
Submitted:
06 June 2024
Posted:
07 June 2024
You are already at the latest version
Abstract
Hypochlorite (ClO−) and viscosity both affect the physiological state of mitochondria, and their abnormal levels are closely related to many common diseases. Therefore, it is of great significance to develop a mitochondria-targeting fluorescent probes for the dual sensing of ClO− and viscosity. Herein, we have explored a new fluorescent probe XTAP−Bn, which responds sensitively to ClO− and viscosity with off-on fluorescence changes at 558 and 765 nm, respectively. Because the emission wavelength gap is more than 200 nm, XTAP−Bn can effectively eliminate the signal crosstalk during the simultaneous detection of ClO− and viscosity. In addition, XTAP−Bn exhibits a series of advantages, including high selectivity, rapid response, good water solubility, low cytotoxicity, and excellent mitochondrial targeting ability. More importantly, probe XTAP−Bn has been successfully employed to monitor the dynamic change of ClO− and viscosity levels in mitochondria of living cells and zebrafish. Taken together, this work not only provides a reliable tool for identifying mitochondrial dysfunction, but also offers a potential approach for the early diagnosis of mitochondrial-related diseases.
Keywords:
Hypochlorite
; Viscosity
; Bioimaging
1. Introduction
Mitochondria, as essential energy-supplying organelles, has played the crucial roles in many cellular processes, including central metabolism, signal transduction, and cell apoptosis [1]. In cellular systems, mitochondrial viscosity was regarded as a crucial parameter for assessing mitochondrial function, because it was closely associated with the mitochondrial respiratory state [2]. Nevertheless, the high level of mitochondrial viscosity would cause mitochondrial swelling and cellular dysfunction, thereby inducing atherosclerosis, diabetes, cell malignancy, and even cancer [3,4]. Meanwhile, hypochlorite (ClO−), as a highly reactive oxygen species (ROS), was mainly produced in mitochondria by the peroxidation of hydrogen peroxide (H2O2) with chloride ion (Cl−) under the catalysis of myeloperoxidase (MPO) [5]. Mitochondrial ClO− played the key roles in fighting against external pathogens and regulating the redox homeostasis [6]. However, the excessive ClO− level could lead to oxidative stress and DNA damage, which would cause some inflammatory diseases, cystic fibrosis, diabetes, and neurodegenerative disease [7,8]. It was worth noting that during the oxidative stress stimulation, the diffusion of ClO− was closely related to the cellular viscosity [8]. Therefore, the simultaneous detection of ClO− and viscosity in mitochondria will be particularly useful for the corresponding biological research and the disease clinical diagnosis.
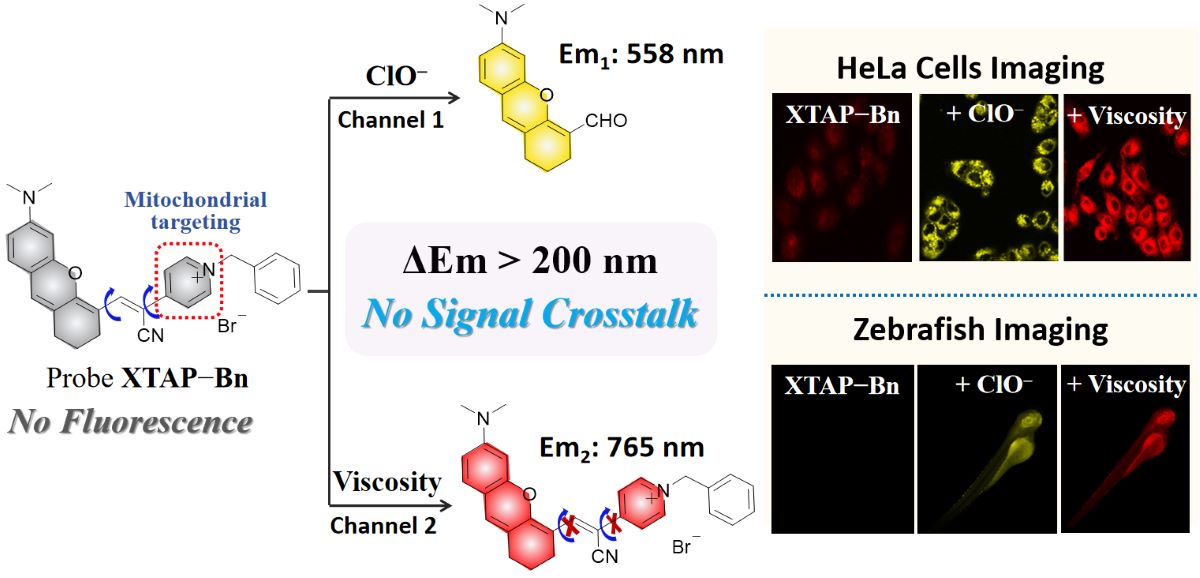
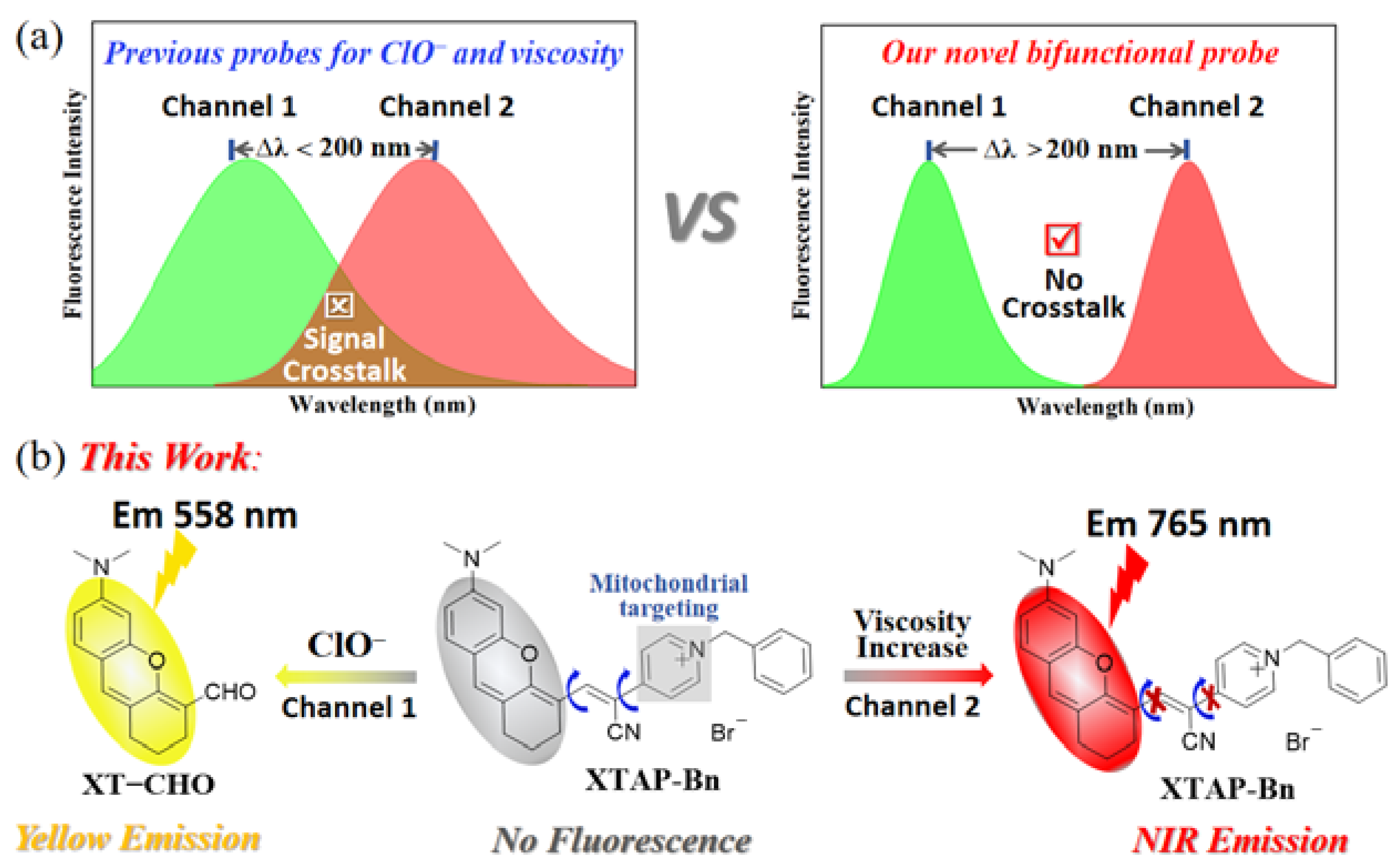
Fluorescence imaging technology has became a powerful tool for biological system monitoring, mainly due to the advantages of simple operation, high spatiotemporal resolution, and noninvasive detection [9]. At present, numerous fluorescent probes have been reported for the independent detection of ClO− [10,11,12,13,14,15,16,17] (Table S1), or viscosity [18,19,20,21,22,23,24] (Table S2). However, many of these probes were severely hydrophobic, as a result, required to use a large amount of toxic organic co-solvents [10,11,13,14,16,17]. Meanwhile, some probes lacked the mitochondrial-targeting function [10,11,12,13,16,17,18,19,21,23], and needed a long response time (several minutes) [10,12,15,16,17]. Obviously, the above problems would seriously hinder the biological applicability of these reported probes. Furthermore, the fluorescent probes for the simultaneous detection of ClO− and viscosity have been rarely reported [25,26,27,28] (Table S3). For difunctional fluorescent probes, they could shorten detection time, reduce tool synthesis costs, and simplify testing procedures. More importantly, the difunctional fluorescent probes were able to effectively avoid the fluorescence interference caused by the combination of two different probes in the complex physiological environments [29]. Unfortunately, the reported difunctional probes for ClO− and viscosity generally suffered from the problem of signal crosstalk during the detection due to the insufficient difference of emission wavelength, which would result in the false positive signals and erroneous judgment (Scheme 1a). To overcome signal crosstalk, the fluorescence wavelength difference (Δλ) between two channels of an ideal difunctional probes should be larger than 200 nm, which was twice the half width of regular fluorescence peak [30]. Taken together, it is of great significance to explore a water-soluble fluorescent probe for the fast and simultaneous detection of ClO− and viscosity in mitochondria without signal crosstalk.
Herein, we have explored a novel difunctional fluorescent probe XTAP−Bn, which could monitor ClO− and viscosity at two emission channels without signal crosstalk (Scheme 1b). Probe XTAP−Bn was designed based a typical D−π−A structure, in which the C=C bond (π linker) bridged the xanthene (XT) skeleton (electron donor, D) and the acrylonitrile-pyridinium (AP) moiety (electron acceptor, A). For XTAP−Bn, its D-π-A structure provided the intramolecular rotors, which caused the fluorescence quenching in PBS buffer. While increasing viscosity, the intramolecular rotation of XTAP−Bn was inhibited, thereby resulting in a bright NIR emission (λem = 765 nm). Meanwhile, the C=C bond of XTAP−Bn could be specifically oxidized by ClO−, which produced the fluorophore XT−CHO and released an intense yellow emission (λem = 558 nm). Given the huge wavelength gap (Δλ = 207 nm), XTAP−Bn could effectively eliminate the signal crosstalk during the simultaneous detection of ClO− and viscosity. In addition, probe XTAP−Bn responded to ClO− rapidly (within 12 s), selectively, and exhibited high sensitivity to viscosity. Owing to the excellent mitochondria-targeting ability, XTAP−Bn could simultaneously detect ClO− and viscosity in mitochondria of living cells. More importantly, probe XTAP−Bn was successfully employed to monitor the dynamic change of ClO− and viscosity levels in zebrafish, indicating its excellent applicability in vivo.
