
Submitted:
31 May 2024
Posted:
31 May 2024
You are already at the latest version
Abstract
Porcine parvovirus 8 (PPV8), a novel virus in the Parvoviridae family, was first identified in 2022 in lung samples of domestic pigs from China. Retrospective analyses showed that it had been circulating in China since 1998, but no other countries had reported its presence so far. A recent study conducted in South America did not detect any PPV8-positive samples in that region. Here we report the detection of PPV8 in Hungarian and Slovakian pig farms and the estimated prevalence of the virus in Hungary. Altogether 2230 serum, 233 oral fluid, and 115 processing fluid samples were systematically collected from 23 Hungarian and 2 Slovakian pig farms between 2020 and 2023. A real-time quantitative PCR method was developed to detect the viral genome. Our results revealed the presence of PPV8 on 65% of the Hungarian farms and on both Slovakian farms included in our study, marking its first detection in Europe. Oral fluid samples showed the highest positivity rates, reaching up to 100% in some herds. The viral genome was successfully detected in serum and processing fluid samples too, but with significantly lower prevalence rates of 4% and 5%, respectively. Genetic analysis of 11 partial VP2 sequences demonstrated high similarity to the original Chinese strain but with unique amino acid mutations, suggesting possible local evolution of the virus. Our study presents the first scientific evidence of PPV8 infection outside of China and offers a comprehensive assessment of its prevalence in the Hungarian pig population. Further research is required to understand its potential impact on swine health.
Keywords:
porcine parvovirus 8
; novel porcine parvoviruses
; first detection
; viral prevalence
; oral fluid
1. Introduction
Porcine parvovirus 8 (PPV8), a novel member of the Parvoviridae family, was first detected in 2022 by high-throughput sequencing (HTS) of lung tissue samples originating from PRRSV-positive pigs in China [1]. Parvoviruses are small, non-enveloped viruses with linear, single-stranded DNA genome, typically ranging from 4 to 6.3 kilobases in length. Their genome contains two major open reading frames (ORFs) that encode for the non-structural proteins (NS) and capsid proteins (VP), crucial for the viral replication and structure, respectively [2].
The Parvoviridae family is divided into three subfamilies: Parvovirinae and Hamaparvovirinae, which infect vertebrates and the Densovirinae, which infect arthropods. To date, eight different parvoviruses have been identified in pigs (PPV1–8), which are classified into the Protoparvovirus, Tetraparvovirus, Copiparvovirus and Chapparvovirus genera [3].
The first known PPV (PPV1) was identified in the 1960s, and remains a major causes of reproductive losses in pigs [4]. Recent advances in metagenomic technology have led to the identification of novel PPVs worldwide [1,5,6,7,8,9,10]. However, unlike PPV1, their pathogenic roles and clinical relevance have not been defined.
Genomic comparisons show that PPV8 shares 44.18% nucleotide identity with PPV1, but only 16.23–24.17% similarity with PPV2–7, indicating significant genetic diversity among novel PPVs. Despite its relatively low sequence similarity to other PPVs, PPV8 retains conserved amino acid sites that are characteristic of parvoviruses [1]. Phylogenetic analysis based on the NS1 gene further supports that PPV8 is significantly distinct from other known novel parvoviruses, as it shares the highest sequence similarity with PPV1 and clusters together with Protoparvoviruses [1]. According to the latest ICTV classification criteria for parvoviruses, it qualifies as a new species within the Protoparvovirus genus [3].
The first retrospective study of PPV8 suggests that the virus has wide geographical distribution and long-term existence in China [1], but it has not been reported outside of this country. A recent study in Columbia found no PPV8-positive cases in 234 serum samples of gilts collected from 40 different, clinically healthy herds [11]. In the present study, our aim was to screen large-scale pig herds in Hungary and Slovakia to detect the presence of PPV8.
