
Submitted:
02 June 2024
Posted:
03 June 2024
You are already at the latest version
Abstract
Cellular senescence has recently been recognized as a hallmark of cancer, reflecting its association with aging and inflammation, its role as a response to deregulated proliferation and oncogenic stress, and its induction by cancer therapies such as radiation, chemotherapy, and targeted agents. While such therapy-induced senescence (TIS) has been linked to resistance, recurrence, metastasis and normal tissue toxicity, TIS has the potential to enhance therapy response and stimulate anti-tumor immunity. In this review, we examine the Jekyll and Hyde nature of senescent cells (SnCs), with a particular focus on how their persistence while expressing the senescence-associated secretory phenotype (SASP) modulates the tumor microenvironment through autocrine and paracrine mechanisms. Via the SASP, the formation of SnCs may alternately mediate resistance or response to conventional therapy. Further, toward fulfilling the unmet potential of cancer immunotherapy, we consider how SnCs may influence tumor inflammation and serve as a source of antigen to potentiate anti-tumor immunity. This new view suggests treatment strategies based on inducing TIS to enhance immune checkpoint blockade. Finally, we describe strategies for mitigating the detrimental effects of senescence, such as modulating the SASP or targeting SnC persistence, toward enhancing the benefits of cancer treatment.
Keywords:
senescence
; SASP (senescence‐associated secretory phenotype)
; immune surveillance
; immunosuppression
; senolytics
; tumor microenvironment
; cancer therapy
; therapy resistance
1. Introduction
Cellular senescence was first described as an in vitro phenomenon, the Hayflick limit [1], recognizing the loss of proliferative potential during serial passage as a form of cellular aging that is now known as replicative senescence (RS). It took several decades for evidence to overcome the argument that senescence was simply a culture artifact, including studies with human tissues establishing a pathway from telomere shortening to RS [2] and demonstrating the accumulation of senescent cells (SnCs) during aging [3]. Beyond proliferation in the absence of telomerase, senescence can be triggered by replication fork collapse, oxidative damage, oncogenic stress and cancer therapies, all potentially leading to irreparable DNA damage (DDR) [4]. Despite entering a similarly prolonged or permanent growth arrest, unlike quiescent cells, SnCs remain metabolically active. SnCs typically express a characteristic range of proteins that decorate the surface or are released into their microenvironment known as the senescence-associated secretory phenotype (SASP) that includes cytokines, damage-associated molecular patterns (DAMPs), and other signals. These SASP factors along with metabolic changes can alter cellular behaviors in an autocrine and/or paracrine manner [5,6,7,8,9]. A striking effect of SnC metabolism and the SASP is to induce a bystander effect in neighboring cells [10], propagating senescence through tissues and providing a cellular basis for organ dysfunction and organismal aging.
Along with its association with aging, senescence has been recognized as a hallmark of cancer [5]. Whether senescence is beneficial or detrimental to cancer formation, progression and treatment has remained controversial over the past decades. On the one hand, the induction of senescence can serve as a barrier against malignant transformation and excessive hyperproliferation due to reduced proliferative capacity [11,12]. On the other hand, the accumulation of SnCs has been regarded as a driver of cancer progression and therapy resistance, primarily mediated by inflammatory factors in the SASP [13]. Along these lines, persistent senescence has been associated with promoting malignant transformation, accelerating tumor growth, inducing cancer stemness, facilitating distant metastasis, maintaining chronic inflammation, and dampening the anti-tumor immune response [14,15,16,17]. Conversely, genetic elimination of SnCs has been shown to delay spontaneous tumorigenesis and reduce cancer-related mortality [18,19,20]. Senolytic drugs that selectively target senescent cells have also demonstrated significant potential in improving cancer therapies [9,21,22]. However, our research [23] and other studies have pointed to an opposing effect due to vaccine-like properties of SnCs in inhibiting tumor growth and boosting radiation therapy. More recently, an increasing number of studies and reviews have discussed anti-tumor effects of SnCs linked to their immunogenicity and immune surveillance mechanisms that normally limit the persistence and accumulation of SnCs as beneficial [24,25,26]. These studies point to upregulation of inflammatory mediators including DAMPs, chemotactic factors, and antigen presentation machinery as features that contribute to activation of both innate and adaptive immune responses. Recent findings that SnCs can express immune checkpoint factors that limit their surveillance suggest them as potential targets for immune checkpoint blockade (ICB) therapies. Such findings underscore the positive aspects of TIS, extending beyond suppression of cell proliferation to significantly enhance anti-tumor immunity.
This review examines cellular senescence in the context of cancer, highlighting diverse roles of SnCs in the tumor microenvironment (TME) and arguing for a broad view of senescence and its functions. Furthermore, we will discuss how the interaction between SnCs and the immune system can lead to either beneficial or detrimental outcomes depending on the specific features of SnCs, particularly the SASP. We will then delve into the potential for modulating the SASP and eliminating SnCs using senolytics. These approaches may be able to limit the adverse effects of senescence while amplifying its beneficial impact, offering a practical strategy to improve cancer therapies.
2. Senescence in the Context of Cancer
Senescence is a normal fate for cells in diverse physiological contexts, including embryonic development [27,28], wound healing [29,30], normal aging [31], viral infection [32], and other stress-related conditions. In cancer, SnCs can originate from different sources and are detected in premalignant lesions as well as within the tumor microenvironment (TME) during different phases of cancer initiation, progression, and post therapies [33,34]. These non-proliferative cells can be triggered by diverse stimuli, including reactive oxygen species (ROS), DNA damage, imbalanced cellular signaling networks, and epigenetic alterations.
One of the core pathways linking DNA damage to senescence is the tumor suppressor and transcription factor p53, whose activation drives the expression of cyclin-dependent kinase (CDK) inhibitors, such as p16INK4a and p21CIP, leading to cell cycle arrest. In SnCs, the pro-apoptotic effects of p53 appear to be offset by other factors that maintain cell survival, such as Bcl-2 family proteins.
2.1. Oncogene-Induced Senescence (OIS)
Oncogene activation is a potent inducer of cellular senescence. Initial studies demonstrated that cultured primary mammalian cells with oncogenic Ras overexpression undergo senescence in a p53-dependent manner in vitro [35]. Subsequent work showed that deregulated Ras activity induces senescence in the mammary gland in vivo [36]. Other oncogenes, such as Mos, Cdc6, and Cyclin E, have also been described as senescence inducers, reinforcing the notion that OIS provides a barrier to malignant progression [37]. The mechanisms of OIS are notably linked to DNA damage signaling [37,38]. Indeed, Ras activation has been shown to trigger DNA hyper-replication, leading to replication fork stalling and collapse [39] and telomere attrition [40], which can elicit the DDR and induce senescence. Oncogene activation and OIS is also associated with increased ROS that may drive cell proliferation but can also cause DNA damage, cell cycle arrest, and ultimately senescence [41,42]. In addition to oncogene activation, the inactivation of tumor suppressors, such as PTEN, can also induce cellular senescence [43,44]. While one study found that PTEN loss induces DNA damage, leading to senescence in mouse prostate epithelial cells [43], other studies suggest that PTEN loss can cause senescence independently of DNA damage, but rather through increased p53 stabilization that consequently enhances transcription of p53 targets like p21CIP [44].
2.2. Therapy-Induced Senescence (TIS)
Cellular senescence that occurs in response to genotoxic stress from radiation and/or chemotherapies is known as therapy-induced senescence (TIS) [14,45]. DNA damaging agents used in cancer treatment can generate DNA single- and/or double-strand breaks on telomeres [46] or other chromosome regions [47]. Such damage initiates the DDR through a kinase cascade involving ATM, ATR, CHK1, and CHK2 [48]. These signaling events converge at the tumor suppressor p53, resulting in cell cycle arrest. While cell proliferation can resume when DNA damage is repaired, irreparable DNA damage induces a prolonged DDR and extended growth arrest that can progress to cellular senescence [49]. Inhibiting DDR kinases such as ATM may allow cells to bypass senescence or even permit SnCs to re-enter the cell cycle [50].
Although a p53-dependent cell cycle checkpoint is often described as an essential mechanism to induce TIS, p53 negative cancer cells can be driven into senescence and genotoxic stress is not required per se. After treatment with spindle poisons or other agents that disrupt mitosis, surviving cells can enter senescence without DNA damage [51]. In turn, blocking cytokinesis by inhibiting Aurora kinases or Polo-like kinase 1 results in mitotic slippage, leading to binucleate G1 cells that progress to senescence [52,53]. Simply enforcing G1 arrest is sufficient, as targeting CDK4/6 kinase with inhibitors such as Palbociclib or Abemaciclib prevents E2F-mediated transcription and indirectly leads to senescence [54]. Targeting chromatin modifications with epigenetic drugs such as the histone deacetylase inhibitors, sodium dibutyrate (SDB) and Trichostatin A (TSA) [55], can also induce senescence, potentially via a similar mitotic slippage-mediated pathway leading to proliferative arrest and progression to senescence [56]. Reexamining how therapy-induced DNA damage may induce senescence similarly implicated mitotic slippage as the relevant outcome of prolonged checkpoint arrest [57]. Cells responding to ROS exposure or telomere erosion also display a similar pattern of mitotic defects and cytokinesis failure followed by cellular arrest and onset of senescence. That so many different senescence-inducing perturbations may converge on this common pathway reinforces the need to consider how senescent cells were formed in predicting the effects of TIS may have on their microenvironment.
3. Hallmarks of Cellular Senescence
Along with activation of the lyosomal enzyme GLB1 that is detected as senescence-associated beta-galactosidase (SA-βGal) [58], SnCs typically display a characteristic enlarged and flattened cell morphology along with a combination of altered metabolism and changes in gene expression and signaling (Figure 1). Some features of SnCs are particularly heterogeneous, likely reflecting the variety of senescence-inducing mechanisms. Additionally, features often described as characteristic of senescence such as SA-βGal and the SASP are quite variable and can be observed in cells that do not otherwise display senescence phenotypes. These uncertainties have led to a literature devoted to how to detect and differentiate SnCs in vitro and in tissue [59,60,61] and provide further challenges in linking senescence to cancer.
3.1. The DNA Damage Response
SnCs often display a persistent DNA damage response (DDR) associated with constitutive staining for nuclear foci of phosphorylated histone H2AX (γH2AX), p53-binding protein (53BP1), and/or phosphorylated ATM [49]. The DDR in these cells can be initiated by cell-intrinsic factors, including telomeric damage, DNA replication errors, or replication fork collapse [38,62]. Genotoxic agents such as radiation and chemotherapeutic drugs that induce single- or double-strand breaks in chromosomal DNA can also serve as potent DDR activators [63,64]. In certain SnCs, prolonged DNA damage foci have been detected in undamaged chromatin that may be attributed to hyperactive ATM activity [57]. A critical component of the DDR signaling cascade is p53, which undergoes activating phosphorylation by DDR-associated kinases including ATM, CHK1 and CHK2, reducing affinity for the MDM2 E3 ubiquitin ligase and stabilizing p53 levels [63]. The transcriptional activity of p53 then drives the expression of genes that initiate and maintain the senescence phenotype [65]. Additionally, CHK1-mediated phosphorylation inhibits CDC25, blocking the G2/M transition to induce G2 arrest [66]. Notably, the DDR kinase cascade also influences other signaling pathways, including NF-κB, STING, MAPK, and STAT, contributing to characteristics of senescence beyond cell cycle arrest [67,68].
3.2. CDK Inhibitors and Cell Cycle Arrest
A DDR-related hallmark of SnCs is the upregulation of CDK inhibitors [69]. In particular, the expression of p16INK4A (encoded by CDKN2A) remains low in healthy, young tissues but increases under senescence-inducing stresses, making it a valuable senescence marker [31]. p16INK4A selectively blocks the activity of cyclin D-dependent kinases CDK4 and CDK6, facilitating Rb-associated cell cycle arrest [70,71]. As demonstrated in the INK-ATTAC and p16-3MR mouse models, targeted removal of p16-positive cells can potentially extend life and health span [30,72,73]. Along with p16INK4A, the CDKN2A gene encodes p14ARF in human or p19ARF in mouse, an alternate reading frame protein which shares p16INK4A exons 2 and 3 [74]. ARF modulates the cell cycle through its interaction with MDM2 to enhance p53-dependent gene transcription [75,76]. Another INK4 family member, p15INK4B (encoded by CDKN2C), may compensate in the absence of p16INK4A [77]. For instance, Akt-mediated p15INK4B expression is required for senescence in response to androgen stimulation in prostate cancer cells [78].
The expression of p21CIP/Waf1, encoded by the p53 target gene CDKN1A, is also increased by senescence-inducing stimuli [79]. While capable of inhibiting multiple cyclin/CDK complexes, p21CIP predominantly induces G1 cell cycle arrest via blocking CDK2 [80]. Beyond p53, p21CIP induction can also result from p53-independent stress signals, including TGF-β, Rb, and integrin, which facilitate Sp1/Sp3 transcription factor recruitment to the p21 promoter and increase p21 expression [81]. However, unlike the consistent upregulation of p16INK4A, p21CIP levels vary in senescence [82], limiting the value of p21 as a senescence marker.
3.3. Chromatin and Nuclear Changes
SnCs often undergo significant changes in chromosome structures and epigenetic modifications. Senescence-associated heterochromatic foci (SAHF), marked by large nucleoli and punctate DNA foci visible after DAPI staining, represent a distinct heterochromatic structure that correlates with the stable repression of E2F-targeted genes [83]. The development of SAHF and associated chromatin structure depends on the histone chaperone proteins ASF1A and HIRA [84], which are influenced by various signaling pathways, such as Rb and the NOTCH-HMGA1 axis [83,85]. Notably, SAHF formation is observed in human cells experiencing accelerated senescence in an ATR-dependent manner and less associated with RS or mouse cells [69,86].
A notable feature of SnCs is a decrease in levels of the nuclear inner membrane protein Lamin B1 [87,88]. This reduction is linked to large-scale chromatin remodeling including SAHF formation [89]. The decline in Lamin B1 also destabilizes the nuclear envelope [90]. This may facilitate the release of cytoplasmic chromatin fragments (CCFs) that trigger the cGMP-AMP synthase stimulator of interferon genes (cGAS-STING) pathway, amplifying the SASP and subsequent inflammation [91].
Epigenetic remodeling impacting senescence-associated gene expression is another hallmark of SnCs. Reduced Lamin B may lead to redistribution of the active mark H3K4me3 and repressive mark H3K27me3 [92]. The silencing mark H3K9me2 may be reduced in SnCs, due to DDR-induced proteasomal degradation of methyltransferases G9a and GLP [93]. Activation of histone acetyltransferase p300 and resulting acetylation may support senescence-specific gene expression [94], potentially linked to engagement of histone acetylation reader protein BRD4 in activating super-enhancers [95].
3.4. Resistance to Apoptosis
While cellular senescence and apoptosis can be triggered by similar stimuli [19], it has been shown that senescent human fibroblasts display resistance to p53-dependent apoptosis [96]. One factor may be persistent expression of the anti-apoptotic protein Bcl-2, providing a direct mechanism for evading apoptosis [97]. In SnCs, the Bcl-2 gene shows enhanced H4K16 acetylation but diminished H4K20Me3, suggesting active transcription, whereas BAX expression is repressed by a reverse modification pattern [98]. Other Bcl-2 family proteins, such as Bcl-w and Bcl-xl, are upregulated in both RS and OIS [99,100]. Additionally, p21CIP may inhibit NF-κB and subsequent JNK cascade dependent cell death in SnCs [101]. FOXO4 is another protein that enhances the survival of SnCs via blocking p53-induced apoptosis [102]. SnCs also display increased levels of other pro-survival proteins, including HSP90, Ephrins (EFNB1 or 3), PI3Kδ, and plasminogen-activator inhibitor-2 (PAI-2) [103,104]. Senolytics can induce apoptosis and selectively eliminate SnCs by targeting these anti-apoptosis mechanisms [100,103,104,105].
3.5. Dysfunctional Mitochondria
Mitochondrial dysfunction is recognized as a hallmark of cellular senescence [106]. SnCs display downregulation of fission protein 1 (Fis1) and dynamin-related protein 1 (Drp1), leading to an imbalance in mitochondrial fission and fusion and formation of hyper-fused mitochondria with altered morphology and function [107]. The accumulation of dysfunctional mitochondria is further compounded by reduced mitophagy, delaying mitochondrial turnover [108]. Mitochondrial dysfunction may be a direct cause of senescence insofar as mitochondrial depletion can prevent senescence in vitro and in vivo [109].
Increased mitochondrial mass and/or persistence correlates with higher oxygen consumption in TIS and OIS [110,111]. Impaired mitochondria are associated with increased ROS, which likely induce DDR and reinforce senescence [112,113]. Depending on how hypoxia or hyperoxia influences ROS, the impact of oxygen tension on senescence induction can vary [114]. Interestingly, both impairing mitochondrial respiration complexes and enhancing respiration via pyruvate dehydrogenase can increase redox stress and senescence [115,116]. Redox stress may also arise from accumulated labile iron, which could further contribute to the SASP in SnCs [117]. Additionally, the loss of antioxidant enzymes, such as superoxide dismutase (SOD), can drive senescence [115].
