
Submitted:
09 June 2024
Posted:
12 June 2024
You are already at the latest version
Abstract
Despite chronic fibrosis and associated excess extracellular matrix (ECM) deposition and crosslinking occurring in many pathological conditions, few in vitro studies examine how fibrosis-induced ECM stiffening impacts lymphatic endothelial cells (LEC) behavior and subsequent new lymphatic vessel growth and function. This study examined the stiffening profile of PhotoCol®—commercially available methacrylated type I collagen—photo-crosslinked with photoinitiators Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP), Irgacure 2959 (IRG), and Ruthenium/Sodium Persulfate (Ru/SPS) prior to evaluating PhotoCol® permeability and LEC response to PhotoCol® at low and high stiffness levels representing normal and fibrotic ECM stiffness. Ru/SPS produced the highest maximum stiffness for photo-crosslinked PhotoCol® hydrogels. While Ru/SPS stiffness values did not differ significantly with increased light exposure, permeability decreased significantly with increasing light exposure for 40 kDa dextran. Finally, LECs on softer PhotoCol® appeared smaller with less prominent VE-Cadherin (cell-cell junction) and F-actin expression compared to cells on stiffer PhotoCol®. Overall, PhotoCol® with Ru/SPS photoinitiator provides the fibrillar microstructure and dynamic stiffness range that enables systematic study of LEC stiffness responses relevant to the stiffened ECM observed in pathologies with significant fibrotic activity. Therefore, this study demonstrates the utility of PhotoCol® with Ru/SPS for disease modeling applications, particularly modeling lymphatic vascular growth and function during fibrosis.
Keywords:
Methacrylated collagen
; mechanical assessment
; photoinitiator
; human dermal lymphatic endothelial cells
; disease modeling
1. Introduction
The lymphatic system has long been viewed as a passive component of the immune and circulatory systems. However, in the last 15 years more evidence has emerged to show that the lymphatic system plays an active role in wound healing, cancer, and fibrosis [1] through regulation and dysregulation of the vasculature. In many of these healing and disease states, the tissue surrounding lymphatic vasculature undergoes dynamic biochemical (e.g., soluble factors) and biophysical (e.g., stiffness) changes that influence cell behavior [2,3,4,5]. One important biophysical change is increased tissue stiffening that is often associated with chronic fibrosis—a pathological wound healing condition marked by excess tissue deposition and scarring [6]. Increased tissue deposition and subsequent tissue densification also alters tissue transport properties, which changes the biochemical environment by altering how signaling molecules move through the tissue to reach cells. However, little is known about how tissue stiffening affects lymphatic vessel growth and function, particularly under disease conditions where tissue stiffness is much higher than what is observed in healthy tissue or during lymphatic development. Thus, it is important to leverage modeling approaches that use materials capable of exhibiting mechanical properties of healthy and fibrotic tissue environments to understand the impact of stiffness on lymphatic vasculature in fibrosis and wound healing.
Over the last several years, there has been more interest in investigating the relationship between tissue stiffness and lymphatic vasculature. To date, these studies primarily focus on lymphatic development and/or employ modeling strategies that approximate tissue stiffness levels observed across developmental and growth stages rather than disease. In work from Frye et al. [7], human dermal lymphatic endothelial cells (HDLEC) were cultured on Softwell™ or Softslip™ dishes (Matrigen) that provided stiff substrates (4 kPa, embryonic cardinal vein; 8 and 12 kPa, muscle; 25 kPa, bone) or soft substrates (0.2 kPa, tissue surrounding cardinal vein) in vitro. After 24 hours, HDLECs on softer substrates showed upregulation of genes for cell–matrix adhesion, cell migration, new lymphatic vessel growth (e.g., metalloproteinases 1, 2, and 10), and vascular development stages such as valve formation (e.g., GATA2), while genes for cell proliferation were downregulated. They related their results to concurrent in vivo studies where increased extracellular matrix (ECM) stiffness within the cardinal vein appeared to support HDLECs that were flat and tightly attached to the underlaying basement membrane. Alternatively, HDLECs that migrated outside of the cardinal vein into a softer ECM were more elongated, spindle shaped as a precursor to network formation. Alderfer et al. chose a different approach to study tissue stiffness effects by using thiol-modified hyaluronic acid (HA) hydrogels conjugated with heparin and thiol-modified gelatin [8]. Tunable matrix stiffness was achieved by varying ratios of polyethylene glycol diacrylate (PEG-DA) to produce soft (30 Pa), medium (300 Pa), and firm (900 Pa) substrates for culturing HDLECs. They identified matrix stiffness as a key player in activating vascular endothelial growth factor receptor (VEGFR)-3, the primary receptor for VEGF-C. On softer matrices, VEGFR-3 activation increased, which enabled more VEGF-C binding and subsequent formation of more extensive cord-like structures when compared to stiffer matrices. Genes for characteristic lymphatic markers (i.e., Prox1) and proteolytic enzymes involved in cell migration and tube formation (i.e., metalloproteinases 2 and 14) were also upregulated on softer matrices, similar to Frye et al. [7]. Collectively, these studies demonstrate mechanosensing capabilities of HDLECs and highlight the importance of ECM stiffness in directing HDLEC behavior (cell spreading, migration, proliferation) and lymphatic capillary tube formation, and both studies suggest that HDLECs prefer softer substrates [7,8]. Stiffer substrates, which appear to be more inhibitory to HDLEC behaviors related to lymphatic capillary growth, have potential to represent stages of fibrosis in vitro where ECM stiffness increases. However, there are in vivo situations where fibrotic conditions and increased ECM stiffness support lymphatic capillary growth rather than hinder (e.g., cancer, renal fibrosis), and questions remain on how in vitro models can be designed to achieve the physical properties necessary to appropriately mimic some of these pathological conditions.
Within the fields of tissue engineering and in vitro disease modeling, there are a myriad of options for substrates with tunable stiffness properties [9,10]. As we consider materials within the context of lymphatic studies, it is also important that they also have physiological relevance to ECM composition to tissues surrounding lymphatic capillaries and higher order lymphatic vessels [4,7,8,11]. In the previously described in vitro studies, substrate (or ECM) stiffness differences were achieved in different ways. Softwell™ or Softslip™ dishes have the advantage of defined stiffness properties across a wide stiffness range 0.1-100 kPa. However, they are only suited for 2D monolayer cultures and are unable to support 3D sprouting into the substrate. Moreover, stiffness cannot be tuned outside what is available through the company. HA composite hydrogels offer more room for stiffness tunability with additional physiological relevance as HA directly interacts with HDLECs in vivo [12,13]. Cells can also be cultured on top or within these hydrogels with the latter approach supporting 3D sprouting studies. However, a 30-300 Pa stiffness range for lymphatic studies [8] is more representative of developmental stages of lymphatic vessel growth, and significantly higher values are needed to study disease states. For instance, in conditions like pancreatic cancer that have intratumoral and peritumoral lymphatic capillaries, tissue stiffness has been measured at 5.46 ± 3.18 kPa compared to 1.06 ± 0.25 kPa for normal pancreatic tissue [14]. When considering other materials with tunable stiffness capabilities, type I collagen emerges as a potential candidate. Type I collagen is a prominent ECM component in fibrotic tissues, as fibroblasts deposit collagen that undergoes enzymatic crosslinking (i.e., lysyl oxidase) to increase overall ECM stiffness during fibrosis [15]. Since lymphatic capillaries are not surrounded by a continuous basement membrane or support cells like blood capillaries, they are directly attached to the underlying ECM through adhesion molecules like integrins that bind collagens and fibronectin, as well as elastin microfibril interfacer 1 [16,17,18]. Therefore, changes in type I collagen structure (e.g., crosslinking) and stiffness that occur during fibrosis can be sensed by HDLECs. Moreover, results from HDLEC stiffness studies showed stiffness-mediated changes in matrix metalloproteinases that target a1(I) and a2(I) peptide chains within the type I collagen triple helix (e.g., MMP-1, -2, -14) during cell migration and lymphatic sprouting [7,8,11,19]. Although these features and outcomes provide rationale for using type I collagen as a substrate for lymphatic studies, traditionally prepared type I collagen hydrogels lack the dynamic stiffness range needed to represent fibrotic tissue stiffness levels.
Methacrylated type I collagen offers an alternative to produce collagen-based hydrogels with a wide dynamic stiffness range that can be defined “on demand” for lymphatic studies. Free amines on the type I collagen molecule are modified with methacrylate groups, and when modified collagen is combined with a photoinitiator, light irradiation promotes crosslinking via methacrylamide polymerization (i.e., photo-crosslinking) [20,21]. With this modification and photo-crosslinking, methacrylated type I collagen stiffness can increase from approximately 0.5 kPa(uncrosslinked) up to 8 kPa [20] while maintaining its higher order fibrillar microstructure, which is advantageous for recreating in vivo-like tissue. Other methacrylated materials such as gelatin, HA, and various polysaccharides (e.g., dextran, alginate, chitosan) can achieve high stiffness and are routinely used cell and tissue culture as substrates, but they lack fibrillar structures on their own [22,23]. Studies have shown that photo-crosslinked methacrylated collagens from human, bovine, and rat tissue sources have a robust mechanical response and support cell viability with photoinitiators lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) and Irgacure 2959 (IRG) [20,21,24,25]. While stiffness and lack of toxicity are important to establish, there has been little work exploring other characteristics that impact cell behavior (e.g., permeability, diffusivity). Studies have been performed to measure transport of soluble factors like dextran and bovine serum albumin through non-methacrylated type I collagens. Hsu et al. showed an inverse relationship between permeability speed and size of soluble factors but not within the context of changing concentration or crosslinking [26]. They also investigated transport across a fibroblast barrier. Chen et al. looked at the interplay between concentration and crosslinking to establish release characteristics of methacrylated collagen hydrogels photo-crosslinked with IRG [25]. Using 70 kDa FITC-labeled dextran, diffusion was observed to be more dependent on collagen concentration rather than photo-crosslinking with the highest diffusion rate observed in uncrosslinked collagen at low concentration. They cited increased fibril density from higher collagen concentration as the driving factor in hindering diffusion rather than increased stiffness from photo-crosslinking (~40 Pa vs. 200 Pa). These studies provide some insight into molecular transport through methacrylated collagen gels, but more information is needed for a broader range of molecular sizes and matrix properties (e.g., stiffness, microstructure) that are more representative of fibrotic tissues. However, they represent progress for establishing potential for methacrylated type I collagen to be used in disease models to study the interplay between cells and their microenvironment.
