
Submitted:
13 June 2024
Posted:
14 June 2024
You are already at the latest version
Abstract
In Cameroon, Aedes mosquitoes transmit various arboviruses, posing significant health risks. We aimed to characterize the Aedes virome in southwestern Cameroon and identify potential core viruses which might be associated with vector competence. A total of 398 Aedes mosquitoes were collected from four locations (Bafoussam, Buea, Edea and Yaounde). Aedes albopictus dominated all sites except for Bafoussam where Aedes africanus prevailed. Metagenomic analyses of mosquitoes grouped per species into 54 pools revealed notable differences in eukaryotic viromes between Ae. africanus and Ae. albopictus, with the former exhibiting greater richness and diversity. Thirty-seven eukaryotic virus species from 16 families were identified, including six novel viruses with near complete genome sequences. Seven viruses were further quantified in individual mosquitoes via qRT-PCR. Although none of them could be identified as core viruses, Guangzhou sobemo-like virus and Bafoussam mosquito solemovirus virus were highly prevalent regionally in Ae. albopictus and Ae. africanus, respectively. This study highlights the diverse eukaryotic virome of Aedes species in southwestern Cameroon. Despite their shared Genus, Aedes species exhibit limited viral sharing, with varying viral abundance and prevalence across locations. Ae. africanus, an understudied vector, harbors a rich and diverse virome, suggesting potential implications for arbovirus vector competence.
Keywords:
Aedes mosquitoes
; Ae. africanus
; Ae. albopictus
; metagenomics
; core virome
; eukaryotic virome
1. Introduction
Mosquitoes (Family: Culicidae) pose a significant threat to global health as they are efficient vectors of major infectious agents [1,2]. Mosquito Genera of medical importance are Anopheles, Culex and Aedes, which are the most efficient pathogen vectors of the class of Arthropods [3,4]. These Genera carry pathogens (parasites, filarial worms, and arboviruses) which are responsible for at least 17% of all human and animal diseases [5,6]. Approximately 73% of these pathogenic agents are arboviruses, a significant part of which is known to originate from wildlife [7]. In Africa, arboviruses have caused over 35 arboviral diseases and at least 26 of these diseases have been detected in Cameroon [6,8]. The most prevalent arboviral diseases in Cameroon include Dengue, Chikungunya, Yellow fever, Zika, and Rift valley fever [9,10].
In addition to arboviruses capable of infecting both vertebrate and invertebrate cells, the mosquito virome (viral part of the microbiota) also contains a large proportion of viruses which can only infect vertebrate cells and are referred to as Insect Specific Viruses (ISVs) [11]. The main mechanism of transmission and maintenance of ISVs is through vertical transmission (from an infected female mosquito to their offspring) [13], although recent research has suggested another potential mechanism for virus transmission, which is through mosquito excreta [12,13,14].. ISVs being transferred from mosquito generation to generation in a particular mosquito population for a period of time are referred to as the “core virome”, a clear quantitative definition is currently lacking [15]. Although the exact mechanism of microbial interactions is not fully understood, ISVs such as Phasi Charoen-like virus (PCLV), Palm Creek virus and bacteria such as Wolbachia have been hypothesized to modulate their mosquito host with respect to vector competence for important arboviruses such as West Nile virus (WNV), Dengue Virus (DENV), Zika virus (ZIKV) [16,17,18,19].
The increased discovery of ISVs can be largely attributed to metagenomic Next-Generation Sequencing (mNGS). This valuable tool has revolutionized the identification of viruses, mosquito species, and endosymbionts, like Wolbachia, from minimal sample quantities [20,21]. One of the significant advantages of mNGS is its ability to detect known and unknown viruses, as most viruses are very challenging to isolate and grow. This capability has benefited mosquito virology, by unveiling a previously unexplored diversity of viruses within mosquito populations. The insights gained from viral profiling via mNGS hold significant implications for disease surveillance and public health.
In Cameroon the most common vector of arboviral diseases is the Aedes mosquito [22]. Previous studies portray Ae. aegypti, and Ae. albopictus as the major vectors of arboviruses, with Ae. aegypti dominating the northern part and Ae. albopictus dominating the southern part of the country [6,23,24,25]. Recent studies also show the circulation of Ae. africanus which is also very prevalent in regions where arboviral diseases have been reported, unfortunately, there is very little known about its vector competence [9,26,27]. Ae. simpsoni was also recently identified circulating abundantly in rural forest settings in southwestern Cameroon and in Maroua, located in North Cameroon [6,28]. The distribution of these mosquitoes varies with the prevailing climatic zones. There are five sub climatic or ecological zones in Cameroon: Equatorial mountain moonsoon, Equatorial guinean, Equatorial moonsoon, Tropical sudanian and Tropical sudano-sahelian (Figure 1). The climatic zones in Cameroon are dominated by the Warm desert and Semi-arid climate in the North, the Tropical Savanna in the Central part, and the Equatorial Monsoon climate in the southern part of the country and along the coast [23,24,28].
This study aims to elucidate the eukaryotic virome composition of the Aedes species prevalent in the southwestern region of Cameroon, specifically the environments of Buea, Edea, Yaoundé and Bafoussam with a focus on identifying core viruses. To achieve this aim, we employed the mNGS technique to characterize the Aedes mosquito virome across these regions. Subsequently, we conducted quantitative analysis using Reverse Transcription Polymerase Chain Reaction (qRT-PCR) to investigate suspected core viruses in individual mosquitoes across different mosquito species and geographical locations. The findings of this study add valuable information on the Aedes eukaryotic virome composition (identity, diversity, and abundance) in southwestern Cameroon. Understanding the composition of the mosquito virome, is essential as it provides fundamental knowledge to comprehending microbial interaction which is an appealing strategy for arboviral disease control.
2. Materials and Methods
2.1. Mosquito Sampling and Processing
Mosquitoes were collected from August to September 2020 (rainy season) from farms and gardens around households at dawn and dusk in four regions in Cameroon: Buea, Edea, Yaounde, and Bafoussam (Table S1). Aedes mosquitoes were trapped using two methods: the BG sentinel trap (supplemented by a BG lure and carbon dioxide made from yeast, water, and sugar) and aspiration using portable aspirators. The mosquitoes were transported in a mini freezer containing ice packs to the Molecular and Cell Biology Laboratory (MCBL) of the University of Buea for characterization.
Species identification and gender determination were carried out based on morphological characteristics using the stereo binocular microscope following Pictoria Keys [29]. A total of 398 mosquitoes were captured in different climatic zones, out of which 216 female mosquitoes of dominant species were grouped into 54 pools (4 mosquitoes per pool) based on species, and capture location (Table 1).
2.2. Viral Enrichment, Amplification, Library Construction and Sequencing
The 54 pools were processed for NGS using the NetoVIR protocol [30]. Each mosquito pool was homogenized in 400 μL PBS using 2.8 mm ceramic beads and centrifuged at 17,000g for 3 min. Supernatant (150 μL) was filtered using a 0.8 μm (PES) filter at 17,000g for 1 min. to remove bacterial and host cells and to enrich Virus-Like Particles (VLPs). Free-floating nucleic acids in the filtrate were subjected to nuclease digestion using a combination of enzymes. Specifically, 2 μL of Benzonase (25-19 units/µL) and 1 μL of Micrococcal nuclease (2,000,000 gel units/mL) were added to the sample, along with a homemade buffer (1 M Tris, 100 mM CaCl2, and 30 mM MgCl2). The mixture was incubated at 37 °C for 2 h. Subsequently, RNA and DNA were extracted using the QIAGEN Viral RNA mini kit without carrier RNA following the manufacturer’s instructions. Random amplification of reverse transcribed RNA and DNA was done using the Complete Whole Transcriptome Amplification Kit (WTA 2) (Sigma Aldrich) for 17 cycles. The WTA2 products were purified using the MSB® Spin PCRapace (Stratec). Libraries for Illumina sequencing were prepared using the Nextera XT DNA Library preparation kit from Illumina and further purified using 1:1 ratio of Agencourt AMPure XP beads (Beckman Coulter, Inc.). Prior to sequencing, DNA library size distribution and quality were determined using the Bioanalyzer and qubit measurements. Sequencing of the samples was performed on a NextSeq500 High throughput platform (Illumina) for 300 cycles (2×150 bp paired ends). The target for each pool was an average of 10 million paired end reads.
2.3. Identification and Annotation Eukaryotic Sequences Retrieved from Sequenced Aedes Mosquito Pools
The sequencing of 54 pools generated an average of 7.3 million raw reads (0.3 to 13.6 million) per pool. The bioinformatic analysis was first conducted using the Virome Paired-End Reads (ViPER v1.1) pipeline (https://github.com/Matthijnssenslab/ViPER). First, adapters and low-quality reads were trimmed from the raw paired end reads using Trimmomatic [31]. Subsequently, reads mapping to contigs which are known to be found in reagents (contaminants) and the host genome (Accession numbers for Aedes aegypti: GCA_002204515.1 and Aedes albopictus: GCA_006496715.1) were removed using Bowtie 2 [32]. The trimmed reads were de novo assembled into 124,398 virus contigs of at least 500bp in length using metaSPAdes and clustered across samples into 57,516 non-redundant (nr) contigs using Blast and CheckV [33,34]. These nr contigs were taxonomically annotated using DIAMOND (based on BLASTx), KronaTools and TaxonKit which classifies based on the lowest common ancestor [35,36,37]. From these nr contigs, only contigs belonging to eukaryotic viruses were extracted. Thereafter, nr contigs were blasted against the mosquito NCBI nr database to filter out Endogenous Viral Elements (EVEs; generally below 1000bp and not abundant but highly prevalent in almost all mosquito samples of the same Aedes species). Nr contigs with at least 70% nt similarity to mosquito sequences in the database were filtered out resulting in 121 eukaryotic virus contigs. Abundance and taxonomy tables of these eukaryotic virus contigs were used for making comparative analysis: alpha diversity (Observed and Simpson indices), beta diversity (Bray-Curtis dissimilarity and ordination analysis with adonis2) and heat maps in R with ComplexHeatmap [38], ggplot2 [39], and phyloseq packages [40]. Also, an abundance correlation analysis was done in R to identify the un-annotated/mis-annotated segments of segmented viruses. This makes use of contigs corresponding to RNA-dependent RNA-polymerase (RdRp) to identify contigs that displayed significant correlations and similar prevalence across samples [21].
2.4. Virus Identification and Phylogenetic Analysis
For each identified eukaryotic virus contigs, ORF Finder was utilized to predict Open Reading Frames (ORFs) and identify contigs with complete coding capacity. To determine the evolutionary history of potential novel viruses, phylogenetic trees were constructed based on amino acid sequences of the RdRp protein. Amino acid sequences from related viruses belonging to the same Family or Order were retrieved from NCBI and included in the analysis. Phylogenetic trees were constructed using the maximum likelihood method with 1000 bootstraps, and pairwise distances (%) were calculated in MEGA11 [41].
