
Submitted:
04 June 2024
Posted:
05 June 2024
You are already at the latest version
Abstract
Gemcitabine (2',2'-difluoro-2'-deoxycytidine), a widely used anticancer drug, is considered as a gold standard in treating aggressive pancreatic cancers. Gamma-proteobacteria that colonize the pancreatic tumors contribute to chemoresistance against gemcitabine by metabolizing the drug to a less active, deaminated, form. The gemcitabine transporters of these bacteria are unknown to date. Furthermore, there is no complete knowledge on the gemcitabine transporters in Escherichia coli or any other related proteobacteria. In this study, we investigate the complement of gemcitabine transporters in E. coli K-12 and two common chemoresistance-related bacteria (Klebsiella pneumoniae, Citrobacter freundii). We find that E. coli K-12 has two high-affinity gemcitabine transporters with distinct specificity properties, namely NupC and NupG, whereas the gemcitabine transporters of C. freundii and K. pneumoniae include the NupC and NupG orthologs, indistinguishable functionally from their counterparts, and, in K. pneumoniae, one additional NupC variant, designated KpNupC2. All these bacterial transporters have higher affinity for gemcitabine than their human counterparts. The highest affinity (KM 2.5-3.0 μΜ) is exhibited by NupGs of the Bacteria-specific Nucleoside-H+ Symporter (NHS) family followed by NupCs (KM 10-13 μΜ) of the Concentrative Nucleoside Transporter (CNT) family, 15-100 times higher than the affinities reported for the human gemcitabine transporter hENT1/SLC29A1 which is primarily associated with gemcitabine uptake in the pancreatic adenocarcinoma cells. Our results offer a basis for further insight into the role of specific bacteria in drug availability within tumors and understanding the structure-function differences of bacterial and human drug transporters.
Keywords:
gemcitabine
; chemoresistance
; gamma-proteobacteria
; Escherichia coli K-12
; nucleoside transporters
1. Introduction
Purine and pyrimidine nucleobases/nucleosides are essential to life as we know it by being the coding building blocks of genetic information and core moieties of molecules with fundamental roles in information flow, signaling and metabolism.
Based on their role as antimetabolites, nucleobase/nucleoside analogs have long been used as antimicrobial, antiviral or anticancer drugs [1]. Such analogs hijack the nucleotide metabolism and inhibit key nucleotide-salvage/interconversion enzymes or become incorporated into DNA or RNA, leading to cytotoxicity.
Despite the plenitude of knowledge on the intracellular antimetabolite metabolism, the cellular uptake of nucleobase/nucleoside antimetabolites at the level of the membrane is still understudied, especially in bacteria. In particular, regarding the cellular uptake of anticancer antimetabolites which is important for understanding variations in the drug effectiveness and chemoresistance, research has focused almost entirely on the human genome-encoded transporters of the tumor cells [2,3] and rarely, if at all, on bacterial drug-transporting counterparts in the associated tumor microenvironment.
Bacteria in the tumor microenvironment can greatly affect the availability and toxicity of antimetabolites to the cancer cells and contribute to chemoresistance. A prominent example concerns gemcitabine (2′,2′-difluoro-2′-deoxycytidine) (dFdC), a cytidine analog commonly used in cancer therapy and especially as a frontline drug for pancreatic cancer [4]. Bacteria-mediated metabolism of gemcitabine has been implicated with enhanced chemoresistance in pancreatic ductal adenocarcinoma [5]. The phenomenon is also linked with active transport of the drug by the bacteria, based on indirect evidence from preincubation of gemcitabine with Escherichia coli K-12 devoid of the nucleoside-related transporter gene nupC and use of the cell-free supernatant in a pancreatic adenocarcinoma cell line culture [5]. However, the transporters responsible for the uptake of gemcitabine by the tumor-associated bacteria have not been elucidated to date.
Bacteria colonizing the pancreatic tumors are diverse, but gamma-proteobacteria of certain genera are common in these populations [5,6] and possess a so-called long isoform of cytidine deaminase (CDDL) which was correlated with the phenomenon of bacteria-mediated chemoresistance [5]. The gemcitabine transporters in these bacteria are unknown but, based on phylogenetic considerations, they might belong to the Concentrative Nucleoside Transporter (CNT) family, which is evolutionarily widespread [7], or the Nucleoside-H+ Symporter (NHS) family which is confined in Bacteria [8]. The known gemcitabine transporters encoded in human fall in either the CNT (SLC28) or the Equilibrative Nucleoside Transporter (ENT) family (SLC29) [9,10] and transport of gemcitabine into the pancreatic tumor cells has been linked primarily with hENT1 [11,12,13] and, to a minor extent, with hCNT1 or hCNT3 [14,15,16]. Functional knowledge of the bacterial gemcitabine transporters of the tumor microbiome would be important to fully understand the involvement of the tumor-associated bacteria to the chemoresistance phenomenon and highlight the specificity differences between the bacterial and the human transporters.
In the present study, we investigate the complement of gemcitabine transporters of the CNT and NHS families in E. coli K-12 and two common gamma-proteobacteria species of the pancreatic tumor microbiome (Klebsiella pneumoniae, Citrobacter freundii). The results show that E coli K-12 contains two efficient high-affinity gemcitabine transporters with distinct specificity properties, namely NupC and NupG, whereas the gemcitabine transporters of C. freundii and K. pneumoniae include the NupC and NupG orthologs, indistinguishable functionally from their counterparts, and, in K. pneumoniae, one additional functional variant of NupC, designated KpNupC2. Our study reveals that these bacterial transporters have higher affinity for gemcitabine than their human counterparts in the tumor cells.
2. Results
2.1. NupC and NupG are Efficient Gemcitabine Transporters of Escherichia coli K-12
The E. coli genome contains six members of the nucleoside transporter families CNT or NHS, namely NupC, NupX and PsuT/YeiM of family CNT and NupG, XapB and YegT of family NHS. The available functional evidence is sparse. NupC has been characterized as a pyrimidine-preferring nucleoside transporter [17] and NupG as a broad-specificity pyrimidine/purine nucleoside transporter [8], whereas PsuT/YeiM and XapB are referred to as pseudouridine and xanthosine transporters, respectively, based on their association with corresponding pseudouridine [18] and xanthosine utilization operons [19], and XapB has been proposed to be a xanthosine-preferring nucleoside transporter [19]. Concerning gemcitabine, only NupC has been pointed out as relevant to gemcitabine transport, based on earlier studies of functional characterization of NupC through heterologous expression in Xenopus laevis oocytes [17], and the effect of nupC deletion mutants on the gemcitabine metabolism [5] or the development of E. coli resistance against gemcitabine [20].
We here examine the gemcitabine transporting potential of the six nucleoside-related gene products of E. coli K-12 after expressing the genes extrachromosomally in the genetic background of Ε. coli JW2389 (ΔnupC) (Figure 1A). We show that [3H]gemcitabine can be transported with high affinity (based on the KM values) and efficiency (based on Vmax/KM values) by NupC and NupG, but not by NupX, PsuT, XapB or YegT (Figure 1B,C). Concerning the related pyrimidine nucleosides cytidine and uridine, we find that NupC, NupG, but also NupX can transport these two nucleosides. However, NupX shows comparatively low efficiency for uridine and cytidine and low affinity for cytidine (Supplementary Figure S1). The gemcitabine uptake activity of NupC or NupG is inhibited competitively by cytidine (Figure 1D) and uridine (Figure 1E); the same is true of the inhibition of NupC or NupG uridine uptake activity by gemcitabine (Figure 1F). Based on the calculated KM and Ki values, NupG exhibits 3.5-fold higher affinity (3.5-fold lower KM) for gemcitabine than NupC but approximately 2-fold lower affinity for cytidine and 4-6-fold lower affinity for uridine. NupC, on the other hand, exhibits similar KM or Ki values for uridine, cytidine, and gemcitabine or uridine and cytidine, respectively (Figure 1G). The ΚΜ values deduced for uridine or gemcitabine uptake by NupC and for uridine uptake by NupG are in the same range with the ones reported previously for NupC [17] and NupG [21].
A specifity profile analysis of NupC and NupG using assays of [3H]-uridine (0.1 μM) uptake and [3H]-gemcitabine (0.1 μM) uptake in the presence of 103-104-fold molar excess of unlebeled nucleosides shows that NupC is inhibited to completeness by all pyrimidine nucleosides (uridine, gemcitabine, cytidine, thymidine) and adenosine but does not recognize guanosine, inosine, or xanthosine, whereas NupG is inhibited to completeness by all nucleosides tested except xanthosine (Supplementary Figure S2), in agreement with previous findings [21]. Of the other CNT and NHS homologs, XapB, which shares high sequence similarity with NupG and does not differ from NupG in the predicted binding-site region [8], exhibits a unique functional profile as it transports xanthosine and its transport activity is not inhibited to a substantial extent by any other nucleoside (Supplementary Figure S3), whereas xanthosine, uridine or cytidine transport activity was not detected in our experiments for PsuT/YeiM or YegT.
