23 May 2024
Posted:
24 May 2024
You are already at the latest version
Abstract
One important goal of research on heterogeneous catalysis for Fischer Tropsch synthesis (FTS) is to understand the connection linking catalyst morphology and its activity. Development of these effective catalysts highlights the potential for understanding the effectiveness of promoted supported catalysts and revealing their complex and dynamic effects in FTS. The metal-support interaction (MSI) is influenced by the physicochemical and textural features provided by the modified support and can directly influence the sizes of active metal crystallites, reactants and/or products mass transfer, dispersion, mechanical strength, stability and consequently the adsorption-desorption characteristics of the catalyst. Herein, this article explores the general strategies for modifying silica supports (achieved through doping, coating, or promoting the support or support precursor) and how these strategies can be manipulated to further alter catalyst performance and product selectivity by enhancing the amount of active metal species and improving the intrinsic turnover frequency (TOF). Furthermore, the review emphasises a new strategy (the inverse model of the silica support modification) aiming at overcoming the challenges posed by active metal coating or coverage, as well as pore blockages by promoters that occur during conventional preparation of promoted silica supported FT catalysts.
Keywords:
support modification
; metal-support interaction
; inverse model
; activity
; selectivity
1.0. Introduction
Current developments of global FT synthesis capability and the advances of scholar research on the subject validate the continuing attention to the synthesis methods of promoted supported catalysts [1,2,3,4,5,6]. The current plentiful of natural gas and elevated market need for clean fuels all around the world has led to a shift in attention to supported Co-based FTS catalysts more especially for the production on long chain hydrocarbons (C5+). These supported catalysts (promoted or unpromoted) have been considered to be ideal for FTS. However, the key barrier in the FTS process is product selectivity, as formation of products is linked to the Anderson-Schulz Flory (ASF) law [7,8,9,10,11,12]. So, to be economically viable, it is important to optimally alter the catalyst composition and structure as well as process parameters as illustrated in Figure 1 below. By so doing, product distribution can be shifted towards target products [13,14]. So, the selection of the optimum factors, whichever individually or in mixture, can sustain the yield, without any need for catalyst replacement in a short time. Different parameters, therefore, (mainly catalyst type), can be manipulated to maximize FT activity coupled with higher product selectivity.
Catalyst formulation plus activation are some of the important factors that can be modified to give the supported Co catalyst the desired characteristics for enhanced selectivity, i.e., development and manipulation of catalyst or support structures of Cobalt-grounded FT catalysts to obtain enhanced pore transfer rates which leads to limited secondary hydrogenation plus increased proportion of liquid olefins. Alternative approaches have demonstrated beneficial aspects that restrict unwanted over-cracking in the FTS process, [15,16]. Both the pore transport improvement and surface modification effects can offer a blueprint regarding eccentric FT product dispersals aiming at high α-olefin product and heavier paraffinic fuel selectivities maintaining the principal zero CO2 by-product characteristic of supported cobalt-based FT catalysts [17].
Early investigations primarily concentrated on synthesizing and characterizing well-defined model catalysts. These studies yielded valuable insights into the influence of factors such as Co particle size [18,19,20,21], crystallographic structure [22], thermal history [23], and the effect of promoters [24,25,26] on catalyst functioning. Nevertheless, they often overlooked the impact of support materials on catalytic behaviour.
Support materials play a pivotal role in cobalt-based catalysts for FTS by providing framework strength and stabilizing the distributed Co particles. Various materials including alumina, (Al2O3) [33,34,35,36,37], silica (SiO2) [27,28,29,30,31,32], silicon carbides (SiC) [45], Titania (TiO2), [38,39,40] mixed oxides [41], zeolites [42,43,44], , and carbon nanostructures [46] have been studied as support candidates. Among these, surface modified SiO2, Al2O3, and TiO2 are predominantly used for Co-based catalysts due to their appropriate porosity, stability and, mechanical features under FTS environment.
Silica, a commonly used support material, offers benefits for instance increased surface area, microporosity and microporosity properties as required [28,47,48,49,50]. However, the limited interaction between metal catalysts and silica can hinder the crystallites dispersion, thereby reducing the quantity of active sites available for catalysis. Conversely, alumina is considered the most desirable catalyst support because of its great surface area likewise excellent mechanical properties. Despite its benefits, alumina faces challenges such as stability issues and the development of tough-to-activate aluminates, particularly in acidic or alcoholic reactions [28,47,48,49,50].
Advancements in the materials design have empowered significant fabrication of very porous supports expanding the range of potential catalyst supports for various processes. Additionally, catalyst support modification has emerged as a approach to boost the performance hence high product selectivity of heterogeneous catalysts [52]. By modifying the support material chemically or physically, it is possible to influence features including pore size distribution, surface area, acidity/basicity, and thermal stability, thereby improving FT synthesis efficacy.
Earlier studies explored the impact of supports on the performance of catalyst, i.e., they examined the effect brought by chemical composition of supports. Reuel and Bartholomew [53] noted a decrease turnover frequency (TOF) following the sequence Co/TiO2 > Co/SiO2 > Co/Al2O3 > Co/MgO. Conversely, Borg et al. [54] observed a significant support effect on hydrocarbon selectivity patterns when comparing Co crystallites of the same size on various Al2O3 supports. However, Iglesia and colleagues [55] reported negligible support effects catalyst performance and product formation on catalysts supported on magnesiochromite (MgCr2O4), SiO2, and Al2O3. Moreover, investigation has been done on the function of rare-earth-metal and alkali-metal oxides. Takahashi et al. [56] found that modifying an Al2O3 support using rare-earth-metal oxides positively impacted catalyst performance and formation of long chain hydrocarbons for CoRu/ Al2O3 catalysts, contrasting with observations by Bertole et al. [57] who realised negative effects on FT performance while using yttrium oxide (Yox) for CoRe/TiO2 and CoRe/Al2O3 catalysts. Thus, despite advanced scientific and technical magnitude, the influence provided by the chemical features of support materials on catalytic activity remains incompletely understood, with contradictory findings in earlier literature.
Modification of the textural features of Al2O3 facilitates the addition of active metals also it enhances accessibility of active metal species. Co/SiO2 FTS catalysts display improved C5+ formation and reduce CH4 compared to Co/ Al2O3. Larger Co crystallites (lower dispersions) that are simpler to reduce are frequently formed during the Co FTS catalyst production process when silica is used as a support in place of alumina [58,59]. Extensive research has been conducted about the impact of silica structure as a cobalt catalyst support. Ernst et al. [60], for instance, looked at the FT performance of cobalt supported by silica in both basic and acidic conditions. They discovered that the support surface area and catalyst activity are directly connected, and that SiO2 supported catalysts having typical pore sizes of less than 4 nm yield the least methane selectivity. According to a study by Saib et al. [61], silica supports with medium pore size (10nm) had the high catalytic performance and heavy hydrocarbon formation.TiO2 displays exceptional chemical and thermal resistance. The intensity of the MSI of Co-TiO2 lies between those of Al2O3 and SiO2 [39,62]. Zeolites are attractive due to their shape-selective properties, which can restrict product development bigger than their pore sizes also limiting higher hydrocarbon selectivity in FTS by constraining chain growth. Carbon materials have also gained attention for their exceptional resistance in both basic and acidic environments [63,64,65,66,67]. The pore structure of carbon materials can be tailored to target specific reactions. Carbon’s fundamental inertness towards producing weak MSI is crucial for achieving a higher reduction degree, making it suitable as an FTS catalyst support [62].
Current understanding points to two primary causes of differences in previous research. First, intra-pellet diffusion rates of reactants and products are impacted by the variation in intrinsic textural qualities across various oxides, which influences both activity and selectivity [68,69]. Second, catalysts with significantly varied metal dispersions exhibit distinct activity and selectivity due to particle size effects when a specific cobalt weight loading is applied to supports with varying specific surface areas. Therefore, it is necessary to precisely control parameters like support texture, the degree of Co reduction, and particle size in order to isolate the inherent impact of the chemical nature of the support material on catalytic performance [70].
In this article, we explored the different and contemporary strategies for catalyst support modification and their effects in terms of active metals (mainly Co) crystallite sizes, degree of reduction, and catalyst activity and selectivity for FTS. Advancement of these capable catalysts draws attention to the possibilities of understanding the efficacy of promoted supported catalysts and illuminating their complex and dynamic effects in FTS.
2.0. Strategies for Support Modification
2.1. Support Doping
Traditionally, choosing the support has concentrated on the non-reactivity factor of the support material (i.e., alumina, silica, titania, polymers, and zeolites) [39]. It is only recently that industry and researchers value that majority of supports influence of the steric and electronic features of the catalyst and can take place in collective or individual reactions. Utilizing the capabilities of these “non-innocent” supports during FTS catalyst preparation, one should develop extensive knowledge of intricate catalyst surface chemistry [71]. This will then allow one to be knowledgeable in model systems which can permit organized exploitation of acid−base characteristics. These can be altered through support doping with compatible M2+ M3+ cations such a Mn2+, Cu2+, Zn2+, Ni2+ etc, [72]. In this case, the electronic properties of such a s catalyst can be altered without affecting catalyst morphology and surface area [71]. Here, support doping entails introducing dopant species into the support material to modify its characteristics and improve the performance of supported catalysts. Various methods commonly employed for support doping in catalysis include:
- Incipient Wetness Impregnation (IWI): This approach involves saturating the support material with a solution containing dopant precursors. Subsequent drying and calcination yield the doped support material, with dopant species dispersed on the support surface or integrated into its structure during calcination [73,74,75,76,77].
- Co-precipitation: In this method, the support material and dopant precursors are blended in a solution, and a precipitating agent is added to prompt the formation of dopant-incorporated support particles. The resulting mixture is then subjected to drying plus calcination to achieve the doped support material [78,79,80].
These methodologies offer flexibility in regulating the doping level, distribution, and composition of dopant species within the support material. This allows for the customization of support properties to meet specific catalytic needs.
Vast research on support doping has been studied to try to boost catalytic features. An example of this was carried out by Wei H and company [86], in which they developed transition metals including Fe, Cu, Mn, Mo, or Co-doped ceria nanozymes. These were found to display superior catalytic activity in comparison to pristine CeO2. These were ascribed to the synergistic result plus surface oxygen compounds [86]. Similar results were obtained when rare-earth elements including Zr, La, Pr, Eu, or Nd were doped in the CeO2 catalyst, i.e., with increased oxygen vacancy imperfections, and high Ce3+ active sites, it led to improved peroxidase-like, oxidase-like, and antioxidant enzyme-imitating performance [87].
Xu. X. L and the company [32] have explored a method of silica support modification by doping. They realized that silica-supported Au on TiO2 support was relatively new for the selective cyclohexane oxidation process. Thus, opted for SiO2 doping with TiO2 and utilized the modified silica (doped) as Au support. This was based on the surface sol–gel (SSG) preparation that was formerly established by Kunitake and company that simplifies the production of thin films supported on solid substrate coupled with wideness regulation of molecular accuracy [88]. Afterall, they realised that the catalytic performance and product composition may be greatly maintained in four recycling oxidation outcomes, indicating an elevated stability of the doped supported gold (Au) catalyst. Furthermore, Yan and company have utilized the above process to alter the silica prior to adding Au elements, [89]. In the initial experiment, the catalytic activity of the doped and un-modified Au/SiO2 catalysts were performed for an hour. The un-modified Au/SiO2 catalyst displayed a clear 0.9 % conversion as compared to the doped catalyst (Au/TiO2/SiO2-1) which demonstrated a 3.2% conversion and enhanced selectivity towards cyclohexanol and cyclohexanone. They concluded that, with a small loading titania doping, catalyst activity increased notably. [32]. Likewise, the Au/TiO2/SiO2-1 catalysts displayed higher selectivities close to 90 %. Therefore, small doping of silica-supported Au catalysts using TiO2 leads to elevated selectivity as compared to non-doped silica-supported catalysts. This corresponds to results by Luo and company who detailed that doping allows for diverse contact of boundary sites among Au and metal oxide, and this is the main effect leading to differences in catalytic performance. Luo et al observed that for doped Au/SS@Cu, the CO conversion reached 80% as compared to 60% for non-doped Au/SS at 300 °C. This is attributed to silica and titania assisting the activation of oxygen (O2) in 2 ways i.e., movement within the metal bulk and water transport around the gold species [90].
Savost’yanov et al [91]researched how alumina dopant addition affects catalyst performance plus selectivity of Co/SiO2 catalyst for FTS. Catalyst preparation was achieved using IWI. They uncovered no apparent promotional effects were observed for alumina doping at atmospheric pressure. As soon as the pressure was increased to 20 Bar, an increase (from 49 to 54%) in CO conversion was noted when dopant loading enlarged from 0 to 1%. Similarly, CH4 selectivity decreased to 10.7% from 15.4%; at the same time, selectivity for higher hydrocarbons (C5+) grew to 80.1% from 77.4%. Still, an additional increase in alumina dopant loading above 1 wt.% gives the reverse outcome. Alumina-doped (1 wt.%) silica-supported cobalt catalyst creates a narrower molecular weight distribution, and this is due to the cumulative growth of C8 – C25 fraction and decreased long-chain hydrocarbons. It is important to note that electronic effects can influence FTS performance alongside structural features. Alumina has been identified to possess an electron-donating atmosphere. These catalyst-surface electron-donating features promote CO adsorption as opposed to hydrogen (H2) adsorption because CO is an π-acceptor. Therefore, alumina as a structural promoter can enhance Co dispersal and be used as an electronic promoter to control Co metal chemisorption effects [91]. The above experimental outcomes are consistent with late stated doping effect of alumina on activated carbon-supported cobalt (Co/AC) [92].
