
Submitted:
12 June 2024
Posted:
13 June 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Vaccine antigens must present the correct conformation of viral fusion glycoproteins to elicit effective immune responses. Virus-like particles (VLPs) serve as promising vaccine platforms because they mimic the membrane-embedded conformations of fusion glycoproteins on native viruses. Here, we employed SARS-CoV-2 VLPs (SMEN) presenting ancestral, Beta, or Omicron spikes to identify the variant that elicits potent and cross-protective immune responses in the highly sensitive K18-hACE2 mouse model. A combined intranasal and intramuscular administration regimen of the SMEN vaccine generated effective immune responses and was predominantly mediated by antibodies with minor contributions from T cells. Immunization with SMEN presenting an ancestral spike resulted in 100, 75, or 0% protection against ancestral, Delta or Beta VOC-induced mortality, respectively, whereas SMEN presenting the most divergent Omicron spike provided only limited protection (50%, 0%, and 25%) against ancestral, Delta, and Beta variants, respectively. By contrast, SMEN with a Beta spike offered 100% protection against the variants used in this study. Thus, the Beta variant not only overcame the immunity produced by other variants, but also elicited diverse and effective immune response. Our findings suggest that leveraging the Beta variant spike protein can enhance SARS-CoV-2 immunity, potentially leading to a more comprehensive vaccine against emerging variants.
Keywords:
SARS-CoV-2
; vaccine
; virus-like particles
; intranasal
; intramuscular
; neutralizing antibodies
; variants of concern
; cross-VOC protection
; beta
; omicron
1. Introduction
The SARS-CoV-2 Spike protein (S) present on the surface of virions is required for engaging the receptor ACE2 to enter cells [1,2,3,4,5]. Neutralizing humoral immune responses directed towards the S protein effectively blocks virus entry and has been correlated with protection from severe disease [4,6]. Several vaccine platforms developed during the COVID-19 pandemic have incorporated various forms of S as target antigen [7,8,9]. Among these, mRNA-based vaccines emerged as the preferred platform in the USA because of their high potency, cost-effectiveness in manufacturing, and rapid large-scale deployment capabilities, which addressed a significant challenge in vaccine development, particularly during the COVID-19 pandemic [10]. Although the efficacy of most vaccines to prevent SARS-CoV-2 infection and transmission has been reduced against variants of concern (VOCs), vaccine-based immunity remains protective against severe disease and hospitalization [11]. In recent years, several studies have highlighted rare but noteworthy adverse effects associated with mRNA vaccines, including cardiovascular complications, thrombosis, thrombocytopenia, and fatigue, which have fueled vaccine hesitancy [12,13,14,15,16,17]. Other side effects, such as Bell’s palsy and transverse myelitis, have been observed in individuals receiving inactivated virus-based vaccines [18]. In addition, adenoviral vector-based vaccines were found to trigger vaccine-induced immune thrombocytopenia and thrombosis (VITT) due to cross-reactive antibodies that target platelet factor 4 [19]. Consequently, the continued development of different vaccine platforms, including traditional protein-based vaccines, is essential for providing additional choices for individuals with comorbidities and safety concerns to improve acceptance rates.
Virus-like particles are a viable vaccine platform that has been evaluated for effectiveness in human clinical trials against respiratory viruses, including influenza and respiratory syncytial virus (RSV). Notably CoV-2 VLP-based vaccines have also been assessed in several clinical trials against SARS-CoV-2, highlighting its potential [20,21]. SARS-CoV-2 virus-like particles (VLPs; SMEN) can be readily generated without any need for proprietary technology by expressing four SARS-CoV-2 structural proteins in cell culture: spike (S), membrane (M), envelope (E), and nucleocapsid (N). Unlike mRNA vaccines that require below freezing temperatures, SMEN VLPs are stable for weeks at 4 °C [22,23]. They are non-infectious as they do not contain any replication-competent genetic material. The high immunogenicity of SMEN particles makes them suitable for a variety of vaccine efficacy experiments and are valuable as research tools [24,25,26]. VLPs have been used to study the impact of mutations in structural proteins and to screen therapeutics [27]. Importantly, their modular nature allows easy adaptation to S proteins from emerging variants, facilitating studies that examine their breadth and protective efficacy. SMEN VLPs can be administered via the traditional intramuscular route to elicit peripheral immunity to control the systemic spread of the virus. In addition, SMEN VLPs can be safely administered intranasally to potentially induce effective immunity in the respiratory mucosa, which is crucial for preventing virus transmission and remains a broad goal in vaccinology [28]. Unlike mRNA or adenovirus-based vaccine platforms, SMEN particles mimic the membrane-embedded conformations of the spike protein on native viruses [29]. Furthermore, the inclusion of additional viral proteins (M, E, and N) in SMEN particles can augment immunity by generating responses to a broad range of viral proteins [10,30,31,32].
Neutralizing antibodies are essential for protection against SARS-CoV-2 infection [33]. Therefore, fundamental investigations comparing the cross-protective neutralizing responses elicited by S proteins from prominent variants of concern (VOCs) are essential for guiding future vaccine development. Additionally, it is crucial to determine whether the elicited humoral immune responses correlate with protection against homologous and heterologous VOCs in vivo, using stringent challenge models. In this study, we employed the SMEN VLP vaccine to assess the efficacy of S protein derived from ancestral, Delta, Beta, and Omicron variants in eliciting robust cross-protective immune responses in the highly sensitive K18-hACE2 mouse challenge model. Our data indicate that a combined intranasal and intramuscular administration regimen of the SMEN vaccine generates the most effective immune response for the virologic control and mitigation of lung inflammation. Protection was largely mediated by antibodies, with T cells contributing towards limiting virus dissemination and inflammation. Notably, the Beta variant not only overcame the immunity produced by other variants, but SMEN bearing S from the Beta VOC also elicited the most diverse and effective humoral immune response. Our results indicate that harnessing the S protein from the Beta variant may contribute to a more comprehensive vaccine targeting emerging variants.
2. Materials and Methods
2.1. Ethics Statement
Animal experiments conducted at Yale University were approved by the Yale Institutional Biosafety Committee (IBC) and Institutional Animal Care and Use Committee (IACUC). Protocols for imaging SARS-CoV-2 infected animals using IVIS under Animal Biosafety Level 3 (ABSL-3) conditions were revised and approved by the IACUC, IBSCYU, and Yale Animal Resources Center (YARC). To alleviate pain and discomfort in the animals, procedures involving virus inoculation, blood drawing, or animal imaging were performed under anesthesia.
2.2. Cell and Viruses
Vero E6 (CRL-1586) human embryonic kidney (HEK-239) was obtained from the American Type Culture Collection (ATCC) and cultured at 37°C in RPMI supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES (pH 7.3), 1 mM sodium pyruvate, 1× non-essential amino acids, and 100 U/ml of penicillin–streptomycin. SARS-CoV-2 (WA1)-expressing NanoLuc luciferase (nLuc) was obtained from Craig Wilen (Yale University). B.1.617.2 (Delta), B.1.351 (Beta), and Omicron (BA.1) VOC were isolated from a patient at the Yale New Haven Hospital and was obtained from Craig Wilen (Yale University). All viruses were authenticated using next generation sequencing (Yale Center for Genomic Analysis). We generated nanoluc luciferase (nLuc)-expressing reporter viruses for Delta, Beta, and Omicron VOC for non-invasive BLI imaging of infected mice using the circular polymerization extension reaction (CPER), as described previously [34,35,36]. Briefly, viral RNA was converted into cDNA using a PrimeScript RT kit (Takara Bio) with a mix (1:4) of random and oligo dT primers. The cDNA was then used as a template to amplify 11 overlapping fragments that were ~2-3 kb long using specific primers and PrimeStar GXL polymerase (Takara Bio). An nLuc-P2A cassette was inserted into the N-terminus of nucleocapsid gene (nLuc-P2A-Nucleocapsid). The gel-purified overlapping fragments were circularized with CPER using a Hepatitis Delta virus ribozyme-spacer-CMV promoter cassette that overlapped the 3′ and 5′ ends of the genome. CPERs were transfected into T-25 flasks of HEK293 cells using polyethyleneimine and cultured under BSL3 conditions. HEK293 cells were resuspended and co-cultured with Vero E6 ACE2/TMPRSS2 (Vero AT) cells for 5-9 days until CPE was clearly visible and nLuc activity was detected in the culture supernatants. The viruses were propagated in Vero AT cells by infecting them in T150 cm2 flasks at a multiplicity of infection (MOI) of 0.1. Culture supernatants were collected after 18-24 h when cytopathic effects were clearly visible. The cell debris was removed by sedimentation and filtered through a 0.45-micron filter to generate virus stocks. Viruses were concentrated by adding one volume of cold (4 °C) 4x PEG-it Virus Precipitation Solution (40 % (w/v) PEG-8000 and 1.2 M NaCl; System Biosciences) to three volumes of virus-containing supernatant. The solution was mixed by inverting the tubes several times and incubating overnight at 4 °C. The precipitated virus was harvested by centrifugation at 1,500 × g for 60 min at 4 °C. The concentrated virus was then resuspended in PBS and aliquoted for storage at −80°C. All work with infectious SARS-CoV-2 was performed in the Institutional Biosafety Committee approved BSL3 and A-BSL3 facilities at the Yale University School of Medicine.
2.3. Mouse Experiments
All animals were maintained in the (SPF-free) barrier facility of the Yale University Animal Resource Centre (YARC) under a 14:10 light: dark cycle. The breeding populations of mice and infected animals were maintained in separate rooms. All SARS-CoV-2-infected animals were housed in an animal room under BSL3 containment. Cages, animal waste, bedding, and animal carcasses were disposed and decontaminated according to the guidelines of the Environmental Health Services at Yale. All replication-competent virus-infected animals were handled under ABSL3 conditions with personnel donning pressurized air purified respirators (PAPR), double gloves, shoe covers, sleeve covers, and disposable gowns. hACE2 transgenic B6 mice (heterozygous) were obtained from the Jackson Laboratory. 6–8-week-old male and female mice were used in all the experiments. Heterozygous mice were crossed and genotyped to select heterozygous mice for experiments using the primer sets recommended by the Jackson Laboratory. Each cohort size was 4–8 to allow statistical testing and was conducted with two to three biological replicates to allow parallel evaluation of different cohorts. The number of animals (n = 4–8 per cohort) required to achieve statistically significant results was calculated based on a priori power analysis. We calculated the required power and sample sizes based on data from pilot experiments and previous studies [37,38,39,40]. Animals with sex- and age-matched littermates were randomly selected for experiments. No animals were excluded because of illness after the experiments. At the time of experimentation, care was taken to include equal numbers of male and female mice whenever possible, to ensure that the sex of the animals did not constitute a biological variable during the analysis.
2.4. Generation of SMEN VLPs
SMEN VLPs were prepared by transfecting HEK293 cells at 60% confluence with individual plasmids expressing SARS-CoV-2 spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins at a ratio of 1:1:5:5 using polyethyleneimine (PEI) as a transfection reagent. Transfected cells were washed 6 h post-transfection and replenished with RPMI supplemented with 10% FBS. The culture supernatant was collected 48 h post-infection, cleared through a 0.45 µm filter, and partially purified by ultracentrifugation after being overlaid with 15% sucrose, followed by storage at -80 °C in aliquots. The SMEN VLPs protein was estimated using PierceTM BCA protein assay kit (ThermoFisher, Cat# 23225) and further characterized by western blot and Cryo-ET analyses (see below)
2.5. Western Blot
To test the incorporation of individual proteins, partially purified SMEN VLPs presenting different spike proteins were solubilized in 1X NuPAGE sample buffer, denatured, and subjected to SDS-PAGE analyses using NuPAGE 4-12% Bis-Tris gels. After transfer, the membranes were probed with antibodies to the SARS-CoV-2 spike, nucleocapsid, membrane, and envelope proteins (Cell Signaling Technology, 1:2000). To test the ability of sera from SMEN-vaccinated mice to recognize S, M, E, and N proteins, SMEN VLPs with different spikes were processed as described above and probed with mouse sera at a 1:2000 dilution. The membranes were probed with appropriate secondary antibodies (1:10,000) conjugated to horseradish peroxidase (HRP), developed using SuperSignal™ West Pico PLUS Chemiluminescent Substrate, and images were acquired using Amersham ImageQuant™ 800 Western blot imaging systems.
2.6. SMEN VLPs Vaccination Regimen and SARS-CoV-2 Challenge
For all in vivo experiments, 6 to 8 weeks male and female mice were used. K18-hACE2 transgenic mice (heterozygous) were administered intranasally (i.n; mucosal vaccination) or intramuscularly (i.m; systemic vaccination) with mix of SMEN VLPs (250 µg) and vaccigrade TLR7/TLR8 agonist R848 (30 ug, vaccigrade was used only i.m.) (InvivoGen US, San Diego, CA, USA) as an adjuvant at each site. Uninfected mice or mice administered R848 alone were used as controls. Intranasal administration was performed under anesthesia at a volume of 25-30 µl. The mice were boosted once with the same amount of VLPs, i. n. and i. m., 14 days after the first challenge. Blood was collected on day 21 to determine antibody titers. The mice were intranasally challenged with 1 × 105 PFU SARS-CoV-2-nLuc viruses in 25-30 µL volume under anesthesia (0.5%–5% isoflurane delivered using a precision Dräger vaporizer with an oxygen flow rate of 1 L/min). The starting body weight was set at 100%. For survival experiments, mice were monitored every 8-12 h starting six days after the virus challenge. Lethargic and moribund mice or mice that had lost more than 20% of their body weight were sacrificed and considered to have succumbed to infection for Kaplan-Meier survival plots. Mice were considered to have recovered if they gained all the lost weight.
2.7. Passive Transfer of Sera
C57BL/6 mice were vaccinated with SMENWA1, as described above. 21 days after the initial vaccination, the mice were anesthetized using isoflurane inhalation (3-5% isoflurane, oxygen flow rate of 1.5 L/min), and blood was collected from either the eye or submandibular vein. Serum was obtained from whole blood by sedimenting at 2,000 x g for 15 min at room temperature. Sera from mice before vaccination (pre-immune sera) or after vaccination (immune sera) was combined, and 500 µl of this serum was administered intraperitoneally to naïve K18-hACE2 mice (B6 background) one day prior to SARS-CoV-2 infection.