2.2. Phylogenetic Analysis of NupC and NupG Homologs in Proteobacteria
We focused our analysis on the phylum of Proteobacteria because proteobacteria because proteobacteria, especially gamma-proteobacteria, are enriched in the pancreatic tumor-associated microbiome relative to the gut microbiome and are common in pancreatic tumors [5,6]. We performed a comprehensive phylogenetic analysis of the families CNT and NHS in the phylum of Proteobacteria to identify major clades relevant to NupC and NupG, respectively, and assign other proteobacterial relatives of NupC and NupG to distinct phylogenetic clades. This strategy allowed us to elucidate multiple closely related homologs of the E. coli gemcitabine-transporting members, including homologs from two species commonly found in pancreatic tumor microbiomes (K. pneumoniae and C. freundii).
The homologs from CNT family comprise two major monophyletic groups, one of which contains NupC and the other contains the other two E. coli members (NupX, PsuT) and the two structurally known CNTs (vcCNT [7,22], CNTnw [23]) in separate subclades (Figure 2A; Supplementary Figure S4). The homologs from K. pneumoniae are distributed in the NupC clade (two homologs) and the NupX/vcCNT clade (one homolog). Of them, the one is closely related (ortholog) to NupC and was named KpNupC, the second one is a NupC paralog (72% identity) and was named KpNupC2, and the third one is distantly related to NupC (30% identity) but is most related to vcCNT (70% identity) and was named KpvcCNT (Figure 2A; Supplementary Figure S5). C. freundii has two homologs, the one closely related to NupC and the other closely related to PsuT, that were named CfNupC and CfPsuT, respectively (Figure 2A).
The homologs from NHS family comprise three major phylogenetic groups, one of which is clearly separated and contains NupG and XapB (Figure 3A; Supplementary Figure S6). C. freundii has five homologs, distributed in the NupG/XapB clade (two homologs) and the other two groups. Three of them are closely related (87–95% identical) to NupG, XapB or YegT, and were named CfNupG, CfXapB and CfYegT, respectively. The remaining two homologs are in distantly related subclades but more related to YegT (36% identity) than to NupG or XapB, and were named Cf-Yeg-1 and Cf-Yeg-x (Figure 3A; Supplementary Figure S7). Only two homologs were found in K. pneumoniae, the one closely related to NupG and the other closely related to Cf-Yeg-1, and were named KpNupG and Kp-Yeg-1, respectively (Figure 3A).
2.3. Functional Characterization of Gemcitabine-Transporting NupC Homologs of K. pneumoniae and C. freundii
Of 262 fully sequenced K. pneumoniae genomes (based on information available in the JGI IMG/M database [24]; Feb. 2022), 221 contain at least one CNT homolog; of them, 201 contain all three CNTs (KpNupC, KpNupC2, KpvcCNT; Figure 2A) and the remaining 20 genomes contain either KpNupC alone (16 strains) or KpNupC and KpvcCNT (4 strains). Of the 54 fully sequenced C. freundii genomes, 32 have at least one CNT homolog; 28 of them contain both CfNupC and CfPsuT and 4 have CfNupC alone (Supplementary Table S1). We mobilized these five CNT genes from the genomes of K. pneumoniae ATCC 25955 and C. freundii ATCC 8090, accordingly, and transferred them to pT7-5/-BAD plasmid vectors for expression in Ε. coli JW2389 (ΔnupC). After confirmation of expression in the E. coli plasma membrane (Supplementary Figure S8), we examined the relevant gene products for transport of [3H]-gemcitabine in cell-based transport assays.
We find that CfNupC, KpNupC and KpNupC2 transport [3H]-gemcitabine (0.1 μM) at rates comparable to NupC, whereas CfPsuT or KpvcCNT does not transport gemcitabine (Figure 2B).
Kinetic analysis shows that CfNupC, KpNupC and KpNupC2 transport [3H]-gemcitabine with KM (10–13 μΜ) and Vmax values (87–102 nmol min−1 mg−1) that are essentially indistinguishable from NupC (Figure 2D; Table 1).
The inhibition profiles of [3H]-gemcitabine transport by other nucleosides show that, similar to NupC, CfNupC, KpNupC and KpNupC2 recognize with high affinity all natural pyrimidine nucleosides (including uridine, cytidine, thymidine (i.e., 2′-deoxy-thymidine)) and the analog 2′-deoxy-uridine (but not 3’-deoxy-uridine) and also recognize with high affinity adenosine (but not any other purine nucleoside) (Figure 2C); the Ki values of CfNupC, KpNupC or KpNupC2 for uridine and cytidine are similar to the ones of NupC (Figure 2E; Table 1). In addition, the Ki values of KpNupC for 2′-deoxy-uridine, thymidine or adenosine are very similar to the ones of NupC, whereas KpNupC2 differs from NupC in having roughly twofold lower affinity for thymidine and 2′-deoxy-uridine (Table 1).
2.4. Functional Characterization of Gemcitabine-Transporting NupG Homologs of K. pneumoniae and C. freundii
Regarding the NHS family, of 262 fully sequenced K. pneumoniae genomes, 218 (83%) contain at least one NHS homolog. Of them, 197 contain two (KpNupG, Kp-Yeg-1) and the remaining 21 genomes contain only KpNupG. Of the 54 fully sequenced C. freundii genomes, 33 contain at least one NHS homolog; 9 of them contain five homologs (as designated in Figure 3A), 19 have four (CfNupG, CfXapB, CfYegT, Cf-Yeg-x) and 5 have three homologs (CfNupG, CfXapB, CfYegT) (Supplementary Table S1). We mobilized the five most relevant NHS genes (except Cf-Yeg-x and Cf-Yeg-1/Kp-Yeg-1) from the genomes of K. pneumoniae ATCC 25955 and C. freundii ATCC 8090, accordingly, and transferred them to pT7-5/-BAD plasmid vectors for expression in Ε. coli JW2389 (ΔnupC). After confirmation of expression in the E. coli plasma membrane (Supplementary Figure S8), we examined the relevant gene products for transport of [3H]-gemcitabine in cell-based transport assays.
We find that CfNupG and KpNupG transport [3H]-gemcitabine (0.1 μM) at rates comparable to NupG, whereas CfXapB and CfYegT do not transport gemcitabine (Figure 3B); the kinetic analysis shows that CfNupG and KpNupG are essentially indistinguishable from NupG in the KM (2.7–2.8 μΜ) and Vmax (45–47 nmol min−1 mg-1) of [3H]-gemcitabine transport (Figure 3D; Table 1).
The inhibition profiles of [3H]-gemcitabine transport show that, similar to NupG, CfNupG and KpNupG recognize with high affinity a wide range of nucleosides, including pyrimidine nucleosides (uridine, cytidine, thymidine (i.e., 2′-deoxy-thymidine), 2′-deoxy-uridine (but not 3′-deoxy-uridine)) and purine nucleosides (adenosine, guanosine, inosine, but not xanthosine) (Figure 3C); the Ki values of CfNupG or KpNupG for uridine and cytidine are very similar to the ones of NupG (Figure 3E; Table 1). In addition, the Ki values of KpNupG for 2’-deoxy-uridine, thymidine, adenosine, guanosine or inosine are indistinguishable from the ones of NupG (Table 1).
2.5. Distinction of the NupG Functional Profile from the NupC Functional Profile
As summarized in Table 1, all four NupC homologs studied here, from three different enterobacterial species, exhibit the same gemcitabine-related profile, distinct from the set of the three NupG homologs, which are also functionally equivalent with each other. The NupG profile is characterized by four- to fivefold higher affinity (lower KM) and twofold higher efficiency (Vmax/KM) for gemcitabine transport relative to NupCs, recognition of both pyrimidine and purine nucleosides including guanosine and inosine which are not ligands for NupCs, but also three- to fivefold lower affinity for adenosine, three- to sixfold lower affinity for uridine and two- to threefold lower affinity for thymidine and cytidine relative to NupCs. Among NupCs, KpNupC2, a paralog of KpNupC, deviates only by having roughly twofold lower affinity for the 2′-deoxy nucleosides (2′-deoxy-uridine and thymidine) relative to the other NupCs.
3. Discussion
Proteobacteria are enriched in the bacterial populations colonizing pancreatic ductal adenocarcinoma tumors relative to the gut microbiome. Gamma-proteobacteria that are common in these microbiome populations contain a >800-nt long isoform of cytidine deaminase (CDDL) which has been correlated with the phenomenon of chemoresistance since it can rapidly convert gemcitabine into the less toxic 2′,2′-difluoro-2′-deoxyuridine (dFdU) [5]. On the other hand, the transmembrane transporters responsible for the uptake of the drug by the bacteria are not fully known. The import of gemcitabine into the bacterial cells has been associated with the nucleoside transporter NupC of the CNT family, based on the fact that only NupC had been linked with gemcitabine transport in the model gamma-proteobacterium E. coli K-12 from earlier studies [17]. However, prior to the present study, no functional knowledge was available for the potential gemcitabine transport systems in K. pneumoniae or C. freundii or any other gamma-proteobacteria, and, even in E. coli, complete knowledge of the potential gemcitabine transporters was missing. In this work, we shift attention to a more systematic investigation of the potential bacterial transporters of the drug. We show that E. coli K-12 contains two high-affinity gemcitabine transporters, one of which (NupC) was known but not studied systematically in this respect in the past and the other (NupG of the NHS family) is shown here as a gemcitabine transporter for the first time. We also show that both NupC and NupG are present as gemcitabine transporters in the two related Enterobacteriaceae species K. pneumoniae and C. freundii which are common and possibly linked with chemoresistance in the pancreatic tumor microbiomes [5,6].