3.6. Deregulated Metabolism
Lipid accumulation, lipid signaling and deregulated lipid metabolism have long been linked to cellular senescence (Figure 2). Nearly three decades ago, Obeid and colleagues found that ceramide levels increase as cells enter RS and that simply feeding cells ceramide was sufficient to induce accelerated senescence in otherwise proliferative fibroblasts [118]. The pro-senescent effects of ceramide may be linked to mitochondrial dysfunction, altered mitophagy and ROS production [119,120]. Cholesterol biosynthetic pathways appear to modulate senescence by similar mechanisms, albeit more indirectly [121,122]. Similarly, exposing proliferative cells to free fatty acids can potentially induce senescence by increasing oxidative stress [123,124]. Along with sphingolipids, multiple lipid metabolic pathways are altered in SnCs [125,126,127]. Our studies noted the activation of diverse lipid metabolism pathways and increased lipid uptake, leading to accumulation of lipid droplets (LDs) in DNA damage-induced SnCs [124]. There is also an increase in lipid oxidative damage that leads to the upregulation of reactive aldehyde and responsive proteins, such as aldehyde dehydrogenases and lysosomal palmitoyl-protein thioesterases [124]. Lipid peroxidation, where unsaturated lipids are oxidized by a free radical-driven chain reaction that yields lipid-derived electrophiles, appears to be a common feature in multiple forms of senescence. Lipid peroxidation-derived aldehydes such as 4-hydroxynonenal (HNE) accumulate and form protein adducts in SnCs [124]. They can induce senescence on their own and have been implicated in mediating bystander senescence in tissues [10,124]. Again, mitochondria may be key players here, serving both as sites of lipid peroxidation via cardiolipin oxidation and as targets for lipid aldehyde-adduction to proteins, nucleic acids and/or lipids, leading to dysfunction [128]. Senescent T cells also demonstrate increased fatty acid oxidation and synthesis, especially cholesterol esters, which contributes to LD accumulation via a cPLA2α-dependent pathway [129]. Inhibition or knockdown of cPLA2α in T cells reduces LD accumulation, prevents senescence induction, and promotes effector T cell functions and anti-tumor immunity [129].
Intracellular accumulation of lipofuscin, a pigmented lipid-protein aggregate, has long been associated with tissue aging and neurodegeneration and may serve as a biomarker for cellular senescence [130,131]. Lipofuscin formation appears to derive from crosslinking of proteins by lipid peroxidation-derived electrophiles [132]. In SnCs, lipofuscin displays autofluorescence between 450-490 nm and co-localizes with SA-β-gal [130,133]. Staining for lipofuscin with Sudan Black B or its analog GLP13 have been applied to detect senescence both in vitro and in vivo [130,134,135].
Cancer cells often fail to display a Pasteur effect and continue to consume glucose consumption in the presence of oxygen, a phenomenon known as the Warburg effect [136]. Similarly, aerobic glycolysis has been observed in SnCs, potentially linked to mitochondrial dysfunction. In radiation-induced senescent human colon cancer cells, glycolysis has been reported as the primary energy source (Figure 2) [137]. While enhanced glucose metabolism can help cancer cells bypass OIS, a hyperglycemic state may accelerate senescence [138]. Notably, 2-deoxy-D-glucose (2DG) induces apoptosis more frequently in cyclophosphamide-induced senescent lymphoma cells than in non-senescent cells, highlighting a senescence-specific vulnerability to glycolysis inhibitors [139]. Mitochondrial dysfunction also affects other metabolic pathways. For instance, an altered NAD+/NADH ratio in SnCs influences the SASP [140,141]. NAD+-dependent protein SIRT1 is also reduced through autophagosome-lysosome degradation during cellular senescence [142].
3.7. Regulation of the SASP
Cells undergoing senescence typically adopt an altered pattern of metabolite release, protein secretion, plasma membrane protein expression, and vesicle release that mediate autocrine, juxtacrine, paracrine and systemic effects. These features are typically described as characteristics of the senescence-associated secretory phenotype (SASP), often comprising a mix of pro-inflammatory factors including cytokines, chemokines, growth factors, proteases, lipids, extracellular components, matrix metalloproteinases (MMPs), nucleic acids, and extracellular vesicles [143]. Notably, along with proteins released by active secretion, the SASP includes macromolecules released via passive leakage through membrane defects, exocytosis of vesicles and shedding of transmembrane protein ectodomains, cleaved by enzymes such as disintegrin and ADAM17 [144,145].
SnCs induced without DNA damage as by ectopic expression of p16INK4A or p21CIP typically do not promote inflammation, suggesting an association of the inflammatory SASP with stress responses [146]. Depleting DDR proteins like ATM, NBS1, or CHK2 [67,147], or inhibiting stress-induced p38 mitogen-activated protein kinases (MAPKs) [148], can diminish inflammatory SASP secretion. These stress signals predominantly activate NF-κB and C/EBP-β, driving the production of proinflammatory cytokines such as IL-1α, IL-6, and IL-8 [18,149,150,151,152]. SASP regulation involves autocrine feedback loops. For instance, IL-6 can reinforce senescence and promote a self-amplifying secretion network [153]. Surface-bound IL-1α can also initiate a positive loop that enhances IL-6 and IL-8 secretion [154]. IL-1 receptor signaling, on the other hand, elevates microRNAs 146a and 146b, creating a negative loop that suppresses excessive SASP expression [155]. Notch1, often upregulated in SnCs, modulates SASP composition by negatively regulating the proinflammatory transcription factor C/EBP-β, shifting the SASP towards a TGF-β-rich secretome [156,157]. A positive feedback loop between TGF-β and Notch further contributes to G1 cell cycle arrest and subsequent senescence [158,159]. TGF-β may also promote paracrine senescence by increasing ROS and DDR via NADPH oxidase 4 (Nox4) and activating p21 signals [28,160].
Mammalian TOR (mTOR) signaling, a key regulator of lifespan and aging [161], also plays critical roles in the production of the SASP [162,163]. During senescence, mTOR activity leads to 4EBP1 phosphorylation and MAPKAPK2 translation. This, in turn, phosphorylates ZFP36L1, preventing the degradation of SASP-related transcripts [164]. Inhibiting mTOR using drugs like Rapamycin effectively reduces inflammatory cytokine expression by suppressing IL-1α translation and diminishing NF-κB activity [165]. Such findings underscore mTOR inhibitors as potential SASP suppressors to alleviate SASP-associated pathologies [166].
Recent studies have identified the cGAS-STING pathway as another central regulator of the SASP. In SnCs, cGAS senses single and double-stranded cytosolic DNA and chromatin fragments, including those derived from retrotransposon reverse transcription, chromosomal DNA damage processing, dysfunctional mitochondria, and micronuclei, and generates the secondary messenger cGAMP to activate the adaptor protein STING [167,168]. STING then recruits TBK1 and IκB kinase, activating IFN regulatory factor 3 (IRF3) [169] and NF-κB [170] and leading to the secretion of type I interferons and inflammatory cytokines [171]. Additionally, STING activation can promote the expression of Toll-like receptor 2 (TLR2) in OIS, further driving SASP via p38-MAPK activity [172]. Inhibiting STING signaling reduces SASP production and its associated functions in SnCs [168,173]. Deficiency of cGAS alters senescence, primarily due to changes in the SASP but also altered oxidative stress response [167,171].
Metabolic processes may also affect the SASP and cellular senescence. During OIS, high mobility group A (HMGA) proteins promote the expression of nicotinamide phosphoribosyl transferase (NAMPT), the primary enzyme for NAD+ biosynthesis. This leads to a high NAD+/NADH ratio, which activates p38-MAPK while inhibiting AMPK, thereby increasing the expression of proinflammatory SASP. Supplementing senescent fibroblasts with nicotinamide mononucleotides (NMNs) further elevates the NAD+/NADH ratio and encourages the secretion of inflammatory cytokines [141]. Conversely, raising NAD+ levels in mouse brains through nicotinamide riboside (NR) administration has been shown to reduce inflammation by suppressing cGAS-STING signaling [174], underscoring the context-dependent effects of NAD metabolism on SASP.
SASP regulation is further influenced by epigenetic modifications [175]. The diminished H3K9Me2 at SASP gene promoters contributes to IL-6 and IL-8 expression [93]. Inhibition of histone deacetylase (HDAC) can activate a proinflammatory SASP even in the absence of DNA damage, potentially due to chromatin hyperacetylation [176]. Similarly, downregulating NAD-dependent deacetylase sirtuin-1 (SIRT1) during senescence results in increased acetylation of H3K9 and H4K16 at SASP promoter regions, facilitating SASP expression [177]. SnCs also show elevated levels of the histone variant macroH2A1, which further stimulates SASP gene expression via positive feedback loops [178]. Other epigenetic regulators, such as BRD4, HMGB2, GATA4, and MLL1, have also been identified as critical modulators of SASP [175].
4. Senescence as an Ally in Cancer Treatment
Due to the limitations of cancer cell proliferation, senescence was initially perceived as a mechanism that suppresses tumors and enhances anti-tumor effects [179] (Figure 3). Recent studies have further revealed the interactions between SnCs and the immune system, highlighting the opportunity to utilize SnCs' immune-activating capabilities to advance cancer therapies (Table 1).
4.1. Senescence as a Defensive Mechanism against Tumorigenesis
Proliferative arrest via OIS has been proposed as a key barrier to carcinogenesis. The common observation of SnCs in premalignant lesions may indicate a tumor suppressive role. Senescence induced by oncogenic H-RasG12V may prevent primary fibroblasts from excessive proliferation and full malignant transformation [35]. Similarly, lymphocytes that fail to undergo N-RasG12D-induced senescence show rapid proliferation and develop into aggressive but apoptosis-competent lymphomas in vivo [179]. Acute inactivation of PTEN in the prostate leads to growth arrest and senescence in a p53-dependent manner, while concurrent inactivation of PTEN and p53 bypasses senescence and results in invasive cancer [180]. In the context of premalignancy, the SASP may serve a beneficial role [181], though resulting inflammation might ultimately have adverse consequences.
4.2. Senescence-Associated Growth Arrest in Cancer
Senescence impacts cancer treatment outcomes. The absence of p53 or INK4a disrupts premature senescence induced by Cyclophosphamide, leading to diminished therapeutic efficacy in murine lymphoma models. Conversely, tumors in mice that undergo cyclophosphamide-induced senescence display a significantly improved prognosis after chemotherapy treatments [182]. In ovarian cancer, Cisplatin resistance has been associated with impaired senescence initiation caused by increased expression of Aurora kinase A [183].
A notable feature of SnCs is their capability to induce senescence in surrounding cells, known as paracrine or bystander senescence, which further enhances tumor-suppressive effects [10,184,185]. Such spread of senescence can be mediated through direct cell-cell contact via gap junctions and increased ROS levels, triggering the DDR in neighboring cells [10]. The SASP can further support paracrine senescence [186,187]. NF-κB and C/EBP-β dependent SASP factors such as IL-8 and GRO create a self-amplifying secretory network by activating CXCR2, which enhances ROS and facilitates senescence induction. TGF-β promotes paracrine senescence through p16INK4a and p21CIP1-dependent mechanisms, with TGFBR1 inhibitors partially mitigating this [184]. IGFBP7, secreted by BRAFV600E-driven SnCs, induces senescence and apoptosis in neighboring cells via MEK-ERK signaling and offers a potential mechanism for melanoma suppression [188]. Other soluble SASP components that have been identified to induce paracrine senescence include IL-1, VEGF, CCL2, and CCL20 [184].
4.3. Innate Immune Responses Activated by SnCs
The role of SnCs in cancer therapy extends beyond proliferation defects, with a growing body of research highlighting their ability to activate immune surveillance against both senescent and non-senescent tumor cells [16,186,189]. Early studies revealed that p53 reactivation in H-RasG12V/p53off liver tumors predominantly induces senescence rather than apoptosis. These p53-dependent SnCs trigger an innate immune response, recruiting neutrophils and natural killer (NK) cells that specifically target tumor cells, representing a potent tumor-suppression mechanism [190]. TIS can also amplify anti-tumor immunity. For instance, senescence in melanoma cells induced by the Aurora kinase A inhibitor Alisertib triggers NF-κB signaling that activates a SASP attracting phagocytes such as neutrophils and macrophages [191]. Depleting macrophages in vivo weakens immune surveillance and diminishes tumor suppression, reinforcing the importance of macrophages in cancer control [191]. In fact, it has been observed that multiple inducers of SnCs, including the alkylating agent Temozolomide, result in the secretion of cytokines that drive macrophages towards an anti-tumor M1 phenotype, elucidating one mechanism through which SnCs contribute to macrophage-mediated tumor surveillance [192,193]. Similarly, exposing non-activated M0 macrophages to extracellular vesicles from radiation-induced senescent lung tumors elevates the expression of M1-associated proinflammatory genes (e.g., IL-1β and IL-6) while simultaneously reducing M2 markers (e.g., Arg1 and Egr2) [194]. Suppressing autophagy in chemotherapy-induced senescence by targeting Arg3 may further augment M1-associated immune surveillance through SASP alterations [193].
Natural Killer (NK) Cells
Along with phagocytes, NK cells play pivotal roles in senescence-associated anti-tumor immunity. Senescence disruption in B cell lymphoma reduces immune cell infiltration, especially of NK cells, leading to increased therapy resistance and reduced survival rates [151]. Liver cancer cells undergoing H-RasG12V-induced senescence promote NK cell activity via engaging the NK activating receptor NKG2D and secreting NK cell-attracting chemokines such as CCL2, thus amplifying NK surveillance of tumors [195]. Treatment of myeloma cells with Doxorubicin or Melphalan induces SnCs with elevated surface expression of NK cell-engaging ligands, including NKG2D ligands MICA/B and DNAM-1 ligand PVR, facilitating NK cell degranulation and tumor cell elimination [196,197,198]. These NK cell activators can be upregulated by DDR enzymes such as ATM and Chk1/2 in an E2F1-dependent manner, highlighting one mechanism by which SnCs promote NK cell activation [196,197].
The SASP factor is another critical element enabling SnCs to boost NK cell functions. Conditioned media from aneuploid SnCs enhances NK cell efficiency against both senescent and proliferating tumor cells [199]. In K-RasG12D/p53null lung cancer models, senescence induction via Trametinib (MEK inhibitor) or Palbociclib (CDK4/6 inhibitor) upregulates immune-modulatory molecule ICAM-1 and the SASP component TNF-α via NF-κB activation, facilitating NK cell-mediated tumor surveillance. Like NK cell depletion, neutralizing TNF-α significantly compromises this anti-tumor response [200].
Epigenetic alteration also influences the immunogenicity of senescence via SASP modulation. The lack of BRD4, a key mediator of epigenetic transcription activation, results in reduced cytokine release and diminished NK cell activation by SnCs [95]. The polycomb-related complex 2 (PRC2) catalytic subunit EZH2, an epigenetic transcriptional repressor, suppresses pro-inflammatory SASP, limiting the ability to stimulate cytotoxic lymphocytes. Combining the EZH2 inhibitor Tazemetostat with senescence-inducing agents such as Trametinib and Palbociclib significantly enhances immune infiltration and activates NK and T cell-mediated tumor control in preclinical models of pancreatic ductal adenocarcinoma (PDAC) [201].
4.4. Adaptive Immune Responses Activated by SnCs
Beyond innate immunity, SnCs have also been proposed to activate adaptive immunity to further limit tumor progression and enhance cancer therapies. Recent studies have confirmed senescence can serve as a form of immunogenic cell stress [202] and defined factors that may potentiate their ability to drive T cell-mediated cytotoxicity [203,204,205].
Hepatocytes undergoing N-RasG12V-driven senescence may enhance CD4+ T cell-mediated immunity, contributing to tumor surveillance [206]. Radiation-induced senescence has been shown to facilitate NKT cell infiltration, providing a protective barrier against tumor development. Conversely, the failure of cells to undergo senescence, due to deficiencies in IL-6 or Rb, has been linked to accelerated osteosarcoma progression [207].
The SASP serves as a vital bridge between SnCs and adaptive immunity. Dexamethasone-induced senescence in lung adenocarcinoma cells leads to the release of chemokines such as CCL2, CCL4, CXCL1, and CXCL2, which are critical for the migration of T and NK cells to the tumor site [208]. SnCs induced by the Aurora kinase A inhibitor Alisertib can secrete CCL5 through an NF-κB-dependent pathway, contributing to T cell-mediated tumor regression [209]. This anti-tumor effect is further enhanced when Alisertib is used in combination with a T cell-activating CD137 agonist antibody, indicating synergistic benefits in melanoma models [209].
Moreover, SnCs induced by either DNA damage agents or CDK4/6 inhibitors express antigen presentation machinery that potentiates T cell responses [210]. Similarly, SnCs induced by Abemaciclib demonstrate increased surface levels of β2M and present tumor antigens to the T cell receptor (TCR) of cytotoxic CD8+ T cells, thereby eliciting T cell dependent anti-tumor responses [211]. Combining Abemaciclib with anti-PD-L1, an immune checkpoint inhibitor, results in further tumor suppression [211]. Senescent AML cells triggered by cytosine arabinoside also display increased MHC expression, effectively activating T cell proliferation [212]. This effect can be enhanced by the anti-PD-1 antibody Nivolumab [212]. These synergies suggest a potential combination strategy of senescence-inducing therapies with immune checkpoint inhibitors to achieve enhanced cancer control.
4.5. Utilizing Senescence to Overcome Resistance to Immune Checkpoint Blockade (ICB)
As a way to leverage the immunostimulatory properties of senescence and the SASP, recent studies have re-examined inducing senescence in tumors to reshape the microenvironment from immune evasion toward immune recognition. Exposing the senescence-inducing properties of N-RasG12D in liver cancer significantly enriches the lymphocyte population while reducing the presence of Gr1+ myeloid-derived suppressor cells (MDSCs), allowing for a shift towards immune activation against tumor progression [204]. These SnCs show increased sensitivity to IFNγ, enhancing antigen processing and presentation capabilities under limited IFN-γ availability. Knocking out IFNGR1 in SnCs to block IFNγ signals results in diminished MHC-I expression and subsequent decline in cytotoxic T cell activity and tumor suppression capability [204]. Doxorubicin-induced senescence modifies the immune landscape by promoting CD8+ T cell recruitment, while removing SnCs with senolytic agents like ABT-263 reverses this infiltration [213].