2.8. Immunodepletion of CD4+ and CD8+ T Cells
To investigate the contribution of T cells to vaccine-mediated protection in mice, CD4+ T and CD8+ T cells were depleted using mouse anti-CD4 (BioX-Cell; clone YTS 191; 12.5 mg/kg body weight) and anti-CD8α (BioX-Cell; clone 53.6.7; 12.5 mg/kg body weight) monoclonal antibodies (mAbs). The mAbs were administered to mice via intraperitoneal (i.p.) injection every two days, starting from -2 days until the experimental endpoint. Rat IgG2b (BioX-Cell; 12.5 mg/kg body weight) was used as a control. Blood samples were collected every 48 h via retroorbital bleeding to isolate peripheral blood mononuclear cells (PBMCs) using the Ficoll method. The cells were then stained with antibodies to mouse CD45 (Alexa Fluor™ 488, clone 30-F11), CD3 (Alexa Fluor™ 647, clone 145-2C11), CD4 (PE-Cy7, clone GK1.5), and CD8 (PE, clone 53-6.7), and samples were acquired using a BD Accuri C6 flow cytometer and C6 sampler software.
2.9. Bioluminescence Imaging (BLI) of SARS-CoV-2 Infection
All procedures and protocols for the In Vivo Imaging System (IVIS) imaging of SARS-CoV-2-infected animals under ABSL-3 conditions were approved by the Institutional Animal Care and Use Committee (IACUC), Institutional Biosafety Committee (IBSCYU), and Yale Animal Research Committee (YARC). Imaging was performed using the IVIS Spectrum® (PerkinElmer) and XIC-3 animal isolation chamber (PerkinElmer), which provided biological isolation of anesthetized mice or individual organs during the imaging procedure. All mice were anesthetized by isoflurane inhalation (3-5% isoflurane, oxygen flow rate of 1.5 L/min) prior to and during bioluminescent imaging (BLI) using the XGI-8 Gas Anesthesia System. Prior to imaging, 100 µL of nanoluc luciferase (nLuc) substrate, furimazine (NanoGloTM, Promega, Madison, WI) diluted 1:40 in endotoxin-free PBS was retro-orbitally administered to mice under anesthesia. The mice were then placed into the XIC-3 animal isolation chamber (PerkinElmer), which was pre-saturated with isoflurane and oxygen mix. The mice were imaged in both the dorsal and ventral positions on the indicated days post-infection. The animals were then imaged again after euthanasia and necropsy by spreading an additional 200 µL of substrate onto the exposed intact organs. Additional droplets of furimazine in PBS (1:40) were added to the organs and soaked for 1-2 minutes before BLI. Images were acquired and analyzed using the Living Image v4.7.3 in vivo software package (Perkin Elmer Inc.). Image acquisition exposures were set to auto, with the imaging parameter preferences set in the following order: exposure time, binning, and f/stop. Images were acquired using a luminescent f/stop of 2 and a photographic f/stop of 8. Binning was set to medium. For comparative analyses, images were compiled and batch-processed using the image browser with collective luminescent scales. Photon flux was measured as luminescent radiance (photons/sec/cm2/sr). During luminescent threshold selection for image display, luminescent signals were considered background when the minimum threshold setting resulted in displayed radiance above non-tissue-containing or known uninfected regions.
2.10. SARS-CoV-2 Neutralization Assay and Calculation of Cross-Reactive Neutralization Index
Serum samples from immunized mice were subjected to heat inactivation at 56°C for 30 minutes and then diluted in 4-fold serial dilutions in serum-free RPMI, starting at a dilution of 1:10. Each serum dilution (50 µl) and 200 PFU of virus (50 µl; WA1, Delta, Beta or Omicron viruses expressing nLuc reporter) were combined and incubated at 37°C for 1 hour. Subsequently, a 96-well tissue culture plate with sub-confluent VeroE6 cells was infected for 4 hours. The cells were washed three times with serum-free RPMI, followed by the addition of fresh RPMI media supplemented with 10% FBS. After 24 hours post-infection, the cells were washed twice more with 1x PBS and lysed in 100 µl of 1x passive lysis buffer. 20 µl of the lysates were transferred to a 96-well solid white plate (Costar Inc) and nLuc activity was measured using the Tristar multiwell Luminometer (Berthold Technology, Bad Wildbad, Germany) for 2.5 seconds by adding 20 µl of Nano-Glo® substrate (diluted in 1x PBS at a 1:40 dilution) in nanoluc assay buffer (Promega Inc, WI, USA). Uninfected monolayer of Vero cells treated identically served as controls to determine basal luciferase activity and obtain normalized relative light units. Cells infected with virus that were treated with sera from non-vaccinated mice were set to 100%. The data were processed and plotted using GraphPad Prism 8 v8.4.3 to calculate IC50 (50% inhibitory concentration) values. Cross-reactive neutralization index (CRNI) was calculated based on the IC50 values using the following formula:
CRNI= (IC50 of sera for heterologous strain/average IC50 for homologous or reference strain) ×100.
A CRNI value approaching 100% signifies a considerable level of cross-reactivity, implying that the antibodies are equally effective against both the strains. Conversely, a value significantly below 100% suggests diminished cross-reactivity, indicating that the antibodies exhibit reduced efficacy against the variant strain compared to the reference strain.
2.11. Fc-Signaling Assay and Calculation of Cross-Reactive Fc-Signaling Index
Mouse FcgRIV (Genbank accession number: NM_144559.2) was synthesized and cloned into a lentivirus packaging construct (pGenlenti) using Genscript service. Jurkat N-FAT Luciferase (JNL) cells (Signosis) were transduced with VSV-G decorated lentivirus and grown under puromycin selection to generate the JNL-mFcgRIV for the FcgR signaling assay. VeroE6 cells (1x 104) were seeded in a 96-well tissue culture plate and infected with WA1, Delta, Beta, and Omicron SARS-CoV-2 variants for 4 h. The levels of infection among all SARS-CoV-2 strains used was normalized based on nLuc signals in infected VeroE6 cells as measured 24 h post-infection (equivalent to ~150-200 PFU). The cells were then washed three times with serum-free RPMI, followed by the addition of 150µl of fresh RPMI media supplemented with 10% FBS. An amount equivalent to IC50 of sera from each immunized mice was mixed with 2x 104 JNL-mFcgRIV cells in a final volume of 50µl and added to mock or virus-infected VeroE6 cells and incubated for 4 h. The cells were lysed with 50µl of Britelite substrate (Perkin Elmer) containing lysis buffer, and luciferase activity was measured after 5 minutes. Mock-infected VeroE6 cells incubated with JNL-mFcgRIV cells and sera were used as a control to normalize the Fc-signaling activity. Cross-reactive Fc-signaling index (CRFSI) was calculated based on the Fc activity measured using IC50 (neutralizing) equivalent sera for each variant used in our study using the following formula:
CRFSI= (Fc signaling activity in IC50 equivalent sera for heterologous strain/ average Fc signaling activity with IC50 equivalent sera for homologous or reference strain) ×100.
2.12. Plaque Forming Assay
Virus stock titers were assessed through a standard plaque assay. To begin, 4 x 10^5 Vero-E6 cells were seeded into a 12-well plate and allowed to incubate for 24 hours. The cells were then infected with serially diluted virus stock, followed by the addition of 1ml of pre-warmed 0.6% Avicel (RC-581 FMC BioPolymer) in complete RPMI medium. After 48 hours, plaques were visualized by fixing the cells in 10% paraformaldehyde for 15 minutes and staining them with 0.2% crystal violet in 20% ethanol for 1 hour. The plates were then rinsed with water to clearly observe the plaques.
2.13. Measurement of Viral Burden
Organs from infected or uninfected mice, including the brain and lungs, were collected, weighed, and homogenized in 1 mL of serum-free RPMI medium containing penicillin-streptomycin. The homogenization was performed using a 2 mL tube containing 1.5 mm Zirconium beads with the BeadBug 6 homogenizer (Benchmark Scientific, TEquipment Inc.). The viral titers were determined using two highly correlated methods. In the first method, total RNA was extracted from the homogenized tissues using the RNeasy Plus Mini kit (Qiagen Cat # 74136), reverse transcribed with the iScript advanced cDNA kit (Bio-Rad Cat #1725036), and then a SYBR Green Real-time PCR assay was performed to determine the copies of SARS-CoV-2 N gene RNA using primers SARS-CoV-2 N F: 5′-ATGCTGCAATCGTGCTACAA-3′ and SARS-CoV-2 N R: 5′-GACTGCCGCCTCTGCTC-3′. All real-time PCR assays based on SYBR Green have built-in melt-curve analyses to ensure the estimation of only specific PCR products and not false positives. In the second method, nLuc activity was used as a surrogate for the plaque assay. Serially diluted clarified tissue homogenates were used to infect Vero-E6 cell culture monolayers. Infected cells were washed with PBS, lysed using 1X Passive lysis buffer, and nLuc activity was measured as described above. The data were processed and plotted using GraphPad Prism 8 v8.4.3.
2.14. mRNA Expression Analyses of Signature Inflammatory Cytokines and Lung Injury/Repair Genes
Brain and lung tissue samples were obtained from mice during necropsy. Approximately 20 mg of tissue was suspended in 500 µL of RLT lysis buffer, and RNA was extracted using the RNeasy plus Mini kit (Qiagen Cat # 74136). The RNA was reverse transcribed into cDNA using the iScript advanced cDNA kit (Bio-Rad Cat #1725036). To determine the mRNA copy numbers of key inflammatory cytokines or lung pathology markers, multiplex qPCR was performed using the iQ Multiplex Powermix (Bio Rad Cat # 1725848) and PrimePCR Probe Assay mouse primers FAM-GAPDH, HEX-IL6, TEX615-CCL2, Cy5-CXCL10, Cy5.5-IFNgamma, HEX-IL1B, Cy5-Krt8, Cy5.5-Krt5, HEX-Adamts4 and TEX615-Itga5. The reaction plate was analyzed using the CFX96 touch real-time PCR detection system, and the scan mode was set to all channels. The PCR conditions included an initial denaturation step at 95 °C for 2 minutes, followed by 40 cycles of 95 °C for 10 seconds and 60 °C for 45 seconds. A melting curve analysis was performed to ensure that each primer pair resulted in the amplification of a single PCR product. The mRNA copy numbers of Il6, Ccl2, Cxcl10, Ifnɣ, and Il1b in the cDNA samples of infected mice were normalized to Gapdh mRNA using the formula ΔCt(target gene)=Ct(target gene)-Ct(Gapdh). The fold increase was determined using the 2-ΔΔCt method, comparing treated mice to uninfected controls.
2.15. Disease Burden and Bliss Index Scores
Computation of disease burden (Figure S1G) was carried out as reported previously [34]. We included a total of nine parameters. They were viral loads [N mRNA expression, titers (nLuc activity)] and inflammation [Ccl2 and Cxcl10 mRNA expression] in both the brain and lung tissues, lung pathology (Krt8 and Adamts4 mRNA expression), mortality, and extent of delay in death. For each parameter, data were normalized by setting the values obtained for animals in the control group (untreated or adjuvant treated) to 100. Total disease burden was calculated by summing the normalized values of each parameter (viral load, inflammation, lung injury, mortality). The total value was divided by 9 for nine parameters analyzed to normalize the data.
To determine if combinatorial vaccine regimen provided benefit over intranasal or intramuscular vaccination regimen, we utilized Bliss index scores (Figure S1G) based on overall disease burden under each condition. Bliss index scores were calculated as done previously [34,41] using the formula below: A Bliss index score of less than -10 was considered antagonistic, an index between -10 and +10 was considered an additive, and an index more than 10 was considered synergistic.
2.16. Cryo-Electron Tomography of SARS-CoV-2 VLPs
6 nm gold tracer was added to the concentrated SARS-CoV-2 VLPs (SMEN particles) at 1:3 ratio, and 5 µl of the mixture was placed onto freshly glow discharged holey carbon grids for 1 min. Grids were blotted with filter paper, and rapidly frozen in liquid ethane using a homemade gravity-driven plunger apparatus. Cryo-grids were imaged on a cryo-transmission electron microscope (Titan Krios, Thermo Fisher Scientific) that was operated at 300 kV, using a Gatan K3 direct electron detector in counting mode with a 20eV energy slit and Volta Phase Plate (VPP). Tomographic tilt series between −51° and +51° were collected by using SerialEM [42] in a dose-symmetric scheme [43] with increments of 3°. The nominal magnification was 64,000 X, giving a pixel size of 1.346 Å on the specimen. The raw images were collected from single-axis tilt series with accumulative dose of ~120 e− per Å2. The defocus was –0.5 μm and 8 frames were saved for each tilt angle. Frames were motion-corrected using Motioncorr2 [44] to generate drift-corrected stack files, which were aligned using gold fiducial makers by IMOD/etomo [45]. Tomograms were reconstructed by weighted back projection and tomographic slices were visualized with IMOD.
2.17. Quantification and Statistical Analysis
Data were analyzed and graphed using the software program GraphPad Prism (La Jolla, CA, USA). Pairwise comparisons were assessed for statistical significance using the non-parametric Mann-Whitney test, with a two-tailed approach. To determine the statistical significance of the survival curves, the log-rank (Mantel-Cox) test was employed. For analyzing grouped data, a two-way ANOVA was conducted, followed by Tukey’s multiple comparison tests. Statistical significance was established at P < 0.05. P values are indicated as: ∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001; ∗∗∗∗, P < 0.0001.
3. Results
3.1. Systemic and Mucosal Vaccination of the SARS-CoV-2 VLPs (SMEN) Vaccine Provides Enhanced Protection in K18-hACE2 Mice against a Lethal Virus Challenge
We sought to investigate the efficacy of SMEN vaccine in providing protection against lethal challenge in the highly susceptible K18-hACE2 mouse model of infection. Towards this end, first evaluated systemic [intramuscular (i.m.; adjuvanted)], mucosal [intranasal (i.n.; unadjuvanted)] or combined (i.m. + i.n.) vaccination regimen to determine if mucosal immunity contributes to protection (Figure 1A). We used the TLR7/8 agonist R848 (Resiquimod; vaccigrade) as an adjuvant for intramuscular administrations. For intranasal route, SMEN particles were administered without any adjuvant to avoid adjuvant-triggered inflammatory responses in the lung. We produced ancestral SARS-CoV-2 VLPs (SMENWA1) by co-transfecting individual plasmids encoding the SARS-CoV-2 spike (SWA1), membrane (M), envelope (E), and nucleocapsid (N) proteins followed by purification via high-speed sedimentation through a sucrose cushion (Figure S1A). Analyses of SMEN particles by western blot confirmed the packaging of the four viral proteins while cryo-electron tomography (cryoET) illustrated the presence of prefusion S protein on the particles (Figure S1B,C). We followed a prime-boost strategy of vaccination an analyzed neutralizing antibody (nAb) titers in the sera and bronchioalveolar lavage fluid (BALF) (Figure 1A). Our analyses revealed distinct patterns of nAb titers. While intramuscular vaccination elicited strong nAb titers in sera but significantly reduced levels in BALF, intranasal vaccination induced robust nAb titers in BALF but diminished titers in the sera. Notably, the combinatorial administration resulted in robust nAb titers in both the sera and BALF of mice (Figure 1B, C and Supplementary Figure S1D,E).
