
Submitted:
25 May 2024
Posted:
27 May 2024
You are already at the latest version
Abstract
Pathogenic adenovirus (Ad) infections are widespread but typically mild and transient, except in the immunocompromised. As vectors for gene therapy, vaccine and oncology applications, Ad based platforms offer advantages including ease of genetic manipulation, scale of production and well established safety profiles, making them attractive tools for therapeutic development. However, the immune system often poses a significant challenge that may need to be overcome for adenovirus based therapies to be truly efficacious. Both pre-existing anti-Ad immunity in the population as well as the rapid development of an immune response against engineered adenoviral vectors can have detrimental effects upon the downstream impact of an adenovirus-based therapeutic. This review focusses on the different challenges posed, including pre-existing natural immunity and anti-vector immunity induced by a therapeutic, both in context of the innate and adaptive immune responses. We summarize different approaches developed with the aim of tackling these problems, as well as their outcomes and potential future applications.
Keywords:
Adenovirus
; Vector
; Immunity
; T-cell
; Antibody
; Seroprevalence
; Evasion
1. Introduction
Adenovirus (Ad) based vectors are popular tools for multiple clinical applications, as they are relatively easy to genetically manipulate, can be produced to high titers, and efficiently transduce cells. There are a broad range of Ad-based applications, with Ads developed as vaccine vectors, gene therapy vectors and oncolytic virotherapy (OV) treatments. One of the most studied Ads in this context is human Ad type 5 (HAdV-C5), a species C Ad. However, to date 7 different species (A-G) and over 50 distinct serotypes have been identified, which differ in structure, cell entry pathways and pathogenesis [1,2,3,4].
There are a multitude of genetic adaptations that can be made to Ads. Deletion of genes essential for replication is one of the most common, since it converts Ads from mildly pathogenic viruses to vectors that can no longer replicate or cause disease. This is particularly useful for vaccine applications, where short term viral persistence is sufficient for the desired vaccine immunogenicity. Conditionally replicating Ads are useful as OVs, as they are designed to replicate only in malignant cells and leave healthy tissues unharmed. Other modifications can be made to retarget viruses to specific cells or tissues. HAdV-C5 uses multiple mechanisms for cell entry, and native Ad receptors often do not exclusively target the desired tissue, if at all [3,5,6,7]. Overcoming these native interactions is complex, although several approaches for Ad retargeting have been successful, through both de-targeting of natural receptors [8] and re-targeting of vectors [5,7,8]. These recent advances make HAdV-C5 and other Ad vectors an even more intriguing tool for the development of precision targeted virotherapies.
The immune system is an additional limitation to effective targeting of Ad vectors in vivo, particularly in systemic applications such as intravenous OV injections or administration of gene therapies. Following systemic application, the viral platform will encounter both the innate and adaptive arms of the patient immune system, which have evolved to be highly effective in clearing pathogens through numerous mechanisms [9].
Consequently, the ideal Ad-based vector will need to evade the patient’s immune system, to permit safe and efficient treatment. In this review, we investigate various pathways through which an immune response against Ad vectors can be mounted, as well as their potential impacts on future treatments. Furthermore, we explore how research has approached these hurdles, and critically appraise the advantages and disadvantages of each.
2. Basic Ad Structure
Although they can differ in immune epitopes, all Ads share the same basic structure. The ~ 35kb double stranded DNA genome and core proteins are surrounded by the major capsid proteins. The trimeric hexon protein is arranged in triangular planes, which form the faces of the icosahedral capsid. The vertices are formed by the integrated penton base, which connects to the trimeric fiber proteins, which consist of a shaft and a knob domain. Minor capsid proteins are integrated into the capsid structure. [10,11] (Figure 1)
Comparing the amino acid sequences, the hexon proteins of different Ad serotypes mainly vary in the hypervariable regions (HVRs), which are structurally located on the outside of the capsid. The fiber differs between serotypes in the knob/receptor-binding regions, as well as in fiber shaft length [10,11,12].
All capsid proteins are involved in natural Ad tropism. The Ad penton base contains an RGD motif which binds integrins αvβ3 and αvβ5 [11]. These have an important role in Ad internalisation [13,14]. Commonly identified Ad receptors, which are usually bound through the trimeric knob domain, include Coxsackie and Adenovirus Receptor (CAR), CD46, Desmoglein 2 (DSG2), Sialic Acid (SA) and Heparan Sulphate Proteoglycans (HSPGs) [15,16].
CAR is the primary receptor for HAdV-C5, HAdV-C2, as well as most species C Ads [8,17,18,19]. As the name suggests, most Ads have some degree of affinity for CAR [18], although interestingly not all Ads use it as their primary receptor. Fiber length, as well as the fiber knob domains differs greatly between species and change CAR binding capabilities [20]. For example HAdV-D30 does not bind CAR, despite having CAR binding residues in the knob domain [21]. HAdV-D26 only weakly binds CAR, which has been attributed to sterical hinderance limiting access to its CAR binding domain [22].
In HAdV-F40 and HAdV-F41, which express fibers of 2 different lengths on the same virion, fiber length has also been connected to CAR binding capacity. While the longer fiber protein is able to bind CAR, the more abundant shorter fiber binds HSPGs instead [16,23].
Species D Ads are highly diverse within their own species in the knob domain [22]. While they may engage CAR with varying affinity [17,24], SA may provide an alternative receptor target for some species D Ads [25,26,27]. Species D Ads may also enter cells via CD46, however recent evidence suggests this to be via a direct interaction with hexon rather than via the fiber knob protein [28,29].
Receptor usage for species B Ads is similarly diverse. Species B Ad fiber knobs commonly bind CD46, for example that of HAdV-B35 [30,31,32,33,34], whilst HAdV-B3, HAdV-B7 and HAdV-B11 appear to be able to bind both CD46 and DSG2 [35,36,37].
Similarly to the fiber, the hexon protein exhibits significant inter-species variation in the amino acid sequence. Most of this variation is restricted to the 7 HVRs located on the outside of the Ad capsid (Figure 2). Intra-species variability is also particularly high in species D Ads [38].
While the HAdV-C5 hexon protein binds blood clotting factor X (FX) in HVRs 5 and 7 [39], other species Ads do not always have this interaction as they lack the critical binding residues. Using the amino acid identified as most critical to FX binding, we can predict this interaction across serotypes and species. From this we can see that, while it is a common feature of Ads, species A and D viruses are not likely to bind FX (Figure 2). Similarly, species C Ads have also been shown to bind lactoferrin via HVR1 to enter CAR-negative cells via an as of yet undetermined mechanism [40]. It has also been demonstrated that ChAdOx1 (which is based on ChAd-Y25 [41,42]), HadV-C5 and HAdV-D26 may also weakly bind platelet factor 4 (PF4) via the hexon [43] in a charge dependent manner.
Virus: host interactions dictate Ad tropism and toxicity. CAR is expressed at tight junctions between epithelial cells throughout the body [44] as well as on erythrocytes [45]. CD46 and SA are ubiquitously expressed. [46]. DSG2 is a transmembrane glycoprotein localised to desmosomes, junctions between epithelial cells of the heart, lymph nodes and spleen [46]. FX binding of selected Ads will lead to liver transduction, as FX bridges the hexon to heparan sulphate proteoglycans (HSPGs) on the surface of hepatocytes [39,47]. There may also be more, yet unaccounted for receptor/host protein interactions, which are related to specific Ad infective patterns and pathogenesis but have yet to be identified and explored.
In addition to these interactions, which lead to cell entry and eventually infection, the virus will also be sensed by both the innate and adaptive immune system, eventually leading to the development of immunological memory.
3. Basic Concepts of Antiviral Immune Responses
As an evolutionary necessity, the human body possesses an innate immune system, which acts as a first responder to any foreign attacks, such as injury or microbial infection. An immediate response initiated by the innate immune system is targeted at a broad range of pathogens. This response can be made more specific through the expression of pathogen induced signalling molecules, such as type I interferons (IFNs). These are secreted by virus infected cells upon the engagement of virus specific pathogen recognition receptors (PRRs), which sense pathogen associated molecular patterns (PAMPs). There are a multitude of cells involved in the innate immune response, which are activated through the secretion of type I IFNs [48,49] (Figure 3).
In the blood, non-cellular components of the innate immune system, such as complement, engage with pathogens to aid cellular components in mounting a response (Figure 3). Complement (C) proteins are found in the blood and activate the innate immune system via three pathways: Classical, alternative and mannan-binding lectin (which is specific to bacterial pathogens). The classical complement cascade requires antibody binding to a target to engage C1q. The alternative pathway does not require antibody binding, but involves binding of C3 to conserved motifs of the pathogen directly. Once activated, complement has various mechanisms of pathogen destruction, such as facilitating destruction or direct lysis of cells through the membrane attack complex [51,52] (Figure 3).
Viral infection of cells also induces the expression of defensins, which are small peptides with anti-viral properties (Figure 3) [53]. Furthermore, upon the death of a virally infected cell, the engagement of PRRs with damage associated molecular patterns (DAMPs) also contribute to the overall systemic state of inflammation [54].
Compared to the innate response, the response of the adaptive immune system is more specific to the pathogen encountered (Figure 4).
Cell activation requires antigen presentation: For CD8+ T cells, this occurs via Major Histocompatibility Complex (MHC) I , for CD4+ T cells, via MHC-II of professional APCs such as DCs. Generally, 3 signals are required for T cell activation and expansion: Interaction of the TCR with a matching MHC and antigen, co-stimulation (for example through CD28-CD80/86 in CD4+ T cells) and cytokine signalling [55,56]. Activation induces different effector functions: CD8+ T cells, also called cytotoxic T cells specialise in either directly killing or inducing apoptosis in infected cells. CD4+ T cells, also called helper T cells, modulate the immune response through the secretion of cytokines. They are also able to activate B cells and induce antibody class switching via CD40/CD40L signalling. Activated B cells turn into plasma cells, which are able to secrete different classes of antibodies, a major component of the adaptive immune response [57,58,59,60,61,62,63]. If B cells are not initially specialised through secretion of antibody types, through class switching the backbone of secreted antibodies can be changed, changing effector function. After secretion of IgM or IgD as the native B cell receptors, class switch can help specialise the immune response through secretion of IgG, IgA or IgE instead [63].
Broadly, there are two categories of cells involved in the adaptive response, though they can be further classified by receptor expression and specific function. These are categorised as responses driven by T cells, which mature in the thymus, and B cells, which mature in the bone marrow. Through maturation, T cell receptors (TCRs) specific to non-self antigens are selected and released to activate the adaptive response. T cell activation requires the presentation of antigens from the encountered pathogen through antigen presenting cells (APCs) to naïve T cells. [57] (Figure 4). Both T cells and B cells are able to differentiate into memory cells after a successfully mounted immune response, which allows for rapid expansion of relevant cell populations upon re-encounter of a pathogen [58].
B cell receptors (BCRs), much like TCRs, are matured in a rigorous selection process. Once formed, naïve B cells leave the bone marrow and await activation in the spleen. From there, they travel to secondary lymphoid organs, such as lymph nodes, where they sample passing antigen and await activation, which in viral infection is T cell dependent. Activated CD4+ T cells recognizing presented antigen on the B cells MHC-II will activate the B cell, causing it to rapidly undergo clonal expansion into plasma cells. These are able to secrete the former BCR as antibodies, a main effector of the adaptive immune response . Furthermore, they are able to specialise these antibodies through CD4+ T-cell help to increase their target binding affinity [59,62] (Figure 4).
One of the most well studied aspects of population anti-Ad immunity are neutralising antibodies (NAbs), due to their ability to directly inhibit Ad reinfection. Additionally, there are several other mechanisms through which antibodies can confer immunity to infection.There are 5 different classes of antibodies: IgM, IgE, IgD, IgA and IgG, which are determined by the heavy chain structural part of the antibody. Plasma cells can be induced to switch antibody classes as needed by helper T cells, however the epitopes recognised will not be affected by class changes. Different classes of antibodies are associated with different functions and different expression levels across tissues [60,62] (Figure 4).
Antibody effector functions include agglutination and opsonisation (crosslinking viral particles into larger aggregates and marking particles for uptake by phagocytes), as well as complement activation and neutralisation. While some of these require the presence of effector cells, many of these mechanisms can be facilitated by the presence of antibodies alone. Without effector cells present, the most common measure of antibody effect on a viral pathogens is through neutralisation: This is not a singular process, but sums up all mechanisms that can be involved in the reduction of infectivity [64,65].
4. Natural Infection
Ad infections usually cause mild and transient disease in healthy individuals. In rare cases, they cause more significant outbreaks (epidemics) in populations. In the mid to late 20th century, acute respiratory disease (ARD) caused by different Ad infections (HAdV-E4 and HAdV-B7) was common in US military recruits, which was successfully curbed by an emergency vaccination program using enteric-coated, live HAdV-E4 and HAdV-B7 as oral vaccines [66]. Severe Ad infections outside of these conditions have been rare, and typically very restricted in the immunocompetent population [67]. In the immunocompromised however, even a usually mild virus can cause significant harm [68]. Consequently, most Ads are originally isolated in hospitalised, immunocompromised individuals. Symptoms depend on the specific serotype, though some symptoms are common among Ads of the same species [1]. HAdV-D53, which was first isolated in 2005, causes keratoconjunctivitis [69]. Keratoconjunctivitis was also observed in HAdV-D37, isolated in 1976, which has also been found to cause rare cases of cervicitis [70]. In 2016 and 2019, HAdV-D8 caused several epidemic outbreaks of keratoconjunctivitis [71,72].
HAdV-D32 has been found to cause hepatitis, gastrointestinal (GI) ulcers and respiratory disease [73]. HAdV-D15, first isolated in 1957, causes pharyngitis, respiratory and GI symptoms in different paediatric patients [74].
HAdV-B3 caused a severe outbreak of respiratory illness in a paediatric care facility in the US in 2005, with a 6% mortality rate [75]. An outbreak of HAdV-B7 was reported with 25% mortality at a paediatric care facility [76], and another outbreak of severe respiratory illness in a rehabilitation facility in 2018 was also attributed to HAdV-B7 [77].
More examples are summarised by Mennechet et al [1].
4.1. Innate Immunity
Innate responses to acute virus infection are hard to measure and do not often appear in literature. Mouse models often do not represent natural infection well as the routes of administration are usually systemic, such as intravenous or intraperitoneal, which will result in less tissue specific and more systemic reposes. There are very few studies on acutely infected patients, which are needed to measure innate immune responses.
As described previously, Ad DNA can be sensed intracellularly once the virus has infected cells, leading to the release of IFNs. This is the main pathway for antiviral response in the innate immune system [78]. Doronin et al. suggest that induction of IFN signaling is dependent of the Ads ability to bind FX, as FX binding was essential for macrophage Ad sensing in the spleen. In this case, Ad binding FX may serve as a PAMP, recognition of which is TLR4 dependent [79]. However, this study was done by intravenous administration of HAdV-C5 to mice and may therefore not be representative of other tissue specific reactions.
Macrophages are tissue resident phagocytes and are very likely to encounter Ad particles upon infection by whichever route. In a macrophage cell line, cyclic GMP-AMT synthase (cGAS) was identified as one of the main drivers initiating the anti-Ad response. cGAS senses intracellular viral DNA and induces a signalling cascade resulting in the production of IFNs. Knockdown of cGAS, or any signalling molecule in the cascade such as STING or IRF3, resulted in loss of this function [80]. Plasmacytoid dendritic cells (pDCs) showed to have a special role in the sensing of species B and other CD46 binding Ads through TLR9 sensing. Iacobelli-Martinez et al. showed pDCs, which are found in secondary lymphoid organs such as lymph nodes, produced large amounts of IFN-α, while myeloid DCs sense Ads through intracellular DNA in a TLR-independent manner in donor peripheral blood mononuclear cells (PBMCs) in vitro [81].
Ads of different species may activate the innate immune system more or less strongly. Teigler et al. found that HAdV-B35 and HAdV-D26 induce much higher levels of pro-inflammatory cytokines, such as IFNs, in PBMC assays. They linked this to differences in endosomal escape: Compared to HAdV-C5, HAdV-B35 and HAdV-D26 release from the endosome much later, allowing for differences in intracellular receptor sensing, such as intraendosomal engagement of TLR9 [82]. Johnson et al. also observed a much increased production of IFN-α by HAdV-D28 and HAdV-B35 in their PBMC assays, compared to HAdV-C5, leading to a stronger activation of the NK response [83].
In a respiratory infection, as commonly found in species B Ads, resident cells in the lungs, respiratory tract and local draining lymph nodes will be more involved in the infection. Fluid from bronchoalveolar lavage (BAL) samples in humans has been shown to contain defensins, which drastically reduced Ad infectivity in vitro [84]. Defensins confer neutralisation to Ad infections through intracellular degradation, by preventing the fiber and pVI from detaching from the capsid and thereby virus escape from the endosome [53,85]. Species D and F Ads show resistance to this pathway, which Smith et al. attributed to specific binding residues required in the penton region, which are common to some but not all Ad species [53]. In serum samples of immunocompetent children in Argentina with acute respiratory HAdV-B infection of unspecified serotype, systemic levels of IL-6 and TNFα levels were associated with more severe disease. The study included 28 children, which were classified according to moderate, severe and deadly infection [86]. Both IL-6 and TNFα are pro-inflammatory cytokines secreted by tissue resident macrophages [87]. From this we can assume that in most people, where Ad infection is transient and mild, the initial immune responses are mainly restricted to the infected tissue.
As Ad infections from various species cause GI symptoms[1], innate immune cells resident at these mucosal sites will be first responders during natural infection. Numerous immune cell types are present in the GI tract, as the body is commonly exposed to pathogens via this route.
4.2. Adaptive Immunity
Many studies into population immunity against Ads use neutralising ability of donor sera as a marker of anti-Ad immunity. In a large cohort of donors, neutralisation assays are relatively cheap and easy to run [89], especially in comparison to assays measuring T cells.
While seroprevalence rates vary by location, such as HAdV-C5 being more prevalent in Europe than Asia, there are also trends that can be observed across all regions. In 2003, Vogels et al. compared a number of rare Ad serotypes for prevalence in 6 different regions across the globe. While Species A and C generally had high seroprevalence, species B was far less common. Species D Ads, which are a much larger and more diverse species, showed lower seroprevalence rates than species A and C though not as low as species B [90]. Another study by Abbink et al. published in 2007 examined seroprevalence in Africa comparing HAdV-C5 to rare serotypes from species B and D found similar results. In their study, species D Ads were found to be less common than species B Ads, though both were significantly more rare than HAdV-C5 seroprevalence, which was observed in all samples [2]. Comparatively, in 2015, Dakin et al. investigated seroprevalence of rare serotype HAdV-D49, a species D Ad, compared to HAdV-C5 in a Scottish cohort. HAdV-C5 neutralisation was found in 31% of samples, whilst NAbs against HAdV-D49 were not detected. [91] Another study by Klann et al. in 2022 tested donor serum of a cohort in Germany for pre-existing anti-Ad antibodies across different species. Surprisingly, 11 out of 39 viruses were found to have higher seroprevalence than HAdV-C5, while the remaining 28 viruses were less seroprevalent. In agreement with previous findings, higher neutralisation was found in species C Ads compared to species B and D, however NAbs against HAdV-B3, HAdV-C2, HAdV-E4, HAdV-C1 and HAdV-G52 were more prevalent than NAbs to HAdV-C5, which is unexpected [92].
NAbs can be directed against all major capsid proteins [93,94,95,96]. In serum samples from the United States and Afrika, Sumida et al. in 2005 showed the presence of both fiber and hexon antibodies, though NAb titers against hexon were much higher [97]. Contrarily , Yu et al. found in a Chinese cohort that natural infection induces a strong anti-fiber response, as over 90% of their NAb serum donors exhibited antibodies binding the fiber, specifically targeting the fiber knob. They however confirmed that anti-hexon antibodies, though only found in about 40% of their donors, induced a much stronger neutralising response [94]. Bradley et al. also found the main epitope targeted by NAbs in a South African cohort was the Hexon protein, though fiber NAbs were also detected [93]. They also narrowed down the binding of hexon specific NAbs to the HVR regions of the hexon protein [98]. The hexon HVRs are structurally located on the outside of the capsid, making them an ideal target for antibody recognition.
Though these studies all look at neutralisation, mechanisms of neutralisation may vary between different antibodies. An experimentally produced anti-HAdV-C5 hexon antibody, 9C12, was unable to prevent virus entry, but did show neutralising activity. As the antibody remained bound after Ad internalisation, the authors proposed interference with Ad uncoating or replication [99]. The same antibody was later confirmed to elicit its neutralisation via blockage of microtubule trafficking after endosomal escape [85]. In another study, anti-HAdV-C5 antibodies protected virions from changes in pH, which are necessary for endosomal escape [100]. Anti-HAdV-C5 fiber antibodies have been shown to act mainly through agglutination[101]. Others showed that IgM anti-HAdV-C5 antibodies increase uptake and destruction by tissue resident macrophages, such as Kupffer cells in the liver [102] as well as in the blood [103]. As Ads are usually passed on through respiratory or GI infection, the mucosal antibody IgA may play a part in natural anti-Ad immunity. IgA can confer neutralisation though agglutination [104].
T cell responses, though just as important as antibody responses in the anti-Ad immune response, are investigated less commonly. PBMC assays are cost- and labour- intensive to run. As a recall response CD4+ T-cells require time for reactivation, CD8+ T-cells can be reactivated within minutes, but only once the virus has entered cells and antigens are expressed in a MHC-I [58].
Olive et al. identified several T cell epitopes in conserved regions of the hexon protein. Epitope H910-924, located on the C-terminus of the protein, was shown to be particularly effective for developing CD4+ T cell responses [105]. H910-924 reactive CD4+ T cells were found in PBMCs of 14 out of 18 healthy volunteers and also predicted to be cross-reactive across a range of human as well as non-human Ads, such as chimpanzee, porcine and bovine Ads [106]. Onion et al. performed PBMC assays on samples from 10 healthy donors and found 100% to have anti-HAdV-C5 specific memory CD4+ T cells. After in vitro stimulation of naïve PBMCs with HAdV-C5 whole virus, hexon, or fiber, responding T cells could be found to whole virus and hexon, but not fiber. This further indicates the importance of the CD4+ T cells in the anti-Ad response, and hexon as an immunodominant epitope for CD4+ T cells. Upon expansion of anti-HAdV-C5 specific CD4+ T cells they could also see cross reactivity to other Ad serotypes and species [107]. Concurrently, a study in a Dutch cohort isolating PBMCs from 10 healthy volunteers found similarly high levels of CD4+ response, as well as cross-reactivity between human species A, B, C and D Ads [108].
In studies into CD8+ T cells using PBMCs from seven donors, memory cytotoxic T cells that were stimulated in vitro were also able to cross react between HAdV-C5 and HAdV-B35. When deployed against cells expressing hexon, penton and fiber antigen, they were most effective in killing cells expressing the hexon antigen, though anti-fiber and penton responses were observed for some donors. Epitopes for CD8+ T cells were mapped to both the N- and C-terminus of the hexon protein [109]. Cross-reactivity of cytotoxic T cells was further established in other PBMC assays, where CD8+ T cells were isolated and successfully able to kill fibroblasts infected with HAdV-B7, HAdV-B11, HAdV-C2, HAdV-C5, HAdV-C6, HAdV-D8 and HAdV-E4, which once more was attributed to conserved hexon epitopes [110]. A more recent and much larger study involving PBMCs from 64 healthy donors and 26 immunocompromised patients with ongoing Ad infection revealed that the penton base, additionally to the hexon, contains immunodominant epitopes for CD8+ T cells. Using selected peptide sequences from both the hexon and penton, strong responses could be elicited in donor PBMCs, with chosen hexon epitopes and whole virus stimulation eliciting stronger responses in CD4+ T cells, and penton epitopes alone leading to stronger CD8+ activation. Reactive cells to both hexon and penton could be found across all donors, though a higher proportion of anti-Penton CD8+ T cells was found in patients with ongoing Ad infection [111]. The authors therefore conclude that higher numbers of anti-penton T cells are required for control of an ongoing infection. However, as their patients are immunocompromised (and infection may not be well controlled at all), there could be other reasons involved, such as differing immune responses in the immunocompetent or less penton specific T cells being retained as memory cells.
As described previously, both CD8+ and CD4+ T-cells cells appear to primarily recognise more conserved regions of the Ads hexon protein [105,107,108,109,110] rather than the fiber and penton protein [109,112] making them potentially cross-reactive between serotypes and species. This is true for memory cells as well [106]. While the response may be skewed towards CD4+ T cells [105,108], both play a critical role in the response to Ad infection. Hutnick et al used PBMCs from 17 healthy donors to investigate pre-existing immunity against HAdV-C5, ChAd7 and ChAd7. Pre-existing CD4+ and CD8+ T cells to HAdV-C5 were detected in all donors, and were also able to cross-react with ChAd6 and ChAd7, with which previous infection is extremely unlikely. Interestingly, while most CD4+ T cells displayed a memory phenotype, most CD8+ T cells displayed an effector phenotype instead. When tested for reactivity against specific peptides, CD4+ T cells were targeted to HVRs as well as conserved regions of the hexon. CD8+ T cells also responded against the hexon, however reactions were of higher magnitude compared to CD4+ T cells. The authors theorise that the high level of reactivity results from common contact with different serotypes of Ad, skewing selection for T cells against conserved epitopes, as well as low level persistence of Ad in lymphatic tissue [113].
However, T cell responses can still vary between different donors and according to serotype. A 2005 study inducing anti-Ad memory in PBMCs found the reactivation response against HAdV-C5 very high across all donors, while responses against HAdV-A12 and HAdV-B11 were very low [112]. Unfortunately, PBMC assays may not always accurately represent in vivo conditions, consequently more studies are needed to give a more in-depth understanding of T cell responses to natural Ad infection.
As re-encounter of relevant antigens leads to an extension of protection by memory cells, continuous low-level or latent infection of wild type Ads below the threshold of clinical detection[114,115,116,117] may be an explanation of why anti-Ad memory cells can be commonly isolated from donor PBMCs [106,108,109,110,118]. Continuous low level Ad infection has ben proposed by Tran et al. to be related to the development of regulatory T-cells (T-regs), which have immunosuppressive functions, allowing for low levels of Ad to evade the immune system [119]. Others have attributed the continuous low level expression of anti-Ad antigens to the innate immune system instead, through successful suppression of Ad replication by type I IFNs [120]. While the suppression of an anti-Ad response vs the development of an effective memory response may seem contradictory at first glance, the immune system is usually in a constant state of balance between pro- and anti-inflammatory responses. If low levels of Ad are able to evade immune evasion through the development of T-regs, the same low levels of antigen may still keep immunological memory sharp.