Our transport kinetic analysis shows that both NupC and NupG have higher kinetic affinities for gemcitabine relative to their human counterparts, i.e., KM of 2–3 μΜ (NupG) or 10–13 μΜ (NupC), compared to 0.2–0.3 mM for hENT1 [25,26,27] and 20–60 μM for hCNT1 or hCNT3 [25,26,27,28,29]. The main transporter shown to mediate gemcitabine uptake in human cell lines is hENT1 (SLC29A1) and clinical correlation studies have shown that low expression of hENT1 in pancreatic adenocarcinoma is linked with poor outcomes of the gemcitabine treatment [11,12,13]. NupG might be an interesting new candidate for further research of its substrate specificity in comparison to hENT1. Unlike NupC which is a member of the evolutionarily widespread CNT family, NupG belongs to the prokaryote-specific NHS family which is structurally distinct from the nucleoside transporter families (CNT, ENT) in human. NHS and ENT are distantly related, both belonging to the Major Facilitator Superfamily [30] sharing the same overall fold and mechanistic motif (rocker-switch mechanism) [31]. However, NupG (NHS) and hENT1 are unrelated in sequence, have different binding site residues and differ in their functional properties. hENT1 is a uniporter and NupG a proton symporter, hENT1 has 100-fold lower affinity for gemcitabine (see above) and at least 10-fold lower affinity for uridine [32,33], both proteins show broad specificity for purine and pyrimidine nucleosides but hENT1 can additionally transport nucleobases, with roughly 10-fold lower affinities [34]. hENT1 has been studied for recognition of a range of antimetabolite nucleoside analogs and shown to have high affinity for several of them (dideoxycytidine, dideoxythymidine, azidothymidine, ribavirin, dideoxyinosine, cladribine, dipyridamole) [32,33,34,35,36]. It would be interesting to assay NupG for recognition of these and other similar analogs to elucidate potential hENT1 substrate motifs that are not recognized by NupG.
Another intriguing feature with respect to NupG derives from the comparison of the binding pocket residues in its recently solved structure [8] with other members of the NHS family. The ribose moiety of the nucleoside (uridine) in the structure of NupG is stabilized with hydrogen bonds from three conserved residues (R136, T140, E264) which are invariable in the proteobacterial NHS transporters. The same is true of the neighboring D323 which is not directly in contact with substrate but is considered crucial for coupling substrate binding with protonation, based on the properties of the D323A and D323N mutants [8]. However, additional residues that interact with the nucleobase moiety through hydrogen bonds (Q225, N228, Q261, Y318) or π-π interactions (F322, F143) are also invariable in the monophyletic cluster containing the NupG and XapB homologs (see Figure S6 and S7). In contrast to NupG which is of broad specificity but does not transport or recognize xanthosine (Figure S2) [8], XapB appears to be selective for xanthosine transport (Figure S3) [19]. Thus, it follows that residues at the periphery of the binding pocket are crucial for the substrate profile of NupG and underlie the functional distinction between NupG and XapB.
Another aspect of our work concerns the phylogenetic analysis of the distribution of NupC, NupG and related transporter homologs among proteobacteria. This is important to investigate to understand the realm of functional transporters that might be relevant to the chemoresistance-related metabolism of gemcitabine in tumor microbiomes. Summarizing our key observations, we have found that NupC orthologs (constituting a subgroup of the NupC clade, sharing 74–99% pairwise sequence identity) are clustered in almost all families of Enterobacterales, whereas the NupG orthologs (subgroup of the NupG/XapB clade, with 78–99% pairwise sequence identity) are mostly confined in Enterobacteriaceae (Supplementary Table S2). All species containing NupC and/or NupG possess the long cytidine deaminase isoform CDDL and the few species of Enterobacterales lacking CDD are devoid of both NupC and NupG (Table S2). In addition to NupC, the paralog NupC2 (which is probably a gemcitabine transporter similar to NupC, based on the results with KpNupC2) is also present in several species, including most Enterobacteriaceae. Interestingly, as well, although most K. pneumoniae strains contain both NupC, NupC2 and NupG, 13% of them lack NupC2, 6% contain only NupC and 5% contain only NupG as a potential gemcitabine transporter (Table S1). Of the other CNTs or NHSs, which show no detectable gemcitabine transport (Figure 1, Figure 2 and Figure 3), KpvcCNT is related in sequence and phylogeny (Figure S6) to vcCNT, which has been characterized as a uridine transporter with high affinity for uridine and cytidine, but very low affinity for gemcitabine (KD about 1.5 mM, 40-fold higher than that of uridine, based on fluorescence anisotropy measurements) [22]. The presence of KpvcCNT orthologs is not correlated with genomes containing the active CDDL isoform in any of the Enterobacterales families (Table S2). Overall, the data imply that either NupC or NupG or both NupC and NupG might be involved in the cellular uptake of gemcitabine depending on the enterobacterial family, strain or species.
Apart from Enterobacterales, other bacteria also contain the CDDL isoform which has been implicated with the gemcitabine resistance [5]. In the analysis of bacterial species from the Kyoto Encyclopedia of Genes and Genomes (KEGG) [37] shown in [5] (Table S5 in [5]), 98.4% of the genomes containing CDDL are gamma-proteobacteria. Enterobacterales constitute two thirds of these genomes. The remaining one third belong to genera that appear in our phylogenetic analysis of nucleoside transporters in clusters that are closely related to CNTnw [23] (Haemophilus, Mannheimia, Aggregatibacter, Pasteurella) or vcCNT [7] (Vibrio, Allivibrio, Aeromonas, Shewanella) (Figure S6). It seems plausible to assume that some of these homologs might be involved in the uptake of gemcitabine in the aforementioned gamma-proteobacteria.
Experimental evidence in E. coli indicates the association of NupC with gemcitabine transport through the properties of nupC-knockout mutants. One piece of this evidence refers to the partial abrogation of gemcitabine metabolism in CDDL-containing E. coli K-12 that lack the nupC gene, as judged from the compromised alleviation of the gemcitabine effect on a human pancreatic adenocarcinoma cell line [5]. This effect of the nupC-knockout is partial and much less pronounced (10-fold higher EC50) than the effect of the CDDL-knockout (Figure 2C in [5]), implying involvement of additional gemcitabine transporters. The second piece of evidence comes from the study of adaptation of E. coli to gemcitabine through an experimental evolution strategy highlighting that nupC loss-of-function mutations correlate with gemcitabine resistance [20]. In the context of this study, the authors also performed a genome-wide screen showing that several different single gene losses can confer resistance and impact the bacterial drug degradation. Apart from NupC, other transporters, as well as metabolic genes and transcription factors were among single-gene knockouts yielding gemcitabine resistance [20]. Some of the resistance effects are complex, as they involve both increased import and increased deamination of gemcitabine. This is the case with the cytR-knockout, since CytR is a repressor of numerous genes, including both gemcitabine transporters nupC and nupG and the gemcitabine deaminating enzyme cdd. Overall, it appears that multiple alternative mutation routes in E. coli could lead to gemcitabine chemoresistance.
In conclusion, the initial characterization of gemcitabine-related transporter properties of NupG in E. coli, C. freundii, K. pneumoniae, and the phylogenetic analysis of NupG, NupC and related nucleoside transporters in the NHS and CNT families might broaden our understanding of the bacterial gemcitabine transporters involved in the phenomenon of bacteria-mediated chemoresistance and encourage experimentation towards analyzing the differences between bacterial and human drug transporters at the molecular level.
4. Materials and Methods
4.1. Phylogenetic Analysis of CNT and NHS Families in Proteobacteria.
As of Feb. 2022, we selected all genomes in phylum Proteobacteria from the IMG/M database at JGI [24] with a genome status marked as 'Finished'. A total of 6662 bacterial strains were recovered and classified according to class (alpha-, beta-, gamma-, delta-, epsilon- proteobacteria). Using E. coli NupC or NupG as a query for CNT or NHS family, respectively, we performed BLAST-p search in each one of the five classes and retrieved all homologous sequences (cutoff E value 1 × 10−5). We identified 6434 sequences belonging to the CNT family and 4560 belonging to the NHS family. The data size underwent an initial reduction to 927 CNT and 388 NHS sequences, by selecting homologs from one strain per species (a strain containing the maximum number of homologs for each species). Subsequently, the data size was reduced further to 275 CNT and 146 NHS sequences, by retaining homologs from one strain per genus (a strain containing the maximum number of homologs for each genus). Both sets of sequences were aligned with Muscle and subjected to Maximum Likelihood (ML) phylogenetic analysis with MEGA7 [38].
