
Submitted:
06 June 2024
Posted:
10 June 2024
You are already at the latest version
Abstract
Cellular senescence accumulates with age and has been shown to impact numerous physiological and pathological processes, including immune function. The role of cellular senescence in cancer is multifaceted, but the impact on immune checkpoint inhibitor response and toxicity has not been fully evaluated. In this review, we evaluate the impact of cellular senescence in various biological compartments, including the tumor, the tumor microenvironment, and the immune system on immune checkpoint inhibitor efficacy and toxicity. We provide an overview of the impact of cellular senescence in normal and pathological contexts and examine recent studies that have connected aging and cellular senescence to immune checkpoint inhibitor treatment in both the pre-clinical and clinical contexts. Overall, senescence plays a multi-faceted, context-specific role, and has been shown to modulate immune-related adverse event incidence as well as immune checkpoint inhibitor response.
Keywords:
immune checkpoint inhibitor
; aging
; cellular senescence
; immune-related adverse events
1. Introduction
Cancer is largely a disease of aging, and older patients are more likely to present with advanced, unresectable cancers [1,2]. The advent of immune checkpoint inhibitor (ICI) therapy has transformed clinical management of unresectable and metastatic solid and hematological malignancies [3,4]. However, the predictive biomarkers for both response and toxicity are scarce [5,6,7]. Widely used biomarkers for response, including PD-L1 expression and tumor mutational burden, achieve an area under the receiver operatic characteristic curve (AUC-ROC) between 0.51 and 0.74 [6,8], suggesting that key aspects of the biology underlying response to ICI are still enigmatic. Many patients on ICI treatment also experience immune-related adverse events (irAEs), where the treatment is believed to incite autoimmune-mediated damages of tissues. Many factors have been shown to influence the incidence of toxicities of ICI treatment, but the exact mechanisms of irAEs remain poorly understood [9,10,11].
Aging and cellular senescence have been shown to play a critical role in modulating the response to anti-cancer therapies in multiple contexts [12,13,14]. The extent of impact that aging and cellular senescence (hereafter referred to as senescence) have on ICI response and toxicity remains poorly understood and is an evolving area of research. Senescence can occur in tumor cells, in tumor-adjacent stromal cells, in normal tissue, and in the immune system, affecting multiple factors that govern ICI response.
In this review, we seek to explore the current understanding of the effect of senescence on immune checkpoint inhibitor response and toxicity. We provide an overview of senescence, review preclinical evidence of the potential mechanisms by which senescence and aging impact ICI response and toxicity, and highlight clinical findings that assess the relationship between aging, senescence, and ICI treatment.
2. Overview of Aging and Senescence
2.1. Aging Is a Result of Senescence
Aging has been implicated in multiple aspects of cancer initiation [15], progression [16], metastasis [17], and treatment [18]. Cellular senescence, a key biological driver of the phenotypes associated with aging, was first described by Weismann in 1881 when evaluating the principles that govern the duration of life and was subsequently re-discovered by Hayflick and Moorhead in the early 1960s [19]. It is defined as a transition to irreversible growth arrest of a cell [20]. Unlike quiescence, which is a reversible transition to the G0 phase of the cell cycle, senescence is believed to be irreversible, although there is some evidence that cells can transition out of senescence [21]. Senescence was initially characterized by the expression of hallmark senescence genes, namely p16INK4A and p14ARF (encoded by CDKN2A), and p21CIP1/WAF1 (encoded by CDKN1A). These hallmark genes are involved in cell cycle regulation and arrest [22,23,24,25] and can be induced by different mechanisms that include oncogene signaling, DNA damage, oxidative stress, and others (Figure 1). The inhibitory activity of p16INK4A and p14ARF leads to elevated p53 (TP53) activity and decreased phosphorylation of Rb (RB1), which results in cell cycle arrest [26,27] and subsequent senescence. Senescence is also characterized by the senescence-associated secretory phenotype (SASP), which involves the production of several secreted cytokines and chemokines that are produced by senescent cells [28,29,30]. Expression of SASP-related genes is at least in part driven by nuclear factor B (NFKB1) signaling. In their study, Chien et al. showed that inhibition of NF-B signaling in vitro in senescent cells decreased the mRNA expression of IL1A, IL6, CXCL1, and ICAM1, known components of the SASP [31]. At the chromatin level, the formation of senescence-associated heterochromatin foci has also been used as a key marker of senescence. These heterochromatin foci are mediated by the recruitment of Rb to E2F promoters [32]. Senescence-associated -galactosidase [33], a lysosomal -galactosidase is a widely used indicator of cellular senescence of cultured cells or senescent cells in tissues. While some studies have called into question the specificity of this indicator [34], other work has shown that selective elimination of cells expressing senescence-associated -galactosidase results in the reversal of senescence-associated phenotypes [35].
2.2. Causes and Consequences of Senescence
Senescence can be brought on by insult to the cell, including DNA damage [29], oncogene activation [36,37], metabolic stress [38,39,40,41], and replication stress [42]. Loss of homeostasis in the context of these cellular stresses leads to cell cycle arrest, driven by p16INK4A and p21CIP1/WAF1. and serves both protective and maladaptive roles in physiological function [43,44,45]. In normal development, p21-driven senescence plays a critical role in tissue remodeling during embryogenesis [46]. Controlled senescence is also required for proper wound-healing [47]. In a study of wound healing, the secretion of PDGF-AA (homodimeric isoform encoded by PDGF), a component of the SASP, was required for wound healing in a preclinical mouse model of senescence [48], and ablation of senescent cells inhibited the wound healing process. Senescence has also been shown to induce angiogenesis through the actions of SASP [21]. Our previous work also showed that angiotensin II was sufficient to induce senescence in kidney endothelial cells, and that selective clearance of senescent cells reverses downstream effects of senescence and prevents senescence-mediated immune infiltration [49]. Senescent cells characterized by p16INK4A expression accumulate during physiologic aging. In addition to physiologic changes, multiple age-related pathologies (or “senopathies”) have been linked to the increased abundance of senescent cells [50], including osteoarthritis [51], pulmonary fibrosis [52], atherosclerosis [53], to name a few.
In the context of cancer, senescence plays a vital role in halting the proliferation of pre-malignant cells. Senescence is induced by oncogenic signaling – for example, BRAF V600 mutations are commonly found in non-malignant melanocytic nevi, which are induced to senesce through activation of oncogenic BRAF signaling [54]. One of the key mediators of cellular senescence, p16INK4A, signals through the tumor suppressor Rb1 (RB1) to halt cell proliferation in cells undergoing potentially tumorigenic stress [55]. Indeed, the evolutionary pressures behind senescence include tumor suppressive function [56]. While senescence is usually beneficial in the prevention of tumorigenesis, some effects of senescence may exacerbate tumorigenesis and enable tumor cell functions that promote malignancy. Trends of cellular senescence with age and related aspects of cancer onset and treatment response and toxicity are outlined in Figure 2. These aspects of cellular senescence are further elaborated on below.
2.3. Therapeutic Modulators of Senescence
In their seminal work, Baker et al. demonstrated in a murine model that age-related phenotypes can be reversed through the clearance of p16-driven senescent cells [57]. This finding spurred a flurry of investigation into drugs that would specifically target and destroy senescent cells but leave normal proliferating or reversibly growth-arrested cells intact. Multiple drug repurposing studies proposed candidate compounds including dasatinib and quercetin [58], fisetin and curcumin [59], anti-apoptotic BCL2/BCL-xL inhibitors such as navitoclax [60,61], and others. These treatments and the history of their development have been extensively reviewed [62]. The results of several clinical trials evaluating senolytic therapies in patients with existing age-related pathologies have shown promising albeit modest results [63,64,65,66]. The potential impact of senolytic or senomodulatory therapy on the safety and efficacy of ICI treatment has not yet been evaluated, but encouraging preclinical evidence [67,68] suggests that this interaction may be worth investigating in a clinical trial.
3. Senescence and the Immune System
3.1. Immune Checkpoint Protein Expression in Different Cell Types
The immune checkpoint protein PD-1 (programmed death receptor 1) is present on exhausted T cells. Binding of PD-1 to its ligands, PD-L1 or PD-L2, suppresses T cell activity and proliferation [69]. PD-L1 is expressed on tumor cells as a mechanism of immune evasion [70]. Therapeutic disruption of the PD-1/PD-L1 interaction promotes PD-1+ T-cell-mediated killing of PD-L1+ tumor cells. However, PD-1 is also expressed in other immune and non-immune cell types (Figure 3). Natural killer (NK) cells also express PD-1, and this expression appears to be age-dependent [71]. PD-1 expression on tumor cells has been highlighted as a mechanism of immunotherapy resistance [72]. PD-L1, similarly, is not only expressed on tumor cells. Immune cells in the tumor microenvironment have been shown to express PD-L1, including CD8+ T cells. The PD-L1+CD8+ T cell population plays a pro-tumorigenic role by suppressing effector T cells, but also promotes self-tolerance [73], which may also partially explain the onset of irAEs in anti-PD1 therapy. At a basal level, other normal cell types, including microvascular endothelial cells and renal tubule epithelial cells also express low levels of PD-L1 [73]. Aging and senescent cells have been observed to express PD-L1 more than younger cells of the same type, leading some to suggest the use of anti-PD1 or anti-PD-L1 ICI treatment as a potential senolytic agent [74,75].
Targeting the cytotoxic T-lymphocyte associated protein 4 (CTLA4) immune checkpoint receptor has also proven to be an effective strategy in reversing T-cell exhaustion. Normally, the interactions between CTLA4 and its binding partner B7 leads to stimulation and activation of lymphocytes [76]. Inactive T-cells express higher levels of CTLA4, and abrogation of the interaction between CTLA4 and B7 resulted in reactivation in T-cells [77,78]. However, like PD-1, CTLA4 is not exclusively expressed in exhausted T-cells. In normal healthy pituitary cells, CTLA4 is expressed widely, leading to hypophysitis as a common immune-related adverse event in anti-CTLA4 treatment [79]. Aging also leads to an increase in the expression of CTLA4 on the surface of T-cells cells, suggesting that T-cell exhaustion and senescence-associated processes may be intertwined [80].