The impact of cellular senescence on the tumor microenvironment presents a promising strategy to overcome resistance and enhance tumor sensitivity to immunotherapies (Table 2). Inducing senescence with Doxorubicin in brain metastases from mammary carcinoma enhances the efficacy of anti-PD-1 immune checkpoint blockade, suggesting a synergistic approach of combining immunotherapy with senescence induction for better outcomes [213]. In PDAC models, combining Trametinib and Palbociclib to induce senescence leads to the secretion of NF-kB-dependent pro-angiogenic SASP factors such as VEGF, PDGFA/B, FGF2, and MMPs, which promote vascular remodeling and enhance the delivery and efficacy of genotoxic agents like gemcitabine. Importantly, this senescence-associated vascular remodeling also facilitates T cell migration into the tumor, priming immunologically cold tumors for increased responses to anti-PD-1 [214]. Melanoma resistance to ICB has been linked to T cell exclusion mediated by inhibited antigen processing/presentation, reduced IFN-γ signaling, and compromised immune modulation. Treatment with Abemaciclib disrupts this mechanism by leveraging the SASP to enhance T cell infiltration, which makes melanomas more responsive to therapies like anti-PD-1 and anti-CTLA-4 [215]. Downregulating p21 expression in melanoma cells decreases the senescence triggered by the Aurora kinase B inhibitor AZD1152 and limits AZD1152-induced T cell cytotoxicity [216]. In ICB-resistant ovarian cancer, utilizing SnCs as a form of cell therapy significantly improves the efficacy of anti-PD-1 treatments by stimulating CD8+ T cell and dendritic cell (DC) activation [217]. Interestingly, IFN-γ generated following ICBs can induce senescence via Stat1 signals and subsequent p21Cip activation. Tumors lacking Stat1 or p21Cip fail to undergo senescence post ICB treatment and display resistance to therapies such as anti-PD-L1 and anti-LAG3 [218]. This marks a pivotal role of IFN-γ-dependent senescence induction for enabling T cells and macrophages to effectively eliminate cancer cells, emphasizing the significance of cellular senescence in immunotherapy responses.
4.6. Immunogenic Senescent Tumor Cells Function as Cancer Vaccines
Building on observations that SnCs left in tumors after genotoxic therapy might be able to contribute to subsequent anti-tumor immune response, a 2012 study directly tested vaccination with senescent tumor cells [23]. Here, SnCs induced by combining ionizing radiation and the PARP inhibitor Veliparib displayed both preventive and therapeutic effects against tumor growth in multiple models, mediated through a T cell-dependent mechanism [23]. Subsequent research from our team and others has further explored the potential of inducing immunogenic senescence to form cancer vaccines. Not only does this apply to combination radiation plus Veliparib-induced senescence, but also to Doxorubicin-induced senescent melanoma cells and Irinotecan-Cisplatin-induced senescent ovarian cancer cells [203,205,217]. Due to enhanced antigenicity and adjuvanticity, these SnCs recruit and activate DCs, which are critical for full activation of T cells and immune surveillance against cancer [203,205,217].
Notably, it is specifically viable SnCs, as opposed to apoptotic or stressed cells, that trigger an effective CD8+ T cell-driven anti-tumor immune responses [203,205]. Such immunogenic SnCs not only serve as an effective monotherapy against primary tumors and metastases, but also synergize with traditional cancer therapies, including irradiation and ICB [23,203,205,217]. SnCs lacking STING fail to activate DCs effectively and lead to a diminished vaccine effect against cancer development [205]. However, treatment with the STING agonist DMXAA alone in non-SnCs does not lead to DC activation nor improve anti-tumor immunity [205,217]. Such findings underscore the necessity of a complete senescence program in achieving effective immune responses in cancer therapy.
5. Senescence as an Enemy in Cancer Treatment
Under normal physiological conditions, timely removal of SnCs is important for tissue health and overall organismal homeostasis, while their accumulation can contribute to inflammaging and age-related disease. Although cellular senescence is an important tumor suppressive mechanism that limits the proliferation of damaged cells, it can paradoxically become an adversary in cancer treatment. Persistent SnCs within the TME promote a chronic inflammatory state that incites tumorigenesis and tumor growth and dampens anti-tumor immunity, exacerbating the adverse effects of cancer therapies [17,219]. The detrimental roles of senescence are linked to therapy resistance, metastasis, relapse, and poor treatment outcomes. Strategies that modulate the effects of the SASP or to eliminate these unwanted SnCs are being explored as avenues to enhance cancer therapies.
5.1. Malignant Transformation and Tumor Growth
SnCs in the stroma are increasingly implicated in cancer initiation and progression. Compared to pre-senescent fibroblasts, co-injecting neoplastic cells with senescent fibroblasts significantly increases tumor formation and growth rates, which suggests SnCs may create a niche that favors cancer development [220]. Similarly, non-malignant epithelial cells exposed to senescent fibroblasts display changes indicative of invasive behavior and early malignant transformation, such as increased migration and nuclear atypia [221,222]. The elimination of SnCs through targeting of p16INK4a-positive cells in transgenic INK-ATTAC mouse models has demonstrated a reduction in age-related tumorigenesis, further supporting the direct link between senescence accumulation and cancer risk [223]. This effect is observed irrespective of the initial senescence trigger, reinforcing the broad nature of SnC-related growth promotion [220,224].
Conditioned medium from SnCs can confer growth advantages on non-senescent recipient cells [221,222], implicating SASP factors as contributors to SnC-related growth acceleration. For example, amphiregulin (AREG), promotes cancer cell proliferation and survival via paracrine activation of the epidermal growth factor receptor (EGFR) pathway in nearby cells [225]. Senescent mesenchymal cells release IL-6, which can drive Stat3 signals and subsequent carcinogenesis by enhancing transformation, proliferation, and invasion. Depleting IL-6 in these SnCs can significantly reduce their ability to stimulate the proliferation and migration of neighboring breast tumor cells [226]. Inhibiting the cGAS-dependent SASP in senescent fibroblasts similarly diminishes their capacity to support tumor growth [224]. Other SASP factors, such as osteopontin and matrix MMPs, have also been implicated in promoting cell proliferation and cancer progression [221,227,228].
5.2. Metastasis
SnCs can contribute to metastasis by promoting the migration, invasion, and extravasation of cancer cells as well as serving as a pre-metastatic niche. As an example, breast cancer cells implanted along with HER2-induced SnCs exhibit a marked increase in metastatic capability via non-cell autonomous mechanisms [229]. This enhancement of metastatic potential is also observed following chemotherapy, where TIS in stromal cells and fibroblasts creates a nurturing environment conducive to tumor colonization in distant organs such as the liver, lungs, and bones [230].
The pro-metastatic effect of senescence is largely attributed to the SASP. The conditioned medium from senescent fibroblasts formed by treatment with bleomycin alters breast cancer cell morphology and migration, advancing them to an aggressive state by affecting microtubule stability and dynamics [231]. Senescent colon cancer cells treated with Fluorouracil also display a SASP that induces epithelial-to-mesenchymal transition (EMT) and enhances tumor cell invasion [232]. Specific SASP factors may play predominant roles in these pro-metastatic effects. CXCL12 in BRAFV600E-induced senescent thyrocytes is essential for their ability to promote thyroid carcinoma cell invasion [233]. IL-6 secreted by senescent osteoblasts contributes to increased osteoclast genesis and bone metastases [234]. MMP-1 and MMP-2 produced by senescent fibroblasts can promote skin carcinoma cell migration and keratinocytes EMT [235]. Chemerin, a newly identified SASP factor, boosts the migration of cutaneous squamous cell carcinoma cells by activating the MAPK signaling pathway [236].
SnCs influence vascular structure remodeling that further facilitates tumor metastasis. Co-transplantation of senescent thyrocytes with thyroid cancer cells has been shown to promote the development of lymphatic vessel networks and metastatic foci in lymph nodes [233]. Additionally, senescent melanoma cells secrete SFRP2, a Wnt antagonist that stimulates angiogenesis and accelerates melanoma metastasis [237]. Other SASP factors implicated in angiogenesis include vascular endothelial growth factor (VEGF) and connective tissue growth factor (CTGF) [238,239]. Suppressing the SASP through anti-inflammatory agents like Metformin has been shown to reduce pathological neovascularization driven by senescence, offering a potential strategy for mitigating cancer metastasis [240].
5.3. Therapy Resistance and Cancer Stemness
The accumulation of SnCs within tumors is identified as a significant mechanism underlying therapy resistance. For instance, Doxorubicin-induced senescent endothelial cells in the thymus contribute to a chemo-resistant microenvironment. This niche supports the survival and eventual relapse of residual cancer cells, facilitated by the release of SASP factors such as IL-6 and Timp-1 [241]. It has been observed that IL-6 secreted by senescent endothelial cells via PI3K/AKT signaling pathways is sufficient to confer chemoprotective effects [242]. In breast cancer, p53-dependent senescence shields cancer cells from apoptosis via the SASP, consequently reducing the efficacy of Doxorubicin [243]. The SASP released by Ras-induced senescent mesothelioma cells activate Stat3 signaling in neighboring cells and fosters the development of an EMT-like, clonogenic, and chemo-resistant subpopulation of cancer cells [244].
Senescence may also contribute to stemness, which furthers tumor recurrence and therapy resistance. Ectopic expression of Yamanaka factors (OCT4, SOX2, KLF4, and cMYC) in mice paradoxically leads to both cell reprogramming and senescence [245]. Key components of the senescence machinery, including p16INK4A, p21CIP1, p53, and H3K9me3, have complementary roles as regulators of stemness [246]. Inhibiting SASP factors such as IL-6 can reduce cell de-differentiation, indicating that SnCs may enhance cellular plasticity via paracrine effects [247]. RASG12V-induced SnCs can secrete a Stat3-dependent SASP that leads to the development of a subpopulation of progenitor-like cancer cells that exhibit resistance to chemotherapy agents like pemetrexed [244]. Doxorubicin-induced senescence in liver cancer increases the expression of genes associated with reprogramming and liver stemness, including c-MYC and EpCAM, as well as the multidrug resistance gene ABCG2 [248]. This enables adjacent tumor cells to upregulate tumor-initiating capabilities [248]. Exposure of breast cancer MCF-7 cells to conditioned medium from SnCs or to key SASP factors like IL-6 and IL-8 induces stemness characteristics such as elevated CD44 expression and self-renewal capabilities [249]. The senescence-mediated promotion of tumor cell reprogramming extends to hematological malignancies as well. DNA damage-induced senescent myeloma cells facilitate the emergence, sustenance, and migration of stem-like cancer cells through a CHK2-dependent SASP [250]. Evidence also points to senescence playing a role in cancer stemness in B-cell lymphoma, B-cell chronic lymphocytic leukemia (B-CLL), and acute myeloid leukemia (AML) [246].
5.4. Senescence Suppresses Anti-Immunity and Strategies to Overcome Senescence-Mediated Immunosuppression for Improved Cancer Treatment
Cellular senescence has been implicated in dampening immune responses (Figure 3 and Table 3). Systemic IR-induced senescence compromises the phagocytic capabilities of DCs and macrophages, thereby diminishing the proliferation of splenic B and T cells. Studies have demonstrated that targeted elimination of SnCs in the p16-3MR mouse model effectively restores the function of antigen-presenting cells (APCs) and leads to the rejuvenation of both T and B cell populations [251]. The removal of p16INK4-high senescent malignant cells similarly boosts T cell infiltration and reduces tumor-promoting macrophages in glioblastoma, thereby aiding in tumor suppression and enhancing survival rates in tumor-bearing mice [252].
The SASP contributes to immunosuppressive effects mediated by SnCs. In prostate tumors lacking PTEN and treated with Docetaxel, SnCs exhibit sustained Jak2/Stat3 activation that induces an immunosuppressive SASP, leading to the accumulation of suppressive myeloid cells while depleting CD4+, CD8+, and NK cells. Genetically or pharmacologically inhibiting Jak2/Stat3 triggers a robust anti-tumor immune response and re-sensitizes tumors to Docetaxel [253]. Conditioned medium from p27Kip-driven senescent fibroblasts encourages the differentiation of bone marrow stem cells into CD11b+Ly6GHi myeloid-derived suppressor cells (MDSCs), suppressing cytotoxic immunity and fostering a pro-tumorigenic environment. Neutralizing IL-6 from these fibroblasts reduces MDSC levels, reactivates T cell function, and delays tumor progression [254]. Injection of Palbociclib-induced senescent fibroblasts also leads to MDSC infiltration through NF-kB pathways and accelerates melanoma growth in immunocompetent hosts [255]. Interestingly, tumor-infiltrating MDSCs can antagonize senescence by secreting IL-1RA that blocks IL-1α-mediated OIS in neighboring cells, further impacting the overall efficacy of cancer treatment. Treatment with a CCR2 antagonist greatly reduces Gr-1+ myeloid cell infiltration, improving chemotherapy in PTEN-null prostate cancer [256]. In hepatocellular carcinoma, CCL2 secreted by N-RasG12V-induced senescent hepatocytes disrupts the maturation of CCR2+ myeloid precursors. As a result, these immature myeloid cells efficiently suppress NK cell activity and significantly facilitate tumor progression [257].
Notch signaling also emerges as a key regulator of immunosuppressive functions of SnCs. Elevated Notch signaling in N-RasG12V-induced senescent hepatocytes leads to a TGF-β-enriched SASP, which dampens T cell-mediated immune surveillance [157]. Radiation-induced senescence in the lung similarly fosters a pro-tumorigenic microenvironment, characterized by increased neutrophil infiltration and amplified Notch signaling, with Notch inhibition markedly reducing radiation-enhanced metastases [258]. Under hypoxic conditions, TGF-β can trigger senescence by repressing E2F targets and inducing a SASP that creates an immunosuppressive environment, which results in elevated myeloid cells and diminished immunotherapy success [259]. Consequently, knockout of TGF-β receptors in lung cancer cells alleviates the senescence phenotype and restores immune balance [259]. In colorectal cancer, senescent tumor cells driven by oxidative stress upregulate CXCL12 and CSF1 secretion. Increased levels of CXCL12 lead to reduced CXCR4 expression on CD8+ T cells and inhibit their chemotactic migration. CSF1 promotes M2 macrophages differentiation that further impedes CD8+ T cell activation. CXCL12/CSF1 neutralization therefore increases T cell infiltration and the efficacy of anti-PD-1 treatment [260].
Metabolites secreted by SnCs can also contribute to immune suppression. Senescent hepatic stellate cells (HSCs) induced by obesity-associated gut microbe upregulate their production of prostaglandins (e.g., PGE2) via TLR2 signaling. The increase in PGE2 promotes regulatory T cells (Tregs) in the liver and diminishes cytotoxic T cell activity. Blocking the PGE2 receptor PTGER4 rejuvenates the anti-tumor immunity by elevating CD103+ DCs and CD8+ T cells while reducing Tregs, thus protecting against hepatocellular carcinoma induced by a high-fat diet [261].
In addition to immunosuppressive effects of the SASP, senescent dermal fibroblasts display increased surface expression of the non-classical MHC molecule HLA-E that contributes to immunosuppressive effects by activating the NKG2A-mediated inhibitory pathway in NK and CD8+ T cells [262]. HLA-E induction can be amplified by IL-6 in both cell autonomous and non-autonomous manner. Blocking the interaction between HLA-E and NKG2A enhances immune surveillance of SnCs in vitro, offering a strategy to alleviate senescence-associated immunosuppression [262]. Additionally, SnCs regardless of DNA damage induced can upregulate the immune checkpoint molecule PD-L1, leading to CD8+ T cell inhibition [263,264]. The mechanisms behind PD-L1's upregulation include enhanced E2F1 and/or NF-kB-mediated transcription, reduced proteasome activity, and cGAS-STING-driven paracrine effects [263,264,265]. Studies on aging have revealed that anti-PD-1 treatments enhance immune surveillance of SnCs and reduce age-related dysfunctions in mice [263]. Similarly, combining senescence-inducing chemotherapy, like Mitoxantrone, with anti-PD-1/PD-L1 therapies leads to tumor regression via cytotoxic T cells [225]. SnCs induced by Doxorubicin or the CDK4/6 inhibitor Palbociclib upregulate PD-L2, another immune checkpoint receptor ligand that disrupts T cell function through PD-1 engagement and facilitates MDSC recruitment [266]. Targeting this immunosuppressive mechanism with a PD-L2 blockade in combination with chemotherapy results in significant tumor reduction in murine syngeneic models [266]. Such findings underscore the potential for immunotherapies to overcome SnC-induced immune suppression.
Figure 3.
Positive and negative effects of cellular senescence in the tumor microenvironment. Left, the anti-tumor effects of cellular senescence. Senescent cells contribute to anti-tumor defenses by facilitating the recruitment and activation of immune cells such as phagocytes (i.e., Mφ), dendritic cells (DCs), T cells, and natural killer (NK) cells. This is achieved through the SASP factors and surface proteins including class I MHC, NKG2D ligands, and ICAM-1. Activation of DCs by senescent cells provides an additional mechanism for cytotoxic lymphocyte-mediated tumor control. Senescence also plays a role in promoting angiogenesis, which contributes to the mobilization of immune cells to the tumor site. Beyond these immune-mediated actions, senescent cells contribute to the maintenance and spread of the senescent state through autocrine and paracrine mechanisms that suppress cell proliferation. Interleukin signals, particularly IL-1, are implicated in paracrine senescence. Right, the pro-tumor effects of cellular senescence. Senescent cells can dampen cytotoxic lymphocyte function by expressing certain surface molecules, including the non-canonical class I MHC molecule HLA-E and immune checkpoints PD-L1/2. Additionally, the SASP facilitates the recruitment and differentiation of immunosuppressive cells, like myeloid-derived suppressor cells (MDSCs) and M2-like macrophages, inhibiting NK and T cell function. Senescent cells may also contribute to tumor progression by promoting non-immunologic processes such as epithelial-mesenchymal transition (EMT), vasculogenesis, cancer cell reprogramming, malignant transformation, and hyperproliferation.
