
Submitted:
13 June 2024
Posted:
14 June 2024
You are already at the latest version
Abstract
The field of epigenetics, which explores how heritable traits or stable alterations of cell functions occur without changes to the DNA sequence and how environmental factors affect gene expression, is rapidly advancing. Epigenetics plays a critical role in memory formation, learning, and cognitive functioning, effectively bridging genetics with environmental influence. Maternal behavior in rats, for instance, can modify the stress response of their offspring through epigenetic mechanisms such as histone acetylation and DNA methylation. Understanding neurodegenerative diseases like Parkinson's disease (PD) depends on comprehending the mechanisms of epigenetic regulation. Epigenetic modifications, which alter proteins and DNA structure without changing the DNA sequence, can activate or suppress gene expression, assisting organisms in adapting to environmental changes. These modifications may influence genes involved in inflammation, synaptic plasticity, and neuroprotection, potentially contributing to cognitive deficits in PD patients. MicroRNAs (miRNAs), histone modifications, and DNA methylation are the epigenetic mechanisms that significantly influence cell differentiation and maintain normal cellular activity. Understanding the intricate molecular processes of epigenetics could lead to more targeted and effective therapies. Despite numerous findings on how epigenetics affects cognitive alterations in PD patients, only some studies coordinate and integrate these results. Here, we summarize recent findings on how epigenetic changes impact gene expression linked to cognitive impairments in PD.
Keywords:
alpha-synuclein
; cognition
; epigenetics
; Parkinson’s disease
; Tau
1. Introduction
Epigenetics, the study of how environmental factors influence gene expression and organismal responses, is rapidly expanding. These intricate mechanisms are pivotal in the development and maintenance of homeostasis. Epigenetic modifications enable organisms to adapt to environmental changes [1]. They entail alterations in DNA structure and proteins without changing the DNA sequence [2], and they are capable of activating or suppressing gene expression through feedback mechanisms [3].
Parkinson’s disease (PD) is a prevalent neurodegenerative disorder, second only to Alzheimer’s disease (AD). It manifests with distinctive symptoms, including bradykinesia, muscular rigidity, unstable posture and gait, hypokinetic movement disorder, and resting tremor [4,5,6]. Lewy bodies (LB) containing α-Synuclein (α-Syn) and other factors may lead to progressive dopaminergic neuron loss in the substantia nigra pars compacta (SNpc) in both sporadic and familial PD [7]. Understanding α-Syn’s pathogenic role is challenging due to its complex interactions with genetic traits, molecular components, and environmental factors contributing to its accumulation [8].
Exploring epigenetics and its intricate molecular mechanisms may elucidate the pathogenesis of neurodegenerative disorders such as PD [9], potentially leading to more effective, targeted treatments. Despite numerous investigations into the impact of epigenetics on cognitive changes in PD patients, there is a need for more studies linking these findings. Here, we present our vision of how epigenetic modifications affect gene expression related to neuroprotection, inflammation, and synaptic plasticity, potentially contributing to PD’s cognitive deficits.
2. Synuclein and Tau: ‘Seeding and Spreading’
Alpha-synuclein (α-Syn), encoded by the SNCA gene, is a 14.5-kilodalton (kD) protein comprising 140 amino acids [10,11]. It constitutes the major component of Lewy bodies (LBs) and Lewy neurites (LNs), the pathological hallmarks of PD [12]. Initially identified as a genetic susceptibility factor for familial autosomal dominant PD, the SNCA gene is associated with a higher PD risk due to pathogenic alterations, including mutations, single nucleotide polymorphisms (SNPs), and multiplications [13,14,15].
SNPs such as rs11931074, rs356165, rs356219, and rs7684318 have been linked to increased PD risk among Caucasians and Asians [16,17,18]. The variants of SNPs in SNCA vary across populations, with different risk variants observed in East Asians compared to European populations [19,20,21,22]. Genome-wide association studies (GWAS) have identified additional SNCA polymorphisms, such as rs8180209, rs3756063, rs356165, and rs7681154, associated with PD age onset and progression [23,24,25,26]. A meta-analysis revealed eight SNPs in SNCA with significant differences among ethnicities affecting PD risk [27]. Previous studies also highlighted the relevance of SNPs such as rs356186, rs356219, rs894278, rs2583988, and rs2619363 in increasing PD risk [28]. These variants with different odds ratios and allele frequencies may guide genetic screening strategies and predict the age of PD onset. Polymorphisms in the SNCA promoter region, including REP1, rs2736990, rs894278, rs356219, and rs356165, are promising predictors of early PD onset [29,30,31,32].
The epigenetic regulation of SNCA among PD patients has been extensively studied [2,32,33,34]. SNPs within the promoter/regulatory regions may influence epigenetic effects and transcription factor bindings [33]. The SNCA gene contains a large CpG island extending from the 5′ end to intron 1 [33]. In PD patients, hypomethylation of the intron1 region of the SNCA CpG islands is more pronounced in various brain regions than in controls [34,35]. Ten-eleven translocation methylcytosine dioxygenase 1 (TET1) acts as a repressor of SNCA, targeting the CpG island binding protein [36]. Postmortem studies on PD tissue samples revealed lower levels of TET1 compared to controls, suggesting a potential therapeutic target [37]. Additionally, two significant linkage disequilibrium (LD) blocks located in intron 4 to downstream of the 3′-untranslated region (3′-UTR) area and the promoter region at the 5′ side of the gene harbor SNPs that play crucial roles in gene processing [38].
Tau, a microtubule-associated protein (MAP) family member, is implicated in various neurodegenerative diseases, including AD and Tauopathies [39,40]. Prion diseases, fatal neurodegenerative disorders found in humans and animals, involve the self-propagation of prions via templated conversion of endogenous prion molecules, leading to infectivity [41,42]. These aggregates can transfer between cells, continuing propagation as new conversion units, thus spreading infection [41,42,43]. α-Syn and Tau proteins exhibit prion-like behavior with a stable spreading pattern, characteristic of prion diseases [44,45]. Table 1 summarizes the results of experiments on α-Syn and Tau seeding and spreading conducted over the past decade.
3. Importance of Epigenetics
Epigenetic mechanisms are particularly significant in cell differentiation, where cells of the same species develop distinct traits and functions despite sharing identical DNA [46]. Epigenetics encompasses three primary categories: 1) DNA methylation, 2) Histone modifications, and 3) microRNAs (miRNAs). Precisely regulating these processes is crucial for maintaining normal cellular function. Conversely, dysregulation or disruption of these mechanisms can contribute to the development of various diseases [47].
Epigenetics in Cognition
Epigenetics plays a pivotal role in memory formation, learning, and cognitive functions by bridging genetics with environmental factors. Weaver et al. found that maternal behavior in rats alters the HPA axis and hippocampal glucocorticoid receptor expression in offspring. This impact on stress response persists into adulthood through epigenetic mechanisms such as histone acetylation and DNA methylation [46].
Cognitive behavioral therapy for anxiety disorders relies on epigenetic mechanisms like histone 4 acetylation to regulate gene expression during conditioned fear extinction. These mechanisms enhance BDNF expression and the elimination of conditioned fear by promoting synaptic plasticity and long-term memory formation [47].
Miller and colleagues’ study demonstrated how epigenetic processes are implicated in memory formation in rats. Their findings revealed that exposure to fear-inducing stimuli increased DNMT expression, resulting in elevated methylation levels that suppressed the memory suppressor gene PP1. Additionally, the same stimuli triggered demethylation and enhanced reelin gene expression, which plays a vital role in facilitating synaptic plasticity [48].
In addition to normal cellular functioning and well-being, epigenetic mechanisms have been linked to cognitive disorders. Rett syndrome, AD, drug addiction, autism spectrum disorders, and schizophrenia are examples [49].
4. Epigenetics in PD
In 2019, global estimates indicated a prevalence of over 8.5 million individuals affected by PD [50]. According to a meta-analysis, there is an age-related increase in the incidence of PD in both genders. However, the rise in incidence is more pronounced in males aged 60-69 and 70-79 [51]. The typical clinical manifestations encompass a movement disorder characterized by bradykinesia, resting tremor, and rigidity, while postural instability tends to manifest at a later phase [52].
Various risk factors, such as smoking, caffeine consumption, and exposure to pesticides, hypothetically influence the development of PD, but their precise impact remains uncertain [52].
PD is primarily an idiopathic disorder, but approximately 10-15% of cases have a family history, and 5% have Mendelian inheritance patterns [116]. There are 23 PARK genes connected to PD, some demonstrating autosomal dominant and others autosomal recessive inheritance [117]. The most significant genetic risk factors for PD are mutations in the GBA1 gene, which codes for β-glucocerebrosidase and is associated with Gaucher disease [118]. Additional genetic risk factors for PD comprise the major histocompatibility complex, class II (HLA-DQB1) [119], and the MAPT gene, which encodes the protein Tau, among various others [120]. Numerous studies have been dedicated to exploring the involvement of epigenetics in PD.
