
Submitted:
05 June 2024
Posted:
06 June 2024
You are already at the latest version
Abstract
Background: The secretion of alarmin cytokines by epithelial cells, including thymic stromal lymphopoietin (TSLP), interleukin (IL)-25, and IL-33, heralds the onset of the inflammatory cascade and immune effector infiltration in asthma. However, alarmin cytokine expression in the upper airways in asthma remains largely unknown.
Methods: We recruited 40 participants with asthma who were categorized into four severity groups as per the Global Initiative for Asthma (GINA) classifications (10 in each group of GINA-1/2, -3, -4, and -5). Cells were derived from buccal, nasal and throat brushings, and intracellular alarmin cytokine expression (TSLP, IL-25, and IL-33) was assessed in cytokeratin 8+ (Ck8+) epithelial cells immediately after collection using flow cytometry with fluorescence minus one (FMO) controls. We assessed differences in alarmin cytokine expressions across asthma severity using quantile regression adjusted for age and sex.
Results: Of all patients, 24 (60%) were females with a mean (standard deviation [SD]) age of 41 (16) years. TSLP levels in Ck8+ epithelial cells in nasal samples of GINA-5 patients were significantly (p=0.03) higher than other GINA groups after adjusting for age and sex but did not differ between patients with and without nasal comorbidities. However, we did not find any significant changes in TSLP levels in Ck8+ epithelial cells in buccal and throat samples across GINA groups. IL-25 or IL-33 (obtained from nasal, buccal, and throat epithelial cell samples) were not significantly different across GINA groups.
Conclusions: Our study demonstrates for the first time that Ck8+ nasal epithelial cells from GINA-5 asthmatics express elevated levels of TSLP.
Keywords:
Nasal brushings
; TSLP
; IL-25
; IL-33
; cytokeratin-8
; epithelial cells
1. Introduction
Asthma affects greater than 300 million people globally, up to 10% of which is considered to fall under the diagnostic criteria for severe asthma [1,2]. Approximately half of severe asthma is poorly controlled, with significant health burden and impact on quality of life despite adherence to standard inhaled therapies including inhaled corticosteroids (ICS) and long-acting β2 agonists (LABA) [3]. The majority of these patients have evidence of type 2 (T2) inflammation, and are candidates for biologic therapies directed against T2 inflammatory mediators (e.g., immunoglobulin E (IgE), interleukin (IL-4)/IL-13, IL-5, and thymic stromal lymphopoietin, (TSLP)); however, it remains a challenge to determine which therapeutic is likely to work for the individual patient [4,5].
Epithelial cell-derived alarmin cytokines, TSLP, IL-25, and IL-33, have recently emerged as playing important roles in activation of inflammatory responses in the airways. These cytokines are released by airway epithelial cells in response to numerous biological and environmental triggers and initiate a cascade of downstream immune responses leading to airway inflammation in asthma patients [6,7]. TSLP is released in response to airborne triggers including allergens, pollutants, cigarette smoke, and viral infection [6,7,8,9,10,11,12]. TSLP plays a role in the activation of a range of immune cells including dendritic cells, T-helper 2 cells, mast cells, eosinophils, and ILC2s [13,14,15,16,17,18]. IL-25 release is induced by exposure to common protease allergens including house dust mite, leading to the induction of allergic inflammation through direct activation of eosinophils and T-helper 2 cells resulting in increased release of IL-4, IL-5 and IL-13 and airway eosinophilia [6,19,20,21]. IL-33 is expressed in a wide variety of tissue cells, including epithelial and endothelial cells, and is released into the airways resulting from cellular damage or following exposure to inhaled allergens; IL-33 synergistically promotes activation and recruitment of type-2 innate lymphoid cells (ILC2) along with TSLP [22,23].
Molecular signatures in buccal, pharyngeal, and nasal samples have been explored as potential surrogates for asthma previously, with varying degrees of success. Previously, buccal brushings have been shown to have detectable changes in both epigenetics and gene expression when comparing subjects with or without asthma [24,25,26]. To our knowledge, pharyngeal epithelial cells have not been directly studied in the context of asthma, although a recent study has identified the pharyngeal microbiota as a potential biomarker for asthma exacerbations despite continued ICS therapy [27]. The nasal epithelium has been more robustly studied in the context of asthma, with nasal microbiome, nasal lavage, and nasal brushings all showing potential as asthma biomarkers [28,29,30,31]. It has previously been suggested that nasal epithelial cells isolated from asthmatic patients and cultured ex vivo express greater levels of TSLP following rhinovirus infection as compared to cells from healthy individuals [12]. However, despite these recent advances, studies measuring in situ expression of alarmin cytokines in nasal, buccal, and pharyngeal epithelia in asthma are lacking. We sought to assess whether the upper airway epithelium of patients with asthma may be an alternative source for the measurement of intracellular alarmin cytokines (TSLP, IL-25, and IL-33) in asthma and whether their expression is altered in association with asthma severity.
2. Materials and Methods
2.1. Study Design and Participants
In this cross-sectional study, we recruited 40 adult (> 18 years) participants with asthma who were naïve to biologic therapy. Asthma was diagnosed and categorized by severity according to the Global Initiatives for Asthma, with 10 participants from each GINA category (GINA-1 and -2 were merged due to overlapping medication levels) [1]. Demographic information, medication use, comorbidities, and emergency department visits in the past 12 months were collected. None of the patients had experienced exacerbations within the previous 7 days before sampling. All patients were on stable maintenance therapy for ≥3 months before sample collection. The study was approved by the Health Research Ethics Board of the University of Alberta (Pro00106537), and all participants provided written informed consent.
2.2. Sample Collection and Processing
Participants in the study were requested to rinse their sinuses using a prewarmed bottle of NeilMed Sinuflow ReadyRinse® solution (400mL, NeilMed® Pharmaceuticals, Santa Rosa, CA, USA), and to rinse out their mouths and throats with 0.9% saline solution (50mL) prior to sample collection. Samples were then collected from the buccal, nasal, and pharyngeal cavities of participants using a cytology brush (Pap-Pak® Cytosoft™ Medical Packaging Corporation, USA). Cytology brushes were immediately inserted into 15 mL conical tubes containing 10 mL complete bronchial epithelial growth media (BEGM™ Lonza, Switzerland). To detach cells, tubes were briefly vortexed on a lower speed before centrifugation at 300 g for 10 min at 4°C. Cytobrushes were removed, and cells were once again pelleted by centrifugation to account for any dislodging of cells during cytobrush removal (300 g for 10 min at 4°C). Supernatants were aspirated and the pellets were resuspended in 1 mL FACS buffer (0.5% BSA and 0.05% sodium azide in 1X PBS) and a 10 μL aliquot was taken for counting using Trypan Blue and an automated cell counter, Countessa 3 (Invitrogen, Waltham, MA).
