
Submitted:
04 June 2024
Posted:
06 June 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Determining the genetic contribution of susceptibility to severe SARS-CoV-2 infection outcomes is important for public health measures and individualized treatment. Through intense research on this topic, several hundred genes have been implicated as possibly contributing to the severe infection phenotype(s); however, the findings are complex and appear to be population-dependent. We aimed to determine the contribution of human rare genetic variants associated with a severe outcome of SARS-CoV-2 infections and their burden in the Slovenian population. A panel of 517 genes associated with severe SARS-CoV-2 infection were obtained through an extensive review of the literature, and additionally included target genes identified by the COVID-19 Host Genetic Initiative, as well as the curated Research COVID-19 associated genes from PanelApp, England Genomics. Whole genome sequencing was performed using PCR-free WGS on DNA from 60 patients hospitalized due to severe COVID-19 disease, and the identified rare genomic variants were analyzed and classified according to the ACMG criteria. Background prevalence in the general Slovenian population was determined by comparison with sequencing data from 8025 individuals included in the Slovenian genomic database (SGDB). Results show that several rare pathogenic/likely pathogenic genomic variants in genes CFTR, MASP2, MEFV, TNFRSF13B, and RNASEL likely contribute to the severe infection outcomes in our patient cohort. These results represent an insight into the Slovenian genomic diversity associated with severe COVID-19 outcome.
Keywords:
severe COVID-19
; severe outcome of SARS-CoV-2 infection
; whole-genome sequencing
; WGS
; genetic susceptibility
; rare variants
; human rare genomic variants
Introduction
The ongoing coronavirus pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), results in coronavirus disease 19 (COVID-19) [1,2,3], that manifests with a wide range of symptoms ranging from asymptomatic to critical. Patients present with symptoms such as fever, cough, fatigue, headache, hemoptysis, diarrhea, dyspnea, lymphocytopenia, pneumonia, acute respiratory distress syndrome, and acute cardiac injury, and present with specific radiological findings of ground-glass opacities. Severe cases require hospitalization and may lead to death [4].
Being an RNA virus, SARS-CoV-2 continuously evolves both by mutation and recombination during replication of the genome, and since its first detection, several lineages, sub-lineages, and variants have evolved. Despite some variants evading immunity from previous infection or vaccination, the variability in clinical manifestation (from asymptomatic to severe) of infection cannot be explained by the viral variability alone, and several human host factors, such as age, sex, and various comorbidities are now known to play a role [5,6]. Similarly, the immunological effects and their role in disease pathogenesis have now also been well characterized [7].
However, the underlying cause remains to be determined, and severe manifestations of infection, including death in otherwise healthy middle-aged individuals, suggest that genetic factors may play a role [8]. Indeed, because of genetics’ potential importance in disease prognosis, prevention, and public health planning measures, the field of COVID-19-host genetics has expanded rapidly resulting in several thousand publications on this topic in the last four years.
The results of this research have been more complex. Following early reports on the association between blood groups and occurrence of infection [9,10], and initial examination of a handful of candidate genes, based on their association with other viral interactions, such as ACE2, CLEC4M, MBL, ACE, CD209, FCER2, OAS-1, TLR4, and TNF-α [11,12,13], in the years since, several hundred genes have been implicated to play a role in the complex host genetic contribution to COVID-19. Genome-wide association studies (GWAS) have shown that many common genomic variants are enriched in cohorts of patients with severe COVID-19 [14,15]. So far, the identified polymorphisms show geographical differences, and were mostly shown to have weak effects and even combining them to assess their polygenic risk score (PRS) so far has not been able to effectively predict disease outcome. Similarly, while it has been proposed that the accumulation of weak effects of many rare functional variants may contribute to the overall risk in patients with severe disease [16,17], the information on such variants is scarce in many populations, including ours.
Therefore, we aimed to determine rare genomic variants and their burden in a comprehensive set of evidence-based genes in well-characterized patients hospitalized due to COVID-19 in Slovenia.
Results
We sequenced the whole genomes of 60 patients, hospitalized during the second pandemic wave of COVID-19 in Slovenia [7] to identify variants of interest in 517 evidence-based genes associated with severe COVID-19. The identified variants were classified according to ACMG Criteria [18] and the model of inheritance. The resulting pathogenic/likely pathogenic variants and risk factors, as well as their Slovenian genomic database (SGDB) background prevalence, are given in Table 1.
In total, we identified variants of interest (pathogenic/likely pathogenic variants or risk factors) in 52 of the 60 hospitalized patients. Of the 52 patients with variants of interest, 25 (48%) presented with a single variant, and 27 (52%) presented with more than one variant (Figure 1), highlighting the complex genetics of immunological response to viral infections and the difficulty in assigning causality.
Despite including hundreds of genes in our evidence-based panel, by adhering to strict ACMG criteria, we finally classified a total of 32 variants as pathogenic/likely pathogenic and 4 as risk factors, in 27 of the genes included in the analysis (Table 1). No variants of interest were identified in the LZTFL1 gene, other than the two previously reported GWAS-associated variants rs17713054 and rs35482426 (Table 1). The estimated burden of all identified pathogenic/likely pathogenic variants of interest in our COVID-19 hospitalized patients compared to the control Slovenian population was statistically significant (p=2.8x10-5).
Of these 36 variants of interest, 9 were previously reported as pathogenic/likely pathogenic in the ClinVar database [19], while 17 were reported with conflicting interpretations (unrelated to COVID-19) that included at least one pathogenic/likely pathogenic report. A further 6 of the variants were unclassified in the ClinVar database and were classified as likely pathogenic for the first time in this study. Of the 35 patients with pathogenic/likely pathogenic variants, variants in 10 patients were consistent with the proposed inheritance model, while 25 patients were heterozygous for variants originally associated with an autosomal-recessive disease.
The 7 out of 32 identified pathogenic/likely pathogenic variants that fit the inheritance model proposed for their respective 5 genes, were identified in CFTR (two variants in possible compound heterozygosity), MASP2 (one homozygous occurrence), MEFV, RNASEL, and TNFRSF13B. Pathogenic/likely pathogenic variants consistent with the proposed inheritance model were identified in 10 patients in total (Table 1, Table 2).
Interestingly, out of the 7 patients who died, 3 patients had pathogenic/likely pathogenic variants, consistent with predicted inheritance, vs. 7 in 53 patients who survived severe COVID-19; however, this was not statistically significant. Two of the deceased patients had likely pathogenic variants in the MEFV gene, and one patient had a homozygous pathogenic variant in MASP2. Of the remaining 4 patients who died, one had a risk factor in MBL2 gene and one patient in PRF1 gene, one had a pathogenic variant in AR gene IL36RN, and no variants of interest were identified in one of the deceased patients.
Of the remaining 7 patients with pathogenic/likely pathogenic variants who were hospitalized but survived, four had likely pathogenic variants in MEFV, one was compound heterozygous for pathogenic/likely pathogenic variants in CFTR, and one each had a pathogenic variant in RNASEL and TNFRSF13B respectively. Interestingly, most had additional risk factors (Table 2). Indeed, most patients had several variants of interest (Figure 1), but interestingly, the absence of such variants of interest did not show a statistically significant association with survival (χ2 test p==0.937137).