Numerous investigations have explored the involvement of epigenetics in PD. Song et al. found that chromatin remodeling, RNA-based processes, DNA methylation, and post-translational histone modifications contribute to PD [121]. In another study, Goers et al. found that dopaminergic neurons from PD patients had higher H2A, H3, and H4 histone acetylation than controls. These histone modifications suggest chromatin remodeling is essential in PD development [122].
MiRNAs also play a significant role in epigenetic regulation in PD pathogenesis [123]. Examples include miR-7 and miR-153 downregulating the SNCA gene. Deregulation of the α-Syn gene expression may be linked to α-Syn accumulation and neuronal cell death in PD patients. Reduced levels of these miRNAs may be diagnostic biomarkers for PD, especially in brain regions associated with disease neuropathology, such as the SNpc [124,125].
PD patients’ peripheral blood leukocytes had lower methylation levels in specific CpG-2 sites of the SNCA gene promoter than in controls, suggesting they could be used as biomarkers. In addition, methylation-mediated inhibition of a CpG island in intron 1 of the SNCA gene upregulated SNCA expression in the putamen and cortex of PD patients, indicating hypo-methylation in these regions. These findings suggest that DNA methylation is crucial to PD pathogenesis and may serve as a biomarker in PD [34,126,127].
The epigenetic aspects implicated in PD pathogenesis may also contribute to cognitive impairment. This rapidly evolving field of epigenetics is important in cognitive and memory impairments in PD and offers many unexplored therapeutic avenues.
4.1. DNA Methylation in PD Cognitive Dysfunction
DNA methyltransferases (DNMTs) add methyl groups to cytosine to make 5-methylcytosine (5-mC) on CpG islands [121]. Feng et al. investigated the association between DNMTs and cognition. They examined how DNMT1 and DNMT3a enzymes affect hippocampal neuroplasticity. These authors showed that genetic mutations in these enzymes exclusively in forebrain excitatory neurons reduced plasticity. Consequently, this impairment in plasticity resulted in deficits in learning and memory [128].
While hypermethylation of some genes leads to gene suppression, DNA hypomethylation can increase gene expression. Masliah et al. found methylation patterns in genes previously linked to PD in brain and blood samples from PD patients, supporting the important role of epigenetic alterations as a significant molecular mechanism in PD pathogenesis [129]. Chuang et al. conducted an epigenome-wide association study (EWAS) on PD patients to analyze methylation markers. These researchers found that 7 CpGs were correlated with cognitive deterioration by losing ≥ 4 MMSE points located in KCNB1, DLEU2, and SATB1 genes [129]. Ohlei et al. found that impairments of DNA methylation may cause AD and PD; they identified disease loci potentially acting via SNP-CpG associations and differential DNA methylation. Thus, this study revealed a likely causal role of differential DNA methylation in AD and PD development in several genomic regions associated with disease risk by GWAS [70].
Our literature review suggests three hypotheses that, despite limited data, may link DNA methylation to cognitive impairment in PD patients.
4.1.1. Brain-Derived Neurotrophic Factor (BDNF)
Our first hypothesis is based on the aberrant expression of the brain-derived neurotrophic factor (BDNF). BDNF is recognized for having a significant role in the neuron’s growth, survival, maintenance, and synaptic plasticity. It also plays a crucial role in activity-dependent neuroplasticity, forming the basis of learning and memory in the hippocampus [130]. Depletion of BDNF levels is reported in various neurodegenerative diseases, including AD, Huntington’s disease, and PD [131]. Expression of BDNF is regulated by epigenetic modifications, including DNA methylation [132]. In a study by Howells et al., in situ hybridization revealed a robust expression of BDNF mRNA in SNpc, which is reduced by 70% in clinically and neuropathologically typical PD. This decline is regarded partially due to the depletion of dopaminergic neurons.
Nevertheless, there was a 20% decrease in BDNF mRNA expression in the remaining dopaminergic PD SNpc compared with their normal counterparts [133]. In 2010, Muñoz et al. demonstrated a link between performance in the novel object recognition task and elevated BDNF expression, along with changes in methylation state at the BDNF promoter I site within the hippocampus of mice [134]. Another study has demonstrated a link between the strengthening of fear-based learning and an upregulation in the activity of BDNF exon IV at the genetic level. In contrast, exposure to the context alone resulted in higher quantities of BDNF exons I and VI. These findings propose varying utilization of genetic segments in the hippocampus as a response to different cognitive activities, like acquiring knowledge about a new environment versus associating an emotion with that same environment. Interestingly, the elevated levels of transcripts containing BDNF exon IV were found to align with a decrease in DNA methylation at the corresponding initiator of gene expression. This reduction was directly associated with an overall rise in total BDNF mRNA (specifically, exon IX) in the hippocampus while consolidating fear-based memories [135].
4.1.2. Peroxisome Proliferator-Activated Receptor Gamma Coactivator−1 α (PGC−1α)
Our other hypothesis involves Peroxisome proliferator-activated receptor gamma coactivator−1 α (PGC−1α). There has been a correlation between mitochondrial dysfunction and PD for a long time. This gene Functions as a transcriptional coactivator for nuclear receptors, playing a crucial role in the transcriptional regulation of cellular energy metabolism, the plasticity of microglial cells, and the functioning of mitochondria [136].
Xiaomin et al. investigated methylation changes in the PGC-1α gene in SNpc of PD patients and found hypermethylation and suppression of PGC-1α [137].
On the other hand, several studies demonstrated the potential role of PGC-1α and cognitive impairment [138,139,140]. For instance, in 2020, Han et al. demonstrated the amelioration of cognitive deficits through PGC-1α overexpression, which reduced the production of reactive oxygen species (ROS) [140]. Although limited data support the relationship between DNA methylation of the PGC-1α gene and cognition, the role of PGC-1α overexpression and its effect on cognitive impairment is worth further studies.
4.1.3. Peroxisome Proliferator-Activated Receptor Gamma Coactivator−1 α (PGC−1α)
Our last hypothesis concerns the microtubule-associated protein Tau (MAPT) gene. Numerous neurodegenerative conditions, including PD, are characterized by the abnormal aggregation and formation of inclusions by the MAPT. This pathological correlation has led to the common categorization of these disorders as tauopathies [140]. In a study conducted by Coupland et al., they found that hypermethylation of MAPT in different parts of the brain could be a neuroprotective compensatory mechanism during the initiation of progression of the disease [141].
Furthermore, many studies demonstrated the robust influences of MAPT on cognition and risk of developing dementia [142,143] as Tunold et al. discovered significant associations between the onset of dementia and the presence of APOE ε4 and MAPT H1-haplotype among PD patients [144]. Also, in a study conducted by Jiskoot et al. during the 4-year follow-up of presymptomatic MAPT and GRN mutation carriers and a healthy control group, notable declines in cognitive aspects, including language skills, attention, executive functioning, and social cognition, were observed in MAPT converters [145]. In a study by Winder-Rhodes et al. MAPT homozygosity was linked to impaired picture recognition memory in PD patients [143].
However, we need new studies targeting the relationship between DNA methylation of the MAPT gene and cognition.
4.2. Histone Modification in PD Cognitive Dysfunction
Histones, essential proteins closely associated with DNA, play a vital role in the formation of nucleosomes, the basic units of chromatin [146,147]. The N-terminal ends of histones undergo methylation, acetylation, phosphorylation, ubiquitylation, and SUMOylation. These modifications affect chromatin structure and nucleus genomic information accessibility, which has epigenetic implications [147,148]. Figure 2 shows the organization of nucleosomes and potential epigenetic pathways implicated in cognition processes in PD.
Histone acetylation has been extensively studied in animal models regarding its involvement in learning, memory, and synaptic plasticity. Lubin et al. found increased H3 acetylation at the BDNF promoter region in adult mice undergoing contextual fear conditioning, which impairs learning and memory. This study emphasizes histone acetylation’s role in cognition and memory formation [135]. Histone acetylation also increases in NF-κB-targeted gene regions linked to memory consolidation [149,150].
Dopaminergic neurons in the SNpc are lost in PD. The control of the SNCA gene has been specifically linked to histone modification, including methylation of histone H3 at lysine 4 (H3K4me3) and lysine 27 (H3K27me3). Studies have demonstrated that postmortem brain tissues from PD patients had higher levels of H3K4me3 at the SNCA promoter. However, PD cell culture models treated with a neurotoxic agent have lower levels of H3K4me3 and H3K27me3. These findings suggest that abnormal histone modification in PD neurons may impact gene expression, including α-Syn. Further research is needed to understand how histone methylation controls gene expression and PD pathogenesis [151,152].