2.3. Flow Cytometry
Cells were briefly washed in FACS buffer prior to staining to remove excess debris and mucus. An Aqua Dead Cell Stain Kit (Life Technologies, Burlington, ON, Canada) was used for dead cell discrimination prior to fixation and permeabilization of cells. Live/dead staining was performed for 25 min at 4°C. Cells were then fixed and permeabilized using BD Cytofix/Cytoperm™ fixation/permeabilization kit (BD Biosciences, Mississauga, ON, Canada), according to manufacturer’s instructions. After blocking, cells were labeled with antibodies to Ck8 to identify epithelial cells and alarmin cytokines to quantify their expression levels according to GINA classifications. Primary antibodies were used as follows: anti-TSLP antibody (rabbit polyclonal antibody, ABT330, Millipore Sigma Mississauga, ON, Canada), Ck8 Alexa Fluor 647 (EP1628Y, Abcam, Waltham, MA), anti-IL-25 antibody (182203, R&D Systems, Minneapolis, MN) and IL-33 conjugated to phycoerythrin (PE, 40015C, R&D Systems). To detect TSLP labelling, cells were incubated with Alexa Fluor 488 F(ab’)2 fragment of goat anti-rabbit IgG (H+L) (A11070, Life Technologies Corporation, Eugene, OR). IL-25 labelling was detected with PE-Cy7 rat anti-mouse IgG (M1-14D12, eBioscience, ThermoFisher Scientific, Mississauga, ON, Canada). Primary and secondary stains were each incubated for 30 min at 4°C. Isotype control antibodies were used as follows: Alexa Fluor 647 mouse IgG1 κ Isotype Control (MA5-18167, BD Biosciences), mouse IgG1 FITC-conjugated negative control antibody (400109, Bio-Rad, Mississauga, ON, Canada), and PE-conjugated goat IgG isotype control (IC108P, R&D Systems). Fluorescence Minus One (FMO) controls were also used to identify and gate positive populations. Immunolabelled cells were acquired on a LSR Fortessa-SORP (BD Biosciences) and analyzed using FlowJo software (version 10, Ashland, OR).
2.4. Statistical Analysis
Descriptive statistics are presented as mean (standard deviation [SD]), median (interquartile range [IQR]), and frequency (%) for continuous, count, and categorical variables, respectively. We used multivariable quantile regression to assess the differences of each cytokine across the GINA subgroups. We tested age, sex, smoking status, and nasal comorbidities as potential confounders; however, we retained only age and sex in the models based on Akaike’s information criterion [32]. We performed exploratory stratification analyses to test the differences in cytokine expression among patients with and without nasal comorbidities (allergic rhinitis and chronic rhinosinusitis with/without nasal polyps). All analyses were performed in a complete case approach using STATA 18 (StataCorp, College Station, TX, USA) and a p-value < 0.05 was considered statistically significant.
3. Results
Of all participants, 63% were female, the mean (SD) age was 41 (16) years, and 85% were non-smokers (Table 1). 36 (90%) were on ICS (as daily maintenance therapy or as needed with a reliever) and 13 (33%) had one or more nasal comorbidities. Six patients without and six with nasal comorbidities were receiving treatment with intranasal corticosteroids.
We processed buccal, pharyngeal, and nasal samples immediately (within 1 h) after their collection from participants and subjected these to flow cytometry analysis without further manipulation. After gating for live cells, singlets, and Ck8 as the epithelial cell marker, we found detectable levels of alarmin cytokines in all samples obtained from patients with GINA 1/2-5 (Figure 1).
The median (IQR) MFIs of TSLP, IL-25, and IL-33 were 6,590 (3,829 – 19,032), 3,936 (3,104 – 8,061), and 923 (548 – 2,010), respectively, for all Ck8+ cells in nasal samples across GINA classifications. In buccal samples, the median (IQR) MFIs of TSLP, IL-25, and IL-33 were 7,766 (3,578 – 15,120), 14,635 (3,702 – 23,095), and 1,065 (438 – 3566), respectively, and in throat samples, the MFIs were 6,615 (3281 – 13,712), 8,109 (4,251 – 13,795), and 1,115 (649 – 3113), respectively (Table 1).
In nasal brushings, TSLP expression in Ck8+ epithelial cells was similar between GINA-1-4 classifications but was significantly elevated in patients with GINA-5 (p = 0.03 after adjusting for age and sex, Figure 2A). In the exploratory stratification analysis, we found a three-fold increase of Ck8+ epithelial cell TSLP expression in nasal brushings from participants with nasal comorbidities (median MFI: 15,774) compared with those lacking nasal comorbidities (median MFI: 5,759), although this was not statistically significant (Figure 2B). The expression of IL-25 and IL-33 was also elevated in Ck8+ epithelial cells from participants with nasal comorbidities; however, none of the measurements differed significantly in terms of the threshold for statistical significance (Figure 2B).
4. Discussion
In this study, we implemented a novel, non-invasive, and rapid method of measuring alarmin cytokine expression in the upper airways of asthma patients. This flow cytometry-based technique enabled clear detection of TSLP in all samples, and we found a significant elevation of TSLP in nasal Ck8+ epithelial cells from patients classified with GINA-5 asthma after adjusting for age and sex. In contrast to nasal brushings, we observed no significant changes in TSLP expression in buccal or pharyngeal epithelia of GINA-5 patients. To our knowledge, this is the first study directly comparing the nasal, buccal, and pharyngeal epithelia of different severities of asthma, with previous work focusing on broader comparisons between healthy and asthmatic subjects [24,25,26,27]. Furthermore, no significant changes were observed in IL-25 and IL-33 levels across all GINA categories, which agrees with a recent study in TSLP and IL-33 serum levels in mild asthmatics, showing a slight reduction in sera in mild asthma compared to healthy controls [33]. Increased TSLP expression in nasal epithelial cells among those with nasal comorbidities, although not significant, is consistent with previous studies in allergic rhinitis and chronic rhinosinusitis [34,35].