In the current study, PhotoCol®, a methacrylated type I collagen formulation (Advanced BioMatrix, Inc., Carlsbad, CA) was characterized on its “on demand” stiffening properties with LAP, IRG, and Ruthenium/Sodium Persulfate (Ru/SPS) photoinitiators. Irradiation with constant light exposure (20 minutes) and burst light exposure (30-, 60-, and 90 seconds) of 395-405 nm was used to measure maximum and intermediate shear storage modulus values (i.e., stiffness). Results showed that Ru/SPS yielded photo-crosslinked samples with the greatest dynamic stiffness range (0.5 - 6 kPa), and stiffness values were similar when exposed to burst light exposures. Permeability studies showed a decrease in overall permeability with 40 kDa dextran for 30- and 90 second photo-crosslinked PhotoCol® compared to uncrosslinked, but no difference was observed with 60-76 kDa dextran. Stiffer hydrogels were hypothesized to induce a phenotype in LECs indicative of fibrosis response, i.e. thicker and zipper like cellular junctions. When HDLECs were cultured on fibronectin-coated PhotoCol® with Ru/SPS at low stiffness (0.5 kPa) and high stiffness (6 kPa), results showed thicker cell-cell junctions (i.e., VE-Cadherin) and increased average cell area and shape irregularity on stiffer samples. Overall, we demonstrated that PhotoCol® with Ru/SPS induces morphological changes in HDLECs when photo-crosslinked to produce stiffness levels that are consistent with fibrotic tissues. Moreover, we established that PhotoCol©, with its “on demand” stiffening capability and fibrillar microstructure, is a viable substrate for in vitro models of lymphatic vasculature that focus on modeling fibrotic disease states.
2. Results
2.1. PhotoCol® Hydrogels with Ruthenium/Sodium Persulfate Achieve a Broad Dynamic Stiffness Range
There are multiple options for photoinitiators for methacrylated materials based on their irradiation wavelength, polymerization mechanisms, and stiffening capacities. We chose photoinitiators that use visible light to facilitate photo-crosslinking— lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP; 405 nm) and Ruthenium/Sodium Persulfate (Ru/SPS; 400-450 nm)—to avoid using ultraviolet (UV) light. These two photoinitiators also have better cytocompatibility than Irgacure 2959 (IRG; 365 nm), which uses UV light for photo-crosslinking. Irgacure was included, because it is a common photo-initiator that has been previously characterized on stiffness, degradation, and cytocompatibility [20].
We first performed rheological testing in oscillatory shear to compare the stiffening profiles of PhotoCol® with IRG, LAP, and Ru/SPS under constant light (20 minutes total light exposure after self-assembly) (Figure 1A). Results showed that PhotoCol® with Ru/SPS had a more predictable stiffening profile with a rapid increase in the shear storage modulus (G’) after photoinitiation (light exposure at 10 minutes after self-assembly) and flat, steady stiffness plateau before and after light exposure. The tests also showed that PhotoCol® reaches the plateau of maximum stiffness with photo-crosslinking in approximately 3 minutes for samples containing IRG and Ru/SPS and approximately 6 minutes for samples containing LAP, and as expected, the shear loss modulus (G”) was much lower than G’ over the entire duration of the test. The Ru/SPS samples had significantly higher stiffness values than LAP and IRG (Figure 1B) at 5.93±2.2 kPa for Ru/SPS compared to 1.96±1.06 kPa and 1.38±0.53 kPa for LAP and IRG, respectively. The higher stiffness values indicated that PhotoCol® samples with Ru/SPS- were more elastic and mechanically sound than samples with LAP or IRG. We also calculated the fold change increase in stiffness before and after photo-crosslinking to determine the dynamic range for each photoinitiator. All photoinitiators yielded significantly different dynamic ranges with the lowest being PhotoCol® with IRG and the highest being PhotoCol® with Ru/SPS (Figure 1C). Overall, the stiffness response for Ru/SPS—highest maximum stiffness and greatest dynamic stiffness range—combined with the use of non-UV light (405-450 nm), informed our choice of using Ru/SPS as the photoinitiator for remaining studies.
2.2. Stiffness of PhotoCol® with Ruthenium/Sodium Persulfate is Unchanged with Burst Light Exposure
Our prior rheological tests used constant light exposure to ensure that maximum stiffness was reached for each sample. However, burst light exposures of 30-, 60-, and 90 seconds after initial self-assembly were used to assess tunability of G’ for PhotoCol® with Ru/SPS (Figure 2A). A dose-dependent trend was observed with increased maximum G’ values with increasing exposure time. However, when we calculated the fold change differences at each light exposure duration, increases in stiffness and dynamic range were not significantly different (Figure 2B). We expected to see significant increases in stiffness with increased exposure time, because a previous study that characterized methacrylated collagen and IRG observed significant differences [20]. However, since that study used 3.75 mg/mL methacrylated collagen versus 7.122 mg/mL PhotoCol® in the current study, our higher working concentration might have contributed to the discrepancy. We chose a high working concentration to maximize overall stiffness values to approximate fibrotic tissue stiffness levels, but other approaches can be taken to generate a more tunable material. Visual assessment of collagen fibril microstructure (confocal reflectance microscopy) of uncrosslinked PhotoCol® with Ru/SPS compared to samples with 90 seconds and 5 minutes of photo-crosslinking showed some qualitative differences in fibril arrangement and uniformity (Figure 2C). Uncrosslinked samples appeared to have the most homogeneous and uniform distribution of fibrils among the three conditions. As exposure time increased, collagen microstructure became less uniform and seemingly less dense, although perceived changes in fibril density may be more related to altered fibril distribution (i.e., fibril heterogeneity). In addition, fibril length appeared slightly longer in crosslinked samples, but no definitive quantitative measurements were taken. To better characterize and understand the implications of changes in the microstructure, quantitative measurements of fibril microstructure (e.g., density, diameter, projected pore size) should be taken.
2.3. PhotoCol® Permeability Decreases with Photo-Crosslinking for 40 kDa Dextran
It is important to consider how molecular transport is affected by photo-crosslinking in methacrylated collagen prepared at higher concentration and stiffness levels. Permeability was impacted by photo-crosslinking for one of the two dextran sizes tested, but the duration of photo-crosslinking did not have an affect (Figure 3). For 40 kDa dextran, permeability coefficients were significantly lower for PhotoCol® samples photo-crosslinked for 30 seconds (7.96×10-7 ± 2.12×10 –7 m/s) and 90 seconds (5.59×10-7 ± 2.40×10 –7 m/s) compared to uncrosslinked samples (2.58×10-6 ± 1.18×10 –6 m/s). Conversely, there was no statistical significance for 60-76 kDa dextran for uncrosslinked groups (1.11×10-6 ± 2.14×10 –7 m/s) and PhotoCol® samples photo-crosslinked for 30 seconds (6.38×10-7 ± 3.91×10 –7 m/s) and 90 seconds (7.40×10-7 ± 1.97×10 –7 m/s). There was also no difference in permeability between dextran sizes at each photo-crosslinking level.
Overall, this result indicates that transport of molecules around 40 kDa was impacted by the degree of crosslinking, whereas movement of larger molecules was more likely to be impeded regardless of crosslinking. We also performed permeability studies with 10 kDa dextran molecules in photo-crosslinked samples, and the associated permeability coefficients were comparable to what was measured for 40 kDa dextran in uncrosslinked samples (data not shown), suggesting that transport of smaller molecules were not affected by crosslinking. Within the microenvironment surrounding lymphatic capillaries, HDLECs will be exposed to soluble factors of varied size.
Therefore, these permeability results reinforce that it is not only important to have a substrate that does not impede molecular transport, but it is also important to consider how mass transport is regulated in fibrotic microenvironments in vitro and in vivo. To deepen understanding of the implications of this work for lymphatic vasculature, additional dextran sizes should be investigated, particularly between 40 and 60 kDa. Inflammatory molecules like tumor necrosis factor (TNF)-α fall into this size range and are known to impact lymphatic function and leukocyte trafficking. In addition, use of pore size as a metric should be tested to gain greater insight into permeability properties to explain this relationship.
2.4. PhotoCol® Stiffness Alters HDLEC Morphology and Cell-Cell Junctions
Qualitative observations of HDLEC cultured on photo-crosslinked PhotoCol® (stiff) showed larger cells with greater cytoplasmic area and more prominent VE-Cadherin signal (Figure 4B) than cells cultured on uncrosslinked PhotoCol® (soft) (Figure 4A). Peripheral F-actin localization was also noted in HDLECs on photo-crosslinked gels, suggesting colocalization with VE-Cadherin.