Below are some of the most significant histone modifications and their target genes and pathways for treating neurodegenerative diseases, especially PD and cognition impairment.
4.2.1. H3K9me2
H3K9me2 suppresses genes associated with memory processes. Consequently, any disturbance in this epigenetic mechanism can impair memory function [153]. Gupta-Agarwal et al.’s study reveals that boosting H3K9me2 levels in the lateral amygdala enhances memory formation [154]. In another study by Gupta-Agarwal, higher H3K9me2 levels were noted in the CA1 of the hippocampus and entorhinal cortex (EC) in rats during memory consolidation [155].
In the context of PD, the effects of α-Syn on chromatin and histone modifications were investigated by Riddle et al. [156]. Overexpression of α-Syn in transgenic Drosophila and inducible SH-SY5Y neuroblastoma cells led to increased histone H3K9 methylations, specifically mono- (H3K9me1) and di- (H3K9me2) methylation. This increase in H3K9me2 was associated with changes in the expression of genes encoding the L1CAM cell adhesion molecule [156]. Histone H3K9me2, known for its involvement in repressed gene transcription, is implicated in memory consolidation and neuronal plasticity [157]. Furthermore, the study revealed the role of euchromatic histone-lysine N-methyltransferase 2 (EHMT2) in catalyzing the formation of H3K9me2, emphasizing its potential significance in PD-associated mechanisms. These findings point to the impact of H3K9 dimethylation in the epigenetic regulation underlying PD pathogenesis.
On the other hand, some studies have shown the possible role of H3K9me2 in cognition. The Gupta et al. study [158], discovered that contextual fear conditioning leads to both H3K4 trimethylation and H3K9 dimethylation in the hippocampus, indicating active gene expression and repression during memory formation. Surprisingly, enhancing histone acetylation through histone deacetylases (HDACs) inhibition with NaB reduces H3K9 dimethylation, suggesting a potential mechanism for memory enhancement. Furthermore, Gupta et al. showed that H3K9 dimethylation increases in the CA1 area of the hippocampus following both fear conditioning and exposure to a novel context, highlighting its role in associative and novel context learning-triggered memory processes [158].
4.2.2. H3K4me3 and H3K27me3
Methylation of histones can alter gene expression, with trimethylation of H3K4 causing transcriptional activation and H3K27 causing transcriptional inhibition. H3K4me3 has a potential role in iron metabolism regulation. Epigenetic modifications, such as 6-OHDA, have been suggested to play a role in PD pathogenesis. Södersten et al. study found that 6-OHDA caused a significant decrease in H3K4me3 and H3K27me3 levels in the striatum. Local changes in H3K27me3 and H3K4me3 have not been established, possibly due to different expression patterns between brain regions. These findings suggest that histone methylation may play a role in the pathogenesis of PD [159,160].
GSK-J4, a histone demethylase inhibitor with cross-blood-brain barrier ability, has been identified as a potent iron suppressor for PD. This compound selectively reduced intracellular labile iron in dopaminergic neurons and suppressed H2O2 and 6-OHDA-induced cell death in vitro. The iron-suppressive effect was mediated by increasing the expression of ferroportin-1, and it rescued dopaminergic neuron loss and motor defects in 6-OHDA-induced PD rats. GSK-J4 also rescued abnormal changes of histone methylation, H3K4me3, and H3K27me3 during 6-OHDA treatment, with the iron-suppressive and neuroprotective effects sensitive to H3K4me3 inhibition only. This compound has a more substantial PD therapeutic potential than conventional iron chelators [161].
4.2.3. K4me3
Histone’s post-translational modifications in the regulatory region of the SNCA gene were studied by Guhathakurta et al. [37]. Notably, the SNCA promoter exhibited elevated histone H3 lysine 4 trimethylation (H3K4me3) in postmortem PD samples. This elevation persisted in neuronal and PD-derived induced pluripotent stem cell lines while lowering H3K4me3 levels decreased α-Syn. These results point to a potential therapeutic avenue to mitigate α-Syn levels. The study explored recruiting JARID1A to the SNCA promoter to modulate expression, resulting in reduced H3K4me3 and lowered α-Syn expression. While global H3K4me3 levels remained unchanged, these findings emphasize the critical role of H3K4me3 in PD pathogenesis and its potential for therapeutic targeting [162,163,164]. However, several studies suggest that H3K4me3 regulates BDNF expression, aiding learning and memory. BDNF affects learning and memory by influencing synaptic plasticity [165].
In the Morse et al. study [153], the link between histone lysine methylation and learning as well as memory formation was explored. The findings demonstrated that learning was associated with an increase in BDNF gene transcription and H3K4me3 level at BDNF promoter 4 in the hippocampus.
A compelling outcome emerged: aged hippocamp exhibited memory reversal after learning. Intriguingly, higher baseline methylated histone lysine levels (H3K9me2 and H3K4me3) were observed in older hippocampi. Manipulating this baseline methylated histone level in young individuals with initially intact memory disrupted the balance between histone acetylation and methylation, leading to memory dysfunction. The study further revealed that the gene Zif265 (Egr1), linked with memory, correlated with changes in methylated histone lysines in aged adults compared to their younger counterparts, marked by significantly increased Zif265 mRNA levels in the aged hippocampus (C1) relative to young rats. An attractive hypothesis suggests that alterations in H3K4me3 could contribute to memory impairment in individuals with PD [166].
4.2.4. H3K27me3
The effects of H3K27me3 on SNCA, a gene associated with PD, were investigated. In the adult human brain, the SNpc region, H3K27me3, H3K4me3, and H3K27ac, were found to be preferentially enriched in the regulatory regions of SNCA. H3K4me3 and H3K27ac are associated with transcription initiation and active enhancer activity, respectively, while H3K27me3 is associated with gene repression. These results suggest that H3K27me3 may play a role in regulating the expression of SNCA. Elevated levels of α-Syn could be a key factor in developing PD and other synucleinopathies [33,164].
Histone H3 lysine 27 trimethylation (H3K27me3) creates a repressive chromatin environment that suppresses gene expression. The demethylase UTX, on the other hand, functions by eliminating H3K27me2/3, effectively triggering a mechanistic shift that activates extensive gene clusters. A study by Tang et al. demonstrated that the deficiency of the H3K27 demethylase enzyme Utx leads to disruptions in neuronal development and synaptic plasticity, leading to mood and cognitive deficits in Utx cKO mice [167].
4.2.5. H3K9ac
Tang et al. examined protein extracts from the SNpc of control, early PD, and late PD patients, determining the amount of acetylation of the histone residue AcH3-Lys9. The findings revealed an increase in AcH3-Lys9 acetylation in PD subjects’ SNpcs compared to controls, with a rise in early PD cases that were barely detectable and an increase in late PD cases that were detectable. Histone acetylation and the progression of PD are associated, as shown by the correlation between the degree of acetylation and the Braak stage of PD patients.
Harrison and coauthors examined the expression of several proteins, including tyrosine hydroxylase (TH), human leukocyte antigen DP-1 (HLA-DP-1), as well as HDACs, and SIRTs. In PD patients, HLA-DP1 expression increased, while TH expression decreased. However, the expression levels of HDACs and SIRTs did not show significant variations over time. The research suggests that histone acetylation and the production of specific proteins may play a role in the initiation and advancement of PD [168].
Another mouse study found that enhancing histone lysine acetylation can effectively regulate memory and learning, specifically in fear extinction tests. The administration of HDAC inhibitors improved memory, increased synaptic plasticity, and facilitated fear extinction. These findings offer promising prospects for developing therapies aimed at addressing anxiety disorders [169].
Peixoto et al.’s results have also indicated elevated histone acetylation, including H3K9 and H4K5, in healthy young mice after contextual fear conditioning. These findings suggest a potential involvement of acetylation in memory function. The enzyme accountable for this acetylation process, HDAC, was investigated to pinpoint the primary contributor. HDAC consists of two highly related proteins, HDAC1 and HDAC2, that form the catalytic core. Interestingly, HDAC2 negatively impacts memory formation and neuronal plasticity, unlike HDAC1 [170].
In a recent study, Huang et al. investigated trained immunity in microglial cells, including innate immune cells’ capacity to store memories of inflammatory environmental stimuli. The memory-priming trigger was lipopolysaccharide (LPS), and the subsequent environmental insult used was manganese (Mn). The authors demonstrated that microglia that had been exposed to LPS had an excessive inflammatory response. They proposed that the persistence of this heightened immune response is mediated by epigenetic reprogramming, especially the accumulation of histone 3 lysine 27 acetylation (H3K27ac) marks. The researchers validated these results in postmortem human PD brains and a mouse model of PD. Inhibiting H3K27ac deposition was also reported to increase mitochondrial performance and minimize inflammatory response. Overall, the research indicates that the memory of microglia and the following inflammatory response are regulated by H3K27ac-driven epigenetic reprogramming [171].