To assess the potential implications of divergent nAbs titers in the two compartments on protective immunity, both control [mock, no treatment and adjuvant alone] and vaccinated groups of mice were challenged intranasally with a lethal dose of SARS-CoV-2WA1 (Figure 1A). Our analyses revealed that all the mice in the control group exhibited severe morbidity and demonstrated up to 20% loss from their starting body weight by day 6 and succumbed to SARS-CoV-2-induced mortality. Notably, the course of infection in mice treated with adjuvant alone was unaltered and similar to untreated mice eliminating the concerns of innate immune memory from our analyses [46,47]. In comparison, cohorts of mice immunized either via systemic or mucosal routes experienced only a transient body weight loss (~5-10%) between days 6 and 10 post-challenge and eventually recovered from infection and survived. Mice in the combined administration group did not experience any weight loss, indicating near-complete protection from infection (Figure 1D,E). These data were corroborated by our assessment of viral loads (Titers and N mRNA expression) in the brain and lung tissues which revealed significantly low or undetectable viral RNA in either tissue for all mice in the SMEN-immunized groups (Figure 1F and Figure S1F). To assess lung pathology, we utilized specific markers for various parameters that were identified in recent transcriptomics studies as substitutes [34,48,49,50,51,52,53]. Specifically, we used mRNA expression of Krt8 (indicative of injury/repair), Krt5 (related to dysfunctional repair), Adamts4 (associated with fibrosis), and Itga5 (indicative of damage-responsive fibroblasts). Our data revealed that mice immunized with SMEN, irrespective of the administration route, exhibited significantly lower mRNA expression of all the analyzed markers compared to those in the control group at the time of necropsy (Figure 1G). Of the vaccinated groups, only mice immunized intramuscularly, exhibited 5-20-fold higher mRNA expression levels of Krt8 and Adamts4 compared to uninfected mice, suggesting relatively lower protection in the lung conferred by systemic vaccination compared to the mucosal route. Moreover, mRNA expression of inflammatory cytokines in the brain were significantly reduced in all three groups of vaccinated mice compared to those in the control group suggesting virologic control in the brain (Figure 1H,I). Although the overall mRNA expression of inflammatory cytokines in the lung was significantly reduced compared to the control groups, we observed a graded reduction in the vaccinated groups, with the combined regimen demonstrating the most substantial decrease (similar to uninfected mice), followed by the mucosal route (9-fold reduction) and then the systemic route (2-fold reduction) (Figure 1H,I). We computed total disease burden for mice under each regimen using eight parameters (titers in brain and lung; injury markers (Krt8, Adamts4), cytokine expression (Ccl2, Cxcl10) in lung and brain) as previously described [34]. Our evaluation revealed a significant overall reduction in total disease burden between the control and vaccinated groups with the combined regimen displaying the highest reduction among all the groups. Additionally, we calculated Bliss index scores to determine whether the combined regimen offered any advantage over the individual regimens alone. Our analysis revealed a Bliss index score of -0.4 (Bliss index score range: -10 to 10 denotes additivity), suggesting that the combined regimen exhibited an additive efficacy in vivo (Figure S1G).
Thus, the data underscored the effectiveness of mucosal versus systemic vaccine regimens in protecting lung from SARS-CoV-2-induced pathology. Our data also demonstrated the superior overall efficacy of the combined regimen in reducing SARS-CoV-2-induced disease burden. Consequently, we adopted the combined vaccination regimen for the remainder of our study.
3.2. Antibodies Play a Major Role in SMEN Vaccine-Mediated Protection
The combined regimen of SMEN vaccine delivery elicited potent nAbs in both BALF and sera (Figure 1B,C). To ascertain if antibodies contributed to vaccine-mediated protection, we investigated whether passive transfer of sera from vaccinated mice can confer protection in naïve mice against SARS-CoV-2-induced mortality. To gain deeper insights into the impact of sera on virus replication and dissemination, we used our bioluminescence imaging (BLI)-guided platform, which enables real-time longitudinal monitoring of virus replication using nanoLuc luciferase (nLuc)-expressing reporter SARS-CoV-2WA1-nLuc [34,37,38]. Pre-immune sera or sera from vaccinated mice (immune sera) were collected and adoptively transferred (i.p.) to naïve K18-hACE2 mice one day before lethal challenge with SARS-CoV-2WA1-nLuc (Figure 2A). BLI and nLuc flux quantification showed that the pre-immune sera-treated groups experienced uncontrolled virus replication in the lungs, which spread to the brain, leading to more than 20% body weight loss and 100% mortality. (Figure 2B–F). In contrast, 75% of mice treated with immune sera exhibited reduced virus replication from 2-6 dpi in the lung followed by its clearance at 8 dpi (Figure 2B,C and Figure S2). Although one mouse in the group receiving immune sera exhibited a below-detection nLuc signal in the lung at 8 dpi, it lost body weight after a brief recovery period from 8-10 dpi. BLI revealed that the virus had spread to the brain, resulting in its death (Figure 2B–F and Figure S2). Viral load and inflammatory cytokine mRNA analyses in the target tissues corroborated our BLI analysis and revealed significant control of virus replication and inflammation in the mice receiving immune sera (Figure 2G–I and Figure S2C). Therefore, our analyses indicate that the humoral immune response elicited by the SMEN vaccine plays a major role in curbing virus replication and establishing immune control.
3.3. CD8+ and CD4+ T Cells Elicited by SMEN Vaccine Play a Minor but Distinct Role in Immunity by Contributing to Diminishing Virus Spread and Reducing Inflammation
We next evaluated the involvement of T cells in contributing to SMEN vaccine-mediated immunity. Towards this end, we immunodepleted subtypes of T cells by treating vaccinated mice with anti-CD4 or anti-CD8+ T cell-depleting antibodies. Mice treated with isotype antibodies served as control. After confirming specific depletion of CD4+ or CD8+ T cells in the blood by flow cytometry (Figure S3A,B), we challenged the groups of mice with lethal dose of SARS-CoV-2WA1-nLuc. Longitudinal imaging and quantification of nLuc signals revealed expansion of virus infection in the lung after its initial appearance at 2 dpi and dissemination to the brain by 4-6 dpi in the adjuvant-treated mice (Figure 3B–D). In comparison, the isotype Ab-treated vaccinated mice demonstrated significantly lower nLuc signals in the lung that was quickly controlled without virus dissemination to the brain. Non-invasive imaging indicated that virologic control remained comparable after T cell depletion, implying a minimal role of T cells in SMEN-mediated protection. However, depletion of CD8+ T cells resulted in detectable and increased nLuc signals in the lungs of the vaccinated groups, suggesting a greater contribution of CD8+ T cells compared to CD4+ T cells in protection (Figure 3B–D). Body weight analyses and survival data confirmed the minimal role of T cells, as all mice in the vaccinated and T cell depleted group survived lethal challenge (Figure 3E,F). Imaging of isolated organs post-necropsy, which allows for improved sensitivity, revealed sustained virologic control in the lungs and was confirmed by quantification of N mRNA expression, despite T cell depletion (Figure S3C,E). However, significantly lower yet detectable nLuc signals were observed in the brain of T cell-depleted mice, in contrast to mice in the isotype-treated groups that demonstrated complete virologic control (Figure S3C). This was also evident in the detectable viral loads and expression of viral N mRNA in the brain following T cell depletion (Figure 3G and Figure S3E). In addition, mRNA expression of inflammatory cytokines Ccl2 and Cxcl10 remained significantly higher than isotype treated vaccine mice in both lung and brain (Figure 3H,I). Thus, considering the less impact on SARS-CoV-2-induced mortality, our data suggest that SMEN vaccine-elicited T cells play a minor yet distinct role in immunity by helping to reduce the spread of virus to the brain and mitigating inflammatory responses in infected tissues.
3.4. Cross-Protective Efficacy of SMENWA1 Vaccine is reduced Against Delta and ineffective against Beta VOC
The development of hybrid immunity through infection and/or vaccination complicates the assessment of cross-protective efficacy solely attributable to vaccine- or variant-specific spike protein-generated immune responses. Lethal mouse models of SARS-CoV-2 infection like K18-hACE2 mice fill this gap and allow in vivo evaluation for cross-protective immunity in a naïve background. For evaluating cross-protective efficacies, we generated SMEN-VLPs carrying spike protein from Delta, Beta and Omicron VOCs in addition to the above described SMENWA1 and validated them using western blot analyses (Figure S4A). As a first step, we evaluated the cross-protective efficacy of SMENWA1 vaccine against Beta and Delta VOC that carried distinctive nAb-resistant mutations in S and arose during the pandemic in separate geographic areas [54]. We challenged adjuvant-treated or SMENWA1-vaccinated K18-hACE2 mice intranasally with lethal dose of Beta or Delta VOC (Figure 4A). Evaluation of physical parameters revealed that adjuvant-treated mice exhibited steady body weight loss and succumbed to infection upon Delta or Beta VOC challenge (Figure 4B). However, 75% of vaccinated mice recovered and survived from lethal Delta VOC challenge despite the 10-15% initial loss in body weight from 2 to 8 dpi. Accordingly, vaccination with SMENWA1 protected 75% of the mice against Delta VOC-induced mortality (Figure 4C,D). The reduction of viral loads (N mRNA expression and titers in the lung and brain of surviving Delta VOC challenged mice was significantly low and near the limit of detection indicating virus clearance (Figure 4E and Figure S4B). Moreover, inflammatory cytokine expression (Ccl2, Cxcl10) in the lung and brain were also significantly reduced (Figure 4F,G). In contrast, Beta VOC was highly resistant to SMENWA1-elicited immunity, causing a steady loss in body weight and resulted in 100% mortality among the vaccinated mice (Figure 4B–D). Only one mouse experienced a 2-day delay in death. In addition, the vaccinated mice showed 4 logs higher Beta VOC viral loads in lung and the brain than uninfected controls. This was despite significantly lower viral loads (1-2 logs) than adjuvant-treated groups indicating impaired virologic control (Figure 4E and Figure S4B). In addition, inflammatory cytokine mRNA expression were also 1-3 logs higher than uninfected mice despite significant reduction compared to adjuvant treated mice (Figure 4F,G). As humoral immune responses played a major role in protection (Figure 2), we analyzed Delta, Beta and Omicron (BA.1) (most divergent in our study) VOC cross-reactive activities in the sera of SMENWA1 vaccinated mice. Western blot analyses showed that sera from SMENWA1-vaccinated mouse detected all the proteins in SMEN particles, including spike proteins from heterologous variants (Figure S4C). These demonstrated successful immunization and generation of expected immune response. We then interrogated both neutralizing and Fc-signaling index as several previous studies have indicated that both neutralizing and Fc-effector functions of antibodies contribute to protection against SARS-CoV-2 (Figure 4H,I and Figure S4D). For our comparative analyses of cross-reactive index, the neutralizing and Fc signaling activity against the homologous WA1 strain was set to 100. The data indicated that while cross-neutralization activity against all VOCs were overall significantly diminished (cross-reactive neutralizing index, CRNI = 26), the loss in cross-neutralizing activity against Beta and Omicron VOCs was severe (CRNI= 2 and 3 respectively) compared to Delta VOC (CRNI = 21) (Figure 4H). A similar decline in the overall cross-reactive Fc-signaling index (CRFSI = 99) was observed for Fc-signaling activity in the SMENWA1-elicited sera. The sera demonstrated better activity against the Delta VOC (CRFSI = 62 vs 100 for WA1) compared to the Beta and Omicron VOCs, where the cross-reactive indices were significantly lower (CRFSI = 21 and 16, respectively (Figure 4I). These data explained the contrasting outcome and higher protection seen against Delta VOC compared to Beta VOC. Together, our data show that the SMENWA1 vaccine has a reduced capacity to provide immunity against Beta and Omicron variants and explained the superior resistance of Beta VOC for causing breakthrough infections as noted in our in vivo experiments in mice.
3.5. SMENOmicron Vaccine Provides Diminished Cross-Protection against Previous Variants of SARS-CoV-2.
The Omicron variants have a constellation of mutations in S (37 mutations in Omicron BA.1 compared to WA1) [55] that allows evasion of vaccine/infection-induced immunity from previous variants. Hence, we explored if SMEN vaccine with the more divergent Omicron spike can elicit broader immunity and demonstrate better protection. Towards this end, we challenged adjuvant-treated and SMENOmicron vaccinated K18-hACE2 mice (combined regimen) with ancestral (WA1), Delta, Beta and Omicron (BA.1) variants. The Omicron VOC does not produce a lethal infection in K18-hACE2 mouse model, and the mice recover in 3-4 days after exhibiting a brief reduction in body weight that peaks at 2 dpi [34]. We therefore terminated the Omicron VOC-challenged cohorts of mice at 3 dpi (Figure 5A). Our data revealed that immunity elicited by the SMENOmicron vaccine was effective against Omicron VOC as immunization prevented body weight loss and significantly reduced viral loads in the lungs compared to adjuvant-treated mice (Figure 5B,C). Surprisingly, the SMENOmicron vaccine did not offer 100% protection against the three strains tested. It provided limited protection, with survival rates of 50%, 0%, and 25% with delayed death against lethal challenges with the ancestral, Delta, and Beta variants, respectively (Figure 5B). The viral loads mirrored the survival data, showing diminished virologic control for all three strains tested, except for the homologous Omicron VOC, where the vaccine demonstrated the highest efficacy (Figure 5C and Figure S5A). Analysis of pro-inflammatory cytokines corroborated these observations and revealed that SMENOmicron vaccine reduced mRNA expression of Ccl2 (1.7-6-fold in brain and 2.5-5.3-fold in lung) and Cxcl10 (1.8-10.1-fold in brain and 1.5-7.4-fold in lung) when challenged with heterologous SARS-CoV-2 strains. However, the SMENOmicron vaccine was significantly more effective in reducing inflammation (22-26-fold reduction in brain; 6.5-14.3-fold in lung) when challenged with homologous strain (Figure S5B,C). Western blot analyses confirmed that sera from SMENOmicron-vaccinated mouse had reactivity against the proteins in SMEN particles, including spike proteins from heterologous variants demonstrating successful immunization and elicitation of the expected immune response (Figure S5D). To understand the limited protection offered by the vaccine, we analyzed the sera from SMENOmicron vaccinated mice for cross-reactive neutralizing and Fc-signaling activity (Figure 5D,E). Indeed, the SMENOmicron vaccine exhibited a low total CRNI of 31. The CRNI was lowest against the Delta VOC (5), followed by Beta (10) and WA1 (16), mirroring the level of protection observed in the survival analyses (Figure 5D). Evaluation of SMENOmicron-elicited sera for cross-reactive Fc-signaling index revealed broad activity against the three strains that totaled to 190 (Figure 5E). The data imply that although the antibodies elicited by the SMENOmicron vaccine may not exhibit high cross-neutralizing activity against previous variants, which is a crucial first line of defense, they were able to bind S on infected cells and mediate Fc receptor signaling. Overall, our data indicate that antibodies elicited by the Omicron Spike had reduced ability to neutralize previous VOCs owing to its extensive divergence, making the SMENOmicron vaccine less effective against these earlier variants in mice.
