
Submitted:
05 June 2024
Posted:
06 June 2024
You are already at the latest version
Abstract
Thanks to several Vitis vinifera backcrosses with an initial V. vinifera L. × V. rotundifolia (previously Muscadinia rotundifolia) interspecific cross, the MrRUN1/MrRPV1 locus (resistance to downy and powdery mildews) was introgressed in genotypes phenotypically close to V. vinifera varieties. To check the consequences of introgressing parts of the V. rotundifolia genome on gene expression during fruit development, we conducted a comparative RNA-seq study on single berries from different V. vinifera cultivars and V. vinifera × V. rotundifolia hybrids, including ‘G5’ and two derivative microvine lines, ‘MV102’ (resistant) and ‘MV32’ (susceptible) segregating for the MrRUN1/RPV1 locus. RNA-Seq profiles were analyzed on a comprehensive set of single berries from the end of the herbaceous plateau to the ripe stage. Pair-end reads were aligned both on V. vinifera PN40024.v4 reference genome, V. rotundifolia cv ‘Trayshed’ and cv ‘Carlos’, and to the few resistance genes from the original V. rotundifolia cv ‘52’ parent available at NCBI. Weighted Gene Co-expression Network Analysis (WGCNA) led to classifying the differentially expressed genes into 15 modules either preferentially correlated with resistance or berry phenology and composition. Resistance positively correlated transcripts predominantly mapped on the 4-5 Mb distal region of V. rotundifolia chromosome 12 beginning with the MrRUN1/MrRPV1 locus, while the negatively correlated ones mapped on the orthologous V. vinifera region, showing this large extremity of LG12 remained recalcitrant to internal recombination during the successive backcrosses. Some constitutively expressed V. rotundifolia genes were also observed at lower density outside this region. Genes overexpressed in developing berries from resistant accessions, either introgressed from V. rotundifolia, or triggered by these in the vinifera genome, spanned various functional groups, encompassing calcium signal transduction, hormone signaling, transcription factors, plant–pathogen-associated interactions, disease resistance proteins, ROS and phenylpropanoid biosynthesis. This transcriptomic insight provides a foundation for understanding the disease resistance inherent in these hybrid cultivars and suggests a constitutive expression of NIR NBS LRR triggering calcium signaling. Moreover, these results illustrate the magnitude of transcriptomic changes caused by the introgressed V. rotundifolia background in backcrossed hybrids, on a large number of functions largely exceeding the ones constitutively expressed in single resistant gene transformants.
Keywords:
Hybrids
; Fruit development
; V. rotundifolia
; V. vinifera
; Transcriptome
; Resistance genes
1. Introduction
Grapevine (Vitis vinifera L.) holds significant agricultural importance as the major species cultivated worldwide within the Vitaceae family, with thousands of cultivars dedicated to wine grapes, table grapes, or raisins production [1]. Grapevine industries face significant challenges due to the prevalence of fungal diseases, particularly powdery and downy mildews, caused by the biotrophic pathogen Erysiphe necator and Plasmopara viticola, respectively [2]. These pathogens, imported from the New World in the 19th [3], and transmitted to the susceptible Eurasian vinifera, became major concerns for European viticulture, necessitating heavy reliance on fungicides for control, posing environmental and health concerns [4]. Interspecific hybridization aims to combine the high-quality fruit characteristics of the Eurasian V. vinifera with the disease resistances of wild Vitis species from other continents, including the more distant V. rotundifolia [5,6]. V. rotundifolia, also known as the muscadine grape, is native to the southeastern United States and has inherent resistance to many pests and diseases. Ongoing grape breeding programs now prioritize traits related to agronomy and production, such as yield, quality, and disease resistance. These characteristics are crucial in determining the acceptance of new grape varieties by farmers and the market value of the grapes [7]. The enhancement of agronomic and production traits in grapes should be facilitated by a deeper understanding of their developmental and physiological characteristics. Over the past 25 years, numerous studies have been conducted to characterize grapevine fruit development at the molecular level. Transcriptomics was used for the characterization of varietal diversity between a cultivated grapevine variety and the PN40024 reference genome [8,9], to characterize the molecular response of berries to water deficit [10,11], temperature [12], combined stress [13], abscisic acid (ABA) [14] and pathogens [15], although these last ones are more frequently studied on leaves [16,17]. Comparative transcriptomic analysis between cultivated and wild species revealed the conservation of expressed genes [18]. Recently, a study explored the genetic diversity of nucleotide-binding leucine-rich repeat receptor (NLR) genes between wild and domesticated grapevine populations [19]. By analyzing 17 genotypes, they identified and classified these NLR genes into eight distinct types, discovering that wild populations generally possess a higher number of these genes compared to cultivated varieties. Additionally, gene ontology (GO) enrichment analysis indicated a reduction in programmed cell death associated gene families, a key immune response, in domesticated grapevines. This suggests that domestication may have led to a decrease in the pool of resistance-related genes, as observed in other crops like tomatoes [20].
Genetic improvement through hybridization, particularly incorporating fungus-tolerance traits, is a key focus for developing new grapevine cultivars [21,22]. The MrRUN1/MrRPV1 locus proved very convenient to confer a high level of tolerance to powdery and downy mildews [23,24,25,26]. MrRUN1/MrRPV1 were introgressed in vinifera according to a pseudo-backcrossing breeding scheme [25,27]. The ‘G5’, ‘G14’ and ‘Artaban’ genotypes, which were obtained after the fourth and fifth backcrosses of a V. vinifera × M. rotundifolia hybrid with V. vinifera exhibit a high level of tolerance against downy and powdery mildews [7,25,28,29]. A study reported the screening of a BAC library from a powdery mildew-resistant plant and narrowed down the Run1/Rpv1 locus to a ∼1 Mbp in the chromosome 12 [25]. A cluster of 11 resistance gene analogs (RGAs) was identified within this locus including seven genes encoding full-length TIR-NBS-LRR resistance proteins. Sequencing of this region ruled out the presence of other kinds of resistance against fungal pathogens [28]. A novel approach for multi-domain and multi-gene family identification provided insights into the evolutionary dynamics of disease resistance genes in core eudicot including grapevine, indicating that R-genes typically show an unusually high turnover rate due to strong selection to keep up in a biological arms race with plant pathogens [30]. The QTL region is usually screened with microsatellites markers, however more than 900 total individuals from two additional progenies were required for the narrowing of this QTL suggesting that many genes outside the MrRUN1/MrRPV1 locus may frequently be also introgressed alongside this locus [26].
In order to address the introgression of additional genes and its consequences on gene expression in ripening berries, we conducted a comprehensive transcriptomic study on 102 single-berries from non-resistant V. vinifera cultivars and resistant or non-resistant V. rotundifolia x V. vinifera derivative hybrids. Fruits were sampled across different stages of berry development. Following reads alignment on both V. vinifera PN400024.V4, V. rotundifolia var. ‘Carlos’ and ‘Trayshed’ genomes, Weighted Gene Co-expression Network Analysis (WGCNA) and Gene Ontology (GO) analysis allowed us to elucidate which LG12 region from V. rotundifolia was introgressed in the hybrids together with Run1 and Rpv1 genes. The multifaceted consequences of such introgression of M. rotundifolia genes on the transcriptome of the grapevine fruit are described.
2. Results
2.1. RNA-seq Reads Statistics
The total number of reads per sample ranged from 35 to 45 million. Low-quality reads excluded from further analysis accounted for 11% of the total reads, reflecting the sequencing quality and preprocessing thresholds. Only 1.8% of high-quality reads did not align with the merged genomes. Among reads mapped on assigned features, 96% mapped in single positions on the PN40024.v4 genome and 90% of these single copy reads still mapped in a unique position on the merged genomes, indicating that they were genetically distant enough, to warrant successful mapping to the right orthologs. (Table S1).
2.2. Principal Component Analysis (PCA) Highlights the Development Program of the Single Berry
RNA expressions from 102 single berries were quantified following the alignment of pair-end reads to the merged Vitis vinifera ‘PN400024’, Muscadinia rotundifolia ‘Trayshed’ and ‘Carlos’ genomes plus the seven R genes available for the pertinent ‘52’ parent. A total of 29516 genes were identified as expressed (to be considered as such, the sum of the normalized read count of a gene must be > 1). A principal component analysis (PCA) was conducted following variance stabilizing transformation (vst) of HTSeq counts, with the first and second components accounting for 43 % of the variance. As much as 37% of the variance of gene expression (PC1) is associated with berry development and ripening. Single berries inside triplicates were generally grouped at each developmental stage. Although V. rotundifolia descendants were clearly resolved from native V. vinifera cultivars on PC2, resistant and non-resistant varieties were not resolved in this PCA, indicating that the number of genes affected by MrRUN1/MrRPV1 introgression should be comparatively low (Figure 1).
2.3. Identification of Differentially Expressed Genes
Transcripts expression values were compared between the resistant cultivars group (G5 and MV102 samples, including three and seven sampling dates, respectively) and the susceptible one (Syrah, Merlot clone1, Merlot clone2 and MV32 samples obtained at 8, 4,5, or 7 dates respectively). Setting the log2 fold change threshold as 1, FDR <0.05 then p-value <0.02 yielded a total of 5211 differentially expressed genes (DEGs) among the 29516 expressed ones, including 2899 up-regulated and 2312 down-regulated ones (Figure 2A). Among the genes overexpressed in berries from resistant accessions, 832, 1003, 1058, and 6 genes were mapped on PN40024.V4 12X2, V. rotundifolia cv ‘Trayshed’ and cv ‘Carlos’ genomes plus seven V. rotundifolia cv ‘52’ RGA genes, respectively. Conversely, regarding down-regulated genes, respectively 937, 660, and 715 genes originated from the V. vinifera PN40024.V4 12X2, V. rotundifolia cv ‘Trayshed’, and ‘Carlos’ genomes (Figure 2B).
2.4. Functional Annotation of the Differentially Expressed Genes
We employed the DIANE bioinformatics package for DEGs functional annotation. The Gene Ontology (GO) enrichment analysis revealed an enrichment in 31 cellular components, 119 molecular functions, and 98 biological processes. In terms of biological processes (BP), the prominent enriched categories included defense responses to nematodes (GO:0002215), mitochondrial mRNA modification (GO:0080156), cell wall modification (GO:0042545), cytidine to uridine editing (GO:0016554), and responses to cytokinin (GO:0009735) etc. For cellular components (CC), the main categories were the apical part of cell (GO:0045177), Casparian strip (GO:0048226), plasmodesma (GO:0009506), vacuolar membrane (GO:0005774), chromosomes (GO:0005694), etc. Lastly, the molecular functions (MF) identified were predominantly associated with Rab geranylgeranyltransferase activity (GO:0004663), G-protein beta/gamma-subunit complex binding (GO:0031683), monoatomic cation transmembrane transporter activity (GO:0008324), acyltransferase activity (GO:0016746), and chitin binding (GO:0008061). The top 10 categorizations and their implications are further illustrated in Figure 3A. We compared the up-DEGs and down-DEGs GO terms in order to get the unique up- and down DEGs. The top 10 GO functional annotation terms were listed (Figure 3 B, C). The up-DEGs unique GO term contain cell wall organization (GO:0071555), tRNA 5’-leader removal (GO:0001682), regulation of photosynthesis (GO:0010109), xyloglucan metabolic process (GO:0010411), response to oxidative stress (GO:0006979). On the pther side, the down-DEGs unique GO term included SCF ubiquitin ligase complex (GO:0019005), response to cadmium ion (GO:0046686), oxidoreductase activity acting on single donors with incorporation of molecular oxygen, incorporation of two atoms of oxygen (GO:0016702), hydrolase activity, acting on esterbonds (GO:0016788), fatty acid biosynthetic process (GO:0006633).
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
DEGs between berries from susceptible genotypes vs those from MrRUN1/MrRPV1 introgressed ones were submitted to WGCNA. All samples were included in the WGCNA because the sample clustering dendrogram (Figure 4A) showed no glaring outliers. Next, before the gene coexpression network was built, the soft threshold power β was determined. The network satisfied the 0.9 scale-free topology threshold (Figure 4B), and with 10 soft-thresholding power, the mean connectedness was almost 0 (Figure 4B). Finally, 15 modules were identified using dynamic tree trimming and average hierarchical clustering, (Figure 4C).
2.6. Identification of Key Modules Associated with Traits
The dendrogram and heatmap analysis (Figure 5) revealed genetic and trait-based relationships among single berries, categorized into distinct groups by the presence/absence of the MrRUN1/MrRPV1 locus, and phenology markers like the concentrations of malic acid, tartaric acid, and glucose + fructose (Table S2). Sample clustering indicates four main branches, mostly associated with developmental stages, as shown by the relative concentrations of malic acid and sugars.
The trait/module relationships are presented in Figure 6. “ME steelblue” and “ME dark olive green” modules exhibited strong positive correlations with malic acid concentration, and a strong negative one with sugar concentration, highlighting a possible role of this module gene in malic acid accumulation and green berry development, before being repressed during ripening (sugar accumulation phase). “MEsteelblue” module displays positive correlations with tartaric acid concentration, indicating its transcripts might be involved in tartaric acid production or general dilution in the expanding berry. Notably, the yellow green module exhibited the most robust positive correlation with the introgression of MrRUN1/MrRPV1 markers in berries (r=0.99, p=1e-80), followed by the green yellow module (r=0.55, p=3e-09) (Figure 6). Gene numbers per module are presented in Figure S1. The green yellow and yellow green modules are found in the same branch of the Eigengene dendrogram and adjacency heatmap (Figure 6B, C). Consequently, both modules were considered for further analysis.
