
Submitted:
28 May 2024
Posted:
29 May 2024
You are already at the latest version
Abstract
Type2 diabetes(T2D) is a pervasive modern disease, classified as a metabolic disorder marked by hyperglycemia, hyperinsulinemia, and life-long threatening cardiovascular morbidities. Subclinical inflammation stands as the primary pathogenetic factor propelling T2D progression, correlated with heightened levels of proinflammatory cytokines. Immune cells serve as the prominent sources of cytokine production, and under diabetic conditions, they tend toward a proinflammatory phenotype, fostering chronic inflammation. This review zeroes in on the disrupted homeostasis characteristic of diabetic conditions, encompassing cytokines, hormones, and their interplay with altered immune function, all contributing to the onset of diabetic cardiovascular complications.
Keywords:
Type2 diabetes
; cardiovascular complications
; cytokines
; insulin
; adiponectins
; immune disorder
1. Introduction
T2D is initially characterized by prolonged elevated blood glucose and often co-exists with a cluster of clinical features known as metabolic syndrome[10]. These features encompass central obesity, hyperlipidemia, hyperglycemia, and hyperinsulinemia[11]. According to latest estimates by the International Diabetes Federation, approximately 463 million individuals worldwide, with 31 million adults in the United States alone, are living with diabetes in 2019, with T2D accounting for about 90% cases[12]. Furthermore, individuals with T2D face a 2 to 4-fold higher risk of developing cardiovascular diseases (CVDs), which include coronary heart disease, cerebrovascular disease, peripheral artery disease, and aortic atherosclerosis[12]. Among T2D patients, 40-50% exhibit chronic kidney disease(stage1-4) and 80.5% had hypertension[13,14,15,16]. These conditions and diseases collectively contributed to the occurrence of heart failure and hemorrhagic stroke, which stand as the leading cause of mortality in individuals with type 2 diabetes[17,18].
The escalating prevalence of T2D is closely linked to the rise in rates of obesity, primarily fueled by overnutrition and sedentary lifestyles[19,20]. Obesity serves as a significant predictive risk factor for T2D development, highlighting the critical role of lifestyle factors in disease prevention and management [21]. Additionally, hyperglycemia emerges as a hallmark clinical feature of T2D, triggering protein hyper-glycosylation and enhanced oxidative stress, which in turn leads to cellular and systemic dysfunction, contributing to the detrimental physiological outcomes of T2D [22,23]. The relationship between T2D, metabolic syndrome, and cardiovascular diseases underscores the urgent need for comprehensive preventive measures and therapeutic interventions targeting lifestyle modifications, glycemic control, and cardiovascular risk reduction strategies [24,25,26,27]. Understanding the intricate interplay between these conditions is essential for addressing the multifaceted challenges of T2D and its associated complications[28,29,30].
2. Effects of Cytokines (IFNγ, TNFα, IL17) on the Development of T2D and Its Related Cardiovascular Complications
Cytokines are professional intercellular communicating tools employed by immune cells. They can communicate with distant immune cells to elicit systematic effects, or they can serve as paracrine and autocrine factors to orchestrate localized inflammation[31]. Interferon gamma (IFNγ), renowned for its diverse roles in immune regulation, extends its influence beyond traditional immunity, impacting the development of hypertension and contributing significantly to T2D pathogenesis [32]. In T2D, IFNγ stimulated pro-inflammatory macrophage accumulation in the adipose tissue (AT), and mice deficient in IFNγ had improved the metabolic signatures and decreased the number of AT-macrophages in the HFD (high-fat diet) mice [33]. Additionally, IFNγ-neutralizing antibody improved endothelial dysfunction in Db/Db mice by curtailing superoxide production[34]. Furthermore, The atherogenic role of IFNγ was evidenced by IFNγ deficient ApoE-KO mice which exhibited a 50% reduction of the lesion size and lipid accumulation [35]. Conversely, ApoE-KO mice injected with IFNγ showed a 2-fold increase in lesion size and T cells infiltration compared to vehicle injection[36]. In vitro mechanistic studies reveal that IFNγ enhances oxidative stress, activates endothelial cells and macrophages into pro-inflammatory state, contribute to metabolic disturbance[37]. Moreover, our research indicates that IFNγ as a hypertensive factor by facilitating the formation of immunological synapses between CD8+ T cells and renal tubular cells, leading to increased sodium retention and hypertension in a classic salt-sensitive hypertension model[38]. Given the co-existence of significant elevation in blood pressure and IFNγ in T2D patients[16,39], further studies are needed to decipher whether IFNγ is the “culprit” that contributes to the progression of T2D-induced cardiovascular disorders.
Tumor necrosis factor alpha (TNFα) was the first ‘adipokine’ discovered in 1980th with multiple functions in both innate and adaptive immunity[40]. Later, it was clarified that TNFα is majorly produced by immune cells such as macrophages [41], and T cells[42]. Elevated level of TNFα was reported in adipose tissue and systemic circulation in obesity and diabetes, contributing to the development of insulin resistance[41,43]. Mechanistic studies have illuminated its role in phosphorylating IRS1 (insulin receptor binding substrate), converting it into an inhibitor of insulin receptor signaling, leading to insulin resistance in metabolic tissue[44]. In addition to its contributing role in insulin resistance, it has been implicated in T2D-related vascular morbidities[45]. TNFα deficiency curtailed atherosclerotic lesion formation, possibly through attenuating macrophage foam cell formation and deposition in the arterial wall intima[46,47,48,49].Anti-TNFα antibody treatment restored the impaired coronary artery dilation in Db/Db mice through dampening NFkB (nuclear factor kappa B) mediated proinflammatory signaling[50]. Additional studies showed that Anti-TNFα ameliorated hypertension induced by a high-fat high-fructose diet in rat[51]. Subsequent mechanistic studies revealed that TNFα impaired the production of NO (nitro oxide) production and increased superoxide generation through NADPH oxidase in various vascular beds [48,52,53]. Moreover, elevated TNFα levels are associated with increased immune cells infiltration in kidney specimens from diabetic patients with renal disorders [54,55,56]. Prospective investigations into TNFα signaling’s involvement in T cell-mediated chronic inflammation in diabetic conditions may unveil novel therapeutic avenues for T2D patients with chronic kidney disease.