2. Materials and Methods
2.1. Sample collection
Serum, oral fluid and processing fluid samples were gathered between 2020 and 2023, from 23 Hungarian and 2 Slovakian pig farms to screen for various pathogens (Table 1.). Samples were systematically collected on farrow-to-finish farms from pigs of different age groups (2-, 4-, 6-, 8-, 10-, 14- and 18-week-old pigs, gilts, and sows of 2 and 4 parities) on farms with sow herd size between 520 and 2200. In total, 2230 serum samples, 233 oral fluid and 115 processing fluid samples were obtained, and all of them were stored at –80 °C until processing and further analysis. The farms participation in this sampling campaign was voluntary, with no significant clinical diseases reported during the period of samplings. More details of the sampling strategy can be found in our previous studies [12,13].
2.2. Sample processing and DNA extraction
Prior to DNA extraction the processing fluid and oral fluid samples were centrifuged and the serum samples belonging to the same age group were pooled by 5. The viral DNA was extracted from oral fluid, processing fluid and serum samples with the QIAmp cador Pathogen Mini Kit (Qiagen, Hilden, Germany) using the QIAcube automatic nucleic acid extractor according to the manufacturer’s protocol. Nucleic acids were stored at –80°C until further analysis.
2.3. qPCR detection and genetic analysis of PPV8
Real-time quantitative PCR (qPCR) was used to detect the presence of the viral DNA in the examined samples. The PCR assays were run on a Q qPCR Machine (Quantabio, Beverly, MA, USA), and each reaction contained 10 µL PerfeCTa qPCR ToughMix (Quantabio Beverly, MA, USA), 2 µL extracted DNA, 900 nM specific primers (forward: 5’-GCATGATGCCATACACACC-3’ and reverse: 5’-TGTCTTGTTGCTTGTCCTTG-3’) and 250 nM probe 5’-HEX-TGGAACCCTTTCGTTCCTCCAATCTACAA-BHQ-3’) in a 20 µL final volume. The primers and probe were designed to detect the VP2 capsid gene using the Eurofins PCR Primer Design Tool (https://eurofinsgenomics.eu/en/ecom/tools/pcr-primer-design/). For the PCR reactions we used the following temperature profile: 95 °C for 3 min followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. Statistical analysis of the qPCR results was conducted with GraphPad Prism 8. Fisher's exact test was used to compare the prevalence of PPV8 across different diagnostic matrices. The Ct values detected in different sample types were analysed with the Mann–Whitney test through pairwise comparison. Statistical significance was set at p < 0.05.
Sequencing PCR reactions were carried out according to the PCR method described by Guo et al. 2022 [1]. In each reaction 5 µl extracted DNA was added to a mix of 5× PhusionTM HF Buffer (Thermo ScientificTM), 200 µM dNTPs, 1 µM of each primer and 0.5 units of PhusionTM High-Fidelity DNA Polymerase. The reactions were run in a Genesy 96T gradient PCR machine (Tianlong, China) with the following temperature profile: 98 °C for 30 s followed by 45 cycles of 98 °C for 10 s, 65 °C for 20 s and 72°C for 45 s followed by a final elongation step at 72°C for 5 min. After agarose gel electrophoresis, the 554 bp long amplicons were manually cut and then purified using Qiagen Gel Extraction Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Capillary electrophoresis was conducted by a commercial provider, Eurofins BIOMI Kft. (Gödöllő, Hungary). All chromatograms were visualized and trimmed using Chromas 2.6.6 software (Technelysium Pty Ltd., South Brisbane, Australia). The obtained sequences were compared with each other, and a reference strain downloaded from GenBank (OP021638). The alignment against the reference PPV8 sequence was performed using MEGAX software [14]. All PPV8 sequences detected in this study have been deposited in the NCBI GenBank under the accession numbers PP781989 – PP781999.
3. Results and Discussion
In recent years, the incidence of emerging infectious diseases in pigs has been increasing, spreading rapidly due to global trade and the movement of people and animals. Although the impact of these newly identified viruses on swine health is still largely unknown, detecting and assessing their prevalence in pig populations can help us understand the significance and epidemiology of emerging viruses. The aim of this study was to investigate the presence of PPV8 infection in our sample collection. For rapid and specific detection of the PPV8 viral DNA, we developed a probe-based qPCR method and examined serum, oral fluid, and processing fluid samples collected from 23 Hungarian and 2 Slovakian pig farms.
