
Submitted:
11 June 2024
Posted:
12 June 2024
You are already at the latest version
Abstract
The most common type of head and neck cancer (HNC) is head and neck squamous cell carcinoma (HNSCC). It describes a heterogenous neoplastic disease that arises from the mucosal epithelium of the of the upper respiratory system and the gastrointestinal tract. It is characterized by high morbidity and mortality, being is the eighth most common cancer worldwide, with 931,931 newly diagnosed cases and 467,125 deaths according to the latest Global Cancer Statistics (GLOBOCAN) data. Despite the use of modern low-invasive surgical techniques and application of promising new chemoradiotherapies or combination treatment, the frequency of recurrences and the five-year survival of these patients remains unsatisfactory. The microenvironment of head and neck cancer comprises various cells that regulate neoplastic expansion. It is believed that the mesenchymal stromal cells (MSCs) present in the tumour milieu play a key role in the modulation of tumour initiation, development and progression, in the regulation of cancer treatment response, survival rates, patient outcomes and long-term prognosis; they also influence the resistance to cisplatin-based chemotherapy, the gold standard for advanced HNSCC. MSCs, are multipotent, heterogeneous, mobile cells, which meet specific stemness criteria defined by the International Society for Cellular Therapy (ISCT). Although no MSC-specific markers exist, they can be recognized based on several others, such as CD73, CD90 and CD105, while lacking the presence of CD45, CD34, CD14 or CD11b, CD79α, or CD19 and HLA-DR antigens; they share phenotypic similarity with stromal cells and their capacity to differentiate into other cell types. In the tumour niche, MSC populations are characterized by cell quiescence, self-renewal capacity, low reactive oxygen species production and the acquisition of epithelial-to-mesenchymal transition properties, they may play a key role in the process of acquiring drug resistance, and thus treatment failure. MSCs must also demonstrate plastic adherence in vitro and be able to differentiate into specific mesodermal cell types after stimulation i.e., adipocytes, chondrocytes, and osteoblasts when cultured under well-defined conditions. The present narrative review examines the links between mesenchymal stromal/stem cells and HNC, as well as the different mechanisms involved in the development of resistance to current chemo- and radiotherapies in HNSCC. It provides an overview of the current literature, including key opinion-forming systematic reviews, as well as cross-sectional, longitudinal, prospective and interventional studies based on cell culture models in vitro, animal or in vivo models of HNSCC; it also examines the possibilities of pharmacological targeting of stemness-related chemoresistance in HNSCC. It describes promising new strategies to optimize chemoradiotherapy with the potential to personalize patient treatment approaches, and highlights future therapeutic perspectives in HNC. The data concerning tumour-associated MSCs is arranged according to increasing clinical credibility.
Keywords:
anti-cancer therapy
; carcinogenesis
; cell plasticity
; chemoresistance
; head and neck cancer (HNC)
; head and neck squamous cell carcinoma (HNSCC)
; mesenchymal stromal cells (MSCs)
; tumour niche
; tumour environment
1. Introduction
Head and neck cancer (HNC) comprises a heterogeneous group of malignant neoplasms originating from the mucosa of the oral cavity, pharynx, larynx, nose, salivary glands and oesophagus, with the former three being the most frequent [1,2]. More than 90% of HNC are squamous cell carcinomas (HNSCC) arise from mucosa of the oral cavity, oropharynx and larynx. All HNSCC demonstrate morphological and molecular heterogeneity, and thus also vary with regard to their clinical course [3]. According to the latest epidemiological data from Global Cancer Statistics (GLOBOCAN 2020), HNSCC is the eighth most common cancer in the world, causing 931,931 newly cases and 467,125 deaths in patients with HNSCC of various origins, accounting for 3% of all cancers and ~1.5% of all cancer deaths. Importantly, analyses clearly indicate that by 2030, the incidence of HNSCC will continue to increase by 30%, i.e., 1.08 million new cases per year [GLOBOCAN; gco.iarc.fr/today (accessed on 30 April 2024)] [4,5]. Unfortunately, despite great advances in surgical techniques and application of modern therapeutic modalities of chemoradiation, this common type of malignancy is associated with high mortality and morbidity due to frequent nodal metastases and local neoplastic recurrences and the development of chemoradioresistance. Importantly, approximately 60-70% of patients are diagnosed at an advanced stage of malignant disease, i.e., WHO classification stages III and IV; as such, the condition is characterised by low global overall survival, with five-year tumour-free survival rates not exceeding 40-60% [6,7,8,9].
HNSCC is most commonly observed in tobacco and alcohol users, and individuals exposed to environmental pollutants and other specific carcinogen-containing products; these have traditionally accounted for 90% of cases of HNSCC according to the National Comprehensive Cancer Network (NCCN) and are usually associated with a poor prognosis [10,11,12,13]. Importantly, research clearly shows that tar and tobacco smoke contain over 7,000 toxic substances, 60% of which are active carcinogens. Tobacco components such as benzo(a)pyrene, a key polycyclic aromatic hydrocarbon (PAH), as well as tobacco-specific nitrosamines (TNA) and N′-nitrosonornicotine (NNN), considerably increase the risk of tumour development and invasion, and the occurrence of lymph node and distant metastases by inducing a phenotype similar to epithelial-mesenchymal transition (EMT) [14,15,16]. Alcohol causes atrophic epithelial changes, acting as a co-carcinogen that increases the risk of HNSCC in concomitant smokers. Moreover, the main metabolite of ethanol, acetaldehyde, is also a strong mutagen. The interaction of these two factors increases the risk of developing HNSCC by up to 35 times [14,15,16]. According to data provided by the Cancer Genome Atlas Network (TCGA), tobacco-related cancers show abnormalities in a number of genes, including cell cycle regulators (CDKN2A and CCND1), regulators of cell proliferation and survival (TP53, HRAS, PIK3CA and EGFR), genes related to cell differentiation (NOTCH1) and Wnt signalling pathway genes [3]. They are also associated with loss of chromosome 9p, responsible for reduced expression of p16 (CDKN2A), and duplication of chromosome 7p, favouring overexpression of the epidermal growth factor receptor (EGFR). In addition, tumours are commonly associated with mutations of proto-oncogenes, i.e., c-MYC, c-KIT, HER-2, RAS, BCL-2, STAT3, and inhibition of anti-oncogenes, i.e., tumour suppressors RB1, P53, INK4, PTEN and CDKN2A, as well as others promoting the pro-inflammatory tumour microenvironment [3,17,18].
In recent years, human papillomavirus (HPV) infection, primarily HPV-16, but also HPV-18, has been observed in the an epithelial of oropharyngeal location (OPSCC). It is more common in middle-aged patients, demonstrating different progression and treatment responses, being much more commonly diagnosed in the earlier stages, and is characterised by better prognosis due to greater sensitivity to chemoradiation and immune checkpoint (ICI) blockade [19,20]. This neoplasm is biologically and clinically unique, and accounts for as much as 38%–80% of new diagnoses of HNSCC [19,21]. OPSCCs contain the HPV genome, which contributes to tumour metaplasia through the activity of viral oncoproteins E6 and E7 [22,23]. OPSCC activity is associated with the integration of HPV genomic DNA with that of host epithelial cells. Increased expression of HPV16/18 virus antigens E5, E6 and E7, which act as oncoproteins/oncogenic factors, induces malignant transformation. HPV16/18 E6 and pRB1 degrade proteins that regulate the cell cycle, e.g., the tumour suppressor protein p53, through HPV16/18 E7; this inactivates key intracellular signalling pathways responsible for cell cycle control [15,24,25,26]. In addition, HPV16/18 E6 protein disturbs the activity of the c-MYC oncogene, increasing the transcription of the human telomerase catalytic subunit (hTERT), which contributes to the immortalization of tumour cells, and disrupts the function of CDK, cyclins and E2F transcription factors. It has also been found that MYC reverses the inhibitory action of CDK p27KIP1 and p21CIP1/WAF1 [25,27]. According to TCGA, typical gene mutations for HPV-associated cancers include PIK3CA, DDX3X, CYLD and FGFR [28]. HPV-induced tumours are also express amplification of chromosome 3q and the loss of chromosomes 16p, 16q, 14q, 13q and 11q [17,18].
In the early clinical stages (I and II), the treatment of HNSCC patients is mostly surgical, being commonly based on modern low-invasive surgical techniques. However, at higher clinical stages, the treatment is more likely to include radiotherapy (RT) or concurrent Cisplatin-based chemotherapy with surgery, especially in patients whose tumours are locoregionally advanced or have positive margins. Unfortunately, as advanced HNSCC frequently demonstrates resistance to conventional treatment, i.e., platinum (CDDP)-based chemotherapy plus 5-fuorouracil (5-FU), extensive research is still being carried out to develop new molecularly-targeted therapies. So far, Cetuximab, a monoclonal antibody against the epidermal growth factor receptor (EGFR), and the PD-1 inhibitors Nivolumab and Pembrolizumab, have been approved for the treatment of clinically-advanced HNSCC. Unfortunately, only cetuximab shows limited effectiveness in patients with unresectable and recurrent HNSCC [9]. Importantly, with the growth in understanding of the biological nature of HPV+ tumours, and its relationship to prognostic indicators of HPV-related OPSCC, the traditional staging of HNSCC using the tumour-node-metastasis system has been supplemented by the new 2017 AJCC/UICC staging system: this eighth edition of the American Joint Committee on Cancer (AJCC) proposes a de-intensified treatment protocol for patients with HPV-related OPSCC to reduce long-term associated morbidity [29,30,31]. However, recent clinical trials (RTOG 1016 and De-ESCALaTE) indicate that patients with HPV-related OPSCC present a more complex therapeutic problem, as a large cohort of patients who received de-escalation, de-intensified treatment had significantly worse results than those who received standard care [32,33]. Moreover, patients with HPV-positive multi-regional primary OPSCC and a second primary cancer at any head and neck location demonstrated inter alia lower pT/pN grade compared to those with a single primary tumour [34].
Regardless of its origin and type, the HNSCC tumour microenvironment (TME) consists of various stromal cells that may regulate neoplastic development, progression and invasiveness. Interestingly, recent studies clearly indicate that the presence of microenvironmental stemness cells in tumour niche, and their potential to modulate carcinogenesis, tumour advancement and aggressiveness, may also influence the response to cisplatin-based chemotherapy and accompanying radiotherapy (RT), as well as new molecular targeted procedures. One component of the tumour microenvironment ecosystem comprises the tumour-associated mesenchymal stromal cells (MSCs): a small but critical subpopulation of the extracellular matrix (ECM) cells capable of self-renewal, multilineage differentiation and regeneration. The growing field of research on their role in relation to tumorigenesis in HNSCC has resulted in a few new and interesting studies over the last decade. Unfortunately, the role of the MSC in the tumour milieu on HNSCC pathogenesis, therapeutic resistance and patient prognosis remains unclear. Even so, the activities of stemness ECM cells present in the tumour microenvironment may influence future therapeutic strategies for HNC. Nevertheless, MSC cells are also reported to exhibit dual roles as both pro- and anti-cancer factors in human cancers, including HNSCC [35,36,37,38,39].
This narrative review aims to present a comprehensive picture of relevant latest literature regarding the importance of tumour-associated mesenchymal stromal cells (MSCs) on the initiation of carcinogenesis, determination of more stemness and invasive phenotype and the activation of regulatory mechanisms involved in the stemness-related development of resistance; the review also covers the chemoresistant effects to cisplatin-based chemotherapeutics contributing to treatment failure, and the potential therapy of HNC of various site origins. The corpus of research comprised a wide range of recent pre- and clinical early trials, experimental and molecular studies, including in vitro cell culture models and animal or in vivo models of HNSCC. The following inclusion criteria were applied: (a) written in English language; (b) studies based on humans or animal tissues (both in vivo and in vitro); (c) cohort studies; (d) retrospective studies; (e) prospective studies; (f) patients with confirmed diagnosis of HNC. This review also presents up-to-date knowledge and focuses on the possibilities of pharmacological targeting of stemness-related chemoradioresistance in HNSCC. The final search (conducted on April 30, 2024) included the most valuable and highest-rated peer-reviewed articles published from the last decade (January 2014 to April 2024), all of which are accessible via the PubMed/Medline/EMBASE/Cochrane Library database. The databases were searched using the following keywords: “head and neck neoplasm or HNSCC”, “squamous cell carcinoma of head and neck”, “head and neck cancer”, “head and neck carcinoma”, “HPV-related tumours”, “mesenchymal stromal cells”, “MSC”, “stem cell resistance”, “therapeutic or drug resistance”, “stemness potential”, “stemness markers”. There was no restriction on language or research group characteristics. The following exclusion criteria applied: (a) unpublished articles or conference proceedings; (b) case reports, case records, and letters to the editor; (c) studies where the patient’s diagnosis is uncertain (e.g., no histopathological confirmation); (e) abstracts. This work discusses the obtained issues in detail, in order of increasing clinical credibility.
2. Characteristics of Mesenchymal Stromal Cells (MSCs)
2.1. The Identification, Distribution and Pathophysiological Role of Mesenchymal Stromal Cells (MSCs)
Mesenchymal stromal cells (MSCs) constitute a small population of multipotent, mobile cells, but an interesting one that has been intensively studied. MSCs participate in tissue homeostasis regulation homeostasis, tissue/organ repair, maintenance of blood vessel integrity and immune system modulation [40,41,42,43]. However, due the lack of MSC-specific markers and their phenotypic similarity to other stromal cells, it can be difficult to accurately characterise MSCs. The latest observations indicate that inflammatory signals at the lesion site promote the migration of MSCs to the site of damage and cancer niche, where the production and secretion of signalling molecules occurs and subsequent differentiation into various cell types. The migration of the MSCs to the injured or inflamed site is controlled by stimulatory signals, which also influence the host response and promote naïve MSC differentiation. The chronic inflammation accompanying the tumour microenvironment is a key condition favouring the colonization of the TME by MSCs; indeed, numerous studies indicate that tumour-associated mesenchymal stromal cells can be recovered from various neoplastic tissues [38,47,48]. The main sources of MSCs are bone marrow, adipose tissue and dental pulp, but they can also be recovered from other organs, i.e., muscular, bone, and cartilage tissue in vivo; however, the most commonly-used tissue sources for obtaining MSCs are BM-derived MSCs (BM-MSCs) and adipose tissue-derived MSCs (A-MSCs) [40,44,45,46]. It is postulated that MSCs are most often located near blood vessels, in permeating arterioles and within the tunica adventitia of arteries. Mesenchymal cells from these tissues have the ability to divide symmetrically (forming two stem cells or two differentiated cells) or asymmetrically (forming one stem cell and one differentiated cell). They are also believed to be able to transform into certain tissue lines and have motility properties [41,46,48].
Since the ability of MSCs to transdifferentiate cells is controversial, the International Society for Cellular Therapy (ISCT) has proposed that the terminology for these cells should only refer to cells meeting specific stemness criteria [49,50,51]. The most important of these are the three minimum criteria: (1) MSCs must demonstrate plastic-adherence when grown in vitro, (2) MSCs must express surface antigens such as CD73, CD90 and CD105, while lacking CD45, CD34, CD14 or CD11b, CD79α, or CD19 and HLA-DR antigens, (3) when cultured under well-defined conditions, MSCs must be able to differentiate into specific mesodermal cell types after stimulation i.e., adipocytes, chondrocytes, and osteoblasts. In addition to the mesodermal lineage, MSCs have high plasticity and be capable of transforming into cells of non-mesodermal origin i.e., ectodermal and endodermal lineages, such as neuronal cells, cardiomyocytes, hepatocytes, or epithelial cells. Further in vitro research of MSCs has allowed their identification based on other features, like the presence of a subset of other characteristic surface markers and the ability to form colonies [40,48,52,53,54]. One set of markers proposed for the identification of MSCs includes the superficial proteins, for instance smooth muscle actin (SMA), Gremlin-1 (GREM1), Meflin (ISLR), PDPN (Podoplanin), STRO-1, and stage-specific embryonic antigen 4 (SSEA-4) [55,56,57,58,59,60,61]. Interestingly, the presence of a key SMA marker allows MSCs to be differentiated from activated fibroblasts/myofibroblasts in the ECM. Moreover, SMA up-regulation promotes the transformation of mesenchymal stromal cells into myofibroblasts and cancer-activated fibroblasts (CAFs) [62,63,64,65]. However, MSC identification can be hampered by the fact that they present similar surface markers and microvascular pericytes to fibroblasts [66,67,68]. Even so, fibroblasts have been differentiated from MSCs by co-expression of fibroblast activation protein alpha (FAP-α) and fibroblast specific protein 1 (FSP, also known as S100A4) [69,70]. Gremlin-1 (GREM1) is also indicated to be an important candidate marker of MSCs [56,57]. Unfortunately, markers such as ISLR, PDPN, STRO-1, and SSEA-4 are also expressed in other cell lineages and hence have limited value.
The microenvironmental conditions present during tissue injury or tumorigenesis induce immune cells, endothelial cells, and fibroblasts to secrete several essential interleukins, chemokines and other cell mediators that promote MSC mobilization. MSCs themselves can produce factors that stimulate tissue repair, modulate the immune response and inflammation, and influence subsequent stages of cancer development. Endothelial cell selectins also are observed to promote MSC homing and differentiation [71,72]. Importantly, it is the CD44 and CD24 antigens present on the MSCs surface may interact with endothelial selectins, such as P-selectin (CD24/CD44 binding) and integrins (α4β1/α6β4) and then contribute to the rolling of MSCs in the microenvironment after chemotaxis [73,74,75,76,77,78]. Both P-selectin and galectin-1 (GAL-1) bind CD44, as well as TNF-α, IL-6, IL-1β and IFN-γ; the latter is also an endogenous glycan-binding protein. Importantly, both influence the interaction between MSCs and endothelial cells, but also immunocompetent cells in the tumour milieu, thus favouring tumour development: they may initiate activation of T cells apoptosis, equip dendritic cells (DCs) with tolerogenic potential, promote IL-10-mediated T cell tolerance, mediate suppression of CD8+ cells and CD4+ Th1/Th17 pro-inflammatory subtype lymphocytes. They also contribute to cancer growth, cell adhesion and migration, thereby affecting the process of tumour-immune escape, cancer transformation, and neoplastic metastases [72,75,76]. Pro-inflammatory cytokines released from damaged tissue, or those produced by immune cells in the tumour TME, may determine the overexpression of extracellular matrix metalloproteinases (MT-MMPs), i.e., MMP-2, MMP-9 and MMP-14, in the tumour-associated mesenchymal stromal cell environment and, consequently, promote cell migration to the stromal tissue and transmigration of into the fenestrated endothelium [78,79]. Similarly, other mediators such as IL-8, insulin-like growth factor (IGF)-1, vascular endothelial growth factor (VEGF), and platelet-derived growth factor-AB (PGDF-AB) can stimulate and facilitate the migration of MSCs into the injured or tumour stroma [80,81,82]. Unfortunately, the particular properties of the released mediators and multiple agents that make MSCs excellent therapeutic agents can also influence tumour progression [52].
2.2. Dual Roles and the Bidirectional Effect of Mesenchymal Stromal Cells in the Tumour Microenvironment
Tumours, including solid tumours in the head and neck region, are associated with a complex microenvironment including heterogeneous cell populations, such as neoplastic cells, transformed stromal cells/cancer stem cells, immunocompetent cells, blood vessels and lymphatic vessels. Highly-specialized communication and modulation can be found between cells present in the tumour ECM, which is maintained through secreted cytokines, chemokines and various other mediators by paracrine signalling or through cell-cell interaction [83]. These paracrine agents can be directly secreted into the tumour milieus or released via extracellular vesicles (MSC-EVs) [52]. Recent studies have shown that after transformation in the neoplastic niche, the activity of naïve chemoattractant-induced MSCs induces production of various pro-/anti-inflammatory cytokines, chemokines or mediators that give plasticity, variability and uniqueness of the tumour milieu. These modulatory cells may enhance resistance to Cisplatin-based chemotherapy, thus favouring cancer progression and therapeutic failure; the activation of pro-tumour mechanisms is a common cause of shortened survival.
Unfortunately, the primary role of MSCs in the initiation of carcinogenesis and subsequent stages of tumour development has not been fully elucidated; despite this, a few studies indicate that MSCs have a positive impact on the inhibition of cancer lesions by activating anti-cancer mechanisms [84,85,86]. Indeed, numerous recent studies indicate that cancer-associated MSCs appear to have dual effects on tumour progression in various in vitro and animal or in vivo neoplastic models, and the pleiotropic effects of MSCs may therefore confer pro- or anti-tumour functions to cells in the tumour microenvironment [38,52,87,88]. The biomolecules secreted from tumour-associated MSCs and cancer cells are often termed, in accordance with the nomenclature from English literature, an active “secretome”; such a mixture includes key immune-modulating agents, pro-/antiangiogenic factors, pro-survival biological agents or soluble factors inhibiting mobility and cancer cell survival and extracellular matrix modulators [74,89,90,91,92]. Crosstalk between tumour cells and mesenchymal MSCs that can alter the behaviour of TME cells and regulate tumorigenesis through the secretion of secretome biomolecules, has also been noted in tumours of the head and neck region. Examples of soluble agents and modulators in HNC include IL-6, IL-8, plated-derived growth factor (PDGF), beta 2 microglobulin (B2M), cellular communication network factor 2 (CCN2), fibroblast growth factor 19 (FGF19), stromal cell growth factor-beta (SCGF), trans forming growth factor-beta 1 (TGF-β1) and stromal cell-derived factor 1 (SDF-1) and matrix metalloproteinases (MT-MMPs); these have been found to directly regulate the epithelial-mesenchymal plasticity, proliferation, invasion and migration and even drug resistance in HNSCC cells [93]. To summarize, the composition of pro- or anti-tumorigenic mediators secreted in the secretome, and their action, may promote, or inhibit, carcinogenesis in the neoplastic niche occupied by tumour-associated mesenchymal MSCs. Moreover, these components of the secretome may constitute an important target of modern anticancer drugs, which will be discussed in detail in the following paragraphs.
2.2.1. Pro-Tumour Activity of MSCs
Like damaged or injured tissues, tumour cells have a chemoattractant effect on mesenchymal tumour stromal cells, activating their recruitment to the neoplastic niche. One of the most characteristic signalling pathways involved in their mobilization to the tumour microenvironment involves the participation of cytokines, chemokines or chemokine receptors, i.e., CXCL motif chemokine ligand 1/2/12 (CXCL1/2/12), chemokine receptor 4 (CXCR4), chemokine ligand 1/5/7/8 (CCL1/5/7/8). The mesenchymal stromal cells first migrate into the blood, thus allowing the binding of the ligand-receptor to endothelial cells, they move on to sites of chronic inflammation or tumorigenesis milieu in response to chemoattractant signals. When near the destination, the MSCs cross the endothelial border and move towards the target cancerous tissue, guided by the chemotactic gradient, where they promote both tumour progression and cancer arrest [94,95,96]. More importantly, MSCs may act directly via cell-to-cell contact or release numerous soluble factors with pro- and anti-tumour effects into the extracellular matrix; these influence survival, proliferation and angiogenesis, as well as metastasis, and can stimulate and control other cellular functions, thus increasing tumour aggressiveness. These paracrine mediators may be secreted directly into the tumour niche or secreted via extracellular vesicles (MSC-EVs) i.e., exosomes, microvesicles, and apoptotic bodies: these are cell-derived membrane-surrounded balls that deliver bioactive molecules to recipient cells [52,97]. A few factors also stimulate MSCs to migrate towards tumour tissue and may inhibit apoptosis in cancer cells. These biomolecules control the chemotaxis and activity of immune and environmental cells in the TME by promoting the secretion of factors which promote immunosuppressive effects and tumour cell survival. These include growth factors such as transforming growth factor beta 1 (TGF-β1), platelet-derived growth factor (PDGF), epidermal growth factor (EGF), basic fibroblast growth factors (bFGF) or vascular endothelial growth factor (VEGF), together with extracellular matrix molecules such as such as matrix metalloproteinase 2/3/7/9 (MMP-2, MMP-3, MMP-7, MMP-9) [52,98]. Numerous studies have also shown that MSCs isolated from squamous cell carcinomas can mediate cancer progression by secreting pro-inflammatory and pro-angiogenic cytokines such as IL-6 and IL8, which also promote recruitment of leukocytes, i.e., tumour-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), T cells and neutrophils: the cells which facilitate tumour initiation and progression. TGF-β1 production also plays an important role as it can promote TAM infiltration at the tumour site and the transition and differentiation into CAFs, and can facilitate the tumour’s escape from immune surveillance. Moreover, it was confirmed that MSCs recruited from human cancers can actively increase the expression of immunosuppressive agents, i.e., IL-10, TGF-β1 and indoleamine 2,3-dioxygenase (IDO), and angiogenic factors, i.e., TGF-β1, VEGF, angiopoetin-1, endothelin-1, and IL-6. These facilitate further tumour progression and growth through enhanced angiogenesis. They also encourage the formation of further vascularization, and the local/regional and general spread of tumour cells [99,100,101]. For instance, a study using isolated human MSCs intravenously injected into mice with carcinoma found increased TGF-β1 expression to be significantly related to a higher density of new microvessels in the extracellular matrix of neoplastic tissue [102]. Similarly, other researchers have observed that MSCs contribute to cancer pathogenesis by secreting factors that promote immunosuppression in the secretome. For example, an in vitro study by Li et al. [103] showed that the progression of MSCs isolated from human tumour tissue is mediated through the production of interleukin IL-8, a pro-inflammatory and angiogenic chemokine that promotes leukocyte recruitment and tumorigenesis. The active presence of leukocytes and immunocompetent cells such as TAMs, CAFs and neutrophils in the tumour environment facilitate initiation and cancer progression [104,105]. Furthermore, the development and invasiveness of neoplastic cells may also be associated with a wide spectrum of cancer-related signalling pathways involved in the crosstalk between MSC and tumour cells, such as phosphatidylinositol 3-kinase/protein kinase B/mammalian target of Rapamycin (PI3K/AKT/mTOR), the Janus kinase/signal transducers and activators of transcription (JAK/STAT), Wnt signalling, the Hippo pathway, MYC and NF-κβ signalling cascades [106]. PI3K/AKT pathway activity appears to be associated with the acquisition of tumorigenic properties by tumours, such as increased rates of cell proliferation, drug resistance, and stem cell-like phenotypes [107]. Moreover, increasing the expression of the PI3K/AKT pathway was found to stimulate placental growth factor (PlGF) and C-X-C motif chemokine ligand 1 (CXCL1) production in neoplastic stem cells and enhance the formation of new vessels in the tumour milieu [108]. Several studies have also confirmed that MSCs interact with tumours via the JAK/STAT pathway via the control of functional molecules such as growth factors and cytokines [109]. Indeed, IL-6 secreted by MSCs can activate JAK2/STAT3 signalling and thus promote cancer progression [110]. In one in vitro analysis, it was reported that the cell culture medium from bone marrow-derived MSC-CM (BM-MSC-conditioned medium), promoted the progression of head and neck cancer by activating the PI3K/AKT signalling pathway [111]. Additionally, MSC-CM co-cultured with HNC tumour cells increased invasiveness by enhancing cell proliferation, migration, epithelial-mesenchymal transformation, and altering the expression of cell cycle regulatory proteins, as well as by inhibiting apoptosis. Furthermore, these phenomena were induced through the activation of PI3K/AKT/mTOR pathway, and they were observed to enhance the expression of Periostin (POSTN) and N-cadherin in the EMT around the tumour tissues [111]. Interestingly, a recent study found that MSC exposure led to the selection of metastatic cancer cells; these were found to be more resistant to apoptotic effects, which was attributed to a shift from the pro-apoptotic, IL-28/STAT1 cascade to the anti-apoptotic IL-28/STAT3 cascade [112]. In addition to the above-mentioned pathways, Wnt/β-catenin signalling was also described as a key factor in regulating tumour development and cancer cell stemness in numerous types of human cancers [113,114]. It was observed that Wnt protein, induces the release of exosomes containing pro-angiogenetic VEGF and IL-6, and up-regulates the expression of various Wnt target genes, including CCND1, c-MYC and the MMP family. Moreover, the human neoplastic cell line QBC939 demonstrated enhanced metastasis and chemoresistance following treatment with human umbilical cord-derived mesenchymal stem cells (hUC-MSCs) in a xenograft carcinoma model [114]. Interestingly, MSCs can also promote increased tumour aggressiveness and the formation of metastases from cancer cells when engulfed by the neoplastic cells. In such cases, this assumption of the MSC can alter the transcriptome profile of the cancer cell, particularly those related to oncogenic pathways, and enhance epithelial-to-mesenchymal transition, stemness, invasion and metastasis. A recent study found MSC-engulfing tumour cells to be characterised by certain upregulated genes encoding cell surface and extracellular proteins: MSR1 (CD204), WNT5A, ELMO1, IL1RL2 (IL-36), ZPLD1, and SIRPB1 (CD172); in addition, high levels of MSR1 and WNT5A were found to be significantly related to worse metastasis-free survival in cancer patients [115].
2.2.2. Anti-Tumour Activity of MSCs
On the other hand, several recent publications have concluded that tumour-associated MSCs may also have inhibitory effects on the tumorigenic cell phenotype. These may be realised through their cytotoxic effects on cancer cells, thus inhibiting the initiation, development and progression of cancer. This can also provide a starting point for new strategies for human cancer treatment [52,84,106,116]. Compelling evidence shows that MSCs utilize various anti-tumour mechanisms to induce apoptosis, repress tumour growth, inhibit angiogenesis and suppress neoplastic cell proliferation. Interestingly, the most important signalling factors found to repress tumour progression and aggressiveness include MSC-related paracrine soluble secreted factors and MSCs-derived molecules released from exosomes; these include proven anti-proliferative factors such as Dickkopf-related protein 1 (Dkk-1), a soluble Wnt antagonist, phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase (PTEN), and bone morphogenetic protein (BMP), as well as cytotoxic agents such as TNF-related apoptosis-inducing ligand (TRAIL) and TNF-α. In addition, anti-angiogenic factors or immunomodulatory agents have been recorded, as well as some known to inhibit cancer-related signalling, such as PI3K/AKT, Wnt/β-catenin and the JAK/STAT pathway [52,84,106,116]. Studies have shown that human MSCs from various sources may release cytotoxic factors, such as TRAIL, which selectively induces apoptosis in various types of cancer by downregulating the PI3K/AKT or ERK1/2 signalling pathway [116,117]. Also, the ability of MSCs to inhibit cancer cell proliferation may be based on their potential to modulate the Wnt signalling pathway. An in vitro study found MSCs to inhibit cancer cells by secreting the Dkk-1 protein, which is believed to block tumour growth by inhibiting the Wnt pathway [118]. MSCs can also express antitumor activity by inhibiting tumour angiogenesis via downregulation of the PDGF/PDGFR axis, thus restricting vascular growth. Studies have shown that MSC treatment reduced PDGF-BB protein concentrations in tumour lysates, with the levels correlating with reduced activation of PDGFR-β and the isoform of the target protein AKT [119]. Also, the multifunctional cytokine TGF-β1, released from MSCs, can also play a dual role in human tumorigenesis. Indeed, studies have shown that the type III TGF-β receptor (TGFBR3) and its shared extracellular domain (sTGFBR3) may maintain epithelial homeostasis by regulating the TGF-β pathway. Restoring TβRIII expression in human cancer cells inhibited tumour invasiveness and the growth of blood vessels from the existing cancer vasculature in vitro, as well as metastasis in vivo, thus inhibiting the development of an immunotolerant tumour microenvironment [120]. Hence, due to the controversial roles of MSCs in human cancers, future extensive studies should focus on the specific molecular mechanisms involved in MSC-mediated interactions between mesenchymal stemness cells and tumour cells.
