
Submitted:
11 June 2024
Posted:
12 June 2024
You are already at the latest version
Abstract
Huntington’s disease (HD) is a rare but progressive and devastating neurodegenerative disease characterized by involuntary movements, cognitive decline, executive dysfunction, and neuropsychiatric conditions such as anxiety and depression. It follows an autosomal dominant inheritance pattern. Thus, a child who has a parent with the mutated huntingtin (mHTT) gene has a 50% chance of developing the disease. Since HTT protein is involved in many critical cellular processes including neurogenesis, brain development, energy metabolism, transcriptional regulation, synaptic activity, vesicle trafficking, cell signaling, and autophagy, its aberrant aggregates lead to disruption of numerous cellular pathways and neurodegeneration. Essential heavy metals are vital at low concentrations, however, at higher concentrations, can exacerbate HD by disrupting the glial-neuronal communication, and/or causing dysbiosis (disturbance in the gut microbiota, GM), both of which can lead to neuroinflammation and further neurodegeneration. Here, we discuss in detail the interactions of iron, manganese and copper with glial-neuron communication and GM and indicate how this knowledge may pave the way for development of a new generation of disease-modifying therapies in HD.
Keywords:
Huntington’s disease
; Heavy metals
; Iron
; Manganese
; Copper
; Glial cells
; Gut microbiota
; Neuroinflammation
; Gut-brain axis
1. Introduction
Huntington's disease (HD) is a relentlessly progressive and debilitating adult-onset neurodegenerative disorder characterized by a well-defined clinical triad: motor dysfunction including choreoathetosis (involuntary twitching, twisting or squirming movements), where severe cases can cause permanent disability, cognitive decline including memory impairment and executive dysfunction, and psychiatric disturbances including anxiety and depression [1,2]. Pneumonia and suicide are common causes of death [1]. HD is an autosomal dominant disease with a characteristic cytosine, adenine, and guanine (CAG) trinucleotide repeats on the short arm of chromosome 4p16.3 within the huntingtin (HTT) gene leading to the production of a mutant huntingtin protein (mHTT) [2,3]. HTT is involved in many critical cellular processes including neurogenesis and brain development, energy metabolism, transcriptional regulation, synaptic activity, vesicle trafficking, cell signaling, and autophagy [2,4]. It is not surprising therefore that aberrant aggregates of this protein lead to disruption of numerous cellular pathways, triggering a cascade of neurodegeneration [1,2,3].
HD is a rare neurodegenerative disorder (about 4 per 100,00 worldwide) with lower prevalence in Asia and higher prevalence in Europe, North America, and Australia, possibly due to the HTT gene haplotypes [2,5,6]. It typically manifests in mid-life, between the ages of 30 to 50 years, but can occur even before the age of 20, where it is termed juvenile HD. Diagnosis is made based on motor, cognitive, and behavioral tests and is confirmed by genetic testing using DNA analysis. Since no cure is available, treatment is aimed at improving the quality of life and decreasing complications.
Recent advances in molecular biology, focusing not only on the cellular pathways dysregulated by mHTT, but also exploring the potential influence of external factors such as heavy metal exposure and gut microbiota (GM), have paved the way for the development of a new generation of disease-modifying therapies (DMTs) for HD [7,8]. In this review, following a brief description of HD pathology, we explore the role of heavy metals in its etiology and focus on the potential manipulation of the GM as a novel therapeutic strategy.
2. HD Pathophysiology
Three significant categories of risk factors associated with CAG repeat have been identified in HD. The first and foremost is the length of the repeat where the longer the repeats (>35), the earlier the onset of symptoms. Indeed, abnormal CAG triplet repeat leads to an abnormally elongated polyglutamine (polyQ) tract which results in neurodegenerative diseases including HD [9]. CAG length is also a significant factor for the progression of the disease, especially in cognitive, motor, and neurological disturbances. The CAG repeats not only provide information on the age of clinical onset but also predict the age of death, where the course of the disease commonly lasts 15 to 20 years [1]. Second is the instability of CAG, and the third is the genetic modifiers that play an essential role in the progression of the disease [1]. The primary pathophysiological features of HD are the degeneration of neurons in the caudate, putamen, and the cerebral cortex. The brain, particularly in the striatum, atrophies showing extensive neuronal loss. It is believed that the choreiform movements, and the development of dystonia and akinesia are due to the degeneration and loss of substance-P in the medium spiny neurons of the basal ganglia, and cognitive and behavioral dysfunctions are due to cortical atrophy [1]. Several theories have been suggested as reasons for the pathogenesis. These include accumulation of mHTT aggregates leading to an impairment of the ubiquitin-proteosome pathway, transcriptional dysregulation, excitotoxicity due to increased release of glutamate and glutamate agonist from the cortical afferents, mitochondrial dysfunction and altered energy metabolism, changes in axonal transport and synaptic dysfunction [1,10].
Moreover, mHTT is a strong activator of glial cells, the brain's immune cells, leading to chronic neuroinflammation [11]. While the initial activation of the glia is for neuroprotection, overstimulation of these cells results in a neuroinflammatory response which can cause neuronal damage and/or cell death, hence contributing to the disease progression [11,12]. Recent research suggests the potential contribution of environmental factors like heavy metals such as iron (Fe), manganese (Mn) and copper (Cu) in HD pathology [11,12,13]. Heavy metal exposure further disrupts post transcriptional mechanisms, exacerbating the problems caused by mHTT and decreasing the clearance rate of misfolded proteins, hence creating a vicious cycle that accelerates the neurodegeneration process [14]. Heavy metals may also indirectly influence the neuroinflammation and/or mHTT clearance, causing further damage via their interaction with the GM, discussed in more detail below.
The characteristic involuntary movements are progressive as they initially begin in the distal extremities and gradually move to proximal and axial muscles with greater amplitude and could extend to facial muscles. Whereas in the early stages the symptoms manifest as hyperkinetic with involuntary chorea, in later stages, hypokinesia and dystonia predominate. In the later stages of the disease the patient becomes bedridden due to severe rigidity and contractures in the extremities. Dysarthria and dysphagia, trouble in speaking and swallowing, respectively, develop during the course of the disease, which could lead to aspiration and pneumonia, the main cause of death in HD. Dystonia, characterized by increased muscle tone with slower movements, leads to abnormal posturing such as torticollis (stiff neck) and is usually the first sign of motor involvement in HD. Tics and ataxia may also develop. The progression of motor disturbances over time can lead to difficulties in walking, standing, and frequent falls [1] .
In addition to the motor symptoms, behavioral and cognitive disturbances manifest early on. Thus, initially, patients may present with impulsivity, poor attention, and irritability leading to outbursts of anger and aggression. Later, emotional blandness with prominent apathy, loss of intuition, and creativity ensues. These are likely due to degeneration in the fronto-striatal pathway. Apathy, which is also progressive, is the most common feature of the disease. Mood disorders including depression are also common, where suicide is the second most common cause of death in HD. Psychosis and cognitive decline to the point of unawareness appear later. Cognitive decline usually manifests before the onset of motor disturbances. The prominent cognitive changes include difficulty in planning, organizing, and multitasking, which may progress to dementia. Interestingly, it is believed that the memory loss in HD is due to an inefficient search of memory (subcortical in nature) rather than a deficient memory formation. In addition, more common features of cortical dementia such as apraxia and aphasia (speech disorders) are spared in HD. Nonetheless, there is severe slowness of the psychomotor processes [1].
3. Current and Prospective Treatments
Beyond symptomatic management with dopaminergic and other medications [1,15,16], evolving therapeutics for HD target the molecular aspects with the intent of developing disease modifying drugs [2,17]. These techniques include direct DNA/gene therapies to manipulate the HTT gene and correct the CAG repeat [18]. Thus, the potential of genome editing such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the CRISPR/Cas9 system have been suggested [19]. RNA modulation may also be a promising approach where antisense oligonucleotide (ASO) therapies and RNA interference (RNAi) therapies are currently undergoing clinical trials [16,20]. Attempts at enhancing neurogenesis are also being considered [2]. Other disease modifying therapies target aberrant downstream pathways such as excitotoxicity, mitochondrial dysfunction, and neuroinflammation [1].
Excitotoxicity, due to an imbalance between excitatory and inhibitory neurotransmitters, has been a subject of intense studies for more than 2 decades [21,22]. Excessive stimulation by glutamate, the excitatory neurotransmitter, can result in cell death via calcium-mediated mitochondrial dysfunction. The increased cytoplasmic calcium directly targets the mitochondria and alters its membrane potential. This compromises the electron transport chain, a vital pathway for energy production within mitochondria. Consequently, the cell experiences reduced ATP synthesis, hindering its ability to maintain essential cellular functions. Furthermore, the mitochondrial dysfunction compromises the anti-oxidative processes and leads to the overproduction of reactive oxygen species (ROS) [16,20]. Calcium dysregulation also activates apoptotic cell death pathways involving caspase-9 and caspase-3, accelerating programmed cell death [23].
