
Submitted:
11 June 2024
Posted:
12 June 2024
Read the latest preprint version here
Abstract
Respiratory infections with RSV account for ¼ of hospital admissions for acute respiratory infections. Nirsevimab has been developed to reduce the hospital burden of these infections: Since it has a long shelf life thanks to its high affinity for FcRn (neonatal receptor for the Fc fragment of IgG), it requires only a single injection and can be administered to all children under 2 years of age, unlike palivizumab, which is reserved for at-risk children and requires several injections. With the reservation of a large or unknown number of excluded treated subjects in both clinical trials and post-marketing observational studies, nirsevimab has been shown to be highly effective in reducing hospitalization rates for RSV infections. In rare cases, however, RSV infections were more severe in the treated group than in the placebo group. The 2023-2024 immunization campaign involved 4 countries (USA, France, Spain, Luxembourg). Analysis of the results of the 2023 immunization campaign does not allow us to conclude on the efficacy of nirsevimab in the USA (coverage being too low at around 20%); in the 3 other countries coverage is ≥ 80%. Neither clinical trials nor observational studies point to a reduction in all-cause hospitalizations in the immunized age group in this same season compared with previous seasons. The rate of hospitalization for RSV in the treated age bracket is significantly reduced compared with previous seasons in France, Spain and Luxembourg, but biases (exclusion of a large or unknown number of subjects, and changes in diagnostic criteria in France) may moderate this reduction. In France, there is a significant signal of an increase in newborn deaths between 2 and 6 days of age during the 2023-2024 immunization campaign. This signal could be attributable to ADE (antibody-dependent-enhancement). ADE has been observed with RSV F-protein antibodies in inactivated vaccine trials. The theoretical risk of ADE with an anti-RSV F-protein antibody such as nirsevimab has been eliminated by the EMA following clinical trials. In vitro evaluation of nirsevimab's effector functions on FcγR (cellular IgG Fc receptors) and the properties of FcRn cannot exclude the possibility of an ADE. This risk has been incompletely assessed in preclinical in vivo trials. In clinical trials, pharmacokinetic studies show the possibility in rare individuals of sub-neutralizing circulating levels of nirsevimab in the blood and pulmonary mucosa, in the days following injection and at longer distances. This could explain the rare cases of aggravated RSV infections in treated subjects. ADE by disruption of the immune system has not been studied, and could explain why the all-cause hospitalization rate has not fallen in treated age groups: mAbs are indeed capable of promoting infections by binding to FcRn. Given the high price of nirsevimab, the cost-effectiveness of mass immunization campaigns may therefore be debated from an economic as well as a scientific point of view.
Keywords:
nirsevimab
; beyfortus
; ADE
; RSV
; bronchiolitis
; mAbs
; therapeutic monoclonal antibody
1. Introduction
Respiratory syncytial virus (RSV) is currently a primary cause of hospitalization in infants and young children globally. Worldwide, it is estimated that 33 million cases of RSV-associated ARI (acute respiratory infection) occur each year in children under 5, resulting in 3.2 million hospital admissions (including 1.4 million in children under 6 months of age) [1]. Bronchiolitis can severely affect some children under the age of two, and can lead to increased susceptibility to asthma. Hospitalization is required in 3% of cases, and admission to intensive care in 2 to 6% of cases [2]. The RSV virus reinfects children annually, and infection occurs despite the presence of maternal antibodies [3]. Vaccination of infants does not appear to be a feasible solution in the current state of knowledge. In infants under 2 months of age, the presence of maternal antibodies and the immaturity of the immune system prevent an effective immune response to an effective RSV vaccine [4]. Infants have a reduced capacity to produce neutralizing antibodies against hRSV, compared with adults, making the former more susceptible to recurrent infections [5]. In infants, the immune response to RSV is polarized towards a Th2 profile, leading to considerable inflammation of the lungs. To date, no research favoring a vaccine with a Th1 response has been successful [6]. The development of RSV vaccines has been hampered by the results of the inactivated vaccine, which caused deaths from vaccine-antibody-enhanced RSV disease (VAERD - Vaccine-associated enhanced respiratory disease, a form of ADE, antibody-dependent enhancement): the mechanism of VAERD is classically associated with an exaggerated Th2 response, high levels of non-neutralizing antibodies, low levels of neutralizing antibodies and the presence of eosinophils in the pulmonary epithelium [7]. Given the low capacity of infants to develop immunity to RSV and the failures in vaccine development, another approach to protecting infants from RSV is the development of monoclonal antibodies: this is passive immunization. Vaccination of mothers has also been shown to be effective: placental transfer of maternal antibodies protects the newborn during the first months of life [8].
Nirsevimab was developed to reduce the burden of RSV infections on hospitals, as its predecessor, palivizumab, which was reserved for at-risk children, required several injections and could not be recommended for all children for economic and logistical reasons. The modification of the Fc fragment of nirsevimab compared with palivizumab enables it to bind more strongly to FcRn, thus prolonging its lifespan and reducing the number of injections to just one. Both monoclonal antibodies are directed against the RSV F protein. Nirsevimab has strong RSV-neutralizing activity. As the search for vaccines has been hampered by the discovery of an ADE effect due to anti-F protein antibodies, the European Medicines Agency (EMA) has taken into account the theoretical possibility of an ADE through increased FcγR-binding efficacy, and has eliminated this risk based on the results of clinical trials and in vitro and animal studies [9].
This review outlines the state of knowledge on the mechanisms of ADE in RSV infections and proposes a review of the results of clinical and preclinical trials concerning the evaluation of this ADE. Analysis of the initial results of the first nirsevimab immunization campaign during the 2023-2024 season is also envisaged from this point of view. Data from phase I and phase II/III clinical trials and the results of the nirsevimab immunization campaign in 2023-2024 are analyzed in this article: the mixed results could be explained by an ADE caused by this monoclonal antibody. Possible mechanisms of ADE are discussed here.
2. Design of Nirsevimab
Anti-RSV mAbs (monoclonal antibodies) are designed to bind to the RSV F (fusion) protein, which is not susceptible to variation, unlike the G (attachment glycoprotein) protein: the F protein enables viral entry and RNA insertion into target cells (Rocca 2021). Palivizumab (a humanized antibody against the RSV F fusion protein) has been used for over 20 years to prevent RSV infections in infants and children at risk of severe disease; palivizumab (code name MEDI-493) targets the F protein and was approved by the FDA-US in 1998 and by the EMA in 1999. According to Rocca [10], palivizumab reduced hospital admissions in observational studies. A Cochrane review [11] showed no beneficial effect of palivizumab compared with placebo. Pavilizumab should be administered once a month during the RSV circulation season, due to its short serum half-life [12].
Manufacturers of anti-RSV mAbs have made efforts to overcome the disadvantage of palivizumab (too short half-life) and increase its efficacy: they have developed antibodies with extended half-life and higher affinity for F protein [4]. One way of increasing the half-life of IgG mAbs is to optimize their affinity for the neonatal Fc crystallizable fragment receptor (FcRn) [13]. FcRn protects IgG from intracellular degradation by a pH-dependent recycling mechanism, and enables its transport across cell barriers. IgG ingested by the cell via pinocytosis binds to FcRn in the acidic environment of the endosome and is transported intact back to the surface of the plasma membrane [14]. A palivizumab derivative (motavizumab, MEDI-524) targeting F with remodeling of palivizumab’s heavy and light chains showed 70 x greater affinity for F than pavilizumab and 20 x greater anti-viral activity, but was not approved due to adverse effects, particularly on the skin. Substitution of 3 amino acids (YTE: M252Y/S254T/T256E) produced MEDI-524-YTE with an extended half-life of 100 days: YTE mutations increase FcRn binding. More recently, it has been shown that F protein in the prefusion conformation has more conserved epitopes than in the fusion conformation (suptavumab REGN-222, which targeted a variable epitope of F, failed to show clinical efficacy, probably due to a circulating RSV B mutant resistant to this mAb). To remedy this escape, nirsevimab (MEDI-8897) targets the highly conserved Ø epitope of the F protein only in prefusion conformation: it has superior neutralizing power to pavilizumab and an extended half-life thanks to YTE mutations (80-120 days) [10]. It should be noted that mAb binding to FcRn should not occur at neutral pH (7.4): IgGs that bind at neutral pH have a significantly reduced lifespan [15]. High affinity at both neutral and acidic pH leads to accelerated clearance, negating the benefit of strong FcRn binding at acidic pH [16]. YTE mutations increase mAb affinity for FcRn at acidic pH [4], and it has been shown that mAbs with YTE mutations, such as nirsevimab, bind FcRn with very low affinity at neutral pH [15].
3. Definition, Role and Localization of FcRn: Based on our Knowledge of the Role of FcRn, what might be the Consequences of a mAb’s Increased Affinity for this Receptor?
The FcRn is a multifunctional atypical Fc-gamma receptor (FcγR). It is a heterodimer composed of major histocompatibility complex (MHC)-I, which binds to both albumin and the Fc portion of IgG. IgG binding to FcRn accounts for its long life: FcRn protects IgG from intracellular degradation via a pH-dependent recycling mechanism. FcRn enables IgG to be transported across cell barriers, including that of the lungs. FcRn binds IgG only in acidic environments (pH between 5.0 and 6.5). FcRn is expressed on cell surfaces and in cell endosomes in mucous membranes (epithelium of placenta, liver, kidneys, genital tract, lungs) and in endothelial and hematopoietic cells [17]. In the lungs, IgG is transcytosed through the respiratory epithelium from the lumen to the serosa by FcRn [14].
3.1. Transport of Non-Antigen-Bound IgG
Conventional IgG has a half-life of 3 weeks, thanks to its binding to FcRn [14,18]. As mentioned in the introduction, manufacturers have sought to increase the half-life of monoclonal antibodies against RSV, as the only one in commercial use (palivizumab) requires one injection per month due to its short half-life [12]. Increasing affinity for FcRn increases this half-life [4].
FcRn is mainly localized inside the cell, but ligand binding to FcRn can take place either on the cell surface or in the acid endosome. FcRn is also expressed in the epithelium lining the lungs of adult mice, humans and non-human primates [19]. In primates, FcRn is expressed mainly in the epithelium of the upper airways. However, given the much larger surface area of the alveolar epithelium compared with the upper airway epithelium, and the fact that alveolar epithelial cells can transcytose IgG in vitro, a role for IgG uptake by the alveolar epithelium cannot be ruled out [13]. At these sites, FcRn ensures IgG transport across the mucosal barrier and may play a role in immune surveillance and host defense [20].
FcRn enables bidirectional transcytosis of IgG in the apical to basal and reverse directions [21]. In an in vitro model of rat lung epithelium, IgG translocation occurs in both directions (but mainly apical to basal), mediated by FcR binding, which was confirmed in 2023 [22,23]. FcRn is involved in the transport of IgG from the lung lumen to the systemic circulation in mice and cynomolgus monkeys [15]. However, to date, little is known about the role of FcRn in a potential mechanism of active IgG transport from the systemic circulation to the lungs.
3.2. Transport of Protein Antigen-Bound IgG
Antigen-bound IgG (IgG-ICs) also bind to FcRn [24] and APCs (antigen presenting cell) and cause IgG-IC complexes to enter cells [14,19]. FcRn-mediated protein transport is demonstrated through mouse bronchial epithelium in vivo. An Fc-bound erythropoietin is specifically transported by FcRn after intranasal administration. While IgG binding to FcRn is pH-sensitive, FcRn can transport IgG across epithelial barriers: this transport does not require transepithelial pH gradients established at epithelial surfaces [19,25]. This is confirmed by Ye [26], who has constructed a vaccine consisting of an antigen bound to the Fc fragment: this construction allows FcRn-specific transport across the mucosal barrier.
3.3. Intracellular Transport of Viruses by FcRn
FcRn is also capable of promoting viral entry into target cells. This is possible because FcRn is itself an enterovirus receptor, and binding occurs at acidic or neutral pH. The virus is captured by the FcRn and enters the cell and the endosome, where the acidic pH allows uncoating of the virus and release of the genome [27]. The interaction between echoviruses and FcRn occurs at neutral pH on the cell surface before virus entry [20,28]. Echoviruses could be transported across the intestinal barrier and also across the placental barrier, which strongly expresses FcRn. The presence of anti-echovirus IgG is not necessary for virus entry. As early as 1983, Burstin [29] showed that a monoclonal antibody against a reovirus could increase virus growth in P388D1 cells (a murine macrophage-like cell line); this increase occurred with sub-neutralizing antibody concentrations and was mediated by the macrophage-like cell Fc receptor. No specific reference was made to FcRn, which was not characterized at the time. FcRn can promote the entry of IgG-virion complexes (CMV cytomegalovirus) by transcytosis across the placenta: cell types resistant to infection by virion alone can be infected by this process. It has been suggested that IgG-virion complexes can be transcytosed in other tissues: intestine, kidney, lung and breast. However, this only occurs with low-affinity antibodies (or low levels of neutralizing antibodies) [30]. Polyclonal antibodies and a monoclonal antibody increase HIV transcytosis on a human cell line (and more strongly at acidic pH), this transcytosis depending on the FcRn of the target cells. These antibodies are highly neutralizing, and monoclonal antibodies increase transcytosis significantly more than polyclonal antibodies [31]. These results were confirmed with neutralizing polyclonal and monoclonal antibodies from SIV-vaccinated macaques. In a human cell line, transcytosis of SIV virions is increased in the presence of simian IgG specific for the viral envelope when the pH of the apical surface is acidic; this increased transcytosis is FcRn-dependent. Transcytosed viruses remain infectious when serum neutralizing antibody levels are low. The ability of the antibody to capture infectious viruses correlates with increased transcytosis. Strongly neutralizing antibodies do not confer protection in these challenge assays. This study shows that ADE may be an FcRn-mediated mechanism [32]. However, another study [33] shows that polyclonal antibodies and a monoclonal antibody against HIV decrease transcytosis of HIV bound to human antibodies in vitro.
3.4. FcRn Expression in Lung Macrophages
3.5. pH-Dependent Binding of IgG to FcRn on Mucosal Surfaces
FcRn binds to IgG on the surface of mucous membranes when these are acidic: this is the case of intestinal mucosa [34]; IgG binds to the apical surface of cells (in the lumen) [13]. FcRn enables newborns to acquire IgG from breast milk. FcRn-dependent uptake has been demonstrated in the lungs and intestines of mice humanized for FcRn. The pH of neonatal lung mucosa (ASL airway surface liquid) is variable and can be acidic in some individuals: it has been measured at between 6.7 and 7.3 [35,36]. It has been proposed that the pH of children with cystic fibrosis is lower than that of healthy children [37,38], but this difference could be due to methodological bias [35], although all authors agree on the variability of this pH and the observation of an acid surface of the pulmonary mucosa in some infants. Since ADE has been identified as a mechanism of failure in trials of inactivated RSV vaccines via RSV F-protein antibodies, the EMA has admitted that it is also a theoretical risk for an anti-F mAb [9].
4. What are the Mechanisms of ADE in Viral Infections and Following Antiviral Vaccinations, and how might an RSV F-Protein mAb with Increased Affinity to FcRn be Involved? Could mAb Binding to other FcγRs be Involved?
Knowledge of ADE (intrinsic or extrinsic [39]) results from the binding of antibodies to cellular FcγRs (FcRn or other Fcγ) and to complement. This binding can specifically aggravate infection by a virus targeted by these antibodies, but can also lead to a more global disruption of the immune system.
4.1. Immune System Disruption
Vogelzang showed in mice that FcRn located in the lungs could cause non-specific aggravation of infections, under inflammatory conditions. FcRn is expressed in lung parenchyma and lung immune cells, and enables preferential secretion of low-affinity IgG into the lumen. IgG1 with high affinity for FcRn could saturate FcRn receptors locally, counter-intuitively allowing preferential passage of low-affinity IgG into the pulmonary lumen. Under these inflammatory conditions, this process could damage lung tissue [40]. The FcRn binding of IgG-IC complexes formed in the presence of excess antibody has an immunosuppressive effect, in contrast to those formed in the presence of excess antigen (which are immunogenic) [34]. IgG-antigen ICs can activate TF (tissue factor) via monocyte FcRn; this activation of TF leads to the activation of FXa, which is involved in thrombotic phenomena. In this process, FcRn and FcγRIIa may cooperate [41].
IgG-IC binding to FcRn has an inflammatory role, through FcRn engagement on the cell surface in conjunction with binding to other FcγRs: this process could contribute to autoimmunity phenomena [42].
4.2. Mechanisms of ADE of Viral Infection by Specific Anti-Viral Antibodies
ADE has been described in numerous viral infections and anti-viral vaccinations (arthropod-borne viruses, alphaviruses, flaviviruses, respiratory viruses as influenza virus, coronaviruses, RSV, Ebola [43] and measles virus [44]), and has been attributed to sub-neutralizing concentrations of antibodies capable, under certain conditions, of enhancing viral infection by facilitating viral entry into target cells. ADE is produced by interaction between immunoglobulin Fc fragment receptors (FcγR) and the virus-antibody complex. Extrinsic ADE promotes infection of myeloid cells (monocytes, macrophages, dendritic cells - DCs - and granulocytes). Internalization of virus-antibody complexes can also modulate the expression of the innate cytokine response to the virus: this is intrinsic ADE [43].
4.3. The Role of FcγRs in ADE
ADE is mediated by the IgG FcR and the complement receptor, among others. In humans, there are 3 types of Fc receptor that bind human IgG: FcγRI, FcγRII and FcγRIII.
FcγRI is present exclusively on monocytes/macrophages and binds human IgG with high avidity. Its specificity is higher for IgG1 and IgG3 isotypes than for IgG2 and IgG4 isotypes (nirsevimab is an IgG1 mAb). The other two receptors, FcγRII and FcγRIII, are found on monocytes, macrophages, eosinophils, neutrophils, natural killer cells, B lymphocytes and T lymphocytes. Both receptors have relatively low specificity and relatively low avidity for IgG compared with FcγRI [45]7
The role of FcγRIIIa is said to be double-edged. For example, FcγRIIIa in NK cells participates in viral clearance by eliminating infected cells through the ADCC phenomenon [46]. However, the role of FcγRIII is poorly understood and results are contradictory. FcγRIIIa expression is increased in NK cells from patients with severe hRSV-associated pathology, thus suggesting that this receptor may contribute to hRSV disease. This receptor may play an anti-inflammatory role during RSV disease, although it contributes to viral replication. In mice, FcγRIII has a pro-inflammatory role in RSV infection but may promote viral clearance. FcγRIII is present on nasal epithelial cells and controls the balance between tolerance and inflammation through its interaction with TLR4 [47]. The binding of FcγRs (type I or II) depends on the conformational state of IgG (open or closed); FcRn does not contribute to CD4+ activation [48].
4.4. Role of Complement
ADE may also be due to complement activation by the virus-antibody complex: entry into the cell is then via the complement receptor. Many cell types express complement receptors on their surface (complement is a group of serum proteins involved in immune reactions) [49,50]. The effector functions of therapeutic monoclonal antibodies also result from recruitment of the C1q component of complement and subsequent activation of the classical complement pathway. Complement activation eliminates target cells in two ways: direct lysis of the cell (complement-dependent cytotoxicity (CDC or CDCC)) and deposition of opsonins, such as C3b, on the cell surface, which are recognized by complement receptors (CRs) on effector cells. Activated CRs trigger complement-dependent cell-mediated cytotoxicity (CDCC) and complement-dependent cell-mediated phagocytosis (CDCP). Since C1q and FcγR binding sites on the Fc domain are proximal and partially overlapping, amino acid substitutions altering FcγR binding also alter C1q recruitment and vice versa [51]. It has been shown that it is necessary to completely (and not partially) eliminate the effector functions of mAbs against Ebola virus to obtain a therapeutic effect: these effector functions could induce an ADE effect which would cancel out the therapeutic effect. In influenza infections, autopsies of fatal cases have shown that C4d deposits in the lungs indicate the presence of large quantities of CI in the virus’ target organ. It is not known to what extent mAb-mediated cytoxicity contributes to protection or pathogenesis [18].
