
Submitted:
10 June 2024
Posted:
12 June 2024
You are already at the latest version
Abstract
MicroRNAs (miRNA) exert regulatory actions via base-pairing with their binding sites on target mRNAs. Cooperative-binding, i.e., synergism, among binding sites on a mRNA is biochemically well-characterized. We studied whether this synergism is reflected in global relationship between miRNA-mediated regulatory activity and miRNA-binding-site count on the target mRNAs, i.e., leading to a non-linear relationship between the two. Recently, using our own and public datasets, we have enquired into miRNA regulatory actions: first, we analyzed the power-law distribution pattern of miRNA binding sites; second, strikingly, mRNAs for core miRNA regulatory apparatus proteins have extra-ordinarily high binding site counts, forming self-feedback-control loops; third, we revealed that tumor suppressor mRNAs generally have more sites than oncogene mRNAs; and fourth, we characterized enrichment of miRNA target mRNAs in translationally less-active polysomes relative to more-active polysomes. In these four studies, obvious correlation was observed qualitatively between the extent to which a mRNA is miRNA-regulated and its binding sites count. This paper summarizes the used datasets. We also re-analyzed the correlation by comparative linear and non-linear regression analyses. The results support a non-linear, instead of linear, relationship between the two parameters, conceivably a transcriptome-level reflection of cooperative-binding/synergism among miRNA binding sites on a target mRNA.
Keywords:
miRNA (microRNA)
; miRNA binding site
; cooperative binding
; synergism
; linear regression
; non-linear regression
; polynomial regression
1. Introduction
MicroRNAs (miRNAs) are short, single-stranded noncoding RNAs typically comprising 22 nucleotides, with lengths ranging between 21 to 25 nucleotides, existing in almost all metazoans including flies, plants and mammals [1]. In the canonical miRNA biogenesis pathway, primary miRNA (pri-miRNA) is transcribed within the nucleus by RNA polymerase II (Pol II), begins with a 7-methylguanosine cap (m7Gppp) and ends with a 3ʹ poly(A) tail [2]. Pri-miRNA possesses a stem–loop structure, subject to cleavage by the endonuclease Drosha in concert with its partner DGCR8 [3]. The resultant precursor miRNA (pre-miRNA) is subsequently exported from the nucleus via exportin 5 and undergoes further cleavage by the endonuclease Dicer, accompanied by its partner TRBP, leading to the liberation of a miRNA guide strand–miRNA passenger strand duplex. This duplex is then assembled into an Argonaute (AGO) protein complex alongside chaperones, forming a double-stranded RNA (dsRNA) configuration [4]. Through subsequent maturation steps, the miRNA passenger strand is expelled, resulting in the formation of a mature single-stranded RNA-induced silencing complex (RISC) [5].
Some miRNA genes are located within the introns of other genes, thereby biosynthesizing the miRNA through non-canonical pathways [6]. In such instances, Pre-miRNAs, termed branched Mirtron, are spliced directly out of introns of the mRNA encoding host genes [7]. Branched Mirtrons undergo lariat debranching, bypass Drosha cleavage, and are exported by Exportin 5 [8]. Following export, they are processed by Dicer and ultimately loaded into RISC. In the specific case of vertebrate miR-451, its unusually short pre-miRNA hairpin can escape Dicer processing after nuclear export and is instead directly loaded into the AGO protein, which triggers its maturation into a single-stranded miRNA RISC [9,10,11,12].
RISC serves as a guide for miRNAs to recognize complementary sequences located in the 3′ untranslated regions (3′-UTR) of target mRNAs. Upon this binding, RISC inhibits the mRNA through two primary modes: decay and translational repression [13].
The miRNA regulatory actions in plants and animals are different. In plants, most miRNAs are loaded into AGO1, a member of the Ago family that has endonuclease activity, and the RISCs target mRNAs containing perfect or near-perfect complementarity sites, leading to direct cleavage of the target mRNA [14,15]. Conversely, in animals, miRNAs loaded into AGO2 protein are typically base pair with imperfect complementarity, particularly via a 2-8 nucleotide sequence situated at their 5′ end to the 3′-UTR (untranslated region) of their target, known as the seed region [16,17]. Animal RISCs induce target mRNA decay not through endonucleolytic cleavage by AGO2 but by guiding the mRNA to the general mRNA degradation machinery [18]. This process entails initiating deadenylation, recruiting the decapping complex, and subsequently degrading the mRNA via exonucleases, unless the miRNA exhibits full complementarity to its target [19,20]. On the other hand, the miRNA-AGO2 complex can repress target mRNA translation activity through various mechanisms, including inhibiting translation initiation, repressing 60S subunit joining, blocking peptide chain elongation, promoting ribosomal drop-off, or facilitating nascent protein proteolysis [13,21,22]. However, the specific mechanism in the miRNA regulated translation repression currently remains a subject of controversy.
As miRNA is vital for a broad range of fundamental processes, such as development, immune and neuronal function, and metabolic homoeostasis, defective miRNA biogenesis and/or function underlies multiple human diseases [23,24,25,26], underscoring significance of miRNA research – a prominent biological and biomedical topic ever since its initial discovery. Yet, key gaps and challenges remain, e.g., the short (6-8 bases) and mismatch-tolerant miRNA binding sites that enable a miRNA to target a huge number of mRNAs and a mRNA be targeted by many miRNAs. This complexity in miRNA-target relationship imposes challenges, even controversies, in experimental result interpretation [27]. And, cooperation/synergism among binding sites, though biochemically well-characterized [28], remains to be studied at whole transcrptome level.
Using both our own and public datasets, we recently enquired into miRNA regulatory actions. We analyzed the power-law distribution pattern of miRNA binding sites [29]. Strikingly, mRNAs for core miRNA regulatory apparatus proteins have extra-ordinarily high binding site counts, forming self-feedback-control loops [30]. Tumor suppressor mRNAs generally have more sites than oncogene mRNAs [31]. And, we characterized enrichment of miRNA target mRNAs in translationally less-active polysomes relative to more-active polysomes [32]. This paper summarizes the datasets used in these four studies.