Therefore, targeting the Glutamate/GABA imbalance may be a viable option in addressing some of HD symptoms. This contention is further supported by the presence of aberrant NMDA receptor distribution in HD pathogenesis. Specifically, a reduction in palmitoylation, a post-translational modification, was observed in striatal NR2B-containing NMDA receptors of YAC128 mice, a model of HD. This decrease in palmitoylation correlated with an increase in extrasynaptic NMDA receptors, signifying a potential mislocalization of these receptors away from their typical synaptic sites, hence contributing to the vulnerability of striatal neurons in HD. These findings highlight the potential of targeting NMDA receptor palmitoylation as a therapeutic strategy for HD [22]. Medications like memantine and amantadine, both NMDA receptor antagonists, have shown effectiveness in at least the motor symptoms in HD [16,24,25].
As mentioned above, glial cells in general, and micro- and astro-glia in particular, are major contributory cells to neuroinflammation which is one of the aberrant pathways involved in the pathophysiology of HD [26,27]. Hence, below, we briefly discuss the potential role of glial cells in HD pathology.
4. Glial Cells – HD
Glial cells, outnumbering the neurons by 10 to 1, were once considered only as structural support for the neurons. However, they are involved in numerous critical brain functions including myelination, formation of the blood-brain barrier (BBB), development and remodeling of synapses, energetic support for neurons, control of metabolism, regulation of neurotransmitters and neuroendocrine function, control of the fluid/electrolyte homeostasis, detoxification, and immune response [28]. Their dysregulation has been associated with neuropsychiatric and neurodegenerative diseases including HD [2,12,29,30]. Recently we proposed that glial nAChRs may be a suitable target for intervention in Parkinson’s disease (PD) [31]. It would be of interest to determine if this hypothesis can extend to HD.
Four major glial cells (microglia, astrocytes, oligodendrocytes and synantocytes or NG2 cells) have been identified to date. We briefly discuss each with their relevance to neurodegenerative diseases in general, and HD, in particular. Moreover, heavy metal interactions with these cells directly or indirectly via GM is also touched upon.
4.1. Microglia - HD
Microglia, constituting 10%–15% of all central nervous system (CNS) cells are considered the resident immune cells as they constantly survey the environment and react quickly to any kind of insult. They play a vital role in maintaining homeostasis of the brain, however, their overactivation leads to neuroinflammation, which as alluded to above, may be responsible for manifestation of neuropsychiatric and/or neurodegenerative diseases [28]. Microglia also regulate the number of neuronal precursor cells, neurogenesis, the formation and elimination of neuronal synapse, and mediate infiltration of the T-cell into the brain [32].
Depending on the status of their activity, microglia are referred to as resting, activated, or phagocytic. Whereas at the resting or inactive state, they are highly ramified, when activated they contract, assume an enlarged cell body, and proliferate. This happens in response to injury or insult, allowing them to carry their phagocytic activity, whereby debris is eliminated, and repair and recovery can ensue. This essential function can become detrimental if microglia are overactivated causing neuroinflammation followed by neurological anomalies [33,34,35,36].
Microglia express various receptors such as calcium-sensing receptor (CASR), low-density lipoprotein receptor-related protein 1 (LRP1), triggering receptor expressed on myeloid cells-2 (TREM2), nicotinic cholinergic receptors (nAChRs), and toll-like receptors 2 and 4 (TLR2 and TLR4) [33]. TLRs are subject of intense investigation as potential targets for neuropsychiatric/neurodegenerative diseases as they facilitate removal of debris or pathogens by initiating the innate immune response [32,37,38,39].
Importantly, heavy metals (discussed in detail below) can activate microglia and trigger neuroinflammation and neuronal death [40].
4.2. Astroglia (Astrocytes) - HD
Astroglia or astrocytes, have a wide distribution in the brain and may constitute up to 60% of the total cells in certain areas of the brain. They provide nutrients for the neurons, remove waste, monitor, and regulate pH homeostasis, and are key components of the BBB. Moreover, they have extensive synaptic connections with the neurons and help maintain neuronal integrity [32,41,42,43]. They are also key mediators of excitotoxic glutamate reuptake [44,45]. More recently it was reported that astrocytes are the necessary source of TNF-α for mediation of homeostatic synaptic plasticity [46]. Astrocytes contain their own neurotrophic factor, referred to as glial cell line derived neurotrophic factor (GDNF), a protein that like brain derived neurotrophic factor (BDNF), provides trophic support for the growth and differentiation of synapses, and promotes cell survival [42,46]. Astrocytes also express high level of glial fibrillary astrocytic protein (GFAP), which is important for astrocyte-neuron communication, and helps maintain the mechanical strength, shape, and the movement of the cell and is commonly used as a marker for their identification [46,47].
Interestingly, astrocytes can become reactive by polarized microglia to help in defense mechanisms and removal of pathogens [32]. However, in this case also, overstimulation of these cells will result in production of proinflammatory cytokines and contribute synergistically to neuronal dysregulation and/or death [48,49,50]. In this regard, heavy metals can cause astrocyte dysfunction, triggering neuronal as well as oligodendrocyte malfunction [51,52,53]. Moreover, as the BBB controls the transport of nutrients and metabolites into the brain and limits the access of harmful substances, its disruption is associated with the pathophysiology of major neurological disorders. For example, lead-induced damage of the BBB has been implicated in autism spectrum disorder (ASD), whereas Cu, Mn, and Fe disruption of BBB have been linked to HD [13,54,55]. Finally, the GM, which via short-chain fatty acids (SCFAs) maintains the integrity of the BBB, may be highly impacted by gut dysbiosis (discussed below).
4.3. Oligodendrocytes - HD
Oligodendrocytes (OLs), constituting 75% of all glial cells, are well recognized as the primary source of myelination in the CNS [56]. They control extracellular potassium concentration, modulate axonal growth, provide metabolic and trophic supply to myelin, secrete GDNF and BDNF, and like microglia and astrocytes express TLRs, which are also necessary for myelin formation [57,58,59]. Myelinated axons, which comprise the white matter, connect various grey matter areas (consisting of neuronal bodies, axon terminals and dendrites) of the brain to each other, and carry nerve impulses between neurons. Abnormality in white matter has been considered as an early indicator in HD [60]. Importantly, heavy metals can cause dysfunction in these cells as well [51,52,61].
4.4. Synantocytes (NG2 cells) - HD
The fourth subset of major glial cells in CNS, synantocytes, are OLs-precursor cells that are almost uniformly distributed in both white and gray matter areas, associate closely with neuronal cell bodies and dendrites, and maintain the ability to keep proliferating in the adult brain [56,62,63]. These cells can also give rise to astrocytes and neurons [56,62,63], and their potential involvement in neurodegenerative diseases is suspected [64,65]. For example, neuroinflammation and increased BBB permeability in experimental autoimmune encephalomyelitis (EAE), were attributed to NG2 cells [66], where it was postulated that NG2 cells via stimulation of reactive T cells, control IL-12 expression [66]. NG2 cells have been implicated in neuroinflammation [67], and neurovascular unit formation during development [68]. Following acute ischemic stroke, NG2 cells play a key role in angiogenesis and generation of OLs [68]. Because of their influence on neuronal plasticity and communication with neurons, OLs may provide a novel target for therapeutic interventions in a variety of neurological diseases [68,69,70]. Whether heavy metals interact with NG2 cells is yet to be determined.
5. Gut Microbiota
GM is a complex and dynamic population of trillions of bacteria, fungi, archaea and eukarya found in the gastrointestinal tract (GI). Microbiome refers to the genetic composition of these cells, which is now estimated to be slightly higher than the human genome [71,72,73]. GM exhibits remarkable diversity that changes over a person's lifespan following a symbiotic relationship with the host. It plays a vital role in brain development, digestion, nutrient absorption, fermentation of undigested carbohydrates, production of essential vitamins and metabolites like SCFAs, regulation of the immune system, maintenance of BBB integrity, and overall health [74,75]. Dysbiosis, referring to an imbalance in the composition and function of the GM, has been implicated in a wide range of pathological processes including digestive, metabolic, autoimmune, and neurological disorders [55,76,77].
The immune system plays a major role in the perpetuation and maintenance of the symbiotic relationship between the host and the beneficial commensal bacterial strains. Due to its substantial influence on physiological processes as well as its wide implication in various pathological states, the GM is considered as a new ‘metabolic organ,’ with major influence not only on the digestive system but also on other organs, notably the CNS [8,76,77,78,79].