Fifteen out of 23 Hungarian farms tested (65%) were positive for PPV8 (Figure 1), indicating that the viral genome was detected in at least one sample from each of these farms. The within-herd prevalences varied significantly among the PPV8-positive farms (Table 1). Comparing the different diagnostic matrices, oral fluid samples exhibited notably high detection rates (Figure 2A), ranging from 33.3% to 100% in positive herds. Overall, 46% (101/218) of the oral fluid samples were PPV8-positive. The high positivity rate in oral fluids can be explained by their pen-representative nature, collected from weaned pigs and fatteners across different production units. Although this sampling method increases the likelihood of contamination, the ease of collection and ability to represent a collective status make oral fluids suitable for herd- or group-level surveillance. Despite representing multiple animals, some PPV8-positive oral fluid samples were found with notably lower Ct values, compared to other samples, indicating higher viral concentrations in these cases (Figure 2/B). The mean Ct values for weaned pigs (8–12 weeks old) were 31.61±3.22, while those for fatteners (18–20 weeks old) were 32.56±3.47. No significant differences were observed between viral prevalence among weaned pigs and fatteners, as 45% (51/114) of weaned pigs and 48% (50/104) of fatteners were PPV8-positive.
We also detected the virus in 4% (15/412) of serum pools, indicating active viraemia in some animals during the sampling. The within-herd prevalences ranged between 5% and 30%, with mean Ct values of 31.79±2.56. The most affected age groups were 10-week-old pigs with 8% PPV8-positivity rate, and 8- and 10-week-old pigs, each with a 7% positivity rate. This pattern aligns with the ages of the animals from which the oral fluid samples were collected. PPV8-positive processing fluids, suggesting congenital infections, were only found in one herd, where all five samples tested positive, resulting in an overall prevalence of 5% (5/106). The Ct values of this sample type were significantly higher (35.87±1.24) than those detected in serum samples or oral fluids (Figure 2/B). In Slovakia, we also detected the PPV8 viral DNA in samples collected from two farms, identifying three PPV8-positive oral fluid samples in total.
According to our previous results, it can be stated that all novel PPVs, including PPV8 are most commonly detected in oral fluid samples and have the lowest prevalence in processing fluids (manuscript under review). These results align with a similar study conducted in Poland, where high detection rates of novel PPVs were observed in oral fluid samples during the examination of large numbers of serum, oral fluid, and fecal samples [15].
We have successfully obtained 554 bp long partial VP2 sequences from four serum and seven oral fluid (six Hungarian and one Slovakian) samples. The initial identification of the obtained sequences using the NCBI BLAST system showed one match, which was the only existing PPV8 sequence to date. All Hungarian sequences displayed 98.02–99.10% nucleotide identity compared to the Chinese PPV8 strain GDJM2021. Similarly, the Slovakian strain showed high similarity to the Chinese (99.1%) and the Hungarian sequences also (97.83–99.64%). Two amino acid changes were identified in the examined section of the capsid gene. Starting from the VP1 gene’s start codon, the K392Q mutation was found in three strains and the V410I mutation was present in two strains (Figure 3). The origins of these sequences are indicated on the map with blue and red circles, respectively (Figure 1). The geographically clustered farms where these mutations were found are located in the southwestern part of Hungary. Since these amino acid mutations occur in the VP2 capsid gene, they might affect the receptor binding and antigenic properties of the virus. Further research is required to determine the virus’ exact structure, virulence and impact on the host organism.
5. Conclusions
Our study confirms the presence of PPV8 in Europe, specifically within Hungarian and Slovakian swine herds. The samples were collected between 2020 and 2023 which means that PPV8 have been circulating in this region for at least four years. Since the examined herds reported no clinical symptoms or major disease outbreaks at the time of the sampling, conclusions regarding the pathogenicity of PPV8 cannot be drawn. Further research is needed to assess the virus's prevalence in Europe and to understand its impact on swine health.
