
Submitted:
05 June 2024
Posted:
06 June 2024
You are already at the latest version
Abstract
In the United States, the Federal Aviation Administration has officially classified flight crew (FC) consisting of commercial pilots, cabin crew or flight attendants as “radiation workers” since 1994 due to the potential for cosmic ionizing radiation (CIR) exposure at cruising altitudes originating from solar activity and galactic sources. Several epidemiological studies have documented elevated incidence and mortality for several cancers in FC, but it has not yet been possible to establish whether this is attributable to CIR. CIR and its constituents are known to cause a myriad of DNA lesions, which can lead to carcinogenesis unless DNA repair mechanisms remove them. But critical knowledge gaps exist with regard to CIR dosimetry, the role of other genotoxic exposures among FC, and possible biological mechanisms underlying higher cancer rates observed in FC exist. This review summarizes our understanding of the role of DNA damage and repair responses relevant to CIR exposure in FC. We aim to stimulate new research directions and provide information that will be useful for guiding regulatory, public health, and medical decision-making to protect and mitigate risks for those who travel by air.
Keywords:
DNA damage
; DNA repair
; flight
; cancer
; cosmic radiation
; flight attendants
; pilots
1. Introduction
Air travel increases yearly in the United States (U.S), exposing approximately 43,000 pilots and 96,900 flight attendants (FA) to cosmic ionizing radiation (CIR) doses much higher than those experienced at sea level. Still, we have yet to fully understand CIR-related health risks. The National Council on Radiation Protection and Measurements (NCRP) has formally classified flight crew (FC), which includes pilots and FA, as radiation workers [1], receiving an average annual effective dose of 3.07mSv. This dose is equivalent to approximately 30 chest X-rays [2], and five times the average dose (0.59 mSv) of U.S Department of Energy radiation workers [1]. Though significant, this exposure falls well within the United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR) definition of low-dose ionizing radiation (IR) (<100mSv) [3]. CIR levels and composition vary depending on both altitude and latitude [4]. Circumpolar flights operating at cruising altitudes of 35,000 feet or above increase the CIR exposure on FC. The earth’s magnetic field and atmosphere attenuate CIR, but to a lesser degree at higher altitudes and more polar latitudes [5]. Solar particle events (SPEs) occur frequently and can modulate the magnitude of CIR exposure to FC [6]. The complex interplay between the many factors influencing CIR underscores a need for detailed dosimetry and further investigation of the biological effects of this exposure.
While several large studies have documented the health risks associated with working as FC [7,8,9,10], they remain an understudied occupational cohort with respect to understanding mechanisms underlying the biological effects of CIR exposure [11,12]. Mounting evidence from several epidemiological studies indicates elevated rates of melanoma skin cancer, non-melanoma skin cancer, brain/central nervous system cancer and breast cancer have been associated with cumulative CIR exposure in FC [8,9,13,14,15], but some studies have not found a relationship between cumulative exposure and cancer risk [8,16,17]. Efforts to link excess cancer risk in FC exposed to CIR have been hampered by a lack of accurate in-flight CIR measurements and the heterogenous composition of CIR, which represents high linear energy transfer (LET) radiation and precludes direct comparison to the low-LET radiation exposure experienced by other radiation workers. Understanding the effect of CIR exposure alone in FC is challenging as CIR is difficult to measure precisely and the exposure occurs in concert with exposure to a complex mixture of stressors and chemicals from the in-flight environment [8,9,12]. In addition, FC generally have higher health standards for employment (in particular, pilots), because of regular “Fitness for Duty” or “Fit to Fly” assessments directed at weeding out individuals with serious health conditions that may jeopardize safety and performance. This constraint, or study bias, is known as the “healthy worker effect” [18]. The existing technological constraint of measuring real-time CIR exposure for an individual FC and difficulty to find a near to similar control individuals for comparison to landscape of FC exposure during air travel can contribute to problems in corroborating data with a biological endpoint.
Compromised genome integrity is a common thread that links many types of exposure encountered by FC, which may explain their excess cancer risk. Among these exposures, CIR is particularly interesting because exposures to this DNA-damaging agent in FC are estimated to be as much as 2.05-fold higher than in the general population (1.0 mSv) [1,19]. Various DNA lesions are anticipated to result during air travel, albeit at very low levels, given the extremely low dose rate of CIR. Further, the extent of genomic damage during air travel increases with altitude, alongside with increase in both dose and composition of CIR [1,20,21]. By contrast, endogenous cellular processes contribute to a baseline level of oxidative DNA damage that includes approximately 70,000 DNA lesions per cell per day [22]. It is important to take into account that while endogenously induced DNA lesions far outnumber CIR induced DNA lesions [23], CIR can induce complex DNA lesions that are more challenging to repair and more likely to cause mutations or cell death [24]. Thus, there is a need to understand CIR-induced DNA damage, its mechanisms of repair, and its biological consequences.
Multiple molecular pathways are involved in the repair of radiation-induced DNA damage. Variation in the efficiency of DNA repair pathways can govern individual’s sensitivity towards a mutagenic exposure explaining cancer susceptibility. Accumulating unrepaired DNA lesions can contribute to mutations and cellular senescence, resulting in various adverse health outcomes [25,26,27]. In this review, we present the current understanding of how DNA damage and repair in the context of CIR exposure may explain, at least in part, the elevated risk for some types of cancer in FC. This review also outlines current knowledge regarding CIR exposures during flights, discusses the available methods for CIR detection, and provides a brief overview of regulatory guidelines. We also present our perspectives on gaps in our understanding of CIR effects and what will be needed to understand the biological mechanisms in the field of DNA damage and repair underlying excess cancer in FC to help develop appropriate radioprotection protocols for air travel [28].
For this review, the methodology is outlined as follow: in section 2, we familiarize the reader with the concept of CIR, detailing its detection methods and current regulatory measures. To collect up-to-date information until July 2023, we performed searches on Google and the PubMed website. Keywords such as “air travel”, “cosmic radiation”, “flight/airline/commercial pilots”, “regulation”, “CIR dosimetry” were used to ensure comprehensive coverage. All relevant and updated data were included. To compile regulatory radiation doses for occupational workers, we referenced to published and publically available policy letters or guidelines from authoritative websites. In section 3 and 4, we have dived into finding the disperse and scare research work carried out by researchers in field of DNA damage and repair on FC cohort. We searched for the following words as “flight attendants”, “flight/airline/commercial pilots”, “DNA damage”, “DNA repair”, “CIR”, “cosmic radiation”, “cosmic ionizing radiation” on PubMed website until July 2023. Additionally, we focused on studies with sample size greater than 15 and supported their conclusions with statistical analyses.
2. Current Understanding of CIR Exposure during Flight Travel
2.1. CIR Exposure during Flight
CIR is comprised of solar cosmic radiation (SCR) created by solar activity and galactic cosmic radiation (GCR) that originates from outer space and is caused by distant events such as supernovas. SCR is characterized by continuously emitted radiation known colloquially as the solar wind and sporadic solar particle events (SPEs) [4]. CIR components also shift with SCR intensity, oscillating on an 11-year solar cycle. SPEs occur frequently but randomly with variable magnitude leading to short-term fluctuations in CIR exposure [6]. For instance, as per U.S National Oceanic and Atmospheric Administration (NOAA), a recent blackout in Southeast Asia and Australia on 17th April 2022 and another in Australia and New Zealand on 7th November 2022 were caused by solar flares leading to significant shortwave radio blackouts of radiofrequency below 30 mega-Hertz. No measurement of CIR (GCR + SCR) dose exposure was made, underscoring the need for accurate dosimetry and experimental data regarding CIR exposure in combination with SPEs. The impact of SPE is yet to be fully understood or accounted for exposure estimations of FC which is beyond the scope of this paper. Neutrons are the predominant component of CIR (~55%) at flight altitudes, with the remainder comprising both low and high linear energy transfer (LET) radiation [29] (Figure 1). Circumpolar flights at cruising altitudes of 35,000 feet or above are significantly impacted by CIR exposure due to lower protection imparted by the Earth’s atmosphere and magnetic field [5]. At each 4,500-foot increase in altitude, there is an estimated doubling of CIR exposure. At the polar latitudes, the radiation levels are approximately twice as high as at the equator [4]. The heliosphere acts as a giant shield, protecting the planets from GCR, but this protection varies inversely with solar activity [30,31,32]. The intensity of the exposure to GCR increases with altitude up to a peak level of approximately 65,000 feet, known as the Regener-Pfotzer maximum, where cosmic rays impact the Earth’s atmosphere and initiate the cascade of secondary particles and photons that contribute to increased CIR levels [33]. However, recent studies using new dosimetry techniques have found evidence of increasing CIR levels beyond this maximum [34] (Figure 2). At flight altitude (35,000 ft and above), in addition to CIR, FC are potentially subject to terrestrial gamma ray flashes associated with thunderstorms and lightning [11]. FC have been classified as “radiation workers” by various regulatory agencies such as International Commission on Radiological Protection (ICRP) in 1990, Federal Aviation Administration (FAA) in 1994, and NCRP in 2009, and some regulatory agencies have imposed limits on CIR exposure Table 1. In contrast with workers exposed to a single radiation source, FC are exposed to CIR, SPEs, and may also be exposed to non-CIR radiation sources such as radioactive cargo; airport security scanners; medical imaging related to occupational medical surveillance requirements; and ultraviolet radiation, but the magnitude and biological impact of these exposures are still unexplored in FC. Regulatory guidelines have frequently been updated as our understanding of the health effects of radiation has advanced (Figure 3). Taken together, the complex factors influencing CIR levels underscore the need for accurate and detailed dosimetry for assessing the health effects of CIR exposure during flight and informing regulatory guidelines.
2.2. Methods for Measuring or Estimating CIR Exposure during Air Travel
Although FC have been exposed to CIR since the advent of commercial flight, several challenges confront efforts to assess CIR exposure and understand the impact of CIR on FC health. Data regarding CIR exposure to FC are limited because CIR measurements are not routinely carried out on commercial airliners. Historically, CIR measurements taken during commercial flights have been primarily done onboard the Concorde aircraft as it was the only commercial aircraft equipped with radiation dosimeters. This was due to its unique supersonic cruising altitude (60,000 feet), which motivated the use of onboard dosimetry to monitor inflight CIR for radio-protective purposes. Early data collected aboard the Concorde yielded CIR dose rates of 12-15 μSv per hour for supersonic flight. Today’s standard commercial aircraft yield measurements of 4-6 μSv per hour on long-haul subsonic flights and 1-3 μSv per hour on short-haul subsonic flights [35]. For comparison, the average U.S. resident receives an estimated annual background radiation exposure of 3.1 mSv from sources of ground-level cosmic radiation, surface soil radionuclides, radon, and radionuclides in the body [2,19]. The average annual dose of CIR exposure in FC ranges from 3.07 to 6.0 mSv based on flying hours and cruising altitude [4,36] with some cases of FC exposure up to 9.0 mSv [37]. Individual lifetime cumulative doses have been estimated at >100 mSv [38]. Notably, cumulative CIR exposure levels calculated using Concorde approaches often exceed FAA or ICRP informed guidelines created to protect workers and the public from possible radiation-induced health effects in other contexts [35,38,39].
Limited direct measurements of CIR exposure create a major barrier to studying its biological effects in FC. The ideal approach would be to take individual measurements during every flight that they take, but this is not currently feasible. However, directly measuring CIR levels at altitude can be difficult. Measurement devices have historically been cumbersome due to the size of dosimeter and limited in their ability to detect the full spectrum of radiation that comprises CIR. Other than in the case of the Concorde, aircraft manufacturers have not prioritized the placement of static radiation dosimeters into aircraft design. Without onboard equipment, some of the experimental measurements have been performed with temporary dosimeters or individual aircraft details that are unable to measure for FC cumulative dose over longer time periods [4,40,41,42].
Although indirect measurements using computational models that estimate CIR exposure based on flight characteristics (origin, destination, route, date, etc.) have been used, these platforms vary in their estimations based on which CIR component is considered [43]. Because CIR exposure depends on the route, duration, latitude and altitude of flight, CIR exposure estimates need to be based on the actual flight mix encountered by FC, but these tend to be highly variable. CIR exposure can also be highly variable due to the solar cycle and SPEs. Together, these factors create a need for additional research aimed at obtaining accurate, individualized dosimetry and the analysis of potential biomarkers of CIR exposure to determine CIR-related health risks in FC.