2.5. Aedes Mosquito Identification and Quantification of Selected Viruses
Following morphological identification, mosquito samples subjected to the qRT-PCR analyses were also molecularly identified via DNA barcoding. For each mosquito, individual homogenization was carried out in 400 μL of PBS, followed by centrifugation at 17,000g for 3 min. From the resulting supernatant, 150 μL was utilized for DNA extraction using the QIAGEN Viral RNA mini kit without carrier RNA following the manufacturer’s instructions and eluted in a volume of 60 μL. The eluted volume (60 μL) was diluted to 120 μL and divided into 10 aliquots of 12 μL each and stored to avoid multiple freeze thawing cycles of large volumes. The first aliquot was used for the amplification of the cytochrome c oxidase subunit I region of Aedes mosquitoes employing AUCOS primers (Table S2) with the QIAGEN One-step RT-PCR kit, following the manufacturer’s protocol [42]. Briefly, the reaction mixture consisted of 10 μL RNase-free water, 5 μL of 5x QIAGEN OneStep RT-PCR Buffer, 1μL of dNTP Mix, 1.5 μL of each Primer (10 μM), 1μL of QIAGEN OneStep RT-PCR Enzyme Mix, and 5 μL of sample, in a total volume of 25 μL [41]. Thermal cycling conditions for 35 cycles were as follows: initial PCR pre-denaturation step at 95°C for 10 min, denaturation at 94°C for 30 s, annealing at 52°C for 40 s, extension at 72°C for 45 s, and a final extension at 72°C for 10 min. PCR products were evaluated for amplification using 1.5% agarose gel electrophoresis in 1x TBE buffer and Midori Green as the intercalating agent and thereafter visualized under UV light to observe a band of ~700 base pairs (bp). Afterward, the amplicons underwent Sanger sequencing at Macrogen (Amsterdam, Netherlands).
From NGS results obtained from the 54 sequenced pools, a total of seven viruses were selected based on abundance across samples and completeness of their genome sequences. Primers and probes were designed for qPCR quantification targeting the most conserved regions using NCBI Primer-BLAST. The remaining aliquots of the individual mosquitoes (182 single mosquitoes) were analyzed using each of the seven developed qRT-PCRs. Each qRT-PCR reaction was done in a total volume of 20 μL, consisting of 5 μL TaqMan Fast Virus 1-Step Master Mix by Thermo-Fisher, 2 μL forward and reverse primers (10 μM each), 1 μL probe of (5 μM), and 5 μL nucleic acid extract. Standards (representing the qRT-PCR target regions) of known concentration (integrated DNA Technologies) were serially diluted in ten folds (103 – 108 copies) to make a standard curve. Quantification of genome copies for each virus was later determined by multiplying the Cq value obtained from qRT-PCR by the dilution factor 64.08. This factor accounts for the initial dilution in PBS (400 µL), followed by extraction (150 µL taken), and elution in 120 µL of elution buffer. The dilution factor was calculated considering the respective dilution factors for PBS (2.67) and elution buffer (24), resulting in an overall dilution factor of 64.08.
3. Results
3.1. Each Sampling Site Is Dominated by a Single Aedes Species
This study focuses on analyzing the eukaryotic virome of the Aedes mosquito from four regions in southwestern Cameroon. In 2020, a total of 398 Aedes mosquitoes were captured from Bafoussam (n=101), Edea (n=96), Buea (n=96) and Yaounde (n=105) (Figure 1). Analyses of mosquito distribution reveled that, Ae. albopictus species predominated in Edea, Buea and Yaounde, while Ae. africanus was the most prevalent in Bafoussam. Small numbers of Ae. simpsoni were also captured, co-existing in regions predominated by Ae. albopictus. Only a single Ae. aegypti mosquito was captured in Edea (Figure 1).
3.2. Distinct Virus Families Identified within Aedes Mosquito Species from Different Sampling Sites
Across all four locations, 54 pools of Aedes mosquitoes were sequenced which yielded 121 eukaryotic viral contigs. These eukaryotic viral contigs were identified and annotated, revealing their closest relatives to be 37 distinct eukaryotic viruses. Eleven out of these 37 eukaryotic viral genomes were closely related to known virus species which belonged to a group without an official family classification while the 26 other viral genome sequences belonged to 16 established virus families (Figure 2a and Table S4). Besides these eukaryotic virus reads belonging to these 16 virus families, a significant proportion, accounting for 26.2% of all eukaryotic virus reads were attributed to viral genomes not classified within established families (Figure 2b and Table S5).
3.3. The Majority of Eukaryotic Viral Genomes Are Found in Aedes Africanus Species Collected in Bafoussam
There was a notable disparity in the distribution of eukaryotic reads across Ae. africanus and Ae. albopictus pools. Approximately 80.8% of all eukaryotic virus reads were identified in Ae. africanus pools and only 19.2% in Ae. albopictus pools (Figure 3a). The 80.8% found in Ae. africanus were all from the Bafoussam sampling site. Ae. albopictus mosquitoes were captured in the three other regions: Yaoundé, Buea and Edea and constitutes 13.0%, 6.2% and 0% of all eukaryotic reads respectively (Figure 3b and Table S7). A significant proportion of these eukaryotic virus reads, belonged to viral genomes which have not yet been assigned to established virus families (26.1% in Ae. africanus pools and 0.07% in Ae. albopictus pools) (Table S6).
3.4. Significant Difference in Eukaryotic Virome Richness and Diversity between Aedes Mosquito Species
The alpha diversity of the eukaryotic viromes of Ae. africanus and Ae. albopictus populations circulating in all four locations showed a significant discrepancy in the richness and diversity (Figure 4a). Specifically, the virome of Ae. africanus was more abundant and diverse compared to that of Ae. albopictus. For the regions where Ae. albopictus was captured and found to contain eukaryotic viruses (Buea and Yaoundé), there was no statistically significant difference in the richness and diversity of these viromes (Figure 4b). Intriguingly, despite Edea sharing the same climatic zone with Buea, eukaryotic virus contigs were not identified in Ae. albopictus mosquito pools from Edea.
3.5. Significant Difference in Eukaryotic Virome Composition and Distribution of Aedes Mosquito Species from Different Sampling Sites
Beta diversity based on Bray-Curtis dissimilarity showed a distinct partitioning of eukaryotic virus communities at the level of mosquito species rather than the sampling sites (Figure 5). This shows that the specificity of the mosquito species is the dominant driving force in shaping the virome composition across different habitats.
3.6. Diverse and Abundant Eukaryotic Virome in Aedes Africanus, Compared to Aedes Albopictus Mosquito Pools
The results obtained from the alpha and beta diversity analyses is also reflected on the log2 normalized abundance heatmap of the eukaryotic virus species in 54 pools (Figure 6). The abundance of viral reads per species is shown by the intensity of the red color on the heatmap. Notably, eukaryotic virus contigs related to 37 known viruses were identified in this study, showing a very distinct virome for each Aedes species. The virome of Ae africanus was observed to be more abundant and diverse (30 unique virus species) compared to that of Ae. albopictus (6 unique virus species). Interestingly, one viral genome was shared between both Aedes species, showing distant (41.7% BLASTx) resemblance with Hattula totivirus 3. For each virus species, the average BLASTx percent is represented by the different shades of blue on the left in Figure 6, indicating that most of the identified viruses, only showed rather low amino acid similarities to viruses present in GenBank. Furthermore, the dendrogram in the heatmap highlights that the clustering of the viromes is mainly driven by mosquito species and to a lesser extent by location.
3.7. Phylogenetic Analysis of Six Novel Viruses Identified in Aedes Mosquito Pools
Near complete genome sequences were identified for six novel viruses (BLASTx < 90% with viruses in databases), and these were used for phylogenetic analyses (Table S8). All of these viruses were found in Ae. africanus samples collected in Bafoussam in 2020. Figure 7 presents a graphical illustration of the RdRp gene of these novel virus genomes, their closest relatives, as well as their pairwise similarities. In addition, Figure 7 shows phylogenetic trees constructed based on the amino acid sequence of the RdRp which is the most conserved region in these viral genomes. The names of the novel viruses are indicated in red while their closest relatives are in blue. Also, each tree depicts the clade to which novel viruses belong, color-coded in red.
3.7.1. Bunyavirales
Bunyavirales is an order encompassing negative sense single-strand, enveloped RNA viruses. Our tree focuses on three families: Phenuivirdae, Peribunyaviridae and Nairoviridae. Two novel viruses with three segments each (encoding for the S, M and L proteins.) were identified and named Bafoussam mosquito bunyavirus 1 (BMBV1) and Bafoussam mosquito bunyavirus 2 (BMBV2). The L sequences of these two viruses (BMBV1 and BMBV2) had a 68% pairwise amino acid similarity to each other. BMBV1 and BMBV2 showed 44% and 45% pairwise similarities respectively with the L sequence of Salarivirus Mos8CM0 (Figure 7a). These similarities are reflected in the phylogenetic tree, where BMBV1 and BMBV2 are both located in a sub-clade distant from the closest relative Salarivirus Mos8CM0. These 3 viruses cluster most closely to members of the family Phenuiviridae, which are known to infect mammals, birds, arthropods, plants, and fungi. A notable member is the arbovirus Rift valley fever virus.
3.7.2. Orthomyxoviridae
Orthomyxoviridae is a family of negative sense single-strand, enveloped RNA viruses with eight segments. Two novel viruses were identified: Bafoussam mosquito orthomyxovirus 1 (BMOV1) with seven segments and Bafoussam mosquito orthomyxovirus 2 (BMOV2) with eight segments. For both viruses, segments encoding key viral proteins: Polymerase basic protein 1 (PB1), Polymerase basic protein 2 (PB2), Polymerase acidic protein (PA), Nucleoprotein (NP), were identified. The PB2 sequences of BMOV1 and BMOV2 were highly distinct from each other, with only 53% pairwise amino acid similarity. However, the PB2 sequences BMOV1 and BMOV2 showed 55% and 78% pairwise similarity to that of Guadeloupe mosquito quaranja-like virus 1 respectively. Phylogenetically, both viruses (BMOV1 and BMOV2) fell in the same clade, although BMOV2 was more closely related to Guadeloupe mosquito quaranja-like virus 1, whereas BMOV1 formed an outgroup (Figure 7b).
3.7.3. Rhabdoviridae
Rhabdoviridae is a family of negative sense single-strand, enveloped RNA viruses. The amino acid sequence of the L segment of the novel virus Bafoussam mosquito Rhabdovirus (BMRV) showed 59% pairwise amino acid similarity to Ohlsdorf ohlsrhavirus, which belongs to the genus Ohlsrhavirus within the family Rhabdoviridae (Figure 7c). Furthermore, the BMRV genomic sequence had five main ORFs, four of which coded for the proteins N, M, G and RdRp. However, ORF2 was very divergent and showed no amino acid similarity to the existing proteins in the database. After aligning this ORF2 amino acid sequence to the P-protein of Ohlsdorf ohlsrhavirus (occupying the same position of the P protein in the genome), we found a pairwise amino acid similarity of only 21%.
