
Submitted:
12 June 2024
Posted:
14 June 2024
You are already at the latest version
Abstract
Drug-target binding is an essential parameter in drug discovery and design to ensure drug efficacy and specificity. In 2022, the concept of a general intermolecular binding affinity calculator (GIBAC) was for the first time coined: $Kd = f(molecules, envPara)$. Technically, GIBAC represents a set of in silico approaches for structural and biophysical data generation towards a paradigm shift in precise drug discovery and design, providing a comprehensive framework for intermolecular binding affinity calculation with adequate accuracy, precision and efficiency. For the first time, this study reports a prototype of GIBAC (semaGIBAC), i.e., a one-dimensional semaglutide-GLP-1R-based mini static GIBAC, based on an experimental complex structure of semaglutide and GLP-1R. Semaglutide is a potent GLP-1 receptor agonist used to treat type 2 diabetes mellitus by regulating blood glucose levels and promoting weight loss. In 2021, a structural modification involving a Val27-Arg28 exchange was manually introduced to enhance semaglutide-GLP-1R binding affinity. This study employs a comprehensive structural and biophysical analysis aimed at thoroughly exploring the sequence space of semaglutide-GLP-1R to design analogues with improved binding affinity, leading to the identification of a promising semaglutide analogue, which binds to GLP-1R with an affinity that is more than two orders of magnitude (113.3 times) higher than native semaglutide. To sum up, this article puts forward a promising structural biophysical approach for developing GLP-1 receptor agonists with enhanced efficacy, and with a GIBAC prototype (semaGIBAC), this article argues again that the time is now ripe for the construction of a real GIBAC to be listed on the agenda of the drug discovery and design community.
Keywords:
GIBAC
; Intermolecular binding affinity (Kd)
; Structural biophysics
; Synthetic data
; Drug discovery \& design
1. Introduction
1.1. Intermolecular Binding Affinity in Drug Discovery and Design
Intermolecular interactions are the fabrics underlying almost all processes in living organisms [1,2,3,4,5,6,7,8], where two cornerstone concepts, intermolecular binding affinity (Kd) and free binding energy (ΔG), have long been established to physically describe the strengths of biomolecular interactions [9,10,11,12,13,14,15]. Intermolecular binding affinity, e.g., drug-target binding affinity (Kd and ΔG), is an essential parameter in drug discovery and design to ensure efficacy and specificity [6,11,16,17,18,19,20,21]. First, high binding affinity typically correlates with increased efficacy, i.e., the drug can effectively modulate the target’s activity at lower concentrations [22,23,24,25,26,27,28]. Second, a structural biophysical understanding of the intermolecular binding affinity helps in optimizing the potency and selectivity of drug candidates, reducing off-target effects and adverse reactions [29,30,31,32,33,34].
In August of 2022, therefore, the concept of a general intermolecular binding affinity calculator (GIBAC) was for the first time coined, proposed and defined as [35]. Last October, GIBAC was for the first time updated, including its inception, definition, construction, practical applications, technical challenges and limitations and future directions [36]. With the conceptual and practical framework of GIBAC in place [36], this article puts forward a hypothesis that a real GIBAC technically makes sense and is feasible, and aims to test this hypothesis through the construction of a prototype of a real GIBAC [4,35,36].
1.2. Building a GIBAC Prototype with Semaglutide as an Example
In principle, any biomolecule inside Protein Data Bank [37,38,39,40,41] is fit to be used to build a prototype of a real GIBAC [4,35,36]. Here, this article chooses semaglutide due to the availability of abundant experimental data and clinical relevance of semaglutide [42,43]. First, semaglutide is a synthetic long-acting analogue of glucagon-like peptide-1 (GLP-1), which has garnered significant attention for its efficacy in treating type 2 diabetes and obesity [44,45]. Structurally, its direct binding and interaction with the GLP-1 receptor involves intricate mechanisms, where the complex structure has already been experimentally determined with Cryo-EM or X-ray diffraction (Table 1 and Table 2, Figure 1) [42]. From a chemical structure point of view, semaglutide is a peptide-based molecule, including a peptide backbone consisting of 31 amino acids (a modified version of human GLP-1, 7-37), a substitution of alanine with 2-aminoisobutyric acid (an unnatural amino acid [46,47,48]) at position 8 to resist enzyme degradation, and an addition of a C-18 fatty acid chain attached via a spacer (γ-Glu-2x-OEG) at lysine 26 for stronger albumin binding [29,49].
In short, semaglutide is neither small molecule compound, nor monoclonal antibody, nor ADC/PDC, nor an entirely peptide-based drug, making it a reasonable choice as an example for the construction of a GIBAC prototype, i.e., a one-dimensional semaglutide-GLP-1R-based mini static GIBAC, abbreviated below as semaGIBAC and described in Equation 1:
where represents the experimental complex structure of and , and represent the amino acid sequences of and , respectively, =, or or both, n represents the number of missense mutations introduced into the sequence of , r represents the number of structural models built by Modeller [50], T represents temperature [51,52,53,54], and represents the site-specific mutations introduced into the backbone of semaglutide (PDB entry 4ZGM [42], Table 2).
2. Materials and Methods
To build a prototype of a real GIBAC, as mentioned above, this article chooses semaglutide due partly to the availability of abundant experimental data of semaglutide [42,43]. Among the three experimental complex structures of semaglutide and GLP-1R (Table 2), there is only one experimental structure (X-ray diffraction) of the semaglutide backbone in complex with the extracellular domain of GLP-1R (Figure 1, PDB ID: 4ZGM [42]).
According to PDB entry 4ZGM [42] (Table 2), the sequences of the two chains of semaglutide backbone and GLP-1R are listed in italics in fasta format as below,
>4ZGM_1_Chain A (Figure 1)
RPQGATVSLWETVQKWREYRRQCQRSLTEDPPPATDLFCNRTFDEYACWPDGEPGSFVNVSCPWYLPWASSVPQGHVYRFCTAEGLWLQKDNSSLPWRDLSECEESKRGERSSPEEQLLFLY
>4ZGM_2_Chain B_a modified version of human GLP-1 (7-37) (Figure 1)
HAEGTFTSDVSSYLEGQAAKEFIAWLVRGRG
In 2021, a Val27-Arg28 exchange (Table 3) was for the first time introduced into the backbone of semaglutide to strengthen the semaglutide-GLP-1R binding affinity to ∼ one-third of the Kd between native semaglutide and GLP-1R [6,55,56].
First, with PDB entry 4ZGM [42] (Table 2) in place, Modeller [50] was employed to build 10000 structural models with 100% homology to PDB ID: 4ZGM [42], the binding affinities between semaglutide and GLP-1R were calculated using Prodigy [57,58] 10000 times [59]. Second, with PDB entry 4ZGM [42] (Table 2) in place as an initial input, the process of the construction of a prototype GIBAC (semaGIBAC, Equation 1) subsequently consists of an automated in silico generation of synthetic structural and Kd data, as illustrated in Figure 2 and described previously in detail [60]. Briefly, Modeller [50] was employed to build a total of 11200 () homology structural models with 95.42% () homology to PDB ID: 4ZGM [42]. Afterwards, the binding affinities between semaglutide analogues and GLP-1R were calculated using Prodigy [57,58] for 11200 times [59].
3. Results
With PDB entry 4ZGM [42] (Table 2) as an initial input and an automated in silico generation of synthetic structural and Kd data [60], this article for the first time puts forward a prototype GIBAC, i.e., semaGIBAC, for which everything is included in Table S1 of supplementary file semaGIBAC.pdf:
where represents PDB entry 4ZGM [42] (Table 2), and represent sequences of semaglutide backbone () and GLP-1R (), respectively, =, n =1, r =20 [50], T = C. Of further note, here, = indicates that site-specific missense mutation is introduced only into the backbone of semaglutide, but not into GLP-1R (PDB entry 4ZGM [42], Table 2), making semaGIBAC a semaglutide-centered GIBAC prototype [35,36].