2.6.1. Yellow Green Module-Positively Associated with the Presence of MrRUN1/MrRPV1 Microsatellites Markers.
Gene Significance (GS) and module membership (MM) in the yellow green module were found to be substantially associated (cor=0.98, p<1e−200) by Pearson’s correlation analysis (Figure 7A). To eliminate module noise and keep only strongly co-expressed genes, we filtered genes with a module membership (MM) greater than the 0.8 threshold. Notably, the 5 R genes previously annotated in the MrRUN1/MrRPV1 locus [30] appeared within the 213 genes (0.8 < MM < -0.8) in the yellow green module (Figure 7A). The yellow green module Eigengene expression is specifically up-regulated in berries from the G5 and MV102 tolerant hybrids and down-regulated in the susceptible ones, including the MV32 sibling of MV102 (Figure 7B). Nineteen genes were negatively correlated with the Eigengene expression pattern in the yellow green module (MM<-0.8), and 193 genes were positively correlated (MM>0.8). According to the functional annotation of the top 40 genes in the yellow green module heatmap (Figure 8), introgressed genes elicit a concerted molecular response associated with plant resistance, wherein the majority of genes within the module exhibit heightened expression levels in MrRUN1/MrRPV1 introgressed varieties. The up-regulated gene encompassed disease resistance (TIR-NBS-LRR class), NAC domain-containing protein, NB-ARC domain-containing disease resistance, LRR and NB-ARC domain disease resistance, heat-inducible transcription repressor (DUF639), transcriptional-regulating factor 1-like, auxin response factor 1, cell division topological specificity factor chloroplastic-like, Translation initiation factor IF-3, probable receptor kinase.
2.6.2. Green Yellow Module: G5 Specific Transcripts, almost Lost in the Next Backcross
Gene Significance (GS) and module membership (MM) were found to be substantially associated in the green yellow module (cor=0.51, p<1e−08) by Pearson’s correlation analysis (Figure 9A). 24 genes with |MM| >0.8 are clustered in this module (Figure 9C). The expression of green yellow module eigengene showed that this module is rather specifically expressed in G5 (Figure 9B), and almost lost in both MV102 and MV32. Annotated genes within the green yellow module noticeably include AP2-like ethylene-responsive transcription factor AIL5, DUF3511 domain, G-type lectin S-receptor-like serine/threonine-protein kinase SD1-1, TIR domain-containing protein, etc (Figure 9C). Most of these transcripts are preferentially mapped to the V. rotundifolia genomes (Figure 10).
2.7. Gene Introgressed with MrRUN1/MrRPV1 on Chromosome 12.
G5 and MV102 share 193 overexpressed genes (MM>0.8) in the yellow green module. 117 genes among them, that’s to say 60% of the entire module, are located in chromosome 12 (65 genes in V. rotundifolia cv ‘Carlos’, 42 genes in V. rotundifolia cv ‘Trayshed’, and 10 genes in PN40024.v4) (Figure 10). G5 overexpressed genes also encompass green yellow module genes (Figure 9), but these genes, which disappeared in MV102 and MV32, are not grouped in chromosome 12 (Figure 10).
The SSR markers VMC4f3.1 and VMC8g9 were located at chromosome 12 positions 11270828-11271019 and 17943471-17943607, in V. rotundifolia cv. ‘Trayshed’, and at 12811273-12811496 and 16731670-16731806 on V. rotundifolia cv. ‘Carlos’ markers, respectively [31]. The coordinates of VMC4f3.1 were used as a common reference, before checking the precise position of the genes overexpressed in resistant and susceptible genotypes respectively (Figure 11). Remarkably, overexpressed genes in MrRUN1/MrRPV1 introgressed genotypes preferentially mapped to the whole distal part of V. rotundifolia LG12 that starts with the introgressed locus, but largely exceeding the VMC4f3.1 boundary. By contrast, the negatively correlated ones, (i.e., overexpressed in susceptible cultivars) clearly mapped to the orthologous LG12 part of V. vinifera PN140024 genome.
3. Discussion
3.1. PCA and Gene Expression Profiles
The selection of single berries at duly characterized phenological stages, based on the evolution of sugar and acids during their developmental program, has proven to be a robust technique for investigating gene expression throughout the berry ripening process [33,34]. In the present work, a meta-analysis of single berry RNAseq data from different genotypes confirmed the clear clustering of triplicates in the PCA. PC1 highlights the variance due to development, while PC2 likely accounted for genetic differences between native vinifera and their rotundifolia hybrids. Sorting synchronized berries can avoid misinterpretation of phenological differences as genotypic differences. However, naive DEG analysis between introgressed and native genotypes was confused by unequal representativity of samples at specific developmental stages among genotypes. Supplied with a range of phenology related quantitative traits, the WGCNA approach eliminated such confusion, and proved successful in the identification of bona fide genotype markers, as confirmed by the constitutive expression in healthy berries from introgressed genotypes of V. rotundifolia ‘52’ MrRUN1/MrRPVgenes, together with other RGA in the same locus. Since the complete BAC sequences described in [33] were absent from public repositories, and in order to check putative introgressions outside them, we attempted to identify the best orthologs of V. rotundifolia ‘52’ genes in others V. rotundifolia genomes. The reads were thus aligned against a merged reference grapevine genome comprising three genomes (PN40024.v4, V. rotundifolia cvs. ‘Trayshed’, and V. rotundifolia cv. ‘Carlos’ + 7 R resistance genes) providing comprehensive information for all varieties [7,35]. The BAM file was checked using IGV [35] to visualize the mapping of reads onto the merged genome. The analysis of random genes revealed no significant bias in the read alignment process, indicating a robust and unbiased alignment of reads (Figure S2).
3.2. Merged V. vinifera and V. rotundifolia Genome
Due to seniority, comprehensive sequencing and well-documented structure, the highly homozygous Vitis vinifera genotype PN40024.v4 (https://grapedia.org/genomes/) provides a foundational framework for grapevine studies, particularly in gene expression and functional genomics [36]. Many RNA-seq studies used this genome as a reference, sometimes when dealing with non vinifera species [12,13,17,33]. Selecting the appropriate reference genome is critical for RNAseq analysis, particularly with genetically distant genotypes such as deriving from V. rotundifolia, previously considered as a distinct, non interfertile genus (Muscadinia rotundifolia). When analyzing viral pathogens, some researchers align RNA-seq data to a combined grapevine-virus reference genome [37].
The obvious limitations of a single reference genome can be overcome by de novo assembly of reference transcriptomes tailored to specific organisms [16,40]. This approach is advantageous in non-model organisms or highly heterozygous varieties, revealing the genomic complexities of different grapevine cultivars or hybrids [41,42].
Our preliminary attempts of de novo assembly (results not shown) were impaired by the multigenicity of the TIR-NBS-LRR family (Toll/Interleukin-1 Receptor, Nucleotide-Binding Site, Leucine-Rich Repeat) [41]. Massonnet compared NLR (Nucleotide-binding site Leucine-rich Repeat) genes across different haplotypes of V. rotundifolia (‘Trayshed’), particularly the Run1.2 and Run2.2 loci. The clustering of Run1/Rpv1 TIR-NBS-LRRs with Run1.2 TIR-NBS-LRR proteins, along with the association of two TIR-NBS-LRRs from Run1.2 haplotypes with MrRPV1 from V. rotundifolia G52, and a varied number of LRR motifs in their LRR domain, revealed an allelic link between Run1.2 and Run1/Rpv1 [42]. Furthermore, variations in LRR domains imply that these TIR-NBS-LRRs may be particular to various infections and/or effectors [43].
In our RNA-seq analysis, we employed the recent V. rotundifolia ‘Carlos’ and V. rotundifolia ‘Trayshed’ genomes to improve the interpretation of RNAseq data on Vinifera x rotundifolia hybrids derived from M. rotundifolia ‘G52’ [44,45]. However, extensive genetic studies on both wild and cultivated varieties highlighted the inherent genetic diversity within V. rotundifolia species [46,47]. Significant genetic variations may exist between the sequenced ‘Carlos’ or ‘Trayshed’ genomes and M. rotundifolia ‘G52’, as the parent of the Muscadinia derivative hybrids used in this study, and an ubiquitous MrRUN1/RPV1 provider in European breeding programs. These variations could blur gene expression profiling. Therefore, our findings should be considered within the context of these potential genomic discrepancies. It is encouraging in this respect that 85% RNAseq reads specifically mapped to one single position in the merged genotypes, either in V. vinifera, Feechan G52 RGAs [30], V. rotundifolia cv.’Trayshed’ or ‘Carlos’.
3.3. Regulation of Genes Related to Pathogen Response
The plant immune system utilizes two main mechanisms for pathogen perception: pattern-triggered immunity (PTI) and effector-triggered immunity (ETI) [48]. PTI involves membrane-bound pattern recognition receptors (PRRs) that detect pathogen-derived molecules externally, while ETI is activated by intracellular receptors that recognize specific pathogen effectors inside the cell. This often leads to a hypersensitive response involving localized cell death [48]. Both PTI and ETI mechanisms related transcripts, were found constitutively expressed in healthy berries introgressed with the MrRUN1/RPV1 locus. The intracellular receptors mainly consist of nucleotide-binding domain leucine-rich repeat (NLR) proteins, which can include either Toll/interleukin-1 receptor/resistance (TIR) or coiled-coil (CC) domains. Structural studies show that upon effector recognition, these last proteins undergo structural changes that enable them to form oligomeric complexes targeted to the plasma membrane, which initiate cell death signaling via calcium channel activity. TIR domain-containing proteins from several bacterial and one archaeal species can remove the nicotinamide moiety from NAD-capped RNAs (NAD-RNAs) [49].
In present study, in a addition to the 6 RGA, 15 genes annotated as TIR, 12 genes annotated as CC-NBS-LRR, 46 genes as LRR receptors and 8 genes belonging to the CC-NBS-LRR class were found among the differentially expressed genes (DEGs) (Table S3). This finding highlights the critical role of oxidoreductase and NAD+ nucleosidase activities, essential for redox reactions and NAD+ metabolism, in enhancing disease resilience in berries [50,51,52]. In Qu’s study, TIR domains from grapevine TIR-NLRs (RPV1) were shown to induce cell death [17]. Further downstream, TIR domains of EDS1 proteins form complexes with PAD4 and SAG101, which interact with NRG1 and ADR1 to propagate immune responses like cell death [51]. Overall, recent research suggests that TIR or additional domains act as integrated decoys recognizing effectors from pathogens. Proteins homologous to integrated decoys are suspected to be effector targets and involved in disease or resistance. A multilayered regulation of pathogen receptors multimerization driven by interactions among nucleotide-binding domains may act as a signal to activate grapevine immune system. ZBED proteins, containing a decoy BED domain, regulate rice’s defense against the blast fungus Magnaporthe oryzae [53]. Ma’s study revealed that the plant TNL receptor RPP1 recognizes the pathogen effector ATR1 via C-JID and LRR domains, triggering tetramer formation that activates NAD+ hydrolysis and subsequent cell death [54]. Multiple organelles are involved in defense response, including chloroplasts and peroxisomes for hormone production as well as the nucleus, endoplasmic reticulum, and Golgi apparatus for antimicrobial protein production. A pair of LRR kinase-like disease resistance genes orthologs regulates rice response to increased temperature [55].
Defense genes triggered by P. viticola infection in MrRPV1 transgenic V. vinifera Syrah leaves [17] were compared with those constitutively expressed in berries from MrRUN1/RPV1 introgressed genotypes (Table S4). The Vitvi03g00882 gene showed a 2.3-fold increase in expression in berries from fungi-tolerant genotypes when compared to susceptible ones. This gene is paralogous to the two Wall-Associated Kinases (WAKs) triggered by Plasmopora viticola infection in RPV1 transgenic Syrah, from 18 to 36 hours post inoculation [17]. WAKs are known to trigger the plant’s innate immune response by acting as receptors for cell wall-associated oligogalacturonides [56]. Wall-associated receptor kinase-like and G-type lectin S-receptor-like proteins contribute to resistance by interacting with leucine-rich repeat (LRR) domains [57,58]. In the present study, 22 G-type lectin S-receptor-like proteins exhibited differential expression between susceptible and fungi-tolerant genotypes. Of these, half showed up-regulation, while the other half demonstrated down-regulation in fungi-tolerant genotypes (Table S3). One up-regulated G-type lectin S-receptor-like protein (Vitvi13g02551/VIT_13s0156g00590) aligns with the one up-regulated in MrRPV1-transgenic V. vinifera leaves following 36 h infection with P. viticola. In contrast, three down-regulated G-type lectin S-receptor-like proteins (Vitvi13g02552/VIT_13s0156g00580, Vitvi13g02553/VIT_13s0156g00550, Vitvi04g02226/VIT_04s0044g00680) in our study differ from the prior study, which reported an increase at 36 hpi in MrRPV1-transgenic plants (Table S4). 4 DUF domain-containing genes (VITMroCarlos_v1.3.g11564, VITMroTrayshed_v2.0.hap1.chr12.ver2.0.g155730, VITMroTrayshed_v2.0.hap1.chr12.ver2.0.g155780, VITMroTrayshed_v2.0.hap1.chr12.ver2.0.g157440) are annotated in yellowgreen module. The DUF642 gene from the Chinese grape species V. quinquangularis accession Danfeng-2 encodes a cell wall protein involved in both berry growth and defense responses to Erysiphe necator and Botrytis cinerea [57].
Additionally, Vitvi16g01485, the homolog of VIT_10s0042g00930 annotated stilbene synthase, is downregulated in berries from fungi tolerant genotypes. It apparently contrasts to the induction of stylbene synthesis in grapevine leaves from resistant cultivars triggered with the pathogen [17,62].