3.7.4. Solemoviridae
Solemoviridae is a family of positive sense single-strand, non-enveloped RNA viruses. Bafoussam mosquito solemovirus (BMSV) had two segments identified with two ORFs each. The first segment had two ORFs coding for a hypothetical protein and the RdRp protein, while the second segment also had two ORFs coding for the Capsid protein and a hypothetical protein. The amino acid sequence of the first segment containing the RdRp protein of BMSV had a 23% pairwise amino acid similarity to that of Pyongtaek culex solemovirus (Figure 7d).
3.8. In Search for an Aedes Mosquito Core Virome Using qRT-PCR
As NGS based approaches provide only relative data rather than absolute quantification of viruses, we employed qRT-PCR to quantify the most abundant and prevalent viruses (identified in our mosquito pools) using individual mosquitoes. Additionally, qRT-PCR is known to be more sensitive compared to NGS in most instances. From Aedes africanus we selected six abundant viruses: BMSV, BMRV, BMOV1, BMOV2, BMBV1 and BMBV2 and also Guangzhou sobemo-like virus which was the most abundant in Aedes albopictus samples (Figure 8). Quantification of these viruses was done in a total number of 182 individual Aedes mosquitoes (128 Ae. albopictus, 36 Ae. africanus, 17 Ae. simpsoni, and 1 Ae. aegypti) from Bafoussam (46 samples), Buea (41 samples), Edea (45 samples) and Yaoundé (50 samples), as summarized in Table 2. Samples with Cycle threshold (Ct) values ≤ 35 indicating approximately 10 virus genome copies per mosquito were considered positive.
Guangzhou sobemo-like virus was detected in samples from all four regions (Figure 8i). Samples from Buea had the highest median values for genome copies of 5.1 × 104 in Ae. albopictus samples and 2.8 × 103 in Ae. simpsoni samples. Guangzhou sobemo-like virus was also prevalent in samples from Yaounde with median genome copies values of 3.7 × 104 genome copies in Ae. albopictus samples and 1.3 × 103 genome copies in Ae. simpsoni samples (Figure 8ai). In a total of 182 samples, 89 Aedes mosquito samples (48.9%) tested positive for the virus, and it was most prevalent in Ae. albopictus samples (78/128), followed by Ae. simpsoni samples (10/17). The virus was detected in only 1/36 Ae. africanus samples and absent in the only Ae. aegypti sample (Figure 8bi).
Bafoussam mosquito rhabdovirus (BMRV) was only detected in a few samples from only two regions: Edea and Yaoundé (Figure 8aii). Out of 182 Aedes mosquito samples, only eight samples tested positive for the virus (4.4%). Compared to other Aedes species, BMRV was most prevalent in Ae. albopictus samples (7/128), followed by Ae. simpsoni samples (1/17) and absent in Ae. africanus and Ae. aegypti samples (Figure 8bii).
Bafoussam mosquito solemovirus (BMSV) was present in samples from all four regions (Figure 8iii). Samples with the highest median viral loads were Ae. africanus samples from Bafoussam (4.5 x 103 genome copies) followed by Ae. simpsoni samples from Edea (1.8 x 103 DNA copies) (Figure 8aiii). BMSV was detected in 49/182 samples (26.9%), with the majority being in Ae. africanus samples (25/36), followed by Ae. albopictus samples (19/128), Ae. simpsoni samples (5/17) and absent in Ae. aegypti samples (Figure 8biii).
Bafoussam mosquito bunyavirus 1 (BMBV1) was only identified in a few samples from Bafoussam (Figure 8iv). BMBV1 was detected in 14/182 samples (7.7%), with the majority being in Ae. africanus samples (13/36). Only 1/128 Ae. albopictus samples tested positive for BMBV1, while Ae. simpsoni and Ae. aegypti samples all tested negative for the virus (Figure 8biv).
Bafoussam mosquito bunyavirus 2 (BMBV2) was detected in samples from three regions: Bafoussam, Buea and Edea (Figure 8v). Only Ae. simpsoni samples from Edea had a median genome copy number above zero (2.0 x 103 genome copies) (Figure 8av). BMBV2 was identified in 17/182 samples (9.3%), most of which were in Ae. albopictus samples (13/128), followed by Ae. simpsoni samples (3/17). The virus was only present in 1/36 Ae. africanus samples and absent in Ae. aegypti (Figure 8bv).
Bafoussam mosquito orthomyxovirus 1 (BMOV1) was only identified in samples from Bafoussam, just like the case of BMBV1 (Figure 8vi). This virus was present in 3/182 samples (1.6%). All three samples which tested positive for BMOV1 were Ae. africanus samples (3/36) (Figure 8bvi).
Bafoussam mosquito orthomyxovirus 2 (BMOV2) was found in samples from three regions: Bafoussam, Buea and Edea (Figure 8vii). BMOV2 was identified in 16/182 samples (8.8%). Most of the samples which tested positive were Ae. africanus samples (9/36), followed by Ae. albopictus samples (7/128). Ae. simpsoni and Ae. aegypti samples both tested negative for BMOV2 (Figure 8bvii).
4. Discussion
Mosquitoes are important vectors for pathogens like arboviruses which greatly influence human and animal health [21]. Our study focuses on the genus Aedes which constitutes one of the main vectors of arboviruses in Cameroon. Ae. albopictus and Ae. aegypti were described for a long time as the major vectors of arboviral diseases circulating in Cameroon, but recently Ae. africanus mosquitoes were also found to be highly prevalent in West Cameroon [24,26]. In addition, Ae. simpsoni was recently identified to be abundant in rural forest settings in southwestern Cameroon and also in Maroua, located in North Cameroon [6,28].
These Aedes species are responsible for the circulation and transmission of arboviruses in Cameroon, causing arboviral diseases such as Dengue, Chikungunya, Yellow Fever and Zika. Serological studies have reported the presence of these arboviruses in Cameroon by testing IgG and IgM antibodies (Dengue, Chikungunya and Zika) in sera of inhabitants of Douala, Yaounde, Dschang, Garoua, Bertoua, Ngaoundere and Graoua [8,43].
In addition to being major vehicles of arboviruses in Cameroon, Aedes mosquitoes are also potentially home to ISVs which are not known to infect vertebrates, and hence do not cause diseases in humans and animals. In the last decade, research on ISVs has greatly increased due to their potential utility in the prevention and control of arboviral diseases. Mounting evidence show that ISVs interact with other components of the mosquito microbiota and influence mosquito susceptibility to arboviral infection [27,43,44]. Unfortunately, very little is known about the virome of the Aedes mosquitoes circulating in Cameroon [26,41,42]
In this study, we employed viral metagenomics to characterize the viral composition of Aedes mosquito pools from four regions in the southwestern part of Cameroon representing three sub-climatic zones (Table S1). In this part of the country, sampling showed that the dominant Aedes species was Ae. albopictus except for Bafoussam (neighboring town to Dschang) which was dominated by Ae. africanus. This invasive species has been reported to be more prevalent in the southern part of Cameroon because of the favorable climatic conditions which permit the proliferation of their eggs [24,27].
Among the 37 eukaryotic viruses identified in this study, 26 belong to established viral families. mNGS, despite its power, does not provide conclusive insights into the hosts of identified viruses in this study, limiting our ability to conclusively determine their origins. However, identified viruses belonging to families known to infect mosquitoes and insects, such as the Xinmoviridae, Iflaviridae, and Phasmaviridae, likely represent true mosquito-infecting viruses [45,46]. Interestingly, we also identified viruses belonging to families containing known arboviruses, such as Flaviviridae (Menghai flavivirus) and Peribunyaviridae (Duke bunyavirus), suggesting a potential transmission risks to both arthropods and vertebrates, including humans. Moving on, we observed viruses from families possibly derived from the diet or the environment. Among these are Anelloviridae, Circoviridae, Solemoviridae, and Totiviridae, which may have been acquired from viremic hosts during blood feeding or from the environment during nectar feeding. Further, we identified eukaryotic viruses belonging to families known for infecting a broader range of hosts, including Sedoreoviridae (mammals, birds, arthropods, plants, algae), Partitiviridae (plants, fungi, protozoa), Phenuiviridae (mammals, birds, insects, plants, fungi), Rhbadoviridae (humans, animals, plants), and Chrysoviridae (fungi, plants, and possibly insects). Additionally, reads were detected that mapped to contigs annotated as Lampyris noctiluca errantivirus 1, a virus in the family Metaviridae, known to infect animals, plants, and fungi. This family contains retrotransposons capable of inducing mutations and replicating via virus-like particles (VLPs). Moreover, eleven eukaryotic viruses not classified at the family level were identified, several of which were previously found in Aedes and Ochlerotatus species from various regions worldwide [47].
NGS data showed a striking difference in the eukaryotic virome of Ae. africanus and Ae. albopictus samples. The virome of Ae. africanus was richer and more diverse than the virome of Ae. albopictus. This could be due to environmental factors (breeding sites, sources of food), or host immune response to microbiota and microbiota interaction [48,49]. The rich and diverse microbiota of Ae. africanus could have a positive or negative effect on its susceptibility to arbovirus infection and transmission. The observation that the first isolation of ZIKV in mosquitoes was made in Ae. africanus [50] coupled with the fact that it is considered to be the main sylvatic vector of yellow fever virus in Africa [51], suggest that Ae. africanus is a competent vector for arboviruses. Unlike in other studies, where the virome of Ae. albopictus is dominated by more than one virus, in this study Ae. albopictus was dominated by one virus, Guangzhou sobemo-like virus [52,53,54,55]. The reason for this could be either methodological (differences in wetlab procedures or bio-informatics methods and used thresholds), or biological (difference in ISV carriage). In case of the latter, this could have potential implications for distinct vector competences of Cameroonian Ae. albopictus mosquitoes versus mosquitoes in other regions.
Among the 37 eukaryotic viruses, we further characterized six novel viruses (BMSV, BMRV, BMOV1, BMOV2, BMBV1 and BMBV2) for which we obtained near complete genome sequences, all identified from Ae. africanus mosquitoes captured in Bafoussam [51,60,61]. Notably, these viruses showed a large genetic variation. Five of these novel viruses (BMSV, BMOV1, BMOV2, BMBV1 and BMBV2) clustered together in clades of unclassified viruses at the genus level indicating their unique evolutionary lineage and awaiting further official classifications. Only BMRV clustered within the established Genus Ohlsrhavirus. Although some of these newly identified viruses were found in families which contain Genera associated with human, animal, or plant diseases (Orthomyxoviridae and Rhabdoviridae), none of their closest relatives have demonstrated the ability to infect humans, animals, or plants, suggesting that they are all specific to insects.
To further investigate the concept of the “core virome” in mosquitoes [11,56,57], BMSV, BMRV, BMOV1, BMOV2, BMBV1 BMBV2 and Guangzhou sobemo-like virus were quantified in 182 individual mosquitoes as they were abundantly present in our pools. The closest relatives of these seven (novel) viruses all been previously detected in mosquitoes [11,52,54,55,57,58].