6. Gut-Brain Axis
A bidirectional communication pathway, termed the gut-brain axis (GBA), that links the GM to CNS is well-recognized [80]. This axis facilitates communication through the vagus nerve, the immune system, and microbial metabolites. Dysbiosis has been increasingly implicated in the pathogenesis of various neurological disorders through several mechanisms, the most prominent being the neuroinflammation. In dysbiosis, there is release of pro-inflammatory mediators such as cytokines (e.g., interleukin-1β, tumor necrosis factor-α) and chemokines from the immune cells which can then migrate to the CNS via the bloodstream or lymphatic system and exacerbate neuroinflammation affecting brain development and behavior [77,81,82,83].
Some of the metabolites produced by the GM such as the SCFA butyrate, contribute to the epithelial defense, and have antioxidant and anti-inflammatory properties [84,85]. Some other metabolites such as lipopolysaccharide (LPS) are pro-inflammatory and are used to mimic inflammatory diseases [86,87]. An imbalance in the GM may also weaken the intestinal barrier allowing bacterial products and toxins to translocate into the bloodstream. This phenomenon known as the leaky gut, highlights the significance of maintaining the integrity of the GM [88,89]. Of direct relevance to the topic of our discussion are the recent reports implicating dysbiosis in HD, which is elaborated below [77,90].
7. Heavy Metals
Heavy metals are essential for a variety of biological functions [31,91,92,93]. For example, iron (Fe) is a critical component of many vital enzymes or coenzymes such as catalases and cytochromes, which mediate cellular processes and drug metabolism. Indeed, catalases by neutralizing hydrogen peroxide are critical in providing protection against oxidative stress [31]. Fe is also an essential component of hemoglobin, where its deficiency leads to Fe-deficiency anemia [94]. Similarly, Mn acts as an activator or cofactor for a variety of metalloenzymes that are essential for normal cell growth and development [95,96,97,98,99]. Moreover, the enzyme or co-enzymes utilizing Mn play key roles in such functions as gluconeogenesis, suppression of oxidative stress (Mn-superoxide dismutase, SOD) and conversion of glutamate into glutamine (glutamine synthetase) [100,101], all of which have critical biological functions. Copper (Cu) is another metal essential for synthesis of red blood cells, collagen, bone, and connective tissue, maintenance of nerve cells and immune system, It is required for adequate growth, cardiovascular integrity, lung elasticity, neovascularization, neuroendocrine function, and Fe metabolism [93,102]. However, at higher concentration, it can contribute to HD pathology. Below we discuss the relevance of each essential heavy metal to HD vis-à-vis their interaction with GM and inflammatory processes.
7.1. Iron (Fe) - HD
Ferroptosis is a newly discovered form of programmed cell death distinct from apoptosis and necrosis. It is considered a key contributor to the pathogenesis of neurodegenerative diseases [103,104]. This section focuses on evidence linking it to neurodegeneration, particularly to HD [105].
Ferroptosis, an Fe-dependent form of regulated cell death, is characterized by the excessive peroxidation of polyunsaturated fatty acids (PUFAs) found within cell membranes. Enzymes like acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are key in the catalyzation of these reactions [106]. Mitochondrial dysfunction is a hallmark of ferroptosis and a necessary condition for the perpetuation of these reactions. In fact, increased Fe uptake promotes the generation of destructive hydroxyl radicals through the Fenton reaction thus perpetuating the chain reaction of lipid peroxidation, ultimately compromising membrane integrity [107], which leads to cell membrane disruption and cell death. Unlike apoptosis and necrosis, ferroptosis presents with its own cellular pathways. While mitochondria are central to the execution of ferroptosis, other organelles contribute through stress-related pathways. The endoplasmic reticulum, Golgi apparatus, and lysosomes can be involved in amplifying the cell death program [108,109].
Ferroptosis is an oxidative process that needs to be controlled via counter regulatory mechanisms. In this context, glutathione, a major antioxidant system in conjunction with its key enzyme glutathione peroxidase 4 (GPX4), plays a major role in protecting cells from uncontrolled ferroptosis by suppressing lipid peroxidation [110]. Fe metabolism, cysteine availability, and lipid homeostasis are tightly intertwined and serve as key regulatory points for ferroptosis induction or inhibition. Unregulated ferroptosis is a major determinant of neuroinflammation and neurodegenerative diseases [105].
Regarding HD, it has been shown that aggregation and accumulation of mHTT increases the susceptibility of basal ganglia neurons to ferroptotic cell death [111]. Fe overload not only disrupts mitochondrial functions leading to impaired energy production and increased oxidative stress, but also generates highly reactive free radicals that damage lipids, proteins, and DNA and disrupt the redox balance making neurons more susceptible to ferroptotic cell death, thus exacerbating HD [13,112,113,114]. Indeed, elevated levels of lipid peroxidation products have been detected in both cellular HD models and HD patients [115].
A direct interaction between Fe and mHTT is also evident whereby Fe enhances mHTT aggregation and its neurotoxic effect. This creates a vicious cycle that accelerates neurodegeneration [116,117]. In the same way, mHTT might interfere with System Xc-, an antiporter that exchanges glutamate (excitatory neurotransmitter) for cystine (precursor for glutathione synthesis), leading to decreased glutathione (GSH) levels, a crucial antioxidant that protects cells from ferroptosis [116,117].
7.2. Manganese (Mn) – HD
Another essential heavy metal implicated in HD pathophysiology is Mn. As alluded to earlier, Mn is a crucial cofactor for many enzymes, and is necessary for amino acid, cholesterol, glucose, and carbohydrate metabolism; reactive oxygen species scavenging; bone formation; reproduction; and the immune response [124,125]. Mn deficiency can lead to weakness, seizures, infertility and bone malformation. Mn overload, on the other hand, concentrates in the brain especially in the basal ganglia resulting in parkinsonism [126]. Early life exposure to high levels of Mn is thought to impact neurodevelopment, especially cognitive behavior in children [127]. Importantly, high Mn exposure and alteration in the GM has been linked to oxidative stress and neuroinflammation, which are implicated in HD [128,129].
The potential link between Mn, Insulin/IGF signaling and HD, whereby Mn deficiency was shown to share cellular consequences such as increased oxidative stress and mitochondrial dysfunction with HD was reviewed recently [130]. t was concluded that Mn can mimic some actions of insulin/IGF signaling in HD models, thereby providing protection in instances where HD symptoms might be precipitated by Mn deficiency [130].
7.3. Copper (Cu) - HD
Cu toxicity has also been linked to HD [131]. Cu, as mentioned earlier, is an essential metal that plays a critical role in various neurochemical processes, where its dysregulation is detrimental [132]. Studies highlight its potential contribution to neurodegeneration in HD via enhancement of the mHTT toxicity [14]. Cu may also disrupt proteostasis, the process of protein folding and degradation, further contributing to cellular dysfunction [133]. It is noteworthy that Wilson’s disease also involves disruption of Cu metabolism and its deposition in the basal ganglia. People suffering from this genetic disorder present with extrapyramidal signs and symptoms ranging from movement disorders (tremor, dystonia, parkinsonism) to cognitive and speech impairment and psychiatric symptoms, similar to what is observed in HD [134]. Thus, like Fe, Cu may promote mHTT aggregation and toxicity. Cu also modulates the interaction between huntingtin inclusions and the autophagy adaptor protein, which is responsible for clearance of the toxic aggregate [14,135,136,137]. A study using the drosophila model of HD showed that D-penicillamine, a Cu chelator, significantly reduced the formation of amyloid-like huntingtin aggregates, suggesting a potential therapeutic avenue for mitigating the toxicity associated with huntingtin aggregation [14].
8. Heavy metals - GM - HD
Building upon the intriguing link between heavy metal dysregulation and HD, recent research is exploring the potential influence of the GM in HD pathophysiology. The GM is in fact increasingly recognized for its role in brain health and disease [87,90]. The GM can both influence and be influenced by heavy metal exposure. Certain gut bacteria can facilitate the absorption and accumulation of heavy metals like Fe, lead (Pb) and Cu in the body [55]. Conversely, heavy metal exposure can disrupt the composition and function of the GM, triggering inflammatory responses that can indirectly impact the basal ganglia, and exacerbate HD pathology [138]. The basal ganglia, a control center for movement, cognition, and emotional regulation, is critically affected in HD and is particularly susceptible to GM-derived neuroinflammation [138]. Thus, by promoting a healthy GM composition through dietary interventions or prebiotics/probiotics, absorption of heavy metals like Fe and Cu may be curtailed, thereby mitigating their potential contribution to HD pathology.
The intricate relationship between the GM and HD pathophysiology is a burgeoning area of research with significant therapeutic potential. Recent studies suggest a multifaceted interplay between gut bacteria, the immune system, and the CNS that may contribute to HD progression [77,139,140,141]. One key mechanism in this scenario involves SCFAs produced by beneficial bacteria like Bifidobacterium and Faecalibacterium prausnitzii that exert neuroprotective effects [55,142,143]. Thus, in a mouse model of HD, SCFA supplementation improved motor function, reduced mHTT aggregation, and mitigated neuroinflammation. Conversely, dysbiosis leading to LPS production has been linked to an increase in mHTT aggregation and neuronal death [144].