3.2. Immune-Related Adverse Events may Occur Systemically
Treatment with immune checkpoint inhibitor therapy can cause immune-related adverse events in nearly half of all patients [81]. These irAEs can affect nearly any tissue in the body, most commonly in skin, endocrine, and GI organs [82]. While the exact mechanisms of irAEs have not been identified, several observations suggest that irAEs occur from a loss of self-tolerance and increased recognition of self-antigen, from immune overstimulation resulting in increased systemic inflammation, from expression of checkpoint proteins in non-tumor tissue, or other yet uncharacterized mechanisms. These and other hypotheses have been comprehensively reviewed elsewhere [9,83,84,85,86].
4. Senescence and the Immune System
4.1. Immune-Mediated Surveillance and Clearance of Senescence
Surveillance and clearance of senescent cells is a normal physiological function of the immune system. Proteins comprising the senescence-associated secretory phenotype act as paracrine and chemotactic signals, attracting circulating immune cells through the vascular endothelium into the tissue stroma [87]. Cytotoxic CD8+ T cells, as well as NK cells have been shown to play a critical role in the clearance of senescent cells [88]. Immune-mediated surveillance of senescent cells is an important component of anti-tumor immunity [89]. Eggert et al. showed that oncogene-induced senescence in hepatocytes prevents hepatocellular tumorigenesis and stimulates the production of CCL2, a component of the SASP [90]. CCL2 stimulates immature circulating myeloid cells to differentiate into infiltrating macrophages that subsequently can clear senescent cells, and ablation of CCL2 led to hepatocellular carcinoma outgrowth.
4.2. Cellular Senescence Is a Component of Immunosenescence
Senescence can occur throughout the body, but of particular interest in the context of ICI therapy is the role of senescence in immune function and activation. As with other somatic cells, immune cells also undergo cellular senescence and other functional changes with age, resulting in an overall age-associated deterioration in immune function, termed immunosenescence [44]. Age-associated immune cell senescence has been noted in multiple immune compartments, with evidence that senescent CD8+ T-cells were more abundant in older individuals, as well as in younger patients infected with human immunodeficiency virus [91]. Senescence in immune cells has been shown to be driven by p16INK4A, as in other somatic cells, which suggests that p16-driven clearance of senescent cells may also be effective [92].
Another key aspect of immunosenescence that is not encompassed by cellular senescence is the potential for immune cell anergy and exhaustion. Immune exhaustion anergy results from reduced co-stimulatory signals. The loss of costimulatory protein CD28 in T-cells may in part explain age-associated reductions in T-cell activity that define immunosenescence [93]. Immune cell exhaustion, on the other hand, is usually defined by an increased expression of immune checkpoint proteins on the surface of immune cells. These include PD-1 (PDCD1), CTLA4, TIM-3 (HAVCR2), LAG3, and TIGIT [94]. Expression of CD57 was initially believed to more specifically mark senescent T cells [95], but recent studies have demonstrated that the CD57+ T cell population is in fact capable of replicating in vivo [96] and in fact may play an important role in the response to anti-PD1 therapy [97].
Interestingly, many markers of T cell exhaustion have also been associated with aging. In a murine model, CD8+ T cells tended to express higher levels of PD1 and LAG3 in mice of advanced age compared to their younger counterparts, and these aged T cells transcriptionally resembled exhausted T cells [98]. An observational study in humans showed that CTLA4 was more expressed in CD4+ T cells from older people, and that expression of CTLA4 was also negatively correlated with costimulatory CD28 expression [80]. Another study showed that TIM-3 expression was observed in CD8+ cells in aged mice, and that these cells produce the anti-inflammatory cytokine IL-10 [99]. The mechanisms that link immune exhaustion, anergy, and senescence have yet to be completely elucidated, but taken together, the observations in these studies suggest that although immune cell senescence, anergy, and exhaustion are discrete processes, they all associate with aging, possibly explaining age-associated immunosenescence.
Cellular senescence in non-immune cell types also plays a strong role on immune function and immunosenescence. In physiological aging, the SASP from stromal senescent cells has been shown to signal to the vascular endothelium to recruit CD4+ and CD8+ T-cells expressing STAT1 [87]. The SASP acts on these cells to induce clearance of senescent cells, which constitutes a core physiological function of T lymphocytes [100]. Senescent cells have been shown to upregulate the expression of MICA and ULBP2, which bind to the NK cell receptor NKG2D [101]. Evasion mechanisms of senescent cells include upregulation of HLA-E expression by senescent cells, which were shown to dampen the ability of NK cells and CD8+ T-cells to perform degranulation and infiltration functions critical for senescent cells clearance [102]. However, senescence in the tumor microenvironment may play an immunosuppressive role. Montes et al. showed that patient-derived T-cells exhibit senescence-like properties after co-culture with a tumor cell line, including increased p16, p21, and p53 expression, shortened telomere lengths, and loss of CD27 and CD28 [103]. These tumor-induced senescent T-cells slowed proliferation of other non-senescent T-cells and produced regulatory cytokines including IL-6 and interferon gamma [103]. To summarize, immunosenescence can be impacted both by direct cellular senescence in the immune cell population, as well as by senescence in non-immune cells capable of modulating immune function.
5. Clinical Evidence of Aging Impacting Immune Checkpoint Inhibitor Safety and Efficacy
5.1. CIinical Observations Regarding the Impact of Aging on Efficacy
While most clinical studies have found that ICI shows similar efficacy in older and younger patients [101,102,103,104], some studies have reported conflicting findings suggesting that elderly patients may fare worse with ICI in specific contexts [105]. A recent meta-analysis [106] surveyed 30 randomized controlled trials to build a meta-cohort of 17,476 patients with various solid tissue malignancies. Analysis of survival showed that across cancer types, there was overall little to no difference in progression-free survival or overall survival in younger (<65 years) or older (≥65 years) patients [106]. The only statistically significant finding was slightly improved progression-free survival in younger patients with melanoma, but older melanoma patients still benefited significantly from ICI [106]. Importantly, although this study demonstrated no clinical difference between patients stratified into discrete age groups, others have shown that patients at the extremes of age demonstrate more variable response patterns [107]. Further evaluation of the impact of age on ICI efficacy is warranted, and consideration of treatment type, cancer type, and dosage may help delineate the impacts of aging and senescence on ICI efficacy.
5.2. CIinical Observations Regarding the Impact of Aging on Adverse Events
Conflicting evidence about the effects of age on immune-related toxicities resulting from irAEs suggests that there may be an impact of age on ICI toxicity, but that this effect may be modest. A retrospective study of 288 patients with multiple cancer types treated with ICI in Japan found that patients with more severe irAEs tended to be on average 2.7 years older, but that patients with irAEs tended to have improved overall response rates [108]. Other studies focused on specific types of cancer also vary in their findings about ICI efficacy in older patients – a study of 227 patients with head/neck squamous cell carcinoma found that patients aged 70 or older responded similarly to ICI compared to their younger counterparts and had a comparable toxicity profile [109]. Another study showed that anti-PD1/anti-PD-L1 ICI treatment induced fewer overall toxicities in patients aged 70 or older, and that while older patients tended to experience more skin toxicities, younger patients tended to suffer from more endocrine toxicities secondary to ICI treatment [110]. In contrast, another study of anti-PD1 treatment in three different cancer types showed that patients aged 70 or more had slightly higher (though not statistically significant) rates of irAEs than patients younger than 65 [111]. Overall, most studies did not find a significantly different rate of severe irAEs in patients by age, and those that found differences did so in a limited set of contexts.
6. Evidence of Interaction between Senescence and ICI Treatment
6.1. Senescence in Tumor Cells
Senescence has been demonstrated to prevent tumorigenesis and stimulate clearance of pre-malignant cells, suggesting a protective role by preventing cancer [90,112,113]. Loss of senescence upon malignant transformation is well-documented [54]. However, induction of senescence in transformed malignant tumor cells has been associated with poor outcomes. Senescent tumor cells have been shown to promote invasion into the lymphovascular system and prevented apoptosis during migration through expression of senescence-associated E-cadherin [114,115]. On the other hand, senescent tumor cells may also stimulate an immunogenic response more than non-tumor cells. One study showed that mice immunized with senescent B16 murine melanoma cells were more likely to successfully mount an immune response against tumorigenic B16 melanoma compared to immunization with non-senescent or even dying cells. In the context of ICI treatment, one study found that PD-L1 expression tend to be elevated more broadly in senescent cells [75]. This finding suggests that senescence in tumor cells may lead to improved outcomes from ICI treatment. On the other hand, another study in diffuse large B-cell lymphoma (DLBCL) showed that senescent DLBCL tumor cells could signal to macrophages which potentiate the senescence phenotype in other non-senescent tumor cells [116]. These senescent DLBCL tumor cells tended to express more PD-L1, which led to increased targeting of this population with anti-PD1 ICI treatment [116]. Thus, while senescence in pre-malignant cells may prevent malignant transformation, senescence in tumor cells may be linked to more aggressive phenotypes and stimulate invasion and migration, although immune response to senescent tumor cells in the context of ICI may be more potent.
6.2. Senescence in the Immune Compartments
As previously discussed, senescence in peripheral immune cells has been demonstrated to be associated with age and diminished immune effector function. The accumulation of senescent immune cells in specific tissues may explain the onset of irAEs – indeed, one study demonstrated that T-cells could be induced to senescence, characterized by loss of CD27 and CD28, after exposure to tumor cells [100]. Interestingly, senescent T-cells driven by p16INK4A expression also tend to express the exhaustion marker PD-1 [92,96], a key target of ICI. Senescent non-immune cells also tend to express more PD-L1 [75], and the production of SASP is sufficient to induce PD-L1 expression in non-senescent cells [75]. Complementary increases in the expression of PD-1 in aged T-cells explains the observation by Wang et al. that the use of anti-PD-1 ICI treatment resulted in increased senescent cell clearance and overall cellular rejuvenation in a mouse model [74]. Wang et al. also found that treatment with anti-PD1 ICI treatment led to CD8+ T-cell-mediated clearance of senescent cells and reversed age-related phenotypes including steatotic liver disease [74]. While this observation does not directly indicate that senescence in the immune compartment was responsible for age-related phenotypes, it does suggest that targeting age-related exhaustion and senescence markers in the immune compartments, including the ICI target PD-1, may reverse age-associated phenotypes, which includes senescence.