It should be emphasized that a number of experimental factors can influence discrepancies in the observations regarding the ability of MSCs to promote or inhibit neoplastic disease development. These may include inter alia differences in the choice of experimental tumour in vitro and the animal or in vivo models, the origin of the cancer or HNSCC cell lines, the varied source of MSC tissue, the dose or duration of MSC treatment, the method of cell delivery, the control group selected, or an insufficient number of studied groups [121,122]. MSC-mediated signalling changes might also play dual roles in obtaining a pro- or anti-cancer cell phenotype; this discrepancy might be attributable to differences in cancer types from the same region and the activation status of key regulators of pathway cascades. The origin of the tumours in the head and neck region is also an important consideration when comparing systems, even within a single squamous type of HNC, as this can have a considerable influence on their cell biology and molecular activity. Moreover, cancer patients are generally subject to a more complex local situation in the tumour microenvironment, and a peripheral one in the bloodstream, and as such, other factors, e.g., local hypoxia and the supply of oxygen and nutrients to tumour cells, must also be taken into account. Such variation may also be the reason for the failure of proposed modern MSC-based therapies, which may induce unforeseen and unknown intracellular and molecular changes in MSCs and tumour cells [123,124,125]. Therefore, researchers into individualized treatment approaches must exercise particular caution when drawing conclusions regarding the therapeutic effects of MSCs on cancer, and should recognize that MSC-based therapies still remain a challenge.
The schema of dual roles of MSCs in TME and their pro- and anti-tumorigenic activity is shown in Figure 1A.
2.3. Modulation of Immune and Inflammatory Cells by Mesenchymal Stromal Cells
Numerous in vitro studies indicate that tumour-associated mesenchymal stromal cells may modulate the immune response in dendritic cells (DCs), tumour-associated macrophages (TAMs), active CD65dim/CD16bright NK cells, activated T CD4+/CD8+(CTL) and B lymphocytes via their secretome; they also confirm that both direct action via cell-to-cell contact or release of numerous soluble factors and MSC-derived exosomes into the extracellular matrix can control the activation, proliferation, differentiation and function of both the immune cells themselves, and the surrounding cells at the lesion site [126,127,128,129,130]. Interestingly, the biologically-active agents and mediators produced and secreted by the MSCs play a dual role in inhibiting the host defence by interfering with the adaptive and innate immune response [126,131].
A number of associated immune agents with innate and adaptive immune reactions are regulated through MSC-derived molecules; these include IDO, NO (nitric oxide), prostaglandin PGE2, TGF-β1, cytokines IL-10 and IL-6, human leukocyte antigen (HLA-G), iNOS, IL-β1, HO1. Most of these are membrane proteins or proteins secreted during the immune response to infection and autoimmunity, or during tumorigenesis; these have been found to proactively regulate the proliferation and action effects of various immune cell subpopulations via the production of modulatory factors determining angiogenesis, cell death (apoptosis) and tissue regeneration in defined medium components and a heterogeneous culture environment in vivo and in vitro [126,127,128,129,130,131,132]. It was reported that they also may secrete trophic factors that can increase cell survival, cell proliferation and tissue fibrosis. Along with the secretion of immunomodulators or immunosuppressive agents, MSCs can also directly inhibit immune cell activation through cell-to-cell interaction. This direct contact between MSCs and T cells can often inhibit immune cell proliferation: the interaction between programmed death-1 (PD-1) molecules and PD-L1 and PD-L2 ligands can induce apoptosis in effector CD4+ and CD8+ T lymphocytes. Moreover, various types of T cell anergy can be provoked by MSCs; in such cases, the cells inhibit the expression of CD86 and CD80 in antigen-presenting cells (APCs) i.e., DCs and TAMs [126,127,132].
2.3.1. The Adaptive Immune Response
A characteristic feature of tumour-associated MSCs is their strong immunosuppressive and immunomodulatory properties, which allow cancer cells to escape immune surveillance. MSCs can be stimulated within the tumour niche by pro-inflammatory cytokines such as TNF-α, IFN-γ or IL-1β, which are secreted by both macrophages and tumour cells. The activity of MSCs in the tumour milieu is driven by immunological stimuli, e.g., IFN-γ and lipopolysaccharide (LPS); their activation was also found to increase under oxidative, heat shock, hypoxic and nutrient-deprived conditions, which can occur in solid tumours. This was associated with elevated expression of cytoprotective genes and increases in MSC potency caused by greater production and secretion of compensating factors [126,127,128,129,133]. It is well known that the active mesenchymal stromal cells in tumour milieu inhibit the adaptive immune response by secreting mediators contained in exosomes (MSCs-EV) and soluble factors, such as IDO, TGFβ1, TNF-α, IFN-γ, PGE2, NO, HLA-G, HGF, IL-1β, IL-1α, IL-4 and IL-6; they can also do so by interacting with various immune cell types, including T cells, B cells, DC cells, NK cells, monocytes and TAMs. This MSC activity constrains dendritic cell maturation, reduces T cell proliferation, enhances macrophage activation and polarization of M1 towards M2 and facilitates neutrophil mobility. It also affects the regulation of CD56dim/16bright NK cells and invariant natural killer T (iNKT) cells, and shifts the balance of T cell differentiation from pro-inflammatory Th1 to anti-inflammatory Th2. Furthermore, it enhances the maturation of T helper cells into CD4+CD25+Foxp3+Treg pathways, that can inhibit effector T cell responses and thus reduce anti-tumour immunity [126,127,128,129,130,131,132]. In vitro studies have shown that this shift can also regulate the proliferation of B cells and their differentiation into mature plasma cells, and can inhibit IgG immunoglobulin secretion from plasma cells, leading to the development of a suppressive phenotype in vitro and in vivo, by cell cycle arrest [134]. For instance, the cellular effects of the IFN-γ ligand, receptor activity and signalling transduction increase leukocyte attraction and favour the growth, maturation and differentiation of many types of immune cells. They can increase the activity of NK cells and regulate B cell functions, such as the production of Ig classes of immunoglobulins (IgG) [135,136]. Interestingly, INF-γ stimulation of adipose-derived mesenchymal cells (A-MSCs) intensified their immunosuppressive effect and IDO-1 expression, resulting in reduced inflammation and anti-inflammatory protein production [137,138]. Moreover, IFN-γ stimulation stimulated galectin-9 secretion by MSCs, which also attenuates antibody production and B cell proliferation [139]. In another study, MB-MSC (hcBMSC) treatment induced immunosuppression, which inhibited the proliferation of both CD4+ and CD8+ T cells; it also drove M1 polarisation of TAMs and NK cells by increasing the secretion and efficiency of IDO-1, IL-10, PD-L1 and PGE2, which was dependent on both the dose of IFN-γ and the duration of action [137,138]. The combination of IFN-γ priming and MSC sheets may present a new strategy to improve the treatment of localized inflammatory diseases, including the tumour microenvironment via MSCs. Another interesting study compared the influence of MSCs of various origins (MSCs originating from bone marrow, adipose tissue and umbilical cord) on proliferation, immunosuppression and migration toward the activated lymphocytes. The migratory potency of the MSCs was evaluated by transwell migration assay. Interestingly, UC-MSCs, BM-MSCs, and AD-MSCs all suppressed the proliferation of activated lymphocytes to a similar degree. Moreover, chemokines in the co-cultured supernatant showed the highest secretion of CCL2 (MCP-1), while the migration of MSCs toward lymphocytes was attenuated by the inhibitor of PDGF, IGF-1, and MMP inhibitor in a dose-dependent manner [140]. Other studies have examined the immunosuppressive nature of MSCs. In addition to maintaining chronic inflammation it was also shown that MSCs inhibited the effector function of cytotoxic T cells (CTLs) in mouse models by increasing the expression of MSC-derived soluble factors, such as arginase 1 (ARG-1) and inducible nitric oxide synthase 2 (iNOS2) [141,142,143]. MSCs also secreted CXCL3, a member of the growth-related oncogenes (GRO-γ), directly inhibiting the differentiation and function of MSCs, promoting their shift to a myeloid-derived suppressor cell phenotype (MDDCs) [144]. MSCs can also induce recruitment of inhibitory immune cells (MDSCs) through CCL2 signalling, which inhibits the anti-tumour activity of T cells [145]. This was accompanied by an increase in the expression of genes related to MSCs, including COX2, IDO-1, programmed death ligand (PD-L) 1 and 2, and matrix metalloproteinase 9 (MMP-9) in human MDDCs. MSC-secreted hepatocyte growth factor (HGF) further determined the expansion of these cells by directly binding the HGF receptor, c-met, and increasing the STAT3 phosphorylation status in MDSCs [142,143]. It has also been found that the soluble PD-1 ligands produced by MSCs effectively inhibit the effect of IL-2 on T cell activation. Soluble PD-L1 and PD-L2 secreted by MSCs blocked the parallel activation of AKT signalling pathways, which also inhibited T cell activation and proliferation. While PD-1 and PD-L1 have also been found to downregulate T cell activity, it has recently been found that the immunosuppressive effect of MSCs is partially dependent on cytotoxic T cell antigen 4 (CTLA-4). CTLA-4 occurs on the surface of T lymphocytes activated by contact with an antigen and inhibits further response of the lymphocyte [146,147]. This expansion process also appears to be supported by IL-6, a cytokine constitutively secreted by MSCs, which was found to inhibit the apoptosis of neutrophils and lymphocytes [126,127,128]. Also, MSC-derived IL-6 also activates neutrophils through the STAT3-ERK1/2 signal transduction pathway, and shifts their immunosuppressive polarization towards tumour facilitation and supporting cancer progression [148]. Also, NO synthesis and secretion is driven by inducible NO synthase (iNOS) following stimulation by various inflammatory factors, such as IFN-γ, IL-1 and TNF-α, which subsequently suppress T cell function [126,149]. IDO is also of key importance, as it determines the maturation of Th-type cells into regulatory suppressor Treg cells; in this way, IDO, as another element of the tumour milieu, thus inhibiting the anti-tumour immune response. Another very important soluble factor is PGE2, which also works by stimulating typical anti-inflammatory cytokines, such as IL-10, and blocking the synthesis of certain interleukins, such as TNF-α, INF-γ and IL-12 in TAMs and DCs. PGE2 also diminishes the expression of cytokines, i.e., INF-γ and IL-4, shifting the immune balance towards Th2 cells; in this way, it contributes to an increase in anti-tumour activity by promoting immunosuppressive cells such as regulatory CD4+CD8+FoxP3+ Tregs [126,131,150]. Another study showed that mesenchymal stem/stromal cells (MSCs) can induce and promote the differentiation of co-called myeloid-derived suppressor cells (MDSCs) in the bone marrow (BM). The mitogen-activated protein kinase (p38MAPK) pathway was found to be activated in MDSCs induced by MSCs, as indicated by RNA-seq analysis. In addition, Western blot analysis found co-culture of BM cells with MSCs also resulted in phosphorylation of c-Jun N-terminal kinase (JNK) following stimulation with granulocyte-macrophage colony-stimulating factor (GMC-SF), while p38MAPK kinase activation remained unchanged; however, the effects of MSCs on TGF-β1, TGF-β2 and IL-10 production in BM cells was abrogated by inhibition of JNK/MAPK signalling [151].
In summary, MSCs have a strong inhibitory effect on adaptive immune cells, and this ability is exploited by cancer cells. It has been also suggested that MSCs may contribute to the formation or inhibition of HNC neoplastic lesions by the promotion of tumour evolution via various mechanisms. Tumour-associated MSCs may determine the growth of HNC by various mechanisms, such as secreting biomolecules, promoting cell-cell contact, facilitating the acquisition of epithelial-to-mesenchymal transcriptional properties, suppressing the protective activities of immune cells, enhancing angiogenesis, or undergoing differentiation into other tumour stroma components such as TAMs and CAFs. The resulting changes can contribute to the acquisition of drug resistance, and thus treatment failure.
2.3.2. The Innate Immune Response
The innate immune response is linked to the stimulation of toll-like receptors (TLRs) in polymorphonuclear cells (PMNs) such as monocytes and macrophages, and in various types of epithelial cells. The TLR family, comprising TLR1 to TLR13, has been recognised in humans, and its members serve as pattern recognition receptors (PRRs). Some of these recognize molecules that are generally shared by pathogens, known as pathogen-associated molecular patterns (PAMPs) such as microbial-associated molecular patterns (MAMPs) expressed by pathogens, and damage or death-associate molecular patterns (DAMPs) released and expressed by damaged or killed host cells. TLR activation begins through the stimulation of specific pathways via adapter molecules such as myeloid differentiation factor-88 (MyD88), a member of the TIR family, and the TIR domain containing an IFN-β inducing adapter (TRIF). Following this, the TLRs, except TLR3, undergo dimerization. MyD88 signalling results primarily in the activation of nuclear factor NF-κB and mitogen-activated protein kinase (p38MAPK). MyD88 then recruits IL-1R-associated kinases (IRAK1-3), which phosphorylate and activate the protein TRAF6; this in turn polyubiquinates the protein TAK1, which phosphorylates IKK-β. This cascade allows NF-κB to diffuse into the cell nucleus, resulting in the transcriptional activation of inflammatory cytokines and their subsequent induction [152,153,154,155].
In injured or tumorigenic tissues, local factors such as cytokine milieu (TNF-α, endotoxin LPS), hypoxia and Toll-like receptor (TLRs) ligands, stimulate MSCs to secrete growth factors such as VEGF, FGF2, IGF-1, or HGF. This stimulation takes place via an NFκB-dependent mechanism that drives tissue regeneration and angiogenesis, reduces anti-tumour immunity and allows the tumour to escape from immune surveillance in a 3-D culture system [156,157,158,159].
It is known that MSCs with mainly pro-inflammatory effects are also activated during infections and early-stage inflammatory conditions. Exposure to TLR2 (peptidoglycan) from Gram-positive bacteria or TLR4 (LPS) from Gram negative bacteria encourages MSCs to migrate to the site of injury and promotes the immunity [160]. Thus, secretion of IL-6, IL-8, IFN-β, MIF and GM-CSF by MSCs may upregulate neutrophil migration to the site of infection or injury, enhancing their activation and phagocytosis whilst promoting their survival [144,161]. For example, the secretion of pro-inflammatory chemokines such as CCL2, CCL3 and CCL12 by MSCs recruits monocytes to the site of injury, where they differentiate into pro-inflammatory M1 macrophages. Subsequent secretion of GM-CSF by MSCs shifts the balance towards macrophages with an M1 phenotype, thereby increasing bacterial clearance and early wound healing responses. Also, the secretion of chemokine (C-X-C motif) ligand CXCL-9, CXCL-10, macrophage inflammatory protein MIP-1α, MIP-1β and RANTES plays an important role, as these also increase lymphocyte recruitment. It has recently been observed that this phenotype can be influenced by the media used for the in vitro expansion of MSCs: media supplemented with platelet lysate promoted a pro-inflammatory MSC phenotype and GM-CSF secretion, which may also result in the recruitment of immune cells and the maintenance of macrophages in the M1 phenotype [144,162].
Many studies have suggested that MSCs may support or inhibit HNC initiation and their evolution through various inflammatory mechanisms. By secreting biomolecules with trophic properties, tumour-associated MSCs may promote cell-cell contact, thus facilitating epithelial-to-mesenchymal transition, suppressing the protective activities of immune cells, enhancing angiogenesis, or undergoing differentiation into other tumour stroma components such as CAFs. Such changes may determine the growth of HNCs, influence the acquisition of drug resistance, and thus treatment failure. Moreover, with increasing evidence indicating the role of MSCs in the direct modulation of the innate and adaptive immune system, MSC therapy may be seen as a new and promising alternative in the treatment of cancers such as HNC, as well as various other diseases. It is clear that although the specific mechanisms of action by which MSCs exert their immunomodulatory effects in vivo remain largely incompletely understood, due to the idiosyncratic nature of the microenvironment and paracrine signals, they are known to exert significant effects on various immune cell subsets. Further understanding of the role of active factors secreted by MSCs in the secretome, as well as their interactions, will be of key importance for improving and developing new clinical protocols for MSC-based cell therapy.
The schema of MSCs immunosuppression mechanisms and their role in immune cell activity is shown in Figure 1B.
3. Studies on the Role of Mesenchymal Stromal Cells (MSCs) in Head and Neck Cancer. Effects of the Secretome on HNC. Therapeutic Potential of MSCs. The Pharmacological Strategies of MSC-Based Therapies in HNC
3.1. The Stemness Phenotype of Mesenchymal Stromal Cells (MSCs) in HNC. The role of MSCs in Tumorigenesis, Progression and Drug-Resistance Mechanisms in HNC
3.1.1. In Vitro Models of HNSCC
When describing MSC cells and cancer cells in the tumour niche, it is necessary to mention the biomolecules present within the “secretome” [74,91,93]. Mesenchymal stem cells (MSCs) are multipotent cells which demonstrate significant potential in human tissue regeneration due to their capability to migrate to sites of injury, inflammation or cancer to suppress the immune response and reduce accessibility. Many of these are derived from the patient’s own bone marrow (BM-MSCs) or fat tissue (A-MSCs). The crosstalk between neoplastic cells, and the proteomic profile of the MSCs, can modify the secretion of key biomolecules influencing neoplastic cell behaviour and other cell populations; these include various lineages of MSC cells, i.e., from bone marrow, adipose tissue and dental pulp, and immune stromal cells. Interestingly, several lines of evidence indicate that MSCs lose their immunosuppressive and regenerative potency after multiple passages in in vitro. Interestingly, various molecules can regulate the progression of HNC by modulating the behaviour of MSCs. For instance, the IL-6, platelet-derived growth factor (PDGF), and matrix metalloproteinases (MT-MMPs) such as MMP-2, MMP-9, MMP-14 and alpha-1 type I collagen (encoded by COLA1) secreted by tumour cells are known to influence MSC activity. Similarly, β-2-Microglobulin (B2M), cell communication network factor 2 (CCN2), vascular endothelial growth factor (VEGF), tumour necrosis factor-beta (TNF-β), stromal cell-derived factor 1 (SDF-1), serum stem cell growth factor-beta (SCGF-β), Periostin, or osteoblast-specific factor 2 (POSTN/OSF-2), CCL5, IL-6, FGF19, miR-8485 may modulate progression and affect cell survival, proliferation, motility, invasion, and also epithelial-to-mesenchymal transition (EMT). Additionally, various lineages of MSCs determine the immune response, and consequently modulate HNC behaviour, by regulating the expression of IDO, CD39, CD73, differentiation into specific cell types, i.e., fibroblasts, chondroblast, adipose tissue and myofibroblasts. Also, other active proteins such as Gremlin-1 (GREM1), bone morphogenetic protein 4 (BMP4), TGF-β, MT-MMPs, laminin-5, integrin, and EGFR promote cancer cell migration and invasion and EMP. Importantly, the fusion of MSCs and cancer cells leads to the secretion of the DUSP family dual specificity phosphatase 6 (DUSP6) that can regulate NO production by MAPK kinases and reduce cancer cell survival [93]. It is important to point out that transplanted MSCs do not always engraft and differentiate at the site of injury but might exert their therapeutic effects through secreted trophic signals. Few reviews to date have discussed existing proteomic techniques, or those with future applications in MSC secretomics; in addition, few have examined secretome sample preparation, protein/peptide separation, mass spectrometry and protein quantification techniques, i.e., analysis of post-translational modifications, or bioinformatics, immunological techniques, isolation and characterization of secreted vesicles and exosomes, the analysis of mRNAs encoding cytokines [74,91,93]. The most commonly-secreted factors regulating tumour niche cell function are well known ones whose functions are linked to the biological effects of MSCs. These include connective tissue growth factor (CTGF), SERPINE1, TGF-β1, Dickkopf-related protein 3 (Dkk-3) and a myeloid derived growth factor (MYD-GF). They also include newly-identified factors whose roles are not well investigated, for example an aminoacyl-tRNA synthetase-interacting multifunctional protein-1 (AIMP1), C-type lectin domain containing 11A (CLEC11A), growth arrest specific 6 (GAS6), which regulates of natural killer cell differentiation and apoptotic cell clearance, and heparin binding growth factor (HDGF). Another compound if inhibin β-A (INHBA), which induces EMT and accelerates the motility of cancer cells by activating the TGF-β and proprotein convertase subtilisin/kexin type 5 (PCSK5) [74,91].
Moravcikova et al. [74] used a proteomic analysis system to distinguish issue variations in cell surface MSC CD45−/CD31−/CD34−/CD73+/CD105+ antigens from native BM-MSCs through serial culture passage. The findings demonstrate that cancer cell-secreted IL-6 and PDGF in the tumour milieu are sufficient to induce MSC migration into the head and neck tumour stroma. The authors observed characteristic changes in adipogenic and osteogenic differentiative potential during the initial expansion and invasion. The most prominent included decreases in FasL, CD98, CD205, and CD106 antigens, accompanied by a gain in the expression of CD49c, CD63, CD98, and class I/II of MHC molecules. These were accompanied by loss of MAC-inhibitory protein/CD59, loss of ICAM-1/CD54, and increase in CDKN2A expression, as well as increased CD10 expression with adipogenic and osteogenic potential. Watts et al. [163] described the secretion profile of HNSCC cells in vitro based on the JHU-011, JHU-012 and JHU-019 cell OSCC lines. The secretome included stromal cell-derived factor 1 (SDF-1 or CXCL-12), growth-regulated protein alpha (Gro-α or CXCL1), VEGF, PDGF, cytokines IL-6 and L-8, as well as PDGF-AA, as inhibitor of the PDGF-AA receptor and PDGFR-α decreased MSCs stromal chemotaxis to the oral cavity and oral pharyngeal squamous cell carcinoma (OPSCC) cells. The presence of BM-MSCs in HNSCC-derived secretory molecules increased the migration of MSCs towards cancer cells and their invasion, while these were reduced by the inhibition of IL-6 and PDGFR-α. Similarly, Kansy et al. [164] showed that when incubated in supernatants obtained from the FaDu (ATCC HTB-43) and UM-SSC-22B HNSCC cell lines, tumour-derived MSCs promote the progression of head and neck cancer stroma. This was ben attributed the production of tumour-derived MSCs containing inter alia IL-1β, IL-2, IL-4, IL-6, IL-8, granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), INF-γ, macrophage inflammatory protein 1β (MIP1β or CCL4), stromal cell-derived factor (SDF)-1α, and TNF-α in the secretome. HNSCC-derived factors stimulated MSCs and enhanced IL-8 secretion and CD54+ expression. The findings confirm that the stromal cells and tumour niche cells engage in cross-talk, resulting in enhanced HNSCC growth when xenografted into recipient animals in vivo. Ji et al. [165] reported a similar secretome profile among MSCs from gingival-derived normal tissue (GMSCs) within the tumour microenvironment from oral cell lines (CAL-27 and WSU-HN6) after in vitro supplementation with anti-inflammatory IL-10. The GMSCs were found to influence the oral cancer cells via direct co-culture and indirect co-culture systems. A direct co-culture cell proliferation assay indicated that GMSCs inhibited the growth and invasion phenotype of oral cancer cells. The conditioned medium derived from the GMSCs (GMSCs-CM) also exerted an anticancer effect, which indicates that soluble factors in GMSCs-CM play a key role in GMSC-induced inhibition of cancer cell growth. Additionally, the study confirmed that GMSCs could act as activators of tumorigenesis through the upregulation of pro-apoptotic and cell death proteins including p-JNK, cleaved PARP, cleaved caspase-3 and Bax, and the downregulation of proliferation- and anti-apoptosis-related proteins such as p-ERK1/2, Bcl-2, CDK4, cyclin D1, PCNA and survivin. Wang et al. [166] also identified B2M in the secretome of BM-MSCs from oesophageal TE1 and Eca109 cell lines. This interesting study used B2M-encoding gene knockdown to demonstrate that the gene played a part in the invasion and migration of HNSCC cells. Scherzed et al. [167] also obtained interesting data on secretome-based pro-cancer mechanisms in an HNSCC HLaC78 cell line. The study investigated whether human mesenchymal stroma cells (hMSC) support cell motility and cytokine secretion. Interestingly, hMSC enhanced FaDu and HLaC78 cell invasiveness. Cancer cell motility was increased by cytokines such as IL-6, IL-8 and VEGF. Moreover, the inhibition of IL-6 in the MSC secretome decreased HNSCC cell proliferation, which was partly dependent on the MAPK/ERK signalling pathway. Similarly, exposure of human tongue squamous cell carcinoma (TSCCa and CAL-27) cell lines to the MSC secretome resulted in a significant increase in CCN2 in BM-MSCs. However, in tumour-derived MSCs, only CCN2 inhibited cancer cell proliferation, mobility and invasion, and decreased the levels of MMP-9, MMP-2 and epithelial-mesenchymal transition markers in vitro. It is also not surprising that higher expression of CCN2, the connective tissue growth factor (CTGF) was noted in HNSCC tissues than in normal adjacent non-cancerous tissues and this may contribute to the higher aggressiveness of TSCC cells via the promotion of tumour development [168].
There is also great interest in the chemopreventive and therapeutic potential of secretome components and MSC cell activity in HNSCC cancers, one of the most common malignancies of the head and neck area [169,170,171,172,173,174]. Importantly, several recent studies have reported that biomolecules released from the secretome to the neoplastic niche may not only determine the proliferation, growth and invasiveness of head and neck tumours, but also death resistance. Most importantly, a therapeutic strategy has been proposed in which MSCs obtained from different tissues can be loaded in vitro with anti-cancer drugs [169]. For example, MSCs have been isolated and expanded from gingival papilla (GinPa-MSCs) and infused with three important anti-neoplastic drugs: Paclitaxel (PTX), Doxorubicin (DXR) and Gemcitabine (GCB) [169]. The results clearly demonstrate that GinPa-MSCs efficiently absorbed these chemotherapeutics and then expelled them into the tumour milieu in their active form. The drugs were delivered in specific amounts intended to produce the stem cell growth factor-beta (SCGF-β), which inhibits proliferation of human SCC154 oral squamous cell carcinoma cell line growth in vitro. Also, Wang et al. [170] found bone-marrow mesenchymal stem/stromal (BM-MSCs) cells to have anti-apoptotic effects when co-cultured with human derived oropharyngeal squamous carcinoma JHU-12 and JHU-019 (OPSSC) cells. This phenomenon was associated with the activation of PDGFR-α/AKT mediated signalling pathways. This paracrine-mediated PDGF-AA/PDGFR-α signalling highlighted the chemotaxis of MSCs in OSCC. Moreover, the enhancement of the PDGFR-α/AKT pathway by MB-MSCs promoted the expression of anti-apoptotic Bcl-2 and decreased sensitivity to Cisplatin. However, OPSCC-derived JHU-012 cells grown in co-culture with MSCs were significantly more susceptible to CDDP following pretreatment with the receptor tyrosine kinase Crenolanib, a PDGFR-α inhibitor, compared to cancer cells grown alone. Another interesting study is by Liu et al. [171], who showed that isolated BM-MSCs actively interacted with HNSCC cancer cells in vitro (SCC-25 cells) and in vivo, and this interaction intensified the key mechanisms responsible for tumour progression and growth, and drug chemoresistance. Parental head and neck cancer cells, either fused with MSCs or exposed to MSCs, were orthotopically transplanted into the tongues of mice. The fused cancer cells demonstrated more intense mesenchymal cell features, i.e., higher expression of POSTN, GDF11, IGFBP5 and CXCL11, and downregulation of DAPK1, as well as greater proliferation and viability. Moreover, the HNSCC cells incubated with MSC were associated with a more aggressive course of neoplastic disease compared to the parental cell line. Interestingly, a key condition for the transmission of signals from growth factor receptors to regulate gene expression and prevent apoptosis was found to be the PI3K/PTEN/AKT signalling cascade. Importantly, all HNSCC cell lines exposed to MSCs developed resistance to Paclitaxel, which persisted for up to 30 days after the initial co-incubation period. Liu et al. [172] analysed the mechanisms of BM-MSCs involved in promoting the development, progression, invasion, and metastasis of head neck cancer cells (CAL-27 and HM-4) and the tumour-promoting role of periostin (POSTN) on HNC. In vitro data derived from on HNC cells cultured in the presence of BMMSC-conditioned medium (MSC-CM) indicated that the stem cell determines cancer progression by increasing cell proliferation, migration, epithelial-mesenchymal transformation (EMT), and by blocking apoptosis and altering the expression of proteins regulating the cell cycle. Most importantly, BM-MSCs promoted HNC aggressiveness through the PI3K/AKT/mTOR signalling pathway, which was mediated by periostin. As previously mentioned, IL-6 present in the MSC secretome, or produced directly in tumour milieu, may promote EMT and the acquisition of epithelial stem-like cell properties in ameloblastoma epithelial cells (AM): an aggressive odontogenic neoplasm [173,175,176]. An in vitro study of an AM carcinogenesis model by Jiang et al. et al. [173] confirmed increased levels of the pro-tumorigenic and pro-angiogenic cytokine IL-6 in supernatants from isolated mesenchymal stromal cell (AM-MSC) culture. The supernatants inhibited cell proliferation, promoted differentiation, and inhibited epithelial differentiation of the epithelial cells (AM-EpiCs) from follicular AM, thus increasing pre-malignant lesions and accelerating the process of carcinogenesis by chemical carcinogenesis. The secretome of the ameloblastoma-derived MSCs, which contained angiogenic IL-8, functioned through MAPK/STAT3/SLUG signalling pathways and SNAIL-l, Vimentin and ZEB1 factors. Another work on oral mucosal MSC-derived exosomes (OM-MSC-EVs) and their potential therapeutic target in oral premalignant lesions and OSCC cancer cells was presented by Li et al. [174]. The findings indicate that the proliferation and migration of oral leukoplakia with dysplasia mesenchymal stromal cells (LK-MSC) were down-regulated compared with normal oral mucosa (N-MSC), and oral carcinoma (Ca-MSC) cells. It can also be seen that the exosomes secreted by LK-MSCs play an important and essential role in promoting proliferation, migration, and invasion in vitro. Interestingly, microarray analyses of MSC-derived exosomes confirm the presence of microRNA-8485 (miR-8485) ni the MSC-derived exosomes. Exosomal miR-8485, present in both leukoplakia and cancer cells, enhanced the proliferation, migration and invasion of cancer cells under in vitro co-culture conditions. Shi et al. [177] analysed the probable factors determining the progression of oropharyngeal premalignant lesion to NPC carcinoma in vitro. It was found that the isolated BM-MSC-EVs significantly regulate fibroblast growth factor-19 (FGF-19); they therefore act as a potent regulators of nasopharyngeal carcinoma cell lines (CNE1, CNE2, 5-8F and 6-10B) via the FGF19-FGFR4-dependent ERK signalling cascade, and by modulating EMT. Another interesting in vitro study by Hong et al. [178] found that Gremlin-1 overexpression markedly promoted the proliferation and invasion of human oesophageal squamous cell carcinoma (OESSC) in ECa109 and TE-1 cell lines and xenograft tumour models. In addition, shRNA silencing of GREM1 mRNA in MSCs (shGREM1-MSCs) inhibited and then reversed the increased malignancy of OSCC, and medium conditioned with shGREM1-MSCs (shGREM1-MSCs-CM) blocked the cell-cycle process and cell invasion in vitro. The experimental shGREM1-MSCs-CM-induced anti-tumour stem cells effects seem to be controlled by the TGF-β/BMP4 (transforming growth factor-β/bone morphogenetic protein-4) signalling pathway, which was also associated with a decrease TGF-β and Smad-2 and Smad-3 activity, and an increase in. BMP4, Smad-1, Smad-5 and Smad-8 expression. Nakayama et al. [179] report an increase in aggressive phenotype and pro-tumorigenic interactions between adipose-derived MSCs (A-MSCs) and OSCC EC-GI-10 (well-differentiated type) and TE-9 (poorly differentiated type) cell lines in vitro. Pro-neoplastic activity was positively associated with the expression of phosphorylated-insulin-like growth factor-1 receptor (p-IGF1R) and negatively associated with the human epidermal growth factor receptor 2 (EGFR-2) in OSCC cancer tissues. The authors suggest that co-culture of A-MSCs and cancer cells may be a pro-tumorigenic factor promoting neoplastic invasion and increasing the level of MMP-9 and laminin. Similar data were also presented by Wang et al. [180], who reported that fusion of human umbilical cord-derived mesenchymal stem cells (UC-MSCs) with human oesophageal carcinoma cell lines (EC9706) noticeably blocked the carcinogenesis of OESCC. A comparison of the gene expression profiles of human mesenchymal stem cells, oesophageal cancer cells and hybrids indicated that the OECs-hMSC fusion induced apoptosis and benign trans-differentiation. Moreover, fusion also strongly increased the activity of dual specificity phosphatase 6 (DUSP6)/mitogen-activated protein kinase phosphatase-3 (MKP-3), the key regulators in p38MAPK pathway, and exogenous overexpression inhibited tumour growth.
