Submitted:
03 June 2024
Posted:
03 June 2024
You are already at the latest version
Abstract
We investigate the possible laboratory origins of the highly pathogenic avian influenza (HPAI) H5N1 clade 2.3.4.4b genotype B3.13, currently affecting various animal species and causing sporadic human infections. The first detection of HPAI H5N1 clade 2.3.4.4b in the Netherlands in 2020 raises concerns about earlier gain-of-function research. The proximal origins of HPAI H5N1 Clade 2.3.4.4b may be the USDA Southeast Poultry Research Laboratory (SEPRL) in Athens, Georgia and the Erasmus Medical Center in Rotterdam, the Netherlands. Genetic analysis indicates that genotype B3.13 emerged in 2024 and exhibits genetic links to genotype B1.2, which was identified to have originated in Georgia in January 2022 after the start of serial passage research with H5Nx clade 2.3.4.4 in mallard ducks at the USDA Southeast Poultry Research Laboratory (SEPRL) in Athens, Georgia in April 2021. Genotype B1.2 was found in a bottlenose dolphin (Tursiops truncatus) in March 2022 in Florida, indicating sudden new adaptations to different animal species. The NP gene of H5N1 clade 2.3.4.4b (genotype B3.13), which is causing outbreaks in cattle, was reported to have likely originated from an avian influenza A virus derived from mallard ducks. Significant mutations found in recent human cases, including PB2 E627K and PB2 M631L, also suggest links to serial passage experiments. However, further investigation is urgently needed to confirm these findings and to identify all H5N1 laboratory leaks that may have occurred with a focus on mallard ducks and other migratory waterfowl, which have the potential to infect a large number of poultry and livestock facilities around the world. A moratorium on gain-of-function research including serial passage of H5N1 is indicated to prevent a man-made influenza pandemic affecting animals and humans.
Keywords:
H5N1
; Clade 2.3.4.4b
; laboratory leak
; gain-of-function
; bird flu
; USDA
; pandemic
Introduction
As of May 29, 2024, there has been significant media coverage concerning a new highly pathogenic variant of H5N1 avian influenza, commonly referred to as “bird flu.” According to a May 16, 2024 advisory issued by the World Health Organization:
"The goose/Guangdong-lineage of H5N1 avian influenza viruses first emerged in 1996 and has been causing outbreaks in birds since then. Since 2020, a variant of these viruses has led to an unprecedented number of deaths in wild birds and poultry in many countries. First affecting Africa, Asia and Europe, in 2021, the virus spread to North America, and in 2022, to Central and South America. From 2021 to 2022, Europe and North America observed their largest and most extended epidemic of avian influenza with unusual persistence of the virus in wild bird populations. Since 2022, there have been increasing reports of deadly outbreaks among mammals also caused by influenza A(H5) – including influenza A(H5N1) – viruses. There are likely to be more outbreaks that have not been detected or reported. Both land and sea mammals have been affected, including outbreaks in farmed fur animals, seals, sea lions, and detections in other wild and domestic animals such as foxes, bears, otters, raccoons, cats, dogs, cows, goats and others. " [1]
The variant referred to in this WHO advisory is known as highly pathogenic avian influenza (HPAI) H5N1 clade 2.3.4.4b. A literature review on this variant revealed that it—and the subtype (H5Nx) from which it emerged—possess conspicuous functions that were not evident in the viruses from which this new clade is alleged to have evolved. These functions include the following:
1) Increased transmissibility leading to markedly faster intercontinental spread.
2) Increased persistence, causing uncharacteristic outbreaks during summer seasons.
3) Increased virulence for both domesticated and wild birds.
Already in 2017, a team of avian influenza specialists stated that “the emergence and intercontinental spread of highly pathogenic avian influenza A(H5Nx) virus clade 2.3.4.4 is unprecedented.” [3] These researchers noted that the emergence of H5Nx clade 2.3.4.4 is accompanied by a change in receptor-binding specificity, though they also claim that “the potential role of altered receptor specificity in extended host range and the contribution to the rapid worldwide spread of influenza viruses is still unknown.” According to a 2017 paper by Imai and Kawaoka titled ‘The role of receptor binding specificity in interspecies transmission of influenza viruses,’ receptor binding specificity does indeed play a key role in the interspecies transmission of influenza A viruses [4]. Kawaoka’s research builds on the research published by a R.A.M. Fouchier’s Dutch team in 2010 in a paper titled ‘In Vitro Assessment of Attachment Pattern and Replication Efficiency of H5N1 Influenza A Viruses with Altered Receptor Specificity’ [5].
According to a 2021 paper titled ‘H5Nx Viruses Emerged during the Suppression of H5N1 Virus Populations in Poultry’ by a research team of the University of Georgia:
"We show that H5Nx viruses emerged during the successful suppression of H5N1 virus populations in poultry [in China], providing an opportunity for antigenically distinct H5Nx viruses to propagate. Avian influenza vaccination programs would benefit from universal vaccines targeting a wider diversity of influenza viruses to prevent the emergence of novel subtypes."[6].
The findings of these researchers present an illustrative case of Dr. Geert Vanden Bossche’s thesis that mass vaccination with non-sterilizing vaccines can result in the emergence of a new, more virulent viral strain [7]. As the University of Georgia team note, “In particular, we show that the widespread use of H5N1 vaccines likely conferred a fitness advantage to H5Nx viruses due to the antigenic mismatch of the neuraminidase genes.” [6].
Currently the world is facing a global pandemic of H5N1 clade 2.3.4.4b—first detected in October 2020 in the Netherlands [8] — that purportedly evolved from H5Nx viruses and possesses even greater pathogenic functions. An especially striking feature of the newly emerged H5N1 clade 2.3.4.4b is how rapidly it spread from birds in Europe to birds in North America. This rapid spread contrasts with the previously slow intercontinental spread of the goose/Guangdong-lineage of H5N1. After emerging in China in 1996, it was first detected in Europe in 2005, and then in the United States in 2014 [9]. While it apparently took nine years for earlier variants to spread from Europe to the United States, H5N1 clade 2.3.4.4b was first detected in the Netherlands in October 2020 [8] and then in the United States in late 2021 [9]. What could account for the new variant’s extraordinarily rapid intercontinental spread?