4.5. The Same Mechanisms are at Play for ADE in RSV Infection
With regard to intrinsic ADE, complement-fixing immune complexes and complement activation play an important role in the pathogenesis of ERD (enhanced respiratory disease) or VAERD (Vaccine-associated enhanced respiratory disease). IgG and the C3 component of complement are colocalized in the alveoli and bronchioles of mice suffering from VAERD [52].
Inflammatory cytokines aggravate RSV infection: infection of a macrophagic cell line (U937) by RSV, albeit limited compared with other cell types, could aggravate clinical disease through an imbalance in inflammatory cytokine production at the expense of protective cytokines [53].
In 2021, Polack [54] re-examined the case of two young children who died of RSV disease after being vaccinated with an inactivated vaccine in trial (80% of children vaccinated and subsequently infected with RSV had to be hospitalized). VAERD-associated bronchiolitis is characterized at autopsy by diffuse inflammation of the alveoli and infiltration by neutrophils and eosinophils. Eosinophilia is not found in the blood. In classical bronchiolitis, inflammation is centred on the bronchioles, and eosinophils are virtually absent. In VAERD, unprotective antibodies form immunocomplexes with complement, triggering a Th2 polarization of the cellular response (Th2 cytokines increase bronchoconstriction and lung pathology).
4.5.1. Involvement of the Monocytic Lineage in ADE
In ADE during dengue virus infection the monocytic lineage is involved, via the FcγRs it expresses. Macrophages may play a beneficial role in viral clearance, or a deleterious one by promoting replication. During ADE, the formation of virus-antibody complexes increases viral replication in macrophages [55]. This has been confirmed for other viruses [56] and more specifically for RSV infections [39,57,58,59,60,53]. The FcγRs of dendritic cells (which also belong to the monocytic lineage) are also involved [61]. Alveolar macrophages are sensitive to RSV replication [60,62,63], but alveolar macrophages are less sensitive to RSV replication than bronchial epithelial cells [64].
4.5.2. ADE with RSV Antibodies May be Mediated by Macrophages and Phagocytic Cells
Comparing monoclonal antibodies against the RSV F protein, Gimenez showed on cultured macrophage cells (U937 carrying the Fc receptor) that only the neutralizing antibody is able to induce ADE (at least one facilitating epitope is present on F). The extent of ADE correlates with complement fixation and the level of neutralizing antibody. This occurs at low antibody levels. The ADE mechanism could involve the macrophage FcγR, which facilitates virus internalization. Gimenez also proposes to study the macrophage receptor for complement and the in vivo significance of these in vitro studies. The severe pulmonary pathology observed following inactivated RSV vaccination could be the result of both an Arthus reaction and increased virus replication in macrophages [58]. This was confirmed by Krilov in 1989 [59], who demonstrated in vitro ADE in a murine line of macrophages by binding monoclonal antibodies (directed against F protein) to the Fc receptor on recipient cells. ADE occurs with sub-neutralizing dilutions of an F protein mAb. The seasonal peak in the incidence of RSV infections occurs when maternal antibodies are still present but begin to decline in titer. Binding and neutralizing antibody levels may vary according to RSV viral subtype following primary infection. In a macrophagic line, sera from immunized children have the capacity to aggravate RSV infection in vitro at concentrations close to physiological levels. This ADE is dependent on the Fc receptor. These sera are rich in RSV F-protein antibodies. The balance between neutralization and ADE is essentially a function of the antibody/virus ratio. This is an extrinsic ADE by aggravating the infection of alveolar macrophages; but there could also be an intrinsic ADE by altering cytokine production in infected macrophages. It is therefore important to test the possibility of an ADE in the presence of antibodies on a macrophagic cell line carrying several Fc receptors (such as the high affinity FcγRI and the low affinity FcγRIIA). The U937 macrophagic cell line has been proposed [60]: it is also useful for investigating the pro-inflammatory role of IgG binding to FcγRI [65]. Gimenez [66] confirmed these results in 1996 with monoclonal antibodies directed against the F protein, and highlighted the importance of antibody mixtures: neutralization and ADE activities were modified when two mAbs were mixed. A synergistic ADE effect may occur in the presence of different antibodies. Thomas [67] also points out the role of immunocomplex deposition with complement and the increased infection of macrophages when mAbs are directed against the F protein. In 2023, Vandervenrecalls the ability of certain anti-RSV mAbs to induce ADE in FcγR-expressing monocytic cell lines; he stresses the need to evaluate possible harmful effects linked to the effector function of FcγRs. Mabs with high anti-viral activity could contribute to inflammation in the advanced stages of RSV disease and in influenza [39]. For Gomez, pulmonary dendritic cells (DCs) are also infected by RSV via virus-bound anti-F antibodies, and FcγRs play an important role in hRSV pathogenesis even in the presence of neutralizing antibodies. Infection of DCs with antibody-bound hRSV leads to an impaired T response [61].
4.5.3. Importance of Antibody Levels and Quality: ADE can Occur in the Presence of Low Levels of Strongly Neutralizing RSV Antibodies
In the case of mAbs capable of neutralizing dengue virus, ADE occurs only with certain epitopes and viral strains. For the same antibody, neutralization occurs at low dilution and ADE at high dilution: neutralization occurs when virions are in the presence of a large excess of antibody [55].
It has been shown that ADE induced by anti-dengue vaccine antibodies occurs only at low antibody levels (: the correlate of risk for severe dengue is distinct from the correlate of protection) [68].
An in vitro ADE effect has been demonstrated on cells expressing certain FcγRs with sub-neutralizing antibody concentrations in certain viral infections [18]. This was shown for palivizumab on a human monocytic cell line. Pavilizumab and the precursor to nirsevimab (D25, lacking the YTE mutation that increases affinity for FcRn), induce ADE at low concentrations, whereas they are strongly neutralizing; at high concentrations, infection is blocked. For D25, maximum infection was observed at a dilution corresponding to a concentration as low as 1 ng/ml [69]. Palivizumab recognizes the post-fusion (and pre-fusion) form of F protein, whereas D25, which is more potently neutralizing, is specific for the post-fusion form, as is nirsevimab [70]. The role of maternal antibodies confirms the importance of antibody quality and quantity in ADE
RSV infections are usually mild, but severe infections are observed in the first 6 months of life when maternal antibodies are still circulating. Infants with maternal antibodies to RSV were not only susceptible to RSV infections, but the rate of severe illness was higher in these infants than in infants without maternal antibodies [45]. Antibody levels do not correlate with protection against RSV infection in challenged animals, and severe infections in newborns occur despite high levels of maternal antibody [69]. However, the severity of RSV infection requiring oxygen therapy is lower in breast-fed infants than in formula-fed infants [71]. Anti-RSV IgG is found in 95.2% of newborns at birth: there is placental transfer of maternal antibodies; maternal antibodies decrease considerably after 2 months of life; the lowest level is reached at 7 months, after which the level rises again with the first infections. The hospitalization rate for newborns peaks at around 6 months of age. Maternal antibodies do not protect against reinfection: this would be due to the short lifespan of the antibodies and also to mutations in the virus, which escapes the antibodies acquired by the previous infection [72].
5. How Were the Factors Likely to Cause ADE with Nirsevimab Assessed?
5.1. Pharmacokinetics
Given the possibility that a low level of neutralizing antibodies may be capable of inducing ADE, it is important to be familiar with the pharmacokinetics of nirsevimab. For the same antibody, neutralization occurs at low dilution and ADE at high dilution: neutralization occurs when virions are in the presence of a large excess of antibody [55]. In 2006 Dall’Acqua [15] studied the comparative pharmacokinetics of two RSV anti-F mAbs (MEDI-524 and MEDI-524-YTE, which differ from nirsevimab in the epitope targeting the F protein) in IV in cynomolgus at doses higher than those used in clinical trials (30mg/kg). Levels were measured at 4 and 24 days post-injection, and were 4 times higher in BAL than in serum (with a decrease observed as early as 24 days). This confirms that YTE mutations increase lung half-life in monkeys, as expected. In a Phase 1 study on 136 healthy adults (81 of whom received an IM injection of nirsevimab), the maximum concentration of binding antibodies was reached within 3 days. All subjects had virus-neutralizing antibodies prior to injection. Peak neutralizing activity is reached 6 days after IM. The most frequent adverse effect was URTI (upper respiratory tract infection) , with an imbalance in the treated group. Unfortunately, no tests for RSV were carried out [73]. In the EPAR (European Public Assessment Report), pharmacokinetics are assessed by simulation based on animal injections [4] and on data obtained in infants treated with palivizumab (which does not have the YTE mutations). No direct measurements have been made in infants injected with nirsevimab [74]. However, when studying pharmacokinetics in cynomolgus monkeys after IV injection, the elimination phase can be poorly assessed, especially at low plasma concentrations [75]. When the results are used to estimate the levels achieved in humans by IM, this extrapolation could lead to clinical inefficiency. In cynomolgus monkeys, doses much higher than those used clinically (300 mg/kg) are injected IV or IM. Nirsevimab was found in nasal fluid at 24 and 72 h and in BAL at 72 h, with considerable individual variability (in only 5 animals). The mean concentration of nirsevimab in the nasal fluid was approximately 1/10,000 of the serum concentration. The EC90 concentration (providing 90% protection, i.e. 3 log reduction in viral load in cotton rat lungs) of MEDI18897* (equivalent to nirsevimab but lacking the YTE mutations) is calculated at 6.8 μg/ml. According to the simulation model, this concentration would be reached with nirsevimab on the first day after injection in infants. The serum concentration in rats required to block replication of mutant strains may be 10,000 times higher than that required against the wild-type strain [4,74]. In 2018, Domachowske explored the pharmacokinetics of nirsevimab injected at different concentrations into 89 preterm infants with a mean age of 6 months. Binding antibody concentrations were measured at 8, 31, 151 and 361 days post-injection, and neutralizing antibody concentrations at 8, 151 and 361 days. Two treated children were lost to follow-up; maximum antibody levels were reached after 8 days. RSV neutralizing activity correlated with concentration, but the progression in the first 8 days was unknown. 5% of children have an antibody level below 4-fold the baseline after 8 days, and 10% after 151 days. Anti-drug antibodies (ADAs - targeting YTE mutations) are detected in 28% of treated subjects: these ADAs could impact pharmacokinetics between days 151 and 361 in some subjects [76]. In rare treated children, maximum concentration is reached only after 150 or even 200 days [77, figure S5].
The EMA (EPAR [74]) estimates the threshold effective concentration on the basis of in vitro simulations and animal studies. EC50 neutralizing activity is achieved at levels of 0.48 to 60 ng/ml in vitro, depending on the viral strains used. In the MEDLEY trial [78], 6% of infants with chronic lung disease and 20% with congenital heart disease failed to reach the target exposure threshold of 12.8 day.mg/ml associated with protection against RSV after a single dose. According to EPAR ([74] page 49) , data from the MEDLEY [78] and MELODY [79] studies show that in rare children (1.5 to 2%), the concentration required for efficacy is not reached (it remains below 6.8 μg/ml on day 151). Maximum concentration is reached in 6 days, with a half-life of 69 days. Nirsevimab immunogenicity may induce the synthesis of anti-nirsevimab antibodies: after 151 days, nisrsevimab concentrations are lower in subjects in whom ADAs (anti-drug antibody) are detected.
5.2. Study of Nirsevimab Binding to FcγR In Vitro and Ex-Vivo and of Possible ADE in Animals by Manufacturers
According to the EMA Risk Management Plan [9, page 20], ADE is a theoretical risk for mAbs through binding of virus-antibody complexes to Fc receptors on target cells. The EMA prescribes the methods to be used to assess the risk of ADE and concludes that ADE has not been observed in clinical trials or in cotton rats, even at sub-effective doses of 0.125 mg/kg. The EMA notes that no cases of ADE have been reported in the post-marketing surveillance of palivizumab. The EMA EPAR [74] recalls that ADE can be antibody-mediated within a narrow range of sub-neutralizing antibody levels. In the case of RSV Fc-mediated effector function may have a role in ADE of RSV disease, through either cytotoxicity (ADCC), phagocytosis (ADCP antibody- dependent cellular phagocytosis) or complement activation. Risk assessment should be carried out according to Munoz [79], who reminds us that all cases of vaccine failure should be investigated for VAERD, viral infection should be confirmed by detection and quantification of the virus in specific sites (blood, upper and lower respiratory tracts, tissues) as well as characterization and sequencing of the virus; the immune response should be assessed and compared (in the present case mAb levels should be measured). Deposits of C4d and the presence of IC-C1q in fluids should be detected, and C3 levels measured to explore complement consumption. Tissues obtained by biopsy or autopsy should be evaluated for evidence of immunopathology [79]; chest computed tomography (CT) has a high sensitivity for diagnosis of lower respiratory tract disease involvement [79]. In vitro data on ADCP antibody-dependent cellular phagocytosis, ADCD antibody-dependent complement deposition, ADNP antibody-dependent neutrophil phagocytosis and ADNKA antibody-dependent NK cell activation were provided for palivizumab and nirsevimab. Nirsevimab slightly induced ADNP and ADNKA and to a greater extent ADCP and ADCD, in a comparable range as palivizumab. The binding of nirsevimab to the various is studied only in vitro. The in vivo study was carried out with another mAb (1G7) in the cotton rat compared with a mAb lacking FcγR binding [76]. Other studies on the effector functions of nirsevimab and related mAbs have been carried out by the manufacturers and are not mentioned in the EPAR. The in vitro study of monoclonal antibody activity in mice may not reflect what happens in humans, as some mAb variants show a parallel increase in binding at pH 7.4 for murine FcRn, but not for human FcRn, whereas affinity at pH 6.0 is comparable. With MEDI-493 mutants (not having the same targeted epitope as nirsevimab), YTE mutations (present on nirsevimab) increase the binding of this antibody to human FcRn at acidic pH (6) but not at neutral pH (7.4) [80]. Studies of FcγR binding and pharmacokinetics in cynomolgus monkeys with mAbs close to but different from nirsevimab (including motavizumab MEDI-524) show that YTE mutations which increase FcRn binding (and half-life in cynomolgus monkeys) do not affect the virus-neutralizing capacity of motavizumab [80]. Motavizumab, which differs by 13 amino acids from palivizumab, has a 100-fold higher neutralizing activity in cotton rats [81]. The mAb with the YTE mutation has a longer persistence time in the BAL of treated monkeys than the mAb without this mutation. FcγRIIIA binding and ADCC assays on human PBMC were carried out with an antibody other than nirsevimab (this antibody also possesses the YTE mutations, but is not directed against the RSV F protein): YTE mutations were shown to reduce FcγRIIIA binding by a factor of 2 (allotype F158), as well as ADCC activity by a factor of 100 [80].
Clinical trials of nirsevimab began in 2016, but the study of its effector functions mediated by the Fc of this mAb was not published until 2023 [82]. Brady [82] confirms that the YTE substitutions introduced on nirsevimab increase, as expected, its half-life, but strongly decrease the ADCC measured in vitro. The results show that nirsevimab binds to both activating and inhibitory FcγRs. The affinity of nirsevimab is slightly reduced in vitro for the various Fcγ compared with that of the surrogate. Effector functions are studied in vitro (on different human cell types) and ex vivo (using sera from children participating in the nirsevimab clinical trial; these sera are collected 15 days after injection; analyses are performed on serum pools (30 sera from treated participants are mixed, representing only 1.5% of the treated cohort). Ex vivo effector activities vary with serum dilutions, and only stand out from controls for certain dilutions. Ex vivo, nirsevimab promotes neutrophil-dependent phagocytosis at certain dilutions; this ADNP is protective in adenovirus-vectored RSV vaccinees. In vaccinees with adenovirus expressing prefused F protein, neutrophil phagocytosis is increased after RSV challenge. At certain dilutions, nirsevimab amplifies ADCP (an assay reporting phagocytosis by macrophages), and Brady et al. stress the need to better understand its in vivo role in protection against RSV. The role of complement is investigated via ADCD. Ex vivo, nirsevimab (at low dilution -high concentration- only) can strongly increase ADCD compared to placebo at certain dilutions. However, the authors do not anticipate any deleterious effect on this effector function in vivo, without further clarification. Regarding CDC Brady [82] and Booth [83] assume that it will not be exacerbated by nirsevimab. CDC is not investigated here, but the authors do not anticipate CDC in vivo, as the YTE modification overlays the FcRn-binding sites, but this is not demonstrated by any experiments. NK ADCC is mediated only by FcγRIIIA, and nirsevimab activates this effect less than palivizumab in vitro; this is confirmed ex vivo: ADCC is identical to placebo. A basal ADCC activity is observed in placebo sera and is thought to be due to maternal antibodies: this binding of maternal antibodies to FcγRIIIA could compete with nirsevimab and prevent it from binding to this Fc.
6. Clinical trial Results and 2023-2024 Campaign
Following its approval by the EMA on October 31, 2022 [74] and by the FDA on July 17, 2023 [84], nirsevimab was recommended for infants and children under 2 years of age in 4 countries for the 2023-2024 season: USA, France, Spain and Luxembourg. In the USA, on August 3, 2023, the Advisory Committee on Immunization Practices recommended nirsevimab for infants aged <8 months born during or entering their first RSV season and for infants and children aged 8-19 months who are at increased risk of severe RSV disease entering their second RSV season. [85].
In France, the HAS (Haute Autorité de Santé) recommends nirsevimab for infants born at a gestational age of 35 weeks or under and under 6 months of age entering their first RSV season, infants under 2 years of age having required treatment for bronchopulmonary dysplasia in the past 6 months, infants under 2 years of age with congenital heart disease with a haemodynamic impact [86]. In Spain, the Spanish Society of Neonatology recommends nirsevimab for all newborns and at-risk children under two years of age [87]. In Luxembourg, the government has recommended immunization for all newborns and children under one year of age [88].
6.1. Clinical Trial Results
6.1.1. Phase 1 and 2a
In the Phase 1b/2a trial on infants, 5 infants in the treatment group (69 children) developed LRTI (low tract respiratory infection) corresponding to the clinical criteria used for evaluation, and 3 developed febrile convulsions (but none in the placebo group of 16 children); one child in the treatment group was hospitalized for LRTI. With regard to LRTI not corresponding to the protocol definition, 10 children treated versus 1 in the placebo group developed LTRI within 151 days of observation, and RSV A was found 8 days after injection in 2 infants treated with the lowest dose. No RSV viruses were found in hospitalized children, but no virus tests were carried out in children with LRTI who did not meet the protocol criteria (only severe LRTI requiring assisted ventilation were included) or in those with URTI: no virus tests were carried out, despite the fact that 69% of reported adverse events concerned URTI and that 14.1% of treated children developed LRTI compared with 5.6% of placebo-treated children [89]. In the phase 1 trial in adults, the most frequent adverse event was URTI, with an imbalance in the treated group. Unfortunately, no search for RSV was carried out [73].