Advancing from conventional catalyst preparation method, the synthesized catalyst with manageable loadings, precise morphology, and crystalline phases can easily increases the catalytic performance of nanoenzymes, and this is attributed to increased surface oxygen as well as enhanced electron transfer among the substrate and the oxidant [93,94,95]. In summary, doping supports with specific elements can boost the catalytic performance for supported metal catalysts. Perhaps, doping silica with transition metal ions like Fe, Co, Ni, or other oxides supports can introduce additional active sites or modify the support electronic features, leading to improved catalytic performance.
2.2. Support Coating
Another support modification that can be implemented in catalyst synthesis include support coating. It is mainly due to the detrimental effects brought by the MSI (whether weak or strong) that had led to studies aiming at overcoming these challenges gaining more attention. Previously, the interactions between metals and supports were frequently disregarded, with only simplistic metal active sites believed to be critical in catalytic reactions. However, supported metal catalysts have demonstrated that metal sites interacting with supports can possess distinct electron cloud densities, geometric structures, particle morphologies, and chemical compositions. Furthermore, alterations in geometric or electronic structures induced by these MSI and/or strong MSI significantly influence the reactivity at the active centres. This phenomenon can be intricate, particularly when multiple factors are intertwined [96,97,98,99]. In this section, the focus is to manipulate MSI via support coating. Coating the catalyst support has been found to generate excellent effects that are beneficial to FT performance.
To achieve support coating in catalysis, it comprises of placing of a light layer of catalytically active material onto a support material to enhance its performance. This includes the used of different methods such as: Here’s an overview of common methods used for coating support in catalysis, along with relevant literature: Impregnation, in which the support is put in a solution comprising the catalytically active material. The solution is then allowed to impregnate the pores of the support [74,76]; Deposition-Precipitation, whereby a precipitant is deposited into a solution having both the support material and the catalytically active material [100,101] ;Chemical Vapor Deposition (CVD), here gas-phase technique is utilised to add thin films of catalytically active substances on the support surface [102,103]; Physical Vapor Deposition (PVD) includes the evaporation or sputtering of the catalytically active material in a vacuum chamber, followed by deposition onto the support material’s surface to make a thin sheet on the support; ALD technique that allows for the controlled deposition of atomic films of catalytically active materials on the support exterior [104].
Research on the abovementioned technique of support modification has been done. In their work, Xu et al coated copper (Cu) species with mesoporous silica, and this was found to enhance the catalytic ester hydrogenation. Results obtained from both experimental and simulation have shown that during H2 dissociation, Cu–Hδ− and SiO–Hδ+ species have developed at the Cu-O-SiOx boundaries. This mechanism through stabilizing transition steps enhanced the hydrogenation of esters. When followed by H2 reduction, this method of coating Cu with silica provided a useful tool for developing applied Cu catalysts with ample Cu-O-SiOx interfaces as well as limiting Cu nanoparticles from sintering [105].
Jiang et al, showed the control of MSI by impregnating cobalt nitrate on a silica support coated with various loading, and contemplating the relevant advantages of both the carbon when loaded on oxides support, and oxides, the MSI is easily regulated as a result of covering the carbon material with oxides or vice versa. They also outlined that further improvements in metal dispersion can be achieved via modification/coating carbon supports with reactive groups which include, N-, O-containing groups as well as MnO or La2O3. The improvement in terms of FT activity was found for Carbon nanotubes-modified MgO catalysts and this was attributed to a decreased MSI [65].
Research conducted by L. C. Almeida and colleagues [106] explored the catalytic activity and product selectivity for Co/γ-Al2O3 catalysts presented as both powder and washcoated monoliths. The findings demonstrate the feasibility of employing washcoated monolithic structures in FTS. Overall performance of the examined catalysts exhibited considerable similarity. However, the structured catalysts exhibit slightly higher catalytic activity compared to the powder form, with the coated foam showing greater activity than the parallel channel monoliths. Regarding selectivity, the observed high values of parameter Į (ranging between 0.73 and 0.82) and the substantial selectivity towards C5+ (between 80% and 85%) indicate a product distribution favoring fuels like gasoline and diesel. Additionally, the selectivity in terms of methane percentages remains relatively low (around 10%) and consistent across all tested samples. Based on these outcomes and considering the characteristic diffusion lengths calculated for the various systems, it can be inferred that all catalytic samples evaluated in FTS under similar operating conditions are devoid of diffusion limitations.
2.3. Support Promotion
Several substances are normally added to the catalyst to modify the turnover rate of such catalysts. This includes an additive that enhances the rate per second per active site (TOF) [3,107]. It is because of this concept that promoters gained attention and led researchers to find convectional ways to establish promoter effects on FT catalysts. Promoters are deposited in small amounts on catalysts to enhance catalyst performance and stability [14,108,109,110,111,112]. In numerous circumstances, the effects of promoters are interconnected creating a tough decision to exactly tell the real role brought by such a promoter. However, the reflective purpose of the promoter is occasionally not easy to explain because of the lapping effects between the support and metal catalyst. [25].
A wide range of promoters, such as noble metals (e.g., Ru, Pt and Re) [113,114,115,116], alkali metals (Na, K, Rb and Li) [117,118,119], transition metal oxides (La, Cu, Gd, Ni, Zr and Mn ) [1,120,121,122], as well as alkaline earth metals including Mg, Ba, and Ca [123,124,125]; have been researched and applied on FTS, but, there is still controversy on the their effect regarding catalyst performance, selectivity, and deactivation. [62]. The way promoters modify the catalyst or support can be tied to the loading of the support, the support, plus the synthesis method [126]. A lot of reviews have been produced investigating the effects of mainly structural promoters and electronic promoters. Structural promoters’ main purpose is to enhance the dispersion of active metal by controlling the interaction between cobalt-support. These promoters do not normally influence the selectivity since they are responsible for increasing the metal active sites which is usually accomplished through:
- Stabilizing the support oxide
- Sticking the cobalt species on the support oxide by acting as an oxidic interface between support and Co particle
Electronic promotion usually happens once a chemical connection among the promoter and Coo surface. They are better understood through the ligand effects [127]. The active Co species’ electronic environment can be manipulated by introducing a promoter. The effect of this scenario is that there will be an electronic donation or withdrawal causing enhanced intrinsic turnover frequency and change in product distribution. There is a synergistic effect brought by promoters as they influenced the catalyst activity indirectly by changing the local feed configuration. Effects such as [25,62] :
- Hydrogenation or dehydrogenation reactions- sometimes promoter elements either affect the hydrogenation or dehydrogenation reactions, hence shifting selectivity.
- Coke burning at the regeneration stage- promoters can lower the oxidative treatment temperature, this limits the clustering of supported Co species.
Table 2 above highlights the promotional type, and mode plus the prompt of different catalyst promoters utilised to enhance FT activity, selectivity towards long chain HCs, and catalyst stability. Despite vast research on catalyst promotion, the matter of the best practice or loading of the promoter to give the highest stability, activity as well as selectivity is still in question.
The promoter integration can control the MSI and thus attain great activity because of the enhanced degree of reduction of Co oxides with limited development of non-reducible compounds. Therefore, accessible inorganic supports like alumina, silica, and/or titania, have been promoted with other oxides i.e., manganese oxides, magnesium oxides, zirconium oxides, and lanthanum oxides to alter the contact between metal and support. For example, alumina supported Co catalyst can be modified using oxides such as La2O3, ZrO2, and TiO2 to prevent the formation of irreducible Co-aluminate species as well as improve the degree of reduction of such a catalyst. [128].
On their experiments, Lu et al. [129] realized a lower reduction for titania-supported platinum catalysts (Pt/TiO2) because of obstruction on Pt metal active species by titanium oxides complexes which did not reduce completely, and this suppressed the catalytic activity hydrogenation of naphthalene. To counteract the above challenge, they promoted the catalyst support with ZrO2. This limited the blockages of Pt limiting the agglomeration of TiOx species and hence improvement in the degree of reduction as well as catalyst performance on Pt/TiO2-ZrO2. Furthermore, Ruppert et al. [130] likewise explored the outcome of ZrO2 on the crotonaldehyde hydrogenation process using Pt/ZrO2-TiO2 catalysts and found out that the ZrO2 incorporated on the supports efficiently repressed the movement of TiOx species thus limiting blockages of Pt and upgraded its catalytic performance. In the cases above, one can deduce that the promotion of TiO2 with ZrO2 can significantly decline the strong MSI and alter the catalyst morphology. But low thermal stability and surface area, SA of the above-promoted support TiO2-ZrO2 was found, and these are undesirable in catalyst development. This has led to some researchers shifting their attention to alleviating TiO2 and ZrO2 on a SiO2 support via the grafting method, producing fresh supports comprising a bulk surface area and great thermal stability jointed with exclusive features of TiO2 and ZrO2 [38].
Recently, ordered mesoporous SiO2 including SBA-15 and MCM-41 has been studied worldwide. The primary benefit of such supports relates to being able to successfully regulate the optimal active metal crystalline size. Nevertheless, the challenge posed is the weak interaction of the tailored SiO2 support and metal catalyst that generally leads to poor metal catalyst dispersion. This is the reason TiO2 and ZrO2 have been used as promoters to improve or increase metal-support interaction [131]. Researchers have found out that the promotion of ZrO2 for silica-supported Co catalyst has boosted the reduction degree as well as improved catalyst conversion, and selectivity of higher hydrocarbons. Ali et al. (All, Chen & Goodwin, 1995) reflected that the enhanced catalytic performance and selectivity towards heavy hydrocarbons were attributed to the development of boundary linking Co metal plus ZrO2, which encouraged carbon monoxide CO dissociation.
In addition, there is still a deficiency in support and promoter impact or benefits for the normal, fairly low dispersion cobalt catalyst i.e., the influence brought by the metal additives and support on CH4 and C5+ selectivity is still debatable. It was found by Iglesia, that a flat CH4 selectivity for catalyst supported on Al2O3, SiO2, and TiO2 having comparable intraparticle transport/site-density appearances [133], while other stated substantial support and metal-promoter influence [134]. Generally, C5+ and CH4 selectivity is also dependent on reaction parameters, mass transfer limitations, and CO dissociation. Regarding the above aspects of promoter addition together with metal-support interaction, to improve our understanding of their influence on FT catalytic performance and selectivity, the following subjects are priority topics in catalysis.
- Examine the influence of support reformation plus promoter addition on the fundamental FT activity by utilizing in-situ studies, hence preventing possible faults from catalyst deactivation.
- Exploring how support modification on the fundamental CH4 selectivity of FT cobalt catalyst happens without process parameters and mass- transport influence.
- Explore how CO adsorption and dissociation are affected by promoter addition and explore their relationships with variations in FT performance and selectivity towards higher hydrocarbons [57].
3.0. Support Modification for FTS
Divergence in the effect brought by supports in FTS regarding activity and hydrocarbon selectivity still exist. Iglesia [55] has conveyed information saying that oxide supports as well as their altered kinds do not affect FTS performance as opposed to the improved activity ascribed to better metal dispersion. Nonetheless, variations in product selectivity are primarily attributed to reactants and products-diffusion effects brought by enhanced secondary reactions i.e., re-adsorption of olefins [135]. Further research has shown that the chemical structure and support permeability are vital in the fundamental product selectivity in the FT reactions.
In addition, it is reflected that the support can influence the FTS activity indirectly. The physicochemical support features, including pore volume and diameter, can effectively alter the particle sizes of Co0 plus its degree of reduction. Despite so much satisfactory research on conventional supports i.e., silica, alumina, and titania on FT catalysts, there is a lot that can be done to discover and develop new support materials with boosted characteristics. Novel carriers including silicon carbides, nanoparticles, and ordered mesoporous supports with advanced chemical compositions have attracted substantial consideration for numerous applications with minimum undesired properties such as extremely weak or strong metal-support interaction. [136,137]
3.1. Effect of Support Modification on Catalyst Reduction for Fischer Tropsch Synthesis
Promoter integration can adjust the MSI as well as accomplish an improved performance because of the effortless activation of cobalt oxides and limitations on the development of non-reducible eliminates and silicates. Hence, commercially accessible inorganic supports [138,139], have been tailored with additional oxides such as MgO, La2O3, ZrO2, and other elements as “promoters” to modify the MSI. In many cases, the incorporation of oxides to modify the support and hence the catalyst has shown signs of an improved degree of reduction and improved performance due to the weakened MSI [140].