Author Contributions
Conceptualization, G.B. and B.I.; methodology and investigation, G.B., B.I, L.D and K.S.; writing—original draft preparation, B.I.; writing—review and editing, G.B. and B.I. All authors have read and agreed to the published version of the manuscript.
Funding
Project no. RRF-2.3.1-21-2022-00001 has been implemented with the support provided by the Recovery and Resilience Facility (RRF), financed under the National Recovery Fund budget estimate, RRF-2.3.1-21 funding scheme. Barbara Igriczi was supported by the ÚNKP-23-3 (ÚNKP-23-3-II-ÁTE-17) New National Excellence Program of the Ministry for Culture and Innovation from the source of the National Research, Development and Innovation Fund. The funding sources had no role in study design, data collection and analysis or the decision to publish the work.
Institutional Review Board Statement
The active surveillance sampling was approved by the National Scientific Ethical Committee on Animal Experimentation (ethical permission number: PE/EA/544-5/2018).
Informed Consent Statement
Not applicable.
Data Availability Statement
Sequence data gathered during the study can be accessed under GenBank accession numbers: PP781989 – PP781999. Data regarding the name and exact location of the farms involved in the study is confidential due to business secret of the owners.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Y. Guo et al., ‘Identification and genomic characterization of a novel porcine parvovirus in China’, Front. Vet. Sci., vol. 9, p. 1009103, Sep. 2022. [CrossRef]
- F. Streck, C. W. Canal, and U. Truyen, ‘Molecular epidemiology and evolution of porcine parvoviruses’, Infection, Genetics and Evolution, vol. 36, pp. 300–306, Dec. 2015. [CrossRef]
- S. F. Cotmore et al., ‘ICTV Virus Taxonomy Profile: Parvoviridae’, Journal of General Virology, vol. 100, no. 3, pp. 367–368, Mar. 2019. [CrossRef]
- W. L. Mengeling, K. M. Lager, and A. C. Vorwald, ‘The effect of porcine parvovirus and porcine reproductive and respiratory syndrome virus on porcine reproductive performance’, Anim Reprod Sci, vol. 60–61, pp. 199–210, Jul. 2000. [CrossRef]