We also observed correlation between how much a mRNA is miRNA-regulated and its binding sites count. Our re-analyses of the correlation in this study support a non-linear, instead of a linear, relationship between the two, conceivably a reflection of cooperation/synergism among miRNA binding sites.
2. Materials and Methods
2.1. RNA-Seq Datasets of Wildtype and miRNA-Biogenesis-Deficient Cells
As discussed in the Introduction, in canonical miRNA biogenesis pathways, Drosha functions as the RNase responsible for cleaving pri-miRNAs into pre-miRNAs, while Dicer1 RNase is pivotal in processing pre-miRNAs into 22-nucleotide miRNA duplexes. Mutations of key miRNA biogenesis enzymes result in deficiency in miRNA biogenesis deficiency. To investigate the effects of this deficiency on the transcriptome, we obtained the RNA-seq dataset published by Kim et al. [33,34], from the NCBI GEO dataset, under accession number GSE80258. Total RNA was isolated from both wild-type and DROSHA knockout HCT116 cells, followed by sequencing on a HiSeq 2500 (Illumina) platform. Nucleotides displaying low-quality values were filtered out. Subsequently, we aligned the sequencing reads to the human reference genome (hg38) using STAR alignment software and determined gene expression levels with HTSeq-count software. The resulting counts were normalized to reads per kilobase of transcript per million mapped reads (RPKM). Our analysis focused solely on identified protein-coding genes for expression profiling in this study.
Zheng et al. (2014)[35] conducted a comparative RNA-seq analysis of transcriptome variations in wild type and Dicer1 deficient mouse embryonic stem cells (mESCs), which can also be applied to study the role of miRNA in regulating transcriptome. The RNA-seq dataset associated with this study is public at the NCBI GEO database under accession number GSE55338. The experimental procedure involved isolating total polyadenylated RNA from both WT (two biological replicates) and Dicer1-KO (three biological replicates) mESCs, followed by sequencing on the Hi-Seq 2000 Illumina platforms. DESeq29 was applied to normalize reads between samples. Upon data retrieval, mRNAs with a minimum of two replicates showing >1 normalized reads were selected, and the log2 fold change between WT and Dicer1-KO cells was calculated. Gene identifiers were converted to gene symbols and annotated with biotype information by accessing the Ensembl database (release 104) through the R package BiomaRt (version 2.46.3) [36]. Only protein-coding genes were included for expression analysis in this investigation.
2.2. Comparative Polysome Profiling Datasets of Wildtype and miRNA-Biogenesis- Deficient Cells
During mRNA translation, multiple ribosomes simultaneously traverse the coding region of mRNA, forming a polysome. The majority of actively translating ribosomes in cells exist as polysomes, with multiple ribosomes loaded onto a single transcript. Polysome profiling is a technique that uses next generation sequencing (NGS) to analyze polysome-associated, and thus translating, mRNAs. In our analysis, we separated the translationally less-actively light polysomes and the more-active polysomes through sucrose gradient centrifugation [37]. We previously cultured wild type and Dicer1 knockout HCT116 cells, conducted polysome separation, and collected the light polysomes (2- to 9-mers) and heavy polysomes (10-mers or more) fractions, respectively [30]. RNA samples were extracted from these fractions, cDNA libraries were constructed, and RNA-seq was performed using the BGI America DNBseq sequencer. Low-quality reads and multiplexing barcode sequences were filtered out. The resulting dataset was deposited into the NCBI GEO database with the accession number GSE134818. For this study, only transcripts of identified protein-coding genes with minimum expression >0 and maximum expression >1 across all samples were retained.
2.3. Evolutionarily Conserved miRNA Binding Sites Count
The compilation of evolutionarily conserved human and mouse miRNA binding sites was described in the study conducted by Agarwal et al. and retrieved from the TargetScan database 7.1 (June 2016 release) [38]. The methodology was previously outlined in our research [31]. For each mRNA, the number of unique conserved miRNA binding sites on its 3′-UTR was calculated, and used as miRNA binding site count in the current research.
2.4. Statistical Analysis
The R open-source software (version 4.0.2) and the MATLAB software were used for data analysis and plotting. R was used for statistical sample normalization, data processing and LOESS regression. Polynomial and exponential regressions as well as plotting were done with MATLAB. Degree 1 polynomial regression is the same as the simple linear regression. Degree 2 and higher degree polynomial regressions are non-linear.
To compare the linear and non-linear regression, we used the anova function, as the linear (degree 1 polynomial) model is nested inside the non-linear models. The function outputs a F-ratio and a p-value, thus statistically quantifying the improvement from the linear to the non-linear models. We found degree 2 polynomial regression was sufficient for all datasets in this study, as increasing the degree to 3 or higher did not lead to significant improvement.
3. Results
3.1. Description of RNA-Seq and Polysome Profiling Datasets Used in the Previous Studies
The RNA-seq datasets used in our previous studies described above is summarized in Table 1. Zheng et al. (2014) [35] collected two replicates from wildtype mESCs and three replicates from Dicer knockout mESCs and analyzed the expression of lncRNAs and mRNAs. A total of 43,956 transcripts were included in the RNA-seq dataset (GSE55338), with 16,730 transcripts identified as protein-coding genes. Among these protein-coding genes, 15,104 transcripts of identified protein-coding genes with at least two replicates had expression levels >1 were retained for further analysis in the current study. Subsequently, the ORF and miRNA-binding site counts dataset was examined, revealing that 13,611 protein-coding genes had available information for these features. Thus, the final analysis included 13,611 protein-coding genes in the current study.