Some in vitro analyses have found mesenchymal stromal cells to have the opposite effects on a number of pathways associated with cancer and processes in the head and neck region [165,181]. For instance, Ji et al. [165] found MSCs derived from normal gingival tissue (G-MSCs) to inhibit the proliferation of oral squamous cancer cells (CAL-27 and HN6 OSCC cell lines) in vitro and in vivo. MSC-EVs from the secretome present in the studied co-culture systems down-regulated OSCC cells by inducing neoplastic cell death and blocking proliferation. Interestingly, the G-MSC secretome down-regulated the expression of genes associated with proliferation and anti-apoptosis activity, such as p-ERK1/2, Bcl-2, CDK4, Cyclin D1, STAT3, PCNA and survivin, and ERK signalling pathways; it also up-regulated JNK cascade and expression of pro-apoptotic genes including JNK, cleaved PARP, cleaved caspase-3, which negatively regulate of the cell cycle and tumour proliferation and increased angiogenesis. Moreover, treatment with MSC secretome blockade and JNK signalling inhibitor increased cancer cell proliferation in vitro. The dual role of human MSCs on tumour cell growth, mainly their anti-cancer effect, was also presented by Li et al. [181]. This in vitro study analysing the effect of the BM-MSC secretome in oesophageal squamous cell carcinoma cell lines (Eca-109) found that hMSC-conditioned medium repressed the proliferation and invasion of Eca-109 cells, arrested cell-cycle in the G1 phase and intensified the apoptosis of OESCC in vitro in a co-culture system. Treatment with the conditioned medium also reduced the expression of PCNA antigen, cyclin E, pRb protein, Bcl-2, Bcl-xL and MMP-2, and blocked the formation of cyclin E-cyclin 2 (CDK2)-dependent kinase complexes.
Several important studies have also found MSC-mediated immunomodulation to have pro- or anti-tumorigenic potential in HNC models [182,183,184,185]. For example, Liotta et al. [182] found that HNSCC-derived MSCs inhibited the proliferation of CD4+ and CD8+ T cells and promoted the downregulation of INF-γ and TNF-α expression. Interestingly, mesenchymal cells isolated from tumours co-expressed CD29, CD105, and CD73, but not CD31, CD45 and CD133; they also presented human epithelial antigen like bone marrow-derived MSCs (BM-MSCs). Furthermore, HNSCC-isolated MSCs were also characterized by significant immunosuppressive activity on in vitro stimulated T cells, mainly mediated by indoelamine 2,3-dioxygenase (IDO) activity. Moreover, the abundance of cancer-derived MSCs was directly correlated with tumour volume and inversely with the frequency of tumour-infiltrating leukocytes (TILs). Similar conclusions were also presented by Mazzoni et al. [183] who highlight the involvement of MSC IDO-1 in the immunosuppression of the proliferation of HNSCC-derived MSC-mediated T cells in an HNC model. Also, MSCs derived from head and neck cancers inhibited the function and proliferation of T lymphocytes and suppressed the T cell immune response via the down-regulation of amino acid oxidase, known as IL-4 induced gene 1 (IL4I1) and the catabolic products such as H2O2, and kynurenines activation detected in various types of cancer cells. The study also demonstrated that neutralization of IL4I1 activity can block tumour cell migration and restore effective anti-tumour immunity. Another interesting study by Schuler et al. [184] investigated the effect of CD39 and CD73 expression in HNSCC-derived MSCs generated from tumour tissue, and autologous MSCs from healthy control tissue. It proposed that the conversion of extracellular ATP (eATP) to immunosuppressive adenosine (ADO) by the functionally-active ectonucleotidases CD39 and CD73 constituted an immunosuppressive mechanism used by hematopoietic immune cells. Furthermore, MSCs from tumours demonstrated lower CD39 and CD73 protein expression compared to non-cancerous tissue, and this expression correlated with decreased ATP metabolism and the suppression of CD4+ T-cell proliferation. CD39 and CD73 may also constitute a potential novel checkpoint inhibitor of targets due to their tumorigenic action [186]. Allard et al. report that in response to conditions typically occurring in neoplastic disease, such as hypoxia, various cells in the tumour microenvironment acquire adenosine-generating capabilities; these include cancer cells, cancer-associated fibroblasts (CAF), endothelial cells, CD4+CD25+Foxp3+ Tregs, Tr1 cells, Th17 cells, γδ T cells, NK cells, invariant cells (i)NKT, effector and memory T cells, B regulatory cells (Breg), myeloid-derived suppressor cells (MDSC), macrophages and neutrophils. In turn, the described molecular mechanism increased survival of tumour cells and metastases, promoted angiogenesis, increased fibrosis, and up-regulated the suppressive function of Tregs, Tr1, macrophages and MDSCs; by doing so, it also promoted antigen tolerance, inhibited the effector function of lymphocytes and prevented the differentiation of memory T cells into effector cells, facilitating tumour growth. Therefore, the authors predict that adenosinergic and other purinergic-targeting therapies may have clinical application, and their development in combination with other anti-cancer modalities may result in promising future therapeutic approaches. Similar conclusions were presented by Rowan et al. [187], who demonstrated that the use of human adipose tissue-derived stromal cells (A-MSCs) promotes the migration and early metastasis of human CAL-27 and SCC-4 head and neck cancer cell lines and NUDE mouse xenografts. The authors observed that MSCs create an inflammation-induced and tumour-friendly microenvironment through downregulated expression of CD73 and metabolism of ATP, which inhibited T cell proliferation and activity among CD4+ and CD8+ lymphocytes, induced TAM M1 polarization and higher Treg cell immunosuppressive function.
Taken together, these findings suggest that selective inhibition of MSC function in the TME, or the blockade of key signalling pathways for their activity may constitute a viable treatment strategy to combat tumorigenesis and chemoradioresistance; however, such development requires further mechanistic and translational research in head and neck cancers.
However, although in vitro models are valuable for obtaining new information, they cannot fully mimic the complex tumour microenvironment. In contrast, in vivo or animal models such as mouse xenograft models have an advantage in that they can mimic the tumour niche and key intercellular interactions, i.e., communication with stromal cells, stem cancer cells and cells of the innate and adaptive immune system. Such limitations of in vitro studies should always be taken into account when drawing final conclusions regarding the role of mesenchymal cells in any form of cancer, including head and neck cancers. Nevertheless, in vitro studies offer the advantage that the analysed cells are exposed to a relatively homogeneous environment. This affords the researcher ample opportunity to study the effects of constant oxygen levels, induced hypoxia, nutrient composition and a conditioned medium of MSCs (MSCs-CM), as well as limited interactions with other cells.
Table 1 summarizes selected in vitro studies regarding the role of MSCs in the tumour microenvironment included in the review, and their findings.
3.1.2. In Vivo and Animal Models of HNSCC
Various animal and in vivo models have also been used to explore the role of MSCs in the development and progression of head and neck squamous cell cancer (HNSCC). Mouse and hamster studies have found mesenchymal stromal cells to regulate the initiation and growth of HNC and its lymph node and distant metastases [168,172,178,189,192,193,194,195,196]. For instance, Liu al. [172] investigated whether bone marrow mesenchymal stem cells (BM-MSC) are recruited to the tumour microenvironment and have tumour-promoting effects in a murine model of HNC carcinogenesis induced with periostin; BM-MSC was found to promote tumour development, invasion, degree of aggressiveness, lymph node metastases and shorter survival. It was related to enhanced expression of POSTN and epithelial-mesenchymal transition (EMT) in cancer tissues. The POSTN mRNA level was also higher in CAL-27 cell lines of BM-MSC-HNSCC tumours, which was associated with high pathological grade and proliferation rate, tumour volume and lymph node metastasis. The researchers suggest that their findings were dependent on the activity of important molecular pathways such as POSTN-mediated PI3K/AKT/mTOR signalling and N-cadherin activity. Similar results regarding the pro-tumorigenic potential of MSCs were presented by Hong et al. who examined the effect of Gremlin-1 (GREM1) and cell communication network factor 2 (CCN2) in ECa109, TE-1 cell lines and xenograft tumour models of oesophageal squamous cell carcinoma (ESCC) [178]. The findings indicate that tumour-derived MSCs demonstrated a strong ability to promote tumour EMT and cell invasion, and that GREM1 and CCN2 were significantly overexpressed in human ESCC tissues. Moreover, the conditioned medium from mesenchymal stromal cells (GREM1-MSCs-CM) also enhanced the malignancy of xenograft oesophageal tumours in vivo, and increased cell-cycle proliferation, viability and invasion of ESCC in vitro, partly through the TGF-β/BMP signalling pathway. Another interesting in vivo study by Meng et al. [196] evaluated the potential of interactions between tumorous cells obtained from surgical resection, normal oral cells and their surrounding stromal microenvironment to induce tumorigenesis and progression for oral squamous cell carcinoma. The study also analysed the potential targets for therapeutic intervention for OSCC. The data indicated that tumour formation in CG2, HSC-2, and Tca8113 cells infected with lentivirus expressed enhanced levels of TGF-β receptor III (TβRIII), and this molecule was an important potential epithelial-mesenchymal common target. A recent study by Jiang et al. [173] determined that MSC-derived IL-6 contribute to the pathogenesis and progression of ameloblastoma (AM). Interestingly, both in vivo and in vitro studies on fresh tumour samples confirmed that AM-MSC-derived IL-6 enhanced the levels of EMT factors and stem cell-related genes in epithelial cells from follicular AM (AM-EpiCs). Furthermore, the biological actions of the mesenchymal stromal cells were stimulated via the STAT3 and ERK1/2-mediated signalling pathways or by the SLUG gene. The researchers additionally noted that the growth of AM was inhibited by a specific inhibitor of STAT3 or ERK1/2, or by knockdown of SLUG gene expression; this appeared to have the effect of downregulating the expression of EMT- and stem cell-related genes in AM-EpiCs. Shi et al. [177] analysed the effect of bone-marrow mesenchymal stem cell-derived exosomes (BM-MSC-EVs) in the development and progression of nasopharyngeal carcinoma (NPC) in a model of female NOD/SCID mice subcutaneously inoculated with NPC CNE1 and CNE2 cells to induce cancer. It was found that activation of the EMT markers, and stimulation of the fibroblast growth factor (FGF19-FGFR4)-dependent ERK signalling cascade resulted in the greatest facilitation of proliferation, migration and tumorigenesis.
Other publications have examined the effects of various carcinogens in animal HNC carcinogenesis models. These have confirmed the modulatory pro-tumorigenic effect of MSCs, which play a significant role in tumour progression, metastasis, and cancer recurrence, further supporting their potential role in targeted cancer prevention [192,197]. For example Chen et al. [192] propose that various mesenchymal stem cells of different origins, such as normal mucosa-derived MSCs (N-OMSC), dysplasia-derived MSCS (D-OMSC), cancer-derived MSCs (C-OMSC), and the corresponding BM-MSCs, may be involved in tumour formation in oral carcinogenesis by inhibiting T CD3+ and CD45+ cell numbers and proliferation. The experimental carcinogen 4-nitroquinoline-1-oxide (4NQO) initiated dysplasia and cancerous lesions in the oral cavity of female Sprague-Dawley rat OSCC model. The suggested cause of the pro-tumour activity was an increase in the proportion and proliferation capacity of oral lesion-derived MSCs, which effectively reduced the proportion of T immune cells and significantly immunosuppressed their activity associated with oral mucosa malignancy. Furthermore, increased expression of chemokines CCL21 and CXCL12, and SDF1 was noted in the secretome from cancer tissue-derived MSCs. Interesting results were also reported by Kumar et al. [197] who analysed the expression of adipokine, chemerin (RARRES-2) and its receptor (ChemR23) in myofibroblasts (CAMs) and other squamous cell oesophageal cancer stromal cells, and examined their role in recruitment of bone marrow-derived MSCs and tumour progression. The results of the in vitro experiment and xenograft model indicated that chemerin stimulation of MSCs enhanced the phosphorylation of p42/44 and p38, as well as JNK-II kinases and their inhibitors, and PKC reversed chemerin-stimulated MSC migration. Moreover, in a xenograft model consisting of OE21 oesophageal cancer cells and oesophageal squamous cancer-associated myofibroblasts, CCX832 was found to inhibit the homing of intravenously-administered MSCs. The researchers conclude that RARRES-2 secreted by CAMs constituted a potential chemoattractant for MSCs, and its inhibition may delay tumour progression.
However, several publications fail to confirm that MSCs have a pro-tumour effect in HNCs [165,194,195,198]. For instance, Ji et al. demonstrated that conditioned medium derived from GMSCs (GMSCs-CM) showed a strong anti-cancer effect through inhibiting the growth of OSCC [165]. The authors analysed the role of MSCs derived from normal gingival tissue (MSCs-GMSCs) in regulating the proliferation and growth of oral cancer cells (OSCC) in an animal model of male BALB/C nude mice and by direct co-culture and indirect co-culture systems in vitro. Furthermore, it was also confirmed that the intracellular mechanisms responsible for inhibiting tumour growth were related to increased levels of pro-apoptotic genes including JNK, cleaved PARP, cleaved caspase-3 and Bax, and decreased proliferation and reduced expression of anti-apoptosis-related genes such as ERK1/2, Bcl-2, CDK4, cyclin D1, PCNA and survivin. Similar conclusions were presented by Bruna et al. [194], who applied multipotent stromal cells at precancerous stage of oral squamous cell carcinoma (OSCC) in Syrian hamsters after topical application of the mutagen 7.12-dimethylbenz-alpha-anthracene (DMBA) in the buccal pouch. The authors noted that the allogeneic bone marrow-hamster-derived MSCs (BM-MSCs) prevented oral carcinogenesis via inhibition of cancer growth and epithelial dedifferentiation. Moreover, the local administration of mesenchymal cells into the hamster oral mucosa reduced tumour mass and volume, showed anti-proliferative (Ki-67) and pro-apoptotic (caspase 3 cleaved) activation, inhibited angiogenesis (ASMA) and decreased local inflammation (leukocyte infiltration) and differentiation (CK1 and CK4) in animals treated with MSCs compared to untreated ones; it also down-regulated the activation of pro-tumoral gene expression in precancerous lesions. Interestingly, the same team also studied the anticancer effect of systemic intracardial administration of allogeneic BM-MSCs with regard to the initiation and further development of precancerous conditions of OSCC; in this case cancer was induced in Syrian golden hamsters by topical application of DMBA in a single buccal pouch [195]. The authors observed that precancerous lesions progressed from hyperplasia to dysplasia, from dysplasia to papilloma, and from papilloma to carcinoma within four weeks; however, in animals injected with low and intermediate MSC doses, this process was not initiated or up-regulated by systemic administration of MSCs at the hyperplasia or dysplasia stages. All animals treated with MSCs developed OSCC after 13 weeks of treatment, and this condition remained dependent on high doses of mesenchymal cells. Moreover, hamsters receiving BM-MSCs at the hyperplasia plus dysplasia lesion stage and the papilloma stage were significantly less likely to develop OSCC than the control animals. The researchers concluded that injection of low and medium systemic doses of allogeneic MSCs, administered in the early stages of oral carcinogenesis, do not aggravate the progression and growth of precancer lesions. However, further tumour growth was associated with high doses of BM-MSCs in the later stages of OSCC, and this was related to the presence of persistent chronic inflammation and the intensification of immunosuppressive phenomena inhibiting antitumor defence mechanisms. Also, Tan et al. [198] analysed the tumorigenic potential in mesenchymal-stem/stromal-cell-derived small extracellular vesicles (MSC-sEV) in athymic nude mice with FaDu human head and neck cancer xenografts using immortalized E1-MYC 16.3 human ESC-derived mesenchymal stem cells. Interestingly, the intraperitoneal injection of immortalized MSCs transformed with a proto-oncogene (MYC) did not appear to have a pro-tumorigenic role in initiation or anchorage-independent growth at pre- or post-exosome production of HNC tumours in an animal model. The findings indicate that MSC transformation did not confer tumorigenicity on the HNC cancer cells. Moreover, the selected exosome production method did not affect cell growth and did not contribute to the generation of tumour-promoting MSC exosomes. Nevertheless, immortalizing MSCs for exosome production may allow the production of safe exosome preparations for therapeutic applications, but further extensive research is needed.
At the end of this chapter it is worth highlighting that in vitro research or in vivo studies on knockout mice and transgenic mice and hamsters provides ongoing important information on the importance of mesenchymal stem cells (MSCs) in classical target tissues. They also highlight the role of MSCs in HNC initiation, growth and development, including their effects on cancer progression, carcinogenesis and immunomodulation. The vast majority of recent data suggest that the interaction between tumour cells and MSCs within the tumour niche plays a significant role in tumour expansion and nodal or distant metastases, and thus might be exploited for therapeutic intervention. However, further studies in larger cohorts with standardized experimental protocols are needed to confirm this. It should be noted that mouse xenograft models can accurately imitate the tumour microenvironment occurring in real conditions. Such a “natural” cancer niche allows us to observe real, important interactions and communication with stromal cells, cancer stem cells and immunocompetent cells, allowing us reliable and practical conclusions to be obtained. Unfortunately, important limitations regarding the observations and results of this type of research must be taken into account. These may result from the use of MSCs from different sources, including HNSCC of different origins, as well as different or heterogeneous experimental protocols. Furthermore, in in vivo studies, bias and alternative conclusions may also arise from inter alia the heterogeneity of patient samples, insufficient sample sizes of patient and control comparison groups, short post-treatment periods or variable follow-up times. Some may also fail to take into account smoking addiction and excessive alcohol consumption in patients with HNSCC. Also, the studies may be based on different populations from heterogeneous ethnic groups with varying degrees of risk of carcinogenesis in the head and neck region, and who may be exposed to different environmental carcinogens. Additionally, many studies use different analytical endpoints, demonstrate fundamental differences in methodological standardization and employ different research methods. Such variation can result in inconsistent data, even when the same mesenchymal cells are used, and can limit the possibility of generalizing the final results.
Table 2 presents selected animal/in vivo studies on MSCs in the tumour microenvironment described in this review, and the data collected from them.
3.2. The Pharmacological Strategies of MSC-Based Treatment for Human Tumours. MSCs as Carriers of Anti-Tumour Therapeutic Biological Compounds and Their Clinical Application for Oncological Therapy
Over the past decade, research has focused on the potential use of pleiotropic mesenchymal stem cells (MSCs) as highly specialized “Trojan horses” that can deliver biological anti-tumorigenic molecules, interleukins and agents of interferons, drugs or prodrugs to primary tumour cells or tumour milieu or metastatic tumours. The research has attracted great interest primarily due to the fact that MSCs have an innate and induced ability to migrate to the cancer environment or convert MSCs to other cancer-associated cells in the tumour niche. The most common and promising strategies in the production of MSCs are based on genetic engineering: such approaches could transfer various types of therapeutics or biological agents to the TME or directly to neoplastic lesions, thus inhibiting early initiation or further development of the tumour. Thanks to the constant improvement in genetically-manipulated MSCs, the last decade has seen a very rapid development of cell therapies using various derived mesenchymal cells for oncological applications. An increasing number of preclinical and realized clinical phase I and II studies in various human cancers indicate that these specific pluripotent cells may have potential importance in personalized cell therapies because they can be easily obtained through minimally-invasive procedures and then rapidly scaled up [52,200,201].
To date, the ClinicalTrials.gov website [data from ClinicalTrials.gov., U.S. National Library of Medicine, 30.04.2024] lists over fifty registered preclinical and clinical trials that have used MSCs in the treatment of various cancer diseases. Among these studies, only one, a phase I study (NCT0207932), analysed the involvement of MSCs as a therapeutic agent for the direct treatment of head and neck cancer. However, it should be clearly emphasized that the remainder of the works, of varying methodologies, focus on the use of MSCs of various origins for treating irradiation-induced salivary dysfunction, such as hyposalivation and xerostomia/dry mouth in patients with head and neck cancers (for instance PROSPERO CRD42021227336, NCT04489732, NCT047765392, NCT03874572, MESRIX-SAFE, MESRIX, MESRIX-II and MESRIX-III studies). The above-mentioned works will be discussed in the next section [202,203,204,205,206,207,208,209,210,211,212,213,214].
Several preclinical studies have been performed on other human cancers and neoplastic lesions. For example, human umbilical cord-derived MSCs (UC-MSCs) transduced with adenoviral vectors expressing IL-18, IFN-β and other key cytokines such as TRAIL (TNFSF10), as well as key anti-angiogenic agents, pro-apoptotic proteins and growth factor antagonists, effectively inhibited tumour cell proliferation, cancer initiation and development and the formation of metastases; they were also found to induce apoptosis [52,215,216]. Similarly, genetically engineered TRAIL-expressing adipose-derived mesenchymal stem cells (A-MSCs-TRAIL+) created by lentiviral transductions have also shown potent anti-tumour effects in various cancer types, such as glioblastoma, hepatocellular carcinoma and haematological malignancies, such as acute lymphocytic leukaemia, or chronic myelogenous leukaemia [217,218,219,220]. In addition to cytokines and suicide proteins, several other proteins that inhibit carcinogenesis have also been used in anticancer engineering of MSCs. For example, MSCs with positive expression of bone morphogenetic protein 4 (BMP-4) and phosphatidylinositol 3,4,5-triphosphate-3-phosphatase (PTEN) also effectively inhibited tumour growth, induced cell cytotoxicity, and significantly prolonged survival in mouse models of various human cancers [219,221,222].
Interestingly, oncolytic adenoviruses (Ads) also have potential applications in cancer therapy due to their ability to replicate and induce programmed cell death in cancers. Unfortunately, their clinical use has been severely limited due to the lack of use of effective cell-based Ads delivery systems that could protect the transferred molecules from attack by immunocompetent cells, thus preventing virus clearance through antibody neutralization [219]. One particularly interesting study reported the use of polymeric nanoparticle-engineered human adipose-derived mesenchymal stem cells (hA-MSCs) overexpressing the cancer-specific TNF-related apoptosis-inducing ligand (TRAIL) to target tumours in mice. The authors observed that following transplantation of patient-derived orthotopic tumour xenografts to a mouse model, engineered MSCs expressing suicide protein TRAI exhibited long-range directional migration toward tumours in patient-derived GBM orthotropic xenografts; they also showed significant inhibition of neoplastic growth, induction of apoptosis, thus reducing the occurrence of microsatellites, and prolonging animal survival [220]. An interesting concept of anticancer therapy is the “loading” of MSC cells with other types of oncolytic viruses, which are chemotactic to various tumour cells. Such oncolytic virotherapy represents another promising alternative and effective anti-tumour therapeutic role for mesenchymal cells. One study examined the use of MSCs infected with an oncolytic adenovirus, e.g., ICOVIR5 (Celyvir) in the treatment of a murine CMT64 cell line, syngeneic for human lung cancer, as a human adenovirus-semi-permissive tumour model. The researchers found mouse Celyvir (mCelyvir) to demonstrate a significant homing capacity to CMT64 tumours. Interestingly, the combined treatment based on mCelyvir and intratumoural injections of ICOVIR5 was found to act by inhibition of neoplastic growth and induction of CD4+ and CD8+ T cell recruitment to the tumour microenvironment [223]. Another interesting study examined the use of menstrual blood-derived mesenchymal stem cells (MenSCs) infected with the CRAd5/F11 chimeric oncolytic Ads and transplanted in a mouse tumour model. This novel virus delivery platform inhibited cancer progression in a subcutaneous mouse xenograft model of human colorectal cancer [224]. Also, stem cell-released variants of the oncolytic herpes simplex virus (MSC-oHSV) demonstrated noticeable therapeutic efficacy in human advanced brain melanomas with metastatic processes in a relevant immunocompromised and immunocompetent mice tumour model. Interestingly, the authors found that intracarotid administration of MSC-oHSV effectively targeted metastatic neoplastic lesions and significantly prolonged the survival of tumour-bearing C57BL/6 mice. Moreover, the combination of MSC-oHSV with anti-PD-L1 immunotherapy also increased the abundance of tumour-infiltrating IFN-γ-producing CD8+ T cell subpopulation and was associated with a significant increase in the median survival of treated animals [225].
Another modern method for treating animal or human cancers is the use of MSCs carrying anticancer payloads and drugs [52,226,227,228,229]. This technique takes advantage of the fact that MSCs have been found to effectively incorporate drugs and then release them in an active form, and in sufficient amounts, to inhibit various squamous cell carcinomas in vitro. For example, conditioned medium with MSCs originating from gingival papillae (GinPa-MSCs) which had been treated with Paclitaxel (PTX), Doxorubicin (DXR) and Gemcitabine (GCB), was found to be effective against a line of tongue squamous cell carcinoma (SCC154). The authors emphasize that compared to other sources of MSCs, acquiring GinPa-MSC is minimally invasive, and the stem cells can be easily expanded and effectively loaded with anti-cancer drugs, establishing an effective “cellular drug delivery system”. Moreover, drug-loaded gingival mesenchymal stromal cells, particularly with a cargo of GCB, significantly hindered growth and showed anti-proliferative effects against a tongue squamous cell carcinoma SCC154 cell line. This was also accompanied by significantly higher expression of hENT1, the main carrier involved in the transport of gemcitabine in cancer cells. The study indicates that such anti-cancer strategies may be used as a base for future application in oral oncology [226].
Recent studies have also examined the development of therapeutic strategies aimed at improving the loading capacity and efficiency of MSCs. One promising approach to increasing the anticancer effectiveness of MSCs loaded with anti-cancer drugs is based on nanoparticles. Recent studies clearly indicate that drug-encapsulated nanoparticles offer many therapeutic benefits, such as the ability to accumulate in tumour tissues or the neoplastic milieu, mitigate the non-specific toxicity of anti-cancer drugs, prevent sudden uncontrolled release and limit side effects [227,228,229]. In one study, engineered mesenchymal stem cells with drug-loaded nanoparticles carrying Paclitaxel (PTX) were tested against an A549 orthotopic lung tumour model. Despite the use of much lower doses of PTX, the nanoengineered MSCs significantly inhibited tumour growth and improved the survival of immunocompetent C57BL/6 albino female mice bearing orthotopic Lewis Lung Carcinoma (LL/2-luc) [227]. Similar observations were obtained from MSCs loaded with poly(D,L-lactide-co-glycolide) (PLGA) nanoparticles encapsulated with Paclitaxel (PTX-PLGA) for orthotopic glioma therapy in male Sprague Dawley rats. It was found that the MSCs initiated with PTX-PLGA nanoparticles demonstrated significantly greater and prolonged release of PTX in the form of free nanoparticles compared to those initiated with PTX. Moreover, transfer of Paclitaxel from MSCs to tumour mass strongly induced neoplastic cell apoptosis in vitro. Further, the animals demonstrated significantly longer survival after implantation of PTX-PLGA nanoparticle-loaded MSCs than in the case of injection of PTX-based MSCs or PTX-PLGA nanoparticles alone [228]. Another interesting study found that transactivation of transcription (TAT) functionalization of paclitaxel-loaded PLGA polymeric nanoparticles reduced the intracellular accumulation and retention of nanoparticles in laboratory-prepared mesenchymal MSCs in both primary tumours and metastases in a mouse orthotopic model of lung cancer. Moreover, the therapeutic use of nanoengineered MSCs increased MSC viability, inhibited cancerogenic growth and improved overall survival in Fox Chase SCID Beige mice compared to the use of the free or nanoparticle-encapsulated drug [229]. In summary, the data suggests that MSCs bearing nanoparticles may represent an effective potential vehicle for tumour-specific delivery of anticancer drugs, resulting in significantly improved therapeutic efficacy.
Another set of molecules that may represent a potential strategy in the therapy of human cancers are the MicroRNAs (miR), due to their ability to modulate post-transcriptional gene expression. In vitro studies and animal models use MSCs that “carry” a variety of miRs packed into extracellular vesicles (MSC-EVs) and deliver them to neoplastic cells or neighbouring tumour niche cells to induce a therapeutic anti-cancer effect. MSC-derived exosomes may thus be used as delivery vehicles to transfer genetic materials and biomolecules, such as mRNA, DNA and non-coding RNAs, as well as oncolytic adenoviruses (Ads), viral vectors and anti-neoplastic drugs homing to specific sites and recipient cells [52,230,231,232,233]. In one preclinical study, researchers used lentiviral vectors to construct ex vivo cultured bone marrow–derived MSCs as natural biofactories for exosomes carrying miR-124a, and these were applied against multiple patient-derived glioma stem cell (GSC) lines and in a male athymic nude mice (nu/nu) model [231]. Furthermore, in vitro therapeutic use of GSCs with exosomes containing miR-124a (Exo-miR124) led to a biologically significant reduction in GSC viability and clonogenicity compared to control systems, and the in vivo treatment of mice with intracranial GSC267 after systemic administration of Exo-miR124 resulted in half of the experimental GSC xenografts demonstrating long-term survival. Interestingly, it appears that miR-124a acted by silencing Forkhead box (FOX)A2, a known target of miR-124a, and that apoptotic cell death correlated with FOXA2-mediated aberrant intracellular lipid accumulation. A similar study examined the in vitro delivery of exogenous miR-124 to glioblastoma multiforme (GBM U87) cells by human umbilical cord Wharton’s jelly MSCs (WJ-MSCs) [232]. The WJ-MSCs were characterized by functionally significant exosome-dependent or -independent anti-cancer effects associated with decreased CDK6 target gene luciferase activity; they also inhibited U87 cell proliferation and neoplastic cell migration, and increased the chemosensitivity of GBM cells to Temozolomide (TMZ) in endometrial cancer treatment. These findings were confirmed in a later study showing that human umbilical cord mesenchymal stem cell (hUC-MSCs)-derived extracellular vesicles inhibited endometrial cancer (EC) cell proliferation and migration by delivering exogenous tumour suppressor miR-302a through a pronounced neoplastic-homing ability [233]. The researchers reported that miR-302a levels were significantly decreased in EC cancer tissues compared to adjacent non-cancerous tissues. It is believed that the miR-302a overexpression in the cancer cells inhibited cell proliferation and migration, both of which were blocked in cancer cell culture with miR-302a-loaded extracellular vesicles derived from hUC-MSCs. Additionally, laboratory-modified miR-302-rich MSC-EVs significantly inhibited pro-tumorigenic cyclin D1 expression and suppressed the AKT signalling pathway in EC cancer cells in vitro. This suggests that exogenous miR-302a delivered by EVs has great potential as an effective anti-cancer therapy.