Li et al performed H2-TPR on their catalysts to evaluate the reduction performance of cobalt oxide Co3O4. Two key peaks were noted in all catalysts. It is well-detailed that the 1st peak corresponds to the reduction of Co3O4 to CoO and the 2nd peak corresponds to the conversion of CoO to metallic Co [28,36]. In their experiment, they found the 2nd peak broader which implies various CoO particles have interacted differently with the support SiC or particles of CoO having broad particle size distribution. For the catalyst promoted with ZrO2 i.e., Co/Zr/SiC, the catalyst was simpler to reduce as compared to the non-promoted Co/SiC as illustrated by the sharp 1st and 2nd peaks activating 319 and 370 oC. The easement in reduction for the catalyst Co/Zr/SiC was mainly due to bigger crystallite size coupled by exterior located cobalt oxide species. Apart from the two main peaks, the small peak was realized at a temperature of 620oC which was the result of better interaction of the Co-Zr particles with SiC. However, these results were contrary to what was obtained for catalysts promoted with Ca and Mn. It was discovered catalyst promotion with Mn and Ca triggers shifts in the activation of Co species together with subsequent degrees of reduction which dropped to 57% and 45% respectively. Therefore, it has been established that putting additives of Zr enhanced Co catalyst reduction while Mn and Ca decreased the reducibility of Co catalyst [141].
For Illoy and Jalam’s experiments, it was found that more than two peaks occurred in the H2-TPR experiment which showed that some of the Co species did not reduce at the same time i.e., as N2 adsorption results indicate some kind of catalyst’s pore impediment which implies that some particles reduced following the ones blocking the pores reduced. By promoting with potassium K, the degree of reduction was decreased further due to the following reasons: (i) The catalyst activation temperatures changed to higher rates, e.g., at the beginning of the reduction procedure, unpromoted catalyst reduction temperature shifted from 170 to 210oC and 255oC for catalyst promoted with 1% and 3-5% K respectively; (ii) As K-addition increased, the area under the TPR outline under 500oC declined, signifying lower reduction degree (iii) the broader peak for catalysts containing 5% K at temperature of 512oC, indicating strengthened support interaction with Co catalyst. This undesirable influence on the K on the degree of reduction agreed to Jacobs et al. [142] experiments who observed that loading (0.5-5%) K lifted reduction temperatures to elevated temperatures and hence lesser reduction degree. So, has been indicated that potassium affects the interaction connecting cobalt particles with the silica support [143].

Table 3.
Summary of modified catalysts and/or supports and their effects on catalyst reducibility.
| Sample | Reduction degree (DOR) | Ref. |
|---|---|---|
| Co/Al | 39 | Garcilaso et_al, 2019 [144] |
| Co/Zr-Al | 40 | Garcilaso et_al, 2019 [144] |
| Co/Ce-Al | 35 | Garcilaso et_al, 2019 [144] |
| Co/Al | 42 | Barrientos, Garcilaso, Venezia, & Aho, 2017 [145] |
| Co/Zr-Al P | 44 | Barrientos et_al, 2017 [145] |
| Co/Zr-Al ME | 40 | Barrientos et_al, 2017 [145] |
| Co/MS | 98.28 | Wu, Yang, Suo, Qing, Yan, Wu, et al., 2015 [146] |
| Co/Zr-MS | 97.55 | Wu, Yang, Suo, Qing, Yan, Wu, et al., 2015 [146] |
| Co/TiZr-MS | 71.17 | Wu, Yang, Suo, Qing, Yan, Wu, et al., 2015 [146] |
| Co/Pt/SiO2 | 96 | Breejen et_al, 2011 [147] |
| Co/MnO/Pt/SiO2 | 94 | Breejen et_al, 2011 [147] |
From Barrientos et al, [145] the approximated (DOR) was done after 16 hours in an H2 environment. The DOR was as little as around 40% for all three catalysts with a slight difference with Zr addition. These findings were anticipated because the TPR results reveal that Co2+ activates at a temperature over 600 °C. However, the little changes in the reduction degree might have developed from instrument and technical errors. With these results, they presumed that adding some Zr to modify the alumina support has an insignificant effect on the catalyst reducibility. Furthermore, varying catalyst particle sizes were observed, which further made it difficult to conclude that the slight difference in DOR was due to Zr modification or the trivial distinctions in Co particle size distribution. This, however, is confusing because normally Co/ZrO2 catalysts show superior DOR as compared to Co/Al2O3 catalysts. This is because alumina surfaces were not coated by the Zr. So, it could be concluded that the Cr modification can effectively improve the DOR when it has fully coated the alumina to avoid the presence of alumina sites that can form aluminates during reduction. This explains the somewhat higher DOR shown by the Co/Zr-Al P catalyst in which the catalyst surface had more Zr islands as compared to Co/Zr-Al ME.
Wu et al [128] have realized with the addition of TiO2 on the mesoporous silica MS, catalyst reducibility improved i.e., the reduction peaks moved to a lesser temperature. Modification of the support using Zr has shown that indeed Zr impacts the catalyst DOR. It was established that by depositing a bit of Zr, a decline in the intensity of reduction peaks leads to improved DOR. However, with more raise in Zr addition to Co/nTiZr–MS catalysts reduction moved to elevated temperatures which lowered the DOR. Nonetheless, there are inadequate investigations on the synthesis of Co supported on TiO2–ZrO2–SiO2 and the modification impacts of ZrO2 on the catalyst morphology FT activity.
From this table, Breejen et al [147] have determined that complete reduction at reduction temperatures of about 450oC was achieved for both catalysts calcined in air and helium, with and without MnO promotion. However, Smaller crystallite sizes were obtained for MnO promoted catalyst (Mn/Co = 0.08) coupled with a decrease in degree of reduction (62%) which only increased after 2 hours of FTS. The lower DOR may be ascribed to the impeding influence of MnO on the magnitude of reduction. This is in agreement with what was obtained by Morales et al for their catalyst Co/MnO/TiO2 [148].
3.2. Effect of Support Modification on Catalyst Particle Size for Fischer Tropsch Synthesis
Promoters on supports have been shown to alter the catalyst support pore size and surface area. The results display that cobalt with increased metal-support strength on silica led to a substantial drop in surface area. This phenomenon is normally justified by the increase in cobalt oxide crystallites inside support pores in the course of catalyst calcination, consequently causing some pore impediment. This is in line with the pore volume results, which displayed a declining size. Adding of promoters including potassium K, ranging over 3% in the supported catalyst, also strengthens this occurrence. Furthermore, the Scherrer equation was applied to approximate the mean Co crystallite sizes. Even though no apparent relation in the data regarding the Co3O4 and Co, it looks like the mean crystallite size for CoO declines with rising K loading. It can then be concluded that the addition of potassium regulates the size of Co oxides [143].
In another work done by Johnson and Bell, the TEM images showed Co particle sizes ranging from 2-20mn for Mn promoted. For loading of Mn/Co = 0.5, there was visible proof of Mn-rich species with a surface diameter larger than quite a few nanometres. The compositions of each nanoparticle were calculated by choosing rounded areas about the Co nanoparticle as well as the neighbouring Mn in the species maps produced by STEM-EDS. As for the Mn/Co atomic ratio, the X-ray totals for Mn and Co were summed and converted for individual quantification areas. It was found that the mean nanoparticle composition was nearly similar to the catalyst containing Mn/Co =0.01 nonetheless catalysts with more promoter amount displayed mean nanoparticle compositions significantly lower as compared to their equivalent bulk compositions [1].
The Scherrer equation was engaged to compute the mean crystallite sizes of Co3O4 as well as the dispersion of Co0 on the catalyst using the peak of 2θ = 36.90. Even though using total crystal sizes is not very accurate, it was convenient for assessment [149]. The calculated mean crystallite size of Co3O4 grew by 48% (i.e., 25 to 37nm) nm concerning increasing Zr promoter, whereas 16 and 18 nm were calculated for Mn and Ca promoted silica supported catalysts respectively. The Co0 dispersion reduced from 8.3 to 5.8, 7.8, and 3.5% for the corresponding catalysts when Ca, Mn, and Zr promoters were loaded, respectively. These outcomes were in line with the previous studies showing that for silica-supported cobalt catalysts, the Co crystallite sizes increased with increasing Zr [150]. A minor reduction in the Co0 dispersion was stated for Co/CNF catalyst promoted with Mn (L. Bezemer, 2006), as well as enhanced Co3O4 crystallite size for Co/Al2O3 promoted with at least 1% Ca. [152] Furthermore, Li et al realized that for SiC-supported catalysts, the Co3O4 particle sizes were equal to or surpassed the mean pore diameter. This showed that bulk of Co3O4 species were situated either on the exterior surface or the support pores entrance. This agrees with the literature previously stated based on the elucidation of the decrease in mean pore size [141].
In his work, Pardo-Tarifa found out that promoting Zr decreases the SBA-15 and MFC silicas’ pore size and, consequently, the Co3O4 crystallite size. This on the other hand leads to strengthened metal-support interaction and low reducibility. Nevertheless, the cobalt dispersions are still higher as compared to the Zr-free promoted catalysts because of the small Co particle sizes [14].
After reporting the effect of promoter on reduction, Breejen et al. further recorded the effect of MnO on particle size. They found that there is an insignificant change in Co3O4 crystallite size for catalysts calcined in air or helium. Nonetheless, a decline in particle size was noted following promotion with a little quantity of MnO (i.e., Mn/Co = 0.06) in comparison with the unpromoted catalyst. Furthermore, they realized that for helium calcined catalysts, however, the particle sizes of the Co/MnO/Pt/SiO2 catalysts are not affected by the quantity of MnO added, while for air calcinated catalyst particle sizes increased with increasing MnO amount [147].
3.3. Effect of Support Modification on Catalytic Activity and Product Selectivity for FTS
FTS has been demonstrated to be a process that can effectively convert syngas attained from non-renewable feedstocks such as natural gas and coal, or renewable feedstocks including biomass as well as municipal solid waste to clean chemicals and fuels using a catalyst in the process. The process performance and product selectivity, however, can be enhanced by altering the physicochemical features of the catalyst via such as support and promoter additions and changing process parameters.
FTS product selectivity can be expressed by polymerization kinetics, in which the various chain propagation or termination possibilities can be established for the individual routes i.e., . These routes are affected in cooperation with operating environments and catalysts [153]. Therefore, by creating modifications, the product selectivity will normally be altered due to kinetic effects and/or structural effects [154].
Furthermore, it was found by Li et al [141] that FTS activity for catalyst promoted with Mn and Zr, the CO conversion improved by approximately 10% from 71% to 81% and 82% respectively. This was attributed to the weaker Co-SiC interactions introduced via adding additives of Mn or Zr as proved by the hydrogen-TPR & XPS study. As for the product selectivity, based on the weight basis, it was revealed that no CO2 formation was observed for all promoted catalysts, and this corresponded to the conclusions done by Khodakov et al. [70]. They also noticed that the Co/Zr/SiC displayed a slight growth in the C5-C22 hydrocarbon selectivity in comparison to the non-promoted Co/SiC catalyst that exhibited a product percentage peak mainly at C12.
Additionally, literature [28,36] have shown that the promotion of Mn or Zr enhanced the catalyst activity and long chain hydrocarbon C5+ production. The increase in the selectivity of C2-C4 given by Mn-promoted catalysts agrees with results obtained for Co/MnOx. This further proves that Mn is an efficient support promoter for boosting light olefins production. Consistently, the chain growth alpha value α obtained was equal for non-promoted Co/SiC and promoted Co/Zr/SiC catalyst (α = 0.84) whereas an upper limit α-value for the promoted Co/Mn/SiC catalyst was 0.88. A low CO conversion value of 13% for promoted Co/Ca/SiC catalyst, and elevated lower molecular weight hydrocarbons (i.e., CH4, C2-C4) of nearly 95%, can be tied to the low DOR. This is comparable with the explanation in the literature that states that the low degree of reduction leads to poorer FT performance and higher CH4 selectivity [70,141].
Dinse et al, demonstrated the effect of silica support modification using Mn. They found that it triggers the CO conversion rate to reach a maximum at the ratio of Co/Mn of 0.05. This results are ascribed to the relation linking the impacts of MnO placed onto the supported Co catalyst surface i.e., the enhancement in the FT activity by Co species positioned side by side with MnO and as such leading to the promotional effect of CO dissociation caused by Mn2+ cations with the O atom; once again, the decline in the amount of exposed Co sites, resulting from a rise in the ratio of Co/Mn. Another pattern that developed was realized when reaction pressure was raised to 10 atm. For Mn loading at Mn/Co equals 0.125, the Co intake rate declined. This outcome is assigned to increased stability in CO adsorption, which limits inhibited hydrogenation of CO. They accurately calculated a declining reaction rate as the CO conversion increased at a lower pressure of 1 atm, but could not explain the practical small growth in conversion degree and conversion-independent for the Mn-modified catalyst and un-modified catalyst respectively at the 10 atm.