In 2011, Adams et al. suggested the CD46 binding capacity of HAdV-B35, while increasing DC engagement, simultaneously protected the virus from CD4+ T-cell response. Direct binding of HAdV-B35 to CD4+ T cells via CD46 reduces the expression of pro-inflammatory cytokines, as has been shown for multiple CD46 binding Ads [121,122]. In naïve T cells, this mechanisms impeded activation. Additionally, in activated and memory CD4+ T cells cytokine production was also reduced as a result of infection. They suggest that this gives HAdV-B35 as well as possibly other CD46 binding Ads an evolutionary advantage [121], however CD46 internalisation due to binding increases the likelihood of complement induced lysis of cell [123]. This effectively trades lessened activation of the adaptive immune system to enhanced susceptibility to the innate immune system.
In conclusion, due to the high level of pre-existing anti-Ad immunity in the population, any prospective Ad based therapy already faces major obstacles. The presence of NAbs is the most immediate concern for IV applications, while applications that require long-term circulation or expression of Ads will also have to face broadly cross-reactive T cell responses. However, knowledge in this area is severely limited by the gap in knowledge between in vitro and in vivo responses. While the investigation of memory cell targets gives a decent picture of active Ad infection, as mentioned previously, PBMC assays may not always be representative of an in vivo system. Furthermore, since most current infections occur in the immunocompromised, measured responses of active infection may be different to how they would be in the general population.
5. Vector/OV-Induced Immunity
Alongside anti-Ad immunity generated via natural Ad infection, anti-Ad immunity is also generated by the current use of Ad vectors in different therapeutic applications, such as recent mass vaccinations against SARS-CoV-2 using adenoviral platforms. Due to scale of production and high replicative ability, the use of these vectors is on the rise.
5.1. Widespread Ad Vector Development, Clinical Evaluation and Use
As a potential vector for cancer and gene therapies, Ads, especially HAdV-C5, have been well investigated. The immunogenic cell death of tumour cells induced by Ad based OVs and release of DAMPs attracts the immune system to the tumour. The ability of the OV to replicate will allow self-amplification within permissive transformed cells over time without further administration. Genetic changes can be introduced into prevent Ad replication in off-target tissues or prevent replication altogether. The deletion of large parts of the genome, which usually completely prevents replication, allows for the insertion of large transgenes which can serve different purposes [124,125].
A very common example of creating conditionally replicating Ads is deletions in the early phase gene region 1 (E1). Whilst deletion of the entire E1 region prevents replication entirely [126], the deletion or loss of function of its genes E1B or E1A separately may be utilised in the context of OV cancer treatment [124,127]. In healthy cells, E1 gene product will repress host genes involved in preventing viral replication, such as p53 and pRB. Both of these are very commonly mutated in cancers, allowing E1-deleted viral platforms to replicate conditionally in cells deficient in these pathways [124]. Additionally, replication as well as transgene expression can be placed under control of a tumour specific promoter [128], whilst structural changes to capsid proteins can be incorporated to retarget Ads from their natural tropism [8,128], making the vector even more tumour selective.
Transgenes often incorporated into oncolytic Ad vectors include immunomodulatory genes to increase the patients anti-tumour immune response, or prodrug converting enzymes to reduce systemic toxicity of traditional chemotherapy drugs [128]. There are a number of oncolytic Ads currently being investigated in clinical trials, most of which are based on HAdV-C5, some of which are summarised in Table 1.
Where long term gene augmentation is required, such as with gene therapy for monogenic disorders, the use of helper dependent or “gutless” Ad vectors, which are devoid of all viral DNA (except for the inverted terminal repeats, ITRs) and require a helper virus for production, has been extensively explored [141]. However, low level contamination of helper Ads presents a problem, leading to the development of better purification methods [142,143] and more recently, helper-free gutless Ads (HF-GLAds) [144]. These HF-GLAds have shown promise in pre-clinical testing, but are yet to reach -human trials [145].
Over the past decade, gene therapy applications have moved significantly towards using Adenovirus-Associated Virus (AAV) vectors instead of helper dependent Ads. AAVs have a smaller transgene capacity, but allow long term gene expression. They also tend to be less immunogenic than Ad vectors, which is desirable for long term gene correction [146,147], though HF-GLAds and replication deficient vectors also generate reduced anti-vector immune response compared to oncolytic or replication competent Ads [145]. Some gene therapy applications of Ad vectors in clinical trials currently and in the past are listed in Table 2.
As vaccine vectors, Ads are generally used in a replication deficient state. The historic vaccines against Ad, which worked extremely well, were live virus vaccines to protect military recruits against ARD caused by AdV-E4 and AdV-B7 [66]. For vaccination for any other purpose than Ad immunity however, production of viral proteins is a disadvantage, as the aim is for the immune response to be directed against the antigen transgene.
An interesting middle ground has been proposed by Crosby and Barry in their development of “single-cycle” (SC) Ads. Their vaccine vector, based on HAdV-C6, has a deletion in protein IIIa which is essential for DNA packaging and virus assembly. The resulting vector, once it inside the cell, is able to replicate its genetic material and therefore its transgene, without producing viable progenitor virus. Some virus protein was still produced by infected cells [156] still engages the immune system, however the transgene/vector protein ratio is still more favourable. In a later in vivo experiment with Syrian hamsters to investigate the vector for influenza vaccinations, the single cycle replication vector required a 33x lower dosage compared to replication deficient vectors to achieve the same transgene levels, which is a drastically lower amount of vector protein patients would be exposed to [157]. Furthermore, the added Ad antigen may act as an adjuvant to recruit the immune system.
The value of Ad-based vaccines was demonstrated during the COVID-19 pandemic. According to ourworldindata.org, an estimated 5.13 billion people worldwide were vaccinated against SARS-CoV-2. In the UK alone, 50.76 million primary doses were administered to >75% of the population. Within the European Union almost 100% of the vaccinated population was covered by 4 vaccine providers: Pfizer/BioNTech (Comirnaty), Moderna (Spikevax), Oxford/AstraZeneca (Covishield) and Janssen (Jcovden) in order of most administered [158] (Figure 5).
Both Pfizer (Comirnaty) and Moderna (Spikevax) are mRNA vaccines. The Janssen (Jcovden) vaccine is a HAdV-D26 vector based vaccine expressing the SARS-CoV-2 spike antigens [159]. Similarly, the Oxford/AstraZeneca vaccine (Covishield) is based on their ChAdOx1 vector derived from ChAd-Y25, also with SARS-CoV-2 spike antigen transgene [42,160]. This means, that in the EU alone, over 85 million people have been immunised with Ad-based replication deficient vaccine vectors [158].
Although the SARS-CoV-2 vaccines were developed for intramuscular administration, different routes of administration have since been investigated for SARS-CoV-2 [161,162], in addition to other pathogens, such as influenza [163] and HIV [164]. Current uses of and clinical trials into Ad based vaccines have been summarised in Table 3.
As broad usage of Ad based vectors becomes increasingly common, we need a solid understanding of administered vectors with patient immune system to tailor the vector to be most effective in different applications.
5.2. Innate Immunity
Overall, due to the limited receptors available, we would expect sensing by the innate immune system to be very similar between natural Ad infection and Ad vector administration, though it is possible that there are tissue specific differences due to the differently localised or more systemic exposure to vectors, compared to natural infection.
In wild type Ads, the E3 protein enhances Ad immune evasion through several mechanisms, such prevention of intrinsic and extrinsic induction of apoptosis of virus infected cells, and the downregulation of MHC-I molecules on the cell surface [176]. While this leads to decreased engagement of cytotoxic T cells, it also means increased susceptibility to killing by NK cells due to the missing-self hypothesis [177]. Intranasal administration of HAdV-C2 and HAdV-C5 vectors with a complete E3 deletion to cotton rats led to an increased macrophage and polymorphonuclear leukocyte response. The same could be observed by only deleting the E3-19kDa region of the gene, making this the most important region in this response [178]. The E3 region is commonly deleted in may Ad vector applications, either fully or partially, for various reasons. Large deletions of the whole E3 gene region increase transgene capacity, while deletions of the E3-19kDa gene specifically can be used to actively increase antigen exposure, both of viral and tumour antigens in oncolytic vectors [124]. Examples of whole deletions include the oncolytic HAdV-C5 based Onyx-015 [179] or the very similar Oncorine, which is approved for oncolytic virotherapy in China [180]. Partial deletions have been made in several pre-clinical Ad vectors [8,181]. Though immune modulation may not always be the reason for an E3 deletion in vector development, it is important to take into consideration.
Fejer et al. showed that, in intraperitoneal injections in mice, virus entry and endosomal escape trigger IFN expression in vivo in myeloid DCs, in a TLR independent manner and heavily reliant on the positive feedback loop of IFN secretion described earlier (Figure 3). They highlight the importance of splenic myeloid DCs in this interaction, as this is where the vast majority of IFNs were produced [182]. Vectors used in this study were based on HAdV-B3, HAdV-C2 and HAdV-C5 with no modifications to structural proteins. It is likely that the interaction of the Ad penton base with integrin of the spleen is responsible for this strong response. Secretion if type I IFNs has been shown to induce the production of other anti-viral proteins in vitro, which inhibit viral DNA-, RNA and protein synthesis [183]. In a vector context, this means reduced expression of transgenes through the induction on type I IFNs by the innate immune system. Di Paolo et al. also investigated accumulation of their HAdV-C5 based vector in the spleen after intravenous injection of mice. They found that uptake by tissue resident macrophages through integrin binding triggers the transcription of Interleukin (IL) 1α within 10 minutes of administration, with high levels of expression induced by 25 minutes after administration through positive feedback signalling. They also investigated the importance of integrin binding to the response and found a marked reduction of cytokine production if αvβ3 integrin cannot be engaged through the vector [184]. In a later paper, they outlined how expression of IL1α leads to expression of more chemokines such as CXCL2 as well as complement proteins, which attract polymorphonuclear leukocytes to the area. They also observed death of splenic macrophages, which unlike the self-induced death of Kupffer cells in the liver, they were able to attribute to recruited targeted killing by recruited Neutrophils instead [185]. This shows the importance of integrin binding in the very early innate responses to intravenously administered Ad vectors, as well as a potential mutation to bypass this mechanism through removal of the integrin binding RGD-motif.
Kupffer cells are tissue-resident liver macrophage. As natural Ad tropism will direct intravenously administered Ad vectors to the liver, they are an essential factor in the innate anti-Ad vector response: While tissue tropism remains, Kupffer cell depletion markedly increases Ad vector gene delivery in intravenous delivered HAdV-C5 based vectors in mice [186]. Similarly, in vectors where Kupffer cell recognition is reduced, FX dependent transduction of hepatocytes is markedly increased, as shown be Khare et al. in both intravenously injected mouse and Syrian hamster models [187]. Only the prevention of both FX binding and Kupffer cell uptake leads to an increase in systemic Ad delivery in vivo [186]. Interestingly, in repeat administrations within 72 hours without prior Kupffer cell depletion, transduction to the liver and Kupffer cells is still reduced, instead higher Ad gene expression is observed in the marginal zone of the spleen. Changes in biodistribution were attributed to Kupffer cell depletion after the initial dose due to immunogenic cell death. While the authors suggest Kupffer cells will have replenished by 72 hours, these newly bone marrow derived cells will have slightly different receptor expression. Their impaired ability to clear Ad vectors leads to increased transduction to the spleen. At later time points of administration, Kupffer cells regain their scavenger receptors and biodistribution is more similar to the initial administration [188]. This may be an important observation that may change considerations when designing repeat administration schedules, such as for oncolytic virotherapies.
Interactions with components of the complement system further activate the anti-vector immune responses. Ad vectors can directly interact with C3. In the absence of C3, intravenously administered HAdV-C5 vectors cannot induce thrombocytopenia in mice, indicating complement is involved in this response. Overall cytokine release and systemic inflammation was also reduced in C3 knockout mice [189], which can be expected as C3 is involved in positive feedback loops activating the innate immune system (Figure 3). Tian et al. observed that in vivo in mice after intravenous application, complement mainly senses Ad vectors when they have already infected cells, as HAdV-C5 vector defective in cell entry had a much reduced complement response. In vitro, complement was able to interact with vector particles directly, causing a much larger response [190]. This is important to note, as it demonstrates that data on complement will likely have to be collected using in vivo experiments to provide accurate results. For example, the Ad hexon has also been implicated in binding IgG, which may further facilitate complement binding and neutralisation via the complement cascade. In vitro, this mechanism reduces Ad transduction via destruction by the classical complement cascade. In vivo, there appear to be other mechanisms involved as well [190]: As described earlier signalling of activated cells of the innate immune system can increase complement activity (Figure 3), which would not be picked up by pure in vitro experiments.
As with natural Ad infection, vectors from different serotypes and species will engage the immune system in different ways, which may make some serotypes more suitable than others. As touched upon before different species activate the innate immune system to different levels due to differences in tricellular trafficking in natural infection. This is also true for Ad vectors. In a rhesus monkeys vaccination study by Teigler et al., low seroprevalence vectors based on HAdV-B35, HAdV-D26 and HAdV-D48, induced a much stronger innate cytokine response that the HAdV-C5 vector [191].
Though the innate immune system presents challenges for the immediate systemic administration of Ad based vectors, the innate and adaptive immune response are intrinsically linked. Differences between vectors in the innate response will likely also translate to differences in the adaptive response.
5.3. Adaptive Immunity
The development of anti-vector NAbs is a concern for Ad use in any vector context. Neutralisation occurs instantly when viral vectors are administered. There are several mechanisms through which anti-vector antibodies can neutralise an administered Ad based therapy. Anti-vector antibodies can redirect Ads to phagocytes, such as macrophages, and inhibit endosomal escape, leading to Ad degradation rather than propagation in mice [103]. Aggregation has been suggested as the main mechanism of anti-hexon neutralisation in intramuscularly vaccinated rabbits, though the same article also confirms post-entry neutralisation of Ads [100]. This is very likely through a similar mechanism as described above, involving intracellular actions of antibodies. For vector specific anti-Ad antibodies, the intracellular receptor TRIM21, which is antibody activated, has been shown to be a major facilitator of neutralisation through recruitment of proteases to digest intracellular vector particles [192].
Using the same HAdV-C5 vaccine vector encoding different transgenes does not give a satisfactory anti-transgene response, as demonstrated by Steffensen et al. which they were able to attribute mainly to the development of anti-vector NAbs. However, priming and boosting against the same antigen using the same vector is possible, likely because a much lower concentration of antigen required to activate the recall response [193].
Intratumoural injection of replication deficient Ad vectors into subcutaneous tumours in mice is still generates a strong systemic anti-vector NAb response, which is an important consideration in potential Ad based cancer therapies [194]. In 1998, Gahéry-Ségard et al. identified in intratumoural injection of replication deficient HAdV-C5 based vectors in 4 lung cancer patients that antibodies against fiber are expressed first, followed by anti-penton and anti-hexon antibodies. While half the patients developed a strong anti-penton IgG response, the other half developed a strong anti-hexon IgG response. They also found that the presence of anti-penton antibodies was indicative of a strongly neutralising anti-vector response [95]. More recent studies into oncolytic vectors based on HAdV-C5 found that, while replication deficient vectors were supressed more by NAbs (in both human and mouse serum), oncolytic vectors still had diminished transduction in cancer cell lines. Cell killing in vitro was also markedly reduced by the presence of high titer NAbs. In the presence of low titer NAbs, cell killing was possible through overcoming of NAbs, though it was inhibited at earlier time points. Consequently, they found that the higher NAb titer, the later (if at all) decrease in cell viability was detected. In subsequent in vivo studies with tumour xenografts in mice, transgene expression was reduced after administration of anti-vector NAbs to nude mice. Tumour shrinkage was still observed in mice with low dose of transferred anti-Ad NAbs, but significantly reduced in mice transferred with high dose of NAbs [195]. Since for xenograft experiments mice need to be immunosuppressed, the authors transplanted high or low amounts of NAb serum from other mice induced by intravenous challenge, then injected Ad therapies intratumorally. This excludes the presence of cellular immunity, so any effect will be due to NAbs only. While informative, that means these results are likely not representative of a full in vivo system.
Intravenously administered replication deficient vectors induce both anti-hexon and anti-fiber antibodies in mice, though anti-hexon antibodies have a much stronger neutralising effect [93]. The main target for this neutralising response are the HVRs of the hexon [98].
A vaccination study in healthy volunteers by Cheng et al. showed that intramuscular vaccination induced NAbs against the capsid proteins other than the fiber [196]. Previous in vitro reports indicate that this may be due to an anti-hexon response [100,197]. In intramuscular vaccinations with chimeric vectors based on HAdV-C5 and HAdV-B35, which previously showed to be non-crossreactive, NAbs against both parent vectors were detected. However, vector-specific anti-hexon NAbs were more common and effective in adoptive transfer studies, using purified antibodies from previously vaccinated mouse serum [97]. Following a Phase II intramuscular vaccination trial using a HAdV-C5 based vector, NAbs to vector pre- and post-vaccinations were measured, only distinguishing between fiber and not fiber. Vaccine induced NAbs were directed against the capsid, and in some cases both capsid and fiber, while fiber NAbs were mostly detected in volunteers that were seropositive for HAdV-C5 pre-vaccination [198].
Steffensen et al. compared subcutaneous and intramuscular routes of administration using the same replication deficient vector expressing different transgenes and found that the route of administration generating the neutralising response did not matter [193]. More recently, the ChAd36 based vaccine vector BB154 (Table 3) showed that after intranasal vaccination in Phase III clinical trials, vector specific neutralising antibody levels were low [162]. On the other hand, a Phase I clinical trial utilising HAdV-C6 and ChAd3 vectors as intramuscular Hepatitis C vaccines in the UK found heterologous prime-boost schedules to be less effective than expected. They attributed this to cross-reactive NAbs, effective against both vectors [199]. This highlights how differences in route of administration and different serotypes can alter immune response to the vector, which if harnessed correctly can work to our advantage.
Another Phase I clinical trial using a ChAd63 vaccine vector intradermally and intramuscularly for malaria measured anti-ChAd63 NAbs pre- and post-vaccination. They concluded that, since participants with pre-existing anti-ChAd63 NAbs were not observed to mount a reduced transgene response, this will likely not prevent future applications [200]. However, antibody titers pre-vaccination were not published and may therefore be much lower than post-vaccination. Furthermore, re-boosting against the same antigen, which has proven successful before, does not prove that the vector can be re-used against other antigens in the presence of heightened anti-vector immunity. Ewer et a.l and Kimani et al. reported on the same ChAd63 based vector in the UK and African cohorts of a PhaseI/II trial. Both measured low levels of pre-existing NAbs to ChAd63 [201,202]. Interestingly while Ewer et al. noted a minor correlation between low levels of anti-vector antibodies and increased anti-transgene T cell responses [201], indicating the Ad vector might work as an adjuvant, Kimani et al. observed the opposite: Higher anti-vector NAbs correlated with a slight reduction in T cell anti-transgene response [202].
Vaccinations with the ChAdOx1 vector described earlier have put the spotlight on reusability of the vector. A Phase2/3 covid vaccine trial intramuscularly administrated ChAdOx1, anti-vector antibodies were induced with the first vaccination, however boosting did not increase NAbs further. Presence of anti-vector NAbs pre-vaccination had a minor negative impact on anti-transgene NAb titers. Anti-vector NAbs generated after boost vaccination however did not negatively impact the cellular anti-transgene response [203]. As mentioned previously, boosting against the same antigen is possible despite the presence of anti-vector immunity. It is likely that repeated boosters using the ChAdOx1 vector against the same SARS-CoV2 antigen will be successful. However comparing the same metrics (prime vs boost NAbs against transgene or prime vs boost cellular response against transgene) might have been more informative. In 68 patients in Korea, no pre-existing anti-Ad NAbs were detected in only 19. This is not surprising, as pre-existing anti-Ad immunity is very commonly found in populations. The transgene response was observed to be much higher overall in patients without pre-existing anti-Ad NAbs, especially after booster vaccine [204]. This is very surprising, given it is unlikely that many people will have encountered a ChAd before. Authors state that this indicates the presence of cross-reactive NAbs. Using a different vector based on HAdV-C5, 346 Mexican subjects were vaccinated intramuscularly with Ad5-nCoV. Hernandez-Bello observed a trend that pre-existing anti-HAdV-C5 NAbs lead to a less effective transgene response, but this result was not statistically significant [205]. This again defies expectation, as anti-HAdV-C5 NAbs are usually very common. HIV vaccine studies tested intramuscularly in rhesus macaques based on HAdV-C5, ChAd6, ChAd7 showed no anti-Ad antibodies before the first dose. Low titer NAbs to ChAd7 were found after the first dose, with a slight increase over time, while titers of NAbs to HAdV-C5 were high even within short time after first administration. Booster with a different Ad serotype did not only produce vector specific NAbs, but also increased the amount of anti-prime vector NAbs [206]. This is a good example of how different types of Ad can be more or less immunogenic.
Overall, anti-vector NAbs pose a problem to both vaccine and OV applications, especially upon repeat administrations, as NAbs often show the strongest activity against vector specific regions such as the hexon HVRs. However, there is also some evidence for NAbs cross-reacting even across species. It is possible that, by only looking at antibody responses, some studies attributed cross-reactivity to NAbs when cellular immunity may have played a part in unexpected responses as well, especially in human subjects with pre-existing anti-Ad immunity.
Trials for a HAdV-C5 based SARS-CoV2 vaccine in China results showed that vaccine efficacy is limited by pre-existing anti-HAdV-C immunity [207], though authors do not expand on whether his effect is largely due to T cell immunity or neutralizing antibody responses. Likely, both play a role. It has been shown, that ross-reactivity of T cells between Ad serotypes can impair generation of target antibodies even in rare Ad based vaccine vectors [208].
Anti-Ad-vector T cell responses and epitopes are not often studied in vivo. Instead, most articles focus on differences in anti-transgene responses as a measure of efficacy for new vaccine vectors [209,210,211,212,213]. Whilst responses to transgene are important, the development of anti-Ad vector responses and how they differ according to route of administration needs to be studied in more detail.
Heemskerk et al. investigated anti-Ad T cells in PBMCs from 25 healthy donors. In response to stimulation with inactivated HAdV-C5, 76% of donor T cells reacted. Newly stimulated T cells were found to be cross reactive to varying degrees: 3 clones were cross-reactive within species, 4 clones between some species, and 5 cross-reacted between all species tested [214]. While T cells can often be widely cross-reactive, this shows that vector specific T cells are also raised in response to vector administration. Schirmbeck et al., similar to other studies, found no response against a new transgene in intramuscularly HAdV-C5 vector vaccinated mice induced with pre-existing Anti-HAdV-C5 immunity. Their experiment showed that viral replication was not required for the development of an anti-vector CD8+ T-cell response, so minimal amounts of antigen are required for an effective response, which was determined to be targeted to highly conserved regions of the hexon [215].
Investigating the low seroprevalence ChAd63 as a vector for intramuscular malaria vaccines in Phase I clinical trial, pre-existing T cell responses to the ChAd63 vector hexon were found in 8 out of 12 volunteers tested, and increased in all participants after vaccination. Pre-vaccination anti-vector T cell responses led to lower anti-transgene responses, with lower final anti-transgene antibody titers. Both CD4+ and CD8+ T cells were found to contribute to this response [208]. As stated previously, it is unlikely that participants encountered ChAd63 through infection, hence the observed response likely resulted from cross-reactive T cells mounted in response to other Ad infections.
Another phase I clinical trial in the UK investigated using HAdV-C6 and ChAd3 vectors as heterologous HepC vaccine vectors. The study excluded volunteers with anti-vector antibody titers of >200, to prevent NAbs interfering with vector efficacy. They also measured anti-vector T cell response against the hexon. Responses were found even in non-primed subjects, and were increased substantially after vector administration. Booster vaccinations induced less transgene response than predicted. The study excluded participants with >200 anti-Ad antibody titer, so author concluded that anti-Hexon T cell responses were however not negatively correlated with transgene response, and instead attributed less-than-expected booster responses to cross-reactive NAbs developed after first administration [199]. Despite measuring a substantial increase in vector targeted T-cells, authors attribute low booster responses to NAbs. It is extremely unlikely that T cells are completely uninvolved in the anti-vector response presented.
A large amount of anti-Ad vector data is available from the STEP trial (NCT00125970) and follow up data analyses. A replication deficient HAdV-C5 vector was used in a phase II clinical trial as a HIV vaccine vector. 480 participants were vaccinated intramuscularly. The trial failed to protect vaccinees from HIV infection, and actually made some recipients of the HAdV-C5 based vector (uncircumcised men) more susceptible to HIV. While this was a devastating result, it led to several more in-depth studies on anti-Ad vector responses, and how these may affect vector applications. Using PBMCs from trial participants, the HAdV-C5 vector was found to induce receptor expression favourable to HIV in CD4+ T cells, as well as inducing a stronger anti-vector CD4+ than CD8+ T cell response, making them more susceptible to HIV infection [216]. In a follow up from STEP trial, Frahm et al. investigated cellular immunity to Ad vectors. Ad specific T cells could commonly be detected even in the absence of anti-Ad NAbs (in 54% of the seronegative/placebo vaccinated group). Pre-existing CD4+ and CD8+ T cell immunity to Ads induced impaired anti-transgene responses in vaccine vectors, regardless of the presence of NAbs. T cell target epitopes map to conserved region of the vector Ads. For CD4+ and CD8+ most these are commonly hexon, but other, non-structural epitopes are also recognised, such as protein (p) V, pVII, E2 terminal protein (E2pTP) for CD4+ and E3 glycoprotein 19 (E3gp19) and E4 open reading frame 6 (E4ORF6) for CD8+. Interestingly, E3gp19 and E4ORF6 were only targeted in the vaccine group, though results were not statistically significant due to the small sample size [217]. Hutnick et al. identified pre-existing anti-Ad CD8+ T cells in 95% of unvaccinated volunteers of the STEP trial. They attribute these high numbers to cross-reactivity rather than natural occurrence rate of HAdV-C5. Differences in CD8+ response were observed between different NAb titer subjects, though anti-vector response overall increased regardless of seropositivity before vaccination, even with replication deficient Ads [218]. Overall, these results emphasize that serotype specific immunity, though important more immediately upon administration, cannot predict later cross-reactivity due to T cells, and resulting loss of efficacy of vector applications. Furthermore, seropositivity as defined by Nab titer is not an indicator that a person is completely naïve to Ad infection. More research into anti-Ad vector responses is required to more accurately predict how a vector is likely to perform in clinic. Furthermore, it is impossible to regard only one side of the anti-vector response, as T cell and B cell responses (as well as the innate immune system) are intrinsically linked.