6.1.2. Results of Phase 2b and 3 Trials
The trials were sometimes carried out during periods of low RSV circulation: the number of cases observed was low, and this may reduce the statistical power of the trials and the ability to assess ADE [77,90]. Phase 2b and 3 clinical trials on nirsevimab [73,77,78,89,91] were analyzed by the EMA and HAS. In the MELODY trial, only LTRIs were evaluated [77]. The 150-day efficacy on hospitalization for LRTI due to RSV is calculated on a very small number of participants (14 hospitalized in all, 0.6% in the treated group and 1.6% in the placebo group, so the ARR (absolute reduction risk) is 1%). Treated participants hospitalized for LRTI due to RSV were hospitalized longer than those in the placebo group, but no significance can be drawn from this, as too few individuals were involved. 8.1% (80/987) of treated children were removed from the analysis of results. In the MEDLEY study [78], which combines 2 studies (one phase 2b [92] and one phase 3 [77] ), nirsevimab reduced the ARR of LRTI hospitalization for all causes by 2% (total number 108/2350) and by 2% for LRTI due to RSV (28/2350); but 2% of participants in the treated group were excluded before 151 days, compared with 1% in the placebo group, which could distort the estimate of efficacy and safety from the ADE point of view. In preterm infants study, nirsevimab was effective against hospitalization for RSV infection and reduced the severity of RSV infections (only 5 participants in the placebo group were hospitalized in ICU or required assisted ventilation -4). The occurrence of LRTI not due to RSV is not affected by treatment. All-cause hospitalizations are not reported in this study. 5.79% (56/968) of treated children were withdrawn from the study [92]. The open-label HARMONY study [91] involved 8058 infants under 1 year of age in Europe in 2022-2023. The investigator was sometimes the treating physician and decided whether or not hospitalization was due to RSV. RSV PCR tests were not routinely performed, but only after hospitalization, and 16 hospitalized children did not undergo RSV PCR tests. Final results have not been published, as 12 months’ follow-up is required for all children. The study showed a 1.2% reduction in the ARR of hospitalization for LRTI due to RSV, and a 0.4% reduction for very severe LRTI. For LRTI of all causes, the ARR of hospitalization was 1.3%. The Kaplan-Meier curves differ from country to country, and no explanation is given: in France, hospitalizations for LRTI due to RSV occur from the first day of the study among placebos, whereas in Germany they begin only 15 days post-injection, and 1 month later in the UK. This may reflect the staggered circulation of the virus in these countries. Among grade 3 serious adverse events, there were as many infections in the treated group as in the placebo group, and more severe infections in the treated group than in the placebo group. Among treated children, only 500 out of 4037 (12%) were less than 7 days old. 0.22% of treated children withdrew from the study.
6.1.3. Deaths in Trials
The FDA notes an imbalance of deaths in favor of the treated group: 12 deaths in the 3,710 treated participants (0.32%) versus 4 in the 1,797 control participants (0.22%), taking into account one death in the placebo group 6 days after the end of the study and without specifying deaths in the treated groups after the end of the study [93]. In the studies listed by the EMA, 8 deaths were recorded (with the same percentage - 0.3% - in the treated and placebo groups). The Domachowske study [89] (which was published as a correspondence, i.e. not peer-reviewed) concerns premature babies and newborns suffering from heart or lung disease, and compares the effect of the monoclonal antibody previously used (Synagis, palivizumab) with Beyfortus (nirsevimab). It provides a full description of the deaths observed: of the 6 deaths listed, 5 were related to pneumonia or bronchiolitis not attributed to treatment (5 babies who died were treated with Beyfortus (5/614 = 0.81%) and 1 (1/304 = 0.32%) with Synagis). Despite the low number of deaths, there was an imbalance against nirsevimab. 6.86% of treated children (63/918) were withdrawn from the study.
6.2. Pharmacovigilance Data
The EudraVigilance database records spontaneously reported adverse events for Beyfortus. As of April 15, 2024, there were 140 reports, mainly from healthcare professionals (138/140), 89 of which were for “bronchiolitis”, 129 for “RSV” and 56 for “drug ineffective”. Only 26 reports did not concern respiratory events [94]. Moreover, according to the Ile de France pharmacovigilance center, there is a theoretical risk of aggravation of RSV infection through non-neutralizing antibodies [95].
6.3. Results of the 2023-2024 Season Immunization Campaign
In the USA, immunization coverage for children under 20 months of age is less than 20% (except in Alaska, 22.2%), but data do not include children born after October 1, 2023 [96].
An exceptional peak in the rate of hospitalizations due to RSV was observed in 2022-2023 in 0-4 year-olds (68.9 hospitalizations/100,000); the rate in 2023-2024 (34.6/100,000) returned to levels comparable with previous seasons (18.4 in 2019 and 26.5 in 2020, 13.3 in 2021-2022 with a more spread-out season) [97]. The results of this campaign are set out in a non-peer-review article by the CDC: a 90% efficacy rate against RSV-associated hospitalizations is announced, but the method of calculation is not set out (this is a case-control study). Participants were infants under 8 months of age hospitalized for ARI, with known nirsevimab status and having been tested for the presence of RSV by PCR [98]. In Luxembourg, Beyfortus immunization began in maternity hospitals on October 1, 2023, with 84% coverage. Children hospitalized with respiratory symptoms are systematically tested for RSV. RSV-positive newborns under 6 weeks of age are routinely hospitalized, so hospitalization is not a criterion of severity for this age group. As in other countries, the number of hospitalizations among young children was high in 2022. However, the 2023 epidemic was later than in 2022, and had not ended by week 52, when the comparison between the years 2022 and 2023 stopped (for the 2022-2023 season, cases occurring up to week 13 of 2023 are taken into account). 65.3% of children under 6 months hospitalized for RSV infection were not immunized. In 2023, there was a 38% decrease in the number of children under 5 hospitalized for RSV infection, and a 69% decrease in those under 6 months. The authors attribute these differences to immunization with nirsevimab [99]. In France, it is recommended to immunize all newborns before discharge from the maternity hospital (all newborns are targeted) [100,101]. It should be noted, however, that the HAS deems that BEYFORTUS provides a minor clinical added value in the prevention of RSV lower respiratory tract disease in neonates and infants with or without risk factors and ineligible for palivizumab, during their first RSV season [102]. According to a DGS (Direction Générale de la Santé, France) document dated December 26, 2023, 173,000 doses of Beyfortus® 50mg and 64,000 doses of Beyfortus® 100mg had been distributed, with the French government ordering a total of 251,800 doses [103]. In France, the number of births is estimated at 678,000 in 2023 [104], and a rough calculation suggests that these doses are sufficient for the majority of newborns during the immunization season (around 220,000 births). We can therefore assume that immunization coverage is high in France for this season. According to Santé Publique France [105], in comparison with previous seasons (with the exception of the 2022-2023 season, which was exceptionally intense), bronchiolitis-related activity was comparable in intensity in primary care, while hospital activity was comparable to or slightly higher than the reference seasons. The proportion of intensive care unit admissions for children under 2 years of age was comparable to the reference seasons. The proportion of hospitalizations for bronchiolitis in children under 2 years and under 3 months was comparable; in infants aged 3 months and over, emergency room visits and hospitalizations were higher than in previous seasons, and close to levels for the 2022-2023 season. Of the 668 severe cases in intensive care units in children under 2 years of age (between the end of August 2023 and the beginning of April 2024), the majority were in children under 6 months of age (73%), and 23% of cases had been treated with nirsevimab or palivizumab. 494 children under 6 months of age were admitted to intensive care for bronchiolitis during the season, of whom 143 (29%) had received nirsevimab and 10 palivizumab. 69% of these bronchiolitis admissions were due to RSV (alone or in association with other pathogens). It is impossible to derive from these data an estimate of the efficacy of nirsevimab on the risk of ICU hospitalization. Unfortunately, the same type of data (exact number of intensive care unit visits for bronchiolitis) is not available for previous seasons (only the number of hospitalizations is provided [106]). But according to the figure on page 13 of Santé Publique France’s 2023-2024 report, the number of hospitalizations in intensive care units for bronchiolitis in children under 2 years of age during the 2023-2024 season is comparable to that of previous seasons (with the exception of the 2022-20233 season, which was exceptional everywhere) and is even higher at the start of the epidemic (weeks 37 to 46: i.e. from September 15, 2023, the start of the immunization campaign). According to SPF, the number of hospitalizations for bronchiolitis in infants under 3 months of age is comparable to that of previous seasons (again, with the exception of the exceptional 22-23 season), and higher for infants over 3 months of age [105]. A case-control study in preprint surveys children under 5 months of age admitted to intensive care for bronchiolitis between September 15, 2023 and January 31, 2024 in France. Data are from Santé Publique France. 238 cases (newborns hospitalized for bronchiolitis with a positive RSV test) were compared with 50 controls (newborns hospitalized with a negative RSV test). It was therefore impossible in this study to match each case with a control, given the disparity in the numbers of cases and controls. The database included 542 children under 2 years of age admitted to intensive care for bronchiolitis during the 2023-2024 season (SPF data count 668 cases under 2 years of age over a wider period from late August 23 to early April 24); over 200 children were excluded for reasons of age, residing outside metropolitan France or admitted outside the study period; 342 infants admitted to intensive care for bronchiolitis during the study period were retained, but 23 were excluded for various missing data. 8 children were admitted to PICU less than 8 days after nirsevimab administration and 30 after an unknown delay. In all, the study of nirsevimab efficacy involved only 319 cases. Efficacy against ICU hospitalization for RSV bronchiolitis was estimated at between 74.4% and 80.6%, depending on the inclusion criteria [107].
According to an October 2023 survey, immunization coverage is above 80% in almost all regions of Spain [108]. This is confirmed by more recent publications (90% in Andalusia [109], 96.5% in Asturias [110], 86% in Madrid [111]. At national level, according to the SIRVA network [112], hospitalization rates for RSV infection in 0-4 year-olds are comparable for the 2022-2023 and 2023-2024 seasons (the peak is 140/100,000). In Galicia, according to the regional Ministry of Health report, epidemic waves of RSV infection are comparable over time between 2022-23 and 2023-24 in terms of positivity to RSV tested in the general population. There has been a drastic reduction in hospitalizations for RSV in both newborns and children born since April 2023, with a high rate of immunization coverage (93% in NN and 86% for catch-up infants born before the campaign). For children entering their 2nd RSV season, hospitalization rates are identical to previous seasons (they have not been immunized) [113]. The first results of the estimation of the efficacy of nirsevimab on newborns, children under 6 months and at-risk children under 2 years of age, in Galicia, Spain between September 25 and December 31, 2023 have been published [114]. This study is scheduled to run until 2026 [115], so the publication only concerns results from the first 3 months of the immunization campaign. Nirsevimab was offered to all newborns from September 25, 2023 unless medically contraindicated, and to all children born after April 1, 2023. Nosocomial infections were excluded. The efficacy of nirsevimab against hospitalization for RSV infection was 82% (on 46 cases) in intention-to-treat analysis on children under 6 years of age with no risk factors. Sensitivity analysis excludes breakthrough infections and gives 87.5% efficacy (on 44 cases). For all-cause hospitalization, efficacy was 66.2% (on 366 cases). The authors compare the results of the 23-24 season with previous seasons since 2016, excluding the 2020-2022 season because the epidemiology of bronchiolitis was affected by the Covid-19 pandemic. According to figure 4 in the appendix, if we exclude the exceptional 22-23 season, the 23-24 hospitalization rate is comparable to previous seasons, albeit slightly lower (since the p-values are calculated including the 22-23 season, we cannot know the significance of this decrease). Hospitalizations for RSV with severity criteria occur only in the treated groups: in infants born before September 25, 2023 (aged 1 to 6 months), there were 3 ICU admissions and 3 cases of mechanical ventilation (versus 0 in untreated infants). In infants born after September 25 (treated on the day of birth), there were 7 ICU admissions and 4 cases of mechanical ventilation (versus 0 in untreated infants). In Navarre, the efficacy of nirsevimab was calculated from epidemiological surveillance of newborns between October 2023 and January 2024 (the RSV circulation season) according to their immunization status (92% of newborns were treated, mostly within the first 7 days of life): only infants who had visited or been in hospital were taken into account. Despite the low number of non-immunized children (94/1177), efficacy could be calculated on emergency room visits for RSV (22 cases - 87.8%), hospitalization for RSV (16 cases - 88.7%) and admission to intensive care (85.9%, involving 5 children in total). Only newborns admitted to hospital were systematically tested for RSV by PCR; subjects who visited the emergency department were tested according to the paediatrician’s criteria. No details are given on the clinical results of the 5 children admitted to intensive care (2 non-immunized and 3 immunized). It should be noted that 9 subjects residing outside the province of Navarre were excluded, and that newborns injected less than 1 day previously are counted as non-immunized. Children admitted for respiratory illness not due to RSV were not counted. 78 hospitalizations due to RSV were observed for children born in 2023 and 2024, including 5 in ICU [116]; this compares with 664 hospitalizations and 50 ICU admissions for RSV observed in 2022-2023 (between October and March) for all ages combined (the only figures available for previous seasons. In this 2022-2023 report (a season without nirsevimab immunization), the hospitalization rate for RSV in children under 6 months of age was 4.9% [117]. For the 2023-2024 season, 8 out of 94 non-immunized children born between October and December 2023 were hospitalized, i.e. 8.5% of those under 3 months of age [116]. This is the only possible comparison between the 2 consecutive seasons (all hospitalized cases have been tested for RSV for 10 years, personal communication from the Navarre Institute of Public Health and Labor).
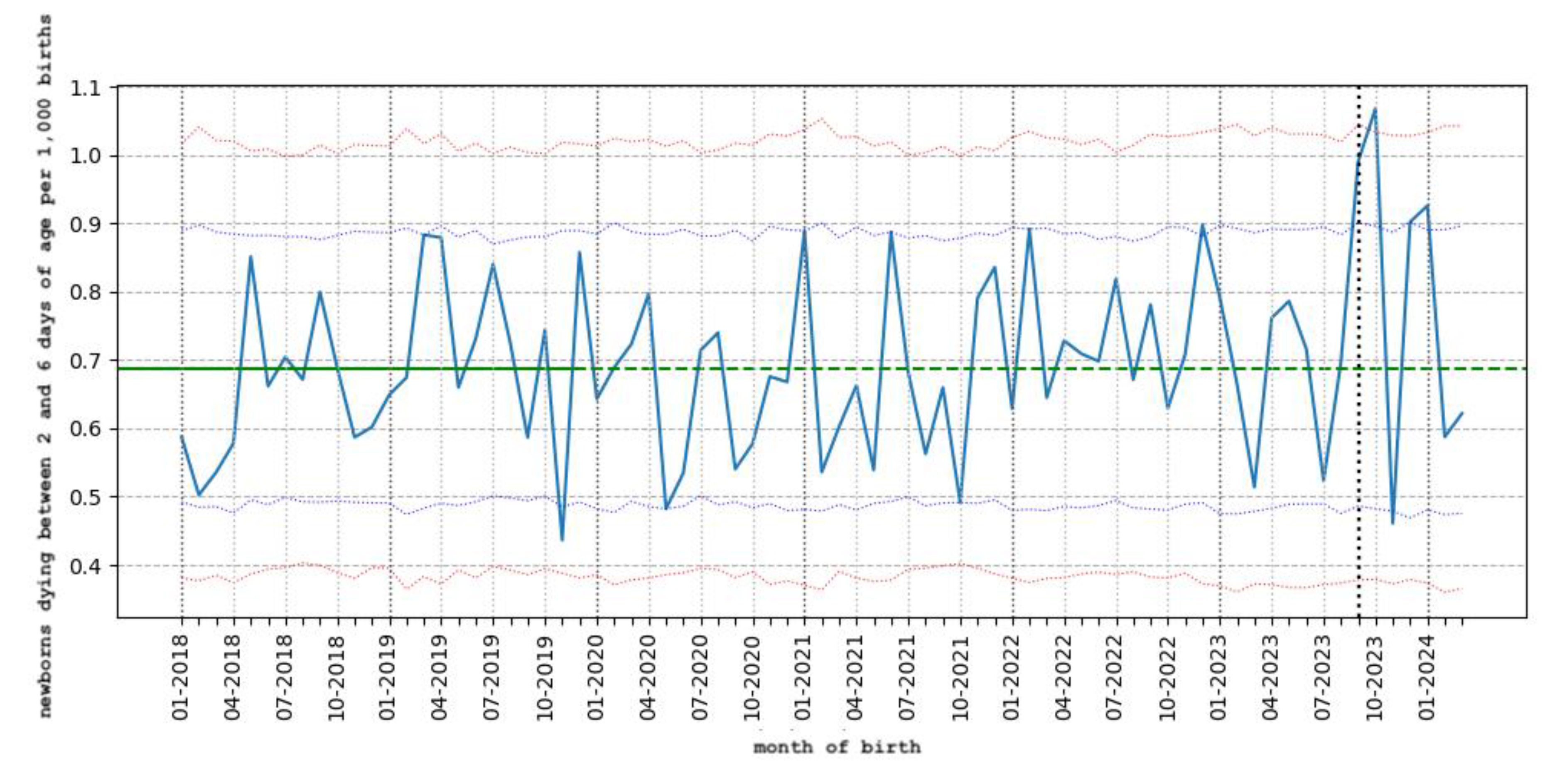
6.4. Stillbirths in France
In France, the government recommends injecting Beyfortus into all newborns leaving the maternity hospital [100]. The average length of stay for childbirth is 4.6 days [118]. Based on official French data on deaths [119] and births [120], it is possible to calculate the mortality rate between 2 and 6 days of life: this makes it possible to monitor the possible immediate impact of an injection of nirsevimab on leaving the maternity hospital, in terms of neonatal mortality. For each month, newborns born during the month and dying between 2 and 6 days of age are counted. The mortality rate is obtained for each month by dividing the number of deaths by the number of births. The reference rate is calculated by dividing the total number of deaths by the total number of births in 2018 and 2019 (before the Covid-19 pandemic): it is 0.69 deaths between 2 and 6 days of life per 1,000 births. This rate is indicated by the green horizontal line. For each month, the expected death rate is calculated by multiplying the reference rate by the number of births. Poisson’s law (a statistical law used for rare events) was used to calculate the different confidence intervals for mortality rates at 95% (classic threshold) and 99.8% (alarming threshold). The 95% confidence interval is marked by the blue dotted lines, with a probability of less than 2.5% for rates below or above the lines at the bottom and top respectively.The 99.8% confidence interval is denoted by the red dotted lines with a probability of less than 1 ‰ for rates below or above the lines at the bottom and top respectively. The black dotted vertical line represents the month of September 2023, with the Beyfortus immunization campaign starting on the 15th of that month). There is a highly significant peak (p=0.006) for babies born in September 2023 and dying between 2 and 6 days with 55 deaths (95% CI 27 - 50). And there is a very highly significant peak (p=0.0007) for babies born in October 2023 with 62 deaths (99.8% CI 22 - 60). In November 2023, after the two highly significant peaks, we observe a significant drop in the number of deaths (p=0.02) with 26 deaths (95% CI 27-50). In December 2023, the number of deaths is high, at the limit of significance at 2.5%, with 50 deaths (95% CI 26-50). And in January 2024 the number of deaths is significant (p=0.022) with 52 deaths (95% CI 27-50). In February and March 2024, mortality rates between 2 and 6 days of life returned to normal.
Figure 1.
Rates of death between 2 and 6 days of life per 1,000 births (France, INSEE data).