In the study by Kim et al. [33,34], one replicate each from wildtype HCT116 cells and Drosha knockout HCT116 cells were included, and the poly(A) enriched RNAs from each sample were subjected to RNA-seq analysis. The expression profiles of these genes were extracted from the RNA-seq dataset (GSE80258), while corresponding ORF lengths and miRNA-binding site counts were summarized according to the methodology outlined in the Materials and Methods section. A total of 18,098 transcript reads were computed from each cell line, with 15,112 transcripts identified as protein-coding genes, whose ORF length and miRNA-binding sites count data are also available.
Finally, in a previous study, we investigated the mRNA expression profiles of light polysome fractions and heavy polysome fractions between HCT116 wildtype cells and Dicer knockout cells (GSE134818). Our findings revealed that miRNAs tend to target mRNAs with longer open reading frames (ORFs), and miRNA-targeted mRNAs exhibit enrichment in translationally less active light polysomes [30]. The polysome profiling dataset utilized in this study comprised a total of 26,281 transcripts, out of which 11,380 transcripts of identified protein-coding genes with minimum expression >0 and maximum expression >1 across all samples were retained in the study.
Here, we used the three datasets to re-analyze the previously observed correlation between the degree to which a mRNA is miRNA-regulated and its miRNA binding sites count and, more importantly, examine whether the synergism among the binding sites is reflected in the relationship.
3.2. The levels of miRNA Regulatory Activity Correlates with the miRNA Binding Site Count
There are two parameters frequently used in assessing miRNA-mediated regulatory activity. The first one is enhanced target-mRNA degradation. The second is inhibition of target-mRNA translation activity. We used both in this study. As in previous studies, we also binned the mRNAs according to the miRNA binding site count, i.e., one bin per binding site count and mRNAs in a bin all have the same count. This binning is necessitated by the binding-site-count power-law distribution pattern, which is followed by many other biological parameters and obscures/hides potential trends without the binning [39].
3.2.1. Target mRNA Degradation
Based on our understanding of miRNA’s regulatory role in mRNA degradation and our previous observations, the deficiency in miRNA production due to Dicer1 or Drosha deletion in mutant cells should lead to an increase in the expression levels of miRNA-targeted mRNAs compared to wildtype cells, and the extent of this expression change should correlate with the count of miRNA binding sites. To test this, we calculated the average log-ratio of expression fold change (log2(KO/WT)) between Dicer1 knockout and wildtype (Figure 1) as well as Drosha knockout and wildtype (Figure 2) cells in each mRNA bin, and analyzed the log-ratios versus the miRNA binding site counts. Not surprisingly, there is an obvious trend shown in the Figure 1 that the degree of expression change induced by Dicer knockout (as indicated by the log2(KO/WT) log-ratio) increases with the number of miRNA binding sites in the mESCs. A similar trend was also observed in the HCT116 cell lines (Figure 2). Thus, miRNAs enhance mRNA degradation, i.e., downregulating target-mRNA abundanec in the transcriptome, and the degree of repression correlates well with the number of miRNA binding sites on the target mRNAs.
3.2.2. Target mRNA Translation Inhibition
Our comparative light- and heavy-polysome profiling datasets, as previously reported, enable examination of the correlation between miRNA binding site count and target-mRNA translation inhibition (Figure 3). We calculated the light- to heavy-polysome log-ratio (log2(Light/Heavy)). As previously described [32], we adjusted the log-ratio with open reading frame (ORF) length by LOESS regressions, removing the effect of ORF length; with all else the same, a longer ORF accommodates more translating ribosomes and miRNA-target-mRNA ORFs tend to be longer [32]. Thus, in this study, the LOESS regression residuals were used as the log-ratio for both WT and Dicer1 knockouit cells. In WT cells, this log-ratio increases along with the binding site count (Figure 3A).
We also re-adjusted this WT cell log-ratio by subtracting that of the Dicer1 knockout mutant cells (log-ratio(WT) – log-ratio(KO)). This subtraction removes the regulatory effects of non-miRNA regulatory elements in the mRNAs, such as those exerted by TRIM71 via binding to 3′-UTR hairpin motifs [40]. The same trend, though weaker, is observed between this adjusted log-ratio and the binding site count (Figure 3B).
3.3. Reflection of the Synergistic, Instead of Additive, Interactions among miRNA Binding Sites
Whether miRNA binding sites synergistically or additively interact with one another has been an active research topic. Currently, the analysis is mostly based on in vitro biochemical analyses, and the consensus is that the interactions are synergistic [28]. We had an opportunity to address this question based on transcriptome-wide datasets. If the interaction among the binding sites is synergistic, the relationship between miRNA-mediated regulatory activity and the binding site count should be non-linear; otherwise, linear relationships should be expected.
Thus, we performed both linear and non-linear weighted regression analyses with the MATLAB software, using the bin-size (mRNA count in the bin) as the weights. As discussed in Materials and Methods, linear regression was performed as degree 1 polynomial regression. Non-linear regressions were performed with degree 2 polynomial regression as well as exponential regression. The anova function was used to quantify how much the non-linear polynomial regression improves over the linear regression, as the latter is nested inside the former.
3.3.1. miRNA-Mediated Target mRNA Degradation
The results are shown visually in Figure 1 and Figure 2. Non-linear regressions gave much better fit then linear regressions for both mESC cells (Figure 1) and the HCT116 cells (Figure 2). Quantitatively, in both types of cells, degree 2 polynomial and exponential regressions result in lower RMSE (residual mean square error) and higher R-square values than the linear degree 1 polynomial regressions. Additionally, comparing the linear and non-linear polynomial regression with the anova function resulted in statistically significant p-values; the mESC cells gave a p-value of 9.063e-5, and the HCT116 cells 3.625e-6.