3. Inter-Individual Variation in Response to Radiation Exposure
Some have postulated that low doses of radiation exposure may have beneficial effects on human health [44,45,46]. Since FC are constantly exposed to chronic low doses of radiation, according to this theory, it is possible that other genotoxic exposures cause the excess cancers seen in this population, and FC are, in fact, less susceptible to health risks from radiation exposures due to an adaptive response (AR) based on epidemiological findings to associate cancer risk in relation to CIR [8,47]. It can further be speculated that there might be heterogeneity in individual responses to CIR due to differences in AR among FC. Such differences have been reported in the response of human lymphocytes to radiation [48], which could further complicate the biological outcomes of CIR exposures in FC [49]. Although some studies have found AR in human subjects, results are inconclusive, debatable, and ambiguous [44,48,50]. Among those that did see AR, the magnitude of the response varied from person-to-person [50], further illustrating the complexity of inter-individual differences and highlighting the challenges of detecting possible AR in FC. The only study investigating AR in FC reported that lymphocytes had higher rates of spontaneous chromosomal aberrations and greater chromosomal sensitivity to bleomycin [51], opposite to what would be expected in the case of an AR. Given the inconsistent findings on AR across human studies, further research is needed to establish the existence of this protective mechanism. Furthermore, assessing risks relevant to the chronic low-dose exposure of mixed (high- and low-LET) radiation in FC are challenging as most of the biological effects have been evaluated on exposure to individual CIR components at much higher doses than are experienced during flight.
During development, many cells are actively dividing and thus more susceptible to DNA damage, raising concerns about the potential health effects of prenatal radiation exposure. The gestational age of the fetus at the time of radiation exposure significantly influences the risk for adverse health effects in later stages of life [52]. Specifically, DNA damage and chromosomal aberrations, changes in ancillary cellular processes, growth deformities, and cognitive defects have been observed upon prenatal radiation exposures [53,54,55,56]. The study by Grajweski et al., incorporated subject occupational and recreational flight history of the 6 months pre-pregnancy and during pregnancy, finding mean effective doses of 1.8 mSv pre-pregnancy and 0.36 mSv during the 1st trimester in the FA cohort. The study also observed a near-significant positive estimate in spontaneous abortion (OR 1.7, 95% CI: 0.95, 3.2) in FA with higher estimated CIR exposure during the 1st trimester, suggesting a 70% increased risk above 0.1 mGy [57]. In a further assessment of the complex factors confounding the flight environment, the study by Grajewski et al., also evaluated other exposures experienced by FAs and found an association of 1st trimester miscarriage with circadian rhythm disruption (OR 1.5, 95% CI: 1.1, 2.2) and high physical job demand (OR 2.5, 95% CI: 1.5, 4.2) [57]. Overall, across reproductive epidemiological studies both the direction of associations and the significance of findings are mixed. However, at the least, there is enough evidence to approach CIR exposure and pregnancy conservatively and there appears to be trending evidence to argue that there is increased risk of adverse reproductive effects in FA associated with employment as FC (and CIR as a proxy), and potentially in pregnant air travelers. Further studies with well-designed CIR estimations are needed to confirm suspected health risks. Limitations of conclusions drawn from these literatures include the small number of studies using a variety of outcomes and studies not accounting for CIR estimation in their designs other than by occupation. Another limitation is that case definitions vary by study, with different thresholds for inclusion of cases for outcomes such as intrauterine fetal demise and spontaneous abortion. Several studies that utilized questionnaire responses were limited by low study response rate [58,59], while other studies were weakened by low numbers of cases in exposed groups [60]. Furthermore, in these analyses study designs and effect estimates are mixed, to include use of small cohorts, different comparison groups and variable statistical reporting methods. The majority of the literature evaluates Western populations, and the one occupational cohort exception that evaluated risk in Chinese FA may be confounded by a Chinese regulation that requires FA to be removed from work upon confirmation of pregnancy [61].
Given the importance of DNA repair in mitigating the biological effects of radiation, inter-individual differences in DNA repair capacity (DRC) may be expected to play a role in susceptibility to the potential health effects of CIR exposure. Several factors govern variation in DRC. These include genetics, sex, age, lifestyle, environmental exposures, etc. [62,63]. Inter-individual variation in DRC for radiation-induced damage has been reported among apparently healthy individuals [48,64,65]. Genome-wide association studies (GWAS) have also linked DNA repair gene variants with various cancer risks in healthy individuals [66]. There is a need for an improved understanding of CIR-induced DNA damage and the interindividual difference in DRC, which could help to explain the elevated cancer risk observed in FC. Identifying biomarkers for environmental exposure and disease is pivotal for population science. Accounting for low level of damage detected upon CIR exposure in contrast to endogenous DNA lesion, there is a need for a more sensitive and specific assays to predict near-to real time biological effect induced by CIR exposure in FC.
4. DNA Damage and Repair Mechanisms Associated with CIR Exposure in FC
Although direct, accurate CIR dosimetry is extremely limited in FC, indirect exposure estimates based on modeling have afforded a strategy for linking CIR with health outcomes and biomarkers of radiation exposure [42,67,68,69,70,71,72,73]. In this section, we discuss the findings of these studies, with an emphasis on DNA damage and repair mechanisms.
4.1. Markers of DNA Damage and Genome Instability in FC that Are Consistent with the Expected Biological Effects of CIR Exposure
This section of the review summarizes available data supporting a model wherein the elevated cancer risk observed in FC can be explained, at least in part, by an underlying molecular mechanism involving CIR-induced DNA damage. Since direct experimentation with actual CIR is not feasible, evidence for potentially harmful occupational exposure to CIR among FC has come from investigations of biomarkers in response to exposure to sources of terrestrial radiation meant to model the qualities of CIR. Although these types of radiation cannot exactly recapitulate the properties of bona fide CIR, they have provided an important model for how CIR may be expected to affect biological systems.
Each day, cells experience >100,000 spontaneous DNA lesions which includes base damages, oxidative damage products such as 8-hydroxy-2-deoxyguanosine (8oxoG), single strand breaks (SSBs) or double strand breaks (DSBs) [74]. These endogenously produced DNA lesions are usually resolved efficiently by cellular DNA repair machinery. The same types of DNA lesions can be induced by IR, including CIR, but they are more likely to occur as clusters of lesions comprised of more than one type of damage [75,76,77]. When two or more isolated lesions occur within 10-20 bp (i.e., ~1-2 helical turns of the DNA), the damage is referred to as a clustered DNA lesion [74,78]. Clustered lesions may include complex DSBs and small DNA fragments arising from multiple closely spaced DSBs [21,79].
An extremely low dose rate relative to background exposure makes it difficult to detect CIR-induced damage. However, this type of radiation has properties that distinguish it from other types of radiation and may enable specialized strategies for detecting its effects on the genome. CIR includes high-LET radiation, which differs from low-LET radiation, such as X-rays, in terms of the ionization density along the radiation track [80] and the relative biological effectiveness (relative measure of DNA damage done by a radiation per unit track of energy deposition in a biological tissue) [81], which govern the types of DNA damage induced [82]. Complex clusters of oxidative DNA lesions are produced to a greater extent by high-LET IR than by low-LET IR and, as a result, have more harmful consequences [76,83]. For instance, human cells exposed to 1.0 Gy 56Fe ions produce 20-50 DNA fragments of <1000base pairs, about 30 times higher than 1.0 Gy of γ rays [79]. DNA damage induced by heavy ions is more efficient in inducing cellular senescence [84], premature chromosomal condensation (biodosimetry to detect interphase chromosomal damage) [85], micronuclei formation (biomarker of IR induced un-repaired DSB during cell cycle) [86], and γ - H2AX (biomarker of DSBs) [87] compared to high doses of low-LET IR. Interestingly, and consistent with the induction of a different spectrum of DNA lesions, mutational signatures induced by high-LET IR are distinct from those induced by low-LET IR [88]. The studies discussed in this section involve much higher levels of DNA damage than would be expected upon CIR exposure in FC. According to the Linear No Threshold (LNT) model, it is assumed that risk associated at higher doses can be extrapolated to lower doses. However, alternative dose response relationships have been proposed, including some that depend upon the details of the of exposure including dose, dose-rate, type of radiation [89].
The biological effects of CIR component particles on cells depend significantly on the dose rate. One study quantitated the level of γ- H2AX foci following exposure to low dose rate (0.015 Gy/min) vs. high dose rate (0.400 Gy/min) neutrons at several total doses (0.125 Gy to 2.0 Gy) in human peripheral blood mononuclear cells (PBMCs). In this study, averaging over all doses, 40% greater induction of γ- H2AX foci was observed after high neutron dose rate exposure compared to low dose rate exposure. The foci levels decreased 24 h after irradiation, and foci remained significantly higher than the background levels irrespective of the neutron dose rate [90]. This indicates that the dose rate is a factor that may need to be considered for cancer risk estimations from neutron exposures, which are the most abundant component of CIR.
A very limited number of studies have directly measured biomarkers of DNA damage in FC. A study on 44 male airline pilots compared to 36 factory workers was conducted to determine whether indirect CIR estimates can correlate with 8oxoG levels in blood and urine samples [30]. The study showed that levels of 8oxoG were higher in pilots, though it was not possible to establish a CIR dose-response relationship. It has also been hypothesized that interactions with non-ionizing radiofrequency (e.g., in-flight Wi-Fi) and low-frequency electromagnetic fields aboard aircraft could augment the deleterious effects of CIR by increasing cellular oxidative stress [91,92,93], however, adverse health effects from non-IR exposure are inconsistent across the literature [94]. A study using comet assays to compare civil FC flying long haul routes versus matched ground staff showed a non-significant trend toward higher levels of basal DNA damage among FC (measure oxidative damage, SSBs and DSBs) [95]. When comparing high LET radiation exposure versus low LET radiation exposure, divergent patterns of DNA damage, gene expression, repair protein mobilization, cytokine activation, and cellular microenvironment remodeling are observed [96]. Furthermore, some particle components of CIR, which have not been studied as thoroughly as photon radiation, also have distinctive properties concerning their biological effects [97], as well as the types of DNA damage they produce, as detailed above. Sensitive assays capable of detecting these rare, distinctive types of DNA damage are needed to investigate the biological effects of CIR.
Several studies using cytogenetic analysis (chromosomal aberrations, formation of micronuclei, sister chromatid exchanges) have yielded mixed results with regard to evidence of CIR-induced damage in FC. Chromosomal aberrations, formed by inaccurately repaired or unrepaired DSBs [98] in human lymphocytes have been considered a reliable indicator of low-dose radiation exposure [3]. They can be observed several years after radiation exposures in PBMCs, such as in atomic bomb survivors [99,100]. Some FC studies have found significantly elevated levels of chromosomal aberrations versus the selected control group [67,68,69] however others found no difference [70,71]. The different findings of these studies may be due to differences in study design including the selection of the control group, and whether a sufficient number of cells were scored to detect an expected small increase in these relatively rare cytological events. Significant higher levels of chromosomal aberrations and micronuclei were observed comparing the Concorde pilot group (estimated mean dose per year from 11 to 37mSv depending on radiation weighting factor of neutron) versus controls who were matched for age, health, and socioeconomic status [67]. Similar statistically significant differences were also observed for civilian pilots and cabin crew of subsonic aircraft (n=192; 120 males and 72 females) compared to non-flying healthy volunteers (n=55; 24 males and 31 females) [68]. On the other hand, a different study showed no difference between 83 airline pilots versus 50 comparison subjects (mean age 47 versus 46 years, respectively) [71]. This underscores the point that the choice of control group is a critical factor. In addition, the parameters used for scoring chromosomal aberrations can play an important role in the outcome. For example, interlaboratory variation in recognizing, rejecting, classifying various type of chromosomal aberrations and setting a different threshold for scoring has been documented [101]. The frequency of chromosomal aberrations is expected to be directly proportional to radiation exposure. Yet, while some FC studies have yielded findings that are consistent with this expectation [70,71] others have not [69]. Significant higher levels of chromosomal translocations per cell was observed in long term male pilots (healthy, non-smoking, aged 40-60 years, recruited by single airline) versus non pilot controls (aged matched, no frequent flying history) [69]. However, the number of translocations per cell showed no dose response relationship among the pilots [69]. Similarly, the study done by Grajewski et al., 2018 [42], observed no association in translocation frequency and estimated absorbed dose from all types of flying male pilots. By contrast, linear relationship between cytokinesis-blocked micronuclei and the average annual effective CIR dose of radiation received or the average annual flying hours in FC was observed in PBMCs [72]. The DNA damage underlying cytogenetic abnormalities in FC could potentially be attributed to other in-flight exposures. Nevertheless, studies that have found elevated levels of chromosomal aberrations and particularly those finding a dose-response relationship with estimated CIR exposure are consistent with genome instability induced by CIR.