7. Discussion
Nirsevimab was approved by the EMA under a fast-track procedure [9, p 7] in accordance with European regulations (article 14(9) of EC-726 of 2004) [121]. This application was prompted by the high estimated burden of RSV LRTI worldwide (3.2 million hospital admissions, including 1.4 million for patients under 6 months of age) [1]. This estimate should be compared with the global burden of ARI in the under-5s, estimated at 12 million hospital admissions [122]. Hospitalizations due to RSV in the under-5s would therefore represent around a quarter of ARI admissions. This proportion is consistent with Del Riccio’s 2023 estimate. According to Del Riccio, admissions for RSV infections account for between 7% and 51% of hospital admissions for acute respiratory infections in Europe (36.1% for France, 33.6% for Luxembourg and 30.6% for Spain). However, admissions for RSV infection account for only 1.8% to 9.9% (3.7% in France) of hospital admissions in the under-5s, which raises the question of the economic benefits of immunizing all infants. It should be noted, however, that Del Riccio’s estimates are based on incomplete data between 1997 and 2018: various acute respiratory pathologies were grouped together, whether or not they were due to RSV [123]. In France, for example, virological diagnosis is not routinely carried out for all cases of bronchiolitis [105]. Prevention of other respiratory infections must therefore also be taken into account if we wish to relieve the burden on paediatric hospitals. The relative risk of hospital admission for RSV infection has increased in the post-pandemic COVID-19 seasons (2021 and especially 2022); this may be due to an increase in RSV testing. Severe cases increased relatively more in older children (> 1 year) during the post-pandemic seasons [124]. If this pattern continues in the future, prevention should target children over one year of age. The aim of this study is to independently assess the benefit of administering nirsevimab to all newborns to prevent hospital overcrowding, particularly in intensive care units. To be of benefit, nirsevimab must reduce the burden of hospitalization and admission to intensive care for all causes.
Analysis of the results of clinical trials and immunization campaigns will have to look for traces of a possible ADE effect, which is part of the EMA’s Risk Management Plan [9]. The facilitation of RSV infections and all respiratory infections should be examined.
7.1. Analysis of the Results of the Clinical Trials and the 2023-2024 Beyfortus Immunization Campaign
7.1.1. Clinical Trials
In the phase 1b/2a infant trial [76], LRTI corresponding to protocol criteria were observed only in the treated group (and not in the placebo group), and RSV A was found 8 days after injection in 2 infants treated with the lowest dose. But no investigation was carried out in participants with LRTI not meeting protocol criteria developed by 14.1% of treated children and 5.6% of placebo. LRTI was more frequent in the treated group than in the placebo group, and RSV was found only in the treated group: these results could be attributed to an ADE. In the phase 1 trial in adults [73], the most frequent adverse event was URTI with imbalance in the treatment group. Unfortunately, the participants were not tested for RSV, which eliminated the possibility of evaluating ADE.
Phase 3 and 2b trials are sometimes carried out outside periods of RSV circulation and therefore only imperfectly assess the possibility of ADE. In the healthy infants study, nirsevimab reduced the absolute risk of hospitalization due to RSV by 1%, but treated participants hospitalized for RSV LRTI were hospitalized longer than those in the placebo group [77]. But the imbalance between placebo and treated groups may lead to a result biased in favor of efficacy [125]. In the Simoes study [78], nirsevimab reduced the ARR of LRTI hospitalization due to all causes by 2% and LRTI due to RSV by 2%; but 2% of participants in the treated group were excluded before 151 days, compared with 1% in the placebo group, which could bias the estimate of efficacy and safety from the ADE point of view. There were more signs of severity (increased respiratory rate, O2<90% and acute hypoxic or ventilatory failure, length of hospital stay, risk of ICU admission and use of assisted ventilation) of all-cause LRTI in participants hospitalized in the treated groups than in the control groups. The same order of magnitude reduction in absolute risk was obtained in the Drysdale Harmonie [91] study on infants aged ≤12 months (1.2% for LRTI due to RSV and 0.4% for very severe LRTI). Among grade 3 serious adverse events, there were as many infections in the treated group as in the placebo group, and more severe infections in the treated group than in the placebo group [90]. In the premature infants study, nirsevimab is effective against hospitalization for LRTI due or not to RSV. However, no details are given on the cause of the 2 deaths in the treated group, nor of the 54 children withdrawn from the study before efficacy calculations were made [92].
In conclusion, in these clinical trials, hospitalizations for LRTI due or not to RSV were rare and very slightly less frequent in the treated groups compared with placebo, but they could be more severe following treatment: in these rare cases, ADE could therefore explain these results. It is worth noting the high rate of children withdrawn from studies before analysis (between 6 and 8%, except in the open-label Drysdale study [90], 0.22%): this could distort efficacy results and the search for ADE.
With regard to deaths, the FDA [93] notes a slight imbalance in favor of the treated groups, whereas the EMA reports the same percentage (some studies are not evaluated by the EMA). In the study of fragile children, 6 deaths were observed, including 5 (0.81%) treated with nirsevimab and 1 (0.32%) with palivizumab; 5 were related to pneumonia or bronchiolitis not attributed to treatment. In the absence of a placebo group, this result may be interpreted in terms of the lower efficacy of nirsevimab compared with palivizumab. It is also possible that nirsevimab facilitates and aggravates bronchiolitis: these injections take place during periods when the virus is circulating. In particular, nirsevimab appears to be more dangerous than its predecessor, palivizumab. RSV testing was not performed except in one case (negative result), which is regrettable in a study of the efficacy of a product against RSV. No autopsies for histopathological analysis of the lungs were reported. It was therefore impossible to look for signs of inflammation and VAERD [89]. However, the EMA recommends ADE risk assessment according to the methods proposed by Munoz: all cases of vaccine failure should be investigated for VAED, viral infection should be confirmed by detection, sequencing and quantification of virus in specific sites; mAb levels should be measured. Immunocomplex deposition and complement consumption should be measured. Tissues obtained by biopsy or autopsy should be evaluated for evidence of immunopathology [79]. In conclusion, it is therefore impossible to exclude ADE as a cause of these 5 deaths. In the European pharmacovigilance data EudraVigilance [94]: the most frequently reported adverse event is bronchiolitis, and could be due to an ADE effect.
7.1.2. Results of the 2023-2024 Season Immunization Campaign
The 2022-2023 season was exceptional in terms of the precocity of virus circulation and the number of hospitalized cases in 3 of the 4 countries where nirsevimab immunization campaigns took place in 2023-2024. This is the case in Luxembourg [99], France [105] and the USA [97]. In Spain, the 22-23 and 23-24 seasons were comparable in terms of hospitalization due to RSV in 0-4 year-olds (peak hospitalization rate of around 140/100,000) and higher than the 21-22 season (peak of 40/100,000) [112]. However, in Galicia [114], the 22-23 season was exceptional in terms of hospitalizations due to RSV (114 figure 2 in the main text and figure 4 in the appendix), as in many regions of the world, and should also have been excluded. The 2023-2024 season was comparable to the seasons preceding Covid-19 in the 4 countries concerned in terms of virus circulation. In the USA, immunization coverage is around 20%, making it unnecessary to compare hospitalization rates for RSV and non-RSV LRTI with previous seasons (hospitalization rates for 0-4 year olds in 2023-2024 are comparable with previous seasons, if we exclude the exceptional 2022-2023 season). A non-peer-review study by the CDC [98] claims 90% efficacy against RSV-associated hospitalizations, but this is a case-control study involving only infants hospitalized for ARI. But it is impossible to match a case with a control given the disproportion of the groups (59 children treated with nirsevimab out of 699). Hospitalizations less than 7 days after nirsevimab injection are excluded. The published figure shows that ARIs are more frequent in the days following injection, and it is unfortunate that the first 7 days have been excluded from the calculation (20 nirsevimab-treated children are excluded for this reason, reducing the number of treated children studied to 59). Of the children hospitalized for ARI due to RSV 6 had received nirsevimab, if we were to add these 20 excluded children, the number would rise to 26, and the product’s efficacy would be less than the advertised 90%. It is therefore impossible to evaluate the possible ADE, but we can suspect that it exists and that it is linked to the maximum circulating antibody level not yet reached, since of the total number of hospitalizations due to RSV, 77% are observed within 7 days of injection.
In Luxembourg, the authors of a study on the results of the immunization campaign attribute to nirsevimab a 38% reduction in the number of children under 5 hospitalized for RSV infection, and a 69% reduction in children under 6 months of age, compared with the previous season. But as in other countries, the 2022-20223 season was exceptional in terms of hospitalization rates, which invalidates the comparison. The immunization status of hospitalized patients has not been studied; the reduction in hospitalizations for RSV in children under 5 years of age is lower than in those under 6 months of age, and the reduction in the burden on hospitals may be lower than hoped [99], since severe cases are more frequent among children over 1 year of age [124]. No data have been published on all-cause hospitalizations, and hence on the potential reduction in hospital burden [99].
In France, we do not have precise estimates of immunization coverage. All newborns have been targeted since September 15, 2023 [100,101], and 251,800 doses have been ordered by the government for the 2023-2024 season (including 64,000 100 mg doses for infants over 5kg and therefore catch-up doses outside maternity wards; 237,000 doses have been distributed in total (94%) [103]. In France, the number of births is estimated at 678,000 in 2023 [104], a rough calculation allows us to estimate that these doses are sufficient for the majority of newborns during the immunization season (around 220,000 births).
According to Santé Publique France [105], hospital activity was comparable to previous seasons (apart from the exceptional season of 22-23), except for infants aged ≥ 3 months, for whom emergency room visits and hospitalizations were higher than in previous seasons, and close to levels for the 2022-2023 season. It should be noted that RSV was only systematically tested on hospitalized patients during the 23-24 season. Despite this uncertainty, the aim of the immunization campaign, which was to avoid overloading these services, has not been achieved.
A case-control study in preprint [107] estimates the efficacy of nirsevimab against hospitalization in intensive care for bronchiolitis due to RSV at between 74.4 and 80.6% (depending on the inclusion criteria) and against hospitalization in intensive care for bronchiolitis due to RSV at between 74.4 and 80.6% (depending on the inclusion criteria). These contradictory results may be explained by the impossibility in this study of matching each case with a control, given the disparity in the numbers of cases and controls, and by the large number of excluded cases (only 48% - 319 out of 668 - of the cases recorded by Santé Publique France were analyzed). 126 cases of admission to intensive care in children under 2 years of age between the end of August 23 and the beginning of April 24 were not included in this study (difference with the complete SPF data - 668 cases - and the 542 cases initially retained in the Paireau study [107]); in the end, the study of the efficacy of nirsevimab concerned only 319 cases out of a total of 668 (48%) following exclusions. As the majority of cases were excluded, it is problematic to calculate the efficacy of nirsevimab, as it is impossible for the excluded cases to have been excluded for reasons of date (only cases occurring between September 15, 2023 and January 31, 2024 were retained): the RSV epidemic began in mid-October 2023 and ended in early January 2024 according to SPF [105]. The majority of these 52% of excluded cases therefore concerned infants with missing data, which is a pity, since according to SPF’s 23-24 report [105], surveillance of RSV infections in patients admitted to the intensive care unit usually concerns only those over 18 years of age. For the 23-24 season, non-exhaustive surveillance is carried out for all cases of bronchiolitis in patients under 2 years of age, regardless of the virus involved (identified or not). It should be noted that for the 2020-21 and 2021-22 seasons described by Vaux [126], a case of bronchiolitis in a hospitalized child under 2 years of age is defined [127] according to the ICD [128] and corresponds to codes J21, J21.0, J21.8, J21.9, which concern all acute bronchiolitis regardless of the virus involved.
It is therefore difficult to draw any conclusion on the efficacy of nirsevimab in preventing ICU hospitalizations for bronchiolitis (whether or not due to RSV), which was the aim of the campaign: to avoid overloading hospitals.
Concerning the possible detection of early ADE among nirsevimab-treated newborns, comparison of the main analysis and sensitivity analysis 2 shows that 45% (17/38) of PICU admissions for LRTI not due to RSV and 27% (14/51) of LRTI cases due to RSV occurred less than 8 days (or after an unknown delay) after nirsevimab injection.
We cannot be certain that newborns over 1 month of age are hospitalized more than 8 days after injection, as they may have received nirsevimab after discharge from the maternity hospital.
Among nirsevimab-treated newborns, a comparison between sensitivity analysis 1 and 2 shows that 8% of PICU admissions for LRTI not due to RSV and 8% for LRTI due to RSV definitely occurred less than 8 days after nirsevimab injection.
These percentages could indicate specific (LRTI due to RSV) and non-specific (LRTI not due to RSV) ADE and should be explored according to Munoz’s criteria [79]. Non-invasive radiological methods are available to assess immunopathological phenomena in the lungs of animals, and could be adapted to children [18].
According to INSEE data (France), there is a significant signal of an increase in newborn deaths between 2 and 6 days of age from September 2023 onwards: Apart from the month of November with a significantly low rate of deaths, the rate of babies dying between 2 and 6 days of age is significantly higher than the rate expected between September 2023 and January 2024, the period of RSV circulation and Beyfortus immunization. Doses of nirsevimab quickly ran out in October [129, p. 52] and dispensing of doses distributed outside maternity wards was drastically reduced in November, only to resume in early December 2023 [130]; it is therefore possible that dispensation in maternity units was also reduced in November 2023, this being in line with the low rate of deaths between 2 and 6 days of life compared with other months. It should be noted that the latest data are not yet fully consolidated. An early ADE of nirsevimab cannot therefore be ruled out to explain these data.
In Spain, immunization coverage is close to 80% nationwide, and hospitalization rates for RSV infection in 0-4 year-olds are comparable for the 2022-2023 and 2023-2024 seasons. The Ares-Gomez study [114], based on registries of all Galician children (immunization status and hospitalization), estimates the efficacy of nirsevimab on RSV hospitalization at 82% (intention-to-treat analysis) in children under 6 months and 66.2% on hospitalization for all causes. Nirsevimab was offered to all newborns unless medically contraindicated, and over 90% of infants under 6 months of age were immunized during the 23-24 season. The reasons for non-immunization (parental refusal or medical contraindication - fragile state of health of the newborn) are not precisely known; some of the non-immunized newborns were certainly more fragile than those immunized, and comparison between the 2 groups will be difficult as they do not involve equivalent populations in terms of health status. Given the small number of non-immunized infants, case-control matching is impossible. Nosocomial infections were excluded. Nirsevimab is administered on the day of birth in Galicia. The study’s lead author confirms that all infections developed in hospital for whatever reason were excluded (personal communication): this means that newborns who acquired the infection during their stay in the maternity hospital were excluded from the study. The estimated efficacy should have taken into account these excluded cases, the number of which is not provided. Moreover, these “nosocomial” cases can be explained by early post-injection ADE of nirsevimab, which their exclusion does not allow us to evaluate. During the crucial 8-day post-injection period, nirsevimab serum levels have not peaked, and are likely to cause ADE, according to what we know. Hospitalization for RSV with severity criteria occurred only in the treated groups. As mentioned above, we can assume that the non-treated group is more fragile overall than the treated group, yet severe cases are found exclusively in the treated group, which may be a sign of treatment-induced ADE. Concerning the efficacy of nirsevimab on all-cause hospitalizations, it should be remembered that the 22-23 season was exceptional in Galicia (figure 4 in the appendix), as in many regions of the world, and should have been excluded from the comparison. If we exclude this exceptional 22-23 season, the 23-24 all-cause hospitalization rate is comparable to previous seasons, albeit slightly lower: this result contradicts the claimed 66.2% effectiveness rate on all-cause hospitalizations in 23-24. In conclusion, the efficacy of nirsevimab in reducing all-cause and RSV hospitalization rates is questionable, and ADE cannot be ruled out in rare cases.
The study carried out in Navarre [116] concerns only hospitalizations due to RSV and cannot therefore estimate the efficacy of nirsevimab on all-cause hospitalizations. It is therefore also impossible to assess any non-specific ADE due to nirsevimab. Newborns injected less than 1 day previously are counted as non-immune: if they develop RSV infection within 24 hours of injection, early ADE cannot be assessed. It is impossible to exclude ADE in very rare cases, since no details are given on the clinical results of the 3 immunized children admitted to intensive care (versus 2 non-immunized). No comparison is made with previous seasons.
From these results, we can conclude that nirsevimab is effective against RSV infections, but that it could in very rare cases cause facilitation/aggravation of RSV infection in the days following injection. On the other hand, nirsevimab does not reduce the rate of all-cause hospitalization in targeted populations, although hospitalization due to RSV is significantly reduced (however, this efficacy may be less than advertised, given the large or unknown number of subjects excluded from clinical trials or observational studies). No ADE was observed by the EMA in clinical trials, despite greater signs of infection severity in treated groups and the frequent exclusion of infected subjects immediately after injection. Preclinical in vitro and in vivo tests did not detect any effector functions of nirsevimab that might give rise to concern about ADE in humans. The analysis below will show why they were incomplete, as they were often carried out on mAbs close to but different from nirsevimab; preclinical trials with nirsevimab were largely published after the product’s approval [82].
7.2. Deficiencies in Preclinical In Vitro Exploration of ADE
Nirsevimab has a strong neutralizing capacity (superior to that of palivizumab) and a high affinity for FcRn, which prolongs its half-life. Following the failure of inactivated RSV vaccines due to ADE caused by anti-RSV F-protein antibodies, the EMA’s risk management plan for nirsevimab mentions the theoretical possibility of nirsevimab binding to FcγRs (to which FcRn belongs) through increased efficacy [9]. No ADE has been observed in humans for palivizumab, which has been in use for 20 years; this mAb recognizes F protein in both postfusion and prefusion conformations, whereas nirsevimab is an IgG1 that targets F protein in prefusion conformation only. It also differs in the domain it targets: the conserved Ø epitope of the F protein. It has a much higher affinity for the F protein than palivizumab, and an increased half-life thanks to YTE mutations that enhance FcRn binding [10]. YTE mutations increase mAb affinity for FcRn at acidic pH [4] and it has been shown that mAbs with YTE mutations, such as nirsevimab, bind human FcRn with very low affinity at neutral pH [15]. This pH-dependent affinity has not been shown specifically for nirsevimab, but simply extrapolated from equivalent mAbs (mAb monoclonal antibody) with YTE mutations. All these modifications compared with palivizumab theoretically open up the possibility of an ADE via binding to FcRn or other FcγRs. FcRn allows the passage of free (non-antigen-bound) IgG across mucosal barriers in both apical to basal and reverse directions [21], particularly in the lungs [13,15,22]. FcRn is also able to bring IgG-antigen complexes into cells such as macrophages [24] and APCs [14,19]. FcRn promotes the entry of free viruses into cells [20,27,28] and also the entry of virion-IgG complexes [30,31,32] with strongly neutralizing antibodies [31]. FcRn is expressed in endothelial cells and epithelium of many tissues, as well as in hematopoietic cells [17]. FcRn is expressed by macrophages [24], including those in the lungs [13]. FcRn binds to IgG on the surface of mucous membranes when these are acidic [34]; this is the case of intestinal mucous membranes, of course, but the pH of neonatal pulmonary mucosa is variable and can be acidic in some individuals [35,36,37,38]. To date little is known about the role of FcRn in a potential IgG active transport mechanism from the general circulation to the lungs [15]. These properties of FcRn and its localization make it capable of facilitating the entry of an IgG antibody-linked virus into the monocytic lineage and lung epithelial cells (extrinsic ADE). In many viral infections, ADE following binding between FcγRs and the virus-antibody complex occurs at sub-neutralizing antibody concentrations. Extrinsic ADE promotes infection of myeloid cells: as early as 1984, Halstead [55] proposed that monocytes were involved in ADE during dengue fever. This has been confirmed for other viruses [56], particularly RSV [39,52,53,54,57,58,59,60,61]. Internalization of virus-antibody complexes can also modulate the expression of the innate cytokine response to the virus (this is intrinsic ADE, dependent on the ratio of activating FcγRs to inhibitors of cytokine production) [43]. The binding of mAbs to complement components and FcγRs can induce ADE, but the therapeutic activity of mAbs also depends on their effector functions (binding to FcγRs) and complement activation [51], but this activation can also induce intrinsic ADE [18,43]. FcγRI binds preferentially to IgG1 with high avidity and is present exclusively on the monocytic lineage; nirsevimab is an IgG1. The other two receptors, FcγRII and FcγRIII, are found on monocytes, macrophages, eosinophils, neutrophils, NK cells, B lymphocytes and T lymphocytes. Both receptors have relatively low specificity and relatively low avidity for IgG compared with FcγRI [45]. All these mechanisms have been proposed to explain ADE during RSV infection [39,52,53,54,57,58,59,60,61] and this ADE has been shown in vitro on a monocyte line in the presence of anti-F antibodies [39,58,66]. ADE has been shown ex vivo with sera from children naturally immunized against RSV when low levels of neutralizing antibodies are present; these sera nevertheless displayed strong anti-F activity [60]. Binding to the FcRn of a mAb injected in large quantities could also disrupt the overall response to infections through an immunosuppressive effect [34], thrombotic [41] and autoimmune phenomena [42], and lead to non-specific worsening of infections. In an inflammatory context (e.g. infection by a respiratory virus), in the presence of high quantities of nirsevimab (IgG1 with high affinity for FcRn), FcRn saturation could lead to a high concentration of low-affinity IgG in the lung lumen, a process that could damage the lungs [40]. mAbs with high anti-viral activity could contribute to inflammation in the advanced stages of the disease, which is probably why they are not used therapeutically [39].