- M. Hijikata, K. Abe, K. M. Win, Y. K. Shimizu, N. Keicho, and H. Yoshikura, ‘Identification of new parvovirus DNA sequence in swine sera from Myanmar’, Jpn J Infect Dis, vol. 54, no. 6, pp. 244–245, 2001. [CrossRef]
- S. K. P. Lau et al., ‘Identification of novel porcine and bovine parvoviruses closely related to human parvovirus 4’, Journal of General Virology, vol. 89, no. 8, pp. 1840–1848, 2008. [CrossRef]
- K. Cheung, G. Wu, D. Wang, D. O. Bayles, K. M. Lager, and A. L. Vincent, ‘Identification and molecular cloning of a novel porcine parvovirus’, Arch Virol, vol. 155, no. 5, pp. 801–806, May 2010. [CrossRef]
- C.-T. Xiao, L. G. Giménez-Lirola, Y.-H. Jiang, P. G. Halbur, and T. Opriessnig, ‘Characterization of a Novel Porcine Parvovirus Tentatively Designated PPV5’, PLoS ONE, vol. 8, no. 6, p. e65312, Jun. 2013. [CrossRef]
- J. Ni et al., ‘Identification and genomic characterization of a novel porcine parvovirus (PPV6) in china’, Virology Journal, vol. 11, no. 1, p. 203, Dec. 2014. [CrossRef]
- R. M. Palinski, N. Mitra, and B. M. Hause, ‘Discovery of a novel Parvovirinae virus, porcine parvovirus 7, by metagenomic sequencing of porcine rectal swabs’, Virus Genes, vol. 52, no. 4, pp. 564–567, 2016. [CrossRef]
- D. S. Vargas-Bermudez, A. Diaz, G. Polo, J. D. Mogollon, and J. Jaime, ‘Infection and Coinfection of Porcine-Selected Viruses (PPV1 to PPV8, PCV2 to PCV4, and PRRSV) in Gilts and Their Associations with Reproductive Performance’, Veterinary Sciences, vol. 11, no. 5, Art. no. 5, May 2024. [CrossRef]
- Igriczi, L. Dénes, I. Biksi, E. Albert, T. Révész, and G. Balka, ‘High Prevalence of Porcine Circovirus 3 in Hungarian Pig Herds: Results of a Systematic Sampling Protocol’, Viruses, vol. 14, no. 6, p. 1219, Jun. 2022. [CrossRef]
- Igriczi, L. Dénes, A. Czétényi, T. Révész, Z. Somogyi, and G. Balka, ‘Prevalence Estimation and Genetic Characterization of Porcine Parainfluenza Virus 1 (PPIV-1) in Hungary and the First Report of the Virus in Slovakia’, Transboundary and Emerging Diseases, vol. 2024, p. e5534854, Jan. 2024. [CrossRef]