For example early gene therapy studies for the treatment of cystic fibrosis in mice using an intranasally administered HAdV-C5 based vector highlighted the importance of CD8+ T cell anti-vector responses, as mice that were challenged pre-treatment and only able to mount CD4+ T cell responses still saw some transgene expression. Additionally, the development of NAbs to the vector they made subsequent administrations much less effective, even in the absence of T cells [219]. Sumida et al. studied intramuscular immunisations with HAdV-C5 based vectors. After immunisation, they transferred splenocytes and serum to naïve mice, to separately test the effects of T cells and NAbs on subsequent vector administration. Both resulted in drastically lower responses to the transgene. Transfer of high levels of NAbs resulted in immediate, but short lived vector inhibition. 53% of mice were still able to induce a transgene response after transfer of splenocytes from double vaccinated mice, though anti-vector antibodies were developed after 2 weeks, either through transferred B or CD4+ T cells. This elegantly shows the synergistic effect of antibodies and T cells: while NAbs confer immediate immunity, long term immunity requires CD4+ T cell help. The immediate vector suppression from transfer of splenocytes was attributed to CD8+ T cells [220]. Similarly, the adaptive immune system will always interact with the innate immune system: For example, complement protein C3 is required to mount a neutralising ant-Ad vector antibody response, as fond by Appledorn et al. [221]. The complement system is an important in the bridge between innate and adaptive immunity, and a lot of the cytokines secreted due to complement activation are essential for the maturation of T and B cells. In the absence of these pro-inflammatory cytokines, it is possible to generate a tolerating anti-Ad response instead of a neutralising one.
As briefly touched upon before, different species and serotypes of Ads are considered to have different immunogenicity. Johnson et al. showed that in PBMCs from healthy donors, HAdV-D28 and HAdV-B35 lead to increased death of APCs. Consequently, less antigen was presented to T cells and response to the transgene was significantly lower than an equivalent HAdV-C5 based vaccine vectors [83]. While in a vaccine context this is not ideal, it also means decreased presentation of vector antigens, leading to reduced anti-vector responses and increasing the potential for effective repeated administration. Similarly, in a study investigating a broad range of ChAd vectors as well as a selection of rare HAdVs for their suitability as vaccine vectors in vivo, found lower doses of species C Ads were required to induce a robust anti-transgene response compared to other human Ads in mice and rhesus macaques [222]. This could be attributed to a similar mechanism as Johnson et al. In the same study, ChAd vectors showed surprisingly effective at inducing anti-transgene responses and additionally were found to be minimally affected by cross-serotype and cross-species anti-vector immunity in mice [222], which indicates no cross-reactive neutralising antibodies are produced. Responses to the same ChAd vector carrying different transgenes however were very limited [222], indicating a strong vector specific response was still induced. For some examples, Ad hexon PF4 binding has been linked to very rare instances of thrombotic thrombocytopenia [43,223,224], which was only apparent due to the wide rollout of the ChAdOx1 nCov-19 and Ad26.COV2.S vaccines during the coronavirus pandemic. Vaccine-induced thrombotic thrombocytopenia (VITT) is hypothesised to driven by autoantibodies against PF4 [224]. It is extremely rare, with roughly one case occurring in 67,000 primary dose vaccinations and one in 500,000 booster dose vaccinations with ChAdOx1 nCoV-19 in the UK according to the National Institute for Care Excellence (NICE) [225,226]. Interestingly, incidence of VITT in the US after administration of the Ad26.COV2.S vaccine was estimated to be 1 in 263,000 administered doses, which is lower than was found with the ChAdOx1 vector [227]. The generation of autoantibodies to PF4 induce aggregation, leading to platelet activation resulting in a thrombotic event [43,228,229].Binding to PF4 to the viral vector, which may well be critical in the generation of anti-PF4 autoantibodies, has been suggested to occur via HVR1[230] and involving interactions at the inter-hexon spaces [231], though neither of these theories have been confirmed. Confirming this mechanism would also allow for predictions of other Ad serotypes to bind PF4, and their consequent prevention.
6. Engineering Ads for Immune Evasion
Mechanisms described previously are important in protection from pathogens. In the case of vectorised Ads however, they might hinder treatment as the immune system is unable to distinguish between the pathogenic virus and the therapeutic vector. Ad vectors are in high demand for multiple clinical applications, and various strategies have been explored to reduce the effect of anti-Ad immunity, both natural and vector induced, on potential future treatments.
In numerous Ad based gene therapy platforms, a decline in gene expression over time is observed [232,233,234] and integration of the target gene needs to be incorporated to increase the duration of gene expression [141]. Natural hepatocyte turnover and expression of vector antigen were suggested as causes for long term failure in a gene therapy study into haemophilia B in mice, which spanned one year [232]. In cystic fibrosis, which was a popular target for initial investigations into vector based gene therapies due to the ease of access to the most affected tissues, initial benefits of vector administration quickly deteriorate due to the strong anti-vector response [235]. This fact was likely aggravated by the fact that the study design called for multiple vector administrations, and the same HAdV-C2 based vector was used for all of them. Brunetti-Pierri et al. achieved transgene expression of a helper dependent Ad vector for as long as 7 years in baboons, though gene expression still declined about 10% per year [236]. In order to not loose efficacy over time, the development of neutralising and memory responses must be minimised.
In cancer therapies, where replicating vectors are used, the problem is not just presented by reaching the target site but also maintaining the levels of the oncolytic vector high enough to have a therapeutic effect. As oncolytic Ad vectors are able to replicate in cancer cells, this increases exposure of the vector the patients immune system. In a single application, the engagement of the immune system can serve the purpose of immunogenic cancer therapy: Release of viral epitopes and PAMPs can serve as an additional activating factor for the anti-cancer immune response desired in oncolytics, and can help flag infected cancer cells for destruction to both the innate and adaptive immune system [237,238]. However, where additional, subsequent doses of oncolytic are required, this response may work against the patient instead, as the development of vector specific neutralising antibodies will prevent effective systemic administration. Consequently, for oncolytic Ad vectors, a fine balance needs to be found between engaging the immune system in a beneficial way and evading anti-vector immunity where it does not serve the patient.
For Ad based vaccines, boosting against the same antigen appears effective, with response to the antigen improved further when using a different vector in a heterologous booster vaccine [239]. This may be due to anti-vector immunity impeding the booster dose. The beneficial effect of heterologous vs homologous vaccine strategies is well established and could be attributed to anti-vector immunity [240,241]. The reduction of protection is shown to be even more pronounced when vaccine recipients were tested for anti-Ad antibodies prior to vaccination: In a trial for an Ad based Ebola vaccine in 2010, seronegative participants had a significantly better antigen specific immune response than those that were seropositive for HAdV-C5[242]. This has been seen in multiple other HAdV-C5 based vaccines since [243,244,245]. However, anti-vector immunity may not always be undesirable: The immune activation against the viral vector may reduce the need for adjuvants and boost the response to the target epitope [240].
To maximise the efficacy of Ad based therapeutics we require Ad vectors that are able to evade aspects all of the presented pathways, as all of them will universally play a role in the response to administered vectors [220]. However, as described there is no one-size-fits-all solution to the issue, as different vector applications may benefit from different solutions.
Here, we summarize these attempts in 3 categories: structural modifications, chemical shielding, and other approaches (Figure 5).
Figure 6.
Overview of Different Strategies for Vector Immune Evasion. To help Ad based vectors evade pre-existing anti-Ad immunity, different solutions have been proposed. These include wholly or partially switching vectors for more rare serotypes, Chemical shielding to physically prevent binding of mediators of an immune response as well as other approaches, such as changes in rote of entry or administration schedules to maximise efficacy despite anti-vector immunity.
Figure 6.
Overview of Different Strategies for Vector Immune Evasion. To help Ad based vectors evade pre-existing anti-Ad immunity, different solutions have been proposed. These include wholly or partially switching vectors for more rare serotypes, Chemical shielding to physically prevent binding of mediators of an immune response as well as other approaches, such as changes in rote of entry or administration schedules to maximise efficacy despite anti-vector immunity.
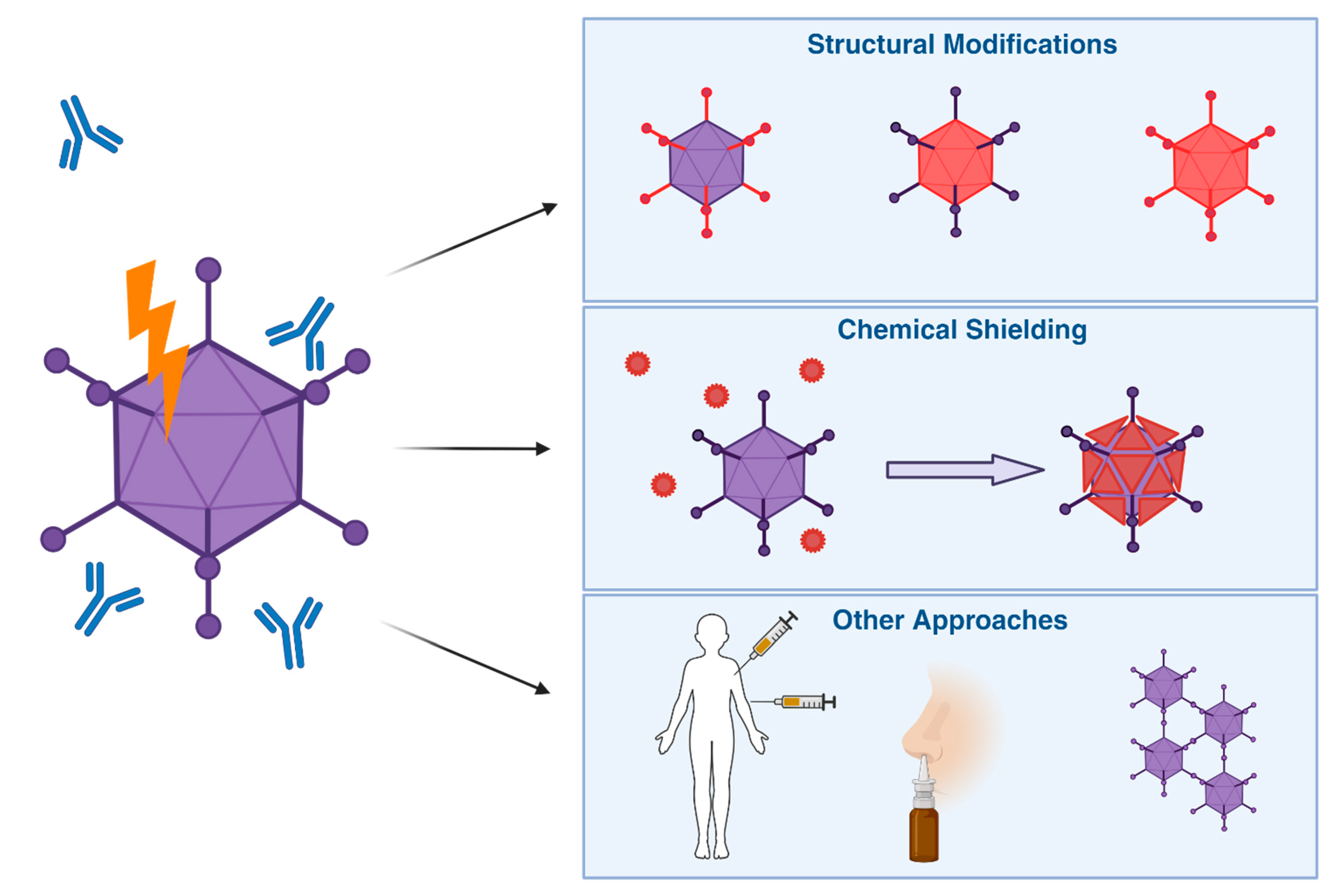
6.1. Structural Modifications
Ad vectors based on different species than HAdV-C5 have been shown to be beneficial in evading pre-existing immunity to HAdV-C5. Some groups have been successful in utilising non-human Ads, such as ChAds, bovine or porcine Ads. As they rare or even absent in the human population, there is less likelihood of specific immunity to whichever Ad was chosen [241,246]. Cross-reactivity between different serotypes, especially when originally targeting different hosts, is also usually low [246,247]. A 2021 study in rhesus macaques using a ChAd36 based vector found single intranasal vaccination dose to confer protection against intranasal and intrabronchial challenge with SARS-CoV2 [248].
Within the human Ads, vectorising alternative species to HAdV-C is a popular strategy [249,250], especially looking at species B[90,251] and D[250,252] Ads due to their particularly low seroprevalence [2,90,91,92].
Vectors that successfully evaded pre-existing anti-Ad immunity have been designed based on HAdV-D26 for the Jcovden SARS-CoV2 vaccine [253], HAdV-B11 [251] and HAdV-B35 [90,254] in various applications. Enadenotucirev, or EnAd (previously called ColoAd1), took the approach of directed evolution to produce a novel tumour selective Ad vector where replication is selective for tumour cells. The result contained parts of HAdV-B3 and HAdV-B11p, and successfully evaded anti-HAdV-C5 antibodies present in human serum neutralising to HAdV-C5 [255]. However, the repeated administration of the same vector still carries the drawbacks of anti-vector immune responses, potentially making this approach self-limiting [246,247].
Penaloza-MacMaster et al. investigated T-cell responses to vectors based on HAdV-D26, HAdV-B35 and HAdV-D48. The advantages of their chosen vectors in vaccine applications included less exhausted T-cell phenotypes and quicker memory reactivation to the transgene [256]. Unfortunately, they do not look into T-cell responses against their vector. If they mirrored the response to the transgene, this would likely heavily impair repeated vector administration.
Investigations into HAdV-D28 and HAdV-B35 as alternative vectors to HAdV-C5 revealed that they induce an increase in NK cell response, reducing their potential for transgene expression in infected cells [83]. This may impair their uses as vaccine vectors, where death of transfected cells is unwanted. However, it may make them well suited to applications as a oncolytics, as they are rare serotypes able to engage killing through the innate immune system, but through killing of APCs may not induce as much of an adaptive anti-vector response.
A comprehensive study by Wang et al. in 2023 evaluated 39 different serotypes covering all the Ad human species, for their potential suitability as vaccine vectors. They specifically focused on CD8+ T cell and DC activation, as well as cytokine expression in their in vitro assays, and predicted HAdV-C1, HAdV-D8, HAdV-B7, HAdV-F41, HAdV-D33, HAdV-C2, HAdV-A31, HAdV-B3 and HAdV-D65 to be particularly useful if vectorised [257]. As they are looking for high immunogenicity in potential vaccine vectors, it will be interesting to see if these predictions hold true, and if viruses they scored with lower immunogenicity ratings may be advantageous in engineering vectors with increased immune evasion in the future.
Several studies have investigated implementing structural changes to the Ad major capsid proteins. Wholly or partially exchanging the hexon protein with the externally exposed HVRs from low seroprevalence has provided these recombinant, chimeric vectors with protection from neutralising anti-HAdV-C5 antibodies.
Whole hexon swaps, especially between Ads of different species, are often structurally incompatible, as shown by Youil et al. Of 18 attempted structural changes, only 4 produced viable vectors. Transduction efficiency in vitro was reduced in HAdV-C5/C1 and HAdV-C5/C2, and drastically reduced in the HAdV-C5/A12 vectors compared to the parental HAdV-C5 vector, though gene expression was only markedly reduced in HAdV-C5/A12. In their in vivo studies, HAdV-C5/A12 proved similarly ineffective in expressing the encoded transgene, and while they concluded that the HAdV-C5/C6 vector was able to evade neutralising antibodies it was still hampered by HAdV-C5 cross-reactive T cells [258].
In 2006, Roberts et al. published their HAdV-C5 based vector, which had all hexon HVRs exchanged for those of HAdV-D48. While it had impressive ability to evade anti-HAdV-C5 neutralising antibodies in vitro [259], there were significant unpredicted dose limiting toxicities in subsequent in vivo experiments [260]. Another attempt was made by Tian et al., where the hexon protein of HAdV-B3 was swapped for that of HAdV-B7. Transduction efficiency was not impacted by the hexon swap, and the vector was able to evade neutralising antibodies targeting parental vectors in vitro. In vivo however, there was cross-reactivity to the HAdV-B3/B7 vector in mice immunized with HAdV-B3, possibly due to humoral immunity as well as anti-fiber antibodies [261].
In 2018, Nguyen et al. replaced the HVR regions of HAdV-C6 with those of HAdV-C57 in an attempt to create an improved oncolytic platform. While they demonstrated an effective anti-tumour response and designed the vector with reduced anti-vector responses in mind, they did not confirm if their approach had any effect on vector immune evasion [262]. The same vector was also evaluated for use in vaccines in 2019 [263], as well as oncolytic applications in 2022 [264] but without evaluation of the effect of HVR swap on anti-vector immunity.
The hexon protein selected in both of the latter cases came from the same species, which potentially made it structurally more permissive compared to swaps involving more distantly related Ads, previously attempted. However, it also means amino acid sequence conversion in some of the HVR sites (as well as the rest of the hexon protein) which may act as T cell epitopes. All these studies also highlight that in vitro experiments are not sufficient to accurately predict the effects of hexon modifications in vivo.
Most recently, Shin et al. published data on their retargeted HAdV-C5 based vector with most or all of hexon HVR regions of HAdV-D43. The HVR swapped vector was able to evade anti-HAdV-C5 neutralising antibodies both in vitro and in vivo and showed no cross-reactivity between neutralising antibodies generated [265]. Unfortunately, administration of the new vector induced a strong anti-vector response, likely making repeated administrations impossible. Furthermore, due to the use of transplanted neutralising serum rather than prime-boost experiments, we currently have no information whether T cell responses between the parental and new vectors might be cross reactive [265].
In theory, the change of hexon epitopes, though difficult, presents an attractive way of increasing vector immune evasion against both natural pre-existing immunity (where anti-hexon antibodies have strong neutralising ability, though they are less common) and previous intramuscular vector exposure (where hexon is the immunodominant epitope). To date, attempts have proven unsuccessful and less impactful than expected, possibly due to T cell immunity mostly targeting conserved parts of the hexon protein, making vectors much less efficient in vivo.
Other studies have investigated whether exchanging the fiber protein may impact upon the evasion of anti-HAdV-C5 immunity. There has been some evidence that this may aid in the evasion of neutralising immunity.
A HAdV-C5 vector pseudotyped with the fiber protein of HAdV-D45 published by Parker et al. in 2009 evaded pre-existing immunity in a proportion of tested serum samples unexpectedly well [47]. Another study by White et al. in 2013 with an HAdV-C5 based vector with an HAdV-B35 fiber showed similar results [266]. Both of these studies were performed in human neutralising serum, therefore not providing information on cellular immunity.
Other studies testing samples in immunized mouse serum could not observe the same effect, such as the HAdV-C5 vector with an HAdV-B7 fiber by Gall et al. [267] and Schoggins et al., who also investigated an HAdV-C5 vector with a HAdV-F41 fiber [268], likely due to previously described differences in responses depending on the mechanism of infection, and the anti-hexon skewed response following intramuscular Ad administration. In 2004, Ophorst et al. investigated an HAdV-C5 vector with an HAdV-B35 fiber for suitability as a vaccine vector in vivo in both mice and non-human primates. The effect of the fiber swap on evading anti-vector immunity was described as minimal [269].
Fiber protein is important in endosomal escape, leading to different routes of trafficking in different Ads [270]. Therefore, fiber swaps can change the intracellular trafficking of vectors and the way an anti-vector immune response is mounted intracellularly through engagement of different PAMPs. Additionally, fiber swaps may have an impact on vectors evading natural pre-existing immunity, due to the common pre-existing anti-fiber response.
Though not a major structural protein, pIX is considered a minor capsid protein that is integrated between hexons on the topical side of the capsid [271]. Seregin et al. showed that a inserting the complement inhibitor DAF (decay-accelerating factor) in pIX successfully prevented C3 binding, effectively shielding the virus from complement engagement [272]. Disengagement from complement could prevent the induction of anti-Ad neutralising antibodies, though the reduction of immunogenicity would likely come with a major trade-off for vaccine and oncolytic applications.
6.2. Chemical Shielding
An advantage of genetic structural alterations, particularly for oncolytic vector use, is the heritability of the modification. Daughter virions produced through replication will all have the same advantages as the parental vector. However, for vectors that do not need to or cannot replicate their immune evasive features, other options are available. A significant body of research into this area concerns the chemical binding and shielding of Ads using hydrophilic polymers.
Traditionally, polyethylene glycol (PEG) derivatives were considered as a good option for chemical shielding of Ads. PEG is a long chain polymer, which when attached to Ads will protrude from the capsid in a hairlike manner [273]. Successful examples include O’Riordan [274], Wortmann et al. [275] and Zeng et al. [276], all of which found that they were able create shielded vectors able to evade neutralising anti-vector responses in vitro and in vivo. In vivo, PEG shielding also successfully prevented complement activation [190].
Unfortunately, PEG itself induces a robust antibody response upon intravenous administration [277]. This limits PEG as a coating option for repeated vector uses. Very high levels of anti-PEG response has been found to be induced by the lipid-nanoparticle based COVID19 mRNA vaccine by Pfizer BioNTech, Comirnaty [278]. According to ourworldindata.org, to date this vaccine has been received by 668 million people across Europe [158]. As a result, at least 73% of the European population (Figure 5) will present anti-PEG neutralising antibodies, and its use is rapidly becoming obsolete: For Ad vectors specifically there is no longer any advantage of using PEG to shield Ads, and we require more modern solutions.
Another historically popular shielding polymer is N-(2-hydroxypropyl)methacrylamide (HPMA). HPMA is able to form a multivalent attachment at the capsid surface, coating it much more closely than PEG [273]. HPMA coating has been successful in retargeting Ad vectors from the natural receptor CAR and prevent neutralisation due to pre-existing antibodies in vitro and in vivo [279,280]. It also prevents binding to blood components such as complement and FX [280]. Coating can increase systemic circulation of Ad vector particles by preventing liver uptake for intravenous administrations [281]. HPMA polymers are less immunogenic when administered intravenously [282] making them a good candidate for viral vector coating.
A current strategy is the use of biodegradable alginate capsules as done for example by Salja et al. Encouragingly, coated vectors were able to evade anti-vector immunity even after HAdV-C5 prime and boost vaccination [283]. An overview of alginate spheres for drug delivery can be found here [284]. A similar strategy employed the use of hyaluronic acid polymers to create vesicles for OV delivery [285]. Though not investigated for immune evasion, packaging of OVs into polymers that the body is already familiar with will very likely be successful in protecting the therapeutic from NAbs. This has also been shown by Zhang et al, who coated their HAdV-C5 based vector in extracellular vesicles and showed protection from anti-Ad NAbs in vitro [286].
Alternatively, coating the virus in non biological materials such as minerals has been investigated, making use of the virus capsid charge to attach differently charged compounds. Wang et al. demonstrated that their vaccine vector was unaffected by anti-vector immunity, while still being able to efficiently deliver its transgene [287].
The Pluckthun lab has developed a specialised shield, which ablates Ad hexon binding blood factors as well as neutralising antibodies. Additionally, they have developed a “cap” that protects the fiber region, which also ablates any native HAdV-C5 binding to CAR. Though it was mostly developed with Ad retargeting in mind, the combination of the cap and shield showed impressive prevention of neutralisation in vitro, though no in vivo data is currently available [288].
An alternative approach combines genetic modifications and chemical shielding. One study demonstrated that the insertion of a cysteine amino acid in HVR5 of the hexon protein, enabled the linkage of polymers via thioether links, examples including HPMA[289] and PEG [290] (which as described is no longer very useful beyond proof of concept). However, the potential in this strategy lies in the versatility: As new polymers emerge in time [291], they could likely all be engineered to attach to these Ad vectors.
6.3. Other Approaches
A number of alternative approaches have been investigated to the problem of reducing anti-vector immunity that do not fit naturally into either of the previous categories, some of which have been summarised here.
Different routes of administration have been proposed as being more or less conducive to Ad transduction in the presence of neutralising antibodies. In 2000, Chen et al. suggested that intramuscular administration of Ad vectors may be able to circumvent anti-Ad immunity when used as a vaccine vector, compared to intranasal and intravenous administration [292]. Controversially, Croyle et al. found that, in the presence of pre-existing anti vector immunity, only vaccination via intranasal route, compared to oral and intramuscular, protected mice from lethal dose vaccine-target challenge [293]. Both cases present in vivo studies performed in mice, with initial vector challenge occurring intramuscular. Wu et al. in 2014 tested their HAdV-C5 based vaccine vector against anti-HAdV-C5 immunity induced intranasally in an effort to emulate natural infection. While they found that their vector had mildly reduced transduction, it still provided protection from vaccine challenge in vivo [294]. Intratumoural injection of Ad based vectors allows for localised therapeutic effect and activation of both anti-tumour and beneficial anti-Ad immune responses [295]. The unique immunological tumour microenvironment, which is usually immunosuppressed due to the expression of immunomodulatory molecules on cancer cells may aid in this. However, dissemination of the vector from the site of injection is prevented by preexisting anti-vector immunity [295]. Intradermal administration of an Ad based vector showed persistent local transgene expression and minimal tissue ad systemic immune engagement [296]. However, very localised transgene expression such as this will likely have very few clinical uses.
Another interesting approach is the packaging of oncolytic Ad vectors into patient cells in a “Trojan Horse” approach to increase delivery to the target tissue. Iscaro et al. coated oncolytic Ads in liposomes specifically for increased macrophage uptake. For their HAdV-C5/C2 based vector, used in intravenous administration, this strategy was designed to circumvent anti-Ad vector NAbs and destruction by phagocytic cells. Both the virus and a plasmid encoding E1A/B are encapsuled in the liposome to allow for replication in macrophages post uptake [297]. Macrophages traffic to hypoxic areas of the TME, which are usually hard to reach for intravenous therapies due to the lack of proper blood supply. Through conditional replication of an oncolytic virus under a hypoxia or cancer specific promoter, OV replication is limited to hard to reach areas of solid tumours while also evading neutralizing immunity. Tumour control was also shown in vivo in mice, using a HAdV-C5 based vector in prostate cancer, and NAb evasion was established in vitro in spheroid tumour models using human neutralising anti-Ad serum. [297,298]. This approach not only offers potential for immune evasion of OVs after intravenous application but also additional advantages in the delivery to the tumour site, providing an attractive novel strategy for OV intravenous delivery.