Animal and cell culture-based models have provided insights into the mechanisms that may be involved in the biological effects of CIR. These models remove the complexity inherent in population settings and enable more detailed investigations of the impact of CIR or its components in a cell- or tissue-specific manner that cannot be achieved using the biological samples usually available from human subjects. CIR exposures are anticipated to have tissue-specific effects on the human body. It is well-established that the biological effects of IR are tissue-dependent (Figure 4) [102,103], partly due to tissue-specific sensitivity to the induction of apoptosis [104]. Thus, there is a need for additional studies to understand the underlying mechanisms. Li et al. reported that exposure to 48Ti ions (one of the components of GCR) induces deletions in lung-derived epithelial cells in vitro when DSBs are induced in regions of the genome that have flanking short microhomologies that promote error-prone alternative end-joining pathways. In addition, in bronchial epithelial cells, they found an increase in chromosomal rearrangements, which are associated with increased lung cancer risk [77]. Persistent epigenetic effects, such as alterations in DNA methylation, have also been reported in lung cells exposed to components of GCR [105].
Despite important advances, our understanding of the impact of CIR exposure on human health remains limited and warrants further investigation. DNA repair mechanisms remove damage induced by CIR. Dysregulation of DNA repair pathways promote genomic instability and may underly the elevated cancer risk observed in FC. The next section delineates the major DNA repair pathways with emphasis on their roles in repair of DNA damage expected to be induced by CIR. The challenge of measuring CIR induced damage has hampered efforts to study it’s health effects, but given the central importance of DNA damage and repair for cancer [106], CIR-induced genomic instability provides a biologically plausible explanation for elevated cancer risk in FC that needs to be further investigated.
4.2. DNA Repair Mechanisms Involved in the Cellular Response to Damage Induced by CIR Components
Genomic instability is a hallmark of cancer and drives radiation-induced carcinogenesis [107,108]. To maintain genome stability, cells employ numerous DNA repair mechanisms [109]. Although it has not been feasible to study these mechanisms directly in the context of CIR, various laboratory-based model systems have been used [110,111,112]. Each component of CIR is expected to induce a distinct spectrum of DNA lesions, each recognized and processed by a specific DNA repair mechanism. To understand the implications of this complexity, many groups have investigated the DNA damage response (DDR) in human PBMCs exposed to various doses of alpha particles, X-rays, and mixed beams of radiation [113]. For example, expression of DDR genes such as ataxia telangiectasia mutated (ATM) protein, tumor protein 53 (P53), and DNA protein kinase catalytic subunit (DNA PKcs) have been reported to be induced to a greater degree when cells are exposed to comparable doses of mixed beam versus pure high-LET alpha particles or pure low-LET X-rays [113]. This suggests that mixed high- and low-LET radiation may present a greater challenge to genomic integrity. Experiments using neutron sources in vitro circumvent some of these challenges and have provided insights into the potential genotoxic effects of CIR. For instance, compared with mock-irradiated controls, a significant increase in micronuclei frequency, DNA damage levels, and ɣ-H2AX foci was observed after neutron irradiation from 241Am-9Be source at doses as low as 9.0 mGy [114]. In addition, consistent with induction of a DDR, increases in GADD45A, CDKN1A, and PARP1 transcript levels were reported at 4 hours post-irradiation with low doses of neutrons in human resting PBMCs [114]. The molecular mechanisms of DNA repair have been extensively studied for higher doses of IR. However, there is a gap in our understanding of the biological effects and health risks associated with exposure to low-dose or low-dose-rate IR and complex mixtures of low- and high-LET radiation exposures, as seen in CIR.
In the remainder of this section, we summarize available literature on the possible role of CIR-induced DNA damage and DNA repair pathways in FC health outcomes. Since very little work has been done with actual CIR-induced DNA damage, most of this work relies on model systems and assumptions based on our understanding of the biological effects of IR. IR produces a variety of types of DNA damage. 1.0 Gy of gamma rays induces thousands of base lesions and SSBs and about 40 DSBs per human cell [23]. Relative to low-LET photon radiation, the high-LET components of CIR may induce a higher proportion of DSBs [115], which are mainly repaired by the non-homologous end joining (NHEJ) and homologous recombination (HR) pathways. Defects in NHEJ and HR activity have been associated with chromosomal aberrations, immune dysfunction, clinical radio-sensitivity, and elevated cancer risk [116,117]. The presence of one or more DNA lesions at or near the ends of DSBs requires additional factors for repair by the NHEJ pathway [118], and the need for DNA end processing increases the possibility of slower and alternative error-prone DNA repair mechanisms [119,120]. A linear dose-response relationship indicates that a single ionization track gives rise to most DSBs for both high- and low-LET radiation [23], but the dense ionization tracks associated with high-LET radiation are more likely to give rise to complex lesions that are repaired slowly compared to those induced by photons [121]. In addition to directly induced DSBs, radiation can lead to DSBs by indirect means that remain incompletely understood. Genome instability can also occur in the progeny of irradiated cells and is termed delayed hyperrecombination [122,123]. Several-fold increases in delayed hyperrecombination that last for multiple weeks have been reported in response to low-LET X-rays or high-LET carbon ion radiation in mice [122]. In further support of the notion that for a more prominent role in DSB repair of DNA damage induced by high-LET radiation, some studies have demonstrated HR-deficient cell lines having greater sensitivity to proton radiation than low-LET photons of the same energy [124]. Microhomology-mediated end joining (MMEJ) (mutagenic DSB repair mechanism that uses microhomologies flanking break the site to guide repair) [125] also contributes to the repair of radiation-induced DSB; the radio-sensitizing effects of MMEJ deficiency depend upon cell type and genetic context [126,127]. However, role of NHEJ, HR and MMEJ still remains unexplored in repair of CIR induced damage in FC cohort.
SSBs, abasic sites, and oxidative DNA damage, the most abundant types of DNA lesions produced by IR, are primarily repaired by the base excision repair (BER) machinery [128]. Several DNA glycosylases including hNTH1(endonuclease III homolog), hOGG1 (8-oxoguanine DNA glycosylase), NEIL1 (Nei like DNA glycosylase 1), initiate repair of oxidative DNA damage induced by IR, and this processing can lead to their conversion to more dangerous SSB and DSB [129]. The essential BER protein apurinic/apyrimidinic endonuclease 1 (APE1) plays a vital role in processing IR-induced oxidative DNA damage and clustered breaks [130]. APE1 deficiency sensitizes cells to IR despite resulting in a smaller number of radiations induced DSBs, presumably arising when replication forks collide with SSB intermediates downstream of APE1. Failure to resolve radiation-induced clustered DNA lesions can result in the accumulation of genetically unstable cells [131]. BER can be presumed to play an important role in CIR induced DNA damage but has not been studied directly in FC.
Bulky DNA adducts, IR induced inter-strand crosslinks, cyclopurines and variety of oxidative lesions are repaired by the nucleotide excision repair (NER) pathway. Although NER-deficient cells are not hypersensitive to IR, genetic polymorphisms in some NER genes have been associated with radiation-related cancers [132]. Some NER proteins are also involved in the repair of inter-strand crosslinks that IR can induce. IR also induces some types of DNA lesions that can be repaired by NER, such as cyclopurines [133], and a variety of oxidative lesions [134]. Little is known about the role of NER in the response to CIR-induced damage in FC. A study evaluating FC cumulative CIR dose exposure (assuming an exposure rate of 6 µSv/hr during flight) showed a significant correlation between CIR dose and NER activity in PBMCs measured by the unscheduled DNA synthesis assay [73]. This could reflect a healthy worker effect [18] but would also be consistent with a radio-adaptive response that has been proposed to underlie radiation hormesis [135] and has been observed in other settings [80,136].
Additional DNA repair pathways including Fanconi Anemia repair, and others are expected to play a role in the response to CIR-induced DNA damage but require further study. The Fanconi Anemia repair pathway eliminates inter-strand DNA crosslinks and protects cells from killing by IR. It is essential for maintaining genome integrity, and defective repair pathway have been associated with increased cancer risk and immune disorders [137]. Based on the complexity of CIR induced DNA damage, multiple DNA repair pathways are expected to be involved in its repair. To address this complexity, there is a need to investigate the role of multiple DNA repair mechanisms for CIR-induced DNA damage in FC. Taken together, these results underscore the importance of the involvement of multiple repair pathways in response to CIR based on the complexity of induced DNA damage and also the need for additional mechanistic studies aimed at understanding the origins of differential sensitivity to high- versus low-LET IR in FC. Such studies are expected to assist in evaluating the potential health effects of in-flight exposure and the eventual design strategies to mitigate them.
5. Discussion and Conclusion
Due to the complex mixture of mutagenic stressors encountered during air travel, there are many challenges in accurately assessing FC exposure to CIR and relating it to associated health risks. CIR exposure changes with altitude and latitude [8,138]. Dosimeters that can provide real-time CIR exposure levels are not regularly used aboard aircraft during flight. Considering the unique characteristics of CIR and the dose and dose-rate exposure for FC, efforts to link flight-related CIR exposure to health are challenging and perhaps rooted in incorrect assumptions based on our understanding of simple photon radiation. CIR at altitude is comprised mainly of neutron particles, but the biological effects of neutrons in combination with other radiation exposures are also understudied. Historically, the overwhelming majority of pilots have been men, but the representation of women has increased drastically in the last decade [139]. Given the risks to reproductive health and fetus development in the case of pregnant FC [10], studies focusing on effects of CIR induced DNA damage in pilots who may become pregnant are particularly needed. Furthermore, although administrative interventions such as schedule rotation can decrease exposure for the individual, the strategy requires a larger pool of workers. As a result, this strategy may not be economically viable.
Radiation-related health outcomes followed in other contexts have often been based on extremely high acute dose exposures due to single events such as nuclear disasters or atomic bomb detonation. This radiation exposure risk differs in particle composition and concerning the chronic low-dose exposure observed in flight. Drawing inferences about the risks associated with flight-related CIR exposure based on studies of naturally occurring background sources of radiation is subject to similar caveats. From a risk mitigation standpoint, the hierarchy of controls [140] generally used to protect workers from occupational hazards may not apply to FC.
Several epidemiological studies investigating whether cancer risk is associated with CIR exposure have yielded varying results that might be explained in partly due to the differences in study design, and the method of estimating exposure [8]. As a result, the biological mechanism underlying excess cancer risk in FC remains to be determined. Laboratory-based studies focused on the biological effects of mixed exposures that more accurately reflect the conditions experienced by FC would be valuable. Future studies will be most impactful if careful consideration is given to the choice of the control population to be compared. Because FC must perform physically and emotionally demanding tasks to remain in the workforce, studies comparing FC to reference populations performing other types of work are likely subject to a form of selection biased known as the “healthy worker effect,” [8,18,141]. The healthy worker effect complicates efforts to estimate the health effects of occupational exposures, because their health status may be higher than that of the reference group. Thus, if is not adequately addressed, the healthy worker effect might be expected to mask adverse health effects of occupational exposures among FC.
Since carcinogenesis is driven by genome instability, we propose a testable model wherein the elevated cancer risk in FC can be explained at least in part by occupational exposure to CIR and other genotoxic agents. CIR exposure may promote genome instability via its direct DNA damaging effects, and the effects may potentially be enhanced by other FC exposures that either also damage DNA or lead to the suppression of DNA repair activity (Figure 5). Given the higher abundance of endogenously produced DNA lesions, identifying the CIR induced DNA damage in FC may require the development of new, highly sensitive, and specific molecular assays. Cytogenetic analysis has provided indirect evidence of CIR induced genome instability and is a valuable approach that should be included in future, larger studies. PBMCs provide invaluable insights into in vivo human responses to environmental and occupational genotoxic exposures. Emerging high throughput technologies to quantify DDR such as the CometChip assay [142], Rapid Automated Biodosimetry Technology (RABiT) [143], γ-H2AX [143], Fluorescence-based multiplexed host cell reactivation (FM-HCR) [26,144,145] and many others can help assess personalized risk and exposures and are compatible with the limited quantity of blood that can usually be obtained from study FC participants [146]. Further improvements to these technologies that enhance the specificity of these assays for CIR induced biological changes will create new opportunities for their application to the answering questions about the biological effects of CIR. These technologies can also potentially identify vulnerable individuals using predictive models or DNA repair biomarkers to assess risk and inform cancer surveillance strategies [26,147]. There is substantial evidence of inter-individual variation in DRC correlating to disease risk and radiation sensitivity [48,148,149]. Additional biomarkers of genome instability such as telomere length [150] and clonal hematopoiesis of indeterminate potential [151] may further clarify the possible role of CIR as an environmental mutagen.
Integrating omics approaches and functional assays together with accurate CIR dosimetry into FC studies has the potential to reveal whether CIR exposure causes cancer in FC. These studies may also enable both global and personalized strategies for cancer prevention [146]. Ultimately, the field will require a multidisciplinary approach incorporating epidemiologists, engineers, and mechanistic biologists to effectively measure and study how complex exposure interactions in the real-world flight environment impact the genomic integrity of the FC [12].