VviWRKY10 and VviWRKY30 have also been shown to play crucial roles in grapevine leaves defense against powdery mildew. VviWRKY10 acts as a negative regulator of salicylic acid (SA)-dependent defense by binding to the W-boxes in the promoters of SA-related genes and inhibiting their transcription. Conversely, VviWRKY30 promotes ethylene (ET)-dependent defense by binding to W-boxes in the promoters of ET-related genes and enhancing their transcription. Additionally, these transcription factors can mutually inhibit each other’s expression, ensuring a balanced defense response [60]. In our study, we identified differentially expressed genes with WRKY domains and W-box motifs, suggesting further layers of complexity in the regulatory networks involving WRKY transcription factors.
Whatever, present results on healthy vines, in the absence of contaminations in their environment, clearly show that the large introgression of muscadinia genes outside the Run1/RPV1 locus triggered huge constitutive transcriptomic changes, when compared to RPV1 single gene transgenic Syrah briefly post inoculation, hence a fortiori, in the absence of pathogen.
4. Materials and Methods
4.1. Grapevine Genotypes
The study encompassed five grapevine genotypes, including the fungus-tolerant G5 and MV102 hybrids, and the susceptible cultivars V. vinifera Syrah, Merlot, and MV32 hybrid. To create a molecular scale of grapevine berry development, we relied on the RNA-sequencing dataset consisting of 102 samples, part of which was published by [55,64]. Samples were collected from before véraison to ripening every 7–10 days in different locations (Table 1). The disease-resistant G5 hybrid, which displays the sugarless trait [61], results from four pseudo-backcrosses of the V. vinifera x M. rotundifolia NC6-15 F1 hybrid with V. vinifera cultivars [7]. G5, also named 3197-81B, was cultivated in the INRAE experimental unit of Pech Rouge, France (43.14° North | 3.14° East). Notably, G5 carries the MrRUN1/RPV1 locus, known for enhancing tolerance to fungal infections [30]. The two hermaphroditic semi-dwarf microvines [62], designated MV032 and MV102 [64], derive from a fifth backcross between the 04c023V0003 female microvine [34] and the G5. The MV102 microvine possesses the MrRUN1/RPV1 locus, contributing to enhanced tolerance to Erysiphe necator and Plasmopara viticola, two major specific fungus diseases in grapevine, while the susceptible MV032 lacks this locus (Figure 12). The two-years-old potted microvines were cultivated in a semi-controlled greenhouse with a temperature range of 25°C during the day and 15°C at night. The greenhouse maintained a vapor pressure deficit of about 1 kPa and a photoperiod of 12 hours of light per day. Syrah and Merlot samples are respectively described in [63] and [64] (Table 1).
4.2. Single Berry Sampling
For sampling, only healthy undamaged bunches were considered for analysis. To avoid circadian cycle influences, berries were sampled at the same time of the day, between 9 AM and 11 AM. Berries were rapidly deseeded and wrapped by tin foil then freezing in liquid N2 and stored at -80 °C. Single berries were cryogenic ground with a mortar and a pestle to a fine powder under liquid N2 manually, around 100mg frozen powder was used for HPLC analysis of soluble sugar and major organic acids to form biological replicates with nearly similar primary metabolite contents and developmental stages. The remaining powder was cryopreserved in liquid nitrogen for subsequent RNA extraction (Table 1).
4.3. HPLC Analysis of Primary Metabolites in Single Berries
One hundred mg of frozen powder sample was subjected to a 6x dilution with a 0.25 N HCl solution. After thorough shaking, the mixture was allowed to stand overnight at room temperature. Subsequently, the samples underwent centrifugation at 13,000 g for 10 minutes, and a supernatant aliquot was further diluted 10x using a solution of 5 mM H2SO4 containing 600 µM acetic acid as an internal standard. The prepared samples were then transferred to high-performance liquid chromatography (HPLC) vials for glucose, fructose, malate and tartrate determination, as in [12].
4.4. RNA Extraction and Sequencing
For every developmental stage, triplicate samples of single berries were chosen, guided by considerations of relative growth, sugars, and organic acids. individual RNA extraction and subsequent library preparation was conducted as in [12]. Sequencing was performed on an NGC Illumina HiSeq3000 in paired-end mode with 2x150 bp reads, at the Genotoul platform of INRAe-Toulouse. A total of 102 samples was obtained, including those of fungus-tolerant G5 and MV102 hybrids, and the susceptible cultivars V. vinifera Syrah [63], Merlot [64], and MV32 hybrid samples.
4.5. Methodology for Transcriptome Analysis
After preprocessing raw reads with fastp (version 0.20.1) to eliminate adaptor sequences and discard low-quality or empty sequences using the following parameters: -q 30 -u 40 -l 36 --cut_tail --cut_tail_window_size 3 --cut_tail_mean_quality 30 --detec t_adapter_for_pe. The resulting high-quality reads were aligned against a merged reference grapevine genome comprising three genomes (V. vinifera PN40024.v4 reference genome, V. rotundifolia cv ‘Trayshed’ and cv ‘Carlos’, and to the few resistance genes from the original V. rotundifolia cv ‘52’ parent available at NCBI) using Hisat2 (version 2.2.1) with default parameters. This yield to an average of 59.221.274 mapped reads per sample (σ = 14.985.062). Subsequently, aligned reads were counted using HTSeq-count (version 0.13.5) using the following options: -mode=union --order=pos --nonunique al -t mRNA -t Parent -s reverse, with the merged annotation between PN40024.v4 (https://integrape.eu/resources/genes-genomes/genome-accessions/), Vitis rotundifolia cv. ‘Carlos’ (https://zenodo.org/record/7944875) and cv ‘Trayshed’ (https://grapegenomics.com/pages/Mrot/download.php)
Analysis and identification of differentially expressed genes
Differentially expressed genes between esistant cultivar and non-resistant cultivar were detected using DIANE [65]. Raw count data have first been normalized using the TMM methodology, and low count genes has been removed. Differential expression analysis have then been performed through DIANE with an FDR <0.05 then p.value <0.02 and a log2 fold change cutoff of 1.
4.6. GO Annotation Analysis
When analyzing differentially expressed genes (DEGs) from genomic data, it’s crucial to understand the biological context of these genes. The DIANE tool, integrated with the clusterProfiler R package, provides a robust framework for this analysis. ClusterProfiler uses Fisher’s exact test based on a hypergeometric distribution to statistically evaluate which Gene Ontology (GO) terms are overrepresented among DEGs. Using R version 4.1.2 (2021-11-01) visualize the barplot of enriched GO terms with associated gene counts and p-values.
4.7. WGCNA Analysis
WGCNA package in R software was utilized to construct a co-expression network [66] for the identify related gene modules and eigengene to traits of interest. According to Pearson’s correlation matrices, we constructed a weighted adjacency matrix by a power function am=|cmn|® (cmn=Pearson’s correlation between gene m and gene n). To emphasize the weak correlations and strong correlations between genes, Module eigengenes were correlated with traits and corresponding p-values were calculated. Coding for the traits see Table S2, For each trait, driver genes within a module were determined based on transcripts with the highest absolute gene significance and module membership, as calculated by WGCNA [66]. Gene significance was evaluated by correlating a transcript’s expression profile with the sample trait, while module membership scores were determined by assessing the correlation between a module eigengene and the expression profile of each transcript. All reported p-values were directly extracted from the WGCNA output. The total connectivity and intramodular connectivity were calculated with weighted and Pearson correlations function.
5. Conclusions
RNA-seq fragments were aligned to a composite genome integrating the Vitis vinifera reference genome, several Muscadine genomes and the RGA inside the homologous MrRUN1/MrRPV1 locus. A continuum of transcripts overexpressed in fungus-tolerant genotypes aligns with the distal 4-5 Mb region of V. rotundifolia chromosome 12, starting with the MrRUN1/MrRPV1 locus, while those preferentially expressed in susceptible varieties align with the orthologous V. vinifera region. This demonstrates that this distal end of chromosome 12 remained recalcitrant to internal recombination during successive backcrosses with V. vinifera. Some V. rotundifolia genes expressed in fungus-tolerant genotypes were also observed outside this region, though to a lesser extent. Overexpressed genes in developing berries, either introgressed from V. rotundifolia, or regulated by them in the V. vinifera genome, span various functional groups, particularly calcium signaling, hormone signaling, transcription factors, plant-pathogen interactions, disease resistance proteins, ROS detoxification, and phenylpropanoid biosynthesis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
“Conceptualization, Charles Romieu; Laurent Torregrosa; and Mengyao Shi; methodology, Charles Romieu, Mengyao Shi; Experimentation, Mengyao shi, Stefania Savoi, Charles Romieu, Audrey Weber software, Mengyao Shi; Alexandre Soriano; Gautier Sarah; writing—original draft preparation, Mengyao Shi; writing—review and editing, Charles Romieu; Laurent Torregrosa; visualization, Mengyao Shi; Alexandre Soriano; Charles Romieu, project administration, Laurent Torregrosa. All authors have read and agreed to the published version of the manuscript.”
Funding
This research was funded by French National Research Agency (G2WAS project, ANR-19-CE20-0024), the Occitanie region and the Institut Agro Montpellier (Preciput ANR).
Data Availability Statement
All Syrah raw transcriptomics reads have been deposited in the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra). The BioProject ID is PRJNA862686. All G5 raw transcriptomics reads have been deposited in the NCBI Sequence Read Archive(https://www.ncbi.nlm.nih.gov/sra/PRJNA1118503). The BioProject ID is SUB14493800. All MV102 and MV32 raw transcriptomics reads have been deposited in the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra). The BioProject ID is PRJN:::::
Acknowledgments
The authors thank Dr Victoria Lesbat-Sichel who provided the Merlot RNA-seq data, Dr Luciana Wilhelm De Almeida who provided G5 single berry samples, and Emmanuelle Garcia and Romain Boulord for the management of the plants.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- P. This, T. Lacombe, and M. R. Thomas, “Historical origins and genetic diversity of wine grapes,” Trends Genet. TIG, vol. 22, no. 9, pp. 511–519, Sep. 2006. [CrossRef]
- R. B. Ferreira, S. S. Monteiro, M. A. Piçarra-Pereira, and A. R. Teixeira, “Engineering grapevine for increased resistance to fungal pathogens without compromising wine stability,” Trends Biotechnol., vol. 22, no. 4, pp. 168–173, Apr. 2004. [CrossRef]
- M. C. Fontaine et al., “Europe as a bridgehead in the worldwide invasion history of grapevine downy mildew, Plasmopara viticola,” Curr. Biol., vol. 31, no. 10, pp. 2155-2166.e4, May 2021. [CrossRef]
- L. Ons, D. Bylemans, K. Thevissen, and B. P. A. Cammue, “Combining Biocontrol Agents with Chemical Fungicides for Integrated Plant Fungal Disease Control,” Microorganisms, vol. 8, no. 12, p. 1930, Dec. 2020. [CrossRef]
- J. Lu, L. Schell, and D. W. Ramming, “Interspecific hybridization between vitis rotundifolia and vitis vinifera and evaluation of the hybrids,” Acta Hortic., no. 528, pp. 481–486, May 2000. [CrossRef]
- V. A. Volynkin et al., “Introgressions of Vitis rotundifolia Michx. to obtain grapevine genotypes with complex resistance to biotic and abiotic stresses,” Vavilov J. Genet. Breed., vol. 25, no. 7, pp. 693–700, Nov. 2021. [CrossRef]
- J.-M. Salmon, H. Ojeda, and J.-L. Escudier, “Disease resistant grapevine varieties and quality: the case of Bouquet varieties,” OENO One, vol. 52, no. 3, Art. no. 3, Aug. 2018. [CrossRef]
- C. Da Silva et al., “The high polyphenol content of grapevine cultivar tannat berries is conferred primarily by genes that are not shared with the reference genome,” Plant Cell, vol. 25, no. 12, pp. 4777–4788, Dec. 2013. [CrossRef]
- L. Venturini et al., “De novo transcriptome characterization of Vitis vinifera cv. Corvina unveils varietal diversity,” BMC Genomics, vol. 14, p. 41, Jan. 2013. [CrossRef]
- L. Ma et al., “Transcriptome analysis of table grapes (Vitis vinifera L.) identified a gene network module associated with berry firmness,” PLoS ONE, vol. 15, no. 8, p. e0237526, Aug. 2020. [CrossRef]
- S. Savoi et al., “Transcriptome and metabolite profiling reveals that prolonged drought modulates the phenylpropanoid and terpenoid pathway in white grapes (Vitis vinifera L.),” BMC Plant Biol., vol. 16, Mar. 2016. [CrossRef]
- M. Rienth, L. Torregrosa, G. Sarah, M. Ardisson, J.-M. Brillouet, and C. Romieu, “Temperature desynchronizes sugar and organic acid metabolism in ripening grapevine fruits and remodels their transcriptome,” BMC Plant Biol., vol. 16, no. 1, p. 164, Jul. 2016. [CrossRef]
- Y. Ju, Z. Min, Y. Zhang, K. Zhang, M. Liu, and Y. Fang, “Transcriptome profiling provide new insights into the molecular mechanism of grapevine response to heat, drought, and combined stress,” Sci. Hortic., vol. 286, p. 110076, Aug. 2021. [CrossRef]
- S. Pilati et al., “Abscisic Acid Is a Major Regulator of Grape Berry Ripening Onset: New Insights into ABA Signaling Network,” Front. Plant Sci., vol. 8, 2017, Accessed: Feb. 05, 2024. [Online]. Available: https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2017.01093.
