Submitted:
26 May 2024
Posted:
13 June 2024
You are already at the latest version
Abstract
Objective To investigate the functional and biological significance of telomere-related active genes in cholangiocarcinoma (CCA) using single-cell sequencing and cell communication analysis. Methods First, we identified and annotated different cell types in CCA samples using single-cell sequencing data. Pseudotime analysis revealed the transcriptional status and differentiation process of CCA cells. We then explored interactions between CCA cells and their regulatory pathways through cell communication analysis. 47 key genes related to telomere activity were identified, and GO and KEGG pathway enrichment analysis was performed. Additionally, we constructed a prognostic model and verified the model's predictive efficacy. Finally, we identified genes associated with drug sensitivity in CCA patients and identified pathways associated with CCA prognosis through tumor TMB analysis. Results We identified 452 active telomere-associated cells. Then, we assessed telomere associated gene activity, obtaining 47 differentially expressed telomere genes by intersecting with CCA differentially expressed genes. Major up-regulated signaling pathways, including MIF-(CD74+CXCR4) and HLA-A-CD8A, were identified through pseudotime analysis. Three telomere-associated prognostic genes were identified using LASSO analysis, and prognostic-related models were built. In addition, PBRM1 mutation load was found to be highest in the high-risk group, making it a potential diagnostic target. Finally, drug sensitivity analysis revealed higher sensitivity to certain therapeutic drugs in the high-risk group, providing guidance for tailored CCA treatment. Conclusion The MIF-(CD74+CXCR4) and HLA-A-CD8A signaling pathways appear to be major pathways for CCA cell proliferation. Prognostic models based on telomere-associated genes show promise for application, indicating the biological and clinical significance of telomere activity genes in CCA.
Keywords:
CCA
; telomere
; single-cell sequencing
; Cell communication
; pseudotime analysis
1. Introduction
CCA is a very deadly form of adenocarcinoma that develops in the epithelial cells of the bile ducts within the hepatobiliary system [1,2,3]. Worldwide, approximately 106,000 people are diagnosed with CCA each year, accounting for 0.3% of all cancers, and the incidence is higher in Asian populations compared to European and North American populations [4]. As early symptoms of bile duct cancer are not obvious, most of the patients have already progressed to advanced stages by the time of diagnosis, losing the opportunity for surgical treatment and ultimately affecting the prognosis [5]. Globally, about 99,000 people die from bile duct cancer every year, accounting for about 0.4% of all cancer mortality. Hence, there is an immediate requirement to develop prognostic models using novel biomarkers in order to enhance the unfavorable prognosis of patients with CCA.
Telomeres, consisting of repetitive DNA sequences found at the tips of chromosomes, are essential for preserving chromosome stability and integrity [6,7]. Telomere shortening and damage are closely related to the development of many diseases such as aging and cancer. Studies have shown that telomere length in malignant cells is usually longer than in normal cells, which is associated with the unlimited proliferative capacity and resistance to apoptosis of tumor cells. Regulating this process is a group of highly conserved regulatory factors, telomere-related genes. telomere-related genes are genes related to telomere structure, function, and regulation of telomerase [6]. Abnormal expression or mutation of these genes will lead to abnormal shortening or damage of telomeres, which will trigger chromosomal instability and abnormal cellular function. In tumor development, the abnormal expression of telomere-related genes is closely related to the shortening of telomeres and chromosomal rearrangements. These abnormalities lead to genomic instability, which promotes the proliferation, metastasis, and drug resistance of tumor cells. Therefore, telomere-associated genes play an important role in regulating tumor development by modulating the stability and function of telomeres [8]. In CCA, several telomere-associated genes are differentially expressed and play a wide range of biological roles, and these genes may be closely related to the treatment and prognosis of CCA. However, the complex role and prognostic significance of telomere-associated genes in CCA are still largely unknown [9,10,11].
The contribution of single-cell sequencing technology to tumor research has been immense. It has enabled researchers to gain insight into the cellular heterogeneity and evolutionary processes of tumors [12]. Single-cell sequencing data are important in identifying cellular heterogeneity, constructing cellular trajectories, and analyzing intracell communication and ligand receptor expression. By analyzing the genomes, transcriptomes, and epigenomes of individual cells, we can reveal the differences between different tumor cells, identify potential therapeutic targets, and provide a basis for personalized therapy. The rapid development of single-cell genomics in recent years has revealed the unknown biological processes and disease causes of CCA [13].
In this study, transcriptomic data, clinical data, and mutation data of CCA were downloaded through TCGA and GEO databases. Single-cell analysis was performed based on single-cell sequencing datasets to identify different cell types and telomere-associated active cells in CCA. The pseudotime analysis to construct cell trajectories and identify branching events, and cell communication analysis to reveal the interactions and signaling pathways between CCA cells. Next, we performed an enrichment analysis of telomere activity-related differential genes and explored the functions of these genes in biological processes and pathways. We constructed prognostic models and column line plots based on the acquired prognostic-related genes, predicted the overall survival of patients, and evaluated the efficacy of the models. Furthermore, we conducted gene set enrichment analysis, gene set variant analysis, immune infiltration analysis, and drug sensitivity analysis to uncover the molecular characteristics and biological pathways underlying CCA. This comprehensive approach offers valuable insights for diagnosing and treating CCA, with the ultimate goal of discovering novel biomarkers and enhancing prognostic prediction for patients with this disease.
Materials and Methods
2.1. Transcriptome Data Download and Preparation
All data used in this study are freely available to the public, mainly from the TCGA (The Cancer Genome Atlas, https://portal.gdc.cancer.gov/) database and GEO (Gene Expression Omnibus, https://www.ncbi. nlm.nih.gov/geo/) databases. CCA expression profiling data, and clinical data were downloaded from the TCGA database via the R package “TCGAbiolinks (version 2.25.0)” [14] as well as by the Single nucleotide mutation (SNV) data predicted by the “VarScan2 Variant Aggregation and Masking” tool were downloaded from the TCGA database. 36 tumor samples and 9 paraneoplastic control samples from TCGA-CHOL and 30 tumor samples from GSE107943 were included in this study. The ComBat method using the R package “sva” [15] corrected for the batch effect of non-biotechnology bias. Principal component analysis (PCA) was used to check the extent of correction.
2.2. Downloading and Processing of Single-Cell Sequencing Data
The GEO database contains a large amount of single-cell sequencing data. In this study, the single-cell sequencing dataset GSE138709 of CCA, which contains five CCA disease samples and three paracancerous controls, was obtained from the GEO database. The single-cell raw data in GSE138709 were imported using the Seurat package (version 4.2.0) [16] for the R programming language. First, low-quality cells and genes were filtered by the following criteria: 1) Cells expressing fewer than 300 genes were removed; 2) Genes expressed in fewer than 1 cell were removed. 3) Cells with fluctuating numbers of expressed genes between 200 and 6,000 were retained. 4) Cells with a percentage of mitochondrial genes of less than 10% were retained. 5) Cells with UMI readings of less than 40,000 were retained. The data were normalized using the “normalizedata” function in the Seurat R package. After data normalization, highly variable genes in single cells were identified by balancing the relationship between average expression and dispersion. Principal component analysis (PCA) was then performed and significant principal components (PCs) were used as input for graph-based clustering. A harmony approach was used to eliminate batch effects across samples. For clustering, we used the function FindClusters, which is based on the Shared Nearest Neighbor (SNN) modular optimized clustering algorithm, to generate 18 clusters on 20 PC components at a resolution of 0.3. The “Runtsne” function is then used for t-distributed random neighbor embedding (t-SNE). Cell aggregation was demonstrated using t-SNE-1 and t-SNE-2. Subsequently, cell clusters were identified by cell type-specific biomarkers, and the proportion of cell types was calculated and evaluated.
2.3. Telomere-Related Gene Scoring
The R package “AUCell” [17] scores each cell based on gene set enrichment analysis. A gene expression ranking for each cell was generated based on the area under the curve (AUC) values of 2089 human telomere-associated genes (Table S1) obtained from the TelNet (http://www.cancertels-ys.org/telnet/) database to estimate the proportion of the gene set with high expression in each cell. Cells that express more genes in the gene set have higher AUC values. The “AUCell_exploreThresholds” function was used to determine a threshold for identifying active cells in the gene set. The AUC score of each cell was then mapped to the TSNE embedding using the “ggplot2” R package (version 3.3.5) to visualize the activated cell clusters.
2.4. Constructing Cellular Trajectories by Pseudotime Analysis
The pseudotime analysis was performed by Monocle 2 [18], which performs reverse graph embedding based on a user-defined list of genes to generate a pseudo-temporal graph that can account for branching and linear differentiation processes. For the proposed temporal analysis of cells with high telomere activity, the raw count data were normalized by estimating a size factor for trajectory inference. Pseudotemporal trajectories were constructed using genes with a high degree of discretization and high expression (discretization estimate ≥ 1 and mean expression ≥ 0.1) [19]. default values were selected for the parameters of the DDRTree algorithm. To further analyze these branching events, we used branching expression analysis modeling (BEAM) implemented in Monocle 2. It helps to identify all genes with significant branching-dependent expression. Branch-dependent expression patterns were visualized as heatmaps using Monocle 2.
2.5. Cell Communication Analysis and Ligand-Receptor Expression
CellChat objects were created by the “CellChat” R package (https://www.github.com/sqjin/CellChat) [20] based on the UMI count matrix of each group (CCA and control). Cell-to-cell communication was analyzed using the “CellChatDB.human” ligand-receptor interaction database as the reference database with default parameters. The “mergeCellChat” function was used to combine the CellChat objects of each group to obtain a comparison of the total number of interactions and the intensity of interactions. The “netVisual_diffInteraction” function was used to visualize the differences in the number or strength of interactions between different cell types between groups. Finally, we used the “netVisual_bubble” and “netVisual_aggregate” functions to visualize the distribution of signaling gene expression between groups.
2.6. GO/KEGG Pathway Enrichment Analysis
Gene Ontology (GO) [21]e nrichment analysis includes biological process (BP), molecular function (MF), and cellular component (CC) analysis. GO enrichment analysis of genes was performed using the R package “clusterProfiler (version 4.2.2)” [22]. Kyoto Encyclopedia of Genes and Genomes (KEGG) [23] is a bioinformatics resource for mining gene lists enriched with significantly altered metabolic pathways. The KEGG enrichment analysis of telomere-related differentially expressed genes (DEGs) in CCA was performed using the R package “clusterProfiler” (p-value <0.05).
2.7. Construction and Validation of Prognostic Models
The genes obtained by taking the intersection with telomere-related genes as key genes were analyzed by the results of the difference analysis of telomere high-activity and low-activity cells in the single-cell dataset, the results of the difference analysis of disease and control, and the results of the difference analysis of the CCA and normal groups in the CCA dataset of TCGA. To assess the prognostic value of the key genes, we used one-way Cox regression analysis to assess the correlation between each gene and survival in the tumor cohort, and genes with P ≤ 0.05 were considered to be significantly associated with survival for further analysis. All CCA tumor samples (n=66) with clinical information were randomly divided into training and validation sets in a 7:3 ratio. The LASSO Cox regression model (R package “glmnet” [24]) was then utilized to narrow down the candidate genes and construct prognostic models. The penalty parameter (λ) was determined by the minimum criterion. Risk scores were calculated using the following formula:
(Coef (genei): risk factor, Expression (genei): gene expression level)
The training group samples were divided into low-risk and high-risk groups based on the optimal cut-off value determined by the surv_cutpoint function. Survival curves were then generated to assess prognostic significance utilizing the Kaplan-Meier method, and statistical significance was evaluated through the log-rank test. A subject operating characteristic curve (ROC) was used to validate the efficacy of the prognostic model. AUC values typically ranged from 0.5 to 1, with closer to 1 indicating better model efficacy. The validation set was also divided into low-risk and high-risk groups for validation of the prognostic model.
2.8. Construction and Validation of Column-Line Diagrams
Clinical information of patients (age, gender and disease stage, etc.) was extracted from the TCGA cohort. A one-way Cox regression analysis was performed combining the clinical information with the risk scores obtained from the prognostic model. In addition, we combined the prognostic and clinical characteristics and constructed a column chart using the R package “RMS” to predict overall survival at 1, 3, and 5 years. The effectiveness of the column plot model was evaluated by ROC curves.