7.2.1. Gaps Remain in the Study of the Effector Functions of Nirsevimab In Vitro
The conclusions of the EMA-EPAR [74] are contradictory when it comes to studying the effector functions of nirsevimab. For the EMA, nirsevimab should exhibit normal Fc-mediated effector functions (complement activation, mediation of phagocytosis, antibody-mediated destruction of virus-infected cells, etc.). Nirsevimab does not prevent virions from attaching to cells. The EMA considers that effector functions are not part of nirsevimab’s mechanism of action (page 18 of EPAR), yet writes that the contribution of Fc-mediated effector functions to protection against RSV disease cannot be excluded (in the rapporteur’s opinion, preclinical data from the cotton rat model seem ambiguous in this respect, [74, page 15]). Mechanisms other than virus neutralization would be necessary for the therapeutic effect, such as viral clearance and killing of infected cells [4]. In rats, these effector functions are not necessary for protection against RSV infection, but higher concentrations of nirsevimab are required in humans for optimal protection [82]. The EMA reiterates the importance of studying them for their involvement in a possible ADE [74, p 29]. However, the EMA does not report any study of ADCC or the role of NK cells, which are nevertheless suspected of playing a primordial role in the balance between protection and pathology following the use of passive immunization [39]. Other study [15] not cited in EPAR-EMA concern FcγR binding with mAbs close to but different from nirsevimab (including motavizumab) and confirm that YTE mutations that increase FcRn binding do not affect the virus’ neutralizing power. However, the clinical trials carried out with this motavizumab have not been published; we don’t know why: were the adverse effects significant? Was the efficacy too low? It is therefore difficult to extrapolate these results to nirsevimab. Dall’Acqua [15] has also shown with a mAb other than nirsevimab that YTE mutations reduce FcγRIIIA binding by a factor of 2, as well as its ADCC activity by a factor of 100. Can we extrapolate this reduction in ADCC to nirsevimab, and what influence will it have on the therapeutic effect? Indeed, the role of FcγRIIIa would be double-edged: it may be pro-inflammatory and contribute to RSV disease, or it may be anti-inflammatory [46,47].
It would therefore have been necessary to study the binding of nirsevimab itself to FcγRIIIA. The assay was performed on the F158 allotype of FcγRIIIA, which is the most common: individuals with the other V158 allotype might react differently to nirsevimab. The presence of Valine increases IgG1 affinity for FcγRIII [138] and there are discrepancies between in vitro (IgG affinity) and in vivo (viral clearance) functional activity according to FcγRIIIa polymorphisms (V/F158). In vivo, however, this difference in affinity would not lead to differences in ADCC activity, but other FcγR receptors could modulate this ADCC capacity of NK cells [131]. In 2023, i.e. after the clinical trials and approval of nirsevimab, Brady [82] published a study of effector functions with this molecule, confirming previous results (on the increased half-life and reduced ADCC in vitro). The in vitro results show that nirsevimab binds to activating and inhibitory FcγRs (the result of these activating and inhibitory functions could vary according to the level of mAb present in the serum); Brady points out that Fc effector functions are linked to the exacerbation of symptoms due to RSV. The ex vivo study (using sera from immunized children) is carried out at a single point in time, without taking into account any variations in concentration over time (maximum levels are known to be reached in infants within 8 days); pooling sera for analysis is likely to mask disparities due to different levels of nirsevimab; effector functions cannot be predicted beyond 150 days post-injection. Nirsevimab promotes beneficial ADNP, but neutrophilic inflammation with infiltration is associated with severe RSV disease. Phagocytosis by macrophages, which can be double-edged, is favored only at certain dilutions. Nirsevimab activates less ADCC in vitro than palivizumab, and in vivo the ADCC of nirsevimab is identical to that of placebo. Complement deposition is strongly increased by nirsevimab at certain dilutions, further underlining the importance of detailed pharmacokinetic studies. Furthermore, no trials have been carried out in the presence of immune complexes of nirsevimab bound to F protein or virus: all trials are performed in the absence of antigen; antigen binding could modify Fc effector functions [18]. CDC is not studied, which is unfortunate as Brady reminds us that complement can have a protective or pathogenic role in viral infections [82]. Lee [51] proposes methods for studying the effector functions of mAbs by complement activation. In a mouse model, certain complement components are required for the development of VAERD and this would be the case for infections aggravated by inactivated RSV vaccine in children. The authors point out that ADCC by NKs could be involved in exaggerating the immune response to influenza virus. The level of ADCC activity in vitro does not correlate with clinical symptoms in primary RSV infection, and the role of ADCC in the protection or pathogenesis of RSV infection remains to be determined (also confirmed by [39]). Brady stresses the importance of phagocytosis by macrophages, which requires further research [82]. Experimental methods have been proposed in the past, but seem to have been overlooked: as early as 1989, Gimenez describes an assay for evaluating ADE, which quantifies it by assessing the amount of virus released by U937 cells (a macrophagic line) [58]. Gimenez [58] and Bournazos [46] also recall the gap between in vitro ADE assays and experimental in vivo systems. Glycosylation of nirsevimab is not described, although it may play a role in mAb binding to FcRγ and C1q [131,132]. In particular, non-fucosylated IgGs bind more strongly to human FcγRIIIA and ADCC assays may show enhanced cytotoxicity [19]. Glycosylation of the Fc region affects mAb stability, and plays a role in CDC and ADCC functions by modulating binding to the Fcγ receptor [133]. The role of glycosylation on FcγR and C1q binding and the consequences thereof have not been studied in detail. Sialylation would not affect FcγRIIIa binding, but afucosylated IgG would have no ADCC activity. Galactosylation of IgG1 positively influences C1q binding and CDC, and sialylation increases C1q binding of galactosylated IgG [131]. Most mAbs have a glycosylation site in the Fc region. The type of glycosylation (mannose, sialic acid, N-acetylneuraminic acid,) can influence pharmacokinetics and in particular binding to FcγIIIa; mAbs produced from Chinese hamster CHO cell can produce afucosylated mAbs with reduced ADCC activity [134]. According to EPAR [74], nirsevimab is produced on CHO and has a glycosylation site in the Fc domain (Asn-306); this glycosylation is of the complex type (EPAR specifies that it has been characterized without further precision). Other specific studies should have been carried out in the presence of other antibodies, neutralization and ADE activities are modified when two mAbs are mixed, and a synergistic ADE effect may occur in the presence of different antibodies) [66]. It is important to study the binding of mAb to FcRn when it is bound to antigen, as this binding is capable of causing structural changes in the region of IgG that binds to FcRn and its interaction with complement [17,135]. The binding of nirsevimab to FcRn should therefore have been studied in the presence of RSV virus or F protein.
Binding to FcγRs depends on the conformational state of IgG (open or closed) [5]: a possible conformational change in nirsevimab when bound to F antigen or virus could alter its binding to FcγRIII and consequently its effector functions and hence its therapeutic effect. mAb D25 is a precursor of nirsevimab (like nirsevimab, it is directed against the Ø epitope of F, unlike palivizumab, which is directed against another epitope). In 2016 Van Mechelen [69] showed in vitro that D25 induced a stronger ADE effect than palivizumab when binding to the FcγR of cells: the ADE effect outweighed the neutralizing power. Van Mechelen’s experiments should have been reproduced with nirsevimab: is nirsevimab capable of inducing an ADE at low concentrations, unlike palivizumab under the conditions of these experiments?
Many unknowns therefore remain, and it’s a pity that some of the preclinical in vitro experiments were carried out only after the clinical trials, and were incomplete.
7.2.2. Animal Studies In Vivo
Assessment of ADE in preclinical trials has been incomplete, using animals that do not allow it to be characterized, with mAbs other than nirsevimab (although close to it) and with doses higher than those used in humans. According to the EMA, no ADE was observed in preclinical trials. However, no histopathological evaluation was carried out in rats in the trials (in particular to look for alveolitis and neutrophilic infiltration, which are the signs of ADE in this animal model). The EMA stresses that as the rats were mature, the translability of the finding to highly immature infants is unknown [74]. The choice of animals to test for ADE is also problematic. The animals usually used to study hRSV (non-human primates, cotton rats, mice and lambs) are semi-permissive to virus replication and experimental infection. Only chimpanzees are completely permissive to hRSV [136]. Cynomolgus macaques do not usually produce signs of disease unless infected with a high titer inoculum. In the VAERD study in cynomolgus monkeys immunized with inactivated RSV virus, pulmonary pathology was not associated with increased viral replication, suggesting that the pathology was due to non-viral antigens. In cotton rats, too, the severity of alveolitis, used as a primary marker of VAERD with inactivated vaccine, depends on the response to non-viral antigens.
Furthermore, in rat trials, it is necessary to use a mAb without YTE mutations, as these increase the affinity of FcRn in rats at neutral pH and suppress the protective effect of FcRn binding [80,82]. It is therefore difficult to extrapolate the results in these models to assess the absence of VAERD in humans. Zhu [4] studied pulmonary viral loads in cotton rats with an antibody almost identical to nirsevimab (lacking the YTE mutations). After viral challenge the day after IM injection, the rats were sacrificed 4 days after challenge. Viral titres in the lungs and nose were greatly reduced after treatment compared with controls who had not received antibodies, and the reduction was dose-dependent, confirming the efficacy of nirsevimab and ruling out ADE a priori. However, we do not know what effect the increased affinity of YTE mutations has on these viral loads. The effect of nirsevimab is being studied in cynomolgus monkeys, which are receiving much higher doses than in clinical trials (300mg/kg IM and IV). The monkeys are not challenged with the virus, so the possible VAERD effect is not studied. A study of VAERD in cotton rats with the nirsevimab substitute lacking YTE mutations is published in 2023 [82]. Viral titer in lung and nasal fluid, measured 4 days after viral challenge, was reduced with the nirsevimab surrogate compared with untreated controls, confirming the efficacy of the product. The authors note that the reduction in viral loads does not depend on Fc effector functions. However, in humans, Fc effector functions are linked to exacerbation of RSV symptoms [5,52,138]. It is difficult to extrapolate these data to humans.
7.2.3. The Pharmacokinetic Study is Incomplete and Shows Periods when Nirsevimab Levels may be Sub-Neutralizing in Some Individuals
The EMA report [74] points out that in ADE trials, rats treated with nirsevimab are exposed to RSV at the time of peak antibody concentration. Indeed, it has been known since 1984 that ADE can occur with a strongly neutralizing antibody if it is present in low concentration in dengue virus infections [55]. Aggravated infections have been reported after immunization with a vaccine containing RSV F protein, when antibody levels were low [82]. For RSV disease, the severity of illness may depend on maternal antibody levels: the variation in levels during the first months of life could explain the contradictory results [57,69,71,72] concerning the protection conferred by these antibodies (as shown for dengue): ADE may be mediated by antibodies within a narrow range of sub-neutralizing antibody levels [18,74]. A rigorous pharmacokinetic study is therefore necessary. In animals, pharmacokinetic studies have been carried out with different antibodies to nirsevimab, using IV doses far higher than those used in humans, with only 2 points of measurement [15], and therefore do not allow extrapolation to humans. The monkey study [4] shows great individual variability, with a nasal fluid level of only 1/10,000 of the serum concentration. The EMA [74] also points to the risk of circulation of a mutant strain of RSV requiring higher serum levels to prevent viral replication. Sub-optimal concentrations of neutralizing antibodies could therefore cause ADE in the lungs, even in the presence of neutralizing serum levels. In adult humans, the maximum concentration of nirsevimab is reached in 3 days, whereas peak neutralizing activity is reached 6 days after IM [73]: sub-neutralizing antibody concentrations could therefore be circulating in the first few days after injection. The pharmacokinetic study in children [78] shows that the maximum level of nirsevimab is reached after 8 days in 95% of subjects. However, 5% of subjects do not reach this level after 8 days, and in 10% of children the level of 4 times the baseline is not maintained at 151 days post-injection. It is therefore possible that low levels of neutralizing antibodies circulate within the first 8 days post-injection (and even for a longer period) in rare subjects. Pharmacokinetic studies have not been carried out in healthy neonates a few days old, who represent the target population for the 2023-2024 campaign. According to the EMA [74], a small proportion of subjects in clinical studies do not reach the concentration required for clinical efficacy. This was confirmed by Hammit [77] in infants a few months old. The pharmacokinetics at birth may differ from those observed in infants a few months old, but the recommendations concern newborns in the first few days of life (on leaving the maternity ward in France [100], on the first day of life in Galicia [114] and in the first 7 days of life in Navarre, Spain [116] and in the USA from birth [137]. As the maximum concentration is reached in infants within a few days, it is possible that in some infants, the protective concentration is not reached as quickly, and that sub-neutralizing doses of antibodies circulate in the blood (or lungs) for some time, thus favoring ADE in epidemic periods in the event of an encounter with the virus. Similarly, neutralizing serum levels may not be maintained in all infants during the epidemic season, due to the waning immunity conferred by nirsevimab. Pharmacokinetic studies should be performed in the presence and absence of viral infection to determine the effect of infection on antibody persistence: The half-life in uninfected and infected animals may differ [18]. No studies of ADE by immune system disruption have been carried out in preclinical trials, and respiratory infections not caused by RSV or not corresponding to the protocol have been neglected in clinical trials [76,90,92]. We found in the results of the immunization campaign that all-cause hospitalizations were not reduced compared with previous seasons.
7.3. Economic Benefits of Nirsevimab: Price and All-Cause Hospitalization Rates
The estimate of the comparative cost of nirsevimab immunization campaigns versus vaccination of mothers, based on product efficacy, does not take into account all-cause hospitalizations. The price estimated to be profitable for society is lower than the price finally applied: Shoukat [139] proposes a price of $290 to make nirsevimab cost-effective, and the Canadian NACI [140] a price of $215 in a program aimed at all infants. However, the price set was higher in the USA ($495) [141] and was raised to $519.75 in April 2024 [142]. In France, the public price of nirsevimab was revealed in spring 2024 at €401.80 [143]. It was not public when the 2023 campaign was launched [144].
Neumann asks that the waning immunity and the possibility of emergence of resistant strains be taken into account in the real cost of nirsevimab (although Neumann, like the other authors, relies solely on hospitalizations due to RSV). Immunized children are protected from RSV for only 6 months; nirsevimab-resistant strains are beginning to emerge [141]. All studies conclude that the immunization program is cost-effective only if applied to at-risk infants. A price reduction of 80-88% has been proposed to make the all-infants seasonal nirsevimab program with catch-up cost-effective [145]. Thus, taking into account only the burden of RSV infections, the price of nirsevimab would be too high to make immunization of all newborns cost-effective. The stated aim of nirsevimab immunization campaigns is to reduce the burden on hospitals. As no reduction in all-cause hospitalizations is observed in the age group concerned by nirsevimab immunization, the cost-effectiveness of mass campaigns will be even more questionable.
8. Conclusions
Nirsevimab has been recommended in 2023 for all newborns and as a catch-up treatment for children during their first season of RSV circulation, with the aim of reducing the hospital burden due to infections. Based on clinical trials and published results from the first immunization campaign, it has been shown to be effective in reducing hospitalizations due to RSV (with the caveat that a large or unknown number of treated subjects were excluded in both clinical trials and post-marketing observational studies). But the overall burden on the hospital may not be reduced (all-cause hospitalizations are not reduced for infants and young children during the 2023-2024 season). This may also be attributed to the low share of RSV admissions in hospital admissions among under-5s. The lack of effect on all-cause hospitalizations could also be attributed to the facilitation/aggravation of RSV infections in rare cases (ADE), and more frequently to increased susceptibility to other infections, particularly respiratory. The evaluation of the ADE effect was incomplete in preclinical trials, as well as in clinical trials and in the analysis of the results of the 2023-2024 campaign. This lack of evaluation is notable both in terms of the facilitation of RSV infections and that of all-cause infections, particularly respiratory. Long-term follow-up of treated children is needed to verify that sub-neutralizing concentrations circulating in the season following injection are not capable of causing ADE in the longer term, even if nirsevimab is protective in the short term. Is it therefore scientifically and economically reasonable to continue recommending immunization of all infants and young children for the following seasons? Shouldn’t this expensive product be reserved for those at risk?