Another strategy involves engineering the vector to infect cells despite the presence of anti-vector immunity by enhancing transduction abilities. In 2023, Yang et al. found that through aggregating their vaccine vector particles and giving them a positive charge through coupling with human serum albumin nanoparticles, transduction in mice was increased both with and without pre-existing vector immunity, compared to the parental vector [299]. Human albumin has been shown previously to protect Ads from NAbs when linked to the hexon protein to coat single virions, in vivo in intraperitoneally pre-immunized and intravenously boosted mice [300] This HAdV-C5 based vector has a mutation in the hexon that allows for independent binding of human serum albumin once in the blood, giving daughter virions the same advantage of biological shielding as parental vectors [301]. This vector is now being investigated in Phase I clinical trials [302,303]. In the case of Yang et al, it is likely that coating with human serum albumin already protected the Ads. However the aggregation observed by Yang et al. gave also the vector increased transduction ability and helped it transduce despite vaccine induced vector immunity, rather than evading pre-existing NAbs.
Generally, replication deficient or empty capsid vectors would be expected to be less immunogenic than replication competent vectors in vivo, due to the reduced amount of viral protein exposed to the body if no virus progeny is produced [304]. Koup et al. proposed that their replication deficient HAdV-C5 vector, which contains deletions in several of the early expression genes, failed to induce a CD4+ T cell response, though anti-vector antibodies were detected [305]. Similarly, the deletion of the E2 gene along with the E1 gene of an HAdV-C5 based cancer vaccine increased transgene expression compared to a vector that only had the E1 deletion in the presence of anti-vector immunity [306]. The deletion of the Ad polymerase (E2) reduces late Ad gene expression, therefore reducing the amount of Ad antigen presented on the surface of infected cells.
However, it has also been shown that even after a single dose of replication deficient HAdV-C5 based vaccine administration, neutralising immunity can be detrimental to repeat administrations [307]. This shows that while deletion of genes for Ad replication likely has some improving effect on generating anti-vector immunity, it will not be enough on its own.
7. Concluding Remarks
Significant progress has been made in the development of more effective Ad based therapies. Genetic modifications and serotype alternatives to HAdV-C5 have shown promise to improve evasion of prevalent pre-existing anti-HAdV-C5 immunity. Chemical shielding provides an attractive opportunity to systemically reach sites of interest in the presence of anti-Ad NAbs. Careful selection of vaccination routes and schedules help optimize immunization strategies for Ad vectors already and close to being in clinic.
However, Ad based vector applications have still to live up to their clinical potential. A part of this may be due to the fact that as of yet, anti-vector responses are not fully defined or understood and research in this area has lagged behind the clinical translation of Ad based therapeutics. As different strategies and routes of administration become more mainstream, our mechanistic knowledge of differences in these routes of administration needs to increase as well. T cell responses to vectors are commonly understudied. While NAb responses are important, ignoring the possibility of cellular anti-vector responses means we do not get the complete picture of what happens in the body upon vector administration. Without this, we will keep seeing misestimations and vectors that work well in an in vitro setting and fail in in vivo systems.
To make better informed choices of modifications to Ad vectors, we need to better define and understand anti-Ad vector responses, overall and to different routes of administration. This understanding can then be applied to the next generation of “smart” Ad based vector platforms, that appropriately evade or harness the immune system where required. This will maximise patient efficacy in the clinic, thus enabling the immune system to function firmly as a friend in the context of Ad based therapeutics.
Author Contributions
Writing—original draft preparation, R.W. with guidance and input from C.M.B. and A.L.P.; writing—review and editing, R.W, C.M.B. and A.L.P. Funding acquisition, A.L.P.
Funding
RW was funded by a Cancer Research UK Biotherapeutic Programme grant to A.L.P. (reference C52915/A29104). A.L.P. and C.M.B are funded by HEFCW.
Conflicts of Interest
ALP is founder and CSO of Trocept Therapeutics. The remaining authors have no conflict of interest to declare.
References
- Mennechet, F.J.D.; Paris, O.; Ouoba, A.R.; Salazar Arenas, S.; Sirima, S.B.; Takoudjou Dzomo, G.R.; Diarra, A.; Traore, I.T.; Kania, D.; Eichholz, K.; et al. A Review of 65 Years of Human Adenovirus Seroprevalence. Expert Rev Vaccines 2019, 18, 597–613. [Google Scholar] [CrossRef] [PubMed]
- Abbink, P.; Lemckert, A.A.C.; Ewald, B.A.; Lynch, D.M.; Denholtz, M.; Smits, S.; Holterman, L.; Damen, I.; Vogels, R.; Thorner, A.R.; et al. Comparative Seroprevalence and Immunogenicity of Six Rare Serotype Recombinant Adenovirus Vaccine Vectors from Subgroups B and D. J Virol 2007, 81, 4654–4663. [Google Scholar] [CrossRef] [PubMed]
- Stasiak, A.C.; Stehle, T. Human Adenovirus Binding to Host Cell Receptors: A Structural View. Med Microbiol Immunol 2020, 209, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, Y.; Nakajima, E.; Ishikawa, M.; Kano, A.; Komane, A.; Fujimoto, T.; Hanaoka, N.; Okabe, N.; Shimizu, H. Construction of New Primer Sets for Corresponding to Genetic Evolution of Human Adenoviruses in Major Capsid Genes through Frequent Recombination. Japanese Journal of Infectious Diseases 2014, 67, 495–502. [Google Scholar] [CrossRef]
- Davies, J.A.; Marlow, G.; Uusi-Kerttula, H.K.; Seaton, G.; Piggott, L.; Badder, L.M.; Clarkson, R.W.E.; Chester, J.D.; Parker, A.L. Efficient Intravenous Tumor Targeting Using the Avβ6 Integrin-Selective Precision Virotherapy Ad5NULL-A20. Viruses 2021, 13, 864. [Google Scholar] [CrossRef]
- Coughlan, L.; Uusi-Kerttula, H.; Ma, J.; Degg, B.P.; Parker, A.L.; Baker, A.H. Retargeting Adenovirus Serotype 48 Fiber Knob Domain by Peptide Incorporation. Hum Gene Ther 2014, 25, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Hiwasa, K.; Nagaya, H.; Terao, S.; Acharya, B.; Hamada, K.; Mizuguchi, H.; Gotoh, A. Improved Gene Transfer into Bladder Cancer Cells Using Adenovirus Vector Containing RGD Motif. Anticancer Research 2012. [Google Scholar]
- Uusi-Kerttula, H.; Davies, J.A.; Thompson, J.M.; Wongthida, P.; Evgin, L.; Shim, K.G.; Bradshaw, A.; Baker, A.T.; Rizkallah, P.J.; Jones, R.; et al. Ad5NULL-A20: A Tropism-Modified, Avβ6 Integrin-Selective Oncolytic Adenovirus for Epithelial Ovarian Cancer Therapies. Clinical Cancer Research 2018, 24, 4215–4224. [Google Scholar] [CrossRef]
- Zaiss, A.K.; Machado, H.B.; Herschman, H.R. TheQ1 Influence of Innate and Pre-Existing Immunity on Adenovirus Therapy. J Cell Biochem 2009, 108, 778. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.C. Adenoviruses: Update on Structure and Function. Journal of General Virology 2009, 90, 1–20. [Google Scholar] [CrossRef]
- Gallardo, J.; Pérez-Illana, M.; Martín-González, N.; San Martín, C. Adenovirus Structure: What Is New? Int J Mol Sci 2021, 22, 5240. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.; Pache, L.; Von Seggern, D.J.; Mullen, T.-M.; Mikyas, Y.; Stewart, P.L.; Nemerow, G.R. Flexibility of the Adenovirus Fiber Is Required for Efficient Receptor Interaction. J Virol 2003, 77, 7225–7235. [Google Scholar] [CrossRef] [PubMed]
- Marttila, M.; Persson, D.; Gustafsson, D.; Liszewski, M.K.; Atkinson, J.P.; Wadell, G.; Arnberg, N. CD46 Is a Cellular Receptor for All Species B Adenoviruses except Types 3 and 7. J Virol 2005, 79, 14429–14436. [Google Scholar] [CrossRef] [PubMed]
- Nestić, D.; Božinović, K.; Pehar, I.; Wallace, R.; Parker, A.L.; Majhen, D. The Revolving Door of Adenovirus Cell Entry: Not All Pathways Are Equal. Pharmaceutics 2021, 13, 1585. [Google Scholar] [CrossRef] [PubMed]
- Arnberg, N. Adenovirus Receptors: Implications for Targeting of Viral Vectors. Trends in Pharmacological Sciences 2012, 33, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Rajan, A.; Palm, E.; Trulsson, F.; Mundigl, S.; Becker, M.; Persson, B.D.; Frängsmyr, L.; Lenman, A. Heparan Sulfate Is a Cellular Receptor for Enteric Human Adenoviruses. Viruses 2021, 13, 298. [Google Scholar] [CrossRef] [PubMed]
- Tsoukas, R.L.; Volkwein, W.; Gao, J.; Schiwon, M.; Bahlmann, N.; Dittmar, T.; Hagedorn, C.; Ehrke-Schulz, E.; Zhang, W.; Baiker, A.; et al. A Human In Vitro Model to Study Adenoviral Receptors and Virus Cell Interactions. Cells 2022, 11, 841. [Google Scholar] [CrossRef] [PubMed]
- Howitt, J.; Anderson, C.W.; Freimuth, P. Adenovirus Interaction with Its Cellular Receptor CAR. In Adenoviruses: Model and Vectors in Virus-Host Interactions: Virion-Structure, Viral Replication and Host-Cell Interactions; Doerfler, W., Böhm, P., Eds.; Current Topics in Microbiology and Immunology; Springer: Berlin, Heidelberg, 2003; pp. 331–364. ISBN 978-3-662-05597-7. [Google Scholar]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a Common Receptor for Coxsackie B Viruses and Adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Shayakhmetov, D.M.; Lieber, A. Dependence of Adenovirus Infectivity on Length of the Fiber Shaft Domain. J Virol 2000, 74, 10274–10286. [Google Scholar] [CrossRef] [PubMed]
- Law, L.K.; Davidson, B.L. Adenovirus Serotype 30 Fiber Does Not Mediate Transduction via the Coxsackie-Adenovirus Receptor. Journal of Virology 2002, 76, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.T.; Greenshields-Watson, A.; Coughlan, L.; Davies, J.A.; Uusi-Kerttula, H.; Cole, D.K.; Rizkallah, P.J.; Parker, A.L. Diversity within the Adenovirus Fiber Knob Hypervariable Loops Influences Primary Receptor Interactions. Nat Commun 2019, 10, 741. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-D.; Liu, X.-L.; Chen, D.-L.; Zou, X.-H.; Wang, M.; Qu, J.-G.; Lu, Z.-Z.; Hung, T. Human Adenovirus Type 41 Possesses Different Amount of Short and Long Fibers in the Virion. Virology 2012, 432, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.T.; Davies, J.A.; Bates, E.A.; Moses, E.; Mundy, R.M.; Marlow, G.; Cole, D.K.; Bliss, C.M.; Rizkallah, P.J.; Parker, A.L. The Fiber Knob Protein of Human Adenovirus Type 49 Mediates Highly Efficient and Promiscuous Infection of Cancer Cell Lines Using a Novel Cell Entry Mechanism. J Virol 2021, 95, e01849–20. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, W.P.; Guilligay, D.; Cusack, S.; Wadell, G.; Arnberg, N. Crystal Structure of Species D Adenovirus Fiber Knobs and Their Sialic Acid Binding Sites. J Virol 2004, 78, 7727–7736. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.T.; Mundy, R.M.; Davies, J.A.; Rizkallah, P.J.; Parker, A.L. Human Adenovirus Type 26 Uses Sialic Acid–Bearing Glycans as a Primary Cell Entry Receptor. Science Advances 2019, 5, eaax3567. [Google Scholar] [CrossRef] [PubMed]
- Mundy, R.M.; Baker, A.T.; Bates, E.A.; Cunliffe, T.G.; Teijeira-Crespo, A.; Moses, E.; Rizkallah, P.J.; Parker, A.L. Broad Sialic Acid Usage amongst Species D Human Adenovirus. npj Viruses 2023, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Persson, B.D.; John, L.; Rafie, K.; Strebl, M.; Frängsmyr, L.; Ballmann, M.Z.; Mindler, K.; Havenga, M.; Lemckert, A.; Stehle, T.; et al. Human Species D Adenovirus Hexon Capsid Protein Mediates Cell Entry through a Direct Interaction with CD46. Proceedings of the National Academy of Sciences 2021, 118. [Google Scholar] [CrossRef]
- Hemsath, J.R.; Liaci, A.M.; Rubin, J.D.; Parrett, B.J.; Lu, S.-C.; Nguyen, T.V.; Turner, M.A.; Chen, C.Y.; Cupelli, K.; Reddy, V.S.; et al. Ex Vivo and In Vivo CD46 Receptor Utilization by Species D Human Adenovirus Serotype 26 (HAdV26). J Virol 2022, 96, e0082621. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 Is a Cellular Receptor for Group B Adenoviruses. Nat Med 2003, 9, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Fleischli, C.; Sirena, D.; Lesage, G.; Havenga, M.J.E.; Cattaneo, R.; Greber, U.F.; Hemmi, S. Species B Adenovirus Serotypes 3, 7, 11 and 35 Share Similar Binding Sites on the Membrane Cofactor Protein CD46 Receptor. J Gen Virol 2007, 88, 2925–2934. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, F.; Kawabata, K.; Koizumi, N.; Inoue, N.; Okabe, M.; Yamaguchi, T.; Hayakawa, T.; Mizuguchi, H. Adenovirus Serotype 35 Vector-Mediated Transduction into Human CD46-Transgenic Mice. Gene Ther 2006, 13, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liaw, Y.-C.; Stone, D.; Kalyuzhniy, O.; Amiraslanov, I.; Tuve, S.; Verlinde, C.L.M.J.; Shayakhmetov, D.; Stehle, T.; Roffler, S.; et al. Identification of CD46 Binding Sites within the Adenovirus Serotype 35 Fiber Knob. J Virol 2007, 81, 12785–12792. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, D.J.; Segerman, A.; Lindman, K.; Mei, Y.-F.; Wadell, G. The Arg279Gln [Corrected] Substitution in the Adenovirus Type 11p (Ad11p) Fiber Knob Abolishes EDTA-Resistant Binding to A549 and CHO-CD46 Cells, Converting the Phenotype to That of Ad7p. J Virol 2006, 80, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Persson, B.D.; Reiter, D.M.; Marttila, M.; Mei, Y.-F.; Casasnovas, J.M.; Arnberg, N.; Stehle, T. Adenovirus Type 11 Binding Alters the Conformation of Its Receptor CD46. Nat Struct Mol Biol 2007, 14, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.; Liu, Y.; Persson, J.; Beyer, I.; Möller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.-B.; et al. Desmoglein 2 Is a Receptor for Adenovirus Serotypes 3, 7, 11, and 14. Nat Med 2011, 17, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Vassal-Stermann, E.; Effantin, G.; Zubieta, C.; Burmeister, W.; Iseni, F.; Wang, H.; Lieber, A.; Schoehn, G.; Fender, P. CryoEM Structure of Adenovirus Type 3 Fibre with Desmoglein 2 Shows an Unusual Mode of Receptor Engagement. Nat Commun 2019, 10, 1181. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Singh, G.; Lee, J.Y.; Dehghan, S.; Rajaiya, J.; Liu, E.B.; Yousuf, M.A.; Betensky, R.A.; Jones, M.S.; Dyer, D.W.; et al. Molecular Evolution of Human Adenoviruses. Sci Rep 2013, 3, 1812. [Google Scholar] [CrossRef] [PubMed]
- Alba, R.; Bradshaw, A.C.; Parker, A.L.; Bhella, D.; Waddington, S.N.; Nicklin, S.A.; van Rooijen, N.; Custers, J.; Goudsmit, J.; Barouch, D.H.; et al. Identification of Coagulation Factor (F)X Binding Sites on the Adenovirus Serotype 5 Hexon: Effect of Mutagenesis on FX Interactions and Gene Transfer. Blood 2009, 114, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Persson, B.D.; Lenman, A.; Frängsmyr, L.; Schmid, M.; Ahlm, C.; Plückthun, A.; Jenssen, H.; Arnberg, N. Lactoferrin-Hexon Interactions Mediate CAR-Independent Adenovirus Infection of Human Respiratory Cells. J Virol 2020, 94, e00542–20. [Google Scholar] [CrossRef]
- Morris, S.J.; Sebastian, S.; Spencer, A.J.; Gilbert, S.C. Simian Adenoviruses as Vaccine Vectors. Future Virol 2016, 11, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Dicks, M.D.J.; Spencer, A.J.; Edwards, N.J.; Wadell, G.; Bojang, K.; Gilbert, S.C.; Hill, A.V.S.; Cottingham, M.G. A Novel Chimpanzee Adenovirus Vector with Low Human Seroprevalence: Improved Systems for Vector Derivation and Comparative Immunogenicity. PLoS One 2012, 7, e40385. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.T.; Boyd, R.J.; Sarkar, D.; Teijeira-Crespo, A.; Chan, C.K.; Bates, E.; Waraich, K.; Vant, J.; Wilson, E.; Truong, C.D.; et al. ChAdOx1 Interacts with CAR and PF4 with Implications for Thrombosis with Thrombocytopenia Syndrome. Science Advances 2021, 7, eabl8213. [Google Scholar] [CrossRef] [PubMed]
- Reeh, M.; Bockhorn, M.; Görgens, D.; Vieth, M.; Hoffmann, T.; Simon, R.; Izbicki, J.R.; Sauter, G.; Schumacher, U.; Anders, M. Presence of the Coxsackievirus and Adenovirus Receptor (CAR) in Human Neoplasms: A Multitumour Array Analysis. Br J Cancer 2013, 109, 1848–1858. [Google Scholar] [CrossRef] [PubMed]
- Asher, D.R.; Cerny, A.M.; Finberg, R.W. The Erythrocyte Viral Trap: Transgenic Expression of Viral Receptor on Erythrocytes Attenuates Coxsackievirus B Infection. Proceedings of the National Academy of Sciences 2005, 102, 12897–12902. [Google Scholar] [CrossRef] [PubMed]
- Hensen, L.C.M.; Hoeben, R.C.; Bots, S.T.F. Adenovirus Receptor Expression in Cancer and Its Multifaceted Role in Oncolytic Adenovirus Therapy. Int J Mol Sci 2020, 21, 6828. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.L.; Waddington, S.N.; Buckley, S.M.K.; Custers, J.; Havenga, M.J.E.; van Rooijen, N.; Goudsmit, J.; McVey, J.H.; Nicklin, S.A.; Baker, A.H. Effect of Neutralizing Sera on Factor X-Mediated Adenovirus Serotype 5 Gene Transfer. J Virol 2009, 83, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral Innate Immunity Pathways. Cell Res 2006, 16, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and Viruses: An Interplay between Induction, Signalling, Antiviral Responses and Virus Countermeasures. J Gen Virol 2008, 89, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Weber, F. Antiviral Innate Immunity: Introduction. Encyclopedia of Virology 2021, 577–583. [Google Scholar] [CrossRef]
- Nesargikar, P.N.; Spiller, B.; Chavez, R. The Complement System: History, Pathways, Cascade and Inhibitors. Eur J Microbiol Immunol (Bp) 2012, 2, 103–111. [Google Scholar] [CrossRef]
- Charles A Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. The Complement System and Innate Immunity. Immunobiology: The Immune System in Health and Disease. 5th edition 2001.
- Smith, J.G.; Silvestry, M.; Lindert, S.; Lu, W.; Nemerow, G.R.; Stewart, P.L. Insight into the Mechanisms of Adenovirus Capsid Disassembly from Studies of Defensin Neutralization. PLoS Pathog 2010, 6, e1000959. [Google Scholar] [CrossRef] [PubMed]
- Land, W.G. Prologue: About DAMPs, PAMPs, and MAMPs. In Damage-Associated Molecular Patterns in Human Diseases: Volume 1: Injury-Induced Innate Immune Responses; Land, W.G., Ed.; Springer International Publishing: Cham, 2018; pp. 191–217. ISBN 978-3-319-78655-1. [Google Scholar]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T Cell Activation. Annu Rev Immunol 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Mescher, M.F. Inflammatory Cytokines as a Third Signal for T Cell Activation. Current Opinion in Immunology 2010, 22, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Hope, J.L.; Bradley, L.M. Lessons in Antiviral Immunity. Science 2021. [Google Scholar] [CrossRef] [PubMed]
- Charles A Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunological Memory. In Immunobiology: The Immune System in Health and Disease. 5th edition; Garland Science, 2001.
- Charles A Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. B-Cell Activation by Armed Helper T Cells. In Immunobiology: The Immune System in Health and Disease. 5th edition; Garland Science, 2001.
- Charles A Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. The Structure of a Typical Antibody Molecule. In Immunobiology: The Immune System in Health and Disease. 5th edition; Garland Science, 2001.