Myeloid-derived suppressor cells (MDSCs) are another important immune population whose frequency drastically changes in the context of aging and senescence. In the context of aging and cancer, myelopoesis has been observed to increase with age in both rodents and humans [117,118], with a particular elevation in patients with a history of cancer [119]. Patients treated with ICI therapy were shown to have a profound clinical response only with a reduction in the peripheral MDSC population, and an elevated MDSC count was predictive of failure to respond to ICI treatment [120,121]. MDSCs have been shown to respond strongly to interferon signaling, with one study showing that ensuring the type I interferon receptor IFNAR1 in MDSCs abrogated any suppressive activity resulting in strong anti-tumor immunity [122]. Another study showed that halting signaling via type I interferons resulted in enhanced suppressive activity of MDSCs, and subsequent resistance to ICI treatment [123]. While the exact impact of cellular senescence within MDSCs has yet to be explored, these cells are highly associated with the onset of age-related immune changes by modulating senescence phenotypes in surrounding cells. For example, one study found that MDSCs prevent senescence in cardiac myofibroblasts, resulting in age-related cardiac fibrosis [124]. Another study demonstrated that a subpopulation of MDSCs could induce senescence in CD8+ T-cells through exosomal transfer of GPR84, a G-protein coupled receptor that signals through p53 to halt cell proliferation [125]. These findings together suggest MDSCs that age-related change in MDSC populations may affect response to ICI.
6.3. Senescence in the Tumor Microenvironment
Stromal senescence may play a pro- or anti-tumor role in cancer development, progression, and treatment response. A breakthrough study by Krtolica et al. showed that co-culture with senescent fibroblasts could stimulate malignant transformation in non-malignant epithelial cells, and hastened tumorigenesis in malignant cells [15]. A more recent study by Haston et al. found in mice that tumor growth and proliferation was attenuated after clearance of senescent tumor-associated macrophages [126]. Clinically, Haston et al. also found senescent macrophages not only in the tumor microenvironments of human lung cancers, but also in pre-cancerous lung lesions, suggesting that senescence does promote tumorigenesis in humans [126]. Cancer-associated fibroblasts in the tumor microenvironment have also been shown to promote invasion and epithelial disruption in oral squamous cell carcinoma [127], suggesting a key role of senescent cells in driving metastatic spread. Another study of numerous prostate and breast cancer cell lines suggested that extracellular vesicles produced by senescent cells promoted resistance to chemotherapeutic agents by upregulating expression of drug transport protein ABCB4 [128]. Taken together, these studies suggest that senescent cells in the tumor microenvironment play a pro-tumorigenic role and may enable tumors to migrate and metastasize, as well as develop resistance to treatment.
In the context of ICI treatment, senescence in the tumor-adjacent stroma or in the tumor immune microenvironment may play a significant immunosuppressive role in response to ICI [129,130]. A recent study directly addressed the question of the impact of senescence on ICI response. Using a murine model that allowed for clearance of senescent cells from the tumor microenvironment, Maggiorani et al. showed that elimination of senescent cells diminished the immunosuppressive activity of myeloid cells and resulted in improved overall survival and response to ICI [131]. In this study, senescence was induced with total body irradiation or by treatment with doxorubicin, and using the p16-3MR transgenic mouse model, senescent cells were cleared upon treatment with ganciclovir, or using the senolytic drug ABT263 (navitoclax) [131]. These results suggest a potential role for senolytic treatments in concert with ICI to improve outcomes. Clinical trials evaluating the safety and efficacy of senolytic drugs are currently underway, although to date, no trials have explored combining senolytic treatments with ICI therapy because of the unpredictable balance between pro- and anti-tumoral effects of senescence.
6.4. The Impact of Senescence on Immune-Related Adverse Events
Senescence may also be linked to the incidence, location, timing and duration of immune-related adverse events (irAEs) secondary to ICI treatment. Clinical observations have indicated that elderly patients may be more susceptible to adverse events [111], especially in specific tissues such as the skin. Senescent cells secrete pro-inflammatory cytokines as part of the SASP, including IL-1, IL-6, IL-8, interferon gamma, and others [28]. Recent clinical studies have shown that blocking IL-6 prevents and ameliorates the onset of irAEs [132,133]. On the other hand, another study found that low serum IL-6 was prognostic of more frequent and severe irAEs [134]. Another attribute that may explain age-associated irAE incidence is the increased expression levels of PD-L1 in senescent tumor microenvironmental cells or in non-tumor stromal cells in patients with high senescent cell burden [75]. These cells may be cleared with ICI treatment, and indeed, one study showed that treatment with ICI is linked to the clearance of senescent cells [74]. Immune-mediated clearance of senescent tissue by the immune system has been shown to be relatively innocuous in transgenic and senolytic-treated mice [57,61,135], but the impact of profound senescence clearance in aged human patients has not been fully explored.
The clearest study linking aging and senescence to the onset of immune-related adverse events is Tsukamoto et al. [136]. In this comprehensive study of the impact of aging on immune checkpoint response and toxicity, the authors develop a murine model for studying the interaction between age and irAEs secondary to anti-PD1. Old (>18 months) and young (<3 months) mice (C57/Bl6-J and Balb-c) were orthotopically injected with melanoma or colon cancer cell lines and subsequently treated with anti-PD1. In this model, older mice appeared to have diminished response to anti-PD1 treatment but tend to have CD4+ T-cell-mediated elevated immune infiltration in non-tumor tissues, including the lung, liver, and kidneys, consistent with the histological hallmarks of irAEs. The authors also emphasize the role of IL-21 and CXCL13 in the onset of irAEs. An important limitation of this study is the sole use of anti-PD1 ICI treatment. However, the authors mention in the supplement and discussion that while addition of anti-CTLA4 to anti-PD1 in their murine model exacerbated the irAE incidence, the underlying biology of CD4+ cell-mediated IL21 production and CXCL13 elevation are not seen in single-agent anti-CTLA4 treatment. This finding may suggest that the mechanism for irAEs on anti-CTLA4 may be distinct and may synergize with anti-PD1 irAEs.
7. Conclusion & Future Directions
The impact of cellular senescence is pleiotropic, and can play multiple roles in the development, progression and spread of cancer. Although the exact roles of senescence on immunotherapy have not been elucidated, evidence suggests that senescence in specific compartments can influence the efficacy and toxicity of immune checkpoint inhibitor treatment. Senescence in tumor cells can result in slowed growth but can also promote invasion and metastasis, as well as confer treatment resistance to anti-cancer therapies, which may include immunotherapy. In stromal cells within the tumor microenvironment, senescence can lead to the production of pro- and anti-inflammatory cytokines, which has been shown to promote resistance to immunotherapy [131]. Senescence in non-tumor tissue may pre-dispose those tissues to the onset of immune-related adverse events [9]. Finally, senescence in immune cells, which express several exhaustion markers including PD-1 [75], may potentiate the effects of immune checkpoint inhibitor treatment, while exacerbating immune-related adverse events [136].
To our knowledge, a few pre-clinical studies have directly addressed the impact of aging and senescence on immune checkpoint inhibitor efficacy and toxicity. Maggiorani et al. [131] showed that senescence elimination could overcome resistance to ICI treatment. Contrastingly, Hao et al. [67] showed that amplifying the SASP could overcome ICI resistance in ovarian cancer. Tsukamoto et al. [136] demonstrated a potential mechanism involving CD4+ T-cells and the cytokines IL-21 and CXCL13, which are notably components of the SASP, through which older individuals would have a higher incidence of irAEs. Further studies are required to evaluate the mechanisms by which senescence can impact immunotherapy treatment. Model organisms used to study p16INK4A-driven senescence include the P16-3MR and INK/ATTAC models [48,57], which enable the clearance of senescent cells induced by p16INK4A. Conditional depletion of cells mediated by alternative forms of senescence, such as p21CIP or p14ARF-mediated senescence, can provide further insights on the effects of different pathways in modulating senescence and effects of ICI treatment. Therapeutic options for senescence clearance are also feasible – senolytic treatments, including navitoclax, dasatinib, quercetin, fisetin, and others have been demonstrated to eliminate senescent cells [63,64,65,66]. Development of the next generation of senolytic and senomodulatory treatments has also begun and holds promise in more specifically and potently targeting senescent cells. These treatments and future directions of senolytic and senomodulatory therapy have been reviewed elsewhere [137]. As of May 2024, no active clinical trials are investigating the impact of senescence clearance or modulation on immunotherapy efficacy or toxicity. Such a clinical trial would inform whether clearance of senescent cells would be beneficial or detrimental for patients receiving immunotherapy such as ICI, and whether senolytic treatments could be safe and effective in combination with ICI treatment.
In summary, aging and cellular senescence may influence immune checkpoint inhibitor response and toxicity, within the tumor, the tumor microenvironment, or the immune system. While clinical evidence regarding the relationship between aging and immune checkpoint inhibitor treatment outcomes are conflicting, preclinical studies have suggested an important role for aging and cellular senescence in modulating the efficacy of immune checkpoint inhibitor therapy and incidence of immune-related adverse events. Further mechanistic studies and clinical trials evaluating the effects of senescent cell clearance are required to evaluate the full scope of interaction between immune checkpoint inhibitor treatment and cellular senescence and establish a refined approach to combinatory treatments.
Author Contributions
Conceptualization: S.S.J, A.W.; Original draft preparation, writing: S.S.J., H.S., and B.N.F.; Original draft preparation, review and editing: S.S.J, G.B.S., H.S., B.N.F., M.E.M., M.O.S., A.W. Supervision: A.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by funding from the National Institutes of Health USA: T32-CA009686 to SSJ, GBS, MEM; T32-GM142520 to SSJ; F30-CA250307 to MEM, R01-CA231291 to AW and P30-CA51008 to AW.