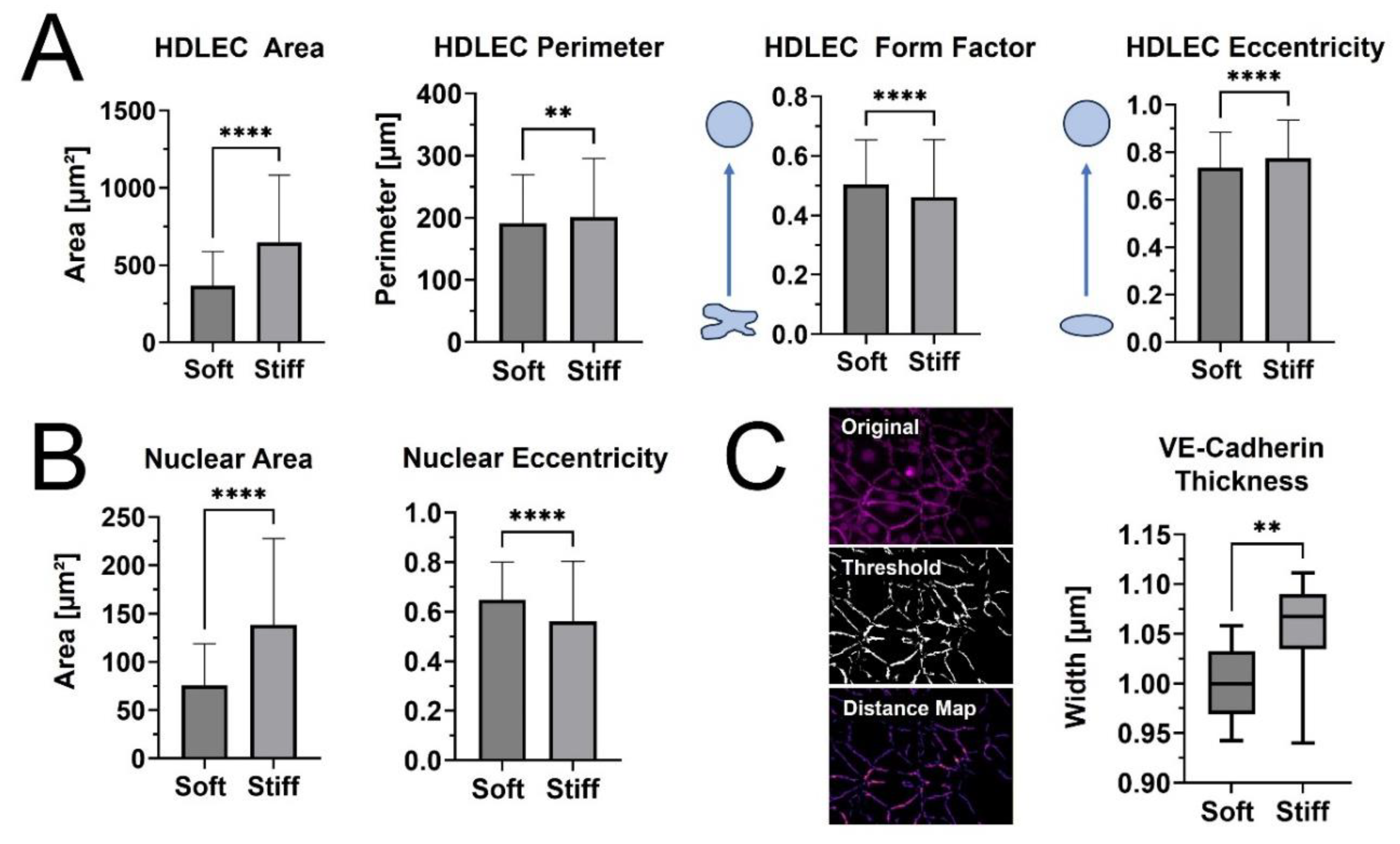
When cell and nuclear shape metrics and VE-Cadherin thickness were measured, differences in morphology were confirmed (Figure 5). The average cellular and nuclear area significantly increased when seeded on stiffer PhotoCol®, while the increase in cell perimeter—an indicator of cell shape irregularity—was significant, yet moderate. Moreover, the decreased form factor observed in HDLECs on stiffer substrates also signaled more irregularly shaped cells, which aligns with the characteristic “oak leaf” shape that is used to describe lymphatic endothelial cells [27,28]. The perimeter result also related to the slight increase in cell eccentricity—an elongation measure— on stiffer substrates because more circular shapes (i.e., higher eccentricity) have a lower perimeter to area ratio (Figure 5A). Smaller and more elongated HDLECs may indicate a migratory and vessel forming morphology [7]. Conversely, even though there was an increase in nuclear area with stiffness, there was a decrease in eccentricity, suggesting more circular nuclei on softer substrates (Figure 5B). HDLEC cultured on photo-crosslinked PhotoCol® also showed thicker VE-Cadherin junctions between cells (Figure 5C). Thicker VE-Cadherin suggested increased zippering of the cell-cell junctions, with thinner VE-Cadherin being closer to discontinuous button junctions. Button like junctions represent a healthy junctional phenotype, whereas zippered junctions are often present in fibrotic environments [28]. The spread of VE-Cadherin thickness data for HDLECs on photo-crosslinked PhotoCol® group overlapped with the uncrosslinked result, which can likely be attributed to artifacts produced in the thresholding process (i.e., numerous small, thin lines not associated with VE-Cadherin) that the software included into the thickness measurements. To remain consistent, the thickness measurement process was kept consistent across groups, thus the artifacts were not removed.
3. Discussion
Recently, there has been increased focus on the relationship between tissues stiffness and lymphatic vessel response, primarily with lymphatic capillaries and LECs [6,7,8]. For our study, we focused on tissue stiffness levels that are found in fibrotic disease conditions, since tissue stiffening from excess ECM deposition and crosslinking are major features of fibrosis that alter the biophysical and biochemical microenvironments surrounding lymphatic vasculature. Methacrylated type I collagen, combined with photoinitiators (LAP, IRG, or Ru/SPs) and light exposure (356-405 nm), was utilized as an “on-demand” stiffened material for fibrotic tissue disease modeling. The three photoinitiations were tested for how they changed the stiffening profiles of PhotoCol®, and Ru/SPS showed the highest average maximum stiffness, highest fold change after photo-crosslinking (i.e., dynamic range), and the quickest time to stiffness plateau. Importantly, the maximum stiffness of Ru/SPS PhotoCol® reached an average of approximately 6 kPa, which is within the stiffness range of some diseased fibrotic tissue [29,30]. When Ru/SPS PhotoCol® was tested for stiffness tunability using burst light exposures, a range of 5-9 kPa was achieved, but the fold change of the burst exposure was not statistically different despite an observable trend of increasing stiffness with light exposure. There was also a visual difference in how the PhotoCol® fibrils formed under different stiffening conditions with longer, less densely packed fibrils with increased photo-crosslinking. Permeability of 40 kDa dextran was statistically different for photo-crosslinked versus uncrosslinked samples, but these effects were not significant for 60-76 kDa dextran. This result demonstrated that as the physical environment changes due to stiffness, the soluble factor signaling may vary depending on the size of the molecule impacting cellular response. LEC morphology was also significantly affected by substrate stiffness with increased cell area and irregularity observed on stiffer PhotoCol©. This results was similar to results found by Frye et.al., where soft substrates induced smaller and more elongated cells, and stiff substrates induced flattened and spread-out morphologies [7]. Notably, our study also quantified the thickness differences in VE-Cadherin cell junctions, which is known to be related to healthy and diseased LEC states [28].
Similar to other studies, our results show that HDLECs are responsive to differences in stiffness of the underlying substrate. However, we add to the field by using methacrylated type I collagen (PhotoCol®) at higher stiffness levels that are more representative of tissues undergoing fibrosis rather than development. Our results also show the impact of photo-crosslinking methacrylated collagen to fibrotic stiffness levels on molecular transport and extend the analysis of HDLEC response to quantify morphological characteristics to include shape metrics for cells and nuclei, along with VE-Cadherin thickness. The morphological assessment is particularly helpful, because morphology is less commonly used to describe changes in lymphatic vasculature outside of flow-based studies that assess changes in elongation and alignment [31,32,33]. Moreover, as we move toward more mechanistic studies, we can relate alterations in mechanosensing pathways to morphological changes for more robust characterization of our model systems. For instance, the YAP/TAZ pathway is notable for its mechanosensing capability LECs when exposed to oscillatory shear and varied ECM stiffness [7,8,34]. Increased substrate stiffness has been shown to promote YAP/TAZ expression and translocation to the nucleus, which blocks Prox-1 and its downstream targets, VEFGR-3 and matrix metalloproteinase-14. Without robust activation of those two targets, lymphatic sprouting and subsequent formation of cord-like structures decreases. However, studies have not investigated whether this signaling pathway has any impact on cell morphology. Less is also known about relationships between VE-Cadherin and either YAP/TAZ or ECM stiffness. Almost all lymphatic studies stain for VE-Cadherin and many assess VE-Cadherin expression via western blot, but very few studies measure VE-Cadherin morphology as we did in the current study [35]. Moreover, the driving factors behind VE-Cadherin expression are not typically investigated. In our study, we not only observed thicker VE-Cadherin junctions formed in HDLECs on stiff photo-crosslinked samples, but we also noted potential association between VE-Cadherin junctions and F-actin formation. A similar observation was noted in HDLECs on viscoelastic substrates [13]. Although neither study investigated the link between the two molecules within the context of HDLECs and stiffness, there are established connections between the VE-Cadherin/Catenin complex with actin [36]. These molecules are integrated through the processes of junction formation and maturation and remain associated during junction remodeling and maintaining junction integrity. Moreover, additional molecules such as the ARP2/3 complex, α-catenin, and p120ctn also help coordinate interactions. It will be important to investigate these mechanisms further within the context of lymphatic vasculature and ECM stiffness. Materials like methacrylated collagen will be helpful in these pursuits, because they retain collagen fibrillar microstructure that is important for cell attachment and appropriate force balance between cells and the surrounding ECM.
Through photo-crosslinking, we produced elastic hydrogels with covalent crosslinks that achieve static stiffness. However, there are temporal elements to the fibrotic process as excess ECM is deposited and crosslinked over time. Beyond stiffness, the biochemical environment also shifts with changes in ECM permeability that affect the movement of soluble factors and altered biodegradation that impacts tissue remodeling. Moreover, temporal changes in tissue stiffness and remodeling also affect ECM viscoelasticity. Fan et al. recently looked beyond static ECM stiffness and identified viscoelasticity as an important ECM property that impacts lymphatic morphogenesis and tube formation [13]. They combined supramolecular and covalent crosslinking to create dynamic HA hydrogels with tunable viscoelasticity that is spatially controlled with UV light exposure. Although the hydrogels exhibit the same maximum shear storage modulus (~1500 Pa) before and after UV irradiation, they differ in stress-relaxation behavior from static stiffness hydrogels (fully covalently crosslinked and elastic). In standard and photo-patterned viscoelastic HA substrates, HDLECs showed evidence of a higher degree of cell spreading and migration with increased F-actin stress fiber formation and focal adhesion assembly. We also observed greater cell area and F-actin formation on our photo-crosslinked samples, even though our stiffness levels were ~4 times higher than Fan et al. HDLECs on viscoelastic HA hydrogels also formed a more extensive and branched lymphatic tube network compared to elastic (static) hydrogels with increased expression of characteristic lymphatic markers (LYVE-1, Prox1, podoplanin, VEGFR-3). Matrix metalloproteinases 1, 2, and 14, which are involved in ECM degradation and remodeling events necessary for tube formation, also increased on viscoelastic hydrogels. Interestingly, these are some of the same markers that others observed as being expressed at higher levels on softer substrates (30-200 Pa) [7,8]. Collectively, these observations and study results provide rationale for why viscoelasticity and temporal stiffening are important factors to consider beyond static stiffness when modeling dynamic processes like fibrosis. These differences may also be a contributing factor to why in vitro lymphatic capillaries do not match the dense, non-functional capillary beds seen in vivo in disease.