- Yan Lei et al., “Transcriptome Analysis of Berries of Spine Grape (Vitis davidii Föex) Infected by Colletotrichum viniferum during Symptom Development,” Horticulturae, vol. 8, no. 9, pp. 843–843, Sep. 2022. [CrossRef]
- N. Vigneron, J. Grimplet, E. Remolif, and M. Rienth, “Claros,” Sci. Rep., vol. 13, no. 1, p. 14664, Sep. 2023. [CrossRef]
- J. Qu, I. Dry, L. Liu, Z. Guo, and L. Yin, “Transcriptional profiling reveals multiple defense responses in downy mildew-resistant transgenic grapevine expressing a TIR-NBS-LRR gene located at the MrRUN1/MrRPV1 locus,” Hortic. Res., vol. 8, no. 1, pp. 1–12, Jul. 2021. [CrossRef]
- T. V. M. Fajardo and V. Quecini, “Comparative transcriptome analyses between cultivated and wild grapes reveal conservation of expressed genes but extensive rewiring of co-expression networks,” Plant Mol. Biol., vol. 106, no. 1–2, pp. 1–20, May 2021. [CrossRef]
- Q. Long et al., “Population comparative genomics discovers gene gain and loss during grapevine domestication,” Plant Physiol., p. kiae039, Jan. 2024. [CrossRef]
- V. Ferrero et al., “Complex patterns in tolerance and resistance to pests and diseases underpin the domestication of tomato,” New Phytol., vol. 226, no. 1, pp. 254–266, Apr. 2020. [CrossRef]
- J. Liu, L. Wang, S. Jiang, Z. Wang, H. Li, and H. Wang, “Mining of Minor Disease Resistance Genes in V. vinifera Grapes Based on Transcriptome,” Int. J. Mol. Sci., vol. 24, no. 20, Art. no. 20, Jan. 2023. [CrossRef]
- T. Possamai, D. Migliaro, M. Gardiman, R. Velasco, and B. De Nardi, “Rpv Mediated Defense Responses in Grapevine Offspring Resistant to Plasmopara viticola,” Plants, vol. 9, no. 6, p. 781, Jun. 2020. [CrossRef]
- M. Park et al., “Chromosome-level genome sequence assembly and genome-wide association study of Muscadinia rotundifolia reveal the genetics of 12 berry-related traits,” Hortic. Res., vol. 9, p. uhab011, Jan. 2022. [CrossRef]
- M. Sargolzaei et al., “Rpv29, Rpv30 and Rpv31: Three Novel Genomic Loci Associated With Resistance to Plasmopara viticola in Vitis vinifera,” Front. Plant Sci., vol. 11, 2020, Accessed: Feb. 14, 2024. [Online]. Available: https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2020.562432.
- J. Pauquet, A. Bouquet, P. This, and A.-F. Adam-Blondon, “Establishment of a local map of AFLP markers around the powdery mildew resistance gene Run1 in grapevine and assessment of their usefulness for marker assisted selection:,” Theor. Appl. Genet., vol. 103, no. 8, pp. 1201–1210, Dec. 2001. [CrossRef]
- C. L. Barker et al., “Genetic and physical mapping of the grapevine powdery mildew resistance gene, Run1, using a bacterial artificial chromosome library,” Theor. Appl. Genet., vol. 111, no. 2, pp. 370–377, Jul. 2005. [CrossRef]
- L. Cadle-Davidson, “Variation Within and Between Vitis spp. for Foliar Resistance to the Downy Mildew Pathogen Plasmopara viticola,” Plant Dis., vol. 92, no. 11, pp. 1577–1584, Nov. 2008. [CrossRef]
- I.B. Dry et al., “Molecular strategies to enhance the genetic resistance of grapevines to powdery mildew,” Aust. J. Grape Wine Res., vol. 16, pp. 94–105, Nov. 2009. [CrossRef]
- A. BOUQUET, J. Pauquet, A.-F. Adam-Blondon, L. Torregrosa, D. Merdinoglu, and S. Wiedemann-Merdinoglu, “Vers l’obtention de variétés de vigne résistantes à l’oïdium et au mildiou par les méthodes conventionnelles et biotechnologiques,” Bull. OIV, vol. 73, pp. 445–452, Jan. 2000.
- A. Feechan et al., “Genetic dissection of a TIR-NB-LRR locus from the wild North American grapevine species Muscadinia rotundifolia identifies paralogous genes conferring resistance to major fungal and oomycete pathogens in cultivated grapevine,” Plant J., vol. 76, no. 4, pp. 661–674, 2013. [CrossRef]
- S. Riaz, A. C. Tenscher, D. W. Ramming, and M. A. Walker, “Using a limited mapping strategy to identify major QTLs for resistance to grapevine powdery mildew (Erysiphe necator) and their use in marker-assisted breeding,” Theor. Appl. Genet., vol. 122, no. 6, pp. 1059–1073, Apr. 2011. [CrossRef]
- S. Savoi, L. Torregrosa, and C. Romieu, “Transcripts repressed at the stop of phloem unloading highlight the energy efficiency of sugar import in the ripening V. vinifera fruit.,” Plant Biology, preprint, Jan. 2021. [CrossRef]
- S. Savoi, L. Torregrosa, and C. Romieu, “Single berry development – a new phenotyping and transcriptomics paradigm,” VITIS – J. Grapevine Res., vol. 62, pp. 49–55, Oct. 2023. [CrossRef]
- J. Chaïb, L. Torregrosa, D. Mackenzie, P. Corena, A. Bouquet, and M. R. Thomas, “The grape microvine - a model system for rapid forward and reverse genetics of grapevines,” Plant J. Cell Mol. Biol., vol. 62, no. 6, pp. 1083–1092, Jun. 2010. [CrossRef]
- J. T. Robinson, H. Thorvaldsdottir, D. Turner, and J. P. Mesirov, “igv.js: an embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV),” Bioinformatics, vol. 39, no. 1, p. btac830, Jan. 2023. [CrossRef]
- A. Velt et al., “An improved reference of the grapevine genome reasserts the origin of the PN40024 highly homozygous genotype,” G3 GenesGenomesGenetics, vol. 13, no. 5, p. jkad067, Mar. 2023. [CrossRef]
- G. Di Gaspero et al., “Evaluation of sensitivity and specificity in RNA-Seq-based detection of grapevine viral pathogens,” J. Virol. Methods, vol. 300, p. 114383, Feb. 2022. [CrossRef]
- C. Jiao, M. Gao, X. Wang, and Z. Fei, “Transcriptome characterization of three wild Chinese Vitis uncovers a large number of distinct disease related genes,” BMC Genomics, vol. 16, no. 1, p. 223, Mar. 2015. [CrossRef]
- C. Rispe et al., “De novo transcriptome assembly of the grapevine phylloxera allows identification of genes differentially expressed between leaf- and root-feeding forms,” BMC Genomics, vol. 17, no. 1, Art. no. 1, Dec. 2016. [CrossRef]
- G. Robertson et al., “De novo assembly and analysis of RNA-seq data,” Nat. Methods, vol. 7, no. 11, pp. 909–912, Nov. 2010. [CrossRef]
- M. G. Claros, R. Bautista, D. Guerrero-Fernández, H. Benzerki, P. Seoane, and N. Fernández-Pozo, “Why Assembling Plant Genome Sequences Is So Challenging,” Biology, vol. 1, no. 2, pp. 439–459, Sep. 2012. [CrossRef]
- M. Massonnet et al., “Ripening Transcriptomic Program in Red and White Grapevine Varieties Correlates with Berry Skin Anthocyanin Accumulation,” Plant Physiol., vol. 174, no. 4, pp. 2376–2396, Aug. 2017. [CrossRef]
- L. McHale, X. Tan, P. Koehl, and R. W. Michelmore, “Plant NBS-LRR proteins: adaptable guards,” Genome Biol., vol. 7, no. 4, p. 212, 2006. [CrossRef]
- N. Cochetel, A. Minio, M. Massonnet, A. M. Vondras, R. Figueroa-Balderas, and D. Cantu, “Diploid chromosome-scale assembly of the Muscadinia rotundifolia genome supports chromosome fusion and disease resistance gene expansion during Vitis and Muscadinia divergence,” G3 GenesGenomesGenetics, vol. 11, no. 4, Apr. 2021. [CrossRef]
- M. Huff et al., “Long-read, chromosome-scale assembly of Vitis rotundifolia cv. Carlos and its unique resistance to Xylella fastidiosa subsp. fastidiosa,” BMC Genomics, vol. 24, no. 1, p. 409, Jul. 2023. [CrossRef]
- K. Buck and M. Worthington, “Genetic Diversity of Wild and Cultivated Muscadine Grapes (Vitis rotundifolia Michx.),” Front. Plant Sci., vol. 13, p. 852130, Mar. 2022. [CrossRef]
- A. Morales-Cruz et al., “Introgression among North American wild grapes (Vitis) fuels biotic and abiotic adaptation,” Genome Biol., vol. 22, no. 1, p. 254, Sep. 2021. [CrossRef]
- S. Hou, Y. Yang, D. Wu, and C. Zhang, “Plant immunity,” Plant Signal. Behav., vol. 6, no. 6, pp. 794–799, Jun. 2011. [CrossRef]
- X. Wang et al., “Toll/interleukin-1 receptor (TIR) domain-containing proteins have NAD-RNA decapping activity,” Nat. Commun., vol. 15, no. 1, Art. no. 1, Mar. 2024. [CrossRef]
- S. Horsefield et al., “NAD+ cleavage activity by animal and plant TIR domains in cell death pathways,” Science, Aug. 2019. [CrossRef]
- J. Chen, X. Zhang, M. Bernoux, J. P. Rathjen, and P. N. Dodds, “Plant Toll/interleukin-1 receptor/resistance protein domains physically associate with enhanced disease susceptibility1 family proteins in immune signaling,” iScience, vol. 27, no. 2, p. 108817, Feb. 2024. [CrossRef]
- J. Ordon et al., “Disentangling cause and consequence: genetic dissection of the DANGEROUS MIX2 risk locus, and activation of the DM2h NLR in autoimmunity,” Plant J., vol. 106, no. 4, pp. 1008–1023, 2021. [CrossRef]
- D. C. J. Wong, P. Ariani, S. Castellarin, A. Polverari, and E. Vandelle, “Co-expression network analysis and cis-regulatory element enrichment determine putative functions and regulatory mechanisms of grapevine ATL E3 ubiquitin ligases,” Sci. Rep., vol. 8, no. 1, p. 3151, Feb. 2018. [CrossRef]
- B. Mauch-Mani and F. Mauch, “The role of abscisic acid in plant-pathogen interactions,” Curr. Opin. Plant Biol., vol. 8, no. 4, pp. 409–414, Aug. 2005. [CrossRef]
- V. Sichel et al., “Intravarietal diversity: an opportunity for climate change adaptation,” 2023, Accessed: Jan. 24, 2024. [Online]. Available: https://agris.fao.org/search/en/providers/122439/records/652f9decc1cd75198696e944.
- A. Brutus, F. Sicilia, A. Macone, F. Cervone, and G. De Lorenzo, “A domain swap approach reveals a role of the plant wall-associated kinase 1 (WAK1) as a receptor of oligogalacturonides,” Proc. Natl. Acad. Sci. U. S. A., vol. 107, no. 20, pp. 9452–9457, May 2010. [CrossRef]
- B. P. H. J. Thomma, T. Nürnberger, and M. H. A. J. Joosten, “Of PAMPs and Effectors: The Blurred PTI-ETI Dichotomy[OA],” Plant Cell, vol. 23, no. 1, pp. 4–15, Jan. 2011. [CrossRef]
- X.-L. Sun et al., “GsSRK, a G-type lectin S-receptor-like serine/threonine protein kinase, is a positive regulator of plant tolerance to salt stress,” J. Plant Physiol., vol. 170, no. 5, pp. 505–515, Mar. 2013. [CrossRef]
- C. Wang, J. Wu, Y. Zhang, and J. Lu, “Muscadinia rotundifolia ‘Noble’ defense response to Plasmopara viticola inoculation by inducing phytohormone-mediated stilbene accumulation,” Protoplasma, vol. 255, no. 1, pp. 95–107, Jan. 2018. [CrossRef]
- M. Zhou et al., “Transcription factors VviWRKY10 and VviWRKY30 co-regulate powdery mildew resistance in grapevine,” Plant Physiol., vol. 195, no. 1, pp. 446–461, May 2024. [CrossRef]
- A. Bigard, C. Romieu, H. Ojeda, and L. J.-M. Torregrosa, “The sugarless grape trait characterised by single berry phenotyping,” OENO One, vol. 56, no. 3, Art. no. 3, Jul. 2022. [CrossRef]
- L. J.-M. Torregrosa, M. Rienth, C. Romieu, and A. Pellegrino, “The microvine, a model for studies in grapevine physiology and genetics,” OENO One, vol. 53, no. 3, Art. no. 3, Jul. 2019. [CrossRef]
- S. Savoi, L. Torregrosa, and C. Romieu, “Transcripts switched off at the stop of phloem unloading highlight the energy efficiency of sugar import in the ripening V. vinifera fruit,” Hortic. Res., vol. 8, no. 1, pp. 1–15, Sep. 2021. [CrossRef]
- V. Sichel et al., “Intravarietal diversity: an opportunity for climate change adaptation,” 2023, Accessed: Jan. 24, 2024. [Online]. Available: https://agris.fao.org/search/en/providers/122439/records/652f9decc1cd75198696e944.
- O. Cassan, S. Lèbre, and A. Martin, “Inferring and analyzing gene regulatory networks from multi-factorial expression data: a complete and interactive suite,” BMC Genomics, vol. 22, no. 1, p. 387, May 2021. [CrossRef]
- P. Langfelder and S. Horvath, “WGCNA: an R package for weighted correlation network analysis,” BMC Bioinformatics, vol. 9, no. 1, p. 559, Dec. 2008. [CrossRef]
Figure 1.