4.2. Materials for Wet-Lab Experiments and General Considerations
[3H]-Gemcitabine ([cytosine-5-3H(N)]-gemcitabine) (20.1 Ci mmol−1), [5,6-3H]-uridine (30.0 Ci mmol-1), [5-3H]-cytidine (27.7 Ci mmol-1) and [8-3H]-xanthosine (17.1 Ci mmol-1) were from Moravek Biochemicals (Brea, CA). Non-radioactive nucleosides and analogs were from Sigma-Aldrich (St. Louis, MO). Nucleosides were prepared in dimethyl sulfoxide (DMSO). Cell cultures were performed in Luria-Bertani broth (LB) or M9 minimal media (M9) in aerobic conditions. For all incubations in liquid media, E. coli cells were grown with shaking at 220 r.p.m. at 37 °C. Oligodeoxynucleotides were synthesized from Eurofins Genomics GmbH. High-fidelity DNA polymerase, restriction endonucleases, alkaline phosphatase and T4 DNA ligase were from Takara Clontech. Horseradish peroxidase (HRP)-conjugated streptavidin was from Millipore. All other reagents were of analytical grade and obtained from commercial sources.
4.3. Bacterial Strains, Coding Sequences and Plasmids
Genomic DNA from Klebsiella pneumoniae DSM 4799 (ATCC 25955) and Citrobacter freundii DSM 30039 (ATCC 8090) were obtained from Eurofins Genomics and used for mobilization of the relevant CNT and NHS sequences. E. coli T184 (lacI+O+Z–Y– (A), prsL, met–, thr–, recA, hsdR/F’, lacIqO+ZD118) was used for mobilization of E. coli CNT and NHS sequences. The genes mobilized correspond to the coding sequences of E. coli NupC (P0AFF2), NupX (P33021), YeiM/PsuT (P33024), NupG (P0AFF4), XapB (P45562), YegT (P76417) (UniProt numbers given in parentheses), K. pneumoniae KpNupC (A0A2W0KM59), KpNupC2 (WP_002898911), KpvcCNT (WP_004146034), KpNupG (WP_038806797), and C. freundii CfNupC (A0A336NW46), CfPsuT (D2TRJ2), CfNupG (A0A7D6VR53), CfXapB (A0A7W3D7V4), CfYegT (A0A7D6VSQ9) (trEMBL or NCBI accession numbers in parentheses). Other sequences of NHS homologs shown in Figure 3A are Cf-Yeg-x (A0A0D7M2I3), Cf-Yeg-1 (A0A7W3HTK5) and Kp-Yeg-1 (J2XAI5). The sequence alignments in Supplementary Figure S5 and S7 were performed using Multalin [39].
For expression in E. coli K-12, the coding sequence of each gene was transferred to a previously described plasmid vector pT7-5 which included the DNA sequence of the biotin-acceptor domain (BAD) of the oxaloacetate decarboxylase from K. pneumoniae as an insert between the ApaI and HindIII sites. This vector was designated pT7-5/-BAD. After insertion of the coding sequence, at the appropriate orientation and frame, the resulting constructs contain the BAD sequence as a C-terminal tag of each CNT or NHS. Following expression, the tagged gene products are biotinylated in vivo during bacterial growth and allow monitoring of the protein level in the E. coli membrane by western blotting [40].
E. coli BW25113 strains with appropriate transporter-gene single knockout (Keio collection) [41] (provided by the Coli Genetic Stock Center Culture Collection) were used for expression of the pT7-5/-BAD-borne CNT or NHS gene from the lacZ promoter/operator and transport assays. In particular, E. coli JW2389 (nupC-knockout) was used for assays of gemcitabine, uridine and cytidine transport and E. coli JW2397 (xapB-knockout) for assays of xanthosine transport. All E. coli strains were transformed according to Inoue et al. [42].
4.4. Molecular Cloning and Bacterial Growth
The coding sequences of the genes were amplified with PCR on the template of genomic DNA and transferred to pT7-5/-BAD by restriction fragment replacement between the BamHI and ApaI sites. The sequences of synthetic oligodeoxynucleotides used as PCR primers are given in Supplementary Table S3. The coding sequence of all constructs was verified by double-strand DNA sequencing (Eurofins Genomics).
E. coli JW2389 or JW2397 harboring given plasmids were grown aerobically at 37 °C in LB containing kanamycin (0.025 mg/ml) and ampicillin (0.1 mg/ml). Fully grown cultures (1 mL) of E. coli JW2389 were transferred to M9 (supplemented with 22.2 mM glucose as a C source and 20 mM NH4Cl as a N source), diluted 10-fold, allowed to grow to mid-logarithmic phase (OD600nm 0.5–0.6), induced with isopropyl-β-D-1-thiogalactopyranoside (IPTG) (0.5 mM) for 105 min at 37 °C, and harvested for use in transport assays or western blotting. Fully grown cultures (1 mL) of E. coli JW2397 were diluted 10-fold in LB, induced with IPTG as above, and harvested for use in the xanthosine transport assays.
4.5. Western Blot Analysis
E. coli JW2397 were washed twice in Tris-HCl (0.05 M), pH 8.0, containing NaCl (0.1 M) and Na2EDTA (1 mM), supplemented with 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF) (0.2 mM), and used to prepare membrane fractions by osmotic shock, treatment with EDTA/lysozyme and sonication [40]. Membrane fractions prepared from 10 mL cell cultures were harvested by ultracentrifugation in an Optima MAX-XP Ultracentrifuge (Beckman Coulter), normalized to a protein concentration of 100 μg per 50 μL in sample loading buffer, and subjected to SDS-PAGE (12%) (25 μg protein per lane). After electrophoresis, proteins were electroblotted to a polyvinylidene difluoride membrane (Parablot PVDF; Macherey Nagel) and the BAD-tagged proteins were probed with HRP-conjugated streptavidin which was used at a dilution of 1:50,000. Signals were developed with enhanced chemiluminescence (ECL).
4.6. Transport Assays and Kinetic Analysis
E. coli JW2389 were washed twice in MK buffer (MES 5 mM, pH 6.5, containing KCl, 0.15 M), normalized to an OD420nm of 10.0 (corresponding to 35 μg of total protein per 50 μL) in the same buffer and assayed for transport of [3H]-gemcitabine, [3H]-cytidine or [3H]-uridine. Before initiating the transport reaction, the cells were energized by addition of glycerol to a final concentration of 20 mM and equilibrated in the assay buffer for 3 min at 25 °C [43]. E. coli JW2397 were prepared and assayed for [3H]-xanthosine transport, in KPi, 0.1 M, pH 7.5. All transport reactions were performed at 25 °C. After termination of reaction, samples were rapidly filtered through Whatman GF/C filters, washed twice immediately with 3 mL of ice-cold KL buffer (KPi, 0.1 M, pH 5.5, LiCl, 0.1 M) and taken for liquid scintillation counting.
To determine KM and Vmax values, data were fitted to the Michaelis-Menten equation using Prism8. To obtain IC50 values in competitive inhibition experiments, data were fitted to the equation y = B + (T − B)/(1 + 10 ((log IC50 − log x) h)) for sigmoidal dose-response (variable slope), using Prism8, where x is the concentration variable, y (the transport rate) ranges from T (top) to B (bottom) and h is the Hill coefficient. The h value was consistently close to −1, indicating competition for a single binding site. Ki values were calculated from the IC50 values, based on the equation: Ki = IC50/[1 + (S/KM)] (where S is the concentration of the radiolabeled substrate used and KM the corresponding value that had been obtained for the relevant transporter in the kinetics assay) [44].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, S.F.; methodology, N.I., E.A., M.B., E.K. and E.T.; validation, E.A., M.B., E.T. and S.F.; formal analysis, N.I., E.A., M.B., E.K., E.T. and S.F.; investigation, N.I., E.A., M.B., E.K., E.T. and S.F.; resources, N.I., E.A., M.B. and S.F.; data curation, N.I., E.A., M.B. and S.F.; writing—original draft preparation, N.I. and S.F.; writing—review and editing, N.I., M.B. and S.F.; supervision, M.B. and S.F.; project administration, S.F.; funding acquisition, S.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by project BIOMED-20 (MIS 5047236) implemented under the action “Reinforcement of Research and Innovation Infrastructure” through the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) co-financed by Greece and EU (European Regional Development Fund), and by a postdoc fellowship from the State Scholarships Foundation (IKY) to M.B. though Operational Programme “Human Resources Development, Education and Lifelong Learning” in the context of the project “Reinforcement of Postdoctoral Reasearchers-2nd Cycle” (MIS 5033021) co-financed by Greece and EU (European Social Fund).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article and supplementary material. Further inquiries can be directed to the corresponding author.