Another critical link in GBA is a bidirectional communication pathway involving the vagus nerve, immune signaling, and the production of neurotransmitters. Dysbiosis can trigger chronic low-grade inflammation in the gut, leading to the activation of immune cells and the release of pro-inflammatory cytokines. These inflammatory signals can then travel up to the brain via the vagus nerve, promoting neuroinflammation and further compromising neuronal health in the basal ganglia [145]. Interestingly, mHTT was shown to be widely expressed in the intestines, which would allow it to interact with the GM, hence affecting the progression of HD [140]. GM involvement in HD pathology has also been verified in several animal models [139,146].
It was mentioned earlier that a leaky BBB, characterized by increased permeability, allows the passage of harmful bacterial products and inflammatory molecules into the brain. It is noteworthy that dysbiosis can also disrupt the tight junctions of the BBB, potentially accelerating neurodegeneration in HD [147].
9. Conclusion
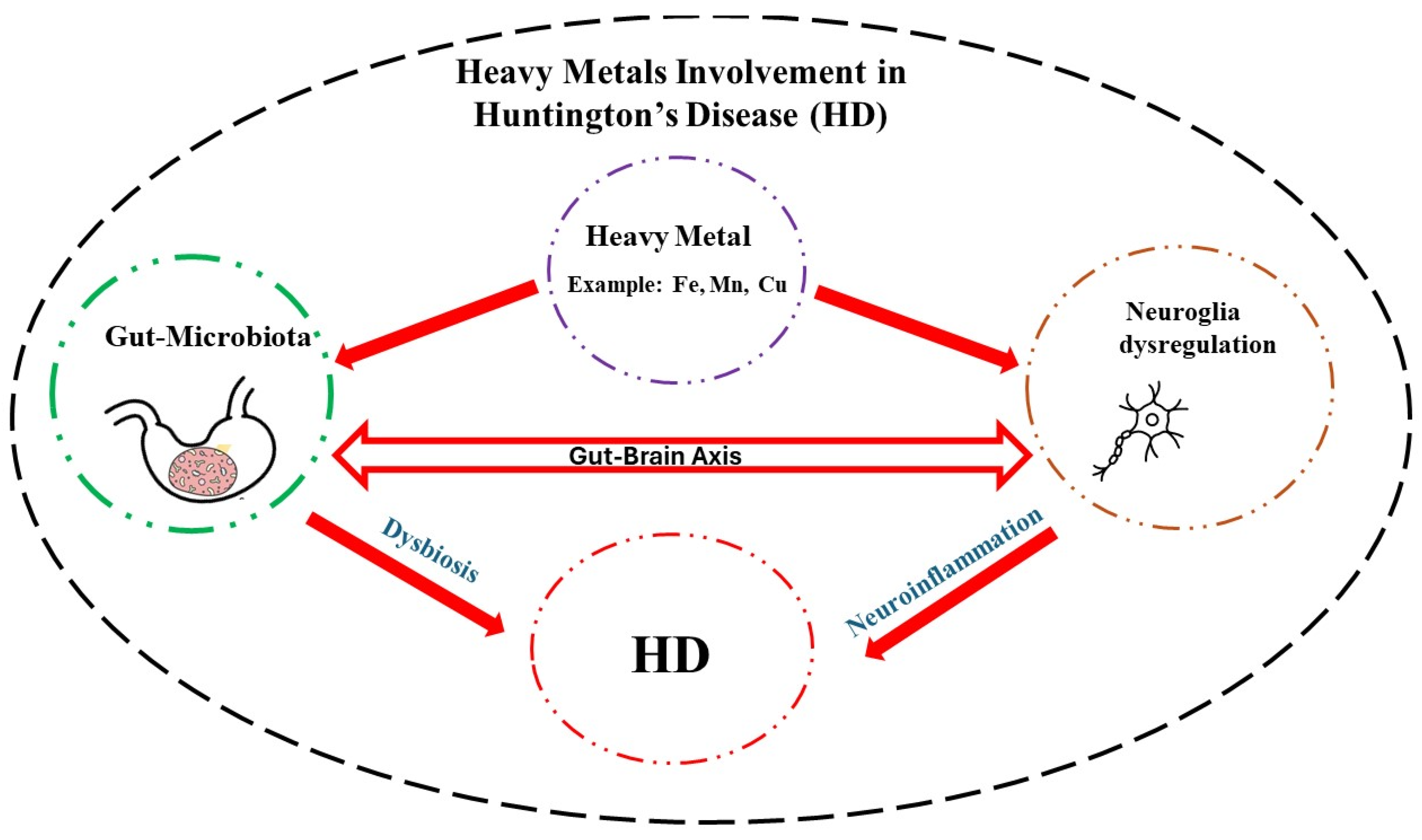
Neurodegenerative diseases exact a tremendous toll on the afflicted and the caregivers. Although in most cases the etiology is unknown, in case of HD, a mutation in huntingtin gene is the main culprit. In this regard, efforts are underway to quantify the mutant protein in the cerebrospinal fluid with the aim of developing effective therapies [148]. In addition, exposure to high levels of essential heavy metals such as Fe, Mn, and Cu may exacerbate HD symptoms by disrupting neuronal communications, particularly, the glial-neuron interaction. High levels of heavy metals via their interaction with the GM and induction of dysbiosis, can also promote neuroinflammation and hence indirectly contribute to HD pathology (Figure 1). Understanding the intricate coordination of the GBA and specific effect of each heavy metal on this axis, may provide further therapeutic intervention in this devastating disease [149,150,151].
Author Contributions
Conceptualization, Y.T., M.A. and B.G.; Writing—original draft preparation, S.B. and N.E.K; Review and editing, Y.T., M.A. and B.G. All authors have read and agreed to the submission of the paper.
Funding
Y.T. was supported in part by NIH/NIAAA R03 AA022479 and NIH/NIGMS (2 SO6 GM08016-39), M.A. was supported in part by grants from the National Institute of Environmental Health Sciences (NIEHS) R01ES10563 and R01ES07331.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ajitkumar: A.; De Jesus, O. Huntington Disease. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2024.
- D’Egidio, F.; Castelli, V.; Lombardozzi, G.; Ammannito, F.; Cimini, A.; d’Angelo, M. Therapeutic Advances in Neural Regeneration for Huntington’s Disease. Neural Regen. Res. 2024, 19, 1991–1997, . [CrossRef]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a007476, . [CrossRef]
- Barron, J.C.; Hurley, E.P.; Parsons, M.P. Huntingtin and the Synapse. Front. Cell. Neurosci. 2021, 15, 689332, . [CrossRef]
- Rawlins, M.D.; Wexler, N.S.; Wexler, A.R.; Tabrizi, S.J.; Douglas, I.; Evans, S.J.W.; Smeeth, L. The Prevalence of Huntington’s Disease. Neuroepidemiology 2016, 46, 144–153, . [CrossRef]
- Pringsheim, T.; Wiltshire, K.; Day, L.; Dykeman, J.; Steeves, T.; Jette, N. The Incidence and Prevalence of Huntington’s Disease: A Systematic Review and Meta-analysis. Mov. Disord. 2012, 27, 1083–1091, . [CrossRef]
- Kim, A.; Lalonde, K.; Truesdell, A.; Gomes Welter, P.; Brocardo, P.S.; Rosenstock, T.R.; Gil-Mohapel, J. New Avenues for the Treatment of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 8363, . [CrossRef]
- Khoshnan, A. Gut Microbiota as a Modifier of Huntington’s Disease Pathogenesis. J. Huntingt. Dis. 2024, 1–15, . [CrossRef]
- Estevam, B.; Matos, C.A.; Nóbrega, C. PolyQ Database—an Integrated Database on Polyglutamine Diseases. Database 2023, 2023, baad060, . [CrossRef]
- Caron, N.S.; Wright, G.E.; Hayden, M.R. Huntington Disease. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle (WA), 1993.
- Crotti, A.; Glass, C.K. The Choreography of Neuroinflammation in Huntington’s Disease. Trends Immunol. 2015, 36, 364–373, . [CrossRef]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258, . [CrossRef]
- Donley, D.W.; Realing, M.; Gigley, J.P.; Fox, J.H. Iron Activates Microglia and Directly Stimulates Indoleamine-2,3-Dioxygenase Activity in the N171-82Q Mouse Model of Huntington’s Disease. PLOS ONE 2021, 16, e0250606, . [CrossRef]
- Lobato, A.G.; Ortiz-Vega, N.; Zhu, Y.; Neupane, D.; Meier, K.K.; Zhai, R.G. Copper Enhances Aggregational Toxicity of Mutant Huntingtin in a Drosophila Model of Huntington’s Disease. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 2024, 1870, 166928, . [CrossRef]
- Paleacu, D. Tetrabenazine in the Treatment of Huntington’s Disease. Neuropsychiatr. Dis. Treat. 2007, 3, 545–551.