Figure 3.
Positive and negative effects of cellular senescence in the tumor microenvironment. Left, the anti-tumor effects of cellular senescence. Senescent cells contribute to anti-tumor defenses by facilitating the recruitment and activation of immune cells such as phagocytes (i.e., Mφ), dendritic cells (DCs), T cells, and natural killer (NK) cells. This is achieved through the SASP factors and surface proteins including class I MHC, NKG2D ligands, and ICAM-1. Activation of DCs by senescent cells provides an additional mechanism for cytotoxic lymphocyte-mediated tumor control. Senescence also plays a role in promoting angiogenesis, which contributes to the mobilization of immune cells to the tumor site. Beyond these immune-mediated actions, senescent cells contribute to the maintenance and spread of the senescent state through autocrine and paracrine mechanisms that suppress cell proliferation. Interleukin signals, particularly IL-1, are implicated in paracrine senescence. Right, the pro-tumor effects of cellular senescence. Senescent cells can dampen cytotoxic lymphocyte function by expressing certain surface molecules, including the non-canonical class I MHC molecule HLA-E and immune checkpoints PD-L1/2. Additionally, the SASP facilitates the recruitment and differentiation of immunosuppressive cells, like myeloid-derived suppressor cells (MDSCs) and M2-like macrophages, inhibiting NK and T cell function. Senescent cells may also contribute to tumor progression by promoting non-immunologic processes such as epithelial-mesenchymal transition (EMT), vasculogenesis, cancer cell reprogramming, malignant transformation, and hyperproliferation.
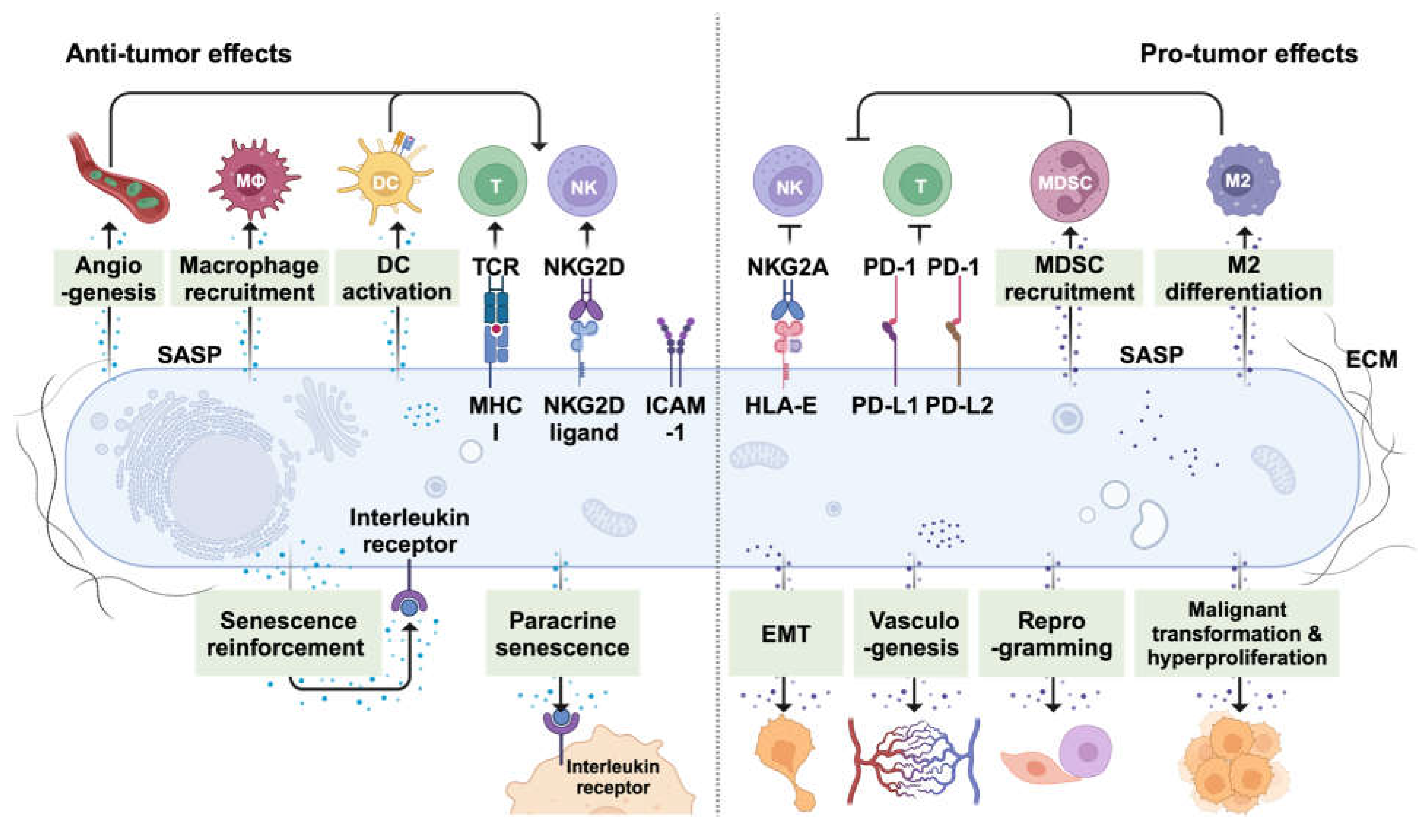

Table 3.
Immunosuppressive senescent cells in cancer.
| Senescence Induction methods | Cancer type | Affected immune cell population | References (PMID) |
|---|---|---|---|
| Doxorubicin | Breast cancer | SASP p16-3MR mice |
27979832 |
| Docetaxel | PTEN loss prostate cancer | Increase Gr1+ MDSCs but decrease T and NK cells | 25263564 |
| p27Kip1 | Squamous cell carcinoma | Increase CD11b+Ly6GHi MDSCs and Tregs | 27272654 |
| Palbociclib | Melanoma | Promote the recruitment of Gr1+ MDCS | 28039358 |
| Pten-loss | PTEN loss prostate cancer | Increase MDSCs | 25156255 |
| N-RasG12V | Liver cancer | Increase MDSCs | 27728804 |
| N-RasG12V | Liver cancer | Reduce CD3+ T cells | 27525720 |
| IR | Lung metastases | Promote Ly6G+ neutrophil recruitment | 35221334 |
| TGF-β |
Lung cancer |
Increase infiltration of immune-suppressive cell types | 36821441 |
| ROS |
Colorectal Cancer | Enhance M2 macrophage polarization | 33643790 |
| Metabolites (DCA and LTA) | Hepatocellular carcinoma | Suppress CD8+ T cells | 28202625 |
| Mitoxantrone |
Prostate cancer | Promote PD-L1 expression in tumors | 31493351 |
| Doxorubicin | Melanoma | PD-L2+ senescent cells dampen T cell activity and promote of CD11b+Gr1+ MDSC recruitment | 38267628 |
| H-RasG12V | Glioblastoma | Decrease T cells and increase tumor promoting macrophages | 36707509 |
5.5. Lifestyle Interventions for Combating Senescence Metabolism
Dietary interventions have long been recognized to extend lifespan and mitigate oxidative stress [267]. Similarly, they can influence the induction and characteristics of cellular senescence [268]. For instance, short-term caloric restriction has been shown to reduce markers of cellular senescence and the SASP in both murine models and middle-aged humans, potentially slowing the progression of aging-related phenotypes [269]. Dietary methionine restriction may also delay senescence and reduce inflammatory SASP factors by enhancing the production of hydrogen sulfide (H2S), an endogenous antioxidant, through the transsulfuration pathway [270,271]. Omega-3 fatty acids, particularly eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) found in fish oil supplements, have been shown to reduce H2O2-induced DNA double-strand breaks (and likely lipid peroxidation) and resulting cellular senescence by promoting the expression of NRF2 and subsequent antioxidant proteins [272]. In addition to dietary control, aerobic exercise has also been found to mitigate senescence and inflammation, protecting naturally aged mice from cancer development [273].
5.6. One-Two Punch" Therapies Using Senolytics against Cancer
Studies have demonstrated that the elimination of p16high SnCs following Doxorubicin chemotherapy enhances breast cancer treatment response, reduces the likelihood of metastasis/relapse, and diminishes chemotherapy-associated toxicity [219]. This approach suggests a potential pharmacological method to mitigate the negative impact of senescence by selectively targeting and eliminating SnCs using selectively toxic compounds, generally known as senolytics (Table 4). Senolytics have shown considerable promise in addressing age-related diseases by eliminating naturally formed SnCs. In the context of cancer, they are often applied in a two-step strategy: initially inducing TIS within the tumor, followed by senolytic treatment targeting the vulnerabilities of the SnCs, referred to as the "one-two-punch" approach [22].
Resistance to apoptosis in SnCs is notably modulated by elevated levels of anti-apoptotic Bcl-2 family proteins. Targeting Bcl-xL with siRNA has been a long-established approach to selectively eliminate SnCs while leaving proliferating or quiescent cells unaffected [104]. BH3 mimetics, such as Navitoclax (ABT-263), which inhibits Bcl-xL, Bcl-2, and Bcl-w, can trigger apoptosis in SnCs effectively via a BAX/BAK-dependent mechanism that contributes to rejuvenation of accelerated or naturally aging mice and elimination of TIS [73,274,275,276]. Applying Navitoclax following the administration of senescence-inducing agents like Doxorubicin or Etoposide leads to substantial tumor suppression in both immune-deficient and -competent models [277,278]. Co-treatment with Navitoclax and Olaparib also displays synergistic effects on ovarian xenografts [279]. Treatment with ABT-737, another Bcl-2 family inhibitor, removes the SnCs induced by K-RasG12D and minimizes the development of premalignant pancreatic lesions before their progression into PDAC [280].
Although Navitoclax has been acknowledged as a potent senolytic agent in various preclinical studies, its clinical application is limited due to significant hematological side effects, including thrombocytopenia. To overcome this limitation, a galacto-conjugate of Navitoclax, Nav-Gal, has been developed. As a prodrug, Nav-Gal's senolytic potential is activated specifically within SnCs upon SA-β-gal cleavage [281]. Nav-Gal can delay tumor progression when combined with Cisplatin [281]. Nav-Gal also parallels Navitoclax in its ability to inhibit lung metastases in a mouse TNBC model, an outcome attributed to the elimination of SnCs induced by Palbociclib from lung endothelial cells [282].
Inhibitors that target specific Bcl-2 family members for eliminating SnCs vary in efficacy, reflecting the diverse role of different Bcl-2 family proteins in providing apoptosis resistance to SnCs. Bcl-xL degraders based on proteolysis-targeting chimera (PROTAC) technology and specific Bcl-xL inhibitors (such as A1331852 or A1155463) display potency in eliminating senescent cancer cells [275,283]. Conversely, the specific Bcl-2 inhibitor ABT-199 shows inconsistent senolytic activity across various models [275,276,278,279]. Nevertheless, ABT-199 can destroy IR-induced senescent sarcoma cells [284]. When combined with the senescence inducers Palbociclib and Fulvestrant, ABT-199 also successfully induces apoptosis and enhances breast cancer responsiveness [285]. The Mcl-1 inhibitor S63845 has been identified as another senolytic [278,286]. The sequential application of Docetaxel followed by S63845 effectively eliminates SnCs and suppresses the growth of prostate cancer. Notably, this combination treatment also revitalizes anti-tumor immunity by diminishing immunosuppressive cells and amplifying cytotoxic immune markers [286].
Beyond agents directly targeting Bcl-2 family members or other survival factors, the best studied senolytic agents are Dasatinib and Quercetin (D+Q), which contribute to extended health and lifespan [104,287]. D+Q has shown potential in preclinical models for controlling age-associated conditions like pulmonary fibrosis and neurodegeneration [288,289]. When used separately, D and Q also demonstrate senolytic properties through targeting senescent ovarian cancer cells triggered by the PARP1/2 inhibitor Olaparib [279]. Other flavonoid derivatives, such as Fisetin and GL-V9, have also been recognized as senolytics [279,290]. While the precise mechanisms of action are still being explored, it has been suggested that senolytic flavonoids may work by blocking Bcl-2 family proteins such as BCL-xL and elevating ROS levels [290]. Despite these promising outcomes, D+Q does not appear to target Doxorubicin-induced senescent hepatocellular carcinoma [291], indicating the need for context-specific applications of this therapeutic strategy.
Compounds such as the BRD4 inhibitor JQ1 and the degrader ARV825 have also emerged as potent senolytics by promoting autophagy-induced cell death. For instance, ARV825 enhances liver cancer therapy outcomes when combined with Doxorubicin [292]. The mTOR inhibitor AZD8055 eradicates SnCs caused by the CDC7 inhibitor XL413, demonstrating a synergistic effect on combating liver cancer. The HDAC inhibitor Panobinostat targets genotoxic TIS primarily via downregulating Bcl-xL expression [293]. Cardiac glycosides such as Digoxin, Digitoxin, and Ouabain, have also been discovered as potential senolytics [294,295]. SnCs exhibit increased susceptibility to the ferroptosis inducer RSL3 compared to non-senescent cells, likely attributed to elevated levels of cytosolic lipid peroxidation [296]. Canagliflozin, an inhibitor of sodium–glucose co-transporter 2 (SGLT2), may indirectly reduce the senescence load induced by a high-fat diet in mouse adipose tissue through activating AMPK signaling and potentiating immune surveillance [297]. These studies have expanded the spectrum of agents for one-two-punch therapy.
Except for small-molecule senolytics, innovative approaches have led to the development of senolytic peptides and cell-based therapies. For example, FOXO4-binding peptide E2 disrupts the interaction between FOXO4 and TP53, leading to reactivation of TP53 transcription that induces apoptosis in SnCs. This intervention has shown promise in eliminating SnCs associated with aging and early-stage melanomas [298]. Furthermore, senolytic chimeric antigen receptor (CAR) T cells targeting a surface marker of SnCs, urokinase plasminogen activator receptor (uPAR), further extend mouse survival in lung adenocarcinoma models treated with a senescence-inducing regimen of Palbociclib and Trametinib [20,299]. These advancements highlight the dynamic and evolving landscape of senolytic therapy, aiming to refine cancer treatment strategies with improved outcomes and reduced toxicity.
6. Conclusions
Here, we highlight how SnCs can display a diverse range of effects on cancer progression, attributed to differences in senescence triggers, genetic backgrounds, tissue-specific contexts, and stages of senescence. Such diversity emphasizes the significant heterogeneity and dynamic nature of SnCs, meriting the discovery of novel cellular markers that reflect the senescence stage and its functional implications. A thorough comprehension of the balance between the beneficial and detrimental effects of senescence on cancer therapy is vital for refining treatment strategies that specifically modulate SnCs to minimize their negative impacts while enhancing their positive therapeutic effects.
Recent technological advancements, including single-cell/nucleus RNA sequencing, have revolutionized our ability to understand the complex regulatory mechanisms behind SnC heterogeneity [300,301,302,303]. These technologies provide insights into gene signatures and potential therapeutic targets for combating senescence-associated diseases. Spatial transcriptomics has emerged as a cutting-edge method that integrates transcriptomic data with histological detail, enabling a comprehensive analysis of the tumor microenvironment (TME) [304]. This technique is particularly valuable for examining the interactions between SnCs and surrounding immune/non-immune cells or the extracellular matrix to identify inflammatory regions within tumors.
The SASP is important for mediating the autocrine and paracrine effects of cellular senescence. Understanding the mechanisms of SASP factors in relation to immune responses and cancer progression, as well as identifying modulators that can redirect the SASP towards aiding cancer therapy, requires further study. Despite the dynamic complexity of the SASP, advancements in proteomics and antibody array technologies have improved our capability to profile SASP components [300]. The development of chemical biology tools for precise protein labeling and enrichment, such as APEX and TurboID [305,306], opens new opportunities for future investigations into SASP characteristics. Collectively, innovative techniques for exploring senescence offer the potential to expand current understandings of how SnCs limit or accelerate cancer. This, in turn, can facilitate the discovery of novel approaches to managing SnCs, thereby improving the efficacy of current cancer therapies.
Although TIS in tumors or normal tissue can serve diverse roles in promoting or antagonizing cancer response and recurrence, an exciting opportunity is available to leverage senescence as an in situ vaccine to boost anti-tumor immune responses and enhance cancer treatment outcomes. This approach could be effective as a monotherapy or in combination therapies. However, the induction of senescence raises safety concerns regarding its long-term effects, especially considering the association between senescence accumulation and aging [307]. The risk that senescent-like cells may re-enter a proliferative state [13,308] and potentially fuel cancer progression underscores the necessity for effective methods to remove persistent SnCs through senolytics. Alternatively, forming SnCs ex vivo and then employing SnC-activated immune cells, such as DCs, as cell-based cancer vaccines presents a potentially a safe avenue for improving the efficacy of current cancer therapies that warrants further exploration.
Author Contributions
Conceptualization: YL, IL, and SJK; Writing & editing: YL, IL, and SJK; Funding acquisition: SJK; Supervision: SJK
Acknowledgments
YL: IL and SJK were supported by NIH R01 grants CA217182, AG069865, CA254047, and CA282781 and CDMRP PRCRP Impact Award CA190982 to SJK. Figures were created with BioRender.com.
Conflicts of Interest
S.J.K. is a co-founder of OncoSenescence and Riptide Therapeutics. Y.L. is an employee of Calico Life Sciences LLC.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work the authors used OpenAI ChatGPT-4 in order to improve readability. After using this tool, the authors reviewed and edited the text as needed and take full responsibility for the content of the publication.