- Koh, C.-H.; Lee, S.; Kwak, M.; Kim, B.-S.; Chung, Y. CD8 T-Cell Subsets: Heterogeneity, Functions, and Therapeutic Potential. Exp Mol Med 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.W.; Cavacini, L. Structure and Function of Immunoglobulins. J Allergy Clin Immunol 2010, 125, S41–S52. [Google Scholar] [CrossRef] [PubMed]
- Stavnezer, J.; Guikema, J.E.J.; Schrader, C.E. Mechanism and Regulation of Class Switch Recombination. Annu Rev Immunol 2008, 26, 261–292. [Google Scholar] [CrossRef] [PubMed]
- Forthal, D.N. Functions of Antibodies. Microbiol Spectr 2014, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Klasse, P.J. Neutralization of Virus Infectivity by Antibodies: Old Problems in New Perspectives. Adv Biol 2014, 2014, 157895. [Google Scholar] [CrossRef]
- Top, F.H. Control of Adenovirus Acute Respiratory Disease in U.S. Army Trainees. Yale J Biol Med 1975, 48, 185–195. [Google Scholar] [PubMed]
- Fox, J.P.; Brandt, C.D.; Wassermann, F.E.; Hall, C.E.; Spigland, I.; Kogon, A.; Elveback, L.R. The Virus Watch Program: A Continuing Surveillance of Viral Infections in Metropolitan New York Families: Observations of Adenovirus Infections: Virus Excretion Patterns, Antibody Response, Efficiency of Surveillance, Patterns of Infection, and Relation to Illness. American Journal of Epidemiology 1969, 89, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Echavarría, M. Adenoviruses in Immunocompromised Hosts. Clin Microbiol Rev 2008, 21, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.P.; Chintakuntlawar, A.; Robinson, C.M.; Madisch, I.; Harrach, B.; Hudson, N.R.; Schnurr, D.; Heim, A.; Chodosh, J.; Seto, D.; et al. Evidence of Molecular Evolution Driven by Recombination Events Influencing Tropism in a Novel Human Adenovirus That Causes Epidemic Keratoconjunctivitis. PLOS ONE 2009, 4, e5635. [Google Scholar] [CrossRef] [PubMed]
- De Jong, J.; Muzerie, C.; Wermenbol, A.; Wigand, R.; Keller, D.; Wadell, G.; Schaap, G. Adenovirus 37: Identification and Characterization of a Medically Important New Adenovirus Type of Subgroup D. Journal of Medical Virology 1981, 7, 105–118. [Google Scholar] [CrossRef]
- Lei, Z.; Zhu, Z.; wang, B. ma ci; mei, H.; Li, H.; ga, D. zeng gong; jie, G.; chi, M. ma bu; Zhang, S.; Ma, C.; et al. Outbreaks of Epidemic Keratoconjunctivitis Caused by Human Adenovirus Type 8 in the Tibet Autonomous Region of China in 2016. PLoS One 2017, 12, e0185048. [Google Scholar] [CrossRef] [PubMed]
- Toovey, O.T.R.; Kulkarni, P.; David, J.; Patel, A.; Lai, F.Y.; Burns, J.; Thompson, C.; Ellis, J.; Tang, J.W. An Outbreak of Adenovirus D8 Keratoconjunctivitis in Leicester, United Kingdom, from March to August 2019. J Med Virol 2021, 93, 3969–3973. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.K.; Caul, E.O.; Porter, H.J.; Oakhill, A. Fatal Adenovirus 32 Infection in a Bone Marrow Transplant Recipient. Journal of Clinical Pathology 1995, 48, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Van Der Veen, J.; Van Der Ploeg, G. Adenovirus Type-15 Infection. The Lancet 1960, 275, 1024. [Google Scholar] [CrossRef]
- James, L.; Vernon, M.O.; Jones, R.C.; Stewart, A.; Lu, X.; Zollar, L.M.; Chudoba, M.; Westercamp, M.; Alcasid, G.; Duffee-Kerr, L.; et al. Outbreak of Human Adenovirus Type 3 Infection in a Pediatric Long-Term Care Facility—Illinois, 2005. Clinical Infectious Diseases 2007, 45, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.I.; Erdman, D.D.; Pur, S.L.; Diaz, P.S.; Segreti, J.; Kajon, A.E.; Belkengren, R.P.; Jones, R.C. Outbreak of Adenovirus Genome Type 7d2 Infection in a Pediatric Chronic-Care Facility and Tertiary-Care Hospital. Clinical Infectious Diseases 2001, 32, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Rozwadowski, F. Notes from the Field: Fatalities Associated with Human Adenovirus Type 7 at a Substance Abuse Rehabilitation Facility — New Jersey, 2017. MMWR Morb Mortal Wkly Rep 2018, 67. [Google Scholar] [CrossRef]
- Atasheva, S.; Shayakhmetov, D.M. Adenovirus Sensing by the Immune System. Curr Opin Virol 2016, 21, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Doronin, K.; Flatt, J.W.; Di Paolo, N.C.; Khare, R.; Kalyuzhniy, O.; Acchione, M.; Sumida, J.P.; Ohto, U.; Shimizu, T.; Akashi-Takamura, S.; et al. Coagulation Factor X Activates Innate Immunity to Human Species C Adenovirus. Science 2012, 338, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.; Stein, S.; Falck-Pedersen, E. Adenovirus Detection by the cGAS/STING/TBK1 DNA Sensing Cascade. J Virol 2014, 88, 974–981. [Google Scholar] [CrossRef]
- Iacobelli-Martinez, M.; Nemerow, G.R. Preferential Activation of Toll-Like Receptor Nine by CD46-Utilizing Adenoviruses. J Virol 2007, 81, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Teigler, J.E.; Kagan, J.C.; Barouch, D.H. Late Endosomal Trafficking of Alternative Serotype Adenovirus Vaccine Vectors Augments Antiviral Innate Immunity. J Virol 2014, 88, 10354–10363. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.J.; Björkström, N.K.; Petrovas, C.; Liang, F.; Gall, J.G.D.; Loré, K.; Koup, R.A. Type I Interferon-Dependent Activation of NK Cells by rAd28 or rAd35, but Not rAd5, Leads to Loss of Vector-Insert Expression. Vaccine 2014, 32, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Bastian, A.; Schäfer, H. Human α-Defensin 1 (HNP-1) Inhibits Adenoviral Infection in Vitro. Regulatory Peptides 2001, 101, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.G.; Cassany, A.; Gerace, L.; Ralston, R.; Nemerow, G.R. Neutralizing Antibody Blocks Adenovirus Infection by Arresting Microtubule-Dependent Cytoplasmic Transport. J Virol 2008, 82, 6492–6500. [Google Scholar] [CrossRef] [PubMed]
- Mistchenko, A.S.; Diez, R.A.; Mariani, A.L.; Robaldo, J.; Maffey, A.F.; Bayley-Bustamante, G.; Grinstein, S. Cytokines in Adenoviral Disease in Children: Association of Interleukin-6, Interleukin-8, and Tumor Necrosis Factor Alpha Levels with Clinical Outcome. J Pediatr 1994, 124, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb Perspect Biol 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.W. Ocular Immune Privilege. Eye 2009, 23, 1885–1889. [Google Scholar] [CrossRef] [PubMed]
- Sprangers, M.C.; Lakhai, W.; Koudstaal, W.; Verhoeven, M.; Koel, B.F.; Vogels, R.; Goudsmit, J.; Havenga, M.J.E.; Kostense, S. Quantifying Adenovirus-Neutralizing Antibodies by Luciferase Transgene Detection: Addressing Preexisting Immunity to Vaccine and Gene Therapy Vectors. J Clin Microbiol 2003, 41, 5046–5052. [Google Scholar] [CrossRef] [PubMed]
- Vogels, R.; Zuijdgeest, D.; van Rijnsoever, R.; Hartkoorn, E.; Damen, I.; de Béthune, M.-P.; Kostense, S.; Penders, G.; Helmus, N.; Koudstaal, W.; et al. Replication-Deficient Human Adenovirus Type 35 Vectors for Gene Transfer and Vaccination: Efficient Human Cell Infection and Bypass of Preexisting Adenovirus Immunity. J Virol 2003, 77, 8263–8271. [Google Scholar] [CrossRef] [PubMed]
- Dakin, R.S.; Parker, A.L.; Delles, C.; Nicklin, S.A.; Baker, A.H. Efficient Transduction of Primary Vascular Cells by the Rare Adenovirus Serotype 49 Vector. Hum Gene Ther 2015, 26, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Klann, P.J.; Wang, X.; Elfert, A.; Zhang, W.; Köhler, C.; Güttsches, A.-K.; Jacobsen, F.; Weyen, U.; Roos, A.; Ehrke-Schulz, E.; et al. Seroprevalence of Binding and Neutralizing Antibodies against 39 Human Adenovirus Types in Patients with Neuromuscular Disorders. Viruses 2022, 15, 79. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.R.; Lynch, D.M.; Iampietro, M.J.; Borducchi, E.N.; Barouch, D.H. Adenovirus Serotype 5 Neutralizing Antibodies Target Both Hexon and Fiber Following Vaccination and Natural Infection. J Virol 2012, 86, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Dong, J.; Wang, C.; Zhan, Y.; Zhang, H.; Wu, J.; Kong, W.; Yu, X. Characteristics of Neutralizing Antibodies to Adenovirus Capsid Proteins in Human and Animal Sera. Virology 2013, 437, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Gahéry-Ségard, H.; Farace, F.; Godfrin, D.; Gaston, J.; Lengagne, R.; Tursz, T.; Boulanger, P.; Guillet, J.-G. Immune Response to Recombinant Capsid Proteins of Adenovirus in Humans: Antifiber and Anti-Penton Base Antibodies Have a Synergistic Effect on Neutralizing Activity. J Virol 1998, 72, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Tsai, V.; Goudreau, A.; Shinoda, J.Y.; Wen, S.F.; Ramachandra, M.; Ralston, R.; Maneval, D.; LaFace, D.; Shabram, P. Specific Depletion of Human Anti-Adenovirus Antibodies Facilitates Transduction in an in Vivo Model for Systemic Gene Therapy. Mol Ther 2001, 3, 768–778. [Google Scholar] [CrossRef]
- Sumida, S.M.; Truitt, D.M.; Lemckert, A.A.C.; Vogels, R.; Custers, J.H.H.V.; Addo, M.M.; Lockman, S.; Peter, T.; Peyerl, F.W.; Kishko, M.G.; et al. Neutralizing Antibodies to Adenovirus Serotype 5 Vaccine Vectors Are Directed Primarily against the Adenovirus Hexon Protein1. The Journal of Immunology 2005, 174, 7179–7185. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.R.; Maxfield, L.F.; Lynch, D.M.; Iampietro, M.J.; Borducchi, E.N.; Barouch, D.H. Adenovirus Serotype 5-Specific Neutralizing Antibodies Target Multiple Hexon Hypervariable Regions. J Virol 2012, 86, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Varghese, R.; Mikyas, Y.; Stewart, P.L.; Ralston, R. Postentry Neutralization of Adenovirus Type 5 by an Antihexon Antibody. J Virol 2004, 78, 12320–12332. [Google Scholar] [CrossRef] [PubMed]
- Wohlfart, C. Neutralization of Adenoviruses: Kinetics, Stoichiometry, and Mechanisms. J Virol 1988, 62, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Everitt, E.; Lutter, L.; Philipson, L. Structural Proteins of Adenoviruses: XII. Location and Neighbor Relationship among Proteins of Adenovirion Type 2 as Revealed by Enzymatic Lodination, Immunoprecipitation and Chemical Cross-Linking. Virology 1975, 67, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Tian, J.; Smith, J.S.; Byrnes, A.P. Clearance of Adenovirus by Kupffer Cells Is Mediated by Scavenger Receptors, Natural Antibodies, and Complement. J Virol 2008, 82, 11705–11713. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, A.K.; Vilaysane, A.; Cotter, M.J.; Clark, S.A.; Meijndert, H.C.; Colarusso, P.; Yates, R.M.; Petrilli, V.; Tschopp, J.; Muruve, D.A. Antiviral Antibodies Target Adenovirus to Phagolysosomes and Amplify the Innate Immune Response1. The Journal of Immunology 2009, 182, 7058–7068. [Google Scholar] [CrossRef] [PubMed]
- de Sousa-Pereira, P.; Woof, J.M. IgA: Structure, Function, and Developability. Antibodies (Basel) 2019, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Olive, M.; Eisenlohr, L.; Flomenberg, N.; Hsu, S.; Flomenberg, P. The Adenovirus Capsid Protein Hexon Contains a Highly Conserved Human CD4+ T-Cell Epitope. Hum Gene Ther 2002, 13, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Olive, M.; Champagne, K.; Flomenberg, N.; Eisenlohr, L.; Hsu, S.; Flomenberg, P. Adenovirus Hexon T-Cell Epitope Is Recognized by Most Adults and Is Restricted by HLA DP4, the Most Common Class II Allele. Gene Ther 2004, 11, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Onion, D.; Crompton, L.J.; Milligan, D.W.; Moss, P.A.H.; Lee, S.P.; Mautner, V. The CD4+ T-Cell Response to Adenovirus Is Focused against Conserved Residues within the Hexon Protein. Journal of General Virology 2007, 88, 2417–2425. [Google Scholar] [CrossRef] [PubMed]
- Veltrop-Duits, L.A.; Heemskerk, B.; Sombroek, C.C.; van Vreeswijk, T.; Gubbels, S.; Toes, R.E.M.; Melief, C.J.M.; Franken, K.L.M.C.; Havenga, M.; van Tol, M.J.D.; et al. Human CD4+ T Cells Stimulated by Conserved Adenovirus 5 Hexon Peptides Recognize Cells Infected with Different Species of Human Adenovirus. European Journal of Immunology 2006, 36, 2410–2423. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Olive, M.; Pulmanausahakul, R.; Schnell, M.; Flomenberg, N.; Eisenlohr, L.; Flomenberg, P. Human CD8+ Cytotoxic T Cell Responses to Adenovirus Capsid Proteins. Virology 2006, 350, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Leen, A.M.; Sili, U.; Vanin, E.F.; Jewell, A.M.; Xie, W.; Vignali, D.; Piedra, P.A.; Brenner, M.K.; Rooney, C.M. Conserved CTL Epitopes on the Adenovirus Hexon Protein Expand Subgroup Cross-Reactive and Subgroup-Specific CD8+ T Cells. Blood 2004, 104, 2432–2440. [Google Scholar] [CrossRef] [PubMed]
- Tischer, S.; Geyeregger, R.; Kwoczek, J.; Heim, A.; Figueiredo, C.; Blasczyk, R.; Maecker-Kolhoff, B.; Eiz-Vesper, B. Discovery of Immunodominant T-Cell Epitopes Reveals Penton Protein as a Second Immunodominant Target in Human Adenovirus Infection. J Transl Med 2016, 14, 286. [Google Scholar] [CrossRef] [PubMed]
- Perreau, M.; Kremer, E.J. Frequency, Proliferation, and Activation of Human Memory T Cells Induced by a Nonhuman Adenovirus. J Virol 2005, 79, 14595–14605. [Google Scholar] [CrossRef] [PubMed]
- Hutnick, N.A.; Carnathan, D.; Ertl, H.; Betts, M.R. Adenovirus-Specific Human T Cells Are Pervasive, Polyfunctional, and Cross Reactive. Vaccine 2010, 28, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Lion, T. Adenovirus Infections in Immunocompetent and Immunocompromised Patients. Clin Microbiol Rev 2014, 27, 441–462. [Google Scholar] [CrossRef]
- Ison, M.G.; Hayden, R.T. Adenovirus. Microbiology Spectrum 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, M.; Li, J.; Yang, G.; Huang, Q.; Li, J.; Wang, H.; He, S.; Li, E. Histone Deacetylase Inhibitors Promote Latent Adenovirus Reactivation from Tonsillectomy Specimens. J Virol 2020, 94, e00100–20. [Google Scholar] [CrossRef] [PubMed]
- Radke, J.R.; Cook, J.L. Human Adenovirus Infections: Update and Consideration of Mechanisms of Viral Persistence. Curr Opin Infect Dis 2018, 31, 251–256. [Google Scholar] [CrossRef] [PubMed]
- King, C.R.; Zhang, A.; Mymryk, J.S. The Persistent Mystery of Adenovirus Persistence. Trends in Microbiology 2016, 24, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.P.; Eichholz, K.; Amelio, P.; Moyer, C.; Nemerow, G.R.; Perreau, M.; Mennechet, F.J.D.; Kremer, E.J. Humoral Immune Response to Adenovirus Induce Tolerogenic Bystander Dendritic Cells That Promote Generation of Regulatory T Cells. PLoS Pathog 2018, 14, e1007127. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Stamminger, T.; Hearing, P. E2F/Rb Family Proteins Mediate Interferon Induced Repression of Adenovirus Immediate Early Transcription to Promote Persistent Viral Infection. PLoS Pathog 2016, 12, e1005415. [Google Scholar] [CrossRef] [PubMed]
- Adams, W.C.; Gujer, C.; McInerney, G.; Gall, J.G.D.; Petrovas, C.; Hedestam, G.B.K.; Koup, R.A.; Loré, K. Adenovirus Type-35 Vectors Block Human CD4+ T-Cell Activation via CD46 Ligation. Proc Natl Acad Sci U S A 2011, 108, 7499–7504. [Google Scholar] [CrossRef] [PubMed]
- Iacobelli-Martinez, M.; Nepomuceno, R.R.; Connolly, J.; Nemerow, G.R. CD46-Utilizing Adenoviruses Inhibit C/EBPβ-Dependent Expression of Proinflammatory Cytokines. J Virol 2005, 79, 11259–11268. [Google Scholar] [CrossRef] [PubMed]
- Membrane Cofactor Protein (CD46) Protects Cells from Complement- Mediated Attack by an Intrinsic Mechanism. J Exp Med 1992, 175, 1547–1551. [CrossRef] [PubMed]
- Baker, A.T.; Aguirre-Hernández, C.; Halldén, G.; Parker, A.L. Designer Oncolytic Adenovirus: Coming of Age. Cancers (Basel) 2018, 10, 201. [Google Scholar] [CrossRef]
- Lin, D.; Shen, Y.; Liang, T. Oncolytic Virotherapy: Basic Principles, Recent Advances and Future Directions. Sig Transduct Target Ther 2023, 8, 1–29. [Google Scholar] [CrossRef]
- He, T.-C.; Zhou, S.; da Costa, L.T.; Yu, J.; Kinzler, K.W.; Vogelstein, B. A Simplified System for Generating Recombinant Adenoviruses. Proc Natl Acad Sci U S A 1998, 95, 2509–2514. [Google Scholar] [CrossRef]
- Heise, C.; Sampson-Johannes, A.; Williams, A.; McCormick, F.; Von Hoff, D.D.; Kirn, D.H. ONYX-015, an E1B Gene-Attenuated Adenovirus, Causes Tumor-Specific Cytolysis and Antitumoral Efficacy That Can Be Augmented by Standard Chemotherapeutic Agents. Nat Med 1997, 3, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Mantwill, K.; Klein, F.G.; Wang, D.; Hindupur, S.V.; Ehrenfeld, M.; Holm, P.S.; Nawroth, R. Concepts in Oncolytic Adenovirus Therapy. Int J Mol Sci 2021, 22, 10522. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Li, G.; Li, P.; Wang, H.; Fang, X.; He, T.; Li, J. Twenty Years of Gendicine® rAd-P53 Cancer Gene Therapy: The First-in-Class Human Cancer Gene Therapy in the Era of Personalized Oncology. Genes & Diseases 2024, 11, 101155. [Google Scholar] [CrossRef] [PubMed]
- Research, C. for B.E. and ADSTILADRIN. FDA 2023.
- Akamis Bio A Multicentre, Open-Label, Non-Randomised First in Human Study of NG-641, a Tumour Selective Transgene Expressing Adenoviral Vector, in Patients With Metastatic or Advanced Epithelial Tumours (STAR); clinicaltrials.gov, 2023.
- Champion, B.R.; Besneux, M.; Patsalidou, M.; Silva, A.; Zonca, M.; Marino, N.; Genova, G. di; Illingworth, S.; Fedele, S.; Slater, L.; et al. Abstract 5013: NG-641: An Oncolytic T-SIGn Virus Targeting Cancer-Associated Fibroblasts in the Stromal Microenvironment of Human Carcinomas. Cancer Research 2019, 79, 5013. [Google Scholar] [CrossRef]
- Orca Therapeutics B.V. A Phase I/IIa Study Evaluating the Safety and Tolerability of Intratumoral Administration of ORCA-010 in Treatment-Naïve Patients With Localized Prostate Cancer.; clinicaltrials.gov, 2023.
- Dong, W.; van Ginkel, J.-W.H.; Au, K.Y.; Alemany, R.; Meulenberg, J.J.M.; van Beusechem, V.W. ORCA-010, a Novel Potency-Enhanced Oncolytic Adenovirus, Exerts Strong Antitumor Activity in Preclinical Models. Hum Gene Ther 2014, 25, 897–904. [Google Scholar] [CrossRef] [PubMed]
- GeoVax, Inc. Phase 1/2, Open-Label Study Evaluating Safety of Repeat Administration of Ad/PNP-F-araAMP (Ad/PNP Administered Intratumorally Followed by Intravenous Fludarabine Phosphate) in Subjects With Recurrent, Local Head and Neck Cancer; clinicaltrials.gov, 2023.
- Rab, R.; Ehrhardt, A.; Achyut, B.R.; Joshi, D.; Gilbert-Ross, M.; Huang, C.; Floyd, K.; Borovjagin, A.V.; Parker, W.B.; Sorscher, E.J.; et al. Evaluating Antitumor Activity of Escherichia Coli Purine Nucleoside Phosphorylase against Head and Neck Patient-Derived Xenografts. Cancer Reports 2023, 6, e1708. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Phase II Window of Opportunity Trial of Stereotactic Body Radiation Therapy and In Situ Oncolytic Virus Therapy in Metastatic Triple Negative Breast Cancer and Metastatic Non-Small Cell Lung Cancer Followed by Pembrolizumab; clinicaltrials.gov, 2022.
- Nasu, Y.; Saika, T.; Ebara, S.; Kusaka, N.; Kaku, H.; Abarzua, F.; Manabe, D.; Thompson, T.C.; Kumon, H. Suicide Gene Therapy With Adenoviral Delivery of HSV-tK Gene for Patients With Local Recurrence of Prostate Cancer After Hormonal Therapy. Molecular Therapy 2007, 15, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Henry Ford Health System Phase I Study of Replication-Competent Adenovirus-Mediated Double Suicide Gene Therapy With Stereotactic Radiosurgery in Patients With Recurrent or Progressive High Grade Astrocytomas; clinicaltrials.gov, 2023.
- Lokon Pharma AB Phase I/II Trial Investigating an Immunostimulatory Oncolytic Adenovirus for Cancer; clinicaltrials.gov, 2023.
- Araújo, N.M.; Rubio, I.G.S.; Toneto, N.P.A.; Morale, M.G.; Tamura, R.E. The Use of Adenoviral Vectors in Gene Therapy and Vaccine Approaches. Genet. Mol. Biol. 2022, 45, e20220079. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.; Ng, P. Improved System for Helper-Dependent Adenoviral Vector Production. Mol Ther 2003, 8, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.; Beauchamp, C.; Evelegh, C.; Parks, R.; Graham, F.L. Development of a FLP/Frt System for Generating Helper-Dependent Adenoviral Vectors. Mol Ther 2001, 3, 809–815. [Google Scholar] [CrossRef]
- Liu, J.; Seol, D.-W. Helper Virus-Free Gutless Adenovirus (HF-GLAd): A New Platform for Gene Therapy. BMB Rep 2020, 53, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Liu, J.; Junn, H.J.; Lee, E.-J.; Jeong, K.-S.; Seol, D.-W. No More Helper Adenovirus: Production of Gutless Adenovirus (GLAd) Free of Adenovirus and Replication-Competent Adenovirus (RCA) Contaminants. Exp Mol Med 2019, 51, 127. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Y.; Peng, P.D.; Ehrhardt, A.; Storm, T.A.; Kay, M.A. Comparison of Adenoviral and Adeno-Associated Viral Vectors for Pancreatic Gene Delivery in Vivo. Hum Gene Ther 2004, 15, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Le, Y.; Zhang, Z.; Nian, X.; Liu, B.; Yang, X. Viral Vector-Based Gene Therapy. Int J Mol Sci 2023, 24, 7736. [Google Scholar] [CrossRef] [PubMed]
- Herantis Pharma Plc. Phase 2, Double-Blind, Placebo-Controlled, Randomized Efficacy, Safety and Tolerability Study of Lymfactin in Combination With Surgical Lymph Node Transfer To Patients With Secondary Lymphedema Associated With the Treatment of Breast Cancer; clinicaltrials.gov, 2023.
- Leppäpuska, I.-M.; Hartiala, P.; Suominen, S.; Suominen, E.; Kaartinen, I.; Mäki, M.; Seppänen, M.; Kiiski, J.; Viitanen, T.; Lahdenperä, O.; et al. Phase 1 Lymfactin® Study: 24-Month Efficacy and Safety Results of Combined Adenoviral VEGF-C and Lymph Node Transfer Treatment for Upper Extremity Lymphedema. Journal of Plastic, Reconstructive & Aesthetic Surgery 2022, 75, 3938–3945. [Google Scholar] [CrossRef] [PubMed]
- XyloCor Therapeutics, Inc. A Phase 1/2 Trial of Direct Administration of AdVEGF-All6A+, a Replication Deficient Adenovirus Vector Expressing a cDNA/Genomic Hybrid of Human VEGF, to the Ischemic Myocardium of Subjects With Angina Pectoris; clinicaltrials.gov, 2024.
- Povsic, T.J.; Nakamura, K.; Henry, T.D.; Traverse, J.H.; Anderson, R.D.; Bakaeen, F.; Latter, D.A.; Ohman, E.M.; Peterson, M.W.; Byrnes, D.; et al. Angiogenic Gene Therapy for Refractory Angina: Results of the Epicardial Delivery of XC001 Gene Therapy for Refractory Angina Coronary Treatment (EXACT) Phase 2 Trial. European Heart Journal 2023, 44, ehad655–1307. [Google Scholar] [CrossRef]
- Stewart, D.J.; Gianchetti, A.; Byrnes, D.; Dittrich, H.C.; Thorne, B.; Manza, L.L.; Reinhardt, R.R. Safety and Biodistribution of XC001 (Encoberminogene Rezmadenovec) Gene Therapy in Rats: A Potential Therapy for Cardiovascular Diseases. Gene Ther 2024, 31, 45–55. [Google Scholar] [CrossRef] [PubMed]
- University of Pennsylvania Phase I Trial to Evaluate the Safety of Platelet Derived Growth Factor B (PDGF-B) and a Limb Compression Bandage in Venous Leg Ulcers; clinicaltrials.gov, 2013.
- Margolis, D.J.; Crombleholme, T.; Herlyn, M. Clinical Protocol: Phase I Trial to Evaluate the Safety of H5.020CMV.PDGF-B for the Treatment of a Diabetic Insensate Foot Ulcer. Wound Repair and Regeneration 2000, 8, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Study Details | Phase I Pilot Study of Ad5-CB-CFTR, an Adenovirus Vector Containing the Cystic Fibrosis Transmembrane Conductance Regulator Gene, in Patients With Cystic Fibrosis | ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/study/NCT00004779 (accessed on 25 March 2024).
- Crosby, C.M.; Barry, M.A. IIIa Deleted Adenovirus as a Single-Cycle Genome Replicating Vector. Virology 2014, 0, 158–165. [Google Scholar] [CrossRef]
- Crosby, C.M.; Matchett, W.E.; Anguiano-Zarate, S.S.; Parks, C.A.; Weaver, E.A.; Pease, L.R.; Webby, R.J.; Barry, M.A. Replicating Single-Cycle Adenovirus Vectors Generate Amplified Influenza Vaccine Responses. J Virol 2017, 91, e00720–16. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, E.; Ritchie, H.; Rodés-Guirao, L.; Appel, C.; Giattino, C.; Hasell, J.; Macdonald, B.; Dattani, S.; Beltekian, D.; Ortiz-Ospina, E.; et al. Coronavirus Pandemic (COVID-19). Our World in Data 2020. [Google Scholar]
- Patel, R.; Kaki, M.; Potluri, V.S.; Kahar, P.; Khanna, D. A Comprehensive Review of SARS-CoV-2 Vaccines: Pfizer, Moderna & Johnson & Johnson. Hum Vaccin Immunother 18, 2002083. [CrossRef]
- Madhi, S.A.; Baillie, V.; Cutland, C.L.; Voysey, M.; Koen, A.L.; Fairlie, L.; Padayachee, S.D.; Dheda, K.; Barnabas, S.L.; Bhorat, Q.E.; et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. New England Journal of Medicine 2021, 384, 1885–1898. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T. Intranasal COVID-19 Vaccine Fails to Induce Mucosal Immunity. Nature Medicine 2022, 28, 2439–2440. [Google Scholar] [CrossRef]
- Singh, C.; Verma, S.; Reddy, P.; Diamond, M.S.; Curiel, D.T.; Patel, C.; Jain, M.K.; Redkar, S.V.; Bhate, A.S.; Gundappa, V.; et al. Phase III Pivotal Comparative Clinical Trial of Intranasal (iNCOVACC) and Intramuscular COVID 19 Vaccine (Covaxin®). NPJ Vaccines 2023, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Migueles, S.A.; Huang, J.; Bolkhovitinov, L.; Stuccio, S.; Griesman, T.; Pullano, A.A.; Kang, B.H.; Ishida, E.; Zimmerman, M.; et al. A Replication-Competent Adenovirus-Vectored Influenza Vaccine Induces Durable Systemic and Mucosal Immunity. J Clin Invest 2021, 131. [Google Scholar] [CrossRef]
- Houser, K.V.; Gaudinski, M.R.; Happe, M.; Narpala, S.; Verardi, R.; Sarfo, E.K.; Corrigan, A.R.; Wu, R.; Rothwell, R.S.; Novik, L.; et al. Safety and Immunogenicity of an HIV-1 Prefusion-Stabilized Envelope Trimer (Trimer 4571) Vaccine in Healthy Adults: A First-in-Human Open-Label, Randomized, Dose-Escalation, Phase 1 Clinical Trial. eClinicalMedicine 2022, 48, 101477. [Google Scholar] [CrossRef] [PubMed]
- Jcovden (Previously COVID-19 Vaccine Janssen) | European Medicines Agency Available online:. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/jcovden-previously-covid-19-vaccine-janssen (accessed on 25 March 2024).
- MHRA Products | Home Available online:. Available online: https://products.mhra.gov.uk/ (accessed on 25 March 2024).
- Research, C. for B.E. and Janssen COVID-19 Vaccine. FDA 2023.
- Vaxzevria (Previously COVID-19 Vaccine AstraZeneca) | European Medicines Agency Available online:. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/vaxzevria-previously-covid-19-vaccine-astrazeneca (accessed on 25 March 2024).
- Bharat Biotech International Limited A Phase III Randomized Open Label Multi-Center Study to Compare Immunogenicity and Safety of BBV154 With COVAXIN®, and to Assess Lot to Lot Consistency of BBV154 in Healthy Volunteers; clinicaltrials.gov, 2022.