- S. Kumar, G. Stecher, M. Li, C. Knyaz, and K. Tamura, ‘MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms’, Molecular Biology and Evolution, vol. 35, no. 6, pp. 1547–1549, Jun. 2018. [CrossRef]
- Miłek, A. Woźniak, M. Guzowska, and T. Stadejek, ‘Detection Patterns of Porcine Parvovirus (PPV) and Novel Porcine Parvoviruses 2 through 6 (PPV2–PPV6) in Polish Swine Farms’, Viruses, vol. 11, no. 5, p. 474, May 2019. [CrossRef]
Figure 1.
The geographical location of the examined farms. The blue and red circles around the farms indicates the origin of strains with K392Q and V410I amino acid mutations, respectively.
Figure 1.
The geographical location of the examined farms. The blue and red circles around the farms indicates the origin of strains with K392Q and V410I amino acid mutations, respectively.
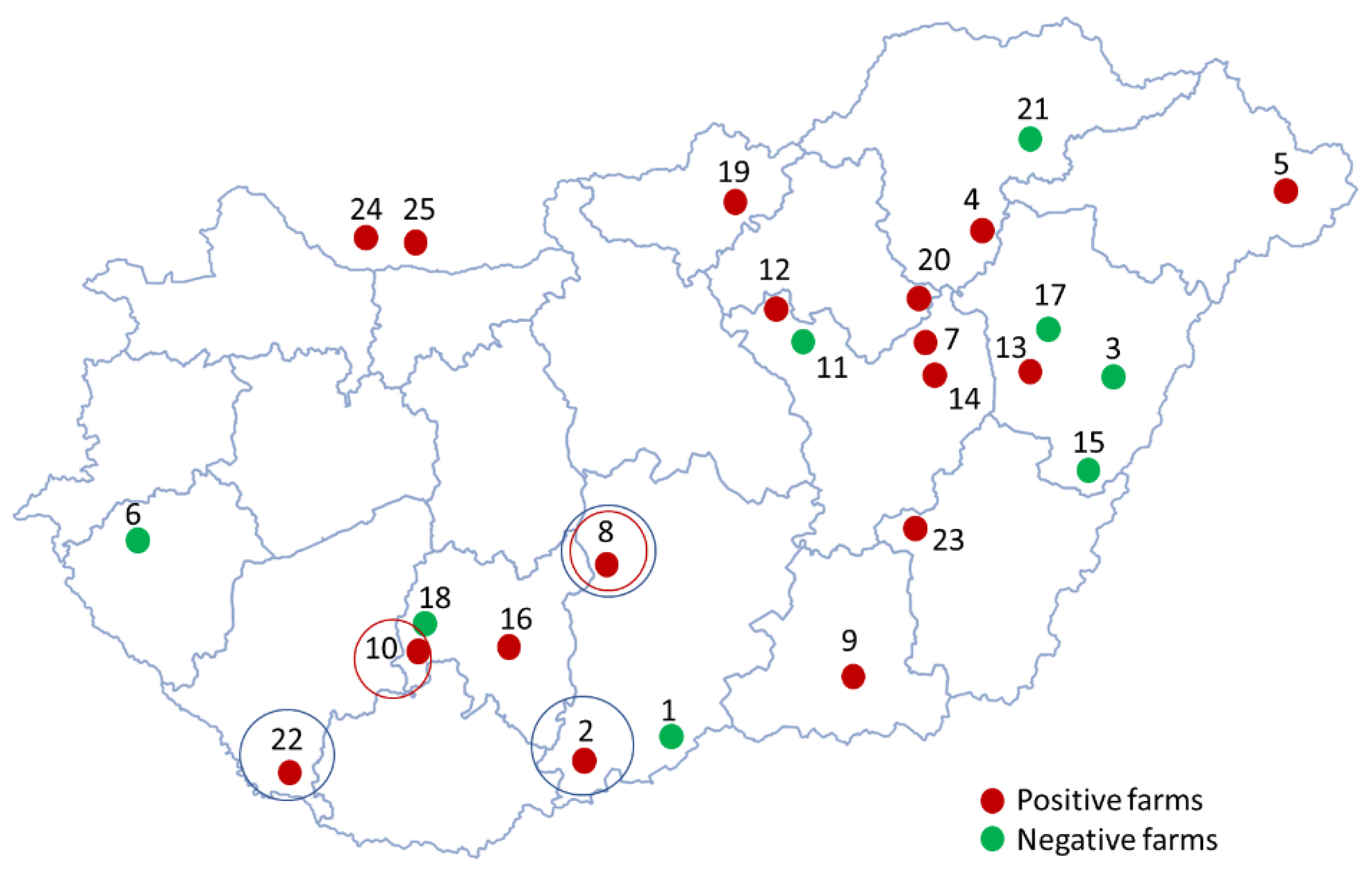
Figure 2.
(a) Percentages of PPV8-positive serum pools, oral fluid and processing fluid samples collected in Hungary (b) Boxplots representing the Ct values of the PPV8-positive serum pools, oral fluid and processing fluid samples collected in Hungary. The whiskers show the minimum and the maximum, and the average values are indicated with “+” signs. The horizontal lines of the box show the first quartile, the median and the third quartile. The asterisks above the columns represent the statistically significant differences (*:p < 0.05; **: p < 0.01).
Figure 2.
(a) Percentages of PPV8-positive serum pools, oral fluid and processing fluid samples collected in Hungary (b) Boxplots representing the Ct values of the PPV8-positive serum pools, oral fluid and processing fluid samples collected in Hungary. The whiskers show the minimum and the maximum, and the average values are indicated with “+” signs. The horizontal lines of the box show the first quartile, the median and the third quartile. The asterisks above the columns represent the statistically significant differences (*:p < 0.05; **: p < 0.01).