Acknowledgments
We thank Andriani Gkartzonika for assistance in some experiments.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Elion, G.B.; Hitchings, G.H. Metabolic basis for the actions of analogs of purines and pyrimidines. Adv. Chemother. 1965, 2, 91–177. [Google Scholar] [CrossRef] [PubMed]
- Girardi, E.; César-Razquin, A.; Lindinger, S.; Papakostas, K.; Konecka, J.; Hemmerich, J.; Kickinger, S.; Kartnig, F.; Gürtl, B.; Klavins, K.; Sedlyarov, V.; Ingles-Prieto, A.; Fiume, G.; Koren, A.; Lardeau, C.-H.; Kandasamy, R.K.; Kubicek, S.; Ecker, G.F.; Superti-Furga, G. A widespread role for SLC transmembrane transporters in resistance to cytotoxic drugs. Nat. Chem. Biol. 2020, 16, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.J.; Mekkawy, A.H.; Morris, D.L. Role of human nucleoside transporters in pancreatic cancer and chemoresistance. World J. Gastroenterol. 2021, 27, 6844–6860. [Google Scholar] [CrossRef]
- Koltai, T.; Reshkin, S.J.; Carvalho, T.M.A.; Di Molfetta, D.; Greco, M.R.; Alfarouk, K.O.; Cardone, R.A. Resistance to Gemcitabine in Pancreatic Ductal Adenocarcinoma: A Physiopathologic and Pharmacologic Review. Cancers 2022, 14, 2486. [Google Scholar] [CrossRef] [PubMed]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; Thaiss, C.A.; Reuben, A.; Livny, J.; Avraham, R.; Frederick, D.T.; Ligorio, M.; Chatman, K.; Johnston, S.E.; Mosher, C.M.; Brandis, A.; Fuks, G.; Gurbatri, C.; Gopalakrishnan, V.; Kim, M.; Hurd, M.W.; Katz, M.; Fleming, J.; Maitra, A.; Smith D., A.; Skalak, M.; Bu, J.; Michaud, M.; Trauger, S.A.; Barshack, I.; Golan, T.; Sandbank, J.; Flaherty, K.T.; Mandinova, A.; Garrett, W.S.; Thayer, S.P.; Ferrone, C.R.; Huttenhower, C.; Bhatia, S.N.; Gevers, D.; Wargo, J.A.; Golub, T.R.; Straussman, R. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; Meltser, A.; Douglas, G.M.; Kamer, I.; Gopalakrishnan, V.; Dadosh, T.; Levin-Zaidman, S.; Avnet, S.; Atlan, T.; Cooper, Z.A.; Arora, R.; Cogdill, A.P.; Khan, M.A.W.; Ologun, G.; Bussi, Y.; Weinberger, A.; Lotan-Pompan, M.; Golani, O.; Perry, G.; Rokah, M.; Bahar-Shany, K.; Rozeman, E.A.; Blank, C.U.; Ronai, A.; Shaoul, R.; Amit, A.; Dorfman, T.; Kremer, R.; Cohen, Z.R.; Harnof, S.; Siegal, T.; Yehuda-Shnaidman, E.; Gal-Yam, E.N.; Shapira, H.; Baldini, N.; Langille, M.G.I.; Ben-Nun, A.; Kaufman, B.; Nissan, A.; Golan, T.; Dadiani, M.; Levanon, K.; Bar, J.; Yust-Katz, S.; Barshack, I.; Peeper, D.S.; Raz, D.J.; Segal, E.; Wargo, J.A.; Sandbank, J.; Shental, N.; Straussman, R. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.L.; Cheong, C.-G.; Lee, S.-Y. Crystal structure of a concentrative nucleoside transporter from Vibrio cholerae at 2.4 Å. Nature 2012, 483, 489–493. [Google Scholar] [CrossRef]
- Wang, C.; Xiao, Q.; Duan, H.; Li, J.; Zhang, J.; Wang, Q.; Guo, L.; Hu, J.; Sun, B.; Deng, D. Molecular basis for substrate recognition by the bacterial nucleoside transporter NupG. J. Biol. Chem. 2021, 296, 100479. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.J.; Lee, S.-Y. Toward a molecular basis of cellular nucleoside transport in humans. Chem. Rev. 2021, 121, 5336–5358. [Google Scholar] [CrossRef]
- Young, J.D. The SLC28 (CNT) and SLC29 (ENT) nucleoside transporter families: a 30-year collaborative odyssey. Biochem. Soc. Trans. 2016, 44, 869–876. [Google Scholar] [CrossRef]
- Nordh, S.; Ansari, D.; Andersson, R. hENT1 expression is predictive of gemcitabine outcome in pancreatic cancer: A systematic review. World J. Gastroenterol. 2014, 20, 8482–8490. [Google Scholar] [CrossRef] [PubMed]
- Greenhalf, W.; Ghaneh, P.; Neoptolemos, J.P.; Palmer, D.H.; Cox, T.F.; Lamb, R.F.; Garner, E.; Campbell, F.; Mackey, J.R.; Costello, E.; Moore, M.J.; Valle, J.W.; McDonald, A.C.; Carter, R.; Tebbutt, N.C.; Goldstein, D.; Shannon, J.; Dervenis, C.; Glimelius, B.; Deakin., M.; Charnley, R.M.; Lacaine, F.; Scarfe, A.G.; Middleton, M.R.; Anthoney, A.; Halloran, C.M.; Mayerle, J.; Oláh, A.; Jackson, R.; Rawcliffe, C.L.; Scarpa, A.; Bassi, C.; Büchler, M.W.; European Study Group for Pancreatic Cancer. Pancreatic cancer hENT1 expression and survival from gemcitabine in patients from the ESPAC-3 trial. J. Natl. Cancer Inst. 2014, 106, djt347. [Google Scholar] [CrossRef] [PubMed]
- Spratlin, J.L.; Mackey, J.R. Human Equilibrative Nucleoside Transporter 1 (hENT1) in Pancreatic Adenocarcinoma: Towards individualized treatment decisions. Cancers 2010, 2, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Skrypek, N.; Duchêne, B.; Hebbar, M.; Leteurtre, E.; van Seuningen, I.; Jonckheere, N. The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the concentrative nucleoside transporter family. Oncogene 2012, 32, 1714–1723. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Hung, S.W.; Patel, B.; Lovin, D.; Govindarajan, R. CNT1 expression influences proliferation and chemosensitivity in drug-resistant pancreatic cancer cells. Cancer Res. 2011, 71, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Hesler, R.A.; Huang, J.J.; Starr, M.D.; Treboschi, V.M.; Bernake, A.G.; Nixon, A.B.; McCall, S.J.; White, R.R.; Blobe, G.C. TGF-β-induced stromal CYR61 promotes resistance to gemcitabine in pancreatic ductal adenocarcinoma through downregulation of the nucleoside transporters hENT1 and hCNT3. Carcinogenesis 2016, 37, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Loewen, S.K.; Yao, S.Y.; Slugoski, M.D.; Mohabir, N.N.; Turner, R.J.; Mackey, J.R.; Weiner, J.H.; Gallagher, M.P.; Henderson, P.J.; Baldwin, S.A.; Cass, C.E.; Young, J.D. Transport of physiological nucleosides and anti-viral and anti-neoplastic nucleoside drugs by recombinant Escherichia coli nucleoside:H+ cotransporter (NupC) produced in Xenopus laevis oocytes. Mol. Membr. Biol. 2004, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Preumont, A.; Snoussi, K.; Stroobant, V.; Collet, J.-F.; Schaftingen, E.V. Molecular identification of pseudouridine-metabolizing enzymes. J. Biol. Chem. 2008, 283, 25238–25246. [Google Scholar] [CrossRef]
- Norholm, M.H.; Dandanell, G. Specificity and topology of the Escherichia coli xanthosine permease, a representative of the NHS sub-family of the major facilitator superfamily. J. Bacteriol. 2001, 183, 4900–4904. [Google Scholar] [CrossRef]
- Saylin, S.; Rosener, B.; Li, C.G.; Ho, B.; Ponomarova, O.; Ward, D.V.; Walhout, A.J.M.; Mitchell, A. Evolved bacterial resistance to the chemotherapy gemcitabine modulates its efficacy in co-cultured cancer cells. eLife 2023, 12, e83140. [Google Scholar] [CrossRef]
- Xie, H.; Patching, S.G.; Galagher, M.P.; Litherland, G.J.; Brough, A.R.; Venter, H.; Yao, S.Y.M.; Ng, A.M.L.; Young, J.D.; Herbert, R.B.; Henderson, P.J.F.; Baldwin, S.A. Purification and properties of the Escherichia coli nucleoside transporter NupG, a paradigm for a major facilitator transporter sub-family. Mol. Membr. Biol. 2004, 21, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.L.; Lee, J.-H.; Lee, K.; Lee, M.; Kwon, D.-Y.; Hong, J.; Lee, S.-Y. Structural basis of nucleoside and nucleoside drug selectivity by concentrative nucleoside transporters. eLife 2014, 3, e03604. [Google Scholar] [CrossRef] [PubMed]