In a July 11, 2022 paper in Nature titled ‘Transatlantic spread of highly pathogenic avian influenza H5N1 by wild birds from Europe to North America in 2021,’ a large international team offered the hypothesis that birds migrating from Europe to Iceland and other North Atlantic islands, and from there to North America in 2021, must have carried the virus across the Atlantic [10]. As they noted in their conclusion:
"The HPAI H5N1 viruses that were detected in Newfoundland in November and December 2021 originated from Northwest Europe and belonged to HPAI clade 2.3.4.4b. Most likely, these viruses emerged in Northwest Europe in winter 2020/2021, dispersed from Europe in late winter or early spring 2021, and arrived in Newfoundland in autumn 2021. The viruses may have been carried across the Atlantic by migratory birds using different routes, including Icelandic, Greenland/Arctic, or pelagic routes. The unusually high presence of the viruses in European wild bird populations in late winter and spring 2021, as well as the greater involvement of barnacle and greylag geese in the epidemiology of HPAI in Europe since October 2020, may explain why spread to Newfoundland happened this winter (2021/2022), and not in the previous winters." [10].
This conclusion contains two implausible elements and a notable omission. First, the hypothetical spread of a new avian influenza variant by migratory birds from Europe to North America by crossing the North Atlantic has never been documented before and therefore appears to be unprecedented. Second, migratory birds in the East Atlantic flyway do not fly from Northwest Europe to North America in the autumn, but the other way around, from North America to Northwest Europe [11]. Third, the paper’s conclusion omits the fact that at the same time (December 2021) the H5N1 clade 2.3.4.4b was purportedly detected in birds in Newfoundland, it was also detected in ducks in South Carolina [12], just two hundred miles east of the USDA’s Southeast Poultry Research Laboratory (SEPRL), which began performing serial passage experiments with H5Nx viruses on mallard ducks in the spring of 2021 [13,14]. H5Nx viruses share the H5 hemagglutinin (HA) protein but have different neuraminidase (NA) proteins, ranging from H5N1 to H5N9.
HPAI H5N1 belonging to clade 2.3.4.4b (genotype B3.13) are currently infecting a large number and variety of animal species in the United States, resulting in sporadic human infections [15]. In the context of modern viral outbreaks involving pathogenic organisms, it is crucial to evaluate the potential origins of the virus, including the possibility of laboratory involvement [16]. In a recent televised interview, former CDC director Dr. Robert Redfield stated:
"In the laboratory, I could make it [H5N1] more infectious to humans in months … it’s been published the four amino acids that I need to change … That’s the real biosecurity threat, that these University labs are doing these bio experiments … Bird Flu, I think, is gonna be the cause of the great pandemic, where they are teaching these viruses how to be more infectious for humans." [17]
The above circumstances prompted us to pose a question—namely, it is possible that HPAI H5N1 clade 2.3.4.4b evolved not in nature, but as a result of serial passage or other Gain-of-Function (GOF) research in a laboratory? We hope the following investigative report will serve as a starting point for further investigation by specialists in the fields of virology, molecular biology, and avian flu epidemiology.
H5N1 Gain-of-Function Research at SEPRL
H5Nx clade 2.3.4.4 serial passage experiments are currently being conducted in mallard ducks at the USDA Southeast Poultry Research Laboratory (SEPRL) in Athens, Georgia since April 2021 [13,14]. A new H5N1 clade 2.3.4.4b genotype (B1) was first detected in Georgia in January 2022, with gene reassortments either in PB2 and NP or in PB2, PB1, and NP. This genotype resulted in a sustained wild bird outbreak in Florida and northern midwestern states, causing a second major wild bird outbreak in Michigan (Figure 1) [18]. The USDA SEPRL in Athens, Georgia has a history of performing GOF research on H5N1 viruses prior to their current serial passage experiments. In 2008, Wasilenko et al. from SEPRL generated recombinant H5N1 viruses by exchanging individual gene segments from two parental H5N1 strains with differing pathogenicity. They specifically exchanged the PB1, PB2, NP, HA, NS, and M genes in these recombinant viruses, which resulted in some mutant viruses exhibiting increased pathogenicity while others showed decreased pathogenicity [19]. Regarding the serial passage experiments being performed at SEPRL, several mutations can arise. Xu et al found that serial passage of H4N6 in mice resulted in PB2 (E158K and E627K) mutations, which significantly increased polymerase activity, leading to the enhanced replication and virulence [20]. Danzy et al mutated H1N1 via 10 serial passages in differentiated human tracheo-bronchial epithelial (HTBE) cells, which resulted in all isolates carrying mutations in the PB2 627 domain and regions of NP thought to interact with PB2 [21]. Zhang et al serially passaged the H10N7 virus (BJ27) in mice and discovered that the well-known mammalian adaptation markers PB2-E627K and PB2-D701N were absent in the mutated strain. Instead, the amino acid substitution PB2-M631L emerged as the dominant contributor to the virus's virulence [22]. Thus, to identify potential laboratory leaks resulting from serial passage experiments, sudden mutations in the PB2 genes and new adaptations to different species should be closely investigated.
Genetic Mutations Raise Suspicion of H5N1 Laboratory Leaks
Genetic analysis of the H5N1 clade 2.3.4.4b (genotype B3.13) from the human case reported in Texas on April 1, 2024, revealed a PB2 E627K mutation [23]. In contrast, the human case reported in Michigan on May 22, 2024 (also genotype B3.13), exhibited a different mutation, PB2 M631L [24]. As we’ve outlined above, these two mutations can be a result of serial passage GOF experiments. Genotype B3.13 emerged in 2024, possibly after reassortment of genotype B3 viruses, and is the current genotype causing widespread H5N1 cattle outbreaks [15]. Elsmo et al indicated that genotype B3.2 was derived from genotype B1.2 [25], which is the strain that exhibited PB2/NP mutations and originated in Georgia in January 2022 [18], shortly after the start of serial passaging experiments at SEPRL in April 2021. Concerningly, genotype B1.2 was found in a bottlenose dolphin (Tursiops truncatus) in March 2022 in Florida, indicating sudden new adaptations to different animal species [26]. The dolphin exhibited neuronal necrosis and inflammation of the brain and meninges, and RT-PCR revealed the brain carried the highest viral load. Furthermore, the NP gene of H5N1 clade 2.3.4.4b (genotype B3.13), which is causing unprecedented cattle infections, was reported to have likely originated from an avian influenza A virus resembling A/mallard/Alberta/567/2021 (H11N9)-like strains [15]. These data indicate that a laboratory leak of H5N1 clade 2.3.4.4b genotype B1 may have occurred and spread via mallard ducks, eventually resulting in the sudden outbreaks among various mammals, cattle, and sporadic human infections (Figure 2). It’s possible that these mutations could have occurred naturally. Nevertheless, the jump from birds to a dolphin with genotype B1.2, first detected in Georgia, is a matter of grave concern.