35 of the 60 patients carried more than 48 risk factor variants in MBL2 (34), PRF1 (7), and APOE (7). The LZTFL1 risk factors rs17713054 and rs35482426 were present in a total of 16 patients, and were the only finding in two patients (Table 2).
In addition to strong variants of interest, we have also identified more than 300 variants of uncertain significance in more than 200 genes from our curated evidence-based panel (Supplementary Table 2). These variants include those already classified as variants of uncertain significance in the ClinVar database or classified as such in this study due to being ultra-rare and having at least a moderate coding impact but lacking other criteria for pathogenicity. Most of these variants were missense variants that would need a functional assessment to reach a pathogenicity classification. Their significance remains to be determined in further studies.
Discussion
The significant effort invested in studying COVID-19-host genetics by research groups worldwide has resulted in numerous insights into the pathogenesis of the disease from different fields, from genetics to immunology, highlighting the complexity of virus-host interactions. It is now known that COVID-19-host genetics is very complex and that disease progression and outcome may correlate with more than one gene or genomic region. Indeed, it is estimated that many hundreds of genes are involved and have a direct or indirect effect on the clinical course of COVID-19 infection. In our study design, we have therefore made a significant effort to include in our analysis all genes for which the highest level of evidence was gathered.
Indeed, a total of 7 pathogenic/likely pathogenic variants consistent with the proposed inheritance model were identified in 10 patients in total, in 5 genes: CFTR (two variants in possible compound heterozygosity), MASP2 (one homozygous occurrence), MEFV, RNASEL, and TNFRSF13B.
The CFTR gene product, cystic fibrosis transmembrane conductance regulator, is an ATP-binding cassette transporter that functions as a ligand-gated anion channel involved in epithelial ion transport. Biallelic pathogenic variants in the CFTR gene cause cystic fibrosis [20], manifesting in chronic bronchopulmonary dysfunction. In our cohort, one patient was found to be a possible compound heterozygote for likely pathogenic variants in the CFTR gene, while 5 patients were found to be carriers of pathogenic/likely pathogenic variants in the heterozygous state. Of note, the intronic CFTR:c.1210-11T>G variant identified in our study, while considered pathogenic, is known to only be so in compound heterozygous state with severe pathogenic CFTR variants, while homozygous individuals are asymptomatic [21]. However, apart from this variant, carriers of single cystic fibrosis-causing variants of the CFTR gene were previously also shown to be more susceptible to the severe form of COVID-19 [22].
The MASP2 gene encodes for a mannan-binding lectin serine protease. While biallelic pathogenic variants in this gene are an established cause for autosomal recessive MASP2 deficiency, manifesting with increased susceptibility to infection due to the defective activation of the complement system, the MASP2 deficiency is common, and many individuals are asymptomatic [23,24]. In our cohort, we identified one likely pathogenic variant in MASP2 in 8 patients, in 7 patients in heterozygous form and in one patient in homozygous form.
Pathogenic and likely pathogenic heterozygous variants in the MEFV gene, which encodes an innate immune sensor, lead to the production of inflammatory mediators during infection [25], and are an established cause for autosomal dominant familial Mediterranean fever. Interestingly, it was shown that infected Mediterranean fever patients do not have a worse outcome of SARS-CoV-2 infection when already hospitalized [26].
A pathogenic variant in the RNASEL gene, encoding a 2-5A-dependent RNase, was found in one patient. RNASEL gene product is involved in the general antiviral interferon response [27] and shows an antiviral role against the Dengue virus [28].
Finally, a pathogenic heterozygous variant in the TNFRSF13B gene, whose product plays a crucial role in humoral immunity [29], was identified in one of our patients. Pathogenic variants in the TNFRSF13B gene are known to lead to common variable immunodeficiency with either autosomal dominant or recessive inheritance model and were previously identified in isolated cases of patients with severe COVID-19 [30,31].
While the gene-associated phenotype and the proposed inheritance model were both considered as parts of the ACMG criteria, it is important to note that the association of a particular variant with SARS-CoV-2 infection outcome may not follow the same inheritance model as the original association of the gene with another disease in which this gene is involved. Therefore, although the 26 heterozygous variants in ADAR, AIRE, ATM, BRCA2, C2, C6, C7, C8B, C9, CFD, CFI, CFTR, CYBA, FANCC, HAVCR2, HPS4, IL36RN, MASP2, PGM3, POLR3A and RNU4ATAC genes cannot be automatically considered to have an effect due to their otherwise autosomal-recessive association with their respective diseases, their contribution to SARS-CoV-2 infection severity would need to be functionally assessed or validated by additional studies. Until such studies provide a final confirmation, their contribution to the severe manifestation of COVID-19 remains unknown (Table 1, Table 2). For example, it is known that pathogenic heterozygous variants in the CFTR gene lead to increased risk for chronic pancreatitis, atypical mycobacterial infections, and bronchiectasis [32], and similar may be shown in the future for COVID-19 also.
Also in line with previous research, which has shown that many different genetic factors can concurrently contribute to the course of COVID-19 infection [16,17], in our study we have also identified more than one pathogenic/likely pathogenic variant or risk factor in almost half of the patient cohort. In particular, pathogenic/likely pathogenic variants that would not be sufficient for clinical manifestation according to the proposed inheritance model were found in 42% of patients, which raises an interesting question on their biological relevance. Indeed, similarly to other groups, we observed a clear burden of rare genomic variants classified as pathogenic/likely pathogenic in genes involved in immunity and host defense, autoinflammation, and autoimmunity were enriched in the selected patient cohort with severe infection outcomes compared to the Slovenian population [33].
Additionally, we identified risk factor variants in 58% (35/60) of our patients, while they were the sole finding in 28% (17/60) of our patient cohort. A total of 4 risk factor variants were identified in the following 3 genes; APOE, MBL2, and PRF1.
The APOE4 risk factor (variant APOE4:c.466T>C) was identified in 7 patients (12%). This risk factor is generally associated with hyperlipoproteinemia, cardiovascular disease, and Alzheimer's disease and has shown an association with COVID-19 disease by several independent studies [34,35,36], however as hyperlipoproteinemia, cardiovascular disease and dementia independently predict a worse outcome, the mechanism remains elusive. In our study, one patient was found to be a homozygote for the APOE4 allele (variant c.466T>C, p.Cys156Arg).
Two common MBL2 risk factors, increasing COVID-19 susceptibility due to MBL deficiency [37,38,39], were identified in 32 patients combined (53%). Although MBL deficiency is considered as an autosomal dominant disease, similar to MASP2 deficiency, it is a common deficiency, and patients may remain asymptomatic. In two of these patients, both variants, MBL2 c.161G>A (rs1800450) and c.154C>T (rs5030737) located in trans, were detected, while two patients were found to be homozygotes for the c.161G>A variant.
Finally, the risk factor PRF1:c.272C>T variant (rs35947132) was detected in 7 patients (12%). PRF1 gene encodes Perforin-1, a pore-forming protein homologous to complement component C9 with a similar mechanism of transmembrane channel formation [40]. While pathogenic variants in the PRF1 gene are associated with aplastic anemia, autosomal recessive familial hemophagocytic lymphohistiocytosis, and non-Hodgkin lymphoma, the c.272C>T risk factor variant (rs35947132) was previously also found to be enriched in several different COVID-19 patient cohorts [41,42,43].