The product selectivity (C5+) increased with raising Mn addition in the entire working environment. The olefin-paraffin (O/P) proportion regarding C2-C4 segment was superior for Mn addition due to a limitation in paraffin formation and enhancement in the olefin formation, i.e., Mn addition repressed secondary reactions responsible for paraffin formation [1,157],
Potassium promotion on SiO2-promoted Co catalyst was performed by Illoy and Jalama. They realized that potassium addition, especially using a small amount of about 1% will lead to a considerable decline in CO2 conversion from 39 to 16%, when it is matched to the unpromoted catalyst. Adding more potassium further worsens this performance but with reduced impact. This behaviour could be assigned to (i) K coating of active sites, even though regarded to occur at a little degree due to lower K-loading, (ii) rise in CO2 adsorption ability i.e., the decline in CO2 conversion versus K promotion that was also stated by Shi et_al [8] and Owen et al. [13] when dealing with CoCu/TiO2 and Co/SiO2 catalysts, respectively. They realized that K addition lowers the catalyst H2 adsorption capability and increases CO2 adsorption. Furthermore, an above 1% addition of K was found to cause an extra drop in CH4 selectivity plus improvement in CO selectivity. On the other, while increasing the K loading, the C2+ hydrocarbons lessened when compared to loading of less than 1% potassium. At K lower loading, the C2+ increase can be credited to a declined surface H/C ratio. This means that the carbon-containing elements from CO2 dissociation will polymerize instead of being hydrogenated which is a norm in a hydrogen-rich reaction [143].
Table 4.
Summary of modified catalysts and/or supports and corresponding influences on C5+selectivity.
Table 4.
Summary of modified catalysts and/or supports and corresponding influences on C5+selectivity.
| Catalysts | Conversion (%) | Main Product | Selectivity (%) | Ref. |
| 12%Co/γ-Al2O3 | 45 | C5+ | 80 | Enger et al., 2011 [154] |
| 12%Co-0.5%Re/γ-Al2O3 | 45 | C5+ | 83.1 | Enger et al., 2011 [154] |
| 12%Co-0.5%Re/5%Mg-γ-Al2O3 | 45 | C5+ | 81.8 | Enger et al., 2011 [154] |
| 12%Co-0.5%Re/5%Zn-γ-Al2O3 | 45 | C5+ | 82.5 | Enger et al., 2011 [154] |
| 9.3%Co/TiO2 | 29.2 | C5+ | 85 | Eschemann, Oenema, & De Jong, 2016 [158] |
| 9.0%Co-0.12%Ag/TiO2 | 33.0 | C5+ | 89 | Eschemann, Oenema, & De Jong, 2016 [158] |
| 8.9%Co-0.11%Pt/TiO2 | 30.4 | C5+ | 83 | Eschemann, Oenema, & De Jong, 2016 [158] |
| 9.4%Co-0.25Re%/TiO2 | 33.1 | C5+ | 88 | Eschemann, Oenema, & De Jong, 2016 [158] |
| 15%Co/SiC | 71 | C5+ | 80 | Li, Wu, & Wu, 2017b [159] |
| 15%Co-5%Zr/SiC | 82 | C5+ | 81 | Li et al., 2017b [159] |
| 15%Co-5%Ca/SiC | 81 | C5+ | 82 | Li et al., 2017b [159] |
| 10%Co/SiO2 | 41.7 | C5+ | 82.5 | Wu, Yang, Suo, Qing, Yan, Wu, et al., 2015 [146] |
| 10%Co/TiO2-SiO2 | 85.9 | C5+ | 85.6 | Wu, Yang, Suo, Qing, Yan, Wu, et al., 2015 [146] |
| 10%Co/TiO2-ZrO2-SiO2 | 80.3 | C5+ | 75.4 | Wu, Yang, Suo, Qing, Yan, Wu, et al., 2015 [146] |
| 20%Co-0.1wt.%Pt/ZSM-5 | 26.8 | C12+ | 52.2 | Subramanian et al., 2016 [160] |
| 20%Co-0.1%Pt/ZSM-5 | 27 | C12+ | 52.0 | Subramanian et al., 2016 [160] |
| 20%Co-0.1wtPt/SiO2-ZSM-5 | 26.4 | C12+ | 60.5 | Subramanian et al., 2016 [160] |
| 20%Co-0.1wtPt/MOR | 40 | C12+ | 60.9 | Subramanian et al., 2016 [160] |
Enger and co-workers illustrated the product selectivity (C5+) at 50% CO conversion for the catalyst loaded with 12 wt% Co. To obtain a constructive effect of catalyst modification using Re, a comparison was done for similar γ-A2O3 supports. For the catalyst modified by adding Mg or Zn, it was realized that the heavy hydrocarbon selectivity is dependent on the amount of modifier added as well as the procedure that was used to load Mg or Zn (whether on the support followed by calcination at a temperature of >900 0C, before impregnating with Co-Re precursor or simultaneous impregnation with Co and Re on the support. Note that, there may be optimal crystallite sizes for the definite Co/Re ratio, in which one can achieve the highest C5+ selectivity due to modification using Re on the catalysts, in which modification of Co using Re generates the highest C5+ selectivity. The improvement in the C5+ fraction could be ascribed to the insertion and re-adsorption into the expanding hydrocarbon chain [133].
Depending on the preparation procedure, variations in olefin-paraffin (o/p) were observed stretching from 1.3 on unpromoted α-Al2O3, possibly indicating a greater degree of re-adsorption to 2.1 on the Zn- and Mg-modified catalysts, conceivably implying limitations of re-adsorption. There was no relationship between the Mg or Zn modified catalysts for o/p ratios of C3 and C4 whereas for C5 and C6 a noticeable increase was found for increase Mg addition. An opposite correlation was found for co-precipitated Re and Zn on the support before calcinating that catalyst at high temperatures [154].
As for Eschemann et al, they carried FTS at 493 K, 20 Bar and recorded almost similar CO conversion ranging between 29.0 to 33.1 for all catalysts. The findings indicated that for the unprompted catalyst, a cobalt-time yield (CTY) of 6.5*10-5 molCO/gCoS coupled with C5+ formation of 85 wt.%. With Ag additives, it was found that the CTY improved to about 12*10-5 molCO/gCoS whereas the C5+ formation considerably increased to 89 wt.%. Enhancement in the catalyst activity was retrieved for Pt loaded catalyst, with an optimal loading realized at 0.21 wt.% Pt. The catalyst performance benefit, however, came with a decrease in C5+ selectivity of 83 wt.%. Hydrogenolysis and methanation reactions have been thought to have led to this decline. Moreover, the turnover frequency (TOF) for catalysts loaded with Pt, or Re were established to have gone up by a factor of 2 above that of unpromoted catalyst, but no significant effect was found for Ag-promoted catalyst [158].
3.4. Effects of Preparation on Activity and Product Selectivity
Catalysts preparation, the underlying/core configuration of the designed catalysts plus the resultant catalytic activity go together in catalysis. The preparation technique decides the structure of the catalyst and, accordingly, is vital for such catalyst’s application. It has been realized that a small change in the catalyst synthesis procedure might poses a substantial effect on the outcome of such a catalyst concerning catalyst structures, consequently having noteworthy differences in catalytic activity and selectivity, [161]. To further emphasize the importance of the catalyst preparation method, more latest experiments by Ai et al. utilizing tungsten-doped Ni/Al2O3 catalysts, has shown that in CO methanation the addition of tungsten enhances reducibility and accordingly improves the number of active sites as well process performance but only when the appropriate preparation procedure is used [162].
Impregnation is one of the commonly utilized methods in the formulation of industrial catalysts. In this case, the primary metal precursor is spread on supports such as silica, titania, alumina, or carbon. Next is drying followed by calcination in nitrogen or air to get a catalyst synthesized by the impregnation method. Oxides support has been identified to be ideal for such catalysts because of their higher surface area and is ideal for stabilizing metal crystallites as well as boosting the mechanical intensity of the formed catalysts. Alternatively, the precipitation method of catalyst synthesis may be used to place the metal catalyst on the support. This is done by putting the appropriate precipitator into the metal precursor solution, which is then dried and calcined. In this preparation method, regulating and monitoring the pH and temperature is key to obtaining a well-defined nanostructured catalyst [163].
Moreover, inverse models [164,165], whereby the support precursor is placed on the active metal catalyst (or its precursor), can be utilized as a way of differentiating the effect at the MSI interface as well as the variations in morphology and reducibility. Petersen and company [165] have demonstrated that when a certain amount of the support precursor (in alkoxide form) is loaded on the metallic cobalt precursor, it leads to the development of nano-sized islands mimicking support-like structures on the active cobalt phase. The importance of controlling these small islands on the active metal phase may simulate MSI, hence providing insights into the influence brought by MSI regarding the performance of the metal active phase [165].
A variety of FTS catalyst modifications have been developed for the manufacture of alcohol, with the Cu–Co bimetallic catalyst being widely used for the generation of mixed alcohols from syngas[166,167,168]. Nevertheless, the products of this catalyst have a negligible C6+OH proportion [169,170,171]. Professor Ding invented a series of cobalt catalysts that are supported by activated carbon and are used to create mixed higher alcohols that have a greater concentration of C6+OH. Pei et al. [172] discovered that although activated carbon-supported cobalt catalysts normally had low activity, adding the right amount of silica to the modification increased both activity and alcohol selectivity. Liu and colleagues observed that the addition of silica species to CuZnAl catalysts enhanced the selectivity of alcohols. While silica is commonly used to directly assist cobalt catalysts in the catalytic conversion of syngas to hydrocarbons with a higher carbon number, these silica-supported cobalt catalysts often exhibit relatively higher catalytic performance but reduced alcohols formation. Recently, Dai et al. [174] reported a 12.7% alcohols selectivity over a sodium-promoted Co/SiO2 catalyst at increased temperatures, while Ribeiro et al. [173] attained a 10.5% alcohols selectivity at 22.7% CO conversion over LaOx-promoted Co/SiO2 catalysts. Sodium addition, however, resulted in a drop in CO conversion, suggesting a trade-off between the selectivity of alcohol and activity loss. In light of these discoveries, cobalt catalysts backed by modified silica have been developed to produce mixed alcohols with enhanced activity and selectivity in addition to premium liquid fuels.
Hong Du and colleagues [50] synthesised a single-metal cobalt catalyst (Co/SiO2-EOAM) by impregnation, using silica pellets modified with ethanolamine as a support for syngas conversion. During catalyst calcination, the combustion of carbon and nitrogen species improved cobalt dispersion while impeding cobalt oxide reduction by promoting cobalt-support contact. The activity and selectivity of the (Co/SiO2-EOAM) catalyst towards alcohols were much higher than those of the unmodified silica-supported cobalt catalysts (Co/SiO2). The enhanced dispersion of cobalt was credited with this increased activity, and the abundance of metallic cobalt and Co2+ atoms enhanced the selectivity towards alcohols.
5.0. Conclusions
Several metals especially from groups 8-10, mainly Fe, Co, or Ni, can aid carbon monoxide and hydrogen adsorption and dissociation, therefore ideal metals for FT catalysts. Owing to its insignificant water-gas shift activity, better stability, plus higher selectivity towards higher hydrocarbons, cobalt has been widely used for FTS. An optimal number of cobalt nanoparticles must be finely spread on the support to enhance catalyst performance and selectivity towards liquid hydrocarbons. However, the effectiveness of the catalyst with respect to activity and product formation is dependent on the support type and the promoter, as well as the preparation method. Therefore, catalyst support selection is an important factor, i.e., support with superior surface area, optimum pore diameter, and well-definite surface chemistry, because such features can substantially modify the catalyst’s activity. As already been stated, active metals are spread on oxides to enhance the surface area of such metals, and subsequently the quantity of active sites. Nevertheless, an optimum contact within the two (oxide and metal) is required to obtain vastly active metal distribution with the best possible strong metal-oxide contact. Lack of optimum metal-oxide interaction has detrimental effects on the entire features of the catalyst that may lead to poor reducibility and or even poor performance due to inactivity. The preparation technique decides the structure of the catalyst and, accordingly, is vital for such catalyst’s application. It has been realized that a small change in the catalyst synthesis might have a substantial effect on the outcome of such a catalyst regarding catalyst structures and, consequently, have noteworthy differences in catalytic activity and selectivity.
This article further stresses the importance of support modification particularly the reverse model which has the potential to over challenges such as coverage of active metal species and pore blockages by promoters during the preparation of conventional supported catalysts. This modern catalyst synthesis technique can provide an effective substitute for nanoparticle preparation with optimum and relevant particle size distributions for ultimate catalytic research specifically for structure-dependent FTS catalysis studies.
6.0. Acknowledgements
The authors of this manuscript would like to acknowledge the financial support from the University of South Africa (UNISA), the Institute of Catalysis and Energy Solutions (ICES), and the National Research Foundation (NRF).
References
- Johnson, G.R.; Werner, S.; Bell, A.T. An Investigation into the Effects of Mn Promotion on the Activity and Selectivity of Co/SiO2 for Fischer–Tropsch Synthesis: Evidence for Enhanced CO Adsorption and Dissociation. ACS Catal. 2015, 5, 5888–5903. [Google Scholar] [CrossRef]
- K. Cheng et al., “Applied Catalysis A : General Sodium-promoted iron catalysts prepared on different supports for high temperature Fischer – Tropsch synthesis,” “Applied Catal. A, Gen., vol. 502, pp. 204–214, 2015.