- Ferguson, M.W.; Kennedy, C.J.; Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Current and Possible Future Therapeutic Options for Huntington’s Disease. J. Cent. Nerv. Syst. Dis. 2022, 14, 11795735221092517, . [CrossRef]
- Sheridan, C. Questions Swirl around Failures of Disease-Modifying Huntington’s Drugs. Nat. Biotechnol. 2021, 39, 650–652, . [CrossRef]
- Dash, D.; Mestre, T.A. Therapeutic Update on Huntington’s Disease: Symptomatic Treatments and Emerging Disease-Modifying Therapies. Neurotherapeutics 2020, 17, 1645–1659, . [CrossRef]
- Alkanli, S.S.; Alkanli, N.; Ay, A.; Albeniz, I. CRISPR/Cas9 Mediated Therapeutic Approach in Huntington’s Disease. Mol. Neurobiol. 2023, 60, 1486–1498, . [CrossRef]
- Tabrizi, S.J.; Estevez-Fraga, C.; van Roon-Mom, W.M.C.; Flower, M.D.; Scahill, R.I.; Wild, E.J.; Muñoz-Sanjuan, I.; Sampaio, C.; Rosser, A.E.; Leavitt, B.R. Potential Disease-Modifying Therapies for Huntington’s Disease: Lessons Learned and Future Opportunities. Lancet Neurol. 2022, 21, 645–658, . [CrossRef]
- Raymond, L.A. Excitotoxicity in Huntington Disease. Clin. Neurosci. Res. 2003, 3, 121–128, . [CrossRef]
- Kang, R.; Wang, L.; Sanders, S.S.; Zuo, K.; Hayden, M.R.; Raymond, L.A. Altered Regulation of Striatal Neuronal N-Methyl-D-Aspartate Receptor Trafficking by Palmitoylation in Huntington Disease Mouse Model. Front. Synaptic Neurosci. 2019, 11, 3, . [CrossRef]
- Gao, J.; Wang, H.; Liu, Y.; Li, Y.-Y.; Chen, C.; Liu, L.-M.; Wu, Y.-M.; Li, S.; Yang, C. Glutamate and GABA Imbalance Promotes Neuronal Apoptosis in Hippocampus after Stress. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2014, 20, 499–512, . [CrossRef]
- Saigoh, K.; Hirano, M.; Mitsui, Y.; Oda, I.; Ikegawa, A.; Samukawa, M.; Yoshikawa, K.; Yamagishi, Y.; Kusunoki, S.; Nagai, Y. Memantine Administration Prevented Chorea Movement in Huntington’s Disease: A Case Report. J. Med. Case Reports 2023, 17, 431, . [CrossRef]
- Verhagen Metman, L.; Morris, M.J.; Farmer, C.; Gillespie, M.; Mosby, K.; Wuu, J.; Chase, T.N. Huntington’s Disease: A Randomized, Controlled Trial Using the NMDA-Antagonist Amantadine. Neurology 2002, 59, 694–699, . [CrossRef]
- Vonsattel, J.P.G.; Keller, C.; Pilar Amaya, M.D. Neuropathology of Huntington’s Disease. In Handbook of Clinical Neurology; Elsevier, 2008; Vol. 89, pp. 599–618 ISBN 978-0-444-51898-9.
- Sapp, E.; Kegel, K.B.; Aronin, N.; Hashikawa, T.; Uchiyama, Y.; Tohyama, K.; Bhide, P.G.; Vonsattel, J.P.; Difiglia, M. Early and Progressive Accumulation of Reactive Microglia in the Huntington Disease Brain. J. Neuropathol. Exp. Neurol. 2001, 60, 161–172, . [CrossRef]
- Tizabi, Y.; Getachew, B.; Hauser, S.R.; Tsytsarev, V.; Manhães, A.C.; Da Silva, V.D.A. Role of Glial Cells in Neuronal Function, Mood Disorders, and Drug Addiction. Brain Sci. 2024, 14, 558, . [CrossRef]
- Saba, J.; Couselo, F.L.; Bruno, J.; Carniglia, L.; Durand, D.; Lasaga, M.; Caruso, C. Neuroinflammation in Huntington’s Disease: A Starring Role for Astrocyteand Microglia. Curr. Neuropharmacol. 2022, 20, 1116–1143, . [CrossRef]
- Sanadgol, N. Editorial: Glial Cells as an Emerging Therapeutic Target in the Pathobiology of Central Nervous System Disorders: Friend or Foe? Front. Cell. Neurosci. 2023, 17, 1191743, . [CrossRef]
- Carvalho, F.V.; Landis, H.E.; Getachew, B.; Diogenes Amaral Silva, V.; Ribeiro, P.R.; Aschner, M.; Tizabi, Y. Iron Toxicity, Ferroptosis and Microbiota in Parkinson’s Disease: Implications for Novel Targets. In Advances in Neurotoxicology; Elsevier, 2024; Vol. 11, pp. 105–132 ISBN 978-0-443-21560-5.
- Pathak, D.; Sriram, K. Neuron-Astrocyte Omnidirectional Signaling in Neurological Health and Disease. Front. Mol. Neurosci. 2023, 16, 1169320, . [CrossRef]
- Soares, É.N.; Costa, A.C.D.S.; Ferrolho, G.D.J.; Ureshino, R.P.; Getachew, B.; Costa, S.L.; Da Silva, V.D.A.; Tizabi, Y. Nicotinic Acetylcholine Receptors in Glial Cells as Molecular Target for Parkinson’s Disease. Cells 2024, 13, 474, . [CrossRef]
- Saitgareeva, A.R.; Bulygin, K.V.; Gareev, I.F.; Beylerli, O.A.; Akhmadeeva, L.R. The Role of Microglia in the Development of Neurodegeneration. Neurol. Sci. 2020, 41, 3609–3615, . [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in Neurodegenerative Diseases: Mechanism and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2023, 8, 359, . [CrossRef]
- De Marchi, F.; Munitic, I.; Vidatic, L.; Papić, E.; Rački, V.; Nimac, J.; Jurak, I.; Novotni, G.; Rogelj, B.; Vuletic, V.; et al. Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines 2023, 11, 2793, . [CrossRef]
- Fatoba, O.; Itokazu, T.; Yamashita, T. Microglia as Therapeutic Target in Central Nervous System Disorders. J. Pharmacol. Sci. 2020, 144, 102–118, . [CrossRef]
- Heidari, A.; Yazdanpanah, N.; Rezaei, N. The Role of Toll-like Receptors and Neuroinflammation in Parkinson’s Disease. J. Neuroinflammation 2022, 19, 135, . [CrossRef]
- Liao, X.; Zhu, W.; Liao, X.; Liu, W.; Hou, Y.; Wan, J. Expression of Toll-like Receptors in the Cerebellum during Pathogenesis of Prion Disease. Front. Behav. Neurosci. 2024, 18, 1341901, . [CrossRef]
- Martínez-Hernández, M.I.; Acosta-Saavedra, L.C.; Hernández-Kelly, L.C.; Loaeza-Loaeza, J.; Ortega, A. Microglial Activation in Metal Neurotoxicity: Impact in Neurodegenerative Diseases. BioMed Res. Int. 2023, 2023, 1–27, . [CrossRef]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154, . [CrossRef]
- Kotliarova, A.; Sidorova, Y.A. Glial Cell Line-Derived Neurotrophic Factor Family Ligands, Players at the Interface of Neuroinflammation and Neuroprotection: Focus Onto the Glia. Front. Cell. Neurosci. 2021, 15, 679034, . [CrossRef]
- Garland, E.F.; Hartnell, I.J.; Boche, D. Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front. Neurosci. 2022, 16, 824888, . [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389, . [CrossRef]
- Stoklund Dittlau, K.; Freude, K. Astrocytes: The Stars in Neurodegeneration? Biomolecules 2024, 14, 289, . [CrossRef]
- Heir, R.; Abbasi, Z.; Komal, P.; Altimimi, H.F.; Franquin, M.; Moschou, D.; Chambon, J.; Stellwagen, D. Astrocytes Are the Source of TNF Mediating Homeostatic Synaptic Plasticity. J. Neurosci. 2024, 44, e2278222024, . [CrossRef]
- Rajkowska, G.; Stockmeier, C. Astrocyte Pathology in Major Depressive Disorder: Insights from Human Postmortem Brain Tissue. Curr. Drug Targets 2013, 14, 1225–1236, . [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional Roles of Reactive Astrocytes in Neuroinflammation and Neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409, . [CrossRef]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819, . [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934, . [CrossRef]
- Pamphlett, R.; Bishop, D.P. The Toxic Metal Hypothesis for Neurological Disorders. Front. Neurol. 2023, 14, 1173779, . [CrossRef]
- Cheli, V.T.; Correale, J.; Paez, P.M.; Pasquini, J.M. Iron Metabolism in Oligodendrocytes and Astrocytes, Implications for Myelination and Remyelination. ASN Neuro 2020, 12, 175909142096268, . [CrossRef]
- Li, B.; Xia, M.; Zorec, R.; Parpura, V.; Verkhratsky, A. Astrocytes in Heavy Metal Neurotoxicity and Neurodegeneration. Brain Res. 2021, 1752, 147234, . [CrossRef]
- Joshi, P.; Bodnya, C.; Ilieva, I.; Neely, M.D.; Aschner, M.; Bowman, A.B. Huntington’s Disease Associated Resistance to Mn Neurotoxicity Is Neurodevelopmental Stage and Neuronal Lineage Dependent. NeuroToxicology 2019, 75, 148–157, . [CrossRef]
- Tizabi, Y.; Bennani, S.; El Kouhen, N.; Getachew, B.; Aschner, M. Interaction of Heavy Metal Lead with Gut Microbiota: Implications for Autism Spectrum Disorder. Biomolecules 2023, 13, 1549, . [CrossRef]
- Michalski, J.-P.; Kothary, R. Oligodendrocytes in a Nutshell. Front. Cell. Neurosci. 2015, 9, . [CrossRef]
- Bsibsi, M.; Nomden, A.; Van Noort, J.M.; Baron, W. Toll-like Receptors 2 and 3 Agonists Differentially Affect Oligodendrocyte Survival, Differentiation, and Myelin Membrane Formation. J. Neurosci. Res. 2012, 90, 388–398, . [CrossRef]
- Kumar, V. Toll-Like Receptors in Adaptive Immunity. In Toll-like Receptors in Health and Disease; Kumar, V., Ed.; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, 2021; Vol. 276, pp. 95–131 ISBN 978-3-031-06511-8.