Even though the current work focuses on a new material approach to investigate the lymphatic response to ECM stiffness, we recognize other work within the field that is making progress toward improved understanding of the lymphatic system. Beyond our work and other work with natural materials that were previously discussed, Hooks et al. recently used a synthetic poly(ethylene glycol) (PEG) hydrogel functionalized with four maleimide groups (PEG-4MAL) and binding arginylglycylaspartic acid (RGD) ligands to observe the relationship between matrix elasticity (stiffness), ligand binding density, and degradability on lymphatic sprouting [37]. The study was designed to target one of the drawbacks of collagen, being that ligand density and stiffness profiles of gels are not independent when using collagen concentration to alter collagen stiffness. They were able to control matrix elasticity via PEG weight percentage without altering the RGD ligand density and generate matrices at 680 Pa with comparable ligand density to 2 mg/mL collagen at 20 Pa. Moreover, they showed successful lymphatic sprouting in vitro and functional grafting into host vasculature in vivo. Although they were able maintain ligand density at increased stiffness levels, methacrylated collagen behaves similarly by achieving multiple stiffness levels via photo-crosslinking at a single collagen concentration [20]. Methacrylated collagens are also capable of being integrated into lymphatic-on-a-chip models, which are popular models for studying lymphatic sprouting and growth [32,38,39,40,41,42]. Lymphatic capillaries sprout into ECM materials from LEC-lined channels, usually guided by a gradient of growth factors such as VEGF-C and sphingosine 1 phosphate [31]. Disease is currently a part of lymphatic-on-a-chip modeling; however, the focus tends to be on using soluble factors and co-cultures to recreate a disease environment. As with most lymphatic capillary models, ECM-based features of fibrosis have not been included in lymphatic-on-a-chip models. Therefore, there is an opportunity to merge the two approaches to advance the field of lymphatic modeling to include more robust disease models with altered stiffness and transport properties that impact LEC behavior.
4. Materials and methods

Table 1.
Materials, Cells, Software, Catalog Number, and Company.
| Materials (Cells and Cell Culture) | Catalog Number | Company |
|---|---|---|
| Human Dermal Lymphatic Endothelial Cells (HDLEC), Adult, Cyropreserved | #C-12217 | PromoCell |
| MV2 Media | #C-39221 | PromoCell |
| Materials (Reagents and Antibodies) | Catalog Number | Company |
| PhotoCol® Methacrylated Collagen | #5198-100MG | Advanced Biomatrix |
| LAP (405nm) | #5269 | Advanced Biomatrix |
| Irgacure 2959 (365nm) | #5200 | Advanced Biomatrix |
| Tuthenium (400-450nm) | #5248 | Advanced Biomatrix |
| Fibronectin Lyophilized (Human) | #5080 | Advanced Biomatrix |
| PBS, Phosphate Buffered Saline, 10X Solution | #BP3991 | Fisher Bioreagents |
| Fluorescein isothiocyanate–dextran, 40kDa | #FD40S-250MG | Sigma Aldrich |
| Fluorescein isothiocyanate–dextran, 60-76kDa | #FD70S-250MG | Sigma Aldrich |
| Pierce™ 16% Formaldehyde (w/v), Methanol-free | #28908 | ThermoFisher Scientific |
| Triton™ X-100 | T8787-50ML | Sigma Aldrigh |
| Alexa Fluor™ 488 Phalloidin | #A12379 | ThermoFisher Scientific |
| DAPI (4',6-diamidino-2-phenylindole, dihydrochloride) | #62247 | ThermoFisher Scientific |
| Bovine Albumin Fraction V (7.5% solution) | #15260037 | ThermoFisher Scientific |
| Rabbit Anti-Human VE-cadherin Polyclonal Antibody | #PA5-19612 | ThermoFisher Scientific |
| Rat Anti-Mouse LYVE1 Monoclonal Antibody (ALY7) | #MA5-32512 | ThermoFisher Scientific |
| Mouse Anti-Human VEGF Receptor 3 Monoclonal Antibody (4H4) | #MA5-15651 | ThermoFisher Scientific |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 647 | # A32795 | ThermoFisher Scientific |
| Materials (Disposables) | Catalog Number | Company |
| Nunc™ Lab-Tek™ II Chambered Coverglass | #155360 | ThermoFisher Scientific |
| Costar® 24-well Clear TC-treated Multiple Well Plates, Individually Wrapped, Sterile | #3524 | Corning |
| Falcon® Permeable Support for 24-well Plate with 8.0 µm Transparent PET Membrane, Sterile | #393097 | Corning |
| Corning® 96-well Flat Clear Bottom Black Polystyrene TC-treated Microplates, Individually Wrapped, with Lid, Sterile | #3603 | Corning |
| Fisherbrand™ Surface Treated Sterile Tissue Culture Flasks, Vented Cap | #FB012937 | Fisher Scientific |
| 96 Well glass bottom plate with high performance #1.5 cover glass | #P96-1.5H-N | Cellvis |
| Materials (Equipment and Software) | Catalog/Part Number | Company |
| MCR 302e WESP rheometer | #241353 | Anton Paar |
| UV Mounted LED | #M365LP1 | Thorlabs |
| UV Mounted LED | #M405LP1 | Thorlabs |
| Leica TCS SP5 Spectral Confocal Microscope | Leica Microsystems | |
| Victor Nivo 6T Multimode Plate Reader | #HH35000500 | Revvity Health Sciences |
| Keyence All-in-One Fluorescence Microscope | #BZ-X810 | KEYENCE Corp. of America |
| GraphPad Prism | GraphPad Software Inc. |
4.1. Preparation of Methacrylated Collagen Hydrogels (PhotoCol®)
Lyophilized methacrylated type I collagen (PhotoCol®, Advanced BioMatrix, Carlsbad, California) was solubilized on a rotator at 4°C in sterile 20 mM acetic acid to a final concentration of 8 mg/mL. Three photoinitiators were prepared according to manufacturer (Advanced BioMatrix) protocols: Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, 17 mg/mL stock volume), Irgacure 2959 (IRG, 10% stock), and Ruthenium (Ru)/Sodium Persulfate (SPS) (Ru at 37.4 mg/mL and SPS at 119mg/mL stock volume). Neutralization solution (Advanced BioMatrix) at 8% (v/v) (7.407 mg/mL) and photoinitiator at 2% (v/v) were mixed with PhotoCol® to make the final hydrogel mixture for each photoinitiator (LAP - 7.262 mg/mL; IRG - 7.334 mg/mL; and Ru/SPS - 7.122 mg/mL). Neutralized PhotoCol® hydrogels with photoinitiator were incubated at 37°C for a minimum of 30 minutes for hydrogel self-assembly prior to photo-crosslinking in subsequent studies.
4.2. Rheological Assessment of PhotoCol® Viscoelasticity
Matrix viscoelastic properties (shear storage modulus, G’; shear loss modulus, G”) were measured using a MCR 302e WESP rheometer (Anton Paar, Graaz, Austria) with a quartz stage to photo-crosslink PhotoCol® hydrogel samples (365-405 nm) during testing. Frequency and strain sweeps were conducted in oscillatory shear on fully crosslinked and un-crosslinked hydrogels to determine the linear viscoelastic region (data not shown). Temporal changes in stiffness in response to photo-crosslinking were conducted in time sweeps (constant: 0.1% strain, 1 hz). Hydrogel samples were allowed to self-assemble for 10 minutes at 37°C before exposure to one of two light exposure conditions using 365 or 405 nm UV mounted LED (Thorlabs, Newton, NJ) —constant and burst expsoure. Constant light was applied for 20 minutes after self-assembly for complete photo-crosslinking and maximum stiffness (G’). Burst light exposures consisted of a single light pulse of 30-, 60-, or 90-seconds after self-assembly with continued measurement for 10 minutes after light exposure.
Using MATLAB (Mathworks, Natick, MA), a locally weighted regression was used on each replicate to create a fitted curve that could be analyzed. The first derivate was taken of these regressions to determine rate of change in stiffness. A threshold of 3 Pa/s rate was used to determine the regions of plateau and regions of stiffening. Average stiffness values of the plateau regions before and after light exposure, within the regions of stiffening, were used to calculate fold change by the ratio of each replicate’s post-stiffening average to the same replicate’s pre-stiffening average. The post-stiffening averages of each replicate were then averaged to give each group’s average maximum stiffness. The average number of seconds the rate of change of stiffening was above the 3 Pa/s threshold was used to calculate the amount of time to stiffness plateau.
4.3. Qualitative Microstructural Assessment of PhotoCol® Hydrogels
Confocal laser scanning microscopy in reflectance mode was used to obtain images of fibrillar collagen microstructure for qualitative assessment. All samples were prepared in Nunc™ Lab-Tek™ II Chambered Coverglass #1.5 slides (ThermoFisher Scientific, Waltham, Massachusetts). Briefly, PhotoCol® hydrogels with Ru/SPS were neutralized and allowed to self-assemble for 30 minutes at 37°C as previously described. Photo-crosslinked samples were exposed to 405 nm light for 90-seconds (longest burst exposure) or 5 minutes (sufficient time to reach maximum G’) using a 405 nm UV mounted LED (Thorlabs). Images were collected with a Leica TCS SP5 Spectral Confocal Microscope (Leica Microsystems, Wetzlar, Germany) using a 40X oil immersion objective (2D scan).