Acknowledgments
Figures made with Biorender.com. Sincere thanks to Dr. Maha Moussa and Dr. Irfan Khan for their discussions on the role of senescence in the immune system.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hudock, N.L.; Mani, K.; Khunsriraksakul, C.; Walter, V.; Nekhlyudov, L.; Wang, M.; Lehrer, E.J.; Hudock, M.R.; Liu, D.J.; Spratt, D.E.; et al. Future Trends in Incidence and Long-Term Survival of Metastatic Cancer in the United States. Commun Med 2023, 3, 1–7. [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA: A Cancer Journal for Clinicians 2023, 73, 17–48. [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annual Review of Pathology: Mechanisms of Disease 2021, 16, 223–249. [CrossRef]
- Lin, E.P.-Y.; Hsu, C.-Y.; Berry, L.; Bunn, P.; Shyr, Y. Analysis of Cancer Survival Associated With Immune Checkpoint Inhibitors After Statistical Adjustment: A Systematic Review and Meta-Analyses. JAMA Network Open 2022, 5, e2227211. [CrossRef]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive Biomarkers for Cancer Immunotherapy with Immune Checkpoint Inhibitors. Biomarker Research 2020, 8, 34. [CrossRef]
- Kao, C.; Powers, E.; Wu, Y.; Datto, M.B.; Green, M.F.; Strickler, J.H.; Ready, N.E.; Zhang, T.; Clarke, J.M. Predictive Value of Combining Biomarkers for Clinical Outcomes in Advanced Non-Small Cell Lung Cancer Patients Receiving Immune Checkpoint Inhibitors. Clinical Lung Cancer 2021, 22, 500–509. [CrossRef]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive Biomarkers for Checkpoint Inhibitor-Based Immunotherapy. The Lancet Oncology 2016, 17, e542–e551. [CrossRef]
- Wang, S.; Zhang, J.; He, Z.; Wu, K.; Liu, X.-S. The Predictive Power of Tumor Mutational Burden in Lung Cancer Immunotherapy Response Is Influenced by Patients’ Sex. International Journal of Cancer 2019, 145, 2840–2849. [CrossRef]
- Les, I.; Martínez, M.; Pérez-Francisco, I.; Cabero, M.; Teijeira, L.; Arrazubi, V.; Torrego, N.; Campillo-Calatayud, A.; Elejalde, I.; Kochan, G.; et al. Predictive Biomarkers for Checkpoint Inhibitor Immune-Related Adverse Events. Cancers 2023, 15, 1629. [CrossRef]
- Shi, W.-J.; Zhao, W. Biomarkers or Factors for Predicting the Efficacy and Adverse Effects of Immune Checkpoint Inhibitors in Lung Cancer: Achievements and Prospective. Chin Med J (Engl) 2020, 133, 2466–2475. [CrossRef]
- Smithy, J.W.; Faleck, D.M.; Postow, M.A. Facts and Hopes in Prediction, Diagnosis, and Treatment of Immune-Related Adverse Events. Clinical Cancer Research 2022, 28, 1250–1257. [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discovery 2017, 7, 165–176. [CrossRef]
- Guillon, J.; Petit, C.; Toutain, B.; Guette, C.; Lelièvre, E.; Coqueret, O. Chemotherapy-Induced Senescence, an Adaptive Mechanism Driving Resistance and Tumor Heterogeneity. Cell Cycle 2019, 18, 2385–2397. [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing Chemotherapy Efficacy in Pten-Deficient Prostate Tumors by Activating the Senescence-Associated Antitumor Immunity. Cell Rep 2014, 9, 75–89. [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.-Y.; Campisi, J. Senescent Fibroblasts Promote Epithelial Cell Growth and Tumorigenesis: A Link between Cancer and Aging. Proc Natl Acad Sci U S A 2001, 98, 12072–12077. [CrossRef]
- Mittmann, L.A.; Haring, F.; Schaubächer, J.B.; Hennel, R.; Smiljanov, B.; Zuchtriegel, G.; Canis, M.; Gires, O.; Krombach, F.; Holdt, L.; et al. Uncoupled Biological and Chronological Aging of Neutrophils in Cancer Promotes Tumor Progression. J Immunother Cancer 2021, 9, e003495. [CrossRef]
- Angelini, P.D.; Fluck, M.F.Z.; Pedersen, K.; Parra-Palau, J.L.; Guiu, M.; Bernadó Morales, C.; Vicario, R.; Luque-García, A.; Navalpotro, N.P.; Giralt, J.; et al. Constitutive HER2 Signaling Promotes Breast Cancer Metastasis through Cellular Senescence. Cancer Research 2013, 73, 450–458. [CrossRef]
- The Importance of Aging in Cancer Research. Nat Aging 2022, 2, 365–366. [CrossRef]
- Kirkwood, T.B.L.; Cremer, T. Cytogerontology since 1881: A Reappraisal of August Weismann and a Review of Modern Progress. Hum Genet 1982, 60, 101–121. [CrossRef]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Experimental Cell Research 1961, 25, 585–621. [CrossRef]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.-A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-Associated Secretory Phenotype Contributes to Pathological Angiogenesis in Retinopathy. Sci Transl Med 2016, 8, 362ra144. [CrossRef]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of Human Cellular Senescence: Roles of the P53 and P16 Pathways. The EMBO Journal 2003, 22, 4212–4222. [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential Roles for Cyclin-Dependent Kinase Inhibitors P21 and P16 in the Mechanisms of Senescence and Differentiation in Human Fibroblasts. Molecular and Cellular Biology 1999, 19, 2109–2117. [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.; Jeganathan, K.B.; Versoza, G.C.; Pezeshki, A.-M.; et al. Naturally Occurring p16Ink4a-Positive Cells Shorten Healthy Lifespan. Nature 2016, 530, 184–189. [CrossRef]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf Expression Is a Biomarker of Aging. J Clin Invest 2004, 114, 1299–1307. [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A Senescence Program Controlled by P53 and p16INK4a Contributes to the Outcome of Cancer Therapy. Cell 2002, 109, 335–346. [CrossRef]
- Leonardo, A.D.; Linke, S.P.; Clarkin, K.; Wahl, G.M. DNA Damage Triggers a Prolonged P53-Dependent G1 Arrest and Long-Term Induction of Cip1 in Normal Human Fibroblasts. Genes Dev. 1994, 8, 2540–2551. [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PLoS Biol 2008, 6, 2853–2868. [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signaling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat Cell Biol 2009, 11, 973–979. [CrossRef]
- Coppé, J.-P.; Kauser, K.; Campisi, J.; Beauséjour, C.M. Secretion of Vascular Endothelial Growth Factor by Primary Human Fibroblasts at Senescence. J Biol Chem 2006, 281, 29568–29574. [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the Senescence-Associated Secretory Phenotype by NF-κB Promotes Senescence and Enhances Chemosensitivity. Genes Dev 2011, 25, 2125–2136. [CrossRef]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell 2003, 113, 703–716. [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Proc Natl Acad Sci U S A 1995, 92, 9363–9367. [CrossRef]
- de Mera-Rodríguez, J.A.; Álvarez-Hernán, G.; Gañán, Y.; Martín-Partido, G.; Rodríguez-León, J.; Francisco-Morcillo, J. Is Senescence-Associated β-Galactosidase a Reliable in Vivo Marker of Cellular Senescence During Embryonic Development? Frontiers in Cell and Developmental Biology 2021, 9.
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of Senescent Cells by β-Galactosidase-Targeted Prodrug Attenuates Inflammation and Restores Physical Function in Aged Mice. Cell Res 2020, 30, 574–589. [CrossRef]
- Pearson, M.; Carbone, R.; Sebastiani, C.; Cioce, M.; Fagioli, M.; Saito, S.; Higashimoto, Y.; Appella, E.; Minucci, S.; Pandolfi, P.P.; et al. PML Regulates P53 Acetylation and Premature Senescence Induced by Oncogenic Ras. Nature 2000, 406, 207–210. [CrossRef]
- Liu, X.; Ding, J.; Meng, L. Oncogene-Induced Senescence: A Double Edged Sword in Cancer. Acta Pharmacol Sin 2018, 39, 1553–1558. [CrossRef]
- Burova, E.; Borodkina, A.; Shatrova, A.; Nikolsky, N. Sublethal Oxidative Stress Induces the Premature Senescence of Human Mesenchymal Stem Cells Derived from Endometrium. Oxidative Medicine and Cellular Longevity 2013, 2013, e474931. [CrossRef]
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA Damage and Senescence of Human Diploid Fibroblast Cells. Proceedings of the National Academy of Sciences 1995, 92, 4337–4341. [CrossRef]
- Burton, D.G.A.; Faragher, R.G.A. Obesity and Type-2 Diabetes as Inducers of Premature Cellular Senescence and Ageing. Biogerontology 2018, 19, 447–459. [CrossRef]
- Obesity Accelerates Age Defects in Human B Cells and Induces Autoimmunity. Immunometabolism 2022. [CrossRef]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication Stress Is a Potent Driver of Functional Decline in Ageing Haematopoietic Stem Cells. Nature 2014, 512, 198–202. [CrossRef]
- Ben-Porath, I.; Weinberg, R.A. The Signals and Pathways Activating Cellular Senescence. Int J Biochem Cell Biol 2005, 37, 961–976. [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging: An Evolutionary Perspective on Immunosenescence. Annals of the New York Academy of Sciences 2000, 908, 244–254. [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat Rev Mol Cell Biol 2014, 15, 482–496. [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [CrossRef]
- Wilkinson, H.N.; Hardman, M.J. Senescence in Wound Repair: Emerging Strategies to Target Chronic Healing Wounds. Frontiers in Cell and Developmental Biology 2020, 8.