- Sanchez-Petidier, M.; Guerri, C.; Moreno-Manzano, V. Toll-like Receptors 2 and 4 Differentially Regulate the Self-Renewal and Differentiation of Spinal Cord Neural Precursor Cells. Stem Cell Res. Ther. 2022, 13, 117, . [CrossRef]
- Sun, Y.; Tong, H.; Yang, T.; Liu, L.; Li, X.-J.; Li, S. Insights into White Matter Defect in Huntington’s Disease. Cells 2022, 11, 3381, . [CrossRef]
- Maiuolo, J.; Macrì, R.; Bava, I.; Gliozzi, M.; Musolino, V.; Nucera, S.; Carresi, C.; Scicchitano, M.; Bosco, F.; Scarano, F.; et al. Myelin Disturbances Produced by Sub-Toxic Concentration of Heavy Metals: The Role of Oligodendrocyte Dysfunction. Int. J. Mol. Sci. 2019, 20, 4554, . [CrossRef]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity From the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51, . [CrossRef]
- Hill, R.A.; Patel, K.D.; Goncalves, C.M.; Grutzendler, J.; Nishiyama, A. Modulation of Oligodendrocyte Generation during a Critical Temporal Window after NG2 Cell Division. Nat. Neurosci. 2014, 17, 1518–1527, . [CrossRef]
- Dimou, L.; Gallo, V. NG 2-glia and Their Functions in the Central Nervous System. Glia 2015, 63, 1429–1451, . [CrossRef]
- Xu, G.; Wang, W.; Zhou, M. Spatial Organization of NG2 Glial Cells and Astrocytes in Rat Hippocampal CA1 Region. Hippocampus 2014, 24, 383–395, . [CrossRef]
- Ferrara, G.; Errede, M.; Girolamo, F.; Morando, S.; Ivaldi, F.; Panini, N.; Bendotti, C.; Perris, R.; Furlan, R.; Virgintino, D.; et al. NG2, a Common Denominator for Neuroinflammation, Blood–Brain Barrier Alteration, and Oligodendrocyte Precursor Response in EAE, Plays a Role in Dendritic Cell Activation. Acta Neuropathol. (Berl.) 2016, 132, 23–42, . [CrossRef]
- Zhang, S.; Wang, Q.; Yang, Q.; Gu, H.; Yin, Y.; Li, Y.; Hou, J.; Chen, R.; Sun, Q.; Sun, Y.; et al. NG2 Glia Regulate Brain Innate Immunity via TGF-Β2/TGFBR2 Axis. BMC Med. 2019, 17, 204, . [CrossRef]
- Hu, X.; Geng, P.; Zhao, X.; Wang, Q.; Liu, C.; Guo, C.; Dong, W.; Jin, X. The NG2-Glia Is a Potential Target to Maintain the Integrity of Neurovascular Unit after Acute Ischemic Stroke. Neurobiol. Dis. 2023, 180, 106076, . [CrossRef]
- Timmermann, A.; Tascio, D.; Jabs, R.; Boehlen, A.; Domingos, C.; Skubal, M.; Huang, W.; Kirchhoff, F.; Henneberger, C.; Bilkei-Gorzo, A.; et al. Dysfunction of NG2 Glial Cells Affects Neuronal Plasticity and Behavior. Glia 2023, 71, 1481–1501, . [CrossRef]
- Vélez-Fort, M.; Maldonado, P.P.; Butt, A.M.; Audinat, E.; Angulo, M.C. Postnatal Switch from Synaptic to Extrasynaptic Transmission between Interneurons and NG2 Cells. J. Neurosci. 2010, 30, 6921–6929, . [CrossRef]
- Shoemaker, W.R.; Chen, D.; Garud, N.R. Comparative Population Genetics in the Human Gut Microbiome. Genome Biol. Evol. 2022, 14, evab116, . [CrossRef]
- Chatterjee, G.; Negi, S.; Basu, S.; Faintuch, J.; O’Donovan, A.; Shukla, P. Microbiome Systems Biology Advancements for Natural Well-Being. Sci. Total Environ. 2022, 838, 155915, . [CrossRef]
- VanEvery, H.; Franzosa, E.A.; Nguyen, L.H.; Huttenhower, C. Microbiome Epidemiology and Association Studies in Human Health. Nat. Rev. Genet. 2023, 24, 109–124, . [CrossRef]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in Health and Diseases. Signal Transduct. Target. Ther. 2022, 7, 135, . [CrossRef]
- Logsdon, A.F.; Erickson, M.A.; Rhea, E.M.; Salameh, T.S.; Banks, W.A. Gut Reactions: How the Blood–Brain Barrier Connects the Microbiome and the Brain. Exp. Biol. Med. 2018, 243, 159–165, . [CrossRef]
- Hrncir, T. Gut Microbiota Dysbiosis: Triggers, Consequences, Diagnostic and Therapeutic Options. Microorganisms 2022, 10, 578, . [CrossRef]
- Sharma, G.; Biswas, S.S.; Mishra, J.; Navik, U.; Kandimalla, R.; Reddy, P.H.; Bhatti, G.K.; Bhatti, J.S. Gut Microbiota Dysbiosis and Huntington’s Disease: Exploring the Gut-Brain Axis and Novel Microbiota-Based Interventions. Life Sci. 2023, 328, 121882, . [CrossRef]
- Badal, V.D.; Vaccariello, E.D.; Murray, E.R.; Yu, K.E.; Knight, R.; Jeste, D.V.; Nguyen, T.T. The Gut Microbiome, Aging, and Longevity: A Systematic Review. Nutrients 2020, 12, 3759, . [CrossRef]
- Tong, H.; Yang, T.; Xu, S.; Li, X.; Liu, L.; Zhou, G.; Yang, S.; Yin, S.; Li, X.-J.; Li, S. Huntington’s Disease: Complex Pathogenesis and Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 3845, . [CrossRef]
- Strandwitz, P. Neurotransmitter Modulation by the Gut Microbiota. Brain Res. 2018, 1693, 128–133, . [CrossRef]
- Li, Y.; Li, Y.-J.; Zhu, Z.-Q. To Re-Examine the Intersection of Microglial Activation and Neuroinflammation in Neurodegenerative Diseases from the Perspective of Pyroptosis. Front. Aging Neurosci. 2023, 15, 1284214, . [CrossRef]
- Buret, A.G.; Motta, J.-P.; Allain, T.; Ferraz, J.; Wallace, J.L. Pathobiont Release from Dysbiotic Gut Microbiota Biofilms in Intestinal Inflammatory Diseases: A Role for Iron? J. Biomed. Sci. 2019, 26, 1, . [CrossRef]
- Follmer, C. Gut Microbiome Imbalance and Neuroinflammation: Impact of COVID -19 on Parkinson’s Disease. Mov. Disord. 2020, 35, 1495–1496, . [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25, . [CrossRef]
- Canani, R.B. Potential Beneficial Effects of Butyrate in Intestinal and Extraintestinal Diseases. World J. Gastroenterol. 2011, 17, 1519, . [CrossRef]
- Skrzypczak-Wiercioch, A.; Sałat, K. Lipopolysaccharide-Induced Model of Neuroinflammation: Mechanisms of Action, Research Application and Future Directions for Its Use. Molecules 2022, 27, 5481, . [CrossRef]
- Mitrea, L.; Nemeş, S.-A.; Szabo, K.; Teleky, B.-E.; Vodnar, D.-C. Guts Imbalance Imbalances the Brain: A Review of Gut Microbiota Association With Neurological and Psychiatric Disorders. Front. Med. 2022, 9, 813204, . [CrossRef]
- Christovich, A.; Luo, X.M. Gut Microbiota, Leaky Gut, and Autoimmune Diseases. Front. Immunol. 2022, 13, 946248, . [CrossRef]
- Camilleri, M. Leaky Gut: Mechanisms, Measurement and Clinical Implications in Humans. Gut 2019, 68, 1516–1526, . [CrossRef]
- Wasser, C.I.; Mercieca, E.-C.; Kong, G.; Hannan, A.J.; McKeown, S.J.; Glikmann-Johnston, Y.; Stout, J.C. Gut Dysbiosis in Huntington’s Disease: Associations among Gut Microbiota, Cognitive Performance and Clinical Outcomes. Brain Commun. 2020, 2, fcaa110, . [CrossRef]
- Witkowska, D.; Słowik, J.; Chilicka, K. Heavy Metals and Human Health: Possible Exposure Pathways and the Competition for Protein Binding Sites. Molecules 2021, 26, 6060, . [CrossRef]