4.3. MicroRNAs in PD Cognitive Dysfunction
MiRNAs are non-coding RNAs that play a pivotal role in modulating biological processes by regulating mRNA expression at the posttranscriptional level [172]. These molecules hold immense significance in biological phenomena, including neuronal differentiation, synaptic plasticity, and the consolidation of memories [173,174].
Ongoing research suggests that miRNAs hold promise as peripheral biomarkers for different types of dementia, such as vascular dementia, AD, and PD [175,176]. These miRNAs also play critical roles in cognitive functions and the regulation of neurodegenerative disease-associated proteins [177,178]. The exact mechanisms that cause cognitive impairment in PD are not fully understood [179]. Table 2 provides an overview of selected relevant studies, which we further examined for a comprehensive understanding.
4.3.1. miR-7
miR-7, predominantly expressed in neurons, plays a crucial role in PD by suppressing α-Syn level through the 3′-UTR of its mRNA. This suppression leads to reduced aggregation in affected brain regions, particularly dopaminergic neurons. This finding suggests that miR-7 could be a potential therapeutic target for PD and other alpha-synucleinopathies by protecting cells against oxidative stress [180]. Yan Zhou et al. identified miR-7 targets Nlrp3, inhibiting inflammasome activation in microglia and showing therapeutic potential for PD. NLRP3 inflammasomes were activated in PD patients and model mice due to α-Syn’s role in microglial endocytosis and cathepsin B release. Caspase-1 deficiency reduced microglia activation and interleukin-1β production, protecting dopaminergic neurons. These findings suggest miR-7 and NLRP3 inflammasomes as potential targets for novel PD therapies [181].
MiR-7 contributes to neuroprotection by downregulating VDAC1 and VDAC3 after ischemic events. Its higher expression in acute ischemic stroke patients suggests its potential as a therapeutic target for stroke, as inhibiting miR-7 preserves neurons and improves neurological outcomes in both global and focal ischemia [182]. MiR-7 suppresses apoptosis in the human DA neuroblastoma cell line SH-SY5Y used as a PD model by directly targeting Bax and Sirt2 [183]. miR-7’s role in PD and neuroprotection may share a common mechanism involving the inhibition of apoptosis-related targets such as Bax and Sirt2. Figure 3 illustrates the mir-7 role in PD.
4.3.2. miR-124-3p
Geng et al. demonstrated that miR-124-3p contributes to neuroprotection in an in vitro PD model by targeting STAT3. Ectopic expression of miR-124-3p reduced neuronal injury, apoptosis, neuroinflammation, and oxidative stress induced by MPP+ treatment, suggesting its potential therapeutic significance in PD [184].
In another study, miR-124-3p was found to be reduced in the perioperative neurocognitive disorder (PND) rat model after a cardiopulmonary bypass (CPB) procedure, and its overexpression showed a protective effect against PND by alleviating cognitive decline, inflammation, and cell apoptosis. Additionally, miR-124-3p was predicted to directly target LPIN1, suggesting a potential therapeutic strategy for preventing and treating PND [185].
Pooling the data together, miR-124-3p appears to play a dual role in both PD and PND since its reduced expression may contribute to neurodegeneration and cognitive impairment. At the same time, its overexpression shows a neuroprotective effect and potential therapeutic significance in both conditions.
4.3.3. miR-124
miR-124 downregulation in PD prompted the development of miR-124-loaded nanoparticles to improve neuroinflammation. The study demonstrated that elevated miR-124 levels reduced pro-inflammatory signaling and mRNA levels of cytokines such as inducible nitric oxide synthase (iNOS), TNF-α, and interleukin 6 (IL-6). Simultaneously, it enhanced neuroprotection and decreased apoptosis, indicating its potential therapeutic utility for PD [186]. In PD, the downregulation of miR-124 in the prefrontal cortex of the left cerebral hemisphere has been identified as a factor related to the activation of the NF-κB signaling pathway, suggesting their potential role in the pathogenesis of PD and offering new targets for PD treatment [187].
Pantano et al. discovered the downregulation of miR-124 in the amygdala in PD patients. Their pipeline for small-RNA (sRNA) quantification identified altered sRNA patterns in premotor stages of PD, suggesting early pathogenic changes. The study emphasizes the tool’s utility in exploring sRNA expression patterns associated with various biological conditions [188].
On the other hand, mir-124 is downregulated in hippocampal neurons due to the Dicer1 gene mutation, leading to enhanced memory strength in mutant mice. The mutant mice exhibited higher efficacy at CA3-to-CA1 synapses, indicating increased synaptic plasticity. These findings suggest that miR-124 plays a significant role in mammals’ learning and memory process in mammals [130].
miR-124 plays a crucial role in memory-related synaptic plasticity by controlling CREB and CREB-mediated signaling during plasticity. It is abundantly present in the brain and constrains serotonin-induced synaptic facilitation, contributing to long-term plasticity in the mature nervous system [189].
Downregulation of miR-124 and abnormal regulation of Zif268 translation due to an EPAC null mutation led to impaired learning and social interactions. EPAC proteins are essential for regulating miR-124 transcription in the brain, impacting spatial learning and social interactions [194].
miR-124 downregulation in the frontal cortex of behavioral variant frontotemporal dementia (FTD) leads to an imbalance in AMPA receptor composition, affecting social behavior. Ectopic miR-124 expression reduces AMPAR levels and partially rescues behavioral deficits, suggesting a potential therapeutic approach for addressing social impairments in FTD [237].
miR-124 downregulation may play a common role in PD and cognition impairment, as it affects neuroinflammation, synaptic plasticity, and social behavior. This suggests its potential as a therapeutic target for these conditions.
4.3.4. miR-195
miR-195 is a potential biomarker that is dysregulated during PD. Downregulation of miR-195 in LPS-stimulated BV2 cells induces microglia activation and the release of pro-inflammatory cytokines. Conversely, miR-195 overexpression inhibits pro-inflammatory cytokines and promotes anti-inflammatory cytokines, indicating its potential as a therapeutic target for PD to modulate neuroinflammation [196].
miR-195 downregulation in rats induced by chronic brain hypoperfusion (CBH) leads to increased expression of amyloid precursor protein (APP) and β-site APP cleaving enzyme 1 (BACE1) proteins, causing memory impairment. Specifically, miR-195 overexpression reduces dementia vulnerability triggered by CBH, suggesting its potential as an anti-dementia approach by regulating APP and BACE1 expression [197].
miR-195 downregulation in CBH promotes Aβ aggregation and Tau hyperphosphorylation, potentially contributing to increased vulnerability to dementia in CBH rats [198].
Pan et al. demonstrated that miR-195 is upregulated in schizophrenia patients and negatively correlates with reduced BDNF protein levels, leading to cognitive impairments in the domains of speed of processing, working memory, and attention/vigilance. Targeting miR-195 as a potential therapeutic approach could enhance cognitive functions in schizophrenia [238].
4.3.5. miR-190
miR-190 downregulation in PD contributes to neuroinflammation and neuronal damage by activating Nlrp3, while miR-190 overexpression inhibits inflammation and protects against neuronal injury, suggesting its potential therapeutic role in PD [201].
MiR-190a-3p is implicated in postoperative cognitive dysfunction (POCD). It may act as a novel predictive indicator for this neurocognitive disorder, with potential roles in pathways related to brain development and psychiatric disorders [202]. miR-190 plays a neuroprotective role by reducing brain ischemia-reperfusion (I/R) injury in rats, suppressing neuronal apoptosis, and modulating the Rho/Rho-kinase signaling pathway, suggesting its potential therapeutic value for ischemic stroke [203].
miR-190 may play a dual role in PD-related cognition, with potential therapeutic implications for neuroinflammation, neuronal damage, and POCD.
4.3.6. miR-146a
miR-146a has been identified as a regulator of GDNF, potentially impacting glia activation and contributing to the progression of PD [239]. PD patients exhibited reduced levels of miRNA-146a compared to healthy controls [240]. A study conducted by Chen et al. showed that miRNA-146a protected against cognitive decline caused by surgical trauma by suppressing hippocampal neuroinflammation [241]. However, miR-146a appears to aggravate cognitive impairment and AD-like pathology by triggering oxidative stress through the mitogen-activated protein kinase (MAPK) signaling pathway [242].