Author Contributions
SMT: Conceived and designed the work that led to the submission; SMT, CS, IM, ZDN: reviewed and acquired information gathered from publications; EM, ZDN: supervised work, acquired funding, and revised and approved the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Acknowledgments
The authors thank Pataje Prasanna, Radiation Research Program, National Cancer Institute, for comments and helpful suggestions. Zachary D. Nagel and Sneh M. Toprani are supported by grants from the National Institutes of Health (R37CA248565, U01ES029520), as well as a pilot grant from the Harvard-NIEHS Center for Environmental Health (P30ES000002).
Conflicts of Interest
The author(s) declare no competing financial interest in relation to the work described.
References
- National Council on Radiation Protection and Measurements Ionizing Radiation Exposure of the Population of the United States. Report No. 160. Council on Radiation Protection and Measurements (NCRP). Bethesda, MD:; 2009.
- Lin, E.C. Radiation Risk From Medical Imaging. Mayo Clin Proc 2010, 85, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- UNSCEAR Annex C. Biological Mechanisms Relevant for the Inference of Cancer Risks from Low-Dose and Low-Dose-RateradiationUNSCEAR 2020/2021 Report(Vienna: UNSCEAR); 2021.
- Aw, J. Cosmic Radiation and Commercial Air Travel. J Travel Med 2003, 10, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Goldhagen, P. Overview of Aircraft Radiation Exposure and Recent ER-2 Measurements. Health Phys 2000, 79, 526–544. [Google Scholar] [CrossRef] [PubMed]
- Lantos, P.; Fuller, N. History of the Solar Particle Event Radiation Doses On-Board Aeroplanes Using a Semi-Empirical Model and Concorde Measurements. Radiat Prot Dosimetry 2003, 104, 199–210. [Google Scholar] [CrossRef] [PubMed]
- McNeely, E.; Gale, S.; Tager, I.; Kincl, L.; Bradley, J.; Coull, B.; Hecker, S. The Self-Reported Health of U. S. Flight Attendants Compared to the General Population. 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- Scheibler, C.; Toprani, S.M.; Mordukhovich, I.; Schaefer, M.; Staffa, S.; Nagel, Z.D.; McNeely, E. Cancer Risks from Cosmic Radiation Exposure in Flight: A Review. Front public Heal 2022, 10. [Google Scholar] [CrossRef]
- Griffiths, R.F.; Powell, D.M.C. The Occupational Health and Safety of Flight Attendants. Aviat Space Environ Med 2012, 83, 514–521. [Google Scholar] [CrossRef]
- McNeely, E.; Mordukhovich, I.; Tideman, S.; Gale, S.; Coull, B. Estimating the Health Consequences of Flight Attendant Work: Comparing Flight Attendant Health to the General Population in a Cross-Sectional Study. BMC Public Health 2018, 18, 1–11. [Google Scholar] [CrossRef]
- Bramlitt, E.T.; Shonka, J.J. Radiation Exposure of Aviation Crewmembers and Cancer. Health Phys 2015, 108, 76–86. [Google Scholar] [CrossRef]
- Toprani, S.M.; Scheibler, C.; Nagel, Z.D. Interplay Between Air Travel, Genome Integrity, and COVID-19 Risk Vis-a-Vis Flight Crew. Front Public Heal 2020, 8, 1–6. [Google Scholar] [CrossRef]
- McNeely, E.; Mordukhovich, I.; Staffa, S.; Tideman, S.; Coull, B. Legacy Health Effects among Never Smokers Exposed to Occupational Secondhand Smoke. 2019, 14. [CrossRef]
- Pinkerton, L.E.; Hein, M.J.; Anderson, J.L.; Little, M.P.; Sigurdson, A.J.; Schubauer-Berigan, M.K. Breast Cancer Incidence among Female Flight Attendants: Exposure–Response Analyses. Scand J Work Environ Heal 2016, 42, 538–546. [Google Scholar] [CrossRef] [PubMed]
- McNeely, E.; Mordukhovich, I.; Staffa, S.; Tideman, S.; Gale, S.; Coull, B. Cancer Prevalence among Flight Attendants Compared to the General Population. Environ Health 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Pukkala, E.; Helminen, M.; Haldorsen, T.; Hammar, N.; Kojo, K.; Linnersjö, A.; Rafnsson, V.; Tulinius, H.; Tveten, U.; Auvinen, A. Cancer Incidence among Nordic Airline Cabin Crew. Int J cancer 2012, 131, 2886–2897. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, L.E.; Hein, M.J.; Anderson, J.L.; Christianson, A.; Little, M.P.; Sigurdson, A.J.; Schubauer-Berigan, M.K. Melanoma, Thyroid Cancer, and Gynecologic Cancers in a Cohort of Female Flight Attendants. 2018, 61, 572–581.
- Schüz, J. Airline Crew Cohorts: Is There More to Learn Regarding Their Cancer Risk? Occup Environ Med 2014, 71, 307. [Google Scholar] [CrossRef] [PubMed]
- Nuclear Regulatory Commission (NRC) Biological Effects of Radiation. U.S. Nuclear Regulatory Commission Backgrounder (2017); 2017.
- ICRP International Commission on Radiological Protection (ICRP). Publication 103: Recommendations of the International Commission on Radiological Protection. Radiat Prot Dosim 2008, 129, 500–507. [Google Scholar]
- Sridharan, D.M.; Asaithamby, A.; Blattnig, S.R.; Costes, S. V.; Doetsch, P.W.; Dynan, W.S.; Hahnfeldt, P.; Hlatky, L.; Kidane, Y.; Kronenberg, A.; et al. Evaluating Biomarkers to Model Cancer Risk Post Cosmic Ray Exposure. Life Sci Sp Res 2016, 9, 19–47. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Barnes, D.E. Repair of Endogenous DNA Damage. Cold Spring Harb Symp Quant Biol 2000, 65, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 7th Edition; Wolters Kluwer Health Adis (ESP), 2012; ISBN 10: 1608311937.
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological Consequences of Radiation-Induced DNA Damage: Relevance to Radiotherapy. Clin Oncol (R Coll Radiol) 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Nagel, Z.D.; Chaim, I.A.; Samson, L.D. Inter-Individual Variation in DNA Repair Capacity: A Need for Multi-Pathway Functional Assays to Promote Translational DNA Repair Research. DNA Repair (Amst) 2014, 19, 199–213. [Google Scholar] [CrossRef]
- Nelson, B.C.; Dizdaroglu, M. Implications of DNA Damage and DNA Repair on Human Diseases. Mutagenesis 2020, 35, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.S.; Jorgensen, T.J.; Kennedy, A.R.; Boice, J.D.; Shapiro, A.; Hu, T.C.C.; Moyer, B.R.; Grace, M.B.; Kelloff, G.J.; Fenech, M.; et al. Mitigating the Risk of Radiation-Induced Cancers: Limitations and Paradigms in Drug Development. J Radiol Prot 2014, 34. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.T. Radiation Protection Aspects of the Cosmic Radiation Exposure of Aircraft Crew. 2004, 109, 349–355.
- Silva, R.; Folgosa, F.; Soares, P.; Pereira, A.S.; Garcia, R.; Gestal-Otero, J.J.; Tavares, P.; Gomes Da Silva, M.D.R. Occupational Cosmic Radiation Exposure in Portuguese Airline Pilots: Study of a Possible Correlation with Oxidative Biological Markers. Radiat Environ Biophys 2013, 52, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Mishev, A. Short- and Medium-Term Induced Ionization in the Earth Atmosphere by Galactic and Solar Cosmic Rays. Int J Atmos Sci 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Usoskin, I.G.; Desorgher, L.; Velinov, P.; Storini, M.; Flückiger, E.O.; Bütikofer, R.; Kovaltsov, G.A. Ionization of the Earth’s Atmosphere by Solar and Galactic Cosmic Rays. Acta Geophys 2009 571 2008, 57, 88–101. [Google Scholar] [CrossRef]
- Phillips, T.; Johnson, S.; Koske-Phillips, A.; White, M.; Yarborough, A.; Lamb, A.; Herbst, A.; Molina, F.; Gilpin, J.; Grah, O.; et al. Space Weather Ballooning. Sp Weather 2016, 14, 697–703. [Google Scholar] [CrossRef]
- Hands, A.D.P.; Ryden, K.A.; Mertens, C.J. The Disappearance of the Pfotzer-Regener Maximum in Dose Equivalent Measurements in the Stratosphere. Sp Weather 2016, 14, 776–785. [Google Scholar] [CrossRef]
- Desmaris, G. Cosmic Radiation in Aviation: Radiological Protection of Air France Aircraft Crew. Ann ICRP 2016, 45, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Blettner, M.; Grosche, B.; Zeeb, H. Occupational Cancer Risk in Pilots and Flight Attendants: Current Epidemiological Knowledge. Radiat Environ Biophys 1998, 37, 75–80. [Google Scholar] [CrossRef]
- Friedberg, W., Faulkner, DN., Snyder, L., Darden, EB Jr., and O’Brien, K. Galactic Cosmic Radiation Exposure and Associated Health Risks for Air Carrier Crewmembers. Aviat Sp Env Med 1989, 60, 1104–1108.
- Dreger, S.; Wollschläger, D.; Schafft, T.; Hammer, G.P.; Blettner, M.; Zeeb, H. Cohort Study of Occupational Cosmic Radiation Dose and Cancer Mortality in German Aircrew, 1960-2014. 2020, 77, 285–291.
- Federal Aviation Administration (FAA) In-Flight Radiation Exposure. US Department of Transportation, FAA Advisory Circular 120–61B.; 2014.
- Bottollier-Depois, J.F.; Chau, Q.; Bouisset, P.; Kerlau, G.; Plawinski, L.; Lebaron-Jacobs, L. Assessing Exposure to Cosmic Radiation on Board Aircraft. Adv Sp Res 2003, 32, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, P.J. Estimated Individual Annual Cosmic Radiation Doses for Flight Crews. Aviat Space Environ Med 1998, 69, 621–625. [Google Scholar] [PubMed]
- Grajewski, B.; Yong, L.C.; Bertke, S.J.; Bhatti, P.; Little, M.P.; Ramsey, M.J.; Tucker, J.D.; Ward, E.M.; Whelan, E.A.; Sigurdson, A.J.; et al. Chromosome Translocations and Cosmic Radiation Dose in Male U.S. Commercial Airline Pilots. Aerosp Med Hum Perform 2018, 89, 616–625. [Google Scholar] [CrossRef] [PubMed]
- EURADOS Comparison of Codes Assessing Radiation Exposure of Aircraft Crew Due to Galactic Cosmic Radiation, EURADOS Report 2012-03.; Braunschweig, 2012.
- Toprani, S.M.S.M.; Das, B. Radio-Adaptive Response of Base Excision Repair Genes and Proteins in Human Peripheral Blood Mononuclear Cells Exposed to Gamma Radiation. Mutagenesis 2015, 30, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.; Koliada, A.; Zabuga, O.; Socol, Y. Health Impacts of Low-Dose Ionizing Radiation: Current Scientific Debates and Regulatory Issues. Dose Response 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Toprani, S.M.; Das, B. Role of Base Excision Repair Genes and Proteins in Gamma-Irradiated Resting Human Peripheral Blood Mononuclear Cells. Mutagenesis 2015, 30, 247–261. [Google Scholar] [CrossRef]
- Calabrese, E.J. The Additive to Background Assumption in Cancer Risk Assessment: A Reappraisal. Environ Res 2018, 166, 175–204. [Google Scholar] [CrossRef]
- Toprani, S.M.; Das, B. Radio-Adaptive Response, Individual Radio-Sensitivity and Correlation of Base Excision Repair Gene Polymorphism (HOGG1, APE1, XRCC1, and LIGASE1) in Human Peripheral Blood Mononuclear Cells Exposed to Gamma Radiation. Environ Mol Mutagen 2020, 61, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Komov, O.; Krasavin, E.; Nasonov, E.; Mel’nikov, L.; Shmakov, N.; Cunh, M.; Testa, E.; Beuve, M. Relationship between Radioadaptive Response and Individual Radiosensitivity to Low Doses of Gamma Radiation: An Extended Study of Chromosome Damage in Blood Lymphocytes of Three Donors. Int J Radiat Biol 2018, 94, 54–61. [Google Scholar] [CrossRef]
- Nenoi, M.; Wang, B.; Vares, G. In Vivo Radioadaptive Response: A Review of Studies Relevant to Radiation-Induced Cancer Risk. Hum Exp Toxicol 2015, 34, 272. [Google Scholar] [CrossRef]
- Bolzán, A.D.; Bianchi, M.S.; Giménez, E.M.; Flaqué, M.C.D.; Ciancio, V.R. Analysis of Spontaneous and Bleomycin-Induced Chromosome Damage in Peripheral Lymphocytes of Long-Haul Aircrew Members from Argentina. Mutat Res 2008, 639, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC) Radiation Emergencies Factsheet – Radiation and Pregnancy: A Fact Sheet for the Public. Department of Health and Human Services, Updated 15 Nov 2011; 2011.