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.T.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Developmental Cell 2014, 31, 722–733. [CrossRef]
- Khan, I.; Schmidt, M.O.; Kallakury, B.; Jain, S.; Mehdikhani, S.; Levi, M.; Mendonca, M.; Welch, W.; Riegel, A.T.; Wilcox, C.S.; et al. Low Dose Chronic Angiotensin II Induces Selective Senescence of Kidney Endothelial Cells. Front Cell Dev Biol 2021, 9, 782841. [CrossRef]
- Lushchak, O.; Schosserer, M.; Grillari, J. Senopathies—Diseases Associated with Cellular Senescence. Biomolecules 2023, 13, 966. [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.-M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local Clearance of Senescent Cells Attenuates the Development of Post-Traumatic Osteoarthritis and Creates a pro-Regenerative Environment. Nat Med 2017, 23, 775–781. [CrossRef]
- Yanai, H.; Shteinberg, A.; Porat, Z.; Budovsky, A.; Braiman, A.; Zeische, R.; Fraifeld, V.E. Cellular Senescence-like Features of Lung Fibroblasts Derived from Idiopathic Pulmonary Fibrosis Patients. Aging 2015, 7, 664–672. [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial Cell Senescence in Human Atherosclerosis. Circulation 2002, 105, 1541–1544. [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-Associated Senescence-like Cell Cycle Arrest of Human Naevi. Nature 2005, 436, 720–724. [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular Senescence and Tumor Suppressor Gene P16. Int J Cancer 2012, 130, 1715–1725. [CrossRef]
- Kowald, A.; Passos, J.F.; Kirkwood, T.B.L. On the Evolution of Cellular Senescence. Aging Cell 2020, 19, e13270. [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-Positive Senescent Cells Delays Ageing-Associated Disorders. Nature 2011, 479, 232–236. [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ Heel of Senescent Cells: From Transcriptome to Senolytic Drugs. Aging Cell 2015, 14, 644–658. [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin Is a Senotherapeutic That Extends Health and Lifespan. EBioMedicine 2018, 36, 18–28. [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a Novel Senolytic Agent, Navitoclax, Targeting the Bcl-2 Family of Anti-Apoptotic Factors. Aging Cell 2016, 15, 428–435. [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of Senescent Cells by ABT263 Rejuvenates Aged Hematopoietic Stem Cells in Mice. Nat Med 2016, 22, 78–83. [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic Drugs: From Discovery to Translation. Journal of Internal Medicine 2020, 288, 518–536. [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics Decrease Senescent Cells in Humans: Preliminary Report from a Clinical Trial of Dasatinib plus Quercetin in Individuals with Diabetic Kidney Disease. EBioMedicine 2019, 47, 446–456. [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in Idiopathic Pulmonary Fibrosis: Results from a First-in-Human, Open-Label, Pilot Study. eBioMedicine 2019, 40, 554–563. [CrossRef]
- Ellison-Hughes, G.M. First Evidence That Senolytics Are Effective at Decreasing Senescent Cells in Humans. eBioMedicine 2020, 56. [CrossRef]
- Nambiar, A.; Kellogg, D.; Justice, J.; Goros, M.; Gelfond, J.; Pascual, R.; Hashmi, S.; Masternak, M.; Prata, L.; LeBrasseur, N.; et al. Senolytics Dasatinib and Quercetin in Idiopathic Pulmonary Fibrosis: Results of a Phase I, Single-Blind, Single-Center, Randomized, Placebo-Controlled Pilot Trial on Feasibility and Tolerability. eBioMedicine 2023, 90. [CrossRef]
- Hao, X.; Zhao, B.; Zhou, W.; Liu, H.; Fukumoto, T.; Gabrilovich, D.; Zhang, R. Sensitization of Ovarian Tumor to Immune Checkpoint Blockade by Boosting Senescence-Associated Secretory Phenotype. iScience 2020, 24. [CrossRef]
- Rutella, S.; Vadakekolathu, J.; Mazziotta, F.; Reeder, S.; Yau, T.-O.; Mukhopadhyay, R.; Dickins, B.; Altmann, H.; Kramer, M.; Knaus, H.; et al. Signatures of Immune Senescence Predict Outcomes and Define Checkpoint Blockade-Unresponsive Microenvironments in Acute Myeloid Leukemia.; February 9 2022.
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, upon Programmed Cell Death. EMBO J 1992, 11, 3887–3895. [CrossRef]
- Blank, C.; Gajewski, T.F.; Mackensen, A. Interaction of PD-L1 on Tumor Cells with PD-1 on Tumor-Specific T Cells as a Mechanism of Immune Evasion: Implications for Tumor Immunotherapy. Cancer Immunol Immunother 2005, 54, 307–314. [CrossRef]
- Deng, M.; Zeng, Y.; Liu, Y.; Wang, X.; Chen, N.; Zhang, M.; Jiang, M.; Zhao, H.; Du, J. Increased PD-1+ NK Cell Subset in the Older Population. IJGM 2024, Volume 17, 651–661. [CrossRef]
- Wang, X.; Yang, X.; Zhang, C.; Wang, Y.; Cheng, T.; Duan, L.; Tong, Z.; Tan, S.; Zhang, H.; Saw, P.E.; et al. Tumor Cell-Intrinsic PD-1 Receptor Is a Tumor Suppressor and Mediates Resistance to PD-1 Blockade Therapy. Proceedings of the National Academy of Sciences 2020, 117, 6640–6650. [CrossRef]
- Wang, T.-W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking PD-L1–PD-1 Improves Senescence Surveillance and Ageing Phenotypes. Nature 2022, 611, 358–364. [CrossRef]
- Onorati, A.; Havas, A.P.; Lin, B.; Rajagopal, J.; Sen, P.; Adams, P.D.; Dou, Z. Upregulation of PD-L1 in Senescence and Aging. Molecular and Cellular Biology 2022. [CrossRef]
- Zhang, Y.Q.; Joost van Neerven, R.J.; Van Gool, S.W.; Coorevits, L.; de Boer, M.; Ceuppens, J.L. B7-CD28 Interaction Is a Late Acting Co-Stimulatory Signal for Human T Cell Responses. Int Immunol 1997, 9, 1095–1102. [CrossRef]
- Lee, K.-M.; Chuang, E.; Griffin, M.; Khattri, R.; Hong, D.K.; Zhang, W.; Straus, D.; Samelson, L.E.; Thompson, C.B.; Bluestone, J.A. Molecular Basis of T Cell Inactivation by CTLA-4. Science 1998, 282, 2263–2266. [CrossRef]
- Fife, B.T.; Bluestone, J.A. Control of Peripheral T-Cell Tolerance and Autoimmunity via the CTLA-4 and PD-1 Pathways. Immunol Rev 2008, 224, 166–182. [CrossRef]
- Iwama, S.; De Remigis, A.; Callahan, M.K.; Slovin, S.F.; Wolchok, J.D.; Caturegli, P. Pituitary Expression of CTLA-4 Mediates Hypophysitis Secondary to Administration of CTLA-4 Blocking Antibody. Sci Transl Med 2014, 6, 230ra45. [CrossRef]
- Leng, Q.; Bentwich, Z.; Borkow, G. CTLA-4 Upregulation during Aging. Mech Ageing Dev 2002, 123, 1419–1421. [CrossRef]
- Thapa, B.; Roopkumar, J.; Kim, A.S.; Gervaso, L.; Patil, P.D.; Calabrese, C.; Khorana, A.A.; Funchain, P. Incidence and Clinical Pattern of Immune Related Adverse Effects (irAE) Due to Immune Checkpoint Inhibitors (ICI). JCO 2019, 37, e14151–e14151. [CrossRef]
- Brown, V.T.; Antol, D.D.; Racsa, P.N.; Ward, M.A.; Naidoo, J. Real-World Incidence and Management of Immune-Related Adverse Events from Immune Checkpoint Inhibitors: Retrospective Claims-Based Analysis. Cancer Investigation 2021, 39, 789–796. [CrossRef]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of Response, Resistance, and Toxicity to Immune Checkpoint Blockade. Cell 2021, 184, 5309–5337. [CrossRef]
- Conroy, M.; Naidoo, J. Immune-Related Adverse Events and the Balancing Act of Immunotherapy. Nat Commun 2022, 13, 392. [CrossRef]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E. Immune-Related Adverse Events of Checkpoint Inhibitors. Nat Rev Dis Primers 2020, 6, 1–21. [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N Engl J Med 2018, 378, 158–168. [CrossRef]