Among the seven viruses quantified in samples, only Guangzhou sobemo-like virus and BMSV were found in samples from all locations (Figure 8; Table S9). Although these viruses were found in samples from all locations, they were more abundant and prevalent in distinct Aedes species. Guangzhou sobemo-like virus, first isolated from Ae. albopictus samples [57], was found in most of our Ae. albopictus and Ae. simpsoni samples while BMSV was found in most Ae. africanus samples (Table S10). In addition to BMSV which was more prevalent in Ae. africanus, BMBV1 was mostly present in Ae. africanus samples. At the moment a clear quantitative definition for a “core virome” is lacking but given the initial qualitative definition of “a set of viruses found in the majority of individuals in a particular mosquito population”, none of our identified viruses seems to meet this criterium for any of the investigated Aedes species across all the investigated sites in Cameroon. However, Guangzhou sobemo-like virus and BMSV could be considered as core viruses in Ae. albopictus, and Ae. africanus, respectively, within particular restricted areas. In a recent study in Belgium we were also unable to identify an abundant core virome in Culex mosquitoes [59]. For future research it would be beneficial to have a more comprehensive and quantitative approach to compare mosquito virome compositions across species, space and time.
Although for our NGS data suggested a minimal overlap in the virome of Ae. africanus and Ae. albopictus, our more sensitive qRT-PCR data showed a larger overlap between the virome of both Aedes species (Figure 8). The qRT-PCR assays also showed a significant overlap in viruses found in Ae. albopictus and Ae. simpsoni (Figure 8).
For future research it would be interesting to isolate the highly abundant and prevalent Guangzhou sobemo-like virus and BMSV for further studies and in vivo vector competence experiments. Understanding the complex interaction between these potential core viruses with the host and/or other components of the host microbiota is essential for gaining insights into the composition, diversity, and dynamics of the mosquito virome. This knowledge does not only enhance our understanding of vector-borne disease dynamics but also helps in the development of more effective strategies for vector control and disease management.
5. Conclusions
Our study shows a striking difference (abundance and diversity) between the eukaryotic viromes of different Aedes species, with the virome of Ae. africanus being richer and more diverse from that of Ae. albopictus. We were unable to identify a true Aedes species specific core virome, although on a local scale Guangzhou sobemo-like virus and Bafoussam mosquito solemovirus virus could be considered as such in Ae. albopictus and Ae. africanus, respectively. Further studies are needed to understand if and how these viruses interact with the rest of the mosquito’s microbiota to influence vector competence.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
Conceptualization, J.M. and S.M.G.; formal analysis K.C.M.D. and L.D.C.; writing—original draft preparation, K.C.M.D.; writing—review and editing, K.C.M.D., J.M., S.M.G., and L.D.C.; supervision; J.M. and S.M.G.; funding acquisition, K.C.M.D., J.M. and S.M.G.
Funding
This research was funded by VLIR-UOS through the Global Minds PhD scholarship, grant number BE2022GMUKULA101 at KU Leuven in Belgium.
Institutional Review Board Statement
Ethical approval (No. 2021/1504-07/UB/SG/IRB/FHS) for the use of mosquito species was obtained from the Faculty of Health Sciences institutional review board, University of Buea, Cameroon, administrative authorization was obtained from the various Reginal Delegations of Health, Research permit (000006/MINRESI/B00/COO/C10/CIP) was obtained from the Ministry of Research and Scientific innovation and the Access to Benefit and Sharing permit (ABS-PERMIT No. 00007/ABS/MINEPDED/PNA/NP-ABS/ABS/FP) following the Nagoya protocol from the Ministry of Environment, Protection of Nature and Sustainable Development (MINEPDED).
Data Availability Statement
The raw sequencing dataset for this study can be found through NCBI’s Sequence Read Archive (SRA) repository (Bioproject PRJNA1089369). Complete viral genome sequences were submitted to GenBank (PP764659-PP764664, PP868493-PP868501, PP898293-PP898302). All R scripts are available at https://github.com/Matthijnssenslab/CameroonMosquitoesVirome.
Acknowledgments
This work was supported VLIR-UOS through Global Minds. Much gratitude goes to the members of the Laboratory of Viral Metagenomics, Rega institute at the KU Leuven as well as to the members of the Molecular and Cell Biology Laboratory, Biotechnology unit at the University of Buea. Many thanks to field assistants Shanui Fritz and Tekoh Terriss.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tandina, F.; Doumbo, O.; Yaro, A.S.; Traoré, S.F.; Parola, P.; Robert, V. Mosquitoes (Diptera: Culicidae) and Mosquito-Borne Diseases in Mali, West Africa. Parasit Vectors 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Nebbak, A.; Almeras, L.; Parola, P.; Bitam, I. Mosquito Vectors (Diptera: Culicidae) and Mosquito-Borne Diseases in North Africa. Insects 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.S.; Higgs, S.; Vanlandingham, D.L. Biological Control Strategies for Mosquito Vectors of Arboviruses. Insects 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Dahmana, H.; Mediannikov, O. Mosquito-Borne Diseases Emergence/Resurgence and How to Effectively Control It Biologically. Pathogens 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Mosquitoes of Public Health Importance and Their Control.
- Bamou, R.; Mayi, M.P.A.; Djiappi-Tchamen, B.; Nana-Ndjangwo, S.M.; Nchoutpouen, E.; Cornel, A.J.; Awono-Ambene, P.; Parola, P.; Tchuinkam, T.; Antonio-Nkondjio, C. An Update on the Mosquito Fauna and Mosquito-Borne Diseases Distribution in Cameroon. Parasit Vectors 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Tajudeen, Y.A.; Oladipo, H.J.; Oladunjoye, I.O.; Yusuf, R.O.; Sodiq, H.; Omotosho, A.O.; Adesuyi, D.S.; Yusuff, S.I.; El-Sherbini, M.S. Emerging Arboviruses of Public Health Concern in Africa: Priorities for Future Research and Control Strategies. Challenges 2022, 13, 60. [Google Scholar] [CrossRef]
- Fokam, E.B.; Levai, L.D.; Guzman, H.; Amelia, P.A.; Titanji, V.P.; Tesh, R.B. , &; Weaver, S.C. Silent Circulation of Arboviruses in Cameroon. East Afr Med J 2010, 87, 262–268. [Google Scholar]
- Alenou, L.D.; Nwane, P.; Mbakop, L.R.; Piameu, M.; Ekoko, W.; Mandeng, S.; Bikoy, E.N.; Toto, J.C.; Onguina, H.; Etang, J. Burden of Mosquito-Borne Diseases across Rural versus Urban Areas in Cameroon between 2002 and 2021: Prospective for Community-Oriented Vector Management Approaches. Parasit Vectors 2023, 16. [Google Scholar] [CrossRef]
- Sadeuh-Mba, S.A.; Yonga Wansi, G.M.; Demanou, M.; Gessain, A.; Njouom, R. Serological Evidence of Rift Valley Fever Phlebovirus and Crimean-Congo Hemorrhagic Fever Orthonairovirus Infections among Pygmies in the East Region of Cameroon. Virol J 2018, 15. [Google Scholar] [CrossRef]
- Shi, C.; Beller, L.; Deboutte, W.; Yinda, K.C.; Delang, L.; Vega-Rúa, A.; Failloux, A.B.; Matthijnssens, J. Stable Distinct Core Eukaryotic Viromes in Different Mosquito Species from Guadeloupe, Using Single Mosquito Viral Metagenomics. Microbiome 2019, 7. [Google Scholar] [CrossRef]
- Ramírez, A.L.; Colmant, A.M.G.; Warrilow, D.; Huang, B.; Pyke, A.T.; McMahon, J.L.; Meyer, D.B.; Graham, R.M.A.; Jennison, A. V.; Ritchie, S.A.; et al. Metagenomic Analysis of the Virome of Mosquito Excreta. mSphere 2020, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- L’Ambert, G.; Gendrot, M.; Briolant, S.; Nguyen, A.; Pages, S.; Bosio, L.; Palomo, V.; Gomez, N.; Benoit, N.; Savini, H.; et al. Analysis of Trapped Mosquito Excreta as a Noninvasive Method to Reveal Biodiversity and Arbovirus Circulation. Mol Ecol Resour 2023, 23, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Narpon, Q.; Serrato-Pomar, I.; Gauliard, C.; Berthomieu, A.; Wichit, S.; Missé, D.; Sofonea, M.T.; Pompon, J. West Nile Virus Is Transmitted within Mosquito Populations through Infectious Mosquito Excreta 1 2 Short Title: Mosquito Excreta-Mediated Transmission 3 4. [CrossRef]
- Shi, C.; Beller, L.; Deboutte, W.; Yinda, K.C.; Delang, L.; Vega-Rúa, A.; Failloux, A.B.; Matthijnssens, J. Stable Distinct Core Eukaryotic Viromes in Different Mosquito Species from Guadeloupe, Using Single Mosquito Viral Metagenomics. Microbiome 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.I.L.; Suzuki, Y.; Carvajal, T.; Muñoz, M.N.M.; Watanabe, K. Intracellular Interactions Between Arboviruses and Wolbachia in Aedes Aegypti. Front Cell Infect Microbiol 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Leitner, M.; Bishop, C.; Asgari, S. Transcriptional Response of Wolbachia to Dengue Virus Infection in Cells of the Mosquito Aedes Aegypti. mSphere 2021, 6. [Google Scholar] [CrossRef]
- Hall-Mendelin, S.; McLean, B.J.; Bielefeldt-Ohmann, H.; Hobson-Peters, J.; Hall, R.A.; Van Den Hurk, A.F. The Insect-Specific Palm Creek Virus Modulates West Nile Virus Infection in and Transmission by Australian Mosquitoes. Parasit Vectors 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Hobson-Peters, J.; Yam, A.W.Y.; Lu, J.W.F.; Setoh, Y.X.; May, F.J.; Kurucz, N.; Walsh, S.; Prow, N.A.; Davis, S.S.; Weir, R.; et al. A New Insect-Specific Flavivirus from Northern Australia Suppresses Replication of West Nile Virus and Murray Valley Encephalitis Virus in Co-Infected Mosquito Cells. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Ren, Q.; Luo, J.; Tian, Z.; Liu, W.; Zhao, B.; Li, J.; Diao, P.; Tan, Y.; Qiu, X.; et al. Analysis of Microorganism Diversity in Haemaphysalis Longicornis From Shaanxi, China, Based on Metagenomic Sequencing. Front Genet 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Batson, J.; Dudas, G.; Haas-Stapleton, E.; Kistler, A.L.; Li, L.M.; Logan, P.; Ratnasiri, K.; Retallack, H. Single Mosquito Metatranscriptomics Identifies Vectors, Emerging Pathogens and Reservoirs in One Assay. Elife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Tedjou, A.N.; Kamgang, B.; Yougang, A.P.; Njiokou, F.; Wondji, C.S. Update on the Geographical Distribution and Prevalence of Aedes Aegypti and Aedes Albopictus (Diptera: Culicidae), Two Major Arbovirus Vectors in Cameroon. PLoS Negl Trop Dis 2018, 13. [Google Scholar] [CrossRef]
- Dé Ric Simard, F.; Nchoutpouen, E.E.; Claude Toto, J.; Fontenille, D. Geographic Distribution and Breeding Site Preference of Aedes Albopictus and Aedes Aegypti (Diptera: Culicidae) in Cameroon, Central Africa; 2005; Vol. 42;
- Tedjou, A.N.; Kamgang, B.; Yougang, A.P.; Njiokou, F.; Wondji, C.S. Update on the Geographical Distribution and Prevalence of Aedes Aegypti and Aedes Albopictus (Diptera: Culicidae), Two Major Arbovirus Vectors in Cameroon. PLoS Negl Trop Dis 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Djiappi-Tchamen, B.; Nana-Ndjangwo, M.S.; Tchuinkam, T.; Makoudjou, I.; Nchoutpouen, E.; Kopya, E.; Talipouo, A.; Bamou, R.; Mayi, M.P.A.; Awono-Ambene, P.; et al. Aedes Mosquito Distribution along a Transect from Rural to Urban Settings in Yaoundé, Cameroon. Insects 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Mercant Osuna, A.; Gidley, A.; Mayi, M.P.A.; Bamou, R.; Dhokiya, V.; Antonio-Nkondjio, C.; Jeffries, C.L.; Walker, T. Diverse Novel Wolbachia Bacteria Strains and Widespread Co-Infections with Asaia Bacteria in Culicine Mosquitoes from Ecologically Diverse Regions of Cameroon. Wellcome Open Res 2023, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Djeunang Dongho, G.B.; Venturi, G.; Fortuna, C.; Paganotti, G.M.; Severini, C.; L’Episcopia, M.; Tsapi, A.T.; Benedetti, E.; Marsili, G.; Amendola, A.; et al. Dengue and Chikungunya Virus Circulation in Cameroon and Gabon: Molecular Evidence among Symptomatic Individuals. Access Microbiol 2022, 4. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, M.C.; Liang, S.; Kracalik, I.T.; Morris, L.; Blackburn, J.K.; Mbam, L.M.; Ba Pouth, S.F.B.; Teboh, A.; Yang, Y.; Arabi, M.; et al. Cholera in Cameroon, 2000-2012: Spatial and Temporal Analysis at the Operational (Health District) and Sub Climate Levels. PLoS Negl Trop Dis 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Rueda, L.M. Pictorial Keys for the Identification of Mosquitoes (Diptera:Culicidae) Associated with Dengue Virus Transmission; Magnolia Press, 2004; ISBN 1877354465.