2.9. Gene Set Enrichment Analysis (GSEA)
The Gene Set Enrichment Analysis (GSEA) algorithm assesses whether a predefined set of genes exhibits statistically significant differences between two biological conditions.GSEA is performed with the R package “clusterProfiler (version 4.2.2)” on an ordered list of all genes aligned according to log2FC, with 1000 gene set alignments performed for each analysis. In this study, c2.cp.kegg.v7.5.1.symbols was used as the reference gene set, which is stored in the Molecular Signatures Database (MSigDB) [25,26,27] database. p < 0.05 was considered to be a gene set with a P < 0.05. 0.05 of the gene set was considered significantly enriched.
2.10. Gene Set Variation Analysis (GSVA)
To investigate the differences in biological function between high and low-risk groups, Gene Set Variation Analysis (GSVA) was performed using the R package “GSVA (version 1.42.0)” based on “c2.cp.kegg.v7.5.1.symbols”. GSVA). The R package “pheatmap (version 1.0.12)” was used to visualize the results.
2.11. Immune Infiltration Analysis
Single Sample Gene Set Enrichment Analysis (ssGSEA) [PMID: 36090900], an extension of Gene Set Enrichment Analysis (GSEA), calculates separate enrichment scores for each pair of samples and gene sets. Each ssGSEA enrichment score represents the extent to which genes in a particular gene set are coordinately up-or down-regulated in the sample. Based on 28 immune cells downloaded from the TISIDB (Tumor and Immune System Interactions Database) (http://cis.hku.hk/TISIDB/index.php) [28] database, including Activated CD8 T cell, Central memory CD8 T cell, Effector memory CD8 T cell, Activated CD4 T cell, Central memory CD4 T cell, Effector memory CD4 T cell, T follicular helper cell, Gamma delta cell, T cell, and T cell. helper cell, Gamma delta T cell, Type 1 T helper cell, Type 17 T helper cell, Type 2 T helper cell, Regulatory T cell, Activated B cell, Immature B cell, Memory B cell, Natural killer cell, CD56bright natural killer cell, CD56dim natural killer cell, Myeloid derived suppressor cell, Natural killer T cell, Activated dendritic cell, Type 1 T helper cell, Type 17 T helper cell, Type 2 T helper cell, Regulatory T cell Activated dendritic cell, Plasmacytoid dendritic cell, Immature dendritic cell, Macrophage, Eosinophil, Mast cell, Monocyte and Neutrophil, Quantification of gene expression profiles for each CCA sample from each CCA disease samples to quantify the relative enrichment score of each immune cell from the gene expression profiles. Differences in immune cell infiltration levels between high and low-risk groups were visualized by the R package “ggplot2 (version 3.3.6)” [29].
2.12. Somatic Mutation Analysis
A landscape of genomic mutational variation was depicted based on mutation data. the R package “maftools” was used to display somatic variants between different subgroups, including single nucleotide polymorphisms (SNPs), insertions and deletions (INDELs), tumor mutational load (TMB), and mutation frequency [30]. Generally, frequently mutated genes (FMGs) with the top 20 mutation frequencies are considered to be the main driver genes of malignant tumors [31].
2.13. Drug Sensitivity Analysis
Based on the half-maximal inhibitory concentration (IC50) data and corresponding gene expression data downloaded from the GDSC database (Genomics of Drug Sensitivity in Cancer, https://www.cancerrxgene.org/) [32], the R package was used “oncoPredict (version 0.2)” [33] to predict potential therapeutic drug sensitivities in patients in the high and low-risk groups of CCA.
2.14. Statistical Analysis
This study was statistically analyzed using R software v4.1.2. Spearman correlation test was used to infer the correlation between two parameters. The Wilcoxon test was employed to assess disparities between two groups, while the Kruskal-Wallis test was applied to evaluate differences among three or more groups. A two-tailed p-value below 0.05 was deemed to indicate statistical significance. The detailed analysis process is shown in Figure 1.
3. Results
3.1. Single-Cell Dimensionality Reduction Clustering and Annotation
To explore the origin of highly expressed genes, we analyzed cell populations from CCA samples using the single-cell sequencing dataset GSE138709. After the initial quality control assessment, we obtained a total of 29,983 cells from the single-cell transcriptome. 8 samples were included in the study, and all cells were aggregated into 18 clusters (Figure 2A), and different cell types were annotated by cell-specific biomarkers based on the gene expression profile of each cluster (Table S2). As shown in Figure 2B, 10 cell types can be found, such as NK cells, malignant cells, and B cells. specific genes for each cell type were visualized by dot plots (Figure 2C). The proportions of different cell types in the control samples (GSM4116579, GSM4116582, GSM4116586) are shown in Figure 2D; the proportions of different cell types in the disease samples (GSM4116580, GSM4116581, GSM4116583, GSM4116584, GSM4116585) are Figure 2E shown.
3.2. Characterization of Telomere-Related Activity
A subpopulation of active cells was utilized to study the expression pattern of telomere-associated genes at the single-cell level. Active cells were identified by the optimal threshold, which showed the presence of 452 telomere-associated active cells, the population of cells with AUC values greater than 0.25 (Figure 3A). Figure 3B highlights the TSNE plot of the active cells and Figure 3C shows the telomere activity of all cells, the darker the color the higher the telomere activity, from Figure 3B and 3C it can be seen that the telomere active cells are mainly malignant cells.
3.3. Pseudotime Analysis
Using telomere-associated active cells, we built a pseudo-temporal cell trajectory to find the important gene expression programs that determine CCA progression. Indeed, the transcriptional states in the trajectory revealed different processes. malignant cells were distributed in different branches, suggesting that malignant cells have different transcriptional states (Figure 4A–D). To elucidate the molecular basis of malignant cells transformation, we explored the genes that determine the branching of malignant cells in CCA. Genes highly expressed in pre-branch were mainly enriched in GO BP pathways such as wound healing and skin development. Genes enriched in response to toxic substance-related pathways were highly expressed in cell branch 2, and genes enriched in T cell activation, antigen processing, and presentation of exogenous peptide antigen via MHC genes in class II were highly expressed in cell branch 1 (Figure 4E).
3.4. Cell Communication Analysis
To further investigate the cellular network of interactions in CCA, we used the R package “Cellchat” to reveal changes in crosstalk between CCA cells. The network plot showed an overall increase in the number of interactions between CCA cell types, an increase in the strength of interactions associated with B cells, fibroblasts, and hepatocytes, and a general decrease in the strength of interactions associated with NK cells (Figure 5A,B). We then further compared the signaling patterns between cells from the CCA group (Figure 5C,D). NK cells were the major signal receivers. Subsequently, from the probability point plots of communication signaling between malignant cells and the ligand-receptor pairs between NK cells and B cells, we could see that the stronger changing communication signaling pairs were MIF - (CD74+CXCR4), HLA-A - CD8A, etc. (Figure 6A). We also explored the interaction of malignant cells with other cell types through the MIF, MHC-I signaling pathway where the above signaling pairs are located (Figure 6B,C). These results preliminarily elucidated the potential interactions between these cell types and helped us to further investigate the integrative role of malignant cells in CCA. As can be seen from Figure 7A–D expression of specific pathway ligand receptors, ligand HLA-A was highly expressed in NK cells.
3.5. Enrichment Analysis of Telomere Activity-Related Differentially Expressed Genes
A total of 805 differentially expressed genes (DEGs 1) were identified by taking the intersection of the results of the differential analysis of telomere high and low activity cells in the single-cell dataset (n=1674, Table S3), and the results of the differential analysis of disease vs. control (n=2225, Table S4), and the differences of these genes between the two groups were statistically significant (p-value < 0.05, | Log2 fold change| > 0.25). We then compared the CCA and normal groups in the TCGA CCA dataset and identified a total of 3658 differentially expressed genes (Table S5), and intersected with telomere-associated genes (n=2089) to obtain 472 telomere-associated differentially expressed genes (DEGs 2), which were statistically different between the two groups (p-value < 0.05, | Log2 fold change| > 2). The distribution of telomere-associated differential genes was demonstrated by volcano plots, and the top 5 up-regulated genes (RALY, LCMT1, CD2BP2, FUS, CCDC115) and top 5 down-regulated genes (LINC01831, C3orf85, LINC02754, AC016682.1, SLC6A13) were illustrated by heat maps (Figure 8A,B). The two sets of differential genes were taken to intersect to obtain 47 key genes (hub genes, Table S6) as shown by the Wayne diagram (Figure 8C).
In order to investigate the biological functions associated with the differential genes, we analyzed the enrichment of telomere-associated differential genes for GO entries (Table S7) and KEGG pathway (Table S8), which are shown by lollipop diagrams. The GO results showed that these genes were mainly involved in “DNA replication”, “positive regulation of DNA metabolic process”, “regulation of DNA metabolic process”, “regulation of DNA metabolic process”, and “DNA metabolism”. “regulation of DNA metabolic process” and “chromosomal region” (BP). Enriched in biological processes such as “chromosomal region”, “chromosome, telomeric region”, “nuclear chromosome”, etc. (BP). Enriched in “chromosomal region”, “chromosome, telomeric region”, “nuclear chromosome” and other cellular components (CC). Enriched in “catalytic activity, acting on DNA”, “single-stranded DNA binding”, “single-stranded DNA helicase”. “single-stranded DNA helicase activity” were enriched in molecular functions (MF) (Figure 8D). KEGG pathway includes “Cell cycle”, “DNA replication”, “Cellular senescence”, etc. (Figure 8E).
3.6. Construction and Validation of Prognostic Models
A univariate Cox analysis was used to identify key genes associated with prognosis (p < 0.05), and disease samples in TCGA and GEO were integrated to finally identify five genes associated with CCA prognosis (DDX39A, SNRPE, TMEM109, ANXA4, ATP1A1). 7/10 CCA samples (66 in total) were selected by random sampling as the training set (n=44) and 3/10 as the validation set (n=22). LASSO regression analysis was performed on the training set to remove redundant genes, and the random number seed was set to 17. A total of 3 genes associated with the prognosis of CCA patients were identified (Table S9), and the results are shown in Figure 9A,B. To determine the robustness of the model constructed using the 3 gene features, the samples were categorized into high-risk and low-risk groups based on the median risk value as a threshold. KM survival curves were constructed for different groups of patients in the training cohort (Figure 9C) and validation cohort (Figure 9D). The results showed that patients in the high-risk group had a significantly worse prognosis compared to patients in the low-risk group in all cohorts. ROC curves were used to determine the efficacy of the model in predicting patient prognosis. In the training cohort, the AUC values for 1, 3, and 5year survival were 0.701, 0.717, and 0.769, respectively (Figure 9E). In the validation cohort, the AUC values for 1, 3, and 5 year survival were 0.779, 0.720, and 0.759, respectively (Figure 9F).
3.7. GSEA and GSVA
To further explore the potential mechanisms of differentially expressed genes, we performed a GSEA enrichment analysis. Using the pathway information in the MsigDB database as a reference, we selected the most significant pathways based on the normalized enrichment score (NES) (Table S10). The results of the GSEA analysis indicated that DRUG METABOLISM CYTOCHROME P450, RETINOL METABOLISM, COMPLEMENT AND COAGULATION CASCADES, TIGHT JUNCTION, ECM RECEPTOR INTERACTION, and FOCAL ADHESION were the most significant pathways. FOCAL ADHESION and other pathways and biological processes were more active in CCA (Figure 10A–F, P< 0.05). In addition, we performed GSVA enrichment analysis using the pathway information in the MsigDB database as a reference and selected the pathways with the most significant differences between the high- and low-risk groups to draw pathway activity maps (Figure 10G, Table S11).
3.8. Immune Infiltration Analysis
We further investigated the level of infiltration of 28 immune cell types in the high- and low-risk groups using the ssGSEA method. The heat map of correlation between immune cells showed that most immune cells were positively correlated with each other (Figure 11A). Immune cells central memory CD4 T cell, Immature B cell, etc. showed significant differences between high and low-risk groups (P value <0.05, Figure 11B). In addition, we examined the significant correlation between prognostic genes and corresponding immune cells and found that ATP1A1 was positively correlated with Central memory CD4 T cell (R=0.327, p < 0.05) and negatively correlated with Immature B cell (R=-0.25, p < 0.05) (Figure 11C,D).