Unfortunately, MSC-based therapies for various human cancer diseases also have significant technical and biological demands. The successful engraftment of MSCs transduced with adenoviral vectors or MSCs “carrying” oncolytic adenoviruses or anticancer payloads and drugs, and their satisfactory survival, remains problematic. One potential solution is to use appropriate biomaterials, such as Gliadel, or a thermos-responsive biodegradable paste, preferably with their own anti-cancer or repair activity, as a scaffold to improve the retention of transplanted stem cells. Such frameworks have been found to demonstrate anti-cancer and repair effects in the tumour niche or in inflamed or damaged tissue in vitro and in vivo [234,235,236,237,238]. A number of studies have described new methods for delivering therapeutic MSCs to biomaterials for the treatment of specific human cancers and the pathological conditions [239,240]. One such study examined the implantation of biodegradable fibrin scaffolds of seeded MSCs into a resection cavity after postoperative brain cancer surgery. The results confirmed the removal of residual tumour cells which could be a cause of later local recurrence, as well as improved anti-cancer MSC persistence as well as longer cancer-free survival [239]. Another study proposed the use of an innovative immunotherapeutic organoid using human mesenchymal stromal cells (hMSCs) genetically modified to secrete bispecific anti-CD33-anti-CD3 antibody (bsAb); these cells were placed in a small biocompatible star-shaped poly(ethylene glycol)-heparin container. The organoid demonstrated slow release of bispecific antibodies and enabled effective minimally-invasive immunotherapy in acute myeloid leukemia (AML). The macroporous biohybrid cryogel platform effectively increased the proliferation and survival of the MSCs, allowing them to release bsAb over a longer period of time in vitro and in vivo. Moreover, the experiment led to sustained active release of bsAb, resulting in high levels capable of inducing a T cell-mediated anti-tumour response and rapid regression of CD33+ blasts in AML [240]. Interstitial implantation of alginate-encapsulated cell expressing a soluble form of leucine-rich repeat and immunoglobulin-like domain 1 (sLrig1) significantly inhibited development and growth of patient-derived glioblastoma multiforme in a mouse orthotopic xenograft model [241]. The usage of Slrig1, a negative regulator of the oncogenic epidermal growth factor receptor (EGFR) family, disrupted downstream signalling in both wild-type and constitutively-active EGFR mutated glioma cells (EGFRvIII) in vitro and in vivo. Interestingly, further noticed effectors included MAP kinase but not AKT signalling.
Hence, initial research suggests that mesenchymal stem cells may be promising therapeutic reference points. In the future, they may be used to treat various diseases, including cancer, due to their ability to inhabit damaged tissues and differentiate into various types of cells and their pleiotropic effect. However, their potential value in the treatment of human cancers is hampered by the fact that preclinical and clinical studies indicate they demonstrate both anticancer and pro-tumour effects, which constitutes important limitations for further research. Despite these significant limitations, current analyses indicate that the latest MSC-based therapies offer considerable anti-tumour potential in human patients, and may represent effective personalized anti-cancer therapies. Among MSC-based therapies, the most promising challenge in developing effective minimally-invasive cancer treatment is the use of MSCs as “Trojan horses” to deliver various therapeutic agents to the tumour niche or neoplastic cells. Another ongoing problem is that the interactions occurring between MSCs and cancer cells are relatively poorly understood, and further knowledge is needed of the activities occurring between them, and thus to improve their safety as therapeutic strategies. In this regard, the use of MSC-derived extracellular vesicles (MSC-EVs) as “cell-free carrier” therapy is becoming an increasingly promising option to remove or mitigate the risks associated with the use of live cells. It is worth noting that in practice, highly-effective MSC-based therapies constitute an acceptable option for known anti-cancer therapeutic procedures, both as mesenchymal cells directly targeting the destruction of the neoplastic lesions and regulating tumour niche remodelling, and as a way of minimizing the side effects of cancer treatments, such as chemoradiotherapy-induced xerostomia (NCT03874572, cardiomyopathy (NCT02509156), Cisplatin-induced acute renal dysfunction (NCT01275612) or radiation-induced haemorrhagic cystitis (NCT0284864), etc. Despite continual progress and the growing body of research on using delivery MSCs, it is difficult to identify clear published clinical studies that could be directly translated into clinical outcomes, which unfortunately hinders further progress in the therapeutic application of MSC-based therapies. Nevertheless, the use of MSC implants is promising and seems to be a safe potential alternative to other therapeutic strategies. Finally, it should be noted that despite the limitations and engineering difficulties that researchers encounter, further in-depth research will hopefully eliminate the complications and symptoms associated with the use of MSC-based for various origin human cancers. An increasing number of cell-free MSC therapy studies indicate that there is a real hope to generate a safe and effective therapeutic product that can inhibit or kill tumour cells, thus improving the survival and quality of life of patients in the advanced stages of this devastating and irreversible disease [52,242].
Most intensive and wide-ranging studies on the influence of MSCs on cancer development and growth, their immunomodulatory abilities and potential anti-cancer therapies are based on in vitro and in vivo 2D human cancer co-culture systems, most of which have employed isolated BM-derived and adipose tissue-derived MSCs. It should be noted, however, that in the last few years, new approaches using more complex 3D in vitro models have also been proposed. Although preclinical 3D dynamic culture systems are still under initial development, they more closely mimic the features of the tumour microenvironment in vivo and interest in them is constantly growing [242,243]. A summary of the various available preclinical studies with 3D models, as well as their limitations, are presented in more detail in a recent publication by Avnet et al. [243].
Another important problem may be the immunogenic phenotype of MSCs, which may have a double-edged effect if the tested cellular systems are not properly controlled in vitro and in vivo. It is believed that resident MSCs can acquire immunosuppressive properties upon exposure to elevated levels of pro-inflammatory cytokines, and then provide support for tissue repair and inhibit tumour initiation and growth through the secretion of TME components. Unfortunately, in the presence of cytokines, i.e., IFN-γ. IL-6, IL-8, SDF-1α and TNF-α, or tumour-derived agents, i.e., IL-10, IDO, TGF-β, and in response to signals generated by direct contact of cancer cells and MSCs, mesenchymal stromal cells may adopt an immunosuppressive phenotype that affects both innate cells, and develop adaptive and facilitates the tumour’s escape from immune surveillance. Moreover, the anti-tumour phenotype of MSCs may also facilitate neoplastic progression via the production of chemokines such as CXCL1 / 5 / 6 / 7 / 8, CCL5 and growth factors such as VEGF, EGF and PDGF; this can result in the acquisition of key genetic and epigenetic changes by the cancer cells that may protect them against cytotoxic cells and drugs, and promote metastasis. However, it is important that, as noted above, genetically-manipulated MSCs can also have significant anti-cancer effects by delivering and expressing various anticancer agents, including type I interferon (IFN-α and IFN-β), CXCL1, IL-2, IL-12, cytokine deaminase, oncolytic virus, TRAIL and nanoparticles, thus reducing the risk of treatment failure. Another important problem is the natural heterogeneity of MSCs, which requires researchers to precisely identify subpopulations of MSC cells in the tumour milieu and determine their mutual relationships to other immune cells and TME compounds. It also remains unsatisfactory to determine whether the derived MSC cell types are stable or transient under experimental conditions and whether one subpopulation can transform into another in response to various microenvironmental stimuli; it is also unclear what proportion of the stromal response to injury or cancer is directed by MSCs rather than other more differentiated stromal cells [52,242].
Also, it must be considered that autologous or allogeneic MSCs actively interact with components of tumour microenvironment, and despite several studies, the therapeutic benefits of autologous or allogeneic MSCs remain inconclusive. This may also indicate that the functional plasticity and heterogeneity typical of MSCs caused by different donors and MSC subpopulations of different origin, as well as interactions with cells present in the niche, may be the reason for the poor results observed in some studies. Allogeneic and autologous MSCs offer many important advantages, e.g., donor selection, unrestrictive cell dose, hypo-immunogenicity, immediate availability, cost effectiveness, better safety and greater suitability for immunocompromised patients. Unfortunately, both MSC selection methods have significant drawbacks: allogeneic transplantation is associated with potential immune rejection, donor-donor heterogeneity, specific immunological memory and quick clearance after infusion, while autologous MSCs are characterised by long-time availability, limited cell dose, donor variability issues and prohibitive cost. However, on balance, despite their own merits and limitations, the allogeneic approach seems to be superior [244,245,246].
Therefore, to optimize anti-cancer treatment, it is necessary to thoroughly understand the mechanisms of action of MSCs, determine their role in new therapies using pleiotropic and multifunctional cells, and define new biomarkers enabling the prediction of response to therapy; these will be of key importance in developing alternative treatment regimens and allowing accurate patient stratification. New research into the use of alternative types of MSCs to deliver therapeutic agents to the tumour niche or neoplastic cells will open up new fields in cancer treatment, including HNC, although the clinical benefits remain unclear.
The current and new discoveries in the field of MSC-based therapy, and potential therapeutic targets in human cancers of various origin, are summarised in Figure 2.
3.3. The Pharmacological Strategies of MSC-Based Treatment for HNC
Most patients with squamous cell head and neck carcinomas (HNSCC) are in an advanced stage (Stage III/IV) due to late detection, and are treated with radiochemo-therapy in this region. This type of complex anti-cancer therapy is frequently associated not only with very failures, resulting in shortened 5-year and overall survival, cancer-free survival and local recurrences and distant metastases, as well as problems with speech and swallowing, oral infection and dental caries. The latter significantly reduces quality of life, and can give rise to complications such as salivary gland hypofunction (MARSH) and xerostomia (RIX), subjectively named dry mouth or Sjögren syndrome following radiotherapy [202,247]. Despite the use of increasingly more precise and effective radiological methods, i.e., intensity-modulated radiation-therapy (IMRT), most patients with HNC experience dose-dependent damage and degeneration of salivary gland parenchyma cells and decreased saliva production [202,247,248,249]. To identify alternative treatments, several studies recorded in ClinicalTrials.gov. have evaluated the use of easily-accessible multipotent and pleiotropic adult progenitor mesenchymal cells in anti-neoplastic therapy and in the preventive treatment of xerostomia [U.S. National Library of Medicine, available on April 30, 2024]. Importantly, most of these studies focus on bone marrow (BM-MSCs) and stromal cells isolated from adipose tissue (A-MSCs), which demonstrate high therapeutic potential due to their specific anti-tumorigenic features, i.e., trophic properties, anti-inflammatory, anti-apoptotic activity, pro-angiogenic characteristics and immunomodulatory effects. These properties make the cells a very promising direction of research for use in anti-cancer therapy and as a source of cells that can promote the regeneration of the salivary gland parenchyma and restore radiation-induced damage. Unfortunately, despite progress in various preclinical in vivo models and human studies, only a few publications have examined the use of MSC transplantation as a potentially curative treatment option in HNC [202,203,204,205,206,207,208,209,210,211,212,213,214].
Most importantly, it should be clearly emphasized that there is currently only a single MSC-application-based research has evaluated the safety and effectiveness of genetically-engineered mesenchymal stem cell therapy as a complement for standard treatment in patients with head and neck cancer (NCT02079324) [52]. A phase I clinical study (NCT02079324), by a team of Korean scientists evaluated the maximum tolerable dose, safety and efficacy of intratumorally injected GX-051 therapeutic agent, a genetically-modified mesenchymal stem cell treatment, against HNC. The clinical study started in 2014 and had only a few secondary outcomes: anti-tumour response by Response Evaluation Criteria in Solid Tumours (RECIST 1.1) on computed tomography, changes of INF-γ and IL-12 levels in blood by ELISA compared to baseline after GX-051 intratumoral injection, assay of immune cells (inter alia CD4+ T cell, CD8+ T cell, NK cell) by FACS on day 1 (baseline), day 29 (end of treatment) and day 57 (follow up); safety profile was examined by vital sign, physical examination, clinical laboratory tests, and CT of NHC region [data from ClinicalTrials.gov., U.S. National Library of Medicine, available on April 30, 2024]. Although the research was planned to be completed in 2015, currently no results have been posted [52].
It should be noted that all existing prospective, randomized and controlled preclinical in vitro and animal models and clinical in vivo or human tissue studies, evaluate the use of MSCs derived from various origins for treating irradiation-induced salivary dysfunction (MARSH) and xerostomia (RIX) in specific head and neck cancers [202,203,204,205,206,207,208,209,210,211,212,213,214]. Many of these are reviewed in the present paper, particularly those concerning radiotherapy-induced xerostomia, e.g., the PROSPERO study (www.crd.ac.uk/prospero), registration number CRD42021227336, and I/II phase clinical studies such as NCT04489732, NCT047765392, NCT03874572 [202,203,204,205,206,207,208,209,210,211,212,213,214]. However, as an analysis of all available publications exceeds the scope of this study, only the most interesting and promising articles were included, based on data obtained from ClinicalTrials.gov., U.S. National Library of Medicine (available on April 30, 2024) and PubMed/Medline/EMBASE Library database and Cochrane Database of Systematic Reviews.
One of the interesting most recent qualitative meta-analyses was study number CRD42021227336 on PROSPERO by Carlander et al. [202]. It examined the effect of mesenchymal stromal/stem cell therapy on Salivary Flow Rate (SFR) in experimentally radiation-induced salivary gland hypofunction in preclinical interventional in vivo models. The analysis included a total of 16 in vivo preclinical studies in animal models and 13 meta-analyses conducted in animal experiments. The researchers used MSCs derived from bone marrow (BM-MSCs), adipose tissue (A-MSCs) and salivary gland tissue (SG-MSCs), which were administered intravenously, intraglandularly or subcutaneously. The summary results of the included studies indicated that MSC-based therapy significantly increased SFR. Furthermore, the results of the preclinical in vivo indicated that treatment restored salivary gland functionality and regenerated tissues following radiotherapy in acinar tissue, vascular areas, and paracrine functioning without reported serious adverse events. Additionally, the most noticeable effect on SFR was observed when MSCs were provided through intraglandular administration compared to systemic transplantation. The meta-analysis also discusses the importance of the time from radiation to administration of MSCs, which was directly related to the number of apoptotic cells [250,251,252,253]. The authors unanimously emphasized that transplantation of isolated tissue-specific human stem cells to radiation-damaged salivary glands rescued hyposalivation, restored acinar and duct cell structure, and decreased the amount of apoptotic cells. These observations indicated that MSC-based intraglandular therapy could be protective, especially in the acute period of radiological treatment [250,251,252,253]. The researchers also discussed two important studies that assessed the effect of intravenously administered MSC therapy on the structure and function of salivary glands after high-dose radiation therapy in head and neck cancer [253,254]. The studies presented similar final conclusions, indicating that transplantation of MSCs by intravenous infusion in the animal models immediately after local radio-irritation significantly increase salivary gland weights, improve SFR and acinic cell function; it also resulted in higher amylase production and micro-vessel densities in MSC-treated salivary glands than in those that only received irradiation. Moreover, in the examined groups of experimental animals, systemic transplantation of mesenchymal cells resulted in increased colonization of salivary glands, which was associated with protection against irradiation-induced cell loss and induction of trans-differentiation into glandular cells. Similar conclusions from the meta-analysis protocol were presented by Jansson et al. [203], conducted in PROSPERO project CRD42021227336, based on data from MEDLINE/PubMed and Embase Library databases and validated according to the peer review of electronic search strategies (PRESS). An objective systematic review including animal intervention studies confirmed the efficacy and safety of MSC implantations in treating post-radiation salivary gland function disturbances and xerostomia. Moreover, multipotent adult MSC-cell therapy based on intra-glandular MSC implantation, intravenous and/or intramuscular injections was shown to enhance the unstimulated SFR, achieving positive changes in salivary gland morphology, cytoprotection, apoptosis and organ vascularity. Importantly, however, the authors of these meta-analyses emphasized the high heterogeneity among the included studies with regard to MSC derivation, species, strains, age, radiation-therapy dose, dosage protocols, administration route of MSC therapy, frequency of treatment and time between radiation and first treatment, as well as frequent insufficient group size; this variation can result in significant limitations. Moreover, it was impossible to analyse the relationship between radiotherapy duration and the effectiveness of MSC therapy due to insufficient data. These limitations should always be taken into account when translating results and conclusions to a clinical setting.
Moreover, and importantly, several in vivo preclinical studies and randomized placebo-controlled phase I/II clinical trials have analysed the safety and efficacy of autologous MSCs treatment for radiation-induced xerostomia (RIX) and restoration of salivary hypofunction (MARSH) in humans, including NCT04489732, NCT047765392 and NCT03874572. The MARSH and RIX symptoms are frequently observed in human cancer patients, and this can be a prognostic factor that can also determine the quality of life for head and neck carcinoma. For example, two studies relating to the use of MSCs as a therapeutic option in the restoration of salivary hypofunction in HNC in humans were recently published by Blitzer et al. [209,211,212]; they were conducted under an approved Food and Drug Administration Investigational New Drug application using an institutional review board–approved protocol (NCT04489732). The study was performed as a pilot, first-in-human study. It described the clinical and morphological effects of a single injection of autologous bone marrow-derived mesenchymal stromal cells (BM-MSCs) into the right submandibular gland stimulated with INF-γ for the treatment of radiation-induced xerostomia [211]. The proposed therapy was found to be safe and well tolerated, and the endpoints analysis showed a clear tendency towards increased saliva production, which persisted in 50% of patients both one and three months after MSC injection; this was accompanied by improved quality of life indicators. The same group of researchers used the same innovative approach to treat RIX and MARSH and improve quality of life in a phase I dose-escalation trial of patients with xerostomia and salivary hypofunction after radiation therapy for head and neck cancer [212]. This analysis, registered as NCT04489732 by the World Health Organization International Clinical Trials Registry Platform, examined the effect of injecting the pro-growth secretome of IFN-γ-stimulated marrow-derived autologous stromal cells (BM-MSCs) taken from patients with HNC who had undergone radiation or chemoradiotherapy into the submandibular gland. A total of 21 to 30 subjects (9 to 18 in phase I study, 12 in expansion cohort) were included in the study group. The study included two endpoints: the dose-limiting toxicities occurring within one month of the submandibular region BM-MSC injection. The outcomes of saliva amounts and composition, ultrasound images of the salivary glands, and the quality of life from 3 to 24 months after treatment were recorded. The researchers indicate that such autotransplantation of BM-MSCs stimulated with IFN-γ into the salivary glands after radiotherapy or chemoradiotherapy could be an innovative method of treating RIX and MARSH, which also translates into restoring a better quality of life. A recent study by Blitzer et al. [209] analysed the functionality of bone marrow mesenchymal stromal cells (BM-MSCs) derived from HNC patients in a FDA-IND enabling study regarding MSC-based treatments for RIX. In this pilot clinical study, bone marrow aspiration was performed in HNC patients who had completed radiotherapy two or more years earlier; the aim was to isolate and culture MSCs after IFN-γ stimulation. The MSCs were additionally implanted in mice with radiation-induced xerostomia and changes in salivary gland histology and saliva production were examined. The results of this preliminary study clearly indicated that autotherapy with IFN-γ-stimulated MSCs in HNC patients led to the acquisition of an immunosuppressive mesenchymal stem cell phenotype and higher protein expression, i.e., GDNF, WNT1, and R-spondin 1 as well as pro-angiogenesis and immunomodulatory cytokine activity. Moreover, in a mouse model, MB-MSC injection after radiation down-regulated the loss of acinar cells, decreased the formation of fibrosis, and increased salivary production. Another noteworthy study weas the analysis performed by Grønhøj et al. [210], who examined the safety and efficacy of MSC-based therapy for radiation-induced xerostomia in a randomized, placebo-controlled phase I/II trial (MESRIX). This interesting randomized trial included 30 patients who had previously received radiotherapy for a T1-2/N0-2A, human papillomavirus-positive (HPV+) oropharyngeal squamous cell carcinoma (OPSCC) and who underwent autologous adipose tissue-derived mesenchymal stem cell (A-MSCs) therapy. In this analysis, the investigators also noted that the use of A-MSC therapy was substantially effective for radiation-induced hypofunction and xerostomia, and led to significantly improved salivary gland secretion, increased whole Salivary Flow Rates (SFR), reduced RIX and MARSH symptoms and improved patient-reported outcomes for months compared to the placebo arm. Interestingly, core-needle biopsies conducted in the trial confirmed up-regulation in serous gland tissue, decreases in adipose and connective tissues in the ASC-arm compared to the placebo-arm; however no differences between groups in gland size or intensity were observed. Importantly, the authors did not observe any adverse events of this therapeutic procedure.
Also very interesting from a clinical point of view are three recent publications on phase I/II randomized trails (MESRIX-I and MESRIX-II) in patients with radiation-induced xerostomia by Lynggaard et al. [207,208]. The first is an investigator-initiated, randomized, single-centre, placebo-controlled trial (MESRIX-I; EudraCT number: 2014-004349-29) involving the injection of allogenic A-MSCs or placebo into both submandibular glands in patients with oropharyngeal squamous cell carcinoma (OPSCC) followed by radiotherapy for a minimum of two years. The study primary endpoint was the observation of serious adverse events after MSC-based treatment. The secondary endpoint was the presence of an entire SFR and local symptoms associated with RIX. The results indicated that during follow-up, no side effects appeared to be related to the MSC treatment. Moreover, the authors observed that whole saliva flow rate increased and OPSCC patient-reported xerostomia symptoms decreased in the patients [208]. In the second study, Lynggaard et al. [207] examined the effectiveness of intraglandular off-the-shelf allogeneic adipose tissue derived mesenchymal stem cell (A-MSCs) therapy in HNC individuals with salivary gland hypofunction (MESRIX-II; EudraCT number: 2018-003856-19). In this safety study, the occurrence of adverse events and the unstimulated and stimulated saliva SFR indicators and composition of saliva were assessed based on the results obtained from the EORTC QLQ-H&N35 and the XQ xerostomia questionnaires, as well as on blood samples and salivary gland scintigraphy. The authors noted no treatment-related serious adverse events during a four-month observation period; they also report a relevant increase in saliva flow rates, a reduction in the feeling of dry mouth and improved swallowing based on patient-related data. These results were presented in another phase I trial using allogeneic MSCs for radiation-induced hyposalivation and xerostomia (MESRIX-SAFE) by the same authors; the study is registered as NCT03874572 according to ClinicalTrials.gov. [U.S. National Library of Medicine, available on April 30, 2024]. This open-label clinical study confirmed the safety and feasibility of local submandibular gland therapy with use of the autologous adipose-derived mesenchymal stem cells from healthy donors in previous oropharynx cancer patients. A nonrandomized, open-label, phase I exploratory study by Strojan et al. also examined the positive impact of MSC-based therapy as a potential medical procedure in patients with head and neck tumours [205]. The researchers present a trial clinical protocol used to evaluate the safety and preliminary efficacy of allogeneic MSCs derived from human umbilical cord tissue (hUC-MSCs). The study itself assessed the clinical effect of hUC-MSC implantation under ultrasound guidance into both parotid glands and both submandibular glands. It included 10 oropharyngeal cancer patients with post-radiation xerostomia and no evidence of disease recurrence during two or more years after chemoradiation (intervention group) and 10 healthy volunteers (control group). The initial data, obtained four months post-procedure, indicated that allogeneic hUC-MSCs treatment yielded positive and expected effects regarding salivary flow and composition, scintigraphic evaluation of MSC grafting, retention and migration, and positive subjective survey data regarding xerostomia and quality of life in the OPSCC patients. The authors confirm that this is a potential and innovative approach to the treatment of RIX syndrome, not only due to the improvement of the functional and morphological features of the salivary tissue, but also to its non-invasive collection procedure, flexibility of cryobanking and biological advantages.
Following from of the MESRIX studies above, Jakobsen et al. [206] present a single-centre, double-blinded, randomized, placebo-controlled, phase II study aimed at investigating the safety and efficacy of mesenchymal stem cell for MARSH and RIX in previous head and neck cancer patients (MESRIX-III), and was registered as NCT04776538 at the ClinicalTrials.gov. [U.S. National Library of Medicine, available on April 30, 2024]. The study began in March 2021 and was completed in September 2023. It included 120 participants who previously had been treated with radiotherapy. The participants were divided into two groups of equal numbers (60:60 in HNC individuals vs. placebo) and received ultrasound-guided injection of either allogeneic adipose-derived mesenchymal stem cell (A-MSCs) or placebo into the submandibular glands. The primary endpoint was the observation of unstimulated whole Saliva Flow Rate (SFR). The secondary endpoints included change in the flow rate of stimulated whole saliva, quality of life based on EORTC QLQ Module for H&N-35 and XQ questionnaires, saliva composition and immune response determined in blood samples (human leukocyte antigen - HLA response) to stem cells, before treatment and at four and twelve-month follow-up. Importantly, the final results and conclusions have already been presented [204]. The analysis of the obtained phase II placebo-controlled trial data clearly indicated that the use of allogeneic A-MSC local submandibular gland therapy resulted in a statistically significant SFR up-regulation, decrease in sticky saliva, fewer notable swallowing difficulties and radiation-induced xerostomia symptoms compared with pretreatment baseline and placebo. Hence, injecting A-MSCs into the submandibular salivary gland may be a promising therapeutic procedure with high effectiveness and safety and may constitute a potential new method of treating hyposalivation and salivary gland regeneration in patients after radiotherapy [204,206].
Other studies have also analysed the possible mechanisms of action of MSCs on salivary gland tissue, favouring higher proliferative activity, greater tissue remodelling, higher density of acinar SG cells and lower post-radiation inflammation and fibrosis [202,256,257,258]. In vitro analysis data confirms that the use of intraglandular MSC transplantation increases the expression of epithelial markers (KRT7 and KRT18) and genes related to organ structure (SMR3A, AMY2A5, PRB1, AMY1, CLDN22, PRPMP, AMY1A, AQP5, α-SMA and CD31), as well as the genes encoding proteins involved in cell migration, survival and differentiation (SDF1-CXCR4 and Bcl-2) [256,257,258]. It also appears that a variety of growth factors and other active biomolecules, including VEGF, HGF, COX-2, BDNF, GDNF, EGF, IGF1, NGF, FGF10 and MMP-2 are responsible for angiogenesis and paracrine function, and promoting tissue repair and restoring the salivary glands [202]. A study of the consequences of intraglandular allogeneic AT-MSC-based therapy and induced changes in the salivary proteome of irradiated HNC patients was performed by Lynggaard et al. [213]. The results indicate the presence of significant differences in over a hundred human proteins associated with post transplantation tissue regeneration between the saliva of the HNC patients with radiation-induced hypofunction and that of healthy controls. Moreover, the authors noted an increase in regenerative effects on the salivary proteome; however, this proteome did not return to a healthy state when compared to control individuals.
In conclusion, numerous studies clearly indicate that MSC transplantation offers various significant benefits in head and neck cancers, leading to better regenerative outcomes by contributing to various aspects of salivary gland tissue repair including their cell proliferation, migration, angiogenesis and weight gain. Unfortunately, it should also be emphasized that large-scale standardized research and further extensive systematic reviews and meta-analyses and meticulous studies on in vitro and animal models, as well as comprehensive, randomized, well-planned clinical trials in humans, are necessary to definitively determine the clinical value and effectiveness of MSC-based therapy methods.
However, it is also necessary to mention the important limitations of these analyses. Although numerous preclinical in vitro and in vivo studies based on various animal models have been carried out to date and promising results have been obtained, it is difficult to indicate clear and conclusive findings regarding the role of MSCs in the stages of carcinogenesis and tumour progression in HNC. The majority of recent research indicates that MSCs have the ability to modulate phenomena directly and indirectly related to cancer occurring in the tumour microenvironment, i.e., apoptosis, differentiation, proliferation, invasion and metastasis, angiogenesis and growth factor signalling in HNC, as well as other cancers. Unfortunately, the ability of active MSCs of various tissue sources to regulate the initiation of cancer or modulate its course often differs from the preclinical data obtained in experimental models. This may be due to many reasons. Firstly, there may not have been any methodological standardization of mesenchymal stem cell activity assessment, translational research model or analytical protocol, or laboratory method. Secondly, many preclinical studies do not take into account certain factors that may have a significant impact on the pro- or anti-tumour role of MSCs, i.e., the use of cells from different regions of the head and neck and different types and origins of tumours. It is also important that the some signalling pathways through which MSCs can interact and determine the function of various cells, such as other populations of stromal cells, stem cancer cells and cells of the innate and adaptive immune system in the cancer microenvironment are not yet fully known and understood. Hence, the preclinical researcher cannot accurately predict the structural and functional differences in metabolic competences and pathways/biomarkers related to the stages of carcinogenesis modulated by MSCs with respect to the clinical analyses and final clinical conclusions.
It is important to emphasizing is that currently, unfortunately, only symptomatic treatment is available for patients suffering from radiation-induced xerostomia and hypofunction, and hyposalivation of salivary gland. Therefore, clinical trials using mesenchymal stromal/stem cells from various sources in modern potential MARSH and RIX treatment strategies in cancer patients are currently of great interest. Existing data from preclinical in vivo studies, systematic reviews, meta-analyses and I/II phase clinical trials clearly confirms that MSC therapy has a significant impact on the restoration of salivary gland function and tissue regeneration after radiotherapy. Moreover, no serious side effects have been observed so far, and intraglandular transplantation was associated with a significantly better therapeutic effect than systemic transplantation. Therefore, in summary, MSC-based therapy shows significant therapeutic potential in the treatment of xerostomia and radiotherapy-induced hyposalivation, but large-scale, sufficiently large, randomized human trials are needed to confirm its effectiveness in clinical settings.
4. Conclusions
The main aim of this review article was to summarize the current state of knowledge regarding the possible importance of mesenchymal stromal/stem cells MSCs in the initiation, development and progression of head and neck cancers. The work serves as a compendium of knowledge on the etiopathogenesis of HNC related to the activity of MSCs, based on the latest publications and available data on the topic, selected by the author of the article. It describes the mechanisms and anti-oncogenic potential of MSC-based therapeutic options, and their strategies; it also provides an overview of the response to chemoradiotherapy associated with the presence of MSCs, their tropism to the tumour microenvironment and the impact on other populations of immune cells present in the tumour niche. This is a very important issue from the clinician’s point of view; to reflect this, it includes a large number of analyses and in vitro and in vivo experimental studies on animal models and humans. Recent evidence indicates that MSCs may be considered as a potential and promising method for the delivery of anti-neoplastic agents to the tumour milieu or as therapeutic onco-modifiers of genes, or as vehicles for transporting chemotherapeutic drugs. Although the satisfying anti-cancer effect of MSCs-based treatment modification has been proven in many studies, several recent findings indicate that within the microenvironment of cancers of various origins, including HNC, the action of native MSCs may not only inhibit neoplastic disease development, but even have a pro-oncogenic effect. Research data confirm that MSC activity may influence key intracellular signalling, i.e., NF-κB, ERK, MAPKp38, and JNK kinases, subfamilies of the mitogen-activated protein kinases (MAPKs) and by modulating the PI3K/AKT pathway and cell cycle progression at crucial transition points. These intracellular pathways also modulate proliferation and angiogenesis upon stimulation by MSCs, thus reducing the increased effectiveness of chemotherapy against HNC. Moreover, MSCS may also act as anti-inflammatory stimulators, thus influencing immunity, oncogenic cellular signalling and apoptosis inhibition, and by regulating the cell cycle and angiogenesis, enhancing the carcinogenic phenomenon of HNC.
Unfortunately, existing data regarding extensive pre- and clinical analyses and the pleiotropic effects of MSCs are often inconclusive, as confirmed by studies in animal models and on human biological material, and in epidemiological studies. Importantly, data from multicentre research, i.e., molecular studies, the latest pre- and early clinical studies and the most important clinical long-term observational and interventional population studies, as well as key opinion-forming systematic reviews, clearly indicate the need for further research; their findings underline the necessity of assessing the effects of MSCs on the evolution of human cancers, including HNC, and the potential benefits of MSCs-based therapeutic approaches that will be safe and effective in inhibiting tumour growth and progression.
It should be also emphasized that the vast majority of publications on the clinical use of MSC-based therapeutic approaches in HNC concern the positive effects of their use against complications of radiotherapy or chemoradiotherapy, i.e., radiation-induced xerostomia (RIX) and restoration of salivary hypofunction (MARSH). Most publications indicate that the autologous mesenchymal stem cells of various origins transplanted into the submandibular salivary glands have promising molecular activity, which contribute to the restoration of salivary gland function and tissue regeneration, reducing subjective xerostomia and improving the quality of life in HNC patients.