Our finding of high upper airway TSLP in GINA-5 patients indicates that this method could potentially be used to identify severe cases in GINA 1-4 patients and allow for immediate treatment escalation. A key feature of severe asthma is persistent, corticosteroid-insensitive airway inflammation. Corticosteroid resistance is thought to be mediated by abundant type 2 cytokine production by group 2 innate lymphoid cells (ILC2) [36], and is dependent on TSLP [37]. Currently, asthma severity is determined post-hoc based on the intensity of treatment required, which can delay necessary therapy in severe disease. The application of upper airway TSLP measurement could therefore provide guidance on appropriate therapy in severe asthma.
The upstream position of alarmin cytokines as orchestrators of allergic airway inflammation has been harnessed as a strategy for asthma therapy [38,39,40]. The anti-TSLP monoclonal antibody, tezepelumab, has been shown to improve exacerbation rates, lung function, symptom control, and quality of life in patients with severe asthma [41,42]. However, severe asthma phenotypes are highly heterogeneous due to inherent differences in etiopathological mechanisms, and thus, it may be valuable to measure the expression of inflammatory mediators prospectively in severe asthma patients to determine which patients are most likely to benefit from this drug. While bronchial brushings and induced sputum may be used for measuring cytokine levels, harvesting cells through these methods is technically challenging, invasive, and not always feasible, particularly in patients with reduced lung function or scant sputum production [5]. Therefore, the upper airways may be an attractive target for study as potential surrogates of the lung.
While this is the first report demonstrating increased upper airway expression of TSLP in severe asthmatics, there are important study limitations. The study was cross-sectional in design and could not provide insight into the effects of asthma exacerbations or treatment on the expression of cytokines. The number of patients within each GINA severity group is small, and GINA severity groups are determined by prescribing physicians, which may not fully reflect disease severity. Analysis of alarmin expression within clinically relevant subgroups such as GINA-5 patients with and without nasal polyposis was thus not possible due to small sample size. For the buccal and pharyngeal brushings, we did not include a medication washout period prior to sample collection. The issue of mouth/throat deposition of inhaled medications is well documented, deposition of ICS may have suppressed the endogenous alterations in alarmin expression in these samples [43,44,45,46]. Although nasal epithelial TSLP may be effective at identifying patients with severe or steroid refractory asthma, it may be a poor biomarker in mild to moderate disease. Compliance with treatment was determined by patient reports, so the severity of disease in noncompliant patients may have been misidentified. We speculate that nasal sampling could be used to identify patients with more severe disease; however, this requires a future prospective study rather than the current cross-sectional study. Additionally, we could not assess TSLP in patients before and after initiation of inhaled steroids. Larger samples sizes are required to fully understand the interplay of these diseases on the expression of alarmin cytokines in the upper airway epithelium.
5. Conclusions
In conclusion, upper airway TSLP expression can be measured using a novel flow cytometric assay using nasal swab samples and is greatly increased in severe asthmatics. The utility of upper airway TSLP as a biomarker of severe asthma and treatment response warrants further study.
Funding
This study was supported by an Externally Sponsored Research Grant from AstraZeneca Ltd. (ESR-20-20575). However, the funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Acknowledgments
This study was supported by an Externally Sponsored Research Grant from AstraZeneca Ltd. (ESR-20-20575). However, the funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflicts of Interest
Irvin Mayers received grants from AstraZeneca Canada and personal fees from AstraZeneca Canada and Sanofi outside this work. AA reports consulting fees, travel support and/or honoraria from BioCryst, Covis Pharma, CSL-Behring, GSK, and Takeda and clinical trial support from AstraZeneca Canada, Astria, BioCryst, Ionis Pharmaceuticals, Kalvista, Octapharma, Pharvaris, and Takeda, outside of the submitted work. Paige Lacy reports grants from AstraZeneca Canada related to this work, grants from Natural Science and Engineering Research Council of Canada, and Synergy Respiratory and Cardiac Care, and personal fees from AstraZeneca Canada, GlaxoSmithKline Canada, and Synergy Respiratory and Cardiac Care, Canada, outside this work. All other authors declare no other competing interests.
References
- GINA Report, Global Strategy for Asthma Management and Prevention 2023 [Available from: https://ginasthma.org/2023-gina-main-report/.
- Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, Adcock IM, Bateman ED, Bel EH, Bleecker ER, Boulet LP, Brightling C, Chanez P, Dahlen SE, Djukanovic R, Frey U, Gaga M, Gibson P, Hamid Q, Jajour NN, Mauad T, Sorkness RL, Teague WG. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. European Respiratory Journal. 2014;43(2):343-73.
- Hekking PW, Wener RR, Amelink M, Zwinderman AH, Bouvy ML, Bel EH. The prevalence of severe refractory asthma. J Allergy Clin Immunol. 2015;135(4):896-902. [CrossRef]
- Jackson DJ, Busby J, Pfeffer PE, Menzies-Gow A, Brown T, Gore R, Doherty M, Mansur AH, Message S, Niven R, Patel M, Heaney LG, Registry UKSA. Characterisation of patients with severe asthma in the UK Severe Asthma Registry in the biologic era. Thorax. 2021;76(3):220-7. [CrossRef]
- Adatia A, Vliagoftis H. Challenges in severe asthma: Do we need new drugs or new biomarkers? Front Med (Lausanne). 2022;9:921967.
- Hong H, Liao S, Chen F, Yang Q, Wang DY. Role of IL-25, IL-33, and TSLP in triggering united airway diseases toward type 2 inflammation. Allergy. 2020;75(11):2794-804. [CrossRef]
- Byers DE. Defining the roles of IL-33, thymic stromal lymphopoietin, and IL-25 in human asthma. Am J Respir Crit Care Med. 2014;190(7):715-6.