- Sunagar, R.; Prasad, S.D.; Ella, R.; Vadrevu, K.M. Preclinical Evaluation of Safety and Immunogenicity of a Primary Series Intranasal COVID-19 Vaccine Candidate (BBV154) and Humoral Immunogenicity Evaluation of a Heterologous Prime-Boost Strategy with COVAXIN (BBV152). Front Immunol 2022, 13, 1063679. [Google Scholar] [CrossRef]
- Albert B. Sabin Vaccine Institute A Phase 2, Randomized, Double-Blind, Placebo-Controlled Trial to Evaluate Safety, Tolerability, and Immune Responses of an Investigational Monovalent Chimpanzee Adenoviral-Vectored Marburg Virus Vaccine in Healthy Adults; clinicaltrials.gov, 2023.
- Ankara City Hospital Bilkent Phase I Randomized Double-Blind Clinical Trial With New SARS-CoV-2 CoVacHGMix Type 5 Adenoviral Vector Vaccine in Healthy Volunteers Aged 18-45 Years; clinicaltrials.gov, 2022.
- McMaster University Phase 1, Open Label Study to Evaluate the Safety and Immunogenicity of ChAd68 and AdHu5 Vector-Based Trivalent COVID-19 Vaccines Delivered Via Inhaled Aerosol; clinicaltrials.gov, 2023.
- Huang, J. A Randomized, Open-Label, Parallel-Controlled Clinical Study on Booster Immunization of Two COVID-19 Vaccines Constructed From Different Technical Routes Among People Aged 18 Years and Older; clinicaltrials.gov, 2023.
- National Institute of Allergy and Infectious Diseases (NIAID) Phase I Open-Label Study of Safety and Immunogenicity of AD4-HIV Envelope Vaccine Vectors in Healthy Volunteers; clinicaltrials.gov, 2024.
- LICHTENSTEIN, D.L.; TOTH, K.; DORONIN, K.; TOLLEFSON, A.E.; WOLD, W.S.M. Functions and Mechanisms of Action of the Adenovirus E3 Proteins. International Reviews of Immunology 2004, 23, 75–111. [Google Scholar] [CrossRef]
- Kumar, V.; McNerney, M.E. A New Self: MHC-Class-I-Independent Natural-Killer-Cell Self-Tolerance. Nat Rev Immunol 2005, 5, 363–374. [Google Scholar] [CrossRef]
- Ginsberg, H.S.; Lundholm-Beauchamp, U.; Horswood, R.L.; Pernis, B.; Wold, W.S.; Chanock, R.M.; Prince, G.A. Role of Early Region 3 (E3) in Pathogenesis of Adenovirus Disease. Proc Natl Acad Sci U S A 1989, 86, 3823–3827. [Google Scholar] [CrossRef]
- Kirn, D. Clinical Research Results with Dl1520 (Onyx-015), a Replication-Selective Adenovirus for the Treatment of Cancer: What Have We Learned? Gene Ther 2001, 8, 89–98. [Google Scholar] [CrossRef]
- Liang, M. Oncorine, the World First Oncolytic Virus Medicine and Its Update in China. Curr Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, X.; Wang, J.; Gao, D.; Li, Y.; Li, H.; Chu, Y.; Zhang, Z.; Liu, H.; Jiang, G.; et al. Re-Designing Interleukin-12 to Enhance Its Safety and Potential as an Anti-Tumor Immunotherapeutic Agent. Nat Commun 2017, 8, 1395. [Google Scholar] [CrossRef] [PubMed]
- Fejer, G.; Drechsel, L.; Liese, J.; Schleicher, U.; Ruzsics, Z.; Imelli, N.; Greber, U.F.; Keck, S.; Hildenbrand, B.; Krug, A.; et al. Key Role of Splenic Myeloid DCs in the IFN-Aβ Response to Adenoviruses In Vivo. PLoS Pathog 2008, 4, e1000208. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, X.; Yang, Y. A Critical Role for Type I IFN–Dependent NK Cell Activation in Innate Immune Elimination of Adenoviral Vectors In Vivo. Molecular Therapy 2008, 16, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, N.C.; Miao, E.A.; Iwakura, Y.; Kaja, M.-K.; Aderem, A.; Flavell, R.A.; Papayannopoulou, T.; Shayakhmetov, D.M. Virus Sensing at the Plasma Membrane Triggers Interleukin-1α–Mediated Pro-Inflammatory Macrophage Response in Vivo. Immunity 2009, 31, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, N.C.; Baldwin, L.K.; Irons, E.E.; Papayannopoulou, T.; Tomlinson, S.; Shayakhmetov, D.M. IL-1α and Complement Cooperate in Triggering Local Neutrophilic Inflammation in Response to Adenovirus and Eliminating Virus-Containing Cells. PLoS Pathog 2014, 10, e1004035. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, E.V.; Doronin, K.; Senac, J.S.; Barry, M.A. Macrophage Depletion Combined with Anticoagulant Therapy Increases Therapeutic Window of Systemic Treatment with Oncolytic Adenovirus. Cancer Res 2008, 68, 5896–5904. [Google Scholar] [CrossRef] [PubMed]
- Khare, R.; May, S.M.; Vetrini, F.; Weaver, E.A.; Palmer, D.; Rosewell, A.; Grove, N.; Ng, P.; Barry, M.A. Generation of a Kupffer Cell-Evading Adenovirus for Systemic and Liver-Directed Gene Transfer. Mol Ther 2011, 19, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Naumenko, V.A.; Vishnevskiy, D.A.; Stepanenko, A.A.; Sosnovtseva, A.O.; Chernysheva, A.A.; Abakumova, T.O.; Valikhov, M.P.; Lipatova, A.V.; Abakumov, M.A.; Chekhonin, V.P. In Vivo Tracking for Oncolytic Adenovirus Interactions with Liver Cells. Biomedicines 2022, 10, 1697. [Google Scholar] [CrossRef]
- Kiang, A.; Hartman, Z.C.; Everett, R.S.; Serra, D.; Jiang, H.; Frank, M.M.; Amalfitano, A. Multiple Innate Inflammatory Responses Induced after Systemic Adenovirus Vector Delivery Depend on a Functional Complement System. Molecular Therapy 2006, 14, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Xu, Z.; Smith, J.S.; Hofherr, S.E.; Barry, M.A.; Byrnes, A.P. Adenovirus Activates Complement by Distinctly Different Mechanisms in Vitro and in Vivo: Indirect Complement Activation by Virions in Vivo. J Virol 2009, 83, 5648–5658. [Google Scholar] [CrossRef] [PubMed]
- Teigler, J.E.; Iampietro, M.J.; Barouch, D.H. Vaccination with Adenovirus Serotypes 35, 26, and 48 Elicits Higher Levels of Innate Cytokine Responses than Adenovirus Serotype 5 in Rhesus Monkeys. J Virol 2012, 86, 9590–9598. [Google Scholar] [CrossRef] [PubMed]
- Bottermann, M.; Foss, S.; van Tienen, L.M.; Vaysburd, M.; Cruickshank, J.; O’Connell, K.; Clark, J.; Mayes, K.; Higginson, K.; Hirst, J.C.; et al. TRIM21 Mediates Antibody Inhibition of Adenovirus-Based Gene Delivery and Vaccination. Proc Natl Acad Sci U S A 2018, 115, 10440–10445. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, M.A.; Jensen, B.A.H.; Holst, P.J.; Bassi, M.R.; Christensen, J.P.; Thomsen, A.R. Pre-Existing Vector Immunity Does Not Prevent Replication Deficient Adenovirus from Inducing Efficient CD8 T-Cell Memory and Recall Responses. PLoS One 2012, 7, e34884. [Google Scholar] [CrossRef] [PubMed]
- Vlachaki, M.T.; Hernandez-Garcia, A.; Ittmann, M.; Chhikara, M.; Aguilar, L.K.; Zhu, X.; The, B.S.; Butler, E.B.; Woo, S.; Thompson, T.C.; et al. Impact of Preimmunization on Adenoviral Vector Expression and Toxicity in a Subcutaneous Mouse Cancer Model. Molecular Therapy 2002, 6, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Ono, R.; Nishimae, F.; Wakida, T.; Sakurai, F.; Mizuguchi, H. Effects of Pre-Existing Anti-Adenovirus Antibodies on Transgene Expression Levels and Therapeutic Efficacies of Arming Oncolytic Adenovirus. Sci Rep 2022, 12, 21560. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Gall, J.G.D.; Nason, M.; King, C.R.; Koup, R.A.; Roederer, M.; McElrath, M.J.; Morgan, C.A.; Churchyard, G.; Baden, L.R.; et al. Differential Specificity and Immunogenicity of Adenovirus Type 5 Neutralizing Antibodies Elicited by Natural Infection or Immunization. J Virol 2010, 84, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Fan, Y.; Li, X.; Gu, S.; Zhou, Z.; Xu, D.; Qiu, S.; Li, C.; Zhou, R.; Tian, X. Identification of Adenovirus Neutralizing Antigens Using Capsid Chimeric Viruses. Virus Research 2018, 256, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Wang, L.; Gall, J.G.D.; Nason, M.; Schwartz, R.M.; McElrath, M.J.; DeRosa, S.C.; Hural, J.; Corey, L.; Buchbinder, S.P.; et al. Decreased Pre-Existing Ad5 Capsid and Ad35 Neutralizing Antibodies Increase HIV-1 Infection Risk in the Step Trial Independent of Vaccination. PLoS One 2012, 7, e33969. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.; Folgori, A.; Capone, S.; Swadling, L.; Aston, S.; Kurioka, A.; Meyer, J.; Huddart, R.; Smith, K.; Townsend, R.; et al. Novel Adenovirus-Based Vaccines Induce Broad and Sustained T Cell Responses to HCV in Man. Sci Transl Med 2012, 4, 115ra1. [Google Scholar] [CrossRef]
- O’Hara, G.A.; Duncan, C.J.A.; Ewer, K.J.; Collins, K.A.; Elias, S.C.; Halstead, F.D.; Goodman, A.L.; Edwards, N.J.; Reyes-Sandoval, A.; Bird, P.; et al. Clinical Assessment of a Recombinant Simian Adenovirus ChAd63: A Potent New Vaccine Vector. The Journal of Infectious Diseases 2012, 205, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Ewer, K.J.; O’Hara, G.A.; Duncan, C.J.A.; Collins, K.A.; Sheehy, S.H.; Reyes-Sandoval, A.; Goodman, A.L.; Edwards, N.J.; Elias, S.C.; Halstead, F.D.; et al. Protective CD8+ T-Cell Immunity to Human Malaria Induced by Chimpanzee Adenovirus-MVA Immunisation. Nat Commun 2013, 4, 2836. [Google Scholar] [CrossRef] [PubMed]
- Kimani, D.; Jagne, Y.J.; Cox, M.; Kimani, E.; Bliss, C.M.; Gitau, E.; Ogwang, C.; Afolabi, M.O.; Bowyer, G.; Collins, K.A.; et al. Translating the Immunogenicity of Prime-Boost Immunization With ChAd63 and MVA ME-TRAP From Malaria Naive to Malaria-Endemic Populations. Mol Ther 2014, 22, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, M.N.; Minassian, A.M.; Ewer, K.J.; Flaxman, A.L.; Folegatti, P.M.; Owens, D.R.; Voysey, M.; Aley, P.K.; Angus, B.; Babbage, G.; et al. Safety and Immunogenicity of ChAdOx1 nCoV-19 Vaccine Administered in a Prime-Boost Regimen in Young and Old Adults (COV002): A Single-Blind, Randomised, Controlled, Phase 2/3 Trial. The Lancet 2020, 396, 1979–1993. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, C.; Lee, J.A.; Lee, S.J.; Lee, K.H.; Kim, J.H.; Ahn, J.Y.; Jeong, S.J.; Ku, N.S.; Yeom, J.-S.; et al. Immunogenicity Differences of the ChAdOx1 nCoV-19 Vaccine According to Pre-Existing Adenovirus Immunity. Vaccines (Basel) 2023, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Bello, J.; Morales-Núñez, J.J.; Machado-Sulbarán, A.C.; Díaz-Pérez, S.A.; Torres-Hernández, P.C.; Balcázar-Félix, P.; Gutiérrez-Brito, J.A.; Lomelí-Nieto, J.A.; Muñoz-Valle, J.F. Neutralizing Antibodies against SARS-CoV-2, Anti-Ad5 Antibodies, and Reactogenicity in Response to Ad5-nCoV (CanSino Biologics) Vaccine in Individuals with and without Prior SARS-CoV-2. Vaccines (Basel) 2021, 9, 1047. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Sandoval, A.; Fitzgerald, J.C.; Grant, R.; Roy, S.; Xiang, Z.Q.; Li, Y.; Gao, G.P.; Wilson, J.M.; Ertl, H.C.J. Human Immunodeficiency Virus Type 1-Specific Immune Responses in Primates upon Sequential Immunization with Adenoviral Vaccine Carriers of Human and Simian Serotypes. J Virol 2004, 78, 7392–7399. [Google Scholar] [CrossRef]
- Zhu, F.-C.; Li, Y.-H.; Guan, X.-H.; Hou, L.-H.; Wang, W.-J.; Li, J.-X.; Wu, S.-P.; Wang, B.-S.; Wang, Z.; Wang, L.; et al. Safety, Tolerability, and Immunogenicity of a Recombinant Adenovirus Type-5 Vectored COVID-19 Vaccine: A Dose-Escalation, Open-Label, Non-Randomised, First-in-Human Trial. The Lancet 2020, 395, 1845–1854. [Google Scholar] [CrossRef]
- Bliss, C.M.; Bowyer, G.; Anagnostou, N.A.; Havelock, T.; Snudden, C.M.; Davies, H.; de Cassan, S.C.; Grobbelaar, A.; Lawrie, A.M.; Venkatraman, N.; et al. Assessment of Novel Vaccination Regimens Using Viral Vectored Liver Stage Malaria Vaccines Encoding ME-TRAP. Sci Rep 2018, 8, 3390. [Google Scholar] [CrossRef]
- Uddbäck, I.; Cartwright, E.K.; Schøller, A.S.; Wein, A.N.; Hayward, S.L.; Lobby, J.; Takamura, S.; Thomsen, A.R.; Kohlmeier, J.E.; Christensen, J.P. Long-Term Maintenance of Lung Resident Memory T Cells Is Mediated by Persistent Antigen. Mucosal Immunol 2021, 14, 92–99. [Google Scholar] [CrossRef]
- Gracht, E.T. van der; Schoonderwoerd, M.J.; Duikeren, S. van; Yilmaz, A.N.; Behr, F.M.; Colston, J.M.; Lee, L.N.; Yagita, H.; Gisbergen, K.P. van; Hawinkels, L.J.; et al. Original Research: Adenoviral Vaccines Promote Protective Tissue-Resident Memory T Cell Populations against Cancer. Journal for Immunotherapy of Cancer 2020, 8. [Google Scholar] [CrossRef]
- Zou, P.; Zhang, P.; Deng, Q.; Wang, C.; Luo, S.; Zhang, L.; Li, C.; Li, T. Two Novel Adenovirus Vectors Mediated Differential Antibody Responses via Interferon-α and Natural Killer Cells. Microbiol Spectr 2023, 11, e0088023. [Google Scholar] [CrossRef] [PubMed]
- Penaloza-MacMaster, P.; Provine, N.M.; Ra, J.; Borducchi, E.N.; McNally, A.; Simmons, N.L.; Iampietro, M.J.; Barouch, D.H. Alternative Serotype Adenovirus Vaccine Vectors Elicit Memory T Cells with Enhanced Anamnestic Capacity Compared to Ad5 Vectors. Journal of Virology 2013, 87, 1373–1384. [Google Scholar] [CrossRef]
- Holst, P.J.; Ørskov, C.; Thomsen, A.R.; Christensen, J.P. Quality of the Transgene-Specific CD8+ T Cell Response Induced by Adenoviral Vector Immunization Is Critically Influenced by Virus Dose and Route of Vaccination. J Immunol 2010, 184, 4431–4439. [Google Scholar] [CrossRef]
- Heemskerk, B.; Veltrop-Duits, L.A.; van Vreeswijk, T.; ten Dam, M.M.; Heidt, S.; Toes, R.E.M.; van Tol, M.J.D.; Schilham, M.W. Extensive Cross-Reactivity of CD4+ Adenovirus-Specific T Cells: Implications for Immunotherapy and Gene Therapy. Journal of Virology 2003, 77, 6562–6566. [Google Scholar] [CrossRef] [PubMed]
- Schirmbeck, R.; Reimann, J.; Kochanek, S.; Kreppel, F. The Immunogenicity of Adenovirus Vectors Limits the Multispecificity of CD8 T-Cell Responses to Vector-Encoded Transgenic Antigens. Molecular Therapy 2008, 16, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Auclair, S.; Liu, F.; Niu, Q.; Hou, W.; Churchyard, G.; Morgan, C.; Frahm, N.; Nitayaphan, S.; Pitisuthithum, P.; Rerks-Ngarm, S.; et al. Distinct Susceptibility of HIV Vaccine Vector-Induced CD4 T Cells to HIV Infection. PLoS Pathog 2018, 14, e1006888. [Google Scholar] [CrossRef] [PubMed]
- Frahm, N.; DeCamp, A.C.; Friedrich, D.P.; Carter, D.K.; Defawe, O.D.; Kublin, J.G.; Casimiro, D.R.; Duerr, A.; Robertson, M.N.; Buchbinder, S.P.; et al. Human Adenovirus-Specific T Cells Modulate HIV-Specific T Cell Responses to an Ad5-Vectored HIV-1 Vaccine. J Clin Invest 2012, 122, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Hutnick, N.A.; Carnathan, D.G.; Dubey, S.A.; Cox, K.S.; Kierstead, L.; Makadonas, G.; Ratcliffe, S.J.; Lasaro, M.O.; Robertson, M.N.; Casimiro, D.R.; et al. Vaccination with Ad5 Vectors Expands Ad5-Specific CD8+ T Cells without Altering Memory Phenotype or Functionality. PLoS One 2010, 5, e14385. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Q.; Ertl, H.C.; Wilson, J.M. Cellular and Humoral Immune Responses to Viral Antigens Create Barriers to Lung-Directed Gene Therapy with Recombinant Adenoviruses. J Virol 1995, 69, 2004–2015. [Google Scholar] [CrossRef] [PubMed]
- Sumida, S.M.; Truitt, D.M.; Kishko, M.G.; Arthur, J.C.; Jackson, S.S.; Gorgone, D.A.; Lifton, M.A.; Koudstaal, W.; Pau, M.G.; Kostense, S.; et al. Neutralizing Antibodies and CD8+ T Lymphocytes Both Contribute to Immunity to Adenovirus Serotype 5 Vaccine Vectors. Journal of Virology 2004, 78, 2666–2673. [Google Scholar] [CrossRef] [PubMed]
- Appledorn, D.M.; McBride, A.; Seregin, S.; Scott, J.M.; Schuldt, N.; Kiang, A.; Godbehere, S.; Amalfitano, A. Complex Interactions with Several Arms of the Complement System Dictate Innate and Humoral Immunity to Adenoviral Vectors. Gene Ther 2008, 15, 1606–1617. [Google Scholar] [CrossRef] [PubMed]
- Colloca, S.; Folgori, A.; Ammendola, V.; Capone, S.; Cirillo, A.; Siani, L.; Naddeo, M.; Grazioli, F.; Esposito, M.L.; Ambrosio, M.; et al. Generation and Screening of a Large Collection of Novel Simian Adenovirus Allows the Identification of Vaccine Vectors Inducing Potent Cellular Immunity in Humans: A Range of Novel Simian Adenoviral Vectors, Which Are Capable of Priming High Levels of T Cell Responses in Man, Has Been Defined. Science translational medicine 2012, 4, 115ra2. [Google Scholar] [CrossRef] [PubMed]
- Muir, K.-L.; Kallam, A.; Koepsell, S.A.; Gundabolu, K. Thrombotic Thrombocytopenia after Ad26.COV2.S Vaccination. N Engl J Med 2021, 384, 1964–1965. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Leunig, A.; Pekayvaz, K.; Esefeld, M.; Anjum, A.; Rath, J.; Riedlinger, E.; Ehreiser, V.; Mader, M.; Eivers, L.; et al. Thrombocytopenia and Splenic Platelet-Directed Immune Responses after IV ChAdOx1 nCov-19 Administration. Blood 2022, 140, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Pavord, S.; Hunt, B.J.; Horner, D.; Bewley, S.; Karpusheff, J. Vaccine Induced Immune Thrombocytopenia and Thrombosis: Summary of NICE Guidance. BMJ 2021, 375, n2195. [Google Scholar] [CrossRef] [PubMed]
- Coronavirus Vaccine - Summary of Yellow Card Reporting Available online:. Available online: https://www.gov.uk/government/publications/coronavirus-covid-19-vaccine-adverse-reactions/coronavirus-vaccine-summary-of-yellow-card-reporting (accessed on 2 May 2024).
- Pai, M. Epidemiology of VITT. Seminars in Hematology 2022, 59, 72–75. [Google Scholar] [CrossRef]
- Nguyen, T.-H.; Medvedev, N.; Delcea, M.; Greinacher, A. Anti-Platelet Factor 4/Polyanion Antibodies Mediate a New Mechanism of Autoimmunity. Nat Commun 2017, 8, 14945. [Google Scholar] [CrossRef]
- Greinacher, A.; Selleng, K.; Palankar, R.; Wesche, J.; Handtke, S.; Wolff, M.; Aurich, K.; Lalk, M.; Methling, K.; Völker, U.; et al. Insights in ChAdOx1 nCoV-19 Vaccine-Induced Immune Thrombotic Thrombocytopenia. Blood 2021, 138, 2256–2268. [Google Scholar] [CrossRef] [PubMed]
- Sallard, E.; Pembaur, D.; Schröer, K.; Schellhorn, S.; Koukou, G.; Schmidt, N.; Zhang, W.; Kreppel, F.; Ehrhardt, A. Adenovirus Type 34 and HVR1-Deleted Adenovirus Type 5 Do Not Bind to PF4: Clearing the Path towards Vectors without Thrombosis Risk 2022, 2022. 11.07.51 5483.
- Baker, A.T.; Boyd, R.J.; Sarkar, D.; Vant, J.; Crespo, A.T.; Waraich, K.; Truong, C.D.; Bates, E.; Wilson, E.; Chan, C.K.; et al. The Structure of ChAdOx1/AZD-1222 Reveals Interactions with CAR and PF4 with Implications for Vaccine-Induced Immune Thrombotic Thrombocytopenia 2021, 2021. 05.19.44 4882.