Equations of the regression models for mESC cells in Figure 1 are listed below:
Linear degree-1 polynomial (linear) fit:
Non-linear degree-2 polynomial (poly2) fit:
Exponential (exp) fit:
For HCT116 cells, the equations of the models in Figure 2 are listed below:
Linear degree-1 polynomial (linear) fit:
Non-linear degree-2 polynomial (poly2) fit:
Exponential (exp) fit:
3.3.2. miRNA-Mediated Target mRNA Translation Inhibition
As shown in Figure 3, the same was observed for miRNA-mediated translation inhibition in HCT116 cells. The trend is clear in WT cells (Figure 3A). Quantitatively, degree 2 polynomial regression results in lower RMSE and higher R-square values than the linear degree 1 polynomial regression. Comparing the linear and non-linear polynomial regressions with anova also resulted in a significant p-value (1.23e-06). However, exponential regression does not improve over the linear regression, actually performing worth. These results indicate a non-linear, but clearly non-exponential, relationship. Equations of the regression models for WT HCT116 cell log-ratio in Figure 3A are listed below:
Linear degree-1 polynomial (linear) fit:
Non-linear degree-2 polynomial (poly2) fit:
Exponential (exp) fit:
The trend becomes weaker upon subtraction of mutant HCT116 cell log-ratios (Figure 3B). However, improvement from linear to non-linear models is statistically significant (anova p-value 0.025). If we remove the data points with > 45 binding sites, which are more scattered due to lower gene counts and thus less statistical power, the p-value lowers to 0.0095. In contrast to results in Figure 3A, exponential and degree-2 polynomial regressions are almost in-distinguishable in terms of regression curves and the RMSE and R-square values, presumably due to removal of non-miRNA-mediated regulatory effects.
Equations of the regression models for adjusted log-ratio in Figure 3B are listed below:
Linear degree-1 polynomial (linear) fit:
Non-linear degree-2 polynomial (poly2) fit:
Exponential (exp) fit:
4. Discussion
In the multi-step process of cellular genetic information flow, mRNAs serve as the carrier of genetic information, while proteins, translated from mRNAs, act as the final effectors to execute cellular functions. Both mRNAs and proteins are dynamically produced and degraded under tight regulation of an intricate regulatory network to maintain the cellular homeostasis. The discrepancy between the abundance of cognate protein and RNA molecules is frequently observed, yet explaining it remains elusive and/or technically challenging [41,42].
MiRNA, one of the key post-transcriptional regulators, might be one critical explanatory factor. It exerts regulation over both the mRNA and protein levels by decreasing target mRNA stability and suppressing their translation activity [43]. There is a paradoxical phenomenon in which miRNAs typically exert a moderate influence on gene expression rather than causing complete silencing of target genes, and the degree of regulation can vary depending on various factors, including the specific miRNA, target mRNA, cellular context, and experimental conditions [44,45,46]. Our recent study elucidated a mechanism to explain the moderate impact of miRNAs on target mRNA translation, whereby miRNA retain the target mRNAs associated with translationally less-active light polysomes, thereby inhibiting translation rather than completely silencing it [32]. Additionally, the predominant mechanism by which miRNAs regulate mRNA repression—whether through promoting mRNA degradation or affecting translation activity—remains unresolved. While some studies emphasize mRNA degradation as the primary mode of regulation [47,48], others highlight translational repression preceding mRNA destabilization [49,50,51,52].
There is a notion that mRNAs harboring higher numbers of miRNA binding sites are under stronger repression by miRNAs, which had been verified in multiple experimental research regarding individual miRNAs or specific functional gene groups [30,53,54]. Our previous studies on the power-law distribution pattern of miRNA binding sites, cancer-related genes (tumor suppressor versus oncogenes), and enrichment of miRNA-target mRNAs in light polysomes all observed an obvious correlation between the capacity of miRNA-mediated mRNA repression and the miRNA binding site count [29,31]. This current study re-investigates the correlation by comparative linear and non-linear regression analyses. Our results favor non-linear relationships between the two for both miRNA-mediated target-mRNA decay and translation inhibition. Conceivably, the non-linear relationship is a reflection of the cooperation/synergism among miRNA binding sites on a target mRNA.
5. Conclusions
This study provides whole-cell transcriptome level evidence for the biochemically well-characterized cooperative-binding/synergism among miRNA binding sites [28]. The significance of our results lies in the ubiquitous phenomenon that multiple miRNA binding sites co-occur on the same target mRNA.
Author Contributions
Conceptualization, B.R. and D.W.; methodology, S.T., Z.Z., B.R. and D.W.; software, S.T. and B.R.; validation, S.T., Z.Z., B.R. and D.W.; formal analysis, S.T., Z.Z., B.R. and D.W.; investigation, S.T., Z.Z., B.R. and D.W.; resources, B.R. and D.W.; data curation, S.T., Z.Z. and D.W.; writing—original draft preparation, S.T. and D.W.; writing—review and editing, S.T., Z.Z., B.R. and D.W.; visualization, S.T. and B.R.; supervision, D.W.; project administration, D.W.; funding acquisition, D.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NIGMS NIH, grant number R15GM147858, to D.W., and by Cancer Prevention and Research Institute of Texas (CPRIT), grant number RP220600, to D.W..