In Figure 3 and Table 4, for PDB entry 4ZGM [42] (Table 2), most of the Kd values are located between 2.5 × 10-6 M and 4.0 × 10-6 M, with an average at 3.278 × 10-6 M, which is rather close to the one Kd (3.4 × 10-6 M) as reported previously [6,59]. In Figure 3 and Table 4, for the 11200 homology structural models (semaGIBAC), most of the Kd values are located between 2.0 × 10-6 M and 4.0 × 10-6 M, with minimum and maximum values at 4.1 × 10-7 M and 9.4 × 10-6 M, respectively. From Figure 3 and Table 4, it can be seen that the the range of the Kd values gets wider and wider as n (Equations 1 and 2) increases from zero to one and to two.
As described above, the semaGIBAC here is built to be a one-dimensional semaglutide-GLP-1R-based semaglutide-centered mini static GIBAC [35,36]:
- semaGIBAC is a semaglutide-centered GIBAC, i.e., missense mutation is introduced only into the backbone of semaglutide (PDB entry 4ZGM [42]).
- semaGIBAC is a mini GIBAC, i.e., semaGIBAC is able to calculate intermolecular Kd only between GLP-1R and semaglutide (analogues) with one mutation.
Last October, GIBAC was for the first time updated, including its inception, definition, construction, practical applications, technical challenges and limitations and future directions, including in particular a set of criteria as listed below [35,36]:
Nonetheless, this article puts forward just only a prototype (i.e., semaGIBAC), instead of a real GIBAC. Specifically,
- semaGIBAC does take one missense mutation into account;
- structural biophysics is at the core of semaGIBAC;
- semaGIBAC takes only some Kd-relevant factors into account, e.g., temperature.
- semaGIBAC does not require an accurate general forcefield for all atoms or a universal linear string/graph-based notation system, as it does not take 8Aib or C-18 fatty acid chain into account;
4. Conclusion
For the first time, this article puts forward a prototype GIBAC, i.e., semaGIBAC, to support the hypothesis that a real GIBAC is technically feasible in practice and of practical use for drug discovery and design:
- semaGIBAC is able to calculate with 95.42% () accuracy the Kd between GLP-1R and any semaglutide analogue with one site-specific missense mutation introduced into its backbone.
- semaGIBAC is able to be used the other way around as a search engine for drug design, i.e., for drug efficacy, semaGIBAC is able to generate a list of semaglutide analogue with one site-specific missense mutation introduced into its backbone, and rank them according to their average Kd (Table 4) values in the range of the minimum and the maximum values of semaGIBAC as included in Table 4, while for drug safety, semaGIBAC is unable to generate a Kd-ranked list of semaglutide analogues with suppressed off-target effects [78,79].
- the construction of semaGIBAC consists of a set of in silico steps of structural and biophysical data generation towards a paradigm shift in precise drug discovery and design, leading to the identification of a promising semaglutide analogue with a Kd of 3.0 × 10-8 M, in contrast to the Kd of 3.278 × 10-6 M for native semaglutide and GLP-1R (PDB entry 4ZGM, Table 2 and Table 4) [42].
5. Discussion
5.1. In Silico Generation of Structural and Biophysical Data with Reasonable Accuracy: Expanding Horizons in Precise Drug Discovery and Design
The past three years saw a big step forward in the use of artificial intelligence (AI) in structural biology for protein structure prediction [63,80,81,82,83,84,85], leading to the generation of computational structural data such as AlphaFold database [63,81,82,83,84]. Nonetheless, to train useful AI models for precise drug discovery and design, a huge number of data is needed with reasonable accuracy, buth experimental and synthetic, both structural and biophysical (Kd and G), where a variety of tools are needed, such as molecular docking tools [86,87,88,89], molecular dynamics simulations tools [69,90], side chain placement and energy minimization algorithms [91] to incorporate structural arrangement information of post-translational modifications (PTMs) [92,93,94], post-expression modifications (PEMs) [6,74] into currently available structural models.
In this regard, this article puts forward a set of in silico steps of structural and biophysical data generation towards a paradigm shift in precise drug discovery and design [59,60]. Take semaglutide for instance, a five-dimensional semaGIBAC requires a total of 314496000000 (Table 5) homology structural models with 82.14% () homology to PDB ID: 4ZGM [42] to be built by Modeller [50], and subsequently a total of 314496000000 (Table 5) times of Prodigy-based [57,58] calculations of the binding affinities between semaglutide analogues and GLP-1R. Take MoleculeX (a protein consisting of 100 amino acids) as another example, the number soars from 314496000000 to 240920064000000 (Table 5).
In short, to build a real GIBAC using an entirely structural biophysics-based approach requires an exhaustive exploration of the entire molecular space (Figure 4) [61,77]. In practice, however, this astranomical task is computationally impossible, which explains partly why this article puts forward just only a structural biophysics-based prototype (i.e., semaGIBAC), instead of a real GIBAC. Therefore, this article here again proposes an AI-based open strategy [95] to make it possible and conceivable to generate a vast amount of data to train a real GIBAC with reasonable accuracy and precision, as openness in data acquisition and generation, and AI algorithms is essential for promoting transparency, reproducibility, and collaboration within the community of drug discovery & design, and for facilitating continued improvement of the performance (accuracy, precision and efficiency) of GIBAC in precise drug discovery & design [35,36] in future.
As mentioned above, this article puts forward a set of in silico steps of structural and biophysical data generation towards a paradigm shift in precise drug discovery and design [59,60]. Specifically, here, the generation of synthetic structural and Kd data is akin to the distribution of the electron cloud of a hydrogen atom (Figure 4), where is the distance between the electron (synthetic data) and the proton (i.e., nucleus, experimental data) of the hydrogen atom. With respect to semaGIBAC, PDB entry 4ZGM (Table 2) is the nucleus, while Table S1 of supplementary file semaGIBAC.pdf is one layer of the electron cloud (Figure 4), while semaGIBAC itself is depicted in Figure 4 as one hydrogen atom. In principle, any biomolecule inside Protein Data Bank [37,39], i.e., experimental structural data, is able to be used as a hydrogen nucleus (Figure 4), around which there is an electron cloud representing a vast set of synthetic structural and biophysical data, while the white region of Figure 4 represents structurally and biophysically uncharted territories, highlighting an astranomical task which is computationally impossible, and calling for an AI-based open approach for the construction of a real GIBAC [35,36].
5.2. Designing Semaglutide Analogues with Elevated Binding Affinity and Efficacy through Continued Exploration of the Uncharted Molecular Space of Semaglutide and GLP-1R
The development of semaglutide analogues with increased GLP-1R binding affinity holds significant clinical relevance, offering the potential for enhanced glucose control, weight loss, and cardiovascular benefits in patients with type 2 diabetes and obesity [42,96,97]. Thanks to the continued development of experimental structural biology and the half-a-century old Protein Data Bank (PDB) [37,38,39,40,98], a comprehensive structural biophysical analysis becomes possible [99,100] for specific ligand-receptor complex structures deposited in PDB, such that our understanding of the structural and biophysical basis of their interfacial stability is able to help us modify the binding affinity of certain drug target and its interacting partners [101,102,103].
With semaglutide as an example here [59], one particular analogue (supplementary file semx.pdb, Table 6) stood out with four missense mutations (G13B_A, I20B_Q, L23B_R and V24B_N) through computational structural modeling and biophysics-based rational design, with a Prodigy-calculated Kd as low as 3.0 × 10-8 M at 37 ∘C (Table 6), while the Kd is 3.278 × 10-6 M (Table 4) for the binding of native semaglutide’s backbone to GLP-1 at 37 ∘C. Technically, the structural biophysics-based rational design of this semaglutide analogue (supplementary file semx.pdb) is a tiny part of the task of the construction of a four-dimensional semaGIBAC, which requires a total of 3276000000 (Table 5) homology structural models with 85.71% () homology to PDB ID: 4ZGM [42] to be built by Modeller [50], and subsequently a total of 3276000000 (Table 5) times of Prodigy-based [57,58] calculations of the binding affinities between semaglutide analogues and GLP-1R. Given the size of the computational task, this article only puts forward one-dimensional semaGIBAC as a GIBAC prototype, making this semaglutide analogue (supplementary file semx.pdb look like a tip of the iceberg floating on the ocean (Figure 4) of a four-dimensional sequence space of semaglutide and GLP-1R (PDB entry 4ZGM [42], Table 2).