3.9. Construction of Nomogram Model
In order to verify whether risk score could be used as a prognostic factor, a one-way Cox regression analysis was performed on the clinical characteristics of patients (e.g., age, disease stage, and gender). The results showed that risk score and disease stage were independent prognostic risk factors for patients (Figure 12A). The results of constructing a nomogram showed that risk score significantly predicted clinical outcomes (Figure 12B). ROC curves were used to determine the efficacy of the nomogram in predicting patient prognosis. The results showed that the nomogram model had good stability and accuracy at 1, 3, and 5 years (Figure 12C).
3.10. TMB and Drug Sensitivity Analysis
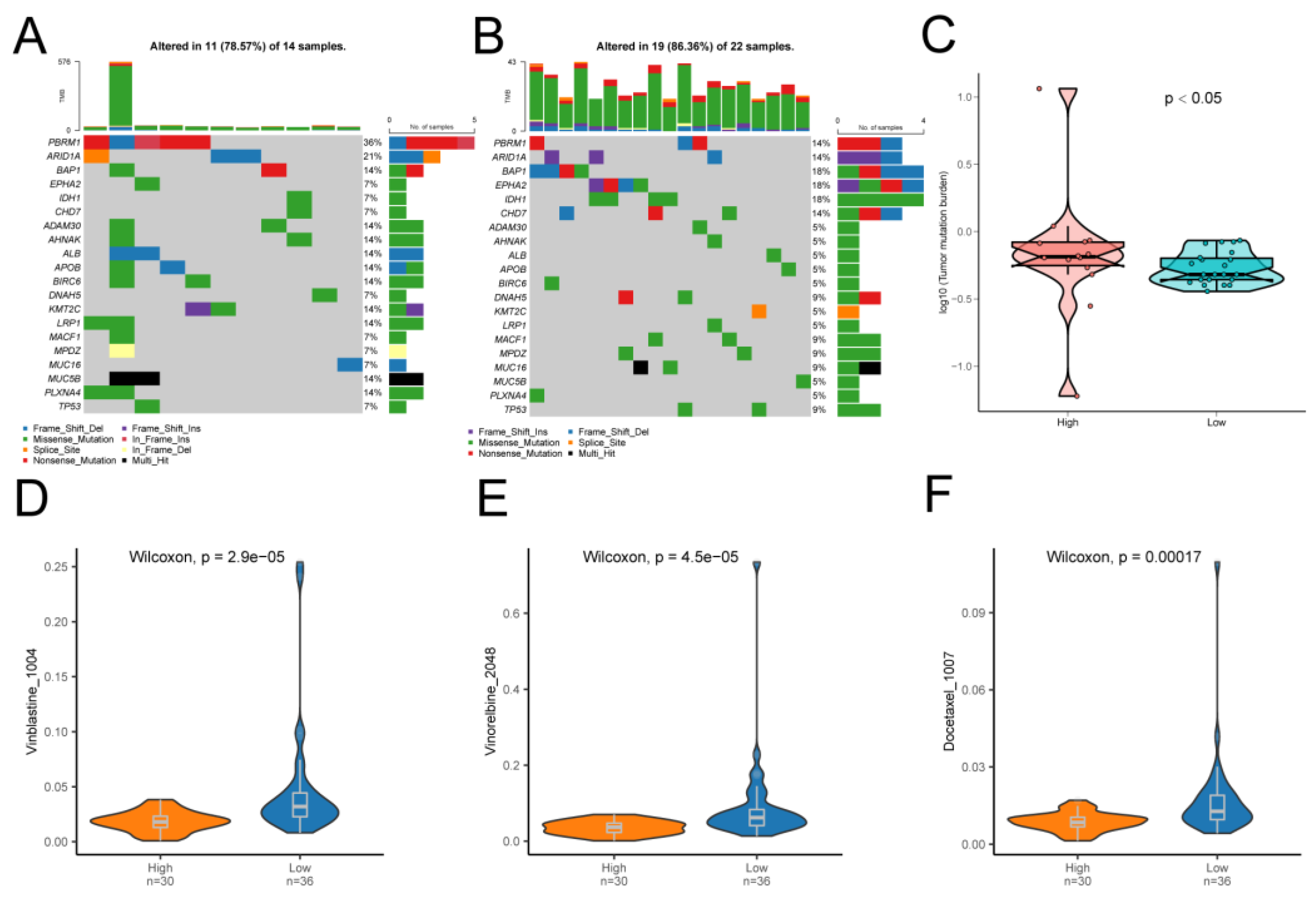
We also assessed specific gene mutations in CCA and visualized the top 20 genes with the highest mutation frequency. PBRM1 had the highest mutation frequency in both high and low-risk groups and was much more frequent in the high-risk group than in the low-risk group (Figure 13A,B). Tumor mutational load (TMB) is a key criterion for the success of immunotherapy. Therefore, somatic mutations associated with CCA were analyzed, and TMB was significantly higher in the high-risk group than in the low-risk group (Figure 13C, P<0.05). We analyzed whether the risk score accurately predicted chemotherapy sensitivity in patients with CCA. The results showed that high-scoring patients were more sensitive to Vinblastine_1004 (Figure 13D), Vinorelbine_2048 (Figure 13E), Docetaxel_1007 (Figure 13F) and other drugs.
Discussion
In order to prevent incomplete replication and instability that can occur at the ends of linear DNA, eukaryotic chromosomes end with characteristic repetitive DNA sequences within special structures called telomeres. Regarding the functions and roles of telomeres, which have attracted the attention of researchers for a long time. Earlier studies have shown a strong relationship between telomeres and human aging. As early as 1990, researchers found that the during aging in vitro and in vivo, the number and length of telomeric DNA in human fibroblasts actually does decrease with successive generations [34]. In 2002, it was demonstrated that aging at the clonal level occurs through telomere shortening (a region of DNA near the end of a chromosome) shortening. Telomere shortening leads to chromosome instability, breakage and mutation, which in turn leads to an increased rate of cancer in people of advanced age [35]. Then, with continuous research, telomeres have been found to play an very important role in tumorigenesis and development. About 85% of tumor cells maintain telomere length by activating telomerase, and about 15% of tumor cells use homologous recombination or other mechanisms to maintain telomere length when telomerase is inactivated or insufficient, which are collectively known as alternative lengthening of telomere (ALT). The presence of ALT enables tumor cells to maintain long telomere length and thus achieve unlimited proliferation [36]. This result was confirmed in different cancers, but it seems that the relationship between telomere length and cancer is not a simple one-dimensional relationship, and different directions of influence were shown between different cancers. For example. For instance, one study analyzed leukocyte telomere length (LTL) in relation to prostate cancer (PCa) using Mendelian randomization and found that longer LTL was associated with a higher risk of PCa [37]. But in another study it was mentioned that sustained cell proliferation or rapid cell renewal through hepatocellular injury leads to a multi-step hepatocellular carcinogenesis process. Therefore, progressive shortening of telomeres and activation of telomerase may be useful markers for early detection of malignant progression of liver disease [38,39]. And a recent study mentioned more frequent telomerase reverse transcriptase (TERT) promoter mutations in HCC with significantly higher TERT expression and longer telomere length. TERT promoter mutations were associated with higher TERT levels and longer telomere length and were also independent predictors of poorer overall survival after hepatectomy [40].
What is puzzling is that cancer cells generally possess long telomeres, which seems to go against conventional knowledge? The plausible answer is that cancer develops through a succession of mutations that bypass cell cycle checkpoints when cell replication is out of control. Thus, shorter telomeres may constitute a fail-safe mechanism against potentially cancerous cells. Most cancers show telomerase activity or employ other mechanisms (ALT pathway) to bypass the telomere barrier [41,42,43]. It follows that the relatively short telomeres in humans are likely to be an evolutionary trade-off, with telomeres becoming shorter as we age, sacrificing the opportunity to further extend our lifespan in exchange for a strong resistance to cancer.
In cholangiocarcinoma, a search of the literature revealed a recent study that investigated telomere length and the risk of developing cholangiocarcinoma, showing an inverse linear relationship. People with the shortest telomeres had a 1.86 times higher risk of developing bile duct cancer compared to those with the longest telomeres [44]. However, until now, no studies have been conducted to investigate the relationship between telomere-related genes and the pathogenesis of cholangiocarcinoma. In this study, we investigated the biological functions of telomere-associated genes in cholangiocarcinoma for the first time.
In our study, we identified different cell types in CCA samples and annotated them by cell communication analysis based on public database gene sequencing as well as single-cell sequencing data. Then, by assessing the activity of telomere-associated genes, telomere-active cells were identified mainly as malignant cells, and 452 telomere-associated active cells were found. Pseudotime analysis was performed to reveal the transcriptional status and differentiation process of CCA cells. In addition, the important gene expression programs in CCA were further explored, and malignant cells were found to have an important role in the proliferation of CCA cells via MIF-(CD74+CXCR4), HLA-A-CD8A, and the ligand HLA-A was highly expressed in NK cells. The next studies targeting the two up-regulated pathways may bring new inspirations and ideas for further mechanistic elaboration and diagnosis of CCA cells.
By taking the intersection of differentially expressed genes with telomere activity genes, 47 key genes related to telomere activity were identified. The enrichment analysis of GO and KEGG pathways for telomere-related differential genes revealed that they were mainly involved in DNA replication, positive regulation of DNA metabolic process, and other biological functions. They are also involved in important cellular metabolic pathways such as “Cell cycle”, “DNA replication” and “Cellular senescence”, suggesting that telomere-associated differential genes are closely related to the cell cycle, cell proliferation, and cellular metabolism. genes are closely related to cell cycle and cell proliferation. The GSEA analysis showed that drug metabolism cytochrome P450, retinol metabolism, complement and coagulation cascades, tight tunction, ECM receptor interaction, focal adhesion and other important cellular metabolic pathways are also relatively active in CCA. Studies targeting these pathways may shed more light on the mechanism of telomere activity genes in CCA.
Next, three genes associated with the prognosis of CCA patients were identified using one-way Cox analysis and LASSO regression analysis. A prognostic model was constructed based on the expression levels of these three genes related to the prognosis of CCA patients and the predictive efficacy of the model was verified. The results showed that the prognostic model showed good predictive efficacy and had the potential for further clinical research. However, further clinical studies are needed to demonstrate this.
Tumor immunity is always an essential topic in oncology research, and this study also addresses tumor immunity in CCA. In the process of exploring tumor immunity, most of the immune cells analyzed among immune cells were positively correlated with each other, suggesting that different immune cells are in a mutually reinforcing relationship. We also observed that the immune cells Central memory CD4 T cell and Immature B cell showed significant differences between high and low-risk groups. The prognostic gene ATP1A1 was positively correlated with Central memory CD4 T cell and Immature B cell, suggesting that the prognostic gene ATP1A1 may be closely related to tumor immunity in CCA.
Finally, we found a very high mutation rate and high TMB of PBRM1 in patients with CCA, which may be the next therapeutic target for CCA to be further explored. The drug sensitivity analysis also revealed that patients in the high-risk group were more sensitive to Vinblastine_1004, Vinorelbine_2048, Docetaxel_1007, etc., this provides new inspiration and reference for the fine-tuned treatment of CCA, which is meaningful and necessary for further investigation.
This study is comprehensive in exploring the telomere-related active genes in CCA, there are limitations in this study as well. Although this study was conducted as scientifically and objectively as possible, there are still some irresistible interfering factors, such as this study is based on data from public databases, and the results are affected by the original data. Secondly, the data of the study came from different organizations, and there are inaccuracies that have an impact on the results.
Conclusions
In summary, this study reveals the functional characteristics and biological mechanisms of telomere-related active genes in CCA through the comprehensive analysis of single-cell transcriptomes and TCGA databases, which provides an important reference for CCA research and treatment.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
Ethics Approval and Consent to Picipate
Not applicable to this study.