Author Contributions
Conceptualization writing, Helene Banoun; Christine Mackoi, Data extraction-methodology-software-data curation.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Acknowledgments
The publication fees were paid by bonsens.org (accessed on 6 June, 2024).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Li Y, Wang X, Blau DM, Caballero MT, Feikin DR, Gill CJ, Madhi SA, Omer SB, Simões EAF, Campbell H, Pariente AB, Bardach D, Bassat Q, Casalegno JS, Chakhunashvili G, Crawford N, Danilenko D, Do LAH, Echavarria M, Gentile A, Gordon A, Heikkinen T, Huang QS, Jullien S, Krishnan A, Lopez EL, Markić J, Mira-Iglesias A, Moore HC, Moyes J, Mwananyanda L, Nokes DJ, Noordeen F, Obodai E, Palani N, Romero C, Salimi V, Satav A, Seo E, Shchomak Z, Singleton R, Stolyarov K, Stoszek SK, von Gottberg A, Wurzel D, Yoshida LM, Yung CF, Zar HJ; Respiratory Virus Global Epidemiology Network; Nair H; RESCEU investigators. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in children younger than 5 years in 2019: A systematic analysis. Lancet. 2022 May 28;399(10340):2047-2064. Epub 2022 May 19. PMCID: PMC7613574. https://pubmed.ncbi.nlm.nih.gov/35598608/. [CrossRef] [PubMed]
- Rocca A, Biagi C, Scarpini S, Dondi A, Vandini S, Pierantoni L, Lanari M. Passive Immunoprophylaxis against Respiratory Syncytial Virus in Children: Where Are We Now? Int J Mol Sci. 2021 Apr 2;22(7):3703. [CrossRef] [PubMed]
- Osiowy C, Horne D, Anderson R. Antibody-dependent enhancement of respiratory syncytial virus infection by sera from young infants. Clin Diagn Lab Immunol. 1994 Nov;1(6):670-7. [CrossRef] [PubMed]
- Zhu Q, McLellan JS, Kallewaard NL, Ulbrandt ND, Palaszynski S, Zhang J, Moldt B, Khan A, Svabek C, McAuliffe JM, Wrapp D, Patel NK, Cook KE, Richter BWM, Ryan PC, Yuan AQ, Suzich JA. A highly potent extended half-life antibody as a potential RSV vaccine surrogate for all infants. Sci Transl Med. 2017 May 3;9(388):eaaj1928. [CrossRef] [PubMed]
- Acevedo OA, Díaz FE, Beals TE, Benavente FM, Soto JA, Escobar-Vera J, González PA, Kalergis AM. Contribution of Fcγ Receptor-Mediated Immunity to the Pathogenesis Caused by the Human Respiratory Syncytial Virus. Front Cell Infect Microbiol. 2019 Mar 29;9:75. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6450440/. [CrossRef] [PubMed]
- Diethelm-Varela B, Soto JA, Riedel CA, Bueno SM, Kalergis AM. New Developments and Challenges in Antibody-Based Therapies for the Respiratory Syncytial Virus. Infect Drug Resist. 2023 Apr 8;16:2061-2074. [CrossRef] [PubMed]
- Rigter A, Widjaja I, Versantvoort H, Coenjaerts FE, van Roosmalen M, Leenhouts K, Rottier PJ, Haijema BJ, de Haan CA. A protective and safe intranasal RSV vaccine based on a recombinant prefusion-like form of the F protein bound to bacterium-like particles. PLoS ONE. 2013 Aug 12;8(8):e71072. [CrossRef] [PubMed]
- Kampmann B, Madhi SA, Munjal I, Simões EAF, Pahud BA, Llapur C, Baker J, Pérez Marc G, Radley D, Shittu E, Glanternik J, Snaggs H, Baber J, Zachariah P, Barnabas SL, Fausett M, Adam T, Perreras N, Van Houten MA, Kantele A, Huang LM, Bont LJ, Otsuki T, Vargas SL, Gullam J, Tapiero B, Stein RT, Polack FP, Zar HJ, Staerke NB, Duron Padilla M, Richmond PC, Koury K, Schneider K, Kalinina EV, Cooper D, Jansen KU, Anderson AS, Swanson KA, Gruber WC, Gurtman A; MATISSE Study Group. Bivalent Prefusion F Vaccine in Pregnancy to Prevent RSV Illness in Infants. N Engl J Med. 2023 Apr 20;388(16):1451-1464. Epub 2023 Apr 5. https://pubmed.ncbi.nlm.nih.gov/37018474/. [CrossRef] [PubMed]
- European Union Risk Management Plan (EU RMP) for Beyfortus® (Nirsevimab) 3 May 2021. Available online: https://www.ema.europa.eu/en/documents/rmp-summary/beyfortus-epar-risk-management-plan_en.pdf (accessed on April 25, 2024).
- Rocca A, Biagi C, Scarpini S, Dondi A, Vandini S, Pierantoni L, Lanari M. Passive Immunoprophylaxis against Respiratory Syncytial Virus in Children: Where Are We Now? Int J Mol Sci. 2021 Apr 2;22(7):3703. [CrossRef] [PubMed]
- Sanders SL, Agwan S, Hassan M, van Driel ML, Del Mar CB. Immunoglobulin treatment for hospitalised infants and young children with respiratory syncytial virus infection. Cochrane Database Syst Rev. 2019. https://doi.org/10.1002/14651858.CD009417.pub2 [PMC free article] [PubMed] [CrossRef] [Google Scholar].
- Resch B. Product review on the monoclonal antibody palivizumab for prevention of respiratory syncytial virus infection. Hum Vaccin Immunother. 2017 Sep 2;13(9):2138-2149. Epub 2017 Jun 12. PMCID: PMC5612471 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5612471/. [CrossRef] [PubMed]
- Roopenian DC, Akilesh S. FcRn: The neonatal Fc receptor comes of age. Nat Rev Immunol. 2007 Sep;7(9):715-25. Epub 2007 Aug 17. https://pubmed.ncbi.nlm.nih.gov/17703228/. [CrossRef] [PubMed]
- Patel DD, Bussel JB. Neonatal Fc receptor in human immunity: Function and role in therapeutic intervention. J Allergy Clin Immunol. 2020 Sep;146(3):467-478. https://pubmed.ncbi.nlm.nih.gov/32896307/. [CrossRef] [PubMed]
- Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2006 Aug 18;281(33):23514-24. Epub 2006 Jun 21. PMID: 16793771 https://pubmed.ncbi.nlm.nih.gov/16793771/. [CrossRef]
- Borrok MJ, Wu Y, Beyaz N, Yu XQ, Oganesyan V, Dall’Acqua WF, Tsui P. pH-dependent binding engineering reveals an FcRn affinity threshold that governs IgG recycling. J Biol Chem. 2015 Feb 13;290(7):4282-90. Epub 2014 Dec 23. PMCID: PMC4326836. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4326836/. [CrossRef] [PubMed]
- Qi T, Cao Y. In Translation: FcRn across the Therapeutic Spectrum. Int J Mol Sci. 2021 Mar 17;22(6):3048. PMCID: PMC8002405. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8002405/. [CrossRef] [PubMed]
- Crowe JE Jr. Human Antibodies for Viral Infections. Annu Rev Immunol. 2022 Apr 26;40:349-386. Epub 2022 Feb 3. PMID: 35113730. https://pubmed.ncbi.nlm.nih.gov/35113730/. [CrossRef] [PubMed]
- Spiekermann GM, Finn PW, Ward ES, Dumont J, Dickinson BL, Blumberg RS, Lencer WI. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life: Functional expression of FcRn in the mammalian lung. J Exp Med. 2002 Aug 5;196(3):303-10. Erratum in: J Exp Med. 2003 Jun 2;197(11):1601. PMCID: PMC2193935 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2193935/. [CrossRef] [PubMed]
- Pyzik M, Sand KMK, Hubbard JJ, Andersen JT, Sandlie I, Blumberg RS. The Neonatal Fc Receptor (FcRn): A Misnomer? Front Immunol. 2019 Jul 10;10:1540. [CrossRef] [PubMed]
- Schlachetzki F, Zhu C, Pardridge WM. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J Neurochem. 2002 Apr;81(1):203-6. [CrossRef] [PubMed]
- Kim KJ, Fandy TE, Lee VH, Ann DK, Borok Z, Crandall ED. Net absorption of IgG via FcRn-mediated transcytosis across rat alveolar epithelial cell monolayers. Am J Physiol Lung Cell Mol Physiol. 2004 Sep;287(3):L616-22. Epub 2004 May 28. https://pubmed.ncbi.nlm.nih.gov/15169676/. [CrossRef] [PubMed]
- Pyzik M, Kozicky LK, Gandhi AK, Blumberg RS. The therapeutic age of the neonatal Fc receptor. Nat Rev Immunol. 2023 Jul;23(7):415-432. Epub 2023 Feb 1. PMCID: PMC9891766. https://pubmed.ncbi.nlm.nih.gov/36726033/. [CrossRef] [PubMed]
- Challa DK, Wang X, Montoyo HP, Velmurugan R, Ober RJ, Ward ES. Neonatal Fc receptor expression in macrophages is indispensable for IgG homeostasis. MAbs. 2019 Jul;11(5):848-860. Epub 2019 Apr 30. PMCID: PMC6601554. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6601554/. [CrossRef] [PubMed]
- Spiekermann GM, Finn PW, Ward ES, Dumont J, Dickinson BL, Blumberg RS, Lencer WI. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life: Functional expression of FcRn in the mammalian lung. J Exp Med. 2002 Aug 5;196(3):303-10. Erratum in: J Exp Med. 2003 Jun 2;197(11):1601. PMCID: PMC2193935 https://pubmed.ncbi.nlm.nih.gov/12163559/. [CrossRef] [PubMed]
- Ye L, Zeng R, Bai Y, Roopenian DC, Zhu X. Efficient mucosal vaccination mediated by the neonatal Fc receptor. Nat Biotechnol. 2011 Feb;29(2):158-63. Epub 2011 Jan 16. PMCID: PMC3197702. https://pubmed.ncbi.nlm.nih.gov/21240266/. [CrossRef] [PubMed]
- Zhao X, Zhang G, Liu S, Chen X, Peng R, Dai L, Qu X, Li S, Song H, Gao Z, Yuan P, Liu Z, Li C, Shang Z, Li Y, Zhang M, Qi J, Wang H, Du N, Wu Y, Bi Y, Gao S, Shi Y, Yan J, Zhang Y, Xie Z, Wei W, Gao GF. Human Neonatal Fc Receptor Is the Cellular Uncoating Receptor for Enterovirus B. Cell. 2019 May 30;177(6):1553-1565.e16. Epub 2019 May 16. PMCID: PMC7111318. https://pubmed.ncbi.nlm.nih.gov/31104841/. [CrossRef] [PubMed]
- Morosky S, Wells AI, Lemon K, Evans AS, Schamus S, Bakkenist CJ, Coyne CB. The neonatal Fc receptor is a pan-echovirus receptor. Proc Natl Acad Sci U S A. 2019 Feb 26;116(9):3758-3763. Epub 2019 Feb 11. PMCID: PMC6397586. https://pubmed.ncbi.nlm.nih.gov/30808762/. [CrossRef] [PubMed]
- Burstin SJ, Brandriss MW, Schlesinger JJ. Infection of a macrophage-like cell line, P388D1 with reovirus; effects of immune ascitic fluids and monoclonal antibodies on neutralization and on enhancement of viral growth. J Immunol. 1983 Jun;130(6):2915-9. [PubMed]
- Maidji E, McDonagh S, Genbacev O, Tabata T, Pereira L. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal Fc receptor-mediated transcytosis. Am J Pathol. 2006 Apr;168(4):1210-26. PMCID: PMC1606573. [CrossRef] [PubMed]
- Gupta S, Gach JS, Becerra JC, Phan TB, Pudney J, Moldoveanu Z, Joseph SB, Landucci G, Supnet MJ, Ping LH, Corti D, Moldt B, Hel Z, Lanzavecchia A, Ruprecht RM, Burton DR, Mestecky J, Anderson DJ, Forthal DN. The Neonatal Fc receptor (FcRn) enhances human immunodeficiency virus type 1 (HIV-1) transcytosis across epithelial cells. PLoS Pathog. 2013;9(11):e1003776. Epub 2013 Nov 21. Erratum in: PLoS Pathog. 2013 Nov;9(11). 10.1371/annotation/31430955-703b-484a-96eb-e180f917d683. PMCID: PMC3836734. [CrossRef] [PubMed]
- Gupta S, Pegu P, Venzon DJ, Gach JS, Ma ZM, Landucci G, Miller CJ, Franchini G, Forthal DN. Enhanced in vitro transcytosis of simian immunodeficiency virus mediated by vaccine-induced antibody predicts transmitted/founder strain number after rectal challenge. J Infect Dis. 2015 Jan 1;211(1):45-52. Epub 2014 May 21. PMCID: PMC4334821. [CrossRef] [PubMed]
- Gonzalez OA, Sagar M. Antibodies and Acidic Environment Do Not Enhance HIV-1 Transcytosis. J Infect Dis. 2016 Oct 15;214(8):1221-4. Epub 2016 Aug 4. PMCID: PMC5034959. [CrossRef] [PubMed]
- Pyzik M, Rath T, Lencer WI, Baker K, Blumberg RS. FcRn: The Architect Behind the Immune and Nonimmune Functions of IgG and Albumin. J Immunol. 2015 May https://pubmed.ncbi.nlm.nih.gov/25934922/ 15;194(10):4595-603. [CrossRef] [PubMed]
- Schultz A, Puvvadi R, Borisov SM, Shaw NC, Klimant I, Berry LJ, Montgomery ST, Nguyen T, Kreda SM, Kicic A, Noble PB, Button B, Stick SM. Airway surface liquid pH is not acidic in children with cystic fibrosis. Nat Commun. 2017 Nov 10;8(1):1409. https://pubmed.ncbi.nlm.nih.gov/29123085/. [CrossRef] [PubMed]
- Fischer, H. Function of Proton Channels in Lung Epithelia. Wiley Interdiscip Rev Membr Transp Signal. 2012 May;1(3):247-258. Epub 2011 Oct 25. PMCID: PMC3362208. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3362208/. [CrossRef] [PubMed]
- Abou Alaiwa MH, Beer AM, Pezzulo AA, Launspach JL, Horan RA, Stoltz DA, Starner TD, Welsh MJ, Zabner J. Neonates with cystic fibrosis have a reduced nasal liquid pH; a small pilot study. J Cyst Fibros. 2014 Jul;13(4):373-7. Epub 2014 Jan 11. PMCID: PMC4060428. https://pubmed.ncbi.nlm.nih.gov/24418186/. [CrossRef] [PubMed]
- Garland AL, Walton WG, Coakley RD, Tan CD, Gilmore RC, Hobbs CA, Tripathy A, Clunes LA, Bencharit S, Stutts MJ, Betts L, Redinbo MR, Tarran R. Molecular basis for pH-dependent mucosal dehydration in cystic fibrosis airways. Proc Natl Acad Sci U S A. 2013 Oct 1;110(40):15973-8. Epub 2013 Sep 16. PMCID: PMC3791714. https://pubmed.ncbi.nlm.nih.gov/24043776/. [CrossRef] [PubMed]
- Vanderven HA, Kent SJ. Fc-mediated functions and the treatment of severe respiratory viral infections with passive immunotherapy - a balancing act. Front Immunol. 2023 Nov 22;14:1307398. PMCID: PMC10710136. https://pubmed.ncbi.nlm.nih.gov/38077353/. [CrossRef] [PubMed]
- Vogelzang A, Lozza L, Reece ST, Perdomo C, Zedler U, Hahnke K, Oberbeck-Mueller D, Dorhoi A, Kaufmann SH. Neonatal Fc Receptor Regulation of Lung Immunoglobulin and CD103+ Dendritic Cells Confers Transient Susceptibility to Tuberculosis. Infect Immun. 2016 Sep 19;84(10):2914-21. PMCID: PMC5038074. https://pubmed.ncbi.nlm.nih.gov/27481246/. [CrossRef] [PubMed]
- Cines DB, Zaitsev S, Rauova L, Rux AH, Stepanova V, Krishnaswamy S, Sarkar A, Kowalska MA, Zhao G, Mast AE, Blumberg LJ, McCrae KR, Poncz M, Hubbard JJ, Pyzik M, Blumberg RS. FcRn augments induction of tissue factor activity by IgG-containing immune complexes. Blood. 2020 Jun 4;135(23):2085-2093. PMCID: PMC7273830. https://pubmed.ncbi.nlm.nih.gov/32187355/. [CrossRef] [PubMed]
- Hubbard JJ, Pyzik M, Rath T, Kozicky LK, Sand KMK, Gandhi AK, Grevys A, Foss S, Menzies SC, Glickman JN, Fiebiger E, Roopenian DC, Sandlie I, Andersen JT, Sly LM, Baker K, Blumberg RS. FcRn is a CD32a coreceptor that determines susceptibility to IgG immune complex-driven autoimmunity. J Exp Med. 2020 Oct 5;217(10):e20200359. PMCID: PMC7537387. https://pubmed.ncbi.nlm.nih.gov/32658257/. [CrossRef] [PubMed]
- Taylor, 2015, Taylor A, Foo SS, Bruzzone R, Dinh LV, King NJ, Mahalingam S. Fc receptors in antibody-dependent enhancement of viral infections. Immunol Rev. 2015 Nov;268(1):340-64. [CrossRef] [PubMed]
- Banoun H. Measles and Antibody-Dependent Enhancement (ADE): History and Mechanisms. Explor Res Hypothesis Med. 2022;7(4):246-252. https://www.xiahepublishing.com/2472-0712/ERHM-2022-00018. [CrossRef]
- Tirado SM, Yoon KJ. Antibody-dependent enhancement of virus infection and disease. Viral Immunol. 2003;16(1):69-86. https://pubmed.ncbi.nlm.nih.gov/12725690/. [CrossRef] [PubMed]
- Bournazos S, Gupta A, Ravetch JV. The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol. 2020 Oct;20(10):633-643. Epub 2020 Aug 11. PMCID: PMC7418887. [CrossRef] [PubMed]
- Golebski K, Hoepel W, van Egmond D, de Groot EJ, Amatngalim GD, Beekman JM, Fokkens WJ, van Drunen CM, den Dunnen J. FcγRIII stimulation breaks the tolerance of human nasal epithelial cells to bacteria through cross-talk with TLR4. Mucosal Immunol. 2019 Mar;12(2):425-433. Epub 2019 Jan 21. https://pubmed.ncbi.nlm.nih.gov/30664707/. [CrossRef] [PubMed]
- Acevedo OA, Díaz FE, Beals TE, Benavente FM, Soto JA, Escobar-Vera J, González PA, Kalergis AM. Contribution of Fcγ Receptor-Mediated Immunity to the Pathogenesis Caused by the Human Respiratory Syncytial Virus. Front Cell Infect Microbiol. 2019 Mar 29;9:75. PMCID: PMC6450440. https://pubmed.ncbi.nlm.nih.gov/30984626/. [CrossRef] [PubMed]
- Xu L, Ma Z, Li Y, Pang Z, Xiao S. Antibody dependent enhancement: Unavoidable problems in vaccine development. Adv Immunol 2021;151:99–133. [CrossRef] [PubMed]
- von Kietzell K, Pozzuto T, Heilbronn R, Grössl T, Fechner H, Weger S. Antibody-mediated enhancement of parvovirus B19 uptake into endothelial cells mediated by a receptor for complement factor C1q. J Virol 2014;88(14):8102–8115. [CrossRef] [PubMed]
- Lee CH, Romain G, Yan W, Watanabe M, Charab W, Todorova B, Lee J, Triplett K, Donkor M, Lungu OI, Lux A, Marshall N, Lindorfer MA, Goff OR, Balbino B, Kang TH, Tanno H, Delidakis G, Alford C, Taylor RP, Nimmerjahn F, Varadarajan N, Bruhns P, Zhang YJ, Georgiou G. IgG Fc domains that bind C1q but not effector Fcγ receptors delineate the importance of complement-mediated effector functions. Nat Immunol. 2017 Aug;18(8):889-898. Epub 2017 Jun 12. Erratum in: Nat Immunol. 2017 Sep 19;18(10 ):1173. PMCID: PMC6015732. [CrossRef] [PubMed]
- Polack FP, Teng MN, Collins PL, Prince GA, Exner M, Regele H; et al. A role for immune complexes in enhanced respiratory syncytial virus disease. J Exp Med (2002) 196(6):859–65. [CrossRef]
- Barr FE, Pedigo H, Johnson TR, Shepherd VL. Surfactant protein-A enhances uptake of respiratory syncytial virus by monocytes and U937 macrophages. Am J Respir Cell Mol Biol. 2000 Nov;23(5):586-92. [CrossRef] [PubMed]