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All datasets used in this study are publicly available at the NCBI GEO databse with the given accession numbers.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- He, L.; Hannon, G.J. MicroRNAs: small RNAs with a big role in gene regulation. Nature reviews genetics 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Denli, A.M.; Tops, B.B.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef]
- MacRae, I.J.; Ma, E.; Zhou, M.; Robinson, C.V.; Doudna, J.A. In vitro reconstitution of the human RISC-loading complex. Proceedings of the National Academy of Sciences 2008, 105, 512–517. [Google Scholar] [CrossRef]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef]
- Berezikov, E.; Chung, W.-J.; Willis, J.; Cuppen, E.; Lai, E.C. Mammalian mirtron genes. Molecular cell 2007, 28, 328–336. [Google Scholar] [CrossRef]
- Okamura, K.; Hagen, J.W.; Duan, H.; Tyler, D.M.; Lai, E.C. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell 2007, 130, 89–100. [Google Scholar] [CrossRef]
- Cifuentes, D.; Xue, H.; Taylor, D.W.; Patnode, H.; Mishima, Y.; Cheloufi, S.; Ma, E.; Mane, S.; Hannon, G.J.; Lawson, N.D. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 2010, 328, 1694–1698. [Google Scholar] [CrossRef]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.; Hannon, G.J. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 2010, 465, 584–589. [Google Scholar] [CrossRef]
- Yang, J.-S.; Maurin, T.; Robine, N.; Rasmussen, K.D.; Jeffrey, K.L.; Chandwani, R.; Papapetrou, E.P.; Sadelain, M.; O’Carroll, D.; Lai, E.C. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proceedings of the National Academy of Sciences 2010, 107, 15163–15168. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jin, D.-Y.; McManus, M.T.; Mourelatos, Z. Precursor microRNA-programmed silencing complex assembly pathways in mammals. Molecular cell 2012, 46, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Qi, Y. RNAi in plants: an argonaute-centered view. The Plant Cell 2016, 28, 272–285. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Schmitter, D.; Filkowski, J.; Sewer, A.; Pillai, R.S.; Oakeley, E.J.; Zavolan, M.; Svoboda, P.; Filipowicz, W. Effects of Dicer and Argonaute down-regulation on mRNA levels in human HEK293 cells. Nucleic acids research 2006, 34, 4801–4815. [Google Scholar] [CrossRef]
- Iwakawa, H.O.; Tomari, Y. The Functions of MicroRNAs: mRNA Decay and Translational Repression. Trends Cell Biol 2015, 25, 651–665. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nature reviews Molecular cell biology 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Huntzinger, E.; Izaurralde, E. Getting to the root of miRNA-mediated gene silencing. Cell 2008, 132, 9–14. [Google Scholar] [CrossRef] [PubMed]
- DeVeale, B.; Swindlehurst-Chan, J.; Blelloch, R. The roles of microRNAs in mouse development. Nature Reviews Genetics 2021, 22, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Nigi, L.; Grieco, G.E.; Ventriglia, G.; Brusco, N.; Mancarella, F.; Formichi, C.; Dotta, F.; Sebastiani, G. MicroRNAs as regulators of insulin signaling: research updates and potential therapeutic perspectives in type 2 diabetes. International journal of molecular sciences 2018, 19, 3705. [Google Scholar] [CrossRef] [PubMed]
- Im, H.-I.; Kenny, P.J. MicroRNAs in neuronal function and dysfunction. Trends in neurosciences 2012, 35, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Rottiers, V.; Näär, A.M. MicroRNAs in metabolism and metabolic disorders. Nature reviews Molecular cell biology 2012, 13, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [PubMed]
- Briskin, D.; Wang, P.Y.; Bartel, D.P. The biochemical basis for the cooperative action of microRNAs. Proc Natl Acad Sci U S A 2020, 117, 17764–17774. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, D. The Pattern of microRNA Binding Site Distribution. Genes (Basel) 2017, 8. [Google Scholar] [CrossRef]
- Wang, D.; Wang, T.; Gill, A.; Hilliard, T.; Chen, F.; Karamyshev, A.L.; Zhang, F. Uncovering the cellular capacity for intensive and specific feedback self-control of the argonautes and MicroRNA targeting activity. Nucleic Acids Res 2020, 48, 4681–4697. [Google Scholar] [CrossRef]