4.4. Permeability Assessment of PhotoCol® Hydrogels
A transwell membrane insert system protocol adapted from Hsu et al. [26] was used to characterize permeability of PhotoCol® hydrogels with Ru/SPS photoinitiator. Neutralized PhotoCol® (60 µL; 7.122mg/mL) was polymerized for 60 min at 37°C on a 8.0 µm PET membrane 24-well transwell insert (Corning, Corning, New York) within a tissue-cultured treated 24-well plate (Corning). For photo-crosslinking, transwell inserts containing PhotoCol® were removed and exposed to 405 nm near UV light for 30- or 90-seconds. Lyophilized human fibronectin (Advanced BioMatrix) was reconstituted in Milli-Q water and diluted in 1X PBS (Fisher Bioreagents, Waltham, Massachusetts) to achieve 5 ug/cm2 (0.03 mg/mL) on the PhotoCol® surface. Samples were then incubated at 37°C for 1 hour to establish a fibronectin coating and submerged in 1X PBS (overnight at 37°C) to remove any excess Ru/SPS from the PhotoCol® gels. A 2 mg/mL solution of fluorescein isothiocyanate (FITC)-labeled dextran at 40 kDa or 60-76 kDa (Sigma Aldrich, St. Louis, Montanna) was added on top of the fibronectin-coated PhotoCol® samples within the inserts as the donor solution, while 1X PBS was pipetted into the outer well to serve as the acceptor solution. Solution volumes were controlled to be at equal heights to minimize flow due to hydrostatic pressure. PhotoCol® with FITC-dextran was incubated at 37°C, and 20 µL was removed from the acceptor solution at one-hour increments for a total of 3-5 hours. The sampled solution was then diluted at 1:25 in 1X PBS and transferred to a black walled polystyrene 96-well plate (Corning) to be read on a PerkinElmer VICTOR Nivo plate reader (Revvity, Waltham, Massachusetts) (480 nm excitation/530 nm emission). A standard curve (0-0.0125 mg/mL of FITC-dextran) was used to calculate the concentration of measured samples. To calculate the permeability coefficient, Fick’s second law was used using the dimensions of the PhotoCol® and the change in concentration of the acceptor solution in the linear region of permeability.
4.5. Lymphatic Endothelial Cell Culture
Human Dermal Lymphatic Endothelial Cells (HDLEC) isolated from adult skin (PromoCell, Heidelberg, Germany) were maintained according to manufacturer instructions in endothelial growth media with MV2 growth supplements (PromoCell): Fetal calf serum (5% v/v), Epidermal growth factor (recombinant human, 5 ng/mL), Basis fibroblast growth factor (recombinant human, 10 ng/mL), Insulin-like growth factor (Long R3 IGF, recombinant human, 20 ng/mL), Vascular endothelial growth factor 165 (recombinant human, 0.5 ng/mL), Ascorbic acid (1 µg/mL), Hydrocortisone (0.2 µg/mL) (PromoCell). All cell culture surfaces were coated with human fibronectin (3.5 µg/cm2; Advanced BioMatrix) prior to HDLEC seeding to promote cell attachment. Cells were passaged and maintained in Fisherbrand™ Surface Treated Sterile Tissue Culture Flasks, Vented Cap (Fisher Scientific, Waltham, Massachusetts) until ready for experimental use. Cells were maintained at 37°C in a humidified incubator (5% CO2) and passaged at 70–90% confluency. All cells were used between passages 6 and 12 for experiments.
4.6. Morphological Assessment of Lymphatic Endothelial Cell Stiffness Response
HDLECs were characterized on their morphological response to hydrogels at different stiffness levels (low and high G'). Cells were seeded in 15 well µ-slide (Ibidi, Gräfelfing, Germany) at 10,000 cells/cm2 on top of fibronectin-coated PhotoCol® prepared with Ru/SPS (photo-crosslinking, 90 seconds of light exposure high G') and without photoinitiator (uncrosslinked, no light exposure, low G') for a total culture time of 3 days (37°C, 5% CO2). To evaluate any impact of light exposure, HDLECs were also seeded on fibronectin-coated 96 well glass bottom plate with high performance #1.5 cover glass plates (Cellvis, Mountain View, California) and exposed to the 90-seconds of light (not shown). Additional HDLECs on coated wells with no light exposure served as 2D controls (not shown). All groups were fixed with 4% paraformaldehyde (ThermoFisher Scientific) in 1X PBS for 15 minutes at 37°C and permeabilized using 0.1% of Triton™ X-100 (Sigma Aldrich) diluted in 1X PBS. Samples were stained with Alexa Fluor™ 488 Phalloidin (1:400, ThermoFisher Scientific) to visualize F-actin and counterstained with 4′,6-diamidino-2-phenylindole (300 nM DAPI; ThermoFisher Scientific) to visualize the nuclei. For immunostaining of characteristic HDLEC markers, fixed samples were blocked with bovine albumin fraction V (7.5% solution; ThermoFisher Scientific) diluted in 1X PBS to 1% and incubated with primary antibodies for VE-Cadherin (1:1000 ThermoFisher Scientific). Samples were then rinsed and incubated with Alexa Fluor™ Plus 647-conjugated secondary antibody donkey anti-rabbit IgG (H+L) (1:200, ThermoFisher Scientific). All samples were imaged with a Keyence BZX810 All-in-One Fluorescence Microscope (KEYENCE Corp. of America, Itasca, IL). Images were collected at 20X magnification for cellular and nuclear area, cellular and nuclear eccentricity, cellular form factor, and VE-Cadherin thickness, utilizing the Keyence BZ-H4A analysis software to create full focus images from z-stacks. The threshold of these images was obtained utilizing CellProfiler™, removing signal from cell nuclei and noise before obtaining the threshold image of just the VE-Cadherin cellular outline. Primary (nuclei) and secondary objects (cell body) were obtained and quantified using CellProfiler™ as well. Threshold images were then quantified using distance map and Vessel Analysis plugins. Due to the visual similarity between threshold images of vasculature and threshold images of cell boundaries, the Vessel Analysis plugin was able to accurately obtain average vessel diameters of four random regions of interest in each image (n=4, N=3).
4.7. Statistical Analysis
Statistical analysis was carried out using GraphPad Prism 10.0 (GraphPad Software Inc., La Jolla, CA). Stiffness data was expressed as mean storage modulus + standard deviation (SD) without removal of outliers. Permeability data was expressed as a mean ± SD after outliers were removed using Grubbs test (α = 0.05). Morphological values were expressed as mean ± SD without removal of outliers. Normality was verified through a Shapiro-Wilk normality test using α = 0.05. Multiple comparisons were performed using one-way or a two-way analysis of variance (ANOVA), and parametric data were analyzed using the Tukey post hoc method (significance: *p<0.05, **p<0.001, ***p<0.0001, ****p<0.00001). Nonparametric data were analyzed with the Mann-Whitney U test (significance: (**p<0.005 and ****p<0.0001).
5. Conclusions
The progressive nature of fibrosis includes dynamic changes in the biophysical and biochemical cues provided to the affected cells. Lymphatic vascular growth and function is heavily affected during the progression of fibrosis and the related stiffening of the surrounding tissue. To prepare an in vitro model that best recapitulates fibrotic stiffening, we characterized the stiffening profiles of methacrylated type I collagen (PhotoCol®) with photo-crosslinking and identified Ru/SPS as the photoinitiator that produced the highest maximum storage modulus and greatest dynamic range of stiffness values that approximate normal to fibrotic tissue stiffness. As others have found, we confirmed that HDLECs are responsive to changes in ECM stiffness. The observed differences in cellular and nuclear area and irregularity, combined with VE-Cadherin thickness measurements show elements of LEC maturation that relate to growth and function. Moreover, they offer more robust morphological characterization for HDLECs that we and others can leverage in future mechanism-based studies to understand specific drivers of stiffness-induced LEC changes. Overall, our focus on considerably higher ECM stiffness values places our work within the disease modeling space rather than lymphatic development. Our findings also establish methacrylated type I collagen as a tunable, natural material that demonstrates the capability to be used for lymphatic disease modeling that will improve our understanding of the biophysical and biochemical effects of fibrosis on lymphatic vasculature.
Author Contributions
Conceptualization, B.N.K.R. and C.F.W.; methodology, B.N.K.R., S.L., D.L.H., E.L.X.; software, B.N.K.R., D.L.H.; validation, B.N.K.R. and C.F.W; formal analysis, B.N.K.R., S.L.; resources, C.F.W.; writing—original draft preparation, B.N.K.R., S.L., C.F.W.; writing—review and editing, B.N.K.R., S.L. C.F.W; visualization, B.N.K.R., S.L., D.L.H.; supervision, C.F.W.; project administration, C.F.W.; funding acquisition, C.F.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Science Foundation Engineering Research Initiation award (2138841 to C.F. Whittington), National Institutes of Health/National Cancer Institute R03 Small Grants Award (5R03CA267449-02 to C.F. Whittington), and the Genentech Research Award from Genentech, a member of the Roche Group (Subaward University of Michigan; awarded to C.F. Whittington).