2. Results and Discussion
2.1. Synthesis and Characterization of Probe XTAP−Bn
In brief, probe XTAP−Bn was synthesized by a two-step reaction (Scheme 2). At first, the Knoevenagel condensation reaction of compound XT−CHO and 2-(pyridin-4-yl)acetonitrile afforded the intermediate XTAP. Subsequently, the following quaternization reaction of XTAP with (bromomethyl)benzene produced probe XTAP−Bn with a 74% yield. The structures of above compounds were confirmed by 1H NMR, 13C NMR, and HRMS, which can be seen in the Supporting Information (Figures S8−S13).
2.2. Spectroscopic Response to Viscosity
To accurately reveal the viscosity-sensitive behaviors of probe XTAP−Bn, its optical properties was investigated in the water−glycerol mixtures with different glycerol contents (VGly %). At first, the UV-absorption spectra and fluorescence emission spectra of probe XTAP−Bn (5 μM) in pure glycerol and water were studied, respectively. As shown in Figure 1a, the maximum absorption peak of XTAP−Bn in pure water was at 638 nm. When using glycerol with high viscosity as a solvent, the maximum absorption peak of XTAP−Bn was red-shifted to 716 nm. Because in high viscosity glycerol medium, the molecular rotation of XTAP−Bn was restricted, and the molecular conjugation was increased, thereby causing the an obvious red-shift in the absorption wavelength [22]. Moreover, it was learned from the fluorescence spectra that XTAP−Bn was almost non-fluorescent in pure water (Figure 1b). But the fluorescence intensity at 765 nm (F765) enhanced almost 105-fold with increasing viscosity (η) from 0.89 cP (H2O, 25 °C) to 945 cP (Glycerol, 25 °C). In a low-viscosity environment, the rotation of probe XTAP−Bn greatly induced nonradiative relaxation of the excitation energy, thereby resulting in a decrease in fluorescence intensity. While with increasing viscosity, the intramolecular rotation of XTAP−Bn was restricted, thus the excited state energy was released as a bright NIR fluorescence. More importantly, on the basis of a Förster−Hoffmann equation [20], the plots of log F765 against log η with the viscosity change from 1.2 cp to 945 cp exhibited a good linear relationship (R2 = 0.9959) (Figure 1c), indicating the high sensitivity of probe XTAP−Bn toward viscosity.
Considering that polarity was an important influencing factor in detecting viscosity, thus the fluorescence spectra of probe XTAP−Bn in some solvents with different polarities were studied. As shown in Figure 1d, the fluorescence intensity of XTAP−Bn at 765 nm did not show significant changes in different polar solutions compared to that in pure glycerol. These results demonstrated that environmental polarity provided negligible interference when detecting viscosity. Moreover, to broaden the applications in a complex environment, the impacts of environmental pH on the fluorescence response of probe XTAP−Bn towards viscosity was then investigated (Figure S1). As the pH varied from 3 to 10, the fluorescence intensity changes could be ignored both in low viscosity (0% glycerol) and high viscosity (90% glycerol) solutions. This confirmed that XTAP−Bn was rather stable to pH, and it could be applied to monitor the intracellular viscosity without the interference of pH.
2.3. Spectroscopic Response to ClO−
Afterward, the spectral response of probe XTAP−Bn (5 μM) toward ClO− in PBS buffer (10 mM, pH 7.4) was studied. With the addition of ClO− (70 μM), the maximum absorption peak of XTAP−Bn was significantly blue-shifted from 638 nm to 452 nm, accompanied by a significant change in the color of solution from dark blue to yellow (Figure 2a). Meanwhile, the fluorescence intensity at 558 nm (F558) exhibited an almost 160-fold enhancement (Figure 2b). These phenomenons were mainly caused by the ClO−−triggered C=C bond breakage of XTAP−Bn, which produced the fluorophore XT−CHO and released a bright yellow emission. Delightfully, the F558 values exhibited an excellent linear relationship (R2 = 0.9993) with the concentrations of ClO− (0–50 μM) (Figure 2c). Based on 3σ/k formula, the limit of detection (LOD) was calculated to be 18 nM (Section 3.5), implying that XTAP−Bn exhibited the much higher sensitivity to ClO− than many reported probes (Table S1).
To evaluate the selectivity, the fluorescence response of probe XTAP−Bn towards various potential interferents, including different active oxygen species (ONOO–, •OH, 1O2, H2O2), some biomolecules (cysteine (Cys), Homocysteine (Hcy), glutathione (GSH)), and some common anions (CO32−, H2PO4−, S2−, SO32−) were investigated, respectively. To our delight, ClO− could trigger a significant enhancement of emission at 558 nm, while all other interfering species caused the negligible changes (Figure S2). To further ascertain the selective response, an anti-interference test of XTAP−Bn toward ClO− was carried out in a competitive environment. As shown in Figure 2d, even under the coexistence with other interfering species, the fluorescence at 558 nm could still be triggered by ClO−. The above results suggested that the sensing behaviors of XTAP−Bn to ClO− were scarcely interfered in the presence of other potential interferents.
The response time was an important parameter for evaluating the feasibility and practicability of a probe. In view of this, the time-dependent fluorescence spectra of probe XTAP−Bn without and with ClO− were studied, respectively (Figure S3). When XTAP−Bn (5 μM) was excited at 620 nm, its fluorescence intensity remained unchanged within 5 min, indicating that XTAP−Bn exhibited an excellent photo-stability. But after treating with ClO− (50 μM), the fluorescence intensity at 558 nm increased rapidly and reached to a plateau within 12 s, suggesting that XTAP−Bn exhibited the much faster response to ClO− than most reported probes (Table S1). Thus, XTAP−Bn was promising to achieve the real-time detection of ClO− in living organism. To expand practicality, the effect of pH on the response of probe XTAP−Bn to ClO− was studied. As shown in Figure S4, the fluorescence intensity of XTAP−Bn (5 μM) were almost unaffected under different pH conditions, indicating the excellent pH-stability of XTAP−Bn. Upon the addition of ClO− (50 μM), the fluorescence intensity (F558) was enhanced significantly in the pH range from 4 to 10, suggesting that XTAP−Bn was capable of ClO− detection in a physiological environment.
2.4. Sensing Mechanism
To study the sensing mechanism of probe XTAP−Bn toward ClO−, the reaction product of probe XTAP−Bn with ClO− was separated, and its structure was then analyzed by HRMS and 1H NMR, respectively. As known from HRMS result (Figure S5), a peak at m/z = 446.22267 attributed to probe XTAP−Bn (calcd for C30H28N3O [M-Br]+ 446.22269) disappeared in the spectrum of XTAP−Bn + ClO−, while a new peak at m/z = 256.13332 was appeared, and this peak was similar with that of compound XT−CHO (calcd for C16H18NO2 [M+H]+ 256.13325). Moreover, according to the 1H NMR results, it was found that the signals of protons on pyridinium ring at 8.72 (Ha) and 7.98 (Hc) ppm as well as the protons on acrylonitrile group (Hb) at 8.43 ppm in XTAP−Bn were all disappeared after the reaction with ClO− (Figure 3). At the same time, a new signal of protons at 10.32 attributed to aldehyde group (Ha’) was emerged. More importantly, the 1H NMR spectra of XTAP−Bn + ClO− presented the same characteristic peaks with that of compound XT−CHO. At last, the fluorescence spectra of probe XTAP−Bn + ClO− were similar with that of compound XT−CHO (Figure S6). The above results provided clear evidence that ClO− induced the oxidation breaks of C=C bond in XTAP−Bn, thereby producing the fluorophores XT−CHO. This proposed sensing mechanism was illustrated as Scheme 1b.
To deeply understand the photophysical properties of XTAP−Bn in detecting ClO−, the electronic structure and the frontier orbital distributions of probe XTAP−Bn and compound XT−CHO were optimized by density functional theory calculation (DFT) using Gaussian 09 programs with B3LYP/6-31+G(d) basis set (Figure 4). For XTAP−Bn, the π electrons on the highest occupied molecular orbital (HOMO) were mainly distributed in the xanthene moiety (electron donor), whereas the electrons on LUMO were primarily arranged in pyridinium terminal (electron acceptor), which indicate a typical intramolecular charge transfer (ICT) effect from the xanthene to pyridinium in XTAP−Bn molecule. But for XT−CHO, the electrons on LUMO were primarily concentrated on the whole molecule. Moreover, the LUMO−HOMO energy gap (ΔE) of XT−CHO (3.058 eV) was larger than that of XTAP−Bn (2.227 eV). The increasing ΔE induced an obvious blue shift of emission wavelength from 765 nm to 558 nm. Notably, these calculated results were well consistent with the experimental data (Table S4).
2.5. Cell Imaging
Prior to cell imaging, the cytotoxicity of probe XTAP−Bn was accessed via MTT assays in HeLa cells. As shown in Figure S7, even after incubating with 25 μM of XTAP−Bn for 10 h, the cell survival rate was more than 90%, indicating the low cytotoxicity of XTAP−Bn. Moreover, owing to the positive charge of pyridinium moiety, XTAP−Bn was supposed to be mitochondria targetable by the electrostatic interaction. In view of this, the mitochondrial targeting ability of XTAP−Bn was estimated by co-incubating with the commercial Mito-Tracker Green in HeLa cells. According to the results from laser confocal microscopy, HeLa cells co-cultured with probe XTAP−Bn exhibited a slight red emission (Figure 5a), mainly due to the high viscosity expression in HeLa cell [20]. Moreover, the red channel of XTAP−Bn and the green channel of Mito-Tracker Green showed a very good overlap, and the Pearson correlation coefficient was 0.92, indicating that XTAP−Bn can specifically localize in the mitochondria of living cells.
Inspired by the excellent viscosity-sensitivity of probe XTAP−Bn in vitro, its application in living cells was then investigated. According to the previous reports, nystatin was able to induce the viscosity change of mitochondria and cause cell apoptosis [20,22]. In this case, HeLa cells were co-incubated with 10 μM and 20 μM nystatin at 37 ℃ for 45 min, respectively. After washing three times with PBS buffer, the cells were then stained with XTAP−Bn (5 μM) for another 30 min. As the concentration of co-cultured nystatin increases, the red intracellular fluorescence became brighter and brighter (Figure 5b,c). These results implied that probe XTAP−Bn could be used as an effective tool to monitor viscosity changes in living cells.
