Submitted:
28 May 2024
Posted:
29 May 2024
You are already at the latest version
Abstract
Monoclonal antibodies (mAbs) have become an increasingly important therapeutic modality in recent years, with applications in oncology, hematology, and immunology. This review discusses the production of therapeutic mAbs, including the various types of mAbs (murine, chimeric, humanized, and fully human), their key applications, and the techniques used for their production, including hybridoma technology, single B cell antibody technology, and phage display technology. The review then proposes advanced techniques to overcome them. These include microfluidic sorting, alternative expression systems (including plants, bacteria, and yeast, and cell-free production systems), and the development of complex antibody formats like bi-specific monoclonal antibodies, antibody fragments, antibody-drug conjugates, and nanobody development. These advanced techniques promise to enhance the efficiency, cost-effectiveness, and scalability of mAb production, paving the way for more effective therapeutic applications. This review highlights the theoretical significance of integrating innovative technologies to overcome existing challenges in mAb production, ultimately advancing the field of precision medicine.
Keywords:
monoclonal antibodies
; therapeutic applications
; hybridoma technology
; single B cell technology
; phage display
1. Introduction to Therapeutic Monoclonal Antibodies
Over the past 20 years, therapeutic monoclonal antibodies (mAbs) have become increasingly important in the fight against a variety of diseases. Monoclonal antibodies have become the preferred therapeutic modality in several therapeutic areas such as oncology, hematology, and immunology [1]. Lab-produced monoclonal antibodies can be called molecules designed to fill in for common antibodies in the immune system, and these proteins are able to recognize and target specific epitopes on antigens [2,3]. These molecules can mimic the immune system’s response to pathogens and cancer cells; mAbs have the ability to search for specific molecules (primarily proteins), whether cell-bound or secreted, and bind to these molecules because they are able to model themselves as the structure of variable regions, resulting in a constant affinity with a single antigenic domain [4].
Monoclonal antibodies (mAbs) can be classified into four types: murine, chimeric, humanized, and fully human antibodies. The first developed mAbs were derived from mice [5], with both variable and constant regions coming from mouse species. Patients receiving initial murine mAb therapies developed human anti-mouse antibodies (HAMAs), leading to hypersensitivity reactions like anaphylaxis, which resulted in the rapid elimination of these antibodies and thus limited their therapeutic effectiveness [3]. Chimeric mAbs are generated by fusing the variable regions of murine mAbs with human antibody constant regions, such as abciximab, used to inhibit platelet aggregation in cardiovascular diseases, and rituximab, indicated for non-Hodgkin’s lymphoma [6]. These structural chimeras reduce immunogenicity and overcome therapeutic issues caused by HAMAs, though chimeric mAbs are often associated with the induction of anti-drug antibody (ADA) reactions [7]. With advances in genetic engineering, humanized mAbs have become predominant; they essentially incorporate murine complementarity-determining regions (CDRs) into human antibody constant regions [7,8], resulting in hybrid immunoglobulins that are 95% human, thus aiding in neutralizing ADAs. Trastuzumab, approved by the FDA in 1998 for breast cancer [9], was the first humanized mAb to be clinically indicated. Fully human mAbs, composed only of human genetic sequences, are typically produced in transgenic mice where endogenous Ig genes have been replaced with human Ig gene clusters. Introduced by Alt et al. in 1985, fully human mAbs like ofatumumab and denosumab offer better-tolerated immune therapies with reduced immunogenicity risks; ofatumumab is used for chronic lymphocytic leukemia, while denosumab is indicated for bone metastasis [10,11] (Figure 1).
The emergence of monoclonal antibody technology gave rise to the development of antibody drugs. In 1986, only 10 years after the birth of monoclonal antibody preparation technology, the US FDA approved the world’s first therapeutic murine monoclonal antibody, an anti-CD3 antibody named muromonab-CD3. It is used to treat allograft rejection after organ transplantation [12]. Since the first therapeutic mAb, muromonab-CD3, was approved in 1992, 119 therapeutic mAbs have been approved in the United States, including two diagnostic mAbs [13]. In the following decades, monoclonal antibody drugs in the field of biomedicine has been a leap forward, and now has a huge market of about 100 billion dollars, and will continue to grow in the foreseeable future, coupled with the difficult to replace the status of antibodies in the diagnosis of disease, the application of monoclonal antibodies in the field of medicine is recognized as a success.
The success of mAbs in therapeutic applications is mainly due to their complex glycoprotein structure, which provides high specificity. mAbs are a type of immunoglobulin (Ig), of which there are five subclasses: IgA, IgD, IgE, IgG, and IgM. IgG is the isotype most relevant to therapeutic use [14]. The molecular weight of IgG is approximately 150 kilodaltons (kDa) and it has a Y-shaped structure composed of three equal-sized segments connected by a flexible hinge. An IgG molecule consists of four polypeptide chains: two heavy (H) chains of about 50 kDa each and two light (L) chains of about 25 kDa each. The two arms of the Y-shaped structure are called fragment antigen-binding (Fab) regions and include the variable regions that differ between different mAbs and are responsible for antigen binding. The stem of the Y is called the constant region, which has less variability and interacts with effector cells and molecules (known as the Fc fragment) (Figure 1a).
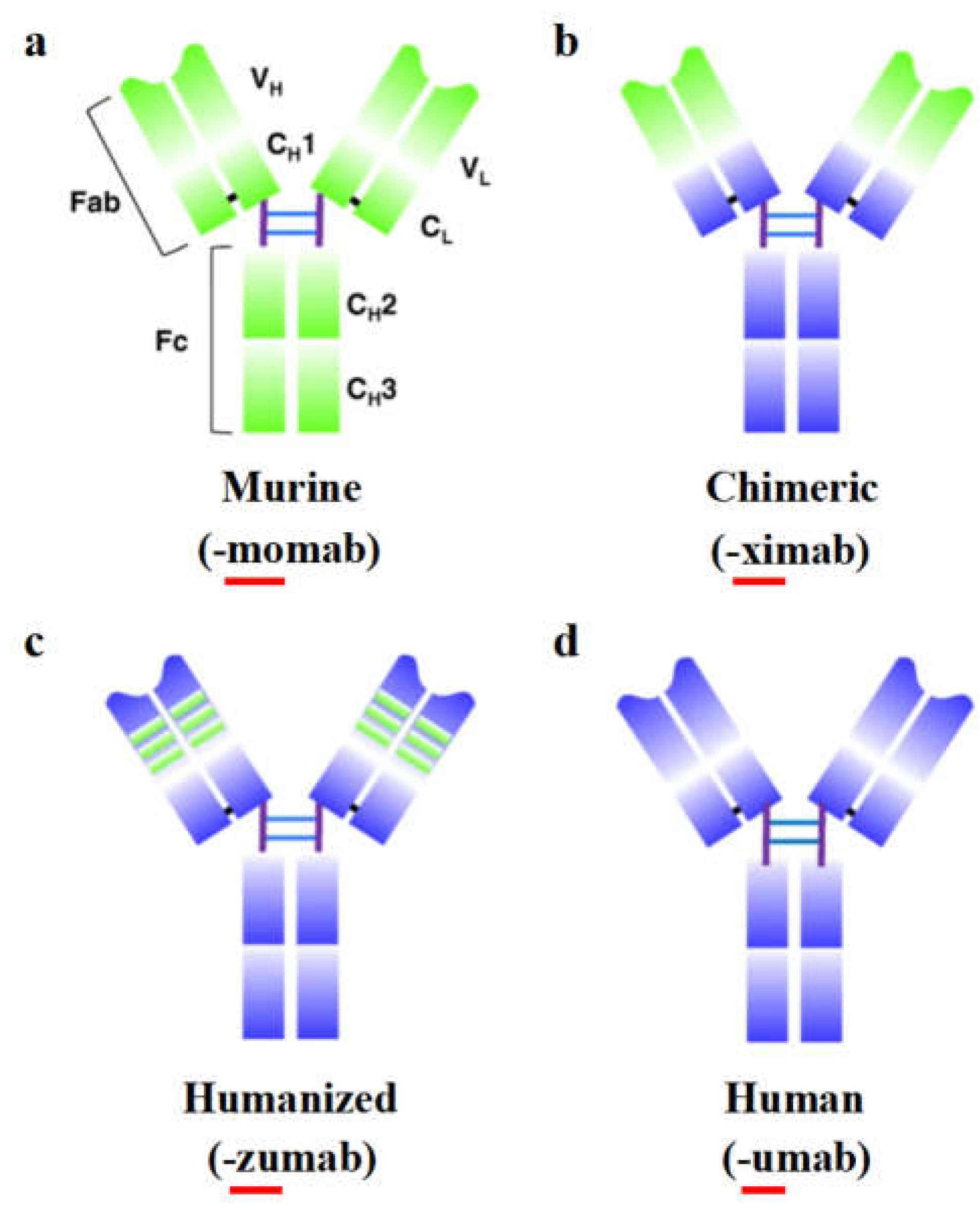
Figure 1.
Overview of IgG antibody, humanization from murine antibodies (green domains) to fully human antibodies (blue domains). (a) The murine monoclonal antibody. (b) The chimeric monoclonal antibody: variable regions are of murine origin, and the rest of the chains are of human origin. (c) Humanized monoclonal antibody: only includes the hypervariable segments of murine origin. (d) Human monoclonal. CH: domains of the constant region of the heavy chain; CL: constant domain of the light chain; Fab and Fc: fragments resulting from proteolysis; VH: variable domain of the heavy chain; VL: variable domain of the light chain. [15] (Copyright 2020, BMC).
Figure 1.
Overview of IgG antibody, humanization from murine antibodies (green domains) to fully human antibodies (blue domains). (a) The murine monoclonal antibody. (b) The chimeric monoclonal antibody: variable regions are of murine origin, and the rest of the chains are of human origin. (c) Humanized monoclonal antibody: only includes the hypervariable segments of murine origin. (d) Human monoclonal. CH: domains of the constant region of the heavy chain; CL: constant domain of the light chain; Fab and Fc: fragments resulting from proteolysis; VH: variable domain of the heavy chain; VL: variable domain of the light chain. [15] (Copyright 2020, BMC).
2. Clinical Applications of Therapeutic mAbs
2.1. mAbs in Viral Infectious Diseases Treatment
Recent advancements in monoclonal antibodies (mAbs) have been predominantly focused on treating oncological diseases. However, significant attention is also being directed towards viral diseases lacking effective vaccines, such as HIV, Lassa fever, and Zika fever, as well as those where antiviral drugs are insufficiently effective, including COVID-19, influenza, and rabies [16]. To date, six antiviral mAb-based drugs have received approval from the WHO and FDA (Table 1). The first approved mAb, Palivizumab [17], targets respiratory syncytial virus. Following this, development efforts shifted towards noncommunicable diseases, resulting in numerous antibody preparations being approved annually, yet no additional antiviral mAbs were approved until recently.
In 2018, the field of antiviral mAbs saw a resurgence with the approval of a mAb drug for HIV treatment. This was followed by the approval of two mAb drugs for Ebola virus treatment in 2020 [18,19,20,21]. Additionally, the FDA granted emergency use authorization in 2020 for Bamlanivimab and the REGEN-COV antibody cocktail against COVID-19. Subsequent studies revealed that Bamlanivimab was not effective as a monotherapy (leading to the FDA’s revocation of its monotherapy authorization in April 2021), but showed efficacy when used in combination with Etesevimab [22]. The rapid development and approval of mAb therapies for SARS-CoV-2 demonstrated the potential for quick and effective mAb responses in emergencies and underscored the utility of mAbs in treating and preventing viral infections.

Table 1.
mAb drugs approved for therapy and prevention of viral infections.
| mAb drug | Target | Antibody variant | Technology | Status |
| Palivizumab (Synagis) |
Glycoprotein F | Humanized (IgG1) | Hybridoma | Approved by FDA in 1998 [23] |
| Ibalizumab (Trogarzo) |
Receptor CD4 | Humanized (IgG4) | Hybridoma | Approved by FDA in 2018 [24] |
| Ansuvimab (Ebanga) |
Glycoprotein (GP) | Human (IgG1) | B cell immortalization | Approved by FDA in 2020 [20] |
| Atoltivimab, Maftivimab and Odesivimab-ebgn (Inmazeb) |
Glycoprotein (GP) | Human (IgG1) | Hybridoma (gene-modified mice) | Approved by FDA in 2020 [25] |
| Etesevimab and Bamlanivimab | Glycoprotein S (RBD) | Human (IgG1) | No data | Emergency use authorization by FDA in 2021 [22,26] |
| Casirivimab and Imdevimab (REGEN-COV) |
Glycoprotein S (RBD) | Human (IgG1) | Single B cell sorting (human and gene-modified mice) |
Emergency use authorization by FDA in 2021 [27] |
The development of mAbs preparations typically involves several key stages: (1) isolating an antibody with the desired properties; (2) employing protein engineering to enhance its target characteristics; (3) generating stable antibody-producing cell lines; and (4) optimizing cultivation conditions and purification processes for the antibody-producing cells.