- Koch, W.; Czop, M.; Iłowiecka, K.; Nawrocka, A.; Wiącek, D. Dietary Intake of Toxic Heavy Metals with Major Groups of Food Products—Results of Analytical Determinations. Nutrients 2022, 14, 1626, . [CrossRef]
- Rieder, G.S.; Duarte, T.; Delgado, C.P.; Rodighiero, A.; Nogara, P.A.; Orian, L.; Aschner, M.; Dalla Corte, C.L.; Da Rocha, J.B.T. Interplay between Diphenyl Diselenide and Copper: Impact on D. Melanogaster Survival, Behavior, and Biochemical Parameters. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2024, 281, 109899, . [CrossRef]
- Anand, I.S.; Gupta, P. Anemia and Iron Deficiency in Heart Failure: Current Concepts and Emerging Therapies. Circulation 2018, 138, 80–98, . [CrossRef]
- Yousefi Babadi, V.; Sadeghi, L.; Shirani, K.; Malekirad, A.A.; Rezaei, M. The Toxic Effect of Manganese on the Acetylcholinesterase Activity in Rat Brains. J. Toxicol. 2014, 2014, 1–4, . [CrossRef]
- Horning, K.J.; Caito, S.W.; Tipps, K.G.; Bowman, A.B.; Aschner, M. Manganese Is Essential for Neuronal Health. Annu. Rev. Nutr. 2015, 35, 71–108, . [CrossRef]
- Andrade, V.; Mateus, M.L.; Batoréu, M.C.; Aschner, M.; Dos Santos, A.M. Toxic Mechanisms Underlying Motor Activity Changes Induced by a Mixture of Lead, Arsenic and Manganese. EC Pharmacol. Toxicol. 2017, 3, 31–42.
- Peres, T.V.; Schettinger, M.R.C.; Chen, P.; Carvalho, F.; Avila, D.S.; Bowman, A.B.; Aschner, M. Manganese-Induced Neurotoxicity: A Review of Its Behavioral Consequences and Neuroprotective Strategies. BMC Pharmacol. Toxicol. 2016, 17, 57, . [CrossRef]
- O’Neal, S.L.; Zheng, W. Manganese Toxicity Upon Overexposure: A Decade in Review. Curr. Environ. Health Rep. 2015, 2, 315–328, . [CrossRef]
- Burton, N.C.; Schneider, J.S.; Syversen, T.; Guilarte, T.R. Effects of Chronic Manganese Exposure on Glutamatergic and GABAergic Neurotransmitter Markers in the Nonhuman Primate Brain. Toxicol. Sci. Off. J. Soc. Toxicol. 2009, 111, 131–139, . [CrossRef]
- Aschner, M.; Gannon, M. Manganese (Mn) Transport across the Rat Blood-Brain Barrier: Saturable and Transferrin-Dependent Transport Mechanisms. Brain Res. Bull. 1994, 33, 345–349, . [CrossRef]
- Aschner, M.; Skalny, A.V.; Martins, A.C.; Sinitskii, A.I.; Farina, M.; Lu, R.; Barbosa, F.; Gluhcheva, Y.G.; Santamaria, A.; Tinkov, A.A. Ferroptosis as a Mechanism of Non-Ferrous Metal Toxicity. Arch. Toxicol. 2022, 96, 2391–2417, . [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285, . [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125, . [CrossRef]
- Ji, Y.; Zheng, K.; Li, S.; Ren, C.; Shen, Y.; Tian, L.; Zhu, H.; Zhou, Z.; Jiang, Y. Insight into the Potential Role of Ferroptosis in Neurodegenerative Diseases. Front. Cell. Neurosci. 2022, 16, 1005182, . [CrossRef]
- Lee, J.-Y.; Kim, W.K.; Bae, K.-H.; Lee, S.C.; Lee, E.-W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184, . [CrossRef]
- Tian, H.-Y.; Huang, B.-Y.; Nie, H.-F.; Chen, X.-Y.; Zhou, Y.; Yang, T.; Cheng, S.-W.; Mei, Z.-G.; Ge, J.-W. The Interplay between Mitochondrial Dysfunction and Ferroptosis during Ischemia-Associated Central Nervous System Diseases. Brain Sci. 2023, 13, 1367, . [CrossRef]
- Feng, S.; Tang, D.; Wang, Y.; Li, X.; Bao, H.; Tang, C.; Dong, X.; Li, X.; Yang, Q.; Yan, Y.; et al. The Mechanism of Ferroptosis and Its Related Diseases. Mol. Biomed. 2023, 4, 33, . [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88, . [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282, . [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P. de L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765, . [CrossRef]
- Johnson, E.B.; Parker, C.S.; Scahill, R.I.; Gregory, S.; Papoutsi, M.; Zeun, P.; Osborne-Crowley, K.; Lowe, J.; Nair, A.; Estevez-Fraga, C.; et al. Altered Iron and Myelin in Premanifest Huntington’s Disease More than 20 Years before Clinical Onset: Evidence from the Cross-Sectional HD Young Adult Study. EBioMedicine 2021, 65, 103266, . [CrossRef]
- Levi, S.; Ripamonti, M.; Moro, A.S.; Cozzi, A. Iron Imbalance in Neurodegeneration. Mol. Psychiatry 2024, . [CrossRef]
- Mancardi, D.; Mezzanotte, M.; Arrigo, E.; Barinotti, A.; Roetto, A. Iron Overload, Oxidative Stress, and Ferroptosis in the Failing Heart and Liver. Antioxid. Basel Switz. 2021, 10, 1864, . [CrossRef]
- Tang, Q.; Liu, H.; Shi, X.-J.; Cheng, Y. Blood Oxidative Stress Marker Aberrations in Patients with Huntington’s Disease: A Meta-Analysis Study. Oxid. Med. Cell. Longev. 2020, 2020, 9187195, . [CrossRef]
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 2019, 12, 68, . [CrossRef]
- Jarosińska, O.D.; Rüdiger, S.G.D. Molecular Strategies to Target Protein Aggregation in Huntington’s Disease. Front. Mol. Biosci. 2021, 8, 769184, . [CrossRef]
- Chen, J.; Marks, E.; Lai, B.; Zhang, Z.; Duce, J.A.; Lam, L.Q.; Volitakis, I.; Bush, A.I.; Hersch, S.; Fox, J.H. Iron Accumulates in Huntington’s Disease Neurons: Protection by Deferoxamine. PLoS ONE 2013, 8, e77023, . [CrossRef]
- Zhu, L.; Li, G.; Liang, Z.; Qi, T.; Deng, K.; Yu, J.; Peng, Y.; Zheng, J.; Song, Y.; Chang, X. Microbiota-Assisted Iron Uptake Promotes Immune Tolerance in the Intestine. Nat. Commun. 2023, 14, 2790, . [CrossRef]
- Correnti, M.; Gammella, E.; Cairo, G.; Recalcati, S. Iron Absorption: Molecular and Pathophysiological Aspects. Metabolites 2024, 14, 228, . [CrossRef]
- Patanè, G.T.; Putaggio, S.; Tellone, E.; Barreca, D.; Ficarra, S.; Maffei, C.; Calderaro, A.; Laganà, G. Ferroptosis: Emerging Role in Diseases and Potential Implication of Bioactive Compounds. Int. J. Mol. Sci. 2023, 24, 17279, . [CrossRef]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting Ferroptosis Opens New Avenues for the Development of Novel Therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372, . [CrossRef]
- Li, Z.; Zhang, Y.; Ji, M.; Wu, C.; Zhang, Y.; Ji, S. Targeting Ferroptosis in Neuroimmune and Neurodegenerative Disorders for the Development of Novel Therapeutics. Biomed. Pharmacother. 2024, 176, 116777, . [CrossRef]
- Aschner, J.L.; Aschner, M. Nutritional Aspects of Manganese Homeostasis. Mol. Aspects Med. 2005, 26, 353–362, . [CrossRef]
- Li, L.; Yang, X. The Essential Element Manganese, Oxidative Stress, and Metabolic Diseases: Links and Interactions. Oxid. Med. Cell. Longev. 2018, 2018, 1–11, . [CrossRef]
- Tizabi, Y.; Getachew, B.; Aschner, M. Butyrate Protects and Synergizes with Nicotine against Iron- and Manganese-Induced Toxicities in Cell Culture. Neurotox. Res. 2024, 42, 3, . [CrossRef]
- Evans, G.R.; Masullo, L.N. Manganese Toxicity. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2024.