4.3.7. miR-155
The regulation of miR-155 was investigated in PD using mouse models lacking the DJ-1 gene. The study revealed that DJ-1 plays a crucial role in controlling the expression of suppressor of cytokine signaling 1 (SOCS1) through miR-155 levels. In DJ-1 knockout cells, increased miR-155 levels led to decreased stability of SOCS1 mRNA, resulting in altered inflammatory responses [207].
The rat model of AD showed increased levels of miR-155 and inflammatory cytokines IL-1β, IL-6, and TNF-α in the hippocampus. An intracerebroventricular infusion of miR-155 inhibitor reduced the inflammatory response and improved learning performance in AD rats, suggesting that miR-155 regulates memory impairment in AD through neuroinflammatory mechanisms and may be a target for preventing or improving cognitive deficits in AD [243]. Another study found that LPS exposure upregulated microglia miR-155 expression, decreasing SOCS-1, an important inflammation inhibitor, and predicted miR-155 target. The study suggests that miR-155 is pro-inflammatory in microglia, and inhibiting it may protect neurons in chronic inflammation [208]. Based on the published data, we hypothesize that miR-155 may serve as a pivotal link connecting cognitive impairments and PD disease.
4.3.8. miR-133b
mir-133b exhibits high levels in the midbrain of healthy controls HC, while it is diminished in PD samples compared to the cerebellum, midbrain, and cerebral cortex [209]. A meta-analysis revealed significant downregulation of miR-133b in both brain and blood specimens of individuals with PD compared to control subjects, indicating a consistent reduction in miR-133b levels across different studies [212]. Schlaudraff and colleagues found that miR-133b was decreased in the midbrain tissue of individuals with PD compared to controls. They suggest that miR-133b may play a role in reducing the expression of TH and, subsequently, dopamine synthesis in PD [213].
miR-133b plays a role in memory, attenuating isoflurane-induced learning and memory impairment in rats. The overexpression of miR-133b promoted the viability of hippocampal neurons and protected them from apoptosis when exposed to isoflurane. These findings points to a potential protective effect of miR-133b against anesthesia-induced cognitive impairment [210]. These results about miR-133b could serve as a critical link connecting cognitive impairments and PD.
4.3.9. miR-126
miR-126 plays a role in PD by disrupting insulin/IGF-1/PI3K signaling in dopamine neurons. Its overexpression impairs IGF-1 signaling, making the neurons more vulnerable to neurotoxins. Enhancing IGF-1 neuroprotection can be achieved by inhibiting miR-126 function, suggesting that elevated miR-126 levels may contribute to PD pathogenesis, and its suppression could serve as a neuroprotective mechanism [215]. miR-126 is dysregulated in PD, particularly in SNpc dopamine neurons. This dysregulation affects gene expression networks and signaling pathways relevant to PD pathogenesis. Interestingly, there are sex-specific differences in miR-126 expression, with more up-regulation in males and more down-regulation in females. These findings suggest that miR-126 may play a functional role in the development of PD, and its impact could vary based on gender [216].
miR-126 plays a significant role in cognition by mediating cognitive impairment in a vascular dementia (VaD) mouse model. Reduction of endothelial miR-126 expression in the model leads to cognitive deficits, decreased cerebral blood flow, white matter damage, inflammation, and glymphatic dysfunction, indicating its involvement in cognitive function and memory [218]. miR-126 exerts neuroprotection in intracerebral hemorrhage by promoting angiogenesis, reducing lesion size, and inhibiting neuronal apoptosis. Overexpression of miR-126 in a rat model of ICH improved behavioral outcomes [217]. Elevated miR-126 levels in PD may impair dopamine neuron signaling, increase neurotoxin vulnerability, and contribute to cognitive impairment.
4.3.10. miR-144
miR-144-3p is consistently down-regulated in early PD and appears to be involved in the regulation of coagulation, suggesting its potential relevance as a biomarker and a key player in PD pathogenesis within the aging framework [244]. The overexpression of miR-144 is implicated in the etiology of PD by potentially modifying the expression of key genes, including PARK2, SNCA, and LRRK2, which are associated with the disease pathology and normal cellular functions [220]. Furthermore, another study by Li et al. showed significant downregulation of miR-144-3p in PD. Its overexpression enhances mitochondrial function by targeting β-amyloid precursor protein (APP). Upregulation of miR-144-3p increases the expression of key genes involved in maintaining mitochondrial function, leading to improved cellular ATP, viability, and mtDNA copy number, while APP overexpression counteracts these effects. This data highlights the crucial role of miR-144-3p in regulating mitochondrial function in PD [245].
MiR-144-3p plays a pivotal role in rescuing impaired fear extinction memory in mice. Murphy et al. suggest that enhancing miR-144-3p expression in the basolateral amygdala can improve fear extinction acquisition and protect against fear renewal, making it a promising target for developing novel treatments for anxiety- and trauma-related disorders that involve cognitive impairment and memory deficits [246]. Another study shows that miR-144 plays a role in cognition by influencing cognitive function and neurological outcomes after traumatic brain injury (TBI). Inhibition of miR-144 improves cognitive deficits, protects neurons against injury, and reduces β-amyloid accumulation through upregulating ADAM10 expression, suggesting a potential therapeutic target for TBI-induced cognitive impairments [221]. Combining the existing data, we hypothesize that miR-144-3p may play a common role in cognition and PD.
4.3.11. miR-148b
MiR-148b is downregulated in PD, a phenomenon linked to its involvement in neurological development and apoptosis [247]. Overexpression of miR-148b in the ischemic brain’s subventricular zone (SVZ) of the ischemic brain has been shown to promote neuroprotection by inhibiting Wnt/β-catenin signaling. This inhibition enhances neural stem cell proliferation and differentiation, indicating its potential as a novel therapeutic strategy for stroke recovery [222].
4.3.12. miR-221
MiR-221 plays a significant role in PD, protecting against oxidative stress and inhibiting Bax/caspase-3 signaling. Moreover, it controls cell viability and apoptosis through the Akt pathway, making it a potential therapeutic target and biomarker for PD [248]. Additionally, miR-221 exhibits promise as a potential biomarker for PD. Its downregulation in serum has been linked to the disease and is correlated with disease severity as assessed by UPDRS scores [249]. Li et al. showed that miR-221 plays a protective role in PD by promoting cell viability and inhibiting apoptosis in 6-OHDA-treated PC12 cells through targeting PTEN and activating the AKT pathway. This data suggests its potential role as a therapeutic target for PD treatment [250].
miR-221 has been implicated in cognitive decline, and its serum levels are associated with the development of dementia. In a prospective study, individuals with higher serum miR-221 levels were found to have significantly increased odds of cognitive decline, suggesting that miR-221 may be a potential biomarker for predicting cognitive impairment and dementia at an early stage [251]. miR-221-3p appears to play a role in cognitive function and may serve as a prognostic marker for cerebrovascular disease, which is known to impact cognitive performance. Elevated levels of miR-221-3p have been observed in stroke patients and individuals with hypertension, hyperlipidemia, and diabetes, suggesting its potential involvement in cognitive impairment related to cerebrovascular conditions [252]. miR-221 may play a dual role in PD-related cognitive decline, acting as a protective factor in PD and a potential biomarker for cognitive impairment.
4.3.13. miR-199a
The role of miR-199a in PD revolves around the regulation of autophagy. In an in vitro model of PD induced by MPP+, increasing miR-199a expression resulted in reduced autophagy, enhanced cellular viability, increased survival rate, and improved cellular pathologies. This effect was achieved by targeting pro-autophagic pathways and GSK3β to activate PTEN/AKT/mTOR signaling. These findings imply that miR-199a might play a neuroprotective role in PD by modulating autophagy-related processes [253]. MiR-199a negatively regulates Neuritin expression, which may cause synaptic damage and cognitive impairment, particularly spatial memory deficits, in AD [227]. According to Nakashima et al., miR-199a plays a role in cognition by regulating neural stem/progenitor cell differentiation. Increased expression of miR-199a ameliorates impaired differentiation of MeCP2-deficient NS/PCs, which is critical for brain development and plasticity.
Furthermore, miR-199a reduces the protein abundance of Smad1, a downstream transcription factor of the BMP signaling pathway. Dysregulation of BMP signaling has been associated with cognitive delay and developmental disorders. The study’s findings indicate that miR-199a may impact cognition by influencing neural cell differentiation and the BMP signaling pathway, which are crucial processes in brain development and function [254]. Our hypothesis is that mir-199a contributes to cognitive impairment in PD.