- Conceição-Neto, N.; Zeller, M.; Lefrère, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Ranst, M. Van; et al. Modular Approach to Customise Sample Preparation Procedures for Viral Metagenomics: A Reproducible Protocol for Virome Analysis. Sci Rep 2015, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A New Versatile Metagenomic Assembler. Genome Res 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV Assesses the Quality and Completeness of Metagenome-Assembled Viral Genomes. Nat Biotechnol 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive Metagenomic Visualization in a Web Browser. BMC Bioinformatics 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Ren, H. TaxonKit: A Practical and Efficient NCBI Taxonomy Toolkit. Journal of Genetics and Genomics 2021, 48, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. 2009.
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol Biol Evol 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.M.; Valentine, M.J.; Kelly, P.J.; Barua, S.; Murillo, D.F.B.; Wang, C. Modification of the Folmer Primers for the Cytochrome c Oxidase Gene Facilitates Identification of Mosquitoes. Parasit Vectors 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.M.; Tchuenkam, V.P.K.; Colton, M.; Stittleburg, V.; Mitchell, C.; Gaither, C.; Thwai, K.; Espinoza, D.O.; Zhu, Y.; Jamal, H.; et al. Arboviruses as an Unappreciated Cause of Non-Malarial Acute Febrile Illness in the Dschang Health District of Western Cameroon. PLoS Negl Trop Dis 2022, 16. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, P.; Lundén, H.; Blomström, A.L. Insect-Specific Virus Evolution and Potential Effects on Vector Competence. Virus Genes 2019, 55, 127–137. [Google Scholar] [CrossRef]
- Lefeuvre, P.; Martin, D.P.; Elena, S.F.; Shepherd, D.N.; Roumagnac, P.; Varsani, A. Evolution and Ecology of Plant Viruses. Nat Rev Microbiol 2019, 17, 632–644. [Google Scholar] [CrossRef]
- Fermin, G. Host Range, Host-Virus Interactions, and Virus Transmission. In Viruses: Molecular Biology, Host Interactions, and Applications to Biotechnology; Elsevier, 2018; pp. 101–134 ISBN 9780128111949.
- Truong Nguyen, P.T.; Culverwell, C.L.; Suvanto, M.T.; Korhonen, E.M.; Uusitalo, R.; Vapalahti, O.; Smura, T.; Huhtamo, E. Characterisation of the RNA Virome of Nine Ochlerotatus Species in Finland. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Jupatanakul, N.; Sim, S.; Dimopoulos, G. Aedes Aegypti ML and Niemann-Pick Type C Family Members Are Agonists of Dengue Virus Infection. Dev Comp Immunol 2014, 43, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Yu, X.; Cheng, G. Impact of the Microbiome on Mosquito-Borne Diseases. Protein Cell 2023, 14, 743–761. [Google Scholar] [CrossRef] [PubMed]
- Epelboin, Y.; Talaga, S.; Epelboin, L.; Dusfour, I. Zika Virus: An Updated Review of Competent or Naturally Infected Mosquitoes. PLoS Negl Trop Dis 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Weetman, D.; Kamgang, B.; Badolo, A.; Moyes, C.L.; Shearer, F.M.; Coulibaly, M.; Pinto, J.; Lambrechts, L.; McCall, P.J. Aedes Mosquitoes and Aedes-Borne Arboviruses in Africa: Current and Future Threats. Int J Environ Res Public Health 2018, 15. [Google Scholar] [CrossRef]
- Li, C.; Liu, S.; Zhou, H.; Zhu, W.; Cui, M.; Li, J.; Wang, J.; Liu, J.; Zhu, J.; Li, W.; et al. Metatranscriptomic Sequencing Reveals Host Species as an Important Factor Shaping the Mosquito Virome. Microbiol Spectr 2023, 11. [Google Scholar] [CrossRef]
- Kubacki, J.; Flacio, E.; Qi, W.; Guidi, V.; Tonolla, M.; Fraefel, C. Viral Metagenomic Analysis of Aedes Albopictus Mosquitos from Southern Switzerland. Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Gómez, M.; Martinez, D.; Muñoz, M.; Ramírez, J.D. Aedes Aegypti and Ae. Albopictus Microbiome/Virome: New Strategies for Controlling Arboviral Transmission? Parasit Vectors 2022, 15. [Google Scholar] [CrossRef]
- Gangopadhayya, A.; Lole, K.; Ghuge, O.; Ramdasi, A.; Kamble, A.; Roy, D.; Thakar, S.; Nath, A.; Sudeep, A.B.; Cherian, S. Metagenomic Analysis of Viromes of Aedes Mosquitoes across India. Viruses 2024, 16. [Google Scholar] [CrossRef]
- Konstantinidis, K.; Dovrolis, N.; Kouvela, A.; Kassela, K.; Rosa Freitas, M.G.; Nearchou, A.; De Courcy Williams, M.; Veletza, S.; Karakasiliotis, I. Defining Virus-Carrier Networks That Shape the Composition of the Mosquito Core Virome of a Local Ecosystem. Virus Evol 2022, 8. [Google Scholar] [CrossRef]
- Shi, C.; Zhao, L.; Atoni, E.; Zeng, W.; Hu, X.; Matthijnssens, J.; Yuan, Z.; Xia, H. Stability of the Virome in Lab- and Field-Collected Aedes Albopictus Mosquitoes across Different Developmental Stages and Possible Core Viruses in the Publicly Available Virome Data of Aedes Mosquitoes. mSystems 2020, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Shahhosseini, N.; Lühken, R.; Jöst, H.; Jansen, S.; Börstler, J.; Rieger, T.; Krüger, A.; Yadouleton, A.; de Mendonça Campos, R.; Cirne-Santos, C.C.; et al. Detection and Characterization of a Novel Rhabdovirus in Aedes Cantans Mosquitoes and Evidence for a Mosquito-Associated New Genus in the Family Rhabdoviridae. Infection, Genetics and Evolution 2017, 55, 260–268. [Google Scholar] [CrossRef] [PubMed]
- De Coninck, L.; Soto, A.; Wang, L.; De Wolf, K.; Smitz, N.; Deblauwe, I.; Mbigha Donfack, K.C.; Müller, R.; Delang, L.; Matthijnssens, J. Lack of Abundant Core Virome in Culex Mosquitoes from a Temperate Climate Region despite a Mosquito Species-Specific Virome. mSystems 2024. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Map showing the climatic zones in Cameroon and the collection sites of Aedes mosquitoes in the Southwestern part of Cameroon. The pie charts show the proportion of the different Aedes mosquito species found in each region.
Figure 1.
Map showing the climatic zones in Cameroon and the collection sites of Aedes mosquitoes in the Southwestern part of Cameroon. The pie charts show the proportion of the different Aedes mosquito species found in each region.
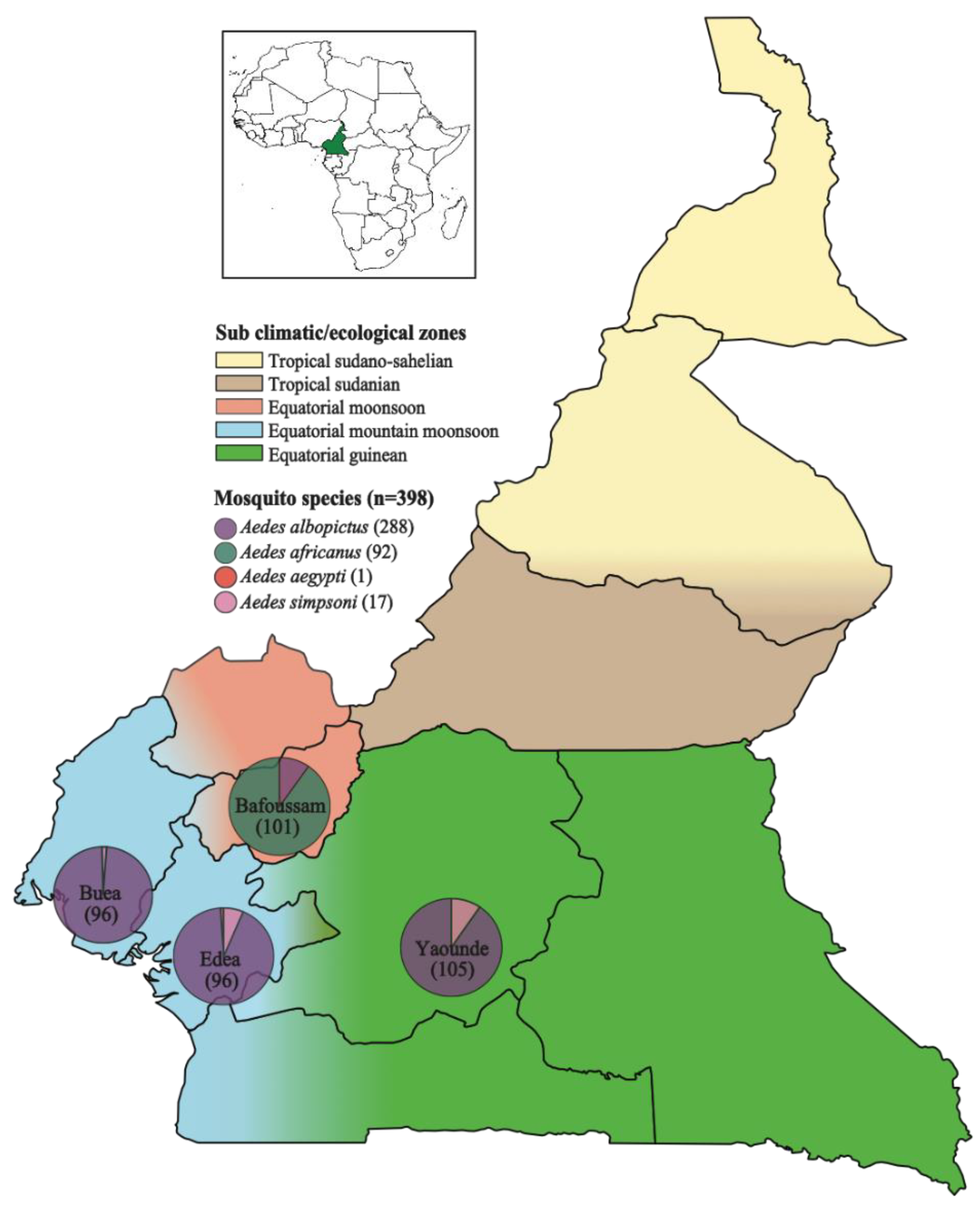
Figure 2.