Interestingly, we did not identify any variants of interest in the LZTFL1 gene that is located within the 3p21.31 risk “Neanderthal” haplotype, identified as important by large GWAS studies [14,44], and carried by approximately 16% of Europeans [45]. Therefore, we have also examined the frequency of the two previously reported intergenic variants surrounding the LZTFL1 gene, rs17713054 [14,44] and rs35482426 [14] in the patient cohort with that of the Slovenian population. There was no difference in the observed allelic frequency of rs17713054 intergenic variant and the rs35482426 intronic between our patient cohort and the general Slovenian population (Table 1).
While our patient cohort was heterogeneous with high comorbidities, this is a typical scenario observed in clinical practice when hospitalization is required for COVID-19. In such patients, by using an evidence-based gene panel and ACMG criteria for classification, pathogenic/likely pathogenic genomic variants were identified in 17% (10/60) of the patients. The presence of pathogenic/likely pathogenic variants consistent with the inheritance model was statistically significantly related to a further negative outcome. However, patients with no identified variants were not less likely to die, limiting our ability to draw any conclusion based on this small number of patients.
Limitations
Our work has the following inherent limitations, which are shared with other similar studies involving control and patient group composition, sample size, and limitations of variant classification. The control group consisted of individuals with unknown COVID-19 infection outcomes. Despite initially aiming to include a control group with mild or absent infection outcome, during the study, the occurrence of multiple infections as well as different vaccination statuses prevented us from obtaining such a group (i.e. vaccinated individuals may have a mild phenotype despite carrying a genetic variant conferring risk for a sever COVID-19 outcome). Similarly, to avoid any inherent bias, the patients hospitalized due to COVID-19 were selected in a blinded fashion regarding their immunological status (as determined in the original immunological study to which they were recruited). Despite these measures, the relatively small sample size of the severe COVID-19 cohort limits our ability to detect all rare variants in our population. Furthermore, the ACMG classification criteria limited our ability to classify variants as pathogenic to mostly exonic variants, where their pathogenicity could be assessed based on their predicted outcome on the protein level without further functional studies. Finally, since we did not perform phasing of variants, compound heterozygosity of the variants in genes with the proposed autosomal recessive inheritance model is assumed but needs additional confirmation. Altogether, genomic analysis has identified pathogenic/likely pathogenic variants in CFTR, MASP2, MEFV, RNASEL, and TNFRSF13B genes in 10 patients included in this study. As these genes are associated with immunodeficiency, susceptibility to infections, and inflammatory disorders, and were consistent with the proposed inheritance model for the gene, these variants could likely be major contributors to the severity of SARS-CoV-2 infection in case of these 10 patients. On the other hand, more than one-third of patients carried a single pathogenic/likely pathogenic variant in at least one COVID-19-associated gene with the proposed autosomal recessive inheritance model. For these variants, there is a possibility that they are additionally contributing to the severe condition of patients with COVID-19, which is a multifactorial disease. While it would be interesting to investigate the pathogenicity of intronic and other non-coding variants in genes with autosomal recessive inheritance model in patients where already one pathogenic/likely pathogenic variant or variant of uncertain significance was identified, this is unfortunately beyond the scope of our current research.
Conclusions
This study is the first to describe the genetic landscape of genomic susceptibility to severe COVID-19 in Slovenia. Several rare genomic variants classified as pathogenic/likely pathogenic were identified as promising candidates related to a higher risk of severe SARS-CoV-2 infection outcome. Our results represent an insight into the Slovenian genomic diversity associated with severe COVID-19 outcomes, with possible implications for future public health preventative measures.
Materials and Methods
Patients
The severe COVID-19 cohort consisted of 60 patients hospitalized during September-December 2020 (the second pandemic wave in Slovenia) at the University Clinic of Respiratory and Allergic Diseases, Golnik, Slovenia, that participated in the Immunological Characteristics of COVID-19 Patients clinical trial (registered at ClinicalTrials.gov (NCT04679428)[7]). Patients or their legally authorized representatives provided informed consent to participate in the study (Slovenian National Medical Ethics Committee Approval No. 0120-201/2020/7 and 0120-333/2020/3).
Hospitalization admission criteria required supplemental oxygen at admission and/or radiological signs of COVID-19 pneumonia. Additional hospitalization criteria included age (above 65), body mass index (above 30), presence of chronic kidney, cardiovascular, or lung disease, diabetes, cancer, advanced liver disease, and presence of immune insufficiency, as described in detail in the original trial publication [7]. Table 3 shows the demographic and clinical data of included hospitalized patients. To exclude unintentional bias, the patients included in the current study belonged to all six immune-phenotype groups identified and described in the original study (i.e. were chosen in a blinded fashion from the original immunological study).
Controls
Controls consisted of whole genome or whole exome (where appropriate) derived variants obtained from 8025 de-identified healthy individuals included in the SGDB at Clinical Institute of Genomic Medicine (CIGM), University Medical Centre Ljubljana (UMCL), Slovenia (Last accessed on 15.05.2024). All included individuals gave informed consent for the de-identified use of their variant information for research purposes. The informed consent statement was prepared according to National Review Board guidelines and approved by the Institutional Ethics Board at the UMCL, Slovenia. Control individuals were used only to determine background variant prevalence. The COVID-19 infection outcome status of the healthy individuals was unknown.
Whole Genome Sequencing
Whole genome sequencing was performed as previously described [46]. Briefly, third-party sequencing center services were used, where a standardized sequence of procedures was performed, consisting of PCR-free WGS library preparation protocol Illumina TrueSeq DNA Nano and sequencing on Illumina NovaSeq 6000 platform (both manufactured by Illumina, San Diego, CA, USA). The mean autosomal depth was greater than 30×. Data analysis, including variant calling, was performed in-house at the CIGM, by the Genome Analysis Toolkit Best Practices workflow from the Broad Institute [46,47,48]. The dbscSNV database of precomputed splice effect predictions and the SpliceAI algorithm were used to detect and annotate splice site variants [49,50].
Gene Panel Selection Strategy
Three strategies were used to select an evidence-based gene panel associated with severe SARS-CoV-2 infection: literature curation of original research publications; high-impact targets identified by the GWAS COVID-19 Host Genetic Initiative (COVID-19 HGI) [51]; and high-support genes from a research COVID-19 associated gene panel (COVID-19 research v1.136 PanelApp), curated by EnglandGenomics [52] (Figure 2).
Literature Curation
To identify human genes and genomic variants associated with SARS-CoV-2 infection, PubMed (www.ncbi.nlm.nih.gov/pubmed), and PubMedCentral (https://www.ncbi.nlm.nih.gov/pmc/) searches were performed from March 2021 to May 2022 by using the search strings: (<((genetic variation*) OR (gene polymorphism*) OR (polymorphism*) OR (gene variant*) OR (allele*) OR(GWAS*) OR (variant*) OR (host genetics*) OR (genetic susceptibility*) OR (genome*wide association study*)) AND ( (COVID-19*) OR (SARS-CoV-2*) OR (coronavirus*)) > [[Title/Abstract]]).