3.6. SMENBeta Vaccine Provides Broad Cross-Protection against Heterlogous Variants of SARS-CoV-2.
The Beta VOC exhibited high resistance to heterologous spike-mediated immunity (0% and 25% survival against SMENWA1 and SMENOmicron vaccine) among the variants we tested in mice (Figure 4 and Figure 5). This prompted us to explore if SMEN vaccine presenting Beta spike can better elicit broadly protective humoral immune responses. To this end, we immunized groups of K18-hACE2 mice with either adjuvant or SMENBeta VLPs using the combined regimen, then challenged them with either the homologous Beta VOC or the heterologous ancestral WA1, Delta, or the Omicron variant (Figure 6A). As expected, the SMENBeta vaccine significantly reduced body weight loss and protected 100% of the mice against the homologous Beta VOC challenge, in contrast to the adjuvant-treated mice that succumbed to the infection. Furthermore, SMENBeta-vaccinated mice showed 100% protection against lethal challenge with the heterologous WA1 or Delta variants, as well as Omicron, where the vaccine-mediated immunity significantly prevented body weight loss (Figure 6B). Evaluation of viral loads in the lung and brain tissues of SMENBeta-vaccinated mice were also consistent with the survival and body weight analyses, showing significant reductions and indicating effective virologic control against all four tested variants (Figure 6C and Figure S6A). Furthermore, the SMENBeta-vaccinated mice displayed a significant reduction in mRNA expression of inflammatory cytokines in both brain and lung tissues during challenge with both homologous or heterologous variants (Figure S6B,C). As was the case with SMENWA1 and SMENOmicron, SMENBeta elicited antibodies against individual proteins in VLPs and cross reacted with heterologous spike proteins, indicating successful seroconversion (Figure S6D). To better understand the breadth of protection, we assessed the cross-reactive neutralization and Fc-signaling activity in the SMENBeta-elicited sera (Figure 6D,E and Figure S6E). The sera had a total CRNI of 159, the highest among the different spike-presenting VLPs tested. Despite low CRNI against Delta VOC (10), IC50 values were still relatively low (2 µl of sera) which likely provided sufficient protection against a lethal Delta challenge (Figure 6D). SMENBeta elicited sera also demonstrated a high cross-reactive Fc-signaling index of 162, second only to SMENOmicron-elicited sera (190) (Figure 6E). These data suggest that the SMENBeta vaccine generated superior cross-reactive neutralizing and Fc-effector potent antibodies among the SMEN variants tested. Thus, the Beta VOC not only resisted immunity produced by other variants but also triggered a diverse and effective humoral immune response. Our study suggests that using the Beta variant spike as a base for vaccine development may lead to better protection against emerging SARS-CoV-2 variants.
4. Discussion
Here we have demonstrated the potential of SMEN VLP vaccine platform in eliciting robust cross-protective immune responses against SARS-CoV-2 variants in vivo using a stringent K18-hACE2 mouse model. By employing S proteins from the ancestral WA1 strain, Beta, and Omicron BA.1 variants, we evaluated the in vivo efficacy of SMEN VLPs as vaccines. Our results highlighted that the combined intranasal and intramuscular administration of the SMEN vaccine generated the most effective immune responses, leading to superior virologic control, reduced lung inflammation, and a lower overall disease burden. These data align with the growing body of evidence highlighting the importance of mucosal immunity in addition to peripheral stimulation for better prognosis following infection by respiratory pathogens including SARS-CoV-2 and influenza [56,57,58,59].
The observed SMEN-mediated immune protection was predominantly antibody-mediated, which is consistent with findings from several other vaccine platforms against SARS-CoV-2 [33,60,61]. SMEN vaccine not only elicited antibodies against the spike protein but also against the M, E, and N proteins. We expect the immune response to the M, E, and N proteins to be consistent across all the analyzed SMEN VLPs and contribute to enhancing overall immunity. Antibodies targeting the N protein has the potential to clear infected cells through Fc-mediated effector functions, as the N protein was shown to be present on infected cell surfaces [62,63]. Additionally, antibodies against the E protein can potentially mitigate E-protein-mediated inflammasome activation by blocking interaction with TLR2 [64,65], contributing to an overall reduction in lung pathology. In addition to antibodies, SMEN vaccine also elicited T cells. We found that vaccine elicited CD4+ and CD8+ T cells played a minor role as their depletion did not compromise protection from virus-induced mortality. Unlike mRNA and adenovirus-based vaccines, SMEN antigens are not expressed within cells but are instead presented exogenously to the immune system. Consequently, diminished CD8+ T cell responses are expected, except for the contributions arising from cross-presentation [66,67]. However, T cells did contribute to limiting virus dissemination to distal organs, such as the brain, and reducing inflammation in the lung. Limiting virus dissemination has important consequences including reduction in multi-organ pathology of COVID-19 [68]. In addition, reducing infection-induced inflammation is known to significantly benefit post-infection recovery and reduce lung pathology [34,69,70].
Notably, SMEN particles presenting Beta spikes elicited diverse and potent humoral immune responses in terms of neutralizing and Fc-signaling activity. The basis for the broad response remains unclear and requires further investigation. The Beta variant was the first VOC that arose mainly due to its capacity for immune evasion [71]. The Beta variant S protein has several key mutations, particularly in the receptor-bind domain (RBD) and the N-terminal domain (NTD) which significantly alter its antigenic profile. The RBD mutations K417N changes the conformation of RBD, whereas E484K substantially enhances nAb escape and N501Y enhances binding affinity to the ACE2 receptor. These alterations also allow Beta VOC to overcome vaccine and infection-induced immunity [71]. We propose that vaccinating with an immune-evasive conformational state such as Beta S, elicits antibodies that are more capable of adapting to common escape mutations, thereby generating a broader immune response. Consistent with our observations, a similar broad Fc-effector activity against heterologous VOCs was also observed in a study that analyzed human sera from Beta VOC-infected individuals [72]. Thus, the unique properties of the Beta spike protein, as previously noted [71,72], may serve as a middle-ground antigenic template in the virus-host co-evolutionary space that provides balanced and broad immunity against both previous and future variants of concern (VOCs). In contrast, the significant antigenic shift and divergence seen in the Omicron variant spike likely led to the overall reduction in nAb-mediated immunity against the ancestral strain and previous VOCs, while broadening the overall reactivity through non-nAb responses. This was evident in our study by the markedly high cross-reactive Fc-signaling index in the sera of SMENOmicron-vaccinated mice.
Given the intense immune pressure elicited by the host, viral fusion glycoproteins display elaborate immune evasion strategies including structural plasticity, sequence variability, conformational flexibility, and glycosylation of vulnerable epitopes [73]. One vaccine strategy to counter immune diversion is to lock fusion glycoproteins in the most vulnerable pre-fusion conformation [73,74,75]. This approach focuses on humoral immune responses to elicit antibodies that are predominantly neutralizing in nature to effectively block virus entry. This strategy was harnessed by various vaccine platforms with the strategic introduction of conformationally rigid proline residues in the S2 region of the spike protein to present the prefusion conformation as vaccine antigens during the COVID-19 pandemic [76]. Restricting conformational flexibility is of paramount importance in designing vaccines against the highly immune-evasive HIV-1 to elicit effective nAbs and remains one of the unachieved goals in HIV-1 vaccinology [77]. However, locking conformations may not necessarily enhance effective immune response for all fusion glycoproteins, as shown for the human cytomegalovirus type III fusion glycoprotein gB [78]. In the case of SARS-CoV-2, which is predominantly controlled through humoral immune responses, stabilizing specific conformations, while tremendously successful, might also limit the breadth of humoral immune responses. In this regard, the SMEN VLP platform has the advantage of presenting an array of membrane-embedded native conformations seen in virions that can potentially contribute to increasing the breadth of humoral immune responses. Overall, our study adds to the utility of the SMEN VLP platform as an additional vaccine option that can complement existing platforms and potentially provide safer alternatives to boost acceptance rates. This versatility and broad-spectrum efficacy make SMEN VLPs a promising tool in the ongoing battle against COVID-19 and future emerging infectious diseases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Production and characterization of the SARS-CoV-2 virus-like particle (SMEN) vaccine and its impact on neutralizing antibody response, Figure S2: Passive transfer of SMEN-vaccine elicited sera provides immunity to naïve mice against lethal SARS-CoV-2 challenge, Figure S3: CD8+ and CD4+ T cells contribute to SMEN Vaccine-mediated protection by diminishing virus spread to the brain, Figure S4: SMENWA1 Vaccine Shows Efficacy Against Delta and Offers Minimal Cross-Protection Against Beta VOC, Figure S5: SMENOmicron Offers Limited Cross-Protection Against Heterologous VOCs, Figure S6: SMENBeta Offers Broad Cross-Protection Against Heterologous VOCs.
Author Contributions
All authors have read and agreed to the published version of the manuscript. Conceptualization, P.D.U. and W.M.; methodology, I.U., K.S., K.K., L.Z., W.L., and M.W.G.; validation, I. U., K.K., W. L.; formal analysis, I.U. and P.D.U.; investigation, I.U., P.D.U., K.S., K.K., L.Z., and W.L.; resources, P.K., W.M.; data curation, I.U., K.K., W.L.; writing—original draft preparation, P.D.U., I.U.; writing—review and editing, P.D.U., W.M. ; supervision, P.D.U., W.M. and P.K.; project administration P.D.U.; funding acquisition W.M., P.D.U., P.K.
Funding
This research was funded by NIH grant R01AI163395 to W.M., a CIHR operating Pandemic and Health Emergencies Research grant #177958 to W.M, CIHR operating grant #487578 to P.D.U, and NIH R24OD026440-03S1 (COVID-19 Administrative Supplement) to P.K. and P.D.U., and NIH grant F31 AI176650 to M.W.G.M.W.G. is a recipient of Gruber Science Fellowship and was supported by NIH T32 AI055403.
Institutional Review Board Statement
All animal experiments described here were reviewed and approved by Yale University (New Haven USA) Institutional Animal Care and Use Committees (IACUC # 2020-10649) as well as SOPs approved by the Yale University (New Haven, USA) Institutional Environmental Health and Biosafety committee (Infectious Agent Protocol #10060).
Informed Consent Statement
Not applicable.
Data Availability Statement
All data supporting the findings of this study are available within the paper and its Supplementary Materials.