Principal component analysis of 102 single berry RNA-seq samples. Gene expression was monitored at 3-10 dates in different genotypes. PCA was performed on variance stabilized transforms of RNA-Seq data on 29516 genes. G represents G5. Me, and M represent two different Merlot clones, Sy represents Syrah genotypes. MV32: G5 descendant devoid of the MrRun1/MrRpv1 locus, MV102: G5 descendant introgressed for the MrRun1/MrRpv1 locus. Within each cultivar, samples are ranked according to their respective sampling dates, indicating that the developmental stage has a major effect on the pattern of gene expression pattern than genotypes.
Figure 1.
Principal component analysis of 102 single berry RNA-seq samples. Gene expression was monitored at 3-10 dates in different genotypes. PCA was performed on variance stabilized transforms of RNA-Seq data on 29516 genes. G represents G5. Me, and M represent two different Merlot clones, Sy represents Syrah genotypes. MV32: G5 descendant devoid of the MrRun1/MrRpv1 locus, MV102: G5 descendant introgressed for the MrRun1/MrRpv1 locus. Within each cultivar, samples are ranked according to their respective sampling dates, indicating that the developmental stage has a major effect on the pattern of gene expression pattern than genotypes.

Figure 2.
Differentially expressed genes (DEGs) between berries of resistant versus non-resistant genotypes. A. X-axis presents down-regulated and up-regulated genes, Y-axis is regulated gene number. B. X-axis presents the reference genome, and 7R presents the resistance genes identified in the RUN1/RPV1 locus of V. rotundifolia 52 [30], which is the right Muscadinia genetic background of G5, MV102 and MV32 genotypes. Trayshed and Carlos stand for V. rotundifolia cv ‘Trayshed’, and ‘Carlos’ respective genomes. The Y-axis presents DEGs number in each genome.
Figure 2.
Differentially expressed genes (DEGs) between berries of resistant versus non-resistant genotypes. A. X-axis presents down-regulated and up-regulated genes, Y-axis is regulated gene number. B. X-axis presents the reference genome, and 7R presents the resistance genes identified in the RUN1/RPV1 locus of V. rotundifolia 52 [30], which is the right Muscadinia genetic background of G5, MV102 and MV32 genotypes. Trayshed and Carlos stand for V. rotundifolia cv ‘Trayshed’, and ‘Carlos’ respective genomes. The Y-axis presents DEGs number in each genome.
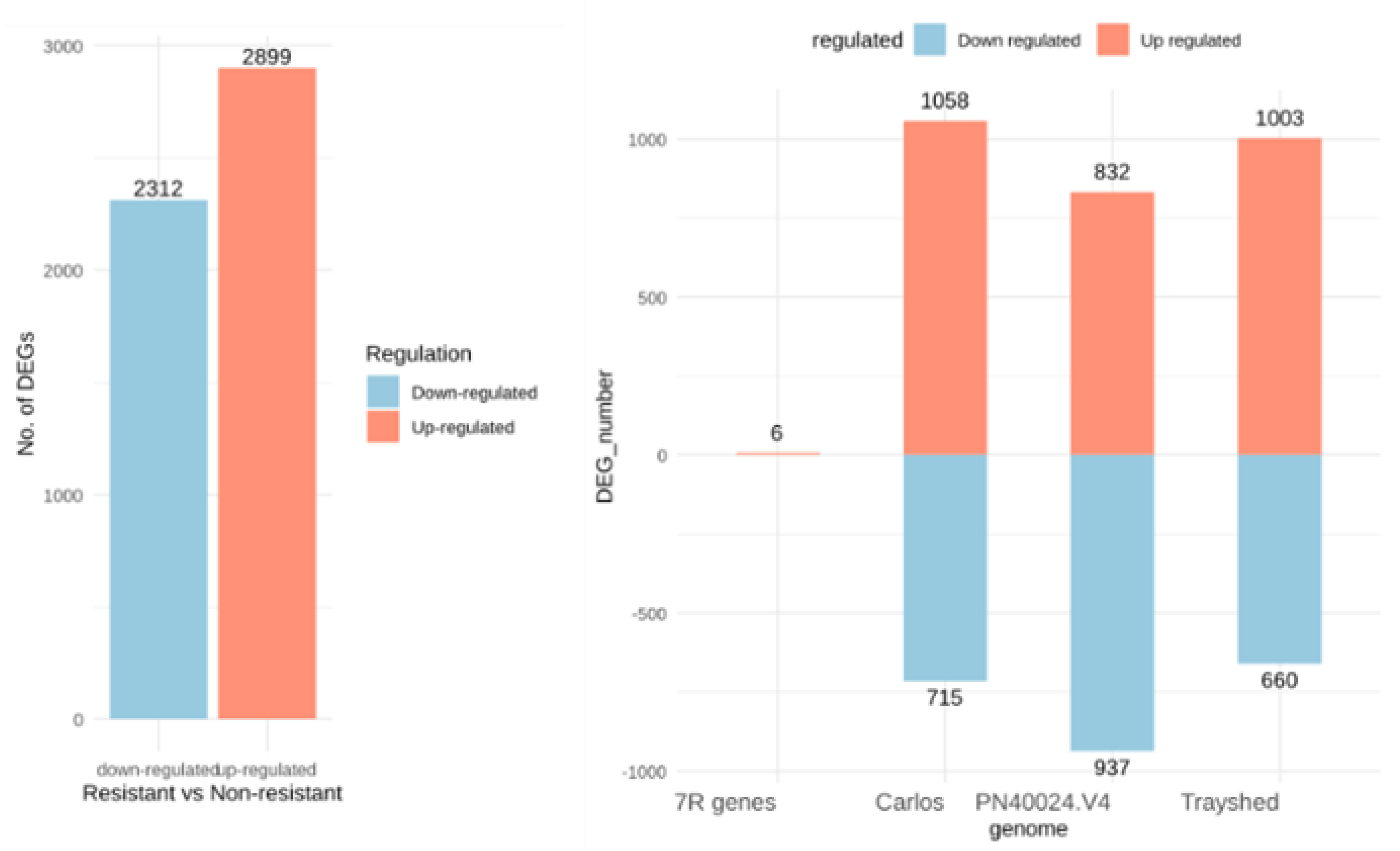
Figure 3.
Gene ontology enrichment analysis of MrRUN1/MrRPV1 introgressed berries versus native vinifera ones. A. Top ten GO terms of DEGs between non-resistant vs resistant berries. B. Top ten uniquely enriched GO terms in up-regulated genes. C. Top ten uniquely enriched GO terms in down-regulated genes. CC, cellular component; MF, molecular function; BP, biological process. Gene Ontology enrichment plots show detected GO terms (under 0.05 in Fischer’s exact tests), color-coded by their adjusted p-value, and shifted in the y-axis depending on the number of genes matching this ontology.
Figure 3.
Gene ontology enrichment analysis of MrRUN1/MrRPV1 introgressed berries versus native vinifera ones. A. Top ten GO terms of DEGs between non-resistant vs resistant berries. B. Top ten uniquely enriched GO terms in up-regulated genes. C. Top ten uniquely enriched GO terms in down-regulated genes. CC, cellular component; MF, molecular function; BP, biological process. Gene Ontology enrichment plots show detected GO terms (under 0.05 in Fischer’s exact tests), color-coded by their adjusted p-value, and shifted in the y-axis depending on the number of genes matching this ontology.
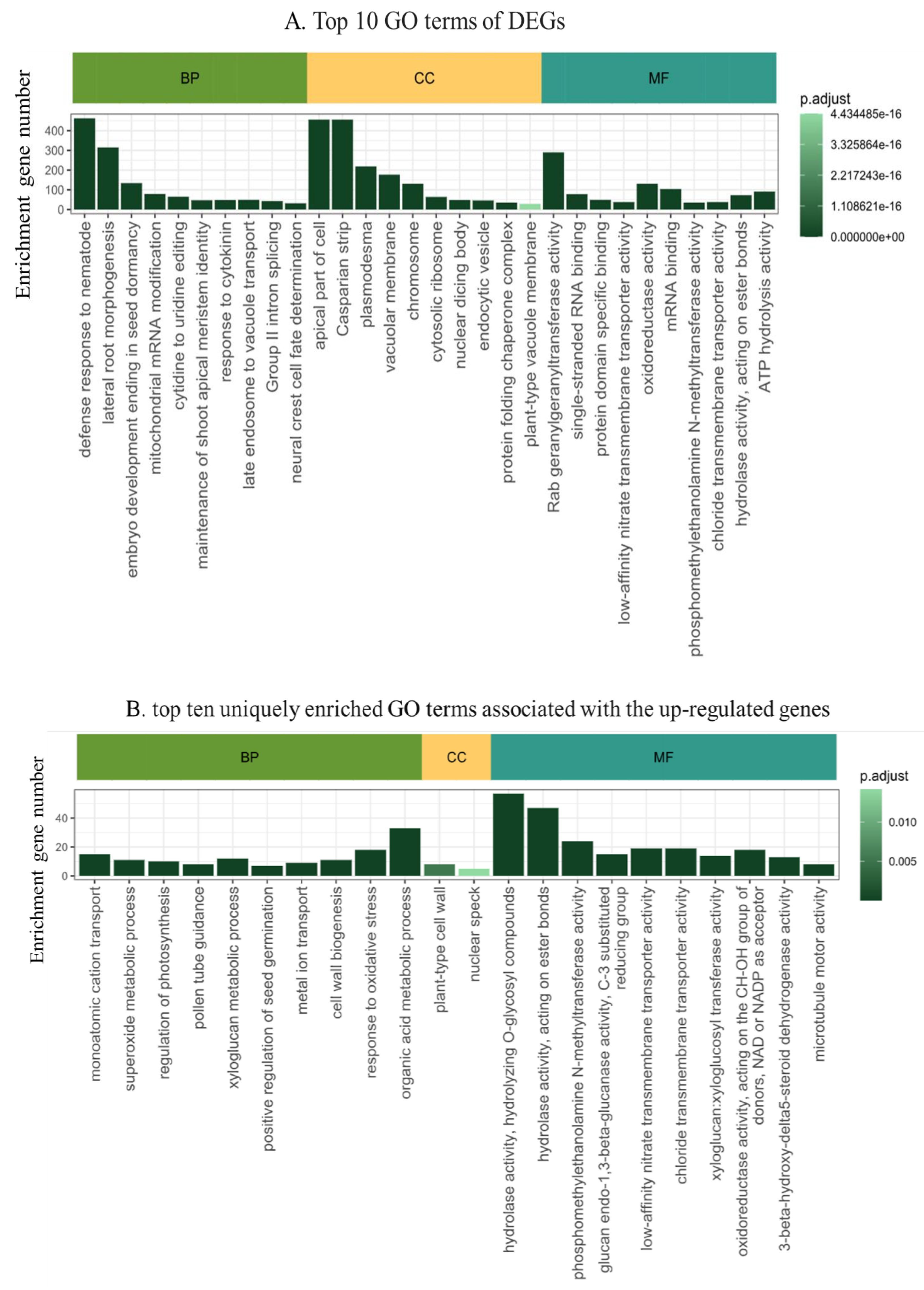
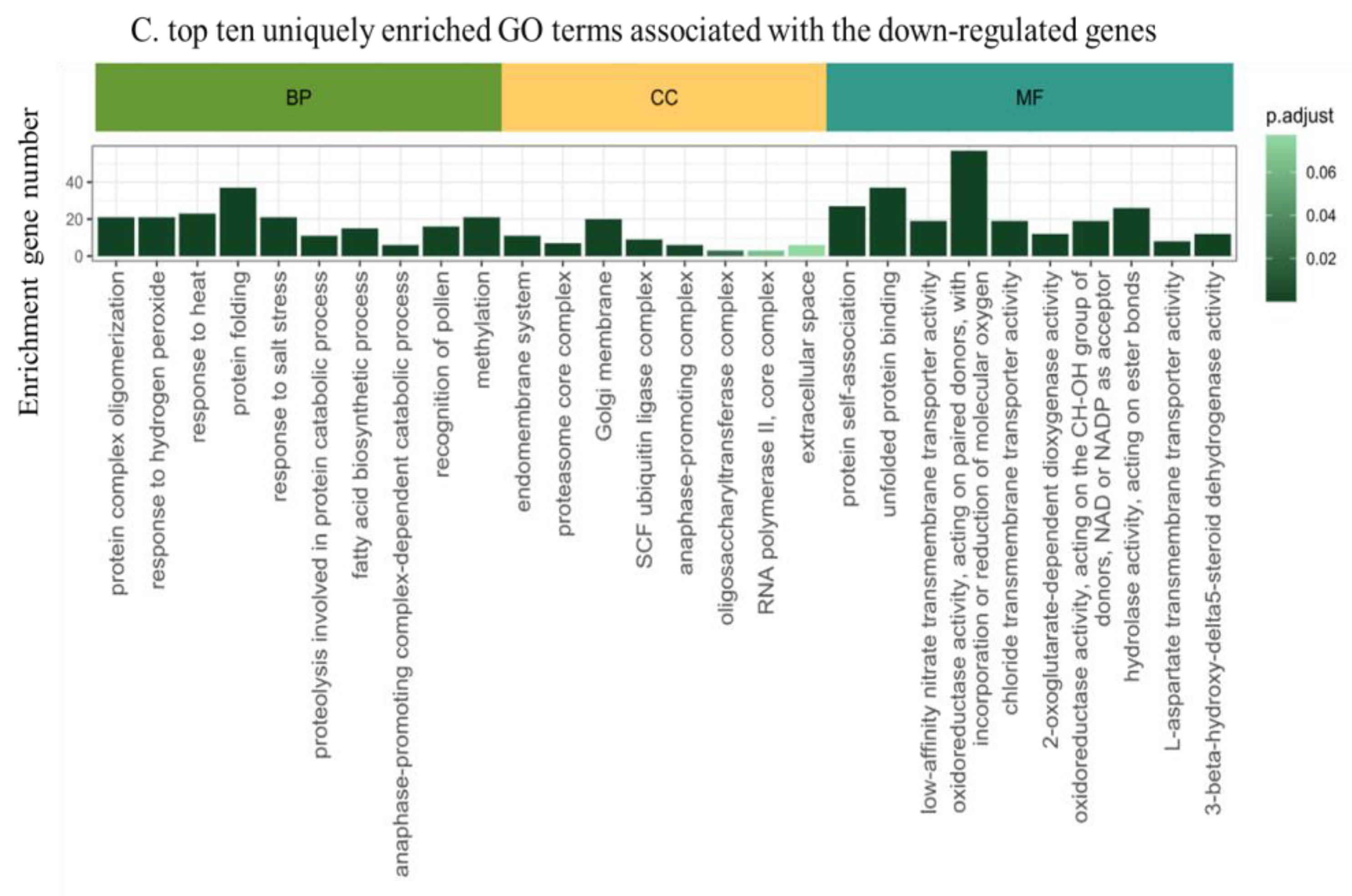
Figure 4.