Afterward, the sensing behavior of probe XTAP−Bn toward ClO− was also investigated in living cells. When the HeLa cells were stained only with 5 μM XTAP−Bn for 30 min, a weak red intracellular fluorescence was observed in red channel, while no fluorescence was observed in yellow channel (Figure 6a,e). However, when the cells were cultivated with XTAP−Bn for 30 min, and then treated with different concentration of ClO− (5 μM, 25 μM, and 50 μM) for another 1 h, respectively, the emission intensities in the yellow channel presented a steady increase (Figure 6f–h), whereas the emission in red channels were dramatically decreased (Figure 6b–d). Because the released ClO− consumed the probe and reduced its concentration, thus an apparent decrease in red fluorescence was observed. Therefore, these results confirmed that probe XTAP−Bn was able to monitor exogenous ClO− with a yellow fluorescence signal change in living cells.
As known from previous reports, the living cells stimulated with lipopolysaccharide (LPS) and N-acetylcysteine (PMA) to generate more ClO− [16,17]. It was should be noted that LPS could also cause inflammation to increase cell viscosity [20,21]. In view of this, the feasibility of probe XTAP−Bn to simultaneously detect ClO− and viscosity was evaluated in HeLa cells after treating with LPS. As shown in Figure 7, upon the treatment of LPS/PMA, the HeLa cells presented significantly increased fluorescence in both yellow and red channels, indicating that more endogenous ClO− was generated, accompanied by an expected increase in viscosity. In addition, the fluorescence enhancement in yellow channel was inhibited distinctly after incubating with N-Acetyl-L-cysteine (NAC) (a ClO− scavenger), but this showed little effect on the fluorescence enhancement of the red channel, further proving that XTAP−Bn can detect endogenous ClO− and viscosity in cells at the same time.
2.6. Zebrafish Imaging
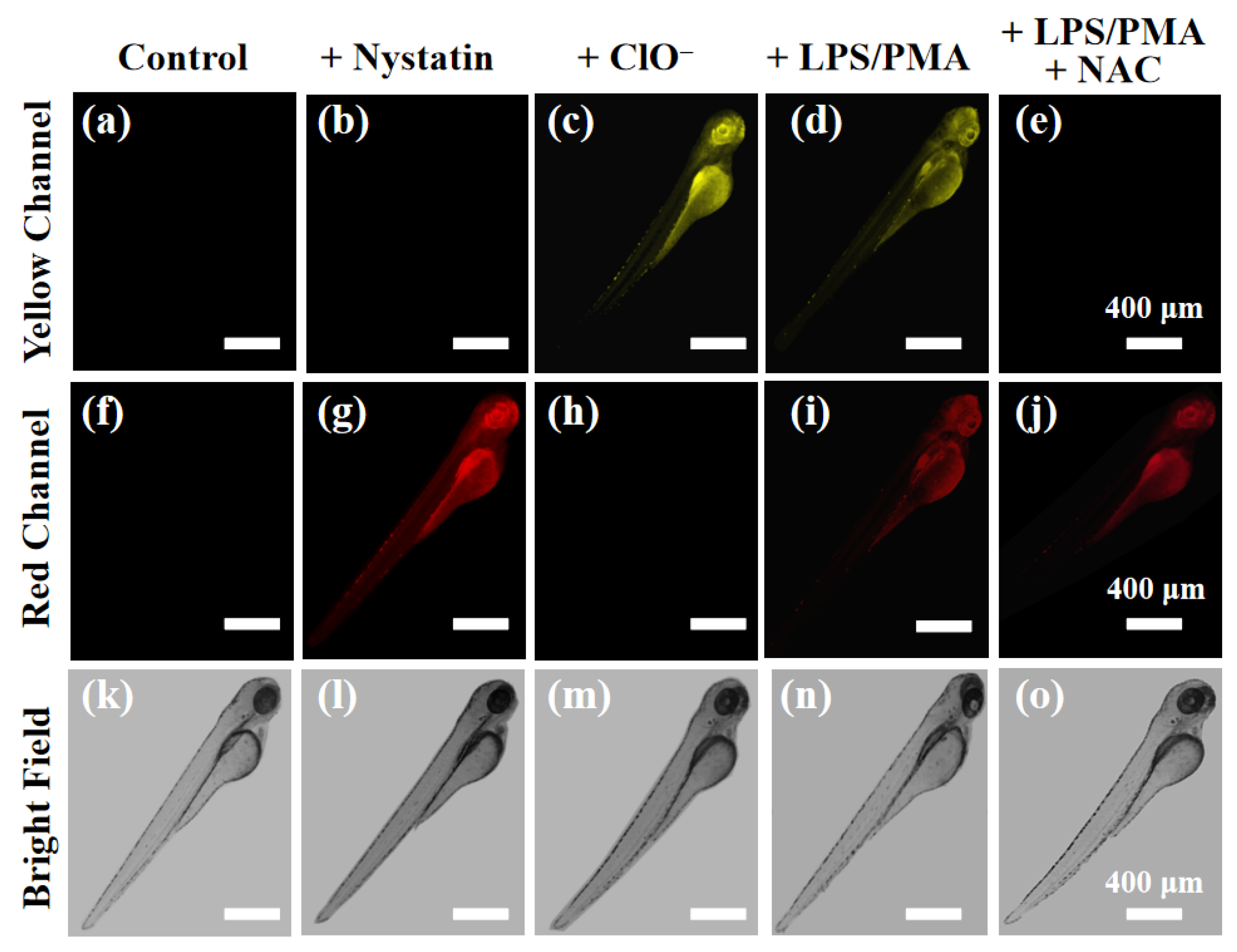
Encouraged by the excellent performance of cell imaging, the feasibility of probe XTAP−Bn to visualize ClO− and viscosity in vivo was studied, and zebrafish larvae was chose as the vertebrate model. As shown in Figure 8a,f, the zebrafish had non-fluorescence in both yellow channel and red channel after treating only with XTAP−Bn (5 μM). However, upon the treatment with nystatin (20 μM) for 2 h, the fluorescence of red channel was noticeable enhanced (Figure 8g). Subsequently, the ability of probe XTAP−Bn to detect ClO− in zebrafish was also investigated. When zebrafish were stained with XTAP−Bn (5 μM) for 1 h, and then incubated with ClO− (50 μM) for another 1 h, the fluorescence of the yellow channel was markedly improved (Figure 8c). More excitingly, after the treatment of LPS/PMA, the HeLa cells presented significantly increased fluorescence in both yellow and red channels (Figure 8d,i), indicating that more endogenous ClO− was generated, accompanied by an expected increase in viscosity. While further incubating with NAC, the fluorescence of yellow channel was disappeared due to the scavenging of ClO− (Figure 8e), but the fluorescence of red channel had a negligible change (Figure 8i). These results were well consistent with that of cell imaging. According to above results, probe XTAP−Bn was capable of simultaneously monitoring the dynamic change of ClO− and viscosity levels in vivo.
3. Materials and Methods
3.1. Materials and Instruments
Unless otherwise stated, all chemicals were purchased from commercial suppliers and used without further purification. Double-distilled water and chromatographic solvents were used for fluorescence tests. The preparation of various active oxygen species (ROS), some biomolecules, and some common anions was described as follows: (a) ONOO–: The stirred solution of NaNO2 (0.6 M, 10 mL) and H2O2 (0.7 M, 10 mL) in deionized H2O was added HCl (0.6 M, 10 mL) at 0 oC, immediately followed by the rapid addition of NaOH (1.5 M, 20 mL). Excess hydrogen peroxide was removed by MnO2. The concentration of ONOO– was determined by UV analysis with the extinction coefficient at 302 nm (ε= 1670 M−1 cm−1), and solution was stored at -20 oC for use; (b) •OH: To a solution of H2O2 (10.0 mM, 1.0 mL) in PBS (10 mM, pH 7.4) was added FeSO4 solution (10.0 mM, 0.1 mL) at room temperature to get the 1 mM stock solution; (c) 1O2: The solution of NaMoO4 (10 mM) and H2O2 (10 mM) was prepared in PBS (10 mM, pH = 7.4) respectively, and mixed the equal aliquots of these solutions to afford the 5 mM stock solution of 1O2; (d) H2O2, Cys (cysteine), GSH (glutathione), Hcy (Homocysteine), and the sodium salts of ClO−, CO32−, H2PO4− , S2− , SO32− were purchased directly from the company, and then diluted with PBS (10 mM, pH = 7.4) to make the 10 mM stock solutions.
For the instruments, a Bruker AV-400 spectrometer was employed to record 1H NMR and 13C NMR spectra. High-resolution mass spectra (HRMS) were obtained with a Thermo Scientific Q Exactive type mass spectrometer. Melting points were taken on with SGW X-4 instrument (Shanghai, China). Elemental Analysis was performed by Elementar Vario EL instrument (Germany). Absorption spectra were determined on TU-1901 UV−vis spectrometer. Fluorescence spectra were collected by Hitachi F-4500 fluorescence spectrometer. The fluorescence images of living cells and zebrafish larva were conducted by Zeiss LSMS880 confocal laser scanning microscope (Germany).
3.2. Synthesis of Compound XTAP
Compound XT−CHO was firstly synthesized according to the reported method [31]. After that, compound XT−CHO (1.02 g, 4.00 mmol) and 4-pyridineacetonitrile (0.54 g, 4.80 mmol) were dissolved in 16 mL ethanol, and then 1 mL piperidine was added into the solution. This mixture was reacted at 70 ℃ for 10 h under N2 atmosphere. After cooling to room temperature, the mixture was concentrated using rotary evaporators, and the remaining solid was purified by column chromatography (DCM/CH3OH as eluent, v/v = 8:1). The final product XTAP was obtained as a dark red solid (0.98 g, 69% yield). m.p. 186.8-188.2 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.58-8.59 (m, 2H, Pyridyl H), 8.23 (s, 1H, vinyl H), 7.57 (d, J = 4.8 Hz, 2H, Pyridyl H), 7.16 (d, J = 8.8 Hz, 1H, -ArH), 6.88 (s, 1H, -ArH), 6.71 (s, 1H, -ArH), 6.54-6.57 (m, 1H, -ArH), 3.00 (m, 6H, -(CH3)2), 2.89 (t, J = 5.6 Hz, 2H, -CH2), 2.53-2.59 (m, 2H, -CH2), 1.75 (t, J = 5.6 Hz, 2H, -CH2); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 155.94, 153.94, 150.17, 150.12, 143.41, 138.06, 128.12, 127.51, 123.22, 118.81, 110.54, 108.71, 107.79,97.72, 44.58, 29.82, 25.63, 22.02. HRMS (ESI+, m/z): Calcd for C23H21N3O [M+H]+, 356.17629; found, 356.17624. Elemental analysis calcd (%) for C23H21N3O: C, 77.72; H, 5.96; O, 4.50; N, 11.82. Found: C, 77.45; H, 5.28; O, 4.88; N, 12.38.