Unfortunately, many of the studies cited in this review present ambiguous results and varying conclusions, which may result from several factors. The inconsistences in assessment and potential systematic bias may be attributed to several clinical limitations, e.g., heterogeneity of clinical material obtained from patients, inadequate sample sizes for comparative groups of patients and controls, as well as insufficiently short or variable observation periods and follow-up times. Also, may studies vary in their use of experimental protocols in in vitro studies, on animal models and in humans, and apply non-standardized questionnaires to assess inter alia the quality of life of cancer-treated patients. Moreover, studies are often conducted between different populations in heterogeneous groups in terms of ethnicity, age and tumour origin; the latter may be characterised by considerable biological differences, and thus ambiguous conclusions. Therefore, further studies with long-term follow-ups, larger study groups, and expanded interventional studies are required to establish definitive conclusions regarding the role of MSCs in carcinogenesis, cancer development and patient prognosis.
One key challenge is to determine the final clinical conditions of nano-engineered MSCs-based therapy as a potentially promising therapeutic method. Such novel virus-delivery and tumour-specific drug delivery platforms offer promise for the effective therapy of various cancers, including HNC, and have been found to significantly improve the anti-cancer efficacy of conventional chemotherapeutic drugs.
Hence, there is as yet no clear indication that treatment with MSC cells has any unequivocal clinical impact. Of course, the best evidence for the benefits of MSC-based therapies in the treatment of cancer would be provided by a randomized controlled trial (RCT), which would be invaluable for providing suitable data and guiding recommendations for MSC therapy in HNC patients. However, to date the numbers of studies and unified experimental and clinical protocols for HNC remain inadequate, and long periods of observation are needed to yield sufficient data.
Author Contributions
Conceptualization, methodology, analysis, investigation, resources, data curation, writing-original draft preparation; writing-review and editing, K.S-K. The author has read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available in a publicly accessible repository.
Conflicts of Interest
The author declare no conflict of interest.
Abbreviations
| A-MSCs | Adipose tissue-derived MSCs |
| B2M | Beta 2 microglobulin |
| BM-MSCs | Bone-marrow-derived MSCs |
| BMP | Bone morphogenetic protein |
| CAFs | Cancer-associated fibroblasts |
| CCN2 | Cellular communication network factor 2 |
| CCL | C-C motif chemokine ligand |
| CDDP | Cisplatin |
| CIS | Carcinoma in situ |
| CSCs | Cancer stem cells |
| CTLs | Cytotxic T cells |
| CXCL | C-X-C motif chemokine ligand |
| Dkk | Dickkopf-related protein |
| DXR | Doxorubicin |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| FGF | Fibroblast growth factors |
| 5-FU | 5-Fluorouracyl |
| GTA | Ganciclovir |
| GCB | Gemcitabine |
| HGF | Hepatocyte growth factor |
| IDO | Indoleamine 2,3-dioxygenase |
| iNOS | Nitric oxide synthase |
| INF-α/β/γ | Type-I interferon alpha/betha/gamma |
| HO1 | Heme oxygenase |
| MARSH | Restoration of salivary hypofunction |
| MAPKs | Mitogen-activated protein kinases |
| MMPs (MT-MMPs) | Matrix metalloproteinases, also known as matrix metallopeptidases |
| MSCs | Mesenchymal stromal/stem cells |
| N-MSC | Naïve MSCs |
| NKs | Activated natural killer cells or CD56dim/CD16bright cells |
| SCGF | Stromal cell growth factor-beta |
| SDF-1 | Stromal cell-derived factor 1 |
| PDGF | Platelet-derived growth factor |
| PGE2 | Prostaglandin E2 |
| PTEN | Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase |
| PTX | Paclitaxel |
| RIX | Radiation-induced xerostomia |
| SFR | Saliva Flow Rate |
| TAMs M1/2 | Tumour-associated macrophages M1/2 |
| TGF-β1 | Transforming growth factor beta 1 |
| TLRs | Toll-like receptors |
| TRAIL | TNF-related apoptosis-inducing ligand |
| Treg | Regulatory T cells, known as suppressor T cells or CD4+CD25+Foxp3+ cells |
| UC-MSCs | Umbilical-cord MSCs |
| VEGF | Vascular endothelial growth factor |
References
- Charap, A.J.; Enokida, T.; Brody, R.; Sfakianos, J.; Miles, B.; Bhardwaj, N.; Horowitz, A. Landscape of natural killer cell activity in head and neck squamous cell carcinoma. J. Immunother. Cancer 2020, 8, e001523. [CrossRef]
- El-Naggar, A.K.; Chan, C.J.; Grandis, J.R.; Takata, T.; Slootweg, P.J. WHO Classification of Head and Neck Tumours, 4th ed.; IARC: Lyon, France, 2017; ISBN 9789283224389.
- The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517(7536), 576–582. [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R. L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71(3), 209–249. [CrossRef]
- Gormley, M.; Creaney, G.; Schache, A; Ingarfield, K.; Conway D.I.. Reviewing the epidemiology of head and neck cancer: Definitions, trends and risk factors. Br. Dent. J. 2022, 233, 780–786. [CrossRef]
- Johnson, D. E.; Burtness, B.; Leemans, C. R.; Lui, V. W. Y.; Bauman, J. E.; Grandis, J. R. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2023, 9(1), 4. [CrossRef]
- 7. Goyal, N.; Day, A.; Epstein, J.; Goodman, J.; Graboyes, E.; Jalisi, S.; Kiess, A. P.; Ku, J. A.; Miller, M. C.; Panwar, A; et al. Head and neck cancer survivorship consensus statement from the American Head and Neck Society. Laryngoscope Investig. Otolaryn. 2021, 7(1), 70–92. [CrossRef]
- Atashi, F.; Vahed, N.; Emamverdizadeh, P.: Fattahi, S.: Paya L. Drug resistance against 5-fluorouracil and cisplatin in the treatment of head and neck squamous cell carcinoma: A systematic review. J Dental Res Dental Clin Dental Prospect. 2021, 15(3), 219–225. [CrossRef]
- Sola, A.M.; Johnson, D.E.; Grandis, J.R. Investigational multitargeted kinase inhibitors in development for head and neck neoplasms. Expert Opin. Investig. Drugs. 2019, 28(4), 351–363. [CrossRef]
- National Comprehensive Cancer Network. Head and neck cancer. 2020. https://www.nccn.org/professionals/ physician_gls/ pdf/head-and-neck.pdf.
- Cancer, I.A.f.R.o. List of Classifications by cancer sites with sufficient or limited evidence in humans, 2019, 1-127. IARC Monographs On The Identification Of Carcinogenic Hazards To Humans. https://monographs.iarc.fr/wp-content/uploads/2019/07/Classification_by_cancer_site_127.pdf.
- Gupta, B.; Johnson N.W.; Kumar, N. Global Epidemiology of Head and Neck Cancers: A Continuing Challenge. Oncol., 2016, 91, 13–23. [CrossRef]
- Ng, J.H.; Iyer, N.G.; Tan M-H.; Edgren, G. Changing epidemiology of oral squamous cell carcinoma of the tongue: A global study, Head Neck. 2017, 39, 297–304. [CrossRef]
- Du, E.; Mazul, A.L.; Farquhar, D.; Brennan, P.; Anantharaman, D.; Abedi-Ardekani, B.; Weissler, M.C.; Hayes, D.N.; Olshan, A.F.; Zevallos, J.P. Long-term Survival in Head and Neck Cancer: Impact of Site, Stage, Smoking, and Human Papillomavirus Status. Laryngoscope 2019, 129(11), 2506–2513. [CrossRef]
- Sun, Z.; Sun, X.; Chen, Z.; Du, J.; Wu, Y. Head and Neck Squamous Cell Carcinoma: Risk Factors, Molecular Alterations, Immunology and Peptide Vaccines. Int. J. Pept. Res. Ther. 2022, 28(1), 19. [CrossRef]
- Miranda-Galvis, M.; Loveless, R.; Kowalski, L.P.; Teng, Y. Impacts of Environmental Factors on Head and Neck Cancer Pathogenesis and Progression. Cells 2021, 10, 389. [CrossRef]
- Saada-Bouzid, E.; Peyrade, F.; Guigay, J. Molecular genetics of head and neck squamous cell carcinoma. Curr. Opin. Oncol. 2019, 31 (3), 131–137. [CrossRef]
- Zhang, Y. X.; Koneva, L. A.; Virani, S.; Arthur, A. E.; Virani, A.; Hall, P. B.; Warden, C. D.; Carey, T. E.; Chepeha, D. B.; Prince, M. E.; et al. Subtypes of HPV-Positive Head and Neck Cancers Are Associated with HPV Characteristics, Copy Number Alterations, PIK3CA Mutation, and Pathway Signatures. Clin. Cancer Res. 2016, 22 (18), 4735–4745. [CrossRef]
- Leemans, C. R.; Snijders, P. J. F.; Brakenhoff, R. H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer. 2018, 18(10), 662. [CrossRef]
- Chow, .LQ.M.; Head and neck cancer. N. Engl. J. Med. 2020, 382, 60–72. [CrossRef]
- Dong, H.; Shu, X.; Xu, Q.; Zhu, C.; Kaufmann, A. M.; Zheng, Z. M.; Albers, A. E.; Qian, X. Current Status of Human Papillomavirus-Related Head and Neck Cancer: From Viral Genome to Patient Care. Virologica Sinica 2021, 36(6), 1284–1302. [CrossRef]
- Wittekindt, C.; Wagner, S.; Bushnak, A.; Prigge, E. S.; von Knebel Doeberitz, M.; Würdemann, N.; Bernhardt, K.; Pons-Kühnemann, J.; Maulbecker-Armstrong, C.; Klussmann, J. P. Increasing Incidence rates of Oropharyngeal Squamous Cell Carcinoma in Germany and Significance of Disease Burden Attributed to Human Papillomavirus. Cancer Prev. Res. (Phila.) 2019, 12(6), 375–382. [CrossRef]
- Zamani, M.; Grønhøj, C.; Jensen, D. H.; Carlander, A. F.; Agander, T.; Kiss, K.; Olsen, C.; Baandrup, L.; Nielsen, F. C.; Andersen, E.; Friborg, J.; von Buchwald, C. The current epidemic of HPV-associated oropharyngeal cancer: An 18-year Danish population-based study with 2,169 patients. Eur J Cancer (Oxford, England: 1990) 2020, 134, 52–59. [CrossRef]
- de Freitas, A. C.; de Oliveira, T. H. A.; Barros, M. R.; Venuti, A. hrHPV E5 oncoprotein: Immune evasion and related immunotherapies. J. Exp. Clin. Cancer Res. 2017, 36. [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2020, 10. [CrossRef]
- Canning, M.; Guo, G.; Yu, M.; Myint, C.; Groves, M. W.; Byrd, J. K.; Cui, Y. Heterogeneity of the Head and Neck Squamous Cell Carcinoma Immune Landscape and Its Impact on Immunotherapy. Front. Cell Dev. Biol. 2019, 7. [CrossRef]
- Lawrence, M. S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S. B.; et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. The Cancer Genome Atlas Network. Nature 2015, 517, 576–582. [CrossRef]
- Seiwert, T. Y.; Zuo, Z. X.; Keck, M. K.; Khattri, A.; Pedamallu, C. S.; Stricker, T.; Brown, C.; Pugh, T. J.; Stojanov, P.; Cho, J.; et al. Integrative and Comparative Genomic Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas. Clin. Cancer Res. 2015, 21 (3), 632-641. [CrossRef]
- Rühle, A.; Grosu, A. L.; Nicolay, N. H. De-Escalation Strategies of (Chemo)Radiation for Head-and-Neck Squamous Cell Cancers-HPV and Beyond. Cancers 2021, 13(9), 2204. [CrossRef]
- Ventz, S.; Trippa, L.; Schoenfeld, J. D. Lessons Learned from Deescalation Trials in Favorable Risk HPV-Associated Squamous Cell Head and Neck Cancer-A Perspective on Future Trial Designs. Clin. Cancer Res. 2019, 25(24), 7281–7286. [CrossRef]
- Amin, M. B.; Greene, F. L.; Edge, S. B.; Compton, C. C.; Gershenwald, J. E.; Brookland, R. K.; Meyer, L.; Gress, D. M.; Byrd, D. R.; Winchester, D. P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA: Cancer J. Clin. 2017, 67(2), 93–99. [CrossRef]
- Gillison, M. L.; Trotti, A. M.; Harris, J.; Eisbruch, A.; Harari, P. M.; Adelstein, D. J.; Jordan, R. C. K.; Zhao, W.; Sturgis, E. M.; Burtness; et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): A randomised, multicentre, non-inferiority trial. Lancet (London, England) 2019, 393(10166), 40–50. [CrossRef]
- Mehanna, H.; Robinson, M.; Hartley, A.; Kong, A.; Foran, B.; Fulton-Lieuw, T.; Dalby, M.; Mistry, P.; Sen, M.; O’Toole, L.; et al. De-ESCALaTE HPV Trial Group. Radiotherapy plus cisplatin or cetuximab in low-risk human papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): An open-label randomised controlled phase 3 trial. Lancet (London, England) 2019, 393(10166), 51–60. [CrossRef]
- Strober, W.; Shishido, S.; Wood, B.; Lewis, J. S.; Jr, Kuhs, K.; Ferris, R. L.; Faden, D. L. Two for the price of one: Prevalence, demographics and treatment implications of multiple HPV mediated Head and Neck Cancers. Oral Oncol. 2020, 100, 104475. [CrossRef]
- Mondino, A.; Vella, G.; Icardi, L. Targeting the tumor and its associated stroma: One and one can make three in adoptive T cell therapy of solid tumors, Cytokine Growth Factor Rev. 2017, 36, 57–65. [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–37. [CrossRef]
- Dogan, V.; Rieckmann, T.; Münscher, A.; Busch, C.J. Current studies of immunotherapy in head and neck cancer. Clin. Otolaryngol. 2018, 43, 13–21. [CrossRef]
- Cuiffo, B.G.; Karnoub, E. Mesenchymal stem cells in tumor development: Emerging roles and concepts. Cell Adh. Migr. 2012, 6, 220–230. [CrossRef]
- Böhrnsen, F.; Fricke, M.; Sander, C.; Leha, A.; Schliephake, H.; Kramer, F.J. Interactions of human MSC with head and neck squamous cell carcinoma cell line PCI-13 reduce markers of epithelia-mesenchymal transition. Clin Oral Investig. 2015, 19(5), 1121–1128. [CrossRef]
- Wei, S.; Li, M.; Wang, Q.; Zhao, Y.; Du, F.; Chen, Y.; Deng, S.; Shen, J.; Wu, K.; Yang, J.; Sun, Y.; Gu, L.; Li, X.; Li, W.; Chen, M.; Ling, X.; Yu, L.; Xiao, Z.; Dong, L.; Wu, X. Mesenchymal Stromal Cells: New Generation Treatment of Inflammatory Bowel Disease. J. Inflamm. Res. 2024, 17, 3307–3334. [CrossRef]
- Bianco, P. Mesenchymal Stem Cells. Annu. Rev. Cell Dev. Biol. 2014, 30, 677–704. [CrossRef]
- Kfoury, Y.; Scadden, D.T. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell. 2015, 16, 239–253. [CrossRef]
- Spees, J.L.; Lee, R.H.; Gregory, C.A. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res. Ther. 2016, 7. [CrossRef]
- Shi, L.; Chen, L.; Gao, X.; Sun, X.; Jin, G.; Yang, Y.; Shao, Y.; Zhu, F.; Zhou, G. Comparison of different sources of mesenchymal stem cells: Focus on inflammatory bowel disease. Inflammopharmacology 2024, 32(3), 1721–1742. [CrossRef]
- Li, C.Y.; Wu, X.Y.; Tong, J.B.; Yang, X.X.; Zhao, J.L.; Zheng, Q.F.; Zhao, G.B.; Ma, Z.J. Comparative analysis of human mesenchymal stem cells from bone marrow and adipose tissue under xeno-free conditions for cell therapy. Stem Cell Res Ther. 2015, 6(1), 55. [CrossRef]
- Takano, T.; Taira, Y.; Suzuki, R.; Matsumoto. H. Immortalized Canine Adipose-Derived Mesenchymal Stem Cells Maintain the Immunomodulatory Capacity of the Original Primary Cells. Int. J. Mol. Sci. 2023, 24(24), 17484. [CrossRef]
- Jin, Q.H.; Kim, H.K.; Na, J.Y.; Jin C.; Seon, J.K. Anti-inflammatory effects of mesenchymal stem cell-conditioned media inhibited macrophages activation in vitro. Sci. Rep. 2022, 12, 4754. [CrossRef]
- Fernández Vallone, V.B.; Romaniuk, M.A.; Choi, H.; Labovsky, V.; Otaegui, J.; Chasseing, N.A. Mesenchymal stem cells and their use in therapy: What has been achieved? Differentiation 2013, 85(1-2), 1-10. [CrossRef]
- Linard, C,; Brachet, M.; Strup-Perrot, C.; Busson, E.; Bonneau, M.; Lataillade, J-J.; Bey, E.; Benderitter, M. Long-term effectiveness of local BMMSCs for skeletal muscle regeneration: A proof of concept obtained on a pig model of severe radiation burn. Stem Cell Res. Ther. 2018; 9, 299. [CrossRef]
- Felker, S.; Shrestha, A.; Bailey, J.; Pillis, D.M.; Siniard, D.; Malik P. Differential CXCR4 expression on hematopoietic progenitor cells versus stem cells directs homing and engraftment. JCI Insight 2022, 7(9), e151847. [CrossRef]
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy 2005, 7, 393–395. [CrossRef]
- Hmadcha, A.; Martin-Montalvo, A.; Gauthier, B.R.; Soria, B.; Capilla-Gonzalez, V. Therapeutic Potential of Mesenchymal Stem Cells for Cancer Therapy. Front. Bioeng. Biotechnol. 2020, 8. [CrossRef]
- Gervois, P.; Struys, T,; Hilkens, P.; Bronckaers, A.; Ratajczak, J.; Politis, C.; Brône, B.; Lambrichts, I.; Martens, W. Neurogenic maturation of human dental pulp stem cells following neurosphere generation induces morphological and electrophysiological characteristics of functional neurons. Stem Cells Dev. 2015, 24(3), 296–311. [CrossRef]
- Bourin, P.; Bunnell, B.; Casteilla, L.; Dominici, M.; Katz, A.; March, K.; Redl, H.; Rubin, J.; Yoshimura, K.; Gimble, J. Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: A joint statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International So, Cytotherapy 2013, 15, 641–648. [CrossRef]
- Dingal, P.C.; Bradshaw, A.M.; Cho, S.; Raab, M.; Buxboim, A.; Swift, J.; Discher, D.E. Fractal heterogeneity in minimal matrix models of scars modulates stiff-niche stem-cell responses via nuclear exit of a mechanorepressor. Nat. Mater. 2015, 14(9), 951–960. [CrossRef]
- Worthley, D.L.; Churchill, M.; Compton, J.T.; Tailor, Y.; Rao, M.; Si, Y.; Levin, D.; Schwartz, M.G.; Uygur, A.; Hayakawa, Y.; Gross, S.; Renz, B.W.; Setlik, W,; Martinez, A.N.; Chen, X.; Nizami, S.; Lee, H.G.; Kang, H.P.; Caldwell, J.M.; Asfaha, S.; Westphalen, C.B.; Graham, T.; Jin, G.; Nagar, K.; Wang, H.; Kheirbek, M.A.; Kolhe, A.; Carpenter, J.; Glaire, M.; Nair, A.; Renders, S.; Manieri, N.; Muthupalani, S.; Fox, J.G.; Reichert, M.; Giraud, A.S.; Schwabe, R.F.; Pradere, J.P.; Walton, K.; Prakash, A.; Gumucio, D.; Rustgi, A.K.; Stappenbeck, T.S.; Friedman, R.A.; Gershon, M.D.; Sims, P.; Grikscheit, T.; Lee, F.Y.; Karsenty, G.; Mukherjee, S.; Wang, T.C. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 2015, 160, 269–284. [CrossRef]
- Smolinska, A.; Chodkowska, M.; Kominek, A.; Janiec, J.; Piwocka, K.; Sulejczak, D.; Sarnowska, A. Stemness properties of SSEA-4+ subpopulation isolated from heterogenous Wharton’s jelly mesenchymal stem/stromal cells. Front Cell Dev Biol. 2024, 12, 1227034. [CrossRef]
- Maeda, K.; Enomoto, A.; Hara, A.; Asai, N.; Kobayashi, T.; Horinouchi, A.; Maruyama, S.; Ishikawa, Y.; Nishiyama, T.; Kiyoi, H;. Kato, T.; Ando, K.; Weng, L.; Mii, S.; Asai, M.; Mizutani, Y.; Watanabe, O.; Hirooka, Y.; Goto, H.; Takahashi, M. Identification of Meflin as a Potential Marker for Mesenchymal Stromal Cells. Sci. Rep. 2016, 6. [CrossRef]
- Aggoune, D.; Sorel, N.; Bonnet, M.L.; Goujon, J.M.; Tarte, K.; Hérault, O.; Domenech, J.; Réa, D.; Legros, L.; Johnson-Ansa, H.; Rousselot, P.; Cayssials, E.; Guerci-Bresler, A.; Bennaceur-Griscelli, A.; Chomel, J.C.; Turhan, A.G. Bone marrow mesenchymal stromal cell (MSC) gene profiling in chronic myeloid leukemia (CML) patients at diagnosis and in deep molecular response induced by tyrosine kinase inhibitors (TKIs). Leuk. Res. 2017, 60, 94–102. [CrossRef]
- Montelatici, E,; Baluce, B.; Ragni, E.; Lavazza, C.; Parazzi, V.; Mazzola, R.; Cantarella, G.; Brambilla, M.; Giordano, R.; Lazzari, L. Defining the identity of human adipose-derived mesenchymal stem cells. Biochem. Cell Biol. 2015, 82, 74–82. [CrossRef]
- Lv, F.J.; Tuan, R.S.; Cheung, K.M.C.; Leung, V.Y.L. Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells. 2014, 32, 1408–1419. [CrossRef]
- Lootens, T.; Roman, B.I.; Stevens, C.V.; De Wever, O.; Raedt, R. Glioblastoma-Associated Mesenchymal Stem/Stromal Cells and Cancer-Associated Fibroblasts: Partners in Crime? Int J Mol Sci. 2024, 25(4), 2285. [CrossRef]
- Mishra, P.J.; Banerjee, D. Activation and differentiation of mesenchymal stem cells. Methods Mol. Biol., Humana Press Inc. 2017, 201–209. [CrossRef]
- Tang, H.; Chu, Y.; Huang, Z.; Cai, J.; Wang, Z. The metastatic phenotype shift toward myofibroblast of adipose-derived mesenchymal stem cells promotes ovarian cancer progression. Carcinogenesis 2019, 41(2), 182-193. [CrossRef]
- Matsumoto, K.; Xavier, S.; Chen, J.; Kida, Y.; Lipphardt, M.; Ikeda, R.; Gevertz, A.; Caviris, M.; Hatzopoulos, A.K.; Kalajzic, I,; Dutton, J.; Ratliff, B.B.; Zhaom, H.; Darzynkiewicz, Z.; Rose-John, S.; Goligorsky, M.S. Instructive Role of the Microenvironment in Preventing Renal Fibrosis. Stem Cells Transl. Med. 2017, 6, 992–1005. [CrossRef]
- Chen, S.; Liang, B.; Xu, J. Unveiling heterogeneity in MSCs: Exploring marker-based strategies for defining MSC subpopulations. J. Transl. Med. 2024, 22(1), 459. [CrossRef]
- Soundararajan, M.; Kannan, S. Fibroblasts and mesenchymal stem cells: Two sides of the same coin?. J Cell Physiol. 2018, 233(12), 9099–9109. [CrossRef]
- Denu, R.A.; Nemcek, S.; Bloom, D.D.; Goodrich, A.D.; Kim, J.; Mosher, D.F.; Hematti, P. Fibroblasts and Mesenchymal Stromal/Stem Cells Are Phenotypically Indistinguishable. Acta Haematol. 2016, 136, 85–97. [CrossRef]
- Kahounová, Z.; Kurfürstová, D.; Bouchal, J.; Kharaishvili, G.; Navrátil, J.; Remšík, J.; Šimečková, Š.; Študent, V.; Kozubík, A.; Souček, K. The fibroblast surface markers FAP, anti-fibroblast, and FSP are expressed by cells of epithelial origin and may be altered during epithelial-to-mesenchymal transition. Cytom. Part A., 2018, 93, 941–951. [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; Deschler, D.G.; Varvares, M.A.; Mylvaganam, R.; Rozenblatt-Rosen, O.; Rocco, J.W.; Faquin, W.C.; Lin, D.T.; Regev, A.; Bernstein, B.E. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [CrossRef]
- Janeczek-Portalska, K., Leferink, A.; Groen, N.; Fernandes, H.; Moroni, L.; van Blitterswijk, C.; de Boer, J. Endothelial differentiation of mesenchymal stromal cells. PLoS ONE 2012, 7(10), e46842. [CrossRef]
- Suila, H.; Hirvonen, T.; Kotovuori, A.; Ritamo, I.; Kerkelä, E.; Anderson, H.; Natunen, S.; Tuimala, J.; Laitinen, S.; Nystedt, J.; Räbinä, J.; Valmu, L. Human umbilical cord blood-derived mesenchymal stromal cells display a novel interaction between P-selectin and galectin-1. Scand. J. Immunol. 2014, 80, 12–21. [CrossRef]
- L Ramos, T.; Sánchez-Abarca, L.I.; Muntión, S.; Preciado, S.; Puig, N.; López-Ruano, G.; Hernández-Hernández, Á.; Redondo, A.; Ortega, R.; Rodríguez, C.; Sánchez-Guijo, F.; del Cañizo, C. MSC surface markers (CD44, CD73, and CD90) can identify human MSC-derived extracellular vesicles by conventional flow cytometry. Cell Commun. Signal. 2016, 14, 2. [CrossRef]
- Moravcikova, E.; Meyer, E.M.; Corselli, M.; Donnenberg, V.S.; Donnenberg, A.D. Proteomic Profiling of Native Unpassaged and Culture-Expanded Mesenchymal Stromal Cells (MSC). Cytom. Part A., 2018, 93, 894–904. [CrossRef]
- Chen, Q.H.; Wu, F.; Liu, L.; Chen, H.; Zheng R.Q.; Wang, H.L.; Yu, L.N. Mesenchymal stem cells regulate the Th17/Treg cell balance partly through hepatocyte growth factor in vitro. Stem Cell Res Ther 2020,11, 91. [CrossRef]
- Ciuculescu, F.; Giesen, M.; Deak, E.; Lang, V.; Seifried, E.; Henschler, R. Variability in chemokine-induced adhesion of human mesenchymal stromal cells. Cytotherapy 2011, 13, 1–8. [CrossRef]
- Guan, X.; Ma, X.; Zhang, L.; Feng, H.; Ma, Z. Evaluation of CD24 as a marker to rapidly define the mesenchymal stem cell phenotype and its differentiation in human nucleus pulposus. Chin. Med. J (Engl) 2014, 127(8), 1474–1481. PMID: 24762592.