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp Cell Res 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Allsopp, R.C.; Vaziri, H.; Patterson, C.; Goldstein, S.; Younglai, E.V.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A 1992, 89, 10114–10118. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu Rev Physiol 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Birch, J.; Gil, J. Senescence and the SASP: many therapeutic avenues. Genes Dev 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; Deursen, J.V.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J Natl Cancer Inst 2021, 113, 1285–1298. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d'Adda di Fagagna, F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat Cell Biol 2019, 21, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J Natl Cancer Inst 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front Oncol 2017, 7, 278. [Google Scholar] [CrossRef]
- Mavrogonatou, E.; Pratsinis, H.; Kletsas, D. The role of senescence in cancer development. Semin Cancer Biol 2020, 62, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Takahashi, A.; Mann, D.J.; Hara, E. Cellular senescence: a double-edged sword in the fight against cancer. Exp Dermatol 2012, 21 Suppl 1, 1–4. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013, 123, 966–972. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep 2014, 15, 1139–1153. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Bousset, L.; Gil, J. Targeting senescence as an anticancer therapy. Mol Oncol 2022, 16, 3855–3880. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat Rev Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Efimova, E.V.; Hamzeh, K.W.; Darga, T.E.; Mauceri, H.J.; Fu, Y.X.; Kron, S.J.; Weichselbaum, R.R. Radiation-inducible immunotherapy for cancer: senescent tumor cells as a cancer vaccine. Mol Ther 2012, 20, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M.; Yoshida, Y.; Ohtani, N. Cellular senescence and the tumour microenvironment. Mol Oncol 2022, 16, 3333–3351. [Google Scholar] [CrossRef] [PubMed]
- Chibaya, L.; Snyder, J.; Ruscetti, M. Senescence and the tumor-immune landscape: Implications for cancer immunotherapy. Semin Cancer Biol 2022, 86, 827–845. [Google Scholar] [CrossRef] [PubMed]
- Piskorz, W.M.; Cechowska-Pasko, M. Senescence of Tumor Cells in Anticancer Therapy-Beneficial and Detrimental Effects. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Canamero, M.; Maraver, A.; Gomez-Lopez, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 2010, 12, 676–685. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M.; et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef]
- Collado, M.; Serrano, M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Sarkisian, C.J.; Keister, B.A.; Stairs, D.B.; Boxer, R.B.; Moody, S.E.; Chodosh, L.A. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol 2007, 9, 493–505. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Mallette, F.A.; Ferbeyre, G. The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle 2007, 6, 1831–1836. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Suram, A.; Kaplunov, J.; Patel, P.L.; Ruan, H.; Cerutti, A.; Boccardi, V.; Fumagalli, M.; Di Micco, R.; Mirani, N.; Gurung, R.L.; et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J 2012, 31, 2839–2851. [Google Scholar] [CrossRef]
- Heeren, G.; Jarolim, S.; Laun, P.; Rinnerthaler, M.; Stolze, K.; Perrone, G.G.; Kohlwein, S.D.; Nohl, H.; Dawes, I.W.; Breitenbach, M. The role of respiration, reactive oxygen species and oxidative stress in mother cell-specific ageing of yeast strains defective in the RAS signalling pathway. FEMS Yeast Res 2004, 5, 157–167. [Google Scholar] [CrossRef]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d'Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ 2014, 21, 998–1012. [Google Scholar] [CrossRef]
- Parisotto, M.; Grelet, E.; El Bizri, R.; Dai, Y.; Terzic, J.; Eckert, D.; Gargowitsch, L.; Bornert, J.M.; Metzger, D. PTEN deletion in luminal cells of mature prostate induces replication stress and senescence in vivo. J Exp Med 2018, 215, 1749–1763. [Google Scholar] [CrossRef]
- Alimonti, A.; Nardella, C.; Chen, Z.; Clohessy, J.G.; Carracedo, A.; Trotman, L.C.; Cheng, K.; Varmeh, S.; Kozma, S.C.; Thomas, G.; et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest 2010, 120, 681–693. [Google Scholar] [CrossRef]
- Wu, P.C.; Wang, Q.; Grobman, L.; Chu, E.; Wu, D.Y. Accelerated cellular senescence in solid tumor therapy. Exp Oncol 2012, 34, 298–305. [Google Scholar]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 2012, 3, 708. [Google Scholar] [CrossRef]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat Rev Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Shay, J.W.; Roninson, I.B. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 2004, 23, 2919–2933. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Mondello, C.; d'Adda di Fagagna, F. Stable cellular senescence is associated with persistent DDR activation. PLoS One 2014, 9, e110969. [Google Scholar] [CrossRef]
- Zhang, X.; Li, J.; Sejas, D.P.; Pang, Q. The ATM/p53/p21 pathway influences cell fate decision between apoptosis and senescence in reoxygenated hematopoietic progenitor cells. J Biol Chem 2005, 280, 19635–19640. [Google Scholar] [CrossRef]
- Mattiello, L.; Soliman Abdel Rehim, S.; Manic, G.; Vitale, I. Assessment of cell cycle progression and mitotic slippage by videomicroscopy. Methods Cell Biol 2024, 181, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Huck, J.J.; Zhang, M.; McDonald, A.; Bowman, D.; Hoar, K.M.; Stringer, B.; Ecsedy, J.; Manfredi, M.G.; Hyer, M.L. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol Cancer Res 2010, 8, 373–384. [Google Scholar] [CrossRef]
- Driscoll, D.L.; Chakravarty, A.; Bowman, D.; Shinde, V.; Lasky, K.; Shi, J.; Vos, T.; Stringer, B.; Amidon, B.; D'Amore, N.; et al. Plk1 inhibition causes post-mitotic DNA damage and senescence in a range of human tumor cell lines. PLoS One 2014, 9, e111060. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Kovatcheva, M.; Davis, L.E.; Tap, W.D.; Koff, A. CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell 2018, 34, 9–20. [Google Scholar] [CrossRef]
- Munro, J.; Barr, N.I.; Ireland, H.; Morrison, V.; Parkinson, E.K. Histone deacetylase inhibitors induce a senescence-like state in human cells by a p16-dependent mechanism that is independent of a mitotic clock. Exp Cell Res 2004, 295, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Stevens, F.E.; Beamish, H.; Warrener, R.; Gabrielli, B. Histone deacetylase inhibitors induce mitotic slippage. Oncogene 2008, 27, 1345–1354. [Google Scholar] [CrossRef]
- Liu, Y.; Efimova, E.V.; Ramamurthy, A.; Kron, S.J. Repair-independent functions of DNA-PKcs protect irradiated cells from mitotic slippage and accelerated senescence. J Cell Sci 2019, 132. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Gonzalez-Gualda, E.; Baker, A.G.; Fruk, L.; Munoz-Espin, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J 2021, 288, 56–80. [Google Scholar] [CrossRef]
- Cohn, R.L.; Gasek, N.S.; Kuchel, G.A.; Xu, M. The heterogeneity of cellular senescence: insights at the single-cell level. Trends Cell Biol 2023, 33, 9–17. [Google Scholar] [CrossRef] [PubMed]
- d'Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, A.; Beausejour, C.; d'Adda di Fagagna, F. A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell 2017, 16, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Furnari, B.; Rhind, N.; Russell, P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science 1997, 277, 1495–1497. [Google Scholar] [CrossRef] [PubMed]
- Malaquin, N.; Carrier-Leclerc, A.; Dessureault, M.; Rodier, F. DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Front Genet 2015, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jonsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol Cell 2018, 71, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat Rev Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Robles, S.J.; Adami, G.R. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 1998, 16, 1113–1123. [Google Scholar] [CrossRef]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Ando, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, "fueling" tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Ko, A.; Han, S.Y.; Song, J. Dynamics of ARF regulation that control senescence and cancer. BMB Rep 2016, 49, 598–606. [Google Scholar] [CrossRef]
- Sherr, C.J. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol 2001, 2, 731–737. [Google Scholar] [CrossRef]
- Sherr, C.J. Tumor surveillance via the ARF-p53 pathway. Genes Dev 1998, 12, 2984–2991. [Google Scholar] [CrossRef]
- Krimpenfort, P.; Ijpenberg, A.; Song, J.Y.; van der Valk, M.; Nawijn, M.; Zevenhoven, J.; Berns, A. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature 2007, 448, 943–946. [Google Scholar] [CrossRef]
- Mirzakhani, K.; Kallenbach, J.; Rasa, S.M.M.; Ribaudo, F.; Ungelenk, M.; Ehsani, M.; Gong, W.; Gassler, N.; Leeder, M.; Grimm, M.O.; et al. The androgen receptor-lncRNASAT1-AKT-p15 axis mediates androgen-induced cellular senescence in prostate cancer cells. Oncogene 2021. [Google Scholar] [CrossRef]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef]
- Dulic, V.; Kaufmann, W.K.; Wilson, S.J.; Tlsty, T.D.; Lees, E.; Harper, J.W.; Elledge, S.J.; Reed, S.I. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 1994, 76, 1013–1023. [Google Scholar] [CrossRef]
- Jung, Y.S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal 2010, 22, 1003–1012. [Google Scholar] [CrossRef]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A 1996, 93, 13742–13747. [Google Scholar] [CrossRef]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell 2005, 8, 19–30. [Google Scholar] [CrossRef]
- Chandra, T.; Kirschner, K.; Thuret, J.Y.; Pope, B.D.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.A.; Salama, R.; Carroll, T.; et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell 2012, 47, 203–214. [Google Scholar] [CrossRef]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d'Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol 2011, 13, 292–302. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr Biol 2017, 27, 2652–2660. [Google Scholar] [CrossRef]
- Sadaie, M.; Salama, R.; Carroll, T.; Tomimatsu, K.; Chandra, T.; Young, A.R.; Narita, M.; Perez-Mancera, P.A.; Bennett, D.C.; Chong, H.; et al. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev 2013, 27, 1800–1808. [Google Scholar] [CrossRef]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J Cell Biol 2013, 202, 129–143. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev 2013, 27, 1787–1799. [Google Scholar] [CrossRef]
- Takahashi, A.; Imai, Y.; Yamakoshi, K.; Kuninaka, S.; Ohtani, N.; Yoshimoto, S.; Hori, S.; Tachibana, M.; Anderton, E.; Takeuchi, T.; et al. DNA damage signaling triggers degradation of histone methyltransferases through APC/C(Cdh1) in senescent cells. Mol Cell 2012, 45, 123–131. [Google Scholar] [CrossRef]
- Sen, P.; Lan, Y.; Li, C.Y.; Sidoli, S.; Donahue, G.; Dou, Z.; Frederick, B.; Chen, Q.; Luense, L.J.; Garcia, B.A.; et al. Histone Acetyltransferase p300 Induces De Novo Super-Enhancers to Drive Cellular Senescence. Mol Cell 2019, 73, 684–698. [Google Scholar] [CrossRef]
- Tasdemir, N.; Banito, A.; Roe, J.S.; Alonso-Curbelo, D.; Camiolo, M.; Tschaharganeh, D.F.; Huang, C.H.; Aksoy, O.; Bolden, J.E.; Chen, C.C.; et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov 2016, 6, 612–629. [Google Scholar] [CrossRef]
- Seluanov, A.; Gorbunova, V.; Falcovitz, A.; Sigal, A.; Milyavsky, M.; Zurer, I.; Shohat, G.; Goldfinger, N.; Rotter, V. Change of the death pathway in senescent human fibroblasts in response to DNA damage is caused by an inability to stabilize p53. Mol Cell Biol 2001, 21, 1552–1564. [Google Scholar] [CrossRef]
- Ryu, S.J.; Oh, Y.S.; Park, S.C. Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ 2007, 14, 1020–1028. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol 2013, 1, 8–16. [Google Scholar] [CrossRef]
- Rochette, P.J.; Brash, D.E. Progressive apoptosis resistance prior to senescence and control by the anti-apoptotic protein BCL-xL. Mech Ageing Dev 2008, 129, 207–214. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 2016, 7, 11190. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J 2017, 36, 2280–2295. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun 2017, 8, 422. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: from discovery to translation. J Intern Med 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett 2019, 593, 1566–1579. [Google Scholar] [CrossRef]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J Cell Sci 2010, 123, 917–926. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Kim, S.J.; Mehta, H.H.; Wan, J.; Kuehnemann, C.; Chen, J.; Hu, J.F.; Hoffman, A.R.; Cohen, P. Mitochondrial peptides modulate mitochondrial function during cellular senescence. Aging (Albany NY) 2018, 10, 1239–1256. [Google Scholar] [CrossRef]
- Takebayashi, S.; Tanaka, H.; Hino, S.; Nakatsu, Y.; Igata, T.; Sakamoto, A.; Narita, M.; Nakao, M. Retinoblastoma protein promotes oxidative phosphorylation through upregulation of glycolytic genes in oncogene-induced senescent cells. Aging Cell 2015, 14, 689–697. [Google Scholar] [CrossRef]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol 2007, 5, e110. [Google Scholar] [CrossRef]
- Otero-Albiol, D.; Carnero, A. Cellular senescence or stemness: hypoxia flips the coin. J Exp Clin Cancer Res 2021, 40, 243. [Google Scholar] [CrossRef]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging (Albany NY) 2012, 4, 3–12. [Google Scholar] [CrossRef]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; van der Burg, S.H.; Verdegaal, E.M.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef]
- Maus, M.; Lopez-Polo, V.; Mateo, L.; Lafarga, M.; Aguilera, M.; De Lama, E.; Meyer, K.; Sola, A.; Lopez-Martinez, C.; Lopez-Alonso, I.; et al. Iron accumulation drives fibrosis, senescence and the senescence-associated secretory phenotype. Nat Metab 2023, 5, 2111–2130. [Google Scholar] [CrossRef]
- Venable, M.E.; Lee, J.Y.; Smyth, M.J.; Bielawska, A.; Obeid, L.M. Role of ceramide in cellular senescence. J Biol Chem 1995, 270, 30701–30708. [Google Scholar] [CrossRef]
- Kogot-Levin, A.; Saada, A. Ceramide and the mitochondrial respiratory chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef]
- Hernandez-Corbacho, M.J.; Salama, M.F.; Canals, D.; Senkal, C.E.; Obeid, L.M. Sphingolipids in mitochondria. Biochim Biophys Acta Mol Cell Biol Lipids 2017, 1862, 56–68. [Google Scholar] [CrossRef]
- Roh, K.; Noh, J.; Kim, Y.; Jang, Y.; Kim, J.; Choi, H.; Lee, Y.; Ji, M.; Kang, D.; Kim, M.S.; et al. Lysosomal control of senescence and inflammation through cholesterol partitioning. Nat Metab 2023, 5, 398–413. [Google Scholar] [CrossRef]
- Ziegler, D.V.; Czarnecka-Herok, J.; Vernier, M.; Scholtes, C.; Camprubi, C.; Huna, A.; Massemin, A.; Griveau, A.; Machon, C.; Guitton, J.; et al. Cholesterol biosynthetic pathway induces cellular senescence through ERRalpha. NPJ Aging 2024, 10, 5. [Google Scholar] [CrossRef]
- Yu, H.; Jiang, X.; Dong, F.; Zhang, F.; Ji, X.; Xue, M.; Yang, F.; Chen, J.; Hu, X.; Bao, Z. Lipid accumulation-induced hepatocyte senescence regulates the activation of hepatic stellate cells through the Nrf2-antioxidant response element pathway. Exp Cell Res 2021, 405, 112689. [Google Scholar] [CrossRef]
- Flor, A.C.; Wolfgeher, D.; Wu, D.; Kron, S.J. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov 2017, 3, 17075. [Google Scholar] [CrossRef]
- Hamsanathan, S.; Gurkar, A.U. Lipids as Regulators of Cellular Senescence. Front Physiol 2022, 13, 796850. [Google Scholar] [CrossRef]
- Millner, A.; Atilla-Gokcumen, G.E. Lipid Players of Cellular Senescence. Metabolites 2020, 10. [Google Scholar] [CrossRef]
- Zeng, Q.; Gong, Y.; Zhu, N.; Shi, Y.; Zhang, C.; Qin, L. Lipids and lipid metabolism in cellular senescence: Emerging targets for age-related diseases. Ageing Res Rev 2024, 97, 102294. [Google Scholar] [CrossRef]
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) oxidation in mitochondria: an emerging target in the ageing process? Biogerontology 2017, 18, 859–879. [Google Scholar] [CrossRef]
- Liu, X.; Hartman, C.L.; Li, L.; Albert, C.J.; Si, F.; Gao, A.; Huang, L.; Zhao, Y.; Lin, W.; Hsueh, E.C.; et al. Reprogramming lipid metabolism prevents effector T cell senescence and enhances tumor immunotherapy. Sci Transl Med 2021, 13. [Google Scholar] [CrossRef]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY) 2013, 5, 37–50. [Google Scholar] [CrossRef]