- Yin, K.; Patten, D.; Gough, S.; Gonçalves, S. de B.; Chan, A.; Olan, I.; Cassidy, L.; Poblocka, M.; Zhu, H.; Lun, A.; et al. Senescence-Induced Endothelial Phenotypes Underpin Immune-Mediated Senescence Surveillance. Genes Dev. 2022, 36, 533–549. [CrossRef]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired Immune Surveillance Accelerates Accumulation of Senescent Cells and Aging. Nat Commun 2018, 9, 5435. [CrossRef]
- Brenner, E.; Schörg, B.F.; Ahmetlić, F.; Wieder, T.; Hilke, F.J.; Simon, N.; Schroeder, C.; Demidov, G.; Riedel, T.; Fehrenbacher, B.; et al. Cancer Immune Control Needs Senescence Induction by Interferon-Dependent Cell Cycle Regulator Pathways in Tumours. Nat Commun 2020, 11, 1335. [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [CrossRef]
- Effros, R.B.; Dagarag, M.; Spaulding, C.; Man, J. The Role of CD8+ T-Cell Replicative Senescence in Human Aging. Immunological Reviews 2005, 205, 147–157. [CrossRef]
- Janelle, V.; Neault, M.; Lebel, M.-È.; De Sousa, D.M.; Boulet, S.; Durrieu, L.; Carli, C.; Muzac, C.; Lemieux, S.; Labrecque, N.; et al. p16INK4a Regulates Cellular Senescence in PD-1-Expressing Human T Cells. Front. Immunol. 2021, 12. [CrossRef]
- Effros, R.B.; Boucher, N.; Porter, V.; Zhu, X.; Spaulding, C.; Walford, R.L.; Kronenberg, M.; Cohen, D.; Schächter, F. Decline in CD28+ T Cells in Centenarians and in Long-Term T Cell Cultures: A Possible Cause for Both in Vivo and in Vitro Immunosenescence. Exp Gerontol 1994, 29, 601–609. [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T Cell Exhaustion.’ Nat Rev Immunol 2019, 19, 665–674. [CrossRef]
- Decman, V.; Laidlaw, B.J.; Doering, T.A.; Leng, J.; Ertl, H.C.J.; Goldstein, D.R.; Wherry, E.J. Defective CD8 T Cell Responses in Aged Mice Are Due to Quantitative and Qualitative Changes in Virus-Specific Precursors. The Journal of Immunology 2012, 188, 1933–1941. [CrossRef]
- Lee, K.; Shin, K.; Kim, G.; Song, Y.C.; Bae, E.; Kim, I.; Koh, C.; Kang, C. Characterization of Age-associated Exhausted CD 8 + T Cells Defined by Increased Expression of Tim-3 and PD -1. Aging Cell 2016, 15, 291–300. [CrossRef]
- Langhi Prata, L.G.P.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent Cell Clearance by the Immune System: Emerging Therapeutic Opportunities. Semin Immunol 2018, 40, 101275. [CrossRef]
- Sagiv, A.; Burton, D.G.A.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D Ligands Mediate Immunosurveillance of Senescent Cells. Aging (Albany NY) 2016, 8, 328–344. [CrossRef]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent Cells Evade Immune Clearance via HLA-E-Mediated NK and CD8+ T Cell Inhibition. Nat Commun 2019, 10, 2387. [CrossRef]
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-Induced Senescent T Cells with Suppressor Function: A Potential Form of Tumor Immune Evasion. Cancer Res 2008, 68, 870–879. [CrossRef]
- Betof, A.S.; Nipp, R.D.; Giobbie-Hurder, A.; Johnpulle, R.A.N.; Rubin, K.; Rubinstein, S.M.; Flaherty, K.T.; Lawrence, D.P.; Johnson, D.B.; Sullivan, R.J. Impact of Age on Outcomes with Immunotherapy for Patients with Melanoma. Oncologist 2017, 22, 963–971. [CrossRef]
- Marrone, K.; Zhang, J.; Feliciano, J.L.; Forde, P.M.; Hann, C.L.; Kelly, R.J.; Ettinger, D.S.; Turner, M.; Rowe, V.; Bonerigo, S.; et al. Immune Checkpoint Inhibition in Elderly Non-Small Cell Lung Cancer Patients. JCO 2018, 36, 137–137. [CrossRef]
- Truong, T.-G.; Yamamoto, C.; Chiu, T.; Niu, F.; Spence, M.M.; Chan, J.; Cooper, R.M.; Moon, H.; Hui, R.L. Immune-Related Adverse Events (IrAE) of Elderly Patients (Pts) Receiving PD-1 or PDL-1 Inhibitors (PDIs) in a Community-Oncology Setting: The Experience at Kaiser Permanente California. JCO 2018, 36, 124–124. [CrossRef]
- Schonfeld, S.J.; Tucker, M.A.; Engels, E.A.; Dores, G.M.; Sampson, J.N.; Shiels, M.S.; Chanock, S.J.; Morton, L.M. Immune-Related Adverse Events After Immune Checkpoint Inhibitors for Melanoma Among Older Adults. JAMA Network Open 2022, 5, e223461. [CrossRef]
- Motzer Robert J.; Escudier Bernard; McDermott David F.; George Saby; Hammers Hans J.; Srinivas Sandhya; Tykodi Scott S.; Sosman Jeffrey A.; Procopio Giuseppe; Plimack Elizabeth R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. New England Journal of Medicine 2015, 373, 1803–1813. [CrossRef]
- Kim, C.M.; Lee, J.B.; Shin, S.J.; Ahn, J.B.; Lee, M.; Kim, H.S. The Efficacy of Immune Checkpoint Inhibitors in Elderly Patients: A Meta-Analysis and Meta-Regression. ESMO Open 2022, 7, 100577. [CrossRef]
- Wong, S.K.; Nebhan, C.A.; Johnson, D.B. Impact of Patient Age on Clinical Efficacy and Toxicity of Checkpoint Inhibitor Therapy. Front Immunol 2021, 12, 786046. [CrossRef]
- Matsuoka, H.; Hayashi, T.; Takigami, K.; Imaizumi, K.; Shiroki, R.; Ohmiya, N.; Sugiura, K.; Kawada, K.; Sawaki, A.; Maeda, K.; et al. Correlation between Immune-Related Adverse Events and Prognosis in Patients with Various Cancers Treated with Anti PD-1 Antibody. BMC Cancer 2020, 20, 656. [CrossRef]
- Saleh, K.; Auperin, A.; Martin, N.; Borcoman, E.; Torossian, N.; Iacob, M.; Ferrand, F.-R.; Khalife, N.; Baste, N.; Guigay, J.; et al. Efficacy and Safety of Immune Checkpoint Inhibitors in Elderly Patients (≥70 Years) with Squamous Cell Carcinoma of the Head and Neck. European Journal of Cancer 2021, 157, 190–197. [CrossRef]
- Paderi, A.; Fancelli, S.; Caliman, E.; Pillozzi, S.; Gambale, E.; Mela, M.M.; Doni, L.; Mazzoni, F.; Antonuzzo, L. Safety of Immune Checkpoint Inhibitors in Elderly Patients: An Observational Study. Curr Oncol 2021, 28, 3259–3267. [CrossRef]
- Singh, H.; Kim, G.; Maher, V.E.; Beaver, J.A.; Pai-Scherf, L.H.; Balasubramaniam, S.; Theoret, M.R.; Blumenthal, G.M.; Pazdur, R. FDA Subset Analysis of the Safety of Nivolumab in Elderly Patients with Advanced Cancers. JCO 2016, 34, 10010–10010. [CrossRef]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence Surveillance of Pre-Malignant Hepatocytes Limits Liver Cancer Development. Nature 2011, 479, 547–551. [CrossRef]
- Lee, S.; Schmitt, C.A. The Dynamic Nature of Senescence in Cancer. Nat Cell Biol 2019, 21, 94–101. [CrossRef]
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.-H.; Park, T.J. Senescent Tumor Cells Lead the Collective Invasion in Thyroid Cancer. Nat Commun 2017, 8, 15208. [CrossRef]
- Kawaguchi, K.; Komoda, K.; Mikawa, R.; Asai, A.; Sugimoto, M. Cellular Senescence Promotes Cancer Metastasis by Enhancing Soluble E-Cadherin Production. iScience 2021, 24, 103022. [CrossRef]
- Reimann, M.; Schrezenmeier, J.; Richter-Pechanska, P.; Dolnik, A.; Hick, T.P.; Schleich, K.; Cai, X.; Fan, D.N.Y.; Lohneis, P.; Maßwig, S.; et al. Adaptive T-Cell Immunity Controls Senescence-Prone MyD88- or CARD11-Mutant B-Cell Lymphomas. Blood 2021, 137, 2785–2799. [CrossRef]
- Heithoff, D.M.; Enioutina, E.Y.; Bareyan, D.; Daynes, R.A.; Mahan, M.J. Conditions That Diminish Myeloid-Derived Suppressor Cell Activities Stimulate Cross-Protective Immunity. Infect Immun 2008, 76, 5191–5199. [CrossRef]