- Tian, S.; Wang, J.; Zhang, F.; Wang, D. Comparative Analysis of microRNA Binding Site Distribution and microRNA-Mediated Gene Expression Repression of Oncogenes and Tumor Suppressor Genes. Genes 2022, 13, 481. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tian, S.; Tikhonova, E.B.; Karamyshev, A.L.; Wang, J.J.; Zhang, F.; Wang, D. The Enrichment of miRNA-Targeted mRNAs in Translationally Less Active over More Active Polysomes. Biology 2023, 12, 1536. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of DROSHA, Exportin 5, and DICER in microRNA biogenesis. Proceedings of the National Academy of Sciences 2016, 113, E1881–E1889. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.; Lim, Y.-H.; Kim, Y.-K. Precise mapping of the transcription start sites of human microRNAs using DROSHA knockout cells. BMC genomics 2016, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Do, B.T.; Webster, D.E.; Khavari, P.A.; Chang, H.Y. Dicer-microRNA-Myc circuit promotes transcription of hundreds of long noncoding RNAs. Nat Struct Mol Biol 2014, 21, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nature protocols 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Gandin, V.; Sikström, K.; Alain, T.; Morita, M.; McLaughlan, S.; Larsson, O.; Topisirovic, I. Polysome fractionation and analysis of mammalian translatomes on a genome-wide scale. Journal of visualized experiments: JoVE 2014. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Padawer, T.; Leighty, R.E.; Wang, D. Duplicate gene enrichment and expression pattern diversification in multicellularity. Nucleic Acids Res 2012, 40, 7597–7605. [Google Scholar] [CrossRef]
- Welte, T.; Goulois, A.; Stadler, M.B.; Hess, D.; Soneson, C.; Neagu, A.; Azzi, C.; Wisser, M.J.; Seebacher, J.; Schmidt, I.; et al. Convergence of multiple RNA-silencing pathways on GW182/TNRC6. Mol Cell 2023, 83, 2478–2492 e2478. [Google Scholar] [CrossRef]
- Wang, D. Discrepancy between mRNA and protein abundance: insight from information retrieval process in computers. Computational biology and chemistry 2008, 32, 462–468. [Google Scholar] [CrossRef]
- Jiang, W.; Guo, Z.; Lages, N.; Zheng, W.J.; Feliers, D.; Zhang, F.; Wang, D. A multi-parameter analysis of cellular coordination of major transcriptome regulation mechanisms. Scientific reports 2018, 8, 5742. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Schwanhäusser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. nature 2008, 455, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, K.; Kim, K.; Sciulli, C.; Dowd, S.R.; Minden, J.S.; Carthew, R.W. Targets of microRNA regulation in the Drosophila oocyte proteome. Proceedings of the National Academy of Sciences 2005, 102, 12023–12028. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.-h.; Ghoshal, K.; Villén, J.; Bartel, D.P. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Molecular cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Béthune, J.; Artus-Revel, C.G.; Filipowicz, W. Kinetic analysis reveals successive steps leading to miRNA-mediated silencing in mammalian cells. EMBO reports 2012, 13, 716–723. [Google Scholar] [CrossRef]
- Djuranovic, S.; Nahvi, A.; Green, R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 2012, 336, 237–240. [Google Scholar] [CrossRef]
- Bazzini, A.A.; Lee, M.T.; Giraldez, A.J. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 2012, 336, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Freimer, J.W.; Hu, T.J.; Blelloch, R. Decoupling the impact of microRNAs on translational repression versus RNA degradation in embryonic stem cells. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Hon, L.S.; Zhang, Z. The roles of binding site arrangement and combinatorial targeting in microRNA repression of gene expression. Genome Biol 2007, 8, R166. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet 2012, 13, 271–282. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The average log-ratio of expression-level fold-of-change between Dicer1-knockout (KO) and wildtype (WT) mESCs (mouse embryonic stem cells) (log2(KO/WT)) versus mRNA miRNA binding site counts (Dataset: GSE 55338). Base-2 logrithm of binding site count is used for the x-axis. The regression curves for linear degree 1 polynomial (linear), non-linear degree 2 polynomial (poly2) and exponential (exp) regressions were super-imposed onto the plotted experimentally measured data points (measured). The RMSE (residual mean sqare error) and R-square values of the regressions are also shown.
Figure 1.
The average log-ratio of expression-level fold-of-change between Dicer1-knockout (KO) and wildtype (WT) mESCs (mouse embryonic stem cells) (log2(KO/WT)) versus mRNA miRNA binding site counts (Dataset: GSE 55338). Base-2 logrithm of binding site count is used for the x-axis. The regression curves for linear degree 1 polynomial (linear), non-linear degree 2 polynomial (poly2) and exponential (exp) regressions were super-imposed onto the plotted experimentally measured data points (measured). The RMSE (residual mean sqare error) and R-square values of the regressions are also shown.
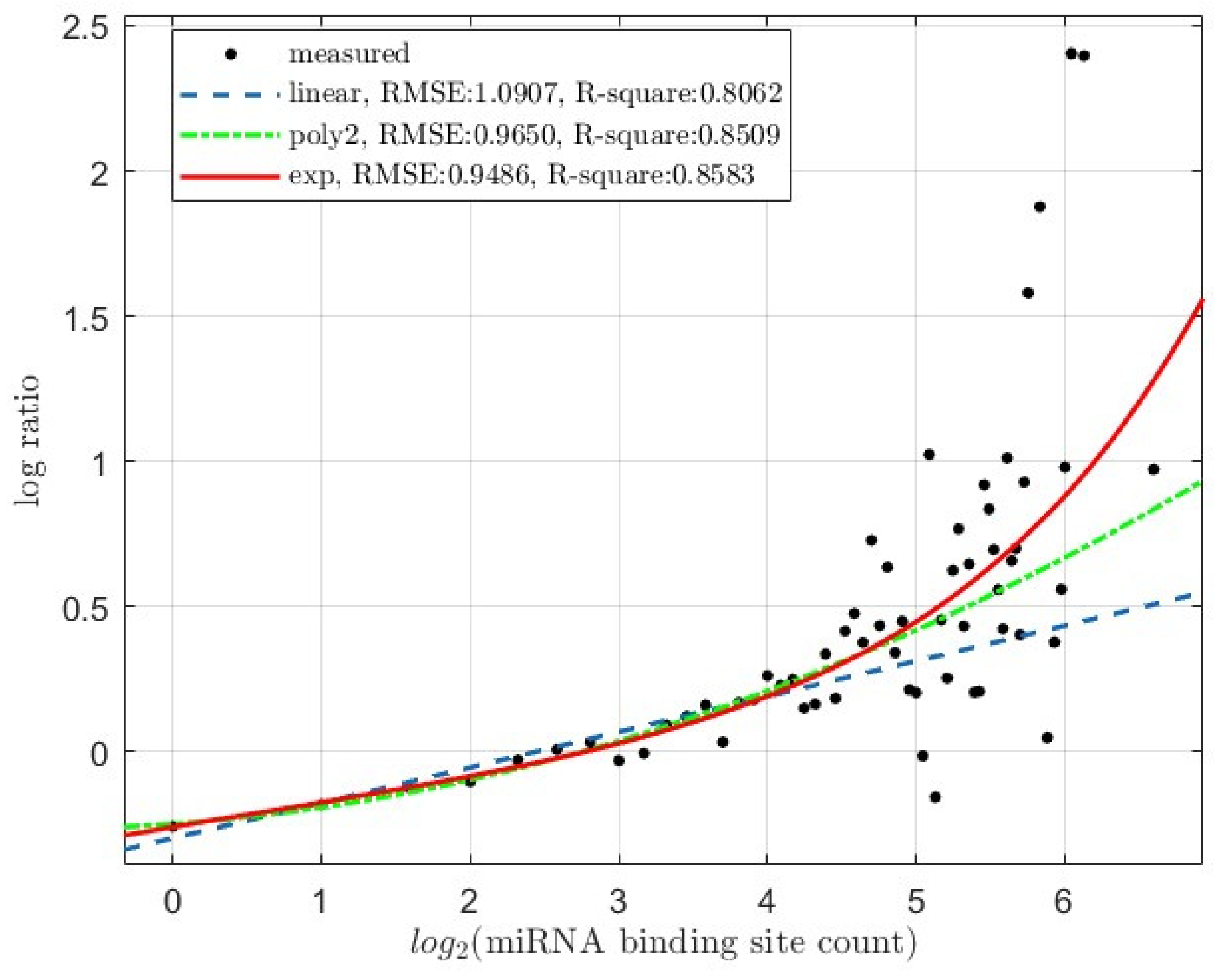
Figure 2.