3.3. Synthesis of Probe XTAP−Bn
Compound XTAP (0.71 g, 2.00 mmol) and (bromomethyl)benzene (0.51 g, 3.00 mmol) were dissolved in 6 mL dry ethanol, and the mixture was then refluxed under N2 atmosphere overnight. After cooling to room temperature, the mixture was concentrated using rotary evaporators, and the obtained residue was then purified using a neutral aluminum oxide column (DCM/CH3OH as eluent, v/v = 10:1). The final product XTAP−Bn was obtained as a dark purple solid (0.78 g, 74% yield). m.p. 221.6-222.4 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.72 (d, J = 6.4 Hz, 2H, Pyridyl H), 8.43 (s, 1H, vinyl H), 7.98 (d, J = 6.8 Hz, 2H, Pyridyl H), 7.41-7.50 (m, 7H, -ArH), 6.96 (s, 1H, -ArH), 6.82-6.84 (m, 1H, -ArH), 5.64 (s, 2H, -CH2), 3.09 (s, 6H, -(CH3)2), 2.92-2.94 (m, 2H, -CH2), 2.66-2.75 (m, 2H, -CH2), 1.82-1.84 (m, 2H, -CH2); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 162.04, 155.07, 153.18, 151.19, 142.74, 139.01, 136.18, 135.05, 129.20, 125.59, 121.52, 119.08, 111.93, 111.30, 110.77, 99.51, 97.12, 67.40, 46.08, 28.53, 25.80, 20.53. HRMS (ESI+, m/z): calcd for C30H28N3O [M-Br]+ 446.22269, found 446.22267. Elemental analysis calcd (%) for C30H28BrN3O: C, 68.44; H, 5.36; N, 7.98; O, 3.04. Found: C, 69.10; H, 4.88; N, 7.72; O, 2.86.
3.4. Optical Study
For viscosity response, probe XTAP−Bn (5 μL, 3 mM in DMSO) was added into 3.0 mL of different viscosity solutions (water/glycerol mixtures with different volume ratios), and the spectra were tested at 20 °C after shaking well. For ClO− detection, the stock solutions of probe XTAP−Bn (5 μM) were prepared in PBS buffer (10 mM, pH 7.4), and the stock solutions of various ROS (ONOO–, •OH, 1O2, H2O2), some biomolecules (cysteine, Homocysteine, glutathione) and some common anions (CO32−, H2PO4−, S2−, SO32−) were prepared as shown in Section 3.3. Unless otherwise stated, all the spectra were recorded at 30 °C for 15 s after treating with any analyte.
3.5. Calculation of the Detection Limit
The detection limit (DL) of probe XTAP−Bn toward ClO− was calculated by following equation: DL = 3σ/k. Where σ was the standard deviation of fluorescence intensity for blank solution, which was measured for eight times. k represented the slope of the linear calibration plot between the fluorescence intensity and ClO− concentration. According to the linear equation:y = 137.6998x + 135.9171, DL = 18 nM.
3.6. Theoretical Calculations
All density functional theory (DFT) calculations were performed with Gaussian 09 program. The ground state (S0) geometries of probe XTAP−Bn and compound XT−CHO were optimized by DFT calculations with B3LYP/6-31G(d) basis set. Moreover, their singlet excited state (S1) geometries were optimized by time-dependent DFT (TDDFT) approaches with the same function program.
3.7. Acytotoxicity Assay
The cytotoxicity was evaluated by MTT assay. HeLa cells were purchased from Wuhan Mingde Biotechnology Co., LTD, and were incubated in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum, and then the cells were kept at 37 °C under the condition of 5% CO2 for 24 h. After that, the cells were incubated with various concentrations of probe XTAP−Bn (5, 10, 15, 20, 25 μM) for 10 h. After washing with PBS, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added and the medium was incubated at 37 °C for 4h. Finally, the absorbance was read at 490 nm using an ELISA reader (Varioskan Flash). The percentage of cell viability was calculated relative to control wells designated as 100% viable cells.
3.8. Living Cell Imaging
All HeLa cells were purchased from Wuhan Mingde Biotechnology Co., LTD. For mitochondria targeting study, HeLa cells were firstly cultivated with probe XTAP−Bn (5 μM) at 37 °C for 30 min in Dulbecco’s Modifed Eagle’s Medium medium containing 10% fetal bovine serum. Afterward, PBS was added for washing three times, and then commercial Mito-Tracker Green and (4 μM) was used to stain the mitochondria. At last, the fluorescence image was completed by a confocal laser scanning microscope. Green Channel: λex = 488 nm; λem =500–550 nm. Red Channel: λex = 633 nm; λem =700–790 nm.
For cellular viscosity imaging, HeLa cells were firstly treated with the different concentration of nystatin (0 μM, 10 μM, 20 μM) for 45 min, respectively, and then incubated with XTAP−Bn (5 μM) at 37 °C for another 30 min. After PBS washing, the image was taken by a confocal fluorescence microscopy (λex = 633 nm, λem = 700−790 nm). For cellular ClO− imaging, HeLa cells were firstly stained with XTAP−Bn (5 μM) for 30 min, and then incubated with different concentration of NaClO (0 μM, 5 μM, 25 μM, and 50 μM) at 37 °C for 1 h, respectively. After PBS washing, the image was taken by a confocal fluorescence microscopy (λex = 458 nm, λem = 520 nm−590 nm).
For endogenous ClO− and viscosity imaging, HeLa cells were divided into three groups. In a control group, the cells were only stained with probe XTAP−Bn (5 μM) at 37 °C for 30 min . In the second group, the cells were firstly treated with 300 ng/mL lipopolysaccharide (LPS) and 300 ng/mL N-acetylcysteine (PMA) for 45 min, and then incubated with XTAP−Bn (5 μM) at 37 °C for another 30 min . In the last group, the cells were treated with 300 ng/mL LPS and 300 ng/mL PMA for 45 min, 50 μM N-acetylcysteine (NAC) for 45 min, and then with XTAP−Bn (5 μM) at 37 °C for another 30 min. After PBS washing, the image was taken by a confocal fluorescence microscopy. Yellow channel: λex = 458 nm, λem = 520 nm−590 nm; Red channel: λex = 633 nm; λem =700–790 nm.
3.9. Zebrafish Imaging
Zebrafish embryos were purchased from Shanghai FishBio Co., Ltd. (Shanghai, China). Larval zebrafish (4 days old) were used for imaging, and they were divided into five groups. In a control group, zebrafish were only cultured with probe XTAP−Bn (5 μM) at 37 ℃ for 1 h. In the second group, zebrafish were grown with nystatin (20 μM) for 2 h, and then stained with XTAP−Bn (5 μM) at 37 ℃ for another 1 h. In the third group, zebrafish were stained with XTAP−Bn (5 μM) for 1 h, and then incubated with NaClO (50 μM) at 37 ℃ for another 1 h. In the fourth group, zebrafish were were firstly treated with 300 ng/mL lipopolysaccharide (LPS) and 300 ng/mL N-acetylcysteine (PMA) for 4 h, and then stained with XTAP−Bn (5 μM) at 37 °C for another 1 h. In the last group, zebrafish were firstly treated with 300 ng/mL LPS and 300 ng/mL PMA for 4 h, and then cultivated with 50 μM N-acetylcysteine (NAC) for 4 h, finally stained with XTAP−Bn (5 μM) at 37 °C for another 1 h. All zebrafish were washed three times with embryo media, and then transferred to a confocal fluorescence microscopy for imaging. Yellow channel: λex = 458 nm, λem = 520 nm−590 nm; Red channel: λex = 633 nm; λem =700–790 nm.
4. Conclusions
In summary, a novel mitochondria-targeting fluorescent probe XTAP−Bn has been reported, which could simultaneously detect ClO− and viscosity by off-on yellow fluorescence (558 nm) and NIR fluorescence (765 nm), respectively. The large wavelength gap of these two channels (207 nm) ensured the detection without signal crosstalk, indicating that XTAP−Bn was obviously superior to previous ClO−/viscosity difunctional probes. Moreover, XTAP−Bn had the advantages of high selectivity, rapid response, good water solubility. More importantly, probe XTAP−Bn displayed the low cytotoxicity and excellent mitochondria-localization ability, and it was successfully employed to monitor the dynamic change of ClO− and viscosity levels in mitochondria of living cells and zebrafish. To sum up, this work not only provides a reliable tool for identifying mitochondrial dysfunction, but also offers a potential approach for the early diagnosis of mitochondrial-related diseases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Summary of the recent single detection probes for ClO−; Table S2: Summary of the recent single detection probes for viscosity; Table S3: Summary of the recent difunctional probes for ClO− and viscosity; Table S4: DFT results for XTAP−Bn and XT−CHO; Figure S1: pH effect on the fluorescence intensity of probe XTAP−Bn (5 μM) at 765 nm in water and glycerol (with 10 % water). λex = 620 nm; Figure S2: Fluorescence spectra of probe XTAP−Bn (5 μM) with ClO− (50 μM) and various other species (100 μM) in PBS buffer (10 mM, pH 7.4); Figure S3: The time-dependent experiments of probe XTAP−Bn (5 μM) without and with ClO− (50 μM); Figure S4: Fluorescence intensity changes of XTAP−Bn (5 μM) without and with ClO− (50 μM) under different pH conditions; Figure S5: The HRMS data of XTAP−Bn without and with ClO−, as well as compound XT−CHO; Figure S6: The fluorescence spectra of XTAP−Bn (5 μM) with ClO− (50 μM) and XT−CHO (5 μM) in PBS buffer; Figure S7: Viability of HeLa cells after the incubation with different concentrations of probe XTAP−Bn; Figure S8: 1H NMR (400 MHz, DMSO-d6) spectrum of XTAP; Figure S9: 13C NMR (100 MHz, DMSO-d6) spectrum of XTAP; Figure S10: 1H NMR (400 MHz, DMSO-d6) spectrum of XTAP−Bn; Figure S11: 13C NMR (100 MHz, DMSO-d6) spectrum of XTAP−Bn; Figure S12: HRMS spectrum of XTAP; Figure S13: HRMS spectrum of XTAP−Bn.