The world’s first detection of H5N1 clade 2.3.4.4b occurred in the Netherlands in October 2020 [8]. Concerningly, Ron Fouchier et al modified H5N1 to become airborne transmissible via HA and PB2 alterations in ferrets at the Erasmus Medical Center in Rotterdam, the Netherlands in 2012 [27]. H5N1 clade 2.3.4.4b, derived from a 2022 farmed mink outbreak in Spain, was recently reported to be airborne transmissible in ferrets due to PB2 T271A mutation [28]. This raises the concern that the original emergence of H5N1 clade 2.3.4.4b may be a result of Fouchier et al’s GOF experiments. Moreover, the primary authors of the Youk et al study work at the SEPRL in Athens, Georgia [18]. It’s a salient fact that these recognized authorities, entrusted with researching H5N1 clade 2.3.4.4b, are known for performing Gain-of-Function experiments on H5N1 viruses [13,14,19]. This raises the concern that—like Drs. Peter Daszak and Ralph Baric in the case of SARS-CoV-2 [29,30,31] —these researchers are unlikely to serve as impartial investigators of possible leaks from their labs. In none of their studies regarding H5N1 clade 2.3.4.4b, do they mention the possibility that human agency may have contributed to its emergence. Instead, they attribute the emergence of these viruses to a reassortment with Eurasian wild bird LPAIVs. They postulate that H5N1 clade 2.3.4.4b genotype A1 first arrived in North America via the East Atlantic flyway in November 2021 [18]. As previously noted, this hypothesis is based on the erroneous proposition that migratory birds departed Europe and flew northwest across the North Atlantic to Newfoundland via Iceland in the autumn of 2021.
It's notable that the Erasmus Medical Center, headed by Ron Fouchier, previously collaborated closely with the SEPRL to develop vaccines against H9 avian influenza viruses, indicating the two laboratories likely share virus samples [32]. This raises the concern that genotype A1 is linked to Fouchier and coworker’s GOF experiments, while genotype B1 is a direct or indirect result of the USDA SEPRL serial passage experiments. However, confirming this requires further detailed investigation. More comprehensive studies and analyses are necessary to determine the precise origins and evolutionary pathways of these genotypes.
H5N1 Gain-of-Function Risks and Biosecurity Concerns
It’s possible that other H5N1 lab leaks could have occurred in laboratories across the world. In 2014, the CDC published a perspective piece that highly advocated for performing GOF experiments on H5N1: “GOF studies are needed to inform our interpretation of genetic data obtained from naturally occurring viruses.” [33]. Not everyone in the virology field agreed with this assertion. Indeed, as noted in the introduction, former CDC director Dr. Robert Redfield recently expressed his grave concern about the danger of conducting GOF studies on H5N1 [17]. Many prominent virologists were appalled that Ron Fouchier published his laboratory methods for genetically manipulating H5N1 to make them transmissible among ferrets, thereby providing a blueprint to lab technicians all over the world who may not be operating in safe conditions, and who may not conduct their laboratory work for benevolent ends [34,35,36,37,38].
Laboratory leaks are far more common than one may realize from news reporting. Since 2001, there have been 309 confirmed and reported lab-acquired infections globally, with a vast majority (78.6%) occurring in the U.S. [39]. This number is expected to be even higher because not all cases are reported to the media or published in peer-reviewed journals. The risk of unreported lab leaks is suspected of being especially high in China, where the Chinese Communist Party (CCP) imposes a high level of secrecy about the activities of Chinese biolabs [40]. In 2022, an illegal biolab tied to the People’s Republic of China was discovered in Reedley, CA. It contained thousands of samples of potential pathogens, including HIV, malaria, tuberculosis, and SARS-CoV-2, as well as nearly a thousand transgenic mice genetically engineered to mimic the human immune system. Lab workers said that the mice were designed "to catch and carry the COVID-19 virus." Additionally, the lab contained a freezer labeled "Ebola" [41].
The most common cause of pathogen escape is "procedural error" [39]. Animal escape is another cause of pathogen escape and could be the culprit behind any possible H5N1 leaks. Mallard ducks, which are used in serial passage experiments at the SEPRL facility, serve as natural reservoirs for many influenza A viruses. The mallard is the most numerous duck species in North America and Eurasia and is known to be an efficient asymptomatic carrier and spreader of H5N1 viruses [42]. As mentioned previously, the NP gene of H5N1 clade 2.3.4.4b (genotype B3.13) was reported to have likely originated from an avian influenza A virus derived from mallard ducks [15].
In January 2014, the CDC experienced an inadvertent cross-contamination incident where a low pathogenic avian influenza (LPAI) A (H9N2) virus culture was contaminated with a HPAI A (H5N1) virus [43]. This contaminated culture was subsequently shipped to the SEPRL in Athens, Georgia, but the issue wasn't identified until May 2014, meaning that unrecognized H5N1 contamination could have been occurred for months. The contamination event revealed gaps in laboratory safety protocols and reporting mechanisms, underscoring the risk of H5N1 escape even within high-containment facilities such as SEPRL. Moreover, in 2012, Kawaoka et al, using GOF techniques, modified H5N1 in the laboratory to better infect ferrets at the University of Wisconsin-Madison by introducing four specific mutations in the viral haemagglutinin (HA) protein within a 2009 pandemic H1N1 virus backbone [44]. These modifications allowed the H5N1 virus to preferentially recognize human-type receptors, replicate efficiently in ferrets, and transmit via respiratory droplets, while causing lung lesions and weight loss without high pathogenicity or mortality.
In November 2013, a researcher at the University of Wisconsin-Madison accidentally pierced their finger with a needle containing this engineered H5N1 virus [45]. The injured researcher was quarantined at home rather than in a specialized facility, raising concerns about the university's preparedness for such incidents. In December 2019, another breach occurred at the same university when a trainee's respirator hose detached during an experiment with a lab-engineered H5N1 virus. The university delayed notifying health officials and federal oversight bodies, raising concerns about inadequate safety measures and reporting practices [45].
These incidents underscore critical lapses in laboratory safety and oversight that can occur at BSL-3 laboratories such as the SEPRL and the Erasmus Medical Center, highlighting the significant risks associated with GOF research. Merler et al estimated that there’s a 5% to 15% chance that an H5N1 lab escape would not be detected at all using model simulations of a lab leak originating in Rotterdam, the Netherlands, the same city were Fouchier and colleagues conducted GOF experiments (Figure 3) [46]. The authors conclude the study by indicating that controlling escape incidents, potentially by mallard ducks or other migratory waterfowl, is not always possible, particularly in highly populated areas. With the rapid proliferation of biosafety laboratories globally, this represents a significant threat to public health.