- Wang W, Li Y, Lv Z, Chen Y, Li Y, Huang KW, Corrigan CJ, Ying S. Bronchial Allergen Challenge of Patients with Atopic Asthma Triggers an Alarmin (IL-33, TSLP, and IL-25) Response in the Airways Epithelium and Submucosa. Journal of Immunology. 2018;201(8):2221-31. [CrossRef]
- Bleck B, Tse DB, Curotto de Lafaille MA, Zhang F, Reibman J. Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturation and polarization via thymic stromal lymphopoietin. J Clin Immunol. 2008;28(2):147-56. [CrossRef]
- Nakamura Y, Miyata M, Ohba T, Ando T, Hatsushika K, Suenaga F, Shimokawa N, Ohnuma Y, Katoh R, Ogawa H, Nakao A. Cigarette smoke extract induces thymic stromal lymphopoietin expression, leading to T. [CrossRef]
- 2-type immune responses and airway inflammation. J Allergy Clin Immun. 2008;122(6):1208-14.
- Calven J, Yudina Y, Hallgren O, Westergren-Thorsson G, Davies DE, Brandelius A, Uller L. Viral stimuli trigger exaggerated thymic stromal lymphopoietin expression by chronic obstructive pulmonary disease epithelium: role of endosomal TLR3 and cytosolic RIG-I-like helicases. J Innate Immun. 2012;4(1):86-99. [CrossRef]
- Reese A, Favoreto S, Quraishi J, Biyasheva A, Shen J, Greiman A, Avila P. Higher Rhinovirus-Induced Production of TSLP in Nasal Epithelial Cells from Asthmatic than Healthy Subjects. J Allergy Clin Immun. 2011;127(2):Ab22-Ab. [CrossRef]
- Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, Ho S, Antonenko S, Lauerma A, Smith K, Gorman D, Zurawski S, Abrams J, Menon S, McClanahan T, de Waal-Malefyt Rd R, Bazan F, Kastelein RA, Liu YJ. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3(7):673-80. [CrossRef]
- Kitajima M, Lee HC, Nakayama T, Ziegler SF. TSLP enhances the function of helper type 2 cells. European Journal of Immunology. 2011;41(7):1862-71. [CrossRef]
- Lai JF, Thompson LJ, Ziegler SF. TSLP drives acute T(H)2-cell differentiation in lungs. J Allergy Clin Immunol. 2020;146(6):1406-18 e7.
- Han NR, Oh HA, Nam SY, Moon PD, Kim DW, Kim HM, Jeong HJ. TSLP Induces Mast Cell Development and Aggravates Allergic Reactions through the Activation of MDM2 and STAT6. J Invest Dermatol. 2014;134(10):2521-30. [CrossRef]
- Wong CK, Hu SQ, Cheung PFY, Lam CWK. Thymic Stromal Lymphopoietin Induces Chemotactic and Prosurvival Effects in Eosinophils Implications in Allergic Inflammation. Am J Resp Cell Mol. 2010;43(3):305-15.
- Camelo A, Rosignoli G, Ohne Y, Stewart RA, Overed-Sayer C, Sleeman MA, May RD. IL-33, IL-25, and TSLP induce a distinct phenotypic and activation profile in human type 2 innate lymphoid cells. Blood Advances. 2017;1(10):577-89. [CrossRef]
- Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, Menon S, Clifford T, Hunte B, Lesley R, Muchamuel T, Hurst SD, Zurawski G, Leach MW, Gorman DM, Rennick DM. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15(6):985-95. [CrossRef]
- Cheung PF, Wong CK, Ip WK, Lam CW. IL-25 regulates the expression of adhesion molecules on eosinophils: mechanism of eosinophilia in allergic inflammation. Allergy. 2006;61(7):878-85. [CrossRef]
- Kouzaki H, Tojima I, Kita H, Shimizu T. Transcription of interleukin-25 and extracellular release of the protein is regulated by allergen proteases in airway epithelial cells. Am J Respir Cell Mol Biol. 2013;49(5):741-50. [CrossRef]
- Kouzaki H, Iijima K, Kobayashi T, O'Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011;186(7):4375-87. [CrossRef]
- Toki S, Goleniewska K, Zhang J, Zhou W, Newcomb DC, Zhou B, Kita H, Boyd KL, Peebles RS, Jr. TSLP and IL-33 reciprocally promote each other's lung protein expression and ILC2 receptor expression to enhance innate type-2 airway inflammation. Allergy. 2020;75(7):1606-17.
- Vyhlidal CA, Riffel AK, Dai H, Rosenwasser LJ, Jones BL. Detecting gene expression in buccal mucosa in subjects with asthma versus subjects without asthma. Pediatr Allergy Immunol. 2013;24(2):138-43. [CrossRef]
- Torrone D, Kuriakose J, Moors K, Jiang H, Niedzwiecki M, Perera F, Miller R. Reproducibility and intraindividual variation over days in buccal cell DNA methylation of two asthma genes, interferon gamma (IFNgamma) and inducible nitric oxide synthase (iNOS). Clin Epigenetics. 2012;4(1):3.
- Murphy TM, Wong CC, Arseneault L, Burrage J, Macdonald R, Hannon E, Fisher HL, Ambler A, Moffitt TE, Caspi A, Mill J. Methylomic markers of persistent childhood asthma: a longitudinal study of asthma-discordant monozygotic twins. Clin Epigenetics. 2015;7:130. [CrossRef]
- Perez-Garcia J, Gonzalez-Carracedo M, Espuela-Ortiz A, Hernandez-Perez JM, Gonzalez-Perez R, Sardon-Prado O, Martin-Gonzalez E, Mederos-Luis E, Poza-Guedes P, Corcuera-Elosegui P, Callero A, Sanchez-Machin I, Korta-Murua J, Perez-Perez JA, Villar J, Pino-Yanes M, Lorenzo-Diaz F. The upper-airway microbiome as a biomarker of asthma exacerbations despite inhaled corticosteroid treatment. J Allergy Clin Immunol. 2023;151(3):706-15. [CrossRef]
- Chen M, Ge Y, Zhang W, Wu P, Cao C. Nasal Lavage Fluid Proteomics Reveals Potential Biomarkers of Asthma Associated with Disease Control. J Asthma Allergy. 2024;17:449-62. [CrossRef]
- Thavagnanam S, Parker JC, McBrien ME, Skibinski G, Shields MD, Heaney LG. Nasal epithelial cells can act as a physiological surrogate for paediatric asthma studies. PLoS One. 2014;9(1):e85802. [CrossRef]
- Hansel TT, Tunstall T, Trujillo-Torralbo MB, Shamji B, Del-Rosario A, Dhariwal J, Kirk PDW, Stumpf MPH, Koopmann J, Telcian A, Aniscenko J, Gogsadze L, Bakhsoliani E, Stanciu L, Bartlett N, Edwards M, Walton R, Mallia P, Hunt TM, Hunt TL, Hunt DG, Westwick J, Edwards M, Kon OM, Jackson DJ, Johnston SL. A Comprehensive Evaluation of Nasal and Bronchial Cytokines and Chemokines Following Experimental Rhinovirus Infection in Allergic Asthma: Increased Interferons (IFN-gamma and IFN-lambda) and Type 2 Inflammation (IL-5 and IL-13). EBioMedicine. 2017;19:128-38.