Data Availability
The data used in this study were obtained from The Cancer Genome Atlas (TCGA, HTTPS:// portal. GDC. cancer. gov/)database and the GEO (https://www.ncbi.nlm.nih.gov/geo/) database, both of which are available in publicly available databases. This study complies with its data use and publication rules.
Conflicts of Interest
We declare that there is no conflict of interest regarding the publication of this paper.
Funding Statement
No funding was used in this study.
Publication Statement
Not Applicable.
References
- Brindley PJ, Bachini M, Ilyas SI, Khan SA, Loukas A, Sirica AE, Teh BT, Wongkham S, Gores GJ: Cholangiocarcinoma. NAT REV DIS PRIMERS 2021, 7(1):65.
- Razumilava N, Gores GJ: Cholangiocarcinoma. LANCET 2014, 383(9935):2168-2179.
- Moeini A, Haber PK, Sia D: Cell of origin in biliary tract cancers and clinical implications. JHEP REP 2021, 3(2):100226.
- Vithayathil M, Khan SA: Current epidemiology of cholangiocarcinoma in Western countries. J HEPATOL 2022, 77(6):1690-1698.
- Rizvi S, Gores GJ: Pathogenesis, diagnosis, and management of cholangiocarcinoma. GASTROENTEROLOGY 2013, 145(6):1215-1229.
- Giardini MA, Segatto M, Da SM, Nunes VS, Cano MI: Telomere and telomerase biology. PROG MOL BIOL TRANSL 2014, 125:1-40.
- Rhodes D, Fairall L, Simonsson T, Court R, Chapman L: Telomere architecture. EMBO REP 2002, 3(12):1139-1145.
- Frias C, Pampalona J, Genesca A, Tusell L: Telomere dysfunction and genome instability. FRONT BIOSCI-LANDMRK 2012, 17(6):2181-2196.
- Londono-Vallejo JA: Telomere instability and cancer. BIOCHIMIE 2008, 90(1):73-82.
- Aragona M, Pontoriero A, Panetta S, La Torre I, La Torre F: [The role of telomere-binding proteins in carcinogenesis]. MINERVA MED 2000, 91(11-12):299-304.
- Lu W, Zhang Y, Liu D, Songyang Z, Wan M: Telomeres-structure, function, and regulation. EXP CELL RES 2013, 319(2):133-141.
- Slovin S, Carissimo A, Panariello F, Grimaldi A, Bouche V, Gambardella G, Cacchiarelli D: Single-Cell RNA Sequencing Analysis: A Step-by-Step Overview. Methods Mol Biol 2021, 2284:343-365.
- Hedlund E, Deng Q: Single-cell RNA sequencing: Technical advancements and biological applications. MOL ASPECTS MED 2018, 59:36-46.
- Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I et al.: TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. NUCLEIC ACIDS RES 2016, 44(8):e71.
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD: The sva package for removing batch effects and other unwanted variation in high-throughput experiments. BIOINFORMATICS 2012, 28(6):882-883.
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R: Integrating single-cell transcriptomic data across different conditions, technologies, and species. NAT BIOTECHNOL 2018, 36(5):411-420.
- Van de Sande B, Flerin C, Davie K, De Waegeneer M, Hulselmans G, Aibar S, Seurinck R, Saelens W, Cannoodt R, Rouchon Q et al.: A scalable SCENIC workflow for single-cell gene regulatory network analysis. NAT PROTOC 2020, 15(7):2247-2276. [CrossRef]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, Trapnell C: Reversed graph embedding resolves complex single-cell trajectories. NAT METHODS 2017, 14(10):979-982.
- Karmaus P, Chen X, Lim SA, Herrada AA, Nguyen TM, Xu B, Dhungana Y, Rankin S, Chen W, Rosencrance C et al.: Metabolic heterogeneity underlies reciprocal fates of T(H)17 cell stemness and plasticity. NATURE 2019, 565(7737):101-105.
- Fang Z, Tian Y, Sui C, Guo Y, Hu X, Lai Y, Liao Z, Li J, Feng G, Jin L et al.: Single-Cell Transcriptomics of Proliferative Phase Endometrium: Systems Analysis of Cell-Cell Communication Network Using CellChat. FRONT CELL DEV BIOL 2022, 10:919731. [CrossRef]
- Gene Ontology Consortium: going forward. NUCLEIC ACIDS RES 2015, 43(Database issue):D1049-D1056.
- Yu G, Wang LG, Han Y, He QY: clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012, 16(5):284-287.
- Kanehisa M, Goto S: KEGG: kyoto encyclopedia of genes and genomes. NUCLEIC ACIDS RES 2000, 28(1):27-30.
- Friedman J, Hastie T, Tibshirani R: Regularization Paths for Generalized Linear Models via Coordinate Descent. J STAT SOFTW 2010, 33(1):1-22.
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al.: Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. P NATL ACAD SCI USA 2005, 102(43):15545-15550. [CrossRef]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P: The Molecular Signatures Database (MSigDB) hallmark gene set collection. CELL SYST 2015, 1(6):417-425.
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP: Molecular signatures database (MSigDB) 3.0. BIOINFORMATICS 2011, 27(12):1739-1740.
- Ru B, Wong CN, Tong Y, Zhong JY, Zhong S, Wu WC, Chu KC, Wong CY, Lau CY, Chen I et al.: TISIDB: an integrated repository portal for tumor-immune system interactions. BIOINFORMATICS 2019, 35(20):4200-4202. [CrossRef]
- Ito K, Murphy D: Application of ggplot2 to Pharmacometric Graphics. CPT-PHARMACOMET SYST 2013, 2(10):e79.
- Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP: Maftools: efficient and comprehensive analysis of somatic variants in cancer. GENOME RES 2018, 28(11):1747-1756.
- Liu Z, Wang L, Guo C, Liu L, Jiao D, Sun Z, Wu K, Zhao Y, Han X: TTN/OBSCN ‘Double-Hit’ predicts favourable prognosis, ‘immune-hot’ subtype and potentially better immunotherapeutic efficacy in colorectal cancer. J CELL MOL MED 2021, 25(7):3239-3251.
- Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR et al.: Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. NUCLEIC ACIDS RES 2013, 41(Database issue):D955-D961. [CrossRef]
- Maeser D, Gruener RF, Huang RS: oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. BRIEF BIOINFORM 2021, 22(6).
- Harley CB, Futcher AB, Greider CW: Telomeres shorten during ageing of human fibroblasts. NATURE 1990, 345(6274):458-460.
- Parwaresch R, Krupp G: Telomere biology and the molecular basis of aging. Arkh Patol 2002, 64(3):37-39.
- Aviv A: Telomeres and human somatic fitness. J GERONTOL A-BIOL 2006, 61(8):871-873.
- Wan B, Lu L, Lv C: Mendelian randomization study on the causal relationship between leukocyte telomere length and prostate cancer. PLOS ONE 2023, 18(6):e286219.
- Miura N, Horikawa I, Nishimoto A, Ohmura H, Ito H, Hirohashi S, Shay JW, Oshimura M: Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet Cytogenet 1997, 93(1):56-62.
- Nishimoto A, Miura N, Oshimura M: [Clinical significance of telomerase activity in precancerous lesion of the liver (adenomatous hyperplasia)]. Nihon Rinsho 1998, 56(5):1244-1247.
- Ningarhari M, Caruso S, Hirsch TZ, Bayard Q, Franconi A, Vedie AL, Noblet B, Blanc JF, Amaddeo G, Ganne N et al.: Telomere length is key to hepatocellular carcinoma diversity and telomerase addiction is an actionable therapeutic target. J HEPATOL 2021, 74(5):1155-1166. [CrossRef]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW: Specific association of human telomerase activity with immortal cells and cancer. SCIENCE 1994, 266(5193):2011-2015.
- Stewart SA: Telomere maintenance and tumorigenesis: an “ALT”ernative road. CURR MOL MED 2005, 5(2):253-257.
- Stewart SA, Hahn WC, O’Connor BF, Banner EN, Lundberg AS, Modha P, Mizuno H, Brooks MW, Fleming M, Zimonjic DB et al.: Telomerase contributes to tumorigenesis by a telomere length-independent mechanism. P NATL ACAD SCI USA 2002, 99(20):12606-12611. [CrossRef]
- Gellert-Kristensen H, Bojesen SE, Tybjaerg-Hansen A, Stender S: Telomere length and risk of cirrhosis, hepatocellular carcinoma, and cholangiocarcinoma in 63,272 individuals from the general population. HEPATOLOGY 2023.
Figure 1.
Flowchart of this study.
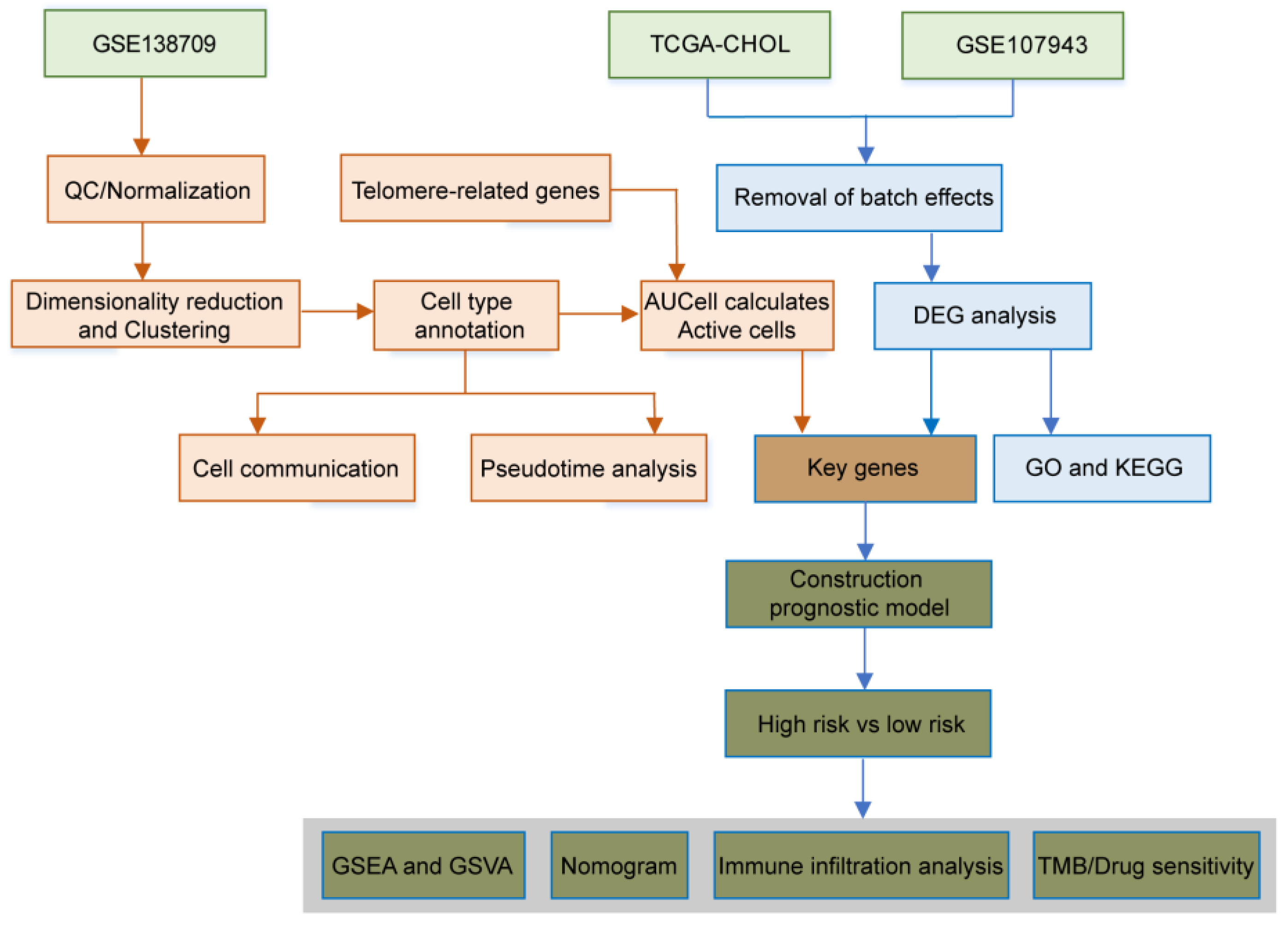
Figure 2.