Of further note, the Kd of this semaglutide analogue (supplementary file semx.pdb is only a result of the structural biophysics-based calculation of Prodigy [57,58]. As a result, its real Kd and efficacy (in both vitro and vivo) need to be experimentally tested in wet-labs (Figure 5), as part of the drug discovery and design process in the early stage of drug R&D, which to date is still a lengthy, costly, difficult and inefficient yet pivotal process [104]. Given this, this article calls again for the construction of a real GIBAC [35,36] with adequate accuracy, precision and efficiency towards a paradigm shift [105] of precise drug discovery & design, until a real GIBAC comes into being and pushing forward the continued development of the industry [106,107,108].
6. Ethical Statement
No ethical approval is required.
7. Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this work, the author used OpenAI’s ChatGPT in order to improve the readability of the manuscript, and to make it as concise and short as possible. After using this tool, the author reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Author Contributions
Conceptualization, W.L.; methodology, W.L.; software, W.L.; validation, W.L.; formal analysis, W.L.; investigation, W.L.; resources, W.L.; data duration, W.L.; writing–original draft preparation, W.L.; writing–review and editing, W.L.; visualization, W.L.; supervision, W.L.; project administration, W.L.; funding acquisition, not applicable.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Murphy, K.M.; Gould, R.J.; Largent, B.L.; Snyder, S.H. A unitary mechanism of calcium antagonist drug action. Proceedings of the National Academy of Sciences 1983, 80, 860–864. [Google Scholar] [CrossRef]
- Zhang, Z.; Palzkill, T. Determinants of binding affinity and specificity for the interaction of TEM-1 and SME-1 β-lactamase with β-lactamase inhibitory protein. J. Biol. Chem. 2003, 278, 45706–45712. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, H.; Wang, H.; Chen, Z.; Zhang, Z.; Chen, X.; Li, Y.; Qi, Y.; Wang, R. PLANET: A Multi-objective Graph Neural Network Model for Protein-Ligand Binding Affinity Prediction. Journal of Chemical Information and Modeling 2023. [Google Scholar] [CrossRef]
- Li, W. High-Throughput Extraction of Interfacial Electrostatic Features from GLP-1-GLP-1R Complex Structures: A GLP-1-GLP-1R-Based Mini GIBAC Perspective 2024. [CrossRef]
- Trosset, J.Y.; Cavé, C. In Silico Drug-Target Profiling. In Target Identification and Validation in Drug Discovery; Springer New York, 2019; pp. 89–103.
- Li, W. Strengthening Semaglutide-GLP-1R Binding Affinity via a Val27-Arg28 Exchange in the Peptide Backbone of Semaglutide: A Computational Structural Approach. Journal of Computational Biophysics and Chemistry 2021, 20, 495–499. [Google Scholar] [CrossRef]
- Noble, D.; Blundell, T.L.; Kohl, P. Progress in biophysics and molecular biology: A brief history of the journal. Progress in Biophysics and Molecular Biology 2018, 140, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.W.; Hess, P.; McCleskey, E.W.; Rosenberg, R.L. Calcium channels: Mechanisms of Selectivity, Permeation, and Block. Annual Review of Biophysics and Biophysical Chemistry 1987, 16, 265–290. [Google Scholar] [CrossRef]
- Greenidge, P.A.; Kramer, C.; Mozziconacci, J.C.; Wolf, R.M. MM/GBSA Binding Energy Prediction on the PDBbind Data Set: Successes, Failures, and Directions for Further Improvement. Journal of Chemical Information and Modeling 2012, 53, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. Journal of Chemical Information and Modeling 2018, 59, 895–913. [Google Scholar] [CrossRef] [PubMed]
- Freire, E. Biophysical methods for the determination of protein-ligand binding constants. Annual review of biophysics 2009, 38, 123–142. [Google Scholar]
- Johnson, C.M. Isothermal Calorimetry. In Protein-Ligand Interactions; Springer US, 2021; pp. 135–159.
- Velázquez-Coy, A.; Ohtaka, H.; Nezami, A.; Muzammil, S.; Freire, E. Isothermal Titration Calorimetry. Current Protocols in Cell Biology 2004, 23. [Google Scholar]
- Sauer, U.G. Surface plasmon resonance-a label-free tool for cellular analysis. Journal of biotechnology 2008, 133, 101–108. [Google Scholar]
- Ernst, R.R.; Bodenhausen, G.; Wokaun, A. Principles of nuclear magnetic resonance in one and two dimensions. Principles of nuclear magnetic resonance in one and two dimensions 1990, 14. [Google Scholar]
- Fuji, H.; Qi, F.; Qu, L.; Takaesu, Y.; Hoshino, T. Prediction of Ligand Binding Affinity to Target Proteins by Molecular Mechanics Theoretical Calculation. Chemical and Pharmaceutical Bulletin 2017, 65, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Zhou, H.X. Calculation of Protein-Ligand Binding Affinities. Annual Review of Biophysics and Biomolecular Structure 2007, 36, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Li, W. Designing rt-PA Analogs to Release its Trapped Thrombolytic Activity. Journal of Computational Biophysics and Chemistry 2021, 20, 719–727. [Google Scholar] [CrossRef]
- Jubb, H.C.; Pandurangan, A.P.; Turner, M.A.; Ochoa-Montaño, B.; Blundell, T.L.; Ascher, D.B. Mutations at protein-protein interfaces: Small changes over big surfaces have large impacts on human health. Progress in Biophysics and Molecular Biology 2017, 128, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni-Kale, U.; Raskar-Renuse, S.; Natekar-Kalantre, G.; Saxena, S.A. Antigen-Antibody Interaction Database AgAbDb: A Compendium of Antigen-Antibody Interactions. In Methods in Molecular Biology; Springer New York, 2014; pp. 149–164.