Conclusions
The proximal origins of highly pathogenic avian influenza H5N1 Clade 2.3.4.4b may be the USDA Southeast Poultry Research Laboratory (SEPRL) in Athens, Georgia and the Erasmus Medical Center in Rotterdam, the Netherlands. Genetic evidence and historical context suggest that laboratory activities, including serial passage and GOF research, could have contributed to the emergence of H5N1 clade 2.3.4.4b. However, definitive causation has not been established, and further investigation is urgently needed to confirm these findings and to identify all H5N1 laboratory leaks that may have occurred with a focus on mallard ducks and other migratory waterfowl, which have the potential to infect a large number of poultry and livestock facilities around the world. A moratorium on GOF research including serial passage of H5N1 is indicated to prevent a man-made influenza pandemic affecting animals and humans.
Ethics statement
Ethical approval was not required as this is a review article.
Funding
This research received no external funding.
Acknowledgments
None
Conflicts of Interest
The authors declare no conflict of interest.
References
- Influenza: A(H5N1). World Health Organization. Published May 16, 2024. Available online: https://www.who.int/news-room/questions-and-answers/item/influenza-h5n1 (accessed on 29 May 2024).
- Graziosi, G.; Lupini, C.; Catelli, E.; Carnaccini, S. Highly Pathogenic Avian Influenza (HPAI) H5 Clade 2.3.4.4b Virus Infection in Birds and Mammals. Animals 2024, 14, 1372. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; de Vries, E.; McBride, R.; Dekkers, J.; Peng, W.; Bouwman, K.M.; Nycholat, C.; Verheije, M.H.; Paulson, J.C.; van Kuppeveld, F.J.; et al. Highly Pathogenic Influenza A(H5Nx) Viruses with Altered H5 Receptor-Binding Specificity. Emerg. Infect. Dis. 2017, 23, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Imai, M.; Kawaoka, Y. The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr. Opin. Virol. 2012, 2, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Chutinimitkul, S.; Van Riel, D.; Munster, V.J.; van den Brand, J.M.A.; Rimmelzwaan, G.F.; Kuiken, T.; Osterhaus, A.D.M.E.; Fouchier, R.A.M.; De Wit, E. In Vitro Assessment of Attachment Pattern and Replication Efficiency of H5N1 Influenza A Viruses with Altered Receptor Specificity. J. Virol. 2010, 84, 6825–6833. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-T.; Su, Y.C.F.; Smith, G.J.D. H5Nx Viruses Emerged during the Suppression of H5N1 Virus Populations in Poultry. Microbiol. Spectr. 2021, 9, e0130921. [Google Scholar] [CrossRef] [PubMed]
- Vanden Bossche G. (Open Letter to WHO) Mass infection prevention and mass vaccination with leaky Covid-19 vaccines in the midst of the pandemic can only breed highly infectious variants. Published March 6, 2021. Accessed June 2, 2024. https://37b32f5a-6ed9-4d6d-b3e1-5ec648ad9ed9.filesusr.com/ugd/28d8fe_266039aeb27a4465988c37adec9cd1dc.pdf.
- Lewis, N.S.; Banyard, A.C.; Whittard, E.; Karibayev, T.; Al Kafagi, T.; Chvala, I.; Byrne, A.; (Akberovna), S.M.; King, J.; Harder, T.; et al. Emergence and spread of novel H5N8, H5N5 and H5N1 clade 2.3.4.4 highly pathogenic avian influenza in 2020. Emerg. Microbes Infect. 2021, 10, 148–151. [Google Scholar] [CrossRef] [PubMed]
- CDC. Emergence and Evolution of H5N1 Bird Flu | Avian Influenza (Flu). www.cdc.gov. Published June 6, 2023. Accessed May 29, 2024. https://www.cdc.gov/flu/avianflu/communication-resources/bird-flu-origin-infographic.html.
- Caliendo, V.; Lewis, N.S.; Pohlmann, A.; Baillie, S.R.; Banyard, A.C.; Beer, M.; Brown, I.H.; Fouchier, R.A.M.; Hansen, R.D.E.; Lameris, T.K.; et al. Transatlantic spread of highly pathogenic avian influenza H5N1 by wild birds from Europe to North America in 2021. Sci. Rep. 2022, 12, 11729. [Google Scholar] [CrossRef]
- BirdLife International. East Atlantic Flyway. Accessed May 30, 2024. http://datazone.birdlife.org/userfiles/file/sowb/flyways/4_East_Atlantic_Factsheet.pdf.
- Bevins, S.N.; Shriner, S.A.; Cumbee, J.C.; Dilione, K.E.; Douglass, K.E.; Ellis, J.W.; Killian, M.L.; Torchetti, M.K.; Lenoch, J.B. Intercontinental Movement of Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4 Virus to the United States, 2021. Emerg. Infect. Dis. 2022, 28, 1006–1011. [Google Scholar] [CrossRef]
- Research Project #440252: US-UK-China Collab: Predictive Phylogenetics For Evolutionary and Transmission Dynamics of Newly Emerging Avian Influenza Viruses. USDA ARS. Accessed May 26, 2024. https://www.ars.usda.gov/research/project/?accnNo=440252.
- Research Project #43962: US-UK-China Collab: Predictive Phylogenetics For Evolutionary and Transmission Dynamics of Newly Emerging Avian Influenza Viruses. USDA ARS. Accessed May 26, 2024. https://www.ars.usda.gov/research/project/?accnNo=439621.
- Hu X, Saxena A, Magstadt DR; et al. Highly Pathogenic Avian Influenza A (H5N1) clade 2.3.4.4b Virus detected in dairy cattle. bioRxiv. 2024. [CrossRef]
- Lipsitch, M. Why Do Exceptionally Dangerous Gain-of-Function Experiments in Influenza?. Methods Mol Biol. 2018;1836:589-608. [CrossRef]
- Declassify the COVID documents: Former CDC director | Vargas Reports. NewsNation. Published May 8, 2024. Accessed May 27, 2024. https://www.newsnationnow.com/video/declassify-the-covid-documents-former-cdc-director-vargas-reports/9678111/.