4.3.14. miR-132
PD patients and PD cell models exhibited increased levels of miR-132-3p and reduced levels of GLRX. The upregulation of miR-132-3p activated microglial cells, but this effect could be reversed by overexpressing GLRX [235].
miR-132 is crucial for temporal memory formation; its increased expression after trace fear conditioning and impaired memory acquisition upon its knockdown in mice emphasize its role in hippocampal-based temporal memory [255]. As a critical activity-dependent regulator of cognition, miR-132 demonstrates its significance by influencing neuronal maturation, dendritic arborization, and spinogenesis within the central nervous system [233]. Upregulation of miR-132 inhibits FOXA1 to mitigate sevoflurane-induced cognitive damage in AD rats [231]. MiR-132 enhances dendrite morphogenesis and plays a crucial role in activity-dependent plasticity by inhibiting p250GAP and activating Rac signaling [230].
5. Therapeutics Perspective of Epigenetics in PD Cognitive Dysfunction
Harvey and colleagues proposed that the distinct neuropsychiatric and cognitive symptoms seen in PD may have a unique epigenetic signature in the brain, allowing for differentiation between individuals with these symptoms and those without [256]. Epigenetic methylation markers linked to accelerated cognitive decline or PD motor progression were found by Chuang et al. They could be useful as biomarkers for PD progression, identifying high-risk patients for early treatment, or drug development targets. Recently, attention has centered on ubiquitin-specific proteases (USPs) in neurodegenerative diseases. USP5 and USP13 degrade ubiquitin chains inside stress granules, clumps of protein or RNA created when cells are stressed. Since Lewy body alpha-synuclein ubiquitination is the pathological hallmark of PD, the possibility of targeting them for treatment has been suggested [257]. Some epigenetic regulation drugs being studied for PD include HDAC inhibitors, DNMT inhibitors, and bromodomain and extra-terminal domain inhibitors.
Histone Deacetylases as Potential Epigenetic Targets
HDACs, an important epigenetic target, have garnered attention in drug discovery for two decades. PD treatment may involve a potent SIRT2 inhibitor, a member of the HDAC family. Inhibition of SIRT2 successfully reduced α-Syn toxicity and altered inclusion morphology in a cellular PD model. Inhibiting SIRT2 with small interfering RNAs has been shown to protect against α-Syn toxicity. The inhibitors also prevented dopaminergic cell death in in vitro and Drosophila PD models. These findings suggest that neurodegeneration and aging are linked, and SIRT2 inhibitors may improve PD-related cognitive and motor symptoms [258].
Sodium butyrate, an HDAC inhibitor, improved cognitive function in PD rats. Like PD patients, the rat model had executive function deficits, particularly in attentional set-shifting. However, sodium butyrate improved attentional set formation and showed promise for treating PD cognitive impairments [259].
Song et al. indicated that dieldrin, a neurotoxic pesticide associated with PD, induced hyperacetylation of core histones in dopaminergic neuronal cells due to proteasomal dysfunction. The inhibition of histone acetyltransferase with anacardic acid attenuated dieldrin-induced histone acetylation, protected dopaminergic cells from degeneration, and induced histone hyperacetylation in relevant brain regions in mouse models. These findings suggest the potential of HDAC-targeted drugs in PD therapy [260].
A signaling cascade is activated during normal learning events, involving Histone Acetyltransferases (HATs) activation and inactivation/removal of HDACs.HATs attach acetyl groups to histone tails, relaxing the nucleosome structure and allowing the binding of DNA-binding transcription factors. However, under usual circumstances, the conditioned response may not persist. By administering an HDAC inhibitor (HDACi), histone tails become hyperacetylated, promoting memory consolidation and strengthening the response. Although this simplified schematic mainly emphasizes the concept of histone acetylation, it is important to acknowledge the involvement of other epigenetic mechanisms that cooperate with HATs and HDACs in this process.
These findings suggest that targeting HDACs, particularly SIRT2, may improve PD-related cognitive and motor symptoms. Potent HDAC inhibitors may be a future target for histone acetylation in PD treatment to improve memory impairment.
6. Conclusions
Treatment of PD patients with cognitive issues is urgent since its progression is disabling. Evidence suggesting reliable methods to control cognitive decline is lacking. Therefore, we reviewed the literature on the role of epigenetics in cognition in PD. The available studies are sometimes contradictory. Hence, we have provided hypotheses about the probable epigenetic mechanisms that may cause cognitive impairment in PD patients in light of the available literature, which is confined and consists of many gaps. To resolve these limitations, researchers need to conduct more comprehensive clinical and trial studies to test these hypotheses. A better understanding of epigenetics will help find new cellular and molecular-based therapies to treat cognitive impairments in PD patients.
Abbreviations
Aβ- amyloid beta; AD -Alzheimer’s disease; α-Syn - α-Synuclein; APP- β-amyloid precursor protein; BDNF - brain-derived neurotrophic factor; CBH - chronic brain hypoperfusion; CPB - cardiopulmonary bypass; EGFR - epidermal growth factor receptor; HATs - Histone Acetyltransferases; HDAC- histone deacetylases; HLA-DP-1 - Human Leukocyte Antigen DP-1; IL-6 – interleukin 6; iNOS - inducible nitric oxide synthase; LB- Lewy bodies; LNs - Lewy neurites; LPS – lipopolysaccharide; MAPK - Mitogen-activated protein kinase; NMDAR - N-methyl-D-aspartate Receptor; MAPs - microtubule-associated proteins; PD- Parkinson’s disease; PFFs - pre-formed fibrils; PND - perioperative neurocognitive disorder; POCD - postoperative cognitive dysfunction; PTMs - post-translational modifications; ROS – reactive oxygen species; SNpc - Substantia Nigra pars compacta; TAT1 - Trans-Activator of Transcription 1; TNF-α - tumor necrosis factor-alpha; SNP - single nucleotide polymorphism.
References
- Moosavi A, Motevalizadeh Ardekani A. Role of Epigenetics in Biology and Human Diseases. Iran Biomed J. 2016;20(5):246-58. [CrossRef]
- Surguchov, A. α-Synuclein and Mechanisms of Epigenetic Regulation. Brain Sci. 2023, 13, 150. [CrossRef]
- Huynh, J.L.; Casaccia, P. Epigenetic mechanisms in multiple sclerosis: implications for pathogenesis and treatment. Lancet Neurol. 2013, 12, 195–206. [CrossRef]
- Doder, M.; Jahanshahi, M.; Turjanski, N.; Moseley, I.F.; Lees, A.J. Parkinson’s syndrome after closed head injury: a single case report. J. Neurol. Neurosurg. Psychiatry 1999, 66, 380–385. [CrossRef]
- Goetz, C.G. The History of Parkinson’s Disease: Early Clinical Descriptions and Neurological Therapies. Cold Spring Harb. Perspect. Med. 2011, 1, a008862. [CrossRef]
- Emamzadeh FN, Surguchov A. Parkinson’s Disease: Biomarkers, Treatment, and Risk Factors. Front Neurosci. 2018;12:612. [CrossRef]
- Nair, V.D.; Ge, Y. Alterations of miRNAs reveal a dysregulated molecular regulatory network in Parkinson’s disease striatum. Neurosci. Lett. 2016, 629, 99–104. [CrossRef]
- Cannon JR, Greenamyre JT. Gene-environment interactions in Parkinson’s disease: specific evidence in humans and mammalian models. Neurobiol Dis. 2013;57:38-46. [CrossRef]
- Zoghbi HY, Beaudet AL. Epigenetics and Human Disease. Cold Spring Harb Perspect Biol. 2016;8(2):a019497. [CrossRef]