- Manso, T.; Folch, G.; Giudicelli, V.; Jabado-Michaloud, J.; Kushwaha, A.; Ngoune, V.N.; Georga, M.; Papadaki, A.; Debbagh, C.; Pégorier, P.; Bertignac, M.; Hadi-Saljoqi, S.; Chentli, I.; Cherouali, K.; Aouinti, S.; Hamwi, A.E.; Albani, A.; Elhassani, M.E.; Viart, B.; Goret, A.; Tran, A.; Sanou, G.; Rollin, M.; Duroux, P.; Kossida, S. IMGT® databases, related tools and web resources through three main axes of research and development. Nucleic Acids Research 2021, 50, D1262–D1272. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, G. How CaV1.2-bound verapamil blocks Ca2+ influx into cardiomyocyte: Atomic level views. Pharmacological Research 2019, 139, 153–157. [Google Scholar] [CrossRef]
- Li, W. Delving deep into the structural aspects of a furin cleavage site inserted into the spike protein of SARS-CoV-2: A structural biophysical perspective. Biophysical Chemistry 2020, 264, 106420. [Google Scholar] [CrossRef]
- Pieber, T.R.; Bode, B.; Mertens, A.; Cho, Y.M.; Christiansen, E.; Hertz, C.L.; Wallenstein, S.O.R.; Buse, J.B.; Akın, S.; Aladağ, N.; Arif, A.A.; Aronne, L.J.; Aronoff, S.; Ataoglu, E.; Baik, S.H.; Bays, H.; Beckett, P.L.; Berker, D.; Bilz, S.; Bode, B.; Braun, E.W.; Buse, J.B.; Canani, L.H.S.; Cho, Y.M.; Chung, C.H.; Colin, I.; Condit, J.; Cooper, J.; Delgado, B.; Eagerton, D.C.; Ebrashy, I.N.E.; Hefnawy, M.H.M.F.E.; Eliaschewitz, F.G.; Finneran, M.P.; Fischli, S.; Fließer-Görzer, E.; Geohas, J.; Godbole, N.A.; Golay, A.; de Lapertosa, S.G.; Gross, J.L.; Gulseth, H.L.; Helland, F.; Høivik, H.O.; Issa, C.; Kang, E.S.; Keller, C.; Khalil, S.H.A.; Kim, N.H.; Kim, I.J.; Klaff, L.J.; Laimer, M.; LaRocque, J.C.; Lederman, S.N.; Lee, K.W.; Litchfield, W.R.; Manning, M.B.; Mertens, A.; Morawski, E.J.; Murray, A.V.; Nicol, P.R.; O’Connor, T.M.; Oğuz, A.; Ong, S.; özdemir, A.; Palace, E.M.; Palchick, B.A.; Pereles-Ortiz, J.; Pieber, T.; Prager, R.; Preumont, V.; Riffer, E.; Rista, L.; Rudofsky, G.; Sarı, R.; Scheen, A.; Schultes, B.; Seo, J.A.; Shelbaya, S.A.; Sivalingam, K.; Sorli, C.H.; Stäuble, S.; Streja, D.A.; T’Sjoen, G.; Tetiker, T.; Gaal, L.V.; Vercammen, C.; Warren, M.L.; Weinstein, D.L.; Weiss, D.; White, A.; Winnie, M.; Wium, C.; Yavuz, D. Efficacy and safety of oral semaglutide with flexible dose adjustment versus sitagliptin in type 2 diabetes (PIONEER 7): a multicentre, open-label, randomised, phase 3a trial. The Lancet Diabetes & Endocrinology 2019, 7, 528–539. [Google Scholar]
- Lingvay, I.; Catarig, A.M.; Frias, J.P.; Kumar, H.; Lausvig, N.L.; le Roux, C.W.; Thielke, D.; Viljoen, A.; McCrimmon, R.J. Efficacy and safety of once-weekly semaglutide versus daily canagliflozin as add-on to metformin in patients with type 2 diabetes (SUSTAIN 8): a double-blind, phase 3b, randomised controlled trial. The Lancet Diabetes & Endocrinology 2019, 7, 834–844. [Google Scholar]
- Li, Y.; Su, M.; Liu, Z.; Li, J.; Liu, J.; Han, L.; Wang, R. Assessing protein-ligand interaction scoring functions with the CASF-2013 benchmark. Nature Protocols 2018, 13, 666–680. [Google Scholar] [CrossRef] [PubMed]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; Tang, A.; Gabriel, G.; Ly, C.; Adamjee, S.; Dame, Z.T.; Han, B.; Zhou, Y.; Wishart, D.S. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Research 2013, 42, D1091–D1097. [Google Scholar] [CrossRef] [PubMed]
- Li, W. Designing Insulin Analogues with Lower Binding Affinity to Insulin Receptor than That of Insulin Icodec 2024. [CrossRef]
- Li, W. A GIBAC-based selectivity strategy for the design of PDE5 inhibitors to minimize visual disturbances 2024. [CrossRef]
- Bischoff, E. Potency, selectivity, and consequences of nonselectivity of PDE inhibition. International Journal of Impotence Research 2004, 16, S11–S14. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Atia, N.N.; Rageh, A.H. Selectivity enhanced cation exchange chromatography for simultaneous determination of peptide variants. Talanta 2019, 199, 347–354. [Google Scholar] [CrossRef]
- Tinberg, C.E.; Khare, S.D.; Dou, J.; Doyle, L.; Nelson, J.W.; Schena, A.; Jankowski, W.; Kalodimos, C.G.; Johnsson, K.; Stoddard, B.L.; Baker, D. Computational design of ligand-binding proteins with high affinity and selectivity. Nature 2013, 501, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Almers, W.; McCleskey, E.W. Non-selective conductance in calcium channels of frog muscle: calcium selectivity in a single-file pore. The Journal of Physiology 1984, 353, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Li, W. Towards a General Intermolecular Binding Affinity Calculator 2022.
- Li, W.; Vottevor, G. Towards a Truly General Intermolecular Binding Affinity Calculator for Drug Discovery & Design 2023.
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nature Structural & Molecular Biology 2003, 10, 980–980. [Google Scholar]
- Li, W. Half-a-century Burial of ρ, θ, and φ in PDB 2021.
- Li, W. Visualising the Experimentally Uncharted Territories of Membrane Protein Structures inside Protein Data Bank 2020.
- Li, W. A Local Spherical Coordinate System Approach to Protein 3D Structure Description 2020.
- Li, W. A Reversible Spherical Geometric Conversion of Protein Backbone Structure Coordinate Matrix to Three Independent Vectors of ρ, θ and φ 2024. [CrossRef]
- Lau, J.; Bloch, P.; Schäffer, L.; Pettersson, I.; Spetzler, J.; Kofoed, J.; Madsen, K.; Knudsen, L.B.; McGuire, J.; Steensgaard, D.B.; Strauss, H.M.; Gram, D.X.; Knudsen, S.M.; Nielsen, F.S.; Thygesen, P.; Reedtz-Runge, S.; Kruse, T. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. Journal of Medicinal Chemistry 2015, 58, 7370–7380. [Google Scholar] [CrossRef]
- Rodbard, H.W.; Rosenstock, J.; Canani, L.H.; Deerochanawong, C.; Gumprecht, J.; Lindberg, S.ø.; Lingvay, I.; Søndergaard, A.L.; Treppendahl, M.B.; Montanya, E. Oral Semaglutide Versus Empagliflozin in Patients With Type 2 Diabetes Uncontrolled on Metformin: The PIONEER 2 Trial. Diabetes Care 2019, 42, 2272–2281. [Google Scholar] [CrossRef] [PubMed]
- Pratley, R.; Amod, A.; Hoff, S.T.; Kadowaki, T.; Lingvay, I.; Nauck, M.; Pedersen, K.B.; Saugstrup, T.; Meier, J.J. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. The Lancet 2019, 394, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Røder, M.E. Clinical potential of treatment with semaglutide in type 2 diabetes patients. Drugs in Context 2019, 8, 1–11. [Google Scholar] [CrossRef]
- Hofmann, D.W.M.; Kuleshova, L.N. A general force field by machine learning on experimental crystal structures. Calculations of intermolecular Gibbs energy with iFlexCryst. Acta Crystallographica Section A Foundations and Advances 2023, 79, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Giannakoulias, S.; Shringari, S.R.; Ferrie, J.J.; Petersson, E.J. Biomolecular simulation based machine learning models accurately predict sites of tolerability to the unnatural amino acid acridonylalanine. Scientific Reports 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zheng, Z.; Dong, L.; Shi, N.; Yang, Y.; Chen, H.; Shen, Y.; Xia, Q. Rational incorporation of any unnatural amino acid into proteins by machine learning on existing experimental proofs. Computational and Structural Biotechnology Journal 2022, 20, 4930–4941. [Google Scholar] [CrossRef] [PubMed]
- Li, W. How Structural Modifications of Insulin Icodec Contributes to Its Prolonged Duration of Action: A Structural and Biophysical Perspective 2023. [CrossRef]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. In Methods in Molecular Biology; Springer US, 2020; pp. 239–255.