Antiviral mAbs are generally derived from the blood of infected or immunized donors, or from synthetic libraries that contain a diverse set of immunoglobulin variable fragments. The immune system of a donor (either human or animal) infected with a pathogen produces a vast array of antibodies targeting all potential epitopes, which undergo affinity maturation in the body to enhance their specificity. Memory B cells from these donors can be isolated to obtain specific antibodies. However, in some cases, a robust immune response does not develop following infection, necessitating the search for more suitable donors or deliberate immunization of donors with the pathogen or its antigens. Booster immunizations can further stimulate the immune response, producing antibodies with higher affinity.
Although immune libraries are generally preferred, a naive antibody repertoire is sometimes used to obtain antiviral mAbs. The primary advantage of this approach is the extensive diversity of antibodies it offers, making it a versatile source for identifying antibodies against any antigen, including those that do not elicit a strong immune response [28]. Synthetic antibody fragment libraries provide unique benefits, such as complete control over the design and selection conditions. These libraries can generate a vast array of antibodies that the donor’s immune system may not naturally produce. However, this method carries the risk of generating antibodies that may be unsafe or ineffective in vivo. Despite these challenges, the feasibility of using mAbs from synthetic libraries to combat viruses and bacterial toxins has been demonstrated [29]. The production methods for antiviral mAbs will be discussed in detail later.
2.2. mAbs in Cancer Treatment
Monoclonal antibodies (mAbs) present a promising avenue for cancer therapy, offering high specificity and efficacy with minimal off-target toxicity in an idealized setting. In cancers where mAbs have shown significant efficacy, such as HER2+ breast cancer, they have become the first-line treatment, surpassing the performance of HER2-specific small-molecule inhibitors like lapatinib and neratinib. These mAbs provide excellent response profiles with relatively low toxicities [30]. Despite this success, only a limited number of cancers, primarily hematologic malignancies, have been effectively treated with mAbs.
Monoclonal antibodies exert their therapeutic effects through various mechanisms, which depend on the location of the target antigen, whether it is on tumor cells, infected cells, cytokines, or growth factors. Understanding these mechanisms is crucial for transforming cancer treatment approaches, as they typically result in a more tolerable toxicity profile compared to traditional therapies [31]. The mechanisms by which mAbs can kill tumor cells include blocking ligand-receptor interactions, blocking signaling pathways, and depletion of targets through Fc interaction.[32,33,34,35,36,37,38,39,40] (Table 2).
First, mAbs can bind to ligands or receptors on cancer cell surfaces, preventing ligand-receptor interactions [41]. Nonconjugated mAbs achieve this through steric hindrance, where the antibody physically blocks the binding region, or by inducing conformational changes in the target cell [42]. Some mAbs can also internalize the antibody-antigen complex, removing the receptor and ligand from contact, and blocking their signaling or biological activity. Conjugated mAbs combine the benefits of mAbs with the ability to transport and release specific therapeutic agents [43,44], such as cytotoxic molecules or drugs, to exert their effects within target cells [45].
Furthermore, mAbs can induce tumor cell death by blocking signaling pathways associated with growth factor receptors [46]. By recognizing the receptors for growth factors, mAbs can inactivate signaling pathways or block ligand binding, interfering with cell proliferation, adhesion, and angiogenesis [47,48]. They can also disrupt important cellular processes required for tumor cell survival and evade immune checkpoints, enhancing the antitumoral immune response [49].
mAbs interact with Fc receptors (FcRs) on effector cells, such as natural killer cells, cytotoxic T cells [50], and macrophages, through their Fc domain. This interaction mediates antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent phagocytosis (ADP), and complement-dependent cytotoxicity (CDC) [51,52]. ADCC triggers the release of lytic compounds from effector cells, leading to target cell lysis [50]. ADP facilitates the recognition and internalization of opsonized cells by phagocytic cells for degradation. CDC involves the recruitment of proteins and the initiation of a proteolytic cascade that induces cell death [53,54].
mAb therapy in cancer treatment combines various mechanisms of action depending on the specific antibody and cancer antigen profile. Understanding these mechanisms is crucial for the development and clinical testing of novel mAb-based therapeutics.
3. Monoclonal Antibody Production Using Various Bioreactor Systems
In vitro technologies for monoclonal antibody (mAb) production typically use small-scale suspension cell culture devices such as T-flasks, Triple flasks, Cellstacks, Hyper flasks, spinner flasks, roller bottles, and shake-flasks [55]. However, these conventional systems yield low antibody concentrations due to limited cell density. To address this limitation, perfusion bioreactor systems have been developed, offering long-term cell culture stability by continuously supplying fresh media and removing spent media along with metabolic waste products [56].
Perfusion bioreactors maintain high cell density and enhance mAb production significantly. They provide various advantages over traditional methods, allowing for much higher yields of mAbs. Different types of perfusion-based high cell density bioreactor systems have been established, including roller bottles, stirred tank bioreactors, and disposable bioreactors. Studies have shown that disposable static membrane bioreactors achieve the highest mAb titers, while stirred tank bioreactors operating in semi-continuous mode with overlay aeration show peak productivity [57].
High cell density bioreactors such as hollow fiber systems, packed bed reactors, and wave bioreactors have been successfully used for cultivating mammalian cells in serum-free media. These systems support the growth of hybridoma and CHO cell lines for therapeutic protein production. High-density culture systems (≤107 cells/ml) are essential for reducing production costs and increasing output in therapeutic protein production, achieving high cell density and viability [58]. For cell lines that struggle to grow in suspension due to shear stress from agitation, improving immobilization techniques for cells on porous beads is crucial. High cell density culture systems often use adsorption or entrapment methods for cell immobilization, with the size, porosity, and surface characteristics of matrices playing significant roles. Packed bed and polymer-based bioreactors are commonly used for cell immobilization and cultivation, offering compact bioreactors with high volumetric production, though operational complexity remains a challenge.
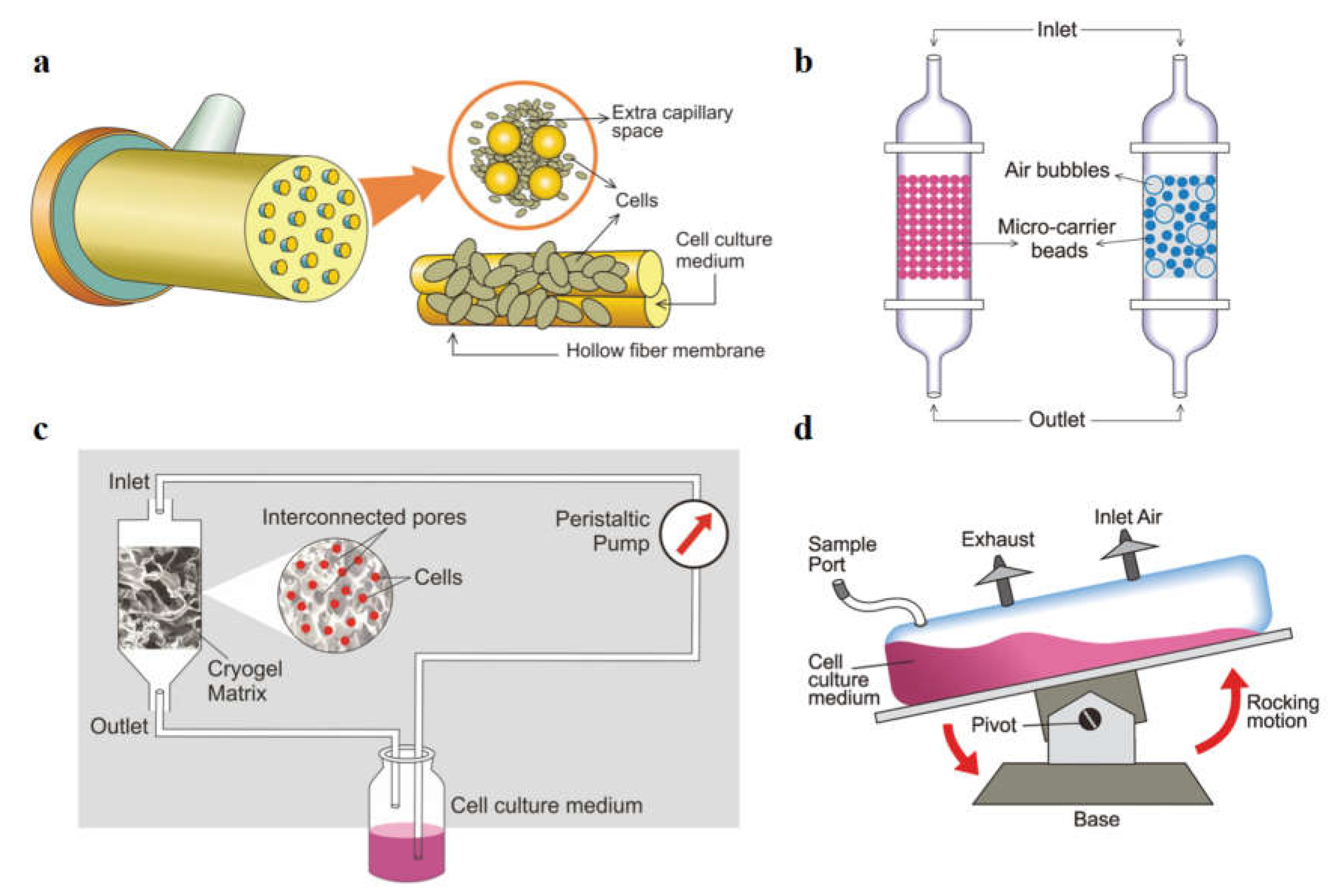
Figure 2.
Schematic representation of producing mAbs using various high cell density bioreators. (a) Hollow fiber bioreactor which consists of a bundle of fibres and media circulated within capillaries. (b) Fixed (left) and fluidized (right) bed reactor systems with cells immobilized over micro-carrier beads. (c) Cryogel bioreactor where cells are entrapped into the pores of the cryogel matrix. (d) Wave bioreactor, tilting along a pivot axis causing the movement of cell culture medium with cells by rocking motion. [59] (Copyright 2019, The Korean Society for Microbiology and Biotechnology).
Figure 2.
Schematic representation of producing mAbs using various high cell density bioreators. (a) Hollow fiber bioreactor which consists of a bundle of fibres and media circulated within capillaries. (b) Fixed (left) and fluidized (right) bed reactor systems with cells immobilized over micro-carrier beads. (c) Cryogel bioreactor where cells are entrapped into the pores of the cryogel matrix. (d) Wave bioreactor, tilting along a pivot axis causing the movement of cell culture medium with cells by rocking motion. [59] (Copyright 2019, The Korean Society for Microbiology and Biotechnology).
The hollow fiber bioreactor (HFBR) (Figure 2a) revolutionizes monoclonal antibody (mAb) production with its unique design. Thousands of semipermeable hollow fibers, each 200 μm in diameter [60], form a continuous perfusion culture system within a cartridge. Cells attach to the outer surface of these fibers, where they thrive in an extra capillary space. Nutrient-rich cell culture medium continuously circulates through the fibers, facilitating optimal growth conditions. The bioreactor’s high surface-to-volume ratio enables the attachment of a large number of cells in a small volume, achieving cell densities of up to 109 cells/ml. With a molecular weight cut-off starting at 5 kDa, a variety of materials like polysulfone and cellulose derivatives are utilized for the hollow fibers, ensuring efficient nutrient exchange. Notably, mAbs can be harvested from a single cartridge for over six months in continuous culture mode, with yields ranging from 137 to 307 mg in crude extract. Further purification protocols, such as hydroxyapatite and ion-exchange chromatography, yield 50 to 150 mg of purified antibody fragments [61]. Additionally, the HFBR allows for culture in reduced serum or serum-free media, facilitating convenient purification. This innovative system offers prolonged production capabilities, enhanced process efficiency, and superior product quality for mAb production.