- Youk, S.; Torchetti, M.K.; Lantz, K.; Lenoch, J.B.; Killian, M.L.; Leyson, C.; Bevins, S.N.; Dilione, K.; Ip, H.S.; Stallknecht, D.E.; et al. H5N1 highly pathogenic avian influenza clade 2.3.4.4b in wild and domestic birds: Introductions into the United States and reassortments, December 2021–April 2022. Virology 2023, 587, 109860. [Google Scholar] [CrossRef]
- Wasilenko, J.L.; Lee, C.W.; Sarmento, L.; Spackman, E.; Kapczynski, D.R.; Suarez, D.L.; Pantin-Jackwood, M.J. NP, PB1, and PB2 Viral Genes Contribute to Altered Replication of H5N1 Avian Influenza Viruses in Chickens. J. Virol. 2008, 82, 4544–4553. [Google Scholar] [CrossRef]
- Xu, G.; Wang, F.; Li, Q.; Bing, G.; Xie, S.; Sun, S.; Bian, Z.; Sun, H.; Feng, Y.; Peng, X.; et al. Mutations in PB2 and HA enhanced pathogenicity of H4N6 avian influenza virus in mice. J. Gen. Virol. 2020, 101, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Danzy, S.; Studdard, L.R.; Manicassamy, B.; Solorzano, A.; Marshall, N.; García-Sastre, A.; Steel, J.; Lowen, A.C. Mutations to PB2 and NP Proteins of an Avian Influenza Virus Combine To Confer Efficient Growth in Primary Human Respiratory Cells. J. Virol. 2014, 88, 13436–13446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, G.; Wang, C.; Jiang, M.; Gao, W.; Wang, M.; Sun, H.; Sun, Y.; Chang, K.-C.; Liu, J.; et al. Enhanced pathogenicity and neurotropism of mouse-adapted H10N7 influenza virus are mediated by novel PB2 and NA mutations. J. Gen. Virol. 2017, 98, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- CDC. Summary Analysis of Genetic Sequences of HPAI A(H5N1) Viruses in Texas. Centers for Disease Control and Prevention. Published April 2, 2024. Accessed May 26, 2024. https://www.cdc.gov/flu/avianflu/spotlights/2023-2024/h5n1-analysis-texas.htm.
- CDC. Technical Update: Summary Analysis of the Genetic Sequence of a Highly. Centers for Disease Control and Prevention. Published May 24, 2024. Accessed May 26, 2024. https://www.cdc.gov/flu/avianflu/spotlights/2023-2024/h5n1-technical-update-may-24-2024.html#_edn1.
- Elsmo, E.J.; Wunschmann, A.; Beckmen, K.B.; Broughton-Neiswanger, L.E.; Buckles, E.L.; Ellis, J.; Fitzgerald, S.D.; Gerlach, R.; Hawkins, S.; Ip, H.S.; et al. Highly Pathogenic Avian Influenza A(H5N1) Virus Clade 2.3.4.4b Infections in Wild Terrestrial Mammals, United States, 2022. Emerg. Infect. Dis. 2023, 29, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Murawski, A.; Fabrizio, T.; Ossiboff, R.; Kackos, C.; Jeevan, T.; Jones, J.C.; Kandeil, A.; Walker, D.; Turner, J.C.M.; Patton, C.; et al. Highly pathogenic avian influenza A(H5N1) virus in a common bottlenose dolphin (Tursiops truncatus) in Florida. Commun. Biol. 2024, 7, 476. [Google Scholar] [CrossRef] [PubMed]
- Herfst, S.; Schrauwen, E.J.A.; Linster, M.; Chutinimitkul, S.; de Wit, E.; Munster, V.J.; Sorrell, E.M.; Bestebroer, T.M.; Burke, D.F.; Smith, D.J.; et al. Airborne Transmission of Influenza A/H5N1 Virus Between Ferrets. Science 2012, 336, 1534–1541. [Google Scholar] [CrossRef]
- Restori, K.H.; Septer, K.M.; Field, C.J.; Patel, D.R.; VanInsberghe, D.; Raghunathan, V.; Lowen, A.C.; Sutton, T.C. Risk assessment of a highly pathogenic H5N1 influenza virus from mink. Nat. Commun. 2024, 15, 4112. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Yount, B.L., Jr.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.-Y.; Donaldson, E.F.; et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513, Erratum in 2020, 16, 1146. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L.; Sims, A.C.; Debbink, K.; Agnihothram, S.S.; Gralinski, L.E.; Graham, R.L.; Scobey, T.; Plante, J.A.; Royal, S.R.; et al. SARS-like WIV1-CoV poised for human emergence. Proc. Natl. Acad. Sci. 2016, 113, 3048–3053. [Google Scholar] [CrossRef]
- DEPARTMENT OF HEALTH & HUMAN SERVICES. Re: Notice of Suspension and Proposed Debarment of Dr. Peter Daszak.; 2024. Accessed June 2, 2024. https://oversight.house.gov/wp-content/uploads/2024/05/HHS-SUSP4D-Notice-of-Dr.-Peter-Daszak_05.21.2024_Redacted.pdf.
- Wang, Y.; Davidson, I.; Fouchier, R.; Spackman, E. Antigenic Cartography of H9 Avian Influenza Virus and Its Application to Vaccine Selection. Avian Dis. 2016, 60, 218–225. [Google Scholar] [CrossRef]
- Davis, C.T.; Chen, L.-M.; Pappas, C.; Stevens, J.; Tumpey, T.M.; Gubareva, L.V.; Katz, J.M.; Villanueva, J.M.; Donis, R.O.; Cox, N.J. Use of Highly Pathogenic Avian Influenza A(H5N1) Gain-Of-Function Studies for Molecular-Based Surveillance and Pandemic Preparedness. mBio 2014, 5, e02431-14. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.I.; Casadevall, A.; Cohen, M.L.; Ehrlich, S.A.; Enquist, L.W.; Fitch, J.P.; Franz, D.R.; Fraser-Liggett, C.M.; Grant, C.M.; Imperiale, M.J.; et al. Adaptations of Avian Flu Virus Are a Cause for Concern. Science 2012, 335, 660–661. [Google Scholar] [CrossRef] [PubMed]
- Keim, P.S. The NSABB Recommendations: Rationale, Impact, and Implications. mBio 2012, 3, e00021-12. [Google Scholar] [CrossRef] [PubMed]
- Osterholm, M.T.; Henderson, D.A. Life Sciences at a Crossroads: Respiratory Transmissible H5N1. Science 2012, 335, 801–802. [Google Scholar] [CrossRef]
- Osterholm, M.T.; Relman, D.A. Creating a Mammalian-Transmissible A/H5N1 Influenza Virus: Social Contracts, Prudence, and Alternative Perspectives. J. Infect. Dis. 2012, 205, 1636–1638. [Google Scholar] [CrossRef]
- Inglesby, T.V. Engineered H5N1: A Rare Time for Restraint in Science. Ann. Intern. Med. 2012, 156, 460–462. [Google Scholar] [CrossRef]
- Blacksell, S.D.; Dhawan, S.; Kusumoto, M.; Le, K.K.; Summermatter, K.; O’keefe, J.; Kozlovac, J.P.; Almuhairi, S.S.; Sendow, I.; Scheel, C.M.; et al. Laboratory-acquired infections and pathogen escapes worldwide between 2000 and 2021: a scoping review. Lancet Microbe 2024, 5, e194–e202. [Google Scholar] [CrossRef] [PubMed]
- Origins Forum Wrap Up: The Science Points to a Lab Leak. United States House Committee on Oversight and Accountability. Published June 29, 2021. Accessed June 2, 2024. https://oversight.house.gov/release/origins-forum-wrap-up-the-science-points-to-a-lab-leak/.