- Teo, G.S.; Ankrum, J.A.; Martinelli, R.; Boetto, S.E.; Simms, K.; Sciuto, T.E.; Dvorak, A.M.; Karp, M.; Carman, C.V. Mesenchymal stem cells transmigrate between and directly through tumor necrosis factor-α-activated endothelial cells via both leukocyte-like and novel mechanisms. Stem Cells 2012, 30(11), 2472–2486. [CrossRef]
- Steingen, C.; Brenig, F.; Baumgartner, L.; Schmidt, J.; Schmidt, A.; Bloch, W. Characterization of key mechanisms in transmigration and invasion of mesenchymal stem cells. J Mol Cell Cardiol. 2008, 44(6), 1072–1084. [CrossRef]
- Hou, Y.; Ryu, C.H.; Jun, J.A.; Kim, S.M.; Jeong, C.H.; Jeun, S.S. IL-8 enhances the angiogenic potential of human bone marrow mesenchymal stem cells by increasing vascular endothelial growth factor. Cell Biol. Int. 2014, 38, 1050–1059. [CrossRef]
- Dreyer, C.H. Jørgensen, .R.; Overgaard, S.; Qin, L.; Ding, M. Vascular Endothelial Growth Factor and Mesenchymal Stem Cells Revealed Similar Bone Formation to Allograft in a Sheep Model. Biomed Res Int. 2021, 2021, 6676609. [CrossRef]
- Scherzed, A.; Hackenberg, S.; Froelich, K.; Rak, K.; Schendzielorz, P.; Gehrke, T.; Hagen, R.; Kleinsasser, N.; The differentiation of hMSCs counteracts their migration capability and pro-angiogenic effects in vitro. Oncol. Rep. 2016, 35, 219–226. [CrossRef]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [CrossRef]
- Li, Y.; Zhong, X.; Zhang, Y.; Lu, X. Mesenchymal stem cells in gastric cancer: Vicious but hopeful. Front. Oncol. 2021, 11, 617677. [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C, Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; Chakravarty, D.; Daian, F.; Gao, Q.; Bailey, M.H.; Liang, W.W.; Foltz, S.M.; Shmulevich, I.; Ding, L.; Heins, Z.; Ochoa, A.; Gross, B.; Gao, J.; Zhang, H.; Kundra, R.; Kandoth, C.; Bahceci, I.; Dervishi, L.; Dogrusoz, U.; Zhou, W.; Shen, H.; Laird, P.W.; Way, G.P.; Greene, C.S.; Liang, H.; Xiao, Y.; Wang, C.; Iavarone, A.; Berger, A.H.; Bivona, T.G.; Lazar, A.J.; Hammer, G.D.; Giordano, T.; Kwong, L.N.; McArthur, G.; Huang, C.; Tward, A.D.; Frederick, M.J.; McCormick, F.; Meyerson, M. Cancer Genome Atlas Research Network; Van Allen, E.M.; Cherniack, A.D.; Ciriello, G.; Sander, C.; Schultz, N. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173(2), 321–337.e10. [CrossRef]
- Torsvik, A.; Bjerkvig, R. Mesenchymal stem cell signaling in cancer progression. Cancer Treat. Rev. 2013, 39(2), 180–188. [CrossRef]
- Cuiffo, B.G.; Campagne, A.; Bell, G.W.; Lembo, A.; Orso, F.; Lien, E.C.; Bhasin, M.K.; Raimo, M.; Hanson, S.E., Marusyk, A.; El-Ashry, D.; Hematti, P.; Polyak, K.; Mechta-Grigoriou, F.; Mariani, O.; Volinia, S.; Vincent-Salomon, A.; Taverna, D.; Karnoub, A.E. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell. 2014, 15, 762–774. [CrossRef]
- Ohkouchi, S.; Block, G.J.; Katsha, A.M.; Kanehira, M.; Ebina, M.; Kikuchi, T.; Saijo, Y.; Nukiwa, T.; Prockop, D.J. Mesenchymal Stromal Cells Protect Cancer Cells From ROS-induced Apoptosis and Enhance the Warburg Effect by Secreting STC1. Mol. Ther. 2012, 20, 417–423. [CrossRef]
- Phinney, D.G.; Pittenger, M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Stem Cells. 2017, 35(4), 851–8. [CrossRef]
- Kupcova Skalnikova, H. Proteomic techniques for characterisation of mesenchymal stem cell secretome. Biochimie 2013, 95, 2196–2211. [CrossRef]
- Tachida ,Y.; Sakurai, H.; Okutsu, J.; Proteomic Comparison of the Secreted Factors of Mesenchymal Stem Cells from Bone Marrow, Adipose Tissue and Dental Pulp. J. Proteomics Bioinform. 2015, 89(12). [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer, 2016, 16, 582–598. [CrossRef]
- de Miranda, M.C.; Melo, M.I.A.; Cunha, P.D.S.; Gentilini, J Júnior.; Faria, J.A.Q.A.; Rodrigues, M.A.; Gomes, D.A. Roles of mesenchymal stromal cells in the head and neck cancer microenvironment. Biomed. Pharmacother. 2021, 144, 112269. [CrossRef]
- Lourenco, S.; Teixeira, V.H.; Kalber, T.; Jose, R.J.; Floto, R.A.; Janes, S.M. Macrophage migration inhibitory factor-CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J. Immunol. 2015, 194(7), 3463–3474. [CrossRef]
- Wobus, M.; List, C.; Dittrich, T.; Dhawan, A.; Duryagina, R.; Arabanian, L. S.; Kast, K.; Wimberger, P.; Stiehler, M.; Hofbauer, L. C.; Jakob, F.; Ehninger, G.; Anastassiadis, K.; Bornhäuser, M. Breast carcinoma cells modulate the chemoattractive activity of human bone marrow-derived mesenchymal stromal cells by interfering with CXCL12. Int. J. Cancer 2015, 136(1), 44–54. [CrossRef]
- Kalimuthu, S.; Oh, J. M.; Gangadaran, P.; Zhu, L.; Lee, H. W.; Rajendran, R. L., Baek, SH.; Jeon, Y.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. In Vivo Tracking of Chemokine Receptor CXCR4-Engineered Mesenchymal Stem Cell Migration by Optical Molecular Imaging. Stem Cells Int. 2017, 8085637. [CrossRef]
- Sheta, M.; Taha, E. A.; Lu, Y.; Eguchi, T. Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology 2023, 12(1), 110. [CrossRef]
- Norozi, F.; Ahmadzadeh, A.; Shahrabi, S.; Vosoughi, T.; Saki, N. Mesenchymal stem cells as a double-edged sword in suppression or progression of solid tumor cells. Tumour Biol. 2016, 37(9), 11679–11689. [CrossRef]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019, 10(4), 273. [CrossRef]
- Barcellos-de-Souza, P.; Comito, G.; Pons-Segura, C.; Taddei, M.L.; Gori, V.; Becherucci, V.; Bambi, F.; Margheri, F.; Laurenzana, A.; Del Rosso, M.; Chiarugi, P. Mesenchymal Stem Cells are Recruited and Activated into Carcinoma-Associated Fibroblasts by Prostate Cancer Microenvironment-Derived TGF-β1. Stem Cells. 2016, 34(10), 2536–2547. [CrossRef]
- Aoto, K.; Ito, K.; Aoki, S. Complex formation between platelet-derived growth factor receptor beta and transforming growth factor beta receptor regulates the differentiation of mesenchymal stem cells into cancer-associated fibroblasts. Oncotarget 2018, 9, 34090–34102. [CrossRef]
- Li, G. C.; Zhang, H. W.; Zhao, Q. C.; Sun, L. I.; Yang, J. J.; Hong, L.; Feng, F.; Cai, Lei. Mesenchymal stem cells promote tumor angiogenesis via the action of transforming growth factor beta1. Oncol. Lett. 2016, 11, 1089–1094. [CrossRef]
- Li, W.; Zhou, Y.; Yang, J.; Zhang, X.; Zhang, H.; Zhang, T.; Zhao, S.; Zheng, P.; Huo, J.; Wu, H. Gastric cancer-derived mesenchymal stem cells prompt gastric cancer progression through secretion of interleukin-8. J. Exp. Clin. Cancer Res. 2015, 34, 52. [CrossRef]
- Guo, X.; Zhao, Y.; Yan, H.; Yang, Y.; Shen, S.; Dai, X.; Ji, X.; Ji, F.; Gong, X.G.; Li, L.; Bai, X.; Feng, X.H.; Liang, T.; Ji, J.; Chen, L.; Wang, H.; Zhao, B. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. 2017, 31, 247–259. [CrossRef]
- Powell, D.; Lou, M.; Barros Becker, F.; Huttenlocher, A. Cxcr1 mediates recruitment of neutrophils and supports proliferation of tumor-initiating astrocytes in vivo. Sci. Rep. 2018, 8, 13285. [CrossRef]
- Lan, T.; Luo, M.; Wei, X. Mesenchymal stem/stromal cells in cancer therapy. J. Hematol. Oncol. 2021, 14, 195. [CrossRef]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47(6), 4587–4629. [CrossRef]
- Li, L.; Li, J.C.; Yang, H.; Zhang, X.; Liu, L.L.; Li Y, Zeng, T.T.; Zhu, Y.H.; Li, X.D.; Li, Y.; Xie, D.; Fu, L.; Guan, X.Y. Expansion of cancer stem cell pool initiates lung cancer recurrence before angiogenesis. Proc. Natl. Acad. Sci. U S A. 2018, 115(38), E8948-E8957. [CrossRef]
- Liongue, C.; Sertori, R.; Ward, A.C. Evolution of cytokine receptor signaling. J. Immunol. 2016, 197(1), 11–8. [CrossRef]
- Zhang, X.; Hu, F.; Li, G.; Li, G.; Yang, X.; Liu, L.; Zhang, R.; Zhang, B.; Feng, Y. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018, 9(2), 25. [CrossRef]
- Liu, C.; Feng, X.; Wang, B.; Wang, X.; Wang, C.; Yu, M.; Cao, G.; Wang, H. Bone marrow mesenchymal stem cells promote head and neck cancer progression through Periostin-mediated phosphoinositide 3-kinase/Akt/mammalian target of rapamycin. Cancer Sci. 2018, 109(3), 688-698. [CrossRef]
- McGuire, J.J.; Frieling, J.S.; Lo, C.H.; Li, T.; Muhammad, A.; Lawrence, H.R.; Lawrence, N.J.; Cook, L.M.; Lynch, C.C. Mesenchymal stem cell-derived interleukin-28 drives the selection of apoptosis resistant bone metastatic prostate cancer. Nat. Commun. 2021, 12(1), 723. [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene, 2017, 36(11), 1461–1473. [CrossRef]
- Wang, W.; Zhong, W.; Yuan, J.; Yan, C.; Hu, S.; Tong, Y.; Mao, Y.; Hu, T.; Zhang, B.; Song G. Involvement of Wnt/β-catenin signaling in the mesenchymal stem cells promote metastatic growth and chemoresistance of cholangiocarcinoma. Oncotarget 2015, 6(39), 42276–42789. [CrossRef]
- Chen, Y. C.; Gonzalez, M. E.; Burman, B.; Zhao, X.; Anwar, T.; Tran, M.; Medhora, N.; Hiziroglu, A.B.; Lee, W.; Cheng, Y.H.; Choi, Y.; Yoon, E.; Kleer, C.G. Mesenchymal Stem/Stromal cell engulfment reveals metastatic advantage in Breast Cancer. Cell Rep. 2019, 27(13), 3916–3926.e5. [CrossRef]
- Lu, L.; Chen, G.; Yang, J.; Ma, Z.; Yang, Y.; Hu, Y.; Lu, Y.; Cao, Z.; Wang, Y.; Wang, X. Bone marrow mesenchymal stem cells suppress growth and promote the apoptosis of glioma U251 cells through downregulation of the PI3K/AKT signaling pathway. Biomed. Pharmacother. 2019, 112, 108625. [CrossRef]
- Akimoto, K.; Kimura, K.; Nagano, M.; Takano, S.; To’a Salazar, G.; Yamashita, T.; Ohneda O. Umbilical cord blood-derived mesenchymal stem cells inhibit, but adipose tissue-derived mesenchymal stem cells promote, glioblastoma multiforme proliferation. Stem Cells Dev. 2013, 22, 1370–1386. [CrossRef]
- Xuan, X.; Tian, C.; Zhao, M.; Sun, Y.; Huang C. Mesenchymal stem cells in cancer progression and anticancer therapeutic resistance. Cancer Cell Int. 2021, 21(1), 595. [CrossRef]
- Ho, I. A., Toh, H. C., Ng, W. H., Teo, Y. L., Guo, C. M., Hui, K. M., Lam, P.Y.P. Human bone marrow-derived mesenchymal stem cells suppress human glioma growth through inhibition of angiogenesis. Stem Cells 2013, 31, 146–155. [CrossRef]
- Hanks, B.A.; Holtzhausen, A.; Evans, K. S.; Jamieson, R.; Gimpel, P.; Campbell, O.M.; Hector-Greene, M.; Sun, L.; Tewari, A.; George, A.; Starr, M.; Nixon, A.B.; Augustine, C.; Beasley, G.; Tyler, D.S.; Osada, T.; Morse, M.A.; Ling, L.; Lyerly, H. K.; Blobe, G. C. J. Clin. Invest. 2020, 123(9), 3925–3940. [CrossRef]
- Bortolotti, F.; Ukovich, L.; Razban, V.; Martinelli, V.; Ruozi, G.; Pelos, B.; Dore, F.; Giacca, M.; Zacchigna, S. In vivo therapeutic potential of mesenchymal stromal cells depends on the source and the isolation procedure. Stem Cell Rep. 2015, 4, 332–339. [CrossRef]
- Bajetto, A.; Pattarozzi, A.; Corsaro, A.; Barbieri, F.; Daga, A.; Bosio, A.; Gatti, M.; Pisaturo, V.; Sirito, R.; Florio T. Different effects of human umbilical cord mesenchymal stem cells on glioblastoma stem cells by direct cell interaction or via released soluble factors. Front. Cell. Neurosci. 2017, 11, 312. [CrossRef]
- Capilla-Gonzalez, V., Lopez-Beas, J., Escacena, N., Aguilera, Y., de la Cuesta, A., Ruiz-Salmeron, R.; Martín, F.; Hmadcha, A.; Soria, B. PDGF restores the defective phenotype of adipose-derived mesenchymal stromal cells from diabetic patients. Mol. Ther. 2018, 26, 2696–2709. [CrossRef]
- Perez, L.M.; de Lucas, B.; Galvez, B.G. Unhealthy stem cells: When health conditions upset stem cell properties. Cell. Physiol. Biochem. 2018, 46, 1999–2016. [CrossRef]
- Rivera, FJ; de la Fuente, A.G.; Zhao C, Silva ME, Gonzalez GA, Wodnar R, Feichtner M, Lange S, Errea O, Priglinger E, O’Sullivan A, Romanelli P, Jadasz, J.J.; Brachtl. G.; Greil, R.; Tempfer. H.; Traweger, A.; Bátiz, L.F, Küry, P.; Couillard-Despres, S.; Franklin, R.J.M.; Aigner, L. Aging restricts the ability of mesenchymal stem cells to promote the generation of oligodendrocytes during remyelination. Glia. 2019, 67(8), 1510–1525. [CrossRef]
- Liang, W.; Chen, X.; Zhang, S; Fang, J.; Chen, M.; Xu, Y.; Chen, X. Mesenchymal stem cells as a double-edged sword in tumor growth: Focusing on MSC-derived cytokines. Cell Mol. Biol. Lett, 2021, 26(1), 3. [CrossRef]
- Timaner, M.; Tsai, K.K.,; Shaked, Y. The multifaceted role of mesenchymal stem cells in cancer. Semin Cancer Biol. 2020, 60, 225–237. [CrossRef]
- Le Naour, A.; Prat, M.; Thibault, B.; Mével, R.; Lemaitre, L.; Leray, H.; Joubert, M.V.; Coulson, K.; Golzio, M.; Lefevre, L.; Mery, E.; Martinez, A.; Ferron, G.; Delord, J.P.; Coste, A.; Couderc, B. Tumor cells educate mesenchymal stromal cells to release chemoprotective and immunomodulatory factors. J Mol Cell Biol. 2020, 12(3), 202–215. [CrossRef]
- Liu, C.; Chu, D.; Kalantar-Zadeh, K.; George, J.; Young, H.A.; Liu, G. Cytokines: From Clinical Significance to Quantification. Adv. Sci. (Weinh). 2021, 8(15), e2004433. [CrossRef]
- Panchalingam, K.M..; Jung, S.; Rosenberg, L.; Behie, L.A. Bioprocessing strategies for the large-scale production of human mesenchymal stem cells: A review. Stem Cell Res. Ther. 2015, 6, 225. [CrossRef]
- Sharma, R.R.; Pollock, K.; Hubel, A.; McKenna, D. Mesenchymal stem or stromal cells: A review of clinical applications and manufacturing practices. Transfusion 2014, 54(5), 1418–1437. [CrossRef]
- Haddad, R.; Saldanha-Araujo, F. Mechanisms of T-cell immunosuppression by mesenchymal stromal cells: What do we know so far? Biomed. Res. Int. 2014, 2014, 216806. [CrossRef]
- Lawson, D. A.; Bhakta, N. R.; Kessenbrock, K.; Prummel, K. D.; Yu, Y.; Takai, K., Zhou A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; Yaswen, P.; Goga, A.; Werb, Z. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526(7571), 131–135. [CrossRef]
- Bhonde, R.R.; Sheshadri, P, Sharma S.; Kumar, A. Making surrogate β-cells from mesenchymal stromal cells: Perspectives and future endeavors. Int. J. Biochem. Cell Biol. 2014, 46, 90–102. [CrossRef]
- López de Padilla, C. M., Niewold, T. B. The type I interferons: Basic concepts and clinical relevance in immune-mediated inflammatory diseases. Gene 2016, 576(1 Pt 1), 14–21. [CrossRef]
- Barrat, F.J.; Crow, M.K.; Ivashkiv, L.B. Interferon target-gene expression and epigenomic signatures in health and disease. Nat, Immunol. 2019, 20(12), 1574–1583. [CrossRef]
- Domenis, R.; Cifù, A.; Quaglia, S.; Pistis, C.; Moretti, M.; Vicario, A.; Parodi, P.C.; Fabris, M.; Niazi, K.R.; Soon-Shiong, P.; Curcio, F. Pro inflammatory stimuli enhance the immunosuppressive functions of adipose mesenchymal stem cells derived exosomes. Sci. Rep. 2018, 8, 1–11. [CrossRef]
- Dunn, C.M.; Kameishi, S.; Cho, Y.K.; Song, S.U.; Grainger, D.W.; Okano, T. (2022). Interferon-Gamma Primed Human Clonal Mesenchymal Stromal Cell Sheets Exhibit Enhanced Immunosuppressive Function. Cells 2022, 11(23), 3738. [CrossRef]
- Ungerer, C.; Quade-Lyssy, P.; Radeke, H.H.; Henschler, R.; Königs, C.; Köhl, U.; Seifried, E.; Schüttrumpf, J. Galectin-9 is a suppressor of T and B cells and predicts the immune modulatory potential of mesenchymal stromal cell preparations. Stem Cells Dev. 2014, 23(7), 755–766. [CrossRef]
- Hori, A.; Takahashi, A.; Miharu, Y.; Yamaguchi, S.; Sugita, M.; Mukai, T.; Nagamura, F.; Nagamura-Inoue, T. Superior migration ability of umbilical cord-derived mesenchymal stromal cells (MSCs) toward activated lymphocytes in comparison with those of bone marrow and adipose-derived MSCs. Front Cell Dev Biol. 2024, 12, 1329218. [CrossRef]
- Chen, H.W.; Chen, H.Y.; Wang, L.T.; Wang, F.H.; Fang, L.W.; Lai, H.Y.; Chen, H.H.; Lu, J.; Hung, M.S.; Cheng, Y.; Chen, M.Y.; Liu, S.J.; Chong, P.; Lee, O.K.; Hsu, S.C. Mesenchymal stem cells tune the development of monocyte-derived dendritic cells toward a myeloid-derived suppressive phenotype through growth-regulated oncogene chemokines. J. Immunol. 2013, 190(10), 5065–5077. [CrossRef]
- Yen, B.L.; Yen, M.L.; Hsu, P.J,; Liu, K.J.; Wang, C.J.; Bai, C,H.; Sytwu, H.K. Multipotent human mesenchymal stromal cells mediate expansion of myeloid-derived suppressor cells via hepatocyte growth factor/c-met and STAT3. Stem Cell Reports. 2013, 1(2), 139–151. [CrossRef]
- Cao, W.; Cao, K.; Cao, J.; Wang, Y.; Shi, Y. Mesenchymal stem cells and adaptive immune responses. Immunol. Lett. 2015, 168(2), 147–153. [CrossRef]
- Le Blanc, K.; Davies, L.C. Mesenchymal stromal cells and the innate immune response. Immunol. Lett. 2015, 68(2), 140–146. [CrossRef]
- Lee, H.J.; Ko, J.H.; Jeong, H.J.; Ko, A.Y.; Kim, M.K.; Wee, W.R,.; Yoon, S.O.; Oh, J.Y. Mesenchymal stem/stromal cells protect against autoimmunity via CCL2-dependent recruitment of myeloid-derived suppressor cells. J. Immunol. 2015, 194(8), 3634–3645. [CrossRef]
- Davies, L.C.; Heldring, N.; Kadri, N.; Le Blanc, K. Mesenchymal Stromal Cell Secretion of Programmed Death-1 Ligands Regulates T Cell Mediated Immunosuppression. Stem Cells. 2017, 35(3), 766–776. [CrossRef]
- Gaber, T.; Schönbeck, K.; Hoff, H.; Tran, C.L.; Strehl, C.; Lang, A.; Ohrndorf, S.; Pfeiffenberger, M.; Röhner, E.; Matziolis, G.; Burmester, G.R.; Buttgereit, F.; Hoff, P. CTLA-4 Mediates Inhibitory Function of Mesenchymal Stem/Stromal Cells. Int. J. Mol Sci. 2018, 19(8), 2312. [CrossRef]
- Zhu, Q.; Zhang, X.; Zhang, L.; Li, W.; Wu, H.; Yuan, X.; Mao, F.; Wang, M.; Zhu, W.; Qian, H.; Xu. W. The IL-6-STAT3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell Death Dis. 2014, 5(6), e1295. [CrossRef]
- Ma, O.K.; Chan, K.H. Immunomodulation by mesenchymal stem cells: Interplay between mesenchymal stem cells and regulatory lymphocytes. World J. Stem Cells 2016, 8(9), 268–278. PMID: 27679683; PMCID: PMC5031888. [CrossRef]
- Wang, M.Y.; Zhou, T.Y.; Zhang, Z.D.; Liu, H.Y.; Zheng, Z.Y.; Xie, H.Q. Current therapeutic strategies for respiratory diseases using mesenchymal stem cells. Med. Comm. 2020, 2(3), 351–380. [CrossRef]
- Lee, H,J.; Oh, J.Y. Mesenchymal Stem/Stromal Cells Induce Myeloid-Derived Suppressor Cells in the Bone Marrow via the Activation of the c-Jun N-Terminal Kinase Signaling Pathway. Int. J. Mol. Sci. 2024, 25(2), 1119. [CrossRef]
- Korneev, K. V.; Atretkhany, K. N.; Drutskaya, M. S.; Grivennikov, S. I.; Kuprash, D. V.; Nedospasov, S. A. TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine 2017, 89, 127–135. [CrossRef]
- Deguine, J.; Barton, G. M. MyD88: A central player in innate immune signaling. F1000 Med. Rep. 2014, 6, 97. [CrossRef]
- Ye, W.; Hu, M. M.; Lei, C. Q.; Zhou, Q.; Lin, H.; Sun, M. S.; Shu, H. B. TRIM8 Negatively Regulates TLR3/4-Mediated Innate Immune Response by Blocking TRIF-TBK1 Interaction. J. Immunol. (Baltimore, Md.:1950) 2017, 199(5), 1856–1864. [CrossRef]
- Shah, P.; Omoluabi, O.; Moorthy, B.; Ghose, R. Role of Adaptor Protein Toll-Like Interleukin Domain Containing Adaptor Inducing Interferon β in Toll-Like Receptor 3- and 4-Mediated Regulation of Hepatic Drug Metabolizing Enzyme and Transporter Genes. Drug Metab. Dispos. 2016, 44(1), 61–67. [CrossRef]
- Affolter, A.; Lammert, A.; Kern, J.; Scherl, C,.; Rotter, N. Precision Medicine Gains Momentum: Novel 3D Models and Stem Cell-Based Approaches in Head and Neck Cancer. Front. Cell Dev. Biol. 2021, 9, 666515. [CrossRef]
- Naaldijk, Y.; Johnson, A.A.; Ishak, S.; Meisel, H.J.; Hohaus, C.; Stolzing, A. Migrational changes of mesenchymal stem cells in response to cytokines, growth factors, hypoxia, and aging. Exp. Cell Res. 2015, 338(1), 97–104. [CrossRef]
- Qiu, X.; Zhang, Y.; Zhao, X.; Zhang, S.; Wu, J.; Guo, H.; Hu, Y. Enhancement of endothelial differentiation of adipose derived mesenchymal stem cells by a three-dimensional culture system of microwell. Biomaterials 2015, 53, 600–608. [CrossRef]
- Kwon, Y.W.; Heo, S.C.; Jeong, G.O.; Yoon, J.W.; Mo, W.M.; Lee, M.J.; Jang, I.H.; Kwon, S.M.; Lee, J.S.; Kim, J.H. Tumor necrosis factor-α-activated mesenchymal stem cells promote endothelial progenitor cell homing and angiogenesis. Biochim. Biophys. Acta. 2013, 1832(12), 2136–2144. [CrossRef]
- Liu, X.; Zhou, Z.; Zeng, W.N.; Zeng, Q.; Zhang, X. The role of toll-like receptors in orchestrating osteogenic differentiation of mesenchymal stromal cells and osteoimmunology. Front. Cell Dev. Biol. 2023, 11, 1277686. [CrossRef]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 10, 1191. [CrossRef]
- Ulivi, V.; Tasso, R.; Cancedda, R.; Descalzi, F. Mesenchymal stem cell paracrine activity is modulated by platelet lysate: Induction of an inflammatory response and secretion of factors maintaining macrophages in a proinflammatory phenotype. Stem Cells Dev. 2014, 23(16), 1858–1869. [CrossRef]
- Watts, T.L.; Cui, R.; Szaniszlo, P.; Resto, V.A.; Powell, D.W.; Pinchuk, I.V.; PDGF-AA mediates mesenchymal stromal cell chemotaxis to the head and neck squamous cell carcinoma tumor microenvironment, J. Transl. Med. 2016, 14(1), 337. [CrossRef]
- Kansy, B.A.; Dißmann, P.A.; Hemeda, H.; Bruderek, K.; Westerkamp, A.M.; Jagalski, V.; Schuler, P.; Kansy, K.; Lang, S.; Dumitru, C.A.; Brandau, S. The bidirectional tumor - mesenchymal stromal cell interaction promotes the progression of head and neck cancer. Stem Cell Res. Ther. 2014, 5, 95. [CrossRef]
- Ji, X.; Zhang, Z.; Han, Y,; Song, J.; Xu, X.; Jin, J.; Su, S.; Mu, D.; Liu, X.; Xu, S.; Cui, H.; Zhao, Z,; Wang, Y.; Liu, H.; Mesenchymal stem cells derived from normal gingival tissue inhibit the proliferation of oral cancer cells in vitro and in vivo. Int. J. Oncol. 2016, 49, 2011–2022. [CrossRef]
- Wang, J.; Yang, W.; Wang, T.; Chen, X.; Wang, J.; Zhang, X.; Cai, C.; Zhong, B.; Wu, J.; Chen, Z.; Xiang, A.P.; Huang, W. Mesenchymal Stromal Cells-Derived β2-Microglobulin Promotes Epithelial-Mesenchymal Transition of Esophageal Squamous Cell Carcinoma Cells. Sci. Rep. 2018, 8(1), 5422. [CrossRef]
- Scherzed, A.; Hackenberg, S.; Radeloff, A. Froelich, K.; Rak, K.; Hagen, R.; Kleinsasser, N. Human mesenchymal stem cells promote cancer motility and cytokine secretion in vitro. Cells Tissues Organs. 2014, 198, 327–337. [CrossRef]
- Wu, Y.L.; Li, H.Y.; Zhao, X.P.; Jiao, J.Y.; Tang, D.X.; Yan, L.J.; Wan, Q.; Bin Pan, C. Mesenchymal stem cell-derived CCN2 promotes the proliferation, migration and invasion of human tongue squamous cell carcinoma cells. Cancer Sci. 2017, 108, 897–909. [CrossRef]
- Coccè, V.; Farronato, D.; Brini, A.T.; Masia, C.; Giannì, A.B.; Piovani, G.; Sisto, F.; Alessandri, G.; Angiero, F.; Pessina, A. Drug Loaded Gingival Mesenchymal Stromal Cells (GinPa-MSCs) Inhibit In Vitro Proliferation of Oral Squamous Cell Carcinoma. Sci. Rep. 2017, 7(1), 9376. [CrossRef]
- Wang, J.; Cui, R.; Clement, C.G.; Nawgiri. R.; Powell. D.W.; Pinchuk, I.V.; Watts, T.L. Activation PDGFR-α/AKT Mediated Signaling Pathways in Oral Squamous Cell Carcinoma by Mesenchymal Stem/Stromal Cells Promotes Anti-apoptosis and Decreased Sensitivity to Cisplatin. Front. Oncol. 2020, 10, 552. [CrossRef]
- Liu, C.; Billet, S.; Choudhury, D.; Cheng, R.; Haldar, S.; Fernandez, A.; Biondi, S.; Liu, Z.; Zhou, H.; Bhowmick, N.A, Bone marrow mesenchymal stem cells interact with head and neck squamous cell carcinoma cells to promote cancer progression and drug resistance. Neoplasia 2021, 23, 118–128. [CrossRef]
- Liu, C.; Feng, X.; Wang, B.; Wang, X.; Wang. C.; Yu, M.; Cao, G.; Wang, H. Bone marrow mesenchymal stem cells promote head and neck cancer progression through Periostin-mediated phosphoinositide 3-kinase/Akt/mammalian target of rapamycin. Cancer Sci. 2018, 109, 688–698. [CrossRef]
- Jiang, C.; Zhang, Q.; Shanti, R.M.; Shi, S.; Chang, T.H.; Carrasco, L.; Alawi, F.; Le, A.D. Mesenchymal Stromal Cell-Derived Interleukin-6 Promotes Epithelial–Mesenchymal Transition and Acquisition of Epithelial Stem-Like Cell Properties in Ameloblastoma Epithelial Cells. Stem Cells. 2017, 35, 2083–2094. [CrossRef]
- Li, W.; Han, Y.; Zhao, Z.; Ji, X.; Wang, X.; Jin, J.; Wang, Q.; Guo, X.; Cheng, Z,; Lu, M.; Wang, G.; Wang, Y.; Liu, H. Oral mucosal mesenchymal stem cell-derived exosomes: A potential therapeutic target in oral premalignant lesions. Int. J. Oncol. 2019, 54, 1567–1578. [CrossRef]
- Almeida, R.D.A.C.; Andrade, E.S.D.S.; Barbalho, J.; Vajgel, A.; Vasconcelos BCDE, Recurrence rate following treatment for primary multicystic ameloblastoma: Systematic review and meta-analysis. Int. J. Oral Maxillofac. Surg. 2016, 45, 359–367. [CrossRef]
- Hendra, F.N.; Van Cann, E.M.; Helder, M.N.; Ruslin, M.; de Visscher, J.G.; Forouzanfar, T.; de Vet, H.C.W. Global incidence and profile of ameloblastoma: A systematic review and meta-analysis. Oral Dis. 2020, 26, 12–21. [CrossRef]
- Shi, S.; Zhang, Q.; Xia, Y.; You, B.; Shan, Y.; Bao, L.; Li, L.; You, Y.; Gu, Z. Mesenchymal stem cell-derived exosomes facilitate nasopharyngeal carcinoma progression. Am. J. Cancer Res. 2016, 6, 459–472. PMID: 27186416, PMCID: PMC4859673.