Construction of the gene coexpression network from DEGs in single berries from resistant vs susceptible genotypes. (A) No glaring outlier emerged from sample clustering (B) Network topology analysis showed that at β = 10, the network satisfied the scale-free topology threshold of 0.9. When β = 10, network topology analysis showed that the mean connectedness was almost zero. (C) Gene dendrogram constructed by clustering dissimilarity (MEDissThres = 0.4). Color-coded modules represented by lines reflecting the consensus topological overlap. The cluster dendrogram at the top shows co-expressed genes. The branches and color bands at the bottom represent the assigned module. Every module has a distinct color that designates a group of co-expressed genes.
Figure 4.
Construction of the gene coexpression network from DEGs in single berries from resistant vs susceptible genotypes. (A) No glaring outlier emerged from sample clustering (B) Network topology analysis showed that at β = 10, the network satisfied the scale-free topology threshold of 0.9. When β = 10, network topology analysis showed that the mean connectedness was almost zero. (C) Gene dendrogram constructed by clustering dissimilarity (MEDissThres = 0.4). Color-coded modules represented by lines reflecting the consensus topological overlap. The cluster dendrogram at the top shows co-expressed genes. The branches and color bands at the bottom represent the assigned module. Every module has a distinct color that designates a group of co-expressed genes.
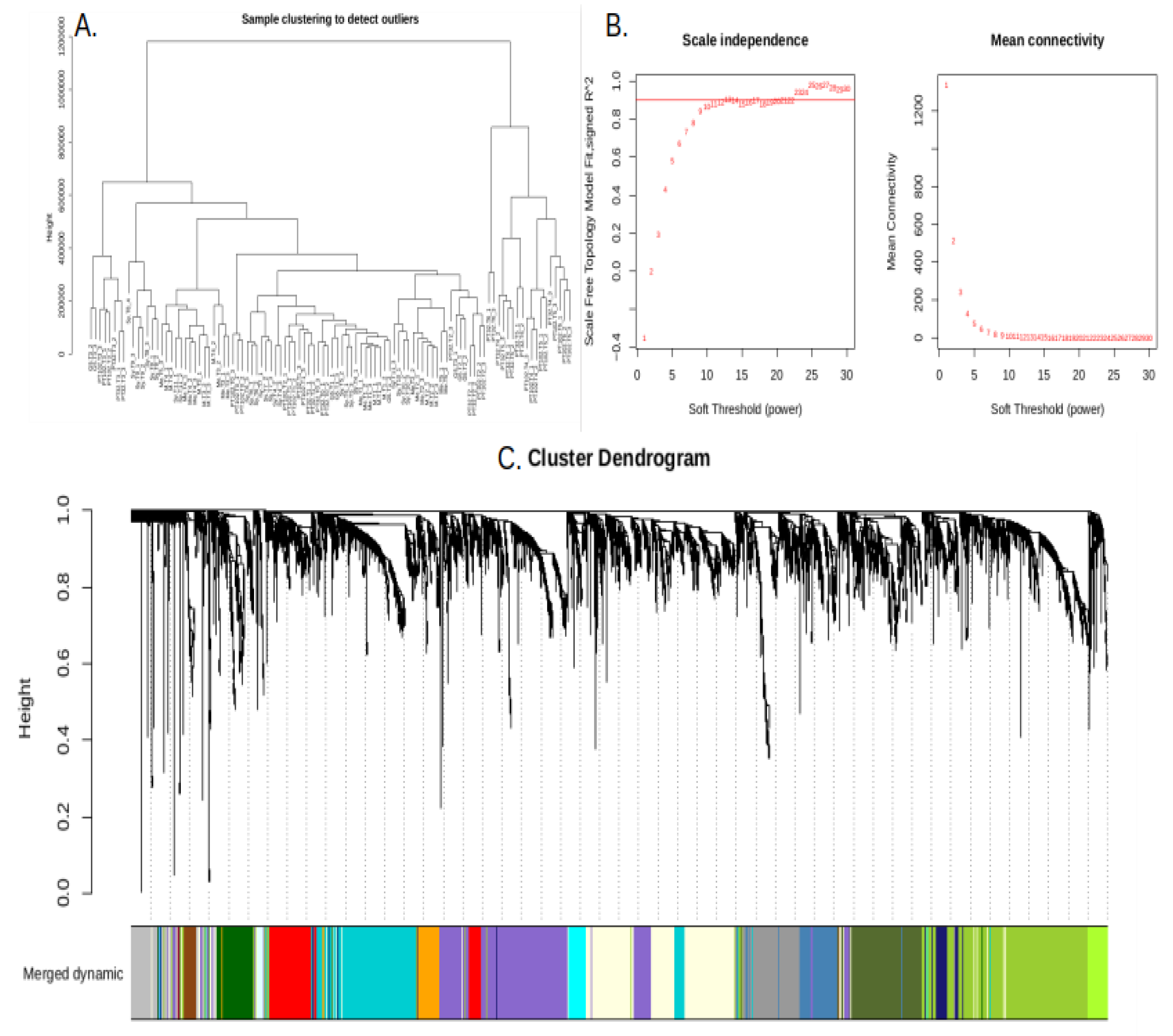
Figure 5.
Sample dendrogram and trait heatmap. The leaves of the tree correspond to samples (method=“average”). The first color band underneath the tree documents the tolerant and susceptible status, in red and white, respectively. The remaining colored bands from top to bottom represent malic, tartaric, and sugar concentrations respectively.
Figure 5.
Sample dendrogram and trait heatmap. The leaves of the tree correspond to samples (method=“average”). The first color band underneath the tree documents the tolerant and susceptible status, in red and white, respectively. The remaining colored bands from top to bottom represent malic, tartaric, and sugar concentrations respectively.
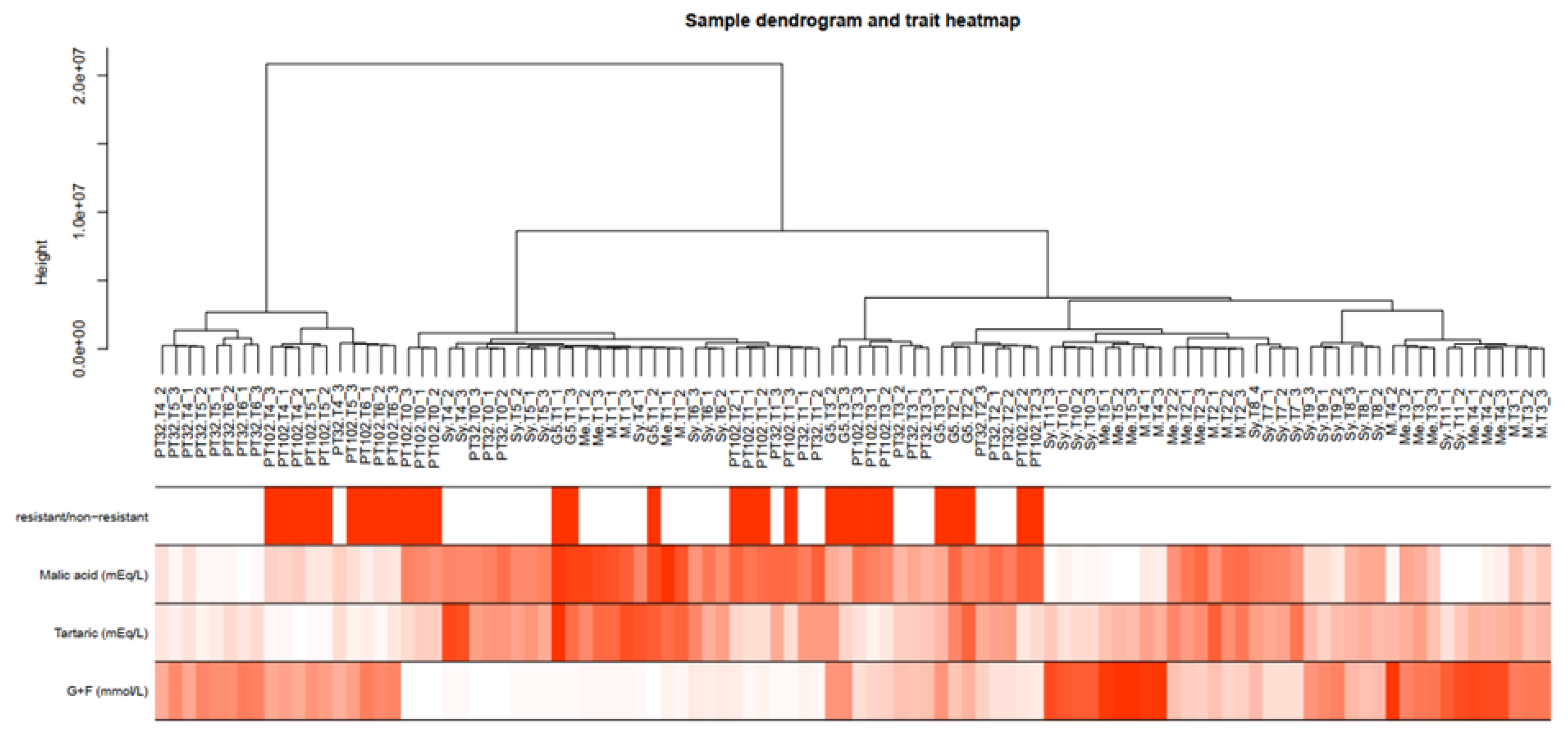
Figure 6.
Module-trait associations, Eigengene dendrogram and Eigengene adjacency heatmap. (A) Heatmap showing associations between traits and gene expression modules. Figures indicate correlations coefficient and (P values). (B) Module eigengenes’ hierarchical clustering (C) Heatmap showing the hub gene network’s adjacency relationships.
Figure 6.
Module-trait associations, Eigengene dendrogram and Eigengene adjacency heatmap. (A) Heatmap showing associations between traits and gene expression modules. Figures indicate correlations coefficient and (P values). (B) Module eigengenes’ hierarchical clustering (C) Heatmap showing the hub gene network’s adjacency relationships.
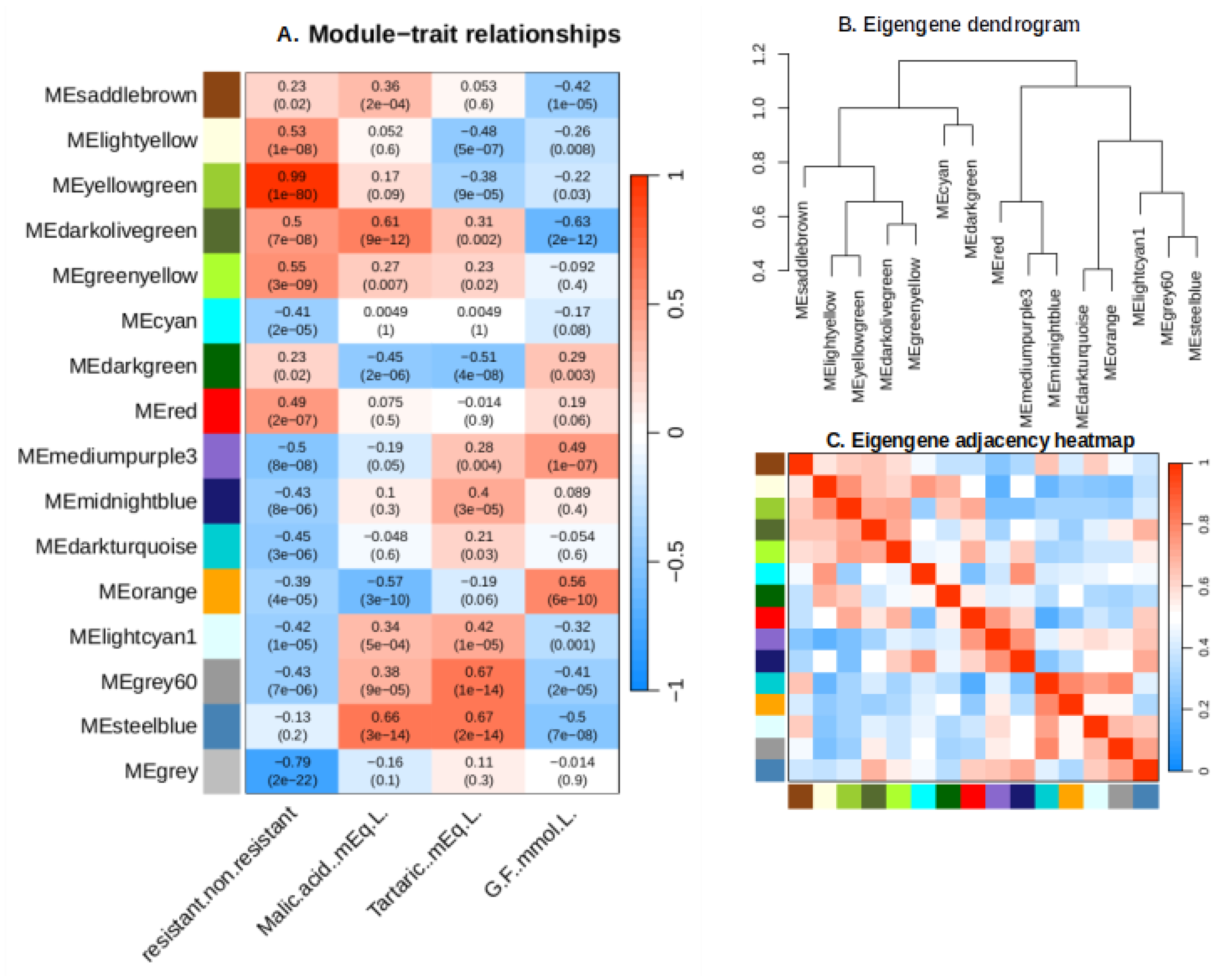
Figure 7.