- Flores, R.R.; Clauson, C.L.; Cho, J.; Lee, B.-C.; McGowan, S.J.; Baker, D.J.; Niedernhofer, L.J.; Robbins, P.D. Expansion of Myeloid-Derived Suppressor Cells with Aging in the Bone Marrow of Mice through a NF-κB-Dependent Mechanism. Aging Cell 2017, 16, 480–487. [CrossRef]
- Verschoor, C.P.; Johnstone, J.; Millar, J.; Dorrington, M.G.; Habibagahi, M.; Lelic, A.; Loeb, M.; Bramson, J.L.; Bowdish, D.M.E. Blood CD33(+)HLA-DR(−) Myeloid-Derived Suppressor Cells Are Increased with Age and a History of Cancer. Journal of Leukocyte Biology 2013, 93, 633–637. [CrossRef]
- Coana, Y.P. de; Wolodarski, M.; Poschke, I.; Yoshimoto, Y.; Yang, Y.; Nystrom, M.; Edback, U.; Brage, S.E.; Lundqvist, A.; Masucci, G.V.; et al. Ipilimumab Treatment Decreases Monocytic MDSCs and Increases CD8 Effector Memory T Cells in Long-Term Survivors with Advanced Melanoma. Oncotarget 2017, 8, 21539–21553. [CrossRef]
- Weber, J.; Gibney, G.; Kudchadkar, R.; Yu, B.; Cheng, P.; Martinez, A.J.; Kroeger, J.; Richards, A.; McCormick, L.; Moberg, V.; et al. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunology Research 2016, 4, 345–353. [CrossRef]
- Alicea-Torres, K.; Sanseviero, E.; Gui, J.; Chen, J.; Veglia, F.; Yu, Q.; Donthireddy, L.; Kossenkov, A.; Lin, C.; Fu, S.; et al. Immune Suppressive Activity of Myeloid-Derived Suppressor Cells in Cancer Requires Inactivation of the Type I Interferon Pathway. Nat Commun 2021, 12, 1717. [CrossRef]
- Chen, J.; Sun, H.-W.; Yang, Y.-Y.; Chen, H.-T.; Yu, X.-J.; Wu, W.-C.; Xu, Y.-T.; Jin, L.-L.; Wu, X.-J.; Xu, J.; et al. Reprogramming Immunosuppressive Myeloid Cells by Activated T Cells Promotes the Response to Anti-PD-1 Therapy in Colorectal Cancer. Sig Transduct Target Ther 2021, 6, 1–14. [CrossRef]
- Sun, S.-N.; Ni, S.-H.; Li, Y.; Liu, X.; Deng, J.-P.; Chen, Z.-X.; Li, H.; Feng, W.-J.; Huang, Y.-S.; Li, D.-N.; et al. G-MDSCs Promote Aging-Related Cardiac Fibrosis by Activating Myofibroblasts and Preventing Senescence. Cell Death Dis 2021, 12, 1–17. [CrossRef]
- Liu, J.; Liu, J.; Qin, G.; Li, J.; Fu, Z.; Li, J.; Li, M.; Guo, C.; Zhao, M.; Zhang, Z.; et al. MDSCs-Derived GPR84 Induces CD8 + T-Cell Senescence via P53 Activation to Suppress the Antitumor Response. J Immunother Cancer 2023, 11, e007802. [CrossRef]
- Haston, S.; Gonzalez-Gualda, E.; Morsli, S.; Ge, J.; Reen, V.; Calderwood, A.; Moutsopoulos, I.; Panousopoulos, L.; Deletic, P.; Carreno, G.; et al. Clearance of Senescent Macrophages Ameliorates Tumorigenesis in KRAS-Driven Lung Cancer. Cancer Cell 2023. [CrossRef]
- Hassona, Y.; Cirillo, N.; Heesom, K.; Parkinson, E.K.; Prime, S.S. Senescent Cancer-Associated Fibroblasts Secrete Active MMP-2 That Promotes Keratinocyte Dis-Cohesion and Invasion. Br J Cancer 2014, 111, 1230–1237. [CrossRef]
- Han, L.; Long, Q.; Li, S.; Xu, Q.; Zhang, B.; Dou, X.; Qian, M.; Jiramongkol, Y.; Guo, J.; Cao, L.; et al. Senescent Stromal Cells Promote Cancer Resistance through SIRT1 Loss-Potentiated Overproduction of Small Extracellular Vesicles. Cancer Research 2020, 80, 3383–3398. [CrossRef]
- Marin, I.; Boix, O.; Garcia-Garijo, A.; Sirois, I.; Caballe, A.; Zarzuela, E.; Ruano, I.; Attolini, C.S.-O.; Prats, N.; López-Domínguez, J.A.; et al. Cellular Senescence Is Immunogenic and Promotes Antitumor Immunity. Cancer Discov 2023, 13, 410–431. [CrossRef]
- Elias, R.; Hartshorn, K.; Rahma, O.; Lin, N.; Snyder-Cappione, J.E. Aging, Immune Senescence, and Immunotherapy: A Comprehensive Review. Seminars in Oncology 2018, 45, 187–200. [CrossRef]
- Maggiorani, D.; Le, O.; Lisi, V.; Landais, S.; Moquin-Beaudry, G.; Lavallée, V.P.; Decaluwe, H.; Beauséjour, C. Senescence Drives Immunotherapy Resistance by Inducing an Immunosuppressive Tumor Microenvironment. Nat Commun 2024, 15, 2435. [CrossRef]
- Dimitriou, F.; Hogan, S.; Menzies, A.M.; Dummer, R.; Long, G.V. Interleukin-6 Blockade for Prophylaxis and Management of Immune-Related Adverse Events in Cancer Immunotherapy. European Journal of Cancer 2021, 157, 214–224. [CrossRef]
- Fa’ak, F.; Buni, M.; Falohun, A.; Lu, H.; Song, J.; Johnson, D.H.; Zobniw, C.M.; Trinh, V.A.; Awiwi, M.O.; Tahon, N.H.; et al. Selective Immune Suppression Using Interleukin-6 Receptor Inhibitors for Management of Immune-Related Adverse Events. J Immunother Cancer 2023, 11, e006814. [CrossRef]
- Valpione, S.; Pasquali, S.; Campana, L.G.; Piccin, L.; Mocellin, S.; Pigozzo, J.; Chiarion-Sileni, V. Sex and Interleukin-6 Are Prognostic Factors for Autoimmune Toxicity Following Treatment with Anti-CTLA4 Blockade. J Transl Med 2018, 16, 94. [CrossRef]
- Casanova, M.; Pitaval, C.; Castrillo, A.; Hidalgo, A. Clearance of Senescent Neutrophils Maintains Homeostatic Levels of Hematopoietic Progenitor Cells in the Circulation. Blood 2011, 118, 863. [CrossRef]
- Tsukamoto, H.; Komohara, Y.; Tomita, Y.; Miura, Y.; Motoshima, T.; Imamura, K.; Kimura, T.; Ikeda, T.; Fujiwara, Y.; Yano, H.; et al. Aging-Associated and CD4 T-Cell-Dependent Ectopic CXCL13 Activation Predisposes to Anti-PD-1 Therapy-Induced Adverse Events. Proc Natl Acad Sci U S A 2022, 119, e2205378119. [CrossRef]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular Senescence and Senolytics: The Path to the Clinic. Nat Med 2022, 28, 1556–1568. [CrossRef]
- Huang, X.; Tian, T.; Zhang, Y.; Zhou, S.; Hu, P.; Zhang, J. Age-Associated Changes in Adverse Events Arising From Anti-PD-(L)1 Therapy. Frontiers in Oncology 2021, 11.
Figure 1.
An overview of senescence causes and effects. Senescence can be induced by several factors, including accumulative DNA damage [27], oncogenic signaling [36], metabolic stress and diseases such as diabetes [40], and therapeutic toxicities from cytotoxic chemotherapy [13]. Senescence is driven at a transcriptional level by several key cell cycle proteins, including p16INK4A, p14ARF, and p21CIP1/WAF1 [25,26]. Senescence induction results in the production of the senescence-associated secretory phenotype (SASP), a collection of chemokines and cytokines expressed in senescent cells including IL-6, IL-8, IL-15, CXCL1, CCL3, and others [28]. Senescence can result in tumor suppression in pre-malignant cells [55], but can also result in treatment resistance [13,131], enhanced metastatic potential [17,115], and suppressed anti-tumor immune function through immunosenescence [89].
Figure 1.
An overview of senescence causes and effects. Senescence can be induced by several factors, including accumulative DNA damage [27], oncogenic signaling [36], metabolic stress and diseases such as diabetes [40], and therapeutic toxicities from cytotoxic chemotherapy [13]. Senescence is driven at a transcriptional level by several key cell cycle proteins, including p16INK4A, p14ARF, and p21CIP1/WAF1 [25,26]. Senescence induction results in the production of the senescence-associated secretory phenotype (SASP), a collection of chemokines and cytokines expressed in senescent cells including IL-6, IL-8, IL-15, CXCL1, CCL3, and others [28]. Senescence can result in tumor suppression in pre-malignant cells [55], but can also result in treatment resistance [13,131], enhanced metastatic potential [17,115], and suppressed anti-tumor immune function through immunosenescence [89].
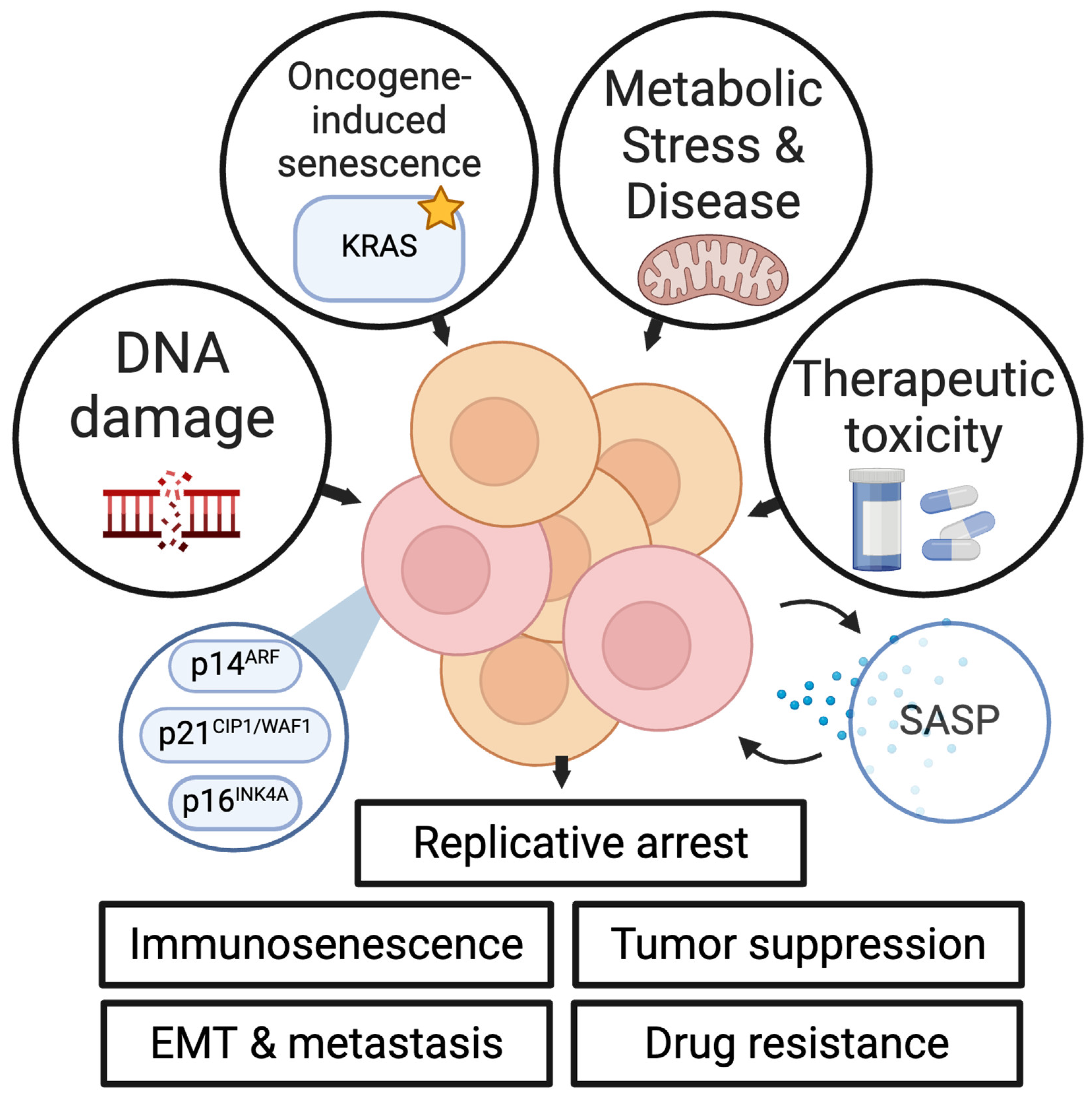
Figure 2.