- INVESTIGATION INTO THE REEDLEY BIOLAB. House Select Committee on the Chinese Communist Party; 2023. Accessed June 2, 2024. https://selectcommitteeontheccp.house.gov/sites/evo-subsites/selectcommitteeontheccp.house.gov/files/evo-media-document/scc-reedley-report-11.15.pdf.
- Kim, J.; Negovetich, N.J.; Forrest, H.L.; Webster, R.G. Ducks: The “Trojan Horses” of H5N1 influenza. Influ. Other Respir. Viruses 2009, 3, 121–128. [Google Scholar] [CrossRef]
- Report on the inadvertent cross-contamination and shipment of a laboratory specimen with influenza virus H5N1. stacks.cdc.gov. Published August 15, 2014. Accessed May 26, 2024. https://stacks.cdc.gov/view/cdc/24766.
- Imai, M.; Watanabe, T.; Hatta, M.; Das, S.C.; Ozawa, M.; Shinya, K.; Zhong, G.; Hanson, A.; Katsura, H.; Watanabe, S.; et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 2012, 486, 420–428. [Google Scholar] [CrossRef]
- Young, A. Lab-created bird flu virus accident shows lax oversight of risky “gain of function” research. USA TODAY. Published April 12, 2023. Accessed May 27, 2024. https://www.usatoday.com/story/opinion/2023/04/11/lab-leak-accident-h-5-n-1-virus-avian-flu-experiment/11354399002/.
- Merler, S.; Ajelli, M.; Fumanelli, L.; Vespignani, A. Containing the accidental laboratory escape of potential pandemic influenza viruses. BMC Med. 2013, 11, 252. [Google Scholar] [CrossRef]
Figure 1.
Spatial diffusion of US H5N1 clade 2.3.4.4b genotype B1.1 and B1.2. Arrows indicate the direction of virus transition as estimated by a Bayesian stochastic search variable selection. The thickness of arrows represents the median Markov jump count/s for all the transitions between geographical location states along phylogenetic branches. The size and intensity of the red circles is proportional to the frequency of detections and number of genotypes found. The temporal formation and extinction of the red circles were superimposed during the surveillance time. The LMRCA (yellow dots) or the location (blue dots) represent the first or second highest mean Markov rewards. *Figure and legend adapted from Youk et al [18]. Permission to use this figure has been granted in accordance with the open access Creative Common CC BY 4.0 license.
Figure 1.
Spatial diffusion of US H5N1 clade 2.3.4.4b genotype B1.1 and B1.2. Arrows indicate the direction of virus transition as estimated by a Bayesian stochastic search variable selection. The thickness of arrows represents the median Markov jump count/s for all the transitions between geographical location states along phylogenetic branches. The size and intensity of the red circles is proportional to the frequency of detections and number of genotypes found. The temporal formation and extinction of the red circles were superimposed during the surveillance time. The LMRCA (yellow dots) or the location (blue dots) represent the first or second highest mean Markov rewards. *Figure and legend adapted from Youk et al [18]. Permission to use this figure has been granted in accordance with the open access Creative Common CC BY 4.0 license.
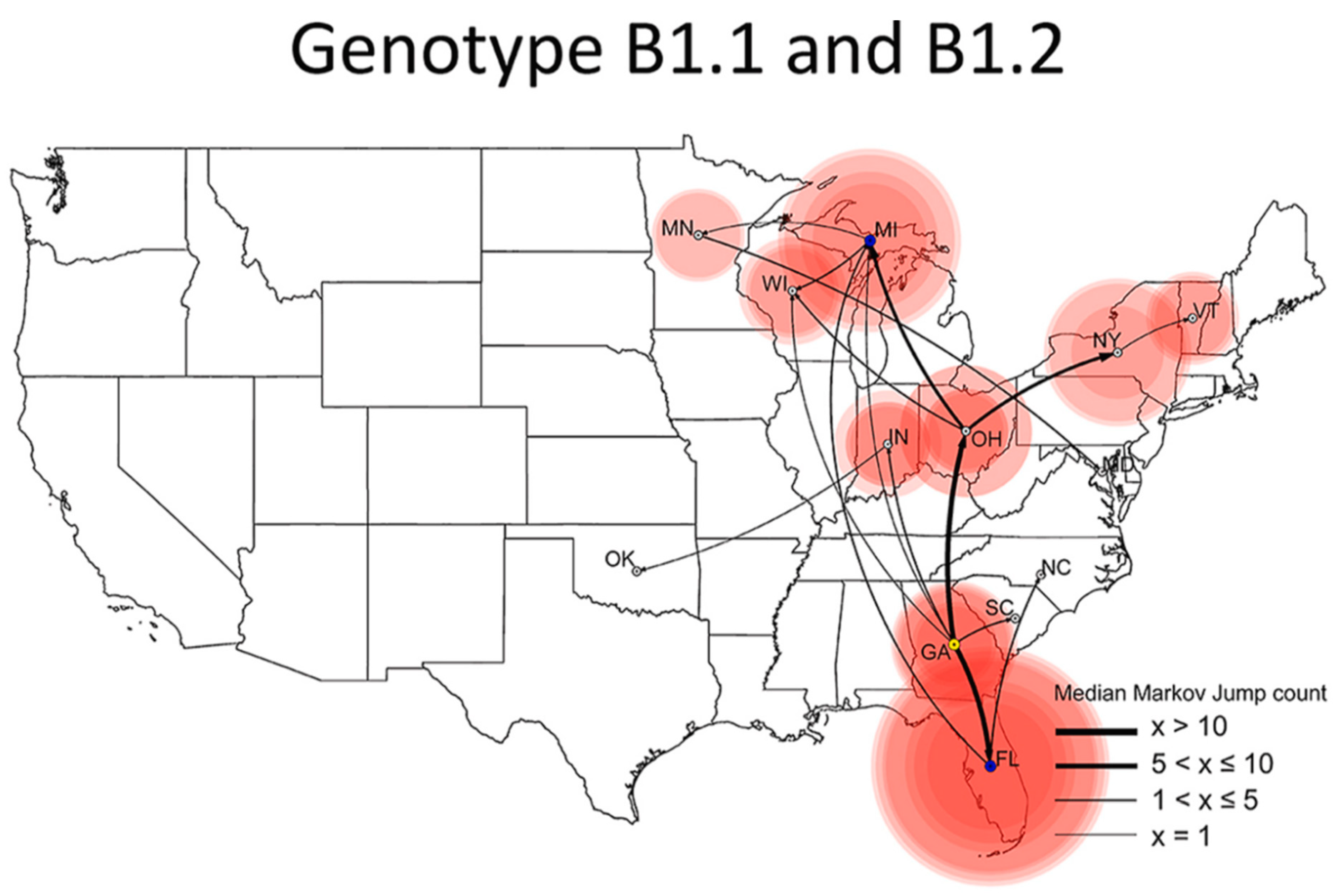
Figure 2.