- de Farias CF, Amorim MM, Dracoulakis M, Caetano LB, Santoro IL, Fernandes AL. Nasal lavage, blood or sputum: Which is best for phenotyping asthma? Respirology. 2017;22(4):671-7.
- Akaike H. New Look at Statistical-Model Identification. Ieee Transactions on Automatic Control. 1974;Ac19(6):716-23.
- Polomska J, Sikorska-Szaflik H, Drabik-Chamerska A, Sozanska B, Debinska A. Exploring TSLP and IL-33 Serum Levels and Genetic Variants: Unveiling Their Limited Potential as Biomarkers for Mild Asthma in Children. J Clin Med. 2024;13(9). [CrossRef]
- Yu HW, Wang WW, Jing Q, Pan YL. TSLP Induces Epithelial–Mesenchymal Transition in Nasal Epithelial Cells From Allergic Rhinitis Patients Through TGF-β1/Smad2/3 Signaling. American Journal of Rhinology & Allergy. 2023;37(6):739-50.
- Kimura S, Pawankar R, Mori S, Nonaka M, Masuno S, Yagi T, Okubo K. Increased Expression and Role of Thymic Stromal Lymphopoietin in Nasal Polyposis. Allergy, Asthma and Immunology Research. 2011;3(3). [CrossRef]
- Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O'Byrne PM, Gauvreau GM, Boulet LP, Lemiere C, Martin J, Nair P, Sehmi R. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. 2016;137(1):75-86 e8. [CrossRef]
- Liu S, Verma M, Michalec L, Liu W, Sripada A, Rollins D, Good J, Ito Y, Chu H, Gorska MM, Martin RJ, Alam R. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: The role of thymic stromal lymphopoietin. J Allergy Clin Immunol. 2018;141(1):257-68.e6. [CrossRef]
- Comeau MR, Ziegler SF. The influence of TSLP on the allergic response. Mucosal Immunol. 2010;3(2):138-47. [CrossRef]
- Theoharides TC, Petra AI, Taracanova A, Panagiotidou S, Conti P. Targeting IL-33 in autoimmunity and inflammation. J Pharmacol Exp Ther. 2015;354(1):24-31. [CrossRef]
- Knolle MD, Rana BM, McKenzie AN. IL-25 as a potential therapeutic target in allergic asthma. Immunotherapy. 2015;7(6):607-10. [CrossRef]
- Menzies-Gow A, Colice G, Griffiths JM, Almqvist G, Ponnarambil S, Kaur P, Ruberto G, Bowen K, Hellqvist A, Mo M, Garcia Gil E. NAVIGATOR: a phase 3 multicentre, randomized, double-blind, placebo-controlled, parallel-group trial to evaluate the efficacy and safety of tezepelumab in adults and adolescents with severe, uncontrolled asthma. Respir Res. 2020;21(1):266. [CrossRef]
- Corren J, Garcia Gil E, Griffiths JM, Parnes JR, van der Merwe R, Sałapa K, O'Quinn S. Tezepelumab improves patient-reported outcomes in patients with severe, uncontrolled asthma in PATHWAY. Annals of Allergy, Asthma & Immunology. 2021;126(2):187-93.
- Corcoran TE. Measurements of deposited aerosol dose in infants and small children. Ann Transl Med. 2021;9(7):595. [CrossRef]
- Newman SP, Chan HK. In vitro-in vivo correlations (IVIVCs) of deposition for drugs given by oral inhalation. Adv Drug Deliv Rev. 2020;167:135-47. [CrossRef]
- Clark AR, Newman SP, Dasovich N. Mouth and oropharyngeal deposition of pharmaceutical aerosols. J Aerosol Med. 1998;11 Suppl 1:S116-21. [CrossRef]
- Svartengren K, Lindestad PA, Svartengren M, Bylin G, Philipson K, Camner P. Deposition of inhaled particles in the mouth and throat of asthmatic subjects. Eur Respir J. 1994;7(8):1467-73. [CrossRef]
Figure 1.
Flow cytometric analysis of alarmin cytokines in upper airway brushings. (A) Sequential gating strategy. Top row: representative nasal brushing sample, single cell selection, live/dead cell gating, and gating for epithelial cells based on Ck8 labelling. Bottom row: Individual alarmin cytokine labelling of epithelial cells. (B) Histograms showing isotype controls and test antibodies for Ck8, TSLP, IL-25 and IL-33 on appropriately gated epithelial cells.
Figure 1.
Flow cytometric analysis of alarmin cytokines in upper airway brushings. (A) Sequential gating strategy. Top row: representative nasal brushing sample, single cell selection, live/dead cell gating, and gating for epithelial cells based on Ck8 labelling. Bottom row: Individual alarmin cytokine labelling of epithelial cells. (B) Histograms showing isotype controls and test antibodies for Ck8, TSLP, IL-25 and IL-33 on appropriately gated epithelial cells.