Summary of the age-associated trends relevant to immune checkpoint inhibitor (ICI) response and toxicity. With age, senescent cell burden and cancer risk increase colinearly. Immune effector function peaks during young adulthood and diminishes in elderly individuals. ICI efficacy has been demonstrated to both increase [67] and decrease [131] with aging and senescence, but in different contexts. Immune-related adverse event incidence either occurs at similar rates [82,101,107] or at elevated rates [108,138] in elderly patients compared to younger patients treated with immune checkpoint inhibitor therapy.
Figure 2.
Summary of the age-associated trends relevant to immune checkpoint inhibitor (ICI) response and toxicity. With age, senescent cell burden and cancer risk increase colinearly. Immune effector function peaks during young adulthood and diminishes in elderly individuals. ICI efficacy has been demonstrated to both increase [67] and decrease [131] with aging and senescence, but in different contexts. Immune-related adverse event incidence either occurs at similar rates [82,101,107] or at elevated rates [108,138] in elderly patients compared to younger patients treated with immune checkpoint inhibitor therapy.
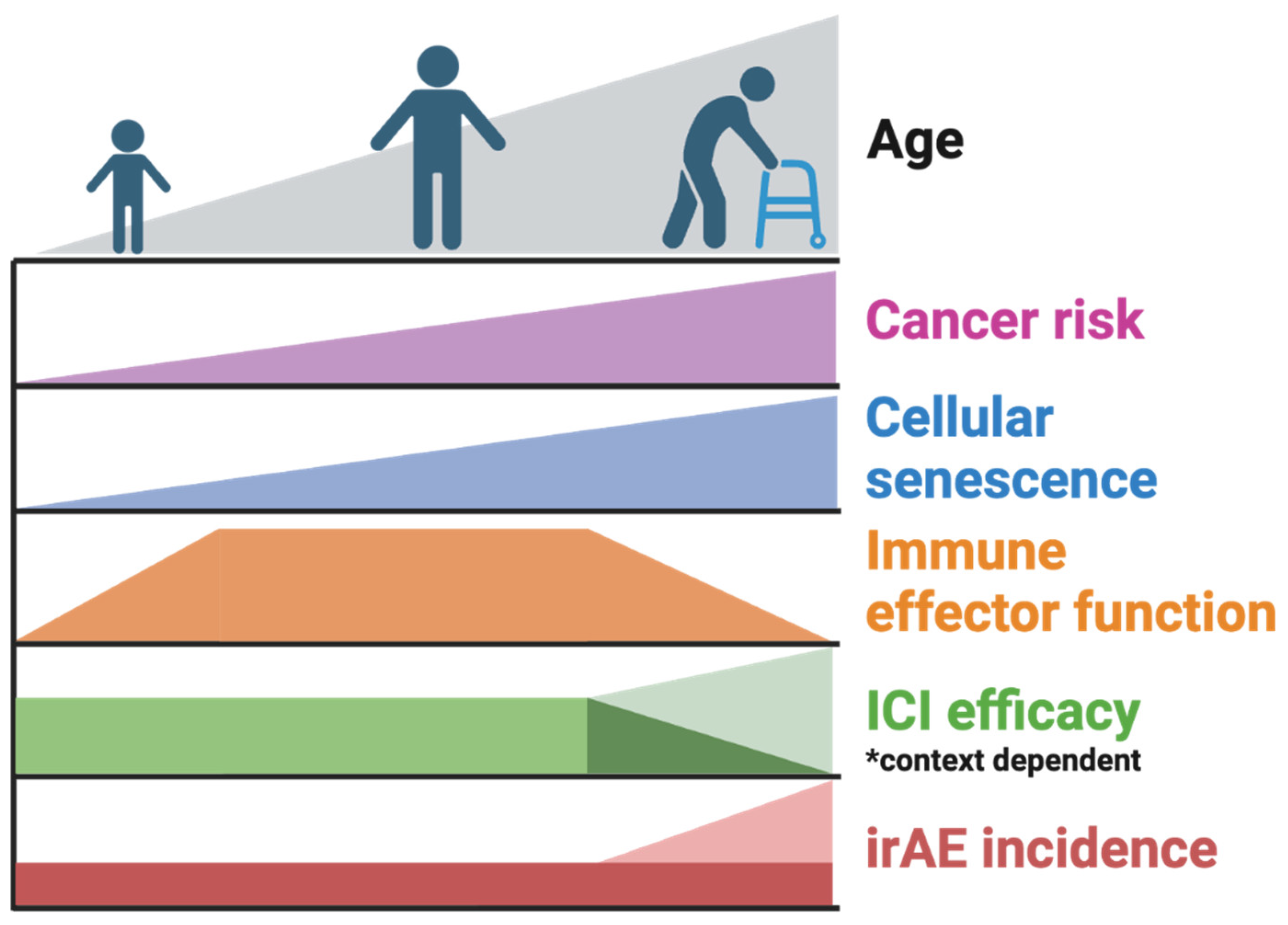
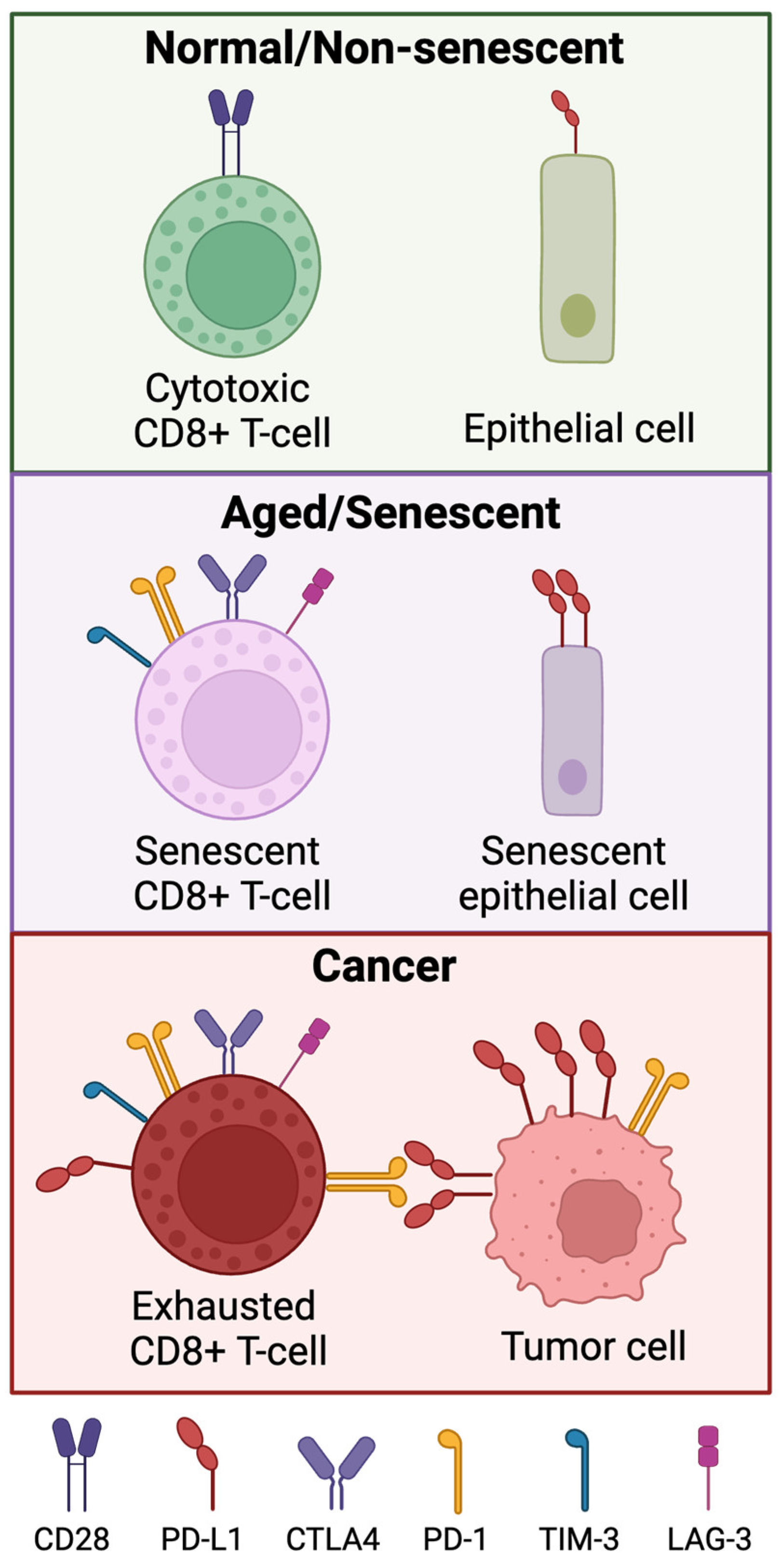
Figure 3.
Expression of immune checkpoint proteins in normal, aged/senescent, cancer cells. Senescent and tumor-associated T-cells have loss of CD27 and CD28. Senescent immune cells express higher levels of PD-1, TIM-3, LAG-3 and CTLA4. Cancer-associated T cells also express these markers, and the interaction between PD-1 and PD-L1 contributes to the exhausted phenotype in tumor-associated T cells. Normal epithelial cells express very low levels of PD-L1, but senescent epithelial cells express PD-L1 at a notably higher level.
Figure 3.
Expression of immune checkpoint proteins in normal, aged/senescent, cancer cells. Senescent and tumor-associated T-cells have loss of CD27 and CD28. Senescent immune cells express higher levels of PD-1, TIM-3, LAG-3 and CTLA4. Cancer-associated T cells also express these markers, and the interaction between PD-1 and PD-L1 contributes to the exhausted phenotype in tumor-associated T cells. Normal epithelial cells express very low levels of PD-L1, but senescent epithelial cells express PD-L1 at a notably higher level.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