- Williams, P.; Fletcher, S. Health Effects of Prenatal Radiation Exposure. Am Fam Physician 2011, 82, 488–493. [Google Scholar]
- United Nations. Scientific Committee on the Effects of Atomic Radiation.; United Nations. General Assembly. Genetic and Somatic Effects of Ionizing Radiation : United Nations Scientific Committee on the Effects of Atomic Radiation : 1986 Report to the General Assembly, with Annexes. 1986, 366.
- Valentin, J.; Cox, R.; Streffer, C. Biological Effects after Prenatal Irradiation (Embryo and Fetus). 2003, 33, 1–206. [CrossRef]
- Saada, M.; Sanchez-Jimenez, E.; Roguin, A. Risk of Ionizing Radiation in Pregnancy: Just a Myth or a Real Concern? Europace 2023, 25, 270. [Google Scholar] [CrossRef] [PubMed]
- Grajewski, B.; Whelan, E.A.; Lawson, C.C.; Hein, M.J.; Waters, M.A.; Anderson, J.L.; Macdonald, L.A.; Mertens, C.J.; Tseng, C.Y.; Cassinelli, R.T.; et al. Miscarriage among Flight Attendants. Epidemiology 2015, 26, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Cone, J.E.; Vaughan, L.M.; Huete, A.; Samuels, S.J. Reproductive Health Outcomes among Female Flight Attendants: An Exploratory Study. J Occup Environ Med 1998, 40, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Lauria, L.; Ballard, T.J.; Caldora, M.; Mazzanti, C.; Verdecchia, A. Reproductive Disorders and Pregnancy Outcomes among Female Flight Attendants. Aviat Sp Env Med 2006, 77, 533–539. [Google Scholar]
- Irgens, Å.; Irgens, L.M.; Reitan, J.B.; Haldorsen, T.; Tveten, U. Pregnancy Outcome among Offspring of Airline Pilots and Cabin Attendants. Scand J Work Environ Health 2003, 29, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, W.; Chan, A.; Li, C.; He, X.; Cui, L.; Lv, Y.; Liu, J.; Guo, X. An Epidemiological Study of Reproductive Health in Female Civil Aviation Employees. Aviat Space Environ Med 2013, 84, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Trzeciak, A.R.; Barnes, J.; Ejiogu, N.; Foster, K.; Brant, L.J.; Zonderman, A.B.; Evans, M.K. Age, Sex, and Race Influence Single-Strand Break Repair Capacity in a Human Population. Free Radic Biol Med 2008, 45, 1631–1641. [Google Scholar] [CrossRef]
- Garm, C.; Moreno-Villanueva, M.; Bürkle, A.; Petersen, I.; Bohr, V.A.; Christensen, K.; Stevnsner, T. Age and Gender Effects on DNA Strand Break Repair in Peripheral Blood Mononuclear Cells. 2013, 12, 58–66. [CrossRef]
- Scott, D.; Barber, J.B.P.; Levine, E.L.; Burrill, W.; Roberts, S.A. Radiation-Induced Micronucleus Induction in Lymphocytes Identifies a High Frequency of Radiosensitive Cases among Breast Cancer Patients: A Test for Predisposition? Br J Cancer 1998, 77, 614–620. [Google Scholar] [CrossRef]
- Distel, L.V.R.; Neubauer, S.; Keller, U.; Sprung, C.N.; Sauer, R.; Grabenbauer, G.G. Individual Differences in Chromosomal Aberrations after in Vitro Irradiation of Cells from Healthy Individuals, Cancer and Cancer Susceptibility Syndrome Patients. Radiother Oncol 2006, 81, 257–263. [Google Scholar] [CrossRef]
- Köberle, B.; Koch, B.; Fischer, B.M.; Hartwig, A. Single Nucleotide Polymorphisms in DNA Repair Genes and Putative Cancer Risk. Arch Toxicol 2016 9010 2016, 90, 2369–2388. [Google Scholar] [CrossRef]
- Heimers, A. Chromosome Aberration Analysis in Concorde Pilots. Mutat Res 2000, 467, 169–176. [Google Scholar] [CrossRef]
- Romano, E.; Ferrucci, L.; Nicolai, F.; Derme, V.; De Stefano, G.F. Increase of Chromosomal Aberrations Induced by Ionising Radiation in Peripheral Blood Lymphocytes of Civil Aviation Pilots and Crew Members. Mutat Res 1997, 377, 89–93. [Google Scholar] [CrossRef]
- Nicholas, J.S.; Butler, G.C.; Davis, S.; Bryant, E.; Hoel, D.G.; Mohr, L.C. Stable Chromosome Aberrations and Ionizing Radiation in Airline Pilots. Aviat Space Environ Med 2003, 74, 953–956. [Google Scholar]
- Cavallo, D.; Marinaccio, A.; Perniconi, B.; Tomao, P.; Pecoriello, V.; Moccaldi, R.; Iavicoli, S. Chromosomal Aberrations in Long-Haul Air Crew Members. Mutat Res 2002, 513, 11–15. [Google Scholar] [CrossRef]
- Yong, L.C.; Sigurdson, A.J.; Ward, E.M.; Waters, M.A.; Whelan, E.A.; Petersen, M.R.; Bhatti, P.; Ramsey, M.J.; Ron, E.; Tucker, J.D. Increased Frequency of Chromosome Translocations in Airline Pilots with Long-Term Flying Experience. Occup Environ Med 2009, 66. [Google Scholar] [CrossRef]
- Chen, W.; Feng, Y.; Liu, J.; Zhang, H.; Guo, Y.; Zhang, Y.; Duan, S.; Peng, X.; Sun, T.; Jia, B.; et al. Influence of Cosmic Radiation on Lymphocyte Micronucleus, Serum Lipid Peroxide and Antioxidation Capacity in Aircrew Members. Chinese Sci Bull 2002 478 2002, 47, 647–653. [Google Scholar] [CrossRef]
- Zwingmann, I.; Welle, I.; van Herwijen, M.; Engelen, J.; Schilderman, P.; Smid, T.; Kleinjans, J. Oxidative DNA Damage and Cytogenetic Effects in Flight Engineers Exposed to Cosmic Radiation - PubMed. Env Mol Mutagen 1998, 32, 121–129. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy. Genes (Basel) 2020, 11. [Google Scholar] [CrossRef]
- Nuszkiewicz, J.; Woźniak, A.; Szewczyk-Golec, K. Ionizing Radiation as a Source of Oxidative Stress—The Protective Role of Melatonin and Vitamin D. Int J Mol Sci 2020, 21, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Hada, M.; Georgakilas, A.G. Formation of Clustered DNA Damage after High-LET Irradiation: A Review. J Radiat Res 2008, 49, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jella, K.K.; Jaafar, L.; Li, S.; Park, S.; Story, M.D.; Wang, H.; Wang, Y.; Dynan, W.S. Exposure to Galactic Cosmic Radiation Compromises DNA Repair and Increases the Potential for Oncogenic Chromosomal Rearrangement in Bronchial Epithelial Cells. Sci Rep 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Asaithamby, A.; Chen, D.J. Mechanism of Cluster DNA Damage Repair in Response to High-Atomic Number and Energy Particles Radiation. Mutat Res 2011, 711, 87. [Google Scholar] [CrossRef]
- Campa, A.; Alloni, D.; Antonelli, F.; Ballarini, F.; Belli, M.; Dini, V.; Esposito, G.; Facoetti, A.; Friedland, W.; Furusawa, Y.; et al. DNA Fragmentation Induced in Human Fibroblasts by 56Fe Ions: Experimental Data and Monte Carlo Simulations. Radiat Res 2009, 171, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Nagamatsu, A.; Nenoi, M.; Fujimori, A.; Kakinuma, S.; Katsube, T.; Wang, B.; Tsuruoka, C.; Shirai, T.; Nakamura, A.J.; et al. Space Radiation Biology for “Living in Space. ” Biomed Res Int 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Juerb, D.; Zwar, M.; Giesen, U.; Nolte, R.; Kriesen, S.; Baiocco, G.; Puchalska, M.; van Goethem, M.J.; Manda, K.; Hildebrandt, G. Comparative Study of the Effects of Different Radiation Qualities on Normal Human Breast Cells. Radiat Oncol 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.; Dai, X. The Relationship between DNA Single-Stranded Damage Response and Double-Stranded Damage Response. Cell Cycle 2018, 17, 73–79. [Google Scholar] [CrossRef]
- Ward, J.F. Some Biochemical Consequences of the Spatial Distribution of Ionizing Radiation-Produced Free Radicals. Radiat Res 1981, 86, 185–195. [Google Scholar] [CrossRef]
- Zhang, X.; Ye, C.; Sun, F.; Wei, W.; Hu, B.; Wang, J. Both Complexity and Location of DNA Damage Contribute to Cellular Senescence Induced by Ionizing Radiation. PLoS One 2016, 11. [Google Scholar] [CrossRef]
- Prasanna, P.G.S.; Escalada, N.D.; Blakely, W.F. Induction of Premature Chromosome Condensation by a Phosphatase Inhibitor and a Protein Kinase in Unstimulated Human Peripheral Blood Lymphocytes: A Simple and Rapid Technique to Study Chromosome Aberrations Using Specific Whole-Chromosome DNA Hybridization Probes for Biological Dosimetry. Mutat Res 2000, 466, 131–141. [Google Scholar] [CrossRef]
- Pujol-Canadell, M.; Perrier, J.R.; Cunha, L.; Shuryak, I.; Harken, A.; Garty, G.; Brenner, D.J. Cytogenetically-Based Biodosimetry after High Doses of Radiation. PLoS One 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Leatherbarrow, E.L.; Harper, J. V.; Cucinotta, F.A.; O’Neill, P. Induction and Quantification of Gamma-H2AX Foci Following Low and High LET-Irradiation. Int J Radiat Biol 2006, 82, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Rose Li, Y.; Halliwill, K.D.; Adams, C.J.; Iyer, V.; Riva, L.; Mamunur, R.; Jen, K.Y.; del Rosario, R.; Fredlund, E.; Hirst, G.; et al. Mutational Signatures in Tumours Induced by High and Low Energy Radiation in Trp53 Deficient Mice. Nat Commun 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, A.; Nasonova, E.; Czerski, K.; Kutsalo, P.; Pereira, W.; Krasavin, E. Production and Distribution of Chromosome Aberrations in Human Lymphocytes by Particle Beams with Different LET. Radiat Environ Biophys 2019, 58, 99. [Google Scholar] [CrossRef]
- Nair, S.; Engelbrecht, M.; Miles, X.; Ndimba, R.; Fisher, R.; du Plessis, P.; Bolcaen, J.; Nieto-Camero, J.; de Kock, E.; Vandevoorde, C. The Impact of Dose Rate on DNA Double-Strand Break Formation and Repair in Human Lymphocytes Exposed to Fast Neutron Irradiation. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Grayson, J.; Lyons, T. Brain Cancer, Flying, and Socioeconomic Status: A Nested Case-Control Study of USAF Aircrew - PubMed. Aviat Sp Env Med 1996, 67, 1152–1154. [Google Scholar]
- Mortazavi, S. The Safety Issues of Onboard Wi-Fi: Possible Interactions of Oxidative Stress-Causing High Altitude, Cosmic Radiation, and Wi-Fi Radiation. React Oxyg Species 2017, 4, 441–444. [Google Scholar] [CrossRef]
- Akdag, M.Z.; Dasdag, S.; Canturk, F.; Karabulut, D.; Caner, Y.; Adalier, N. Does Prolonged Radiofrequency Radiation Emitted from Wi-Fi Devices Induce DNA Damage in Various Tissues of Rats? J Chem Neuroanat 2016, 75, 116–122. [Google Scholar] [CrossRef]
- Bodewein, L.; Schmiedchen, K.; Dechent, D.; Stunder, D.; Graefrath, D.; Winter, L.; Kraus, T.; Driessen, S. Systematic Review on the Biological Effects of Electric, Magnetic and Electromagnetic Fields in the Intermediate Frequency Range (300 Hz to 1 MHz). Environ Res 2019, 171, 247–259. [Google Scholar] [CrossRef]
- Cavallo, D.; Tomao, P.; Marinaccio, A.; Perniconi, B.; Setini, A.; Palmi, S.; Iavicoli, S. Evaluation of DNA Damage in Flight Personnel by Comet Assay. 2002, 516, 148–152. [CrossRef]
- Nelson, G. Fundamental Space Radiobiology. Gravit Sp Biol Bull 2003, 16, 29–36. [Google Scholar]
- Seth, I.; Schwartz, J.L.; Stewart, R.D.; Emery, R.; Joiner, M.C.; Tucker, J.D. Neutron Exposures in Human Cells: Bystander Effect and Relative Biological Effectiveness. PLoS One 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Wang, H.; Perrault, A.R.; Boecker, W.; Rosidi, B.; Windhofer, F.; Wu, W.; Guan, J.; Terzoudi, G.; Panteliasc, G. Mechanisms of DNA Double Strand Break Repair and Chromosome Aberration Formation. Cytogenet Genome Res 2004, 104, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Bauchinger, M. Quantification of Low-Level Radiation Exposure by Conventional Chromosome Aberration Analysis. Mutat Res Genet Toxicol 1995, 339, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Cologne, J.; Sugiyama, H.; Hamasaki, K.; Tatsukawa, Y.; French, B.; Sakata, R.; Misumi, M. Chromosome Aberrations among Atomic-Bomb Survivors Exposed in Utero: Updated Analysis Accounting for Revised Radiation Doses and Smoking. Radiat Environ Biophys 2022, 61, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Ainsbury, E.A.; Livingston, G.K.; Abbott, M.G.; Moquet, J.E.; Hone, P.A.; Jenkins, M.S.; Christensen, D.M.; Lloyd, D.C.; Rothkamm, K. Interlaboratory Variation in Scoring Dicentric Chromosomes in a Case of Partial-Body x-Ray Exposure: Implications for Biodosimetry Networking and Cytogenetic “Triage Mode” Scoring. Radiat Res 2009, 172, 746–752. [Google Scholar] [CrossRef] [PubMed]
- CA, K.; Heintz, P.; Sandoval, D.; Chambers, G.; Adolphi, N.; Paffett, K. Radiation Effects on Tissues and Organs; John Wiley & Sons, Ltd., 2014.