Author Contributions
Conceptualization and data curation, C.G.; formal analysis, D.C.; visualization, L.Z.; investigation, M.M.; validation, resources, and funding acquisition, H.L.; software, writing—review and editing, and Supervision, H.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of China (No. 22174100).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
Thanks for the support and assistance from Wuchang University of Technology during the research process.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bazhin, A.V. Mitochondria and cancer. Cancers. 2020, 12, 2641. [Google Scholar] [CrossRef] [PubMed]
- Katherine, L.P. Cytoarchitecture and physical properties of cytoplasm: Volume, viscosity, diffusion, intracellular surface area. Int. Rev. Cytol. 1999, 192, 189–221. [Google Scholar]
- Tang, L.J.; Zhou, L.; Yan, X.M.; Zhong, K.L.; Gao, X.; Liu, X.Y.; Li, J.R. A simple benzothiazole-based mitochondrial-targeting fluorescent probe for visualizing and monitoring viscosity in living cell, lung organ tissue, and living mice. Dye. Pigment. 2022, 182, 108644. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef]
- Li, H.; Cao, Z.; Moore, D.R.; Jackson, P.L.; Barnes, S.; Lambeth, J.D.; Thannickal, V.J.; Cheng, G. Microbicidal activity of vascular peroxidase in human plasma via generation of hypochlorous acid. Infect. Immun. 2012, 80, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence. Nat. Med. 2004, 10, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, M.R.; Sharpless, N.E. ROS as a tumour suppressor. Nat. Cell. Biol. 2006, 8, 1213–1215. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Huang, Y.; Tao, Y.; Fan, L.; Zhang, Y. Mitochondria-targetable small molecule fluorescent probes for the detection of cancer-associated biomarkers: A review. Anal. Chim. Acta. 2024, 1289, 342060. [Google Scholar] [CrossRef]
- Fan, G.; Zhang, B.; Wang, J.; Wang, N.; Qin, S.; Zhao, W.; Zhang, J. Accurate construction of NIR probe for visualizing HClO fluctuations in type I, type II diabetes and diabetic liver disease assisted by theoretical calculation. Talanta. 2024, 268, 125298. [Google Scholar] [CrossRef]
- Yuan, F.; Wang, B.; Hou, J.T.; Li, J.; Shen, J.; Duan, Y.; Ren, W.X.; Wang, S. Demonstrating HOCl as a potential biomarker for liver fibrosis using a highly sensitive fluorescent probe. Sens. Actuators B Chem. 2023, 378, 133219. [Google Scholar] [CrossRef]
- Shao, S.; Yang, T.; Han, Y. A TICT-based fluorescent probe for hypochlorous acid and its application to cellular and zebrafish imaging. Sens. Actuators B Chem. 2023, 392, 134041. [Google Scholar] [CrossRef]
- Liang, F.; Jiang, J.; Yang, X.; Zhang, G.; Zhou, J.; Han, J.; Geng, Y.; Wang, Z. Si-rhodamine fluorescent probe for monitoring of hypochlorous acid in the brains of mice afflicted with neuroinflammation. Chem. Commun. 2023, 59, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Chen, Y.; Geng, S.; Yao, S.; Guo, Z.; He, W. Super-resolution imaging of mitochondrial HClO during cell ferroptosis using a near-infrared fluorescent probe. Anal. Chem. 2022, 94, 17904–17912. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, L.; Wang, J.; Qian, X.; Wu, Y.; Liu, Y. Mitochondria-targeted ratiometric chemdosimeter to detect hypochlorite acid for monitoring the drug-damaged liver and kidney. Anal. Chem. 2022, 94, 11881–11888. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, S.J.; Singha, S.; Yang, Y.J.; Park, S.K.; Ahn, K.H. Ratiometric detection of hypochlorous acid in brain tissues of neuroinflammation and maternal immune activation models with a deep-red/near-infrared emitting probe. ACS Sens. 2021, 6, 3253–3261. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhou, Y.; Li, Z.; Zhou, Y.; Liu, X.; Peng, X. Rational design of AIE-based fluorescent probes for hypochlorite detection in real water samples and live cell imaging. J. Hazard. Mater. 2021, 418, 126243. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yuan, S.; Qiao, M.; Jin, X.; Chen, J.; Guo, L.; Su, J.; Qu, D.H.; Zhang, Z. Exploring the depth-dependent microviscosity inside a micelle using butterfly-motion-based fluorescent probes. J. Am. Chem. Soc. 2023, 145, 26494–26503. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Cai, W.; Niu, N.; Wen, Y.; Wu, Q.; Wang, L.; Wang, D.; Tang, B.Z.; Zhang, R. Viscosity-responsive NIR-II fluorescent probe with aggregation-induced emission features for early diagnosis of liver injury. Biomaterials. 2023, 300, 122190. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Guan, X.; Chen, Y.; Tan, X.; Zhang, S.; Feng, G. Mitochondrial membrane potential independent near-infrared mitochondrial viscosity probes for real-time tracking mitophagy. Anal. Chem. 2023, 95, 5687–5694. [Google Scholar] [CrossRef]
- Guo, Y.; Leng, H.; Chen, Q.; Su, J.; Shi, W.; Xia, C.; Zhang, L.; Yan, J. Development of novel near-infrared GFP chromophore-based fluorescent probes for imaging of amyloid-β plaque and viscosity. Sens. Actuators B Chem. 2022, 372, 132648. [Google Scholar] [CrossRef]
- Wu, Y.; Yin, C.; Zhang, W.; Zhang, Y.; Huo, F. Mitochondrial-targeting near-infrared fluorescent probe for visualizing viscosity in drug-induced cells and a fatty liver mouse model. Anal. Chem. 2022, 94, 5069–5074. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, S.; Chen, Z.; Wu, F.; Cao, W.; Tian, Y.; Xiong, H. Bichromatic imaging with hemicyanine fluorophores enables simultaneous visualization of non-alcoholic fatty liver disease and metastatic intestinal cancer. Anal. Chem. 2022, 94, 13556–13565. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Zhang, W.; Zhang, Y.; Yin, C.; Huo, F. Viscosity activated NIR fluorescent probe for visualizing mitochondrial viscosity dynamic and fatty liver mice. Chem. Eng. J. 2022, 445, 136448. [Google Scholar] [CrossRef]
- Xu, S.L.; Guo, F.F.; Xu, Z.H.; Wang, Y.; James, T.D. A hemicyanine-based fluorescent probe for ratiometric detection of ClO− and turn-on detection of viscosity and its imaging application in mitochondria of living cells and zebrafish. Sens. Actuators B Chem. 2023, 383, 133510. [Google Scholar] [CrossRef]
- Chai, L.; Li, Y.; Yang, H.; Wang, Y.; Huang, R.; Wei, Z.; Zhan, Z. pH-triggered fluorescent probe for sensing of hypochlorite and viscosity in live cells and chronic wound diabetic mice. Sens. Actuators B Chem. 2023, 393, 134345. [Google Scholar] [CrossRef]
- Huang, X.; Luo, T.; Zhang, C.; Li, J.; Jia, Z.; Chen, X.; Hu, Y.; Huang, H. Dual-ratiometric fluorescence probe for viscosity and hypochlorite based on AIEgen with mitochondria-targeting ability. Talanta. 2022, 241, 123235. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Sun, Y.; Liu, C.; Jiao, X.; Shang, Y.; Zeng, X.; Zhao, L.; Zhao, J. Highly selective turn-on fluorescent probe for hypochlorite and viscosity detection. J. Mol. Struct. 2021, 1227, 129523. [Google Scholar] [CrossRef]
- Liu, T.Z.; Wang, S.; Xu, J.R.; Miao, J.Y.; Zhao, B.X.; Lin, Z.M. FRET-based fluorescent probe with favorable water solubility for simultaneous detection of SO2 derivatives and viscosity. Talanta 2023, 256, 124302. [Google Scholar] [CrossRef]
- Liu, Y.; Feng, S.; Gong, S.; Feng, G. Dual-channel fluorescent probe for detecting viscosity and ONOO− without signal crosstalk in nonalcoholic fatty liver. Anal. Chem. 2022, 94, 17439–17447. [Google Scholar]
- Chao, J.J.; Zhang, H.; Wang, Z.Q.; Liu, Q.R.; Mao, G.J.; Chen, D.H.; Li, C.Y. A near-infrared fluorescent probe for monitoring abnormal mitochondrial viscosity in cancer and fatty-liver mice model. Anal. Chim. Acta. 2023, 1242, 340813. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
(a) The illustration of dual-response fluorescent probe with signal crosstalk and without signal crosstalk. (b) The proposed mechanism of probe XTAP−Bn toward ClO− and viscosity.
Scheme 1.
(a) The illustration of dual-response fluorescent probe with signal crosstalk and without signal crosstalk. (b) The proposed mechanism of probe XTAP−Bn toward ClO− and viscosity.
Scheme 2.
The synthetic route of probe XTAP−Bn.
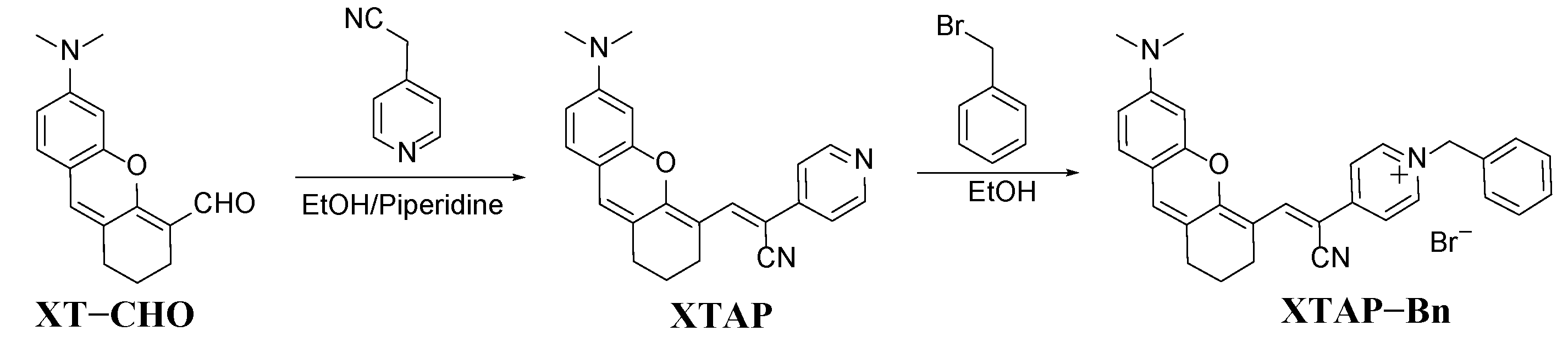
Figure 1.
(a) Absorption spectra of XTAP−Bn (5 μM) in water and glycerol, respectively. Insert: the corresponding photos taken under sunlight. (b) Fluorescence spectra of XTAP−Bn (5 μM) in different ratios of a water−glycerol mixture (glycerol from 0 to 100%). λex = 620 nm (c) Linear relationship between logF765 and logη in the water−glycerol mixture. (d) Fluorescence intensity of XTAP−Bn (5 μM) at 765 nm in different solvents. Excitation was at the maximum absorption wavelength of each one.
Figure 1.
(a) Absorption spectra of XTAP−Bn (5 μM) in water and glycerol, respectively. Insert: the corresponding photos taken under sunlight. (b) Fluorescence spectra of XTAP−Bn (5 μM) in different ratios of a water−glycerol mixture (glycerol from 0 to 100%). λex = 620 nm (c) Linear relationship between logF765 and logη in the water−glycerol mixture. (d) Fluorescence intensity of XTAP−Bn (5 μM) at 765 nm in different solvents. Excitation was at the maximum absorption wavelength of each one.