Another immune cell-derived cytokine that attracts our attention is interleukin 17(IL-17), which modulates immune response by cooperating with other cytokines such as TNFα to amplify the pro-inflammatory signaling in both immune and nonimmune cells[57]. IL-17 family comprises six members (IL17-A to F), and for our purpose, IL-17 refers to IL-17A here. IL-17 also called cytotoxic T-lymphocyte-associated antigen 8 (CTLA-8), is a well-recognized factor to drive an inflammatory cascade through binding to IL-17 receptors[58]. IL-17 is mainly produced from the Th17 cells-subset of CD4+ T cells, however, other immune cells including CD8+ T cells, γδ T cells, and macrophages can produce it and are referred to as type 17 cells[59]. Clinically, both obesity and T2D patients have elevated plasma levels of IL-17 and an increased number of circulating Th17 cells[60,61]. Interestingly, the level of IL-17 in T2D patients with complications was higher than the patients without[62]. The direct evidence of IL-17 as a pathogenic factor of diabetes was provided by Dr. Ohshima, who showed that anti-IL-17 antibodies significantly improved glucose tolerance in KK-Ay mice [63]. Later Jin reported that IL17-deficient mice were protected against diabetic renal fibrosis, glomerular injury, and albuminuria, and the administration of anti-IL-17 antibodies to WT diabetic mice showed similar results[64]. In vitro mechanistic studies revealed IL-17 and hyperglycemia synergistically upregulated the expression of proinflammatory and pro-fibrotic genes including IL-6, TNFα and TGFβ in the renal epithelial cells [64]. Beyond its role in precipitating diabetic nephropathy, IL-17 signaling was atherogenic, which was supported by smaller lesion formation in both anti-IL17 Adenovirus transfected ApoE-KO mice[65] and IL-17 deficient LdlR(low density lipoprotein receptor)-KO mice under western diet treatment[66]. Further mechanistic studies found IL-17 stimulated the production of macrophage-derived pro-inflammatory cytokines (TNFα, IL6, IL1β) and facilitated macrophage infiltration into the adipose tissue and vasculature[67,68,69]. Moreover, IL17 infusion is associated with increased cardiac fibrosis and collagen deposition in the left ventricle[70]. It is indispensable for AngII-induced blood pressure elevation by activation of renal sodium transporters[71,72]. Based on all the previous reports, delineating IL17’s role in diabetes will open avenues for alternative therapeutic targets for T2D patients with cardiovascular complications.
Collectively, cytokines serve as critical mediators in both the induction and progression of T2D and its associated cardiovascular complications. Understanding their complex roles and interactions is essential for developing targeted therapeutic interventions to address these multifaceted pathologies. Further research is needed to elucidate the intricate mechanisms underlying cytokine involvement in T2D and cardiovascular diseases, ultimately improving outcomes for individuals affected by these conditions[73,74].
3. Insulin and Altered Immune Response in the Development of T2D and CVDs
Insulin, an important hormone developed during evolution, plays a central role in storing nutrients within our body cells to mitigate food scarcity [75]. Upon the breakdown of food into small molecules such as glucose by the digestive system, plasma glucose levels reach a threshold that triggers insulin release from pancreatic β-cells[76]. Released insulin exerts its effects by binding to Insulin receptors (InsR) on the target cells[77]. Its primary anabolic effect includes promoting glycogen and protein synthesis while inhibiting lipolysis in metabolic tissue such as the liver, skeletal muscle, and adipose tissue[78]. In times of nutrient surplus, beta cells release high amounts of insulin to store glucose as glycogen in our body[79]. However, prolonged nutrient surplus eventually results in the failure of insulin signaling in major metabolic tissues, which leads to systematic hyperglycemia and hyperinsulinemia in obesity-T2D patients[80].
In addition to the anabolic effect of insulin, the Insulin/Insulin receptor (InsR) is crucial in regulating glucose availability inside the cell. This phenomenon was elucidated in immune cells through the Radio labeled Insulin technique in the 1970s[81,82]. While naïve T cells initially display relatively low expression of InsR, their membrane InsR levels increase following T cell priming or activation[82].This upregulation can stimulate their proliferation and enhance the production of IFNγ[83,84]. Conversely, CD4+ T cells deficient in InsR exhibited reduced IFNγ and proliferation upon antigen stimulation. Furthermore, exogenous insulin boosted ECAR and OCR which indicated increased ATP production to support the demands for T cell’s activation and effector function. These in vitro findings suggest that insulin/InsR signaling induces T cell activation with heightened production of pro-inflammatory cytokines[85]. Similarly, an experiment utilizing a mouse colitis model demonstrated that InsR-deficient T cells were less effective in inducing chronic colonic inflammation[85].
Besides the effects in T cells, the expression of InsR was also observed in tumor-derived macrophage cells [86]. Insulin amplified LPS-mediated cytokines production (TNFα and IL6) in bone marrow-derived macrophages (BMDM) and human monocytes [87,88]. Chronic insulin treatment was found to enhance macrophage migration towards inflammatory chemokines (MCP-1) from adipose tissue[89]. Moreover, a myeloid lineage-specific InsR-/- mice(MphIRKO) exhibited reduced atherosclerotic lesion size in the descending aorta compared to control mice, indicating that Insulin/InsR signaling in macrophage promotes obesity-induced atherosclerotic lesion formation[90].
Despite its atherogenic effect, chronic insulin administration was found to increase mean arterial blood pressure in animal models[91,92,93]. And intravenous insulin infusion in healthy volunteers led to a 50% reduction in urinary sodium excretion[94]. Given that sodium reabsorption is paramount to regulating blood volume and pressure[95], chronic hyperinsulinemia-mediated sodium retention may contribute to the development of hypertension in T2D patients. Additionally, recent clinical studies reported that insulin prescription in T2D patients was associated with a higher risk of all-cause mortality [96,97]. Therefore, further mechanistic study on uncovering the detrimental effects of insulin in T2D-induced cardiovascular complications is desired, offering valuable insights for designing appropriate treatments for T2D patients.
4. Association of Adipocyte Derived Hormone, the Immune System, and T2D-CVDs
Adipose tissue serves as the site of triglyceride storage and free fat acid release in response to food availability[98]. Moreover, it functions as an endocrine organ, actively regulating circulating glucose/insulin homeostasis[99]. Increased adipose tissue mass indicates a heightened risk of metabolic disorders such as T2D and its associated complications[100]. Apart from adipocytes, the predominant cell type in AT is macrophage, constituting up to 40% of the tissue[101,102]. The function of AT is intricately regulated by localized cell-cell interactions, which can be dysregulated during obesity[103]. Notably, AT resident M1 macrophage play a crucial role in sustaining the pro-inflammatory state, contributing to hyperglycemia, hyperlipidemia, and a cluster of metabolic disorders[104,105]. In addition to cytokines, Adipokines are bioactive factors secreted from AT, acting either as endocrine hormones to regulate distant organs or as paracrine factors affecting localized tissue/cells[106]. Among these, adiponectin and leptin are the two significant adipokines.