- Hirschi, M.; Johnson, Z.L.; Lee, S.-Y. Visualizing multistep elevator-like transitions of a nucleoside transporter. Nature 2017, 545, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntermann, M.; Hajek, P.; Ritter, S.J.; Webb, C.; Wu, D.; Varghese, N.J.; Reddy, T. B. K.; Mukherjee, S.; Ovchinnikova, G.; Nolan, M.; Seshardi, R.; Roux, S.; Visel, A.; Woyke, T.; Eloe-Fadrosh, E.A.; Kyprides, N.C.; Ivanova, N.N. The IMG/M data management and analysis system v.7: content updates and new features. Nucleic Acids Res. 2023, 51, D723–D732. [Google Scholar] [CrossRef]
- Mackey, J.R.; Yao, S.Y.M.; Smith, K.M.; Karpinski, E.; Baldwin, S.A.; Cass, C.E.; Young, J.D. Gemcitabine transport in Xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J. Natl. Cancer Inst. 1999, 21, 1876–1881. [Google Scholar] [CrossRef] [PubMed]
- Mackey, J.R.; Baldwin, S.A.; Young, J.D.; Cass, C.E. Nucleoside transport and its significance for anticancer drug resistance. Drug Resist. Updat. 1998, 1, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Mackey, J.R.; Mani, R.S.; Selner, M.; Mowles, D.; Young, J.D.; Belt, J.A.; Crawford, C.R.; Cass, C.E. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998, 58, 4349–4357. [Google Scholar] [PubMed]
- García-Manteiga, J.; Molina-Arcas, M.; Casado, F.J.; Mazo, A.; Pastor-Anglada, M. Nucleoside transporter profiles in human pancreatic cancer cells: role of hCNT1 in 2',2'-difluorodeoxycytidine- induced cytotoxicity. Clin. Cancer Res. 2003, 9, 5000–5008. [Google Scholar]
- Hu, H.; Endres, C.J.; Chang, C.; Umapathy, N.S.; Lee, E.W.; Fei, Y.J.; Itagaki, S.; Swaan, P.W.; Ganapathy, V.; Unadkat, J.D. Electrophysiological characterization and modeling of the structure activity relationship of the human concentrative nucleoside transporter 3 (hCNT3). Mol. Pharmacol. 2006, 69, 1542–1553. [Google Scholar] [CrossRef]
- Drew, D.; Boudker, O. Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem. 2016, 85, 543–572. [Google Scholar] [CrossRef]
- Yan, N. Structural biology of the major facilitator superfamily transporters. Annu. Rev. Biophys. 2015, 44, 257–283. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, M.; Beaumont, M.; Yao, S.Y.; Sundaram, M.; Boumah, C.E.; Davies, A.; Kwong, F.Y.; Coe, I.; Cass, C.E.; Young, J.D.; Baldwin, S.A. Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat. Med. 1997, 3, 89–93. [Google Scholar] [CrossRef]
- Ward, J.L.; Sherali, A.; Mo, Z.P.; Tse, C.M. Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J. Biol. Chem. 2000, 275, 8375–8381. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.Y.; Ng, A.M.L.; Cass, C.E.; Baldwin, S.A.; Young, J.D. Nucleobase transport by human equilibrative nucleoside transporter 1 (hENT1). J. Biol. Chem. 2011, 286, 32552–32562. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zeng, X.; Shi, Y.; Liu, M. Functional characterization of human equilibrative nucleoside transporter 1. Protein Cell 2017, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.J.; Lee, S.-Y. Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat. Struct. Mol. Biol. 2019, 26, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef]
- Karatza, P.; Frillingos, S. Cloning and functional characterization of two bacterial members of the NAT/NCS2 family in Escherichia coli. Mol Membr Biol. 2005, 22, 251–261. [Google Scholar] [CrossRef]
- Baba, T.; Ara, T.; Hasegawa, M.; Takai, Y.; Okumura, Y.; Baba, M.; Datsenko, K.A.; Tomita, M.; Wanner, B.L.; Mori, H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006, 2, 2006–0008. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Nojima, H.; Okayama, H. High efficiency transformation of Escherichia coli with plasmids. Gene 1990, 96, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Botou, M.; Lazou, P.; Papakostas, K.; Lambrinidis, G.; Evangelidis, T.; Mikros, E.; Frillingos, S. Insight on specificity of uracil permeases of the NAT/NCS2 family from analysis of the transporter encoded in the pyrimidine utilization operon of Escherichia coli. Mol Microbiol. 2018, 108, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Gemcitabine-related transport properties of E. coli CNTs and NHSs. E. coli JW2389 expressing the indicated CNT or NHS homologs from pT7-5/-BAD were analyzed for protein levels in the membrane (A) and transport of [3H]-gemcitabine (B–E) or [3H]-uridine (F), as indicated. Panel (A): Membrane fractions (25 μg of membrane protein per lane) were subjected to SDS-PAGE (12%) and western blotting using HRP-conjugated streptavidin. Molecular mass standards (in kDa) were run in parallel, as indicated on the left. Panels (B,C): [3H]-Gemcitabine (dFdC) uptake was assayed at 0.1 μM ((C), time course) or at 5 sec to measure transport rate at a range of concentrations ((B), kinetics). Panels (D,E): Kinetics of inhibition of [3H]-gemcitabine (0.1 μM) uptake by cytidine (D) or uridine (E), based on measurements of rates at 5 sec. Panel (F): Kinetics of inhibition of [3H]-uridine (0.1 μM) uptake by gemcitabine, based on measurements of rates at 5 sec. Panel (G): KM, Vmax and Ki values deduced from experiments shown in (B–E) and in Supplementary Figure S1. Values presented in panels (B–G) are the means of 3–5 determinations with SD shown. Values obtained with vector alone were subtracted from the measurements in all cases (except in the time course shown in panel (C)); the values obtained with vector alone (indicated as pT7-5) are also given in the kinetics of panel B for comparison reasons.
Figure 1.
Gemcitabine-related transport properties of E. coli CNTs and NHSs. E. coli JW2389 expressing the indicated CNT or NHS homologs from pT7-5/-BAD were analyzed for protein levels in the membrane (A) and transport of [3H]-gemcitabine (B–E) or [3H]-uridine (F), as indicated. Panel (A): Membrane fractions (25 μg of membrane protein per lane) were subjected to SDS-PAGE (12%) and western blotting using HRP-conjugated streptavidin. Molecular mass standards (in kDa) were run in parallel, as indicated on the left. Panels (B,C): [3H]-Gemcitabine (dFdC) uptake was assayed at 0.1 μM ((C), time course) or at 5 sec to measure transport rate at a range of concentrations ((B), kinetics). Panels (D,E): Kinetics of inhibition of [3H]-gemcitabine (0.1 μM) uptake by cytidine (D) or uridine (E), based on measurements of rates at 5 sec. Panel (F): Kinetics of inhibition of [3H]-uridine (0.1 μM) uptake by gemcitabine, based on measurements of rates at 5 sec. Panel (G): KM, Vmax and Ki values deduced from experiments shown in (B–E) and in Supplementary Figure S1. Values presented in panels (B–G) are the means of 3–5 determinations with SD shown. Values obtained with vector alone were subtracted from the measurements in all cases (except in the time course shown in panel (C)); the values obtained with vector alone (indicated as pT7-5) are also given in the kinetics of panel B for comparison reasons.
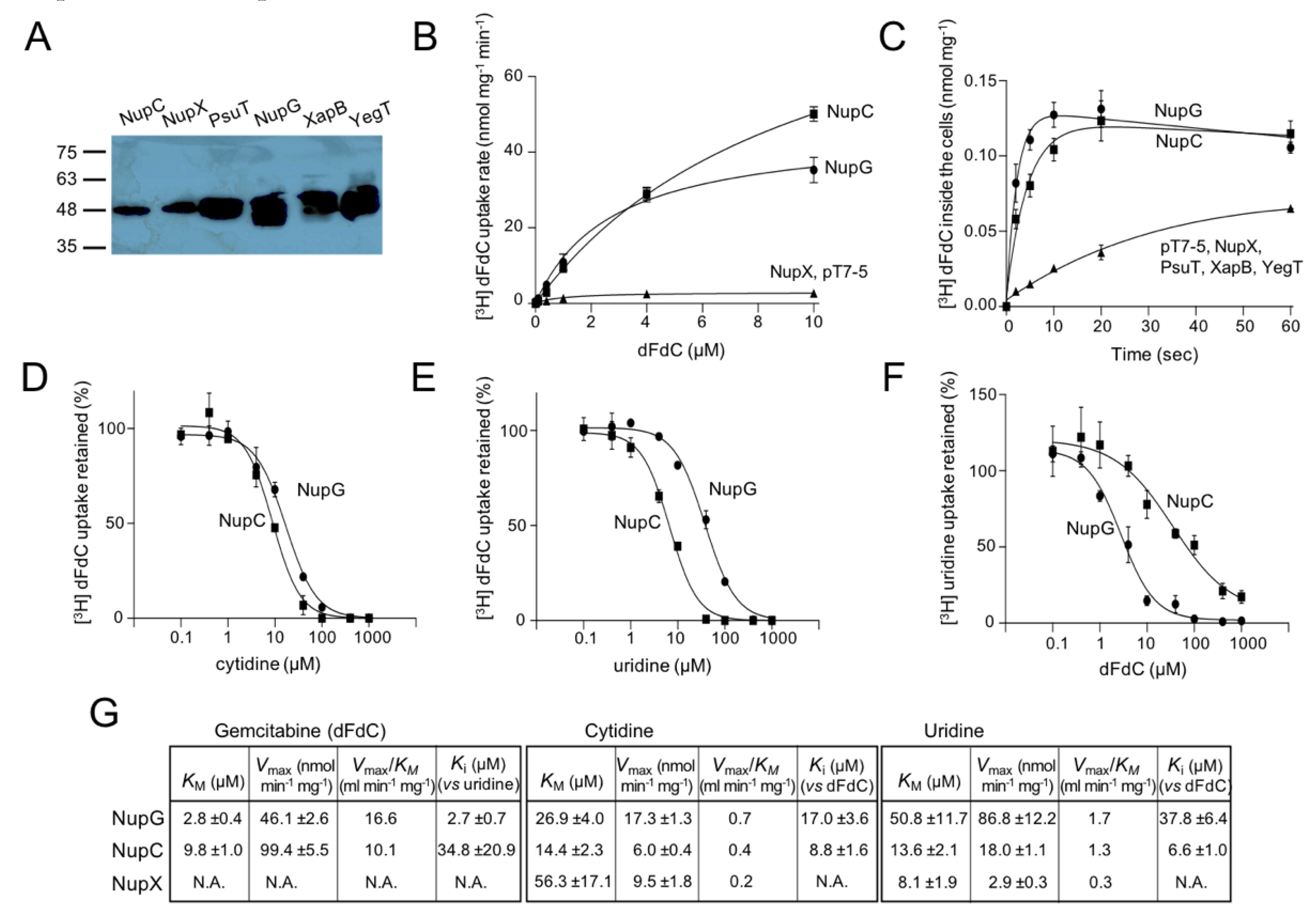
Figure 2.