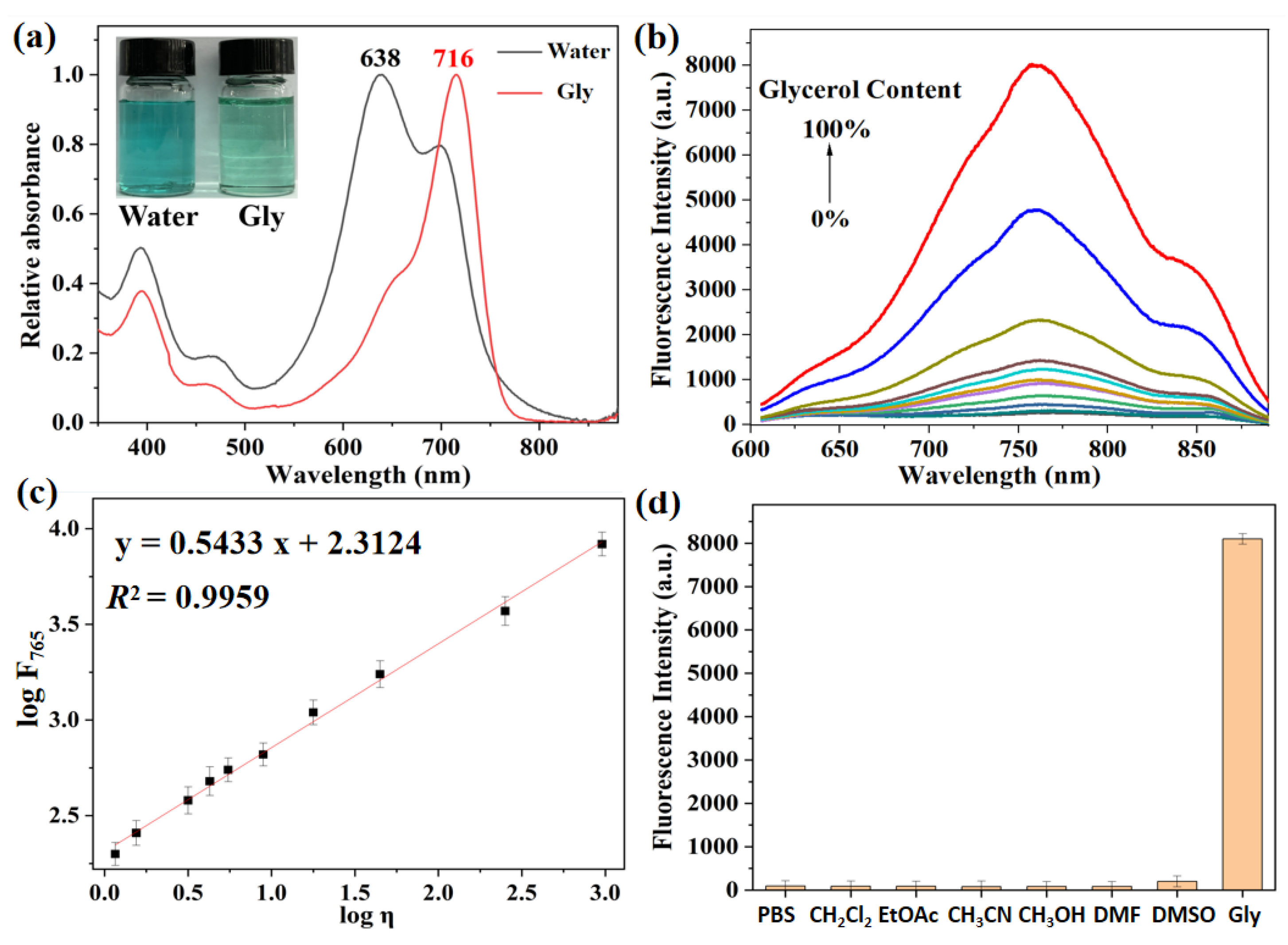
Figure 2.
(a) Absorption spectra of XTAP−Bn (5 μM) in PBS buffer without and with ClO− (70 μM). Insert: the corresponding photos taken under sunlight. (b) Fluorescence spectra of XTAP−Bn (5 μM) in PBS buffer after treating with different concentration of ClO−, λex = 482 nm. Insert: the corresponding photos taken under 365 nm light irradiation. (c) Linear fitting graph of fluorescence intensity at 558 nm with ClO− concentrations from 0 μM to 50 μM. (d) The fluorescence intensity of XTAP−Bn (5 μM) at 558 nm in the presence of various analytes (100 μM) without and with ClO− (50 μM). 1: Blank; 2: ONOO–; 3: •OH; 4: 1O2; 5: H2O2; 6: Cys; 7: Hcy; 8: GSH; 9: CO32−; 10: H2PO4−; 11: S2−; 12: SO32−. λex = 482 nm, error bars are ± SD (n = 3).
Figure 2.
(a) Absorption spectra of XTAP−Bn (5 μM) in PBS buffer without and with ClO− (70 μM). Insert: the corresponding photos taken under sunlight. (b) Fluorescence spectra of XTAP−Bn (5 μM) in PBS buffer after treating with different concentration of ClO−, λex = 482 nm. Insert: the corresponding photos taken under 365 nm light irradiation. (c) Linear fitting graph of fluorescence intensity at 558 nm with ClO− concentrations from 0 μM to 50 μM. (d) The fluorescence intensity of XTAP−Bn (5 μM) at 558 nm in the presence of various analytes (100 μM) without and with ClO− (50 μM). 1: Blank; 2: ONOO–; 3: •OH; 4: 1O2; 5: H2O2; 6: Cys; 7: Hcy; 8: GSH; 9: CO32−; 10: H2PO4−; 11: S2−; 12: SO32−. λex = 482 nm, error bars are ± SD (n = 3).
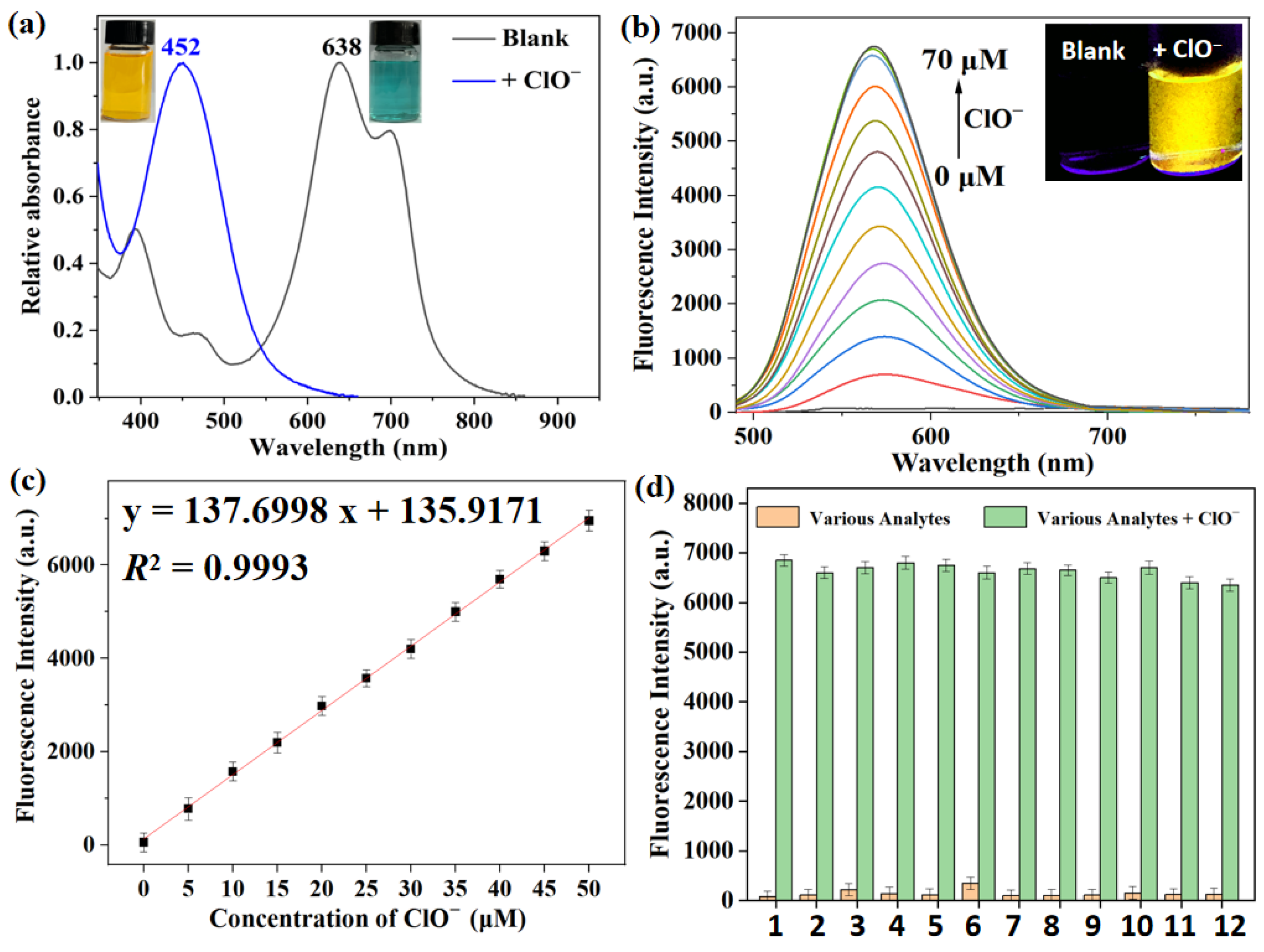
Figure 3.
1H NMR spectra of compound XT−CHO, probe XTAP−Bn, and the isolated product of XTAP−Bn + ClO− conducted in DMSO-d6.
Figure 3.
1H NMR spectra of compound XT−CHO, probe XTAP−Bn, and the isolated product of XTAP−Bn + ClO− conducted in DMSO-d6.
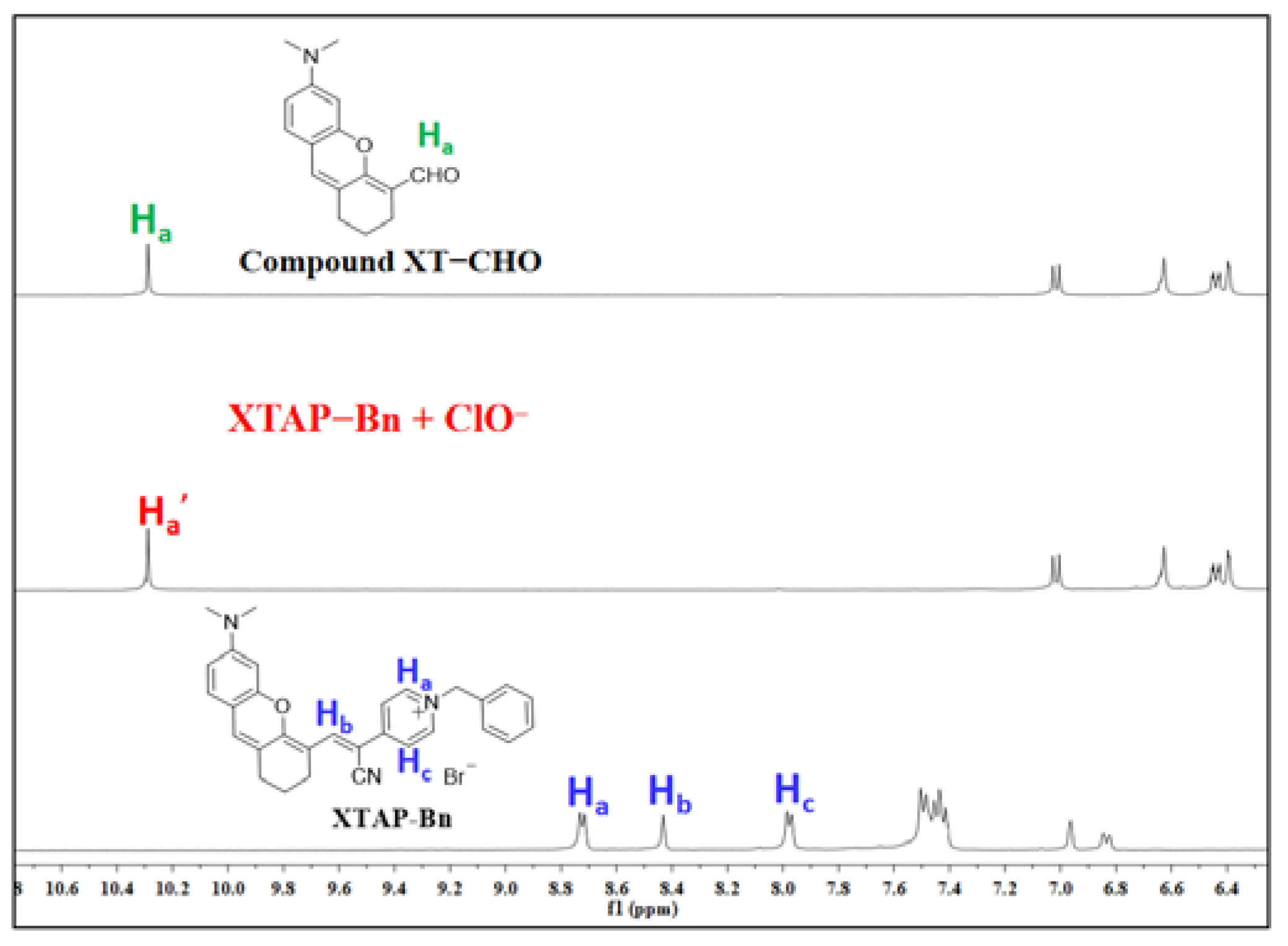
Figure 4.