Adiponectin, the most abundant adipokine in plasma, negatively correlated with T2D and insulin resistance[107]. It exists as three oligomeric complexes: trimer, hexamer, and multimer, with the latter known as high molecular weight (HMW) adiponectin[108]. HMW adiponectin can be cleaved by leukocyte elastase to the globular adiponectin, the most bioactive isoform[109,110]. Adiponectin exerts biological effects by binding with ADR1, ADR2, or T cadherin[111,112]. Following its discovery in 1995, experiments injecting purified adiponectin into WT mice revealed a significant reduction of blood glucose within two hours, observed both in WT and Ob/Ob diabetic mice[113,114]. Similar results replicated in other independent studies, demonstrating that continuous infusion of low-dose recombinant adiponectin mitigated hyperglycemia and hyperinsulinemia by a high-fat diet (HFD) in WT mice[107]. Conversely, adiponectin KO mice exhibited severe insulin resistance and elevated serum TNFα following a two-week HFD challenge[115]. These findings collectively suggest that reduced adiponectin levels contribute to the pathogenesis of metabolic disorders in T2D.
Low levels of adiponectin may also be associated with altered immune function in T2D patients, perpetuating chronic inflammation and exacerbating T2D-related cardiovascular complications[116]. Researchers using transgenic mice (ADR1-TG) with adiponectin overexpression in macrophages found that these mice had lower body weight despite no difference in food consumption, indicating adiponectin metabolic stimulatory effects through enhanced utilization of glucose and fatty acids in various tissues. Furthermore, ADR1-TG/LdlR mice showed reduced atherosclerotic lesion size and foam cell infiltration following an HFD challenge[117]. Similarly, the protective effect of adiponectin against atherosclerotic lesion formation was observed in gAd-TG/ApoE-KO mice, characterized by the over-expression of globular adiponectin[118]. Mechanistic studies proposed that adiponectin reduced levels of TNFα and IL-6, consequently mitigating the development of macrovascular morbidities[115,119]. Additionally, by activating PPARγ, adiponectin reduces oxLDL uptake in macrophages and diminishes foam cell formation, highlighting its anti-atherosclerotic effect[120]. Recently, it has been reported that adiponectin as an immunosuppressor, negatively regulate T cell activation and production of inflammatory cytokines-IFNγ and IL-17[121,122]. Further investigation is imperative to explicit whether the deficiency of adiponectin in diabetic conditions unleashes an inflammatory response that exacerbates the progression of T2D-related cardiovascular complications.
Leptin, an adipocytokine, is encoded by obese (Ob) gene and produced by adipose tissue[123]. The cloning and identification of the leptin gene occurred in 1995[124,125]. However, the remarkable phenotypes of severe obesity and hyperphagia observed in leptin knock-out (Ob/Ob) mice were recognized much earlier in the 1950s[126]. The anorexigenic(appetite-suppressing) function of leptin primarily occurs through its binding to the leptin receptor (LepR), which is abundantly expressed in the hypothalamus[127,128]. Elevated circulating leptin levels have been associated with a higher prevalence of obesity and T2D [129,130,131]. Additionally, hypertensive, overweight females have been reported to exhibit higher plasma levels of leptin than their normotensive counterparts[132,133]. Those clinical observations have spurred significant interest in uncovering the pathogenic role of leptin in accentuating T2D-induced CVDs.
Given that atherosclerosis is a leading cause of CVDs, researchers have examined the effects of leptin on atherosclerosis in the classic atherosclerotic-prone mouse model. Studies demonstrated that leptin-deficient ApoE-KO mice develop smaller atherosclerotic areas compared to leptin-intact ApoE-KO mice on an atherogenic diet, and exogenous leptin administration increases the atherosclerotic area in ApoE-KO mice[134]. Similar findings were reported by other researchers utilizing different atherosclerotic models [134,135,136,137]. One mechanistic study reported that transferring Tregs isolated from Ob/Ob mice, which exhibit a significant reduction in IFNγ production to ApoE/Rag2-KO mice resulted in a marked decrease in lesion size compared to those receiving WT Tregs[136]. Moreover, studies have consistently shown that leptin stimulates T cell proliferation, IFNγ production, glucose uptake, and aerobic glycolysis[138,139,140,141,142,143]. Investigating whether leptin-immune activation contributes to the onset of T2D, and its associated CVDs represents a promising new area for research.
5. Summary
In this review, we highlighted significant discoveries from the past century that underscore the pivotal role of immune cell-derived cytokines in perpetuating the progression of T2D and its associated CVDs [74]. Specifically, cytokines such as IFNγ, TNFα and IL-17, released from immune cells, have been identified as critical pathogenic factors in T2D and T2D-induced vascular morbidities[144]. Substantial evidence collectively supports this notion from studies utilizing specific cytokine-deficient ApoE-KO mice, all exhibiting decreased formation of microvascular morbidities[35,46,69,145].
Given that obesity-T2D represents a metabolic disorder characterized by disturbed hormone balance, recent reports have shed light on the roles of insulin, adiponectin, and leptin in immune activation, which may contribute to the accelerated progression of T2D and subsequent CVDs. [146,147,148]. Insulin and leptin have been identified as pro-atherogenic agents, stimulating pro-inflammatory phenotype of both T cells and macrophages[142,149]. Conversely, adiponectin is an immune suppressor, limiting T cell activation and IFNγ production[122,150].
Taken together, diabetic conditions characterized by elevated circulating insulin and leptin levels and reduced adiponectin levels create an environment conducive to producing immune-derived pro-inflammatory cytokines (IFNγ, TNFα and IL-17). This chronic inflammation sustains and exacerbates the progression of cardiovascular complications, including atherosclerosis, hypertension, chronic kidney diseases.
6. Perspective
The “Thrift gene hypothesis” and extensive research have established that the central roles of insulin, leptin and adiponectin in maintaining adequate energy storage, reducing energy intake, and enhancing glucose and fatty acid metabolism, respectively[78,127,151]. These mechanisms contribute to the successful survival of metazoans. However, over the last decades, overnutrition resulting from highly palatable foods and sedentary lifestyles has led to chronic dysregulation of insulin and leptin secretion. This dysregulation may serve as a trigger for immune activation and subsequent release of pro-inflammatory cytokines [152,153].
Cytokines, crucial messengers employed by immune system, plays a key role in this process. Excessive production of pro-inflammatory cytokines such as IFNγ from T cells can recruit circulating monocytes to adipose tissue or other tissues like vasculature, leading to localized inflammation[154,155]. This localized inflammation contributes to the widespread tissue-specific damage under diabetic conditions[28,156,157,158].