Fixed and fluidized bed reactors (Figure 2b) offer viable alternatives to hollow fiber bioreactors for producing therapeutic proteins. Microcarrier beads support cell immobilization within a column, enabling continuous perfusion. Common microcarriers like Fibra-Cel and SIRAN achieve high cell densities and productivity, even up to 108 cells/ml in serum-free media. Cytoline from GE Healthcare is popular in fluidized systems, reaching cell densities of 2×108 cells/ml [62]. Recent advancements aim to minimize void volume, enhancing viability and productivity. Centrifugal bioreactors, utilizing centrifugal force for cell immobilization, exhibit promising results, with cell density increasing approximately 1.7-fold with doubled centrifugal force. These systems represent pioneering solutions in bioprocessing technology.
Disposable cryogel bioreactors (Figure 2c) offer an economically favorable solution for mAb production, combining high cell density with extended cell lifetime. These bioreactors utilize polymeric cryogels as matrices for cell cultivation, alongside a medium reservoir and peristaltic pump. Cryogels, formed at subzero temperatures, feature an interconnected network of macropores, facilitating efficient mass transfer. They offer simple synthesis, aqueous solvent usage, and high porosity with mechanical strength. Hybridoma cell lines are immobilized within cryogel matrices, enhancing adherence and ensuring unhindered diffusion of substrates and metabolites. Coating with gelatin further improves cell-matrix interaction. Notably, continuous operation of a polyacrylamide cryogel bioreactor yielded monoclonal antibody productivity of 130 μg/ml on day 36, showcasing its potential for long-term production [63].
Wave bioreactors, pioneered by Singh, are efficient and cost-effective systems for bioprocessing, suitable for various cell lines including hybridoma, CHO, NSO, and insect cells (Figure 2d) [64]. These disposable reactors feature a cell bag chamber containing culture medium, cells, and an air circulation port. Mixing and agitation occur through a rocking motion, facilitating efficient mass transfer and creating an optimal environment for cell growth, achieving densities exceeding 108 cells/ml. Scalable up to 1,000 L, these bioreactors allow separate monitoring of cell growth and antibody production in batch and perfusion cultures. Recent studies demonstrate their suitability for virus cultivation, insect cell cultivation, and embryonic cell expansion. Wave bioreactors hold promise for producing high-value proteins from challenging virus sources, offering significant potential for upstream processing.
4. Current Methods of Producing Therapeutic mAbs
To date, various well-established monoclonal antibody (mAb) preparation technologies are available, including the hybridoma technique, phage display technique, and single B cell antibody preparation techniques. The choice of method can be tailored to the specific characteristics of the desired antibody.
4.1. Hybridoma Technology for mAb Production
4.1.1. Introduction to Hybridoma Technology
Hybridoma technology, described by Georges Köhler and Cesar Milstein in 1975, involves immunizing animals with a desired antigen and fusing specific B lymphocytes with „immortal” myeloma cells [65]. The resulting hybrid cells, called hybridomas, are cloned to create stable monoclonal cell lines. These clones, which secrete the antibody of interest, are then cultured on a large scale to produce the antibody in the desired quantities [66]. The fusion of B lymphocytes and myeloma cells is commonly achieved using polyethylene glycol (PEG), which can be cytotoxic and cause non-specific membrane fusion [67,68]. Fusogenic viruses, such as Sendai and vesicular stomatitis viruses, are alternatives that avoid PEG’s cytotoxic effects [68]. Another method is the pearly chain technique, where fusion occurs with the help of an electric field and laser radiation. This method irradiates the contact cell surface with pulsed laser beams to create a small perforation in the cell membrane, promoting cell fusion [67,69]. Despite its advantages over PEG-mediated fusion, the pearly chain method still cannot selectively control the fusion of a specific B lymphocyte with a myeloma cell [67].
Muromonab-CD3, also known as orthoclone (OKT3), was the first monoclonal antibody (mAb) approved by the FDA in 1986 for human therapeutic use [70]. This murine hybridoma-derived mAb targets CD3 on mature peripheral T cells to prevent organ allograft rejection [71]. However, human anti-mouse immune responses limited the clinical use of murine mAbs [72]. Human hybridomas were initially unsuccessful due to genetic instability. Technological advances led to chimeric mAbs, combining murine variable regions with human constant regions. The first FDA-approved chimeric mAb, abciximab, was approved in 1994 [73,74]. Humanization techniques later allowed grafting non-human antibody CDRs into human antibody scaffolds [75], leading to the approval of the first humanized antibody, daclizumab, in 1997. In the following decade, transgenic animals were developed to produce fully human mAbs by integrating human immunoglobulin genes into animal genomes. The first fully human mAb from transgenic animals, panitumumab, was approved in 2006 [76].
4.1.2. Procedures of Using Hybridoma to Produce mAbs
Below is a general process for developing monoclonal antibodies (mAbs) using mouse and rat myeloma cells, which is currently the mainstream method for mAb development. The first step involves injecting laboratory animals, such as rabbits or mice, with a selected antigen through a series of injections over several weeks. This stimulates B cell differentiation into plasma B cells and memory B cells. Once a sufficient number of antibodies are created in the animal serum, the animal is sacrificed [77]. Following sacrifice, the spleen is removed under aseptic conditions to isolate the activated B-cells using density gradient centrifugation. The presence of antibodies in the serum is identified using methods like ELISA [77] or flow cytometry. The activated B lymphocytes are then fused with myeloma cells.
A few weeks before cell fusion, metastatic tumor cells are incubated in 8-azaguanine to make the HGPRT genes in the myeloma cells non-functional. This makes the myeloma cells sensitive to HAT media, which is preferred in hybridoma technology. Activated B lymphocytes are fused with HAT-sensitive myeloma cells using polyethylene glycol (PEG), which promotes the fusion of the plasma membranes. Electrofusion, using an electric field, is another method that is more efficient [77,78].
Then, Cells are cultured in HAT media for 10–14 days to select fused hybridoma cells. Unfused B cells die quickly, and unfused myeloma cells die due to lack of HGPRT genes, leaving only viable hybrid cells [79]. HAT-selected hybridoma cells are transferred to ELISA plates, each containing a single hybridoma cell [80]. Screening identifies hybridomas producing desired antibodies specific to an antigen’s epitope.
Finally, selected hybridomas are transferred to large culture vessels for in vivo or in vitro propagation [77]. In vivo involves injecting hybridoma cells into mice, while in vitro involves culturing cells in lab conditions for high-purity antibody production.
4.1.3. Challenges and Difficulties in Hybridoma Technology
One of the most critical steps in mAb production is the choice of a cell line. The cells must propagate well, be highly stable in culture, and produce high mAb titres in its active form, meaning the protein is folded correctly, glycosylated, and not aggregated. Chinese hamster ovary cell line (CHO) is the most preferred cell line used for large-scale production of mAbs. However, the development of stable cell lines is a tedious process that sometimes takes months to years, where the success of this process depends on the random genome integration of transgenes [81].
Another major challenge associated with hybridoma development is the requirement of purified antigen to generate a specific immune response. In some cases, it is a challenging task to purify the antigen. The lack of specific immune reagents for characterization and monitoring of these numerous proteins limits the overall time process for the production of hybridoma. Expression and purification of recombinant protein are time-consuming and sometimes not cost-effective [82]. Additionally, immunization of animals with these purified recombinant proteins in formulation with adjuvants sometimes leads to alteration of the native conformation of these proteins, which finally leads to an undesired immune response in animals.
The efficiency of hybridoma technology also depends heavily on animal immunization. Factors affecting the efficiency of immunization include the route of administration of antigen, dose, choice of adjuvant, number of boosts, and immunization protocol. DNA-based immunizations are generally preferred where it is difficult to express full-length proteins, and immune responses are mainly targeted towards native or conformational epitopes because the structural integrity of the protein is critical for the induction of functional mAbs.
A significant challenge in the field of human hybridomas is the requirement of lymphocytes from actively infected patients or those exposed to the antigen. If the active immune response is absent or insufficient, the probability of circulating B cells is very poor or negligible. Due to ethical issues, it is generally not possible to immunize humans [83].
Finally, after cell fusions between B cells and myeloma cells, hybridoma technology protocols include multistep screening and cloning processes to identify antigen-specific hybridomas, which is labor-intensive and time-consuming. To address the limitations of hybridoma technology in mAb production, new techniques such as single B cell antibody technology and antibody phage display technology have been developed and widely adopted [84].
4.2. Single B Cell Antibody Technology
4.2.1. Introduction to Single B Cell Antibody Technology
Single B cell antibody technology represents a significant advancement in generating monoclonal antibodies (mAbs). Unlike traditional methods that require fusing B lymphocytes with myeloma cells [85], this technology enables the detection and isolation of individual functional B lymphocytes from heterogeneous primary cell populations. It also allows for antibody gene amplification and cloning without the need to immortalize the selected antibody-secreting cell. This approach, collectively known as single B cell antibody technology, [86] facilitates the rapid generation of neutralizing mAbs for various applications [87], including emerging infectious diseases. Notably, this technology has been instrumental in developing mAbs against viral infections such as HIV [88], Dengue [89], and SARS-CoV-2 [90]. The following sections briefly outline the basic concepts and benefits of single B cell antibody technology.
4.2.2. Procedures of Using Single B Cell Antibody Technology to Produce mAbs
The first step is the identification and isolation of single B cells. The screening and isolation of ASCs can be performed in either a random or antigen-specific manner from peripheral blood or lymphoid tissue samples. Random selection can involve recovering B cells by flow cytometry [91] or micromanipulation [92] from tissues. For antigen-specific selection, multi-parameter flow cytometry or other fluid-based approaches are typically employed [93]. Flow cytometry systems are efficient for recovering single cells and have been used successfully to isolate IgG+ memory B lymphocytes reactive to specific antigens, such as those from donors with HIV [94]. Antigen-specific IgG+ B cells represent a small percentage of circulating cells, and reagents targeting B cell surface markers are useful for their identification and isolation [95].
After isolating single B cells, the next step is the amplification, cloning, and expression of the immunoglobulin genes. This involves lysing the cells, synthesizing cDNA from total mRNA via reverse transcription, and amplifying the full-length immunoglobulin genes for the variable and constant regions of the light and heavy chains using PCR [96]. The amplified fragments are cloned into linear expression cassettes to generate the immunoglobulin domains in cell-based expression systems, such as mammalian or bacterial cells. Typically, antibodies are expressed in Fab form [97], but they can also be produced in other formats like full-length IgG and single-chain variable fragments (scFv) [98]. Additionally, the “single-cell RT-PCR-linked in vitro expression” (SICREX) platform allows for antibody expression outside a cell unit, using a mixture containing the transcription/translation machinery from E. coli, significantly reducing the time required to generate antibodies [99].
4.2.3. Challenges and Difficulties in Single B Cell Antibody Technology
While single B cell antibody technology offers several advantages, it also has notable disadvantages and challenges. Antigen-specific IgG+ B cells are rare in the circulation, and identifying and isolating them requires reagents targeting B cell surface markers [95]. Although a variety of antibodies are available to detect human B lymphocytes, allowing for the distinction of cells at different developmental stages, isolating B cells from non-human species is more difficult. For example, despite the availability of antibodies against mouse B lymphocyte markers such as CD45R and CD19, sorting B cells from species like rabbits and guinea pigs is challenging due to the limited repertoire of appropriate targeting antibodies [94].
The cost of expensive sorting devices is another significant consideration, as they are integral to isolating antigen-specific single B lymphocytes from polyclonal mixtures. Alternative strategies, such as antigen-coated magnetic beads, cell-based microarrays, and soft lithographic methods for micro engraving, can be employed but are also costly and require extensive expertise [97].
Additionally, although the single-cell RT-PCR-linked in vitro expression (SICREX) platform accelerates antibody production by eliminating the need for gene-cloning, transformation, and cultivation, it has its own drawbacks. Specifically, the synthesis of proteins in this system can result in incorrect folding of antibody domains, potentially compromising the functionality of the generated antibodies [99].
4.3. Antibody Phage Display Technology
4.3.1. Introduction to Antibody Phage Display Technology
Phage display technology provides a robust alternative to conventional hybridoma methods for monoclonal antibody (mAb) production, eliminating the risk of Human Anti-Mouse Antibody (HAMA) responses commonly associated with antibodies produced by hybridoma platforms. First demonstrated by George P. Smith in 1985, phage display involves altering the phage genome to fuse the coat protein genes with those of the desired peptide, allowing the peptide to be displayed on the phage surface [100,101]. Smith and Sir Gregory P. Winter received the Nobel Prize in Chemistry in 2018 for their pioneering work in this field [102].