We aimed to include only original research reports, such as observational, cross-sectional, and/or cohort studies exploring the link between the disease progression and or severity and their connection to the genetic variations found in the affected population. The individuals included in these studies have had to have a clinical diagnosis of COVID-19 infections (positive PCR or antigen test) and their symptoms had to be professionally assessed. Patient outcomes had to be defined in the studies (mortality, severity, disease symptoms, etc.). The genomic markers had to be defined according to the European Medicine Agency definition [53] and included single-nucleotide polymorphisms (SNPs), short sequence repeat variants, haplotypes, alternative alleles, and other variants.
We only included articles in the English language. The following exclusion criteria were used to screen the publications’ title/abstracts: duplicates, clinical drug trials, in vitro assays, in silico studies, animal studies, population studies, reviews, and meta-analyses were excluded, as were studies reporting results from less than 30 human subjects/cases. In cases where reviews referenced original work missed by our search string strategy, these publications were also included in the analysis (Figure 2).
Next, the publications were analyzed if they qualified for inclusion regarding participants, interventions, controls, and outcomes (PICO) [54], as detailed in Supplementary Table S1. The selected articles had to meet at least three of the four PICO criteria. Due to the volume of literature regarding COVID-19 since 09.05.2022, to cross-check that no important studies were missed, top-journal review references after 09.05.2022 were examined for additional original functional studies. Only genes/variants replicated by at least one independent study, or results confirmed functionally were included in the final selection of genes/genomic variants. In this way, 170 original studies of human genetic variation related to SARS-CoV-2 were included in the screening (Figure 2, Supplementary Table S2).
COVID-19 Human Genetics Initiative
The COVID-19 HGI was established in 2020 to facilitate the collaboration of the human genetics scientific community in COVID-19 host-genetics research [51]. The initiative has so far generated important results supporting the role of several human genetic variants in COVID-19 disease [14,55,56,57,58,59,60,61]. We have therefore also included the results generated by the COVID-19 HGI in our final gene panel selection (Figure 2, Supplementary Table S2).
Genomics England PanelApp
The Genomics England PanelApp is a crowdsourcing tool for gene panels, where they can be shared, downloaded, and evaluated by the scientific community [52]. Their COVID-19 research panel (current version 1.136) currently contains a set of 697 entities, of which 461 (459 genes and 2 regions) have been curated by expert reviewers, as having good evidence for association with COVID-19. The criteria for this curation include, but are not limited to: there being multiple original studies demonstrating such association; a clinical study of an association that is supported by additional validation (in vitro/ in vivo / in silico/ other evidence); Research and/or Clinical community consensus on the likely importance of this gene; previous strong evidence of the involvement of the gene in a primary immunodeficiency disorder; and the gene being on the current list of known human genes associated with inborn errors of immunity by the International Union of Immunological Societies (IUIS)[52,62]. These 459 genes were also included in our final gene panel selection (Figure 2, Supplementary Table S2).
Genes and Interpretation
The final selection of genes in our SARS-CoV-2 gene panel included 517 genes. Of these genes, the following 79 were identified from at least two independent sources, were functionally confirmed, and/or were identified through the joint efforts of the COVID-19 HGI: ABO, ACE2, ACSL6, AGT, AGTR1, AK5, APOE, ARHGEF38, ATP11A, BCL11A, CASC20, CCHCR1, CCR2, CCR5, CCR9, CFTR, CXCR6, DPP9, EFNA4, ELF5, FBRSL1, FOXP4, FURIN, FUT2, FYCO1, HCN3, HIP1, HLA-DRB1, IFITM3, IFNA10, IFNAR1, IFNAR2, IFNL3, IFNL4, IL10RB, IL6, IRF1, IRF3, IRF7, JAK1, KANSL1, LTA, LZTFL1, MBL2, MUC5B, NOS3, NR1H2, NXPE3, OAS1, OAS2, OAS3, PLEKHA4, PLSCR1, PNPLA3, PRF1, RAB2A, RAVER1, RGMA, SFTPD, SLC22A31, SLC2A5, SLC6A20, SRRM1, TBK1, THBS3, TICAM1, TLL1, TLR3, TLR4, TLR7, TMEM65, TMPRSS2, TNF, TRIM46, TYK2, VDR, XCR1, ZGLP1 and ZKSCAN1. The genes in bold are those identified by international GWAS and multiple independent publications. The full list of the 517 genes is given in Supplementary Table S2.
The background population frequency of the identified variants of interest was determined from the GnomAD [63] and the SGDB (CIGM, UMCL). Genomic data interpretation was performed according to the ACMG/AMP standards and guidelines [64] and was performed by at least two analysts of which at least one was a specialist in clinical and/or laboratory genetics, as previously described [48].
The proposed inheritance model and disease mechanisms of each gene were taken into account in the interpretation. Finally, the previously reported clinical manifestations associated with each identified variant were assessed in regard to their relevance to COVID-19-associated disease. All classified variants of interest are listed in Supplementary Table S3.
Statistical Data Analysis
Statistical data analysis was performed using version 4.1.3 of the R computing environment [65]. A p-value of <0.05 was considered statistically significant. Genotype and allele frequencies were compared between the case and control group using the chi-square test or Fisher's exact test when the expected frequency was less than 5. In controlling the false discovery rate (FDR) for multiple testing, the Benjamini and Hochberg adjustment of p-values was employed [66]. A burden analysis was conducted to determine if there was a higher prevalence of all identified pathogenic/likely pathogenic variants of interest in our COVID-19 hospitalized patients compared to the general Slovenian population. We estimated the burden using the chi-square test.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Ethics statement
All included individuals gave informed consent for the de-identified research use of their variant information. The informed consent statement was prepared according to Slovenian National Review Board guidelines. Patients or their legally authorized representatives provided informed consent to participate in the study (Study registration nr. NCT04679428, Slovenian National Medical Ethics Committee Approval No. 0120-201/2020/7 and 0120-333/2020/3).
Author Contributions
AK: AM, and BP designed the study. AK, HV, MČS, and TL performed extensive literature searches and curation. JŠ, MR, UBS, BB, BR, and PK provided samples for analysis together with the demographic and clinical data of patients. TL, AM, and AK collected, analyzed, and interpreted data and prepared figures and tables. MČS performed the statistical analyses. AK, TL, and BP wrote the manuscript. All authors contributed to the article, critically revised it and have approved the final submitted version. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Slovenian Research and Innovation Agency (Programme P3-0326, Programme P3-0360, ARRS-RPROG-JP-COVID19-Prijava/2020/091). This work was supported by the Slovenian Research Agency (Programme P3-0326, ARRS-RPROG-JP-COVID19).