Proportion of eukaryotic virus reads assembled from 54 mosquito pools for each virus family. a. Proportion of eukaryotic virus reads for each viral family. b. Proportion of eukaryotic virus reads for viruses unclassified at family level.
Figure 2.
Proportion of eukaryotic virus reads assembled from 54 mosquito pools for each virus family. a. Proportion of eukaryotic virus reads for each viral family. b. Proportion of eukaryotic virus reads for viruses unclassified at family level.
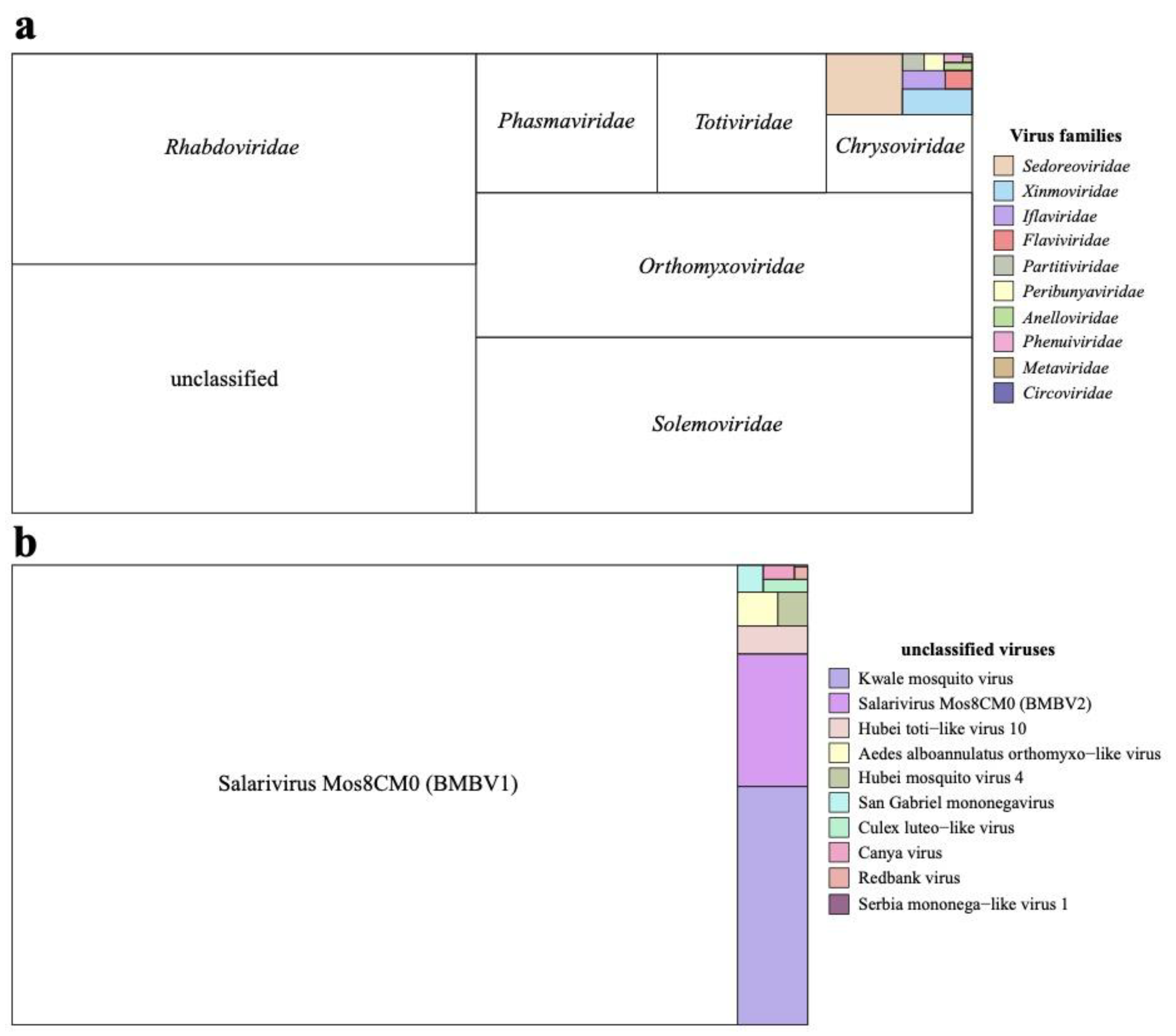
Figure 3.
Proportion eukaryotic virome reads of Aedes mosquito species from the southwestern part of Cameroon. a. Eukaryotic virome reads of Ae. africanus and Ae. albopictus b. Eukaryotic virome reads of Aedes mosquitoes from Yaoundé, Buea and Edea.
Figure 3.
Proportion eukaryotic virome reads of Aedes mosquito species from the southwestern part of Cameroon. a. Eukaryotic virome reads of Ae. africanus and Ae. albopictus b. Eukaryotic virome reads of Aedes mosquitoes from Yaoundé, Buea and Edea.
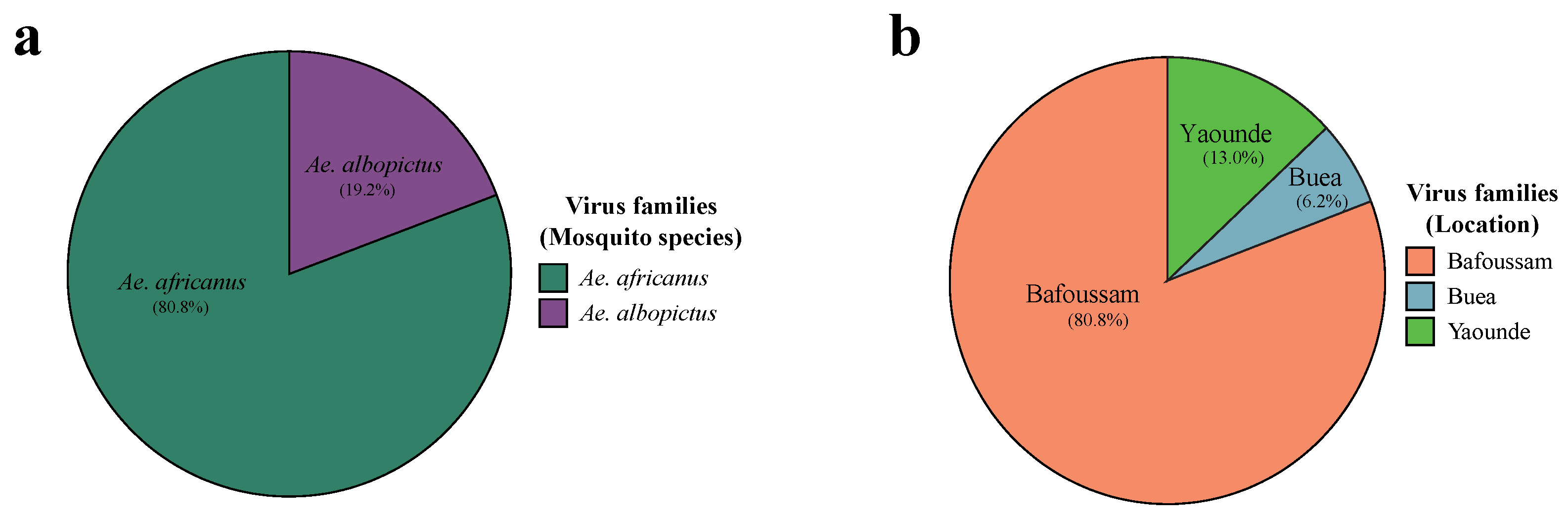
Figure 4.
Alpha diversity of eukaryotic viruses from pools of Aedes mosquito. a. Alpha diversity comparison between Ae. africanus and Ae. albopictus. b. Alpha diversity comparison between Bafoussam, Buea and Yaoundé. Wilcoxon test: p < 0.0001 (****), p < 0.001 (***).
Figure 4.
Alpha diversity of eukaryotic viruses from pools of Aedes mosquito. a. Alpha diversity comparison between Ae. africanus and Ae. albopictus. b. Alpha diversity comparison between Bafoussam, Buea and Yaoundé. Wilcoxon test: p < 0.0001 (****), p < 0.001 (***).