- Polack FP, Alvarez-Paggi D, Libster R, Caballero MT, Blair RV, Hijano DR, de la Iglesia Niveyro PX, Menendez DR, Gladwell W, Avendano LM, Velozo L, Wanek A, Bergel E, Prince GA, Kleeberger SR, Johnson J, Pociask D, Kolls JK. Fatal enhanced respiratory syncytial virus disease in toddlers. Sci Transl Med. 2021 Oct 20;13(616):eabj7843. Epub 2021 Oct 20. PMCID: PMC10712289 https://pubmed.ncbi.nlm.nih.gov/34669442/. [CrossRef] [PubMed]
- Halstead SB, Venkateshan CN, Gentry MK, Larsen LK. Heterogeneity of infection enhancement of dengue 2 strains by monoclonal antibodies. J Immunol. 1984 Mar;132(3):1529-32. https://pubmed.ncbi.nlm.nih.gov/6607288/. [PubMed]
- Thomas S, Smatti MK, Ouhtit A, Cyprian FS, Almaslamani MA, Thani AA, Yassine HM. Antibody-Dependent Enhancement (ADE) and the role of complement system in disease pathogenesis. Mol Immunol. 2022 Dec;152:172-182. Epub 2022 Nov 10. PMCID: PMC9647202 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9647202/. [CrossRef] [PubMed]
- Tirado SM, Yoon KJ. Antibody-dependent enhancement of virus infection and disease. Viral Immunol. 2003;16(1):69-86. https://pubmed.ncbi.nlm.nih.gov/12725690/. [CrossRef] [PubMed]
- Gimenez HB, Keir HM, Cash P. In vitro enhancement of respiratory syncytial virus infection of U937 cells by human sera. J Gen Virol. 1989 Jan;70 ( Pt 1):89-96. https://pubmed.ncbi.nlm.nih.gov/2732688/. [CrossRef] [PubMed]
- Krilov LR, Anderson LJ, Marcoux L, Bonagura VR, Wedgwood JF. Antibody-mediated enhancement of respiratory syncytial virus infection in two monocyte/macrophage cell lines. J Infect Dis. 1989 Nov;160(5):777-82. https://pubmed.ncbi.nlm.nih.gov/2809253/. [CrossRef] [PubMed]
- Osiowy C, Horne D, Anderson R. Antibody-dependent enhancement of respiratory syncytial virus infection by sera from young infants. Clin Diagn Lab Immunol. 1994 Nov;1(6):670-7. [CrossRef] [PubMed]
- Gómez RS, Ramirez BA, Céspedes PF, Cautivo KM, Riquelme SA, Prado CE, González PA, Kalergis AM. Contribution of Fcγ receptors to human respiratory syncytial virus pathogenesis and the impairment of T-cell activation by dendritic cells. Immunology. 2016 Jan;147(1):55-72. Epub 2015 Nov 6. PMCID: PMC4693880 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4693880/. [CrossRef] [PubMed]
- Cirino NM, Panuska JR, Villani A, Taraf H, Rebert NA, Merolla R, Tsivitse P, Gilbert IA. Restricted replication of respiratory syncytial virus in human alveolar macrophages. J Gen Virol. 1993 Aug;74 ( Pt 8):1527-37. [CrossRef] [PubMed]
- Wang Y, Zheng J, Wang X, Yang P, Zhao D. Alveolar macrophages and airway hyperresponsiveness associated with respiratory syncytial virus infection. Front Immunol. 2022 Oct 20;13:1012048. [CrossRef] [PubMed]
- Makris S, Bajorek M, Culley FJ, Goritzka M, Johansson C. Alveolar Macrophages Can Control Respiratory Syncytial Virus Infection in the Absence of Type I Interferons. J Innate Immun. 2016;8(5):452-63. Epub 2016 Jul 16. PMCID: PMC5322584. [CrossRef] [PubMed]
- Kecse-Nagy C, Szittner Z, Papp K, Hegyi Z, Rovero P, Migliorini P, Lóránd V, Homolya L, Prechl J. Characterization of NF-κB Reporter U937 Cells and Their Application for the Detection of Inflammatory Immune-Complexes. PLoS ONE. 2016 May 27;11(5):e0156328. [CrossRef] [PubMed]
- Gimenez HB, Chisholm S, Dornan J, Cash P. Neutralizing and enhancing activities of human respiratory syncytial virus-specific antibodies. Clin Diagn Lab Immunol. 1996 May;3(3):280-6. PMCID: PMC170331. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC170331/. [CrossRef] [PubMed]
- Thomas S, Smatti MK, Ouhtit A, Cyprian FS, Almaslamani MA, Thani AA, Yassine HM. Antibody-Dependent Enhancement (ADE) and the role of complement system in disease pathogenesis. Mol Immunol. 2022 Dec;152:172-182. Epub 2022 Nov 10. PMCID: PMC9647202 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9647202/. [CrossRef] [PubMed]
- Katzelnick LC, Gresh L, Halloran ME, Mercado JC, Kuan G, Gordon A, Balmaseda A, Harris E. Antibody-dependent enhancement of severe dengue disease in humans. Science. 2017 Nov 17;358(6365):929-932. Epub 2017 Nov 2. PMCID: PMC5858873. https://pubmed.ncbi.nlm.nih.gov/29097492/. [CrossRef] [PubMed]
- van Mechelen L, Luytjes W, de Haan CA, Wicht O. RSV neutralization by palivizumab, but not by monoclonal antibodies targeting other epitopes, is augmented by Fc gamma receptors. Antiviral Res. 2016 Aug;132:1-5. Epub 2016 May 13. https://pubmed.ncbi.nlm.nih.gov/27185625/. [CrossRef] [PubMed]
- Rigter A, Widjaja I, Versantvoort H, Coenjaerts FE, van Roosmalen M, Leenhouts K, Rottier PJ, Haijema BJ, de Haan CA. A protective and safe intranasal RSV vaccine based on a recombinant prefusion-like form of the F protein bound to bacterium-like particles. PLoS ONE. 2013 Aug 12;8(8):e71072. PMCID: PMC3741363. https://pubmed.ncbi.nlm.nih.gov/23951084/. [CrossRef] [PubMed]
- Jang MJ, Kim YJ, Hong S, Na J, Hwang JH, Shin SM, Ahn YM. Positive association of breastfeeding on respiratory syncytial virus infection in hospitalized infants: A multicenter retrospective study. Clin Exp Pediatr. 2020 Apr;63(4):135-140. Epub 2019 Nov 12. PMCID: PMC7170789. [CrossRef] [PubMed]
- Pasittungkul S, Thongpan I, Vichaiwattana P, Thongmee T, Klinfueng S, Suntronwong N, Wanlapakorn N, Vongpunsawad S, Poovorawan Y. High seroprevalence of antibodies against human respiratory syncytial virus and evidence of respiratory syncytial virus reinfection in young children in Thailand. Int J Infect Dis. 2022 Dec;125:177-183. Epub 2022 Nov 1. [CrossRef] [PubMed]
- Griffin MP, Khan AA, Esser MT, Jensen K, Takas T, Kankam MK, Villafana T, Dubovsky F. Safety, Tolerability, and Pharmacokinetics of MEDI8897, the Respiratory Syncytial Virus Prefusion F-Targeting Monoclonal Antibody with an Extended Half-Life, in Healthy Adults. Antimicrob Agents Chemother. 2017 Feb 23;61(3):e01714-16. [CrossRef] [PubMed]
- EMA-EPAR Beyfortus-Nirsevimab, Available on line : https://www.ema.europa.eu/en/medicines/human/EPAR/beyfortus#assessment-history, https://web.archive.org/web/20240105135355/https://www.ema.europa.eu/en/medicines/human/EPAR/beyfortus#assessment-history (accessed on April 15, 2024).
- Haraya, K., Tachibana, T. Translational Approach for Predicting Human Pharmacokinetics of Engineered Therapeutic Monoclonal Antibodies with Increased FcRn-Binding Mutations. BioDrugs 37, 99–108 (2023). [CrossRef]
- Domachowske JB, Khan AA, Esser MT, Jensen K, Takas T, Villafana T, Dubovsky F, Griffin MP. Safety, Tolerability and Pharmacokinetics of MEDI8897, an Extended Half-life Single-dose Respiratory Syncytial Virus Prefusion F-targeting Monoclonal Antibody Administered as a Single Dose to Healthy Preterm Infants. Pediatr Infect Dis J. 2018 Sep;37(9):886-892. [CrossRef] [PubMed]
- Hammitt LL, Dagan R, Yuan Y, Baca Cots M, Bosheva M, Madhi SA, Muller WJ, Zar HJ, Brooks D, Grenham A, Wählby Hamrén U, Mankad VS, Ren P, Takas T, Abram ME, Leach A, Griffin MP, Villafana T; MELODY Study Group. Nirsevimab for Prevention of RSV in Healthy Late-Preterm and Term Infants. N Engl J Med. 2022 Mar 3;386(9):837-846. https://pubmed.ncbi.nlm.nih.gov/35235726/. [CrossRef] [PubMed]
- Simões EAF, Madhi SA, Muller WJ, Atanasova V, Bosheva M, Cabañas F, Baca Cots M, Domachowske JB, Garcia-Garcia ML, Grantina I, Nguyen KA, Zar HJ, Berglind A, Cummings C, Griffin MP, Takas T, Yuan Y, Wählby Hamrén U, Leach A, Villafana T. Efficacy of nirsevimab against respiratory syncytial virus lower respiratory tract infections in preterm and term infants, and pharmacokinetic extrapolation to infants with congenital heart disease and chronic lung disease: A pooled analysis of randomised controlled trials. Lancet Child Adolesc Health. 2023 Mar;7(3):180-189. Epub 2023 Jan 9. PMCID: PMC9940918. https://pubmed.ncbi.nlm.nih.gov/36634694/. [CrossRef] [PubMed]
- Munoz FM, Cramer JP, Dekker CL, Dudley MZ, Graham BS, Gurwith M, Law B, Perlman S, Polack FP, Spergel JM, Van Braeckel E, Ward BJ, Didierlaurent AM, Lambert PH; Brighton Collaboration Vaccine-associated Enhanced Disease Working Group. Vaccine-associated enhanced disease: Case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine. 2021 May 21;39(22):3053-3066. Epub 2021 Feb 23. PMCID: PMC7901381. https://pubmed.ncbi.nlm.nih.gov/33637387/. [CrossRef] [PubMed]
- Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: Biological consequences. J Immunol. 2002 Nov 1;169(9):5171-80. https://pubmed.ncbi.nlm.nih.gov/12391234/. [CrossRef] [PubMed]
- Cingoz O. Motavizumab. MAbs. 2009 Sep-Oct;1(5):439-42. https://doi.org/10.4161/mabs.1.5.9496. Epub 2009 Sep 10. Erratum in: MAbs. 2010 Sep-Oct;2(5):591. PMID: 20065632; PMCID: PMC2759493.
- Brady T, Cayatte C, Roe TL, Speer SD, Ji H, Machiesky L, Zhang T, Wilkins D, Tuffy KM, Kelly EJ. Fc-mediated functions of nirsevimab complement direct respiratory syncytial virus neutralization but are not required for optimal prophylactic protection. Front Immunol. 2023 Oct 11;14:1283120. https://doi.org/10.3389/fimmu.2023.1283120. PMID: 37901217; PMCID: PMC10600457 https://pubmed.ncbi.nlm.nih.gov/37901217/.
- Booth BJ, Ramakrishnan B, Narayan K, Wollacott AM, Babcock GJ, Shriver Z, Viswanathan K. Extending human IgG half-life using structure-guided design. MAbs. 2018 Oct;10(7):1098-1110. https://doi.org/10.1080/19420862.2018.1490119. Epub 2018 Jul 26. PMID: 29947573; PMCID: PMC6204840. https://pubmed.ncbi.nlm.nih.gov/29947573/.
- US-FDA FDA Approves New Drug to Prevent RSV in Babies and Toddlers, Available on line : https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-prevent-rsv-babies-and-toddlers. https://web.archive.org/web/20230801224229/https://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-prevent-rsv-babies-and-toddlers accessed April 15,2024.
- Jones JM, Fleming-Dutra KE, Prill MM; et al. Use of Nirsevimab for the Prevention of Respiratory Syncytial Virus Disease Among Infants and Young Children: Recommendations of the Advisory Committee on Immunization Practices — United States, 2023. MMWR Morb Mortal Wkly Rep 2023;72:920–925. https://doi.org/.
- HAS nirsevimab Transparency Committee, 19 July 2023, accessed April 22, 2024. https://www.has-sante.fr/plugins/ModuleXitiKLEE/types/FileDocument/doXiti.jsp?id=p_3476336. https://web.archive.org/web/20240104100601/https://www.has-sante.fr/upload/docs/application/pdf/2023-11/beyfortus_19072023_summary_ct20356_en.pdf.
- Sánchez Luna M, Fernández Colomer B, Couce Pico ML; en representación de la Junta Directiva de la Sociedad española de Neonatología SENEO Comisión de Infecciones SENEO y Comisión de Estándares de SENEO. Recommendations of the Spanish Society of Neonatology for the prevention of severe respiratory syncytial virus infections with nirsevimab, for the 2023-2024 season. An Pediatr (Engl Ed). 2023 Oct;99(4):264-265. https://doi.org/10.1016/j.anpede.2023.09.005. Epub 2023 Sep 20. PMID: 37739825. https://pubmed.ncbi.nlm.nih.gov/37739825/.
- New immunisation to protect newborns and young children against bronchiolitis, The Luxembourg Governement press release, September 22, 2023 accessed May 17, 2024, https://gouvernement.lu/en/actualites/toutes_actualites/communiques/2023/09-septembre/22-immunisation-bronchiolite-nourrissons.html. https://web.archive.org/web/20230922222249/https://gouvernement.lu/en/actualites/toutes_actualites/communiques/2023/09-septembre/22-immunisation-bronchiolite-nourrissons.html.
- Domachowske JB, Khan AA, Esser MT, Jensen K, Takas T, Villafana T, Dubovsky F, Griffin MP. Safety, Tolerability and Pharmacokinetics of MEDI8897, an Extended Half-life Single-dose Respiratory Syncytial Virus Prefusion F-targeting Monoclonal Antibody Administered as a Single Dose to Healthy Preterm Infants. Pediatr Infect Dis J. 2018 Sep;37(9):886-892. https://doi.org/10.1097/INF.0000000000001916. PMID: 29373476; PMCID: PMC6133204.
- Domachowske J, Madhi SA, Simões EAF, Atanasova V, Cabañas F, Furuno K, Garcia-Garcia ML, Grantina I, Nguyen KA, Brooks D, Chang Y, Leach A, Takas T, Yuan Y, Griffin MP, Mankad VS, Villafana T; MEDLEY Study Group. Safety of Nirsevimab for RSV in Infants with Heart or Lung Disease or Prematurity. N Engl J Med. 2022 Mar 3;386(9):892-894. https://doi.org/10.1056/NEJMc2112186. PMID: 35235733. https://pubmed.ncbi.nlm.nih.gov/35235733/.
- Drysdale SB, Cathie K, Flamein F, Knuf M, Collins AM, Hill HC, Kaiser F, Cohen R, Pinquier D, Felter CT, Vassilouthis NC, Jin J, Bangert M, Mari K, Nteene R, Wague S, Roberts M, Tissières P, Royal S, Faust SN; HARMONIE Study Group. Nirsevimab for Prevention of Hospitalizations Due to RSV in Infants. N Engl J Med. 2023 Dec 28;389(26):2425-2435. https://doi.org/10.1056/NEJMoa2309189. PMID: 38157500. https://pubmed.ncbi.nlm.nih.gov/38157500/.
- Griffin MP, Yuan Y, Takas T, Domachowske JB, Madhi SA, Manzoni P, Simões EAF, Esser MT, Khan AA, Dubovsky F, Villafana T, DeVincenzo JP; Nirsevimab Study Group. Single-Dose Nirsevimab for Prevention of RSV in Preterm Infants. N Engl J Med. 2020 Jul 30;383(5):415-425. https://doi.org/10.1056/NEJMoa1913556. Erratum in: N Engl J Med. 2020 Aug 13;383(7):698. PMID: 32726528. https://pubmed.ncbi.nlm.nih.gov/32726528/.
- FDA Biologics License Application (BLA) 761328 Nirsevimab Antimicrobial Drugs Advisory Committee Meeting June 8, 2023 Division of Antivirals, Office of Infectious Diseases Center for Drug Evaluation and Research, accessed April 13,2024 https://www.fda.gov/media/169322/download. https://web.archive.org/web/20230613110427/.
- EudraVigilance European database of suspected adverse drug reaction reports. https://www.adrreports.eu/fr/eudravigilance.html accessed 15 April 2024.
- Pharmacovigilance d'Île de France, campagne d'immunisation contre le VRS-Beyfortus, 31 janvier 2024, accessed April 22,2024 https://www.pharmacovigilance-iledefrance.fr/d%C3%A9tails-dune-br%C3%A8ve/campagne-dimmunisation-contre-le-vrs-beyfortus-nirs%C3%A9vimab. https://web.archive.org/web/20240223011807/https://www.pharmacovigilance-iledefrance.fr/détails-dune-brève/campagne-dimmunisation-contre-le-vrs-beyfortus-nirsévimab.
- CDC, Nirsevimab Coverage, Children 0 to 19 months, United States. Data are current through February 29, 2024.accessed April 15,2024 https://www.cdc.gov/vaccines/imz-managers/coverage/rsvvaxview/nirsevimab-coverage-children-0-19months.html.
- RSV-NET Interactive Dashboard, CDC, accessed April15,2024 https://www.cdc.gov/rsv/research/rsv-net/dashboard.html.
- Moline HL et al., Early Estimate of Nirsevimab Effectiveness for Prevention of Respiratory Syncytial Virus–Associated Hospitalization Among Infants Entering Their First Respiratory Syncytial Virus Season — New Vaccine Surveillance Network, October 2023–February 2024 https://www.cdc.gov/mmwr/volumes/73/wr/mm7309a4.htm?s_cid=mm7309a4_w.
- Ernst C, Bejko D, Gaasch L, Hannelas E, Kahn I, Pierron C, Del Lero N, Schalbar C, Do Carmo E, Kohnen M, Andlauer E, Hublart P, Masi S, de la Fuente Garcia I, Vergison A, Mossong J. Impact of nirsevimab prophylaxis on paediatric respiratory syncytial virus (RSV)-related hospitalisations during the initial 2023/24 season in Luxembourg. Euro Surveill. 2024 Jan;29(4):2400033. https://doi.org/10.2807/1560-7917.ES.2024.29.4.2400033. PMID: 38275017; PMCID: PMC10986653.
- DGS-URGENT 9, 08/24/2023, DGS-URGENT N°2023_14, PREVENTION MEDICAMENTEUSE DES BRONCHIOLITES A VRS A PARTIR DE SEPTEMBRE accessed August 30, 2023, available on line :.https://sante.gouv.fr/IMG/pdf/dgs-urgent_2023-14_-_traitement_preventif_vrs.pdf. https://web.archive.org/web/20240202032041/https://sante.gouv.fr/IMG/pdf/dgs-urgent_2023-14_-_traitement_preventif_vrs.pdf.
- HAS Bronchiolite : La HAS publie des réponses rapides pour accompagner l’administration du Beyfortus® Communiqué de presse - 14 sept. 2023, available on line: https://www.has-sante.fr/jcms/p_3461146/fr/bronchiolite-la-has-publie-des-reponses-rapides-pour-accompagner-l-administration-du-beyfortus accessed April 22,2024. https://web.archive.org/web/20240104084538/https://www.has-sante.fr/jcms/p_3461146/fr/bronchiolite-la-has-publie-des-reponses-rapides-pour-accompagner-l-administration-du-beyfortus.
- HAS Assessing health technologies, summary, nirsevimab, Original French opinion adopted by the Transparency Committee on 19 July 2023, available on line : https://www.has-sante.fr/plugins/ModuleXitiKLEE/types/FileDocument/doXiti.jsp?id=p_3476336. https://web.archive.org/web/20240104100601/https://www.has-sante.fr/upload/docs/application/pdf/2023-11/beyfortus_19072023_summary_ct20356_en.pdf accessed april 22, 2024.