The average log-ratio of expression-level fold-of-change between Drosha knockout (KO) and wildtype (WT) HCT116 cells (log2(KO/WT)) versus mRNA miRNA binding site counts (Dataset: GSE 80258). Base-2 logrithm of binding site count is used for the x-axis. The regression curves for linear degree 1 polynomial (linear), non-linear degree 2 polynomial (poly2) and exponential (exp) regressions were super-imposed onto the plotted experimentally measured data points (measured). The RMSE (residual mean sqare error) and R-square values of the regressions are also shown.
Figure 2.
The average log-ratio of expression-level fold-of-change between Drosha knockout (KO) and wildtype (WT) HCT116 cells (log2(KO/WT)) versus mRNA miRNA binding site counts (Dataset: GSE 80258). Base-2 logrithm of binding site count is used for the x-axis. The regression curves for linear degree 1 polynomial (linear), non-linear degree 2 polynomial (poly2) and exponential (exp) regressions were super-imposed onto the plotted experimentally measured data points (measured). The RMSE (residual mean sqare error) and R-square values of the regressions are also shown.
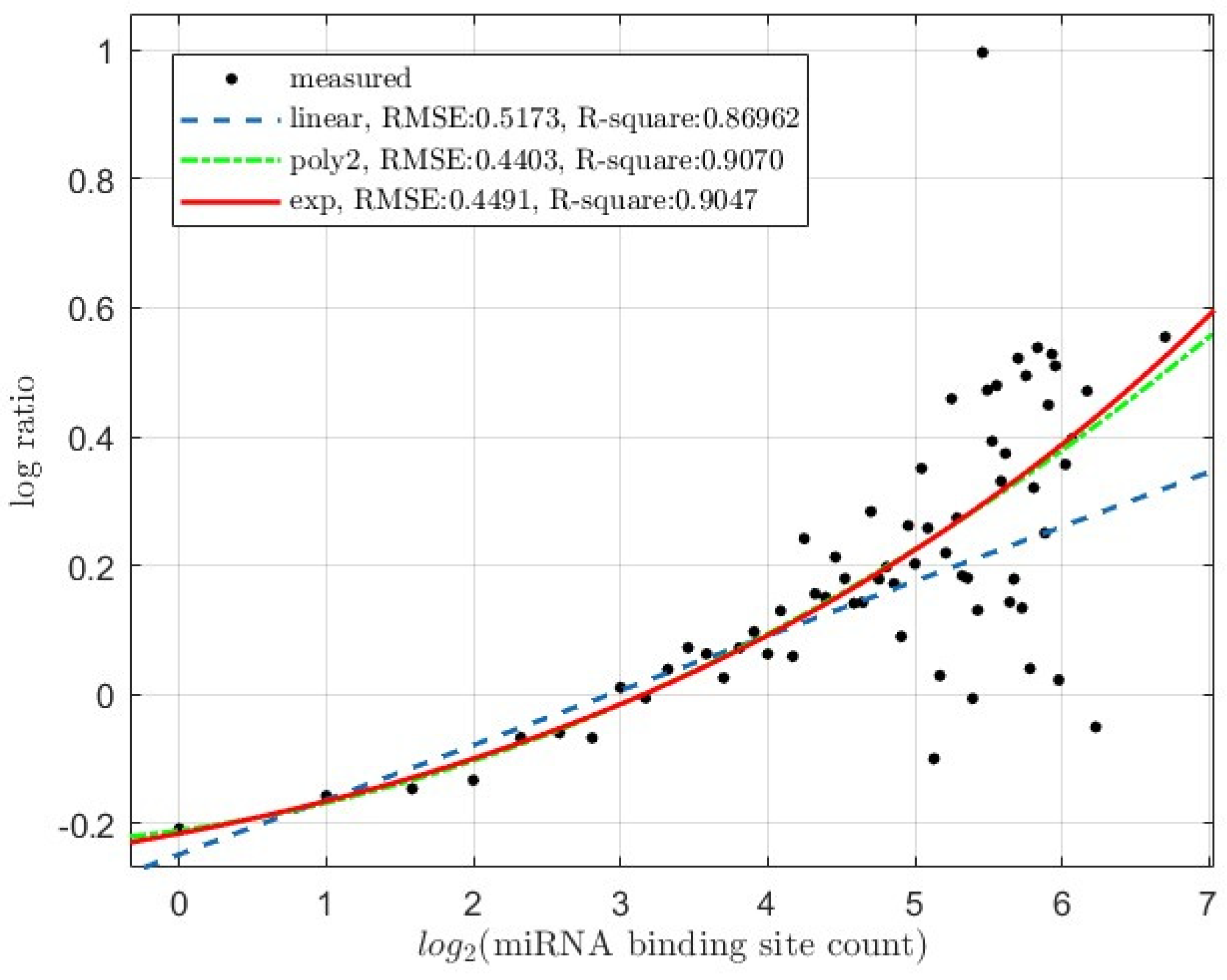
Figure 3.
Scatter-plots of the light- to heavy-polysome log-ratio of wildtype (WT) HCT116 cells (A) and the adjusted light- to heavy-polysome log-ratio (B) versus mRNA miRNA binding site counts (Dataset: GSE134818). Base-2 logrithm of binding site count is used for the x-axis. In B, the adjusted log-ratio is calculated as the difference between WT cell log-ratio and Dicer1 knockout (KO) cell log-ratio (log-ratio(WT) – log-ratio(KO)); please see text for details. The regression curves for linear degree 1 polynomial (linear), non-linear degree 2 polynomial (poly2) and exponential (exp) regressions were super-imposed onto the plotted experimentally measured data points (measured). The RMSE (residual mean sqare error) and the R-square values of the regressions are also shown.