- Li, W. Gravity-driven pH adjustment for site-specific protein pKa measurement by solution-state NMR. Measurement Science and Technology 2017, 28, 127002. [Google Scholar] [CrossRef]
- Webb, H.; Tynan-Connolly, B.M.; Lee, G.M.; Farrell, D.; O’Meara, F.; Søndergaard, C.R.; Teilum, K.; Hewage, C.; McIntosh, L.P.; Nielsen, J.E. Remeasuring HEWL pKa values by NMR spectroscopy: Methods, analysis, accuracy, and implications for theoretical pKa calculations. Proteins: Structure, Function, and Bioinformatics 2010, 79, 685–702. [Google Scholar] [CrossRef]
- Hansen, A.L.; Kay, L.E. Measurement of histidine pKa values and tautomer populations in invisible protein states. Proceedings of the National Academy of Sciences 2014, 111. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. Journal of Chemical Theory and Computation 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Ahrén, B.; Atkin, S.L.; Charpentier, G.; Warren, M.L.; Wilding, J.P.H.; Birch, S.; Holst, A.G.; Leiter, L.A. Semaglutide induces weight loss in subjects with type 2 diabetes regardless of baseline BMI or gastrointestinal adverse events in the SUSTAIN 1 to 5 trials. Diabetes, Obesity and Metabolism 2018, 20, 2210–2219. [Google Scholar] [CrossRef] [PubMed]
- Aroda, V.R.; Rosenstock, J.; Terauchi, Y.; Altuntas, Y.; Lalic, N.M.; Villegas, E.C.M.; Jeppesen, O.K.; Christiansen, E.; Hertz, C.L.; Haluzík, M. PIONEER 1: Randomized Clinical Trial of the Efficacy and Safety of Oral Semaglutide Monotherapy in Comparison With Placebo in Patients With Type 2 Diabetes. Diabetes Care 2019, 42, 1724–1732. [Google Scholar] [CrossRef]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein–protein complexes. eLife 2015, 4. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics, 2016; btw514. [Google Scholar]
- Li, W. An Exhaustive Exploration of the Semaglutide-GLP-1R Sequence Space towards the Design of Semaglutide Analogues with Elevated Binding Affinity to GLP-1R 2024. [CrossRef]
- Li, W. In Silico Generation of Structural and Intermolecular Binding Affinity Data with Reasonable Accuracy: Expanding Horizons in Drug Discovery and Design 2024. [CrossRef]
- Wang, T.; He, X.; Li, M.; Shao, B.; Liu, T.Y. AIMD-Chig: Exploring the conformational space of a 166-atom protein Chignolin with ab initio molecular dynamics. Scientific Data 2023, 10. [Google Scholar] [CrossRef] [PubMed]
- Shulman, R.; Wüthrich, K.; Yamane, T.; Patel, D.J.; Blumberg, W. Nuclear magnetic resonance determination of ligand-induced conformational changes in myoglobin. Journal of Molecular Biology 1970, 53, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.B.; Burch, J.D.; McKay, D.; Bustamante, C.; Crackower, M.A.; Wu, H. Could AlphaFold revolutionize chemical therapeutics? Nature Structural & Molecular Biology 2021, 28, 771–772. [Google Scholar]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. Journal of Molecular Biology 2021, 433, 167208. [Google Scholar] [CrossRef]
- Higgins, M.K. Can We AlphaFold Our Way Out of the Next Pandemic? Journal of Molecular Biology 2021, 433, 167093. [Google Scholar] [CrossRef]
- Platzer, G.; Okon, M.; McIntosh, L.P. pH-dependent random coil 1H, 13C, and 15N chemical shifts of the ionizable amino acids: a guide for protein pKa measurements. Journal of Biomolecular NMR 2014, 60, 109–129. [Google Scholar] [CrossRef]
- Yang, A.S.; Honig, B. On the pH Dependence of Protein Stability. Journal of Molecular Biology 1993, 231, 459–474. [Google Scholar] [CrossRef]
- Harris, T.K.; Turner, G.J. Structural Basis of Perturbed pKa Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life (International Union of Biochemistry and Molecular Biology: Life) 2002, 53, 85–98. [Google Scholar] [CrossRef]
- Li, W. Characterising the interaction between caenopore-5 and model membranes by NMR spectroscopy and molecular dynamics simulations. PhD thesis, University of Auckland, 2016.
- Hansen, A.L.; Kay, L.E. Measurement of histidine pKa values and tautomer populations in invisible protein states. Proceedings of the National Academy of Sciences 2014, 111, E1705–E1712. [Google Scholar] [CrossRef] [PubMed]
- Box, K.; Bevan, C.; Comer, J.; Hill, A.; Allen, R.; Reynolds, D. High-Throughput Measurement of pKa Values in a Mixed-Buffer Linear pH Gradient System. Analytical Chemistry 2003, 75, 883–892. [Google Scholar] [CrossRef]
- Petukh, M.; Stefl, S.; Alexov, E. The Role of Protonation States in Ligand-Receptor Recognition and Binding. Current Pharmaceutical Design 2013, 19, 4182–4190. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. Journal of Chemical Theory and Computation 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M. Design of ultra-stable insulin analogues for the developing world. Journal of Health Specialties 2013, 1, 59. [Google Scholar] [CrossRef]
- Nuhoho, S.; Gupta, J.; Hansen, B.B.; Fletcher-Louis, M.; Dang-Tan, T.; Paine, A. Orally Administered Semaglutide Versus GLP-1 RAs in Patients with Type 2 Diabetes Previously Receiving 1–2 Oral Antidiabetics: Systematic Review and Network Meta-Analysis. Diabetes Therapy 2019, 10, 2183–2199. [Google Scholar] [CrossRef]
- Bucheit, J.D.; Pamulapati, L.G.; Carter, N.; Malloy, K.; Dixon, D.L.; Sisson, E.M. Oral Semaglutide: A Review of the First Oral Glucagon-Like Peptide 1 Receptor Agonist. Diabetes Technology & Therapeutics 2020, 22, 10–18. [Google Scholar]
- Müller, C.E.; Hansen, F.K.; Gütschow, M.; Lindsley, C.W.; Liotta, D. New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science. ACS Pharmacology & Translational Science 2021, 4, 1712–1713. [Google Scholar]
- He, Y.; Su, J.; Lan, B.; Gao, Y.; Zhao, J. Targeting off-target effects: endoplasmic reticulum stress and autophagy as effective strategies to enhance temozolomide treatment. OncoTargets and Therapy 2019, Volume 12, 1857–1865. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nature Methods 2017, 14, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Shen, Y.; Wang, Y.; Yuan, H.; Wu, Y.; Gu, Q. Protein Conformation Generation via Force-Guided SE(3) Diffusion Models, 2024. [CrossRef]
- Callaway, E. The entire protein universe: AI predicts shape of nearly every known protein. Nature 2022. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; Lepore, R.; Schwede, T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Hasani, H.J.; Barakat, K. Homology Modeling: an Overview of Fundamentals and Tools. International Review on Modelling and Simulations (IREMOS) 2017, 10, 129. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. Journal of Computational Chemistry 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Gircha, A.I.; Boev, A.S.; Avchaciov, K.; Fedichev, P.O.; Fedorov, A.K. Hybrid quantum-classical machine learning for generative chemistry and drug design. Scientific Reports 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Roggia, M.; Natale, B.; Amendola, G.; Maro, S.D.; Cosconati, S. Streamlining Large Chemical Library Docking with Artificial Intelligence: the PyRMD2Dock Approach. Journal of Chemical Information and Modeling 2023. [Google Scholar] [CrossRef] [PubMed]
- Agu, P.C.; Afiukwa, C.A.; Orji, O.U.; Ezeh, E.M.; Ofoke, I.H.; Ogbu, C.O.; Ugwuja, E.I.; Aja, P.M. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Scientific Reports 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Merz, K.M. Calculating protein-ligand binding affinities with MMPBSA: Method and error analysis. Journal of chemical theory and computation 2017, 13, 4751–4767. [Google Scholar]
- Deng, N.J.; Zheng, Q.; Liu, J.; Hao, G.F. Predicting protein–ligand binding affinity with a random matrix framework. PLoS computational biology 2012, 8, e1002775. [Google Scholar]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nature structural biology 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Canzar, S.; Toussaint, N.C.; Klau, G.W. An exact algorithm for side-chain placement in protein design. Optimization Letters 2011, 5, 393–406. [Google Scholar] [CrossRef]
- Herget, S.; Ranzinger, R.; Maass, K.; Lieth, C.W. GlycoCT—a unifying sequence format for carbohydrates. Carbohydrate Research 2008, 343, 2162–2171. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.M.; Moreno, P.; Fabregat, A.; Hermjakob, H.; Steinbeck, C.; Apweiler, R.; Wakelam, M.J.O.; Vizcaíno, J.A. LipidHome: A Database of Theoretical Lipids Optimized for High Throughput Mass Spectrometry Lipidomics. PLoS ONE 2013, 8, e61951. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Subramaniam, S. Template-based combinatorial enumeration of virtual compound libraries for lipids. Journal of Cheminformatics 2012, 4. [Google Scholar] [CrossRef]
- Hanson, B.; Stall, S.; Cutcher-Gershenfeld, J.; Vrouwenvelder, K.; Wirz, C.; Rao, Y.; Peng, G. Garbage in, garbage out: mitigating risks and maximizing benefits of AI in research. Nature 2023, 623, 28–31. [Google Scholar] [CrossRef]
- Blüher, M.; Rosenstock, J.; Hoefler, J.; Manuel, R.; Hennige, A.M. Dose–response effects on HbA1c and bodyweight reduction of survodutide, a dual glucagon/GLP-1 receptor agonist, compared with placebo and open-label semaglutide in people with type 2 diabetes: a randomised clinical trial. Diabetologia 2023, 67, 470–482. [Google Scholar] [CrossRef]
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Frontiers in Endocrinology 2019, 10, 1–32. [Google Scholar]
- Li, W. Structurally Observed Electrostatic Features of the COVID-19 Coronavirus-Related Experimental Structures inside Protein Data Bank: A Brief Update 2020.