The process begins with mRNA from human B cells, which is converted into cDNA and amplified using PCR to generate antibody fragments. These fragments, which include heavy chains (HCs) and light chains (LCs), can be combined to form single-chain fragment variable (scFv) or Fab fragments, resulting in an antibody library where each antibody is displayed as a fusion protein with a phage coat protein [103]. Parmley and Smith developed a selection process called „biopanning,” where phages displaying antibodies with the highest affinity for the target antigen are selected and subsequently cloned into a prokaryotic expression system [101,102]. However, this method produces non-glycosylated antibodies, which may affect their pharmacokinetics and distribution [101].
Phage display technology is advantageous for producing mAbs as it allows for the rapid generation of large antibody libraries and the development of specific therapeutic antibodies. For instance, Adalimumab (Humira®) is a humanized anti-TNF antibody derived from phage display that prevents TNF from binding to its receptors, thus suppressing immunoinflammatory responses in autoimmune diseases [104]. Another example is Namilumab, an IgG1-κ anti-GM-CSF antibody, which was obtained by phage display-guided humanization of a rat scFv and is indicated for rheumatoid arthritis, psoriasis, and psoriatic arthritis [101,104].
4.3.2. Procedures of Using Phage Display Technology to Produce mAbs
To generate an antibody phage library, antibody gene fragments are first cloned into vectors. Filamentous M13 phage and phagemid, which combine the characteristics of plasmids and phages, can be used as vectors [105]. While M13 phage can independently produce phage particles and display antibodies, phagemids require infection with a helper phage to package the phagemid as single-strand DNA into a virion particle [106,107,108]. These vectors transform E. coli by electroporation. Once the phage display library is obtained, the antibodies displayed on the vector surface are screened through biopanning [109]. Antibodies are commonly displayed in single-chain variable fragment (scFv) or antigen-binding fragment (Fab) forms.
There are four types of antibody display libraries: immune, naïve, semisynthetic, and synthetic. Immune libraries are derived from immunized animals or humans, primarily used to discover antibodies against infectious pathogens or cancer antigens [110]. These libraries contain a restricted repertoire that has undergone antigen-driven in vivo selection [111]. Naïve libraries, generated from B lymphocytes of non-immunized donors, like the scFv library from Cambridge Antibody Technology, provide a natural antibody gene repertoire [112]. Synthetic libraries are created entirely in silico, bypassing the need to isolate antibody genes. Semisynthetic libraries use both natural and synthetically randomized CDRs, improving the chances of finding antibodies with high specificity and affinity [110,112].
Constructing the phage display library is crucial, with the size of the library directly proportional to the likelihood of finding a specific antibody [111]. Next-Generation Sequencing (NGS) analyzes the variability, sequence composition, and size of the libraries [113]. Although building a phage display library is more expensive than generating hybridomas, the antibody screening step is faster and cheaper [114].
4.3.3. Challenges and Difficulties in Phage Display Technology
Despite its promise, phage display technology for antibody development has certain limitations. The diversity of the phage library is constrained by bacterial transformation efficiency, capping at a maximum repertoire of 1010-1011 variant antibodies. This limitation can be addressed by mRNA and ribosome display techniques, which are cell-free methods that offer larger library sizes and greater antibody diversity, up to 1014 variants [115,116].
Additionally, since antibodies selected through phage display are produced in E. coli, they lack glycosylation. Using eukaryotic display platforms, such as yeast [117] or mammalian expression systems [118], can help overcome this issue. Other drawbacks of phage display include the potential for generating biased repertoires and the loss of natural antibody pairing information [119].
5. Advanced Techniques for the Improvement of Therapeutic mAbs Production
Several established technologies are utilized in the production of monoclonal antibodies (mAbs), each with distinct principles and drawbacks. Hybridoma technology, pioneered by Köhler and Milstein, involves fusing B lymphocytes with myeloma cells to create hybrid cells that produce specific antibodies and proliferate indefinitely. However, it is dependent on successful animal immunization, is slow, labor-intensive, and faces ethical constraints with human lymphocytes. Single B cell antibody technology isolates individual B cells using techniques like flow cytometry, lyses them, and clones their immunoglobulin genes for expression. This method quickly generates mAbs but requires costly equipment and reagents, and isolating antigen-specific B cells is challenging. Phage display technology uses bacteriophages to display antibody fragments, screening libraries to identify high-affinity binders. While efficient, it is limited by bacterial transformation efficiency and lacks post-translational modifications, affecting therapeutic efficacy. In summary, each technology has significantly advanced antibody production, despite their respective limitations.
To address these limitations, several advanced techniques and alternative production platforms have been proposed, including microfluidic sorting, nanobody utilization, alternative expression systems, cell-free production, and the development of complex antibody formats.
5.1. Microfluidic Sorting Technique in mAb Screening
Microfluidic sorting techniques have emerged as crucial tools in monoclonal antibody (mAb) screening, particularly for hybridoma cells. These techniques streamline the separation and analysis of fused cells post-electrofusion at the single-cell level. High-throughput microarray-based methods and CD microfluidic devices, like those developed by Lai et al., automate ELISA processes, significantly reducing reagent consumption and assay time compared to traditional methods [120]. These devices utilize centrifugal and capillary forces for fluid flow control, effectively detecting antibodies with lower reagent volumes and faster processing times.
Moreover, microsphere-based assays and microfluidic chip electrophoresis have shown superior performance in detecting and analyzing antibodies compared to standard immunoassays and SDS-PAGE. Droplet-based microfluidics have also been pivotal, encapsulating single hybridoma cells in microdroplets to achieve rapid antibody detection within small volumes. For instance, Joensson et al. utilized fluorescently labeled peptides in droplet-based systems for fluorescent polarization analysis, achieving high accuracy in antibody detection [121].
For sorting, flow cytometry and fluorescence-activated cell sorting (FACS) are widely used but require fluorescent labeling of cell surface proteins, which is less efficient for detecting secreted antibodies. Droplet-based microfluidic devices enhance this process by combining high-throughput screening with flow cytometry’s sensitivity. These devices generate microdroplets using a two-phase system, encapsulating cells in aqueous droplets surrounded by immiscible oil with biocompatible surfactants. This setup maintains cell viability and facilitates various follow-up manipulations, such as live-cell monitoring and long-term experiments [122].
The integration of droplet-based sorting platforms with fluorescence-activated droplet sorting (FADS) systems has shown remarkable efficiency. For example, hybridoma cells producing inhibitory antibodies against ACE-1 were sorted based on low fluorescence intensity, indicating effective ACE-1 inhibition [123]. This system achieved a sorting rate of 50,000 cells per hour with significant enrichment of desired hybridoma cells. Additionally, protocols developed by Mazutis et al. enabled binding assays without washing steps, using fluorescent signals to sort antibodies efficiently, achieving sorting rates exceeding 1 kHz and high enrichment accuracy [124].
5.2. Alternative Expression Systems of mAbs
5.2.1. Plant Cells
Plant cells have emerged as a promising alternative to traditional mammalian and microbial expression systems for the production of monoclonal antibodies (mAbs). The use of plant-based platforms offers several advantages, including their ability to perform human-like post-translational modifications, cost-effectiveness, and scalability.
One of the main plant cell lines used for mAb production is tobacco (Nicotiana benthamiana). This plant-based system has demonstrated the capacity to produce a wide range of antibody formats, including full-length IgG, Fab, and single-chain variable fragments (scFv). The glycosylation patterns of plant-produced mAbs are generally similar to their human counterparts, with the exception of the absence of core fucose residues, which can enhance antibody-dependent cellular cytotoxicity (ADCC) activity [125].
Another plant-based expression system is the moss Physcomitrella patens, which has been successfully employed for the production of various therapeutic proteins, including mAbs. Moss cells can be cultivated in large-scale bioreactors and possess the ability to perform complex post-translational modifications, ensuring the proper folding and functionality of the expressed mAbs [126].
The use of plant cells for mAb production offers several advantages, such as the ease of genetic manipulation, the absence of mammalian pathogens, and the potential for cost-effective large-scale manufacturing. Additionally, plant-based mAbs have demonstrated comparable or even superior efficacy and safety profiles compared to their mammalian-derived counterparts in various pre-clinical and clinical studies.
5.2.2. Bacterial Cells
Bacterial expression systems, particularly Escherichia coli, have been extensively explored as an alternative to mammalian and plant-based platforms for the production of monoclonal antibodies (mAbs). Bacteria offer several advantages, including rapid growth, well-established genetic manipulation techniques, and the ability to achieve high-yield protein production.
While bacteria do not naturally possess the machinery to perform complex post-translational modifications essential for the proper folding and functionality of mAbs, significant progress has been made in engineering bacterial strains to address this limitation. Strategies such as the introduction of eukaryotic chaperones and the co-expression of enzymes responsible for glycosylation have enabled the production of mAbs with human-like glycosylation patterns [127].
One of the key challenges in using bacterial cells for mAb production is the formation of inclusion bodies, which can lead to the accumulation of insoluble, inactive protein. To overcome this issue, researchers have explored various approaches, including the use of fusion tags, optimized expression conditions, and the development of periplasmic expression systems.
Despite the inherent challenges, bacterial expression systems have shown promise for the production of antibody fragments, such as Fab and scFv, which do not require extensive post-translational modifications. These smaller antibody formats can be effectively produced in E. coli and have been utilized in various therapeutic and diagnostic applications.
5.2.3. Yeast Cells
Yeast cells have also been explored as an alternative expression system for the production of monoclonal antibodies (mAbs). Yeast, such as Saccharomyces cerevisiae and Pichia pastoris, offer several advantages, including the ability to perform eukaryotic post-translational modifications, high-level protein expression, and the potential for cost-effective large-scale manufacturing.
Yeast cells can effectively assemble and secrete full-length IgG antibodies, as well as various antibody fragments, including Fab and scFv. The glycosylation patterns of yeast-produced mAbs are generally more similar to those of mammalian cells compared to bacterial systems, though some differences may still exist [128].
One of the key advantages of using yeast cells for mAb production is their scalability and ease of genetic manipulation. Yeast can be cultivated in large-scale bioreactors, and the introduction of recombinant genes encoding the antibody of interest is relatively straightforward.
Additionally, yeast cells offer the potential for cost-effective production due to their faster growth rates, simpler media requirements, and the ability to utilize inexpensive carbon sources. This makes yeast-based platforms an attractive option for the large-scale manufacturing of mAbs, particularly for developing regions or settings with limited resources.
Overall, the use of plant, bacterial, and yeast cells for mAb production represents a diverse landscape of expression systems, each with its own set of advantages and challenges. The selection of the appropriate cell line will depend on various factors, including the specific requirements of the mAb, the targeted application, and the desired characteristics of the final product.
5.2.4. Cell-Free Production
Cell-free (CF) systems are emerging as a flexible alternative for protein production, eliminating cell membrane constraints and enabling batch-to-batch consistency. These systems, based on cell lysates, accelerate the synthesis of monoclonal antibodies (mAbs) and antibody-drug conjugates (ADCs), crucially bypassing the need for transfection, selection, and clone expansion. This approach is particularly advantageous for high-throughput screening and early-stage antibody evaluation, significantly reducing the time from discovery to production. For example, CF technology allows for the rapid synthesis of antibodies and the creation of large antibody libraries using ribosome and mRNA display systems, which support higher library sizes than traditional phage and yeast display methods. Successful CF production of various antibody formats, including scFvs, Fab fragments, and complete IgGs, has been demonstrated in E. coli, reticulocyte, wheat germ, and CHO systems. Advances in bioorthogonal chemistries further enhance ADC development, enabling site-specific modifications. Notably, bispecific antibodies, which can target two different epitopes simultaneously, benefit from CF systems for precise assembly and functional testing. These innovations highlight the potential of CF systems to streamline antibody production and enhance therapeutic development [129].
5.3. Development of Complex Antibody Formats
5.3.1. Production of Bi-Specific Monoclonal Antibodies (BsAbs)
BsAbs, including BiTEs and IgG-like formats, offer enhanced specificity and targeting capabilities compared to traditional antibodies. By simultaneously binding two different antigens, they facilitate immune cell recruitment or delivery of cytotoxic payloads, improving therapeutic efficacy. BiTEs, lacking an Fc region, directly engage T cells for tumor cell killing. Approved BsAbs like blinatumomab demonstrate efficacy in hematological malignancies. Additionally, BsAbs targeting immune checkpoint molecules or oncogenic signaling pathways show promise in cancer therapy, with candidates like erfornilimab and cadonilimab in late-phase clinical trials, illustrating the expanding landscape of BsAbs in oncology [130].