- Ministry of the Environment Government of Japan (JCN1000012110001) Radiosensitivity of Organs and Tissues [MOE]. In Booklet to Provide Basic Information Regarding Health Effects of Radiation; 2013.
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of Apoptosis in Health and Disease: The Balancing Act of BCL-2 Family Proteins. Nat Rev Mol Cell Biol 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Powell, D.R.; Li, Z.; Bell, J.S.K.; Barwick, B.G.; Feng, H.; McCrary, M.R.; Dwivedi, B.; Kowalski, J.; Dynan, W.S.; et al. Galactic Cosmic Radiation Induces Persistent Epigenome Alterations Relevant to Human Lung Cancer. Sci Rep 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Wilson, D.M. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging; Am J Hum Genet, 2019; Vol. 105, pp. 237–257.
- Limoli, C.L.; Ponnaiya, B.; Corcoran, J.J.; Giedzinski, E.; Kaplan, M.I.; Hartmann, A.; Morgan, W.F. Genomic Instability Induced by High and Low LET Ionizing Radiation. Adv Space Res 2000, 25, 2107–2117. [Google Scholar] [CrossRef]
- Y, Y.; Dai, W. Genomic Instability and Cancer. J Carcinog Mutagen 2015, 5, 1000165. [Google Scholar]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ Mol Mutagen 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- IARC (International Agency for Research on Cancer) IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Tobacco Smoke and Involuntary Smoking. Lyon (FR): International Agency for Research on Cancer; 2004. (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 83.); 2004.
- IARC (International Agency for Research on Cancer) IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Painting, Firefighting, and Shiftwork. Lyon (FR): International Agency for Research on Cancer; 2010. (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 98.); 2010.
- da Silva, J. DNA Damage Induced by Occupational and Environmental Exposure to Miscellaneous Chemicals. Mutat Res Rev Mutat Res 2016, 770, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Brzozowska, B.; Sollazzo, A.; Lundholm, L.; Lisowska, H.; Haghdoost, S.; Wojcik, A. Simultaneous Induction of Dispersed and Clustered DNA Lesions Compromises DNA Damage Response in Human Peripheral Blood Lymphocytes. PLoS One 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Soren, D.C.; Toprani, S.M.; Jain, V.; Saini, D.; Das, B. Quantitation of Genome Damage and Transcriptional Profile of DNA Damage Response Genes in Human Peripheral Blood Mononuclear Cells Exposed in Vitro to Low Doses of Neutron Radiation. Int J Radiat Res 2019, 17, 1–14. [Google Scholar]
- Roobol, S.J.; van den Bent, I.; van Cappellen, W.A.; Abraham, T.E.; Paul, M.W.; Kanaar, R.; Houtsmuller, A.B.; van Gent, D.C.; Essers, J. Comparison of High- and Low-LET Radiation-Induced DNA Double-Strand Break Processing in Living Cells. Int J Mol Sci 2020, 21, 1–19. [Google Scholar] [CrossRef]
- Slatter, M.A.; Gennery, A.R. Update on DNA-Double Strand Break Repair Defects in Combined Primary Immunodeficiency. Curr Allergy Asthma Rep 2020, 20. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Lewis, S.; Wlodarski, M.W. DNA Repair Syndromes and Cancer: Insights Into Genetics and Phenotype Patterns. Front Pediatr 2020, 8. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, B.P.C.; Chen, D.J. DNA-PK: A Dynamic Enzyme in a Versatile DSB Repair Pathway. DNA Repair (Amst) 2014, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Pang, D.; Winters, T.A.; Jung, M.; Purkayastha, S.; Cavalli, L.R.; Chasovkikh, S.; Haddad, B.R.; Dritschilo, A. Radiation-Generated Short DNA Fragments May Perturb Non-Homologous End-Joining and Induce Genomic Instability. J Radiat Res 2011, 52, 309–319. [Google Scholar] [CrossRef]
- Yuan, Y.; Britton, S.; Delteil, C.; Coates, J.; Jackson, S.P.; Barboule, N.; Frit, P.; Calsou, P. Single-Stranded DNA Oligomers Stimulate Error-Prone Alternative Repair of DNA Double-Strand Breaks through Hijacking Ku Protein. Nucleic Acids Res 2015, 43, 10264–10276. [Google Scholar] [CrossRef]
- Schmid, T.E.; Dollinger, G.; Beisker, W.; Hable, V.; Greubel, C.; Auer, S.; Mittag, A.; Tarnok, A.; Friedl, A.A.; Molls, M.; et al. Differences in the Kinetics of Gamma-H2AX Fluorescence Decay after Exposure to Low and High LET Radiation. Int J Radiat Biol 2010, 86, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.P.; Hirakawa, H.; Nakajima, N.I.; Moore, S.; Nie, J.; Sharma, N.; Sugiura, M.; Hoki, Y.; Araki, R.; Abe, M.; et al. Low- and High-LET Ionizing Radiation Induces Delayed Homologous Recombination That Persists for Two Weeks before Resolving. Radiat Res 2017, 188, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Grim, S.; Smith, L.E.; Kim, P.M.; Nickoloff, J.A.; Goloubeva, O.G.; Morgan, W.F. Ionizing Radiation Induces Delayed Hyperrecombination in Mammalian Cells. Mol Cell Biol 2004, 24, 5060–5068. [Google Scholar] [CrossRef] [PubMed]
- Grosse, N.; Fontana, A.O.; Hug, E.B.; Lomax, A.; Coray, A.; Augsburger, M.; Paganetti, H.; Sartori, A.A.; Pruschy, M. Deficiency in Homologous Recombination Renders Mammalian Cells More Sensitive to Proton versus Photon Irradiation. Int J Radiat Oncol Biol Phys 2014, 88, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-Mediated End Joining: Good, Bad and Ugly. Mutat Res - Fundam Mol Mech Mutagen 2018.
- Wood, R.D.; Doublié, S. DNA Polymerase θ (POLQ), Double-Strand Break Repair, and Cancer. DNA Repair (Amst) 2016, 44, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Smith, C.M.; Simpson, D.A.; Gupta, G.P. Targeting Non-Homologous and Alternative End Joining Repair to Enhance Cancer Radiosensitivity. Semin Radiat Oncol 2022, 32, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb Perspect Biol 2013, 5, 1–22. [Google Scholar] [CrossRef]
- Yang, N.; Galick, H.; Wallace, S.S. Attempted Base Excision Repair of Ionizing Radiation Damage in Human Lymphoblastoid Cells Produces Lethal and Mutagenic Double Strand Breaks. DNA Repair (Amst) 2004, 3, 1323–1334. [Google Scholar] [CrossRef]
- Fung, H.; Demple, B. Distinct Roles of Ape1 Protein in the Repair of DNA Damage Induced by Ionizing Radiation or Bleomycin. J Biol Chem 2011, 286, 4968–4977. [Google Scholar] [CrossRef]
- Eccles, L.J.; O’Neill, P.; Lomax, M.E. Delayed Repair of Radiation Induced Clustered DNA Damage: Friend or Foe? Mutat Res 2011, 711, 134–141. [Google Scholar] [CrossRef]
- Rajaraman, P.; Bhatti, P.; Doody, M.M.; Simon, S.L.; Weinstock, R.M.; Linet, M.S.; Rosenstein, M.; Stovall, M.; Alexander, B.H.; Preston, D.L.; et al. Nucleotide Excision Repair Polymorphisms May Modify Ionizing Radiation-Related Breast Cancer Risk in US Radiologic Technologists. Int J cancer 2008, 123, 2713–2716. [Google Scholar] [CrossRef] [PubMed]
- Terzidis, M.A.; Ferreri, C.; Chatgilialoglu, C. Radiation-Induced Formation of Purine Lesions in Single and Double Stranded DNA: Revised Quantification. Front Chem 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Raja, S.; Van Houten, B. The Involvement of Nucleotide Excision Repair Proteins in the Removal of Oxidative DNA Damage. Nucleic Acids Res 2020, 48, 11227–11243. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Mattson, M.P. How Does Hormesis Impact Biology, Toxicology, and Medicine? NPJ aging Mech Dis 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Latimer, J.J.; Alhamed, A.; Sveiven, S.; Almutairy, A.; Klimas, N.G.; Abreu, M.; Sullivan, K.; Grant, S.G. Preliminary Evidence for a Hormetic Effect on DNA Nucleotide Excision Repair in Veterans with Gulf War Illness. Mil Med 2020, 185, E47–E52. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Smogorzewska, A. Fanconi Anaemia and the Repair of Watson and Crick DNA Crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Sanlorenzo, M.; Wehner, M.R.; Linos, E.; Kornak, J.; Kainz, W.; Posch, C.; Vujic, I.; Johnston, K.; Gho, D.; Monico, G.; et al. The Risk of Melanoma in Airline Pilots and Cabin Crew: A Meta-Analysis. JAMA Dermatology 2015, 151, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Institute, P. Women Pilot Statistics: Female Representation in Aviation - Pilot Institute. Available online: https://pilotinstitute.com/women-aviation-statistics/#:~:text=This is a massive 151,compared to 6.03%25 in 2021. (accessed on 3 January 2023).
- National Institute for Occupational Safety and Health (NIOSH) Hierarchy of Controls. NIOSH Workplace Safety and Health Topics; 2015.