- Ehrhardt, A.; Kay, M.A. A New Adenoviral Helper–Dependent Vector Results in Long-Term Therapeutic Levels of Human Coagulation Factor IX at Low Doses in Vivo. Blood 2002, 99, 3923–3930. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, D.B.; Himeda, C.L.; Winther, B.L.; Wilson, C.B.; Kay, M.A. Implication of Interfering Antibody Formation and Apoptosis as Two Different Mechanisms Leading to Variable Duration of Adenovirus-Mediated Transgene Expression in Immune-Competent Mice. Journal of Virology 1999, 73, 4755–4766. [Google Scholar] [CrossRef] [PubMed]
- Balagué, C.; Zhou, J.; Dai, Y.; Alemany, R.; Josephs, S.F.; Andreason, G.; Hariharan, M.; Sethi, E.; Prokopenko, E.; Jan, H.; et al. Sustained High-Level Expression of Full-Length Human Factor VIII and Restoration of Clotting Activity in Hemophilic Mice Using a Minimal Adenovirus Vector. Blood 2000, 95, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Ramsey, B.W.; Meeker, D.P.; Aitken, M.L.; Balfour, R.P.; Gibson, R.L.; Launspach, J.; Moscicki, R.A.; Richards, S.M.; Standaert, T.A. Repeat Administration of an Adenovirus Vector Encoding Cystic Fibrosis Transmembrane Conductance Regulator to the Nasal Epithelium of Patients with Cystic Fibrosis. J Clin Invest 1996, 97, 1504–1511. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Ng, T.; Iannitti, D.; Cioffi, W.; Stapleton, G.; Law, M.; Breinholt, J.; Palmer, D.; Grove, N.; Rice, K.; et al. Transgene Expression up to 7 Years in Nonhuman Primates Following Hepatic Transduction with Helper-Dependent Adenoviral Vectors. Human Gene Therapy 2013, 24, 761. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front Immunol 2018, 9, 866. [Google Scholar] [CrossRef] [PubMed]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic Viruses as Engineering Platforms for Combination Immunotherapy. Nat Rev Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shaw, R.H.; Stuart, A.S.V.; Greenland, M.; Aley, P.K.; Andrews, N.J.; Cameron, J.C.; Charlton, S.; Clutterbuck, E.A.; Collins, A.M.; et al. Safety and Immunogenicity of Heterologous versus Homologous Prime-Boost Schedules with an Adenoviral Vectored and mRNA COVID-19 Vaccine (Com-COV): A Single-Blind, Randomised, Non-Inferiority Trial. The Lancet 2021, 398, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liang, B.; Wang, W.; Li, L.; Feng, N.; Zhao, Y.; Wang, T.; Yan, F.; Yang, S.; Xia, X. Viral Vectored Vaccines: Design, Development, Preventive and Therapeutic Applications in Human Diseases. Sig Transduct Target Ther 2023, 8, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Torres, J.; Cabello-Gutiérrez, C.; Ayón-Núñez, D.-A.; Soldevila, G.; Olguin-Alor, R.; Diaz, G.; Acero, G.; Segura-Velázquez, R.; Huerta, L.; Gracia-Mora, I.; et al. Caveats of Chimpanzee ChAdOx1 Adenovirus-Vectored Vaccines to Boost Anti-SARS-CoV-2 Protective Immunity in Mice. Appl Microbiol Biotechnol 2024, 108, 179. [Google Scholar] [CrossRef] [PubMed]
- Ledgerwood, J.E.; Costner, P.; Desai, N.; Holman, L.; Enama, M.E.; Yamshchikov, G.; Mulangu, S.; Hu, Z.; Andrews, C.A.; Sheets, R.A.; et al. A Replication Defective Recombinant Ad5 Vaccine Expressing Ebola Virus GP Is Safe and Immunogenic in Healthy Adults. Vaccine 2010, 29, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-X.; Hou, L.-H.; Meng, F.-Y.; Wu, S.-P.; Hu, Y.-M.; Liang, Q.; Chu, K.; Zhang, Z.; Xu, J.-J.; Tang, R.; et al. Immunity Duration of a Recombinant Adenovirus Type-5 Vector-Based Ebola Vaccine and a Homologous Prime-Boost Immunisation in Healthy Adults in China: Final Report of a Randomised, Double-Blind, Placebo-Controlled, Phase 1 Trial. Lancet Glob Health 2017, 5, e324–e334. [Google Scholar] [CrossRef] [PubMed]
- Gray, G.; Buchbinder, S.; Duerr, A. Overview of STEP and Phambili Trial Results: Two Phase IIb Test-of-Concept Studies Investigating the Efficacy of MRK Adenovirus Type 5 Gag/Pol/Nef Subtype B HIV Vaccine. Current Opinion in HIV and AIDS 2010, 5, 357. [Google Scholar] [CrossRef] [PubMed]
- Travieso, T.; Li, J.; Mahesh, S.; Mello, J.D.F.R.E.; Blasi, M. The Use of Viral Vectors in Vaccine Development. npj Vaccines 2022, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, S.; Hays, J.; HogenEsch, H.; Mittal, S.K. Circumvention of Vector-Specific Neutralizing Antibody Response by Alternating Use of Human and Non-Human Adenoviruses: Implications in Gene Therapy. Virology 2000, 272, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Mastrangeli, A.; Harvey, B.-G.; Yao, J.; Wolff, G.; Kovesdi, I.; Crystal, R.G.; Falck-Pedersen, E. “Sero-Switch” Adenovirus-Mediated In Vivo Gene Transfer: Circumvention of Anti-Adenovirus Humoral Immune Defenses Against Repeat Adenovirus Vector Administration by Changing the Adenovirus Serotype. Human Gene Therapy 1996, 7, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.O.; Feldmann, F.; Zhao, H.; Curiel, D.T.; Okumura, A.; Tang-Huau, T.-L.; Case, J.B.; Meade-White, K.; Callison, J.; Chen, R.E.; et al. A Single Intranasal Dose of Chimpanzee Adenovirus-Vectored Vaccine Protects against SARS-CoV-2 Infection in Rhesus Macaques. Cell Rep Med 2021, 2, 100230. [Google Scholar] [CrossRef] [PubMed]
- Uchino, J.; Curiel, D.T.; Ugai, H. Species D Human Adenovirus Type 9 Exhibits Better Virus-Spread Ability for Antitumor Efficacy among Alternative Serotypes. PLOS ONE 2014, 9, e87342. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Senac, J.S.; Weaver, E.A.; May, S.M.; Jelinek, D.F.; Greipp, P.; Witzig, T.; Barry, M.A. Species D Adenoviruses as Oncolytics Against B Cell Cancers. Clin Cancer Res 2011, 17, 6712–6722. [Google Scholar] [CrossRef] [PubMed]
- Holterman, L.; Vogels, R.; van der Vlugt, R.; Sieuwerts, M.; Grimbergen, J.; Kaspers, J.; Geelen, E.; van der Helm, E.; Lemckert, A.; Gillissen, G.; et al. Novel Replication-Incompetent Vector Derived from Adenovirus Type 11 (Ad11) for Vaccination and Gene Therapy: Low Seroprevalence and Non-Cross-Reactivity with Ad5. Journal of Virology 2004, 78, 13207–13215. [Google Scholar] [CrossRef] [PubMed]
- Bates, E.A.; Davies, J.A.; Váňová, J.; Nestić, D.; Meniel, V.S.; Koushyar, S.; Cunliffe, T.G.; Mundy, R.M.; Moses, E.; Uusi-Kerttula, H.K.; et al. Development of a Low-Seroprevalence, Avβ6 Integrin-Selective Virotherapy Based on Human Adenovirus Type 10. Molecular Therapy - Oncolytics 2022, 25, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Sadoff Jerald; Gray Glenda; Vandebosch An; Cárdenas Vicky; Shukarev Georgi; Grinsztejn Beatriz; Goepfert Paul A; Truyers Carla; Fennema Hein; Spiessens Bart. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19. New England Journal of Medicine 2021, 384, 2187–2201. [Google Scholar] [CrossRef] [PubMed]
- Havenga, M.; Vogels, R.; Zuijdgeest, D.; Radosevic, K.; Mueller, S.; Sieuwerts, M.; Weichold, F.; Damen, I.; Kaspers, J.; Lemckert, A.; et al. Novel Replication-Incompetent Adenoviral B-Group Vectors: High Vector Stability and Yield in PER.C6 Cells. Journal of General Virology 2006, 87, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Di, Y.; Seymour, L.; Fisher, K. Activity of a Group B Oncolytic Adenovirus (ColoAd1) in Whole Human Blood. Gene Ther 2014, 21, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Penaloza-MacMaster, P.; Provine, N.M.; Ra, J.; Borducchi, E.N.; McNally, A.; Simmons, N.L.; Iampietro, M.J.; Barouch, D.H. Alternative Serotype Adenovirus Vaccine Vectors Elicit Memory T Cells with Enhanced Anamnestic Capacity Compared to Ad5 Vectors. J Virol 2013, 87, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hetzel, M.; Zhang, W.; Ehrhardt, A.; Bayer, W. Comparative Analysis of the Impact of 40 Adenovirus Types on Dendritic Cell Activation and CD8+ T Cell Proliferation Capacity for the Identification of Favorable Immunization Vector Candidates. Frontiers in Immunology 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Youil, R.; Toner, T.J.; Su, Q.; Chen, M.; Tang, A.; Bett, A.J.; Casimiro, D. Hexon Gene Switch Strategy for the Generation of Chimeric Recombinant Adenovirus. Hum Gene Ther 2002, 13, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Nanda, A.; Havenga, M.J.E.; Abbink, P.; Lynch, D.M.; Ewald, B.A.; Liu, J.; Thorner, A.R.; Swanson, P.E.; Gorgone, D.A.; et al. Hexon-Chimaeric Adenovirus Serotype 5 Vectors Circumvent Pre-Existing Anti-Vector Immunity. Nature 2006, 441, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, L.; Bradshaw, A.C.; Parker, A.L.; Robinson, H.; White, K.; Custers, J.; Goudsmit, J.; Van Roijen, N.; Barouch, D.H.; Nicklin, S.A.; et al. Ad5:Ad48 Hexon Hypervariable Region Substitutions Lead to Toxicity and Increased Inflammatory Responses Following Intravenous Delivery. Mol Ther 2012, 20, 2268–2281. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Su, X.; Li, H.; Li, X.; Zhou, Z.; Liu, W.; Zhou, R. Construction and Characterization of Human Adenovirus Serotype 3 Packaged by Serotype 7 Hexon. Virus Research 2011, 160, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Crosby, C.M.; Heller, G.J.; Mendel, Z.I.; Barry, M.E.; Barry, M.A. Oncolytic Adenovirus Ad657 for Systemic Virotherapy against Prostate Cancer. Oncolytic Virother 2018, 7, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Matchett, W.E.; Anguiano-Zarate, S.S.; Nehete, P.N.; Shelton, K.; Nehete, B.P.; Yang, G.; Dorta-Estremera, S.; Barnette, P.; Xiao, P.; Byrareddy, S.N.; et al. Divergent HIV-1-Directed Immune Responses Generated by Systemic and Mucosal Immunization with Replicating Single-Cycle Adenoviruses in Rhesus Macaques. J Virol 2019, 93, e02016–18. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-C.; Hansen, M.J.; Hemsath, J.R.; Parrett, B.J.; Zell, B.N.; Barry, M.A. Modulating Oncolytic Adenovirus Immunotherapy by Driving Two Axes of the Immune System by Expressing 4-1BBL and CD40L. Hum Gene Ther 2022, 33, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Jiang, H.; Gillard, A.; Kim, D.; Fan, X.; Singh, S.; Nguyen, T.T.; Sohoni, S.; Lopez-Rivas, A.; Parthasarathy, A.; et al. Chimeric Oncolytic Adenovirus Evades Neutralizing Antibodies from Human Patients and Exhibits Enhanced Anti-Glioma Efficacy in Immunized Mice. Molecular Therapy 2024. [Google Scholar] [CrossRef] [PubMed]
- White, K.M.; Alba, R.; Parker, A.L.; Wright, A.F.; Bradshaw, A.C.; Delles, C.; McDonald, R.A.; Baker, A.H. Assessment of a Novel, Capsid-Modified Adenovirus with an Improved Vascular Gene Transfer Profile. J Cardiothorac Surg 2013, 8, 183. [Google Scholar] [CrossRef] [PubMed]
- Gall, J.; Kass-Eisler, A.; Leinwand, L.; Falck-Pedersen, E. Adenovirus Type 5 and 7 Capsid Chimera: Fiber Replacement Alters Receptor Tropism without Affecting Primary Immune Neutralization Epitopes. J Virol 1996, 70, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Nociari, M.; Philpott, N.; Falck-Pedersen, E. Influence of Fiber Detargeting on Adenovirus-Mediated Innate and Adaptive Immune Activation. J Virol 2005, 79, 11627–11637. [Google Scholar] [CrossRef] [PubMed]
- Ophorst, O.J.A.E.; Kostense, S.; Goudsmit, J.; de Swart, R.L.; Verhaagh, S.; Zakhartchouk, A.; van Meijer, M.; Sprangers, M.; van Amerongen, G.; Yüksel, S.; et al. An Adenoviral Type 5 Vector Carrying a Type 35 Fiber as a Vaccine Vehicle: DC Targeting, Cross Neutralization, and Immunogenicity. Vaccine 2004, 22, 3035–3044. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, N.; Leopold, P.L.; Hackett, N.R.; Ferris, B.; Worgall, S.; Falck-Pedersen, E.; Crystal, R.G. Fiber Swap between Adenovirus Subgroups B and C Alters Intracellular Trafficking of Adenovirus Gene Transfer Vectors. J Virol 1999, 73, 6056–6065. [Google Scholar] [CrossRef] [PubMed]
- Parks, R.J. Adenovirus Protein IX: A New Look at an Old Protein. Molecular Therapy 2005, 11, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Seregin, S.S.; Aldhamen, Y.A.; Appledorn, D.M.; Hartman, Z.C.; Schuldt, N.J.; Scott, J.; Godbehere, S.; Jiang, H.; Frank, M.M.; Amalfitano, A. Adenovirus Capsid-Display of the Retro-Oriented Human Complement Inhibitor DAF Reduces Ad Vector–Triggered Immune Responses in Vitro and in Vivo. Blood 2010, 116, 1669–1677. [Google Scholar] [CrossRef]
- Kreppel, F.; Kochanek, S. Modification of Adenovirus Gene Transfer Vectors With Synthetic Polymers: A Scientific Review and Technical Guide. Molecular Therapy 2008, 16, 16–29. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, C.R.; Lachapelle, A.; Delgado, C.; Parkes, V.; Wadsworth, S.C.; Smith, A.E.; Francis, G.E. PEGylation of Adenovirus with Retention of Infectivity and Protection from Neutralizing Antibody in Vitro and in Vivo. Hum Gene Ther 1999, 10, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, A.; Vöhringer, S.; Engler, T.; Corjon, S.; Schirmbeck, R.; Reimann, J.; Kochanek, S.; Kreppel, F. Fully Detargeted Polyethylene Glycol-Coated Adenovirus Vectors Are Potent Genetic Vaccines and Escape from Pre-Existing Anti-Adenovirus Antibodies. Mol Ther 2008, 16, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Han, J.; Zhao, D.; Gong, T.; Zhang, Z.; Sun, X. Protection of Adenovirus from Neutralizing Antibody by Cationic PEG Derivative Ionically Linked to Adenovirus. Int J Nanomedicine 2012, 7, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Ichihara, M.; Yoshioka, Y.; Ishida, T.; Nakagawa, S.; Kiwada, H. Intravenous Administration of Polyethylene Glycol-Coated (PEGylated) Proteins and PEGylated Adenovirus Elicits an Anti-PEG Immunoglobulin M Response. Biol Pharm Bull 2012, 35, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y.; Yuan, C.; Xu, X.; Zhou, W.; Huang, Y.; Lu, H.; Zheng, Y.; Luo, G.; Shang, J.; et al. Polyethylene Glycol (PEG)-Associated Immune Responses Triggered by Clinically Relevant Lipid Nanoparticles in Rats. npj Vaccines 2023, 8, 1–13. [Google Scholar] [CrossRef]
- Fisher, K.D.; Stallwood, Y.; Green, N.K.; Ulbrich, K.; Mautner, V.; Seymour, L.W. Polymer-Coated Adenovirus Permits Efficient Retargeting and Evades Neutralising Antibodies. Gene Ther 2001, 8, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Subr, V.; Kostka, L.; Selby-Milic, T.; Fisher, K.; Ulbrich, K.; Seymour, L.W.; Carlisle, R.C. Coating of Adenovirus Type 5 with Polymers Containing Quaternary Amines Prevents Binding to Blood Components. Journal of Controlled Release 2009, 135, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Green, N.K.; Herbert, C.W.; Hale, S.J.; Hale, A.B.; Mautner, V.; Harkins, R.; Hermiston, T.; Ulbrich, K.; Fisher, K.D.; Seymour, L.W. Extended Plasma Circulation Time and Decreased Toxicity of Polymer-Coated Adenovirus. Gene Ther 2004, 11, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Ríhová, B.; Kovár, M. Immunogenicity and Immunomodulatory Properties of HPMA-Based Polymers. Adv Drug Deliv Rev 2010, 62, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Sailaja, G.; HogenEsch, H.; North, A.; Hays, J.; Mittal, S.K. Encapsulation of Recombinant Adenovirus into Alginate Microspheres Circumvents Vector-Specific Immune Response. Gene Ther 2002, 9, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Dhamecha, D.; Movsas, R.; Sano, U.; Menon, J.U. Applications of Alginate Microspheres in Therapeutics Delivery and Cell Culture: Past, Present and Future. Int J Pharm 2019, 569, 118627. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Iscaro, A.; Zambito, G.; Mijiti, Y.; Minicucci, M.; Essand, M.; Lowik, C.; Muthana, M.; Censi, R.; Mezzanotte, L.; et al. Development of a New Hyaluronic Acid Based Redox-Responsive Nanohydrogel for the Encapsulation of Oncolytic Viruses for Cancer Immunotherapy. Nanomaterials 2021, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, J.; Zhang, H.; Wei, J.; Wu, J. Extracellular Vesicles-Mimetic Encapsulation Improves Oncolytic Viro-Immunotherapy in Tumors With Low Coxsackie and Adenovirus Receptor. Front Bioeng Biotechnol 2020, 8, 574007. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, C.; Li, P.; Wu, T.; Zhou, H.; Yang, D.; Liu, Y.; Ma, X.; Song, Z.; Nian, Q.; et al. Vaccine Engineering with Dual-Functional Mineral Shell: A Promising Strategy to Overcome Preexisting Immunity. Advanced Materials 2016, 28, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Ernst, P.; Honegger, A.; Suomalainen, M.; Zimmermann, M.; Braun, L.; Stauffer, S.; Thom, C.; Dreier, B.; Eibauer, M.; et al. Adenoviral Vector with Shield and Adapter Increases Tumor Specificity and Escapes Liver and Immune Control. Nat Commun 2018, 9, 450. [Google Scholar] [CrossRef] [PubMed]
- Prill, J.-M.; Šubr, V.; Pasquarelli, N.; Engler, T.; Hoffmeister, A.; Kochanek, S.; Ulbrich, K.; Kreppel, F. Traceless Bioresponsive Shielding of Adenovirus Hexon with HPMA Copolymers Maintains Transduction Capacity In Vitro and In Vivo. PLoS One 2014, 9, e82716. [Google Scholar] [CrossRef]
- Krutzke, L.; Prill, J.M.; Engler, T.; Schmidt, C.Q.; Xu, Z.; Byrnes, A.P.; Simmet, T.; Kreppel, F. Substitution of Blood Coagulation Factor X-Binding to Ad5 by Position-Specific PEGylation: Preventing Vector Clearance and Preserving Infectivity. Journal of Controlled Release 2016, 235, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Alaswad, S.O.; Mahmoud, A.S.; Arunachalam, P. Recent Advances in Biodegradable Polymers and Their Biological Applications: A Brief Review. Polymers (Basel) 2022, 14, 4924. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Kovesdi, I.; Bruder, J.T. Effective Repeat Administration with Adenovirus Vectors to the Muscle. Gene Ther 2000, 7, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Croyle, M.A.; Patel, A.; Tran, K.N.; Gray, M.; Zhang, Y.; Strong, J.E.; Feldmann, H.; Kobinger, G.P. Nasal Delivery of an Adenovirus-Based Vaccine Bypasses Pre-Existing Immunity to the Vaccine Carrier and Improves the Immune Response in Mice. PLoS One 2008, 3, e3548. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Z.; Yu, R.; Zhang, J.; Liu, Y.; Song, X.; Yi, S.; Liu, J.; Chen, J.; Yin, Y.; et al. Intramuscular Delivery of Adenovirus Serotype 5 Vector Expressing Humanized Protective Antigen Induces Rapid Protection against Anthrax That May Bypass Intranasally Originated Preexisting Adenovirus Immunity. Clin Vaccine Immunol 2014, 21, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Bramson, J.L.; Hitt, M.; Gauldie, J.; Graham, F.L. Pre-Existing Immunity to Adenovirus Does Not Prevent Tumor Regression Following Intratumoral Administration of a Vector Expressing IL-12 but Inhibits Virus Dissemination. Gene Ther 1997, 4, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.G.; Worgall, S.; Ely, S.; Leopold, P.L.; Crystal, R.G. Cellular Immune Responses of Healthy Individuals to Intradermal Administration of an E1-E3- Adenovirus Gene Transfer Vector. Hum Gene Ther 1999, 10, 2823–2837. [Google Scholar] [CrossRef] [PubMed]
- Iscaro, A.; Jones, C.; Forbes, N.; Mughal, A.; Howard, F.N.; Janabi, H.A.; Demiral, S.; Perrie, Y.; Essand, M.; Weglarz, A.; et al. Targeting Circulating Monocytes with CCL2-Loaded Liposomes Armed with an Oncolytic Adenovirus. Nanomedicine: Nanotechnology, Biology and Medicine 2022, 40, 102506. [Google Scholar] [CrossRef]
- Muthana, M.; Giannoudis, A.; Scott, S.D.; Fang, H.-Y.; Coffelt, S.B.; Morrow, F.J.; Murdoch, C.; Burton, J.; Cross, N.; Burke, B.; et al. Use of Macrophages to Target Therapeutic Adenovirus to Human Prostate Tumors. Cancer Research 2011, 71, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, S.; Wang, Y.; Shao, F.; Lv, P.; Li, R.; Zhao, X.; Zhang, J.; Zhang, X.; Li, J.; et al. Lung-Targeted Transgene Expression of Nanocomplexed Ad5 Enhances Immune Response in the Presence of Preexisting Immunity. Engineering (Beijing) 2023. [Google Scholar] [CrossRef] [PubMed]
- Rojas, L.A.; Condezo, G.N.; Moreno, R.; Fajardo, C.A.; Arias-Badia, M.; San Martín, C.; Alemany, R. Albumin-Binding Adenoviruses Circumvent Pre-Existing Neutralizing Antibodies upon Systemic Delivery. Journal of Controlled Release 2016, 237, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Mato-Berciano, A.; Morgado, S.; Maliandi, M.V.; Farrera-Sal, M.; Gimenez-Alejandre, M.; Ginestà, M.M.; Moreno, R.; Torres-Manjon, S.; Moreno, P.; Arias-Badia, M.; et al. Oncolytic Adenovirus with Hyaluronidase Activity That Evades Neutralizing Antibodies: VCN-11. Journal of Controlled Release 2021, 332, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Bazan-Peregrino, M.; Garcia-Carbonero, R.; Laquente, B.; Álvarez, R.; Mato-Berciano, A.; Gimenez-Alejandre, M.; Morgado, S.; Rodríguez-García, A.; Maliandi, M.V.; Riesco, M.C.; et al. VCN-01 Disrupts Pancreatic Cancer Stroma and Exerts Antitumor Effects. J Immunother Cancer 2021, 9, e003254. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carbonero, R.; Bazan-Peregrino, M.; Gil-Martín, M.; Álvarez, R.; Macarulla, T.; Riesco-Martinez, M.C.; Verdaguer, H.; Guillén-Ponce, C.; Farrera-Sal, M.; Moreno, R.; et al. Phase I, Multicenter, Open-Label Study of Intravenous VCN-01 Oncolytic Adenovirus with or without Nab-Paclitaxel plus Gemcitabine in Patients with Advanced Solid Tumors. J Immunother Cancer 2022, 10, e003255. [Google Scholar] [CrossRef] [PubMed]
- Muruve, D.A.; Cotter, M.J.; Zaiss, A.K.; White, L.R.; Liu, Q.; Chan, T.; Clark, S.A.; Ross, P.J.; Meulenbroek, R.A.; Maelandsmo, G.M.; et al. Helper-Dependent Adenovirus Vectors Elicit Intact Innate but Attenuated Adaptive Host Immune Responses In Vivo. Journal of Virology 2004, 78, 5966–5972. [Google Scholar] [CrossRef] [PubMed]
- Koup, R.A.; Lamoreaux, L.; Zarkowsky, D.; Bailer, R.T.; King, C.R.; Gall, J.G.D.; Brough, D.E.; Graham, B.S.; Roederer, M. Replication-Defective Adenovirus Vectors with Multiple Deletions Do Not Induce Measurable Vector-Specific T Cells in Human Trials. J Virol 2009, 83, 6318–6322. [Google Scholar] [CrossRef] [PubMed]
- Gabitzsch, E.S.; Xu, Y.; Balint, J.P.; Hartman, Z.C.; Lyerly, H.K.; Jones, F.R. Anti-Tumor Immunotherapy despite Immunity to Adenovirus Using a Novel Adenoviral Vector Ad5 [E1-, E2b-]-CEA. Cancer Immunol Immunother 2010, 59, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Santra, S.; Seaman, M.S.; Xu, L.; Barouch, D.H.; Lord, C.I.; Lifton, M.A.; Gorgone, D.A.; Beaudry, K.R.; Svehla, K.; Welcher, B.; et al. Replication-Defective Adenovirus Serotype 5 Vectors Elicit Durable Cellular and Humoral Immune Responses in Nonhuman Primates. J Virol 2005, 79, 6516–6522. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structural schematic and species classification of selected adenovirus serotypes. Species: A=orange, B=purple, C=blue, D=green, E=red, F=pink, G=yellow. Schematic created using Biorender. Species tree was generated using PhyloT and ITOL, using NCBI TaxIDs. All human Ads relevant to this review, as well as chimpanzee (Ch) Ad-Y25 were included in the selection.
Figure 1.
Structural schematic and species classification of selected adenovirus serotypes. Species: A=orange, B=purple, C=blue, D=green, E=red, F=pink, G=yellow. Schematic created using Biorender. Species tree was generated using PhyloT and ITOL, using NCBI TaxIDs. All human Ads relevant to this review, as well as chimpanzee (Ch) Ad-Y25 were included in the selection.
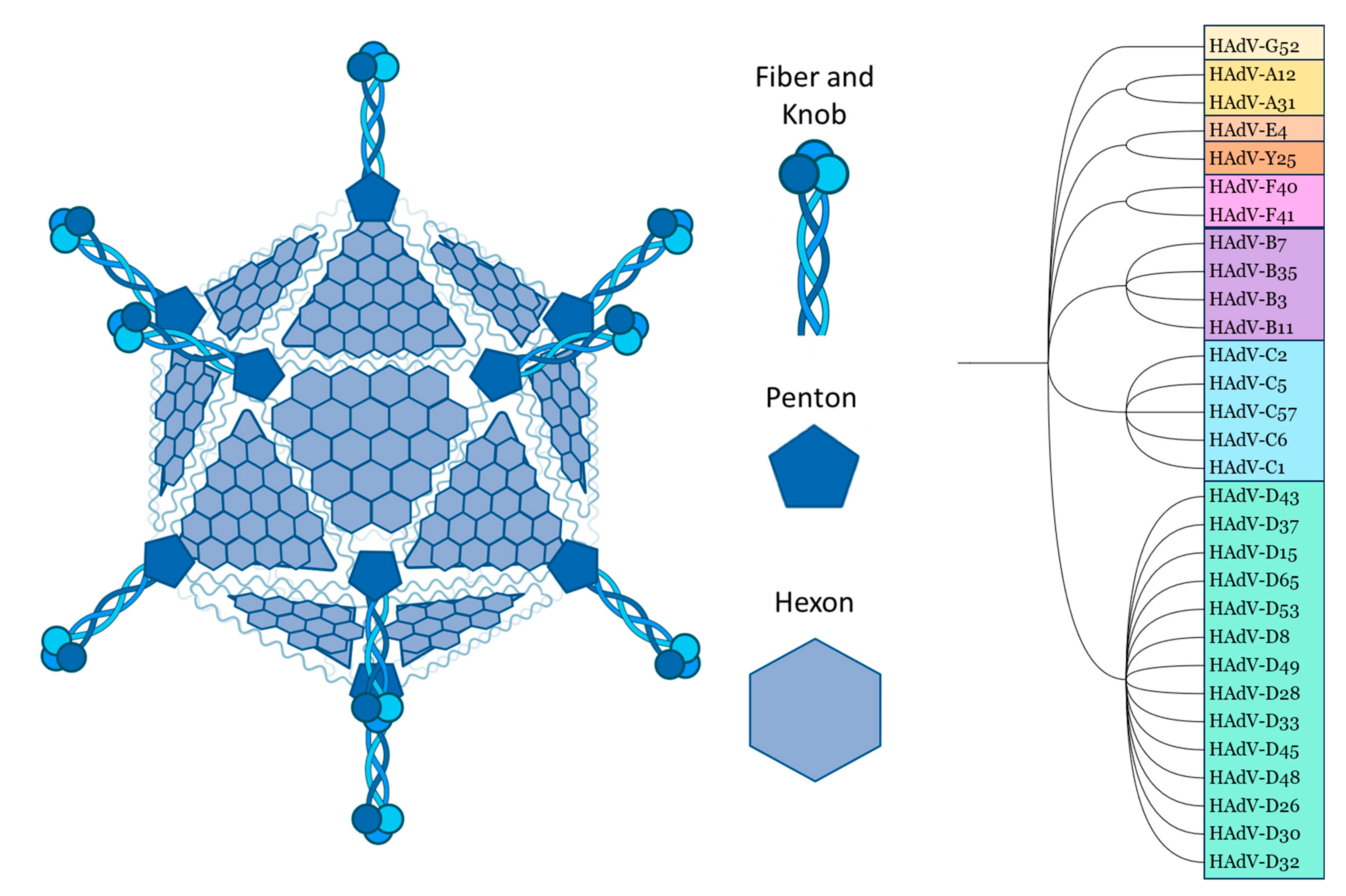
Figure 2.