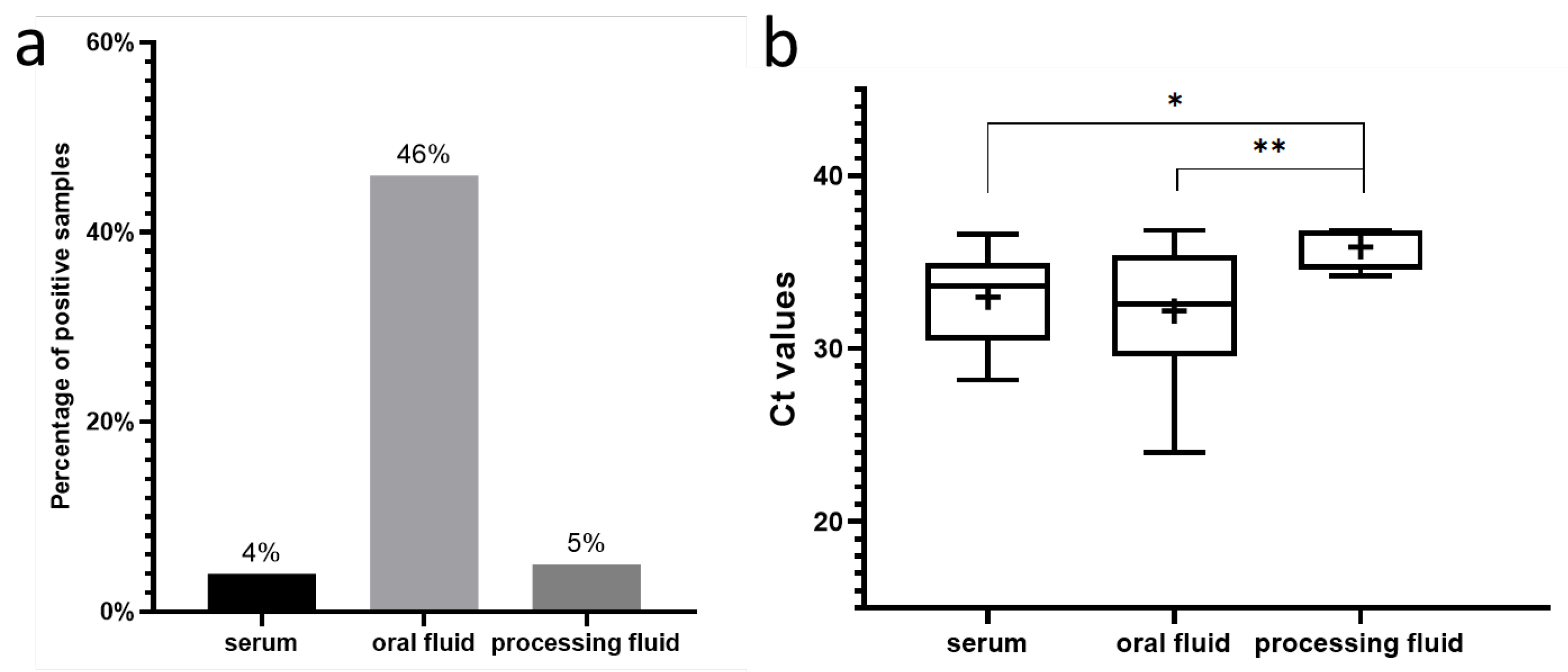
Figure 3.
Amino acid order of the PPV8 strains detected in this study, highlighting the two amino acid mutations, compared to the PPV8-GDJM2021 strain.
Figure 3.
Amino acid order of the PPV8 strains detected in this study, highlighting the two amino acid mutations, compared to the PPV8-GDJM2021 strain.
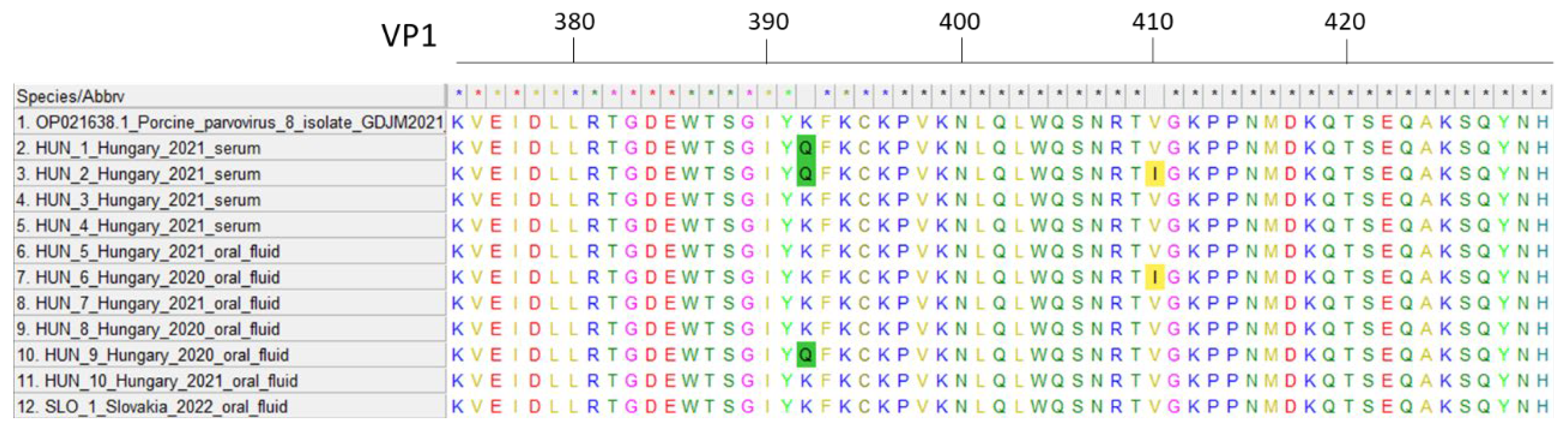

Table 1.