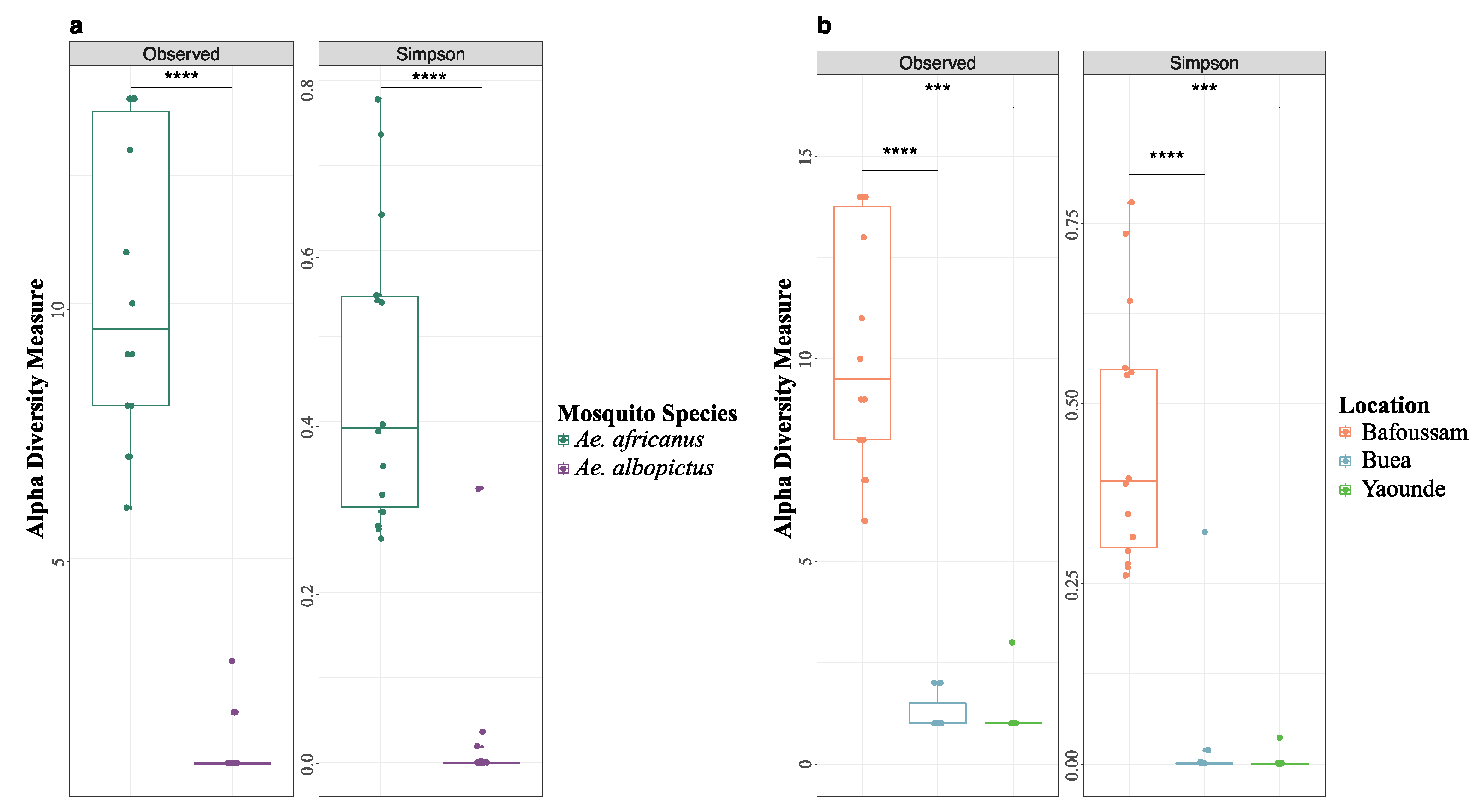
Figure 5.
Principal Coordinates Analysis of eukaryotic viruses in Aedes mosquito species across locations (Bafoussam, Buea, Yaoundé). Adonis test: R2 (Mosquito species) = 0.294, R2 (Locations) = 0.006, p = 0.001.
Figure 5.
Principal Coordinates Analysis of eukaryotic viruses in Aedes mosquito species across locations (Bafoussam, Buea, Yaoundé). Adonis test: R2 (Mosquito species) = 0.294, R2 (Locations) = 0.006, p = 0.001.
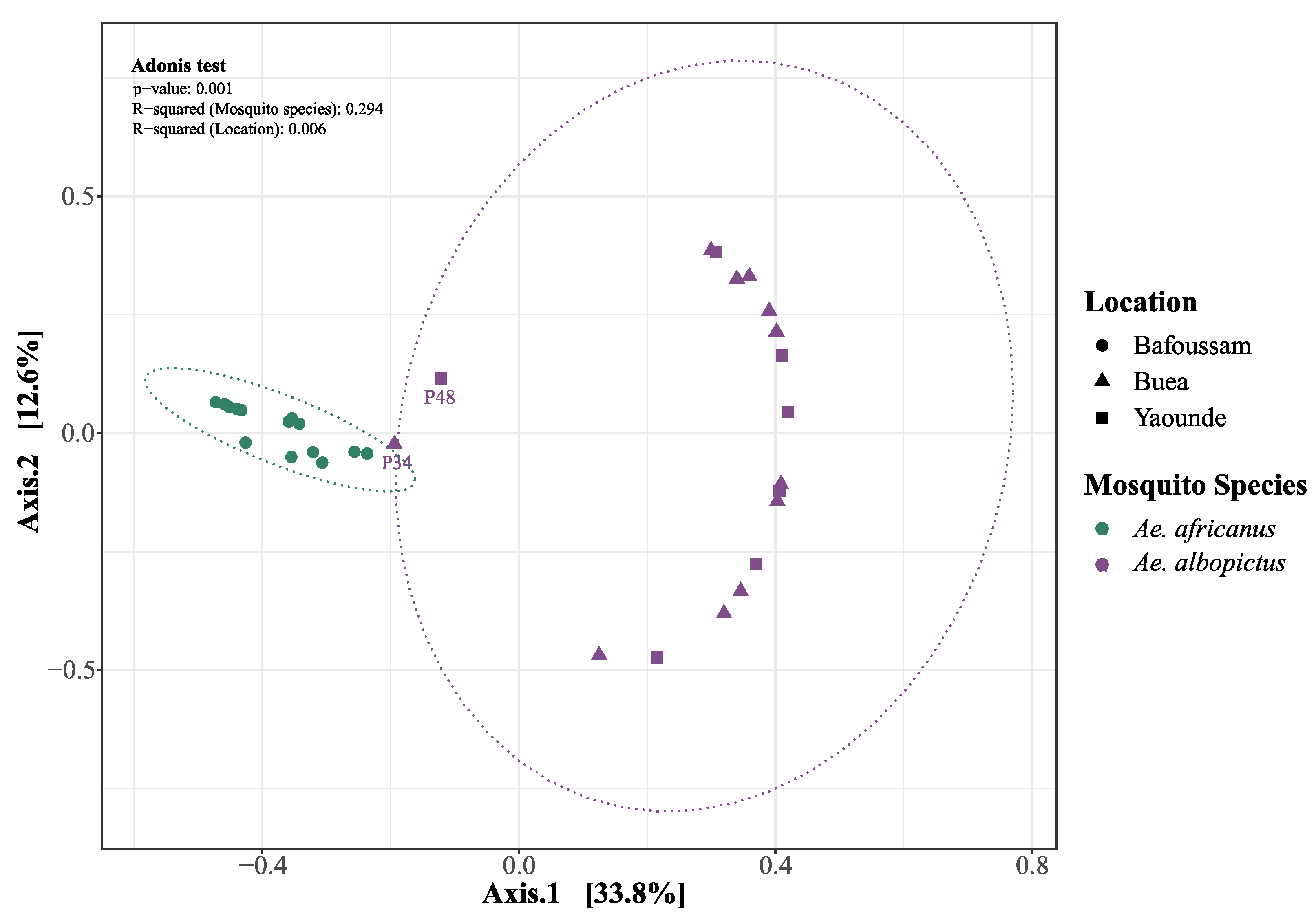
Figure 6.
Read count of eukaryotic viral species on log2 scale. BLASTx percent identity to the most closely related reference sequence is shown in the shaded blue boxes. The virus name in green is the only virus species found in both Aedes species. Viruses in red were selected for qRT-PCR analysis and the abbreviations of novel viruses with near complete genomes (BLASTx < 90%) are shown between brackets.
Figure 6.
Read count of eukaryotic viral species on log2 scale. BLASTx percent identity to the most closely related reference sequence is shown in the shaded blue boxes. The virus name in green is the only virus species found in both Aedes species. Viruses in red were selected for qRT-PCR analysis and the abbreviations of novel viruses with near complete genomes (BLASTx < 90%) are shown between brackets.
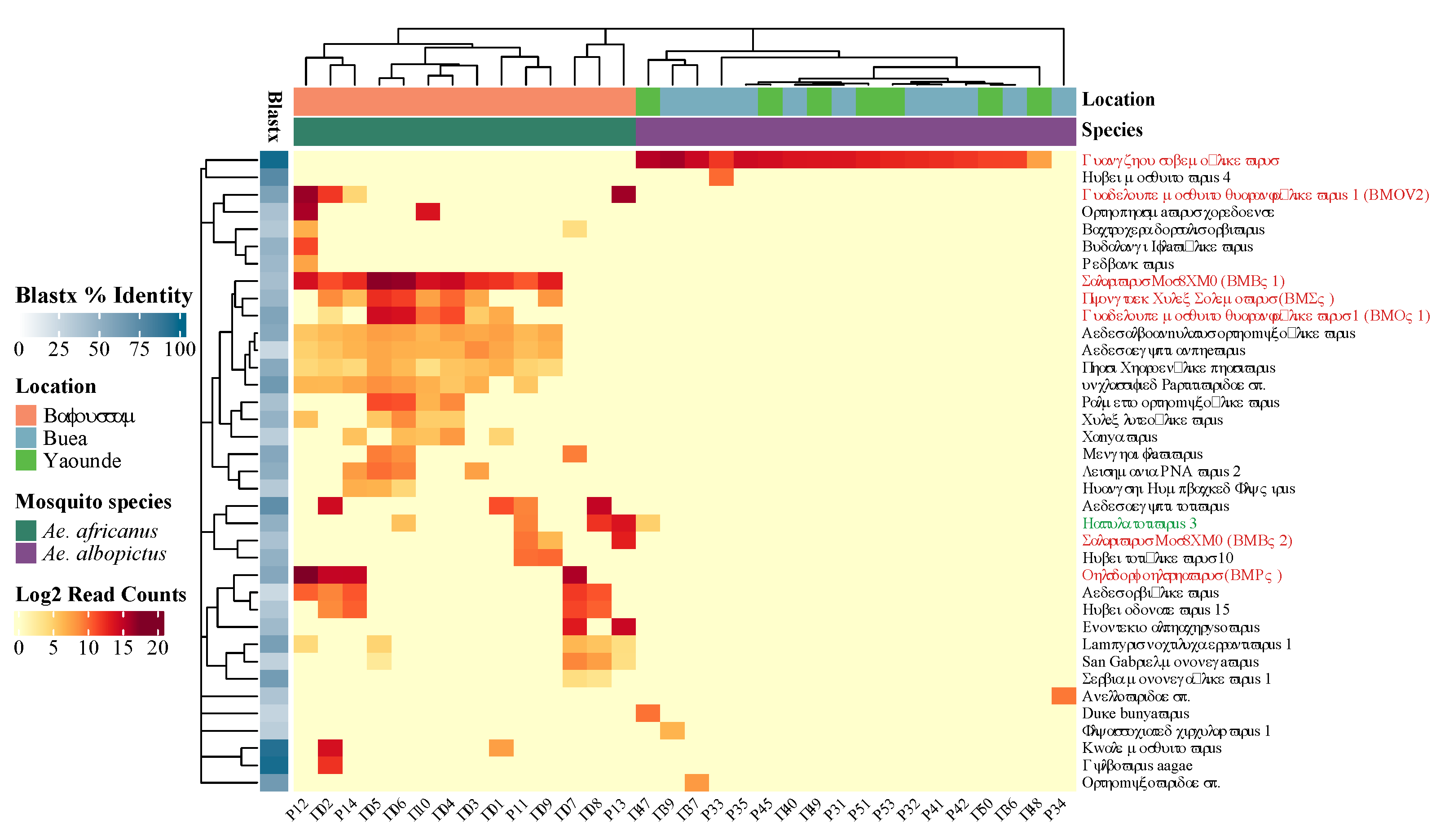
Figure 7.