- DGS Beyfortus, fin de la campagne de distribution, 12/26/2023, reference MARS n° 2023-23, limited diffusion. https://sante.gouv.fr/professionnels/article/dgs-urgent accessed January 12,2024.
- INSEE Bilan démographique 2023, January 16,2024, available on line https://www.insee.fr/fr/statistiques/7750004 accessed on June 4, 2024.
- Bulletin Infections Respiratoires Aigües, publié le 17 avril 2024, https://www.santepubliquefrance.fr/maladies-et-traumatismes/maladies-et-infections-respiratoires/grippe/documents/bulletin-national/infections-respiratoires-aigues-grippe-bronchiolite-covid-19-.-bilan-de-la-saison-2023-2024, accessed April 22,2024.
- Bronchiolite : Bilan de la surveillance hivernale 2022-2023, Santé Publique France, July 19, 2023, accessed May 1, 2024. https://www.santepubliquefrance.fr/les-actualites/2023/bronchiolite-bilan-de-la-surveillance-hivernale-2022-2023.
- Paireau J., Durand C., Raimbault S., Cazaubon J., Mortamet G.; et al.. Nirsevimab effectiveness against cases of respiratory syncytial virus bronchiolitis hospitalised in pediatric intensive care units in France, September 2023 -January 2024. 2024. pasteur-04501286 https://hal.science/pasteur-04501286 March 12, 2024.
- La sanidad pública paga 209 euros por cada vacuna infantil contra la bronquiolitis, Civio, October 30, 2023, available on line : https://civio.es/medicamentalia/2023/10/30/nirsevimab-beyfortus-precio-virus-respiratorio-sincitial/. https://web.archive.org/web/20231103052634/https://civio.es/medicamentalia/2023/10/30/nirsevimab-beyfortus-precio-virus-respiratorio-sincitial/ accessed April 23,2024.
- Servicio Andaluz de Salud, El Distrito Serranía recibe un reconocimiento por la cobertura de la vacuna combinada frente a difteria, tétanos, tosferina y poliomielitis 30 January 2024, Available on line : https://www.sspa.juntadeandalucia.es/servicioandaluzdesalud/todas-noticia/el-distrito-serrania-recibe-un-reconocimiento-por-la-cobertura-de-la-vacuna-combinada-frente https://web.archive.org/web/20240602170353/https://www.sspa.juntadeandalucia.es/servicioandaluzdesalud/todas-noticia/el-distrito-serrania-recibe-un-reconocimiento-por-la-cobertura-de-la-vacuna-combinada-frente accessed February 10 2024.
- Administradas 452.000 vacunas por virus respiratorios, Ondacero.es ; April 8,2024, Available on line :. https://www.ondacero.es/emisoras/asturias/noticias/administradas-452000-vacunas-virus-respiratorios_202404086613a4ec099903000111cfb3.html. https://web.archive.org/web/20240602170546/https://www.ondacero.es/emisoras/asturias/noticias/administradas-452000-vacunas-virus-respiratorios_202404086613a4ec099903000111cfb3.html accessed April 23, 2024.
- La Comunidad de Madrid reduce un 90% los ingresos hospitalarios de menores de un año tras incorporar la vacuna contra la bronquiolitis, Communiqué de presse de la Comunidad de Madrid, April 16,2024, Available on line : https://www.comunidad.madrid/notas-prensa/2024/04/16/comunidad-madrid-reduce-90-ingresos-hospitalarios-menores-ano-incorporar-vacuna-bronquiolitis. https://web.archive.org/web/20240602170719/https://www.comunidad.madrid/notas-prensa/2024/04/16/comunidad-madrid-reduce-90-ingresos-hospitalarios-menores-ano-incorporar-vacuna-bronquiolitis accessed April 23,2024.
- Ministerio de ciencia, innovacion y universidades, Gobierno de Espana, Informes semanales de vigilancia centinela de IRAs y de IRAG: Gripe, Covid-19 y otros virus respiratorios, Available on line : https://www.isciii.es/QueHacemos/Servicios/VigilanciaSaludPublicaRENAVE/EnfermedadesTransmisibles/Paginas/VIGILANCIA-CENTINELA-DE-INFECCION-RESPIRATORIA-AGUDA.aspx. https://web.archive.org/web/20240122052000/https://www.isciii.es/QueHacemos/Servicios/VigilanciaSaludPublicaRENAVE/EnfermedadesTransmisibles/Documents/GRIPE/Informes%20semanales/Temporada_2023-24/Informe%20semanal_SiVIRA_022024.pdf. https://web.archive.org/web/20230316142333/https://www.isciii.es/QueHacemos/Servicios/VigilanciaSaludPublicaRENAVE/EnfermedadesTransmisibles/Documents/GRIPE/Informes%20semanales/Temporada_2022-23/Informe%20semanal_SiVIRA_102023.pdf accessed April 23, 2024.
- FOLLOW-UP REPORT ON IMMUNIZATION WITH NIRSEVIMAB IN GALICIA Dirección Xeral de Saúde Pública Data up to week 13, 2024 (31-03-2024), Available on line : https://www.nirsegal.es/en. https://web.archive.org/web/20240409062929/https://assets-global.website-files.com/65774b0d3a50ee58b24dba82/660e95b8142542e4cae669d2_Report_RSV_week13.pdf accessed April 23, 2024.
- Ares-Gómez S, Mallah N, Santiago-Pérez MI, Pardo-Seco J, Pérez-Martínez O, Otero-Barrós MT, Suárez-Gaiche N, Kramer R, Jin J, Platero-Alonso L, Alvárez-Gil RM, Ces-Ozores OM, Nartallo-Penas V, Mirás-Carballal S, Piñeiro-Sotelo M, Malvar-Pintos A, González-Pérez JM, Rodríguez-Tenreiro-Sánchez C, Rivero-Calle I, Salas A, Durán-Parrondo C, Martinón-Torres F; NIRSE-GAL study group. Effectiveness and impact of universal prophylaxis with nirsevimab in infants against hospitalisation for respiratory syncytial virus in Galicia, Spain: Initial results of a population-based longitudinal study. Lancet Infect Dis. 2024 Apr 30:S1473-3099(24)00215-9. https://doi.org/10.1016/S1473-3099(24)00215-9. Epub ahead of print. PMID: 38701823. https://pubmed.ncbi.nlm.nih.gov/38701823/.
- Evaluation of the Effectiveness and Impact of Nirsevimab Administered as Routine Immunization (NIRSE-GAL), accessed May 10, 2024, https://classic.clinicaltrials.gov/ct2/show/NCT06180993. https://web.archive.org/web/20240602172604/https://classic.clinicaltrials.gov/ct2/show/NCT06180993.
- Ezpeleta G, Navascués A, Viguria N, Herranz-Aguirre M, Juan Belloc SE, Gimeno Ballester J, Muruzábal JC, García-Cenoz M, Trobajo-Sanmartín C, Echeverria A, Martínez-Baz I, Vera-Punzano N, Casado I, López-Mendoza H, Ezpeleta C, Castilla J. Effectiveness of Nirsevimab Immunoprophylaxis Administered at Birth to Prevent Infant Hospitalisation for Respiratory Syncytial Virus Infection: A Population-Based Cohort Study. Vaccines (Basel). 2024 Apr 4;12(4):383. https://doi.org/10.3390/vaccines12040383. PMID: 38675765; PMCID: PMC11054679. https://pubmed.ncbi.nlm.nih.gov/38675765/.
- Boletin de Salud Publica de Navarra, n°126, September 2023, Available on line http://www.navarra.es/home_es/Gobierno+de+Navarra/Organigrama/Los+departamentos/Salud/Organigrama/Estructura+Organica/Instituto+Navarro+de+Salud+Publica/Publicaciones/Publicaciones+profesionales/Epidemiologia/Boletin+ISP.htm. https://web.archive.org/web/20231129071427/http://www.navarra.es/NR/rdonlyres/AECCD760-AB2A-4841-818A-FA53478FD6DC/488194/BOL126INT1.pdf accessed May 1, 2024.
- DREES, La Naissance : Caractéristiques des accouchements, Available on line: https://drees.solidarites-sante.gouv.fr/sites/default/files/2021-07/Fiche%2024%20-%20La%20naissance%20-%20caract%C3%A9ristiques%20des%20accouchements.pdf. https://web.archive.org/web/20231206003500/https://drees.solidarites-sante.gouv.fr/sites/default/files/2021-07/Fiche%2024%20-%20La%20naissance%20-%20caractéristiques%20des%20accouchements.pdf accessed April 23, 2024.
- INSEE Décès et Mortalité, Available on line : https://www.insee.fr/fr/statistiques/7767420?sommaire=7764286. et https://www.insee.fr/fr/statistiques/6959517?sommaire=4487854 , accessed April 30, 2024.
- Nombre mensuel de naissances (de janvier 2015 à octobre 2023), Available on line : https://www.insee.fr/fr/statistiques/7758827?sommaire=5348638 et https://www.insee.fr/fr/statistiques/8064935?sommaire=7944361 , accessed April 30,2024.
- Article 14(9) EC-726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency, Available on line : http://data.europa.eu/eli/reg/2004/726/oj. https://web.archive.org/web/20231030191458/https://eur-lex.europa.eu/eli/reg/2004/726/oj accessed May 15,2024.
- Nair H, Simões EA, Rudan I, Gessner BD, Azziz-Baumgartner E, Zhang JSF, Feikin DR, Mackenzie GA, Moiïsi JC, Roca A, Baggett HC, Zaman SM, Singleton RJ, Lucero MG, Chandran A, Gentile A, Cohen C, Krishnan A, Bhutta ZA, Arguedas A, Clara AW, Andrade AL, Ope M, Ruvinsky RO, Hortal M, McCracken JP, Madhi SA, Bruce N, Qazi SA, Morris SS, El Arifeen S, Weber MW, Scott JAG, Brooks WA, Breiman RF, Campbell H; Severe Acute Lower Respiratory Infections Working Group. Global and regional burden of hospital admissions for severe acute lower respiratory infections in young children in 2010: A systematic analysis. Lancet. 2013 Apr 20;381(9875):1380-1390. https://doi.org/10.1016/S0140-6736(12)61901-1. Epub 2013 Jan 29. PMID: 23369797; PMCID: PMC3986472. https://pubmed.ncbi.nlm.nih.gov/23369797/.
- Del Riccio M, Spreeuwenberg P, Osei-Yeboah R, Johannesen CK, Fernandez LV, Teirlinck AC, Wang X, Heikkinen T, Bangert M, Caini S, Campbell H, Paget J; RESCEU Investigators. Burden of Respiratory Syncytial Virus in the European Union: Estimation of RSV-associated hospitalizations in children under 5 years. J Infect Dis. 2023 Nov 28;228(11):1528-1538. https://doi.org/10.1093/infdis/jiad188. PMID: 37246724; PMCID: PMC10681872 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10681872/.
- Suss RJ, Simões EAF. Respiratory Syncytial Virus Hospital-Based Burden of Disease in Children Younger Than 5 Years, 2015-2022. JAMA Netw Open. 2024 Apr 1;7(4):e247125. https://doi.org/10.1001/jamanetworkopen.2024.7125. PMID: 38635270. https://pubmed.ncbi.nlm.nih.gov/38635270/.
- Sullivan K, Sullivan B. Does nirsevimab prevent lower respiratory infections caused by respiratory syncytial virus? J Perinatol. 2024 May;44(5):767-769. https://doi.org/10.1038/s41372-024-01970-y. Epub 2024 Apr 18. PMID: 38637681; PMCID: PMC11090781. https://pubmed.ncbi.nlm.nih.gov/38637681/.
- Vaux S, Viriot D, Forgeot C, Pontais I, Savitch Y, Barondeau-Leuret A, Smadja S, Valette M, Enouf V, Parent du Chatelet I. Bronchiolitis epidemics in France during the SARS-CoV-2 pandemic: The 2020-2021 and 2021-2022 seasons. Infect Dis Now. 2022 Sep;52(6):374-378. https://doi.org/10.1016/j.idnow.2022.06.003. Epub 2022 Jun 23. PMID: 35753628; PMCID: PMC9222408. https://pubmed.ncbi.nlm.nih.gov/35753628/.
- Delestrain C, Danis K, Hau I, Behillil S, Billard MN, Krajten L, Cohen R, Bont L, Epaud R. Impact of COVID-19 social distancing on viral infection in France: A delayed outbreak of RSV. Pediatr Pulmonol. 2021 Dec;56(12):3669-3673. Epub 2021 Sep 2. PMID: 34473914; PMCID: PMC8662089. https://pubmed.ncbi.nlm.nih.gov/34473914/. [CrossRef]
- International Classification of Diseases, Tenth Revision, International Statistical Classification of Diseases and Related Health Problems 10th Revision latest version 2019, WHO, Available on line : https://icd.who.int/browse10/2019/en accessed May 12, 2024.
- Journal Officiel, France, 12/231/2023, available on line : https://www.senat.fr/questions/jopdf/2023/2023-12-21_seq_20230050_0001_p000.pdf, accessed on June 3, 2024.
- EPI-PHARE, Epidémiologie des produits de santé, GIS ANSM-CNAM, Rapport d’étude d’utilisation du Nirsévimab (Beyfortus®) en ville en France lors de la première campagne de prévention (saison 2023/2024) 8 avril 2024, available on line : https://www.epi-phare.fr/rapports-detudes-et-publications/utilisation-beyfortus/ https://web.archive.org/web/20240603171635/https://www.epi-phare.fr/rapports-detudes-et-publications/utilisation-beyfortus/ accessed on June 3, 2024.
- Dekkers G, Treffers L, Plomp R, Bentlage AEH, de Boer M, Koeleman CAM, Lissenberg-Thunnissen SN, Visser R, Brouwer M, Mok JY, Matlung H, van den Berg TK, van Esch WJE, Kuijpers TW, Wouters D, Rispens T, Wuhrer M, Vidarsson G. Decoding the Human Immunoglobulin G-Glycan Repertoire Reveals a Spectrum of Fc-Receptor- and Complement-Mediated-Effector Activities. Front Immunol. 2017 Aug 2;8:877. https://doi.org/10.3389/fimmu.2017.00877. PMID: 28824618; PMCID: PMC5539844.
- Hiatt A, Bohorova N, Bohorov O, Goodman C, Kim D, Pauly MH, Velasco J, Whaley KJ, Piedra PA, Gilbert BE, Zeitlin L. Glycan variants of a respiratory syncytial virus antibody with enhanced effector function and in vivo efficacy. Proc Natl Acad Sci U S A. 2014 Apr 22;111(16):5992-7. https://doi.org/10.1073/pnas.1402458111. Epub 2014 Apr 7. PMID: 24711420; PMCID: PMC4000855.
- Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs. 2011 Nov-Dec;3(6):568-76. https://doi.org/10.4161/mabs.3.6.17922. Epub 2011 Nov 1. PMID: 22123061; PMCID: PMC3242843.
- Liu L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J Pharm Sci. 2015 Jun;104(6):1866-1884. https://doi.org/10.1002/jps.24444. Epub 2015 Apr 14. PMID: 25872915.
- Mokhtary 2022 Mokhtary P, Pourhashem Z, Mehrizi AA, Sala C, Rappuoli R. Recent Progress in the Discovery and Development of Monoclonal Antibodies against Viral Infections. Biomedicines. 2022 Aug 2;10(8):1861. PMCID: PMC9405509. https://pubmed.ncbi.nlm.nih.gov/36009408/. [CrossRef] [PubMed]
- Taylor G. Animal models of respiratory syncytial virus infection. Vaccine. 2017 Jan 11;35(3):469-480. https://doi.org/10.1016/j.vaccine.2016.11.054. Epub 2016 Nov 29. PMID: 27908639; PMCID: PMC5244256.
- FDA full prescribing information Beyfortus, Reference ID : 5203445, Available on line : https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/761328s000lbl.pdf. https://web.archive.org/web/20230804203015/https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/761328s000lbl.pdf accessed May 28, 2024.
- van Erp EA, Luytjes W, Ferwerda G, van Kasteren PB. Fc-Mediated Antibody Effector Functions During Respiratory Syncytial Virus Infection and Disease. Front Immunol. 2019 Mar 22;10:548. https://doi.org/10.3389/fimmu.2019.00548. PMID: 30967872; PMCID: PMC6438959. https://pubmed.ncbi.nlm.nih.gov/30967872/.
- Shoukat A, Abdollahi E, Galvani AP, Halperin SA, Langley JM, Moghadas SM. Cost-effectiveness analysis of nirsevimab and maternal RSVpreF vaccine strategies for prevention of Respiratory Syncytial Virus disease among infants in Canada: A simulation study. Lancet Reg Health Am. 2023 Nov 9;28:100629. PMID: 38026446; PMCID: PMC10663690. https://pubmed.ncbi.nlm.nih.gov/38026446/. [CrossRef]
- Advisory Committee Statement (ACS) National Advisory Committee on Immunization (NACI) Statement on the prevention of respiratory syncytial virus (RSV) disease in infants: Supplementary systematic review of economic evidence, Available on line : https://www.canada.ca/content/dam/phac-aspc/documents/services/publications/vaccines-immunization/national-advisory-committee-immunization-statement-prevention-respiratory-syncytial-virus-disease-infants-supplementary-systematic-review-economic-evidence/naci-appendix-2024-05-17.pdf. https://web.archive.org/web/20240521111714/https://www.canada.ca/content/dam/phac-aspc/documents/services/publications/vaccines-immunization/national-advisory-committee-immunization-statement-prevention-respiratory-syncytial-virus-disease-infants-supplementary-systematic-review-economic-evidence/naci-appendix-2024-05-17.pdf accessed May 26, 2024.
- Neumann S, Alverson B. Nirsevimab: The Hidden Costs. Hosp Pediatr. 2024 May 9;14(6):e2024007739. PMID: 38721666. https://pubmed.ncbi.nlm.nih.gov/38721666/. [CrossRef]
- AAP news, Sanofi raising price of RSV immunization nirsevimab April 12, 2024, Available on line : https://publications.aap.org/aapnews/news/28657/Sanofi-raising-price-of-RSV-immunization. https://web.archive.org/web/20240530142639/https://publications.aap.org/aapnews/news/28657/Sanofi-raising-price-of-RSV-immunization accessed April 20, 2024.
- BEYFORTUS 50 mg sol inj ser préremplie, Vidal, Available on line : https://www.vidal.fr/medicaments/beyfortus-50-mg-sol-inj-ser-preremplie-242583.html. https://web.archive.org/web/20240526204604/https://www.vidal.fr/medicaments/beyfortus-50-mg-sol-inj-ser-preremplie-242583.html accessed May 26, 2024.
- Circuit de prescription du Nirsevimab (BEYFORTUS ®) ARS de Bretagne, Available on line: https://www.bretagne.ars.sante.fr/system/files/2023-09/Presentation%20Dr%20Lefevre.pdf. https://web.archive.org/web/20240329150151/https://www.bretagne.ars.sante.fr/system/files/2023-09/Presentation%20Dr%20Lefevre.pdf accessed October 10, 2023.
- Gebremedhin B. Gebretekle, Man Wah Yeung, Raphael Ximenes, Alexandra Cernat, Alison E. Simmons, April Killikelly, Winnie Siu, Ellen Rafferty, Nicholas Brousseau, Matthew Tunis, Ashleigh R. Tuite medRxiv 2024.03.21.24304675. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