Phylogeny and gemcitabine transport properties of C. freundii and K. pneumoniae CNTs. Panel (A): Phylogenetic analysis of 275 CNT homologs representing one fully sequenced genome per genus for all Proteobacteria, as retrieved from the IGM/M database at JGI (the complete phylogenetic tree is given in Supplementary Figure S4). The evolutionary history was inferred by the Maximum Likelihood method based on the Jones-Taylor-Thornton model as implemented in MEGA7. The tree with the highest log likelihood is shown. The percentage of trees in which the associated taxa clustered together is shown (as decimal) next to the indicated major branches. The outgroup (shown on top) consists of E. coli members of families NHS (NupG, XapB, YegT) and NCS1 (YbbW, and CodB, appearing as a separate clade). Clades shown as cuneiforms consist of multiple homologs. Different colors indicate different classes of Proteobacteria, including alpha- (green), beta- (yellow), gamma- (red, for Enterobacteriaceae; orange, for all others), delta- (purple) or epsilon- (dark purple) proteobacteria or clades with homologs from more than one class (gray). The homologs from E. coli K-12, K. pneumoniae ATCC 25955 and C. freundii ATCC 8090, as well as the structurally known homologs (with the corresponding PDB accession numbers) are indicated. Panel (B): [3H]-Gemcitabine (dFdC) transport rates (0.1 μΜ) by E. coli JW2389 expressing the indicated homologs from pT7-5/-BAD. Panel (C): Inhibition of [3H]-gemcitabine (0.1 μΜ) uptake rate of E. coli JW2389 expressing NupC, CfNupC, KpNupC or KpNupC2 (darker to lighter gray in the histogram) by the indicated unlabeled nucleosides (0.1 mM). Panel (D): Kinetics of [3H]-gemcitabine transport. Panel (E): Dose-response inhibition curves of the [3H]-gemcitabine (0.1 μΜ) uptake rate by cytidine (above) and uridine (below). The data in (D,E) for NupC, CfNupC, KpNupC and KpNupC2 are given as open rectangles (and interrupted lines), closed circles, closed rectangles, and open inverted triangles, respectively. Transport rates are deduced from measurements at 5 sec and given as the means of 3–5 determinations with SD shown. Values obtained with vector alone were subtracted from the measurements in all cases; the values obtained with vector alone (indicated as pT7-5) are also given in the kinetics plot (D) for comparison.
Figure 2.
Phylogeny and gemcitabine transport properties of C. freundii and K. pneumoniae CNTs. Panel (A): Phylogenetic analysis of 275 CNT homologs representing one fully sequenced genome per genus for all Proteobacteria, as retrieved from the IGM/M database at JGI (the complete phylogenetic tree is given in Supplementary Figure S4). The evolutionary history was inferred by the Maximum Likelihood method based on the Jones-Taylor-Thornton model as implemented in MEGA7. The tree with the highest log likelihood is shown. The percentage of trees in which the associated taxa clustered together is shown (as decimal) next to the indicated major branches. The outgroup (shown on top) consists of E. coli members of families NHS (NupG, XapB, YegT) and NCS1 (YbbW, and CodB, appearing as a separate clade). Clades shown as cuneiforms consist of multiple homologs. Different colors indicate different classes of Proteobacteria, including alpha- (green), beta- (yellow), gamma- (red, for Enterobacteriaceae; orange, for all others), delta- (purple) or epsilon- (dark purple) proteobacteria or clades with homologs from more than one class (gray). The homologs from E. coli K-12, K. pneumoniae ATCC 25955 and C. freundii ATCC 8090, as well as the structurally known homologs (with the corresponding PDB accession numbers) are indicated. Panel (B): [3H]-Gemcitabine (dFdC) transport rates (0.1 μΜ) by E. coli JW2389 expressing the indicated homologs from pT7-5/-BAD. Panel (C): Inhibition of [3H]-gemcitabine (0.1 μΜ) uptake rate of E. coli JW2389 expressing NupC, CfNupC, KpNupC or KpNupC2 (darker to lighter gray in the histogram) by the indicated unlabeled nucleosides (0.1 mM). Panel (D): Kinetics of [3H]-gemcitabine transport. Panel (E): Dose-response inhibition curves of the [3H]-gemcitabine (0.1 μΜ) uptake rate by cytidine (above) and uridine (below). The data in (D,E) for NupC, CfNupC, KpNupC and KpNupC2 are given as open rectangles (and interrupted lines), closed circles, closed rectangles, and open inverted triangles, respectively. Transport rates are deduced from measurements at 5 sec and given as the means of 3–5 determinations with SD shown. Values obtained with vector alone were subtracted from the measurements in all cases; the values obtained with vector alone (indicated as pT7-5) are also given in the kinetics plot (D) for comparison.
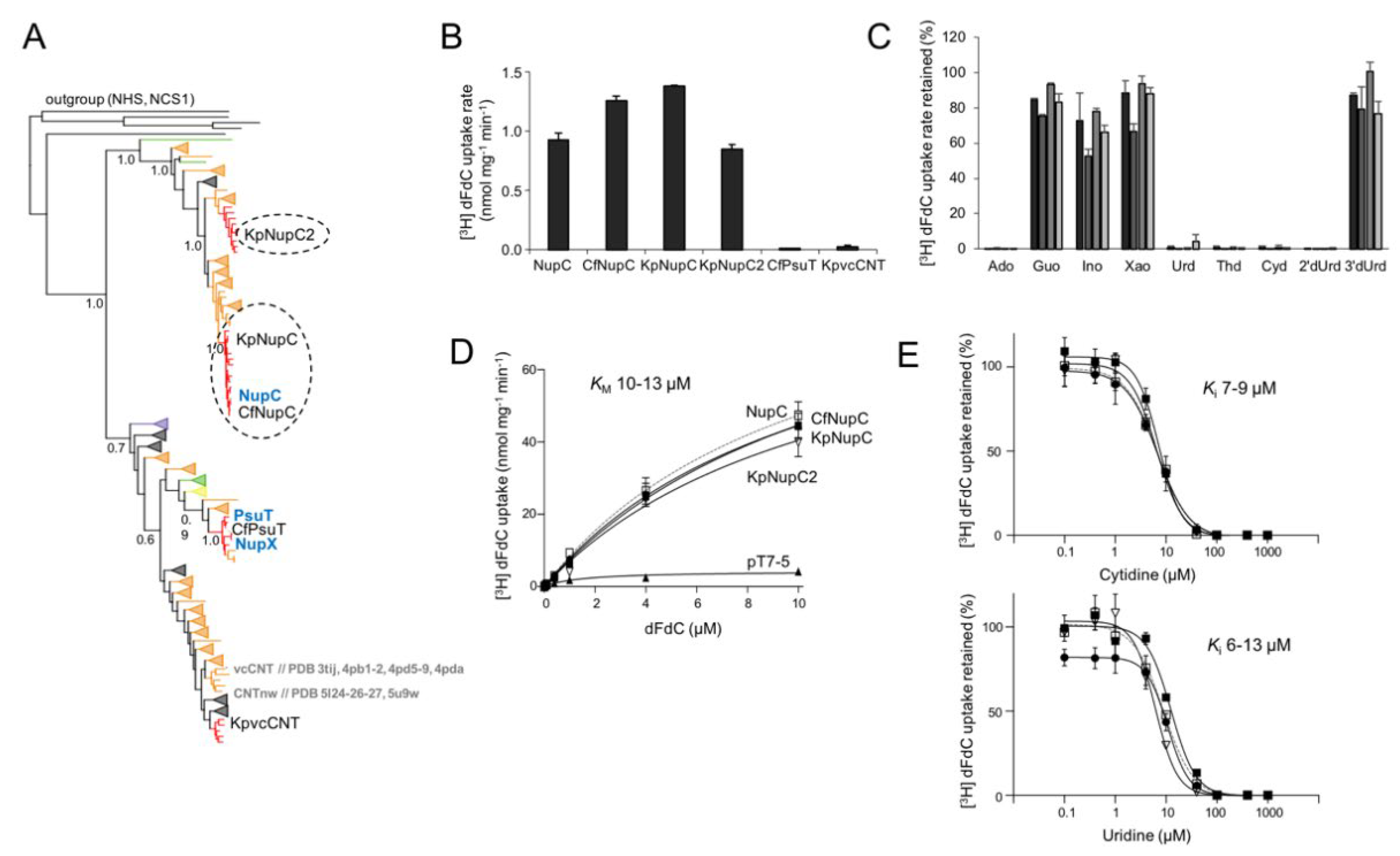
Figure 3.