References
- Petrova, T.V.; Koh, G.Y. Organ-specific lymphatic vasculature: From development to pathophysiology. J Exp Med 2018, 215, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Avraham, T.; Daluvoy, S.; Zampell, J.; Yan, A.; Haviv, Y.S.; Rockson, S.G.; Mehrara, B.J. Blockade of transforming growth factor-β1 accelerates lymphatic regeneration during wound repair. Am J Pathol 2010, 177, 3202–3214. [Google Scholar] [CrossRef] [PubMed]
- Avraham, T.; Zampell, J.C.; Yan, A.; Elhadad, S.; Weitman, E.S.; Rockson, S.G.; Bromberg, J.; Mehrara, B.J. Th2 differentiation is necessary for soft tissue fibrosis and lymphatic dysfunction resulting from lymphedema. FASEB J 2013, 27, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Ogata, F.; Fujiu, K.; Matsumoto, S.; Nakayama, Y.; Shibata, M.; Oike, Y.; Koshima, I.; Watabe, T.; Nagai, R.; Manabe, I. Excess Lymphangiogenesis Cooperatively Induced by Macrophages and CD4+ T Cells Drives the Pathogenesis of Lymphedema. J Invest Dermatol 2016, 136, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Avraham, T.; Yan, A.; Zampell, J.C.; Daluvoy S, V.; Haimovitz-Friedman, A.; Cordeiro, A.P.; Mehrara, B.J. Radiation therapy causes loss of dermal lymphatic vessels and interferes with lymphatic function by TGF-β1-mediated tissue fibrosis. Am J Physiol - Cell Physiol 2010, 299, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Ruliffson, B.N.K.; Whittington, C.F. Regulating Lymphatic Vasculature in Fibrosis: Understanding the Biology to Improve the Modeling. Adv Biol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Taddei, A.; Dierkes, C.; et al. Matrix stiffness controls lymphatic vessel formation through regulation of a GATA2-dependent transcriptional program. Nat Commun 2018, 9, 1511. [Google Scholar] [CrossRef] [PubMed]
- Alderfer, L.; Russo, E.; Archilla, A.; Coe, B.; Hanjaya-Putra, D. Matrix stiffness primes lymphatic tube formation directed by vascular endothelial growth factor-C. FASEB J 2021, 35, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Baruffaldi, D.; Palmara, G.; Pirri, C.; Frascella, F. 3D Cell Culture: Recent Development in Materials with Tunable Stiffness. ACS Appl Bio Mater 2021, 4, 2233–2250. [Google Scholar] [CrossRef]
- AhmadianKia, N.; Goli-Malekabadi, Z.; Pournaghmeh, S. Application of cell laden hydrogels with temporally tunable stiffness in biomedical research. J Biomater Appl 2023, 38, 179–193. [Google Scholar] [CrossRef]
- Detry, B.; Erpicum, C.; Paupert, J.; et al. Matrix metalloproteinase-2 governs lymphatic vessel formation as an interstitial collagenase. Blood 2012, 119, 5048–5056. [Google Scholar] [CrossRef]
- Saha, S.; Fan, F.; Alderfer, L.; Graham, F.; Hall, E.; Hanjaya-Putra, D. Synthetic hyaluronic acid coating preserves the phenotypes of lymphatic endothelial cells. Biomater Sci 2023, 11, 7346–7357. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Su, B.; Kolodychak, A.; Ekwueme, E.; Alderfer, L.; Saha, S.; Webber, M.J.; Hanjaya-Putra, D. Hyaluronic Acid Hydrogels with Phototunable Supramolecular Cross-Linking for Spatially Controlled Lymphatic Tube Formation. Cite This ACS Appl Mater Interfaces 2023, 15, 58195. [Google Scholar] [CrossRef] [PubMed]
- Rubiano, A.; Delitto, D.; Han, S.; Gerber, M.; Galitz, C.; Trevino, J.; Thomas, R.M.; Hughes, S.J.; Simmons, C.S. Viscoelastic properties of human pancreatic tumors and in vitro constructs to mimic mechanical properties. Acta Biomater 2018, 67, 331–340. [Google Scholar] [CrossRef]
- Pehrsson, M.; Mortensen, J.H.; Manon-Jensen, T.; Bay-Jensen, A.C.; Karsdal, M.A.; Davies, M.J. Enzymatic cross-linking of collagens in organ fibrosis – resolution and assessment. Expert Rev Mol Diagn 2021, 21, 1049–1064. [Google Scholar] [CrossRef]
- Danussi, C.; Spessotto, P.; Petrucco, A.; Wassermann, B.; Sabatelli, P.; Montesi, M.; Doliana, R.; Bressan, G.M.; Colombatti, A. Emilin1 Deficiency Causes Structural and Functional Defects of Lymphatic Vasculature. Mol Cell Biol 2008, 28, 4026–4039. [Google Scholar] [CrossRef]
- Wiig, H.; Keskin, D.; Kalluri, R. Interaction between the extracellular matrix and lymphatics: Consequences for lymphangiogenesis and lymphatic function. Matrix Biol 2010, 29, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Alexander, J.S.; Orr, A.W. Integrins and Their Extracellular Matrix Ligands in Lymphangiogenesis and Lymph Node Metastasis. Int J Cell Biol 2012, 2012, 1–12. [Google Scholar] [CrossRef]
- Amar, S.; Smith, L.; Fields, G.B. Matrix metalloproteinase collagenolysis in health and disease. Biochim Biophys Acta - Mol Cell Res 2017, 1864, 1940–1951. [Google Scholar] [CrossRef]
- Gaudet, I.D.; Shreiber, D.I. Characterization of methacrylated Type-I collagen as a dynamic, photoactive hydrogel. Biointerphases 2012, 7, 1–9. [Google Scholar] [CrossRef]
- Brinkman, W.T.; Nagapudi, K.; Thomas, B.S.; Chaikof, E.L. (2003) Photo-Cross-Linking of Type I Collagen Gels in the Presence of Smooth Muscle Cells: Mechanical Properties, Cell Viability, and Function. [CrossRef]
- Tabatabaei, F.; Moharamzadeh, K.; Tayebi, L. Fibroblast encapsulation in gelatin methacryloyl (GelMA) versus collagen hydrogel as substrates for oral mucosa tissue engineering. J Oral Biol Craniofacial Res 2020, 10, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Li, Y.; Xu, K.; Yin, J. Advantages of photo-curable collagen-based cell-laden bioinks compared to methacrylated gelatin (GelMA) in digital light processing (DLP) and extrusion bioprinting. Mater Today Bio 2023, 23, 100799. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.M.; Patrawalla, N.Y.; Kajave, N.S.; Brown, A.B.; Kishore, V. Species-Based Differences in Mechanical Properties, Cytocompatibility, and Printability of Methacrylated Collagen Hydrogels. Biomacromolecules 2022, 23, 5137–5147. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Wei, S.Y.; Chen, W.L.; Hsu, T.L.; Chen, Y.C. (2023) Reconstructing vascular networks promotes the repair of skeletal muscle following volumetric muscle loss by pre-vascularized tissue constructs. J Tissue Eng. [CrossRef]
- Hsu, H.H.; Kracht, J.K.; Harder, L.E.; Rudnik, K.; Lindner, G.; Schimek, K.; Marx, U.; Pörtner, R. (2018) A Method for Determination and Simulation of Permeability and Diffusion in a 3D Tissue Model in a Membrane Insert System for Multi-well Plates. J Vis Exp. [CrossRef]
- Baluk, P.; Fuxe, J.; Hashizume, H.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med 2007, 204, 2349–2362. [Google Scholar] [CrossRef]
- Baluk, P.; McDonald, D.M. Buttons and Zippers: Endothelial Junctions in Lymphatic Vessels. Cold Spring Harb Perspect Med 2022, 12, a041178. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.R.; Brugge, J.S. A Stiff Blow from the Stroma: Collagen Crosslinking Drives Tumor Progression. Cancer Cell 2009, 16, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Samani, A.; Zubovits, J.; Plewes, D. Elastic moduli of normal and pathological human breast tissues: An inversion-technique-based investigation of 169 samples. Phys Med Biol 2007, 52, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.-C.; Yeh, Y.-T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.-L.; Li, Y.-S.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc Natl Acad Sci 2016, 113, 11525–11530. [Google Scholar] [CrossRef]
- Akbari, E.; Spychalski, G.B.; Rangharajan, K.K.; Prakash, S.; Song, J.W. Flow dynamics control endothelial permeability in a microfluidic vessel bifurcation model. Lab Chip 2018, 18, 1084–1093. [Google Scholar] [CrossRef]
- Choi, D.; Park, E.; Jung, E.; et al. ORAI1 Activates Proliferation of Lymphatic Endothelial Cells in Response to Laminar Flow Through Krüppel-Like Factors 2 and 4. Circ Res 2017, 120, 1426–1439. [Google Scholar] [CrossRef]
- Cho, H.; Kim, J.; Ahn, J.H.; Hong, Y.K.; Mäkinen, T.; Lim, D.S.; Koh, G.Y. YAP and TAZ Negatively Regulate Prox1 during Developmental and Pathologic Lymphangiogenesis. Circ Res 2019, 124, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zaw, A.M.; Anderson, D.E.J.; Jeong, Y.; Kunihiro, J.; Hinds, M.T.; Yim, E.K.F. Fucoidan and topography modification improved in situ endothelialization on acellular synthetic vascular grafts. Bioact Mater 2023, 22, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Abu Taha, A.; Schnittler, H.-J. Dynamics between actin and the VE-cadherin/catenin complex. Cell Adh Migr 2014, 8, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Hooks, J.S.T.; Bernard, F.C.; Cruz-Acuña, R.; Nepiyushchikh, Z.; Gonzalez-Vargas, Y.; García, A.J.; Dixon, J.B. Synthetic hydrogels engineered to promote collecting lymphatic vessel sprouting. Biomaterials 2022, 284, 121483. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chung, M.; Jeon, N.L. Three-dimensional biomimetic model to reconstitute sprouting lymphangiogenesis in vitro. Biomaterials 2016, 78, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, N.; Robering, J.W.; Kengelbach-Weigand, A.; Al-Abboodi, M.; Beier, J.P.; Horch, R.E.; Boos, A.M. Human adipose-derived stem cells support lymphangiogenesis in vitro by secretion of lymphangiogenic factors. Exp Cell Res 2020, 388, 111816. [Google Scholar] [CrossRef] [PubMed]
- Song, J.W.; Munn, L.L. Fluid forces control endothelial sprouting. Proc Natl Acad Sci 2011, 108, 15342–15347. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.R.; Ilan, I.S.; Lee, E. A bioengineered lymphatic vessel model for studying lymphatic endothelial cell-cell junction and barrier function. Microcirculation 2021, 28, e12730. [Google Scholar] [CrossRef]
- Henderson, A.R.; Choi, H.; Lee, E. Blood and Lymphatic Vasculatures On-Chip Platforms and Their Applications for Organ-Specific In Vitro Modeling. Micromachines 2020, 11, 147. [Google Scholar] [CrossRef]
Figure 1.
Viscoelastic properties of PhotoCol® with photoinitiators and constant light exposure. (A) Shear storage modulus (G') and shear loss modulus (G") versus time for PhotoCol® combined with one of three photoinitiators: IRG, LAP, and Ru/SPS. Samples self-assembled for 600 seconds (10 minutes) prior to photo-crosslinking with 365-405 nm light for 20 minutes (constant light exposure). Curves represent G’ and G” (mean± SD; N=3-4) (B) Maximum G’ calculated from G’ vs time curves for PhotoCol® samples with photoinitiators. Bars represent mean+SD (N=3-4). (C) Calculated fold-change (mean+SD) before and after photo-crosslinking for PhotoCol® samples with photoinitiators. Significance for B and C was determined with a one-way ANOVA and a Tukey's test post-hoc (ns = no significance, *p<0.05, **p<0.001, ***p<0.0001, ****p<0.00001).
Figure 1.