Acknowledgments
We thank Tina Izard, Michael Farzan and Hyeryun Choe for sharing plasmids expressing SARS-CoV-2 M, E and N proteins and Sri Lakshmi Tejaswi Boodapati for help maintaining mouse colony.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nature Reviews Molecular Cell Biology 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef]
- Montgomerie, I.; Bird, T.W.; Palmer, O.R.; Mason, N.C.; Pankhurst, T.E.; Lawley, B.; Hernández, L.C.; Harfoot, R.; Authier-Hall, A.; Anderson, D.E.; et al. Incorporation of SARS-CoV-2 spike NTD to RBD protein vaccine improves immunity against viral variants. iScience 2023, 26, 106256. [Google Scholar] [CrossRef]
- Kumari, M.; Su, S.-C.; Liang, K.-H.; Lin, H.-T.; Lu, Y.-F.; Chen, K.-C.; Chen, W.-Y.; Wu, H.-C. Bivalent mRNA vaccine effectiveness against SARS-CoV-2 variants of concern. Journal of Biomedical Science 2023, 30, 46. [Google Scholar] [CrossRef]
- Meyer Zu Natrup, C.; Tscherne, A.; Dahlke, C.; Ciurkiewicz, M.; Shin, D.L.; Fathi, A.; Rohde, C.; Kalodimou, G.; Halwe, S.; Limpinsel, L.; et al. Stabilized recombinant SARS-CoV-2 spike antigen enhances vaccine immunogenicity and protective capacity. J Clin Invest 2022, 132. [Google Scholar] [CrossRef]
- Ao, D.; Lan, T.; He, X.; Liu, J.; Chen, L.; Baptista-Hon, D.T.; Zhang, K.; Wei, X. SARS-CoV-2 Omicron variant: Immune escape and vaccine development. MedComm (2020) 2022, 3, e126. [Google Scholar] [CrossRef]
- Lau, J.J.; Cheng, S.M.S.; Leung, K.; Lee, C.K.; Hachim, A.; Tsang, L.C.H.; Yam, K.W.H.; Chaothai, S.; Kwan, K.K.H.; Chai, Z.Y.H.; et al. Real-world COVID-19 vaccine effectiveness against the Omicron BA.2 variant in a SARS-CoV-2 infection-naive population. Nature Medicine 2023, 29, 348–357. [Google Scholar] [CrossRef]
- Mohsin, M.; Mahmud, S.; Uddin Mian, A.; Hasan, P.; Muyeed, A.; Taif Ali, M.; Faysal Ahmed, F.; Islam, A.; Maliha Rahman, M.; Islam, M.; et al. Side effects of COVID-19 vaccines and perceptions about COVID-19 and its vaccines in Bangladesh: A Cross-sectional study. Vaccine X 2022, 12, 100207. [Google Scholar] [CrossRef]
- Schäfer, I.; Oltrogge, J.H.; Nestoriuc, Y.; Warren, C.V.; Brassen, S.; Blattner, M.; Lühmann, D.; Tinnermann, A.; Scherer, M.; Büchel, C. Expectations and Prior Experiences Associated With Adverse Effects of COVID-19 Vaccination. JAMA Network Open 2023, 6, e234732–e234732. [Google Scholar] [CrossRef]
- Solís Arce, J.S.; Warren, S.S.; Meriggi, N.F.; Scacco, A.; McMurry, N.; Voors, M.; Syunyaev, G.; Malik, A.A.; Aboutajdine, S.; Adeojo, O.; et al. COVID-19 vaccine acceptance and hesitancy in low- and middle-income countries. Nature Medicine 2021, 27, 1385–1394. [Google Scholar] [CrossRef]
- Bozkurt, B.; Kamat, I.; Hotez, P.J. Myocarditis With COVID-19 mRNA Vaccines. Circulation 2021, 144, 471–484. [Google Scholar] [CrossRef]
- Bekal, S.; Husari, G.; Okura, M.; Huang, C.A.; Bukari, M.S. Thrombosis Development After mRNA COVID-19 Vaccine Administration: A Case Series. Cureus 2023, 15, e41371. [Google Scholar] [CrossRef]
- SeyedAlinaghi, S.; Karimi, A.; Pashaei, Z.; Afzalian, A.; Mirzapour, P.; Ghorbanzadeh, K.; Ghasemzadeh, A.; Dashti, M.; Nazarian, N.; Vahedi, F.; et al. Safety and Adverse Events Related to COVID-19 mRNA Vaccines; a Systematic Review. Arch Acad Emerg Med 2022, 10, e41. [Google Scholar] [CrossRef]
- Hosseini, R.; Askari, N. A review of neurological side effects of COVID-19 vaccination. Eur J Med Res 2023, 28, 102. [Google Scholar] [CrossRef]
- Wang, J.J.; Schonborn, L.; Warkentin, T.E.; Chataway, T.; Grosse, L.; Simioni, P.; Moll, S.; Greinacher, A.; Gordon, T.P. Antibody Fingerprints Linking Adenoviral Anti-PF4 Disorders. N Engl J Med 2024, 390, 1827–1829. [Google Scholar] [CrossRef]
- Chu, K.B.; Quan, F.S. Respiratory Viruses and Virus-like Particle Vaccine Development: How Far Have We Advanced? Viruses 2023, 15. [Google Scholar] [CrossRef]
- Gao, X.; Xia, Y.; Liu, X.; Xu, Y.; Lu, P.; Dong, Z.; Liu, J.; Liang, G. A perspective on SARS-CoV-2 virus-like particles vaccines. Int Immunopharmacol 2023, 115, 109650. [Google Scholar] [CrossRef]
- Yilmaz, I.C.; Ipekoglu, E.M.; Bulbul, A.; Turay, N.; Yildirim, M.; Evcili, I.; Yilmaz, N.S.; Guvencli, N.; Aydin, Y.; Gungor, B.; et al. Development and preclinical evaluation of virus-like particle vaccine against COVID-19 infection. Allergy 2022, 77, 258–270. [Google Scholar] [CrossRef]
- Naskalska, A.; Dabrowska, A.; Szczepanski, A.; Jasik, K.P.; Gromadzka, B.; Pyrc, K. Functional Severe Acute Respiratory Syndrome Coronavirus 2 Virus-Like Particles From Insect Cells. Front Microbiol 2021, 12, 732998. [Google Scholar] [CrossRef]
- Tariq, H.; Batool, S.; Asif, S.; Ali, M.; Abbasi, B.H. Virus-Like Particles: Revolutionary Platforms for Developing Vaccines Against Emerging Infectious Diseases. Front Microbiol 2021, 12, 790121. [Google Scholar] [CrossRef]
- Nooraei, S.; Bahrulolum, H.; Hoseini, Z.S.; Katalani, C.; Hajizade, A.; Easton, A.J.; Ahmadian, G. Virus-like particles: preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J Nanobiotechnology 2021, 19, 59. [Google Scholar] [CrossRef]
- Comas-Garcia, M.; Colunga-Saucedo, M.; Rosales-Mendoza, S. The Role of Virus-Like Particles in Medical Biotechnology. Mol Pharm 2020, 17, 4407–4420. [Google Scholar] [CrossRef]
- Syed, A.M.; Taha, T.Y.; Tabata, T.; Chen, I.P.; Ciling, A.; Khalid, M.M.; Sreekumar, B.; Chen, P.Y.; Hayashi, J.M.; Soczek, K.M.; et al. Rapid assessment of SARS-CoV-2-evolved variants using virus-like particles. Science 2021, 374, 1626–1632. [Google Scholar] [CrossRef]
- Tan, C.W.; Valkenburg, S.A.; Poon, L.L.M.; Wang, L.F. Broad-spectrum pan-genus and pan-family virus vaccines. Cell Host Microbe 2023, 31, 902–916. [Google Scholar] [CrossRef]
- Lu, M. Single-Molecule FRET Imaging of Virus Spike-Host Interactions. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nature Reviews Microbiology 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nature Reviews Microbiology 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Zhou, L.; Zhou, Y.; Yan, H.; Lan, K. Emerging SARS-CoV-2 variants: Why, how, and what’s next? Cell Insight 2022, 1, 100029. [Google Scholar] [CrossRef]
- Goldblatt, D.; Alter, G.; Crotty, S.; Plotkin, S.A. Correlates of protection against SARS-CoV-2 infection and COVID-19 disease. Immunol Rev 2022, 310, 6–26. [Google Scholar] [CrossRef]
- Ullah, I.; Escudie, F.; Scandale, I.; Gilani, Z.; Gendron-Lepage, G.; Gaudette, F.; Mowbray, C.; Fraisse, L.; Bazin, R.; Finzi, A.; et al. Bioluminescence imaging reveals enhanced SARS-CoV-2 clearance in mice with combinatorial regimens. iScience 2024, 27, 109049. [Google Scholar] [CrossRef]
- Torii, S.; Ono, C.; Suzuki, R.; Morioka, Y.; Anzai, I.; Fauzyah, Y.; Maeda, Y.; Kamitani, W.; Fukuhara, T.; Matsuura, Y. Establishment of a reverse genetics system for SARS-CoV-2 using circular polymerase extension reaction. Cell Rep 2021, 35, 109014. [Google Scholar] [CrossRef]
- Amarilla, A.A.; Sng, J.D.J.; Parry, R.; Deerain, J.M.; Potter, J.R.; Setoh, Y.X.; Rawle, D.J.; Le, T.T.; Modhiran, N.; Wang, X.; et al. A versatile reverse genetics platform for SARS-CoV-2 and other positive-strand RNA viruses. Nat Commun 2021, 12, 3431. [Google Scholar] [CrossRef]
- Ullah, I.; Prevost, J.; Ladinsky, M.S.; Stone, H.; Lu, M.; Anand, S.P.; Beaudoin-Bussieres, G.; Symmes, K.; Benlarbi, M.; Ding, S.; et al. Live imaging of SARS-CoV-2 infection in mice reveals that neutralizing antibodies require Fc function for optimal efficacy. Immunity 2021, 54, 2143–2158. [Google Scholar] [CrossRef]
- Ullah, I.; Beaudoin-Bussieres, G.; Symmes, K.; Cloutier, M.; Ducas, E.; Tauzin, A.; Laumaea, A.; Grunst, M.W.; Dionne, K.; Richard, J.; et al. The Fc-effector function of COVID-19 convalescent plasma contributes to SARS-CoV-2 treatment efficacy in mice. Cell Rep Med 2023, 4, 100893. [Google Scholar] [CrossRef]
- Papini, C.; Ullah, I.; Ranjan, A.P.; Zhang, S.; Wu, Q.; Spasov, K.A.; Zhang, C.; Mothes, W.; Crawford, J.M.; Lindenbach, B.D.; et al. Proof-of-concept studies with a computationally designed M(pro) inhibitor as a synergistic combination regimen alternative to Paxlovid. Proc Natl Acad Sci U S A 2024, 121, e2320713121. [Google Scholar] [CrossRef]
- Grandi, A.; Tomasi, M.; Ullah, I.; Bertelli, C.; Vanzo, T.; Accordini, S.; Gagliardi, A.; Zanella, I.; Benedet, M.; Corbellari, R.; et al. Immunogenicity and Pre-Clinical Efficacy of an OMV-Based SARS-CoV-2 Vaccine. Vaccines (Basel) 2023, 11. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Mastronarde, D.N. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 2005, 152, 36–51. [Google Scholar] [CrossRef]
- Hagen, W.J.H.; Wan, W.; Briggs, J.A.G. Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. J Struct Biol 2017, 197, 191–198. [Google Scholar] [CrossRef]
- Zheng, S.Q.; Palovcak, E.; Armache, J.P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 2017, 14, 331–332. [Google Scholar] [CrossRef]
- Mastronarde, D.N.; Held, S.R. Automated tilt series alignment and tomographic reconstruction in IMOD. J Struct Biol 2017, 197, 102–113. [Google Scholar] [CrossRef]
- Sherwood, E.R.; Burelbach, K.R.; McBride, M.A.; Stothers, C.L.; Owen, A.M.; Hernandez, A.; Patil, N.K.; Williams, D.L.; Bohannon, J.K. Innate Immune Memory and the Host Response to Infection. J Immunol 2022, 208, 785–792. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Netea, M.G.; Chavakis, T. Innate immune memory, trained immunity and nomenclature clarification. Nat Immunol 2023, 24, 1393–1394. [Google Scholar] [CrossRef]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.E.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.C.; Looney, M.R.; et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 2015, 517, 621–625. [Google Scholar] [CrossRef]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun 2020, 11, 3559. [Google Scholar] [CrossRef]
- Ray, S.; Chiba, N.; Yao, C.; Guan, X.; McConnell, A.M.; Brockway, B.; Que, L.; McQualter, J.L.; Stripp, B.R. Rare SOX2(+) Airway Progenitor Cells Generate KRT5(+) Cells that Repopulate Damaged Alveolar Parenchyma following Influenza Virus Infection. Stem Cell Reports 2016, 7, 817–825. [Google Scholar] [CrossRef]
- Kumar, P.A.; Hu, Y.; Yamamoto, Y.; Hoe, N.B.; Wei, T.S.; Mu, D.; Sun, Y.; Joo, L.S.; Dagher, R.; Zielonka, E.M.; et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 2011, 147, 525–538. [Google Scholar] [CrossRef]
- Choi, J.; Park, J.E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.K.; Han, N.; Lee, J.H. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 2020, 27, 366–382. [Google Scholar] [CrossRef]
- Boyd, D.F.; Allen, E.K.; Randolph, A.G.; Guo, X.J.; Weng, Y.; Sanders, C.J.; Bajracharya, R.; Lee, N.K.; Guy, C.S.; Vogel, P.; et al. Exuberant fibroblast activity compromises lung function via ADAMTS4. Nature 2020, 587, 466–471. [Google Scholar] [CrossRef]
- Chen, K.K.; Tsung-Ning Huang, D.; Huang, L.M. SARS-CoV-2 variants—Evolution, spike protein, and vaccines. Biomed J 2022, 45, 573–579. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Xing, M.; Hu, G.; Wang, X.; Wang, Y.; He, F.; Dai, W.; Wang, X.; Niu, Y.; Liu, J.; Liu, H.; et al. An intranasal combination vaccine induces systemic and mucosal immunity against COVID-19 and influenza. NPJ Vaccines 2024, 9, 64. [Google Scholar] [CrossRef]
- Oh, J.E.; Song, E.; Moriyama, M.; Wong, P.; Zhang, S.; Jiang, R.; Strohmeier, S.; Kleinstein, S.H.; Krammer, F.; Iwasaki, A. Intranasal priming induces local lung-resident B cell populations that secrete protective mucosal antiviral IgA. Sci Immunol 2021, 6, eabj5129. [Google Scholar] [CrossRef]
- Mao, T.; Israelow, B.; Pena-Hernandez, M.A.; Suberi, A.; Zhou, L.; Luyten, S.; Reschke, M.; Dong, H.; Homer, R.J.; Saltzman, W.M.; et al. Unadjuvanted intranasal spike vaccine elicits protective mucosal immunity against sarbecoviruses. Science 2022, 378, eabo2523. [Google Scholar] [CrossRef]
- Iwasaki, A. Exploiting Mucosal Immunity for Antiviral Vaccines. Annu Rev Immunol 2016, 34, 575–608. [Google Scholar] [CrossRef]
- Gilbert, P.B.; Montefiori, D.C.; McDermott, A.B.; Fong, Y.; Benkeser, D.; Deng, W.; Zhou, H.; Houchens, C.R.; Martins, K.; Jayashankar, L.; et al. Immune correlates analysis of the mRNA-1273 COVID-19 vaccine efficacy clinical trial. Science 2022, 375, 43–50. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; Astudillo, M.G.; Yang, D.; Miller, T.E.; Feldman, J.; Hauser, B.M.; Caradonna, T.M.; Clayton, K.L.; Nitido, A.D.; et al. COVID-19-neutralizing antibodies predict disease severity and survival. Cell 2021, 184, 476–488. [Google Scholar] [CrossRef]
- Lopez-Munoz, A.D.; Kosik, I.; Holly, J.; Yewdell, J.W. Cell surface SARS-CoV-2 nucleocapsid protein modulates innate and adaptive immunity. Sci Adv 2022, 8, eabp9770. [Google Scholar] [CrossRef]
- Lopez-Munoz, A.D.; Santos, J.J.S.; Yewdell, J.W. Cell surface nucleocapsid protein expression: A betacoronavirus immunomodulatory strategy. Proc Natl Acad Sci U S A 2023, 120, e2304087120. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Ambrozek-Latecka, M.; Kozlowski, P.; Hoser, G.; Bandyszewska, M.; Hanusek, K.; Nowis, D.; Golab, J.; Grzanka, M.; Piekielko-Witkowska, A.; Schulz, L.; et al. SARS-CoV-2 and its ORF3a, E and M viroporins activate inflammasome in human macrophages and induce of IL-1alpha in pulmonary epithelial and endothelial cells. Cell Death Discov 2024, 10, 191. [Google Scholar] [CrossRef]
- Helft, J.; Manicassamy, B.; Guermonprez, P.; Hashimoto, D.; Silvin, A.; Agudo, J.; Brown, B.D.; Schmolke, M.; Miller, J.C.; Leboeuf, M.; et al. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J Clin Invest 2012, 122, 4037–4047. [Google Scholar] [CrossRef]
- Zinkernagel, R.M. On cross-priming of MHC class I-specific CTL: rule or exception? Eur J Immunol 2002, 32, 2385–2392. [Google Scholar] [CrossRef]
- Stein, S.R.; Ramelli, S.C.; Grazioli, A.; Chung, J.Y.; Singh, M.; Yinda, C.K.; Winkler, C.W.; Sun, J.; Dickey, J.M.; Ylaya, K.; et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature 2022, 612, 758–763. [Google Scholar] [CrossRef]
- Mohandas, S.; Jagannathan, P.; Henrich, T.J.; Sherif, Z.A.; Bime, C.; Quinlan, E.; Portman, M.A.; Gennaro, M.; Rehman, J.; Force, R.M.P.T. Immune mechanisms underlying COVID-19 pathology and post-acute sequelae of SARS-CoV-2 infection (PASC). Elife 2023, 12. [Google Scholar] [CrossRef]
- Mourad, A.; Thibault, D.; Holland, T.L.; Yang, S.; Young, A.R.; Arnold Egloff, S.A.; Thomas, L.E. Dexamethasone for Inpatients With COVID-19 in a National Cohort. JAMA Netw Open 2023, 6, e238516. [Google Scholar] [CrossRef]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Oosthuysen, B.; Lambson, B.E.; de Oliveira, T.; Vermeulen, M.; van der Berg, K.; et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat Med 2021, 27, 622–625. [Google Scholar] [CrossRef]
- Richardson, S.I.; Manamela, N.P.; Motsoeneng, B.M.; Kaldine, H.; Ayres, F.; Makhado, Z.; Mennen, M.; Skelem, S.; Williams, N.; Sullivan, N.J.; et al. SARS-CoV-2 Beta and Delta variants trigger Fc effector function with increased cross-reactivity. Cell Rep Med 2022, 3, 100510. [Google Scholar] [CrossRef]
- Graham, B.S.; Gilman, M.S.A.; McLellan, J.S. Structure-Based Vaccine Antigen Design. Annu Rev Med 2019, 70, 91–104. [Google Scholar] [CrossRef]
- Sanders, R.W.; Moore, J.P. Virus vaccines: proteins prefer prolines. Cell Host Microbe 2021, 29, 327–333. [Google Scholar] [CrossRef]
- Byrne, P.O.; McLellan, J.S. Principles and practical applications of structure-based vaccine design. Curr Opin Immunol 2022, 77, 102209. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505. [Google Scholar] [CrossRef]
- Lu, M.; Ma, X.; Castillo-Menendez, L.R.; Gorman, J.; Alsahafi, N.; Ermel, U.; Terry, D.S.; Chambers, M.; Peng, D.; Zhang, B.; et al. Associating HIV-1 envelope glycoprotein structures with states on the virus observed by smFRET. Nature 2019, 568, 415–419. [Google Scholar] [CrossRef]
- Sponholtz, M.R.; Byrne, P.O.; Lee, A.G.; Ramamohan, A.R.; Goldsmith, J.A.; McCool, R.S.; Zhou, L.; Johnson, N.V.; Hsieh, C.-L.; Connors, M.; et al. Structure-based design of a soluble human cytomegalovirus glycoprotein B antigen stabilized in a prefusion-like conformation. bioRxiv 2024. [Google Scholar]
Figure 1.