Identification of cellular subpopulations from single-cell sequencing data. (A) TSNE plot showing the distribution of single-cell subpopulations. (B) TSNE plot showing the annotation results of single-cell cell subpopulations. (C) Expression of marker genes in each cell type. (D) Fan plot showing the distribution of different cell types in control samples. (E) Fan plot showing the distribution of different cell types in CCA samples.
Figure 2.
Identification of cellular subpopulations from single-cell sequencing data. (A) TSNE plot showing the distribution of single-cell subpopulations. (B) TSNE plot showing the annotation results of single-cell cell subpopulations. (C) Expression of marker genes in each cell type. (D) Fan plot showing the distribution of different cell types in control samples. (E) Fan plot showing the distribution of different cell types in CCA samples.
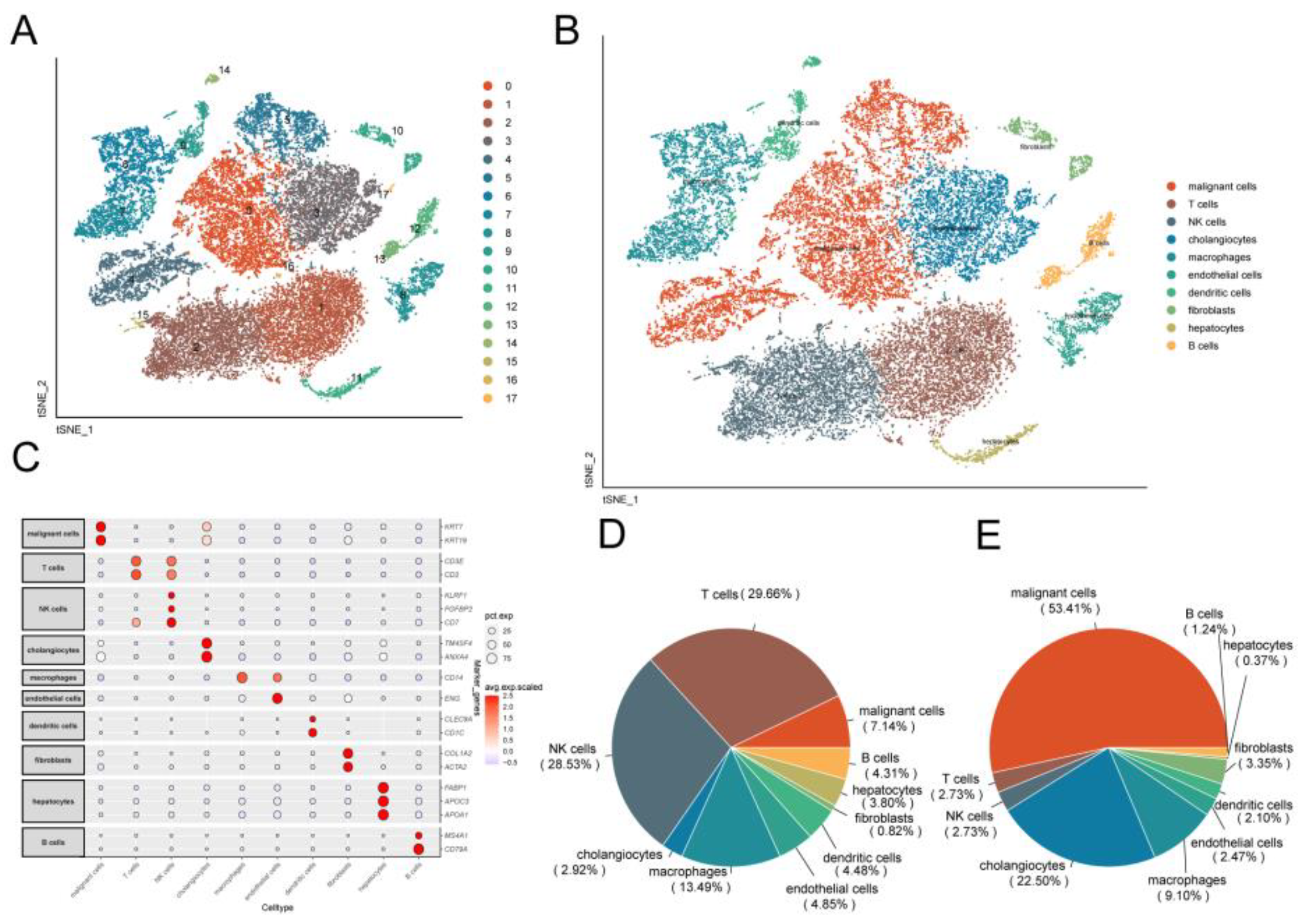
Figure 3.
Identification of telomere-associated activity. (A) AUC score of telomere-associated genes with a threshold value of 0.25. (B) TSNE plot of the distribution of cells with AUC values greater than the threshold, with screened cells in blue. (C) TSNE chromaticity plot showing the scoring of cell activity, the brighter the color, the higher the activity.
Figure 3.
Identification of telomere-associated activity. (A) AUC score of telomere-associated genes with a threshold value of 0.25. (B) TSNE plot of the distribution of cells with AUC values greater than the threshold, with screened cells in blue. (C) TSNE chromaticity plot showing the scoring of cell activity, the brighter the color, the higher the activity.
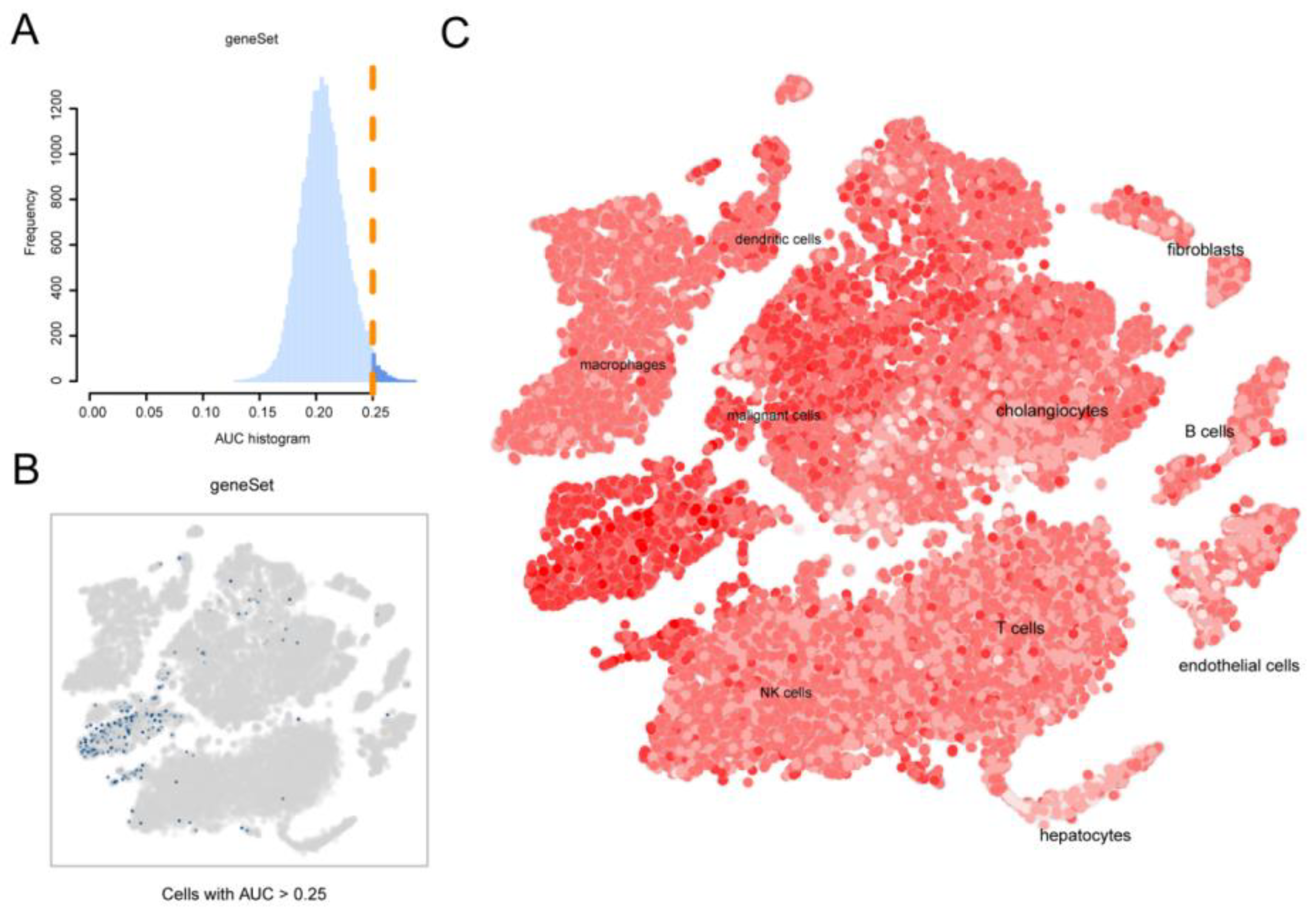
Figure 4.
pseudotime analysis reveals cellular transcription patterns. (A) Pseudotime trajectories showing cell distribution based on cell type. (B) Pseudo-time traces were categorized by Monocle2 into three different states. (C) Pseudotime color gradient transitions from dark blue to light blue. (D) Stacked histogram showing the distribution of cell types in different states. (E) Heatmap showing different branches. (cell fate) of differentially expressed genes (DEGs). GO pathways significantly enriched in different gene clusters in the heatmap are shown to the left.
Figure 4.
pseudotime analysis reveals cellular transcription patterns. (A) Pseudotime trajectories showing cell distribution based on cell type. (B) Pseudo-time traces were categorized by Monocle2 into three different states. (C) Pseudotime color gradient transitions from dark blue to light blue. (D) Stacked histogram showing the distribution of cell types in different states. (E) Heatmap showing different branches. (cell fate) of differentially expressed genes (DEGs). GO pathways significantly enriched in different gene clusters in the heatmap are shown to the left.
Figure 5.
Results of intercell communication analysis. (A) Network diagram demonstrating the number of interactions between cells. (B) Network diagram showing the strength of interactions between cells. Heatmap visualizing the signals sent (C) and received (D) by cells in the CCA group.
Figure 5.
Results of intercell communication analysis. (A) Network diagram demonstrating the number of interactions between cells. (B) Network diagram showing the strength of interactions between cells. Heatmap visualizing the signals sent (C) and received (D) by cells in the CCA group.