5.3.2. Antibody Fragments (Fab, scFv, VHH) and Analogs
Antibody fragments like Fab, scFv, and VHH, along with analogs such as Anticalin, Centyrin, DARPins, Affibody, and Knottin, play pivotal roles in drug development. These fragments offer advantages like small size, deep tissue penetration, and cost-effectiveness. ScFv, composed of VH and VL linked by a peptide chain, facilitates diverse applications. Centyrins, based on type-III fibronectin domains, and DARPins, derived from ankyrin repeats, exhibit robustness and versatility comparable to antibodies. Affibodies, originating from Staphylococcus protein-A Z-domains, demonstrate improved hydrophilicity and stability. Knottins, with excellent stability and small size, are prominent in research. VHH, or nanobodies, derived from camelids, are gaining prominence, with examples like caplacizumab and envafolimab showing therapeutic efficacy in various diseases, including aTTP and advanced solid tumors [131].
5.3.3. Creating Antibody–Drug Conjugates (ADC)
ADCs, a rapidly expanding segment of antibody drugs, combine a monoclonal antibody, cytotoxic payload, and linker. They selectively target tumor cells expressing specific antigens, delivering cytotoxic drugs intracellularly via receptor-mediated endocytosis, leading to tumor cell death. Payloads, including tubulin polymerization inhibitors like DM1 and auristatins, and DNA-damaging agents like calicheamicin and SN-38, contribute to their efficacy. FDA-approved ADCs like Gemtuzumab ozogamicin, Brentuximab vedotin, and Trastuzumab emtansine have revolutionized cancer treatment. Ongoing developments, such as DS-8201a for HER-2-expressing tumors and sacituzumab govitecan for breast cancer, promise further therapeutic advancements [132].
5.3.4. Nanobody for the Therapeutic Development
Nanobodies, also known as single domain antibodies, are derived from naturally occurring heavy chain antibodies in camelids or sharks. They are the smallest antigen-binding units of antibodies, characterized by their nanometer size (2.5 nm × 4 nm × 3 nm). Nanobodies exhibit high specificity and affinity for antigens, making them suitable for therapeutic applications. Their small size allows easy tissue penetration and effective targeting of solid tumors and brain therapy due to their ability to cross the blood-brain barrier.
Nanobodies are also more easily engineered into multivalent or multispecific forms for enhanced therapeutic functions. They can be produced in Escherichia coli or yeast, significantly reducing manufacturing costs. With a stable structure and high solubility, nanobodies retain their antigen-binding ability under harsh conditions, including high temperatures and chemical denaturants. Lacking an Fc domain, they have low immunogenicity and high homology with human VH domains, making them safe for long-term treatments. Nanobodies have shown promise in tumor diagnosis and therapy, particularly when conjugated with functional moieties for improved imaging and drug delivery [133].
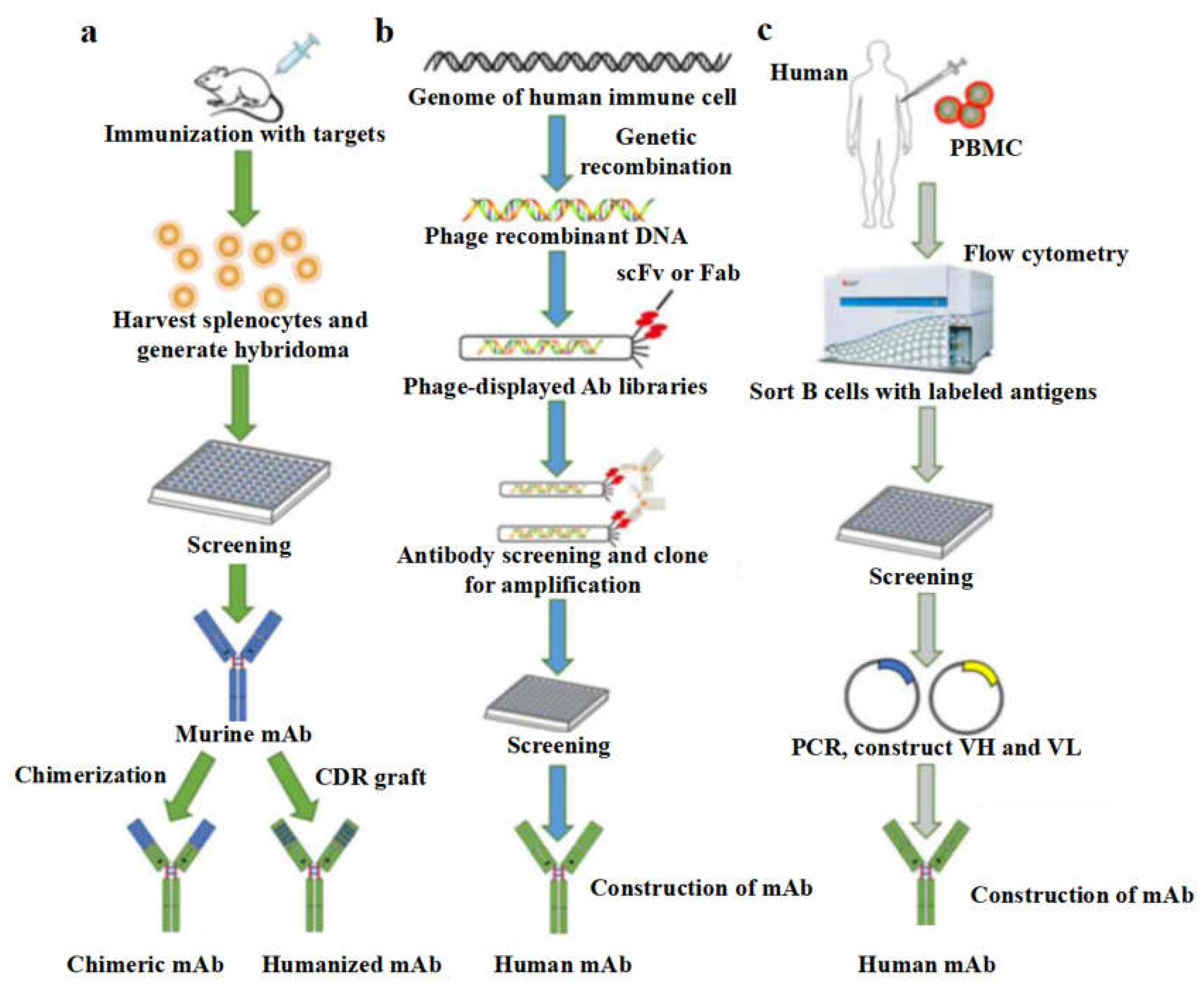
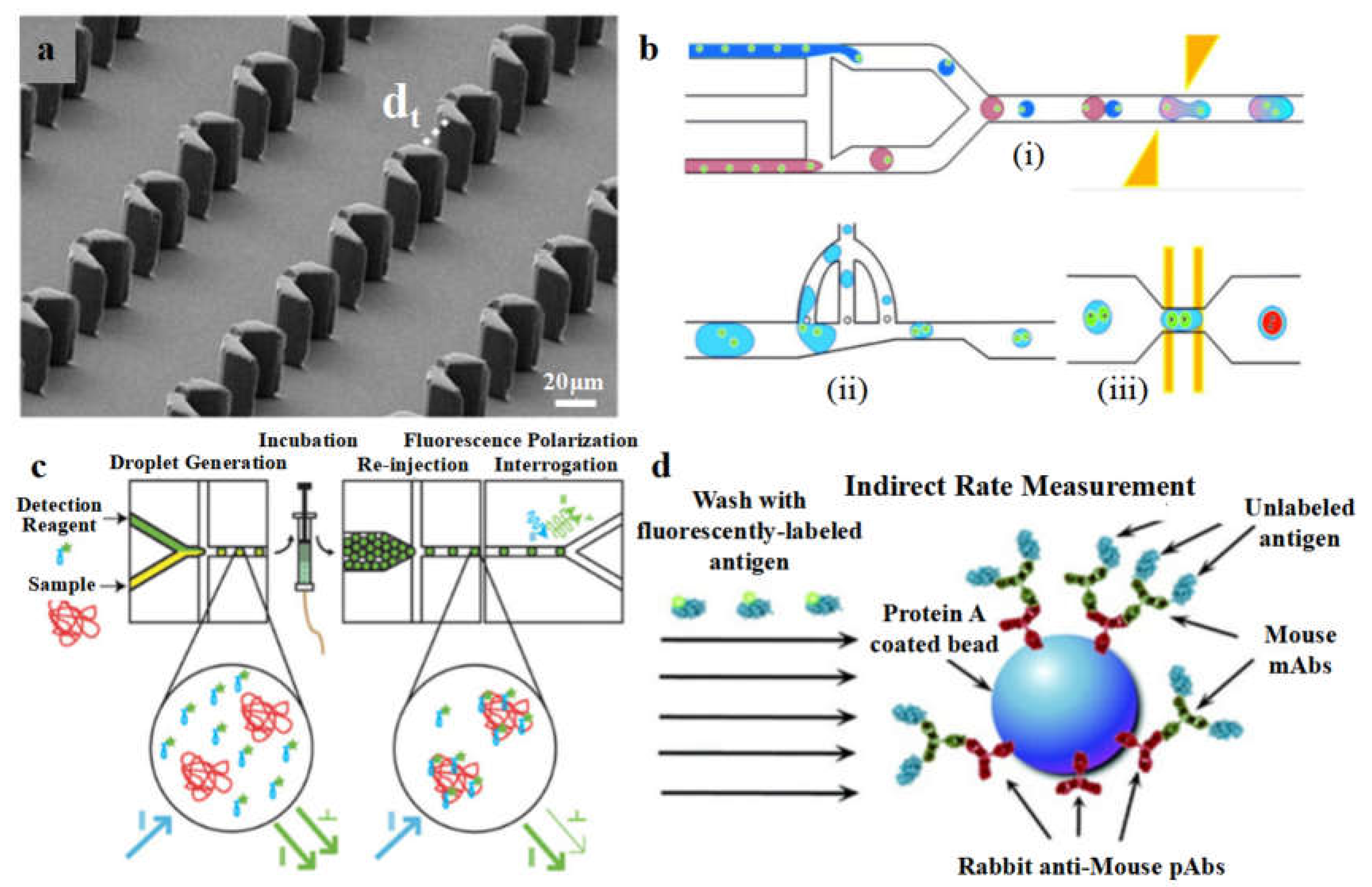
Figure 3.
Approaches for the preparation of therapeutic monoclonal antibodies (mAbs). (a) Hybridoma technique. The traditional murine hybridoma technique starts by the immune response of mice triggered with the desired antigens. After that, splenocytes are harvested and fused with myeloma cells to produce hybridoma cells. After the screening, selected hybridoma cells are used to persistently generate chimeric or humanized monoclonal antibodies. (b) Phage display. A human phage-displayed antibody library is used to select the antigens of interest. After immuno-positive phage clones screened by ELISA and DNA sequences, construction of the mAb is made to help express humanized mAb. (c) The single B cell technique. peripheral blood mononuclear cells (PBMCs) are prepared from infected or vaccinated donors so as to isolate suitable B cells by flow cytometry. After that, VH and VL information of each B cell informs the generation of human mAbs by RT-PCR. [134] (Copyright 2022, Lu et al.).
Figure 3.
Approaches for the preparation of therapeutic monoclonal antibodies (mAbs). (a) Hybridoma technique. The traditional murine hybridoma technique starts by the immune response of mice triggered with the desired antigens. After that, splenocytes are harvested and fused with myeloma cells to produce hybridoma cells. After the screening, selected hybridoma cells are used to persistently generate chimeric or humanized monoclonal antibodies. (b) Phage display. A human phage-displayed antibody library is used to select the antigens of interest. After immuno-positive phage clones screened by ELISA and DNA sequences, construction of the mAb is made to help express humanized mAb. (c) The single B cell technique. peripheral blood mononuclear cells (PBMCs) are prepared from infected or vaccinated donors so as to isolate suitable B cells by flow cytometry. After that, VH and VL information of each B cell informs the generation of human mAbs by RT-PCR. [134] (Copyright 2022, Lu et al.).