During evolution, our body have developed a brake system, including anti-inflammatory cytokines and Tregs cells, to resolve the inflammation. Future investigations focusing on suppressing inappropriate activation of immune cells by activating our self-anti-inflammatory mechanism hold promise for developing therapies for diabetic patients[159,160].
Figure 1.
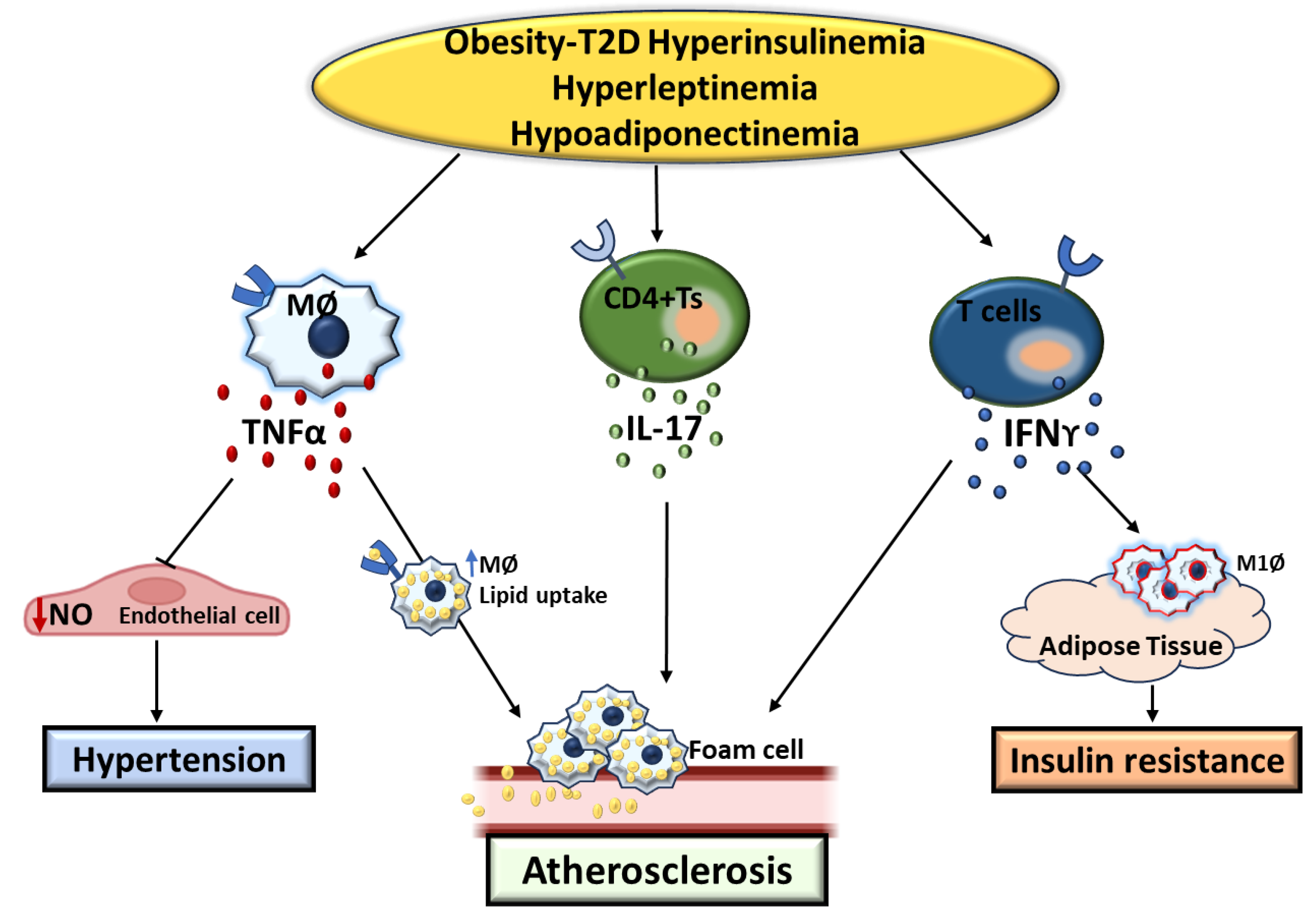
Summary Figure: key pathogenic factors involved in the development of T2D and its vascular complications. NO: Nitro Oxide, MØ: Macrophage, M1Ø: Type1 Macrophage, CD4+Ts: CD4+ lymphocytes.
Obesity diabetic condition is characterized by elevated levels of insulin (hyperinsulinemia), reduced levels of adiponectin (hypoadiponectinemia), and increased levels of leptin (hyperleptinemia), all of which contribute to dysregulation of cytokine production from immune cells[85,87,88,116,119,122,130,140,142,143]. Specifically, Tumor necrosis factor (TNFα), exacerbates endothelium dysfunction and enhances Lipid uptake by macrophage, thereby promoting the development of atherosclerosis and hypertension[46,48,49]. IFNγ triggers the infiltration of macrophage into adipose tissue and vasculature, resulting in insulin resistance and microvascular damage[34,35,36]. Additionally, Interleukin-17 (IL-17) in T2D can stimulate the formation of atherosclerotic lesions by activating MØ and facilitating their infiltration to various vascular beds[65,68,69].
References
- DeFronzo, R.A., et al., Type 2 diabetes mellitus. Nature Reviews Disease Primers, 2015. 1(1): p. 15019.
- Olokoba, A.B., O.A. Obateru, and L.B. Olokoba, Type 2 diabetes mellitus: a review of current trends. Oman Med J, 2012. 27(4): p. 269-73. [CrossRef]
- Francisco, C.O., et al., Cytokine profile and lymphocyte subsets in type 2 diabetes. Braz J Med Biol Res, 2016. 49(4): p. e5062. [CrossRef]
- Stentz, F.B. and A.E. Kitabchi, Activated T lymphocytes in Type 2 diabetes: implications from in vitro studies. Curr Drug Targets, 2003. 4(6): p. 493-503. [CrossRef]
- Touch, S., K. Clément, and S. André, T Cell Populations and Functions Are Altered in Human Obesity and Type 2 Diabetes. Curr Diab Rep, 2017. 17(9): p. 81. [CrossRef]
- Cortez-Espinosa, N., et al., CD39 expression on Treg and Th17 cells is associated with metabolic factors in patients with type 2 diabetes. Hum Immunol, 2015. 76(9): p. 622-30. [CrossRef]
- Martinez, N., et al., Chromatin decondensation and T cell hyperresponsiveness in diabetes-associated hyperglycemia. J Immunol, 2014. 193(9): p. 4457-68. [CrossRef]
- Menart-Houtermans, B., et al., Leukocyte Profiles Differ Between Type 1 and Type 2 Diabetes and Are Associated With Metabolic Phenotypes: Results From the German Diabetes Study (GDS). Diabetes Care, 2014. 37(8): p. 2326-2333. [CrossRef]
- van Beek, L., et al., Increased systemic and adipose tissue inflammation differentiates obese women with T2DM from obese women with normal glucose tolerance. Metabolism, 2014. 63(4): p. 492-501.