Acknowledgments
We thank all patients and their relatives who participated in the study.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet. 2020, 395, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. Journal of Autoimmunity. 2020, 109, 102433. [Google Scholar] [CrossRef] [PubMed]
- Baradaran, A.; Ebrahimzadeh, M.H.; Baradaran, A.; Kachooei, A.R. Prevalence of Comorbidities in COVID-19 Patients: A Systematic Review and Meta-Analysis. ABJS 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.D.; Lone, N.I.; Baillie, J.K. Comorbidities, multimorbidity and COVID-19. Nat Med. 2023, 29, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Šelb, J.; Bitežnik, B.; Bidovec Stojković, U.; Rituper, B.; Osolnik, K.; Kopač, P.; et al. Immunophenotypes of anti-SARS-CoV-2 responses associated with fatal COVID-19. ERJ Open Res. 2022, 8, 00216–02022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; Zhang, Q.; Casanova, J.-L.; Su, H.C.; COVID Team. Severe COVID-19 in the young and healthy: monogenic inborn errors of immunity? Nat Rev Immunol. 2020, 20, 455–456. [Google Scholar] [CrossRef] [PubMed]
- Golinelli, D.; Boetto, E.; Maietti, E.; Fantini, M.P. The association between ABO blood group and SARS-CoV-2 infection: A meta-analysis. PLoS One 2020, 15, e0239508. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, T.; Ma, L.; Zhang, H.; Wang, H.; Wei, W.; et al. The impact of ABO blood group on COVID-19 infection risk and mortality: A systematic review and meta-analysis. Blood Rev. 2020, 100785. [Google Scholar] [CrossRef]
- Di Maria, E.; Latini, A.; Borgiani, P.; Novelli, G. Genetic variants of the human host influencing the coronavirus-associated phenotypes (SARS, MERS and COVID-19): rapid systematic review and field synopsis. Hum Genomics. 2020, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Elhabyan, A.; Elyaacoub, S.; Sanad, E.; Abukhadra, A.; Elhabyan, A.; Dinu, V. The role of host genetics in susceptibility to severe viral infections in humans and insights into host genetics of severe COVID-19: A systematic review. Virus Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Lopez, O.; Daimiel, L.; Ramírez de Molina, A.; Martínez-Urbistondo, D.; Vargas, J.A.; Martínez, J.A. Exploring Host Genetic Polymorphisms Involved in SARS-CoV Infection Outcomes: Implications for Personalized Medicine in COVID-19. Int J Genomics. 2020, 2020, 6901217. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Rawlik, K.; Bretherick, A.D.; Qi, T.; Wu, Y.; Nassiri, I.; et al. GWAS and meta-analysis identifies 49 genetic variants underlying critical COVID-19. Nature. 2023, 617, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; et al.; Severe Covid-19 GWAS Group Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N Engl J Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Fang, M.; Luo, Y.; Zheng, F.; Jin, Y.; Cheng, F.; et al. Rare Variants in Inborn Errors of Immunity Genes Associated With Covid-19 Severity. Front Cell Infect Microbiol. 2022, 12, 888582. [Google Scholar] [CrossRef] [PubMed]
- Khadzhieva, M.B.; Gracheva, A.S.; Belopolskaya, O.B.; Kolobkov, D.S.; Kashatnikova, D.A.; Redkin, I.V.; et al. COVID-19 severity: does the genetic landscape of rare variants matter? Front Genet. 2023, 14, 1152768. [Google Scholar] [CrossRef] [PubMed]
- The ACMG Laboratory Quality Assurance Committee; Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Wang, Y.; Wrennall, J.A.; Cai, Z.; Li, H.; Sheppard, D.N. Understanding how cystic fibrosis mutations disrupt CFTR function: From single molecules to animal models. The International Journal of Biochemistry & Cell Biology. 2014, 52, 47–57. [Google Scholar] [CrossRef]
- Nykamp, K.; Truty, R.; Riethmaier, D.; Wilkinson, J.; Bristow, S.L.; Aguilar, S.; et al. Elucidating clinical phenotypic variability associated with the polyT tract TG repeats in, C.F.T.R. Hum Mutat. 2021, 42, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Baldassarri, M.; Fava, F.; Fallerini, C.; Daga, S.; Benetti, E.; Zguro, K.; et al. Severe COVID-19 in Hospitalized Carriers of Single CFTR Pathogenic Variants. J Pers Med. 2021, 11, 558. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kumari, B.; Ali, A.; Patel, P.K.; Sharma, A.K.; Nair, R.; et al. Mannose-binding lectin gene 2 variant DD (rs 5030737) is associated with susceptibility to COVID-19 infection in the urban population of Patna City (India). Mol Genet Genomics. 2023, 298, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Thiel, S.; Vorup-Jensen, T.; Stover, C.M.; Schwaeble, W.; Laursen, S.B.; Poulsen, K.; et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997, 386, 506–510. [Google Scholar] [CrossRef]
- Masters, S.L.; Lagou, V.; Jéru, I.; Baker, P.J.; Van Eyck, L.; Parry, D.A.; et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci Transl Med. 2016, 8, 332ra45. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, F.; Caporilli, C.; Titolo, A.; Rigante, D.; Esposito, S. Clinical impact and disease evolution of SARS-CoV-2 infection in familial Mediterranean fever. Pharmacol Res. 2022, 182, 106293. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Mukherjee, S.; Manivannan, P.; Malathi, K. RNase L Cleavage Products Promote Switch from Autophagy to Apoptosis by Caspase-Mediated Cleavage of Beclin-1. Int J Mol Sci. 2015, 16, 17611–17636. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-J.; Yu, H.-P.; Chang, B.-L.; Tang, W.-C.; Liao, C.-L.; Lin, Y.-L. Distinct antiviral roles for human 2’,5’-oligoadenylate synthetase family members against dengue virus infection. J Immunol. 2009, 183, 8035–8043. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Bressette, D.; Carrell, J.A.; Kaufman, T.; Feng, P.; Taylor, K.; et al. Tumor Necrosis Factor (TNF) Receptor Superfamily Member TACIIs a High Affinity Receptor for TNFFamily Members, A. P.R.I.L.; BLyS Journal of Biological Chemistry 2000, 275, 35478–35485. [Google Scholar] [CrossRef]
- Schmidt, A.; Peters, S.; Knaus, A.; Sabir, H.; Hamsen, F.; Maj, C.; et al. TBK1 and TNFRSF13B mutations and an autoinflammatory disease in a child with lethal COVID-19. NPJ Genom Med. 2021, 6, 55. [Google Scholar] [CrossRef]
- Kinoshita, H.; Durkee-Shock, J.; Jensen-Wachspress, M.; Kankate, V.V.; Lang, H.; Lazarski, C.A.; et al. Robust Antibody and T Cell Responses to SARS-CoV-2 in Patients with Antibody Deficiency. J Clin Immunol. 2021, 41, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Polgreen, P.M.; Comellas, A.P. Clinical Phenotypes of Cystic Fibrosis Carriers. Annu Rev Med. 2022, 73, 563–574. [Google Scholar] [CrossRef]
- Tangye SG, COVID Human Genetic Effort consortium. Impact of SARS-CoV-2 infection and COVID-19 on patients with inborn errors of immunity. J Allergy Clin Immunol. 2023, 151, 818–831. [Google Scholar] [CrossRef]