Genome organization, amino acid sequence similarity between novel virus and their closest relatives (novel virus in red and reference in blue), and maximum likelihood phylogeny based on amino acid sequence of PB2 or RdRp/L protein. a. Bafoussam mosquito bunyavirus 1 (BMBV1) and Bafoussam mosquito bunyavirus 2 (BMBV2) b. Bafoussam mosquito orthomyxovirus 1 (BMOV1) and Bafoussam mosquito orthomyxovirus 2 (BMOV2) c. Bafoussam mosquito Rhabdovirus (BMRV) d. Bafoussam mosquito solemovirus (BMSV).
Figure 7.
Genome organization, amino acid sequence similarity between novel virus and their closest relatives (novel virus in red and reference in blue), and maximum likelihood phylogeny based on amino acid sequence of PB2 or RdRp/L protein. a. Bafoussam mosquito bunyavirus 1 (BMBV1) and Bafoussam mosquito bunyavirus 2 (BMBV2) b. Bafoussam mosquito orthomyxovirus 1 (BMOV1) and Bafoussam mosquito orthomyxovirus 2 (BMOV2) c. Bafoussam mosquito Rhabdovirus (BMRV) d. Bafoussam mosquito solemovirus (BMSV).
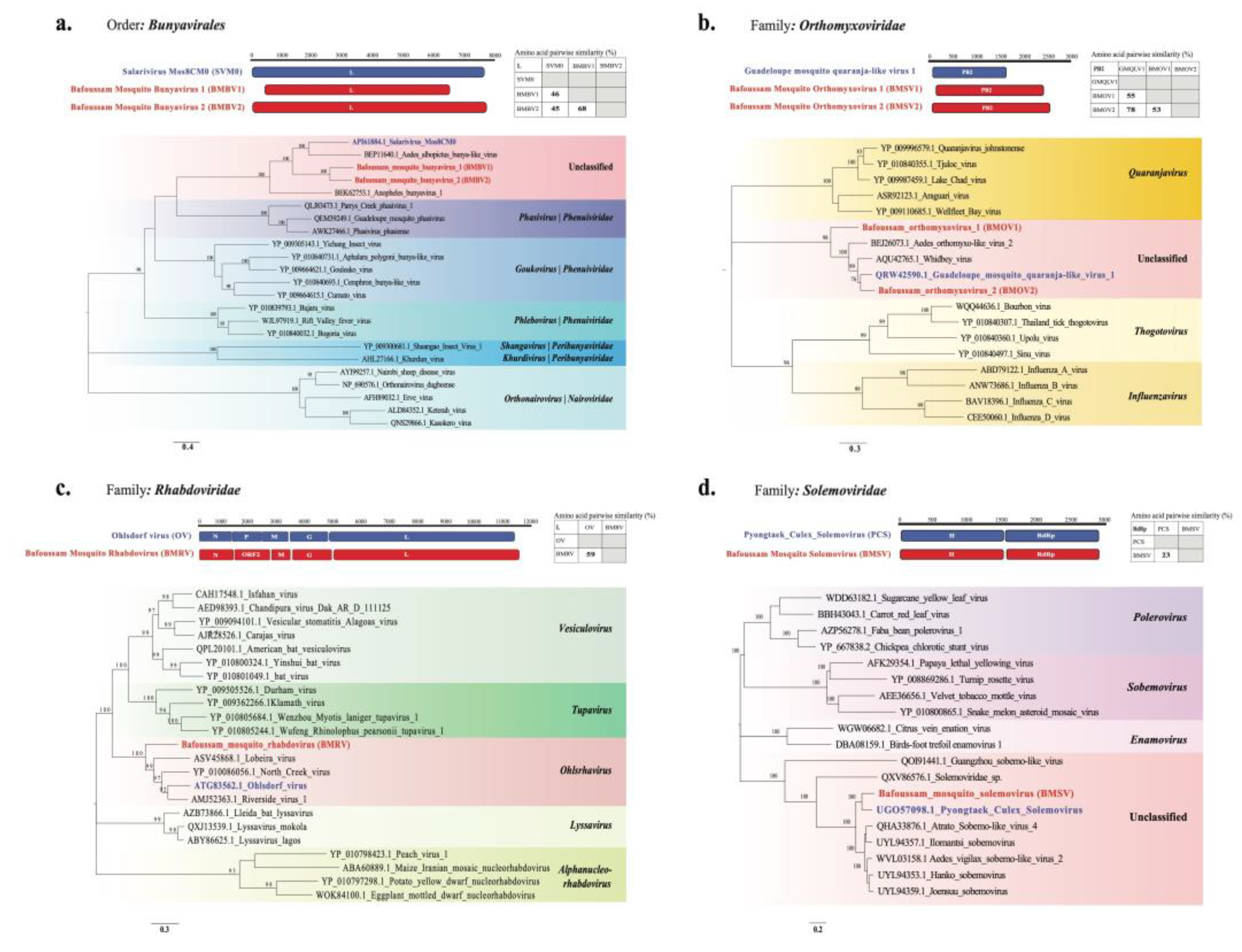
Figure 8.
Quantification of seven abundant viruses. a. Genome copy numbers of viruses in 182 individual mosquito samples. The red dot in the violin plot represents the median. The samples above the dotted line had Ct ≤ 35 and are thus considered positive. b. Prevalence (%) of selected viruses across individual Aedes species is shown by intensity of red color. The values in the circles represent the number of mosquito samples that tested positive for the virus.
Figure 8.
Quantification of seven abundant viruses. a. Genome copy numbers of viruses in 182 individual mosquito samples. The red dot in the violin plot represents the median. The samples above the dotted line had Ct ≤ 35 and are thus considered positive. b. Prevalence (%) of selected viruses across individual Aedes species is shown by intensity of red color. The values in the circles represent the number of mosquito samples that tested positive for the virus.
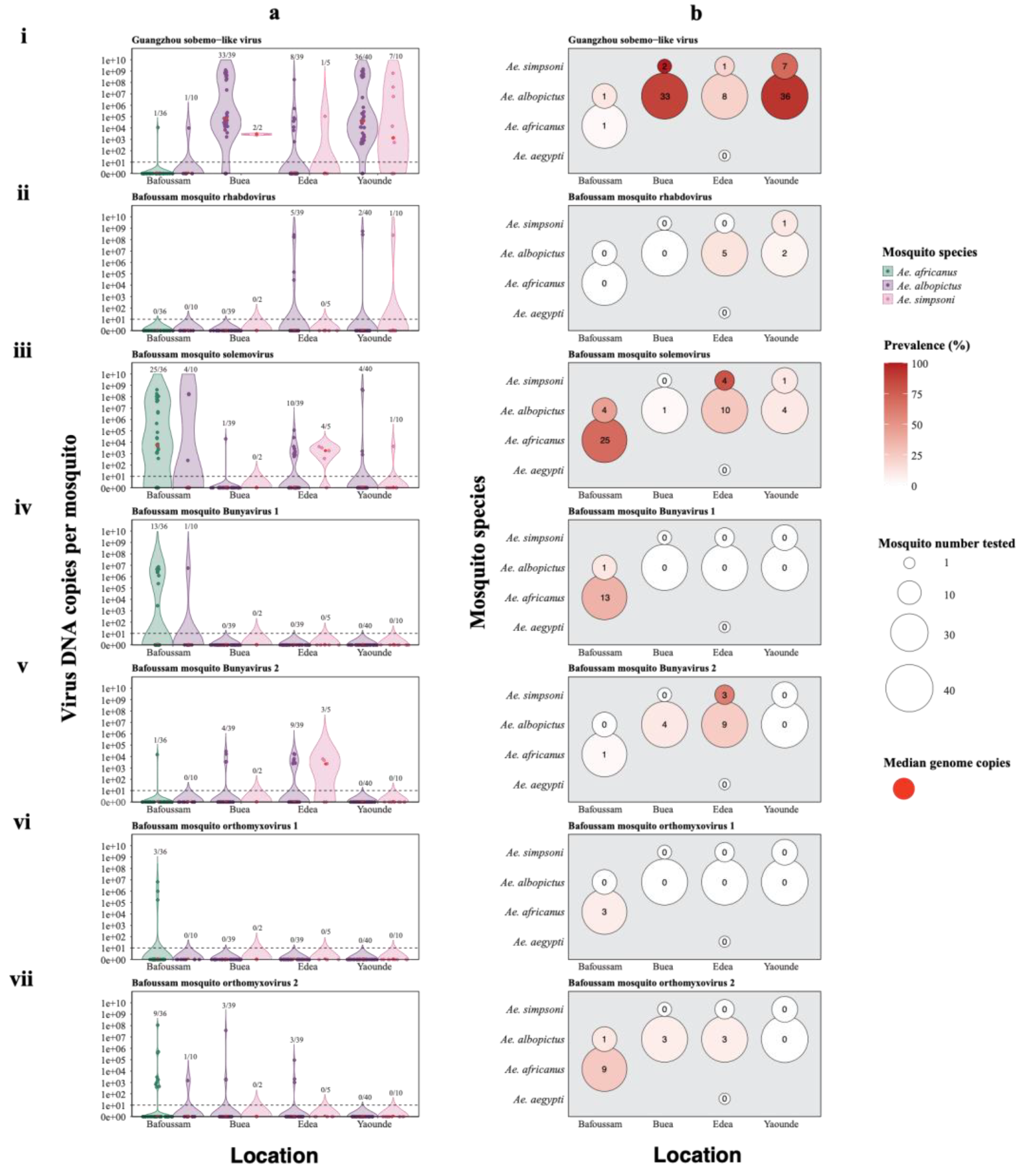

Table 1.
Data on mosquito pools before Illumina sequencing.
| Location | Mosquito species | Number of Pools of 4 mosquitoes |
|---|---|---|
| Bafoussam | Aedes africanus | 14 |
| Buea | Aedes albopictus | 14 |
| Edea | Aedes albopictus | 12 |
| Yaoundé | Aedes albopictus | 14 |
Table 2.
Data on individual mosquitoes for quantification of selected viruses.
| Location | Mosquito species | Individual mosquitoes tested |
|---|---|---|
| Bafoussam | Ae. africanus | 36 |
| Ae. albopictus | 10 | |
| Buea | Ae. albopictus | 39 |
| Ae. simpsoni | 2 | |
| Edea | Ae. albopictus | 39 |
| Ae. simpsoni | 5 | |
| Ae. aegypti | 1 | |
| Yaoundé | Ae. albopictus | 40 |
| Ae. simpsoni | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