Summary of the examined Hungarian and Slovakian farms, sample types and sample sizes and the number of PPV8-positive samples.
Table 1.
Summary of the examined Hungarian and Slovakian farms, sample types and sample sizes and the number of PPV8-positive samples.
| Hungarian farms | serum samples | serum pools | PPV8- positive |
oral fluids |
PPV8-positive | processing fluids | PPV8- positive |
|---|---|---|---|---|---|---|---|
| Farm 1 | 100 | 20 | 0 | 10 | 0 | 5 | 0 |
| Farm 2 | 100 | 20 | 3 | 10 | 9 | 4 | 0 |
| Farm 3 | 100 | 20 | 0 | 12 | 0 | 10 | 0 |
| Farm 4 | 0 | 0 | 0 | 10 | 9 | 3 | 0 |
| Farm 5 | 100 | 20 | 0 | 10 | 10 | 5 | 0 |
| Farm 6 | 100 | 20 | 0 | 10 | 0 | 8 | 0 |
| Farm 7 | 90 | 18 | 0 | 10 | 9 | 1 | 0 |
| Farm 8 | 130 | 26 | 2 | 10 | 5 | 2 | 0 |
| Farm 9 | 100 | 20 | 1 | 10 | 5 | 5 | 0 |
| Farm 10 | 100 | 20 | 1 | 10 | 8 | 5 | 0 |
| Farm 11 | 80 | 16 | 0 | 8 | 0 | 5 | 0 |
| Farm 12 | 100 | 20 | 0 | 9 | 3 | 5 | 0 |
| Farm 13 | 100 | 20 | 2 | 10 | 6 | 5 | 5 |
| Farm 14 | 60 | 12 | 0 | 10 | 10 | 5 | 0 |
| Farm 15 | 100 | 20 | 0 | 10 | 0 | 5 | 0 |
| Farm 16 | 70 | 14 | 0 | 5 | 2 | 0 | 0 |
| Farm 17 | 100 | 20 | 0 | 10 | 0 | 5 | 0 |
| Farm 18 | 60 | 12 | 0 | 4 | 0 | 4 | 0 |
| Farm 19 | 100 | 20 | 0 | 10 | 5 | 5 | 0 |
| Farm 20 | 100 | 20 | 0 | 10 | 10 | 3 | 0 |
| Farm 21 | 70 | 14 | 0 | 10 | 0 | 2 | 0 |
| Farm 22 | 100 | 20 | 0 | 10 | 5 | 5 | 0 |
| Farm 23 | 100 | 20 | 6 | 10 | 5 | 9 | 0 |
| Total | 2060 | 412 | 15 | 218 | 101 | 106 | 5 |
| Slovakian farms | |||||||
| Farm 24 | 100 | 20 | 0 | 10 | 1 | 5 | 0 |
| Farm 25 | 70 | 14 | 0 | 5 | 2 | 4 | 0 |
| Total | 170 | 34 | 0 | 15 | 3 | 9 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