- Moreno-Garcia, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front Neurosci 2018, 12, 464. [Google Scholar] [CrossRef]
- Kikugawa, K.; Kato, T.; Beppu, M.; Hayasaka, A. Fluorescent and cross-linked proteins formed by free radical and aldehyde species generated during lipid oxidation. Adv Exp Med Biol 1989, 266, 345–356. [Google Scholar] [CrossRef]
- Flor, A.; Pagacz, J.; Thompson, D.; Kron, S. Far-red Fluorescent Senescence-associated beta-Galactosidase Probe for Identification and Enrichment of Senescent Tumor Cells by Flow Cytometry. J Vis Exp 2022. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Kouroupi, M.; Balaska, K.; Koukourakis, M.I. A Novel Lipofuscin-detecting Marker of Senescence Relates With Hypoxia, Dysregulated Autophagy and With Poor Prognosis in Non-small-cell-lung Cancer. In Vivo 2020, 34, 3187–3193. [Google Scholar] [CrossRef]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Munoz-Espin, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Nagineni, C.N.; Naz, S.; Choudhuri, R.; Chandramouli, G.V.R.; Krishna, M.C.; Brender, J.R.; Cook, J.A.; Mitchell, J.B. Radiation-Induced Senescence Reprograms Secretory and Metabolic Pathways in Colon Cancer HCT-116 Cells. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Blazer, S.; Khankin, E.; Segev, Y.; Ofir, R.; Yalon-Hacohen, M.; Kra-Oz, Z.; Gottfried, Y.; Larisch, S.; Skorecki, K.L. High glucose-induced replicative senescence: point of no return and effect of telomerase. Biochem Biophys Res Commun 2002, 296, 93–101. [Google Scholar] [CrossRef]
- Dorr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Dabritz, J.H.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Nacarelli, T.; Lau, L.; Fukumoto, T.; Zundell, J.; Fatkhutdinov, N.; Wu, S.; Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Schultz, D.; et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol 2019, 21, 397–407. [Google Scholar] [CrossRef]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat Cell Biol 2020, 22, 1170–1179. [Google Scholar] [CrossRef]
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat Commun 2017, 8, 15729. [Google Scholar] [CrossRef]
- Morancho, B.; Martinez-Barriocanal, A.; Villanueva, J.; Arribas, J. Role of ADAM17 in the non-cell autonomous effects of oncogene-induced senescence. Breast Cancer Res 2015, 17, 106. [Google Scholar] [CrossRef]
- Effenberger, T.; von der Heyde, J.; Bartsch, K.; Garbers, C.; Schulze-Osthoff, K.; Chalaris, A.; Murphy, G.; Rose-John, S.; Rabe, B. Senescence-associated release of transmembrane proteins involves proteolytic processing by ADAM17 and microvesicle shedding. FASEB J 2014, 28, 4847–4856. [Google Scholar] [CrossRef]
- Coppe, J.P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.Y.; Campisi, J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem 2011, 286, 36396–36403. [Google Scholar] [CrossRef]
- Rodier, F.; Munoz, D.P.; Teachenor, R.; Chu, V.; Le, O.; Bhaumik, D.; Coppe, J.P.; Campeau, E.; Beausejour, C.M.; Kim, S.H.; et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci 2011, 124, 68–81. [Google Scholar] [CrossRef]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 2011, 30, 1536–1548. [Google Scholar] [CrossRef]
- Huggins, C.J.; Malik, R.; Lee, S.; Salotti, J.; Thomas, S.; Martin, N.; Quinones, O.A.; Alvord, W.G.; Olanich, M.E.; Keller, J.R.; et al. C/EBPgamma suppresses senescence and inflammatory gene expression by heterodimerizing with C/EBPbeta. Mol Cell Biol 2013, 33, 3242–3258. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Mongi-Bragato, B.; Grondona, E.; Sosa, L.D.V.; Zlocowski, N.; Venier, A.C.; Torres, A.I.; Latini, A.; Leal, R.B.; Gutierrez, S.; De Paul, A.L. Pivotal role of NF-kappaB in cellular senescence of experimental pituitary tumours. J Endocrinol 2020, 245, 179–191. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A 2009, 106, 17031–17036. [Google Scholar] [CrossRef]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging (Albany NY) 2009, 1, 402–411. [Google Scholar] [CrossRef]
- Ito, Y.; Hoare, M.; Narita, M. Spatial and Temporal Control of Senescence. Trends Cell Biol 2017, 27, 820–832. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol 2016, 18, 979–992. [Google Scholar] [CrossRef]
- Valdez, J.M.; Zhang, L.; Su, Q.; Dakhova, O.; Zhang, Y.; Shahi, P.; Spencer, D.M.; Creighton, C.J.; Ittmann, M.M.; Xin, L. Notch and TGFbeta form a reciprocal positive regulatory loop that suppresses murine prostate basal stem/progenitor cell activity. Cell Stem Cell 2012, 11, 676–688. [Google Scholar] [CrossRef]
- Kagawa, S.; Natsuizaka, M.; Whelan, K.A.; Facompre, N.; Naganuma, S.; Ohashi, S.; Kinugasa, H.; Egloff, A.M.; Basu, D.; Gimotty, P.A.; et al. Cellular senescence checkpoint function determines differential Notch1-dependent oncogenic and tumor-suppressor activities. Oncogene 2015, 34, 2347–2359. [Google Scholar] [CrossRef]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFbeta-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine 'bystander senescence'. Aging (Albany NY) 2012, 4, 932–951. [Google Scholar] [CrossRef]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Lee, M.Y.; Zhang, X.; Hasan, S.; Desta, H.; Tenenbaum, S.A.; Melendez, J.A. Redox and mTOR-dependent regulation of plasma lamellar calcium influx controls the senescence-associated secretory phenotype. Exp Biol Med (Maywood) 2020, 245, 1560–1570. [Google Scholar] [CrossRef]
- van Vliet, T.; Varela-Eirin, M.; Wang, B.; Borghesan, M.; Brandenburg, S.M.; Franzin, R.; Evangelou, K.; Seelen, M.; Gorgoulis, V.; Demaria, M. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol Cell 2021, 81, 2041–2052. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Tomimatsu, K.; Narita, M. Translating the effects of mTOR on secretory senescence. Nat Cell Biol 2015, 17, 1230–1232. [Google Scholar] [CrossRef]
- Gluck, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Loo, T.M.; Miyata, K.; Tanaka, Y.; Takahashi, A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci 2020, 111, 304–311. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A 2017, 114, E4612–E4620. [Google Scholar] [CrossRef]
- Hari, P.; Millar, F.R.; Tarrats, N.; Birch, J.; Quintanilla, A.; Rink, C.J.; Fernandez-Duran, I.; Muir, M.; Finch, A.J.; Brunton, V.G.; et al. The innate immune sensor Toll-like receptor 2 controls the senescence-associated secretory phenotype. Sci Adv 2019, 5, eaaw0254. [Google Scholar] [CrossRef]
- Takahashi, A.; Loo, T.M.; Okada, R.; Kamachi, F.; Watanabe, Y.; Wakita, M.; Watanabe, S.; Kawamoto, S.; Miyata, K.; Barber, G.N.; et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 2018, 9, 1249. [Google Scholar] [CrossRef]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer's disease via cGAS-STING. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Nacarelli, T.; Liu, P.; Zhang, R. Epigenetic Basis of Cellular Senescence and Its Implications in Aging. Genes (Basel) 2017, 8. [Google Scholar] [CrossRef]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS One 2015, 10, e0116480. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol Cell 2015, 59, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.; Schlegelberger, B.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and cancer - role and therapeutic opportunities. Nat Rev Clin Oncol 2022, 19, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, H.; Wang, X.; Aoki, Y.; Wang, X.; Yang, Y.; Cheng, X.; Wang, Z.; Wang, X. Aurora-A/SOX8/FOXK1 signaling axis promotes chemoresistance via suppression of cell senescence and induction of glucose metabolism in ovarian cancer organoids and cells. Theranostics 2020, 10, 6928–6945. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-kappaB signalling. Mech Ageing Dev 2018, 170, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Lecot, P.; Alimirah, F.; Desprez, P.Y.; Campisi, J.; Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br J Cancer 2016, 114, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Verzella, D.; Tessitore, A.; Alesse, E.; Capalbo, C.; Zazzeroni, F. Cancer secretome and inflammation: The bright and the dark sides of NF-kappaB. Semin Cell Dev Biol 2018, 78, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef] [PubMed]
- von Kobbe, C. Cellular senescence: a view throughout organismal life. Cell Mol Life Sci 2018, 75, 3553–3567. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- Liu, Y.; Hawkins, O.E.; Su, Y.; Vilgelm, A.E.; Sobolik, T.; Thu, Y.M.; Kantrow, S.; Splittgerber, R.C.; Short, S.; Amiri, K.I.; et al. Targeting aurora kinases limits tumour growth through DNA damage-mediated senescence and blockade of NF-kappaB impairs this drug-induced senescence. EMBO Mol Med 2013, 5, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Peng, N.; Kang, H.H.; Feng, Y.; Minikes, A.M.; Jiang, X. Autophagy inhibition signals through senescence to promote tumor suppression. Autophagy 2023, 19, 1764–1780. [Google Scholar] [CrossRef]
- Tesei, A.; Arienti, C.; Bossi, G.; Santi, S.; De Santis, I.; Bevilacqua, A.; Zanoni, M.; Pignatta, S.; Cortesi, M.; Zamagni, A.; et al. TP53 drives abscopal effect by secretion of senescence-associated molecular signals in non-small cell lung cancer. J Exp Clin Cancer Res 2021, 40, 89. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med 2013, 210, 2057–2069. [Google Scholar] [CrossRef]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [PubMed]
- Soriani, A.; Iannitto, M.L.; Ricci, B.; Fionda, C.; Malgarini, G.; Morrone, S.; Peruzzi, G.; Ricciardi, M.R.; Petrucci, M.T.; Cippitelli, M.; et al. Reactive oxygen species- and DNA damage response-dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1. J Immunol 2014, 193, 950–960. [Google Scholar] [CrossRef]
- Antonangeli, F.; Soriani, A.; Ricci, B.; Ponzetta, A.; Benigni, G.; Morrone, S.; Bernardini, G.; Santoni, A. Natural killer cell recognition of in vivo drug-induced senescent multiple myeloma cells. Oncoimmunology 2016, 5, e1218105. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.W.; Vigano, S.; Ben-David, U.; Amon, A.; Santaguida, S. Aneuploid senescent cells activate NF-kappaB to promote their immune clearance by NK cells. EMBO Rep 2021, 22, e52032. [Google Scholar] [CrossRef]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.C.; Ho, Y.J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef]
- Chibaya, L.; Murphy, K.C.; DeMarco, K.D.; Gopalan, S.; Liu, H.; Parikh, C.N.; Lopez-Diaz, Y.; Faulkner, M.; Li, J.; Morris, J.P.t.; et al. EZH2 inhibition remodels the inflammatory senescence-associated secretory phenotype to potentiate pancreatic cancer immune surveillance. Nat Cancer 2023, 4, 872–892. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Guilbaud, E.; Schmidt, D.; Kroemer, G.; Marincola, F.M. Targeting immunogenic cell stress and death for cancer therapy. Nat Rev Drug Discov 2024. [Google Scholar] [CrossRef] [PubMed]
- Marin, I.; Boix, O.; Garcia-Garijo, A.; Sirois, I.; Caballe, A.; Zarzuela, E.; Ruano, I.; Attolini, C.S.; Prats, N.; Lopez-Dominguez, J.A.; et al. Cellular Senescence Is Immunogenic and Promotes Antitumor Immunity. Cancer Discov 2023, 13, 410–431. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.A.; Ho, Y.J.; Mezzadra, R.; Adrover, J.M.; Smolkin, R.; Zhu, C.; Woess, K.; Bernstein, N.; Schmitt, G.; Fong, L.; et al. Senescence Rewires Microenvironment Sensing to Facilitate Antitumor Immunity. Cancer Discov 2023, 13, 432–453. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pagacz, J.; Wolfgeher, D.J.; Bromerg, K.D.; Gorman, J.V.; Kron, S.J. Senescent cancer cell vaccines induce cytotoxic T cell responses targeting primary tumors and disseminated tumor cells. J Immunother Cancer 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Leong, H.S.; Lin, D.M.; Popkiss, S.; Pang, P.; Garsed, D.W.; Walkley, C.R.; Cullinane, C.; Ellul, J.; Haynes, N.M.; et al. Immune response to RB1-regulated senescence limits radiation-induced osteosarcoma formation. J Clin Invest 2013, 123, 5351–5360. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, P.; Rosati, R.; Mamdani, H.; Wright, R.E., 3rd; Hussain, Z.; Naeem, A.; Dzinic, S.; Polin, L.; Gavande, N.S.; Ratnam, M. Senescence-associated secretory proteins induced in lung adenocarcinoma by extended treatment with dexamethasone enhance migration and activation of lymphocytes. Cancer Immunol Immunother 2023, 72, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Johnson, C.A.; Prasad, N.; Yang, J.; Chen, S.C.; Ayers, G.D.; Pawlikowski, J.S.; Raman, D.; Sosman, J.A.; Kelley, M.; et al. Connecting the Dots: Therapy-Induced Senescence and a Tumor-Suppressive Immune Microenvironment. J Natl Cancer Inst 2016, 108, djv406. [Google Scholar] [CrossRef]
- Lee, D.H.; Imran, M.; Choi, J.H.; Park, Y.J.; Kim, Y.H.; Min, S.; Park, T.J.; Choi, Y.W. CDK4/6 inhibitors induce breast cancer senescence with enhanced anti-tumor immunogenic properties compared with DNA-damaging agents. Mol Oncol 2024, 18, 216–232. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Gilioli, D.; Fusco, S.; Tavella, T.; Giannetti, K.; Conti, A.; Santoro, A.; Carsana, E.; Beretta, S.; Schönlein, M.; Gambacorta, V. Therapy-induced senescence upregulates antigen presentation machinery and triggers anti-tumor immunity in Acute Myeloid Leukemia. bioRxiv, 2011; 2022.2011. 2017.515658. [Google Scholar]
- Uceda-Castro, R.; Margarido, A.S.; Cornet, L.; Vegna, S.; Hahn, K.; Song, J.Y.; Putavet, D.A.; van Geldorp, M.; Citirikkaya, C.H.; de Keizer, P.L.J.; et al. Re-purposing the pro-senescence properties of doxorubicin to introduce immunotherapy in breast cancer brain metastasis. Cell Rep Med 2022, 3, 100821. [Google Scholar] [CrossRef]
- Ruscetti, M.; Morris, J.P.t.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.J.; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2020, 181, 424–441. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997 e924. [Google Scholar] [CrossRef] [PubMed]
- Punt, S.; Malu, S.; McKenzie, J.A.; Manrique, S.Z.; Doorduijn, E.M.; Mbofung, R.M.; Williams, L.; Silverman, D.A.; Ashkin, E.L.; Dominguez, A.L.; et al. Aurora kinase inhibition sensitizes melanoma cells to T-cell-mediated cytotoxicity. Cancer Immunol Immunother 2021, 70, 1101–1113. [Google Scholar] [CrossRef]
- Hao, X.; Zhao, B.; Zhou, W.; Liu, H.; Fukumoto, T.; Gabrilovich, D.; Zhang, R. Sensitization of ovarian tumor to immune checkpoint blockade by boosting senescence-associated secretory phenotype. iScience 2021, 24, 102016. [Google Scholar] [CrossRef]
- Brenner, E.; Schorg, B.F.; Ahmetlic, F.; Wieder, T.; Hilke, F.J.; Simon, N.; Schroeder, C.; Demidov, G.; Riedel, T.; Fehrenbacher, B.; et al. Cancer immune control needs senescence induction by interferon-dependent cell cycle regulator pathways in tumours. Nat Commun 2020, 11, 1335. [Google Scholar] [CrossRef]
- Demaria, M.; O'Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A 2001, 98, 12072–12077. [Google Scholar] [CrossRef]
- Parrinello, S.; Coppe, J.P.; Krtolica, A.; Campisi, J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci 2005, 118, 485–496. [Google Scholar] [CrossRef]
- Lawrenson, K.; Grun, B.; Benjamin, E.; Jacobs, I.J.; Dafou, D.; Gayther, S.A. Senescent fibroblasts promote neoplastic transformation of partially transformed ovarian epithelial cells in a three-dimensional model of early stage ovarian cancer. Neoplasia 2010, 12, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Zhao, B.; Towers, M.; Liao, L.; Monteiro, E.L.; Xu, X.; Freeman, C.; Peng, H.; Tang, H.-Y.; Havas, A. TXNRD1 drives the innate immune response in senescent cells with implications for age-associated inflammation. Nature Aging 2024, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Long, Q.; Zhu, D.; Fu, D.; Zhang, B.; Han, L.; Qian, M.; Guo, J.; Xu, J.; Cao, L.; et al. Targeting amphiregulin (AREG) derived from senescent stromal cells diminishes cancer resistance and averts programmed cell death 1 ligand (PD-L1)-mediated immunosuppression. Aging Cell 2019, 18, e13027. [Google Scholar] [CrossRef] [PubMed]
- Di, G.H.; Liu, Y.; Lu, Y.; Liu, J.; Wu, C.; Duan, H.F. IL-6 secreted from senescent mesenchymal stem cells promotes proliferation and migration of breast cancer cells. PLoS One 2014, 9, e113572. [Google Scholar] [CrossRef] [PubMed]