- Mazurova, K.; Miyassarova, A.; Eliseev, O.; Stytsenko, V.; Glotov, A.; Stavitskaya, A. Fischer–Tropsch Synthesis Catalysts for Selective Production of Diesel Fraction. Catalysts 2023, 13, 1215. [Google Scholar] [CrossRef]
- Gupta, P.K.; Mahato, A.; Gupta, G.K.; Sahu, G.; Maity, S. Fischer–Tropsch synthesis over Pd promoted cobalt based mesoporous supported catalyst. Oil Gas Sci. Technol. – Rev. d’IFP Energies Nouv. 2021, 76, 21. [Google Scholar] [CrossRef]
- V. Vosoughi, A. K. Dalai, N. Abatzoglou, and Y. Hu, “Applied Catalysis A, General Performances of promoted cobalt catalysts supported on mesoporous alumina for Fischer-Tropsch synthesis,” Appl. Catal. A, Gen., vol. 547, no. May, pp. 155–163, 2017.
- Cano, L.; Blanco, A.G.; Lener, G.; Marchetti, S.; Sapag, K. Effect of the support and promoters in Fischer-Tropsch synthesis using supported Fe catalysts. Catal. Today 2017, 282, 204–213. [Google Scholar] [CrossRef]
- Hillestad, M. Modeling the Fischer–Tropsch Product Distribution and Model Implementation. Chem. Prod. Process. Model. 2015, 10, 147–159. [Google Scholar] [CrossRef]
- Kuipers, E.; Scheper, C.; Wilson, J.; Vinkenburg, I.; Oosterbeek, H. Non-ASF Product Distributions Due to Secondary Reactions during Fischer–Tropsch Synthesis. J. Catal. 1996, 158, 288–300. [Google Scholar] [CrossRef]
- V. A. Online, K. D. Kruit, D. Vervloet, and F. Kapteijn, “Catalysis Science & Technology,” pp. 2210–2213, 2013.
- B. Todic, T. Olewski, N. Nikacevic, and D. B. Bukur, “Modeling of Fischer-Tropsch Product Distribution over Fe-based Catalyst,” vol. 32, no. 2007, pp. 793–798, 2013.
- Förtsch, D.; Pabst, K.; Groß-Hardt, E. The product distribution in Fischer–Tropsch synthesis: An extension of the ASF model to describe common deviations. Chem. Eng. Sci. 2015, 138, 333–346. [Google Scholar] [CrossRef]
- Patzlaff, J.; Liu, Y.; Graffmann, C.; Gaube, J. Studies on product distributions of iron and cobalt catalyzed Fischer–Tropsch synthesis. Appl. Catal. A: Gen. 1999, 186, 109–119. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the Development of Novel Cobalt Fischer−Tropsch Catalysts for Synthesis of Long-Chain Hydrocarbons and Clean Fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef]
- F. Pardo-tarifa, Cobalt catalyst supports for Fischer-Tropsch synthesis. 2017.
- Duyckaerts, N.; Bartsch, M.; Trotuş, I.; Pfänder, N.; Lorke, A.; Schüth, F.; Prieto, G. Intermediate Product Regulation in Tandem Solid Catalysts with Multimodal Porosity for High-Yield Synthetic Fuel Production. Angew. Chem. Int. Ed. 2017, 56, 11480–11484. [Google Scholar] [CrossRef]
- Duyckaerts, N.; Trotuş, I.-T.; Swertz, A.-C.; Schueth, F.; Prieto, G. In Situ Hydrocracking of Fischer–Tropsch Hydrocarbons: CO-Prompted Diverging Reaction Pathways for Paraffin and α-Olefin Primary Products. ACS Catal. 2016, 6, 4229–4238. [Google Scholar] [CrossRef]
- R. Syngas et al., “Design of Cobalt Fischer − Tropsch Catalysts for the Combined Production of Liquid Fuels and Ole fi n Chemicals from Hydrogen- Rich Syngas,” 2021.
- Bezemer, G.L.; Bitter, J.H.; Kuipers, H.P.C.E.; Oosterbeek, H.; Holewijn, J.E.; Xu, X.; Kapteijn, F.; van Dillen, A.J.; de Jong, K.P. Cobalt Particle Size Effects in the Fischer−Tropsch Reaction Studied with Carbon Nanofiber Supported Catalysts. J. Am. Chem. Soc. 2006, 128, 3956–3964. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.; Prieto, G.; Rollán, J. Nanofibrous γ-Al2O3 as support for Co-based Fischer–Tropsch catalysts: Pondering the relevance of diffusional and dispersion effects on catalytic performance. J. Catal. 2009, 263, 292–305. [Google Scholar] [CrossRef]
- Fischer, N.; van Steen, E.; Claeys, M. Structure sensitivity of the Fischer–Tropsch activity and selectivity on alumina supported cobalt catalysts. J. Catal. 2013, 299, 67–80. [Google Scholar] [CrossRef]
- Tuxen, A.; Carenco, S.; Chintapalli, M.; Chuang, C.-H.; Escudero, C.; Pach, E.; Jiang, P.; Borondics, F.; Beberwyck, B.; Alivisatos, A.P.; et al. Size-Dependent Dissociation of Carbon Monoxide on Cobalt Nanoparticles. J. Am. Chem. Soc. 2013, 135, 2273–2278. [Google Scholar] [CrossRef] [PubMed]
- Karaca, H.; Safonova, O.V.; Chambrey, S.; Fongarland, P.; Roussel, P.; Griboval-Constant, A.; Lacroix, M.; Khodakov, A.Y. Structure and catalytic performance of Pt-promoted alumina-supported cobalt catalysts under realistic conditions of Fischer–Tropsch synthesis. J. Catal. 2011, 277, 14–26. [Google Scholar] [CrossRef]
- G. Prieto, P. Concepción, R. Murciano, and A. Martínez, “The impact of pre-reduction thermal history on the metal surface topology and site-catalytic activity of Co = SiO 2 Fischer – Tropsch catalysts,” J. Catal., vol. 302, pp. 37–48, 2013.
- T. Jermwongratanachai et al., “Fischer – Tropsch synthesis : TPR and XANES analysis of the impact of simulated regeneration cycles on the reducibility of Co / alumina catalysts with different promoters ( Pt, Ru, Re, Ag, Au, Rh, Ir ),” Catal. Today, vol. 228, pp. 15–21, 2014.
- F. Morales and B. M. Weckhuysen, “Promotion Effects in Co-Based Fischer—Tropsch Catalysis,” ChemInform, vol. 38, no. 2, 2007.
- K. M. Cook, S. Poudyal, J. T. Miller, C. H. Bartholomew, and W. C. Hecker, “Applied Catalysis A : General Reducibility of alumina-supported cobalt Fischer – Tropsch catalysts : Effects of noble metal type, distribution, retention, chemical state, bonding, and influence on cobalt crystallite size,” “Applied Catal. A, Gen., vol. 449, pp. 69–80, 2012.
- Garbarino, G.; Cavattoni, T.; Riani, P.; Busca, G. Support effects in metal catalysis: a study of the behavior of unsupported and silica-supported cobalt catalysts in the hydrogenation of CO2 at atmospheric pressure. Catal. Today 2019, 345, 213–219. [Google Scholar] [CrossRef]
- H. K. Æ. B. J. Æ. J. T. Wolan, “Silica-Supported Cobalt Catalysts for Fischer – Tropsch Synthesis : Effects of Calcination Temperature and Support Surface Area on Cobalt Silicate Formation,” pp. 72–78, 2009.
- G. Okoye-chine, C. O. L. Mbuya, T. S. Ntelane, M. Moyo, and D. Hildebrandt, “The effect of silanol groups on the metal-support interactions in silica- supported cobalt Fischer-Tropsch catalysts. A temperature programmed surface reaction,” J. Catal., vol. 381, pp. 121–129, 2020.
- Uchisawa, J.; Tango, T.; Caravella, A.; Hara, S.; Haneda, M.; Murakami, T.; Nakagawa, H.; Nanba, T.; Obuchi, A. Effects of the Extent of Silica Doping and the Mesopore Size of an Alumina Support on Activity as a Diesel Oxidation Catalyst. Ind. Eng. Chem. Res. 2014, 53, 7992–7998. [Google Scholar] [CrossRef]
- Min, B.; Santra, A.; Goodman, D. Understanding silica-supported metal catalysts: Pd/silica as a case study. Catal. Today 2003, 85, 113–124. [Google Scholar] [CrossRef]
- Xu, L.-X.; He, C.-H.; Zhu, M.-Q.; Wu, K.-J.; Lai, Y.-L. Silica-Supported Gold Catalyst Modified by Doping with Titania for Cyclohexane Oxidation. Catal. Lett. 2007, 118, 248–253. [Google Scholar] [CrossRef]
- Rytter, E.; Eri, S.; Skagseth, T.H.; Schanke, D.; Bergene, E.; Myrstad, R.; Lindvåg, A. Catalyst Particle Size of Cobalt/Rhenium on Porous Alumina and the Effect on Fischer−Tropsch Catalytic Performance. Ind. Eng. Chem. Res. 2007, 46, 9032–9036. [Google Scholar] [CrossRef]
- Fischer-Tropsch reaction over alumina-supported cobalt catalyst : activation using H 2 and CO Master ’ s Dissertation Report submitted in partial fulfilment of Magister Technologiae In Chemical Engineering In the FACULTY OF EGINEERING AND THE BUILT ENVIRONMENT Of the University of Johannesburg Compiled by Student Number Supervised by Co-Supervised by : Phathutshedzo Rodney Khangale : Prof Kalala Jalama : Prof Reinout Meijboom,” 2012.
- Rane, S.; Borg. ; Rytter, E.; Holmen, A. Relation between hydrocarbon selectivity and cobalt particle size for alumina supported cobalt Fischer–Tropsch catalysts. Appl. Catal. A: Gen. 2012, 437-438, 10–17. [Google Scholar] [CrossRef]
- Shimura, K.; Miyazawa, T.; Hanaoka, T.; Hirata, S. Fischer–Tropsch synthesis over alumina supported cobalt catalyst: Effect of promoter addition. Appl. Catal. A: Gen. 2015, 494, 1–11. [Google Scholar] [CrossRef]
- Jabbari, A.; Ghasemzadeh, K.; Khajavi, P.; Assa, F.; Abdi, M.; Babaluo, A.; Basile, A. Surface modification of α-alumina support in synthesis of silica membrane for hydrogen purification. Int. J. Hydrogen Energy 2014, 39, 18585–18591. [Google Scholar] [CrossRef]
- Castillo, R.; Koch, B.; Ruiz, P.; Delmon, B. Influence of the Amount of Titania on the Texture and Structure of Titania Supported on Silica. J. Catal. 1996, 161, 524–529. [Google Scholar] [CrossRef]
- S. Storsæter, B. Tøtdal, J. C. Walmsley, B. S. Tanem, and A. Holmen, “Characterization of alumina-, silica-, and titania-supported cobalt Fischer-Tropsch catalysts,” J. Catal., vol. 236, no. 1, pp. 139–152, Nov. 2005.
- Morales, F.; Desmit, E.; Degroot, F.; Visser, T.; Weckhuysen, B.M. Effects of manganese oxide promoter on the CO and H2 adsorption properties of titania-supported cobalt Fischer–Tropsch catalysts. J. Catal. 2007, 246, 91–99. [Google Scholar] [CrossRef]
- Di Fronzo, A.; Pirola, C.; Comazzi, A.; Galli, F.; Bianchi, C.; Di Michele, A.; Vivani, R.; Nocchetti, M.; Bastianini, M.; Boffito, D. Co-based hydrotalcites as new catalysts for the Fischer–Tropsch synthesis process. Fuel 2013, 119, 62–69. [Google Scholar] [CrossRef]
- Concepción, P.; López, C.; Martínez, A.; Puntes, V.F. Characterization and catalytic properties of cobalt supported on delaminated ITQ-6 and ITQ-2 zeolites for the Fischer–Tropsch synthesis reaction. J. Catal. 2004, 228, 321–332. [Google Scholar] [CrossRef]
- Sartipi, S.; Alberts, M.; Meijerink, M.J.; Keller, T.C.; Pérez-Ramírez, J.; Gascon, J.; Kapteijn, F. Towards Liquid Fuels from Biosyngas: Effect of Zeolite Structure in Hierarchical-Zeolite-Supported Cobalt Catalysts. ChemSusChem 2013, 6, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-C.; Lee, S.; Cho, K.; Na, K.; Lee, C.; Ryoo, R. Mesoporous MFI Zeolite Nanosponge Supporting Cobalt Nanoparticles as a Fischer–Tropsch Catalyst with High Yield of Branched Hydrocarbons in the Gasoline Range. ACS Catal. 2014, 4, 3919–3927. [Google Scholar] [CrossRef]
- Solomonik, I.G.; Gorshkov, A.S.; Mordkovich, V.Z. Fischer-Tropsch Synthesis over a Cobalt Catalyst Supported on Titania-Doped Silicon Carbide. Catal. Ind. 2020, 12, 235–243. [Google Scholar] [CrossRef]
- Xiong, H.; Motchelaho, M.A.; Moyo, M.; Jewell, L.L.; Coville, N.J. Correlating the preparation and performance of cobalt catalysts supported on carbon nanotubes and carbon spheres in the Fischer–Tropsch synthesis. J. Catal. 2011, 278, 26–40. [Google Scholar] [CrossRef]
- J. Andrew, “Catalysis Science & Technology Fischer – Tropsch synthesis of hydrocarbons from,” pp. 2210–2229, 2014.