- Hong, D.; Liu, T.; Huang, W.; Liao, Y.; Wang, L.; Zhang, Z.; Chen, H.; Zhang, X.; Xiang, Q. Gremlin1 Delivered by Mesenchymal Stromal Cells Promoted Epithelial-Mesenchymal Transition in Human Esophageal Squamous Cell Carcinoma. Cell Physiol. Biochem. 2018, 47(5), 1785–1799. [CrossRef]
- Nakayama, A.; Aoki, S.; Uchihashi, K.; Nishijima-Matsunobu, A.; Yamamoto, M.; Kakihara, N.; Iwakiri, R.; Fujimoto, K.; Toda, S. Interaction between Esophageal Squamous Cell Carcinoma and Adipose Tissue in Vitro. Am. J. Pathol. 2016, 186, 1180–1194. [CrossRef]
- Wang, Y.; Fan, H.; Zhou, B.; Ju, Z.; Yu, L.; Guo, L.; Han, J.; Lu, S. Fusion of human umbilical cord mesenchymal stem cells with esophageal carcinoma cells inhibits the tumorigenicity of esophageal carcinoma cells. Int. J. Oncol. 2012, 40, 370–377. [CrossRef]
- Li, L.; Tian, H.; Yue, W.; Zhu, F.; Li, S,; Li, W. Human mesenchymal stem cells play a dual role on tumor cell growth in vitro and in vivo. J. Cell. Physiol. 2011, 226, 1860–1867. [CrossRef]
- Liotta, F.; Querci, V.; Mannelli, G.; Santarlasci, V.; Maggi, L.; Capone, M.; Rossi, M.C.; Mazzoni, A.; Cosmi, L.; Romagnani, S.; Magg, E.; Gallo, O. Annunziato F, Mesenchymal stem cells are enriched in head neck squamous cell carcinoma, correlates with tumour size and inhibit T-cell proliferation. Br. J. Cancer. 2015, 112, 745–754. [CrossRef]
- Mazzoni, A.; Capone, M.; Ramazzotti, M.; Vanni, A.; Locatello, L.G.; Gallo, O.; De Palma, R.; Cosmi, L.; Liotta, F.; Annunziato, F.; Maggi, L. IL4I1 Is Expressed by Head–Neck Cancer-Derived Mesenchymal Stromal Cells and Contributes to Suppress T Cell Proliferation. J. Clin. Med. 2021, 10, 2111. [CrossRef]
- Schuler, P.J.; Westerkamp, A.M.; Kansy, B.; Bruderek, K.; Dissmann, P.A.; Dumitru, C.A.; Lang, S.; Jackson, E.K.; Brandau, S. Adenosine metabolism of human mesenchymal stromal cells isolated from patients with head and neck squamous cell carcinoma. Immunobiology 2017, 222, 66–74. [CrossRef]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28 (9), 984–999. [CrossRef]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [CrossRef]
- Rowan, B.G.; Lacayo, E.A.; Sheng, M.; Anbalagan, M.; Gimble, J.M.; Jones, R.K.; Joseph, W.J.; Friedlander, P.L.; Chiu, E.S.; Human Adipose Tissue-Derived Stromal/Stem Cells Promote Migration and Early Metastasis of Head and Neck Cancer Xenografts. Aesthetic Surg. J. 2015, 36, 93–104. [CrossRef]
- Salo, S.; Bitu, C.; Merkku, K.; Nyberg, P.; Bello, I.O.; Vuoristo, J.; Sutinen, M.; Vähänikkilä, H.; Costea, D.E.; Kauppila, J.; Lehenkari, P.; Dayan, D.; Vered, M.; Risteli, J.; Salo, T. Human Bone Marrow Mesenchymal Stem Cells Induce Collagen Production and Tongue Cancer Invasion. PLoS ONE. 2013, 8(10), e77692. [CrossRef]
- Hong, D.; Liu, T.; Huang, W.; Liao, Y.; Wang, L.; Zhang, Z.; Chen, H.; Zhang, X.; Xiang, Q. Gremlin1 delivered by mesenchymal stromal cells promoted epithelial-mesenchymal transition in human esophageal squamous cell carcinoma. Cell. Physiol. Biochem. 2018, 47, 1785–1799. [CrossRef]
- Castellone, M.D.; Laatikainen, L.E.; Laurila, J.P.; Langella, A.; Hematti, P.; Soricelli, A.; Salvatore, M.; Laukkanen, M.O. Brief Report: Mesenchymal Stromal Cell Atrophy in Coculture Increases Aggressiveness of Transformed Cells. Stem Cells. 2013, 31, 1218–1223. [CrossRef]
- Mazzoni, A.; Maggi, L.; Montaini, G.; Ramazzotti, M.; Capone, M.; Vanni, A.; Locatello, L.G.; Barra, G.; De Palma, R.; Gallo, O.; Cosmi, L.; Liotta, F.; Annunziato, F. Human T cells interacting with HNSCC-derived mesenchymal stromal cells acquire tissue-resident memory like properties. Eur. J. Immunol. 2020, 50(10), 1571–1579. [CrossRef]
- Chen, Y.; Wang, X.; Fang, J.; Song, J.; Ma, D.; Luo, L.; He, B.; Xia, J.; Lui, V.W.Y.; Cheng, B.; Wang, Z. Mesenchymal stem cells participate in oral mucosa carcinogenesis by regulating T cell proliferation. Clin. Immunol. 2019, 198, 46–53. [CrossRef]
- Ziebart, A.; Huber, U.; Jeske, S.S.; Laban, S.; Doescher, J.; Hoffmann, T.K.; Brunner, C.; Jackson, E.K.; Schuler, P.J. The influence of chemotherapy on adenosine-producing B cells in patients with head and neck squamous cell carcinoma. Oncotarget 2017, 9, 5834–5847. [CrossRef]
- Bruna, F.; Arango-Rodríguez, M.; Plaza, A.; Espinoza, I.; Conget, P. The administration of multipotent stromal cells at precancerous stage precludes tumor growth and epithelial dedifferentiation of oral squamous cell carcinoma. Stem Cell Res. 2017, 18, 5–13. [CrossRef]
- Bruna, F.; Plaza, A.; Arango, M.; Espinoza, I.; Conget, P. Systemically administered allogeneic mesenchymal stem cells do not aggravate the progression of precancerous lesions: A new biosafety insight. Stem Cell Res. Ther. 2018, 9, 137. [CrossRef]
- Meng, W.; Wu, Y.; He, X.; Liu, C.; Gao, Q.; Ge, L.; Wu, L.; Liu, Y.; Guo, Y.; Li, X.; Liu, Y.; Chen, S.; Kong, X.; Liang, Z.; Zhou, H. A systems biology approach identifies effective tumor-stroma common targets for oral squamous cell carcinoma. Cancer Res. 2014, 74, 2306–2315. [CrossRef]
- Kumar, J.D.; Holmberg, C.; Kandola, S.; Steele, I.; Hegyi, P.;p Tiszlavicz, L.; Jenkins, R.; Beynon, R.J.; Peeney, D.; Giger, O.T.; Alqahtani, A.; Wang, T.C.; Charvat, T.T.; Penfold, M.; Dockray, G.J.; Varro, A. Increased expression of chemerin in squamous esophageal cancer myofibroblasts and role in recruitment of mesenchymal stromal cells. PLoS ONE 2014, 9(7), e104877. [CrossRef]
- Tan, T.T.; Lai, R.C.; Padmanabhan, J.; Sim, W.K.; Choo, A.B.H.; Lim, S.K. Assessment of Tumorigenic Potential in Mesenchymal-Stem/Stromal-Cell-Derived Small Extracellular Vesicles (MSC-sEV). Pharm. 2021, 14, 345. [CrossRef]
- Zielske, S.P.; Livant, D.L.; Lawrence, T.S. Radiation Increases Invasion of Gene-Modified Mesenchymal Stem Cells into Tumors. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 843–853. [CrossRef]
- Escacena, N.; Quesada-Hernandez, E.; Capilla-Gonzalez, V.; Soria, B.; Hmadcha, A. Bottlenecks in the efficient use of advanced therapy medicinal products based on mesenchymal stromal cells. Stem Cells Int. 2015, 2015, 895714. [CrossRef]
- Melen, G. J.; Franco-Luzon, L.; Ruano, D.; Gonzalez-Murillo, A.; Alfranca, A.; Casco, F., Lassaletta, Á.; Alonso, M.; Madero, L.; Alemany, R.; García-Castro, J.; Ramírez, M. Influence of carrier cells on the clinical outcome of children with neuroblastoma treated with high dose of oncolytic adenovirus delivered in mesenchymal stem cells. Cancer Lett. 2016, 371(2), 161–170. [CrossRef]
- Carlander, A.F.; Gundestrup, A.K.; Jansson, P.M.; Follin, B.; Hoeeg, C.; Kousholt, B.S.; Larsen, R.T.; Jakobsen, K.K.; Rimborg, S.; Fischer-Nielsen, A.; Grønhøj, C.; Buchwald, C.V.; Lynggaard, C.D. Mesenchymal Stromal/Stem Cell Therapy Improves Salivary Flow Rate in Radiation-Induced Salivary Gland Hypofunction in Preclinical in vivo Models: A Systematic Review and Meta-Analysis. Stem Cell Rev. Rep. 2024, 20(4), 1078–1092. [CrossRef]
- Jansson, P.M.; Lynggaard, C.D.; Carlander, A.F.; Jensen, S.B.; Follin, B.; Hoeeg, C.; Kousholt, B.S.; Larsen, R.T.; Grønhøj, C.; Jakobsen, K.K.; Rimborg, S.; Fischer-Nielsen, A.; Menon, J.M.L.; on Buchwald, C. Mesenchymal stromal/stem cell therapy for radiation-induced salivary gland hypofunction in animal models: A protocol for a systematic review and meta-analysis. Systematic reviews 2022, 11(1), 72. [CrossRef]
- Jakobsen, K.K.; Carlander, A.F.; Todsen, T.; Melchiors, J.; Paaske, N.; Østergaard Madsen, A.K.; Kloch Bendtsen, S.; Mordhorst, C.; Stampe, H.; Kastrup, J.; Ekblond, A.; Haack-Sørensen, M.; Farhadi, M.; Maare, C.; Friborg, J.; Lynggaard, C.D.; Werner Hauge, A.; Christensen, R.; Grønhøj, C.; von Buchwald, C. Mesenchymal Stem/Stromal Cell Therapy for Radiation-Induced Xerostomia in Previous Head and Neck Cancer Patients: A Phase II Randomized, Placebo-Controlled Trial. Clin. Cancer Res: An official journal of the American Association for Cancer Research 2024, 30(10), 2078–2084. [CrossRef]
- Strojan, P.; Plavc, G.; Kokalj, M.; Mitrovic, G.; Blatnik, O.; Lezaic, L.; Socan, A.; Bavec, A.; Tesic, N.; Hartman, K.; Svajger, U. Post-radiation xerostomia therapy with allogeneic mesenchymal stromal stem cells in patients with head and neck cancer: Study protocol for phase I clinical trial. Radiol Oncol. 2023, 57(4), 538–549. [CrossRef]
- Jakobsen, K.K.; Carlander, A.F.; Grønhøj, C.; Todsen, T.; Melchiors, J.; Paaske, N.; Madsen, A K.Ø.; Kastrup, J.; Ekblond, A.; Haack-Sørensen, M.; Farhadi, M.; Maare, C.; Friborg, J.; Lynggard, C. D.; von Buchwald, C. Effectiveness and safety of mesenchymal stem/stromal cell for radiation-induced hyposalivation and xerostomia in previous head and neck cancer patients (MESRIX-III): A study protocol for a single-centre, double-blinded, randomised, placebo-controlled, phase II study. Trials 2023, 24(1), 567. [CrossRef]
- Lynggaard, C. D., Grønhøj, C., Christensen, R., Fischer-Nielsen, A., Melchiors, J., Specht, L., Andersen, E., Mortensen, J., Oturai, P., Barfod, G. H., Haastrup, E. K., Møller-Hansen, M., Haack-Sørensen, M., Ekblond, A., Kastrup, J., Jensen, S. B., & von Buchwald, C. Intraglandular Off-the-Shelf Allogeneic Mesenchymal Stem Cell Treatment in Patients with Radiation-Induced Xerostomia: A Safety Study (MESRIX-II). Stem Cells Transl Med. 2022, 11(5), 478–489. [CrossRef]
- Lynggaard, C.D.; Grønhøj, C.; Jensen, S.B.; Christensen, R.; Specht, L.; Andersen, E.; Andersen, T.T.; Ciochon, U.M.; Rathje, G.S.; Hansen, A.E.; Stampe, H.; Fischer-Nielsen, A.; von Buchwald, C. Long-term Safety of Treatment with Autologous Mesenchymal Stem Cells in Patients with Radiation-Induced Xerostomia: Primary Results of the MESRIX Phase I/II Randomized Trial. Clin Cancer Res. 2022, 28(13), 2890–2897. [CrossRef]
- Blitzer, G. C., Paz, C., Glassey, A., Ganz, O. R., Giri, J., Pennati, A., Meyers, R. O., Bates, A. M., Nickel, K. P., Weiss, M., Morris, Z. S., Mattison, R. J., McDowell, K. A., Croxford, E., Chappell, R. J., Glazer, T. A., Rogus-Pulia, N. M., Galipeau, J., & Kimple, R. J. Functionality of bone marrow mesenchymal stromal cells derived from head and neck cancer patients - A FDA-IND enabling study regarding MSC-based treatments for radiation-induced xerostomia. Radiother Oncol. 2024, 192, 110093. [CrossRef]
- Grønhøj, C.; Jensen, D.H.; Vester-Glowinski, P.; Jensen, S.B.; Bardow, A.; Oliveri, R.S.; Fog, L.M.; Specht, L.; Thomsen, C.; Darkner, S.; Jensen, M.; Müller, V.; Kiss, K.; Agander, T.; Andersen, E.; Fischer-Nielsen, A.; von Buchwald, C. Safety and Efficacy of Mesenchymal Stem Cells for Radiation-Induced Xerostomia: A Randomized, Placebo-Controlled Phase 1/2 Trial (MESRIX). Int. J. Radiat. Oncol. Biol. Phys. 2018, 101(3), 581–592. [CrossRef]
- Blitzer, G.C.; Rogus-Pulia, N. M.; Mattison, R.J.;Varghese, T.; Ganz, O.; Chappell, R.; Galipeau, J.; McDowell, K.A.; Meyers, R.O.; Glazer, T.A.; Kimple, R.J. Marrow-Derived Autologous Stromal Cells for the Restoration of Salivary Hypofunction (MARSH): A pilot, first-in-human study of interferon gamma-stimulated marrow mesenchymal stromal cells for treatment of radiation-induced xerostomia. Cytotherapy 2023, 25(11), 1139–1144. [CrossRef]
- Blitzer, G.C.; Rogus-Pulia, N. M.; Mattison, R.J.;Varghese, T.; Ganz, O.; Chappell, R.; Galipeau, J.; McDowell, K.A.; Meyers, R.O.; Glazer, T.A.; Kimple, R.J. Marrow-Derived Autologous Stromal Cells for the Restoration of Salivary Hypofunction (MARSH): Study protocol for a phase 1 dose-escalation trial of patients with xerostomia after radiation therapy for head and neck cancer: MARSH: Marrow-Derived Autologous Stromal Cells for the Restoration of Salivary Hypofunction. Cytotherapy 2022, 24(5), 534–543. [CrossRef]
- Lynggaard, C.D.; Jersie-Christensen, R.; Juhl, M.; Jensen, S.B.; Grønhøj, C.; Melchiors, J.; Jacobsen, S.; Møller-Hansen, M.; Herly, M.; Ekblond, A.; Kastrup, J.; Fischer-Nielsen, A.; Belstrøm, D.; von Buchwald, C. Intraglandular mesenchymal stem cell treatment induces changes in the salivary proteome of irradiated patients. Commun. Med. (Lond.) 2022, 2(1), 160. [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Tazetdinova, L.G.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Application of Mesenchymal Stem Cells for Therapeutic Agent Delivery in Anti-tumor Treatment. Front. Pharmacol. 2018, 9, 259. [CrossRef]
- Shen, C.J.; Chan, T.F.; Chen, C.C.; Hsu, Y.C.; Long, C.Y.; Lai, C.S. Human umbilical cord matrix-derived stem cells expressing interferon-beta gene inhibit breast cancer cells via apoptosis. Oncotarget 2018, 7, 34172–34179. [CrossRef]
- Liu, X., Hu, J., Li, Y., Cao, W., Wang, Y., Ma, Z., Li, F. Mesenchymal stem cells expressing interleukin-18 inhibit breast cancer in a mouse model. Onco. Lett. 2018, 15(5), 6265–6274. [CrossRef]
- Ciavarella, S.; Grisendi, G.; Dominici, M.; Tucci, M.; Brunetti, O.; Dammacco, F., Silvestris, F. In vitro anti-myeloma activity of TRAIL-expressing adipose-derived mesenchymal stem cells. Br. J. Haematol. 2012, 157, 586–598. [CrossRef]
- Fakiruddin, K.S.; Baharuddin, P.; Lim, M.N.; Fakharuzi, N.A.; Yusof, N.A.; Zakaria, Z. Nucleofection optimization and in vitro anti-tumourigenic effect of TRAIL-expressing human adipose-derived mesenchymal stromal cells. Cancer Cell Int. 2014, 14, 122. [CrossRef]
- Guo, X.R.; Yang, Z.S.; Tang, X.J.; Zou, D.D.; Gui, H.; Wang, X.L., Ma, S.N.; Yuan, Y.H.; Fang, J.; Wang, B.; Zhang, L.; Sun, X.Y.; Warnock, G.L.; Dai, L.J.; Tu, H.J. The application of mRNA-based gene transfer in mesenchymal stem cell-mediated cytotoxicity of glioma cells. Oncotarget 2016, 7, 55529–55542. [CrossRef]
- Jiang, X.; Fitch, S.; Wang, C.; Wilson, C.; Li, J.; Grant, G.A., Yang, F. Nanoparticle engineered TRAIL-overexpressing adipose-derived stem cells target and eradicate glioblastoma via intracranial delivery. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 13857–13862. [CrossRef]
- Li, G.C.; Zhang, H.W.; Zhao, Q.C.; Sun, L.I.; Yang, J.J.; Hong, L., Feng F.; Cai, L. Mesenchymal stem cells promote tumor angiogenesis via the action of transforming growth factor beta1. Oncol. Lett. 2016, 11, 1089–1094. [CrossRef]
- Mangraviti, A.; Tzeng, S.Y.; Gullotti, D.; Kozielski, K.L.; Kim, J.E.; Seng, M.; Abbadi, S.; Schiapparelli, P.; Sarabia-Estrada, R.; Vescovi, A.; Brem, H.; Olivi, A.; Tyler, B.; Green, J.J.; Quinones-Hinojosa, A. Non-virally engineered human adipose mesenchymal stem cells produce BMP4, target brain tumors, and extend survival. Biomaterials 2016, 100, 53–66. [CrossRef]
- Rincon, E.; Cejalvo, T.; Kanojia, D.; Alfranca, A.; Rodriguez-Milla, M.A.; Gil Hoyos, R.A.; Han, Y.; Zhang, L.; Alemany, R.; Lesniak, M. S.; García-Castro, J. Mesenchymal stem cell carriers enhance antitumor efficacy of oncolytic adenoviruses in an immunocompetent mouse model. Oncotarget 2017, 8(28), 45415–45431. [CrossRef]
- Guo, Y.; Zhang, Z.; Xu, X.; Xu, Z.; Wang, S.; Huang, D.; Li, Y.; Mou, X.; Liu, F.; Xiang, C. Menstrual blood-derived stem cells as delivery vehicles for oncolytic adenovirus virotherapy for Colorectal Cancer. Stem Cells Dev. 2019, 28, 882–896. [CrossRef]
- Du, W.; Seah, I.; Bougazzoul, O.; Choi, G.; Meeth, K.; Bosenberg, M.W.; et al. Stem cell-released oncolytic herpes simplex virus has therapeutic efficacy in brain metastatic melanomas. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E6157–E6165. [CrossRef]
- Brini, A.T.; Coccè, V.; Ferreira, L.M.; Giannasi, C.; Cossellu, G.; Giannì, A.B.; Angiero, F.; Bonomi, A.; Pascucci, L.; Falchetti, M.L.; Ciusani, E.; Bondiolotti, G.; Sisto, F.; Alessandri, G.; Pessina, A., Farronato, G. Cell-mediated drug delivery by gingival interdental papilla mesenchymal stromal cells (GinPa-MSCs) loaded with paclitaxel. Expert Opin Drug Deliv. 2016, 13(6), 789–798. [CrossRef]
- Layek, B.; Sadhukha, T.; Panyam, J.; Prabha, S. Nano-engineered mesenchymal stem cells increase therapeutic efficacy of anticancer drug through true active tumor targeting. Mol. Cancer Ther. 2018, 17, 1196–1206. [CrossRef]
- Wang, X.; Gao, J.; Ouyang, X.; Wang, J.; Sun, X.; and Lv, Y; Mesenchymal stem cells loaded with paclitaxel-poly(lactic-co-glycolic acid) nanoparticles for glioma-targeting therapy. Int. J. Nanomed. 2018, 13, 5231–5248. [CrossRef]
- Moku, G.; Layek, B.; Trautman, L.; Putnam, S.; Panyam, J.; and Prabha, S. Improving payload capacity and anti-tumor efficacy of mesenchymal stem cells using TAT peptide functionalized polymeric nanoparticles. Cancers 2019, 1, E491. [CrossRef]
- Collino, F.; Bruno, S.; Lindoso; R. S.; Camussi, G. miRNA expression in mesenchymal stem cells. Curr. Pathobiol. Rep. 2014, 2, 101–107. [CrossRef]
- Lang, F.M.; Hossain, A.; Gumin, J.; Momin, E.N.; Shimizu, Y.; Ledbetter, D.; Shahar, T.; Yamashita, S.; Parker Kerrigan, B.; Fueyo, J.; Sawaya, R., Lang, F.F. Mesenchymal stem cells as natural biofactories for exosomes carrying miR-124a in the treatment of gliomas. Neuro. Oncol. 2018, 20(3), 380–390. [CrossRef]
- Sharif, S.; Ghahremani, M.H.; Soleimani, M. Delivery of exogenous miR-124 to glioblastoma multiform cells by wharton’s jelly mesenchymal stem cells decreases cell proliferation and migration, and confers chemosensitivity. Stem Cell Rev. 2018, 14, 236–246. [CrossRef]
- Sharif, S.; Ghahremani, M.H.; Soleimani, M. Delivery of exogenous miR-124 to glioblastoma multiform cells by wharton’s jelly mesenchymal stem cells decreases cell proliferation and migration, and confers chemosensitivity. Stem Cell Rev. 2018, 14, 236–246. [CrossRef]
- Bregy, A.; Shah, A.H.; Diaz, M.V.; Pierce, H.E.; Ames, P.L.; Diaz, D.; Komotar, R.J. The role of Gliadel wafers in the treatment of high-grade gliomas. Expert Rev. Anticancer Ther. 2013, 13(12), 1453–1461. [CrossRef]
- Smith, S.J.; Tyler, B.M.; Gould, T.; Veal, G.J.; Gorelick, N.; Rowlinson, J.; Serra, R.; Ritchie, A.; Berry, P.; Otto, A.; Choi, J.; Skuli, N.; Estevez-Cebrero, M.; Shakesheff, K.M.; Brem, H.; Grundy, R.G.; Rahman, R. Overall survival in malignant glioma is significantly prolonged by neurosurgical delivery of etoposide and temozolomide from a thermo-responsive biodegradable paste. Clin. Cancer Res. 2019, 25(16), 5094–5106. [CrossRef]
- Zeng, X.; Qiu, X.C.; Ma, Y.H.; Duan, J.J.; Chen, Y.F.; Gu, H.Y., Wang, J.M.; Ling, E.A.; Wu, J.L.; Wu, W.; Zeng, Y.S. Integration of donor mesenchymal stem cell-derived neuron-like cells into host neural network after rat spinal cord transection. Biomaterials 2015, 53, 184–201. [CrossRef]
- Wang, Q.L.; Wang, H.J.; Li, Z.H.; Wang, Y.L.; Wu, X.P.; Tan, Y.Z. Mesenchymal stem cell-loaded cardiac patch promotes epicardial activation and repair of the infarcted myocardium. J. Cell Mol. Med. 2017, 21, 1751–1766. [CrossRef]
- Diomede, F.; Gugliandolo, A.; Cardelli, P.; Merciaro, I.; Ettorre, V.; Traini, T.; Bedini, R.; Scionti, D.; Bramanti, A.; Nanci, A.; Caputi, S.; Fontana, A.; Mazzon, E.; Trubiani, O. Three-dimensional printed PLA scaffold and human gingival stem cell-derived extracellular vesicles: A new tool for bone defect repair. Stem Cell Res. Ther. 2018, 9(1), 104. [CrossRef]
- Sheets, K.T.; Bago, J.R.; Hingtgen, S.D. Delivery of Cytotoxic Mesenchymal Stem Cells with Biodegradable Scaffolds for Treatment of Postoperative Brain Cancer. Methods Mol. Biol. 2018, 1831, 49–58. [CrossRef]
- Aliperta, R.; Welzel, P.B.; Bergmann, R.; Freudenberg, U.; Berndt, N.; Feldmann, A.; Arndt, C.; Koristka, S.; Stanzione, M.; Cartellieri, M.; Ehninger, A.; Ehninger, G.; Werner, C.; Pietzsch, J.; Steinbach, J.; Bornhäuser, M.; Bachmann, M.P. Cryogel-supported stem cell factory for customized sustained release of bispecific antibodies for cancer immunotherapy. Sci Rep. 2017, 7, 42855. [CrossRef]
- Johansson, M.; Oudin, A.; Tiemann, K.; Bernard, A.; Golebiewska, A.; Keunen, O.; Fack, F.; Stieber, D.; Wang, B.; Hedman, H.; Niclou, S.P. The soluble form of the tumor suppressor Lrig1 potently inhibits in vivo glioma growth irrespective of EGF receptor status. Neuro. Oncol. 2013, 15(9), 1200–1211. [CrossRef]
- Galland, S.; Stamenkovic, I. Mesenchymal stromal cells in cancer: A review of their immunomodulatory functions and dual effects on tumor progression. J. Pathol. 2020, 250(5), 555–572. [CrossRef]
- Avnet, S.; Lemma, S.; Cortini, M.; Di Pompo, G.; Perut, F., Baldini N.. Pre-clinical models for studying the interaction between mesenchymal stromal cells and cancer cells and the induction of stemness. Front. Oncol. 2019, 9. 305. [CrossRef]
- Li, C.; Zhao, H;.; Cheng, L.; Wang, B. Allogeneic vs. autologous mesenchymal stem/stromal cells in their medication practice. Cell Biosci. 2021, 11(1), 187. [CrossRef]
- Wang, J.; Duan, X.; Yang, L.; Liu, X.; Hao, C.; Dong, H.; Gu, H.; Tang, H.; Dong, B.; Zhang, T.; Gao, G.; Liang, R. Comparison of Survival Between Autologous and Allogeneic Stem Cell Transplantation in Patients with Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma: A Meta-Analysis. Cell Transplant. 2020;29, 963689720975397. [CrossRef]
- Durand, N.; Zubair, A.C. Autologous versus allogeneic mesenchymal stem cell therapy: The pros and cons. Surgery 2022, 171(5), 1440–1442. [CrossRef]
- Jensen, S.B.; Vissink, A.; Limesand, K.H.; Reyland, M.E. Salivary gland hypofunction and Xerostomia in Head and Neck Radiation patients. J. Natl. Cancer Inst. Monogr. 2019, 2019(53), lgz016. [CrossRef]
- Vissink, A.; van Luijk, P.; Langendijk, J.A.; Coppes, R.P. Current ideas to reduce or salvage radiation damage to salivary glands. Oral Diseases. 2015;21, e1–10. [CrossRef]
- Nutting, C.M.; Morden, J.P.; Harrington, K.J.; Urbano, T.G.; Bhide, S.A.; Clark, C.; Miles, E.A.; Miah, A.B.; Newbold, K.; Tanay, M.; Adab, F.; Jefferies, S.J.; Scrase, C.; Yap, B.K.; A’Hern, R.P.; Sydenham, M.A.; Emson, M.; Hall, E.; & PARSPORT trial management group. Parotid-sparing intensity modulated versus conventional radiotherapy in head and neck cancer (PARSPORT): A phase 3 multicentre randomised controlled trial. The Lancet Oncology 2011, 12, 127–136. [CrossRef]
- Jeong, J.; Baek, H.; Kim, Y.J.; Choi, Y.; Lee, H.; Lee, E.; Kim, E.S.; Hah, J.H.; Kwon, T.K.; Choi, I.J.; Kwon, H. Human salivary gland stem cells ameliorate hyposalivation of radiation-damaged rat salivary glands. Exp. Mol. Med. 2013, 45(11), e58. [CrossRef]
- Lim JY, Yi T, Choi JS, Jang YH, Lee S, Kim HJ; et al. Intraglandular transplantation of bone marrow-derived clonal mesenchymal stem cells for amelioration of post-irradiation salivary gland damage. Oral Oncology 2013, 49, 136–143. [CrossRef]
- Lim, J.Y.; Ra, J.C.; Shin, I.S.; Jang, Y.H.; An, H.Y.; Choi, J.S.; Kim, W.C.; Kim, Y.M. Systemic transplantation of human adipose tissue-derived mesenchymal stem cells for the regeneration of irradiation-induced salivary gland damage. PLoS ONE 2013, 8(8), e71167. [CrossRef]
- Wang, Z.; Xing, H.; Hu, H.; Dai, T.; Wang, Y.; Li, Z.; An, R.; Xu, H.; Liu, Y.; Liu, B. Intraglandular transplantation of adipose-derived stem cells combined with platelet-rich fibrin extract for the treatment of irradiation-induced salivary gland damage. Exp. Ther. Med. 2018, 15(1), 795–805. [CrossRef]
- Li, Z.; Wang, Y.; Xing, H.; Wang, Z.; Hu, H.; An, R.; Xu, H.; Liu, Y.; Liu, B. Protective efficacy of intravenous transplantation of adipose-derived stem cells for the prevention of radiation-induced salivary gland damage. Arch. Oral Biol. 2015, 60(10), 1488–1496. [CrossRef]
- Shin, H.S.; Lee, S.; Hong, H.J.; Lim, Y.C.; Koh, W.G.; Lim, J.Y. Stem cell properties of human clonal salivary gland stem cells are enhanced by three-dimensional priming culture in nanofibrous microwells. Stem. Cell Res. Ther. 2018, 9(1), 74. [CrossRef]
- Choi, J.S.; An, H.Y.; Shin, H.S.; Kim, Y.M.; Lim, JY. Enhanced tissue remodelling efficacy of adipose-derived mesenchymal stem cells using injectable matrices in radiation-damaged salivary gland model. J. Tissue Eng. Regen. Med. 2018, 12(2), e695–e706. [CrossRef]
- Shin, H.S.; Lee, S.; Kim, Y.M.; Lim, J.Y. Hypoxia-activated adipose mesenchymal stem cells prevents Irradiation-Induced Salivary Hypofunction by enhanced paracrine effect through fibroblast growth factor 10. Stem Cells. 2018, 36, 1020–1032. [CrossRef]
- Mulyani, S.W.M.; Astuti, E.R.; Wahyuni, O.R.; Ernawati, D.S.; Ramadhani, N.F. Xerostomia Therapy due to Ionized Radiation using preconditioned bone marrow-derived mesenchymal stem cells. Eur. J. Dent. 2019, 13, 238–242. [CrossRef]
Figure 1.
(A) Dual effect of tumour-associated mesenchymal stromal cells (MSCs) on head and neck cancer (HNC). MSCs can affect both tumour progression or tumorigenesis inhibition with a tendency to promote the former. MSCs in TME show pro-tumorigenic activity on neoplastic cells directly via cell-to-cell contact or extracellular vesicles (MSC-EVs). MSC-EVs include key immune-modulating agents, proangiogenic factors, pro-survival biological agents or soluble factors stimulating cell mobility and extracellular matrix modulators, which favour higher tumour aggressiveness. MSCs isolated from squamous cell carcinomas can mediate cancer progression by secreting pro-inflammatory and pro-angiogenic cytokines such as IL-6 and IL8, which also promotes the recruitment of TAMs, CAFs, T cells, neutrophils and other MSCs. In addition, TGF-β1,VEGF, EGF, PDGF and cytokines IL-6 and IL-8, as well as B2M, CCN2, SCGF, SDF-1 and chemokines such as CXCL 1/5/7/8 are all primarily responsible for tissue remodelling and growth. The formation of new vessels in the tumour niche, enabling the initial growth of neoplastic lesions and then tumour metastasis, is associated with the action of VEGF, IGF-1, TGF-β1, FGF, angiopoetin-1, endothelin-1, and cytokines IL-6 and IL-8, which facilitate further tumour progression and growth by enhancing angiogenesis, increasing vascularization and the local/regional and general spread of tumour cells. Most of these MSC-derived factors directly regulate the epithelial-mesenchymal plasticity, proliferation, invasion and migration and even drug resistance in HNSCC cells. MSCs can also activate other key signalling pathways, soluble agents and modulators, giving an opposite effect on tumour progression and aggressiveness. The anti-tumour MSC-related paracrine soluble secreted factors and MSC-derived molecules released from exosomes which inhibit tumorigenesis include anti-proliferative factors such as Dkk-1, oncostatin-M, a soluble Wnt antagonist, PTEN, BMP, cytotoxic agents such as TRAIL, IFN-α, IGFBP and TNF-α, anti-angiogenic factors or immunomodulatory agents. MSCs may also actively inhibiting further tumour development by blocking cancer-related signalling, such as PI3K/AKT, Wnt/β-catenin, JAK/STAT pathway. (B) The immunomodulation mechanisms of MSCs and their role in immune cell activity. Adaptive immune system: The active MSCs in the tumour milieu inhibit the adaptive immune response through the secretion of mediators contained in exosomes (MSCs-EV) and soluble factors, such as IDO, TGFβ1, TNF-α, IFN-γ, PGE2, NO, HLA-G, HGF, IL-1β, IL-1α, IL-4 and IL-6; they also interact with various immune cell types, including T cells, B cells, DC cells, NK cells, monocytes and TAMs. This MSC activity constrains dendritic cell maturation, reduces T cell proliferation, enhances macrophage activation and polarizes them from M1 towards M2; it also facilitates neutrophil mobility and affects the regulation of CD56dim/16bright NK cells and invariant natural killer T (iNKT) cells. In addition, it can shift the balance of T cell differentiation from the Th1 to an anti-inflammatory Th2 phenotype and enhance the maturation of T helper cells into CD4+CD25+Foxp3+Treg pathways, which can inhibit effector T cell responses and thus reduce anti-tumour immunity. The innate immune system: In tumorigenic tissues, local factors, such as cytokine milieu TNF-α and endotoxin LPS, hypoxia and Toll-like receptor (TLRs) ligands, stimulate MSCs, promoting the large-scale secretion of growth factors such as VEGF, FGF2, IGF-1, or HGF by an NFκB-dependent mechanism with the effect of driving tissue regeneration, angiogenesis, reducing anti-tumour immunity and allowing the effective escape of the tumour from immune surveillance. Abbreviations: MSCs: Mesenchymal stromal cells; CSCs: Cancer stem cells; BM-MSCs: Bone-marrow-derived MSCs; A-MSCs: Adipose tissue-derived MSCs; N-MSC: Naïve MSCs; CAFs: Cancer-associated fibroblasts; ECM: Extracellular matrix; CIS: Carcinoma in situ; MMPs (MT-MMPs): Matrix metalloproteinases, also known as matrix metallopeptidases; TGF-β1: Transforming growth factor beta 1; CXCL1/2/12: C-X-C motif chemokine ligand 1/2/12; CCL5: C-C motif chemokine ligand 5; VEGF: Vascular endothelial growth factor; EGF: Epidermal growth factor; PDGF: Platelet-derived growth factor; FGF: Fibroblast growth factors; TRAIL: TNF-related apoptosis-inducing ligand; INF-α: Type-I interferon alpha; IGFBP: Insulin-like growth factor-binding protein; Dkk-1: Dickkopf-related protein 1; HGF: Hepatocyte growth factor; PTEN: Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase; BMP: Bone morphogenetic protein; TRAIL: TNF-related apoptosis-inducing ligand; PGE2: Prostaglandin E2; B2M: Beta 2 microglobulin; CCN2: Cellular communication network factor 2; SCGF: stromal cell growth factor-beta; SDF-1: stromal cell-derived factor 1; IDO: Indoleamine 2,3-dioxygenase; iNOS: Nitric oxide synthases; HO1: Heme oxygenase 1; CTLs: Cytotxic T cells; CD4+CD25+Foxp3+Treg: Regulatory T cells, known as suppressor T cells; TAMs M1/2: Tumour-associated macrophages M1/2, CD56dim/CD16bright NK cells: Activated natural killer cells, TLRs: Toll-like receptors; → activation mechanisms; ¦ inhibitory mechanisms.