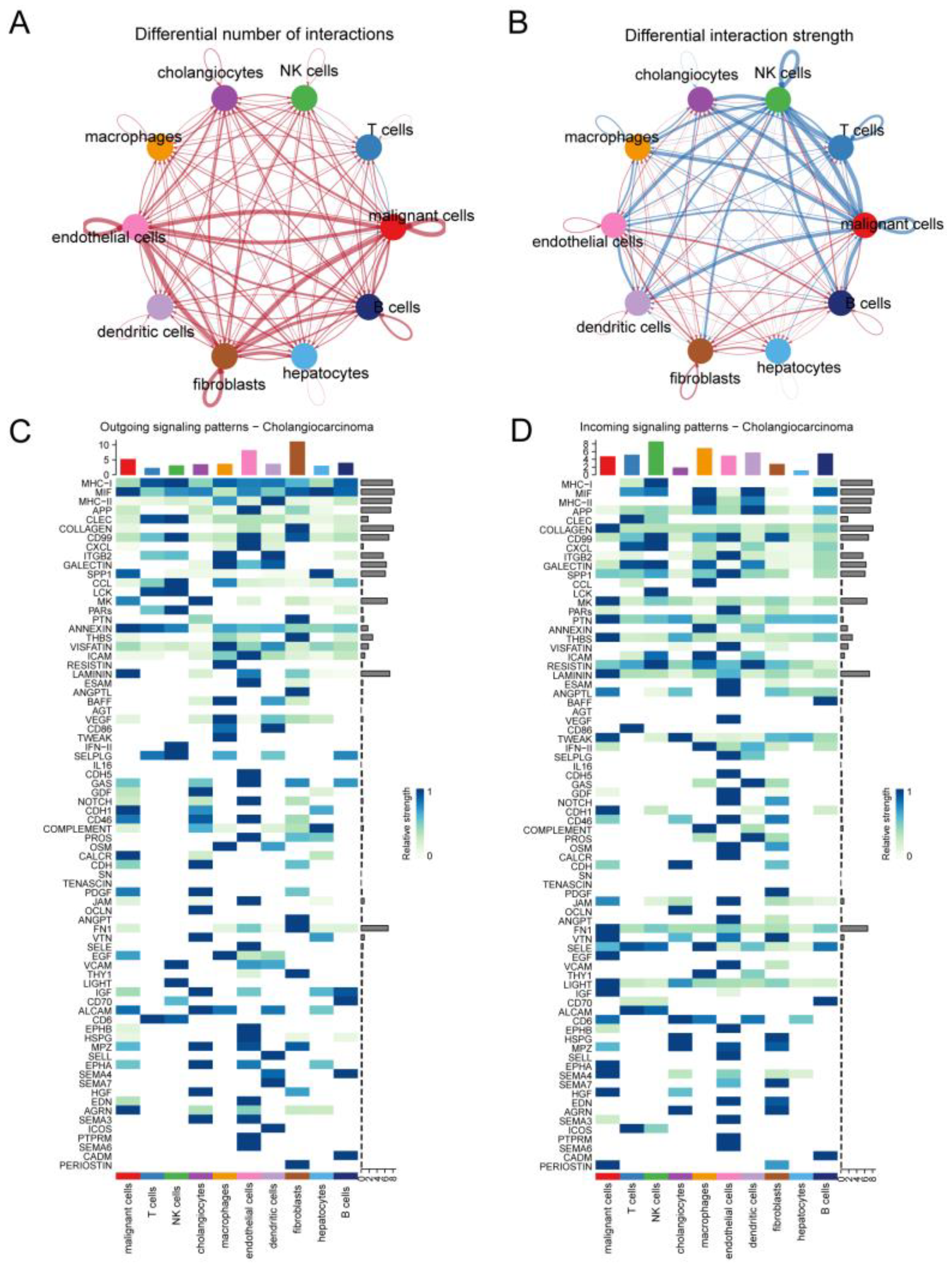
Figure 6.
Changes in intercell communication in CCA. (A) Dot plots showing signaling pathways in which malignant cells communicate with NK cells and B cells with increased and decreased strength. String diagrams show malignant cells interacting with other cell types through specific signaling pathways MIF (B), and MHC-I (C).
Figure 6.
Changes in intercell communication in CCA. (A) Dot plots showing signaling pathways in which malignant cells communicate with NK cells and B cells with increased and decreased strength. String diagrams show malignant cells interacting with other cell types through specific signaling pathways MIF (B), and MHC-I (C).
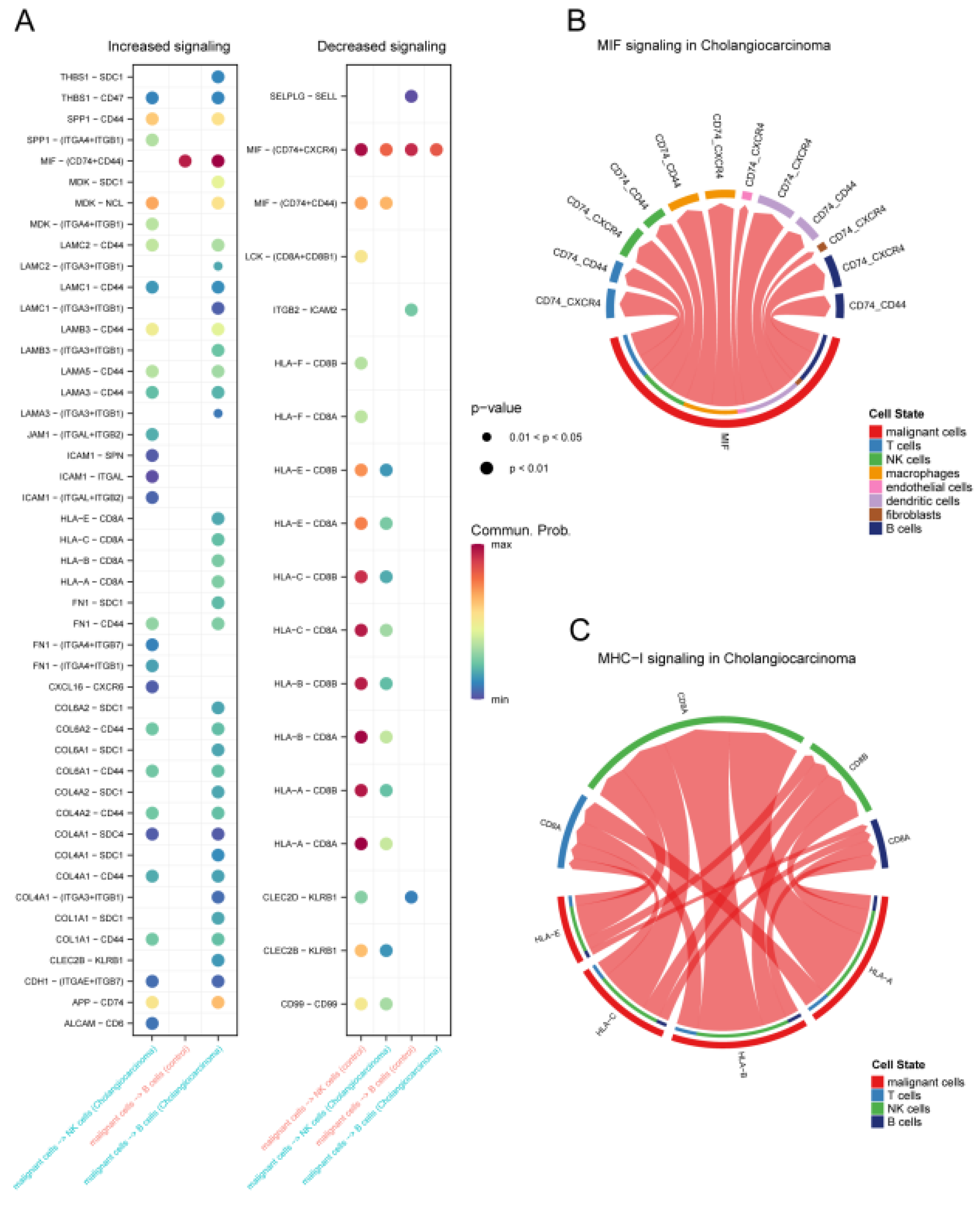
Figure 7.
Expression of specific pathway ligand receptors in different cells of CCA. Dot plots (A) and violin plots (B) of ligand receptors of the MIF pathway in CCA expressed in different cells. Dot plots (C) and violin plots (D) of ligand receptors of the MHC-I pathway in CCA expressed in different cells.
Figure 7.
Expression of specific pathway ligand receptors in different cells of CCA. Dot plots (A) and violin plots (B) of ligand receptors of the MIF pathway in CCA expressed in different cells. Dot plots (C) and violin plots (D) of ligand receptors of the MHC-I pathway in CCA expressed in different cells.
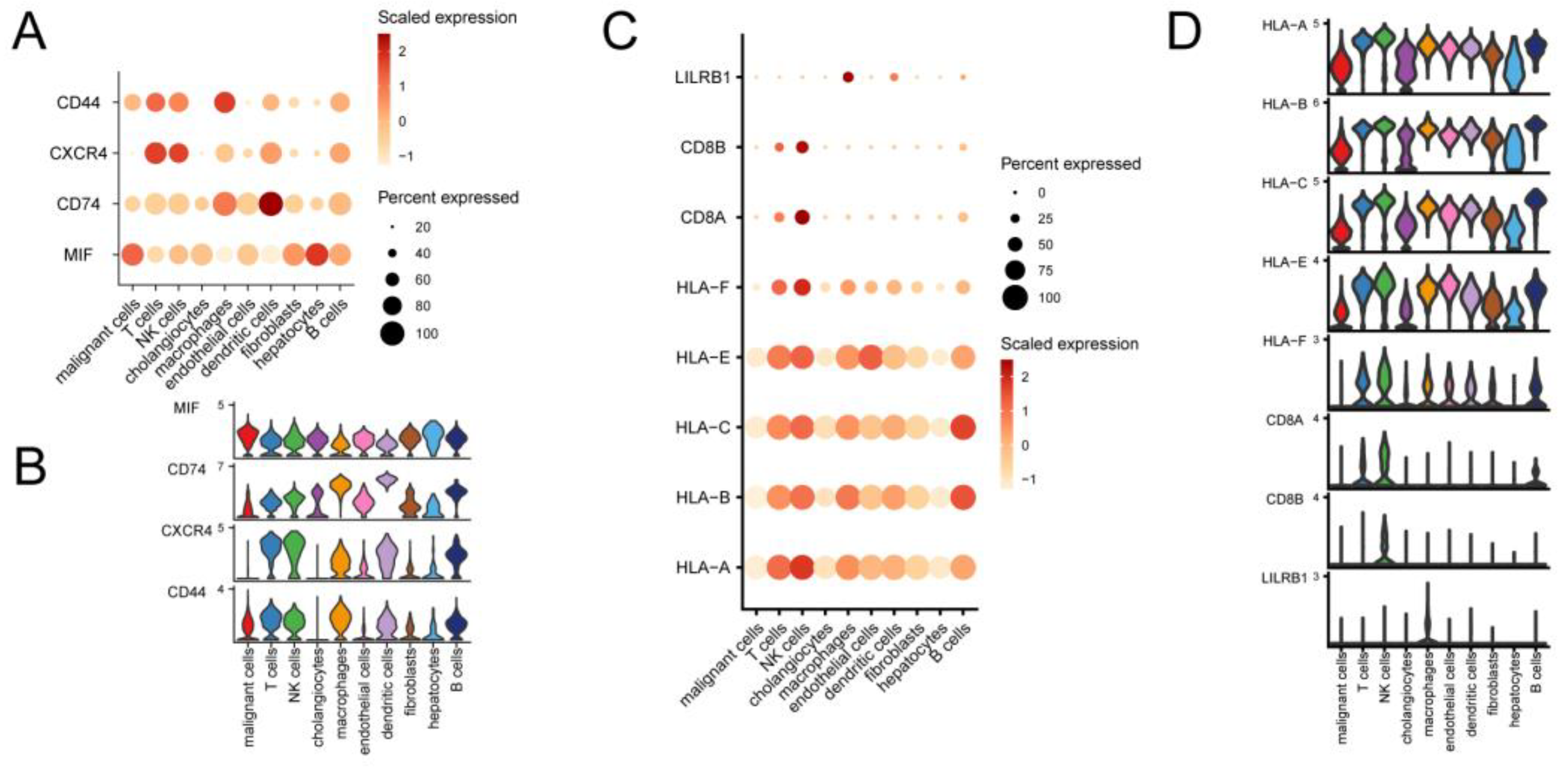
Figure 8.
Enrichment analysis of telomere activity-related differentially expressed genes in CCA. (A) Volcano plot depicting genes significantly differentially expressed between CCA and normal control. (B) Heatmap of the top 5 significantly differentially expressed genes in up- and down-regulation. (C) Wayne plots of the intersection of differential genes with transcriptional differential genes and telomere-associated differential genes between different subgroups of cells and different telomere-active cells in single cells. (D) GO-enriched lollipop plot showing the top three pathways with the highest significance for Biological process (BP), Cellular component (CC), and Molecular function (MF). (E) KEGG enrichment analysis.
Figure 8.
Enrichment analysis of telomere activity-related differentially expressed genes in CCA. (A) Volcano plot depicting genes significantly differentially expressed between CCA and normal control. (B) Heatmap of the top 5 significantly differentially expressed genes in up- and down-regulation. (C) Wayne plots of the intersection of differential genes with transcriptional differential genes and telomere-associated differential genes between different subgroups of cells and different telomere-active cells in single cells. (D) GO-enriched lollipop plot showing the top three pathways with the highest significance for Biological process (BP), Cellular component (CC), and Molecular function (MF). (E) KEGG enrichment analysis.