- Li, W. How do SMA-linked mutations of SMN1 lead to structural/functional deficiency of the SMA protein? PLOS ONE 2017, 12, e0178519. [Google Scholar] [CrossRef]
- Li, W. Extracting the Interfacial Electrostatic Features from Experimentally Determined Antigen and/or Antibody-Related Structures inside Protein Data Bank for Machine Learning-Based Antibody Design 2020.
- Li, W. Calcium Channel Trafficking Blocker Gabapentin Bound to the α-2-δ-1 Subunit of Voltage-Gated Calcium Channel: A Computational Structural Investigation 2020.
- Li, W. Inter-Molecular Electrostatic Interactions Stabilizing the Structure of the PD-1/PD-L1 Axis: A Structural Evolutionary Perspective 2020.
- Li, W. Designing Nerve Growth Factor Analogues to Suppress Pain Signal Transduction Mediated by the p75NTR-NGF-TrkA Complex: A Structural and Biophysical Perspective 2024. [CrossRef]
- Zhou, S.F.; Zhong, W.Z. Drug Design and Discovery: Principles and Applications. Molecules 2017, 22, 279. [Google Scholar] [CrossRef]
- Subramaniam, S.; Kleywegt, G.J. A paradigm shift in structural biology. Nature Methods 2022, 19, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, A.M.J.J. 50 years of PDB: a catalyst in structural biology. Nature Methods 2021, 18, 448–449. [Google Scholar] [CrossRef] [PubMed]
- Vakili, M.G.; Gorgulla, C.; Nigam, A.; Bezrukov, D.; Varoli, D.; Aliper, A.; Polykovsky, D.; Das, K.M.P.; Snider, J.; Lyakisheva, A.; Mansob, A.H.; Yao, Z.; Bitar, L.; Radchenko, E.; Ding, X.; Liu, J.; Meng, F.; Ren, F.; Cao, Y.; Stagljar, I.; Aspuru-Guzik, A.; Zhavoronkov, A. Quantum Computing-Enhanced Algorithm Unveils Novel Inhibitors for KRAS, 2024. [CrossRef]
- Gupta, R.; Srivastava, D.; Sahu, M.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Molecular Diversity 2021, 25, 1315–1360. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Crystal structure of semaglutide backbone in complex with the GLP-1 receptor extracellular domain.
Figure 1.
Crystal structure of semaglutide backbone in complex with the GLP-1 receptor extracellular domain.
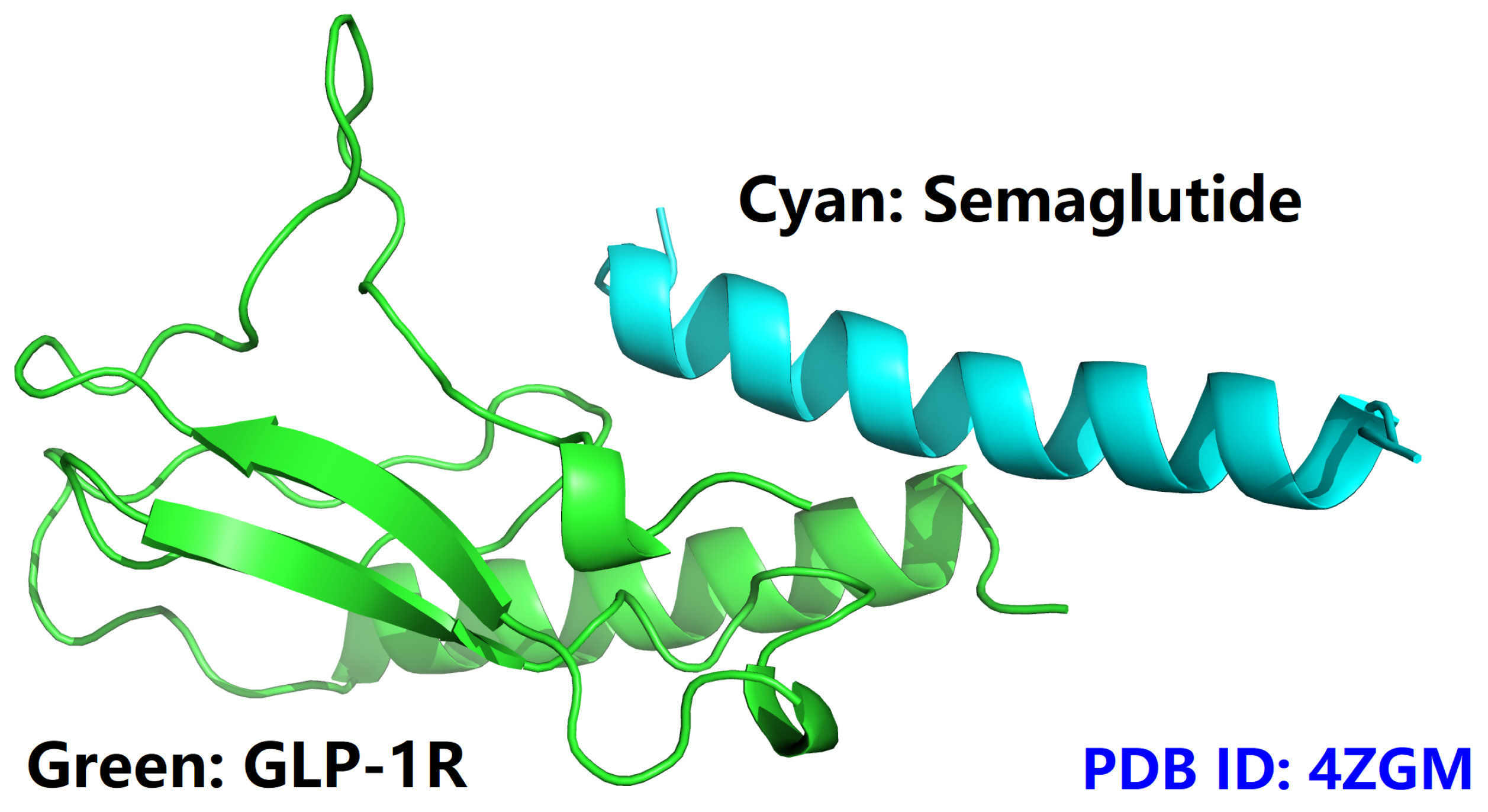
Figure 2.