- C. G. Okoye-chine, M. Moyo, and D. Hildebrandt, “Journal of Industrial and Engineering Chemistry Fischer – Tropsch synthesis : The effect of hydrophobicity on silica-supported iron catalysts,” J. Ind. Eng. Chem., vol. 97, pp. 426–433, 2021.
- J. Jung, S. Woo, and D. Ju, “Fischer – Tropsch Synthesis over cobalt based catalyst supported on different mesoporous silica,” Catal. Today, vol. 185, no. 1, pp. 168–174, 2012.
- H. Du, M. Jiang, M. Zhao, X. Ma, and Z. Xu, “ScienceDirect Activity and selectivity enhancement of silica supported cobalt catalyst for alcohols production from syngas via Fischer-Tropsch synthesis,” Int. J. Hydrogen Energy, vol. 47, no. 7, pp. 4559–4567, 2021.
- H. Singleton, R. Oukaci, and J. G. Goodwin, “Fischer-Tropsch Activity for Non Promoted Cobalton-Alumina Catalysts,” vol. 1, no. 12, 2001.
- Bezemer, G.; Radstake, P.; Falke, U.; Oosterbeek, H.; Kuipers, H.; Vandillen, A.; Dejong, K. Investigation of promoter effects of manganese oxide on carbon nanofiber-supported cobalt catalysts for Fischer–Tropsch synthesis. J. Catal. 2006, 237, 152–161. [Google Scholar] [CrossRef]
- C. H. Bartholomew, “Effects of Support and Dispersion on the CO Hydrogenation Activity / Selectivity Properties of Cobalt,” vol. 88, 1984.
- Z. Yu, Ø. Borg, D. Chen, C. Enger, V. Frøseth, and E. Rytter, “Carbon Nanofiber Supported Cobalt Catalysts for Fischer – Tropsch Synthesis with High Activity and Selectivity Carbon nanofiber supported cobalt catalysts for Fischer – Tropsch synthesis with high activity and selectivity,” no. June, pp. 1–6, 2006.
- Iglesia, E. Design, synthesis, and use of cobalt-based Fischer-Tropsch synthesis catalysts. Appl. Catal. A: Gen. 1997, 161, 59–78. [Google Scholar] [CrossRef]
- E. S. P. B. V, “Design of Fischer-Tropsch Catalysts by Pulse Surface Reaction Rate Analysis II. Selective Production of Liquid Fuel Fraction on Ruthenium / Alumina Catalyst Promoted by Rare Earth Oxides,” vol. 38, pp. 61–69, 1988.
- Bertole, C.J.; A Mims, C.; Kiss, G. Support and rhenium effects on the intrinsic site activity and methane selectivity of cobalt Fischer–Tropsch catalysts. J. Catal. 2004, 221, 191–203. [Google Scholar] [CrossRef]
- K. Jalama et al., “Applied Catalysis A : General A comparison of Au / Co / Al 2 O 3 and Au / Co / SiO 2 catalysts in the Fischer – Tropsch reaction,” vol. 395, pp. 1–9, 2011.
- Martínez, C. López, F. Márquez, and I. Díaz, “Fischer-Tropsch synthesis of hydrocarbons over mesoporous Co/SBA-15 catalysts: The influence of metal loading, cobalt precursor, and promoters,” J. Catal., vol. 220, no. 2, pp. 486–499, 2003.
- Ernst, S. Libs, P. Chaumette, and A. Kiennemann, “Preparation and characterization of Fischer – Tropsch active Co / SiO 2 catalysts,” vol. 186, pp. 145–168, 1999.
- Saib, A.; Claeys, M.; van Steen, E. Silica supported cobalt Fischer–Tropsch catalysts: effect of pore diameter of support. Catal. Today 2002, 71, 395–402. [Google Scholar] [CrossRef]
- Z. Gholami, Z. Tišler, and V. Rubáš, “Recent advances in Fischer-Tropsch synthesis using cobalt-based catalysts : a review on supports, promoters, and reactors Recent advances in Fischer-Tropsch synthesis using,” Catal. Rev., vol. 63, no. 3, pp. 512–595, 2021.
- Fu, T.; Li, Z. Review of recent development in Co-based catalysts supported on carbon materials for Fischer–Tropsch synthesis. Chem. Eng. Sci. 2015, 135, 3–20. [Google Scholar] [CrossRef]
- Y. Chen, J. Wei, M. S. Duyar, V. V Ordomsky, A. Y. Khodakov, and J. Liu, “To cite this version : Chem Soc Rev,” 2022.
- Jiang, Z.-S.; Zhao, Y.-H.; Huang, C.-F.; Song, Y.-H.; Li, D.-P.; Liu, Z.-T.; Liu, Z.-W. Metal-support interactions regulated via carbon coating – A case study of Co/SiO2 for Fischer-Tropsch synthesis. Fuel 2018, 226, 213–220. [Google Scholar] [CrossRef]
- Cydric, A. Nzihou, P. Serp, K. Soulantica, and D. Pham, “Applied Catalysis A, General Cobalt catalysts on carbon-based materials for Fischer-Tropsch synthesis : a review,” Appl. Catal. A, Gen., vol. 609, no. 20, p. 117906, 2021. 20 November.
- Iqbal, S.; Davies, T.E.; Morgan, D.J.; Karim, K.; Hayward, J.S.; Bartley, J.K.; Taylor, S.H.; Hutchings, G.J. Fischer Tropsch synthesis using cobalt based carbon catalysts. Catal. Today 2015, 275, 35–39. [Google Scholar] [CrossRef]
- Iglesia, E.; Reyes, S.C.; Madon, R.J. Transport-enhanced $alpha;-olefin readsorption pathways in Ru-catalyzed hydrocarbon synthesis. J. Catal. 1991, 129, 238–256. [Google Scholar] [CrossRef]
- C. S. Catalysts, J. Bao, J. He, Y. Zhang, Y. Yoneyama, and N. Tsubaki, “A Core / Shell Catalyst Produces a Spatially Confined Effect and Shape Selectivity in a Consecutive Reaction,” pp. 353–356, 2008.
- Khodakov, A.Y.; Griboval-Constant, A.; Bechara, R.; Zholobenko, V.L. Pore Size Effects in Fischer Tropsch Synthesis over Cobalt-Supported Mesoporous Silicas. J. Catal. 2002, 206, 230–241. [Google Scholar] [CrossRef]
- Wang, K.; Horlyck, J.; Finn, M.T.; Mesa, M.G.; Voutchkova-Kostal, A. Electronic Effects of Support Doping on Hydrotalcite-Supported Iridium N-Heterocyclic Carbene Complexes. ACS Omega 2022, 7, 24705–24713. [Google Scholar] [CrossRef] [PubMed]
- Yaseneva, P.; An, N.; Finn, M.; Tidemann, N.; Jose, N.; Voutchkova-Kostal, A.; Lapkin, A. Continuous synthesis of doped layered double hydroxides in a meso-scale flow reactor. 2019. [Google Scholar] [CrossRef]
- V. K. Yadav and T. Das, “Reaction Chemistry & Engineering the FeMnO δ / MgO – Al 2 O 3 catalyst used for the,” pp. 2298–2312, 2022.
- Kraum, M.; Baerns, M. Fischer–Tropsch synthesis: the influence of various cobalt compounds applied in the preparation of supported cobalt catalysts on their performance. Appl. Catal. A: Gen. 1999, 186, 189–200. [Google Scholar] [CrossRef]
- Y. Lee, B. Kim, H. Park, S. Ahn, K. Kim, and H. Roh, “Customized Ni – MgO – Al 2 O 3 catalyst for carbon dioxide reforming of coke oven gas : Optimization of preparation method and co-precipitation pH,” vol. 42, no. September, 2020.
- Nakanishi, M.; Uddin, A.; Kato, Y.; Nishina, Y.; Hapipi, A.M. Effects of preparation method on the properties of cobalt supported β-zeolite catalysts for Fischer-Tropsch synthesis. Catal. Today 2017, 291, 124–132. [Google Scholar] [CrossRef]
- M. Arsalanfar, A. A. Mirzaei, H. R. Bozorgzadeh, and A. Samimi, “CHEMISTRY A Review of Fischer-Tropsch Synthesis on the Cobalt Based Catalysts,” 2014.
- Mohammadi, H.; Nekobahr, E.; Akhtari, J.; Saeedi, M.; Akbari, J.; Fathi, F. Synthesis and characterization of magnetite nanoparticles by co-precipitation method coated with biocompatible compounds and evaluation of in-vitro cytotoxicity. Toxicol. Rep. 2021, 8, 331–336. [Google Scholar] [CrossRef]
- Kirichenko, G. W. Graham, W. Chun, and R. W. Mccabe, Effect of coprecipitation conditions on the surface area, phase composition, and reducibility of CeO2-ZrO2-Y2O3 materials for automotive three-Way catalysts, vol. 118. Elsevier Masson SAS, 1998.
- M. Kipnis, E. Volnina, I. Belostotsky, R. Galkin, and N. Zhilyaeva, “Effective Cu / ZnO / Al 2 O 3 Catalyst for Methanol Production : Synthesis, Activation, Catalytic Performance, and Regeneration.
- Dubey, R.; Rajesh, Y.; More, M. Synthesis and Characterization of SiO2 Nanoparticles via Sol-gel Method for Industrial Applications. Mater. Today: Proc. 2015, 2, 3575–3579. [Google Scholar] [CrossRef]
- M. Parashar, V. Kumar, and S. Ranbir, “Metal oxides nanoparticles via sol – gel method : a review on synthesis, characterization and applications,” J. Mater. Sci. Mater. Electron., vol. 31, no. 5, pp. 3729–3749, 2020.
- Bokov, D.; Jalil, A.T.; Chupradit, S.; Suksatan, W.; Ansari, M.J.; Shewael, I.H.; Valiev, G.H.; Kianfar, E. Nanomaterial by Sol-Gel Method: Synthesis and Application. Adv. Mater. Sci. Eng. 2021, 2021, 1–21. [Google Scholar] [CrossRef]
- Lu, J.; Elam, J.W.; Stair, P.C. Synthesis and Stabilization of Supported Metal Catalysts by Atomic Layer Deposition. Accounts Chem. Res. 2013, 46, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.J.; Mackus, A.J.M.; Verheijen, M.A.; van der Marel, C.; Kessels, W.M.M. Supported Core/Shell Bimetallic Nanoparticles Synthesis by Atomic Layer Deposition. Chem. Mater. 2012, 24, 2973–2977. [Google Scholar] [CrossRef]
- M. Zhang, Z. Lou, M. Zhou, and H. Wei, “Accepted Article.
- G. Vinothkumar, S. Rengaraj, P. Arunkumar, S. W. Cha, and K. S. Babu, “and La 3 + ) - Doped Cerium Oxide Nanoparticles for Enhanced Multienzyme-Mimetic and Hydroxyl Radical Scavenging Activity,” 2019.
- Ichinose and, H. Senzu, “A Surface Sol - Gel Process of TiO 2 and Other Metal Oxide Films with Molecular,” vol. 4756, no. 10, pp. 1296–1298, 1997.
- Yan, W.; Mahurin, S.M.; Overbury, S.H.; Dai, S. Nanoengineering catalyst supports via layer-by-layer surface functionalization. Top. Catal. 2006, 39, 199–212. [Google Scholar] [CrossRef]
- Luo, J.; Ersen, O.; Chu, W.; Dintzer, T.; Petit, P.; Petit, C. Anchoring and promotion effects of metal oxides on silica supported catalytic gold nanoparticles. J. Colloid Interface Sci. 2016, 482, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Savost’yanov, A.P.; Yakovenko, R.E.; Sulima, S.I.; Bakun, V.G.; Narochnyi, G.B.; Chernyshev, V.M.; Mitchenko, S.A. The impact of Al 2 O 3 promoter on an efficiency of C 5+ hydrocarbons formation over Co/SiO 2 catalysts via Fischer-Tropsch synthesis. Catal. Today 2017, 279, 107–114. [Google Scholar] [CrossRef]
- Ma, W.-P.; Ding, Y.-J.; Lin, L.-W. Fischer−Tropsch Synthesis over Activated-Carbon-Supported Cobalt Catalysts: Effect of Co Loading and Promoters on Catalyst Performance. Ind. Eng. Chem. Res. 2004, 43, 2391–2398. [Google Scholar] [CrossRef]
- Y. Huang, W. Li, J. Ren, and X. Qu, “Accepted Article.