The Proximal Origin of Highly Pathogenic Avian Influenza H5N1 Clade 2.3.4.4b. The left side of the figure depicts H5Nx Clade 2.3.4.4 serial passage experiments at SEPRL (1) resulting in the detection of H5N1 clade 2.3.4.4b genotype B1.1/B1.2 in Georgia (2), which then spread throughout the U.S. via mallard ducks (orange dotted lines). Light red shaded states indicate genotype B1 spread as estimated by Youk et al [18]. A few months later, genotype B1.2 was detected in bottlenose dolphins in Florida (3). After several more mutations, H5N1 clade 2.3.4.4b (genotype 3.13b) was detected in Texas cattle for the first time in history (4). The right side of the figure depicts the serial passage gain-of-function experiments conducted with H5N1 clade 2.3.4.4 in mallard ducks. Initially, natural H5N1 clade 2.3.4.4 strains are injected into ducks and assessed for pathogenicity (1). The virus is then serially passaged from one duck to another to induce mutations (2). After multiple passages, the mutated H5N1 clade 2.3.4.4 strains are collected (3) and subsequently tested for pathogenicity (4). Abbreviations: SEPRL; USDA Southeast Poultry Research Laboratory, GA; Georgia, TX; Texas, FL; Florida. *Created with Biorender.com.
Figure 2.
The Proximal Origin of Highly Pathogenic Avian Influenza H5N1 Clade 2.3.4.4b. The left side of the figure depicts H5Nx Clade 2.3.4.4 serial passage experiments at SEPRL (1) resulting in the detection of H5N1 clade 2.3.4.4b genotype B1.1/B1.2 in Georgia (2), which then spread throughout the U.S. via mallard ducks (orange dotted lines). Light red shaded states indicate genotype B1 spread as estimated by Youk et al [18]. A few months later, genotype B1.2 was detected in bottlenose dolphins in Florida (3). After several more mutations, H5N1 clade 2.3.4.4b (genotype 3.13b) was detected in Texas cattle for the first time in history (4). The right side of the figure depicts the serial passage gain-of-function experiments conducted with H5N1 clade 2.3.4.4 in mallard ducks. Initially, natural H5N1 clade 2.3.4.4 strains are injected into ducks and assessed for pathogenicity (1). The virus is then