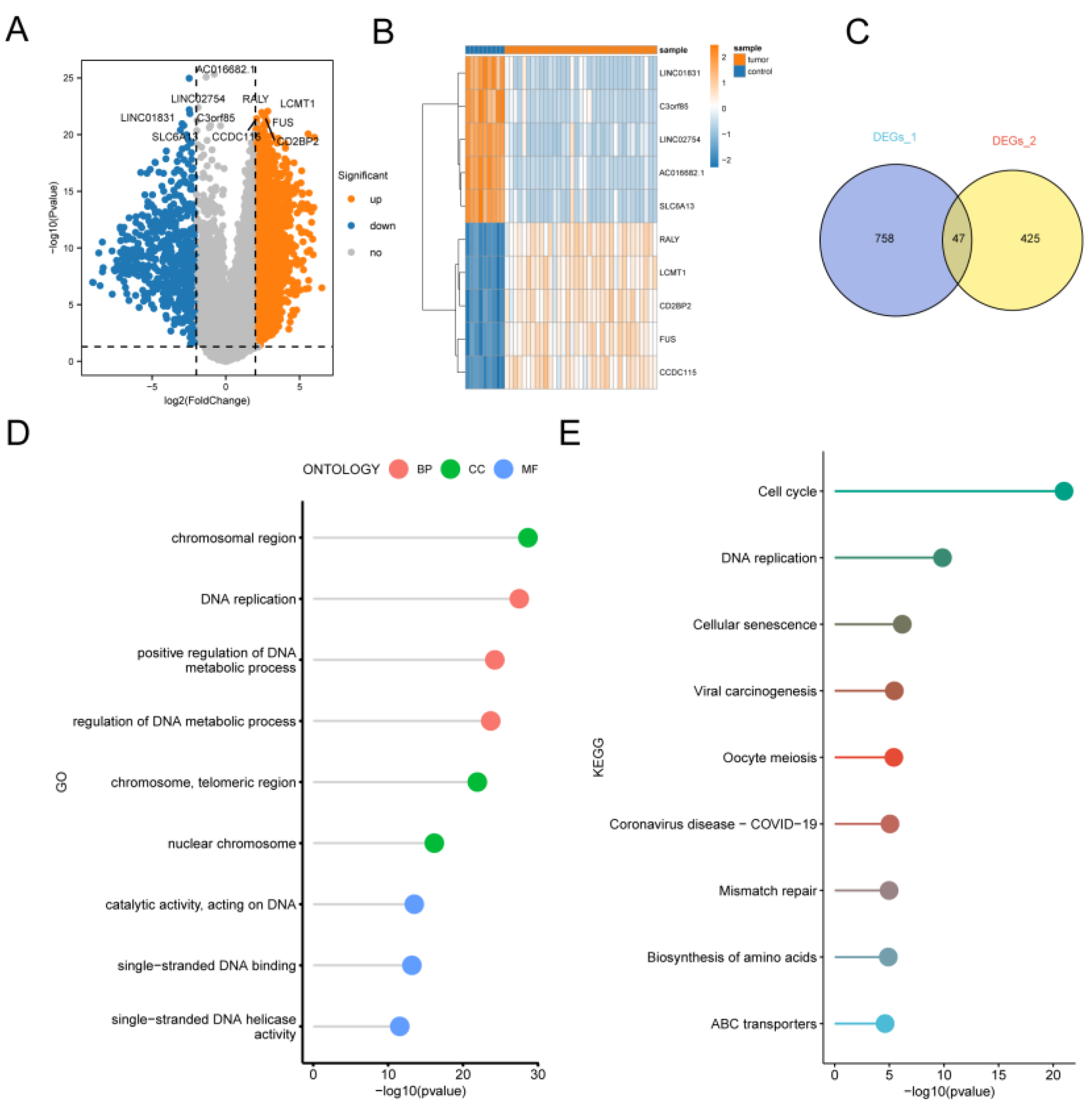
Figure 9.
Cox and LASSO regression analysis of the bile duct cancer dataset. (A) LASSO regression trajectory of the independent variables, with the horizontal coordinate indicating the logarithm of the lambda of the independent variables and the vertical coordinate indicating the independently accessible coefficients. (B) Confirmation intervals under each lambda in the LASSO regression. (C) Survival curves for patients in the high- and low-risk groups from the training cohort, respectively. (D) Survival curves for patients in the high- and low-risk groups from the validation cohort, respectively. The yellow color represents the high-risk group and the blue color represents the low-risk group. (E) Time-dependent ROC curves for 1, 3, and 5 year survival for the model used for the training cohort. (F) Time-dependent ROC curves for 1, 3, and 5 year survival for the validation cohort model.
Figure 9.
Cox and LASSO regression analysis of the bile duct cancer dataset. (A) LASSO regression trajectory of the independent variables, with the horizontal coordinate indicating the logarithm of the lambda of the independent variables and the vertical coordinate indicating the independently accessible coefficients. (B) Confirmation intervals under each lambda in the LASSO regression. (C) Survival curves for patients in the high- and low-risk groups from the training cohort, respectively. (D) Survival curves for patients in the high- and low-risk groups from the validation cohort, respectively. The yellow color represents the high-risk group and the blue color represents the low-risk group. (E) Time-dependent ROC curves for 1, 3, and 5 year survival for the model used for the training cohort. (F) Time-dependent ROC curves for 1, 3, and 5 year survival for the validation cohort model.
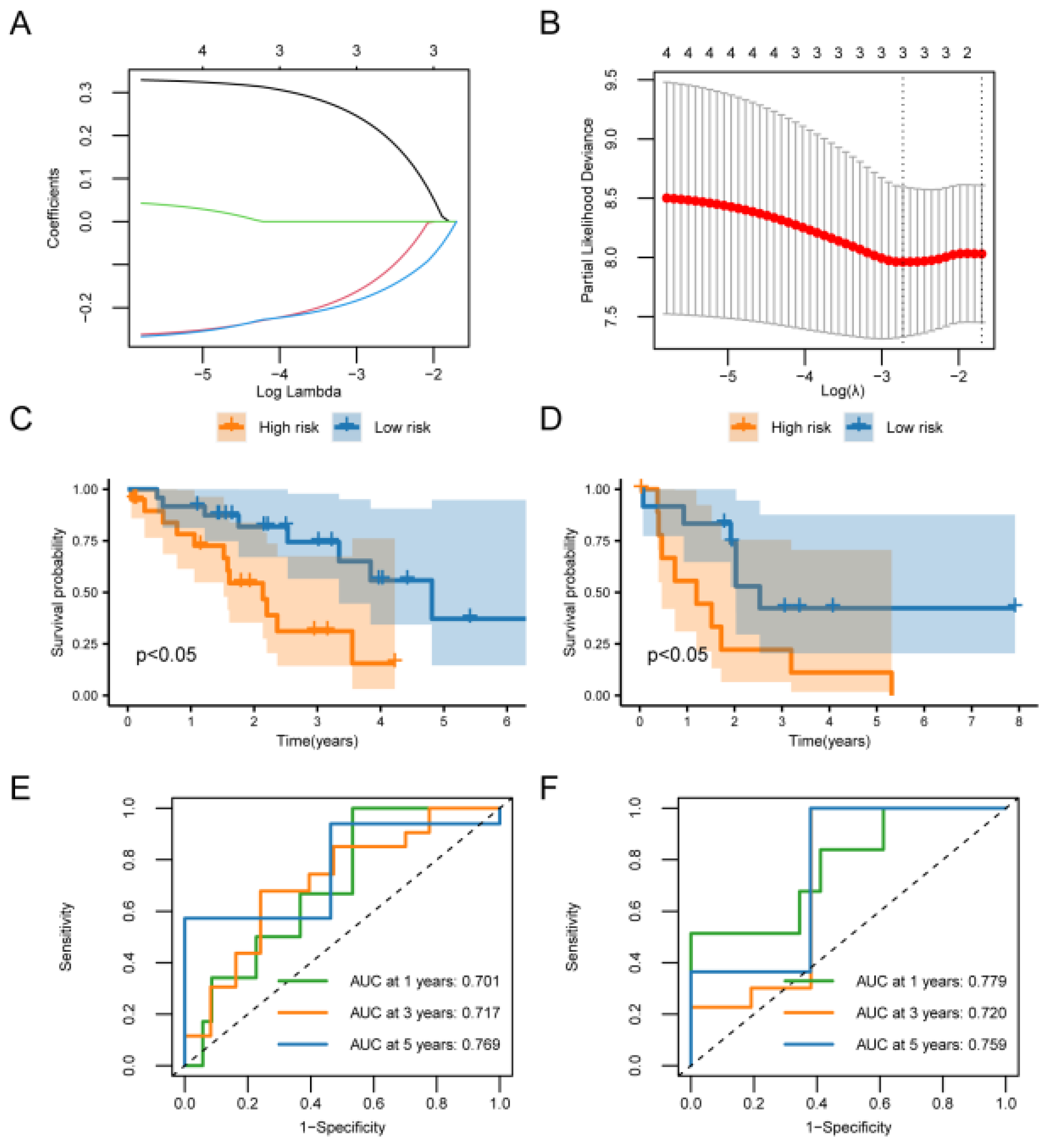
Figure 10.
Pathways enriched for GSEA and GSVA. (A) DRUG METABOLISM CYTOCHROME P450.(B) RETINOL METABOLISM.(C) COMPLEMENT AND COAGULATION CASCADES.(D) TIGHT JUNCTION. (E) ECM RECEPTOR INTERACTION. (F) FOCAL ADHESION.(G) GSVA ENRICHMENT ANALYSIS of pathway maps differentially enriched between high and low-risk groups.
Figure 10.
Pathways enriched for GSEA and GSVA. (A) DRUG METABOLISM CYTOCHROME P450.(B) RETINOL METABOLISM.(C) COMPLEMENT AND COAGULATION CASCADES.(D) TIGHT JUNCTION. (E) ECM RECEPTOR INTERACTION. (F) FOCAL ADHESION.(G) GSVA ENRICHMENT ANALYSIS of pathway maps differentially enriched between high and low-risk groups.