- Pazolli, E.; Luo, X.; Brehm, S.; Carbery, K.; Chung, J.J.; Prior, J.L.; Doherty, J.; Demehri, S.; Salavaggione, L.; Piwnica-Worms, D.; et al. Senescent stromal-derived osteopontin promotes preneoplastic cell growth. Cancer Res 2009, 69, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hornsby, P.J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res 2007, 67, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Angelini, P.D.; Zacarias Fluck, M.F.; Pedersen, K.; Parra-Palau, J.L.; Guiu, M.; Bernado Morales, C.; Vicario, R.; Luque-Garcia, A.; Navalpotro, N.P.; Giralt, J.; et al. Constitutive HER2 signaling promotes breast cancer metastasis through cellular senescence. Cancer Res 2013, 73, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.W.; Steiner, I.; Haider, S.; Robertson, D.; Buus, R.; O'Leary, L.; Isacke, C.M. Therapy-induced normal tissue damage promotes breast cancer metastasis. iScience 2024, 27, 108503. [Google Scholar] [CrossRef]
- Aifuwa, I.; Giri, A.; Longe, N.; Lee, S.H.; An, S.S.; Wirtz, D. Senescent stromal cells induce cancer cell migration via inhibition of RhoA/ROCK/myosin-based cell contractility. Oncotarget 2015, 6, 30516–30531. [Google Scholar] [CrossRef]
- Tato-Costa, J.; Casimiro, S.; Pacheco, T.; Pires, R.; Fernandes, A.; Alho, I.; Pereira, P.; Costa, P.; Castelo, H.B.; Ferreira, J.; et al. Therapy-Induced Cellular Senescence Induces Epithelial-to-Mesenchymal Transition and Increases Invasiveness in Rectal Cancer. Clin Colorectal Cancer 2016, 15, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.H.; Park, T.J. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat Commun 2017, 8, 15208. [Google Scholar] [CrossRef]
- Luo, X.; Fu, Y.; Loza, A.J.; Murali, B.; Leahy, K.M.; Ruhland, M.K.; Gang, M.; Su, X.; Zamani, A.; Shi, Y.; et al. Stromal-Initiated Changes in the Bone Promote Metastatic Niche Development. Cell Rep 2016, 14, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Malaquin, N.; Vercamer, C.; Bouali, F.; Martien, S.; Deruy, E.; Wernert, N.; Chwastyniak, M.; Pinet, F.; Abbadie, C.; Pourtier, A. Senescent fibroblasts enhance early skin carcinogenic events via a paracrine MMP-PAR-1 axis. PLoS One 2013, 8, e63607. [Google Scholar] [CrossRef] [PubMed]
- Farsam, V.; Basu, A.; Gatzka, M.; Treiber, N.; Schneider, L.A.; Mulaw, M.A.; Lucas, T.; Kochanek, S.; Dummer, R.; Levesque, M.P.; et al. Senescent fibroblast-derived Chemerin promotes squamous cell carcinoma migration. Oncotarget 2016, 7, 83554–83569. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H., 3rd; Dang, V.M.; Appleton, J.; O'Connell, M.P.; Cheng, P.; et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 2016, 532, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Kauser, K.; Campisi, J.; Beausejour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem 2006, 281, 29568–29574. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tuxhorn, J.A.; Ressler, S.J.; McAlhany, S.J.; Dang, T.D.; Rowley, D.R. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res 2005, 65, 8887–8895. [Google Scholar] [CrossRef] [PubMed]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci Transl Med 2016, 8, 362ra144. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Hemann, M.T. DNA damage-mediated induction of a chemoresistant niche. Cell 2010, 143, 355–366. [Google Scholar] [CrossRef]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev 2016, 30, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintas-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, J.; Yamada, Y. Unveiling the Role of Senescence-Induced Cellular Plasticity. Cell Stem Cell 2017, 20, 293–294. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marion, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Munoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354. [Google Scholar] [CrossRef] [PubMed]
- Karabicici, M.; Alptekin, S.; Firtina Karagonlar, Z.; Erdal, E. Doxorubicin-induced senescence promotes stemness and tumorigenicity in EpCAM-/CD133- nonstem cell population in hepatocellular carcinoma cell line, HuH-7. Mol Oncol 2021, 15, 2185–2202. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Montero, P.; Londono-Vallejo, A.; Vernot, J.P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal 2017, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Cahu, J.; Bustany, S.; Sola, B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis 2012, 3, e446. [Google Scholar] [CrossRef]
- Palacio, L.; Goyer, M.L.; Maggiorani, D.; Espinosa, A.; Villeneuve, N.; Bourbonnais, S.; Moquin-Beaudry, G.; Le, O.; Demaria, M.; Davalos, A.R.; et al. Restored immune cell functions upon clearance of senescence in the irradiated splenic environment. Aging Cell 2019, 18, e12971. [Google Scholar] [CrossRef]
- Salam, R.; Saliou, A.; Bielle, F.; Bertrand, M.; Antoniewski, C.; Carpentier, C.; Alentorn, A.; Capelle, L.; Sanson, M.; Huillard, E.; et al. Cellular senescence in malignant cells promotes tumor progression in mouse and patient Glioblastoma. Nat Commun 2023, 14, 441. [Google Scholar] [CrossRef] [PubMed]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep 2014, 9, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun 2016, 7, 11762. [Google Scholar] [CrossRef]
- Guan, X.; LaPak, K.M.; Hennessey, R.C.; Yu, C.Y.; Shakya, R.; Zhang, J.; Burd, C.E. Stromal Senescence By Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol Cancer Res 2017, 15, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Di Mitri, D.; Toso, A.; Chen, J.J.; Sarti, M.; Pinton, S.; Jost, T.R.; D'Antuono, R.; Montani, E.; Garcia-Escudero, R.; Guccini, I.; et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Nolan, E.; Bridgeman, V.L.; Ombrato, L.; Karoutas, A.; Rabas, N.; Sewnath, C.A.N.; Vasquez, M.; Rodrigues, F.S.; Horswell, S.; Faull, P.; et al. Radiation exposure elicits a neutrophil-driven response in healthy lung tissue that enhances metastatic colonization. Nat Cancer 2022, 3, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Revandkar, A.; Dubash, T.D.; Ravi, A.; Wittner, B.S.; Lin, M.; Morris, R.; Burr, R.; Guo, H.; Seeger, K.; et al. TGF-beta in the microenvironment induces a physiologically occurring immune-suppressive senescent state. Cell Rep 2023, 42, 112129. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.W.; Kim, Y.H.; Oh, S.Y.; Suh, K.W.; Kim, Y.S.; Lee, G.Y.; Yoon, J.E.; Park, S.S.; Lee, Y.K.; Park, Y.J.; et al. Senescent Tumor Cells Build a Cytokine Shield in Colorectal Cancer. Adv Sci (Weinh) 2021, 8, 2002497. [Google Scholar] [CrossRef]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE(2)-Mediated Suppression of Antitumor Immunity. Cancer Discov 2017, 7, 522–538. [Google Scholar] [CrossRef]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8(+) T cell inhibition. Nat Commun 2019, 10, 2387. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 2022, 611, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Onorati, A.; Havas, A.P.; Lin, B.; Rajagopal, J.; Sen, P.; Adams, P.D.; Dou, Z. Upregulation of PD-L1 in Senescence and Aging. Mol Cell Biol 2022, 42, e0017122. [Google Scholar] [CrossRef] [PubMed]
- Majewska, J.; Agrawal, A.; Mayo, A.; Roitman, L.; Chatterjee, R.; Kralova, J.; Landsberger, T.; Katzenelenbogen, Y.; Salame, T.M.; Hagai, E. p16-dependent upregulation of PD-L1 impairs immunosurveillance of senescent cells. bioRxiv 2001. [Google Scholar]
- Chaib, S.; López-Domínguez, J.A.; Lalinde-Gutiérrez, M.; Prats, N.; Marin, I.; Boix, O.; García-Garijo, A.; Meyer, K.; Muñoz, M.I.; Aguilera, M. The efficacy of chemotherapy is limited by intratumoral senescent cells expressing PD-L2. Nature Cancer 2024, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat Metab 2021, 3, 1290–1301. [Google Scholar] [CrossRef]
- Diwan, B.; Sharma, R. Nutritional components as mitigators of cellular senescence in organismal aging: a comprehensive review. Food Sci Biotechnol 2022, 31, 1089–1109. [Google Scholar] [CrossRef]
- Fontana, L.; Mitchell, S.E.; Wang, B.; Tosti, V.; van Vliet, T.; Veronese, N.; Bertozzi, B.; Early, D.S.; Maissan, P.; Speakman, J.R.; et al. The effects of graded caloric restriction: XII. Comparison of mouse to human impact on cellular senescence in the colon. Aging Cell 2018, 17, e12746. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Wang, W.J.; Liu, J.Q.; Song, Y.H.; Li, P.; Sun, X.F.; Cai, G.Y.; Chen, X.M. Methionine restriction delays senescence and suppresses the senescence-associated secretory phenotype in the kidney through endogenous hydrogen sulfide. Cell Cycle 2019, 18, 1573–1587. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Ogura, Y.; Monno, I.; Xu, J.; Koya, D. Effect of Methionine Restriction on Aging: Its Relationship to Oxidative Stress. Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Sakai, C.; Ishida, M.; Ohba, H.; Yamashita, H.; Uchida, H.; Yoshizumi, M.; Ishida, T. Fish oil omega-3 polyunsaturated fatty acids attenuate oxidative stress-induced DNA damage in vascular endothelial cells. PLoS One 2017, 12, e0187934. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.I.; Bourgeois, J.M.; Nederveen, J.P.; Leite, M.R.; Hettinga, B.P.; Bujak, A.L.; May, L.; Lin, E.; Crozier, M.; Rusiecki, D.R.; et al. Lifelong aerobic exercise protects against inflammaging and cancer. PLoS One 2019, 14, e0210863. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Rahman, M.; Olson, I.; Mansour, M.; Carlstrom, L.P.; Sutiwisesak, R.; Saber, R.; Rajani, K.; Warrington, A.E.; Howard, A.; Schroeder, M.; et al. Selective Vulnerability of Senescent Glioblastoma Cells to BCL-XL Inhibition. Mol Cancer Res 2022, 20, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Leite de Oliveira, R.; Wang, C.; Fernandes Neto, J.M.; Mainardi, S.; Evers, B.; Lieftink, C.; Morris, B.; Jochems, F.; Willemsen, L.; et al. High-Throughput Functional Genetic and Compound Screens Identify Targets for Senescence Induction in Cancer. Cell Rep 2017, 21, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Carpenter, V.J.; Tyutyunyk-Massey, L.; Murray, G.; Leverson, J.D.; Souers, A.J.; Alotaibi, M.R.; Faber, A.C.; Reed, J.; Harada, H.; et al. Clearance of therapy-induced senescent tumor cells by the senolytic ABT-263 via interference with BCL-X(L) -BAX interaction. Mol Oncol 2020, 14, 2504–2519. [Google Scholar] [CrossRef]
- Shahbandi, A.; Rao, S.G.; Anderson, A.Y.; Frey, W.D.; Olayiwola, J.O.; Ungerleider, N.A.; Jackson, J.G. BH3 mimetics selectively eliminate chemotherapy-induced senescent cells and improve response in TP53 wild-type breast cancer. Cell Death Differ 2020, 27, 3097–3116. [Google Scholar] [CrossRef] [PubMed]
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.A.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat Commun 2019, 10, 2556. [Google Scholar] [CrossRef] [PubMed]
- Kolodkin-Gal, D.; Roitman, L.; Ovadya, Y.; Azazmeh, N.; Assouline, B.; Schlesinger, Y.; Kalifa, R.; Horwitz, S.; Khalatnik, Y.; Hochner-Ger, A.; et al. Senolytic elimination of Cox2-expressing senescent cells inhibits the growth of premalignant pancreatic lesions. Gut 2022, 71, 345–355. [Google Scholar] [CrossRef]
- Gonzalez-Gualda, E.; Paez-Ribes, M.; Lozano-Torres, B.; Macias, D.; Wilson, J.R., 3rd; Gonzalez-Lopez, C.; Ou, H.L.; Miron-Barroso, S.; Zhang, Z.; Lerida-Viso, A.; et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 2020, 19, e13142. [Google Scholar] [CrossRef]
- Estepa-Fernández, A.; Alfonso, M.; Morellá-Aucejo, Á.; García-Fernández, A.; Lérida-Viso, A.; Lozano-Torres, B.; Galiana, I.; Soriano-Teruel, P.M.; Sancenon, F.; Orzaez, M. Senolysis reduces senescence in veins and cancer cell migration. Advanced Therapeutics 2021, 4, 2100149. [Google Scholar] [CrossRef]
- He, Y.; Zhang, X.; Chang, J.; Kim, H.N.; Zhang, P.; Wang, Y.; Khan, S.; Liu, X.; Zhang, X.; Lv, D.; et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat Commun 2020, 11, 1996. [Google Scholar] [CrossRef]
- Lafontaine, J.; Cardin, G.B.; Malaquin, N.; Boisvert, J.S.; Rodier, F.; Wong, P. Senolytic Targeting of Bcl-2 Anti-Apoptotic Family Increases Cell Death in Irradiated Sarcoma Cells. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Whittle, J.R.; Vaillant, F.; Surgenor, E.; Policheni, A.N.; Giner, G.; Capaldo, B.D.; Chen, H.R.; Liu, H.K.; Dekkers, J.F.; Sachs, N.; et al. Dual Targeting of CDK4/6 and BCL2 Pathways Augments Tumor Response in Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res 2020, 26, 4120–4134. [Google Scholar] [CrossRef] [PubMed]
- Troiani, M.; Colucci, M.; D'Ambrosio, M.; Guccini, I.; Pasquini, E.; Varesi, A.; Valdata, A.; Mosole, S.; Revandkar, A.; Attanasio, G.; et al. Single-cell transcriptomics identifies Mcl-1 as a target for senolytic therapy in cancer. Nat Commun 2022, 13, 2177. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat Med 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Novais, E.J.; Tran, V.A.; Johnston, S.N.; Darris, K.R.; Roupas, A.J.; Sessions, G.A.; Shapiro, I.M.; Diekman, B.O.; Risbud, M.V. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat Commun 2021, 12, 5213. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Tian, X.; Ye, Y.; Liang, Y.; Zhao, J.; Wu, T.; Lu, N. Identification of GL-V9 as a novel senolytic agent against senescent breast cancer cells. Life Sci 2021, 272, 119196. [Google Scholar] [CrossRef]
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic Cocktail Dasatinib+Quercetin (D+Q) Does Not Enhance the Efficacy of Senescence-Inducing Chemotherapy in Liver Cancer. Front Oncol 2018, 8, 459. [Google Scholar] [CrossRef]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat Commun 2020, 11, 1935. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019, 574, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Triana-Martinez, F.; Picallos-Rabina, P.; Da Silva-Alvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreiros, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat Commun 2019, 10, 4731. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat Metab 2019, 1, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.M.; Wulfmeyer, V.C.; Chen, R.; Erlangga, Z.; Sinning, J.; von Massenhausen, A.; Sorensen-Zender, I.; Beer, K.; von Vietinghoff, S.; Haller, H.; et al. Induction of ferroptosis selectively eliminates senescent tubular cells. Am J Transplant 2022, 22, 2158–2168. [Google Scholar] [CrossRef] [PubMed]
- Katsuumi, G.; Shimizu, I.; Suda, M.; Yoshida, Y.; Furihata, T.; Joki, Y.; Hsiao, C.L.; Jiaqi, L.; Fujiki, S.; Abe, M.; et al. SGLT2 inhibition eliminates senescent cells and alleviates pathological aging. Nat Aging 2024. [Google Scholar] [CrossRef]
- Le, H.H.; Cinaroglu, S.S.; Manalo, E.C.; Ors, A.; Gomes, M.M.; Duan Sahbaz, B.; Bonic, K.; Origel Marmolejo, C.A.; Quentel, A.; Plaut, J.S.; et al. Molecular modelling of the FOXO4-TP53 interaction to design senolytic peptides for the elimination of senescent cancer cells. EBioMedicine 2021, 73, 103646. [Google Scholar] [CrossRef]
- Amor, C.; Fernández-Maestre, I.; Chowdhury, S.; Ho, Y.-J.; Nadella, S.; Graham, C.; Carrasco, S.E.; Nnuji-John, E.; Feucht, J.; Hinterleitner, C. Prophylactic and long-lasting efficacy of senolytic CAR T cells against age-related metabolic dysfunction. Nature Aging 2024, 1–14. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Uyar, B.; Palmer, D.; Kowald, A.; Murua Escobar, H.; Barrantes, I.; Moller, S.; Akalin, A.; Fuellen, G. Single-cell analyses of aging, inflammation and senescence. Ageing Res Rev 2020, 64, 101156. [Google Scholar] [CrossRef]
- Chen, W.; Wang, X.; Wei, G.; Huang, Y.; Shi, Y.; Li, D.; Qiu, S.; Zhou, B.; Cao, J.; Chen, M.; et al. Single-Cell Transcriptome Analysis Reveals Six Subpopulations Reflecting Distinct Cellular Fates in Senescent Mouse Embryonic Fibroblasts. Front Genet 2020, 11, 867. [Google Scholar] [CrossRef] [PubMed]
- Tabula Muris, C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Gurkar, A.U.; Gerencser, A.A.; Mora, A.L.; Nelson, A.C.; Zhang, A.R.; Lagnado, A.B.; Enninful, A.; Benz, C.; Furman, D.; Beaulieu, D.; et al. Spatial mapping of cellular senescence: emerging challenges and opportunities. Nat Aging 2023, 3, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Sanwald, J.L.; Poschmann, G.; Stuhler, K.; Behrends, C.; Hoffmann, S.; Willbold, D. The GABARAP Co-Secretome Identified by APEX2-GABARAP Proximity Labelling of Extracellular Vesicles. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.E.; Park, I.; Kim, J.; Kang, M.G.; Choi, W.G.; Shin, H.; Kim, J.S.; Rhee, H.W.; Suh, J.M. Dynamic tracking and identification of tissue-specific secretory proteins in the circulation of live mice. Nat Commun 2021, 12, 5204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular senescence: a key therapeutic target in aging and diseases. J Clin Invest 2022, 132. [Google Scholar] [CrossRef]
- Truskowski, K.; Amend, S.R.; Pienta, K.J. Dormant cancer cells: programmed quiescence, senescence, or both? Cancer Metastasis Rev 2023, 42, 37–47. [Google Scholar] [CrossRef]
Figure 1.