- B. Wang, F. Liu, Y. Wu, Y. Chen, B. Weng, and C. M. Li, “Sensors and Actuators B : Chemical Synthesis of catalytically active multielement-doped carbon dots and application for colorimetric detection of glucose,” Sensors Actuators B. Chem., vol. 255, pp. 2601–2607, 2018.
- M. Gao, X. Lu, M. Chi, S. Chen, and C. Wang, “INORGANIC CHEMISTRY FRONTIERS nano fi bers and their sensitive colorimetric detection of sul fi te and L -cysteine †,” pp. 1862–1869, 2017.
- M. P. C. Van Etten, M. E. De Laat, E. J. M. Hensen, and I. A. W. Filot, “Unraveling the Role of Metal − Support Interactions on the Structure Sensitivity of Fischer − Tropsch Synthesis,” 2023.
- Y. Zhang et al., “Ru/TiO 2 Catalysts with Size-Dependent Metal/Support Interaction for Tunable Reactivity in Fischer − Tropsch Synthesis,” 2020.
- Q. Cheng, Y. Liu, S. Lyu, Y. Tian, Q. Ma, and X. Li, “Chinese Journal of Chemical Engineering Manipulating metal-support interactions of metal catalysts for Fischer-Tropsch synthesis,” Chinese J. Chem. Eng., vol. 35, pp. 220–230, 2021.
- Macheli, L.; Carleschi, E.; Doyle, B.P.; Leteba, G.; van Steen, E. Tuning catalytic performance in Fischer-Tropsch synthesis by metal-support interactions. J. Catal. 2020, 395, 70–79. [Google Scholar] [CrossRef]
- Zanella, R.; Giorgio, S.; Henry, C.R.; Louis, C. Alternative Methods for the Preparation of Gold Nanoparticles Supported on TiO2. J. Phys. Chem. B 2002, 106, 7634–7642. [Google Scholar] [CrossRef]
- L. A. Calzada et al., “Applied Catalysis B : Environmental Au-Ru / TiO 2 prepared by deposition-precipitation with urea : Relevant synthesis parameters to obtain bimetallic particles,” Appl. Catal. B Environ., vol. 264, no. January 2019, p. 118503, 2020.
- Manawi, Y.M.; Ihsanullah; Samara, A. ; Al-Ansari, T.; Atieh, M.A. A Review of Carbon Nanomaterials’ Synthesis via the Chemical Vapor Deposition (CVD) Method. Materials 2018, 11, 822. [Google Scholar] [CrossRef]
- K. Dahmen, “Chemical Vapor Deposition,” vol. 2, 2002.
- Yang, N.; Bent, S.F. Investigation of inherent differences between oxide supports in heterogeneous catalysis in the absence of structural variations. J. Catal. 2017, 351, 49–58. [Google Scholar] [CrossRef]
- Xu, C.; Chen, G.; Zhao, Y.; Liu, P.; Duan, X.; Gu, L.; Fu, G.; Yuan, Y.; Zheng, N. Interfacing with silica boosts the catalysis of copper. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- L. C. Almeida et al., “Fischer-tropsch catalyst deposition on metallic structured supports,” Stud. Surf. Sci. Catal., vol. 167, pp. 79–84, 2007.
- Shimura, K.; Miyazawa, T.; Hanaoka, T.; Hirata, S. Fischer–Tropsch synthesis over TiO2 supported cobalt catalyst: Effect of TiO2 crystal phase and metal ion loading. Appl. Catal. A: Gen. 2013, 460-461, 8–14. [Google Scholar] [CrossRef]
- N. C. Shiba, X. Liu, H. Mao, X. Qian, D. Hildebrandt, and Y. Yao, “Effect of Ru-promotion on the catalytic performance of a cobalt-based Fischer-Tropsch catalyst activated in syngas or H 2,” Fuel, vol. 320, no. December 2021, p. 123939, 2022.
- Tucker, C.L.; Ragoo, Y.; Mathe, S.; Macheli, L.; Bordoloi, A.; Rocha, T.C.; Govender, S.; Kooyman, P.J.; van Steen, E. Manganese promotion of a cobalt Fischer-Tropsch catalyst to improve operation at high conversion. J. Catal. 2022, 411, 97–108. [Google Scholar] [CrossRef]
- Khasu, M.; Marquart, W.; Kooyman, P.J.; Drivas, C.; Isaacs, M.A.; Mayer, A.J.; Dann, S.E.; Kondrat, S.A.; Claeys, M.; Fischer, N. Empowering Catalyst Supports: A New Concept for Catalyst Design Demonstrated in the Fischer–Tropsch Synthesis. ACS Catal. 2023, 13, 6862–6872. [Google Scholar] [CrossRef]
- Sukkathanyawat, H.; Tungkamani, S.; Phongaksorn, M.; Rattana, T.; Narataruksa, P.; Yoosuk, B. Promoter Effect on the Physico-chemical Properties of Cobalt Based Catalyst for CO Hydrogenation. Energy Procedia 2015, 79, 372–377. [Google Scholar] [CrossRef]
- van Koppen, L.M.; Dugulan, A.I.; Hensen, E.J.; Bezemer, G.L. Tuning stability of titania-supported Fischer-Tropsch catalysts: impact of surface area and noble metal promotion. Catal. Today 2023. [Google Scholar] [CrossRef]
- Diehl, F.; Khodakov, A.Y. Promotion of Cobalt Fischer-Tropsch Catalysts with Noble Metals: a Review. Oil Gas Sci. Technol. – Rev. d’IFP Energies Nouv. 2008, 64, 11–24. [Google Scholar] [CrossRef]
- T. O. Eschemann, J. Oenema, and K. P. De Jong, “Effects of noble metal promotion for Co / TiO 2 Fischer-Tropsch catalysts,” Catal. Today, vol. 261, pp. 60–66, 2016.
- M. De Beer, A. Kunene, D. Nabaho, M. Claeys, and E. Van Steen, “Technical and economic aspects of promotion of cobalt-based Fischer-Tropsch catalysts by noble metals-a review,” J. South. African Inst. Min. Metall., vol. 114, no. 2, pp. 157–165, 2014.
- W. Ma, G. Jacobs, R. A. Keogh, D. B. Bukur, and B. H. Davis, “Applied Catalysis A : General Fischer – Tropsch synthesis : Effect of Pd, Pt, Re, and Ru noble metal promoters on the activity and selectivity of a 25 % Co / Al 2 O 3 catalyst,” “Applied Catal. A, Gen., vol. 437–438, pp. 1–9, 2012.
- H. Xiong, M. A. Motchelaho, M. Moyo, L. L. Jewell, and N. J. Coville, “Effect of Group I alkali metal promoters on Fe / CNT catalysts in Fischer – Tropsch synthesis,” FUEL, vol. 150, pp. 687–696, 2015.
- Y. Yang, H. Zhang, H. Ma, W. Qian, Q. Sun, and W. Ying, “Effect of alkalis ( Li, Na, and K ) on precipitated iron-based catalysts for high-temperature Fischer-Tropsch synthesis,” Fuel, vol. 326, no. June, p. 125090, 2022.
- Zheng, Y.; Xu, C.; Zhang, X.; Wu, Q.; Liu, J. Synergistic Effect of Alkali Na and K Promoter on Fe-Co-Cu-Al Catalysts for CO2 Hydrogenation to Light Hydrocarbons. Catalysts 2021, 11, 735. [Google Scholar] [CrossRef]
- Johnson, G.R.; Bell, A.T. Effects of Lewis acidity of metal oxide promoters on the activity and selectivity of Co-based Fischer–Tropsch synthesis catalysts. J. Catal. 2016, 338, 250–264. [Google Scholar] [CrossRef]
- S. Saheli, A. Reza, R. Azim, M. Michal, and D. Vaclav, “Effect of Synthetic Route and Metal Oxide Promoter on Cobalt-Based Catalysts for Fischer – Tropsch Synthesis,” Catal. Letters, vol. 148, no. 11, pp. 3557–3569, 2018.
- Wang, D.; Chen, L.; Li, G.; Wang, Z.; Li, X.; Hou, B. Cobalt-based Fischer-Tropsch synthesis: Effect of the catalyst granule thermal conductivity on the catalytic performance. Mol. Catal. 2021, 502, 111395. [Google Scholar] [CrossRef]
- J. Li, C. Zhang, X. Cheng, M. Qing, J. Xu, and B. Wu, “Applied Catalysis A : General Effects of alkaline-earth metals on the structure, adsorption and catalytic behavior of iron-based Fischer – Tropsch synthesis catalysts,” “Applied Catal. A, Gen., vol. 464–465, pp. 10–19, 2013.
- H. Ma, Y. Yang, H. Fu, H. Zhang, W. Qian, and Q. Sun, “Effect of alkaline-earth metals ( Mg, Ca, Sr, and Ba ) on precipitated iron-based catalysts for high-temperature Fischer-Tropsch synthesis of light olefins,” Fuel, vol. 357, no. PA, p. 129605, 2024.
- M. Z. Leguizam et al., “Fischer – Tropsch Synthesis : Effect of the Promoter ’ s Ionic Charge and Valence Level Energy on Activity,” pp. 408–426, 2021.
- Horáček, J. Fischer-Tropsch synthesis, the effect of promoters, catalyst support, and reaction conditions selection. Monatshefte Fuer Chemie/chemical Mon. 2020, 151, 649–675. [Google Scholar] [CrossRef]
- Y. Suo, Y. Yao, Y. Zhang, S. Xing, and Z. Yuan, “Journal of Industrial and Engineering Chemistry Recent advances in cobalt-based Fischer-Tropsch synthesis catalysts,” J. Ind. Eng. Chem., vol. 115, pp. 92–119, 2022.
- H. Wu, Y. Yang, H. Suo, M. Qing, L. Yan, and B. Wu, “Journal of Molecular Catalysis A : Chemical Effects of ZrO 2 promoter on physic-chemical properties and activity of Co / TiO 2 – SiO 2 Fischer – Tropsch catalysts,” vol. 396, pp. 108–119, 2015.
- C. Lu, Y. Lin, and I. Wang, “Naphthalene hydrogenation over Pt / TiO 2 – ZrO 2 and the behavior of strong metal – support interaction ( SMSI ),” vol. 198, pp. 223–234, 2000.
- M. Ruppert and T. Paryjczak, “Pt / ZrO 2 / TiO 2 catalysts for selective hydrogenation of crotonaldehyde : Tuning the SMSI effect for optimum performance,” vol. 320, pp. 80–90, 2007.
- Damyanova, S.; Grange, P.; Delmon, B. Surface Characterization of Zirconia-Coated Alumina and Silica Carriers. J. Catal. 1997, 168, 421–430. [Google Scholar] [CrossRef]
- s2.0-S002195178571113X-main.pdf.”.
- E. Iglesia, “Fischer-Tropsch Synthesis on Cobalt Catalysts: Structural Requirements and Reaction Pathways,” vol. 107, pp. 153–162, 1997.
- Corma and, F. V Melo, “Studies in Surface Science and Catalysis 130 A. Corma, F.V. Melo, S.Mendioroz and J.L.G. Fierro (Editors) 9 2000 Elsevier Science B.V. All rights reserved. 1097,” pp. 1097–1102, 2000.
- F. Tropsch and L. U. Okonye, “Sustainable Energy & Fuels Contributing to energy sustainability : a review of mesoporous material supported catalysts for,” pp. 79–107, 2021.
- Vosoughi, V.; Badoga, S.; Dalai, A.K.; Abatzoglou, N. Modification of mesoporous alumina as a support for cobalt-based catalyst in Fischer-Tropsch synthesis. Fuel Process. Technol. 2017, 162, 55–65. [Google Scholar] [CrossRef]
- M. Lualdi, Fischer-Tropsch Synthesis over Cobalt-based Catalysts for BTL Applications. 2012.
- Jacobs, G.; Das, T.K.; Zhang, Y.; Li, J.; Racoillet, G.; Davis, B.H. Fischer–Tropsch synthesis: support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl. Catal. A: Gen. 2002, 233, 263–281. [Google Scholar] [CrossRef]
- et al. , “Fischer-Tropsch synthesis over γ-alumina-supported cobalt catalysts: Effect of support variables,” J. Catal., vol. 248, no. 1, pp. 89–100, May 2007.
- Cai, Z.; Li, J.; Liew, K.; Hu, J. Effect of La2O3-dopping on the Al2O3 supported cobalt catalyst for Fischer-Tropsch synthesis. J. Mol. Catal. A: Chem. 2010, 330, 10–17. [Google Scholar] [CrossRef]
- Z. Li, J. Wu, and L. Wu, “Effect of Zr, Ca and Mn as promoters on the Co/SiC catalysts for the Fischer–Tropsch synthesis,” React. Kinet. Mech. Catal., vol. 122, no. 2, pp. 887–900, 2017.