- Regufe, V.M.G., C. Pinto, and P. Perez, Metabolic syndrome in type 2 diabetic patients: a review of current evidence. Porto Biomed J, 2020. 5(6): p. e101. [CrossRef]
- Gonzalez, L.L., K. Garrie, and M.D. Turner, Type 2 diabetes - An autoinflammatory disease driven by metabolic stress. Biochim Biophys Acta Mol Basis Dis, 2018. 1864(11): p. 3805-3823. [CrossRef]
- Saeedi, P., et al., Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract, 2019. 157: p. 107843. [CrossRef]
- Al-Azzam, N., et al., Hypertension prevalence and associated factors among patients with diabetes: A retrospective cross-sectional study from Jordan. Ann Med Surg (Lond), 2021. 61: p. 126-131. [CrossRef]
- Almalki, Z.S., et al., Prevalence, risk factors, and management of uncontrolled hypertension among patients with diabetes: A hospital-based cross-sectional study. Prim Care Diabetes, 2020. 14(6): p. 610-615. [CrossRef]
- Alsaadon, H., et al., Hypertension and its related factors among patients with type 2 diabetes mellitus - a multi-hospital study in Bangladesh. BMC Public Health, 2022. 22(1): p. 198. [CrossRef]
- Sun, D., et al., Type 2 Diabetes and Hypertension. Circ Res, 2019. 124(6): p. 930-937.
- Schernthaner, G., Cardiovascular mortality and morbidity in type-2 diabetes mellitus. Diabetes Res Clin Pract, 1996. 31 Suppl: p. S3-13. [CrossRef]
- Organization, W.H., Global health estimates: deaths by cause, age, sex and country, 2000-2012. Geneva, WHO, 2014. 9: p. c2014.
- Al-Goblan, A.S., M.A. Al-Alfi, and M.Z. Khan, Mechanism linking diabetes mellitus and obesity. Diabetes Metab Syndr Obes, 2014. 7: p. 587-91.
- Ismail, L., H. Materwala, and J. Al Kaabi, Association of risk factors with type 2 diabetes: A systematic review. Comput Struct Biotechnol J, 2021. 19: p. 1759-1785. [CrossRef]
- Galicia-Garcia, U., et al., Pathophysiology of Type 2 Diabetes Mellitus. Int J Mol Sci, 2020. 21(17). [CrossRef]
- Singh, R., et al., Advanced glycation end-products: a review. Diabetologia, 2001. 44(2): p. 129-46. [CrossRef]
- Yan, S.F., R. Ramasamy, and A.M. Schmidt, Mechanisms of Disease: advanced glycation end-products and their receptor in inflammation and diabetes complications. Nature Clinical Practice Endocrinology & Metabolism, 2008. 4(5): p. 285-293. [CrossRef]
- Winer, D.A., et al., B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med, 2011. 17(5): p. 610-7. [CrossRef]
- Berbudi, A., et al., Type 2 Diabetes and its Impact on the Immune System. Curr Diabetes Rev, 2020. 16(5): p. 442-449.
- Donath, M.Y. and S.E. Shoelson, Type 2 diabetes as an inflammatory disease. Nature Reviews Immunology, 2011. 11(2): p. 98-107. [CrossRef]
- Nikolajczyk, B.S., et al., State of the union between metabolism and the immune system in type 2 diabetes. Genes & Immunity, 2011. 12(4): p. 239-250. [CrossRef]
- Girard, D. and C. Vandiedonck, How dysregulation of the immune system promotes diabetes mellitus and cardiovascular risk complications. Front Cardiovasc Med, 2022. 9: p. 991716. [CrossRef]
- Rohm, T.V., et al., Inflammation in obesity, diabetes, and related disorders. Immunity, 2022. 55(1): p. 31-55.
- Strissel, K.J., G.V. Denis, and B.S. Nikolajczyk, Immune regulators of inflammation in obesity-associated type 2 diabetes and coronary artery disease. Curr Opin Endocrinol Diabetes Obes, 2014. 21(5): p. 330-8. [CrossRef]
- Altan-Bonnet, G. and R. Mukherjee, Cytokine-mediated communication: a quantitative appraisal of immune complexity. Nature Reviews Immunology, 2019. 19(4): p. 205-217. [CrossRef]
- Ivashkiv, L.B., IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nature Reviews Immunology, 2018. 18(9): p. 545-558. [CrossRef]
- Rocha, V.Z., et al., Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res, 2008. 103(5): p. 467-76.
- Zhang, H., et al., Interferon-gamma induced adipose tissue inflammation is linked to endothelial dysfunction in type 2 diabetic mice. Basic Res Cardiol, 2011. 106(6): p. 1135-45. [CrossRef]
- Gupta, S., et al., IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest, 1997. 99(11): p. 2752-61. [CrossRef]
- Whitman, S.C., et al., Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E-/- mice. Am J Pathol, 2000. 157(6): p. 1819-24.
- Lee, L.Y., et al., Interferon-γ Impairs Human Coronary Artery Endothelial Glucose Metabolism by Tryptophan Catabolism and Activates Fatty Acid Oxidation. Circulation, 2021. 144(20): p. 1612-1628.
- Benson, L., et al., The IFNγ-PDL1 Pathway Enhances the Interaction Between CD8+ T Cells and Distal Convoluted Tubules to Promote Salt-Sensitive Hypertension. The FASEB Journal, 2022. 36(S1).
- Abdel-Moneim, A., et al., Association of glycemic status and interferon-γ production with leukocytes and platelet indices alterations in type2 diabetes. Diabetes Metab Syndr, 2019. 13(3): p. 1963-1969. [CrossRef]
- Kalliolias, G.D. and L.B. Ivashkiv, TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nature Reviews Rheumatology, 2016. 12(1): p. 49-62. [CrossRef]
- Beutler, B., et al., Purification of cachectin, a lipoprotein lipase-suppressing hormone secreted by endotoxin-induced RAW 264.7 cells. J Exp Med, 1985. 161(5): p. 984-95. [CrossRef]
- Borsotti, C., et al., Absence of donor T-cell-derived soluble TNF decreases graft-versus-host disease without impairing graft-versus-tumor activity. Blood, 2007. 110(2): p. 783-6. [CrossRef]
- Hotamisligil, G.S., N.S. Shargill, and B.M. Spiegelman, Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science, 1993. 259(5091): p. 87-91.