Figure 1.
(A) Dual effect of tumour-associated mesenchymal stromal cells (MSCs) on head and neck cancer (HNC). MSCs can affect both tumour progression or tumorigenesis inhibition with a tendency to promote the former. MSCs in TME show pro-tumorigenic activity on neoplastic cells directly via cell-to-cell contact or extracellular vesicles (MSC-EVs). MSC-EVs include key immune-modulating agents, proangiogenic factors, pro-survival biological agents or soluble factors stimulating cell mobility and extracellular matrix modulators, which favour higher tumour aggressiveness. MSCs isolated from squamous cell carcinomas can mediate cancer progression by secreting pro-inflammatory and pro-angiogenic cytokines such as IL-6 and IL8, which also promotes the recruitment of TAMs, CAFs, T cells, neutrophils and other MSCs. In addition, TGF-β1,VEGF, EGF, PDGF and cytokines IL-6 and IL-8, as well as B2M, CCN2, SCGF, SDF-1 and chemokines such as CXCL 1/5/7/8 are all primarily responsible for tissue remodelling and growth. The formation of new vessels in the tumour niche, enabling the initial growth of neoplastic lesions and then tumour metastasis, is associated with the action of VEGF, IGF-1, TGF-β1, FGF, angiopoetin-1, endothelin-1, and cytokines IL-6 and IL-8, which facilitate further tumour progression and growth by enhancing angiogenesis, increasing vascularization and the local/regional and general spread of tumour cells. Most of these MSC-derived factors directly regulate the epithelial-mesenchymal plasticity, proliferation, invasion and migration and even drug resistance in HNSCC cells. MSCs can also activate other key signalling pathways, soluble agents and modulators, giving an opposite effect on tumour progression and aggressiveness. The anti-tumour MSC-related paracrine soluble secreted factors and MSC-derived molecules released from exosomes which inhibit tumorigenesis include anti-proliferative factors such as Dkk-1, oncostatin-M, a soluble Wnt antagonist, PTEN, BMP, cytotoxic agents such as TRAIL, IFN-α, IGFBP and TNF-α, anti-angiogenic factors or immunomodulatory agents. MSCs may also actively inhibiting further tumour development by blocking cancer-related signalling, such as PI3K/AKT, Wnt/β-catenin, JAK/STAT pathway. (B) The immunomodulation mechanisms of MSCs and their role in immune cell activity. Adaptive immune system: The active MSCs in the tumour milieu inhibit the adaptive immune response through the secretion of mediators contained in exosomes (MSCs-EV) and soluble factors, such as IDO, TGFβ1, TNF-α, IFN-γ, PGE2, NO, HLA-G, HGF, IL-1β, IL-1α, IL-4 and IL-6; they also interact with various immune cell types, including T cells, B cells, DC cells, NK cells, monocytes and TAMs. This MSC activity constrains dendritic cell maturation, reduces T cell proliferation, enhances macrophage activation and polarizes them from M1 towards M2; it also facilitates neutrophil mobility and affects the regulation of CD56dim/16bright NK cells and invariant natural killer T (iNKT) cells. In addition, it can shift the balance of T cell differentiation from the Th1 to an anti-inflammatory Th2 phenotype and enhance the maturation of T helper cells into CD4+CD25+Foxp3+Treg pathways, which can inhibit effector T cell responses and thus reduce anti-tumour immunity. The innate immune system: In tumorigenic tissues, local factors, such as cytokine milieu TNF-α and endotoxin LPS, hypoxia and Toll-like receptor (TLRs) ligands, stimulate MSCs, promoting the large-scale secretion of growth factors such as VEGF, FGF2, IGF-1, or HGF by an NFκB-dependent mechanism with the effect of driving tissue regeneration, angiogenesis, reducing anti-tumour immunity and allowing the effective escape of the tumour from immune surveillance. Abbreviations: MSCs: Mesenchymal stromal cells; CSCs: Cancer stem cells; BM-MSCs: Bone-marrow-derived MSCs; A-MSCs: Adipose tissue-derived MSCs; N-MSC: Naïve MSCs; CAFs: Cancer-associated fibroblasts; ECM: Extracellular matrix; CIS: Carcinoma in situ; MMPs (MT-MMPs): Matrix metalloproteinases, also known as matrix metallopeptidases; TGF-β1: Transforming growth factor beta 1; CXCL1/2/12: C-X-C motif chemokine ligand 1/2/12; CCL5: C-C motif chemokine ligand 5; VEGF: Vascular endothelial growth factor; EGF: Epidermal growth factor; PDGF: Platelet-derived growth factor; FGF: Fibroblast growth factors; TRAIL: TNF-related apoptosis-inducing ligand; INF-α: Type-I interferon alpha; IGFBP: Insulin-like growth factor-binding protein; Dkk-1: Dickkopf-related protein 1; HGF: Hepatocyte growth factor; PTEN: Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase; BMP: Bone morphogenetic protein; TRAIL: TNF-related apoptosis-inducing ligand; PGE2: Prostaglandin E2; B2M: Beta 2 microglobulin; CCN2: Cellular communication network factor 2; SCGF: stromal cell growth factor-beta; SDF-1: stromal cell-derived factor 1; IDO: Indoleamine 2,3-dioxygenase; iNOS: Nitric oxide synthases; HO1: Heme oxygenase 1; CTLs: Cytotxic T cells; CD4+CD25+Foxp3+Treg: Regulatory T cells, known as suppressor T cells; TAMs M1/2: Tumour-associated macrophages M1/2, CD56dim/CD16bright NK cells: Activated natural killer cells, TLRs: Toll-like receptors; → activation mechanisms; ¦ inhibitory mechanisms.
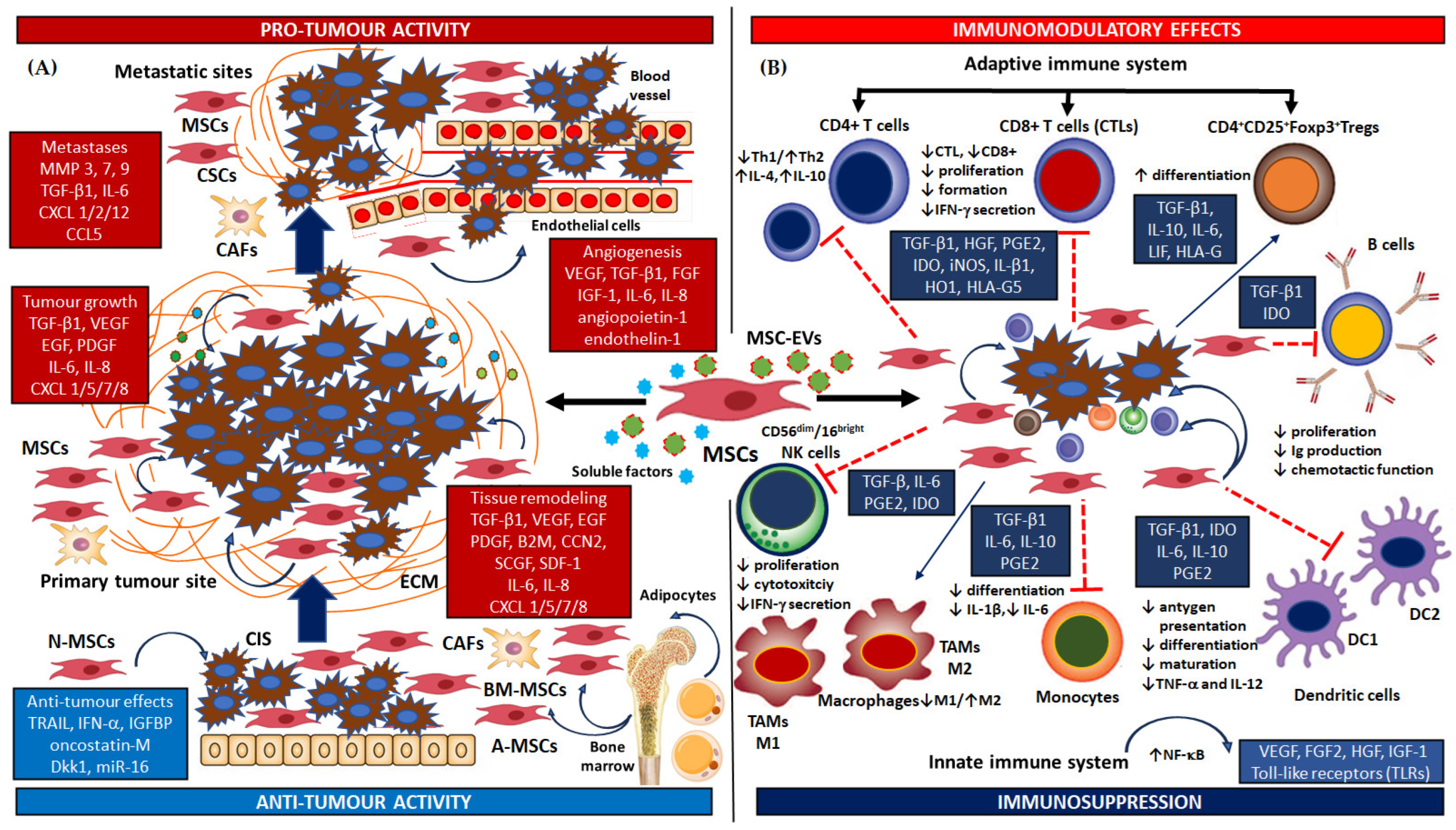
Figure 2.
Current studies and potential targets for MSC-based therapies of human cancer of various origin. MSCs and tumour cells may interact, thus influencing the MSC-based approaches for preventing tumour initiation and growth (A) The chemotactic movement of MSCs toward a tumour environment and chemoattractant-induces infiltration of tumour niche by naive MSCs (N-MSCs), and adipose- or bone marrow-derived mesenchymal cells (A-MSCS, BM-MSCs); in addition, the paracrine effects of MSCs secretome and stimulation of activation cancer stem cells (CSCs) is driven by soluble factors such as growth factors and and chemokines/cytokines, i.e., VEGF, PDGF, IL-8, IL-6, EGF or FGF2, SDF-1, G-CSF, GM-CSF, MCP1, HGF, and TGF-β. (B) MSCs have ability to deliver therapeutic drugs such as Doxorubicin (DOX), Paclitaxel (PTX), Gemcitabine (GCB), 5-Fluorouracil (5-FU) and Cisplatin (CDDP) directly to the tumour microenvironment. In addition to the direct use of MSCs in drug loading and priming, the secreted extracellular vesicles (MSC-EVs) isolated from MSCs represent an alternative approach to delivery (C) MSCs also have a high regulatory potential regarding the role of immunocompetent cells present in the tumour niche. MSCs by secreting or provoking the synthesis of bioactive molecules and immune-modulating agents such as INF-γ, INF-α, IL-2, IL-12, IL-15, IL-18, CXC3L1, TRAIL, NK4, VEGFR1 may promote the regulation of immune cell activation/homing and immune cell differentiation and increase the activation of anti-tumorigenic innate immune response, among others by promoting the anti-cancer activity of cells NK, CAFs, CTLs, TAM2 immune cells. (D) Genetically-modified MSCs containing inter alia suicide genes, DNA, miRNA, non-coding RNAs and other therapeutic proteins such as growth factors, transcription factors, cytokines or viral plasmids, or nano-engineered MSCs carrying nanoparticles, can also be used to deliver a range of tumour-suppressing cargos directly into the neoplastic milieu. These cargos include tumour suppressors, i.e., PTEN, NK4, TRAIL, CXCLs, INF-γ, INF-α, IL-2, IL-21 which can inhibit the formation of tumour cell culture, increase apoptosis and neoplastic expansion. They also be used in cell biobanking. (E) MSCs can also be used in oncolytic virus transduction in constructed preventive and therapeutic cancer vaccines. MSC-cloaked oncolytic viruses such as adenoviruses, herpes simplex viruses, measles viruses, myxoma viruses hold great potential as a novel virus-delivery platform for the therapy of various cancers. Replication of viral particles in MSCs and subsequent release of them not only promotes apoptosis and lysis of cancer cells, but also may be a potential effective engineered method of activating anti-cancer immune cells, i.e., cytotoxic T cells (CTLs), CD56dim/CD16bright NK cells (activated NK cells) and tumour-associated macrophages (TAMs M2). MSCs also have great potential for possible advanced cell therapies in situ, e.g., in the pre-cancerous stage, or as an adjuvant therapy to reduce toxicity associated with systemic treatment, to prevent tumour recurrence after surgery, to reduce the effects of radiotherapy, etc. Numerous studies show that nanotechnology MSCs or virus-carrying MSCs into the neoplastic lesions can serve as an effective vehicle for tumour-specific drug delivery; they can also significantly improve the antitumor efficacy of conventional chemotherapy and promote tumour lysis through the activity of oncolytic viruses. However, the MSCs recruited in the tumour microenvironment can exhibit both pro- and anti-oncogenic properties. Therefore, in order to develop new cancer therapy methods using MSCs, it is necessary to further deepen knowledge leading to understanding of the molecular and cellular interactions between MSCs and the tumour niche; Abbreviations: MSCs: Mesenchymal stromal cells; CSCs: Cancer stem cells; BM-MSCs: Bone-marrow-derived MSCs; A-MSCs: Adipose tissue-derived MSCs; N-MSC: Naïve MSCs; CAFs: Cancer-associated fibroblasts; EVs: MSC-derived extracellular vesicles; CTLA-4: Cytotoxic T-lymphocyte-associated protein 4; TCR: T-cell receptor; PD1/PD-1L: Programmed death receptor 1/ Programmed death receptor 1 ligand; MHC: Major histocompatibility complex; TGF-β1: Transforming growth factor beta 1; CXCL3L1: C-X-C motif chemokine ligand 3/ C-X-C motif chemokine ligand 3 like 1; TRAIL: TNF-related apoptosis-inducing ligand CCL5: C-C motif chemokine ligand 5; VEGF: Vascular endothelial growth factor; EGF: Epidermal growth factor; PDGF: Platelet-derived growth factor; VEGFR1: Vascular endothelial growth factor receptor 1; NK4: HGF-antagonist/angiogenesis inhibitor; TK: Herpes simplex virus (HSV)-1 thymidine kinase; CD: Herpes simplex virus (HSV)-1 cytosine deaminase; PTEN: Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase; CXCLs: The chemokine (C-X-C motif) ligands; GTA: Ganciclovir; CDDP: Cisplatin; DXR: Doxorubicin; PTX: Paclitaxel; GCB: Gemcitabine; 5-FU: 5-Fluorouracyl; CTLs: Cytotxic T cells; CAFs: Cancer-associated fibroblasts; Mϕ2: Tumour-associated macrophages TAMs M2; CD56dim/CD16bright NK cells: Activated natural killer cells; → activation mechanisms; ¦ inhibitory mechanisms.
Figure 2.
Current studies and potential targets for MSC-based therapies of human cancer of various origin. MSCs and tumour cells may interact, thus influencing the MSC-based approaches for preventing tumour initiation and growth (A) The chemotactic movement of MSCs toward a tumour environment and chemoattractant-induces infiltration of tumour niche by naive MSCs (N-MSCs), and adipose- or bone marrow-derived mesenchymal cells (A-MSCS, BM-MSCs); in addition, the paracrine effects of MSCs secretome and stimulation of activation cancer stem cells (CSCs) is driven by soluble factors such as growth factors and and chemokines/cytokines, i.e., VEGF, PDGF, IL-8, IL-6, EGF or FGF2, SDF-1, G-CSF, GM-CSF, MCP1, HGF, and TGF-β. (B) MSCs have ability to deliver therapeutic drugs such as Doxorubicin (DOX), Paclitaxel (PTX), Gemcitabine (GCB), 5-Fluorouracil (5-FU) and Cisplatin (CDDP) directly to the tumour microenvironment. In addition to the direct use of MSCs in drug loading and priming, the secreted extracellular vesicles (MSC-EVs) isolated from MSCs represent an alternative approach to delivery (C) MSCs also have a high regulatory potential regarding the role of immunocompetent cells present in the tumour niche. MSCs by secreting or provoking the synthesis of bioactive molecules and immune-modulating agents such as INF-γ, INF-α, IL-2, IL-12, IL-15, IL-18, CXC3L1, TRAIL, NK4, VEGFR1 may promote the regulation of immune cell activation/homing and immune cell differentiation and increase the activation of anti-tumorigenic innate immune response, among others by promoting the anti-cancer activity of cells NK, CAFs, CTLs, TAM2 immune cells. (D) Genetically-modified MSCs containing inter alia suicide genes, DNA, miRNA, non-coding RNAs and other therapeutic proteins such as growth factors, transcription factors, cytokines or viral plasmids, or nano-engineered MSCs carrying nanoparticles, can also be used to deliver a range of tumour-suppressing cargos directly into the neoplastic milieu. These cargos include tumour suppressors, i.e., PTEN, NK4, TRAIL, CXCLs, INF-γ, INF-α, IL-2, IL-21 which can inhibit the formation of tumour cell culture, increase apoptosis and neoplastic expansion. They also be used in cell biobanking. (E) MSCs can also be used in oncolytic virus transduction in constructed preventive and therapeutic cancer vaccines. MSC-cloaked oncolytic viruses such as adenoviruses, herpes simplex viruses, measles viruses, myxoma viruses hold great potential as a novel virus-delivery platform for the therapy of various cancers. Replication of viral particles in MSCs and subsequent release of them not only promotes apoptosis and lysis of cancer cells, but also may be a potential effective engineered method of activating anti-cancer immune cells, i.e., cytotoxic T cells (CTLs), CD56dim/CD16bright NK cells (activated NK cells) and tumour-associated macrophages (TAMs M2). MSCs also have great potential for possible advanced cell therapies in situ, e.g., in the pre-cancerous stage, or as an adjuvant therapy to reduce toxicity associated with systemic treatment, to prevent tumour recurrence after surgery, to reduce the effects of radiotherapy, etc. Numerous studies show that nanotechnology MSCs or virus-carrying MSCs into the neoplastic lesions can serve as an effective vehicle for tumour-specific drug delivery; they can also significantly improve the antitumor efficacy of conventional chemotherapy and promote tumour lysis through the activity of oncolytic viruses. However, the MSCs recruited in the tumour microenvironment can exhibit both pro- and anti-oncogenic properties. Therefore, in order to develop new cancer therapy methods using MSCs, it is necessary to further deepen knowledge leading to understanding of the molecular and cellular interactions between MSCs and the tumour niche; Abbreviations: MSCs: Mesenchymal stromal cells; CSCs: Cancer stem cells; BM-MSCs: Bone-marrow-derived MSCs; A-MSCs: Adipose tissue-derived MSCs; N-MSC: Naïve MSCs; CAFs: Cancer-associated fibroblasts; EVs: MSC-derived extracellular vesicles; CTLA-4: Cytotoxic T-lymphocyte-associated protein 4; TCR: T-cell receptor; PD1/PD-1L: Programmed death receptor 1/ Programmed death receptor 1 ligand; MHC: Major histocompatibility complex; TGF-β1: Transforming growth factor beta 1; CXCL3L1: C-X-C motif chemokine ligand 3/ C-X-C motif chemokine ligand 3 like 1; TRAIL: TNF-related apoptosis-inducing ligand CCL5: C-C motif chemokine ligand 5; VEGF: Vascular endothelial growth factor; EGF: Epidermal growth factor; PDGF: Platelet-derived growth factor; VEGFR1: Vascular endothelial growth factor receptor 1; NK4: HGF-antagonist/angiogenesis inhibitor; TK: Herpes simplex virus (HSV)-1 thymidine kinase; CD: Herpes simplex virus (HSV)-1 cytosine deaminase; PTEN: Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase; CXCLs: The chemokine (C-X-C motif) ligands; GTA: Ganciclovir; CDDP: Cisplatin; DXR: Doxorubicin; PTX: Paclitaxel; GCB: Gemcitabine; 5-FU: 5-Fluorouracyl; CTLs: Cytotxic T cells; CAFs: Cancer-associated fibroblasts; Mϕ2: Tumour-associated macrophages TAMs M2; CD56dim/CD16bright NK cells: Activated natural killer cells; → activation mechanisms; ¦ inhibitory mechanisms.
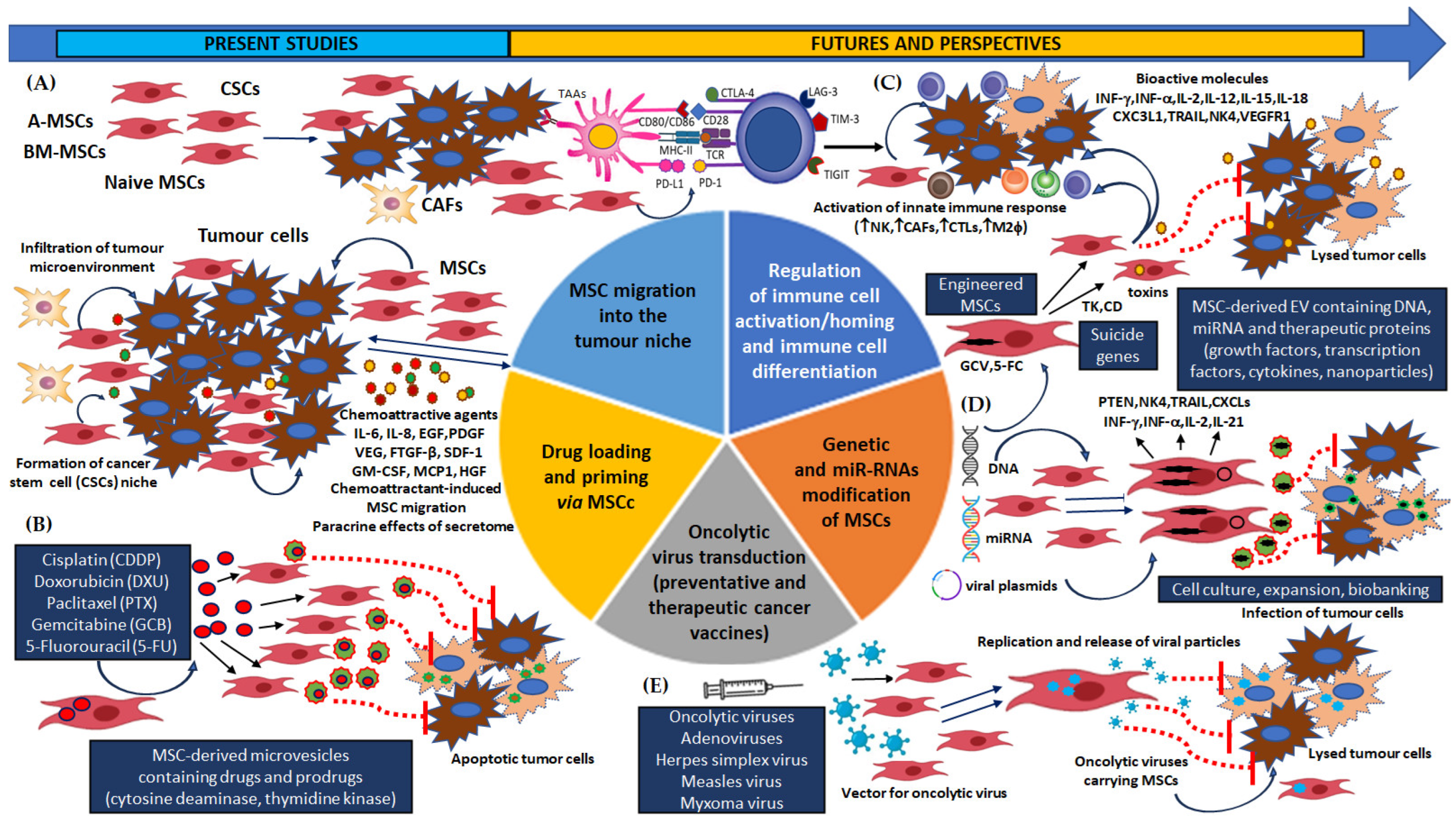

Table 1.
Interplay between MSCs in the tumour microenvironment and cancer cells in the selected in vitro models of HNSCC.
Table 1.
Interplay between MSCs in the tumour microenvironment and cancer cells in the selected in vitro models of HNSCC.
| Author | MSC in in vitro models of HNSCC | |
| Study design | Mechanisms/ Underlying signalling pathway/ Results | |
| Wats et al. [163] |
|
|
| Liu et al. [172] |
|
|
| Salo et al. [188] |
|
|
| Kansy et al. [164] |
|
|
| Ji et al. [165] |
|
|
| Wang et al. [166] |
|
|
| Wu et al. [168] |
|
|
| Coccè et al. [169] |
|
|
| Wang et al. [170] |
|
|
| Liu et al. [171] |
|
|
| Li et al. [174] |
|
|
| Shi et al. [177] |
|
|
| Tian et al. [181] |
|
|
| Hong et al. [189] |
|
|
| Nakayama et al. [179] |
|
|
| Castellone et al. [190] |
|
|
| Wang et al. [180] |
|
|
| Liotta et al. [182] |
|
|
| Mazzoini et al. [183] |
|
|
| Mazzoini et al. [191] |
|
|
| Rowan et al. [187] |
|
|
HNSCC: head and neck squamous cell carcinoma, MSCs: mesenchymal stromal cells, BM-MSCs: bone-marrow derived mesenchymal stem cells, OSCC: oral squamous cell carcinoma, OPSSC: oropharyngeal squamous cell carcinoma, OTSCC/TSCC: oral tongue squamous cell carcinoma, ESCC: oesophageal squamous cell carcinoma, NPC: nasopharyngeal carcinoma, CA-MSC/Tu-MSC: cancer stem cells/MSCs isolated form tumour tissues, GMSCs: MSCs from normal gingival tissue, CAFs: cancer-associated fibroblasts, EMT: epithelial-mesenchymal transition, IHC: immunohistochemistry staining, PINP: type I collagen N-terminal propeptide, POSTN: periostin/osteoblast-specific factor-2, DOK: dysplastic oral keratinocytes, PI3K/Akt/mTOR: phosphoinositide 3-kinase/mammalian target of rapamycin, PDGF-AA: platelet derived growth factor, CAL-27 and HN4: human HNC cell lines, SDF-1α: stromal cell-derived factor-1α, TNF-α: tumour necrosis factor alpha, CD54/ ICAM-1: intercellular adhesion molecule 1, MMP-2/9: metalloproteinases 2 and 9, JNK: c-Jun N-terminal kinases, PARP: Poly (ADP-ribose) polymerase 1, Bax: apoptosis regulator, a member of the Bcl-2 gene family, STAT3: signal transducer and activator of transcription 3, ERK1/2: extracellular signal-regulated kinases 1 and 2, CDK4: cyclin-dependent kinase 4, PCNA: proliferating cell nuclear antigen, B2M: β2-microglobulin, CCN2/CTGF: cysteine rich protein/connective tissue growth factor, GinPa-MSCs: MSCs isolated and expanded from gingival papilla, PDGF-AA/PDGFR-α: platelet-derived growth factor/platelet-derived growth factor receptor, N-MSC: MSCs derived from normal oral mucosa, LK-MSC: MSCs derived from oral premalignant lesion, Ca-MSC: MSCs derived from oral squamous cell carcinoma; PCNA: proliferating cell nuclear antigen, pRb: phospho-retinoblastoma protein, Bcl-2: B-cell lymphoma/leukemia-2, MMP-2: matrix metalloproteinase 2, CDK2: Cyclin E-cyclin-dependent kinase 2, TGF-β/BMP: transforming growth factor-beta/bone morphogenetic protein, IDO: indoelamine 2,3 dioxygenase, Trm: tissue-resident memory cells phenotype.
Table 2.
Interaction between MSCs in the tumour microenvironment and cancer cells in the selected animal/in vivo models of HNSCC.
Table 2.
Interaction between MSCs in the tumour microenvironment and cancer cells in the selected animal/in vivo models of HNSCC.
| Author | MSCs in animal and in vivo models of HNSCC | |
| Study design | Mechanisms/ Underlying signalling pathway/Results | |
| Liu et al. [172] |
|
|
| Kansy et al. [164] |
|
|
| Salo et al. [188] |
|
|
| Ji et al. [165] |
|
|
| Wang et al. [166] |
|
|
| Wu et al. [168] |
|
|
| Liu et al. [171] |
|
|
| Shi et al. [177] |
|
|
| Tian et al. [181] |
|
|
| Hong et al. [189] |
|
|
| Castellone et al. [190] |
|
|
| Wang et al. [180] |
|
|
| Liotta et al. [182] |
|
|
| Chen et al.[192] |
|
|
| Rowan et al. [187] |
|
|
| Zielske et al. [199] |
|
|
| Tan et al. [198] |
|
|
| Bruna et al. [194] |
|
|
| Bruna et al. [195] |
|
|
HNSCC: head and neck squamous cell carcinoma, MSCs: mesenchymal stromal cells, BMSSCs: bone-marrow derived mesenchymal stem cells, OSCC: oral squamous cell carcinoma, OPSSC: oropharyngeal squamous cell carcinoma, OTSCC/TSCC: oral tongue squamous cell carcinoma, ESCC: oesophageal squamous cell carcinoma, NPC: nasopharyngeal carcinoma, CA-MSC/Tu-MSC: cancer stem cells/MSCs isolated form tumour tissues, GMSCs: MSCs from normal gingival tissue, CAFs: cancer-associated fibroblasts, EMT: epithelial-mesenchymal transition, IHC: immunohistochemistry staining, PINP: type I collagen N-terminal propeptide, POSTN: periostin/osteoblast-specific factor-2, PI3K/Akt/mTOR: phosphoinositide 3-kinase/mammalian target of rapamycin, PDGF-AA: platelet derived growth factor, CAL-27 and HN4: human HNC cell lines, SDF-1α: stromal cell-derived factor-1α, TNF-α: tumour necrosis factor alpha, CD54/ ICAM-1: intercellular adhesion molecule 1, MMP-2/9: metalloproteinases 2 and 9, JNK: c-Jun N-terminal kinases, PARP: Poly (ADP-ribose) polymerase 1, Bax: apoptosis regulator, a member of the Bcl-2 gene family, STAT3: signal transducer and activator of transcription 3, ERK1/2: extracellular signal-regulated kinases 1 and 2, CDK4: cyclin-dependent kinase 4, PCNA: proliferating cell nuclear antigen, B2M: β2-microglobulin, CCN2/CTGF: cysteine rich protein/connective tissue growth factor, GinPa-MSCs: MSCs isolated and expanded from gingival papilla, PDGF-AA/ PDGFR-α: platelet-derived growth factor/: platelet-derived growth factor receptor, PCNA: proliferating cell nuclear antigen, pRb: phospho-retinoblastoma protein, Bcl-2: B-cell lymphoma/leukemia-2, MMP-2: matrix metalloproteinase 2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