- Al-Jaf, S.M.A.; Niranji, S.S.; Ali, H.N.; Mohammed, O.A. Association of Apolipoprotein e polymorphism with SARS-CoV-2 infection. Infect Genet Evol. 2021, 95, 105043. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; et al. APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. J Gerontol A Biol Sci Med Sci. 2020, 75, 2231–2232. [Google Scholar] [CrossRef]
- del Ser, T.; Fernández-Blázquez, M.A.; Valentí, M.; Zea-Sevilla, M.A.; Frades, B.; Alfayate, E.; et al. Residence, Clinical Features, and Genetic Risk Factors Associated with Symptoms of COVID-19 in a Cohort of Older People in Madrid. Gerontology 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Medetalibeyoglu, A.; Bahat, G.; Senkal, N.; Kose, M.; Avci, K.; Sayin, G.Y.; et al. Mannose binding lectin gene 2 (rs1800450) missense variant may contribute to development and severity of COVID-19 infection. Infect Genet Evol. 2021, 89, 104717. [Google Scholar] [CrossRef]
- Speletas, M.; Dadouli, K.; Syrakouli, A.; Gatselis, N.; Germanidis, G.; Mouchtouri, V.A.; et al. MBL deficiency-causing B allele (rs1800450) as a risk factor for severe COVID-19. Immunobiology. 2021, 226, 152136. [Google Scholar] [CrossRef]
- Yilmaz, D.; Soyoz, M.; Sahin, A.; Cerci-Alkac, B.; Karahan-Coven, H.I.; Ekemen-Keles, Y.; et al. Association between mannose binding lectin gene polymorphisms and clinical severity of COVID-19 in children. Mol Biol Rep. 2023, 50, 5871–5877. [Google Scholar] [CrossRef]
- Podack, E.R.; Lowrey, D.M.; Lichtenheld, M.; Olsen, K.J.; Aebischer, T.; Binder, D.; et al. Structure, function and expression of murine and human perforin 1 (P1). Immunol Rev. 1988, 103, 203–211. [Google Scholar] [CrossRef]
- Cabrera-Marante, O.; de Frías, E.R.; Pleguezuelo, D.E.; Allende, L.M.; Serrano, A.; Laguna-Goya, R.; et al. Perforin gene variant A91V in young patients with severe COVID-19. Haematologica 2020, 105, 2844–2846. [Google Scholar] [CrossRef] [PubMed]
- Zanchettin, A.C.; Barbosa, L.V.; Dutra, A.A.; Prá, D.M.M.; Pereira, M.R.C.; Stocco, R.B.; et al. Role of Genetic Polymorphism Present in Macrophage Activation Syndrome Pathway in Post Mortem Biopsies of Patients with COVID-19. Viruses 2022, 14, 1699. [Google Scholar] [CrossRef] [PubMed]
- Gelzo, M.; Castaldo, A.; Giannattasio, A.; Scalia, G.; Raia, M.; Esposito, M.V.; et al. MIS-C: A COVID-19-as sociated condition between hypoimmunity and hyperimmunity. Front Immunol. 2022, 13, 985433. [Google Scholar] [CrossRef] [PubMed]
- Downes, D.J.; Cross, A.R.; Hua, P.; Roberts, N.; Schwessinger, R.; Cutler, A.J.; et al. Identification of LZTFL1 as a candidate effector gene at a COVID-19 risk locus. Nat Genet. 2021, 53, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Zeberg, H.; Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature. 2020, 587, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Bergant, G.; Maver, A.; Peterlin, B. Whole-Genome Sequencing in Diagnostics of Selected Slovenian Undiagnosed Patients with Rare Disorders. Life (Basel) 2021, 11, 205. [Google Scholar] [CrossRef] [PubMed]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. CP in Bioinformatics. 2013, 43. [Google Scholar] [CrossRef] [PubMed]
- Ales, M.; Luca, L.; Marija, V.; Gorazd, R.; Karin, W.; Ana, B.; et al. Phenotype-driven gene target definition in clinical genome-wide sequencing data interpretation. Genet Med. 2016, 18, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Research 2014, 42, 13534–13544. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019, 176, 535–548. [Google Scholar] [CrossRef]
- The COVID-19 Host Genetics Initiative. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur J Hum Genet. 2020, 28, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Williams, E.; Foulger, R.E.; Leigh, S.; Daugherty, L.C.; Niblock, O.; et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet. 2019, 51, 1560–1565. [Google Scholar] [CrossRef] [PubMed]
- CH Topic E15 Definitions for genomic biomarkers, pharmacogenomics, pharmacogenetics, genomic data and sample coding categories. EMEA/CHMP/ICH/437986/2006. Available: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-e-15-definitions-genomic-biomarkers-pharmacogenomics-pharmacogenetics-genomic-data-sample-coding_en.
- Rathbone, J.; Albarqouni, L.; Bakhit, M.; Beller, E.; Byambasuren, O.; Hoffmann, T.; et al. Expediting citation screening using PICo-based title-only screening for identifying studies in scoping searches and rapid reviews. Syst Rev. 2017, 6, 233. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.H.L.; Park, D.S.; Coignet, M.V.; McCurdy, S.R.; Knight, S.C.; Partha, R.; et al. AncestryDNA COVID-19 Host Genetic Study Identifies Three Novel Loci. Epidemiology 2020. [Google Scholar] [CrossRef]
- Roberts, G.H.L.; Partha, R.; Rhead, B.; Knight, S.C.; Park, D.S.; Coignet, M.V.; et al. Expanded COVID-19 phenotype definitions reveal distinct patterns of genetic association and protective effects. Nat Genet. 2022, 54, 374–381. [Google Scholar] [CrossRef] [PubMed]
- The GenOMICC Investigators; The ISARIC4C Investigators; The COVID-19 Human Genetics Initiative; 23andMe Investigators; BRACOVID Investigators; Gen-COVID Investigators; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, J.E.; Kosmicki, J.A.; Damask, A.; Sharma, D.; Roberts, G.H.L.; Justice, A.E.; et al. Genome-wide analysis provides genetic evidence that ACE2 influences COVID-19 risk and yields risk scores associated with severe disease. Nat Genet. 2022, 54, 382–392. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Host Genetics Initiative; COVID-19 Host Genetics InitiativeLeadership; Niemi MEK; Karjalainen, J.; Liao, R.G.; Neale, B.M.; et al. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.F.; Shastri, A.J.; Ye, C.; Weldon, C.H.; Filshtein-Sonmez, T.; Coker, D.; et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat Genet. 2021, 53, 801–808. [Google Scholar] [CrossRef]
- Kousathanas, A.; Pairo-Castineira, E.; Rawlik, K.; Stuckey, A.; Odhams, C.A.; Walker, S.; et al. Whole-genome sequencing reveals host factors underlying critical COVID-19. Nature 2022, 607, 97–103. [Google Scholar] [CrossRef]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022, 42, 1473–1507. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Francioli, L.C.; Goodrich, J.K.; Collins, R.L.; Kanai, M.; Wang, Q.; et al. A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. Genetics 2022. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. A language and environment for statistical computing [Internet]. Vienna, Austria: R Foundation for Statistical Computing. 2022. Available from: https://www.R-project.org/.
- Dudbridge, F.; Gusnanto, A.; Koeleman, B.P. Detecting multiple associations in genome-wide studies. Hum Genomics. 2006, 2, 310–317. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Distribution of pathogenic variants, likely pathogenic variants, and risk factors in COVID-19-associated genes among 60 patients and population controls. B) Prevalence and classification of 36 variants of interest in 60 patients. C) Prevalence of 36 variants of interest in 8025 control population individuals.