Hallmarks of cellular senescence. Cellular senescence is characterized by an exit from proliferation to enter a persistent, G1-like cell cycle arrest and adopt a host of other cellular changes. In culture, senescent cells typically display an enlarged and flattened morphology with altered nuclear shape and prominent intracellular vesicles that allow for their identification in phase or brightfield. The increased vesicular content likely underlies the apparent activation of the GLB1 lysosomal β-galactosidase, a biochemical marker of senescence known as senescence-associated β-galactosidase (SA-β-gal), which is readily detected with X-Gal or other chromogenic and fluorogenic β-galactosidase reporters. Other features include a chronically active DNA damage response (DDR), which can be detected as persistent nuclear foci of phosphorylated histone H2AX using anti-γH2AX antibodies. Senescent cells display other nuclear changes, including altered lamins, chromatin remodeling and the formation of senescence-associated heterochromatin foci (SAHF). The persistence of senescent cells despite these indications of cell stress likely reflects resistance to apoptosis, linked to overexpression of anti-apoptotic proteins such as Bcl-2 family members. A related hallmark of senescence is the accumulation of dysfunctional mitochondria, which may underlie the increased levels of reactive oxygen species (ROS) and oxidative damage. Despite their permanently arrested growth and, senescent cells remain metabolically active, albeit in an altered state. The senescence-associated secretory phenotype (SASP) is a key feature of senescent cells, characterized by the release of a wide range of bioactive molecules and extracellular vesicles.
Figure 1.
Hallmarks of cellular senescence. Cellular senescence is characterized by an exit from proliferation to enter a persistent, G1-like cell cycle arrest and adopt a host of other cellular changes. In culture, senescent cells typically display an enlarged and flattened morphology with altered nuclear shape and prominent intracellular vesicles that allow for their identification in phase or brightfield. The increased vesicular content likely underlies the apparent activation of the GLB1 lysosomal β-galactosidase, a biochemical marker of senescence known as senescence-associated β-galactosidase (SA-β-gal), which is readily detected with X-Gal or other chromogenic and fluorogenic β-galactosidase reporters. Other features include a chronically active DNA damage response (DDR), which can be detected as persistent nuclear foci of phosphorylated histone H2AX using anti-γH2AX antibodies. Senescent cells display other nuclear changes, including altered lamins, chromatin remodeling and the formation of senescence-associated heterochromatin foci (SAHF). The persistence of senescent cells despite these indications of cell stress likely reflects resistance to apoptosis, linked to overexpression of anti-apoptotic proteins such as Bcl-2 family members. A related hallmark of senescence is the accumulation of dysfunctional mitochondria, which may underlie the increased levels of reactive oxygen species (ROS) and oxidative damage. Despite their permanently arrested growth and, senescent cells remain metabolically active, albeit in an altered state. The senescence-associated secretory phenotype (SASP) is a key feature of senescent cells, characterized by the release of a wide range of bioactive molecules and extracellular vesicles.
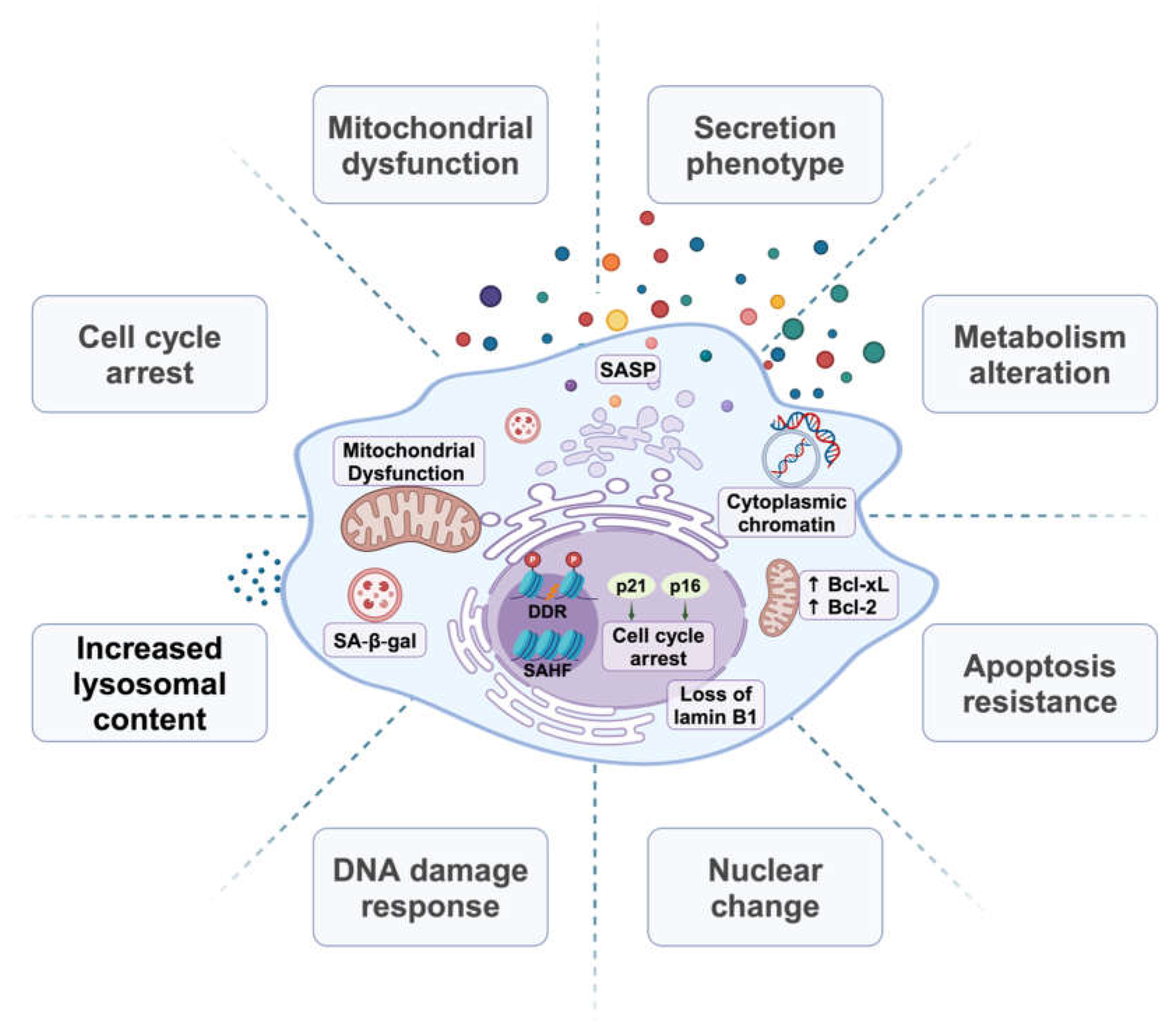
Figure 2.
Metabolic changes in senescent cells. Senescent cells can display a Warburg effect-like pattern by continuing glycolysis regardless of oxygen availability, and this shift is often accompanied by glycolytic enzyme upregulation. Senescent cells may also display increased beta-oxidation and oxygen consumption. Lipid metabolism shifts towards increased fatty acid synthesis, which along with increased lipid uptake, results in accumulation of lipid droplets. Overall, the metabolic changes in senescent cells contribute to elevated ROS levels. Labile iron pools and the Fenton reaction exacerbate oxidative damage by facilitating the conversion of relatively mild oxidants into highly reactive free radicals. As ROS levels rise, oxidative damage occurs across various cellular components, causing depletion of antioxidants, lipid peroxidation, DNA damage, and protein carbonylation. Furthermore, oxidative stress compromises the lysosomal and proteasomal systems. Together, these changes contribute to formation of lipofuscin, a complex aggregate of highly cross-linked non-degradable proteins, carbohydrates, lipids, and transition metals.
Figure 2.
Metabolic changes in senescent cells. Senescent cells can display a Warburg effect-like pattern by continuing glycolysis regardless of oxygen availability, and this shift is often accompanied by glycolytic enzyme upregulation. Senescent cells may also display increased beta-oxidation and oxygen consumption. Lipid metabolism shifts towards increased fatty acid synthesis, which along with increased lipid uptake, results in accumulation of lipid droplets. Overall, the metabolic changes in senescent cells contribute to elevated ROS levels. Labile iron pools and the Fenton reaction exacerbate oxidative damage by facilitating the conversion of relatively mild oxidants into highly reactive free radicals. As ROS levels rise, oxidative damage occurs across various cellular components, causing depletion of antioxidants, lipid peroxidation, DNA damage, and protein carbonylation. Furthermore, oxidative stress compromises the lysosomal and proteasomal systems. Together, these changes contribute to formation of lipofuscin, a complex aggregate of highly cross-linked non-degradable proteins, carbohydrates, lipids, and transition metals.
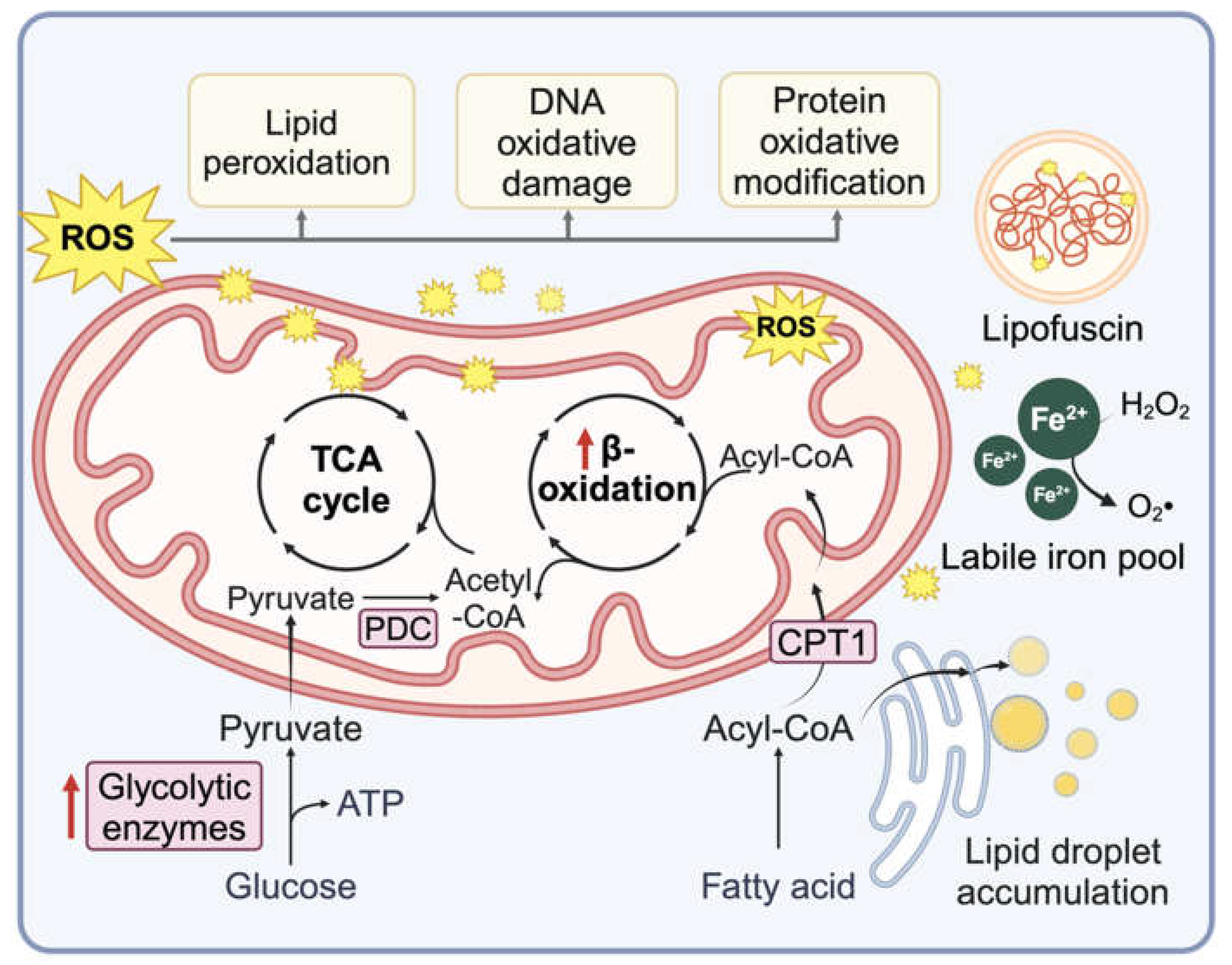
Table 1.
Immunostimulatory senescent cells in cancer.
| Senescence Induction methods | Cancer type | Affected immune cell population | References (PMID) |
|---|---|---|---|
| H-RasG12V and p53 reactivation | Liver cancer |
PMN | 17251933 |
| Alisertib | Melanoma | PMN and Mφ | 23180582 |
| Carbon tetrachloride | Hepatocellular carcinoma | M1 Mφ | 23562644 |
| Temozolomide | Glioblastoma | Increased M1 Mφ and decreased MDSCs | 36472478 |
| IR | Lung cancer | M1 Mφ | 33673859 |
| Cyclophosphamide | B cell Lymphoma | NK cell | 21979375 |
| H-RasG12V | Liver cancer | NK cell | 24043758 |
| Chemo agents | multiple myeloma | NK cell | 19098271 |
| Doxorubicin | multiple myeloma | NK cell | 24913980 |
| Trametinib + Palbociclib | KP lung cancer |
NK cell | 30573629 |
| N-RasG12D | Liver cancer | NK cell | 27099234 |
| Trametinib + Palbociclib + Tazemetostat | KPC PDAC model | NK and T cells | 37142692 |
| N-RasG12V | Liver cancer | CD4+ T cells | 25621566 |
| IR | Osteosarcomas | NKT cells | 24231354 |
| Alisertib | Melanoma | CD8+ T cell | 26719346 |
| Dexamethasone | Lung adenocarcinoma | NK and T cell | 36434273 |
| Abemaciclib | mammary carcinoma | T cell | 28813415 |
| Cytarabine or Palbociclib | AML | T cells | 10.1101/2022.11.17.515658 |
| Doxorubicin | Melanoma | DCs and T cells | 36302218 |
| IR+veliparib | Multiple cancers | DCs, NK, and T cells | 22334019, 36792123 |
| N-RasG12D | Liver cancer | Increased CD8+ T cells and decreased MDSCs | 36302222 |
| Doxorubicin | Metastatic breast cancer | CD8+ T cell | 36384097 |
| Trametinib + Palbociclib | PDAC KPC model | CD8+ T cells | 32234521 |
| Abemaciclib | Melanoma | T cells | 30388455 |
| AZD1152 | Melanoma | T cells | 33123754 |
| Irinotecan + Cisplatin | ovarian cancer | DCs and T cells | 33490922 |
| IFN-γ | Multiple cancers | T cells | 32165639 |
Table 2.
Therapy induced senescence potentiates immunotherapy.
| Senescence induction methods | Cancer type | Immunotherapy agent | PMID (PMID) |
|---|---|---|---|
| Mitoxantrone | prostate cancer | αPD-1/PD-L1 Ab | 31493351 |
| Doxorubicin | melanoma | αPD-L2 Ab | BiorXiv |
| Alisertib | Melanoma | αCD137 Ab | 26719346 |
| Abemaciclib | mammary carcinoma | αPD-L1 Ab | 28813415 |
| IR + Veliparib | Multiple cancers | αPD-L1 Ab | 36792123 |
| Doxorubicin | Metastatic breast cancer | αPD-1 Ab | 36384097 |
| Trametinib + Palbociclib | PDAC (KPC model) | αPD-1 Ab | 32234521 |
| Abemaciclib | Melanoma | αCTLA4 Ab | 30388455 |
| AZD1152 | Melanoma | αCTLA4 Ab | 33123754 |
| Irinotecan + Cisplatin | ovarian cancer | αPD-1 Ab | 33490922 |
Table 4.
Senolytics in cancer.
| MOA | Senolytic agent | Senescence Induction methods | Cancer type | References (PMID) |
|---|---|---|---|---|
| Tyrosine kinase inhibitor + flavonoid derivative | Dasatinib and quercetin | Doxorubicin | Liver Cancer |
30425964 |
| Flavonoid derivative | Fisetin | Olaparib | Ovarian cancer | 31186408 |
| GL-V9 | Doxorubicin | Breast cancer | 33617857 | |
| Bcl-2/Bcl-xL/Bcl-w inhibitor | ABT263 | Doxorubicin or etoposide | Lung and breast cancer | 32652830 |
| ABT263 | SMARCB1 inhibition | Multiple cancer cell lines | 29045843 |
|
| ABT263 |
Olaparib | Ovarian | 31186408 | |
| ABT263 |
Doxorubicin | Breast cancer | 32457483 | |
| ABT263 | Radiation and temozolomide | Glioblastoma |
35191501 | |
| ABT737 | K-RasG12V | Pancreatic cancer | 33649045 | |
| Bcl-xL degrader | PZ15227 | 32332723 | ||
| Selective BCL-xL inhibitor | A1331852, A1155463 | Radiation and temozolomide | Glioblastoma cell lines | 35191501 |
| Selective Bcl-2 inhibitor | ABT199 | IR | Sarcoma cell lines | 33494434 |
| ABT199 |
Palbociclib + fulvestrant | Breast cancer | 32245900 | |
| MCL-1 inhibitor | S63845 | Doxorubicin or etoposide | Breast cancer | 32457483 |
| S63845 | Docetaxel | Prostate cancer | 35449130 | |
| Galacto-conjugation of ABT263 | Nav-Gal | Cisplatin | A549 xenograft | 32233024 |
| Nav-Gal | Palbociclib | Breast cancer lung metastasis | 36566002 | |
| BET degrader | ARV825 | High fat diet or doxorubicin | Liver cancer | 32321921 |
| Cardiac glycosides | Digoxin Digitoxin Proscillaridin A Ouabain |
Therapy induced senescence | Multiple cancer types | 31636264 31799499 34714358 |
| mTOR inhibitor | AZD8055 | CDC7 inhibitor XL413 | Hepatocellular carcinoma | 31578521 |
| HDAC inhibitor | Panobinostat Decrease Bcl-xl |
Taxol, cisplatin | NSCLC and HNSCC | 28507307 |
| Senolytic peptide | FOXO4 binding peptide ES2 |
BRAF inhibitor Dabrafenib | Melanoma | 34689087 |
| Senolytic CAR-T | urokinase-type plasminogen (uPAR)-specific CAR T cells) | Trametinib + Palbociclib | Lung cancer | 32555459 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