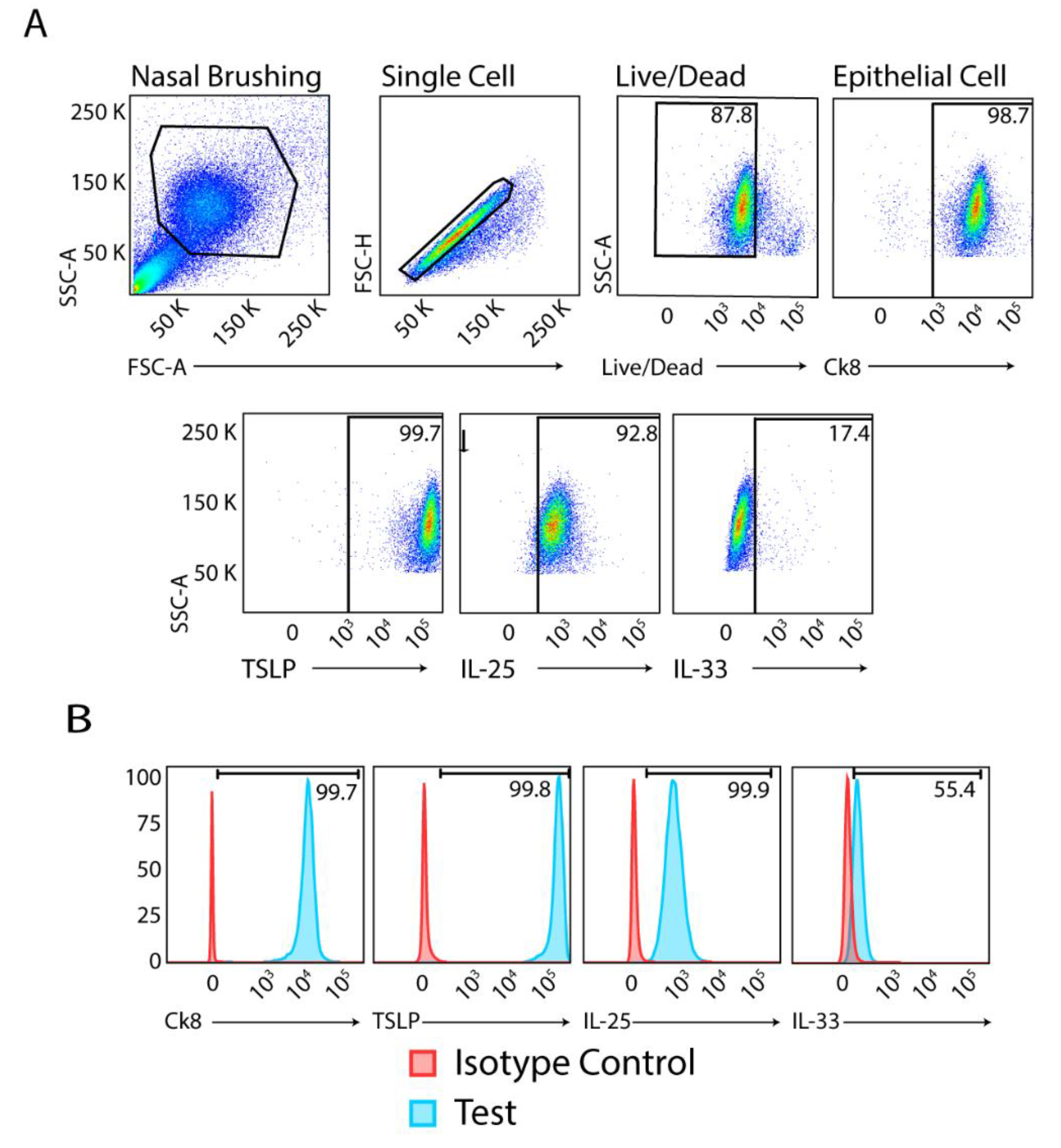
Figure 2.
Expression of alarmin cytokines in nasal Ck8+ epithelial samples in patients with varying GINA classifications. (A) Expression profile of alarmin cytokines (TSLP, IL-25, and IL-33) in Ck8+ nasal epithelial samples from asthma patients according to GINA subgroups. Data are shown as median (IQR) (box and whisker) and 95% confidence intervals (error bars) unless otherwise stated, and p-values were obtained from quantile regression models adjusted for age and sex. (B) Stratification analysis of alarmin cytokine expression among all asthma patients stratified by nasal comorbidities. P-values were obtained from quantile regression adjusted for age and sex.
Figure 2.
Expression of alarmin cytokines in nasal Ck8+ epithelial samples in patients with varying GINA classifications. (A) Expression profile of alarmin cytokines (TSLP, IL-25, and IL-33) in Ck8+ nasal epithelial samples from asthma patients according to GINA subgroups. Data are shown as median (IQR) (box and whisker) and 95% confidence intervals (error bars) unless otherwise stated, and p-values were obtained from quantile regression models adjusted for age and sex. (B) Stratification analysis of alarmin cytokine expression among all asthma patients stratified by nasal comorbidities. P-values were obtained from quantile regression adjusted for age and sex.
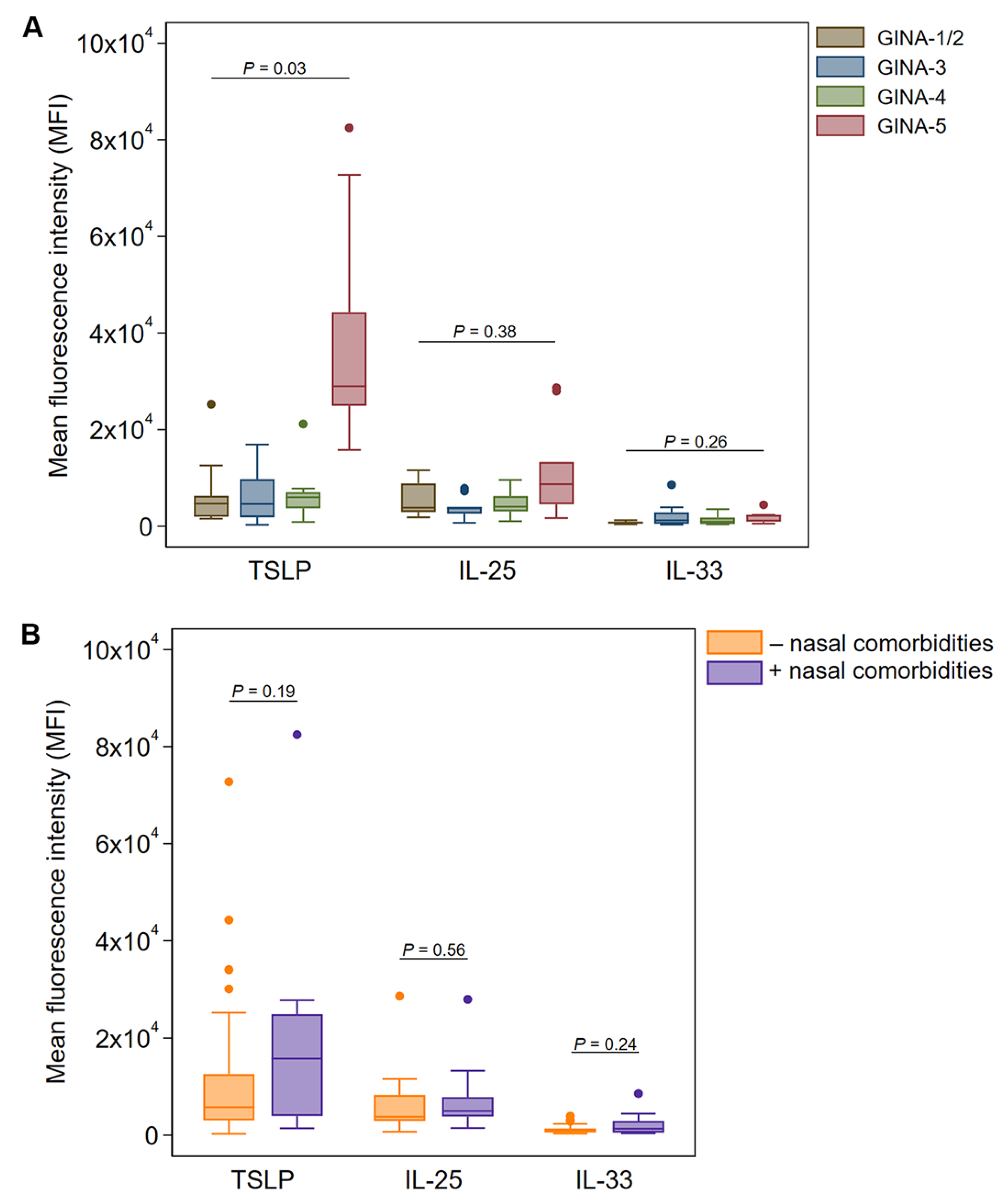
Figure 3.