Figure 1.
Distribution of pathogenic variants, likely pathogenic variants, and risk factors in COVID-19-associated genes among 60 patients and population controls. B) Prevalence and classification of 36 variants of interest in 60 patients. C) Prevalence of 36 variants of interest in 8025 control population individuals.
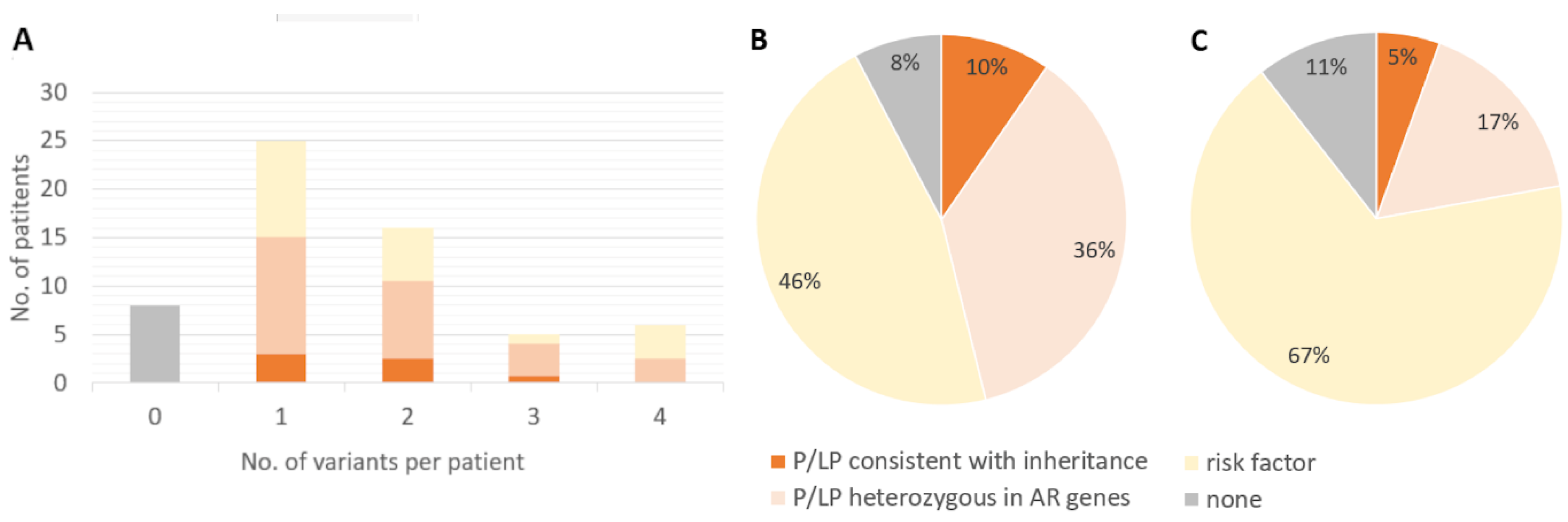
Figure 2.
Evidence-based gene panel design.


Table 1.
Pathogenic variants, likely pathogenic variants, and risk factors in COVID-19-associated genes.
Table 1.
Pathogenic variants, likely pathogenic variants, and risk factors in COVID-19-associated genes.
| Gene | Transcript | Variant | Protein change | No. of patients a | Patients allelic freq. | SGDB allelic freq. | P-value (allelic freq) b | P-value adjusted (allelic freq) d | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|
| ADAR | NM_001365045.1 | c.604C>G | p.Pro202Ala | 1 | 0.0083 | 0.0019 | 0.211 | 0.329 | P |
| AIRE | NM_000383.4 | c.769C>T | p.Arg257* | 2 | 0.0169 | 0.0033 | 0.062 | 0.152 | P |
| APOE | NM_001302688.2 | c.466T>C | p.Cys156Arg | 7 | 0.0667 | 0.1059 | 0.164 c | 0.280 | R |
| ATM | NM_000051.3 | c.6095G>A | p.Arg2032Lys | 1 | 0.0083 | 0.0003 | 0.039 | 0.120 | LP |
| BRCA2 | NM_000059.3 | c.7806-2A>G | splice variant | 1 | 0.0083 | 0.0007 | 0.091 | 0.196 | P |
| C2 | NM_001282459.2.7 | c.841_868del | p.Val281fs | 1 | 0.0085 | 0.0114 | 1 | 1 | P |
| C6 | NM_000065.4 | c.2381+2T>C | splice variant | 1 | 0.0083 | 0.0054 | 0.480 | 0.590 | P |
| C7 | NM_000587.4 | c.1561C>A | p.Arg521Ser | 1 | 0.0083 | 0.0082 | 0.627 | 0.731 | P |
| C7 | NM_000587.4 | c.1924_1925del | p.His643fs | 1 | 0.0083 | 0.0003 | 0.046 | 0.134 | P |
| C8B | NM_000066.4 | c.1282C>T | p.Arg428* | 1 | 0.0083 | 0.0072 | 0.579 | 0.679 | P |
| C9 | NM_001737.5 | c.162C>A | p.Cys54* | 1 | 0.0083 | 0.0061 | 0.522 | 0.630 | P |
| CFD | NM_001317335.2 | c.286delG | p.Glu96fs | 1 | 0.0083 | 0.0006 | 0.076 | 0.177 | LP |
| CFI | NM_001318057.2 | c.111dupA | p.Tyr38fs | 1 | 0.0083 | 0.0011 | 0.133 | 0.246 | LP |
| CFTR | NM_000492.4 | c.1210-11T>G | intronic | 1 | 0.0089 | 0.0107 | 1 | 1 | P |
| CFTR | NM_000492.4 | c.1727G>C | p.Gly576Ala | 1 (CH) | 0.0083 | 0.0038 | 0.370 | 0.482 | LP |
| CFTR | NM_000492.4 | c.2002C>T | p.Arg668Cys | 1 (CH) | 0.0083 | 0.0058 | 0.503 | 0.613 | LP |
| CFTR | NM_000492.4 | c.2991G>C | p.Leu997Phe | 1 | 0.0088 | 0.0026 | 0.262 | 0.380 | LP |
| CFTR | NM_000492.4 | c.3154T>G | p.Phe1052Val | 1 | 0.0083 | 0.0008 | 0.098 | 0.205 | P |
| CFTR | NM_000492.4 | c.3485G>T | p.Arg1162Leu | 2 | 0.0167 | 0.0070 | 0.211 | 0.329 | LP |
| CYBA | NM_000101.4 | c.222delC | p.Ala75fs | 1 | 0.0083 | 0 | 0.142 | 0.255 | LP |
| FANCC | NM_000136.3 | c.487_490del | p.Glu163fs | 1 | 0.0083 | 0 | 0.142 | 0.255 | LP |
| HAVCR2 | NM_032782.5 | c.291A>G | p.Ile97Met | 4 | 0.0333 | 0.0179 | 0.171 | 0.287 | LP |
| HPS4 | NM_001349900.2 | c.649C>T | p.Arg217* | 1 | 0.0083 | 0.0011 | 0.133 | 0.246 | P |
| IL36RN | NM_012275.3 | c.338C>T | p.Ser113Leu | 1 | 0.0083 | 0.0039 | 0.379 | 0.488 | P |
| MASP2 | NM_006610.4 | c.359A>G | p.Asp120Gly | 8 (1 hom) | 0.0750 | 0.0490 | 0.189 c | 0.308 | LP |
| MBL2 | NM_000242.2 | c.154C>T | p.Arg52Cys | 13 | 0.1083 | 0.0724 | 0.131 c | 0.246 | R |
| MBL2 | NM_000242.2 | c.161G>A | p.Gly54Asp | 21 | 0.1917 | 0.1422 | 0.123 c | 0.233 | R |
| MEFV | NM_000243.2 | c.2084A>G | p.Lys695Arg | 5 | 0.0417 | 0.0266 | 0.256 | 0.377 | LP |
| MEFV | NM_000243.2 | c.2230G>T | p.Ala744Ser | 1 | 0.0083 | 0.0036 | 0.353 | 0.468 | LP |
| PGM3 | NM_001199917.2 | c.463delA | p.Arg155fs | 1 | 0.0083 | 0.0002 | 0.041 | 0.124 | LP |
| POLR3A | NM_007055.4 | c.1771-7C>G | intronic | 1 | 0.0083 | 0.0001 | 0.023 | 0.090 | P |
| PRF1 | NM_001083116.3 | c.272C>T | p.Ala91Val | 7 | 0.0583 | 0.0564 | 0.928 | 1 | R |
| RNASEL | NM_021133.4 | c.1567-11_1574del | p.Asp523fs | 1 | 0.0083 | 0.0005 | 0.061 | 0.152 | LP |
| RNU4ATAC | NR_023343.1 | n.8C>T | ncRNA | 1 | 0.0083 | 0.0015 | 0.180 | 0.299 | LP |
| RNU4ATAC | NR_023343.1 | n.40C>T | ncRNA | 1 | 0.0083 | 0.0005 | 0.082 | 0.187 | P |
| TNFRSF13B | NM_012452.3 | c.310T>C | p.Cys104Arg | 1 | 0.0083 | 0.0029 | 0.300 | 0.412 | P |
| rs17713054 | NA | rs17713054-A | intergenic | 14 | 0.1250 | 0.1055 | 0.522 c | NA | RL |
| rs35482426 | NM_001276378.2 | c.-137-18021_-137-18020del | intronic | 15 | 0.1333 | 0.1441 | 0.753 c | NA | RL |
LP = likely pathogenic, P = pathogenic, R = risk factor, RL = risk locus, CH = in compound heterozygous state, hom = in homozygous state. Variants, for which the genotype was consistent with the proposed inheritance model for the gene, are indicated in bold. a In heterozygous state, if not indicated otherwise. b Fisher's exact test for comparison between patients’ and SGBD allelic frequency or c χ2 test if expected frequency was ≥5. d p values adjusted for 689 variants in 517 genes.