- Hotamisligil, G.S., et al., IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science, 1996. 271(5249): p. 665-8.
- Barath, P., et al., Tumor necrosis factor gene expression in human vascular intimal smooth muscle cells detected by in situ hybridization. Am J Pathol, 1990. 137(3): p. 503-9.
- Ohta, H., et al., Disruption of tumor necrosis factor-alpha gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis, 2005. 180(1): p. 11-7.
- Persson, J., J. Nilsson, and M.W. Lindholm, Interleukin-1beta and tumour necrosis factor-alpha impede neutral lipid turnover in macrophage-derived foam cells. BMC Immunol, 2008. 9: p. 70. [CrossRef]
- Zhang, H., et al., Role of TNF-alpha in vascular dysfunction. Clin Sci (Lond), 2009. 116(3): p. 219-30.
- Picchi, A., et al., Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res, 2006. 99(1): p. 69-77.
- Gao, X., et al., Tumor necrosis factor-alpha induces endothelial dysfunction in Lepr(db) mice. Circulation, 2007. 115(2): p. 245-54.
- Hassan, N.F., A.H. Hassan, and M.R. El-Ansary, Cytokine modulation by etanercept ameliorates metabolic syndrome and its related complications induced in rats administered a high-fat high-fructose diet. Sci Rep, 2022. 12(1): p. 20227. [CrossRef]
- Yoshizumi, M., et al., Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ Res, 1993. 73(1): p. 205-9.
- Chen, X., et al., Role of Reactive Oxygen Species in Tumor Necrosis Factor-alpha Induced Endothelial Dysfunction. Curr Hypertens Rev, 2008. 4(4): p. 245-255.
- Awad, A.S., et al., Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int, 2015. 88(4): p. 722-33.
- El-Edel, R., et al., Role of tumor necrosis factor alpha in type 2 diabetic nephropathy. Menoufia Medical Journal, 2020. 33(3): p. 920-925.
- Navarro, J.F. and C. Mora-Fernández, The role of TNF-alpha in diabetic nephropathy: pathogenic and therapeutic implications. Cytokine Growth Factor Rev, 2006. 17(6): p. 441-50.
- Kouri, V.-P., et al., IL-17A and TNF synergistically drive expression of proinflammatory mediators in synovial fibroblasts via IκBζ-dependent induction of ELF3. Rheumatology, 2022. 62(2): p. 872-885.
- Mills, K.H.G., IL-17 and IL-17-producing cells in protection versus pathology. Nature Reviews Immunology, 2023. 23(1): p. 38-54.
- Huangfu, L., et al., The IL-17 family in diseases: from bench to bedside. Signal Transduction and Targeted Therapy, 2023. 8(1): p. 402. [CrossRef]
- Chang, Y.-C., S.-W. Hee, and L.-M. Chuang, T helper 17 cells: A new actor on the stage of type 2 diabetes and aging? Journal of Diabetes Investigation, 2021. 12(6): p. 909-913.
- Chang, Y.C., S.W. Hee, and L.M. Chuang, T helper 17 cells: A new actor on the stage of type 2 diabetes and aging? J Diabetes Investig, 2021. 12(6): p. 909-913.
- Wang, J., et al., Clinical significance of Interleukin 17 receptor E in diabetic nephropathy. Int Immunopharmacol, 2023. 120: p. 110324.
- Ohshima, K., et al., Roles of interleukin 17 in angiotensin II type 1 receptor-mediated insulin resistance. Hypertension, 2012. 59(2): p. 493-9.
- Ma, J., et al., Interleukin 17A promotes diabetic kidney injury. Sci Rep, 2019. 9(1): p. 2264.
- Smith, E., et al., Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation, 2010. 121(15): p. 1746-55.
- van Es, T., et al., Attenuated atherosclerosis upon IL-17R signaling disruption in LDLr deficient mice. Biochem Biophys Res Commun, 2009. 388(2): p. 261-5.
- Chen, S., et al., IL-17A is proatherogenic in high-fat diet-induced and Chlamydia pneumoniae infection-accelerated atherosclerosis in mice. J Immunol, 2010. 185(9): p. 5619-27.
- Eid, R.E., et al., Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation, 2009. 119(10): p. 1424-32.
- Erbel, C., et al., Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J Immunol, 2009. 183(12): p. 8167-75.
- Chang, S.L., et al., Interleukin-17 enhances cardiac ventricular remodeling via activating MAPK pathway in ischemic heart failure. J Mol Cell Cardiol, 2018. 122: p. 69-79. [CrossRef]
- Kamat, N.V., et al., Renal transporter activation during angiotensin-II hypertension is blunted in interferon-γ-/- and interleukin-17A-/- mice. Hypertension, 2015. 65(3): p. 569-76.
- Norlander, A.E., et al., Interleukin-17A Regulates Renal Sodium Transporters and Renal Injury in Angiotensin II–Induced Hypertension. Hypertension, 2016. 68(1): p. 167-174.
- Amin, M.N., et al., Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med, 2020. 8: p. 2050312120965752.
- Mehra, V.C., V.S. Ramgolam, and J.R. Bender, Cytokines and cardiovascular disease. J Leukoc Biol, 2005. 78(4): p. 805-18. [CrossRef]
- Hales, C.N. and D.J.P. Barker, The thrifty phenotype hypothesis: Type 2 diabetes. British Medical Bulletin, 2001. 60(1): p. 5-20.
- Campbell, J.E. and C.B. Newgard, Mechanisms controlling pancreatic islet cell function in insulin secretion. Nature Reviews Molecular Cell Biology, 2021. 22(2): p. 142-158.
- Haeusler, R.A., T.E. McGraw, and D. Accili, Biochemical and cellular properties of insulin receptor signalling. Nature Reviews Molecular Cell Biology, 2018. 19(1): p. 31-44. [CrossRef]
- Ludwig, D.S., et al., Competing paradigms of obesity pathogenesis: energy balance versus carbohydrate-insulin models. European Journal of Clinical Nutrition, 2022. 76(9): p. 1209-1221.
- Ludwig, D.S., et al., The carbohydrate-insulin model: a physiological perspective on the obesity pandemic. Am J Clin Nutr, 2021. 114(6): p. 1873-1885.
- James, D.E., J. Stöckli, and M.J. Birnbaum, The aetiology and molecular landscape of insulin resistance. Nature Reviews Molecular Cell Biology, 2021. 22(11): p. 751-771.
- Cruz-Pineda, W.D., et al., The regulatory role of insulin in energy metabolism and leukocyte functions. J Leukoc Biol, 2022. 111(1): p. 197-208. [CrossRef]
- Helderman, J.H. and T.B. Strom, Emergence of insulin receptors upon alloimmune T cells in the rat. J Clin Invest, 1977. 59(2): p. 338-44.