Buccal expression of alarmin cytokines in Ck8+ epithelial samples of asthma patients according to GINA subgroups. Data are shown as median (IQR) (box and whisker) and 95% confidence intervals (error bars) unless otherwise stated, and p-values were obtained from quantile regression models adjusted for age and sex.
Figure 3.
Buccal expression of alarmin cytokines in Ck8+ epithelial samples of asthma patients according to GINA subgroups. Data are shown as median (IQR) (box and whisker) and 95% confidence intervals (error bars) unless otherwise stated, and p-values were obtained from quantile regression models adjusted for age and sex.
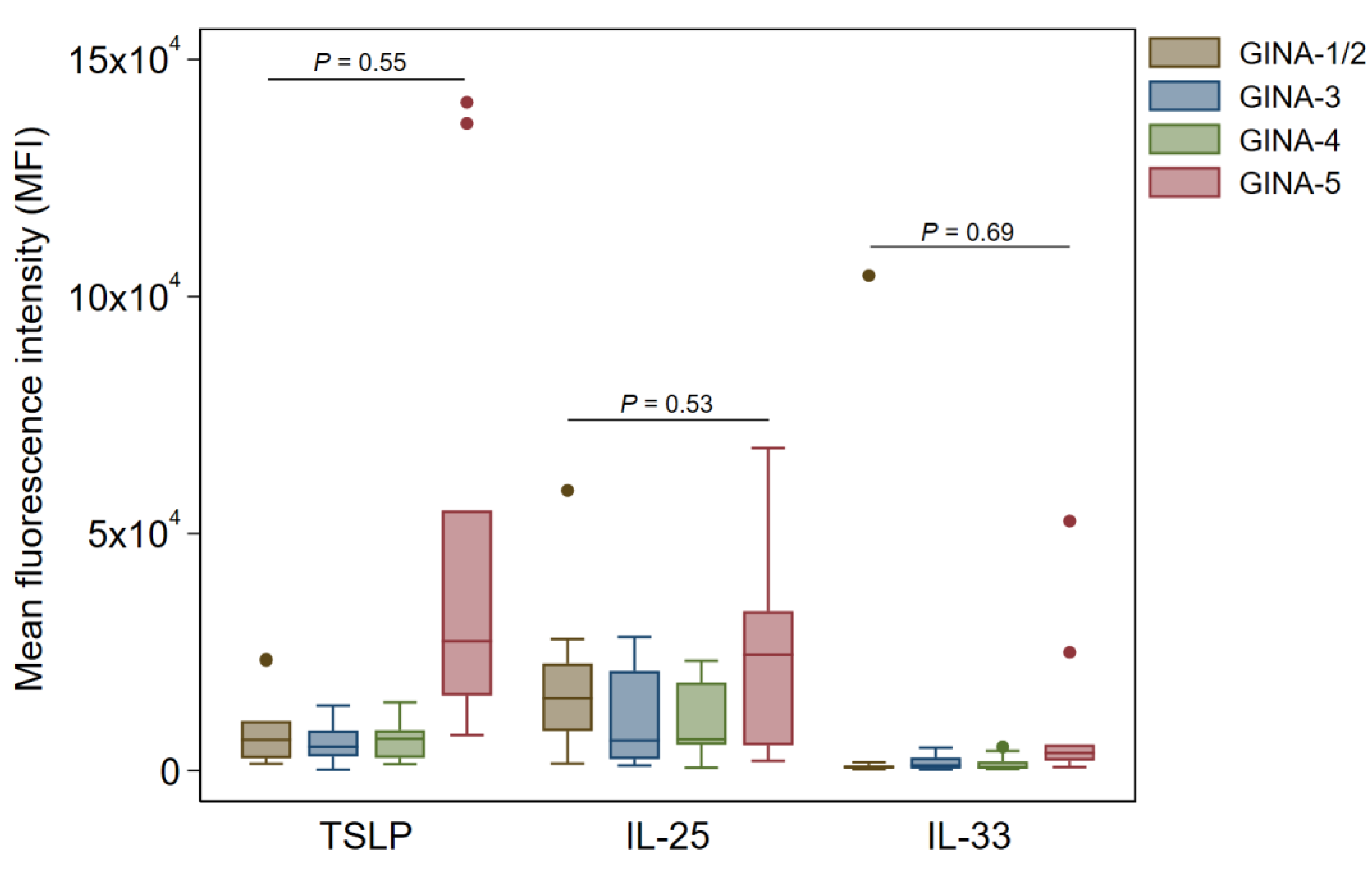
Figure 4.