serially passaged from one duck to another to induce mutations (2). After multiple passages, the mutated H5N1 clade 2.3.4.4 strains are collected (3) and subsequently tested for pathogenicity (4). Abbreviations: SEPRL; USDA Southeast Poultry Research Laboratory, GA; Georgia, TX; Texas, FL; Florida. *Created with Biorender.com.
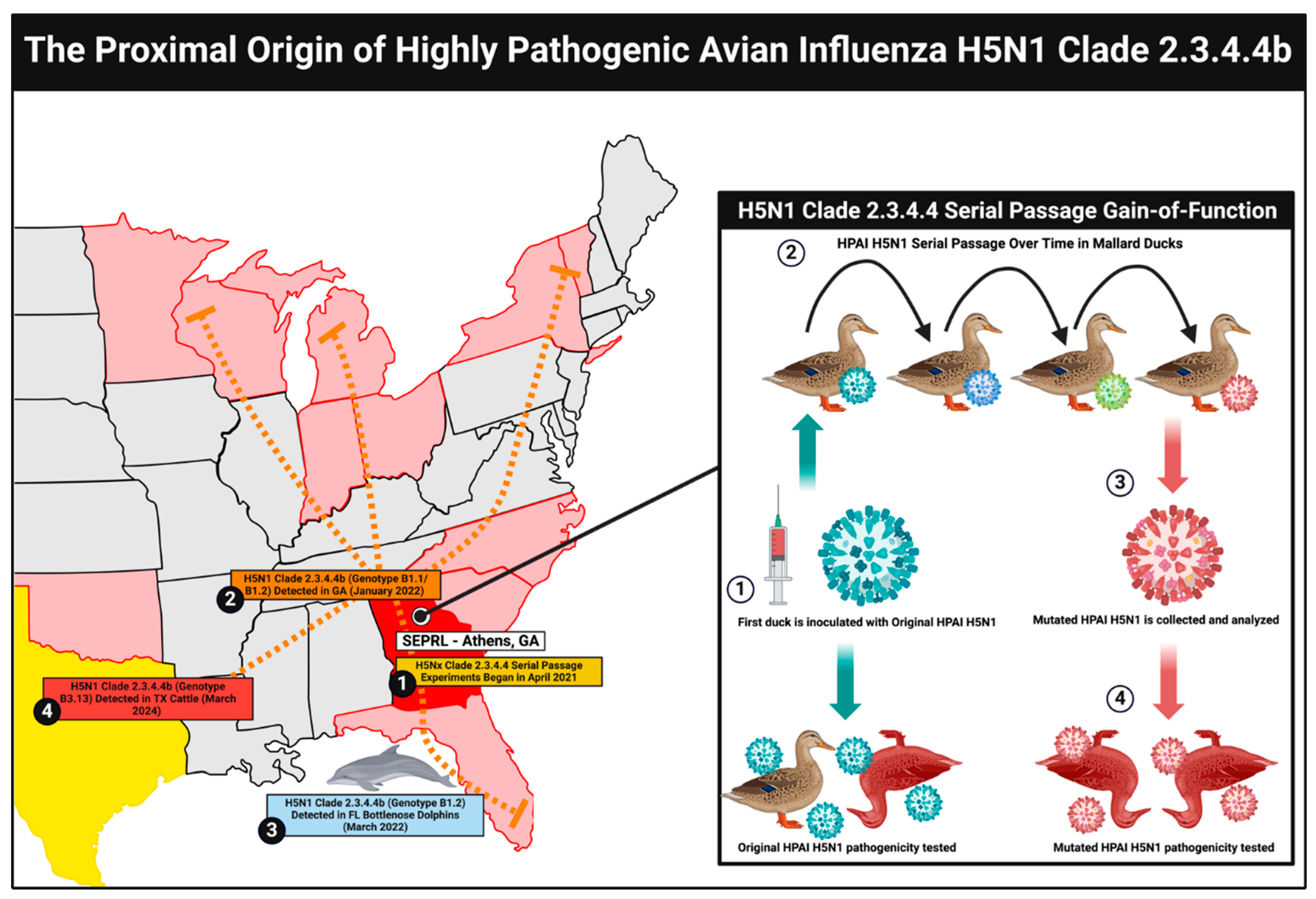
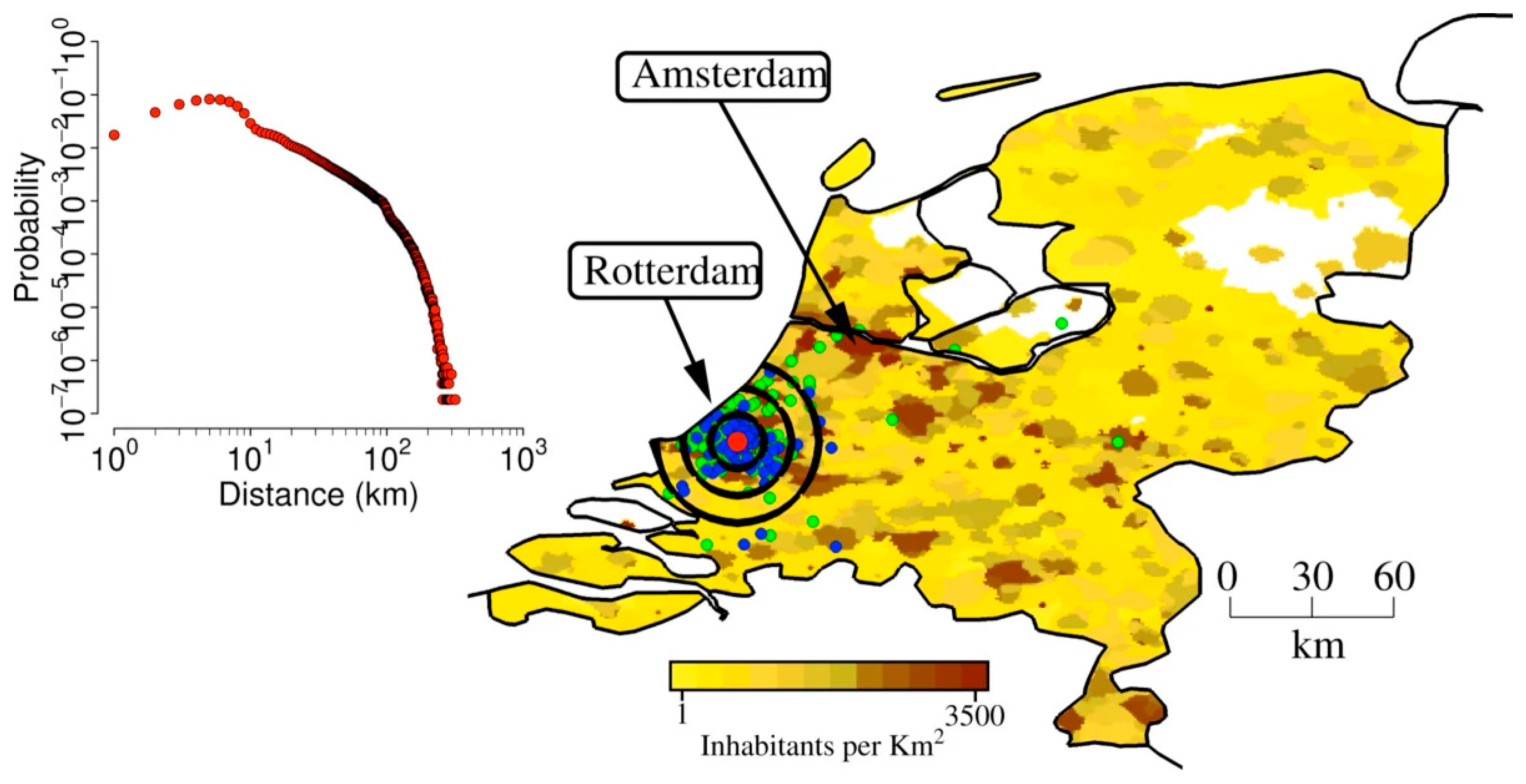
Figure 3.
Area of laboratory escape model simulation. The map shows population density of the Netherlands (colors from yellow to dark brown indicate increasing densities, from 1 to 3,500 inhabitants per km2), the location of the laboratory in a randomly chosen simulation (in Rotterdam, red point), the location of the workers houses (blue points), the location of workplaces and schools attended by household members of laboratory workers (green). Black concentric circles indicate distances of 10 km, 20 km, 30 km from the laboratory. The inset shows the probability of commuting to (at) a certain distance by laboratory workers.*Figure and legend reprinted from Merler et al [46]. The legend title has been adapted. Permission to use this figure has been granted in accordance with the open access Creative Common CC BY 2.0 license.
Figure 3.
Area of laboratory escape model simulation. The map shows population density of the Netherlands (colors from yellow to dark brown indicate increasing densities, from 1 to 3,500 inhabitants per km2), the location of the laboratory in a randomly chosen simulation (in Rotterdam, red point), the location of the workers houses (blue points), the location of workplaces and schools attended by household members of laboratory workers (green). Black concentric circles indicate distances of 10 km, 20 km, 30 km from the laboratory. The inset shows the probability of commuting to (at) a certain distance by laboratory workers.*Figure and legend reprinted from Merler et al [46]. The legend title has been adapted. Permission to use this figure has been granted in accordance with the open access Creative Common CC BY 2.0 license.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