Figure 3.
Scatter-plots of the light- to heavy-polysome log-ratio of wildtype (WT) HCT116 cells (A) and the adjusted light- to heavy-polysome log-ratio (B) versus mRNA miRNA binding site counts (Dataset: GSE134818). Base-2 logrithm of binding site count is used for the x-axis. In B, the adjusted log-ratio is calculated as the difference between WT cell log-ratio and Dicer1 knockout (KO) cell log-ratio (log-ratio(WT) – log-ratio(KO)); please see text for details. The regression curves for linear degree 1 polynomial (linear), non-linear degree 2 polynomial (poly2) and exponential (exp) regressions were super-imposed onto the plotted experimentally measured data points (measured). The RMSE (residual mean sqare error) and the R-square values of the regressions are also shown.
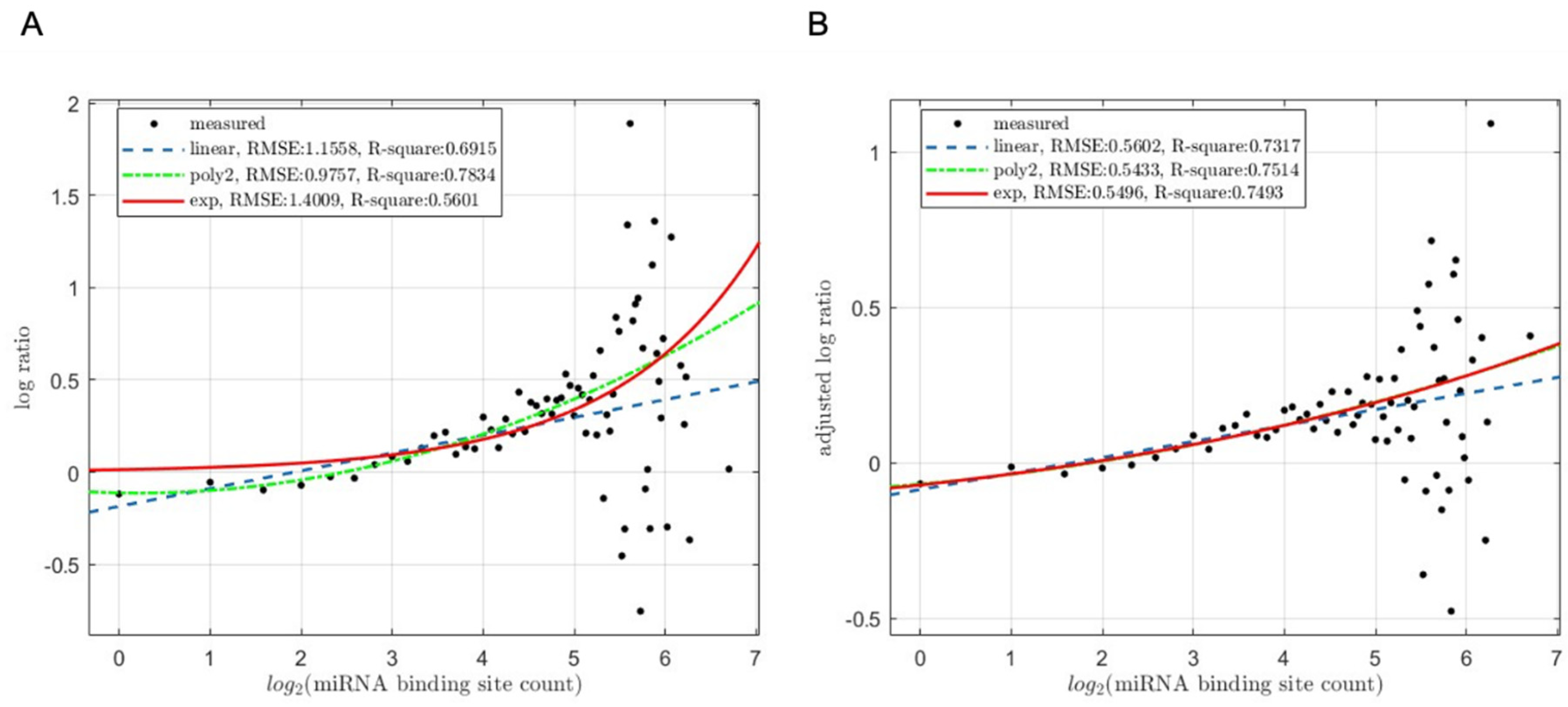

Table 1.
Summary of sequencing reads and transcriptome statistics.
| Samples | Total Reads | NCBI SRA ID | NCBI GEO ID | ||
|---|---|---|---|---|---|
| mESCs GSE55338 |
Dcr KO1 | 185,933,955 | SRR1176702 | GSM1334360 | |
| Dcr KO3 | 207,509,665 | SRR1176703 | GSM1334361 | ||
| Dcr KO4 | 186,427,767 | SRR1176704 | GSM1334362 | ||
| Dcr WT rep1 | 89,965,743 | SRR1176705 | GSM1334363 | ||
| Dcr WT rep1 | 170,831,915 | SRR1176706 | GSM1334364 | ||
| HCT116 GSE80258 |
WT | 198,937,194 | SRR3380994 | GSM2122814 | |
| DroKO | 196,007,170 | SRR3380995 | GSM2122815 | ||
| HCT116 GSE134818 |
WT-Heavy | 35,352,261 | SRR9829887 | GSM3972384 | |
| WT-Light | 35,351,828 | SRR9829888 | GSM3972385 | ||
| KO-Heavy | 35,272,114 | SRR9829889 | GSM3972386 | ||
| KO-Light | 37,403,191 | SRR9829890 | GSM3972387 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