Automated in silico generation of synthetic structural and Kd data.
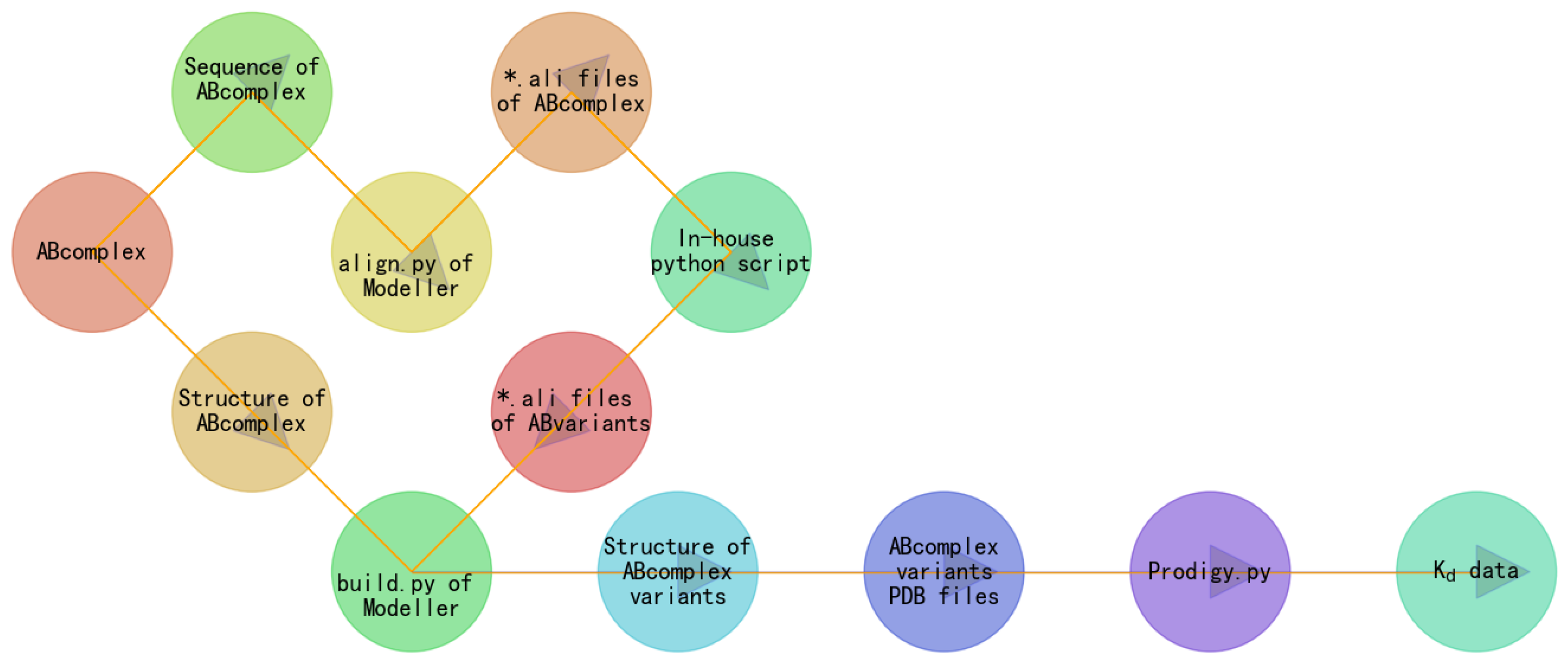
Figure 3.
Distribution of the binding affinities between native semaglutide and GLP-1R (10000 structural models) and between semaglutide mutants/analogues (with one missense mutation) and GLP-1R (11200 structural models).
Figure 3.
Distribution of the binding affinities between native semaglutide and GLP-1R (10000 structural models) and between semaglutide mutants/analogues (with one missense mutation) and GLP-1R (11200 structural models).
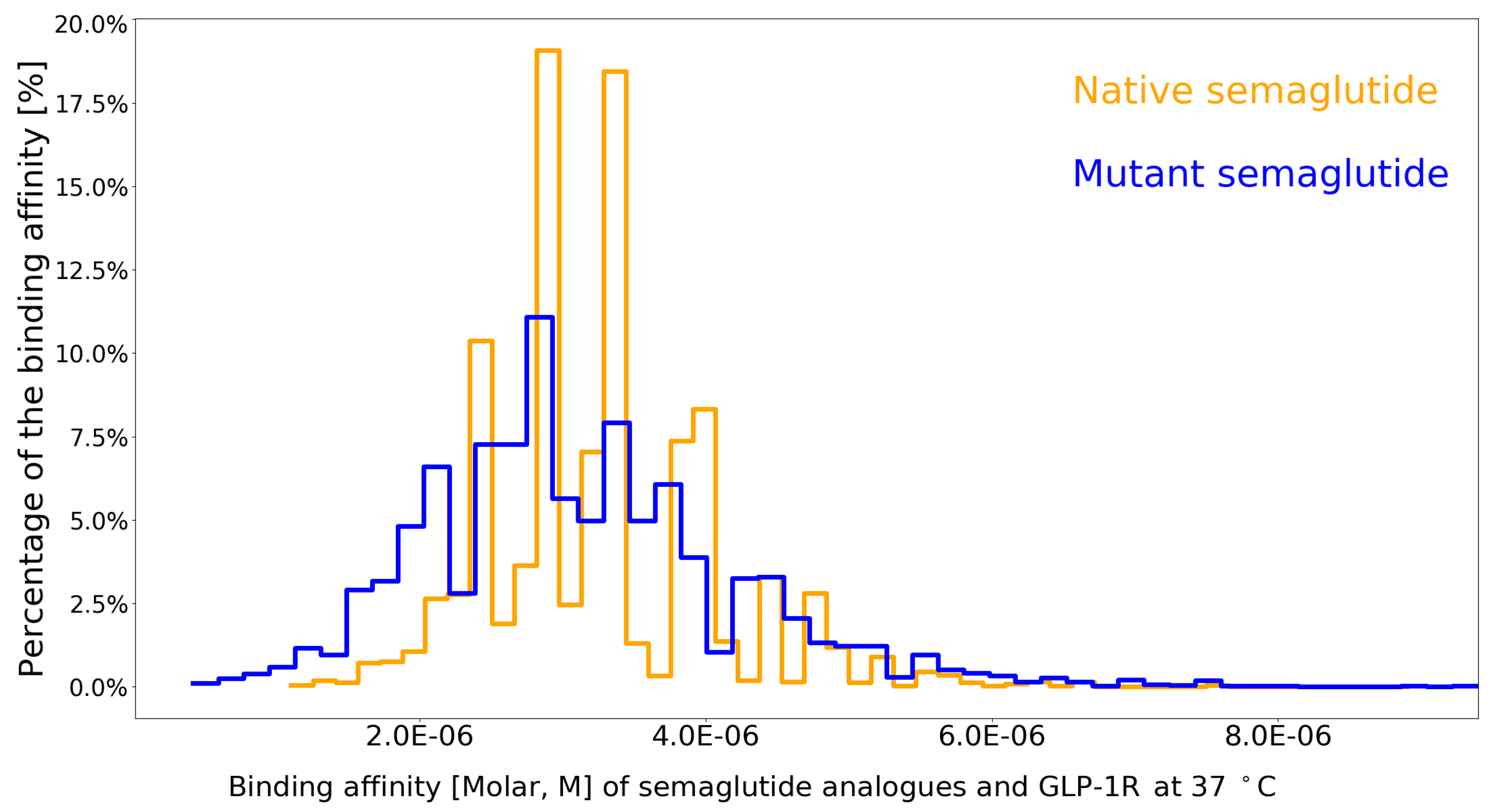
Figure 4.
A partial sketch of the molecular space. In this figure, suppose the molecular space is an ocean with a series of islands, semaGIBAC is one of the island (hydrogen atom), consisting of experimental (nucleus) and computational (electron cloud) structural and biophysical data for any type of (bio)molecule within PDB.