Figure 4.
The microfluidic device based on hydrodynamic trapping for hybridoma cell fusion and characterization. (a) The structure of the hydrodynamic trap; adapted from Ref [135]. (b) The working procedure of multifunction-integrated microdroplet platform for hybridoma formation (including: (i) single-cell encapsulation and droplet pairing, (ii) droplet shrinkage, and (iii) cell electrofusion; adapted from Ref [136].) (c) The schematic diagram of fluorescence polarization analysis of protein contents; adapted from Ref [137]. (d) Schematic diagrams for indirect measurement of the association and dissociation kinetics between the antibodies and antigens; adapted from Ref [138].
Figure 4.
The microfluidic device based on hydrodynamic trapping for hybridoma cell fusion and characterization. (a) The structure of the hydrodynamic trap; adapted from Ref [135]. (b) The working procedure of multifunction-integrated microdroplet platform for hybridoma formation (including: (i) single-cell encapsulation and droplet pairing, (ii) droplet shrinkage, and (iii) cell electrofusion; adapted from Ref [136].) (c) The schematic diagram of fluorescence polarization analysis of protein contents; adapted from Ref [137]. (d) Schematic diagrams for indirect measurement of the association and dissociation kinetics between the antibodies and antigens; adapted from Ref [138].
Conclusion
Monoclonal antibodies (mAbs) have become an increasingly valuable therapeutic modality for treating a variety of diseases, particularly in the fields of oncology, hematology, and immunology. The development of different antibody formats, from murine to fully human, has helped overcome issues of immunogenicity and improve tolerability. Technological advancements in areas like hybridoma, phage display, and single B cell technologies have expanded the pool of available mAb candidates against diverse targets.
Despite their successes, these traditional methods have inherent limitations such as limited diversity, glycosylation issues, and labor-intensive processes. The emergence of advanced techniques such as microfluidics for cell sorting, the use of nanobodies, and the development of alternative expression systems in plants, microalgae, and yeast offer promising solutions. Additionally, the integration of cell-free synthesis platforms and innovative approaches like bispecific antibodies, antibody fragments, and antibody-drug conjugates (ADCs) are pushing the boundaries of what is achievable with mAb technology.
Future directions in mAb production will likely focus on combining these advanced techniques to overcome existing limitations and improve efficiency, specificity, and scalability. The continued evolution of these technologies holds great potential for the development of novel therapeutics and the enhancement of our understanding of the immune system. Ongoing efforts to further optimize mAb production, engineering, and purification processes will be key to realizing the full potential of this versatile class of biologics. As the field of mAb therapeutics continues to evolve, researchers and clinicians are poised to harness their specificity and potency to tackle an ever-widening range of diseases.
References
- H.M. Abdelaziz, M. Gaber, M.M. Abd-Elwakil, M.T. Mabrouk, M.M. Elgohary, N.M. Kamel, D.M. Kabary, M.S. Freag, M.W. Samaha, S.M. Mortada, K.A. Elkhodairy, J.-Y. Fang, A.O, Elzoghby: Inhalable particulate drug delivery systems for lung cancer therapy: Nanoparticles, microparticles, nanocomposites and nanoaggregates, J. Control. Release, 269 (2018), pp. 374-392. [CrossRef]
- V. Bayer. An overview of monoclonal antibodies. Semin. Oncol. Nurs., 35 (5) (2019), Article 150927. [CrossRef]
- J. Posner, P. Barrington, T. Brier, A. Datta-Mannan. Monoclonal antibodies: past, present and future. J.E. Barrett, C.P. Page, M.C. Michel (Eds.), Concepts and Principles of Pharmacology: 100 Years of the Handbook of Experimental Pharmacology, Springer International Publishing (2019), pp. 81-141.
- U. Hafeez, H.K. Gan, A.M. Scott. Monoclonal antibodies as immunomodulatory therapy against cancer and autoimmune diseases. Curr. Opin. Pharmacol., 41 (2018), pp. 114-121. [CrossRef]
- D.D., Banker, Monoclonal antibodies. A review, in: Current Clinical Pharmacology, Vol. 13, Issue 2, 2018, pp. 85–99. [CrossRef]
- R.-M. Lu, Y.-C. Hwang, I.-J. Liu, C.-C. Lee, H.-Z. Tsai, H.-J. Li, H.-C. Wu, Development of therapeutic antibodies for the treatment of diseases, J. Biomed. Sci., 27 (1) (2020), p. 1. [CrossRef]
- K.-T. Jin, B. Chen, Y.-Y. Liu, H.R. Lan, J.-P. Yan, Monoclonal antibodies and chimeric antigen receptor (CAR) T cells in the treatment of colorectal cancer, Cancer Cell Int., 21 (1) (2021), p. 83. [CrossRef]
- H. Waldmann, Human monoclonal antibodies: the benefits of humanization, M. Steinitz (Ed.), Human Monoclonal Antibodies: Methods and Protocols, Springer, New York (2019), pp. 1-10.
- D.P. Clark, N.J. Pazdernik, Immune technology, in: Biotechnology, Academic Cell, 2016, pp. 181–217.
- V. Bayer, An overview of monoclonal antibodies, Semin. Oncol. Nurs., 35 (5) (2019), Article 150927. [CrossRef]
- C. Kang, H.A. Blair, Ofatumumab: a review in relapsing forms of multiple sclerosis, Drugs, 82 (1) (2022), pp. 55-62. [CrossRef]
- Smith S. Ten years of orthoclone OKT3 (muromonab-CD3): A review. J TransPlant Coord, 1996, 6: 109-121. [CrossRef]
- Cai, H.H. Therapeutic Monoclonal Antibodies Approved by FDA in 2020.
- Awwad, S., Angkawinitwong, U., 2018. Overview of antibody drug delivery. Pharmaceutics 10, 1–24. [CrossRef]
- Lu, RM., Hwang, YC., Liu, IJ. et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci 27, 1 (2020). [CrossRef]
- Zeitlin, L., Cone, R.A., Moench, T.R., and Whaley, K.J., Microbes Infect., 2000, vol. 2, pp. 701–708.
- Pollack, P. and Groothuis, J.R., J. Infect. Chemother., 2002, vol. 8, pp. 201–206.
- Rizza, S.A., Bhatia, R., Zeuli, J., and Temesgen, Z., Drugs Today (Barc.), 2019, vol. 55, pp. 25–34.
- Corti, D., Misasi, J., Mulangu, S., Stanley, D.A., Kanekiyo, M., Wollen, S., Ploquin, A., Doria-Rose, N.A., Staupe, R.P., Bailey, M., Shi, W., Choe, M., Marcus, H., Thompson, E.A., Cagigi, A., Silacci, C., Fernandez-Rodriguez, B., Perez, L., Sallusto, F., Vanzetta, F., Agatic, G., Cameroni, E., Kisalu, N., Gordon, I., Ledgerwood, J.E., Mascola, J.R., Graham, B.S., Muyembe-Tamfun, J.J., Trefry, J.C., Lanzavecchia, A., and Sullivan, N.J., Science, 2016, vol. 351, pp. 1339–1342.
- Lee, A., Drugs, 2021, vol. 81, pp. 595–598.
- Sivapalasingam, S., Kamal, M., Slim, R., Hosain, R., Shao, W., Stoltz, R., Yen, J., Pologe, L.G., Cao, Y., Partridge, M., Sumner, G., and Lipsich, L., Lancet Infect. Dis., 2018, vol. 18, pp. 884–893.
- Gottlieb, R.L., Nirula, A., Chen, P., Boscia, J., Heller, B., Morris, J., Huhn, G., Cardona, J., Mocherla, B., Stosor, V., Shawa, I., Kumar, P., Adams, A.C., Van Naarden, J., Custer, K.L., Durante, M., Oakley, G., Schade, A.E., Holzer, T.R., Ebert, P.J., Higgs, R.E., Kallewaard, N.L., Sabo, J., Patel, D.R., Klekotka, P., Shen, L., and Skovronsky, D.M., JAMA, 2021, vol. 325, pp. 632–644.
- Pollack, P. and Groothuis, J.R., J. Infect. Chemother., 2002, vol. 8, pp. 201–206.
- Rizza, S.A., Bhatia, R., Zeuli, J., and Temesgen, Z., Drugs Today (Barc.), 2019, vol. 55, pp. 25–34.
- Corti, D., Misasi, J., Mulangu, S., Stanley, D.A., Kanekiyo, M., Wollen, S., Ploquin, A., Doria-Rose, N.A., Staupe, R.P., Bailey, M., Shi, W., Choe, M., Marcus, H., Thompson, E.A., Cagigi, A., Silacci, C., Fernandez-Rodriguez, B., Perez, L., Sallusto, F., Vanzetta, F., Agatic, G., Cameroni, E., Kisalu, N., Gordon, I., Ledgerwood, J.E., Mascola, J.R., Graham, B.S., Muyembe-Tamfun, J.J., Trefry, J.C., Lanzavecchia, A., and Sullivan, N.J., Science, 2016, vol. 351, pp. 1339–1342.
- Sivapalasingam, S., Kamal, M., Slim, R., Hosain, R., Shao, W., Stoltz, R., Yen, J., Pologe, L.G., Cao, Y., Partridge, M., Sumner, G., and Lipsich, L., Lancet Infect. Dis., 2018, vol. 18, pp. 884–893.
- Weinreich, D.M., Sivapalasingam, S., Norton, T., Ali, S., Gao, H., Bhore, R., Musser, B.J., Soo, Y., Rofail, D., Im, J., Perry, C., Pan, C., Hosain, R., Mahmood, A., Davis, J.D., Turner, K.C., Hooper, A.T., Hamilton, J.D., Baum, A., Kyratsous, C.A., Kim, Y., Cook, A., Kampman, W., Kohli, A., Sachdeva, Y., Graber, X., Kowal, B., DiCioccio, T., Stahl, N., Lipsich, L., Braunstein, N., Herman, G., and Yancopoulos, G.D., N. Engl. J. Med., 2021, vol. 384, pp. 238–251.
- Chan, S.K., Rahumatullah, A., Lai, J.Y., and Lim, T.S., Adv. Exp. Med. Biol., 2017, vol. 1053, pp. 35–59.
- Wu, Y., Li, S., Du, L., Wang, C., Zou, P., Hong, B., Yuan, M., Ren, X., Tai, W., Kong, Y., Zhou, C., Lu, L., Zhou, X., Jiang, S., and Ying, T., Emerg. Microbes Infect., 2017, vol. 6, art. ID e89.
- Oh DY, Bang YJ. HER2 - targeted therapies - a role beyond breast cancer. Nat Rev Clin Oncol, 2020; 17:33–48. [CrossRef]
- Tsao LC, Force J, Hartman ZC. Mechanisms of therapeutic anti-tumor monoclonal antibodies. Cancer Res. 2021;81:4641-51. [CrossRef]
- Han B, Chen J, Wang Z, Li X, Wang L, Wu L, et al. Penpulimab in combination with anlotinib as first-line treatment in advanced nonsquamous non-small-cell lung cancer. J Clin Oncol. 2021;39:e21072. [CrossRef]
- Liu C, Liao J, Wu X, Zhao X, Sun S, Wang H, et al. A phase II study of anlotinib combined with etoposide and platinum-based regimens in the first-line treatment of extensive-stage small cell lung cancer. Thorac Cancer. 2022;13:1463-70. [CrossRef]
- Clinicaltrials.gov. A phase I study to investigate the safety, tolerability, pharmacokinetics and antitumor activities of the recombinant human anti-PD-1 monoclonal antibody for subjects with advanced solid tumors. Bethesda:National Library of Medicine;2021. Clinical trial registration NCT03713905.