Gene module membership (MM) vs. gene significance (GS) in the yellow green module. MM represents the correlation between each gene expression profile and that of the module eigengene. GS represents the association between gene expression and resistance. GS and MM are exceptionally well correlated in this module (0.98, P<1e-200). Resistance gene analogs (RGA) sequences available at NCBI in the true parental MrRUN1/MrRPV1 locus [30] are indicated in red. (B) Eigengene expression pattern in 102 single berries.
Figure 7.
Gene module membership (MM) vs. gene significance (GS) in the yellow green module. MM represents the correlation between each gene expression profile and that of the module eigengene. GS represents the association between gene expression and resistance. GS and MM are exceptionally well correlated in this module (0.98, P<1e-200). Resistance gene analogs (RGA) sequences available at NCBI in the true parental MrRUN1/MrRPV1 locus [30] are indicated in red. (B) Eigengene expression pattern in 102 single berries.
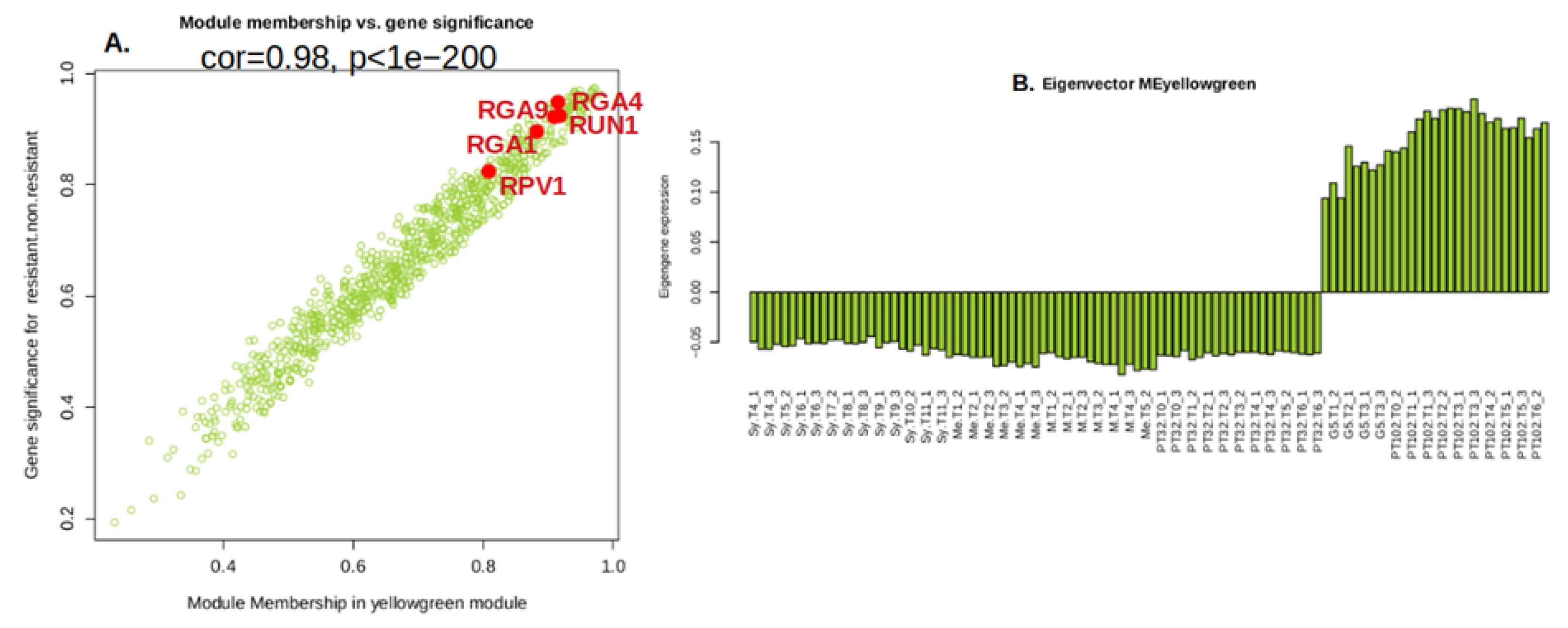
Figure 8.
Heatmap of the expression levels of top 40 annotated genes in the yellowgreen module (Module Membership>0.8) in different samples. Rows: single gene expression and function. Columns: susceptible and tolerant samples. Side Dendrogram: gene clustered according to their expression patterns (clustering_method=”complete”).
Figure 8.
Heatmap of the expression levels of top 40 annotated genes in the yellowgreen module (Module Membership>0.8) in different samples. Rows: single gene expression and function. Columns: susceptible and tolerant samples. Side Dendrogram: gene clustered according to their expression patterns (clustering_method=”complete”).
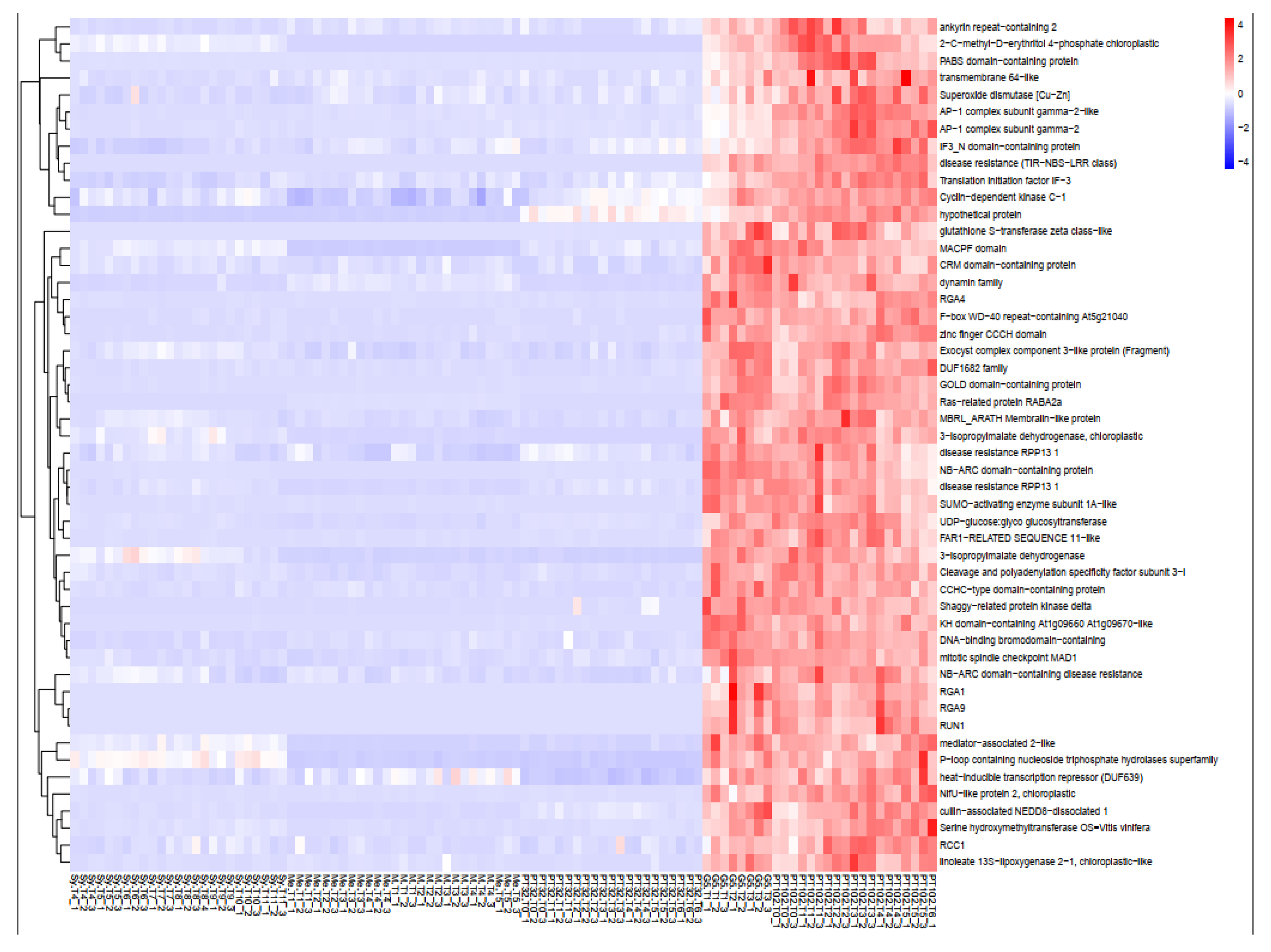
Figure 9.
Scatter plot of green yellow module membership (MM) vs. gene significance (GS) (cor=0.51, p<1e−08). MM represents the correlation between gene expression and that of module eigengene. GS represents the association between gene expression and treatment. (B) Eigengene expression pattern. (C) Heatmap of the expression levels of green yellow module genes across various samples (MM>0.8). Rows: represents a single gene. Columns: represent different samples. clustering_method=”complete”.
Figure 9.
Scatter plot of green yellow module membership (MM) vs. gene significance (GS) (cor=0.51, p<1e−08). MM represents the correlation between gene expression and that of module eigengene. GS represents the association between gene expression and treatment. (B) Eigengene expression pattern. (C) Heatmap of the expression levels of green yellow module genes across various samples (MM>0.8). Rows: represents a single gene. Columns: represent different samples. clustering_method=”complete”.
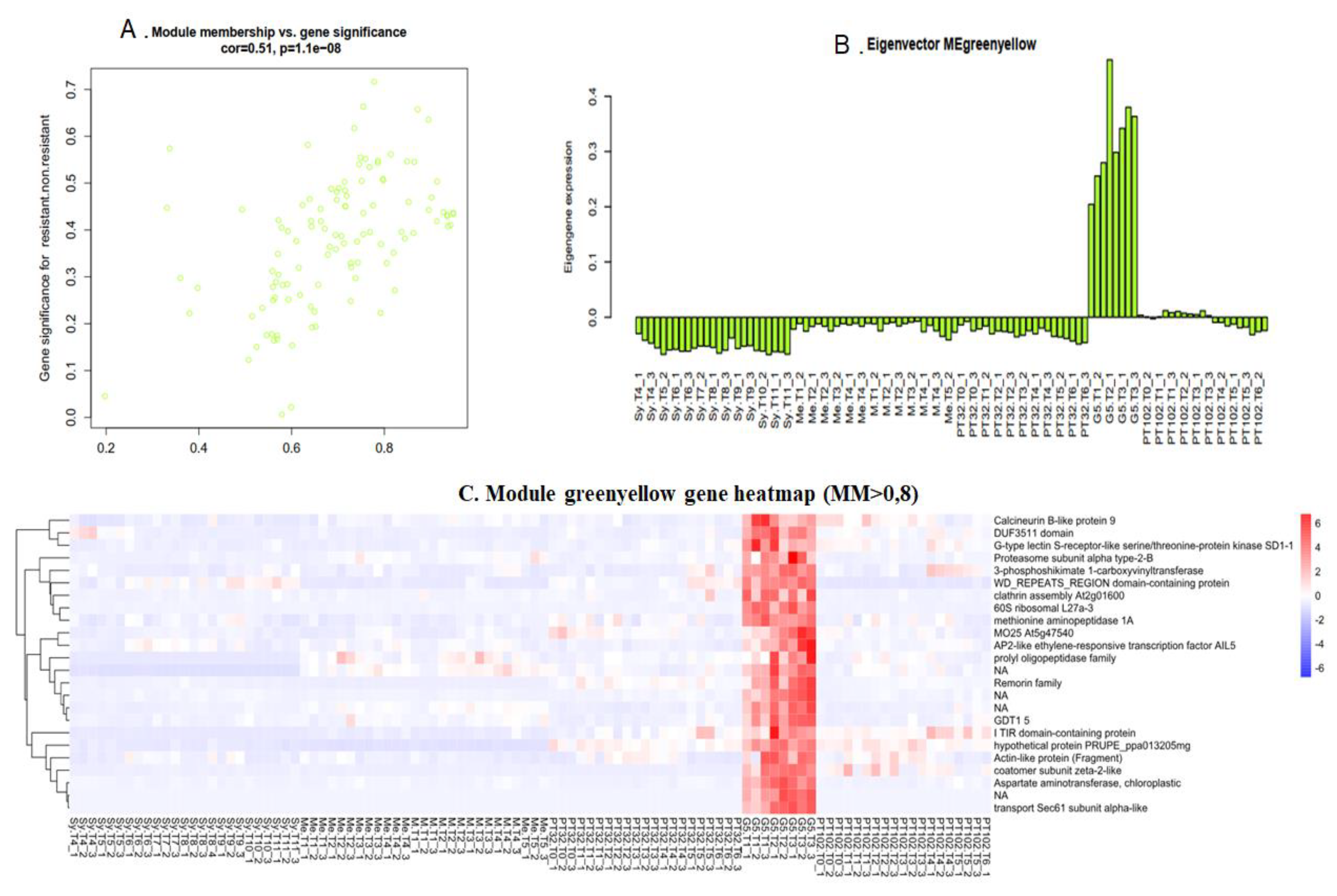
Figure 10.