Figure 4.
A partial sketch of the molecular space. In this figure, suppose the molecular space is an ocean with a series of islands, semaGIBAC is one of the island (hydrogen atom), consisting of experimental (nucleus) and computational (electron cloud) structural and biophysical data for any type of (bio)molecule within PDB.
Figure 5.
A future direction of in silico generation of structural and intermolecular Kd data in precise drug discovery & design. This figure is an extention of Figure 2
Figure 5.
A future direction of in silico generation of structural and intermolecular Kd data in precise drug discovery & design. This figure is an extention of Figure 2
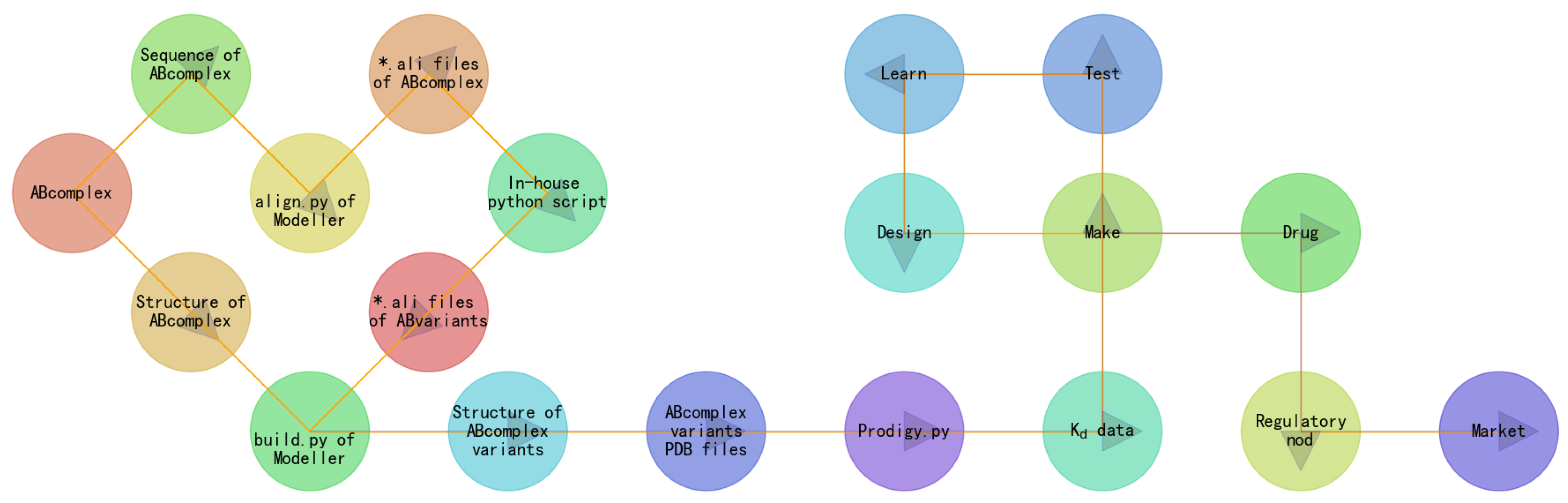

Table 1.
Experimentally determined semaglutide-related structures in Protein Data Bank (QUERY: Polymer Entity Description = "Semaglutide").
Table 1.
Experimentally determined semaglutide-related structures in Protein Data Bank (QUERY: Polymer Entity Description = "Semaglutide").
| PDB ID | Structure Title |
|---|---|
| 7KI0 | Semaglutide-bound Glucagon-Like Peptide-1 (GLP-1) Receptor in Complex with Gs protein |
Table 2.
Experimentally determined semaglutide-related structures in the Protein Data Bank (QUERY: Full Text = "Semaglutide").
Table 2.
Experimentally determined semaglutide-related structures in the Protein Data Bank (QUERY: Full Text = "Semaglutide").
| PDB ID | Structure Title (release date from newest to oldest) |
|---|---|
| 7KI0 | Semaglutide-bound Glucagon-Like Peptide-1 (GLP-1) Receptor in Complex with Gs protein |
| 7KI1 | Taspoglutide-bound Glucagon-Like Peptide-1 (GLP-1) Receptor in Complex with Gs Protein |
| 4ZGM | Crystal structure of semaglutide backbone in complex with the GLP-1 receptor extracellular domain |
Table 3.
Strengthening semaglutide-GLP-1R binding affinity via a Val27-Arg28 exchange in the peptide backbone of semaglutide.
Table 3.
Strengthening semaglutide-GLP-1R binding affinity via a Val27-Arg28 exchange in the peptide backbone of semaglutide.
| PDB file | Protein-Protein Complex | G (kcal/mol) | Kd (M) at 37 ∘C | Fold |
|---|---|---|---|---|
| 4ZGM [42] | semaglutide-GLP-1R [42] | -7.8 | 3.4 × 10-6 | 1 |
| sema.pdb [6] | Val27-Arg28 exchange [6] | -8.4 | 1.1 × 10-6 | 3.09 |
Table 4.
Statistics of the intermolecular binding affinities of PDB entry 4ZGM (Table 2), semaGIBAC and an incomplete two-dimensional semaGIBAC.
Table 4.
Statistics of the intermolecular binding affinities of PDB entry 4ZGM (Table 2), semaGIBAC and an incomplete two-dimensional semaGIBAC.
| Model | Repeat | Mean | Std | Min | Max |
|---|---|---|---|---|---|
| 4ZGM [42] | 10000 | 3.278E-06 | 7.800E-07 | 1.100E-06 | 8.900E-06 |
| semaGIBAC | 11200 | 3.134E-06 | 1.091E-06 | 4.100E-07 | 9.400E-06 |
| 2D semaGIBAC | 154055 | 3.402E-06 | 1.400E-06 | 1.400E-07 | 1.500E-05 |
Table 5.
The Size () of the synthetic structural data set based on semaglutide-GLP-1R complex structure. where k represents the length of semaglutide backbone, n represents the number of missense mutations introduced into semaglutide backbone, where the value of is key to ensure the overall reasonable accuracy of the synthetic structural data.
Table 5.
The Size () of the synthetic structural data set based on semaglutide-GLP-1R complex structure. where k represents the length of semaglutide backbone, n represents the number of missense mutations introduced into semaglutide backbone, where the value of is key to ensure the overall reasonable accuracy of the synthetic structural data.
| 6|cSize (s) of the synthetic structural and biophysical data set | |||||
|---|---|---|---|---|---|
| Semaglutide backbone (28 Aa) | Molecule X (100 Aa) | ||||
| g(28,1) | 560 | g(100,1) | 2000 | ||
| g(28,2) | 151200 | g(100,2) | 1980000 | ||
| g(28,3) | 26208000 | g(100,3) | 1293600000 | ||
| g(28,4) | 3276000000 | g(100,4) | 627396000000 | ||
| g(28,5) | 314496000000 | g(100,5) | 240920064000000 | ||
Table 6.
The binding affinities of native semaglutide (4ZGM), semaglutide with a Val27-Arg28 exchange and semaglutideX (supplementary file semx.pdb, Table 6) to GLP-1R as calculated by Prodigy.
Table 6.
The binding affinities of native semaglutide (4ZGM), semaglutide with a Val27-Arg28 exchange and semaglutideX (supplementary file semx.pdb, Table 6) to GLP-1R as calculated by Prodigy.
| PDB file | Protein-Protein Complex | G (kcal/mol) | Kd (M) at 37 ∘C | Fold |
|---|---|---|---|---|
| 4ZGM [42] | semaglutide-GLP-1R [42] | -7.8 | 3.4 × 10-6 | 1 |
| sema.pdb [6] | Val27-Arg28 exchange [6] | -8.4 | 1.1 × 10-6 | 3.09 |
| semx.pdb [59] | G13B_A I20B_Q L23B_R V24B_N [59] | -10.7 | 3.0 × 10-8 | 113.33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