- Jacobs, G.; Das, T.K.; Zhang, Y.; Li, J.; Racoillet, G.; Davis, B.H. Fischer–Tropsch synthesis: support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl. Catal. A: Gen. 2002, 233, 263–281. [Google Scholar] [CrossRef]
- R. A. Iloy and K. Jalama, “E ff ect of Operating Temperature, Pressure and Silica-Supported Cobalt Catalyst in CO 2,” 2019.
- V. Garcilaso, J. Barrientos, L. F. Bobadilla, O. H. Laguna, and M. Boutonnet, “Promoting effect of CeO 2, ZrO 2 and Ce / Zr mixed oxides on Co / g -Al 2 O 3 catalyst for Fischer-Tropsch synthesis,” vol. 132, pp. 1141–1150, 2019.
- J. Barrientos, V. Garcilaso, B. Venezia, and A. Aho, “Fischer – Tropsch Synthesis Over Zr-Promoted Co / γ-Al 2 O 3 Catalysts,” Top. Catal., vol. 60, no. 17, pp. 1285–1298, 2017.
- Wu, H.; Yang, Y.; Suo, H.; Qing, M.; Yan, L.; Wu, B.; Xu, J.; Xiang, H.; Li, Y. Effects of ZrO 2 promoter on physic-chemical properties and activity of Co/TiO 2 –SiO 2 Fischer–Tropsch catalysts. J. Mol. Catal. A: Chem. 2015, 396, 108–119. [Google Scholar] [CrossRef]
- Breejen, J.P.D.; Frey, A.M.; Yang, J.; Holmen, A.; van Schooneveld, M.M.; de Groot, F.M.F.; Stephan, O.; Bitter, J.H.; de Jong, K.P. A Highly Active and Selective Manganese Oxide Promoted Cobalt-on-Silica Fischer–Tropsch Catalyst. Top. Catal. 2011, 54, 768–777. [Google Scholar] [CrossRef]
- F. Morales et al., “In Situ X-ray Absorption of Co / Mn / TiO 2 Catalysts for Fischer - Tropsch Synthesis,” pp. 16201–16207, 2004.
- Monshi, A.; Foroughi, M.R.; Monshi, M.R. Modified Scherrer Equation to Estimate More Accurately Nano-Crystallite Size Using XRD. World J. Nano Sci. Eng. 2012, 02, 154–160. [Google Scholar] [CrossRef]
- G. R. Moradi, “Promotion of Co = SiO 2 Fischer – Tropsch catalysts with zirconium,” vol. 4, pp. 27–32, 2003.
- L. Bezemer, Cobalt supported on carbon nanofibers as catalysts for the Fischer-Tropsch synthesis.
- Bao, K. Liew, and J. Li, “Journal of Molecular Catalysis A : Chemical Fischer – Tropsch synthesis on CaO-promoted Co / Al 2 O 3 catalysts,” vol. 0, pp. 47–51, 2009.
- S. C. Reyes, R. J. Madon, and S. L. Soled, “Selectivity Control and Catalyst Design in the Fischer-Tropsch Synthesis : Sites, Pellets, and Reactors,” vol. 39, 1993.
- Enger, B.C.; Fossan. -L.; Borg,.; Rytter, E.; Holmen, A. Modified alumina as catalyst support for cobalt in the Fischer–Tropsch synthesis. J. Catal. 2011, 284, 9–22. [Google Scholar] [CrossRef]
- M. Nurunnabi and S. Q. Turn, “Pore size effects on Ru / SiO 2 catalysts with Mn and Zr promoters for Fischer – Tropsch synthesis,” Fuel Process. Technol., vol. 130, pp. 155–164, 2015.
- F. Gu, “La, Mn and Zn promoted microporous iron catalysts with high productivity and stability,” pp. 147–159, 2016.
- Dinse, M. Aigner, M. Ulbrich, G. R. Johnson, and A. T. Bell, “Effects of Mn promotion on the activity and selectivity of Co / SiO 2 for Fischer – Tropsch Synthesis,” vol. 288, pp. 104–114, 2012.
- Eschemann, T.O.; Oenema, J.; de Jong, K.P. Effects of noble metal promotion for Co/TiO2 Fischer-Tropsch catalysts. Catal. Today 2016, 261, 60–66. [Google Scholar] [CrossRef]
- Z. Li, J. Wu, and L. Wu, “Effect of Zr, Ca and Mn as promoters on the Co / SiC catalysts for the Fischer – Tropsch synthesis,” React. Kinet. Mech. Catal., vol. 122, no. 2, pp. 887–900, 2017.
- Subramanian, V.; Zholobenko, V.L.; Cheng, K.; Lancelot, C.; Heyte, S.; Thuriot, J.; Paul, S.; Ordomsky, V.V.; Khodakov, A.Y. The Role of Steric Effects and Acidity in the Direct Synthesis of iso-Paraffins from Syngas on Cobalt Zeolite Catalysts. ChemCatChem 2015, 8, 380–389. [Google Scholar] [CrossRef]
- Kang, S.-H.; Bae, J.W.; Prasad, P.S.S.; Park, S.-J.; Woo, K.-J.; Jun, K.-W. Effect of Preparation Method of Fe–based Fischer–Tropsch Catalyst on their Light Olefin Production. Catal. Lett. 2009, 130, 630–636. [Google Scholar] [CrossRef]
- Gac, W.; Zawadzki, W.; Greluk, M.; Słowik, G.; Rotko, M.; Kuśmierz, M. The Effects of Ce and W Promoters on the Performance of Alumina-Supported Nickel Catalysts in CO2 Methanation Reaction. Catalysts 2021, 12, 13. [Google Scholar] [CrossRef]
- P. Zhai, G. Sun, Q. Zhu, and D. Ma, “Fischer-tropsch synthesis nanostructured catalysts: Understanding structural characteristics and catalytic reaction,” Nanotechnol. Rev., vol. 2, no. 5, pp. 547–576, 2013.
- Macheli, L.; Roy, A.; Carleschi, E.; Doyle, B.P.; van Steen, E. Surface modification of Co3O4 nanocubes with TEOS for an improved performance in the Fischer-Tropsch synthesis. Catal. Today 2018, 343, 176–182. [Google Scholar] [CrossRef]
- P. Petersen, M. Claeys, P. J. Kooyman, and E. Van Steen, “Cobalt-Based Fischer – Tropsch Synthesis : A Kinetic Inverse Model System,” 2019.
- Sun, K.; Tan, M.; Bai, Y.; Gao, X.; Wang, P.; Gong, N.; Zhang, T.; Yang, G.; Tan, Y. Design and synthesis of spherical-platelike ternary copper-cobalt-manganese catalysts for direct conversion of syngas to ethanol and higher alcohols. J. Catal. 2019, 378, 1–16. [Google Scholar] [CrossRef]
- C. Göbel et al., “Structural evolution of bimetallic Co-Cu catalysts in CO hydrogenation to higher alcohols at high pressure,” vol. 383, pp. 33–41, 2020.
- L. Zhao, W. Li, J. Zhou, X. Mu, and K. Fang, “ScienceDirect One-step synthesis of Cu e Co alloy / Mn 2 O 3 e Al 2 O 3 composites and their application in higher alcohol synthesis from syngas,” Int. J. Hydrogen Energy, vol. 42, no. 27, pp. 17414–17424, 2017.
- Z. Zhao et al., “Tuning the Fischer – Tropsch reaction over Co x Mn y La / AC catalysts toward alcohols : Effects of La promotion,” J. Catal., vol. 361, pp. 156–167, 2018.
- Z. Zhao et al., “Ziang Zhao, †, ‡, ¶ Wei Lu, †, ¶ Ruoou Yang, ‡, ∥ Hejun Zhu, *, † Wenda Dong, † Fanfei Sun, ‡, ∥ Zheng Jiang, *, ∥ Yuan Lyu, † Tao Liu, † Hong Du, † and Yunjie Ding *, †, § †,” 2018.
- Z. Shi, H. Yang, P. Gao, X. Chen, H. Liu, and L. Zhong, “Effect of alkali metals on the performance of CoCu / TiO 2 catalysts for CO 2 hydrogenation to long - chain hydrocarbons,” Chinese J. Catal., vol. 39, no. 8, pp. 1294–1302, 2018.
- Pei, Y.; Ding, Y.; Zhu, H.; Du, H. One-step production of C1–C18 alcohols via Fischer-Tropsch reaction over activated carbon-supported cobalt catalysts: Promotional effect of modification by SiO2. Chin. J. Catal. 2015, 36, 355–361. [Google Scholar] [CrossRef]
- M. C. Ribeiro et al., “Tailoring the product selectivity of Co / SiO 2 Fischer-Tropsch synthesis catalysts by lanthanide doping,” Catal. Today, vol. 343, no. October 2018, pp. 80–90, 2020.
- Y. Dai, F. Yu, Z. Li, Y. An, T. Lin, and Y. Yang, “Effect of Sodium on the Structure-Performance Relationship of Co / SiO 2 for Fischer-Tropsch Synthesis,” 2017.
Figure 1.
Factors that affect catalyst performance and product selectivity for FTS [14].
Figure 1.
Factors that affect catalyst performance and product selectivity for FTS [14].
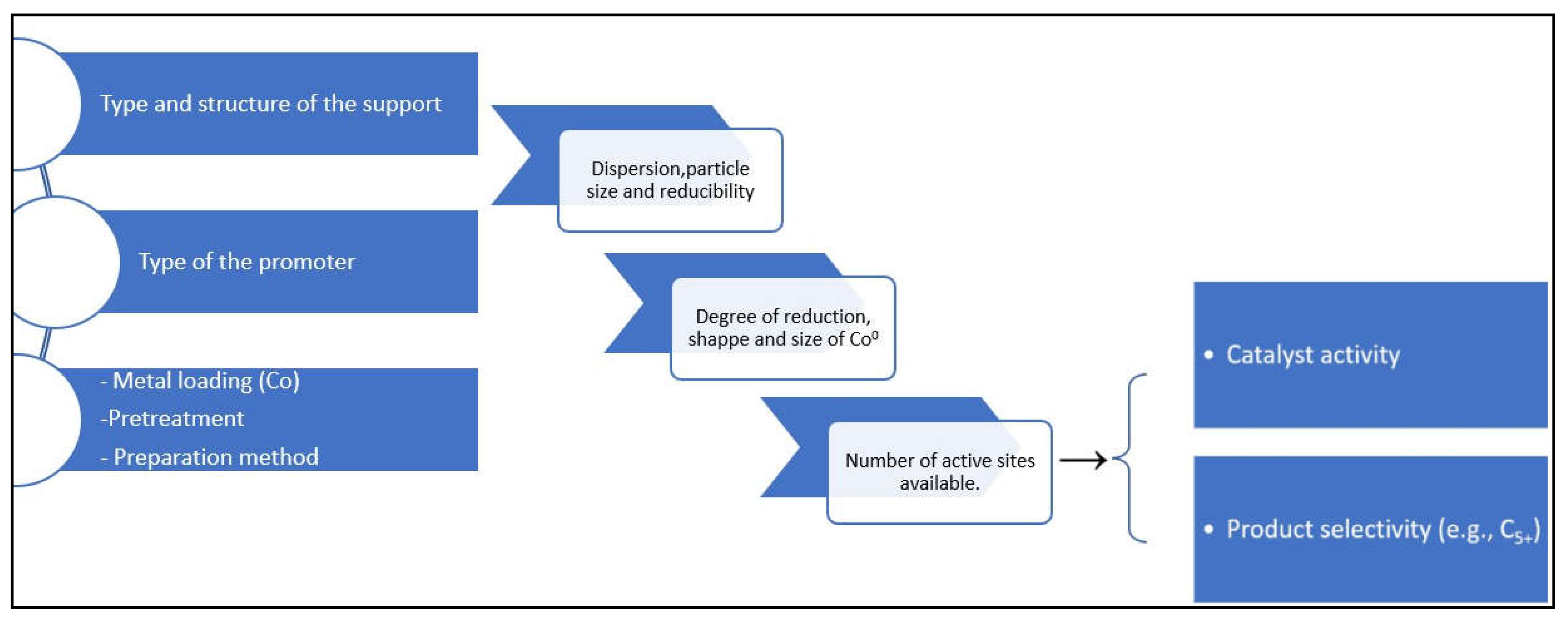
Table 2.
The mode and effect of different elements during catalyst development [25].
Table 2.
The mode and effect of different elements during catalyst development [25].
| Effect on the support and/or catalyst | Promoter elements | ||||
|---|---|---|---|---|---|
| Type of promotion | Mode | Activity | Selectivity | Stability | |
| Structural | Support stabilization | ✓ | ✓ | Mg, Zr, Nb, Rh, Si, Re, Pt, La | |
| Increasing metal dispersion | ✓ | ✓ | Mn, Zr, Ti, Cr, Pd, Ce, Re, Ru, Th | ||
| Electronic | Metal catalyst alloying | ✓ | ✓ | ✓ | Ni, Re, Pt, Ir, Cu, Pd |
| Synergistic | Water-gas Shift | ✓ | ✓ | Ce, Cu, Mn, B | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