Comparison of Hexon HVRs and FX Binding Sites on Selected Ads of Different Species. Alignment created using NCBI COBALT multiple protein sequence alignment, using NCBI protein IDs. Amino acids in black are highly conserved, in yellow are partially conserved, and in red are hypervariable. The amino acid highlighted green is most important in FX binding [39]. This model predicts no FX binding across species A and D. Large stretches of conserved sequence have been abbreviated (in brackets). All human Ads relevant to this review, as well as chimpanzee (Ch) Ad-Y25 were included in the selection.
Figure 2.
Comparison of Hexon HVRs and FX Binding Sites on Selected Ads of Different Species. Alignment created using NCBI COBALT multiple protein sequence alignment, using NCBI protein IDs. Amino acids in black are highly conserved, in yellow are partially conserved, and in red are hypervariable. The amino acid highlighted green is most important in FX binding [39]. This model predicts no FX binding across species A and D. Large stretches of conserved sequence have been abbreviated (in brackets). All human Ads relevant to this review, as well as chimpanzee (Ch) Ad-Y25 were included in the selection.
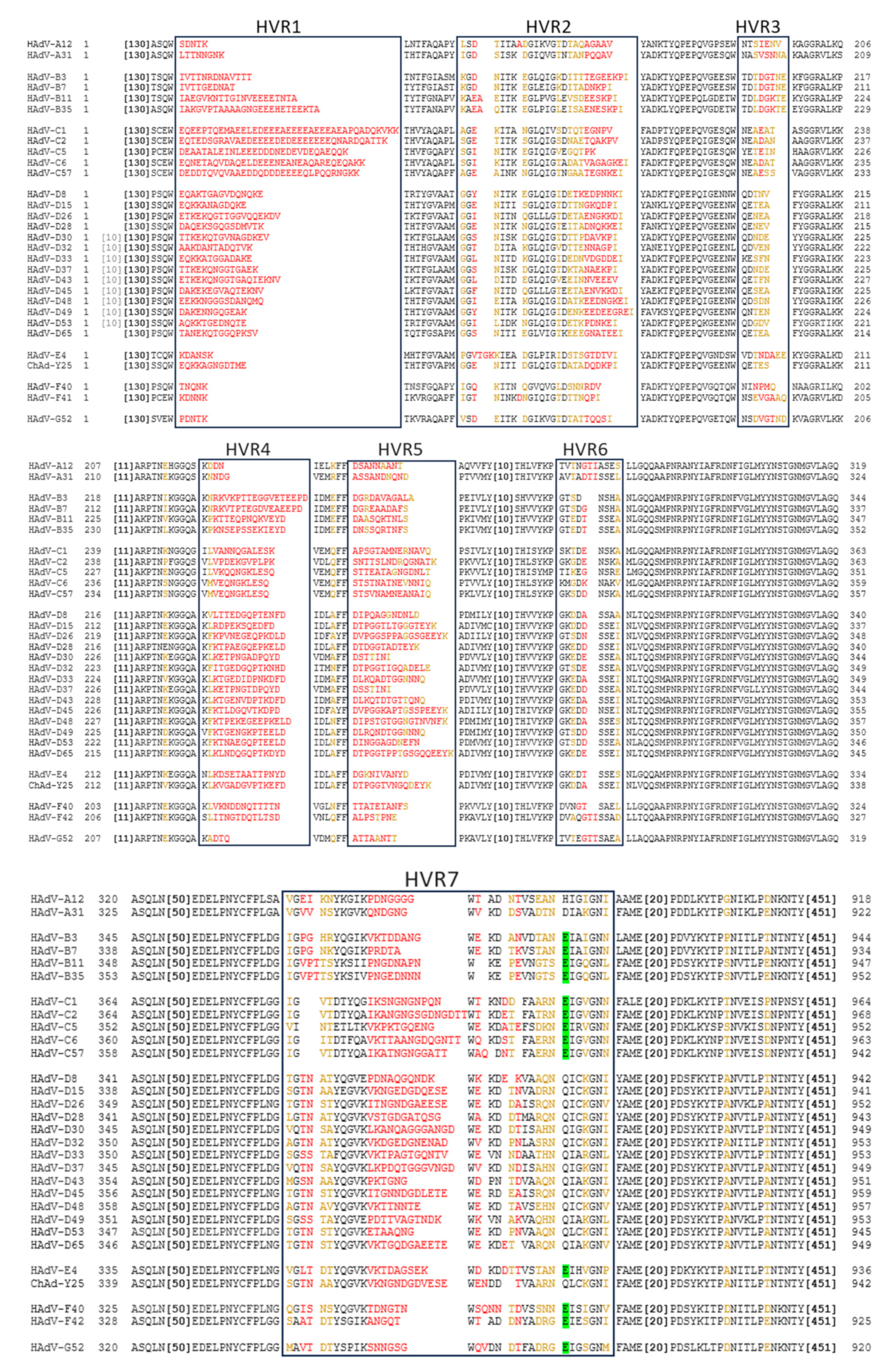
Figure 3.
Summary of innate antiviral immune responses. Through sensing via Toll Like Receptors (TLRs), all cells can secrete IFNs [49]. TLR7/8 sense viral single stranded RNA, TLR3 senses intracellular viral double stranded RNA and TLR9 senses viral double stranded DNA. Through activation of nuclear factor (NF) κB and IFN regulating factor (IRF) 3 signalling, the cell induces production of IFNs by infected cells. This leads to a positive feedback loop through the auto-activation of IRF7 and the production of more IFN by the cells, which in leads to a pro-inflammatory response in the infected tissue [48,50]. The secretion of type I IFNs leads to the recruitment of innate immune cells, which have a range of different functions including destruction of infected cells and processing of viral antigens for presentation to the adaptive immune system [50]. The secretion of cytokines serves to further enhance their function and response: Type I IFNs induce upregulation of MHC-I in tissues, as well as APC maturation and increased NK and monocyte/macrophage activation [49]. NK cell recognise downregulation of MHC-I, which is common in virally infected cells.. IFN-γ and IL-15 enhance CD8+ memory responses and upregulate NK cells in a positive feedback loop [49,50]. TNFα induces apoptosis in cells [49]. Blood components also engage the viral pathogen in a multitude of ways, often to enhance the response of innate immune cells. Engagement of Complement or defensins may also increase the secretion of IFNs [50]. *ADCC: Antibody dependent cellular cytotoxicity. **IL: Interleukin. ***TNF: Tumour necrosis factor.
Figure 3.
Summary of innate antiviral immune responses. Through sensing via Toll Like Receptors (TLRs), all cells can secrete IFNs [49]. TLR7/8 sense viral single stranded RNA, TLR3 senses intracellular viral double stranded RNA and TLR9 senses viral double stranded DNA. Through activation of nuclear factor (NF) κB and IFN regulating factor (IRF) 3 signalling, the cell induces production of IFNs by infected cells. This leads to a positive feedback loop through the auto-activation of IRF7 and the production of more IFN by the cells, which in leads to a pro-inflammatory response in the infected tissue [48,50]. The secretion of type I IFNs leads to the recruitment of innate immune cells, which have a range of different functions including destruction of infected cells and processing of viral antigens for presentation to the adaptive immune system [50]. The secretion of cytokines serves to further enhance their function and response: Type I IFNs induce upregulation of MHC-I in tissues, as well as APC maturation and increased NK and monocyte/macrophage activation [49]. NK cell recognise downregulation of MHC-I, which is common in virally infected cells.. IFN-γ and IL-15 enhance CD8+ memory responses and upregulate NK cells in a positive feedback loop [49,50]. TNFα induces apoptosis in cells [49]. Blood components also engage the viral pathogen in a multitude of ways, often to enhance the response of innate immune cells. Engagement of Complement or defensins may also increase the secretion of IFNs [50]. *ADCC: Antibody dependent cellular cytotoxicity. **IL: Interleukin. ***TNF: Tumour necrosis factor.
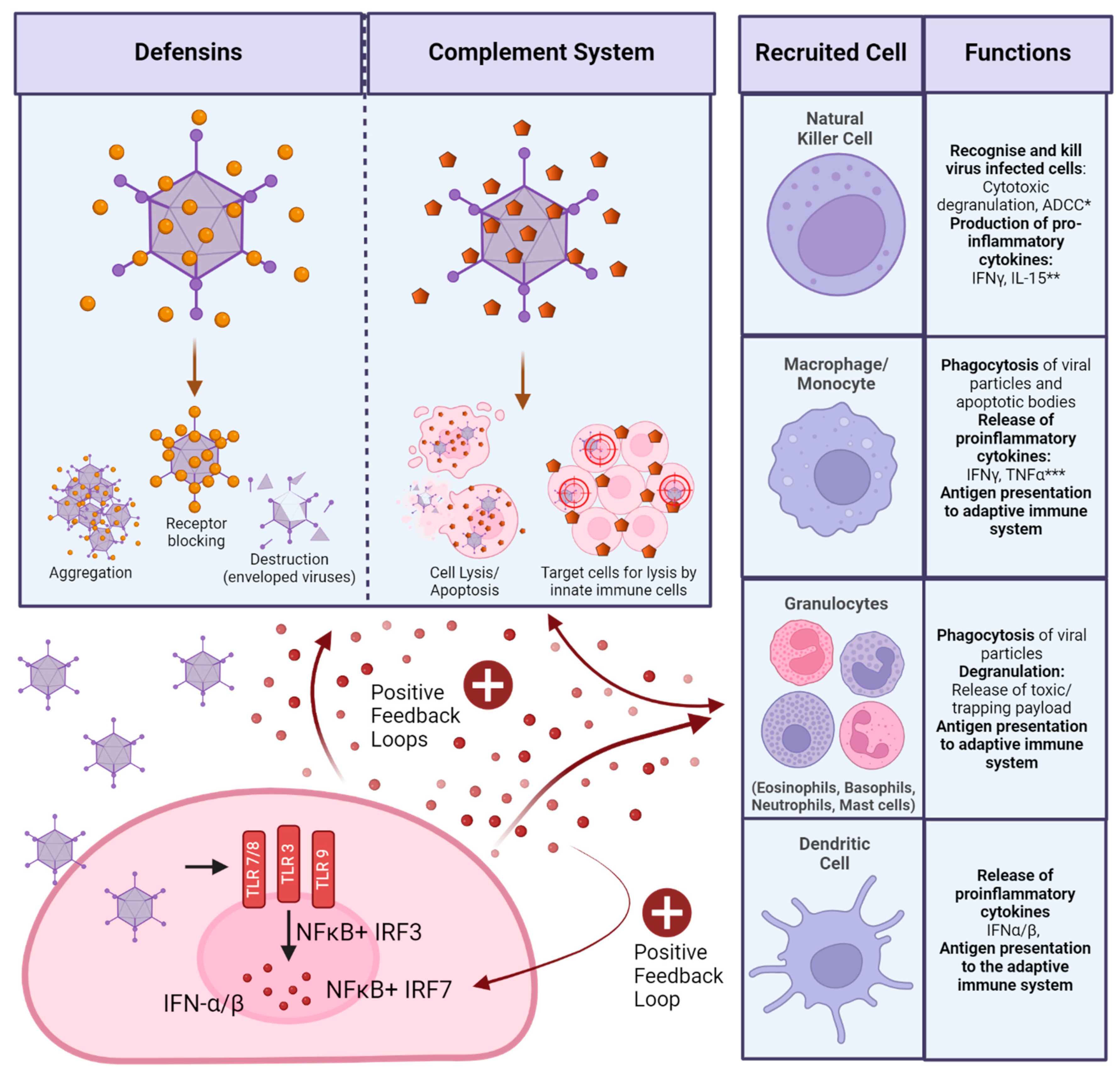
Figure 4.
Summary of adaptive antiviral immune responses.
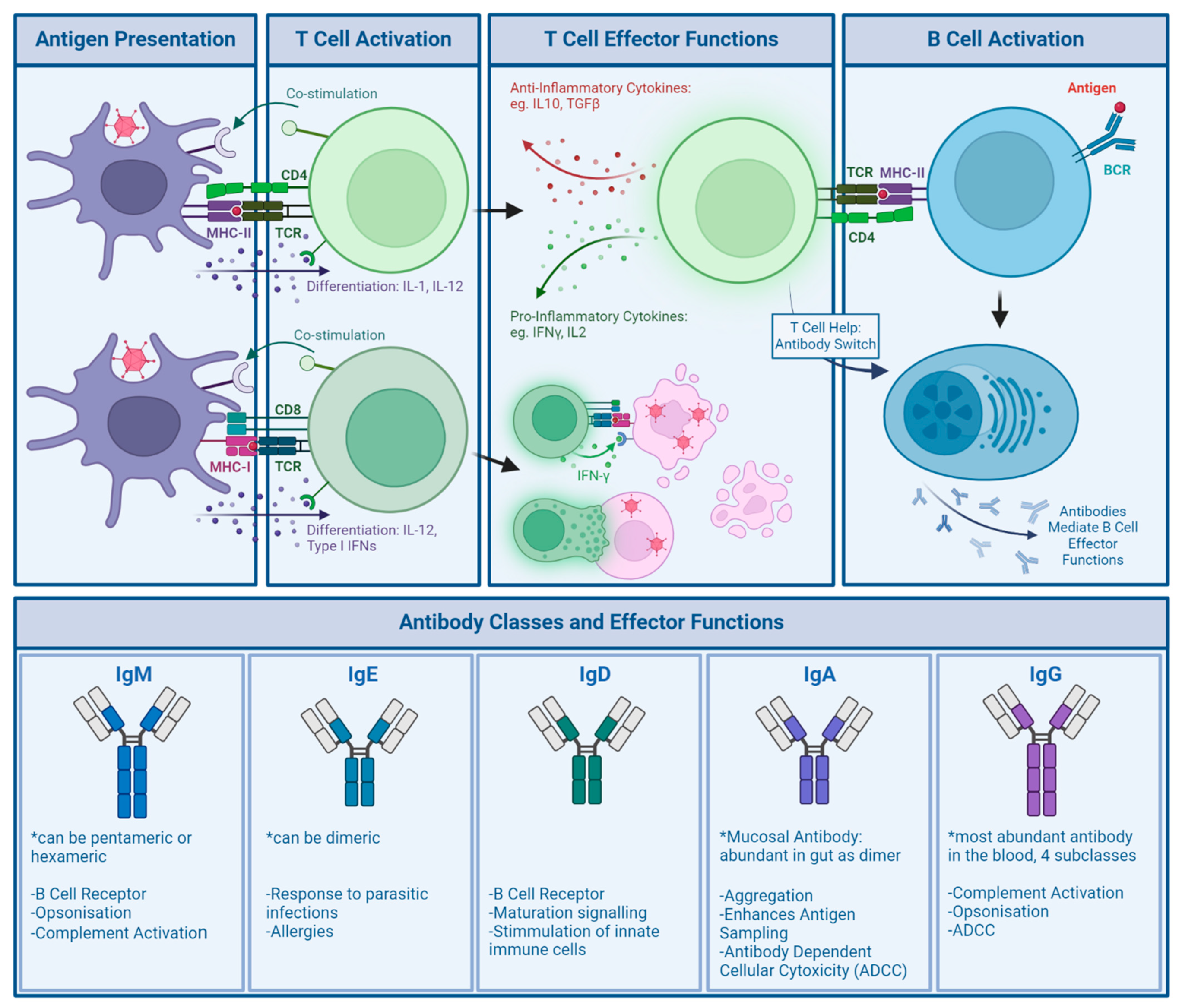
Figure 5.
EU COVID-19 Vaccination Data. Data obtained from ourworldindata.org, last accessed 25.03.2024 [158].
Figure 5.
EU COVID-19 Vaccination Data. Data obtained from ourworldindata.org, last accessed 25.03.2024 [158].
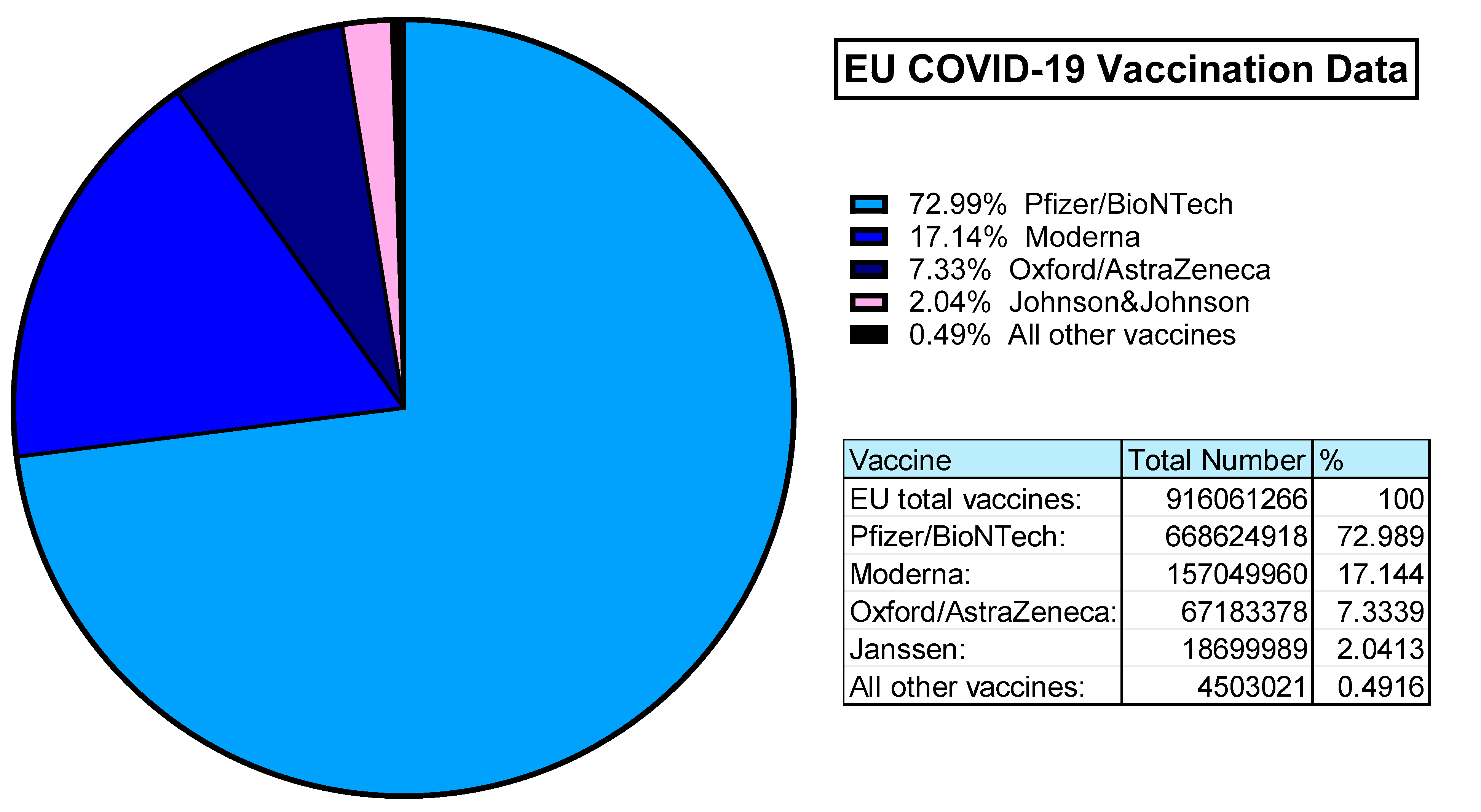

Table 1.
Examples of approved oncolytic and replication deficient (rep def, E1 deleted) Ad based cancer therapies, as well as current and future clinical trials. Information on clinical trials obtained from ClinicalTrials.gov.
Table 1.
Examples of approved oncolytic and replication deficient (rep def, E1 deleted) Ad based cancer therapies, as well as current and future clinical trials. Information on clinical trials obtained from ClinicalTrials.gov.
| Study Title/Drug Name | Status | Mechanism of Action | Vector based on | Targeted Cancer |
|---|---|---|---|---|
| Gendicine[129] | CFDA* approved (2003) |
Oncolytic, Suicide Gene Therapy (p53 restorative gene therapy) | HAdV-C5 | Head and Neck Squamous Cell Carcinoma |
| Adstiladrin[130] | FDA** approved (2022) | Immunomodulatory: IFN-α2b Transgene expression | HAdV-C5 (rep def) | Bladder Cancer Unresponsive to First Line Treatment |
| First in Human Study With NG-641, a Tumour Selective Transgene Expressing Adenoviral Vector (STAR)[131] | Phase I, recruiting |
Oncolytic, Immunostimulatory: FAP-directed bi-specific T-cell activator, Ifα2, CXC9 and CXC10 | HAdV-B11pHAdV-B3(EnAd)[132] | Metastatic or Advanced Epithelial Tumours |
| First in Man Clinical Study to Evaluate Safety and Tolerability of an Oncolytic Adenovirus in Prostate Cancer Patients.[133] | Phase I/IIa, active |
Oncolytic | HAdV-C5[134] | Prostate Cancer |
| Safety and Efficacy of Repeat Administration of Ad/PNP and Fludarabine Phosphate in Patients With Local Head/Neck Cancer[135] | Phase I/II, | Prodrug Conversion | HAdV-C5[136] (rep def) | Recurrent Local Head and Neck Cancer |
| SBRT and Oncolytic Virus Therapy Before Pembrolizumab for Metastatic TNBC and NSCLC (STOMP)[137] | Phase II, active |
Prodrug Conversion | HAdV-C5[138] | Metastatic Triple Negative Breast Cancer, Metastatic Non-Small Cell Lung Cancer |
| Adenovirus Mediated Suicide Gene Therapy With Radiotherapy in Progressive Astrocytoma [139] | Phase I, recruiting |
Prodrug Conversion | HAdV-C5 (rep def) | Different Brain Cancers |
| Trial Investigating an Immunostimulatory Oncolytic Adenovirus for Cancer [140] | Phase I/II, active |
Oncolytic, Immunostimulatory |
HAdV-C5/HAdV-B35 | Pancreatic Adenocarcinoma, Ovarian Cancer, Biliary Carcinoma, Colorectal Cancer |
*CFDA: State Food and Drug Administration of China. ** FDA: U. S. Food and Drug Administration.
Table 2.
Examples of some current and concluded gene therapy trials using Ad based vectors. Information obtained from ClinicalTrials.gov.
Table 2.
Examples of some current and concluded gene therapy trials using Ad based vectors. Information obtained from ClinicalTrials.gov.
| Study Title | Status | Mechanism of Action | Vector based on | Targeted Disease |
|---|---|---|---|---|
| Clinical Study With Lymfactin® in the Treatment of Patients With Secondary Lymphedema (AdeLE)[148] | Phase II, not yet recruiting |
Transgene: Vascular Endothelial growth Factor C (VEGF-C) |
HAdV-C5 (Lymfactin)[149] (rep def) | Secondary Lymphedema (after radiotherapy for breast cancer) |
| Epicardial Delivery of XC001 Gene Therapy for Refractory Angina Coronary Treatment (The EXACT Trial) (EXACT)[150] | Phase I/II, completed (2023) |
Transgene: VEGF |
HAdV-C5[151,152] (rep def) | Angina caused by Coronary Artery Disease |
| Preliminary Testing of New Treatment for Chronic Leg Wounds[153] | Phase I, completed (2011) |
Transgene: Platelet-Derived Growth Factor B (PDGF-B) |
HAdV-C5 (rep def (E1+E3 deleted[154])) | Chronic wounds of the leg/Venous Ulcers |
| Phase I Pilot Study of Ad5-CB-CFTR, an Adenovirus Vector Containing the Cystic Fibrosis Transmembrane Conductance Regulator Gene, in Patients With Cystic Fibrosis[155] | Phase I, completed (2001) |
Transgene: Cystic Fibrosis Transmembrane conductance Regulator (CFTR) |
HAdV-C5 (rep def) | Cystic Fibrosis |
Table 3.
Examples of some vaccines in clinic as well as currently and coming vaccine trials using Ad based vectors. Information obtained from ClinicalTrials.gov.
Table 3.
Examples of some vaccines in clinic as well as currently and coming vaccine trials using Ad based vectors. Information obtained from ClinicalTrials.gov.
| Study Title/Drug Name | Status | Route of Administration | Vector based on | Disease |
|---|---|---|---|---|
| Jcovden | Approved (EMA* March 2021 [165]) Approved (MHRA** December 2022 [166])Revoked (FDA*** March 2023 [167]) |
Intramuscular | HAdV-D26 | SARS-CoV-2 |
| Covishield (UK), Vaxzevria(EU) | Approved (EMA* January 2021[168])Approved (MHRA** July 2021 [166]) |
Intramuscular | ChAdOx1 (ChAd-Y25) | SARS-CoV-2 |
| Phase III Study of BBV154 Intranasal Vaccine in Healthy Volunteers (Nasal154PH3)[169] | Phase III, active |
Intranasal | ChAd36[170] | SARS-CoV-2 |
| Monovalent Chimpanzee Adenoviral-Vectored Marburg Virus Vaccine in Healthy Adults[171] | Phase II, not yet recruiting |
Intramuscular | ChAd-3 | Marburg Virus |
| Phase I Clinical Trial With New SARS-CoV-2 CoVacHGMix Type 5 Adenoviral Vector Vaccine[172] | Phase I, recruiting |
Intramuscular | HAdV-C5 | SARS-CoV-2 |
| Phase 1 Trial of ChAd68 and Ad5 Adenovirus COVID-19 Vaccines Delivered by Aerosol[173] | Phase I, recruiting |
Intranasal | ChAd-68HAdV-C5 | SARS-CoV-2 |
| A Clinical Trial on Booster Immunization of Two COVID-19 Vaccines Constructed From Different Technical Routes[174] | Phase I, recruiting |
Intranasal | HAdV-C5 | SARS-CoV-2 |
| Safety and Immunogenicity of Ad4-HIV Envelope Vaccine Vectors in Healthy Volunteers[175] | Phase I, recruiting |
Intranasal, Intramuscular (boost) | HAdV-E4 | HIV |
| Monovalent Chimpanzee Adenoviral-Vectored Marburg Virus Vaccine in Healthy Adults[171] | Phase II, not yet recruiting |
Intramuscular | ChAd-3 | Marburg Virus |
*EMA: European Medicines Agency. **MHRA: Medicines and Healthcare products Regulatory Agency. ***FDA: U. S. Food & Drug Administration.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