Genomic positions of yellow green and green yellow module genes (|MM|>0.8) Red label: genes from the yellow green module, blue label: genes from green yellow module, specifically expressed in G5.
Figure 10.
Genomic positions of yellow green and green yellow module genes (|MM|>0.8) Red label: genes from the yellow green module, blue label: genes from green yellow module, specifically expressed in G5.
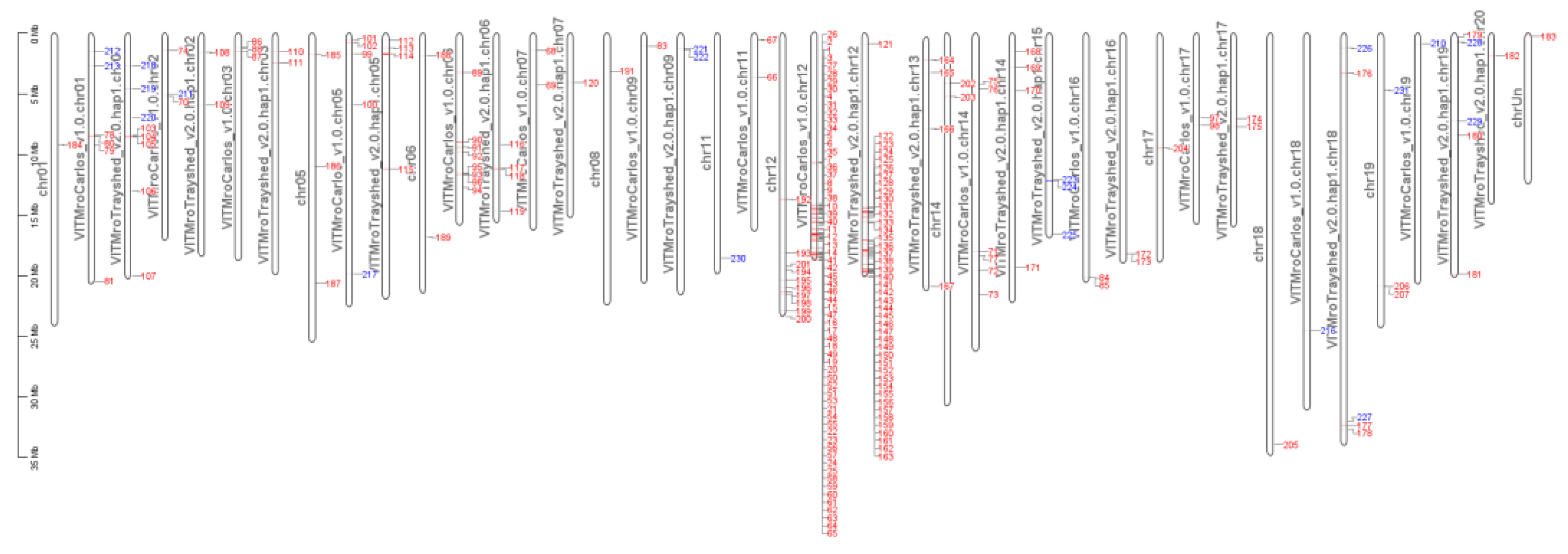
Figure 11.
Genomic positions of yellow green and green yellow module genes associated with Run1/Rpv1 locus. X-axis: gene position in chr12. Y-axis: gene MM. Red point in black circle represents the genes mapped to PN40024.v4; Green point in black circle represents the genes identified in V. rotundifolia ‘Carlos’; Yellow point in black circle represents the genes identified in V. rotundifolia ‘Trayshed’; Yellow point in red circle represents the VMC4f3.1 microsatellite marker. Green point in red circle represents the VMC8g9 marker.
Figure 11.
Genomic positions of yellow green and green yellow module genes associated with Run1/Rpv1 locus. X-axis: gene position in chr12. Y-axis: gene MM. Red point in black circle represents the genes mapped to PN40024.v4; Green point in black circle represents the genes identified in V. rotundifolia ‘Carlos’; Yellow point in black circle represents the genes identified in V. rotundifolia ‘Trayshed’; Yellow point in red circle represents the VMC4f3.1 microsatellite marker. Green point in red circle represents the VMC8g9 marker.
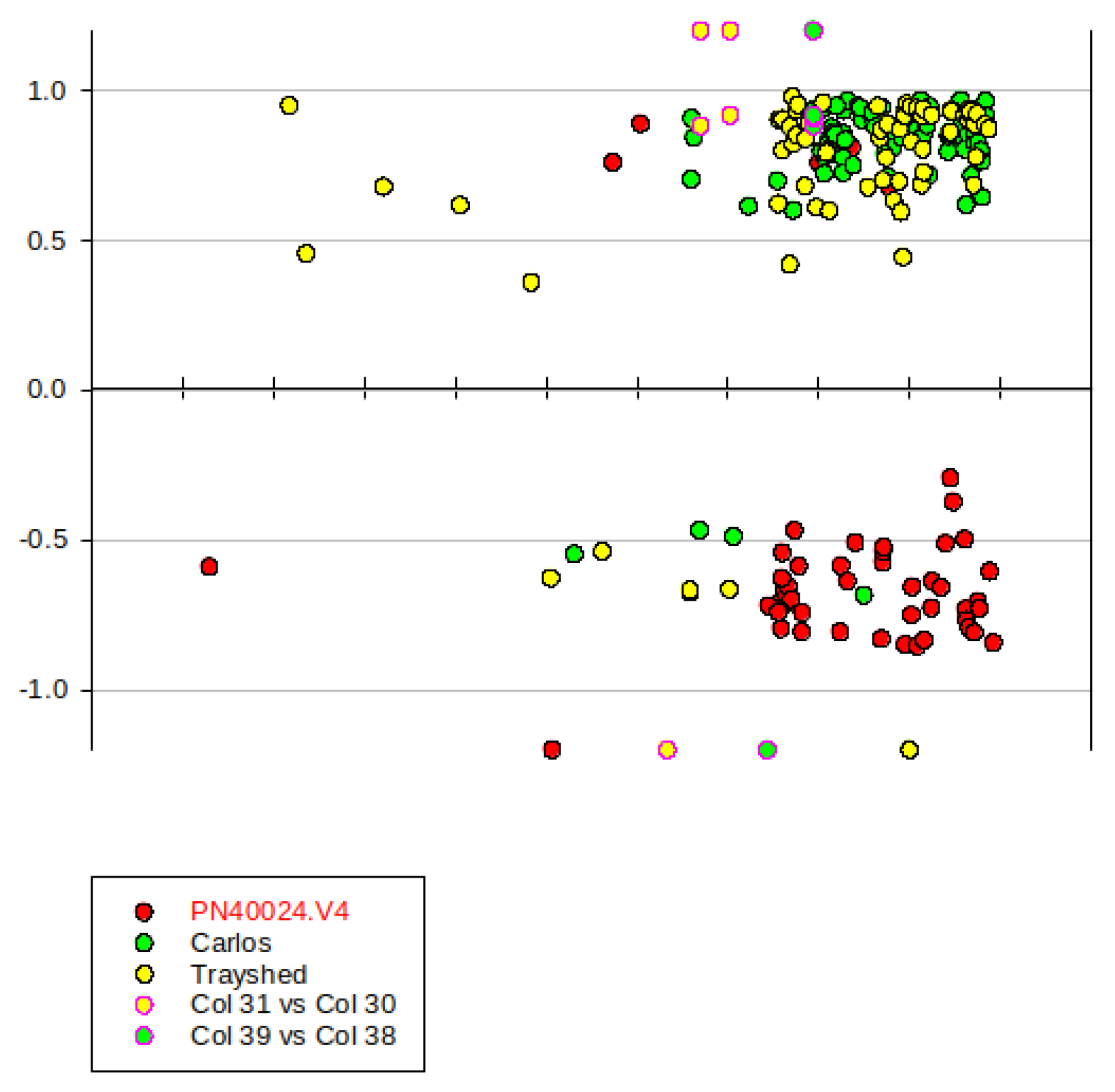
Figure 12.
Pedigree of the fungus-tolerant genotypes (adapted from Ojeda et al., 2017. G5 is a macrovine phenotype and MV102 and MV32 two microvine lines, the first one carrying the RUN1/RPV1 locus and the second without it..
Figure 12.
Pedigree of the fungus-tolerant genotypes (adapted from Ojeda et al., 2017. G5 is a macrovine phenotype and MV102 and MV32 two microvine lines, the first one carrying the RUN1/RPV1 locus and the second without it..
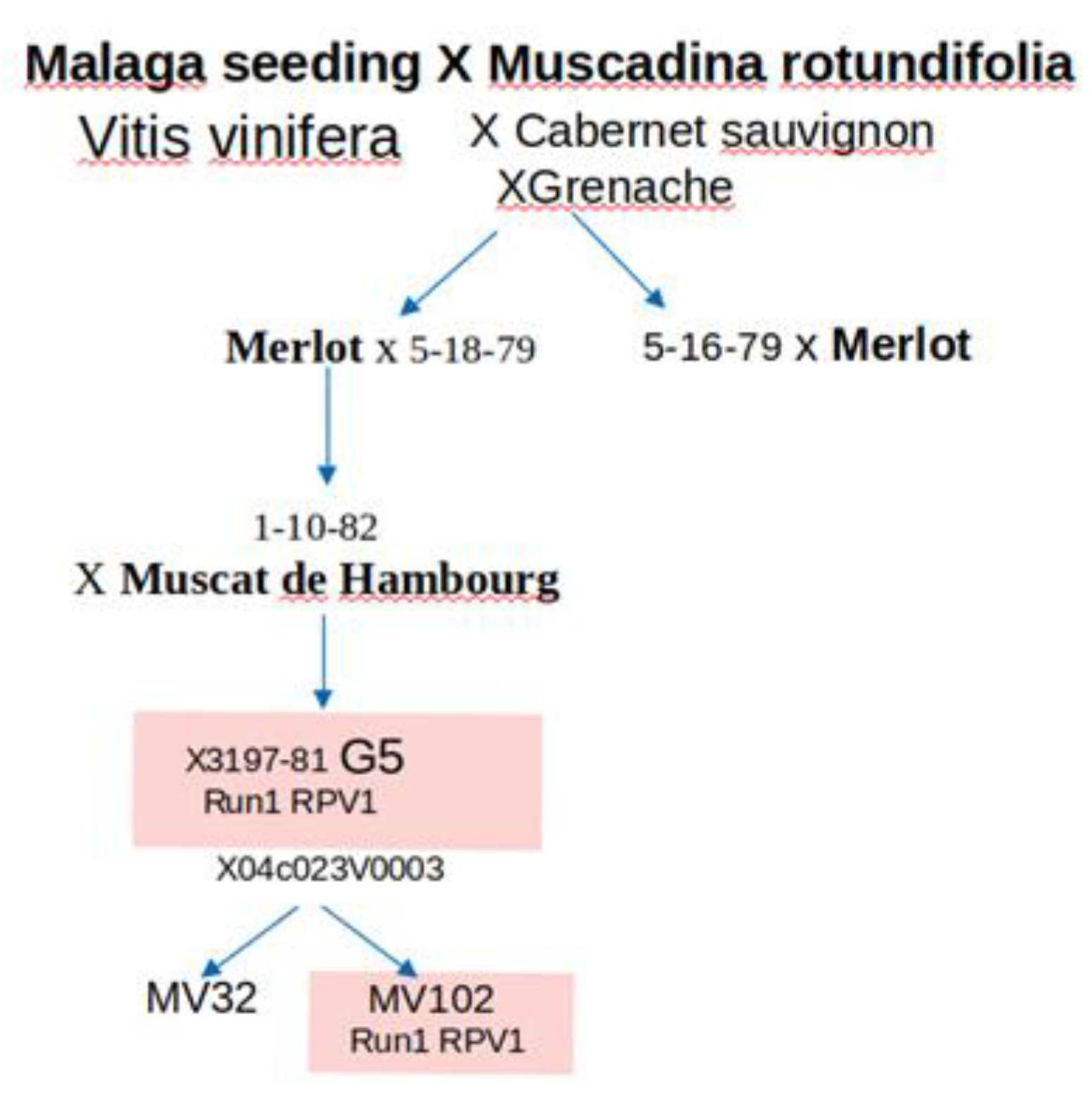

Table 1.
RNA-seq sample.
| Genotype | Traits | Sample Year | Location | Dates of Sampling | Sample Number |
| G5 | resistant | 2021 | Pech Rouge | 3 dates | 8 |
| MV102 (PT102) | resistant | 2018 | Greenhouse | 7 dates | 21 |
| MV32 (PT32) |
non-resistant | 2018 | Greenhouse | 7 dates | 21 |
| Syrah | non-resistant | 2018/2019 | SupAgro campus | 11 dates | 25 |
| Merlot clone1 | non-resistant | 2022 | Bordeaux | 4 dates | 12 |
| Merlot clone2 | non-resistant | 2022 | Bordeaux | 5 dates | 15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