Table 2.
Distribution of pathogenic variants, likely pathogenic variants (in red), and risk factors (in yellow) in COVID-19-associated genes among 60 patients.
Table 2.
Distribution of pathogenic variants, likely pathogenic variants (in red), and risk factors (in yellow) in COVID-19-associated genes among 60 patients.
| Patients | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant | Class | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | 36 | 37 | 38 | 39 | 40 | 41 | 42 | 43 | 44 | 45 | 46 | 47 | 48 | 49 | 50 | 51 | 52 | 53 | 54 | 55 | 56 | 57 | 58 | 59 | 60 | SUM |
| ADAR c.604C>G | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AIRE c.769C>T | P | 1 | 1 | 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| APOE c.466T>C | R | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||
| ATM c.6095G>A | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BRCA2 c.7806-2A>G | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C2 c.841_868del | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C6 c.2381+2T>C | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C7 c.1561C>A | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C7 c.1924_1925del | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C8B c.1282C>T | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C9 c.162C>A | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CFD c.286del | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CFI c.111dup | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CFTR c.1210-11T>G | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CFTR c.1727G>C | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CFTR c.2002C>T | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CFTR c.2991G>C | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CFTR c.3154T>G | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CFTR c.3485G>T | LP | 1 | 1 | 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CYBA c.222del | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FANCC c.487_490del | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HAVCR2 c.291A>G | LP | 1 | 1 | 1 | 1 | 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HPS4 c.649C>T | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IL36RN c.338C>T | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MASP2 c.359A>G hom | LP | 1 | 7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MASP2 c.359A>G het | LP | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||
| MBL2 c.154C>T | R | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 13 | |||||||||||||||||||||||||||||||||||||||||||||||
| MBL2 c.161G>A | R | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 21 | |||||||||||||||||||||||||||||||||||||||
| MEFV c.2084A>G | LP | 1 | 1 | 1 | 1 | 1 | 5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MEFV c.2230G>T | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PGM3 c.463del | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| POLR3A c.1771-7C>G | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PRF1 c.272C>T | R | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||
| RNASEL c.1567-11_1574del | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RNU4ATAC n.40C>T | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RNU4ATAC n.8C>T | LP | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TNFRSF13B c.310T>C | P | 1 | 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LZTFL1 rs17713054 | RL | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 14 | ||||||||||||||||||||||||||||||||||||||||||||||
| LZTFL1 rs35482426 | RL | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 15 | |||||||||||||||||||||||||||||||||||||||||||||
| SUM | 4 | 1 | 2 | 1 | 3 | 2 | 2 | 3 | 2 | 1 | 1 | 1 | 1 | 2 | 1 | 0 | 1 | 2 | 1 | 1 | 1 | 2 | 1 | 2 | 0 | 1 | 0 | 4 | 3 | 2 | 4 | 2 | 1 | 1 | 4 | 2 | 0 | 1 | 2 | 1 | 2 | 0 | 1 | 4 | 1 | 0 | 1 | 3 | 2 | 2 | 1 | 2 | 1 | 1 | 0 | 4 | 1 | 3 | 1 | 0 | 96 | |
Deceased patients are underlined. Variants, for which the genotype was consistent with the proposed inheritance model for the gene, are indicated in bold. LP = likely pathogenic, P = pathogenic, R = risk factor, RL = risk locus, hom = homozygous, het = heterozygous
Table 3.
Demographic and clinical data of included hospitalized patients.
| TOTAL | |
|---|---|
| Subjects n or n (%) | 60 |
| Age mean±sd years | 77 ± 9.9 |
| Male sex n (%) | 31 (52) |
| Previous coexisting disease n (%) | |
| Type 2 diabetes | 17 (28) |
| Heart disease* | 28 (47)) |
| Hypertension | 38 (63 |
| Chronic lung disease | 16 (27) |
| Rheumatic diseases | 5 (8) |
| Cancer | 10 (17) |
| Chronic kidney disease | 7 (12) |
| Number of coexisting diseases n (%) | |
| None | 4 (7) |
| One | 18 (30) |
| Two or more | 38 (63) |
| Died (%) | 7 (12) |
*Excluding hypertension.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