- Vasili E, Dominguez-Meijide A, Outeiro TF. Spreading of α-Synuclein and Tau: A Systematic Comparison of the Mechanisms Involved. Front Mol Neurosci. 2019;12:107. [CrossRef]
- Chen X, de Silva HAR, Pettenati MJ, Rao PN, George-Hyslop PS, Roses AD, et al. The human NACP/α-synuclein gene: chromosome assignment to 4q21.3–q22 and TaqI RFLP analysis. Genomics. 1995;26(2):425-7. [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. et Biophys. Acta (BBA) - Proteins Proteom. 2019, 1867, 890–908. [CrossRef]
- Coppedè, F. Genetics and Epigenetics of Parkinson’s Disease. Sci. World J. 2012, 2012, 1–12. [CrossRef]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364(9440):1167-9. [CrossRef]
- Deng, H.; Yuan, L. Genetic variants and animal models in SNCA and Parkinson disease. Ageing Res. Rev. 2014, 15, 161–176. [CrossRef]
- Myhre R, Toft M, Kachergus J, Hulihan M, Aasly J, Klungland H, et al. Multiple alpha-synuclein gene polymorphisms are associated with Parkinson’s disease in a Norwegian population. Acta Neurologica Scandinavica. 2008;118(5):320-7. [CrossRef]
- Mizuta, I.; Satake, W.; Nakabayashi, Y.; Ito, C.; Suzuki, S.; Momose, Y.; Nagai, Y.; Oka, A.; Inoko, H.; Fukae, J.; et al. Multiple candidate gene analysis identifies α-synuclein as a susceptibility gene for sporadic Parkinson’s disease. Hum. Mol. Genet. 2006, 15, 1151–1158. [CrossRef]
- Ross, O.A.; Gosal, D.; Stone, J.T.; Lincoln, S.J.; Heckman, M.G.; Irvine, G.B.; Johnston, J.A.; Gibson, J.M.; Farrer, M.J.; Lynch, T. Familial genes in sporadic disease: Common variants of α-synuclein gene associate with Parkinson’s disease. Mech. Ageing Dev. 2007, 128, 378–382. [CrossRef]
- Chung, S.J.; Armasu, S.M.; Biernacka, J.M.; Lesnick, T.G.; Rider, D.N.; Lincoln, S.J.; Ortolaza, A.I.; Farrer, M.J.; Cunningham, J.M.; Rocca, W.A.; et al. Common variants in PARK loci and related genes and Parkinson’s disease. Mov. Disord. 2010, 26, 280–288. [CrossRef]
- Trotta, L.; Guella, I.; Soldà, G.; Sironi, F.; Tesei, S.; Canesi, M.; Pezzoli, G.; Goldwurm, S.; Duga, S.; Asselta, R. SNCA and MAPT genes: Independent and joint effects in Parkinson disease in the Italian population. Park. Relat. Disord. 2012, 18, 257–262. [CrossRef]
- Wu-Chou, Y.-H.; Chen, Y.-T.; Yeh, T.-H.; Chang, H.-C.; Weng, Y.-H.; Lai, S.-C.; Huang, C.-L.; Chen, R.-S.; Huang, Y.-Z.; Chen, C.-C.; et al. Genetic variants of SNCA and LRRK2 genes are associated with sporadic PD susceptibility: A replication study in a Taiwanese cohort. Park. Relat. Disord. 2012, 19, 251–255. [CrossRef]
- Chen, Y.; Wei, Q.-Q.; Ou, R.; Cao, B.; Chen, X.; Zhao, B.; Guo, X.; Yang, Y.; Chen, K.; Wu, Y.; et al. Genetic Variants of SNCA Are Associated with Susceptibility to Parkinson’s Disease but Not Amyotrophic Lateral Sclerosis or Multiple System Atrophy in a Chinese Population. PLOS ONE 2015, 10, e0133776. [CrossRef]
- Davis, A.A.; Andruska, K.M.; Benitez, B.A.; Racette, B.A.; Perlmutter, J.S.; Cruchaga, C. Variants in GBA , SNCA , and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol. Aging 2016, 37, 209.e1–209.e7. [CrossRef]
- Wei, Y.; Yang, N.; Xu, Q.; Sun, Q.; Guo, J.; Li, K.; Liu, Z.; Yan, X.; Zhu, X.; Tang, B. The rs3756063 polymorphism is associated with SNCA methylation in the Chinese Han population. J. Neurol. Sci. 2016, 367, 11–14. [CrossRef]
- Foo, J.N.; Tan, L.C.; Irwan, I.D.; Au, W.-L.; Low, H.Q.; Prakash, K.-M.; Ahmad-Annuar, A.; Bei, J.; Chan, A.Y.; Chen, C.M.; et al. Genome-wide association study of Parkinson’s disease in East Asians. Hum. Mol. Genet. 2016, 26, 226–232. [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [CrossRef]
- Zhang, Y.; Shu, L.; Sun, Q.; Pan, H.; Guo, J.; Tang, B. A Comprehensive Analysis of the Association Between SNCA Polymorphisms and the Risk of Parkinson’s Disease. Front. Mol. Neurosci. 2018, 11, 391. [CrossRef]
- Chartier-Harlin, M.-C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [CrossRef]
- Huang Y, Wang G, Rowe D, Wang Y, Kwok JB, Xiao Q, et al. SNCA gene, but not MAPT, influences onset age of Parkinson’s disease in Chinese and Australians. BioMed Research International. 2015;2015. [CrossRef]
- Cardo, L.F.; Coto, E.; De Mena, L.; Ribacoba, R.; Lorenzo-Betancor, O.; Pastor, P.; Samaranch, L.; Mata, I.F.; Díaz, M.; Morís, G.; et al. A Search for SNCA 3′ UTR Variants Identified SNP rs356165 as a Determinant of Disease Risk and Onset Age in Parkinson’s Disease. J. Mol. Neurosci. 2012, 47, 425–430. [CrossRef]
- Elbaz, A.; Ross, O.A.; Ioannidis, J.P.A.; Soto-Ortolaza, A.I.; Moisan, F.; Aasly, J.; Annesi, G.; Bozi, M.; Brighina, L.; Chartier-Harlin, M.; et al. Independent and joint effects of the MAPT and SNCA genes in Parkinson disease. Ann. Neurol. 2010, 69, 778–792. [CrossRef]
- Brockmann, K.; Schulte, C.; Hauser, A.; Lichtner, P.; Huber, H.; Maetzler, W.; Berg, D.; Gasser, T. SNCA: Major genetic modifier of age at onset of Parkinson’s disease. Mov. Disord. 2013, 28, 1217–1221. [CrossRef]
- Guhathakurta, S.; Bok, E.; Evangelista, B.A.; Kim, Y.-S. Deregulation of α-synuclein in Parkinson’s disease: Insight from epigenetic structure and transcriptional regulation of SNCA. Prog. Neurobiol. 2017, 154, 21–36. [CrossRef]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wüllner, U. Methylation Regulates Alpha-Synuclein Expression and Is Decreased in Parkinson’s Disease Patients’ Brains. J. Neurosci. 2010, 30, 6355–6359. [CrossRef]
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Iwata, A. CpG Demethylation Enhances Alpha-Synuclein Expression and Affects the Pathogenesis of Parkinson’s Disease. PLOS ONE 2010, 5, e15522. [CrossRef]
- Wu, H.; D’alessio, A.C.; Ito, S.; Xia, K.; Wang, Z.; Cui, K.; Zhao, K.; Sun, Y.E.; Zhang, Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 2011, 473, 389–393. [CrossRef]
- Guhathakurta, S.; Song, M.K.; Basu, S.; Je, G.; Cristovao, A.C.; Kim, Y.-S. Regulation of Alpha-Synuclein Gene (SNCA) by Epigenetic Modifier TET1 in Parkinson Disease. Int. Neurourol. J. 2022, 26, S85–93. [CrossRef]
- Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, Illig T, et al. Multiple regions of α-synuclein are associated with Parkinson’s disease. Annals of Neurology. 2005;57(4):535-41. [CrossRef]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and Pathology of Tau Protein in Alzheimer Disease. Int. J. Alzheimer’s Dis. 2012, 2012, 1–13. [CrossRef]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [CrossRef]
- Aguzzi, A.; Montrasio, F.; Kaeser, P.S. Prions: health scare and biological challenge. Nat. Rev. Mol. Cell Biol. 2001, 2, 118–126. [CrossRef]
- Lewis, V.; Hooper, N.M. The role of lipid rafts in prion protein biology. Front. Biosci. 2011, 16, 151–168. [CrossRef]
- Costanzo, M.; Zurzolo, C. The cell biology of prion-like spread of protein aggregates: mechanisms and implication in neurodegeneration. Biochem. J. 2013, 452, 1–17. [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [CrossRef]
- Holmes, B.B.; Devos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [CrossRef]
- Weaver, I.C. Epigenetic Programming by Maternal Behavior and Pharmacological InterventionNature Versus Nurture: Let’s Call The Whole Thing Off. Epigenetics 2007, 2, 22–28. [CrossRef]
- Bredy, T.W.; Wu, H.; Crego, C.; Zellhoefer, J.; Sun, Y.E.; Barad, M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn. Mem. 2007, 14, 268–276. [CrossRef]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53(6):857-69. [CrossRef]
- Day, J.J.; Sweatt, J.D. Epigenetic Mechanisms in Cognition. Neuron 2011, 70, 813–829. [CrossRef]
- Parkinson disease 2022 [Available from: https://www.who.int/news-room/fact-sheets/detail/parkinson-disease.