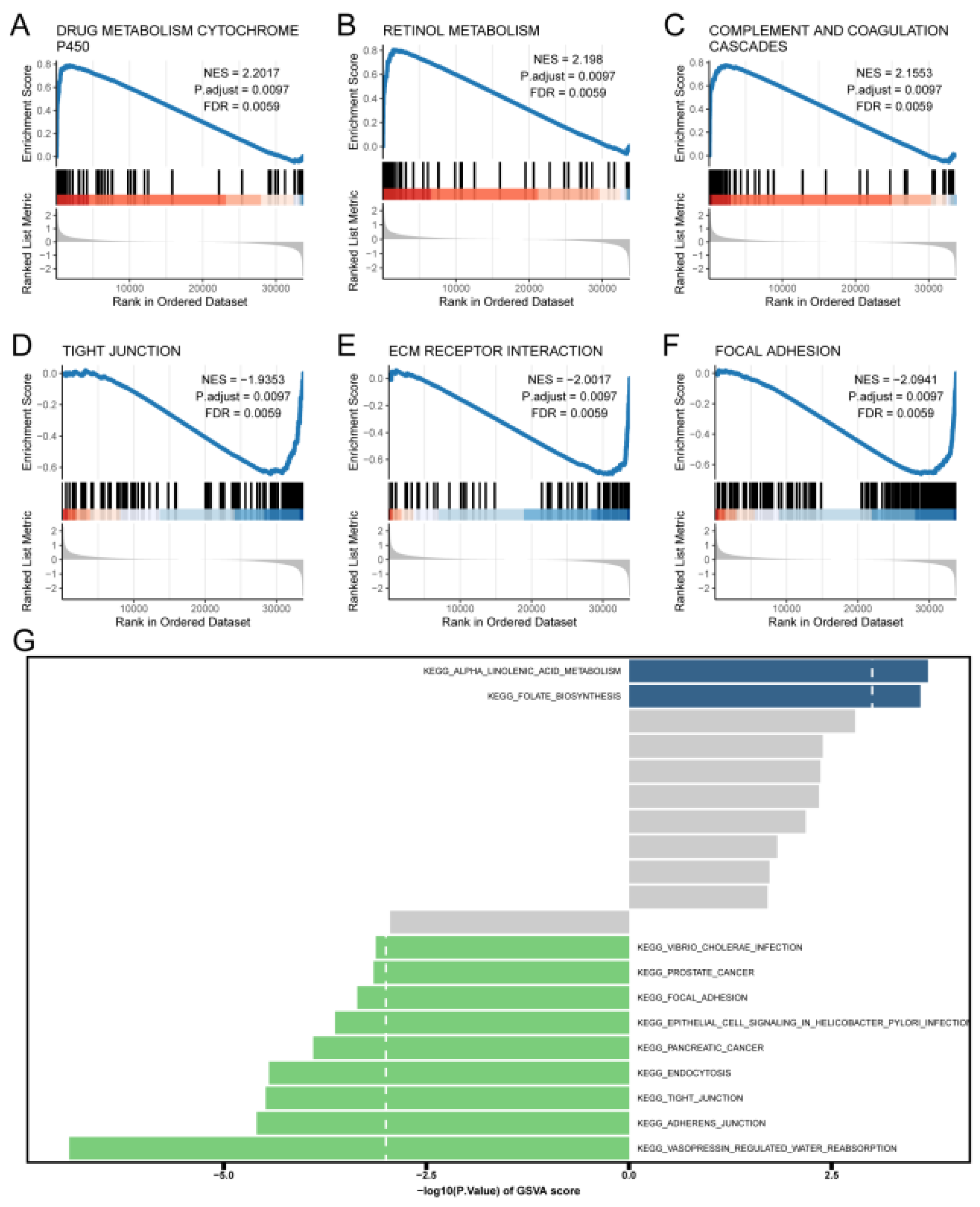
Figure 11.
Levels of immune infiltration between high and low-risk groups. (A) Heatmap of correlation between immune cells. (B) Box line plot of the estimated proportion of immune cells between high and low-risk groups. Correlation between ATP1A1 and Central memory CD4 T cell (C), and Immature B cell (D) immune cells. Asterisks indicate P values : ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05.
Figure 11.
Levels of immune infiltration between high and low-risk groups. (A) Heatmap of correlation between immune cells. (B) Box line plot of the estimated proportion of immune cells between high and low-risk groups. Correlation between ATP1A1 and Central memory CD4 T cell (C), and Immature B cell (D) immune cells. Asterisks indicate P values : ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05.
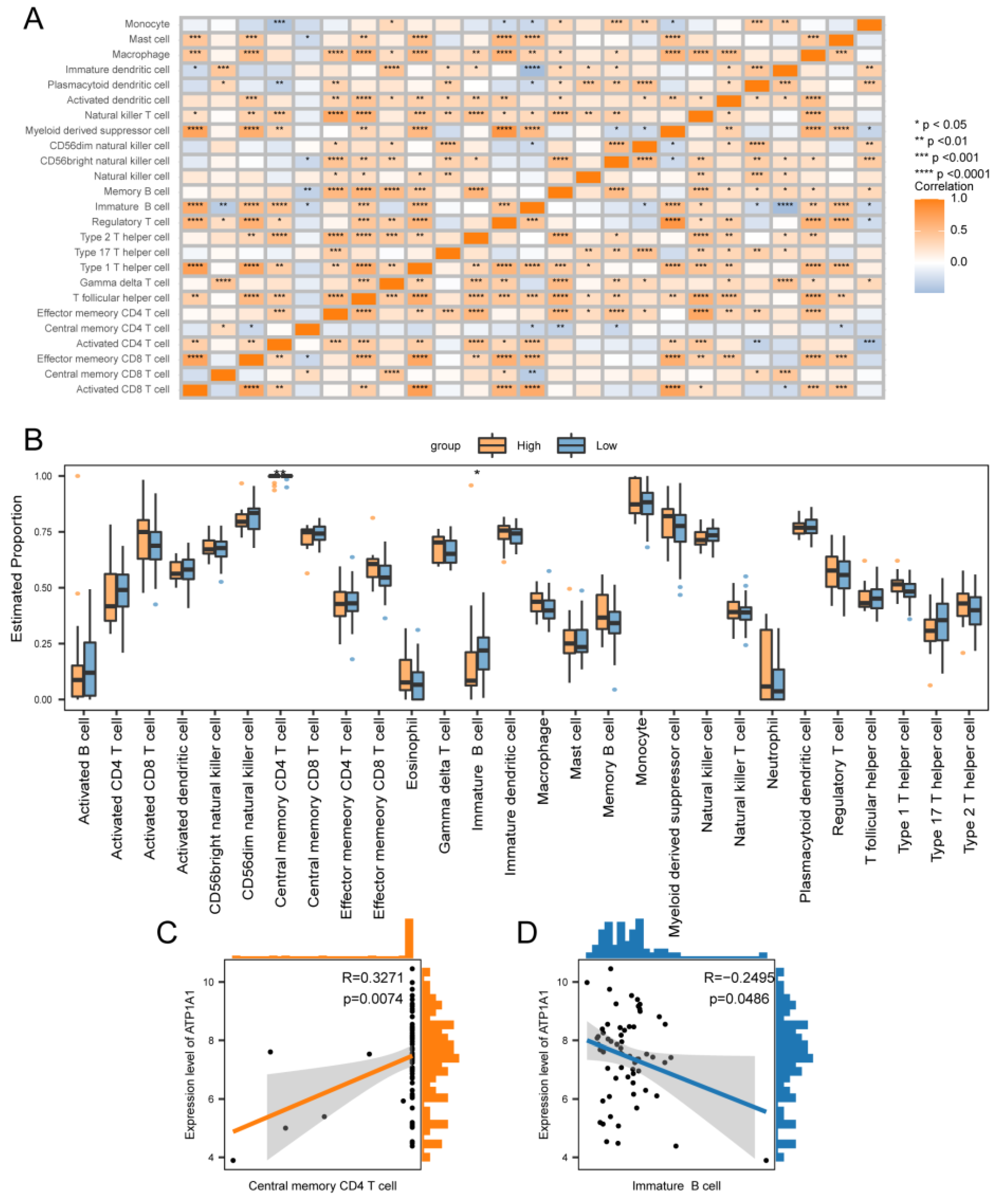
Figure 12.
Risk score as an independent prognostic factor. (A) Results of univariate Cox regression analysis of clinical characteristics. (B) Predictive model of the nomogram. line segments indicate the contribution of clinical factors to outcome events, the total score indicates the sum of the corresponding individual scores for all variable values, and the bottom three lines indicate the prognosis for 1, 3, and 5-year survival corresponding to each value point. (C) Nomogram model ROC curves for 1, 3, and 5 years.
Figure 12.
Risk score as an independent prognostic factor. (A) Results of univariate Cox regression analysis of clinical characteristics. (B) Predictive model of the nomogram. line segments indicate the contribution of clinical factors to outcome events, the total score indicates the sum of the corresponding individual scores for all variable values, and the bottom three lines indicate the prognosis for 1, 3, and 5-year survival corresponding to each value point. (C) Nomogram model ROC curves for 1, 3, and 5 years.
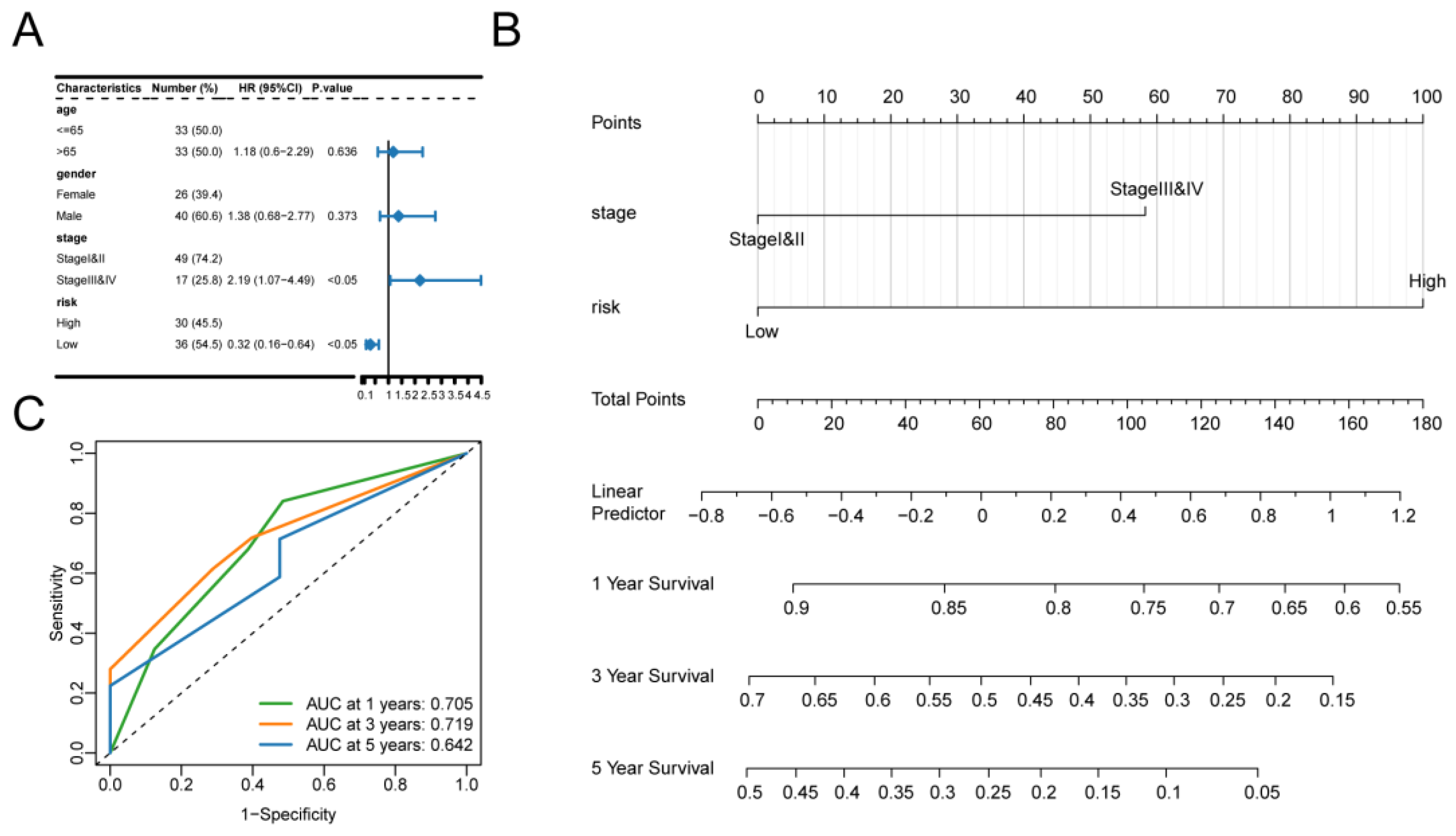
Figure 13.
Differences in TMB and drug sensitivity between high- and low-risk groups. (A) Top 20 genes with the highest mutation frequency in the high-risk and (B) low-risk groups. (C) TMB violin maps for the high and low-risk groups. (D) Differences in drug sensitivity of Vinblastine_1004 between high and low-risk groups. (E) Drug sensitivity difference of Vinorelbine_2048 between high and low-risk groups. (F) The difference in drug sensitivity of Docetaxel_1007 between high and low-risk groups.
Figure 13.
Differences in TMB and drug sensitivity between high- and low-risk groups. (A) Top 20 genes with the highest mutation frequency in the high-risk and (B) low-risk groups. (C) TMB violin maps for the high and low-risk groups. (D) Differences in drug sensitivity of Vinblastine_1004 between high and low-risk groups. (E) Drug sensitivity difference of Vinorelbine_2048 between high and low-risk groups. (F) The difference in drug sensitivity of Docetaxel_1007 between high and low-risk groups.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