Enhanced Protection Against Lethal SARS-CoV-2 Challenge in K18-hACE2 Mice via Systemic and Mucosal SMEN Vaccine Administration (A) A scheme showing experimental design for comparing protective efficacy of SAR-CoV-2 VLP (SMENWA1) vaccine using various routes. 250 µg of SMEN vaccine were used to immunize K18-hACE2 mice as depicted intramuscularly (i.m.) or i.n. using the prime-boost strategy. 30 μg of vaccigrade TLR7/8 agonist R848 was used as adjuvant for the i.m. route. Adjuvant and untreated mice served as control groups. 21 days after immunization, sera/ bronchioalveolar lavage fluids (BALF) were harvested or mice challenged i.n. with 1 x 105 PFU of SARS-CoV-2WA1 and indicated parameters were analyzed to assess protection. (B, C) Neutralizing titers (IC50) in the sera and BALF from indicated groups of vaccinated as in A. (D) A plot showing temporal body weight changes of K18-hACE2 mice at indicated days post infection (dpi) for an experiment shown in (A). Each curve represents one animal. The body weight at the start of the experiment was set to 100%. (E) Kaplan-Meier survival curves for experiment as in A. (F) Viral loads (nLuc activity/mg of tissue) from indicated organs at time of necropsy using Vero-E6 as targets. (G) Fold change in mRNA expression of indicated lung pathology markers, Krt8, Krt5, Adamts4 and Itga5 in the lung tissue after necropsy. (H, I) Fold changes in indicated cytokine mRNA expression in brain and lung tissues of mice under specified treatment regimens after necropsy upon death or at 16 dpi in the surviving animals. Data were normalized to Gapdh mRNA in the same sample and that in non-infected mice after necropsy in the experiments shown in (G-I). Data in (B, C) were analyzed by nonparametric Mann-Whitney T test. Grouped data in (D) and (F-I) were analyzed using 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for the group comparisons to mock are shown in black, with adjuvant shown in light dark, with SMENWA1 i.m. shown in red, and SMENWA1 i.n. shown in magenta. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; ns, not significant; Mean values ± SD are depicted.
Figure 1.
Enhanced Protection Against Lethal SARS-CoV-2 Challenge in K18-hACE2 Mice via Systemic and Mucosal SMEN Vaccine Administration (A) A scheme showing experimental design for comparing protective efficacy of SAR-CoV-2 VLP (SMENWA1) vaccine using various routes. 250 µg of SMEN vaccine were used to immunize K18-hACE2 mice as depicted intramuscularly (i.m.) or i.n. using the prime-boost strategy. 30 μg of vaccigrade TLR7/8 agonist R848 was used as adjuvant for the i.m. route. Adjuvant and untreated mice served as control groups. 21 days after immunization, sera/ bronchioalveolar lavage fluids (BALF) were harvested or mice challenged i.n. with 1 x 105 PFU of SARS-CoV-2WA1 and indicated parameters were analyzed to assess protection. (B, C) Neutralizing titers (IC50) in the sera and BALF from indicated groups of vaccinated as in A. (D) A plot showing temporal body weight changes of K18-hACE2 mice at indicated days post infection (dpi) for an experiment shown in (A). Each curve represents one animal. The body weight at the start of the experiment was set to 100%. (E) Kaplan-Meier survival curves for experiment as in A. (F) Viral loads (nLuc activity/mg of tissue) from indicated organs at time of necropsy using Vero-E6 as targets. (G) Fold change in mRNA expression of indicated lung pathology markers, Krt8, Krt5, Adamts4 and Itga5 in the lung tissue after necropsy. (H, I) Fold changes in indicated cytokine mRNA expression in brain and lung tissues of mice under specified treatment regimens after necropsy upon death or at 16 dpi in the surviving animals. Data were normalized to Gapdh mRNA in the same sample and that in non-infected mice after necropsy in the experiments shown in (G-I). Data in (B, C) were analyzed by nonparametric Mann-Whitney T test. Grouped data in (D) and (F-I) were analyzed using 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for the group comparisons to mock are shown in black, with adjuvant shown in light dark, with SMENWA1 i.m. shown in red, and SMENWA1 i.n. shown in magenta. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; ns, not significant; Mean values ± SD are depicted.
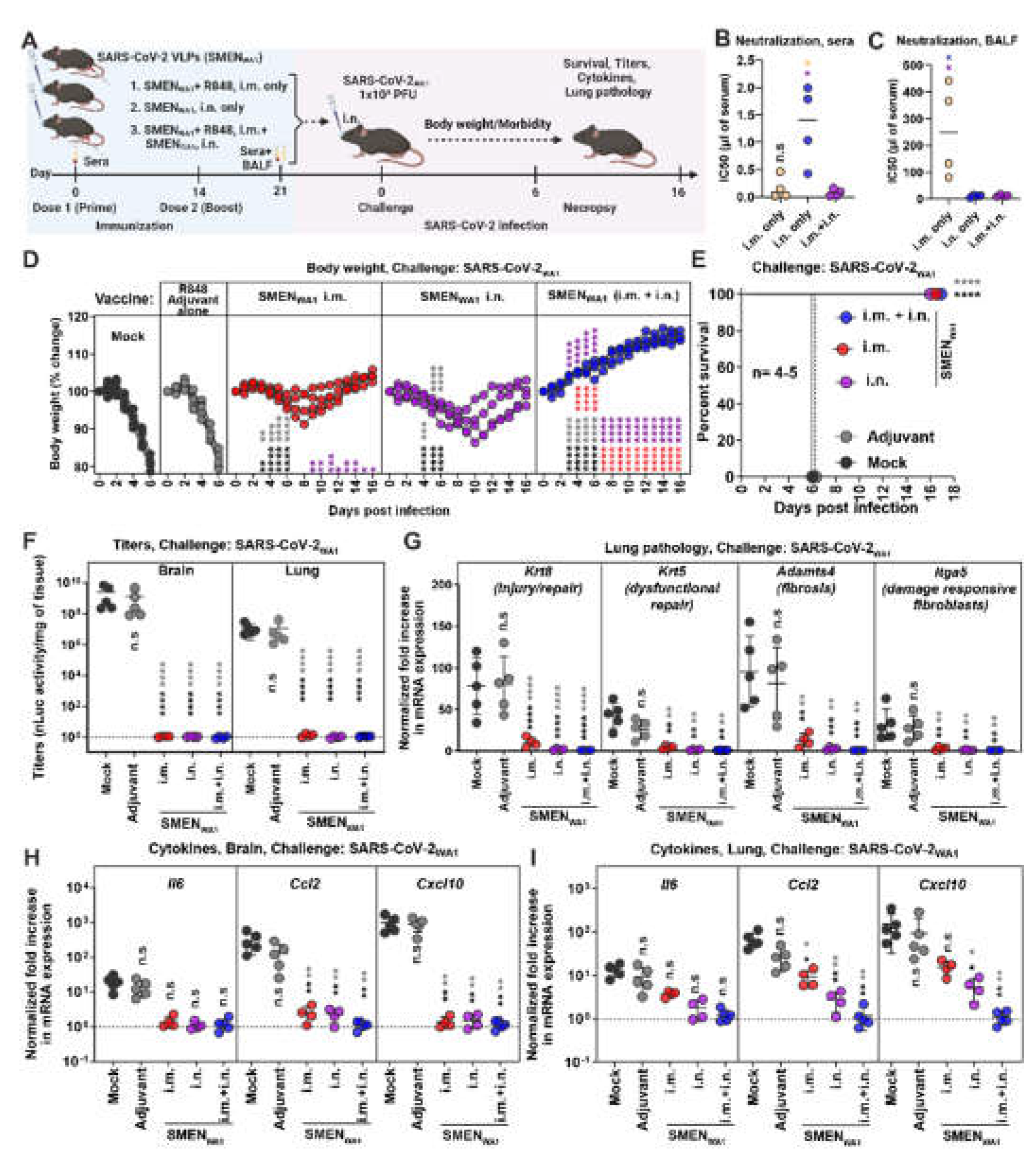
Figure 2.
SMEN Vaccine-Elicited Antibodies Can Protect Naïve Mice from SARS-CoV-2-Induced Mortality (A) Experimental design for testing the contribution of immune sera to conferring protection. Pre-immune sera (500 µL) or sera from vaccinated mice (immune sera) were passively transferred into naïve K18-hACE2 mice one day before challenge with SARS-CoV-2WA1-nLuc (i.n., 1 × 105 PFU). (B) Representative BLI images of SARS-CoV-2WA1-nLuc-infected mice in ventral (v) and dorsal (d) positions. (C-D) Temporal quantification of nLuc signal as flux (photons/sec) computed non-invasively. (E) A plot showing temporal body weight changes of K18-hACE2 mice at indicated dpi for an experiment shown in A. Each curve represents one animal. The body weight at the start of the experiment was set to 100%. (F) Kaplan-Meier survival curves of K18-hACE2 for experiment as in A. (G) Viral loads (nLuc activity/mg of tissue) from indicated organs at time of necropsy using Vero-E6 as targets. (H-I) Fold changes in indicated cytokine mRNA expression in brain and lung tissues of mice under specified treatment regimens after necropsy upon death or at 14 dpi in the surviving animals. Data were normalized to Gapdh mRNA in the same sample and that in non-infected mice after necropsy. Grouped data in (C-E), and (G-I) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for the groups comparisons to pre-immune sera are shown in black ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; ns, not significant; Mean values ± SD are depicted.
Figure 2.
SMEN Vaccine-Elicited Antibodies Can Protect Naïve Mice from SARS-CoV-2-Induced Mortality (A) Experimental design for testing the contribution of immune sera to conferring protection. Pre-immune sera (500 µL) or sera from vaccinated mice (immune sera) were passively transferred into naïve K18-hACE2 mice one day before challenge with SARS-CoV-2WA1-nLuc (i.n., 1 × 105 PFU). (B) Representative BLI images of SARS-CoV-2WA1-nLuc-infected mice in ventral (v) and dorsal (d) positions. (C-D) Temporal quantification of nLuc signal as flux (photons/sec) computed non-invasively. (E) A plot showing temporal body weight changes of K18-hACE2 mice at indicated dpi for an experiment shown in A. Each curve represents one animal. The body weight at the start of the experiment was set to 100%. (F) Kaplan-Meier survival curves of K18-hACE2 for experiment as in A. (G) Viral loads (nLuc activity/mg of tissue) from indicated organs at time of necropsy using Vero-E6 as targets. (H-I) Fold changes in indicated cytokine mRNA expression in brain and lung tissues of mice under specified treatment regimens after necropsy upon death or at 14 dpi in the surviving animals. Data were normalized to Gapdh mRNA in the same sample and that in non-infected mice after necropsy. Grouped data in (C-E), and (G-I) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for the groups comparisons to pre-immune sera are shown in black ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; ns, not significant; Mean values ± SD are depicted.
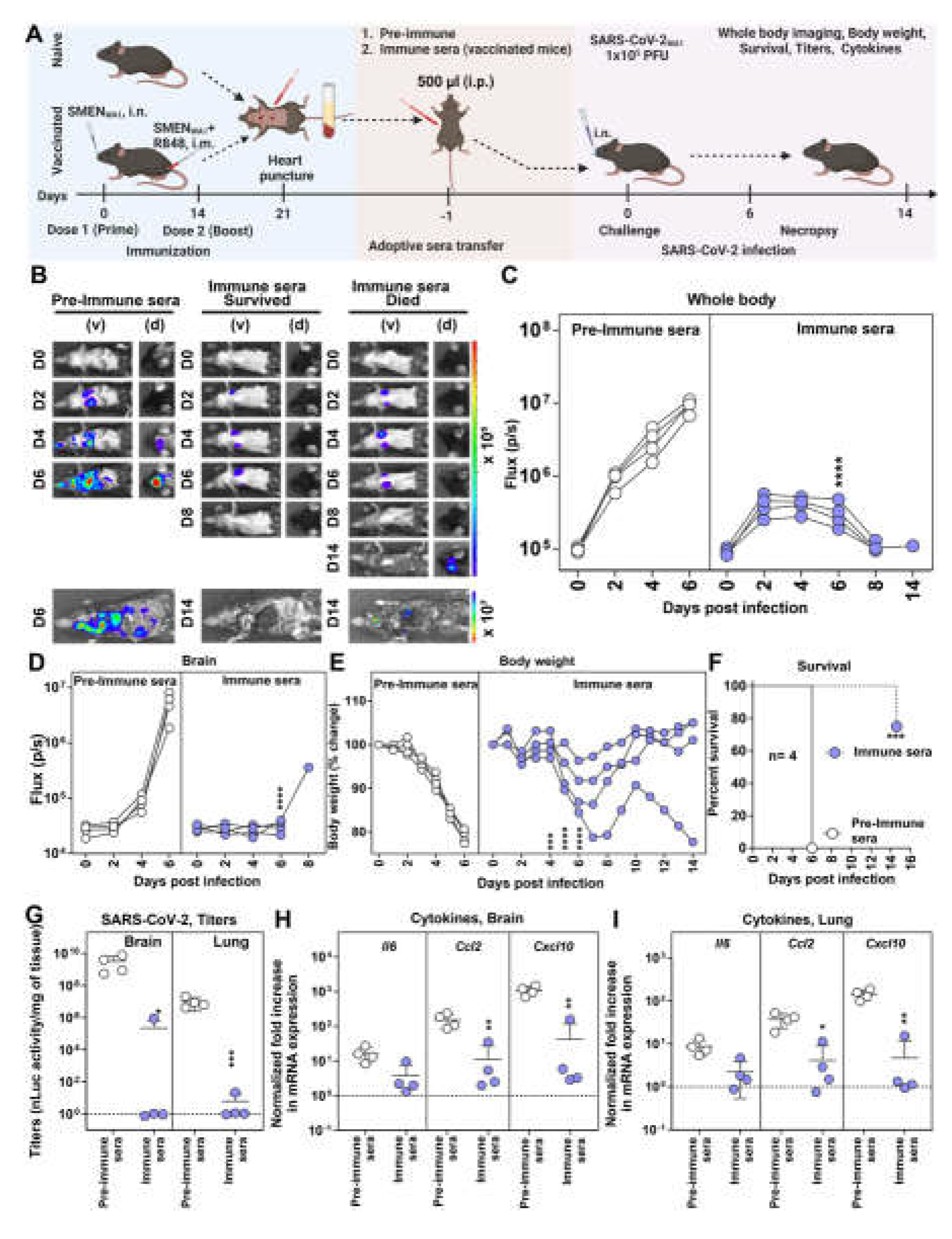
Figure 3.