- Radon, K.; Goldberg, M.; Becklake, M. Healthy Worker Effect in Cohort Studies on Chronic Bronchitis. Scand J Work Environ Health 2002, 28, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Prasongtanakij, S.; Wood, D.K.; Weingeist, D.M.; Fessler, J.; Navasummrit, P.; Ruchirawat, M.; Engelward, B.P. Cometchip: A High-Throughput 96-Well Platform for Measuring DNA Damage in Microarrayed Human Cells. J Vis Exp 2014. [Google Scholar] [CrossRef]
- Garty, G.; Chen, Y.; Salerno, A.; Turner, H.; Zhang, J.; Lyulko, O.; Bertucci, A.; Xu, Y.; Wang, H.; Simaan, N.; et al. The RABIT: A Rapid Automated Biodosimetry Tool for Radiological Triage. Health Phys 2010, 98, 209–217. [Google Scholar] [CrossRef]
- Nagel, Z.D.; Margulies, C.M.; Chaim, I.A.; McRee, S.K.; Mazzucato, P.; Ahmad, A.; Abo, R.P.; Butty, V.L.; Forget, A.L.; Samson, L.D. Multiplexed DNA Repair Assays for Multiple Lesions and Multiple Doses via Transcription Inhibition and Transcriptional Mutagenesis. Proc Natl Acad Sci U S A 2014. [Google Scholar] [CrossRef] [PubMed]
- Nagel, Z.D.; Beharry, A.A.; Mazzucato, P.; Kitange, G.J.; Sarkaria, J.N.; Kool, E.T.; Samson, L.D. Fluorescent Reporter Assays Provide Direct, Accurate, Quantitative Measurements of MGMT Status in Human Cells. PLoS One 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Nagel, Z.D.; Engelward, B.P.; Brenner, D.J.; Begley, T.J.; Sobol, R.W.; Bielas, J.H.; Stambrook, P.J.; Wei, Q.; Hu, J.J.; Terry, M.B.; et al. Towards Precision Prevention: Technologies for Identifying Healthy Individuals with High Risk of Disease. Mutat Res - Fundam Mol Mech Mutagen 2017, 800–802, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Cheong, A.; Nagel, Z.D. Human Variation in DNA Repair, Immune Function, and Cancer Risk. Front Immunol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Hornhardt, S.; Rößler, U.; Sauter, W.; Rosenberger, A.; Illig, T.; Bickeböller, H.; Wichmann, H.E.; Gomolka, M. Genetic Factors in Individual Radiation Sensitivity. DNA Repair (Amst) 2014, 16, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.C.; Kehm, R.; Santella, R.M.; Brenner, D.J.; Terry, M.B. DNA Repair Phenotype and Cancer Risk: A Systematic Review and Meta-Analysis of 55 Case–Control Studies. Sci Reports 2022 121 2022, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Luxton, J.J.; McKenna, M.J.; Lewis, A.; Taylor, L.E.; George, K.A.; Dixit, S.M.; Moniz, M.; Benegas, W.; Mackay, M.J.; Mozsary, C.; et al. Telomere Length Dynamics and DNA Damage Responses Associated with Long-Duration Spaceflight. Cell Rep 2020, 33. [Google Scholar] [CrossRef]
- Marnell, C.S.; Bick, A.; Natarajan, P. Clonal Hematopoiesis of Indeterminate Potential (CHIP): Linking Somatic Mutations, Hematopoiesis, Chronic Inflammation and Cardiovascular Disease. J Mol Cell Cardiol 2021, 161, 98–105. [Google Scholar] [CrossRef]
Figure 1.
Components of CIR: Approximate proportions of the main CIR components that occur in the atmosphere at aircraft altitudes and temperate latitudes. However, the composition of radiation inside the aircraft is expected to differ [31].
Figure 1.
Components of CIR: Approximate proportions of the main CIR components that occur in the atmosphere at aircraft altitudes and temperate latitudes. However, the composition of radiation inside the aircraft is expected to differ [31].
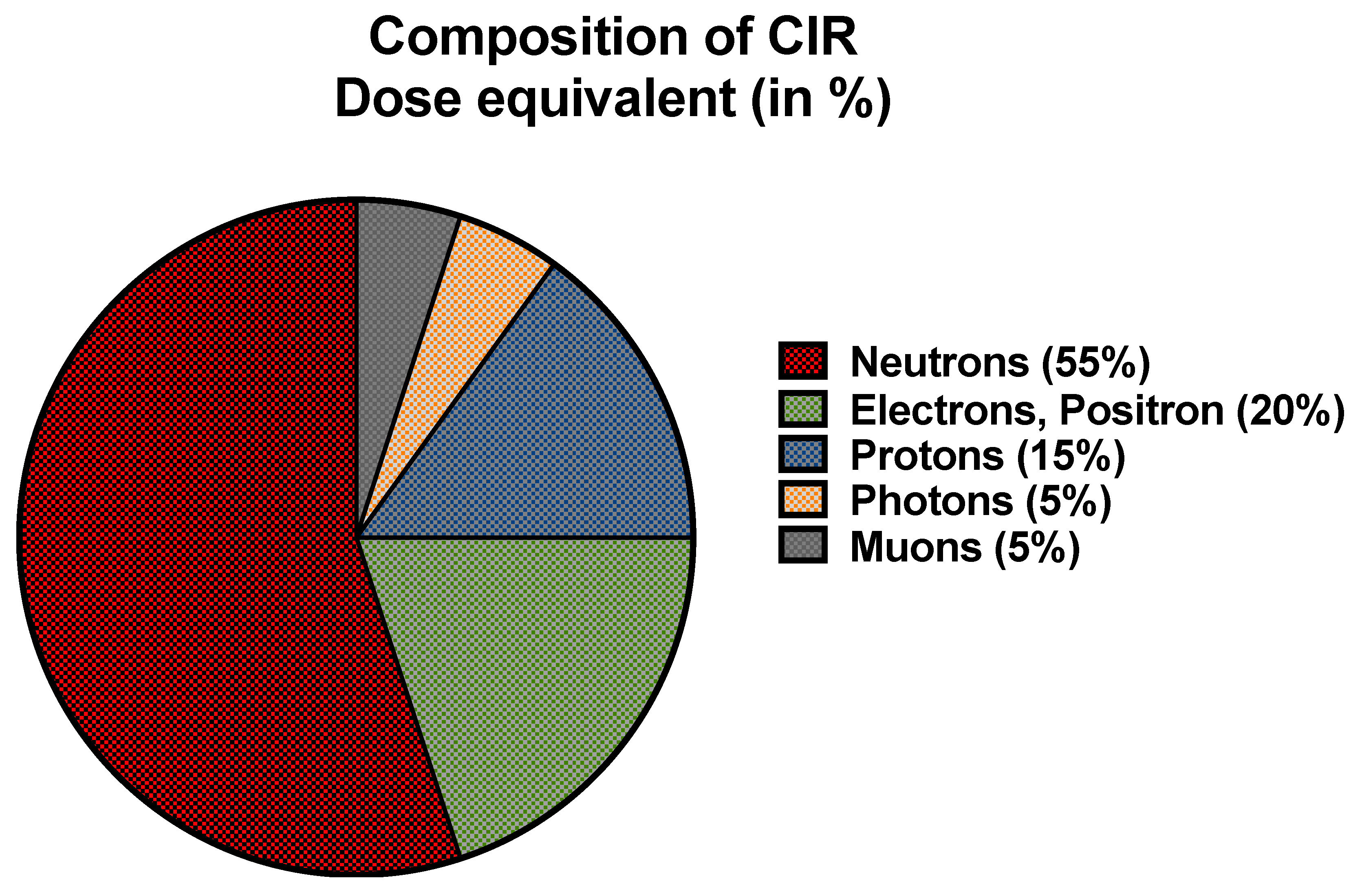
Figure 2.
Cosmic ionizing radiation (CIR) exposures that occur at aircraft cruising altitudes. The heliosphere, earth’s atmosphere, and magnetic field provide some protection (approx. 90%) from CIR exposure. On an average, circumpolar flights (shown in green plane structure) operate at cruising aviation altitude of 35,000 feet or above. The effective dose rate from exposure to the remaining CIR that passes through the stratosphere (including the peak level of approx. 65,000 feet known as the Regener-Pfotzer maximum) is directly proportional to the cruising altitude of the aircraft. [Figure created in BioRender.com].
Figure 2.
Cosmic ionizing radiation (CIR) exposures that occur at aircraft cruising altitudes. The heliosphere, earth’s atmosphere, and magnetic field provide some protection (approx. 90%) from CIR exposure. On an average, circumpolar flights (shown in green plane structure) operate at cruising aviation altitude of 35,000 feet or above. The effective dose rate from exposure to the remaining CIR that passes through the stratosphere (including the peak level of approx. 65,000 feet known as the Regener-Pfotzer maximum) is directly proportional to the cruising altitude of the aircraft. [Figure created in BioRender.com].
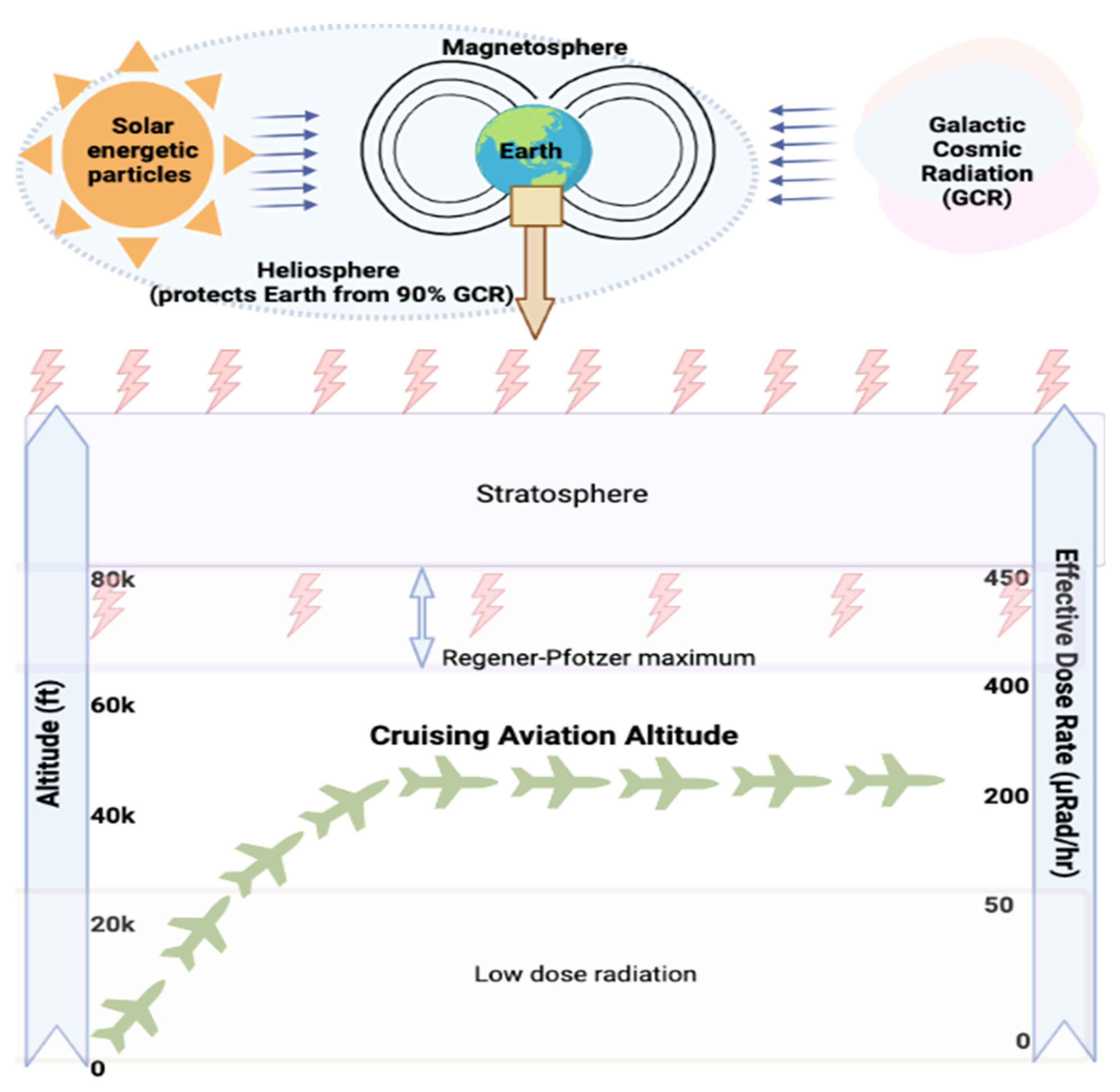
Figure 3.
Regulatory oversight on cosmic ionizing radiation exposure in flight crews. Timeline of considerations and amendments made by various regulatory agencies for occupational radiation exposures in flight crews.
Figure 3.
Regulatory oversight on cosmic ionizing radiation exposure in flight crews. Timeline of considerations and amendments made by various regulatory agencies for occupational radiation exposures in flight crews.