- Porru, S.; Esplugues, A.; Llop, S.; Delgado-Saborit, J.M. The Effects of Heavy Metal Exposure on Brain and Gut Microbiota: A Systematic Review of Animal Studies. Environ. Pollut. 2024, 348, 123732, . [CrossRef]
- Aschner, M.; Martins, A.C.; Oliveira-Paula, G.H.; Skalny, A.V.; Zaitseva, I.P.; Bowman, A.B.; Kirichuk, A.A.; Santamaria, A.; Tizabi, Y.; Tinkov, A.A. Manganese in Autism Spectrum Disorder and Attention Deficit Hyperactivity Disorder: The State of the Art. Curr. Res. Toxicol. 2024, 6, 100170, . [CrossRef]
- Bryan, M.R.; Bowman, A.B. Manganese and the Insulin-IGF Signaling Network in Huntington’s Disease and Other Neurodegenerative Disorders. In Neurotoxicity of Metals; Aschner, M., Costa, L.G., Eds.; Advances in Neurobiology; Springer International Publishing: Cham, 2017; Vol. 18, pp. 113–142 ISBN 978-3-319-60188-5.
- Cordeiro, L.M.; Soares, M.V.; Da Silva, A.F.; Dos Santos, L.V.; De Souza, L.I.; Da Silveira, T.L.; Baptista, F.B.O.; De Oliveira, G.V.; Pappis, C.; Dressler, V.L.; et al. Toxicity of Copper and Zinc Alone and in Combination in Caenorhabditis Elegans Model of Huntington’s Disease and Protective Effects of Rutin. NeuroToxicology 2023, 97, 120–132, . [CrossRef]
- Royer, A.; Sharman, T. Copper Toxicity. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2024.
- Opazo, C.M.; Lotan, A.; Xiao, Z.; Zhang, B.; Greenough, M.A.; Lim, C.M.; Trytell, H.; Ramírez, A.; Ukuwela, A.A.; Mawal, C.H.; et al. Nutrient Copper Signaling Promotes Protein Turnover by Allosteric Activation of Ubiquitin E2D Conjugases 2021.
- Immergluck, J.; Anilkumar, A.C. Wilson Disease. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2024.
- Xiao, G.; Fan, Q.; Wang, X.; Zhou, B. Huntington Disease Arises from a Combinatory Toxicity of Polyglutamine and Copper Binding. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 14995–15000, . [CrossRef]
- Pfalzer, A.C.; Yan, Y.; Kang, H.; Totten, M.; Silverman, J.; Bowman, A.B.; Erikson, K.; Claassen, D.O. Alterations in Metal Homeostasis Occur Prior to Canonical Markers in Huntington Disease. Sci. Rep. 2022, 12, 10373, . [CrossRef]
- Rosas, H.D.; Chen, Y.I.; Doros, G.; Salat, D.H.; Chen, N.; Kwong, K.K.; Bush, A.; Fox, J.; Hersch, S.M. Alterations in Brain Transition Metals in Huntington Disease: An Evolving and Intricate Story. Arch. Neurol. 2012, 69, 887–893, . [CrossRef]
- Suganya, K.; Koo, B.-S. Gut-Brain Axis: Role of Gut Microbiota on Neurological Disorders and How Probiotics/Prebiotics Beneficially Modulate Microbial and Immune Pathways to Improve Brain Functions. Int. J. Mol. Sci. 2020, 21, 7551, . [CrossRef]
- Love, C.J.; Masson, B.A.; Gubert, C.; Hannan, A.J. The Microbiota-Gut-Brain Axis in Huntington’s Disease. In International Review of Neurobiology; Elsevier, 2022; Vol. 167, pp. 141–184 ISBN 978-0-323-99176-6.
- Wronka, D.; Karlik, A.; Misiorek, J.O.; Przybyl, L. What the Gut Tells the Brain—Is There a Link between Microbiota and Huntington’s Disease? Int. J. Mol. Sci. 2023, 24, 4477, . [CrossRef]
- Ekwudo, M.N.; Gubert, C.; Hannan, A.J. The Microbiota–Gut–Brain Axis in Huntington’s Disease: Pathogenic Mechanisms and Therapeutic Targets. FEBS J. 2024, febs.17102, . [CrossRef]
- Getachew, B.; Csoka, A.B.; Bhatti, A.; Copeland, R.L.; Tizabi, Y. Butyrate Protects Against Salsolinol-Induced Toxicity in SH-SY5Y Cells: Implication for Parkinson’s Disease. Neurotox. Res. 2020, 38, 596–602, . [CrossRef]
- Getachew, B.; Csoka, A.B.; Garden, A.R.; Copeland, R.L.; Tizabi, Y. Sodium Butyrate Protects Against Ethanol-Induced Toxicity in SH-SY5Y Cell Line. Neurotox. Res. 2021, 39, 2186–2193, . [CrossRef]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; de Oliveira, A.C.P. Lipopolysaccharide-Induced Neuroinflammation as a Bridge to Understand Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293, . [CrossRef]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain-Gut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44, . [CrossRef]
- Gubert, C.; Love, C.J.; Kodikara, S.; Mei Liew, J.J.; Renoir, T.; Lê Cao, K.-A.; Hannan, A.J. Gene-Environment-Gut Interactions in Huntington’s Disease Mice Are Associated with Environmental Modulation of the Gut Microbiome. iScience 2022, 25, 103687, . [CrossRef]
- Tang, W.; Zhu, H.; Feng, Y.; Guo, R.; Wan, D. The Impact of Gut Microbiota Disorders on the Blood–Brain Barrier. Infect. Drug Resist. 2020, Volume 13, 3351–3363, . [CrossRef]
- Vauleon, S.; Schutz, K.; Massonnet, B.; Gruben, N.; Manchester, M.; Buehler, A.; Schick, E.; Boak, L.; Hawellek, D.J. Quantifying mutant huntingtin protein in human cerebrospinal fluid to support the development of huntingtin-lowering therapies. Sci Rep. 2023, 13, 5332, . [CrossRef]
- Juarez, D.; Handal-Silva, A.; Morán-Perales, J.L.; Torres-Cifuentes, D.M.; Flores, G.; Treviño, S.; Moreno-Rodriguez, A.; Guevara, J.; Diaz, A. New insights into sodium phenylbutyrate as a pharmacotherapeutic option for neurological disorders. Synapse. 2024, Jul;78(4):e22301. PMID: 38819491. [CrossRef]
- Khoshnan, A. Gut Microbiota as a Modifier of Huntington's Disease Pathogenesis. J Huntingtons Dis. 2024, E-Pub ahead of print, doi: 10.3233/JHD-240012.
- Ma, Y.Y.; Li, X.; Yu, J.T.; Wang, Y.J. Therapeutics for neurodegenerative diseases by targeting the gut microbiome: from bench to bedside. Transl Neurodegener. 2024, 13(1):12. [CrossRef]
Figure 1.
Schematic diagram depicting how heavy metals via their interactions with the neuroglia and gut microbiota may contribute to Huntington’s disease (HD) pathology. Detail understanding of this interaction can pave the way for novel therapeutic interventions in this rare but devastating neurological disease.
Figure 1.
Schematic diagram depicting how heavy metals via their interactions with the neuroglia and gut microbiota may contribute to Huntington’s disease (HD) pathology. Detail understanding of this interaction can pave the way for novel therapeutic interventions in this rare but devastating neurological disease.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