Energy-minimized structures and HOMO/LUMO of probe XTAP−Bn and compound XT−CHO by DFT calculations.
Figure 4.
Energy-minimized structures and HOMO/LUMO of probe XTAP−Bn and compound XT−CHO by DFT calculations.
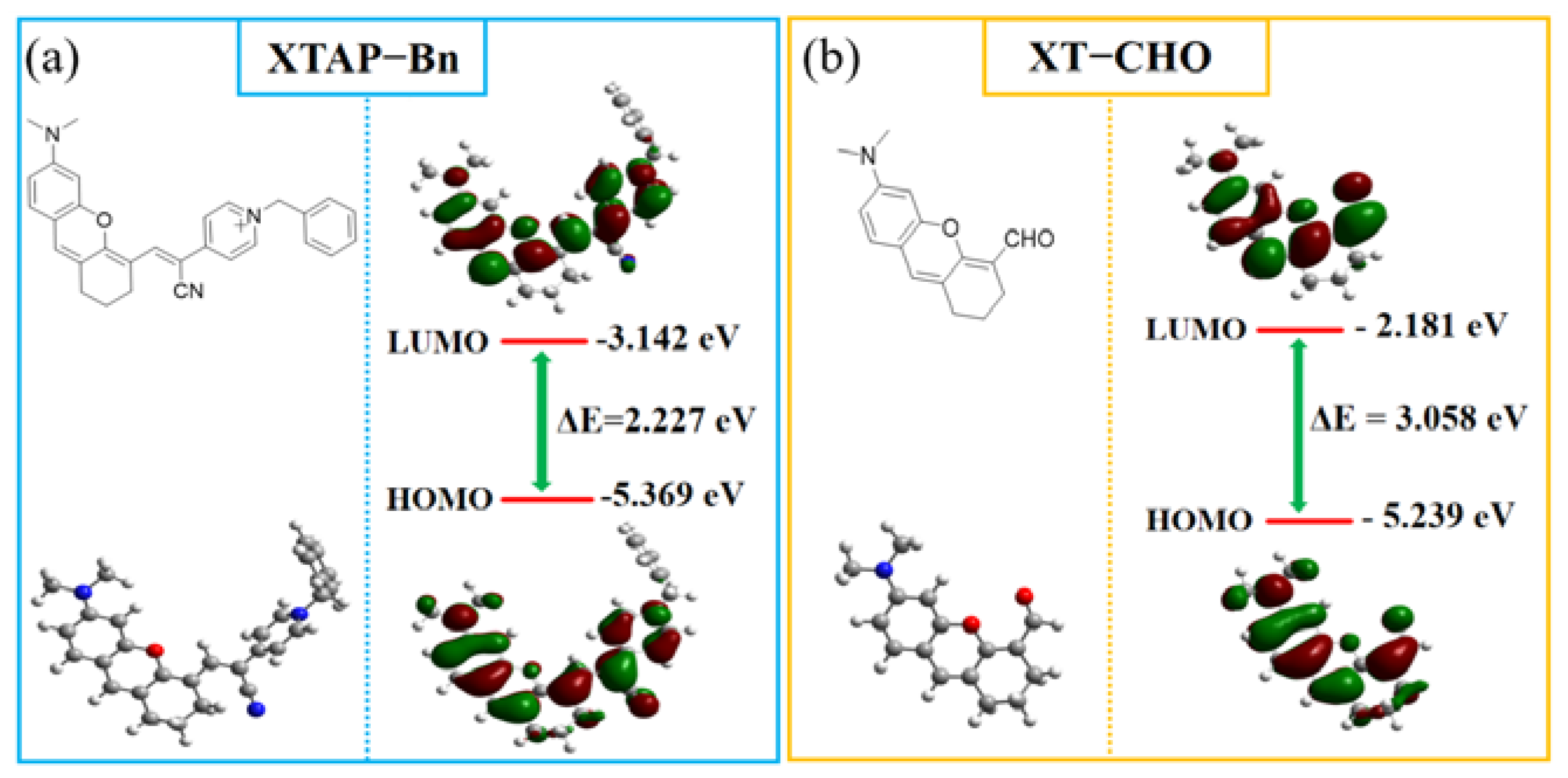
Figure 5.
(a) Confocal fluorescence image of HeLa cells stained with probe XTAP−Bn (red channel), commercial dye Mito-Tracker Green (green channel), overlap image and Pearson correlation coefficient. Green Channel: λex = 488 nm; λem =500–550 nm. Red Channel: λex = 633 nm; λem =700–790 nm. (b) Confocal imaging of viscosity in living HeLa cells. HeLa cells were pretreated with different concentration of nystatin (0 μM, 10 μM, 20 μM) at 37 ℃ for 45 min, and then incubated with XTAP−Bn (5 μM) at 37 ℃ for another 30 min. λex = 633 nm; λem =700–790 nm (c) Relative intensities of cell imaging. Error bars are ± SD (n = 3).
Figure 5.
(a) Confocal fluorescence image of HeLa cells stained with probe XTAP−Bn (red channel), commercial dye Mito-Tracker Green (green channel), overlap image and Pearson correlation coefficient. Green Channel: λex = 488 nm; λem =500–550 nm. Red Channel: λex = 633 nm; λem =700–790 nm. (b) Confocal imaging of viscosity in living HeLa cells. HeLa cells were pretreated with different concentration of nystatin (0 μM, 10 μM, 20 μM) at 37 ℃ for 45 min, and then incubated with XTAP−Bn (5 μM) at 37 ℃ for another 30 min. λex = 633 nm; λem =700–790 nm (c) Relative intensities of cell imaging. Error bars are ± SD (n = 3).
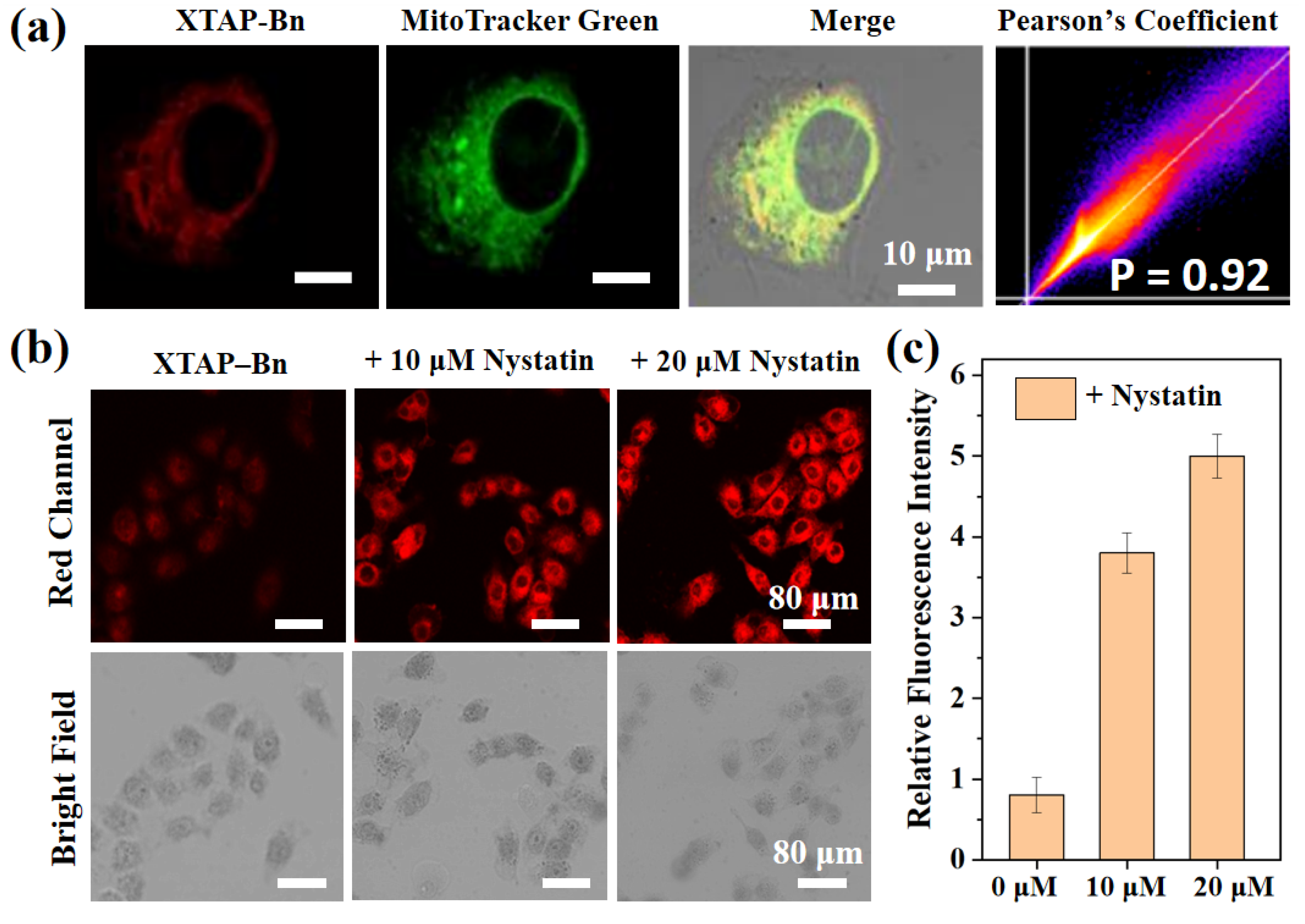
Figure 6.