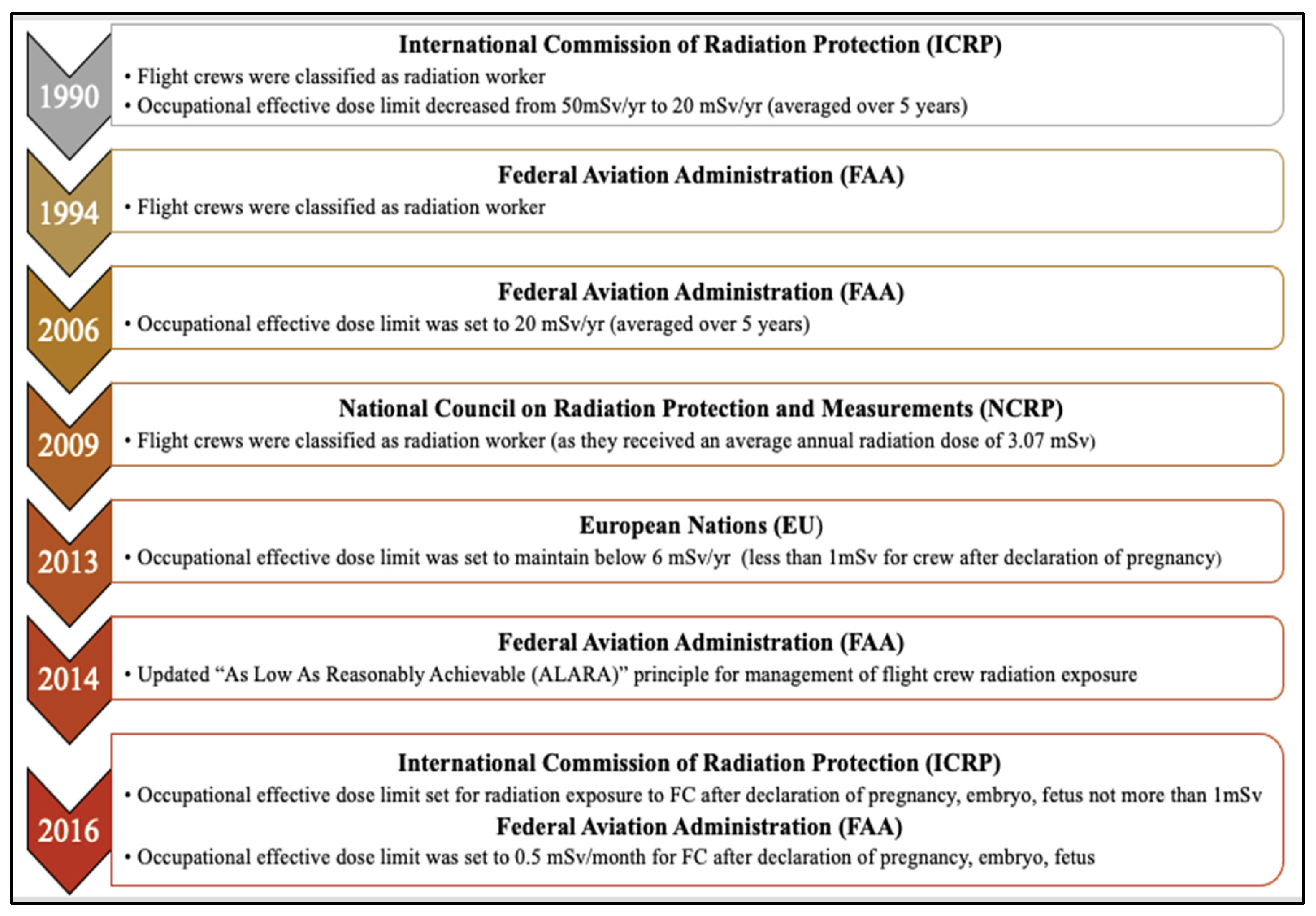
Figure 4.
Radiosensitivity of human tissue and organs. Cell proliferation is generally inversely related to radiation sensitivity in human tissues (this figure is adapted from [65]).
Figure 4.
Radiosensitivity of human tissue and organs. Cell proliferation is generally inversely related to radiation sensitivity in human tissues (this figure is adapted from [65]).
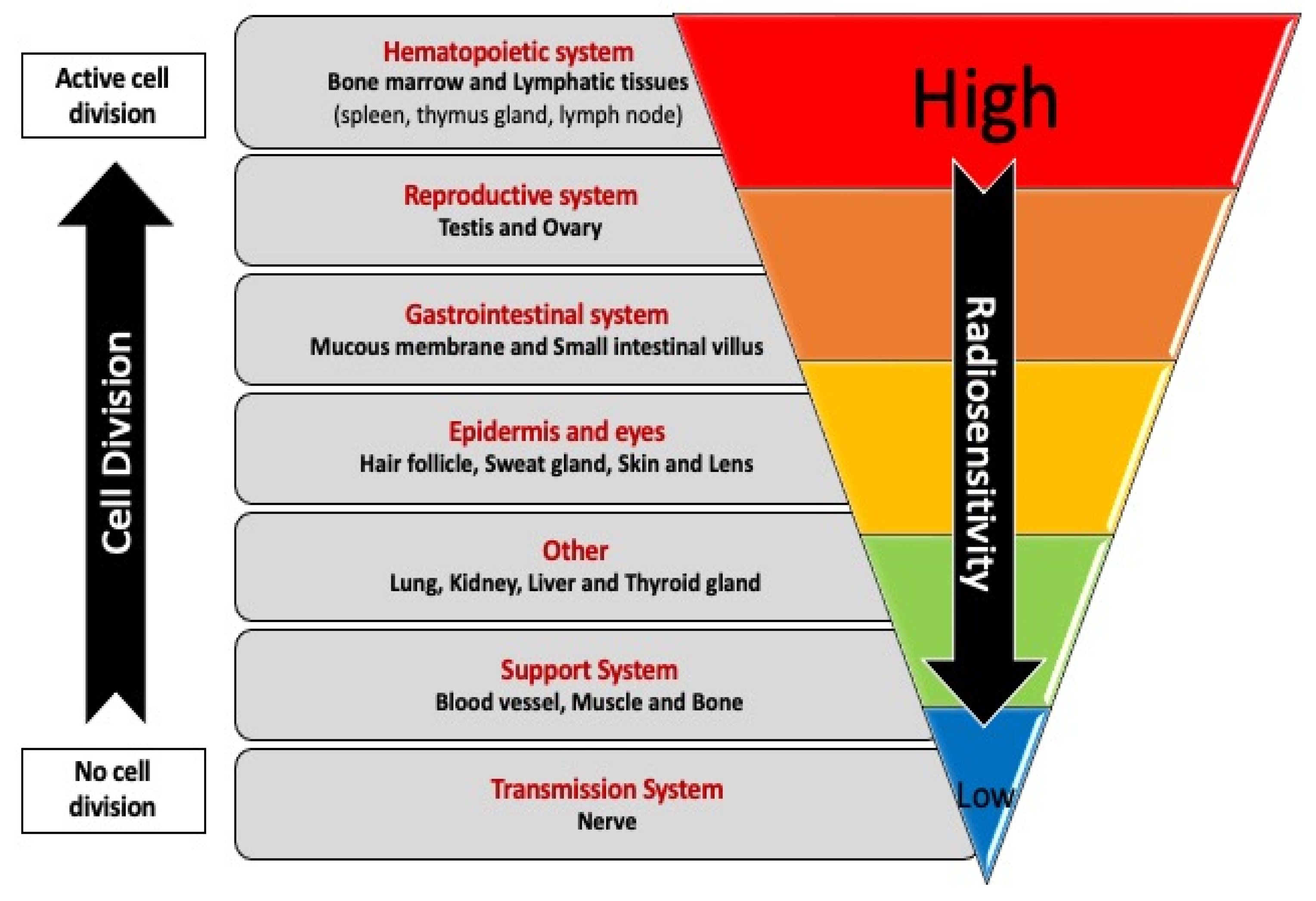
Figure 5.
Proposed model relating air travel, DNA damage and repair, and cancer risk. FC can be exposed to various DNA-damaging agents during air travel, such damage arising directly from flight related exposures (radiation and chemicals) and damage arising indirectly from biological processes that are disrupted during flight (metabolism, respiration, oxidative damage, inflammation). Unrepaired DNA damage leads to genome instability in cells that escape from cell death.
Figure 5.
Proposed model relating air travel, DNA damage and repair, and cancer risk. FC can be exposed to various DNA-damaging agents during air travel, such damage arising directly from flight related exposures (radiation and chemicals) and damage arising indirectly from biological processes that are disrupted during flight (metabolism, respiration, oxidative damage, inflammation). Unrepaired DNA damage leads to genome instability in cells that escape from cell death.
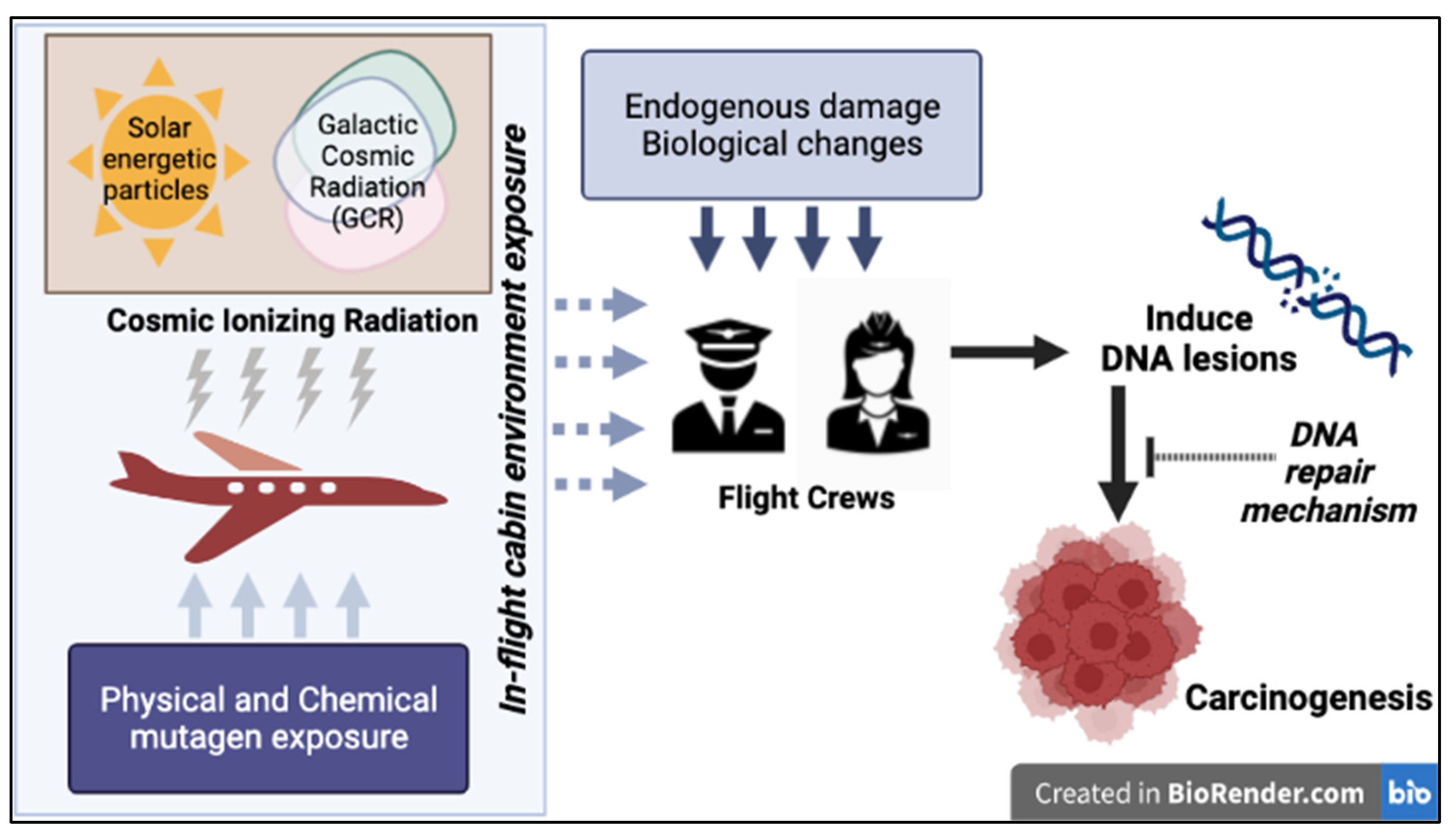

Table 1.
Maximum mean effective ionizing radiation dose limits by various regulatory bodies.
| Exposure Limits | International Commission of Radiation Protection (ICRP) [20] |
European Nations (EU) [43] |
U.S, National Council on Radiation Protection and Measurements (NCRP) [1] | U.S, Nuclear Regulatory Commission, (NRC) [19] |
U.S, Federal Aviation Administration (FAA) [39] |
|---|---|---|---|---|---|
| General Public | 1 mSv/y | 1 mSv/y | 1 mSv/y | 1 mSv/y | 1 mSv/y |
|
Pregnant Women and Unborn Fetus |
1 mSv after declaration of pregnancy | 1 mSv after declaration of pregnancy | 5 mSv and no more than 0.5 mSv in any month |
5 mSv and no more than 0.5 mSv in any month |
1 mSv and no more than 0.5 mSv in any month |
| Occupational Exposure | 20 mSv/y* | 20 mSv/y* employer required monitoring and administrative controls to maintain <6 mSv/y |
50 mSv/y | 50 mSv/y | 20 mSv/y* recommendation for FC to self-monitor without requirements for the employer |
*Averaged over five years (but not more than 50 mSv in 1 year).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