- Kintscher, U., et al., T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol, 2008. 28(7): p. 1304-10.
- Cham, C.M. and T.F. Gajewski, Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol, 2005. 174(8): p. 4670-7.
- Tsai, S., et al., Insulin Receptor-Mediated Stimulation Boosts T Cell Immunity during Inflammation and Infection. Cell Metab, 2018. 28(6): p. 922-934.e4.
- Bar, R.S., C.R. Kahn, and H.S. Koren, Insulin inhibition of antibody-dependent cytoxicity and insulin receptors in macrophages. Nature, 1977. 265(5595): p. 632-635. [CrossRef]
- Tessaro, Fernando H.G., et al., Insulin Influences LPS-Induced TNF-α and IL-6 Release Through Distinct Pathways in Mouse Macrophages from Different Compartments. Cellular Physiology and Biochemistry, 2017. 42(5): p. 2093-2104.
- Tsiotra, P.C., et al., High insulin and leptin increase resistin and inflammatory cytokine production from human mononuclear cells. Biomed Res Int, 2013. 2013: p. 487081. [CrossRef]
- Ratter, J.M., et al., Insulin acutely activates metabolism of primary human monocytes and promotes a proinflammatory phenotype. Journal of Leukocyte Biology, 2021. 110(5): p. 885-891.
- Baumgartl, J., et al., Myeloid lineage cell-restricted insulin resistance protects apolipoproteinE-deficient mice against atherosclerosis. Cell Metab, 2006. 3(4): p. 247-56.
- Manhiani, M.M., M.T. Cormican, and M.W. Brands, Chronic sodium-retaining action of insulin in diabetic dogs. Am J Physiol Renal Physiol, 2011. 300(4): p. F957-65.
- Brands, M.W., et al., Sustained hyperinsulinemia increases arterial pressure in conscious rats. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 1991. 260(4): p. R764-R768. [CrossRef]
- Bursztyn, M., et al., Chronic exogenous hyperinsulinaemia without sugar supplementation: acute salt-sensitive hypertension without changes in resting blood pressure. Journal of hypertension, 1993. 11(7): p. 703-707. [CrossRef]
- DeFronzo, R.A., et al., The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest, 1975. 55(4): p. 845-55.
- Bie, P., Blood volume, blood pressure and total body sodium: internal signalling and output control. Acta Physiol (Oxf), 2009. 195(1): p. 187-96.
- Gamble, J.-M., et al., Insulin use and increased risk of mortality in type 2 diabetes: a cohort study. Diabetes, Obesity and Metabolism, 2010. 12(1): p. 47-53.
- Yu, B., et al., Insulin Treatment Is Associated with Increased Mortality in Patients with COVID-19 and Type 2 Diabetes. Cell Metab, 2021. 33(1): p. 65-77.e2. [CrossRef]
- Scheja, L. and J. Heeren, The endocrine function of adipose tissues in health and cardiometabolic disease. Nature Reviews Endocrinology, 2019. 15(9): p. 507-524.
- Smith, U. and B.B. Kahn, Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. J Intern Med, 2016. 280(5): p. 465-475.
- Grundy, S.M., Adipose tissue and metabolic syndrome: too much, too little or neither. Eur J Clin Invest, 2015. 45(11): p. 1209-17.
- Weisberg, S.P., et al., Obesity is associated with macrophage accumulation in adipose tissue. The Journal of clinical investigation, 2003. 112(12): p. 1796-1808.
- Keuper, M., On the role of macrophages in the control of adipocyte energy metabolism. Endocr Connect, 2019. 8(6): p. R105-r121.
- Chavakis, T., V.I. Alexaki, and A.W. Ferrante, Macrophage function in adipose tissue homeostasis and metabolic inflammation. Nature Immunology, 2023. 24(5): p. 757-766.
- Boutens, L. and R. Stienstra, Adipose tissue macrophages: going off track during obesity. Diabetologia, 2016. 59(5): p. 879-94.
- Li, H., et al., Macrophages, Chronic Inflammation, and Insulin Resistance. Cells, 2022. 11(19).
- Khan, M. and F. Joseph, Adipose tissue and adipokines: the association with and application of adipokines in obesity. Scientifica (Cairo), 2014. 2014: p. 328592.
- Yamauchi, T., et al., The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med, 2001. 7(8): p. 941-6. [CrossRef]
- Wang, Z.V. and P.E. Scherer, Adiponectin, the past two decades. Journal of Molecular Cell Biology, 2016. 8(2): p. 93-100.
- Almer, G., et al., Globular domain of adiponectin: promising target molecule for detection of atherosclerotic lesions. Biologics, 2011. 5: p. 95-105.
- Liu, X., et al., Globular adiponectin ameliorates insulin resistance in skeletal muscle by enhancing the LKB1-mediated AMPK activation via SESN2. Sports Med Health Sci, 2023. 5(1): p. 34-41. [CrossRef]
- Yamauchi, T., et al., Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature, 2003. 423(6941): p. 762-9.
- Denzel, M.S., et al., T-cadherin is critical for adiponectin-mediated cardioprotection in mice. The Journal of Clinical Investigation, 2010. 120(12): p. 4342-4352.
- Berg, A.H., et al., The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med, 2001. 7(8): p. 947-53.
- Combs, T.P., et al., Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J Clin Invest, 2001. 108(12): p. 1875-81.
- Maeda, N., et al., Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med, 2002. 8(7): p. 731-7.
- Hong, X., et al., Association between adiponectin and newly diagnosed type 2 diabetes in population with the clustering of obesity, dyslipidaemia and hypertension: a cross-sectional study. BMJ Open, 2023. 13(2): p. e060377.
- Luo, N., et al., AdR1-TG/TALLYHO mice have improved lipid accumulation and insulin sensitivity. Biochem Biophys Res Commun, 2013. 433(4): p. 567-72.
- Yamauchi, T., et al., Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem, 2003. 278(4): p. 2461-8.
- Salvator, H., et al., Adiponectin Inhibits the Production of TNF-α, IL-6 and Chemokines by Human Lung Macrophages. Front Pharmacol, 2021. 12: p. 718929.
- Tian, L., et al., Adiponectin reduces lipid accumulation in macrophage foam cells. Atherosclerosis, 2009. 202(1): p. 152-61.
- Zhang, Y., et al., Adiponectin's globular domain inhibits T cell activation by interacting with LAIR-1. Biochem Biophys Res Commun, 2021. 573: p. 117-124.