Confocal imaging of ClO− in living HeLa cells. HeLa cells were stained with XTAP−Bn (5 μM) only (a, e, i), with 5 μM ClO− (b, f, j), with 25 μM ClO− (c, g, k), and with 50 μM ClO− (d, h, l), respectively. Red Channel: λex = 633 nm; λem =700–790 nm. Yellow Channel: λex = 458 nm, λem = 520 nm−590 nm.
Figure 6.
Confocal imaging of ClO− in living HeLa cells. HeLa cells were stained with XTAP−Bn (5 μM) only (a, e, i), with 5 μM ClO− (b, f, j), with 25 μM ClO− (c, g, k), and with 50 μM ClO− (d, h, l), respectively. Red Channel: λex = 633 nm; λem =700–790 nm. Yellow Channel: λex = 458 nm, λem = 520 nm−590 nm.
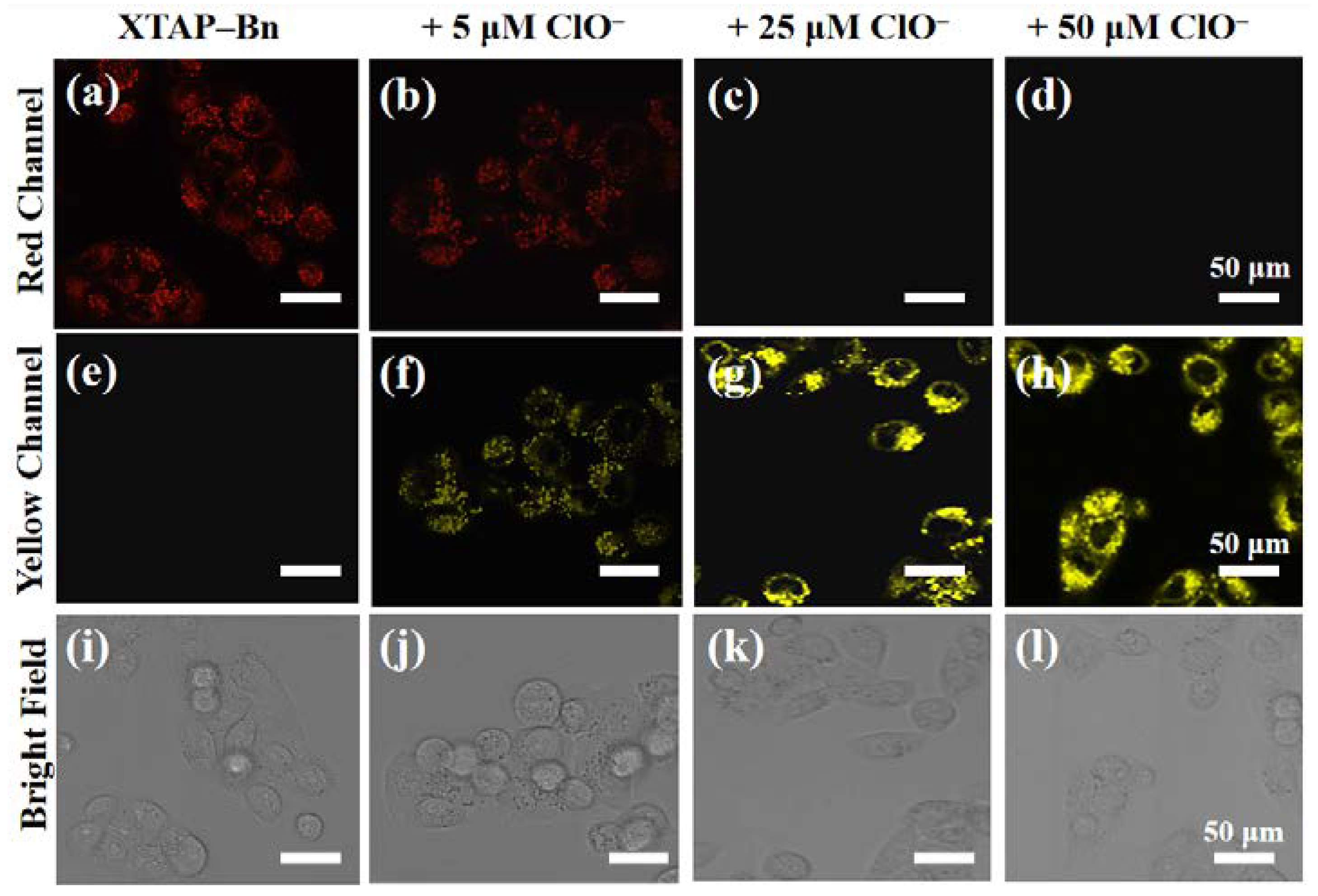
Figure 7.
The simultaneous imaging of endogenous ClO− and viscosity in HeLa cells. (a−d) Cells were stained with XTAP−Bn (5 μM) for 30 min as a control. (e−h) Cells were incubated with LPS (300 ng/mL) and PMA (300 ng/mL) for 45 min, and then stained with XTAP−Bn (5 μM) for another 30 min. (i−l) Cells were incubated with LPS (300 ng/mL) and PMA (300 ng/mL) for 45 min, then NAC (50 μM) for 45 min, and finally stained with XTAP−Bn (5 μM) for another 30 min. Yellow Channel: λex = 458 nm, λem = 520 nm−590 nm; Red Channel: λex = 633 nm; λem =700–790 nm.
Figure 7.
The simultaneous imaging of endogenous ClO− and viscosity in HeLa cells. (a−d) Cells were stained with XTAP−Bn (5 μM) for 30 min as a control. (e−h) Cells were incubated with LPS (300 ng/mL) and PMA (300 ng/mL) for 45 min, and then stained with XTAP−Bn (5 μM) for another 30 min. (i−l) Cells were incubated with LPS (300 ng/mL) and PMA (300 ng/mL) for 45 min, then NAC (50 μM) for 45 min, and finally stained with XTAP−Bn (5 μM) for another 30 min. Yellow Channel: λex = 458 nm, λem = 520 nm−590 nm; Red Channel: λex = 633 nm; λem =700–790 nm.
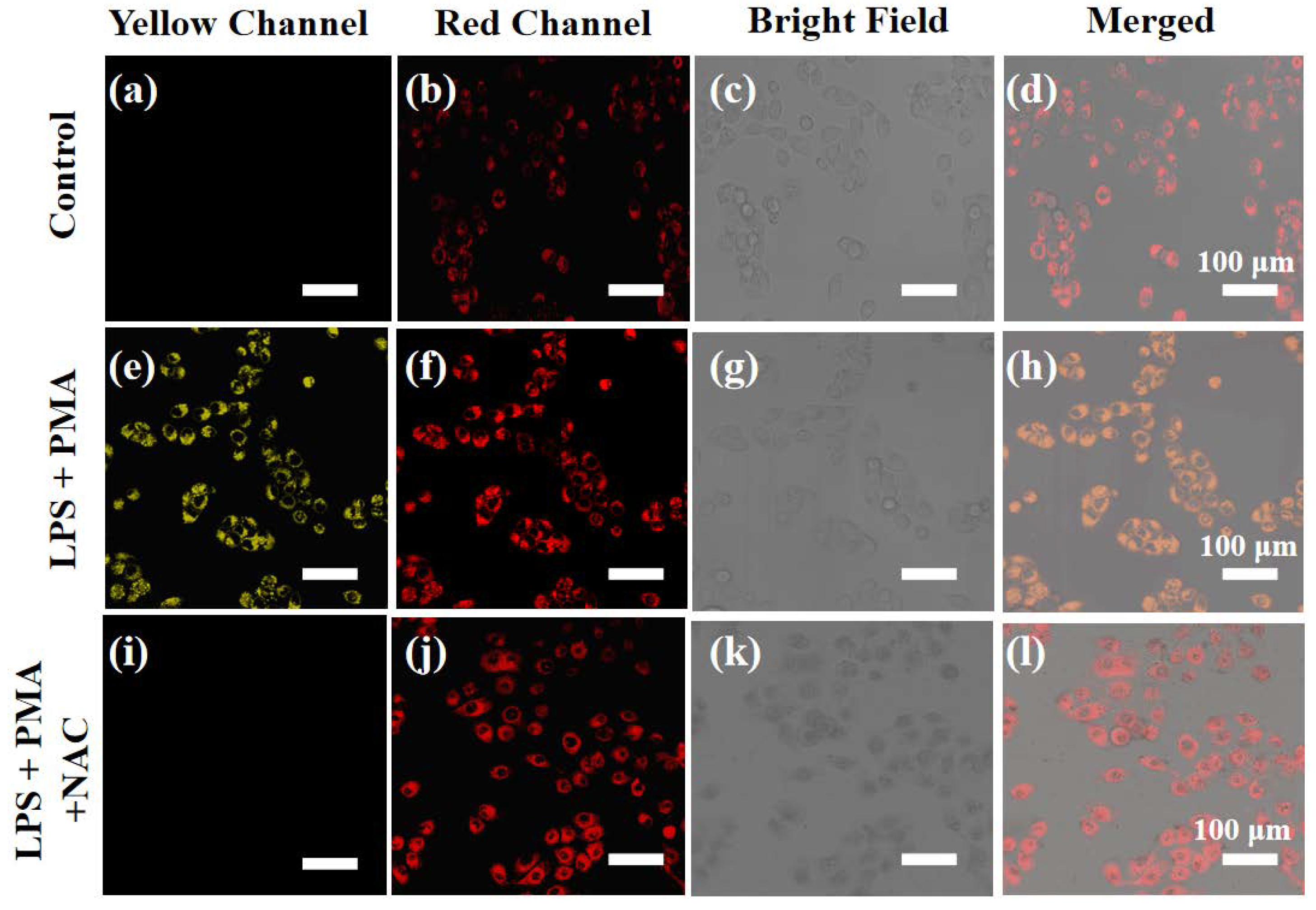
Figure 8.
Imaging the dynamic change of ClO− and viscosity levels in living zebrafish. Cells were stained with XTAP−Bn (5 μM) only as a control (a, f, k), and with 20 μM nystatin (b, g, l), with 50 μM ClO− (c, h, m), with LPS (300 ng/mL) and PMA (300 ng/mL) (d, i, n), with LPS (300 ng/mL), PMA (300 ng/mL) and NAC (50 μM) (e, j, o), respectively. Yellow Channel: λex = 458 nm, λem = 520 nm−590 nm; Red Channel: λex = 633 nm; λem =700–790 nm.
Figure 8.
Imaging the dynamic change of ClO− and viscosity levels in living zebrafish. Cells were stained with XTAP−Bn (5 μM) only as a control (a, f, k), and with 20 μM nystatin (b, g, l), with 50 μM ClO− (c, h, m), with LPS (300 ng/mL) and PMA (300 ng/mL) (d, i, n), with LPS (300 ng/mL), PMA (300 ng/mL) and NAC (50 μM) (e, j, o), respectively. Yellow Channel: λex = 458 nm, λem = 520 nm−590 nm; Red Channel: λex = 633 nm; λem =700–790 nm.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