- Lin S, Gong J, Xu Y, Zhang X, Peng Z, Qi C, et al. Gls-010, a novel anti-PD-1 MAb in Chinese advanced gastrointestinal tumor:result of a phase Ib clinical trial. J Clin Oncol. 2019;37:125. [CrossRef]
- Markham A, Keam SJ. Camrelizumab:first global approval. Drugs. 2019;79:1355-61. [CrossRef]
- Lee A, Keam SJ. Tislelizumab:first approval. Drugs. 2020;80:617-24. [CrossRef]
- . [CrossRef]
- Zhang L, Hao B, Geng Z, Geng Q. Toripalimab:the first domestic anti-tumor PD-1 antibody in China. Front Immunol. 2022;12:730666. [CrossRef]
- Tjulandin S, Demidov L, Moiseyenko V, Protsenko S, Semiglazova T, Odintsova S, et al. Novel PD-1 inhibitor prolgolimab:expanding non-resectable/metastatic melanoma therapy choice. Eur J Cancer. 2021;149:222-32. [CrossRef]
- Manso, T.; Kushwaha, A.; Abdollahi, N.; Duroux, P.; Giudicelli, V.; Kossida, S. Mechanisms of Action of Monoclonal Antibodies in Oncology Integrated in IMGT/MAb-DB. Front. Immunol. 2023, 14, 1129323. [CrossRef]
- Saldarriaga-Valiente, T. Monoclonal Antibodies: Mechanisms of Actions. Diagnostico 2021, 60, 213–217. [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [CrossRef]
- Tsao, L.; Force, J.; Hartman, Z. Mechanisms of Therapeutic Antitumor Monoclonal Antibodies. Cancer Res. 2021, 81, 4641–4651. [CrossRef]
- Rodríguez-Nava, C.; Ortuño-Pineda, C.; Illades-Aguiar, B.; Flores-Alfaro, E.; Leyva-Vázquez, M.; Parra-Rojas, I.; Moral-Hernández, O.; Vences-Velázquez, A.; Cortés-Sarabia, K.; Alarcón-Romero, O. Mechanisms of Action and Limitations of Monoclonal Antibodies and Single Chain Fragment Variable (ScFv) in the Treatment of Cancer. Biomedicines 2023, 11, 1610. [CrossRef]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging New Therapeutic Antibody Derivatives for Cancer Treatment. Nature 2022, 7, 39. [CrossRef]
- Bayer, V. An Overview of Monoclonal Antibodies. Semin. Oncol. Nurs. 2019, 35, 15092. [CrossRef]
- Yip, H.; Papa, A. Signaling Pathways in Cancer: Therapeutic Targets, Combinatorial Treatments, and New Developments. Cells 2021, 10, 659. [CrossRef]
- Tay, M.; Wuihe, K.; Pollara, J. Antibody-Dependent Cellular Phagocytosis in Antiviral Immune Responses. Front. Immunol. 2019, 10, 332. [CrossRef]
- Fishelson, Z.; Kirschfink, M. Complement C5b-9 and Cancer: Mechanisms of Cell Damage, Cancer Counteractions, and Approaches for Intervention. Front. Immunol. 2019, 10, 752. [CrossRef]
- Mezynski, M.; Farrelly, A.; Cremona, M.; Carr, A.; Morgan, C.; Workman, J.; Armstrong, P.; McAuley, J.; Madden, S.; Fay, J.; et al. Targeting the PI3K and MAPK Pathways to Improve Response to HER2-Targeted Therapies in HER2-Positive Gastric Cancer. J. Transl. Med. 2021, 19, 184. [CrossRef]
- Costa, R.; Czerniecki, B. Clinical Development of Immunotherapies for HER2+ Breast Cancer: A Review of HER2-Directed Monoclonal Antibodies and Beyond. NPJ Breast Cancer 2020, 6, 10. [CrossRef]
- Min, H.; Lee, H. Molecular Targeted Therapy for Anticancer Treatment. Exp. Mol. Med. 2022, 54, 1670–1694. [CrossRef]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [CrossRef]
- Baron D. 1990. Industrial production of monoclonal antibodies. Naturwissenschaften 77: 465-471.
- Bartley A, Macleod A J. 1992. A comparative study of monoclonal antibody yield using batch, continuous or perfusion suspension culture techniques. pp. 376-378.
- Goey C H, Bell D, Kontoravdi C. 2019. CHO cell cultures in shake flasks and bioreactors present different host cell protein profiles in the supernatant. Biochem. Eng. J. 144: 185-192. [CrossRef]
- Zheng C, Zhuang C, Chen Y, Fu Q, Qian H, Wang Y. 2017. Improved process robustness, product quality and biological efficacy of an anti-CD52 monoclonal antibody upon pH shift in Chinese hamster ovary cell perfusion culture. Process Biochem. 17: 1359-5113. [CrossRef]
- Jyothilekshmi I, Jayaprakash NS. Trends in Monoclonal Antibody Production Using Various Bioreactor Syst. J Microbiol Biotechnol. 2021 Mar 28;31(3):349-357. [CrossRef]
- Era Jain, Ashok Kumar. 2008. Upstream processes in antibody production: evaluation of critical parameters. Biotechnol. Adv. 26: 46-72. [CrossRef]
- Marya IC, Julio AL, Richard JG. 2001. Solving design equation for a hollow fiber bioreactor with arbitrary kinetics. Chem. Eng. J. 84: 445-461. [CrossRef]
- Lu J, Zhang X, Li J, Yu L, Chen E, Zhu D, et al. 2016. A new fluidized bed bioreactor based on diversion-type microcapsule suspension for bioartificial liver systems. PLoS One 11: e0147376. [CrossRef]
- Nilsang S, Nandakumar KS, Galaev IY, Rakshit SK, Holmdahl R, Mattiason B, et al. 2007. Monoclonal antibody production using a new supermacroporous cryogel perfusion reactor. Biotechnol. Prog. 23: 932-939.
- Irons SL, Chambers AC, Lissina O, King LA, Posse RD. 2018. Protein production using baculovirus expression system. Curr. Prooc. Protein Sci. 91: 1-22. [CrossRef]
- G. Köhler, C. Milstein. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature, 256 (5517) (1975), pp. 495-497. [CrossRef]
- P. Holzlöhner, K. Hanack. Generation of murine monoclonal antibodies by hybridoma technology. JoVE: JoVE, 119 (2017), p. 54832.
- M. Tomita, K. Tsumoto. Hybridoma technologies for antibody production. Immunotherapy, 3 (3) (2011), pp. 371-380. [CrossRef]
- S.A. Smith, J.E. Crowe Jr. Use of human hybridoma technology to isolate human monoclonal antibodies. Microbiol. Spectr., 3 (1) (2015). [CrossRef]
- N. Ohkohchi, H. Itagaki, H. Doi, Y. Taguchi, S. Satomi, S. Satoh. New technique for producing hybridoma by using laser radiation. Laser Surg. Med., 27 (3) (2000), pp. 262-268.
- D.M. Ecker, S.D. Jones, H.L. Levine. The therapeutic monoclonal antibody market. mAbs, 7 (1) (2015), pp. 9-14.
- R.B. Colvin, F.I. Preffer. Laboratory monitoring of therapy with OKT3 and other murine monoclonal antibodies. Clin. Lab. Med., 11 (3) (1991), pp. 693-714. [CrossRef]
- N.R. Gonzales, R. De Pascalis, J. Schlom, S.V. Kashmiri. Minimizing the immunogenicity of antibodies for clinical application. Tumour Biol.: J. Int. Soc. Oncodevelop. Biol. Med., 26 (1) (2005), pp. 31-43. [CrossRef]
- S.A. Smith, J.E. Crowe Jr. Use of human hybridoma technology to isolate human monoclonal antibodies. Microbiol. Spectr., 3 (1) (2015). [CrossRef]
- P.T. Jones, P.H. Dear, J. Foote, M.S. Neuberger, G. Winter. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature, 321 (6069) (1986), pp. 522-525. [CrossRef]
- A.P. Kim, D.E. Baker. Daclizumab. Hospital Pharm., 51 (11) (2016), pp. 928-939.
- Jakobovits, R.G. Amado, X. Yang, L. Roskos, G. Schwab. From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat. Biotechnol., 25 (10) (2007), pp. 1134-1143. [CrossRef]
- Ganguly S, Wakchaure R. Hybridoma technology: a brief review on its diagnostic and clinical significance. Pharmaceut Biol Eval, 3 (issue 6) (2016), pp. 554-555.
- Buck DW, Larrick JW, Raubitschek A, Truitt KE, Senyk G, Wang J, Dyer B(1984) Production of human monoclonal antibodies. In. Kennett RH, Bechtol KB and McKearn TJ (ed) Monoclonal Antibodies and Functional Cell Lines. Progress and Applications. New York: Plenum Press; pp 275-309.
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature, 256 (1975), pp. 495-497. [CrossRef]
- Luckenbach GA. Some recent aspect on hybridoma technology. Mayr WR (Ed.), Advances in Forensic Haemogenetics. Advances in Forensic Haemogenetics, Springer, Berlin, Heidelberg (1988).
- J.Y. Kim, Y.G. Kim, G.M. Lee. CHO cells in biotechnology for production of recombinant proteins: current state and further potential. Appl. Microbiol. Biotechnol., 93 (3) (2012), pp. 917-930. [CrossRef]
- M.K. Gorny, J.Y. Xu, S. Karwowska, A. Buchbinder, S. Zolla-Pazner. Repertoire of neutralizing human monoclonal antibodies specific for the V3 domain of HIV-1 gp120. J. Immunol., 150 (2) (1993), pp. 635-643. [CrossRef]
- J. Li, T. Sai, M. Berger, Q. Chao, D. Davidson, G. Deshmukh, B. Drozdowski, W. Ebel, S. Harley, M. Henry, et al. Human antibodies for immunotherapy development generated via a human B cell hybridoma technology. Proc. National Acad. Sci. USA, 103 (10) (2006), pp. 3557-3562. [CrossRef]
- K.H. Ching, E.J. Collarini, Y.N. Abdiche, D. Bedinger, D. Pedersen, S. Izquierdo, R. Harriman, L. Zhu, R.J. Etches, M.C. van de Lavoir, et al. Chickens with humanized immunoglobulin genes generate antibodies with high affinity and broad epitope coverage to conserved targets. MAbs, 10 (1) (2018), pp. 71-80. [CrossRef]
- G. Köhler, C. Milstein. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature, 256 (5517) (1975), pp. 495-497. [CrossRef]
- J.S. Babcook, K.B. Leslie, O.A. Olsen, R.A. Salmon, J.W. Schrader. A novel strategy for generating monoclonal antibodies from single, isolated lymphocytes producing antibodies of defined specificities. Proc. Natl. Acad. Sci. U. S. A, 93 (15) (1996), pp. 7843-7848. [CrossRef]
- T. Tiller, E. Meffre, S. Yurasov, M. Tsuiji, M.C. Nussenzweig, H. Wardemann. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods, 329 (1–2) (2008), pp. 112-124. [CrossRef]
- J.F. Scheid, H. Mouquet, N. Feldhahn, B.D. Walker, F. Pereyra, E. Cutrell, M.S. Seaman, J.R. Mascola, R.T. Wyatt, H. Wardemann, M.C. Nussenzweig. A method for identification of HIV gp140 binding memory B cells in human blood. J. Immunol. Methods, 343 (2) (2009), pp. 65-67.
- N.D. Durham, A. Agrawal, E. Waltari, D. Croote, F. Zanini, M. Fouch, E. Davidson, O. Smith, E. Carabajal, J.E. Pak, B.J. Doranz, M. Robinson, A.M. Sanz, L.L. Albornoz, F. Rosso, S. Einav, S.R. Quake, K.M. McCutcheon, L. Goo. Broadly neutralizing human antibodies against dengue virus identified by single B cell transcriptomics. eLife, 8 (2019), Article e52384.
- Y. Cao, B. Su, X. Guo, W. Sun, Y. Deng, L. Bao, Q. Zhu, X. Zhang, Y. Zheng, C. Geng, X. Chai, R. He, X. Li, Q. Lv, H. Zhu, W. Deng, Y. Xu, Y. Wang, L. Qiao, Y. Tan, X.S. Xie. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell, 182 (1) (2020), pp. 73-84.