Viscoelastic properties of PhotoCol® with photoinitiators and constant light exposure. (A) Shear storage modulus (G') and shear loss modulus (G") versus time for PhotoCol® combined with one of three photoinitiators: IRG, LAP, and Ru/SPS. Samples self-assembled for 600 seconds (10 minutes) prior to photo-crosslinking with 365-405 nm light for 20 minutes (constant light exposure). Curves represent G’ and G” (mean± SD; N=3-4) (B) Maximum G’ calculated from G’ vs time curves for PhotoCol® samples with photoinitiators. Bars represent mean+SD (N=3-4). (C) Calculated fold-change (mean+SD) before and after photo-crosslinking for PhotoCol® samples with photoinitiators. Significance for B and C was determined with a one-way ANOVA and a Tukey's test post-hoc (ns = no significance, *p<0.05, **p<0.001, ***p<0.0001, ****p<0.00001).
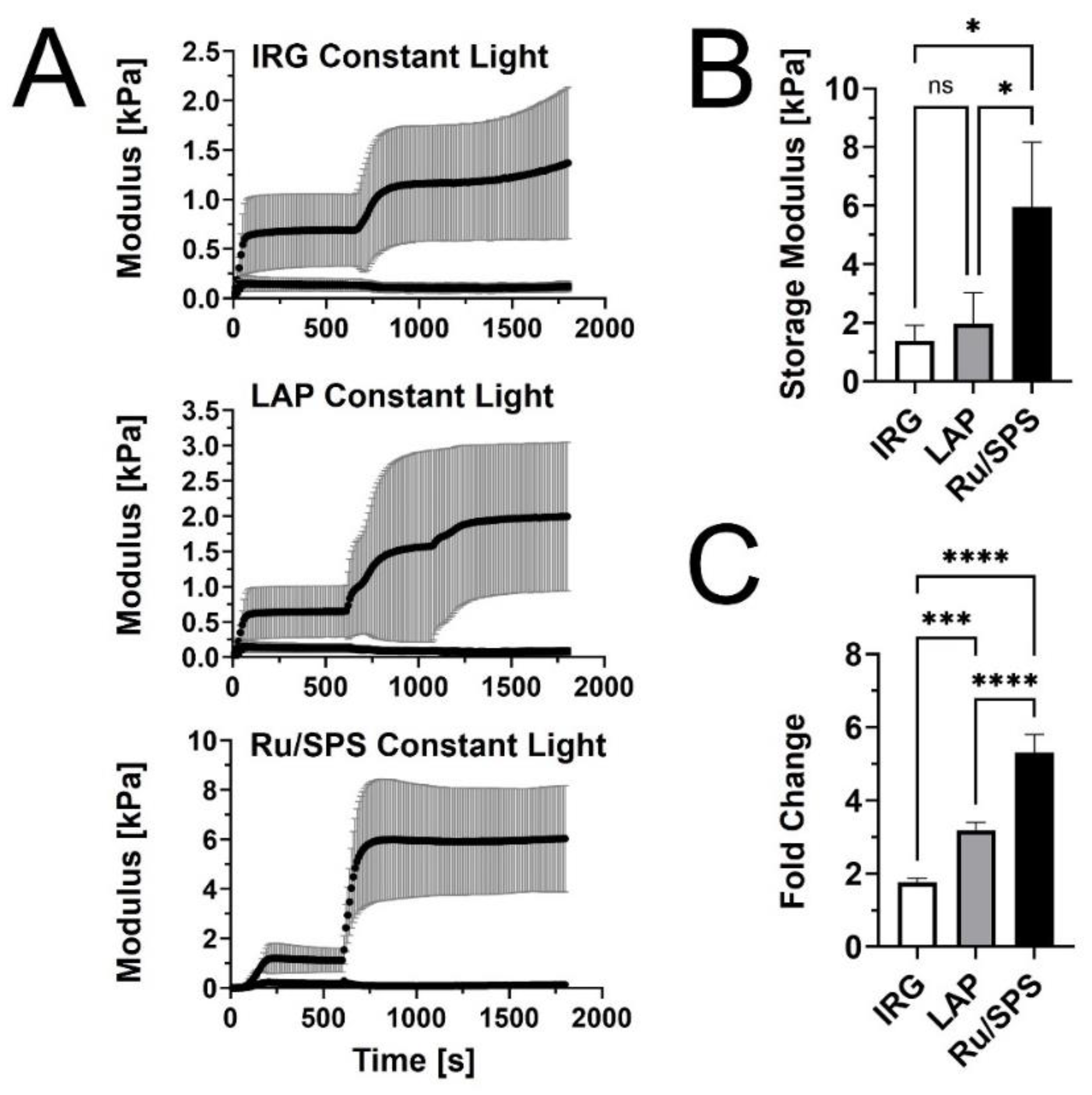
Figure 2.
Viscoelastic response and microstructure of PhotoCol® with Ru/SPS and burst light exposure. (A) Representative curves for shear storage modulus (G’) versus time for PhotoCol® with Ru/SPS with burst light exposure (405 nm; 30s, 60s, and 90s) after 10 minutes of self-assembly. (B) Calculated fold-change + SD before and after photo-crosslinking PhotoCol® samples with Ru/SPS (ns = no significance) (C) Confocal reflection microscopy images for uncrosslinked PhotoCol® (no light exposure) and photo-crosslinked PhotoCol® (90 seconds and 5 minutes) (white fibrils = collagen; Scale bar = 150 µm).
Figure 2.
Viscoelastic response and microstructure of PhotoCol® with Ru/SPS and burst light exposure. (A) Representative curves for shear storage modulus (G’) versus time for PhotoCol® with Ru/SPS with burst light exposure (405 nm; 30s, 60s, and 90s) after 10 minutes of self-assembly. (B) Calculated fold-change + SD before and after photo-crosslinking PhotoCol® samples with Ru/SPS (ns = no significance) (C) Confocal reflection microscopy images for uncrosslinked PhotoCol® (no light exposure) and photo-crosslinked PhotoCol® (90 seconds and 5 minutes) (white fibrils = collagen; Scale bar = 150 µm).
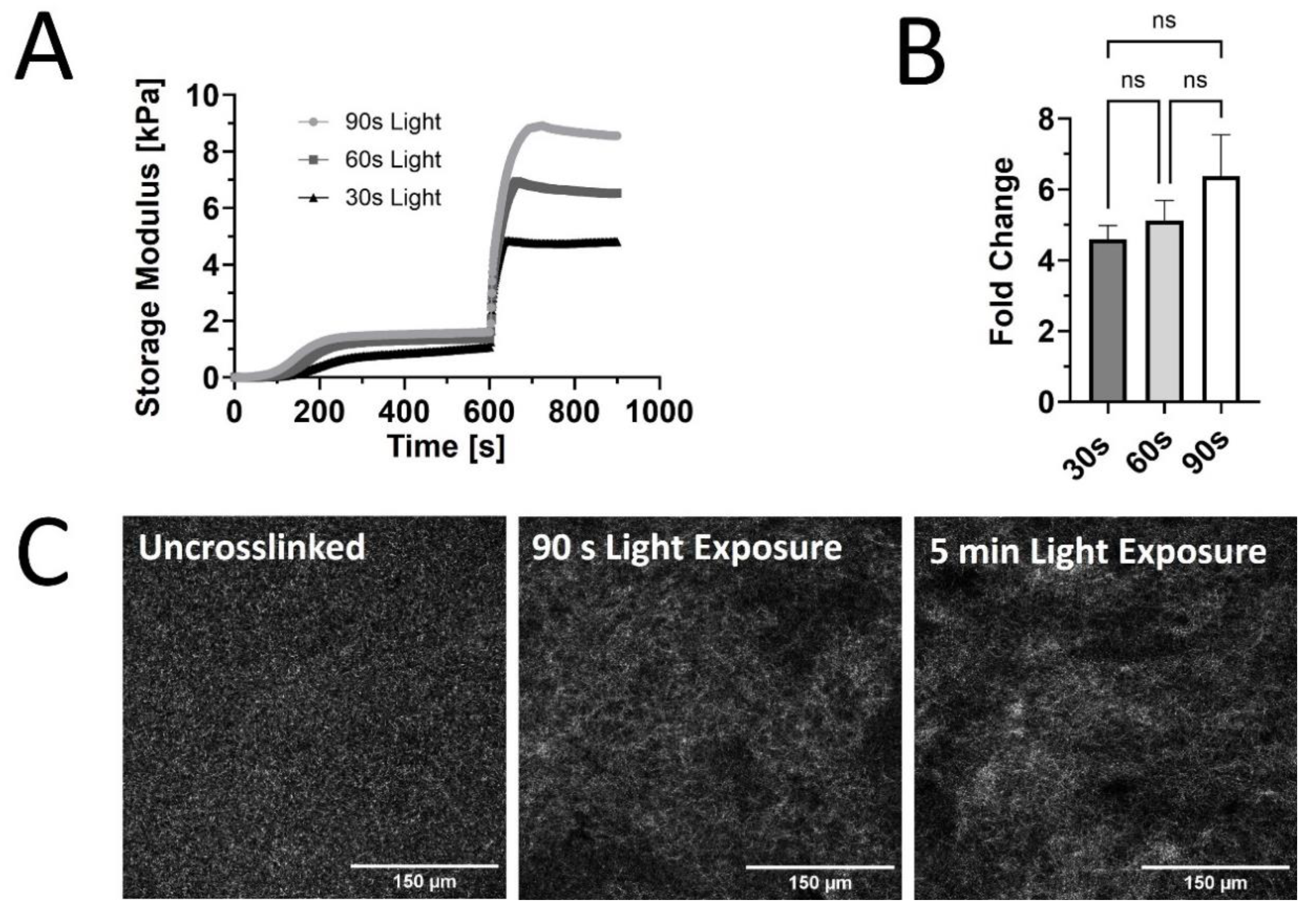
Figure 3.
Permeability coefficients of PhotoCol® with FITC-labeled dextran. Permeability coefficients were measured for PhotoCol® with Ru/SPS and burst light exposure (0 sec – uncrosslinked; 30 sec and 90 sec – photo-crosslinked). Bars represent mean+SD. Significance was determined with ANOVA was used to compare differences between photo-crosslinking duration and dextran size (***p< 0.001, N=3-4) and an unpaired student t-test was used to compare differences between the two dextran sizes at each light duration.
Figure 3.
Permeability coefficients of PhotoCol® with FITC-labeled dextran. Permeability coefficients were measured for PhotoCol® with Ru/SPS and burst light exposure (0 sec – uncrosslinked; 30 sec and 90 sec – photo-crosslinked). Bars represent mean+SD. Significance was determined with ANOVA was used to compare differences between photo-crosslinking duration and dextran size (***p< 0.001, N=3-4) and an unpaired student t-test was used to compare differences between the two dextran sizes at each light duration.