Phylogeny and gemcitabine transport properties of C. freundii and K. pneumoniae NHSs. Panel (A): Phylogenetic analysis of 146 NHS homologs representing one fully sequenced genome per genus for all Proteobacteria, as retrieved from the IGM/M database at JGI (the complete phylogenetic tree is given in Supplementary Figure S5). The outgroup (shown on top) consists of E. coli members of families CNT (NupC, NupX, PsuT) and NCS1 (YbbW, and CodB, appearing as a separate clade). Other methodological details are as in Figure 2A. The homologs from E. coli K-12, K. pneumoniae ATCC 25955 and C. freundii ATCC 8090, as well as the PDB accession number of the structurally known NupG are indicated. Panel (B): [3H]-Gemcitabine (dFdC) transport rates (0.1 μΜ) by E. coli JW2389 expressing the indicated homologs from pT7-5/-BAD. Panel (C): Inhibition of [3H]-gemcitabine (0.1 μΜ) uptake rate of E. coli JW2389 expressing NupG, CfNupG or KpNupG (darker to lighter gray in the histogram) by the indicated unlabeled nucleosides (0.1 mM). Panel (D): Kinetics of [3H]-gemcitabine transport. Panel (E): Dose-response inhibition curves of the [3H]-gemcitabine (0.1 μΜ) uptake rate by cytidine (above) and uridine (below). The data in (D,E) for NupG, CfNupG and KpNupG are given as open rectangles (and interrupted lines), closed circles, and closed rectangles, respectively. Transport rates are deduced from measurements at 5 sec and given as the means of 3–5 determinations with SD shown. Values obtained with vector alone were subtracted from the measurements in all cases; the values obtained with vector alone (indicated as pT7-5) are also given in the kinetics plot (in (B)) for comparison.
Figure 3.
Phylogeny and gemcitabine transport properties of C. freundii and K. pneumoniae NHSs. Panel (A): Phylogenetic analysis of 146 NHS homologs representing one fully sequenced genome per genus for all Proteobacteria, as retrieved from the IGM/M database at JGI (the complete phylogenetic tree is given in Supplementary Figure S5). The outgroup (shown on top) consists of E. coli members of families CNT (NupC, NupX, PsuT) and NCS1 (YbbW, and CodB, appearing as a separate clade). Other methodological details are as in Figure 2A. The homologs from E. coli K-12, K. pneumoniae ATCC 25955 and C. freundii ATCC 8090, as well as the PDB accession number of the structurally known NupG are indicated. Panel (B): [3H]-Gemcitabine (dFdC) transport rates (0.1 μΜ) by E. coli JW2389 expressing the indicated homologs from pT7-5/-BAD. Panel (C): Inhibition of [3H]-gemcitabine (0.1 μΜ) uptake rate of E. coli JW2389 expressing NupG, CfNupG or KpNupG (darker to lighter gray in the histogram) by the indicated unlabeled nucleosides (0.1 mM). Panel (D): Kinetics of [3H]-gemcitabine transport. Panel (E): Dose-response inhibition curves of the [3H]-gemcitabine (0.1 μΜ) uptake rate by cytidine (above) and uridine (below). The data in (D,E) for NupG, CfNupG and KpNupG are given as open rectangles (and interrupted lines), closed circles, and closed rectangles, respectively. Transport rates are deduced from measurements at 5 sec and given as the means of 3–5 determinations with SD shown. Values obtained with vector alone were subtracted from the measurements in all cases; the values obtained with vector alone (indicated as pT7-5) are also given in the kinetics plot (in (B)) for comparison.
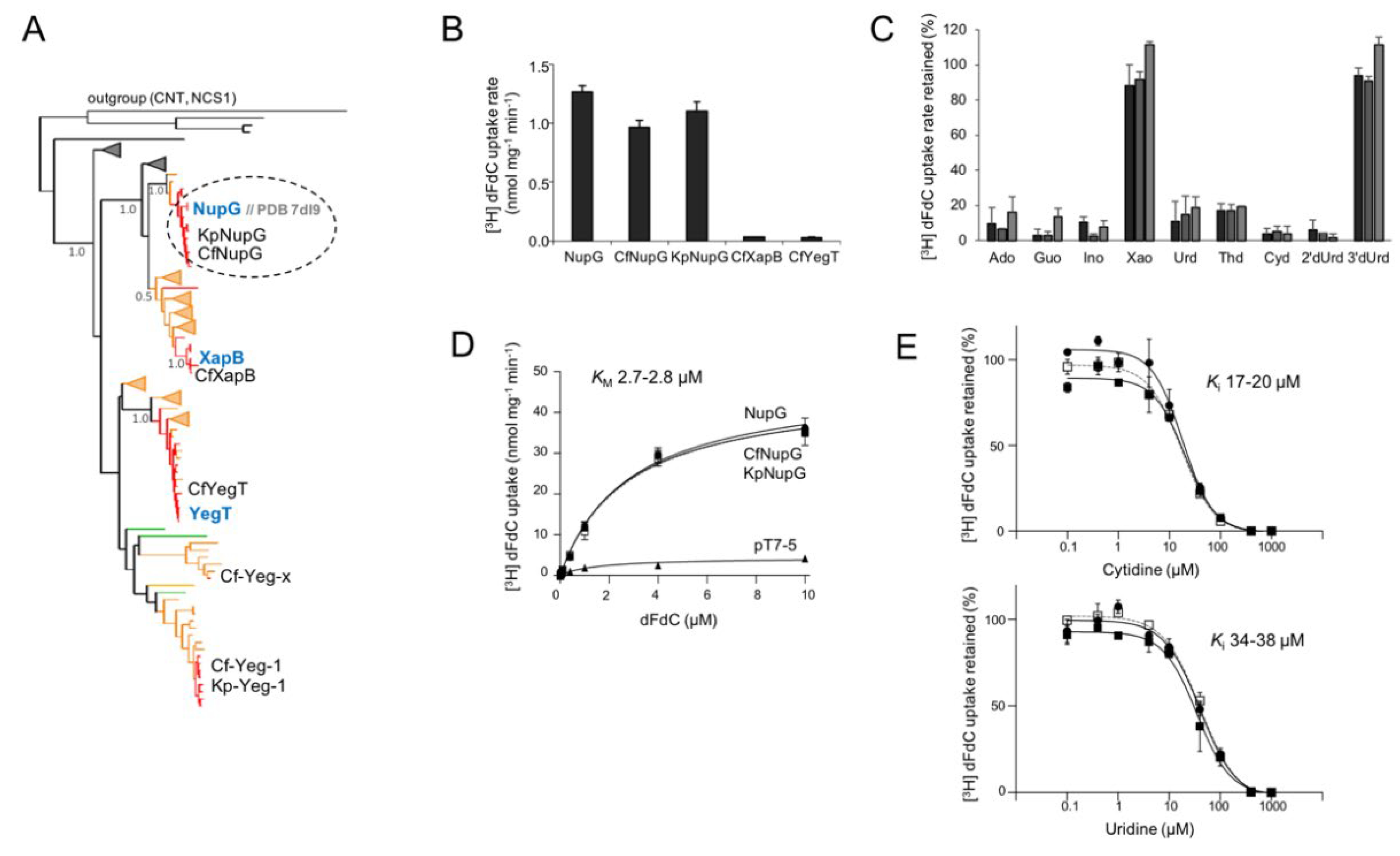

Table 1.
Kinetics and specificity of [3H]-gemcitabine transport.
 |
Data obtained from kinetics of [3H]-gemcitabine (0.01–10 μM) transport and inhibition of [3H]-gemcitabine (0.1 μM) transport by the indicated non-labeled nucleosides (0.1–1000 μΜ), based on rate measurements at 5 sec with E. coli JW2389 expressing the corresponding constructs. N.I. no inhibition in any of the concentrations tested. Inhibited: Transport activity inhibited to completeness by 1000-fold excess (0.1 mM) of the indicated nucleoside.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