- W. Meng, L. Li, W. Xiong, X. Fan, H. Deng, A.J. Bett, Z. Chen, A. Tang, K.S. Cox, J.G. Joyce, D.C. Freed, E. Thoryk, T.M. Fu, D.R. Casimiro, N. Zhang, K. A Vora, Z. An. Efficient generation of monoclonal antibodies from single rhesus macaque antibody secreting cells. mAbs, 7 (4) (2015), pp. 707-718. [CrossRef]
- S. Rajan, M.R. Kierny, A. Mercer, J. Wu, A. Tovchigrechko, H. Wu, W.F. Dall Acqua, X. Xiao, P.S. Chowdhury. Recombinant human B cell repertoires enable screening for rare, specific, and natively paired antibodies. Communications Biol., 1 (2018), p. 5. [CrossRef]
- D.O. Starkie, J.E. Compson, S. Rapecki, D.J. Lightwood. Generation of recombinant monoclonal antibodies from immunised mice and rabbits via flow cytometry and sorting of antigen-specific IgG+ memory B cells. PloS One, 11 (3) (2016), Article e0152282. [CrossRef]
- L. Lei, K. Tran, Y. Wang, J.J. Steinhardt, Y. Xiao, C.I. Chiang, R.T. Wyatt, Y. Li. Antigen-specific single B cell sorting and monoclonal antibody cloning in Guinea pigs. Front. Microbiol., 10 (2019), p. 672. [CrossRef]
- A.S. Adler, R.A. Mizrahi, M.J. Spindler, M.S. Adams, M.A. Asensio, R.C. Edgar, J. Leong, R. Leong, L. Roalfe, R. White, D. Goldblatt, D.S. Johnson. Rare, high-affinity anti-pathogen antibodies from human repertoires, discovered using microfluidics and molecular genomics. mAbs, 9 (8) (2017), pp. 1282-1296. [CrossRef]
- W. Meng, L. Li, W. Xiong, X. Fan, H. Deng, A.J. Bett, Z. Chen, A. Tang, K.S. Cox, J.G. Joyce, D.C. Freed, E. Thoryk, T.M. Fu, D.R. Casimiro, N. Zhang, K. A Vora, Z. An. Efficient generation of monoclonal antibodies from single rhesus macaque antibody secreting cells. mAbs, 7 (4) (2015), pp. 707-718. [CrossRef]
- S. Rajan, M.R. Kierny, A. Mercer, J. Wu, A. Tovchigrechko, H. Wu, W.F. Dall Acqua, X. Xiao, P.S. Chowdhury. Recombinant human B cell repertoires enable screening for rare, specific, and natively paired antibodies. Communications Biol., 1 (2018), p. 5. [CrossRef]
- T. Ojima-Kato, D. Hashimura, T. Kojima, S. Minabe, H. Nakano. In vitro generation of rabbit anti-Listeria monocytogenes monoclonal antibody using single cell based RT-PCR linked cell-free expression systems. J. Immunol. Methods, 427 (2015), pp. 58-65. [CrossRef]
- Z. Shi, Q. Zhang, H. Yan, Y. Yang, P. Wang, Y. Zhang, Z. Deng, M. Yu, W. Zhou, Q. Wang, X. Yang, X. Mo, C. Zhang, J. Huang, H. Dai, B. Sun, Y. Zhao, L. Zhang, Y.G. Yang, X. Qiu. More than one antibody of individual B cells revealed by single-cell immune profiling. Cell Discov., 5 (1) (2019), p. 64. [CrossRef]
- G. Fermin, S. Rampersad, P. Tennant. Viruses as tools of biotechnology: therapeutic agents, carriers of therapeutic agents and genes, nanomaterials, and more. Viruses: Mol. Biol. Host Interact. Appl. Biotechnol. (2018), pp. 291-316.
- R. Reljic, S. George, V. Greiff, F. Ferreira, A.M. Hashem, M.A. Alfaleh, H.O. Alsaab, A.B. Mahmoud, A.A. Alkayyal, M.L. Jones, S.M. Mahler. Phage display derived monoclonal antibodies: from bench to bedside. Front. Immunol., 1 (2020). [CrossRef]
- R. Kumar, H.A. Parray, T. Shrivastava, S. Sinha, K. Luthra. Phage display antibody libraries: a robust approach for generation of recombinant human monoclonal antibodies. Int. J. Biol. Macromol., 135 (2019), pp. 907-918. [CrossRef]
- R.V. Goering, H.M. Dockrell, M. Zuckerman, P.L. Chiodini, Active, passive and adoptive immunotherapy, in: Mims’ Medical Microbiology and Immunology, 6th ed., 2019, pp. 505–510.
- Frenzel, T. Schirrmann, M. Hust. Phage display-derived human antibodies in clinical development and therapy. MAbs, 8 (7) (2016), pp. 1177-1194. [CrossRef]
- N.V. Tikunova, V.V. Morozova. Phage display on the base of filamentous bacteriophages: application for recombinant antibodies selection. Acta naturae, 1 (3) (2009), pp. 20-28. [CrossRef]
- C.F. Barbas 3rd, A.S. Kang, R.A. Lerner, S.J. Benkovic. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc. Natl. Acad. Sci. U. S. A, 88 (18) (1991), pp. 7978-7982.
- H. Lowman. Phage Display for Protein Binding. (2013).
- J.C. Almagro, M. Pedraza-Escalona, H.I. Arrieta, S.M. Pérez-Tapia. Phage display libraries for antibody therapeutic discovery and development. Antibodies, 8 (3) (2019), p. 44. [CrossRef]
- C.H. Wu, I.J. Liu, R.M. Lu, H.C. Wu. Advancement and applications of peptide phage display technology in biomedical science. J. Biomed. Sci., 23 (2016), p. 8. [CrossRef]
- Karlsson, R., Pol, E., & Frostell, Å. (2016). Comparison of surface plasmon resonance binding curves for characterization of protein interactions and analysis of screening data. Analytical Biochemistry, 502, 53–63. [CrossRef]
- Cohen, N., Sabhachandani, P., Golberg, A., & Konry, T. (2015). Approaching near real-time biosensing: Microfluidic microsphere based biosensor for real-time analyte detection. Biosensors, & Bioelectronics, 66, 454–460. [CrossRef]
- Dippong, M., Carl, P., Lenz, C., Schenk, J. A., Hoffmann, K., Schwaar, T., Schneider, R. J., & Kuhne, M. (2017). Hapten-specific single-cell selection of hybridoma clones by fluorescence-activated cell sorting for the generation of monoclonal antibodies. Analytical Chemistry, 89(7), 4007–4012. [CrossRef]
- Debs, B. E., Utharala, R., Balyasnikova, I. V., Griffiths, A. D., & Merten, C. A. (2012). Functional single-cell hybridoma screening using droplet-based microfluidics. Proceedings of the National Academy of Sciences of the United States of America, 109(29), 11570–11575.
- Brouzes, E., Medkova, M., Savenelli, N., Marran, D., Twardowski, M., Hutchison, J. B., Rothberg, J. M., Link, D. R., Perrimon, N., & Samuels, M. L. (2009). Droplet microfluidic technology for single-cell high-throughput screening. Proceedings of the National Academy of Sciences of the United States of America, 106(34), 14195–14200.
- Lomonossoff GP, D’Aoust MA. Plant-produced biopharmaceuticals: A case of technical developments driving clinical deployment. Science. 2016;353(6305):1237-40. [CrossRef]
- Donini M, Marusic C. Current state-of-the-art in plant-based antibody production systems. Biotechnol Lett. 2019;41(3):335-46. [CrossRef]
- Walsh G. New Biopharmaceuticals: a review of new biologic drug approvals over the years, featuring highlights from 2010 and 2011. Biopharm (Int 2012, 256, June), P. 34. Gale OneFile: Health and Medicine.
- . [CrossRef]
- Gregorio NE, Levine MZ, Oza JP. A user’s guide to cell-free protein synthesis. Methods Protoc. 2019;2:24. [CrossRef]
- Li H, Er Saw P, Song E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol Immunol. 2020;17(5):451–61. [CrossRef]
- Stumpp MT, Dawson KM, Binz HK. Beyond antibodies: the DARPin® drug platform. BioDrugs. 2020;34(4):423–33. [CrossRef]
- Hartley JA. Antibody-drug conjugates (ADCs) delivering pyrrolobenzodiazepine (PBD) dimers for cancer therapy. Expert Opin Biol Ther. 2021;21(7):931–43. [CrossRef]
- Bathula, N. V., Bommadevara, H., & Hayes, J. M. (2020). Nanobodies: The future of antibody-based immune therapeutics. Cancer Biotherapy and Radiopharmaceuticals. [CrossRef]
- Frenzel, T. Schirrmann, M. Hust. Phage display-derived human antibodies in clinical development and therapy. mAbs, 8 (7) (2016), pp. 1177-1194. [CrossRef]
- J.C. Almagro, M. Pedraza-Escalona, H.I. Arrieta, S.M. Pérez-Tapia. Phage display libraries for antibody therapeutic discovery and development. Antibodies, 8 (3) (2019), p. 44. [CrossRef]
- C. Hentrich, F. Ylera, C. Frisch, A. Ten Haaf, A. Knappik. Monoclonal antibody generation by phage display: history, state-of-the-art, and future. Handbook of Immunoassay Technologies, Academic Press (2018), pp. 47-80.
- G.R. Burmester, R. Panaccione, K.B. Gordon, M.J. McIlraith, A.P. Lacerda. Adalimumab: long-term safety in 23 458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn’s disease. Ann. Rheum. Dis., 72 (4) (2013), pp. 517-524. [CrossRef]
- K.P. Machold, J.S. Smolen. Adalimumab - a new TNF-alpha antibody for treatment of inflammatory joint disease. Expet Opin. Biol. Ther., 3 (2) (2003), pp. 351-360.
- P.J. Hudson, C. Souriau. Engineered antibodies. Nat. Med., 9 (1) (2003), pp. 129-134.
- Kunamneni, C. Ogaugwu, S. Bradfute, R. Durvasula. Ribosome display technology: applications in disease diagnosis and control. Antibodies, 9 (3) (2020), p. 28. [CrossRef]
- Doerner, L. Rhiel, S. Zielonka, H. Kolmar. Therapeutic antibody engineering by high efficiency cell screening. FEBS Lett., 588 (2) (2014), pp. 278-287. [CrossRef]
- J. Zhu, D. Hatton. New mammalian expression systems. New Bioprocessing Strategies: Development and Manufacturing of Recombinant Antibodies and Proteins, Springer, Cham (2017), pp. 9-50.
- Saggy, Y. Wine, L. Shefet-Carasso, L. Nahary, G. Georgiou, I. Benhar. Antibody isolation from immunized animals: comparison of phage display and antibody discovery via V gene repertoire mining. Protein Eng. Des. Sel.: PEDS, 25 (10) (2012), pp. 539-549. [CrossRef]
- . [CrossRef]
- Kemna, E. W., Wolbers, F., Vermes, I., & van den Berg, A. (2011). On chip electrofusion of single human B cells and mouse myeloma cells for efficient hybridoma generation. Electrophoresis, 32(22), 3138–3146. [CrossRef]
- Schoeman, R. M., Kemna, E. W., Wolbers, F., & van den Berg, A. (2014). High-throughput deterministic single-cell encapsulation and droplet pairing, fusion, and shrinkage in a single microfluidic device. Electrophoresis, 35(2-3), 385–392.
- Joensson, H. N., Zhang, C., Uhlen, M., & Andersson-Svahn, H. (2012). A homogeneous assay for protein analysis in droplets by fluorescence polarization. Electrophoresis, 33(3), 436–439. [CrossRef]
- Singhal, A., Haynes, C. A., & Hansen, C. L. (2010). Microfluidic measurement of antibody antigen binding kinetics from low-abundance samples and single cells. Analytical Chemistry, 82(20), 8671–8679. [CrossRef]
Table 2.
mAb drugs approved for therapy of tumor and their mechanisms.
| mAbs | Target | Antibody variant | Type | Mechanism of action |
| Penpulimab | PD-1/tumor | IgG1 | Humanized | Blocks PD-1 binding, eliminating FcγR binding and completely preventing ADCC/ADCP [32,33]. |
| Zinberelimab | PD-1/tumor | IgG4 | Humanized | Binds to and inhibits PD-1, restoring immune function by activating T-cell responses against tumor cells [34,35]. |
| Camrelizumab | PD-1/tumor | IgG4-k | Humanized | PD-1 receptors on T cells bind to their ligands PD-L1 and PD-L2, inhibiting T cell proliferation and cytokine production [36]. |
| Tislelizumab | PD-1/tumor | IgG4 | Humanized | The interaction between PD-1 and its ligands, PD-L1 and PD-L2, leads to T-cell exhaustion, weakening the immune response against cancer [37]. |
| Sintilimab | PD-1/tumor | IgG4 | Humanized | Blocks the interaction of PD-1 with its ligands (PD-L1 and PD-L2), thereby restoring the endogenous T-cell antitumor response [38]. |
| Toripalimab | PD-1/tumor | IgG4 | Humanized | Binds to PD-1, preventing its interaction with programmed death ligands PD-L1 and PD-L2 [39]. |
| Prolgolimab (Forteca) | PD-1/tumor | IgG1 | Chimeric | Exhibits significant antitumor activity and maintains a manageable safety profile in patients with advanced melanoma [40]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