Expression profile of alarmin cytokines in pharyngeal Ck8+ epithelial samples across GINA classifications. Data are shown as median (IQR) (box and whisker) and 95% confidence intervals (error bars), and p-values were obtained from quantile regression models adjusted for age and sex.
Figure 4.
Expression profile of alarmin cytokines in pharyngeal Ck8+ epithelial samples across GINA classifications. Data are shown as median (IQR) (box and whisker) and 95% confidence intervals (error bars), and p-values were obtained from quantile regression models adjusted for age and sex.
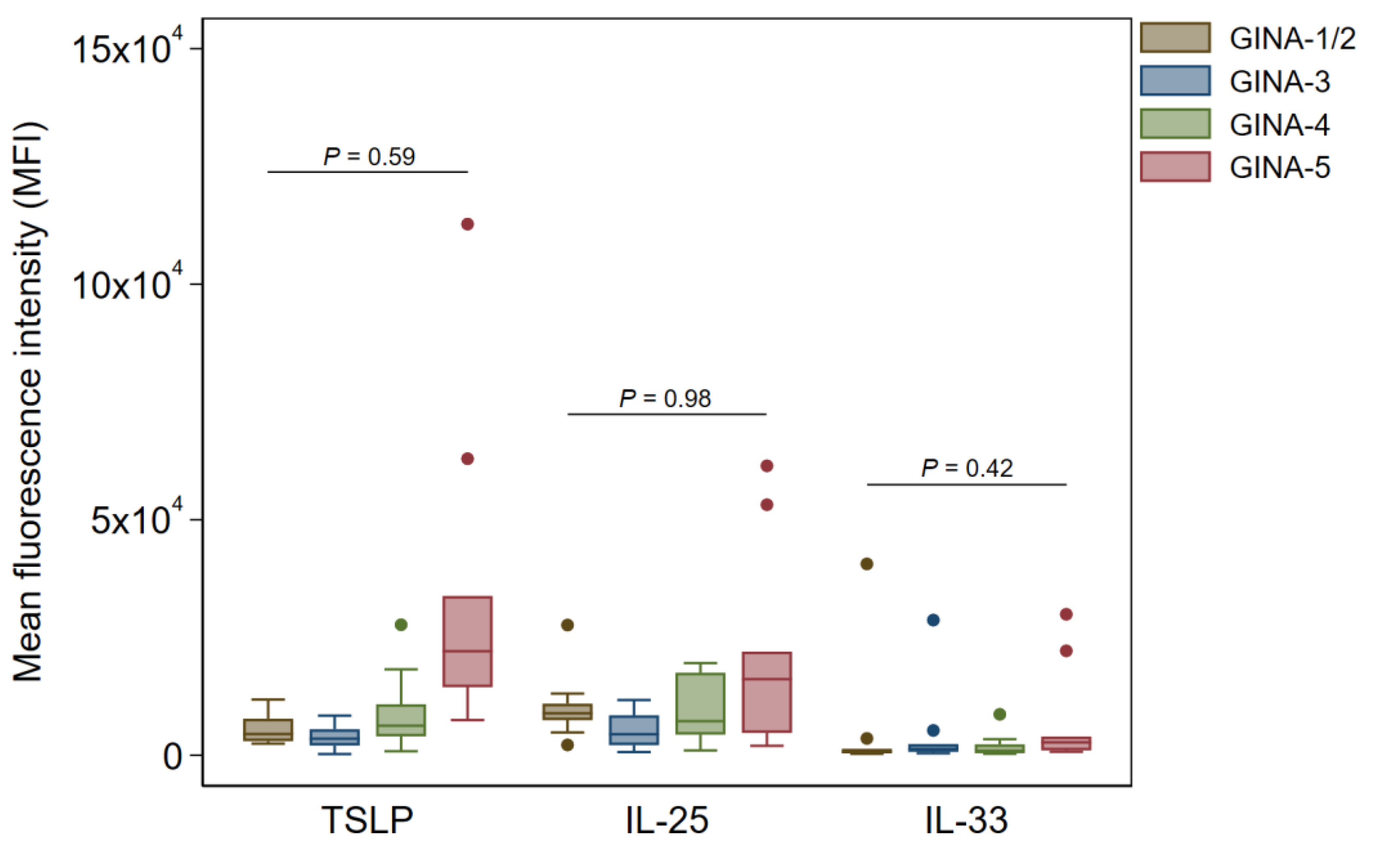

Table 1.
Participant Demographics and clinical characteristics.
| All Patients (n=40) | |
| Demographics | |
| Age, yr, mean (SD) | 40.85 (15.8) |
| Female, n (%) | 24 (60.0) |
| Smoking History, n (%) | |
| Never smoker | 35 (87.5) |
| Clinical Characteristics | |
| GINA classification, n (%) | |
| GINA-1/2 | 10 (25.0) |
| GINA-3 | 10 (25.0) |
| GINA-4 | 10 (25.0) |
| GINA-4 | 10 (25.0) |
| Nasal Polyps, n (%) | 4 (10.0) |
| Medications used, n (%) | |
| ICS/LABA maintenance | 31 (77.5) |
| SABA reliever | 17 (42.5) |
| Nasal steroid spray | 12 (30.0) |
| Anti-allergic* | 11 (27.5) |
| Oral corticosteroid maintenance | 1 (2.5) |
| At least one exacerbation, past 12 months, n (%) | 4 (10.0) |
| Laboratory Investigations | |
| Nasal samples | |
| TSLP (MFI), median (IQR) | 6,590 (3,829 – 19,032) |
| IL-25 (MFI), median (IQR) | 3,936 (3,104 – 8,061) |
| IL-33 (MFI), median (IQR) | 923 (548 – 2,010) |
| Buccal samples | |
| TSLP (MFI), median (IQR) | 7,766 (3,578 – 15,120) |
| IL-25 (MFI), median (IQR) | 14,635 (3,702 – 23,095) |
| IL-33 (MFI), median (IQR) | 1,065 (438 – 3566) |
| Throat samples | |
| TSLP (MFI), median (IQR) | 6,615 (3281 – 13,712) |
| IL-25 (MFI), median (IQR) | 8,109 (4,251 – 13,795) |
| IL-33 (MFI), median (IQR) | 1,115 (649 – 3113) |
* anti-allergic drugs include antihistamines and leukotriene modifiers.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