Minor Yet Distinct Role of CD8+ and CD4+ T Cells in SMENWA1 Vaccine-mediated Protection. (A) Experimental design to test the contribution of CD4+ and CD8+ T cells in SMEN-vaccine meditated protection. K18-hACE2 mice were immunized as shown using the combined regimen with SMENWA1. Adjuvant treated mice served as control. αCD4 and αCD8alpha mAbs (12.5 mg/kg body weight, i.p.) were used to deplete CD4+ and CD8+ T cells every 48-h starting at 19 days post vaccination. Isotype mAb treated cohorts served as controls (Isotype). The mice (n=4 each group) were challenged with 1 x 105 PFU of SARS-CoV-2WA1-nLuc and followed by non-invasive BLI every 2 days from the start of infection till 8 dpi. (B) Representative BLI images of SARS-CoV-2-WA1-nLuc-infected mice in ventral (v) and dorsal (d) positions. Scale bars denote radiance (photons/s/cm2/steradian). (C-D) Temporal quantification of nLuc signal as flux (photons/sec) computed non-invasively in indicated regions. (E) Temporal body weight changes of K18-hACE2 mice at indicated dpi for an experiment shown in A. Each curve represents one animal. The body weight at the start of the experiment was set to 100%. (F) Kaplan-Meier survival curves of K18-hACE2 for experiment as in A. (G) Viral loads (nLuc activity/mg of tissue) from indicated organs at time of necropsy using Vero-E6 as targets. Uninfected cells were used to normalize the signals. (H-I) Fold changes in indicated cytokine mRNA expression in brain and lung tissues of mice under specified treatment regimens after necropsy upon death or at 14 dpi in the surviving animals. Data were normalized to Gapdh mRNA in the same sample and that in non-infected mice after necropsy. Grouped data in (C-E), and (G-I) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for the groups comparisons to adjuvant are shown in black, with CD4 T-cells depleted group are shown as magenta and with CD8 T-cells depleted group are shown as blue. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; ns, not significant; Mean values ± SD are depicted.
Figure 3.
Minor Yet Distinct Role of CD8+ and CD4+ T Cells in SMENWA1 Vaccine-mediated Protection. (A) Experimental design to test the contribution of CD4+ and CD8+ T cells in SMEN-vaccine meditated protection. K18-hACE2 mice were immunized as shown using the combined regimen with SMENWA1. Adjuvant treated mice served as control. αCD4 and αCD8alpha mAbs (12.5 mg/kg body weight, i.p.) were used to deplete CD4+ and CD8+ T cells every 48-h starting at 19 days post vaccination. Isotype mAb treated cohorts served as controls (Isotype). The mice (n=4 each group) were challenged with 1 x 105 PFU of SARS-CoV-2WA1-nLuc and followed by non-invasive BLI every 2 days from the start of infection till 8 dpi. (B) Representative BLI images of SARS-CoV-2-WA1-nLuc-infected mice in ventral (v) and dorsal (d) positions. Scale bars denote radiance (photons/s/cm2/steradian). (C-D) Temporal quantification of nLuc signal as flux (photons/sec) computed non-invasively in indicated regions. (E) Temporal body weight changes of K18-hACE2 mice at indicated dpi for an experiment shown in A. Each curve represents one animal. The body weight at the start of the experiment was set to 100%. (F) Kaplan-Meier survival curves of K18-hACE2 for experiment as in A. (G) Viral loads (nLuc activity/mg of tissue) from indicated organs at time of necropsy using Vero-E6 as targets. Uninfected cells were used to normalize the signals. (H-I) Fold changes in indicated cytokine mRNA expression in brain and lung tissues of mice under specified treatment regimens after necropsy upon death or at 14 dpi in the surviving animals. Data were normalized to Gapdh mRNA in the same sample and that in non-infected mice after necropsy. Grouped data in (C-E), and (G-I) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. Statistical significance for the groups comparisons to adjuvant are shown in black, with CD4 T-cells depleted group are shown as magenta and with CD8 T-cells depleted group are shown as blue. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; ns, not significant; Mean values ± SD are depicted.
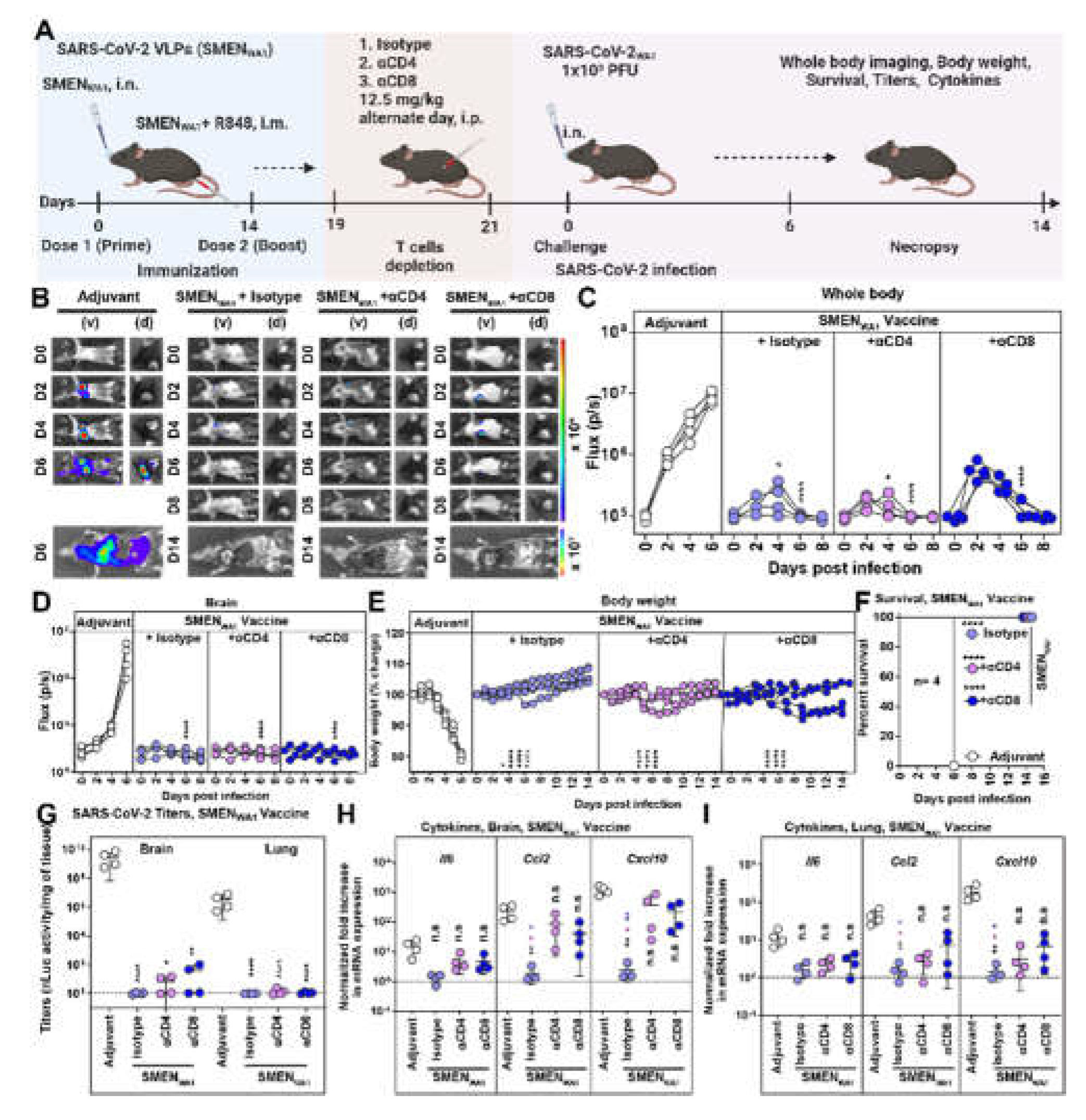
Figure 4.
SMENWA1 Vaccine is Effective Against Delta and Provides Limited Cross-protection Against Beta VOC. (A) Experimental design for testing cross protection efficacy of SMENWA1vaccine against Delta and Beta VOCs. The mice were challenged with 1 x 105 PFU of Delta and Beta SARS-CoV-2 nLuc to SMENWA1 vaccinated mice. (B) A plot showing temporal body weight changes of K18-hACE2 mice at indicated dpi for an experiment shown in A. Each curve represents one animal. The body weight at the start of the experiment was set to 100%. (C, D) Kaplan-Meier survival curves of K18-hACE2 for experiment as in A. (E) Viral loads (nLuc activity) in indicated organs from mice under specified treatment regimens. The titers were evaluated using Vero E6 cells as targets when mice died from infection or at 22 dpi for surviving mice. Uninfected cells were used to normalize the data. (F-G) Fold changes in indicated cytokine mRNA expression in brain and lung tissues of mice under specified treatment regimens after necropsy upon death or at 22 dpi in the surviving animals. Data were normalized to Gapdh mRNA in the same sample and that in non-infected mice after necropsy. (H) Plot showing neutralizing IC50 (µl of serum) values in the sera from SMENWA1 vaccinated mice (n=4; each dot represents one mouse) against WA1, Delta and Beta, Omicron variants. Cross-reactive neutralization index (CRNI) values shown below the plot were calculated (see Methods) by setting the homologous WA1 strain to 100. (E) Fold change in FcγR signaling measured using Jurkat NFAT-luciferase (JNL) cells expressing mFcγRIV, co-cultured with Vero E6 cells infected with the indicated SARS-CoV-2 variants and sera (IC50 equivalent) from SMENWA1-vaccinated mice. Data were normalized to luciferase activity measured in the absence of sera. To estimate the cross-reactive Fc-signaling index (CRFSI), the Fc-signaling activity measured using Vero E6 cells infected with the homologous WA1 strain was set to 100. Grouped data in (B), and (E-G) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. The data in (H, I) were analyzed by nonparametric Mann-Whitney T test. Statistical significance for the group comparisons in (B-G) to adjuvant with Delta VOC infected mice are shown in black, to adjuvant with beta VOC infected mice are shown in light black. Statistical significance for the group comparisons in (H-I) to WA1 are shown in black, to Delta VOC are shown in red.∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; ns, not significant; Mean values ± SD are depicted.
Figure 4.
SMENWA1 Vaccine is Effective Against Delta and Provides Limited Cross-protection Against Beta VOC. (A) Experimental design for testing cross protection efficacy of SMENWA1vaccine against Delta and Beta VOCs. The mice were challenged with 1 x 105 PFU of Delta and Beta SARS-CoV-2 nLuc to SMENWA1 vaccinated mice. (B) A plot showing temporal body weight changes of K18-hACE2 mice at indicated dpi for an experiment shown in A. Each curve represents one animal. The body weight at the start of the experiment was set to 100%. (C, D) Kaplan-Meier survival curves of K18-hACE2 for experiment as in A. (E) Viral loads (nLuc activity) in indicated organs from mice under specified treatment regimens. The titers were evaluated using Vero E6 cells as targets when mice died from infection or at 22 dpi for surviving mice. Uninfected cells were used to normalize the data. (F-G) Fold changes in indicated cytokine mRNA expression in brain and lung tissues of mice under specified treatment regimens after necropsy upon death or at 22 dpi in the surviving animals. Data were normalized to Gapdh mRNA in the same sample and that in non-infected mice after necropsy. (H) Plot showing neutralizing IC50 (µl of serum) values in the sera from SMENWA1 vaccinated mice (n=4; each dot represents one mouse) against WA1, Delta and Beta, Omicron variants. Cross-reactive neutralization index (CRNI) values shown below the plot were calculated (see Methods) by setting the homologous WA1 strain to 100. (E) Fold change in FcγR signaling measured using Jurkat NFAT-luciferase (JNL) cells expressing mFcγRIV, co-cultured with Vero E6 cells infected with the indicated SARS-CoV-2 variants and sera (IC50 equivalent) from SMENWA1-vaccinated mice. Data were normalized to luciferase activity measured in the absence of sera. To estimate the cross-reactive Fc-signaling index (CRFSI), the Fc-signaling activity measured using Vero E6 cells infected with the homologous WA1 strain was set to 100. Grouped data in (B), and (E-G) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. The data in (H, I) were analyzed by nonparametric Mann-Whitney T test. Statistical significance for the group comparisons in (B-G) to adjuvant with Delta VOC infected mice are shown in black, to adjuvant with beta VOC infected mice are shown in light black. Statistical significance for the group comparisons in (H-I) to WA1 are shown in black, to Delta VOC are shown in red.∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; ns, not significant; Mean values ± SD are depicted.
Figure 5.
SMENOmicron Provides Weak Cross-protection Against Heterologous VOCs. (A) Experimental design for testing cross protection efficacy of SMENOmicron vaccine against WA1, Delta, Beta and Omicron VOCs. Indicated groups of mice (control and vaccinated) were challenged with 1 x 105 PFU of SARS-CoV-2 nLuc variants (WA1, Delta, Beta and Omicron) (B) A plot showing temporal body weight changes (top panel) and Kaplan-Meier survival curves (bottom panel) of K18-hACE2 mice for an experiment shown in A. Each curve in top panel represents one animal. The body weight at the start of the experiment was set to 100%. (C) Viral loads (nLuc activity) in indicated organs from mice under specified conditions. The titers were evaluated using Vero E6 cells as targets when mice died from infection or at 16 dpi for surviving mice. (D) Plot showing neutralizing IC50 (µl of serum) values in the sera from SMENOmicron vaccinated mice (n=4, each dot represents one mouse) against Omicron, WA1, Delta and Beta variants. Cross-reactive neutralization index (CRNI) values shown below the plot were calculated (see Methods) by normalizing homologous Omicron variant to 100. (E) Fold change in FcγR signaling measured using Jurkat NFAT-luciferase (JNL) cells expressing mFcγRIV, co-cultured with Vero E6 cells infected with the indicated SARS-CoV-2 variants and sera (IC50 equivalent) from SMENOmicron-vaccinated mice. Data were normalized to luciferase activity measured in the absence of sera. To estimate the cross-reactive Fc-signaling index (CRFSI), the Fc-signaling activity measured using Vero E6 cells infected with the homologous Omicron strain was set to 100. Grouped data in (B, C) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. The data in (D, E) were analyzed by pairwise nonparametric Mann-Whitney test. Statistical significance for the group comparisons to adjuvant with each cohort are shown in black. Statistical significance for the group comparisons in (D, E) to Omicron are shown in black, to WA1 are shown in red, to beta are shown as blue. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗∗, p < 0.0001; ∗∗∗, p < 0.001; ns, not significant; Mean values ± SD are depicted.