- Surendar, J., et al., Adiponectin Limits IFN-γ and IL-17 Producing CD4 T Cells in Obesity by Restraining Cell Intrinsic Glycolysis. Front Immunol, 2019. 10: p. 2555. [CrossRef]
- Picó, C., et al., Leptin as a key regulator of the adipose organ. Rev Endocr Metab Disord, 2022. 23(1): p. 13-30.
- Halaas, J.L., et al., Weight-Reducing Effects of the Plasma Protein Encoded by the <i>obese</i> Gene. Science, 1995. 269(5223): p. 543-546.
- Zhang, Y., et al., Positional cloning of the mouse obese gene and its human homologue. Nature, 1994. 372(6505): p. 425-32.
- Ingalls, A.M., M.M. Dickie, and G.D. Snell, Obese, a new mutation in the house mouse. J Hered, 1950. 41(12): p. 317-8.
- Kelesidis, T., et al., Narrative review: the role of leptin in human physiology: emerging clinical applications. Ann Intern Med, 2010. 152(2): p. 93-100.
- Flak, J.N. and M.G. Myers, Jr., Minireview: CNS Mechanisms of Leptin Action. Molecular Endocrinology, 2016. 30(1): p. 3-12. [CrossRef]
- Brydon, L., et al., Circulating leptin and stress-induced cardiovascular activity in humans. Obesity (Silver Spring), 2008. 16(12): p. 2642-7.
- Obradovic, M., et al., Leptin and Obesity: Role and Clinical Implication. Front Endocrinol (Lausanne), 2021. 12: p. 585887.
- Sims, E.D., et al., Circulating leptin levels are associated with adiposity in survivors of childhood brain tumors. Scientific Reports, 2020. 10(1): p. 4711. [CrossRef]
- Ma, D., et al., Leptin Is Associated With Blood Pressure and Hypertension in Women From the National Heart, Lung, and Blood Institute Family Heart Study. Hypertension, 2009. 53(3): p. 473-479. [CrossRef]
- Simonds, S.E., et al., Leptin mediates the increase in blood pressure associated with obesity. Cell, 2014. 159(6): p. 1404-16.
- Chiba, T., et al., Leptin deficiency suppresses progression of atherosclerosis in apoE-deficient mice. Atherosclerosis, 2008. 196(1): p. 68-75.
- Bodary, P.F., et al., Recombinant Leptin Promotes Atherosclerosis and Thrombosis in Apolipoprotein E–Deficient Mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 2005. 25(8): p. e119-e122.
- Taleb, S., et al., Defective leptin/leptin receptor signaling improves regulatory T cell immune response and protects mice from atherosclerosis. Arterioscler Thromb Vasc Biol, 2007. 27(12): p. 2691-8.
- Beltowski, J., Leptin and atherosclerosis. Atherosclerosis, 2006. 189(1): p. 47-60. [CrossRef]
- Lord, G.M., et al., Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature, 1998. 394(6696): p. 897-901.
- De Rosa, V., et al., A key role of leptin in the control of regulatory T cell proliferation. Immunity, 2007. 26(2): p. 241-55.
- De Rosa, V., et al., Leptin neutralization interferes with pathogenic T cell autoreactivity in autoimmune encephalomyelitis. J Clin Invest, 2006. 116(2): p. 447-55.
- Gerriets, V.A., et al., Leptin directly promotes T-cell glycolytic metabolism to drive effector T-cell differentiation in a mouse model of autoimmunity. Eur J Immunol, 2016. 46(8): p. 1970-83. [CrossRef]
- La Cava, A. and G. Matarese, The weight of leptin in immunity. Nat Rev Immunol, 2004. 4(5): p. 371-9.
- Saucillo, D.C., et al., Leptin Metabolically Licenses T Cells for Activation To Link Nutrition and Immunity. The Journal of Immunology, 2014. 192(1): p. 136-144.
- Tedgui, A. and Z. Mallat, Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev, 2006. 86(2): p. 515-81.
- Koga, M., et al., Postnatal blocking of interferon-gamma function prevented atherosclerotic plaque formation in apolipoprotein E-knockout mice. Hypertens Res, 2007. 30(3): p. 259-67.
- Rahman, M.S., et al., Role of Insulin in Health and Disease: An Update. Int J Mol Sci, 2021. 22(12).
- Luo, L. and M. Liu, Adiponectin: friend or foe in obesity and inflammation. Medical Review, 2022. 2(4): p. 349-362.
- Fernández-Riejos, P., et al., Role of leptin in the activation of immune cells. Mediators Inflamm, 2010. 2010: p. 568343.
- Makhijani, P., et al., Regulation of the immune system by the insulin receptor in health and disease. Front Endocrinol (Lausanne), 2023. 14: p. 1128622.
- Gao, M., D. Cui, and J. Xie, The role of adiponectin for immune cell function in metabolic diseases. Diabetes, Obesity and Metabolism, 2023. 25(9): p. 2427-2438.
- Yanai, H. and H. Yoshida, Beneficial Effects of Adiponectin on Glucose and Lipid Metabolism and Atherosclerotic Progression: Mechanisms and Perspectives. Int J Mol Sci, 2019. 20(5).
- Chen, L., et al., Mechanisms Linking Inflammation to Insulin Resistance. International Journal of Endocrinology, 2015. 2015: p. 508409.
- Kawai, T., M.V. Autieri, and R. Scalia, Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol, 2021. 320(3): p. C375-c391. [CrossRef]
- Rocha, V.Z., et al., Interferon-γ, a Th1 Cytokine, Regulates Fat Inflammation. Circulation Research, 2008. 103(5): p. 467-476.
- Shirai, T., et al., Macrophages in vascular inflammation--From atherosclerosis to vasculitis. Autoimmunity, 2015. 48(3): p. 139-51.
- Tomic, D., J.E. Shaw, and D.J. Magliano, The burden and risks of emerging complications of diabetes mellitus. Nature Reviews Endocrinology, 2022. 18(9): p. 525-539.
- Lamharzi, N., et al., Hyperlipidemia in Concert With Hyperglycemia Stimulates the Proliferation of Macrophages in Atherosclerotic Lesions: Potential Role of Glucose-Oxidized LDL. Diabetes, 2004. 53(12): p. 3217-3225.
- Cervantes, J. and J.E. Kanter, Monocyte and macrophage foam cells in diabetes-accelerated atherosclerosis. Front Cardiovasc Med, 2023. 10: p. 1213177. [CrossRef]
- Schett, G. and M.F. Neurath, Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nature Communications, 2018. 9(1): p. 3261.
- Deckers, J., et al., Engineering cytokine therapeutics. Nature Reviews Bioengineering, 2023. 1(4): p. 286-303.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