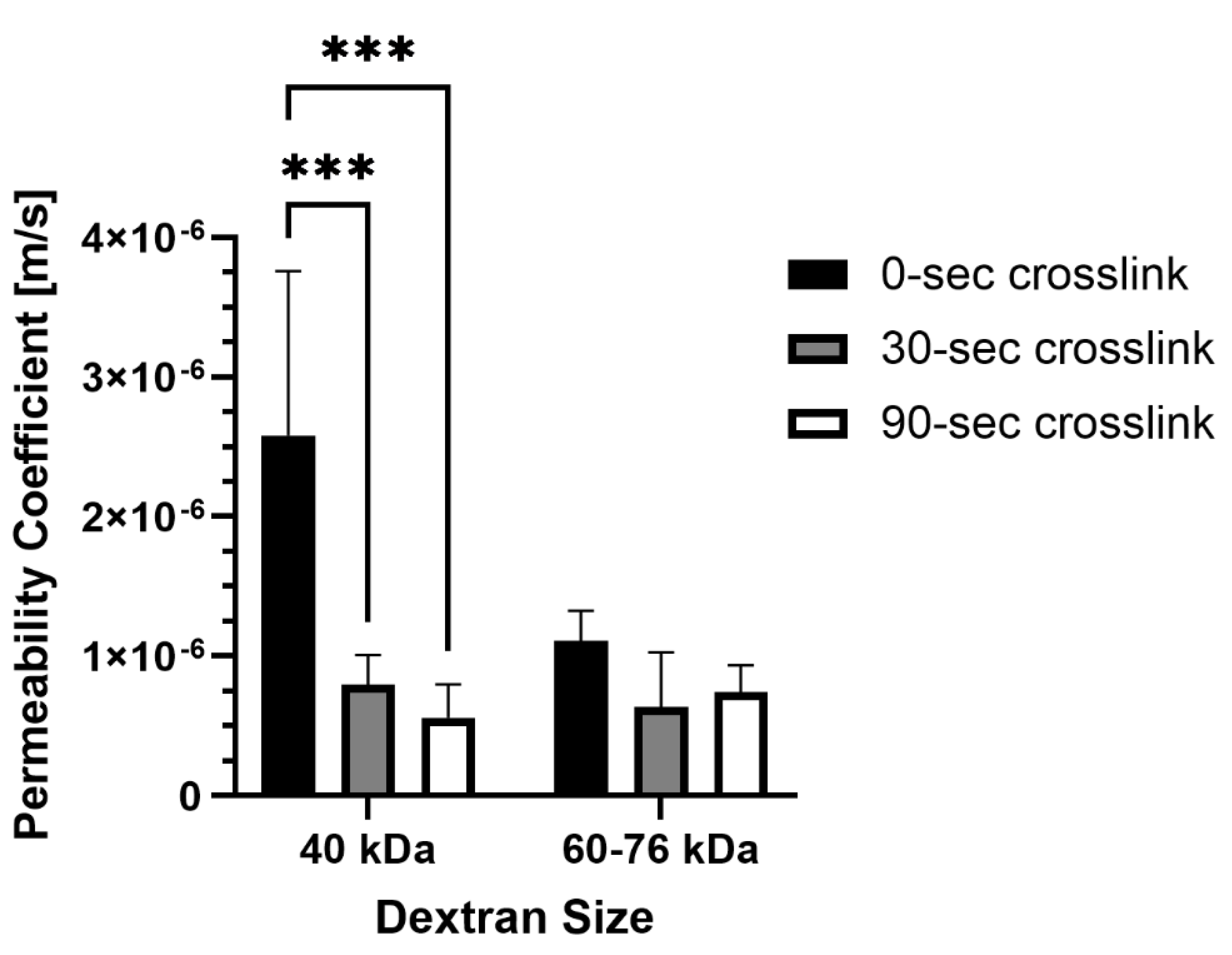
Figure 4.
Qualitative morphological assessment of HDLEC on PhotoCol®. Representative images of human dermal lymphatic endothelial cells (HDLEC) seeded on (A) uncrosslinked (soft, 0.5 kPa) and (B) photo-crosslinked (90 seconds light exposure; stiff, 6 kPa) PhotoCol® with Ru/SPS (fibronectin coated). Samples were stained to visualize F-actin (green) and VE-Cadherin (purple) with a DAPI nuclear counterstain (blue). Images were collected on the Keyence BZ-X series All-in-One Fluorescence Microscope (full focus z-stacks). Scale bar = 150 µm.
Figure 4.
Qualitative morphological assessment of HDLEC on PhotoCol®. Representative images of human dermal lymphatic endothelial cells (HDLEC) seeded on (A) uncrosslinked (soft, 0.5 kPa) and (B) photo-crosslinked (90 seconds light exposure; stiff, 6 kPa) PhotoCol® with Ru/SPS (fibronectin coated). Samples were stained to visualize F-actin (green) and VE-Cadherin (purple) with a DAPI nuclear counterstain (blue). Images were collected on the Keyence BZ-X series All-in-One Fluorescence Microscope (full focus z-stacks). Scale bar = 150 µm.
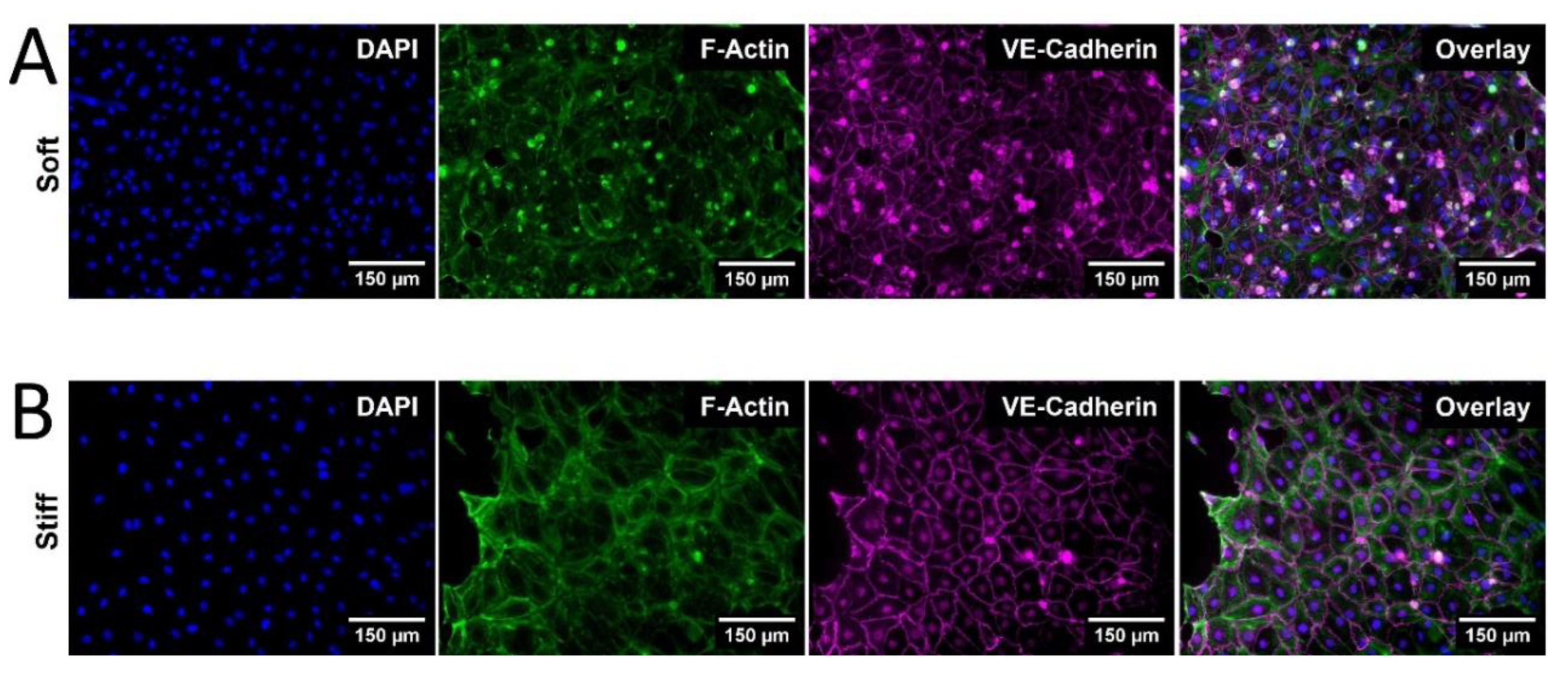
Figure 5.
Qualitative morphological assessment of HDLEC on PhotoCol®. (A) Cell shape metrics: Surface area, Perimeter, Form Factor, and Eccentricity and (B) Nuclear shape metrics: Surface area, Eccentricity for HDLECs on uncrosslinked (0.5 kPa) and photo-crosslinked (6 kPa) PhotoCol® were calculated using CellProfiler™. Bars represent mean+SD. (C) VE-Cadherin thickness (represented by width) for HDLECs on soft and stiff PhotoCol®. Images show steps to thickness quantification: original image, thresholded image (Cell Profiler™), and distance map for quantification (ImageJ Vessel Analysis plug-in). Box and whisker plots represent minimum, maximum, and median widths across upper and lower quartiles (n = 300–700 cells across 3 images per group). Metrics were compared with Mann-Whitney u test (**p<0.005 and ****p<0.0001).
Figure 5.
Qualitative morphological assessment of HDLEC on PhotoCol®. (A) Cell shape metrics: Surface area, Perimeter, Form Factor, and Eccentricity and (B) Nuclear shape metrics: Surface area, Eccentricity for HDLECs on uncrosslinked (0.5 kPa) and photo-crosslinked (6 kPa) PhotoCol® were calculated using CellProfiler™. Bars represent mean+SD. (C) VE-Cadherin thickness (represented by width) for HDLECs on soft and stiff PhotoCol®. Images show steps to thickness quantification: original image, thresholded image (Cell Profiler™), and distance map for quantification (ImageJ Vessel Analysis plug-in). Box and whisker plots represent minimum, maximum, and median widths across upper and lower quartiles (n = 300–700 cells across 3 images per group). Metrics were compared with Mann-Whitney u test (**p<0.005 and ****p<0.0001).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