Figure 5.
SMENOmicron Provides Weak Cross-protection Against Heterologous VOCs. (A) Experimental design for testing cross protection efficacy of SMENOmicron vaccine against WA1, Delta, Beta and Omicron VOCs. Indicated groups of mice (control and vaccinated) were challenged with 1 x 105 PFU of SARS-CoV-2 nLuc variants (WA1, Delta, Beta and Omicron) (B) A plot showing temporal body weight changes (top panel) and Kaplan-Meier survival curves (bottom panel) of K18-hACE2 mice for an experiment shown in A. Each curve in top panel represents one animal. The body weight at the start of the experiment was set to 100%. (C) Viral loads (nLuc activity) in indicated organs from mice under specified conditions. The titers were evaluated using Vero E6 cells as targets when mice died from infection or at 16 dpi for surviving mice. (D) Plot showing neutralizing IC50 (µl of serum) values in the sera from SMENOmicron vaccinated mice (n=4, each dot represents one mouse) against Omicron, WA1, Delta and Beta variants. Cross-reactive neutralization index (CRNI) values shown below the plot were calculated (see Methods) by normalizing homologous Omicron variant to 100. (E) Fold change in FcγR signaling measured using Jurkat NFAT-luciferase (JNL) cells expressing mFcγRIV, co-cultured with Vero E6 cells infected with the indicated SARS-CoV-2 variants and sera (IC50 equivalent) from SMENOmicron-vaccinated mice. Data were normalized to luciferase activity measured in the absence of sera. To estimate the cross-reactive Fc-signaling index (CRFSI), the Fc-signaling activity measured using Vero E6 cells infected with the homologous Omicron strain was set to 100. Grouped data in (B, C) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. The data in (D, E) were analyzed by pairwise nonparametric Mann-Whitney test. Statistical significance for the group comparisons to adjuvant with each cohort are shown in black. Statistical significance for the group comparisons in (D, E) to Omicron are shown in black, to WA1 are shown in red, to beta are shown as blue. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗∗, p < 0.0001; ∗∗∗, p < 0.001; ns, not significant; Mean values ± SD are depicted.
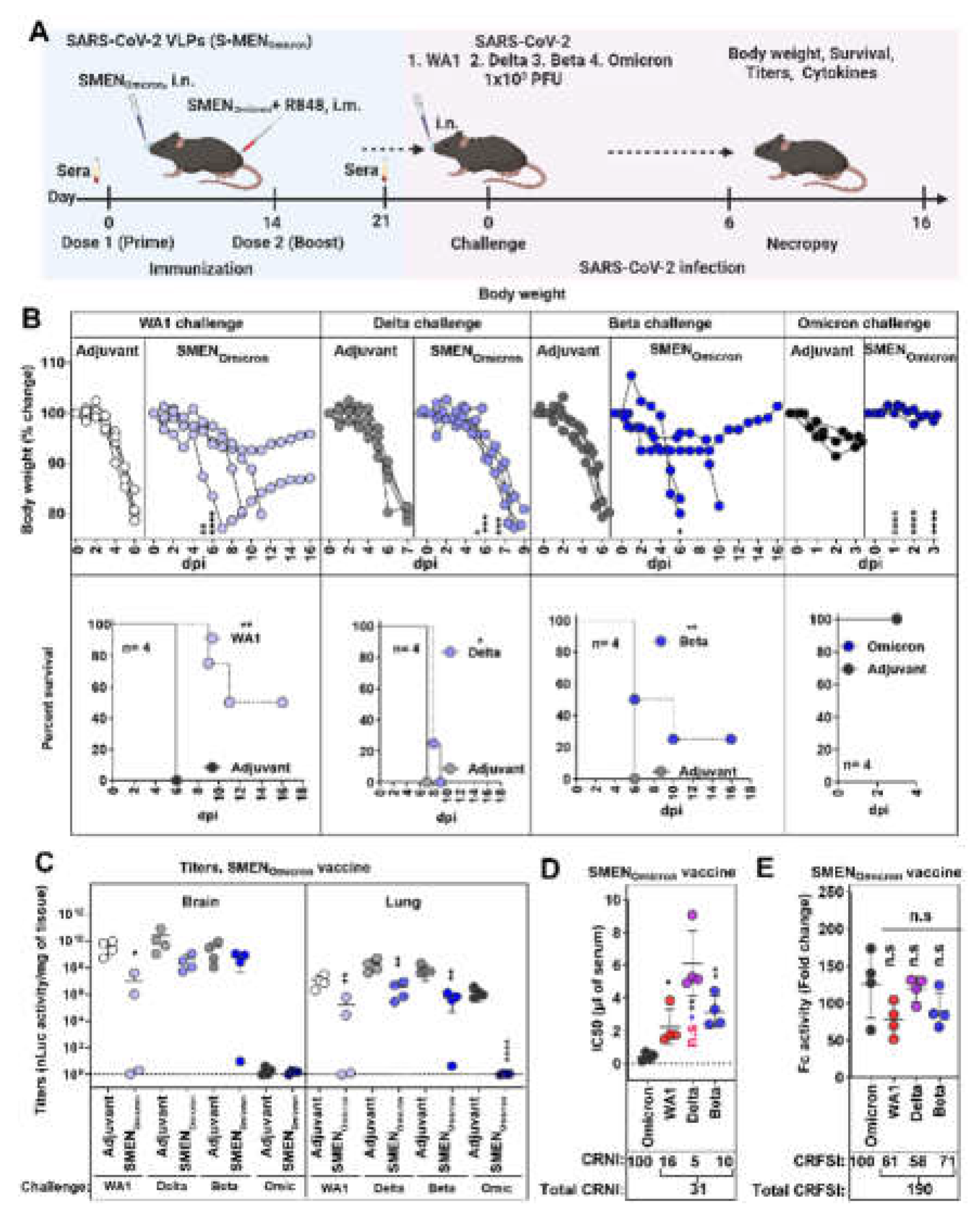
Figure 6.
SMENBeta Vaccination Confers Cross-protection Against Ancestral Strain and Heterologous VOCs. (A) Experimental design for testing protection of SMENBeta vaccine against nanoluc expressing Beta, WA1, Delta, and Omicron reporter VOCs. SMENBeta vaccinated K18-hACE2 mice (combined regimen; n=4) were challenged intranasally with 1 x 105 PFU of indicated SARS-CoV-2 variants and subjected to indicated multiparametric analyses to determine efficacy (B) Temporal body weight changes (top panel) and Kaplan-Meier survival curves (bottom panel) for experiment as in A. Each curve in the top panel represents one animal. The body weight at the start of the experiment was set to 100%. (C) Viral loads (nLuc activity) in indicated organs from mice under specified treatment regimens. The titers were evaluated using Vero E6 cells as targets at the time of death or at 16 dpi for surviving mice. Data were normalized to nLuc activity uninfected Vero E6 cells. (D) Plot showing neutralizing IC50 (µl of serum) values in the sera from SMENBeta vaccinated mice (n=4) against Beta, WA1, Delta and Omicron variants. Cross-reactive neutralization index (CRNI) values shown below the plot were calculated (see Methods) by normalizing the mean IC50 values in the sera for the homologous Beta variant to 100. (E) Fold change in FcγR signaling measured using Jurkat NFAT-luciferase (JNL) cells expressing mFcγRIV, co-cultured with Vero E6 cells infected with the indicated SARS-CoV-2 variants and sera (IC50 equivalent) from SMENBeta-vaccinated mice. Data were normalized to luciferase activity measured in the absence of sera. To estimate the cross-reactive Fc-signaling index (CRFSI), the Fc-signaling activity measured using Vero E6 cells infected with the homologous Beta strain was set to 100. Grouped data in (B, and C) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. The data in (D and E) were analyzed by pair-wise nonparametric Mann-Whitney T test. Statistical significance for the group comparisons to adjuvant with each cohort are shown in black. Statistical significance for the group comparisons in (D, E) to beta are shown in black. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗∗, p < 0.0001; ∗∗∗, p < 0.001; ns, not significant; Mean values ± SD are depicted.
Figure 6.
SMENBeta Vaccination Confers Cross-protection Against Ancestral Strain and Heterologous VOCs. (A) Experimental design for testing protection of SMENBeta vaccine against nanoluc expressing Beta, WA1, Delta, and Omicron reporter VOCs. SMENBeta vaccinated K18-hACE2 mice (combined regimen; n=4) were challenged intranasally with 1 x 105 PFU of indicated SARS-CoV-2 variants and subjected to indicated multiparametric analyses to determine efficacy (B) Temporal body weight changes (top panel) and Kaplan-Meier survival curves (bottom panel) for experiment as in A. Each curve in the top panel represents one animal. The body weight at the start of the experiment was set to 100%. (C) Viral loads (nLuc activity) in indicated organs from mice under specified treatment regimens. The titers were evaluated using Vero E6 cells as targets at the time of death or at 16 dpi for surviving mice. Data were normalized to nLuc activity uninfected Vero E6 cells. (D) Plot showing neutralizing IC50 (µl of serum) values in the sera from SMENBeta vaccinated mice (n=4) against Beta, WA1, Delta and Omicron variants. Cross-reactive neutralization index (CRNI) values shown below the plot were calculated (see Methods) by normalizing the mean IC50 values in the sera for the homologous Beta variant to 100. (E) Fold change in FcγR signaling measured using Jurkat NFAT-luciferase (JNL) cells expressing mFcγRIV, co-cultured with Vero E6 cells infected with the indicated SARS-CoV-2 variants and sera (IC50 equivalent) from SMENBeta-vaccinated mice. Data were normalized to luciferase activity measured in the absence of sera. To estimate the cross-reactive Fc-signaling index (CRFSI), the Fc-signaling activity measured using Vero E6 cells infected with the homologous Beta strain was set to 100. Grouped data in (B, and C) were analyzed by 2-way ANOVA followed by Tukey’s multiple comparison tests. The data in (D and E) were analyzed by pair-wise nonparametric Mann-Whitney T test. Statistical significance for the group comparisons to adjuvant with each cohort are shown in black. Statistical significance for the group comparisons in (D, E) to beta are shown in black. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗∗, p < 0.0001; ∗∗∗, p < 0.001; ns, not significant; Mean values ± SD are depicted.
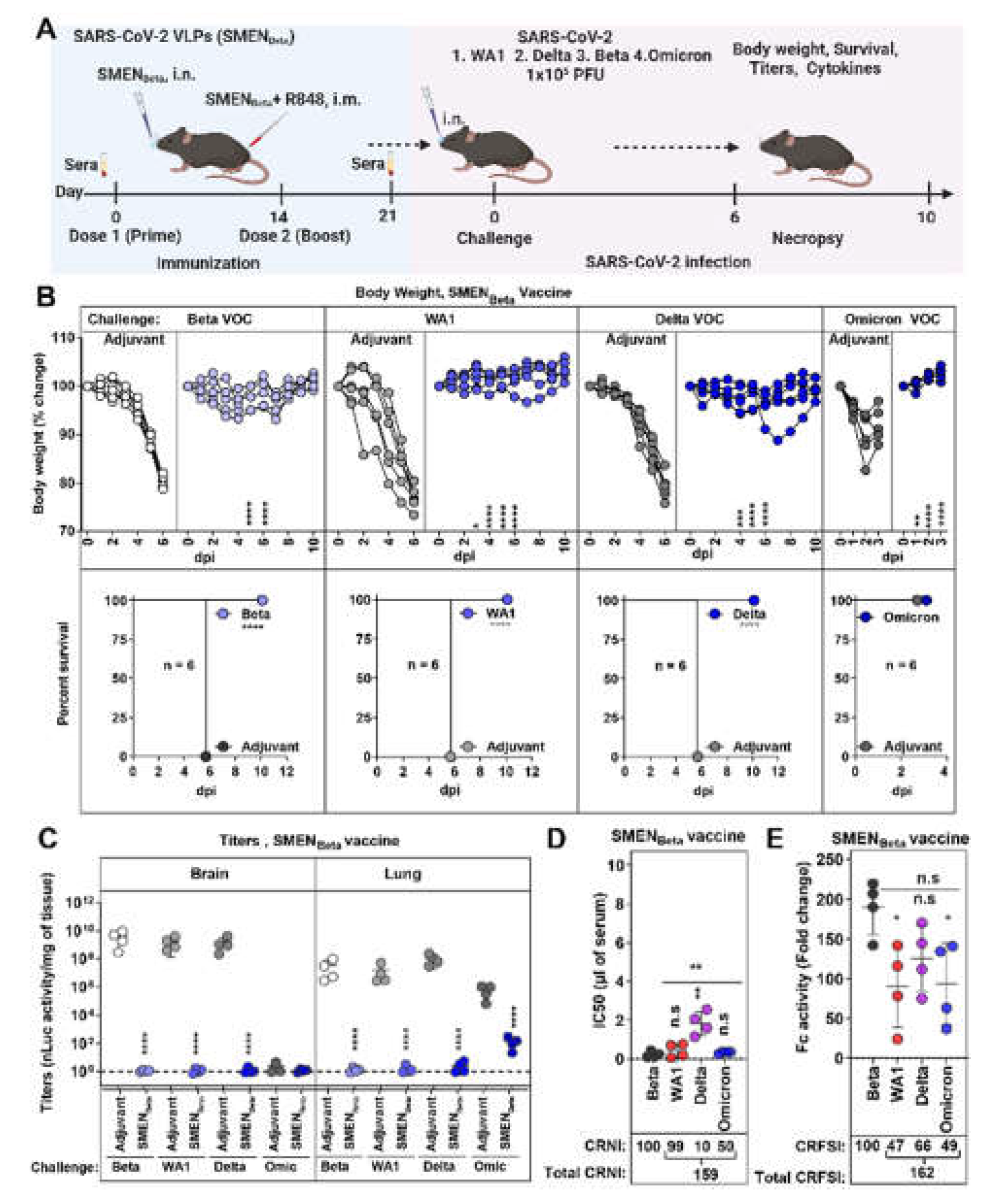
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