- Hirsch, L.; Jette, N.; Frolkis, A.; Steeves, T.; Pringsheim, T. The Incidence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology 2016, 46, 292–300. [CrossRef]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [CrossRef]
- Caillierez, R.; Bégard, S.; Lécolle, K.; Deramecourt, V.; Zommer, N.; Dujardin, S.; Loyens, A.; Dufour, N.; Aurégan, G.; Winderickx, J.; et al. Lentiviral Delivery of the Human Wild-type Tau Protein Mediates a Slow and Progressive Neurodegenerative Tau Pathology in the Rat Brain. Mol. Ther. 2013, 21, 1358–1368. [CrossRef]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [CrossRef]
- Holmes, B.B.; Devos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [CrossRef]
- Levarska, L.; Zilka, N.; Jadhav, S.; Neradil, P.; Novak, M. Of Rodents and Men: The Mysterious Interneuronal Pilgrimage of Misfolded Protein Tau in Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 37, 569–577. [CrossRef]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.A.; Hasegawa, M. Prion-like spreading of pathological α-synuclein in brain. Brain 2013, 136, 1128–1138. [CrossRef]
- Oliveras-Salvá M, Van der Perren A, Casadei N, Stroobants S, Nuber S, D’Hooge R, et al. rAAV2/7 vector-mediated overexpression of alpha-synuclein in mouse substantia nigra induces protein aggregation and progressive dose-dependent neurodegeneration. Mol Neurodegener. 2013;8:44. [CrossRef]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. Embo Rep. 2013, 14, 389–394. [CrossRef]
- Sacino, A.N.; Brooks, M.; McGarvey, N.H.; McKinney, A.B.; A Thomas, M.; Levites, Y.; Ran, Y.; E Golde, T.; I Giasson, B. Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol. Commun. 2013, 1, 38–38. [CrossRef]
- Ulusoy A, Rusconi R, Pérez-Revuelta BI, Musgrove RE, Helwig M, Winzen-Reichert B, et al. Caudo-rostral brain spreading of α-synuclein through vagal connections. EMBO Mol Med. 2013;5(7):1119-27. [CrossRef]
- Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. 2013, 110, 19555–19560. [CrossRef]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small Misfolded Tau Species Are Internalized via Bulk Endocytosis and Anterogradely and Retrogradely Transported in Neurons. J. Biol. Chem. 2013, 288, 1856–1870. [CrossRef]
- Ahmed, Z.; Cooper, J.; Murray, T.K.; Garn, K.; McNaughton, E.; Clarke, H.; Parhizkar, S.; Ward, M.A.; Cavallini, A.; Jackson, S.; et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014, 127, 667–683. [CrossRef]
- Clavaguera, F.; Hench, J.; Lavenir, I.; Schweighauser, G.; Frank, S.; Goedert, M.; Tolnay, M. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 2013, 127, 299–301. [CrossRef]
- Dujardin, S.; Lécolle, K.; Caillierez, R.; Bégard, S.; Zommer, N.; Lachaud, C.; Carrier, S.; Dufour, N.; Aurégan, G.; Winderickx, J.; et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2013, 2, 14–14. [CrossRef]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. 2014, 111, E4376–85. [CrossRef]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.-Y.; Roybon, L.; Melki, R.; Li, J.-Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [CrossRef]
- Recasens A, Dehay B. Alpha-synuclein spreading in Parkinson’s disease. Front Neuroanat. 2014;8:159.
- Reyes, J.F.; Rey, N.L.; Bousset, L.; Melki, R.; Brundin, P.; Angot, E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 2013, 62, 387–398. [CrossRef]
- Rotermund C, Truckenmüller FM, Schell H, Kahle PJ. Diet-induced obesity accelerates the onset of terminal phenotypes in α-synuclein transgenic mice. J Neurochem. 2014;131(6):848-58. [CrossRef]
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; Lee, S.; Regenhardt, R.W.; McGarvey, N.H.; Ayers, J.I.; Notterpek, L.; Borchelt, D.R.; et al. Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10732–10737. [CrossRef]
- Sacino AN, Brooks M, Thomas MA, McKinney AB, McGarvey NH, Rutherford NJ, et al. Amyloidogenic α-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol. 2014;127(5):645-65. [CrossRef]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct Tau Prion Strains Propagate in Cells and Mice and Define Different Tauopathies. Neuron 2014, 82, 1271–1288. [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [CrossRef]
- Bernis, M.E.; Babila, J.T.; Breid, S.; Wüsten, K.A.; Wüllner, U.; Tamgüney, G. Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol. Commun. 2015, 3, 1–18. [CrossRef]
- Bourdenx M, Dovero S, Engeln M, Bido S, Bastide MF, Dutheil N, et al. Lack of additive role of ageing in nigrostriatal neurodegeneration triggered by α-synuclein overexpression. Acta Neuropathol Commun. 2015;3:46. [CrossRef]
- Daher, J.P.; Abdelmotilib, H.A.; Hu, X.; Volpicelli-Daley, L.A.; Moehle, M.S.; Fraser, K.B.; Needle, E.; Chen, Y.; Steyn, S.J.; Galatsis, P.; et al. Leucine-rich Repeat Kinase 2 (LRRK2) Pharmacological Inhibition Abates α-Synuclein Gene-induced Neurodegeneration. J. Biol. Chem. 2015, 290, 19433–19444. [CrossRef]
- Abounit, S.; Wu, J.W.; Duff, K.; Victoria, G.S.; Zurzolo, C. Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 2016, 10, 344–351. [CrossRef]
- D’Abramo, C.; Acker, C.M.; Schachter, J.B.; Terracina, G.; Wang, X.; Forest, S.K.; Davies, P. Detecting tau in serum of transgenic animal models after tau immunotherapy treatment. Neurobiol. Aging 2015, 37, 58–65. [CrossRef]
- Domert, J.; Sackmann, C.; Severinsson, E.; Agholme, L.; Bergström, J.; Ingelsson, M.; Hallbeck, M. Aggregated Alpha-Synuclein Transfer Efficiently between Cultured Human Neuron-Like Cells and Localize to Lysosomes. PLOS ONE 2016, 11, e0168700. [CrossRef]
- Helwig, M.; Klinkenberg, M.; Rusconi, R.; Musgrove, R.E.; Majbour, N.K.; El-Agnaf, O.M.A.; Ulusoy, A.; Di Monte, D.A. Brain propagation of transduced α-synuclein involves non-fibrillar protein species and is enhanced in α-synuclein null mice. Brain 2015, 139, 856–870. [CrossRef]
- Koprich JB, Johnston TH, Reyes G, Omana V, Brotchie JM. Towards a Non-Human Primate Model of Alpha-Synucleinopathy for Development of Therapeutics for Parkinson’s Disease: Optimization of AAV1/2 Delivery Parameters to Drive Sustained Expression of Alpha Synuclein and Dopaminergic Degeneration in Macaque. PLoS One. 2016;11(11):e0167235. [CrossRef]
- Aulić, S.; Masperone, L.; Narkiewicz, J.; Isopi, E.; Bistaffa, E.; Ambrosetti, E.; Pastore, B.; De Cecco, E.; Scaini, D.; Zago, P.; et al. α-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci. Rep. 2017, 7, 1–12. [CrossRef]
- Cavaliere, F.; Cerf, L.; Dehay, B.; Ramos-Gonzalez, P.; De Giorgi, F.; Bourdenx, M.; Bessede, A.; Obeso, J.A.; Matute, C.; Ichas, F.; et al. In vitro α-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol. Dis. 2017, 103, 101–112. [CrossRef]
- DeVos, S.L.; Miller, R.L.; Schoch, K.M.; Holmes, B.B.; Kebodeaux, C.S.; Wegener, A.J.; Chen, G.; Shen, T.; Tran, H.; Nichols, B.; et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 2017, 9. [CrossRef]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. α-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017, 134, 789–808. [CrossRef]
- Ngolab, J.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Trejo, M.; Masliah, D.; Adame, A.; Masliah, E.; Rissman, R.A. Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol. Commun. 2017, 5, 1–10. [CrossRef]
- Shimozawa, A.; Ono, M.; Takahara, D.; Tarutani, A.; Imura, S.; Masuda-Suzukake, M.; Higuchi, M.; Yanai, K.; Hisanaga, S.-I.; Hasegawa, M. Propagation of pathological α-synuclein in marmoset brain. Acta Neuropathol. Commun. 2017, 5, 12. [CrossRef]
- Ulusoy, A.; Phillips, R.J.; Helwig, M.; Klinkenberg, M.; Powley, T.L.; Di Monte, D.A. Brain-to-stomach transfer of α-synuclein via vagal preganglionic projections. Acta Neuropathol. 2016, 133, 381–393. [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 1–25. [CrossRef]
- Polanco, J.C.; Li, C.; Durisic, N.; Sullivan, R.; Götz, J. Exosomes taken up by neurons hijack the endosomal pathway to spread to interconnected neurons. Acta Neuropathol. Commun. 2018, 6, 1–14. [CrossRef]
- Rusconi R, Ulusoy A, Aboutalebi H, Di Monte DA. Long-lasting pathological consequences of overexpression-induced α-synuclein spreading in the rat brain. Aging Cell. 2018;17(2). [CrossRef]
- Vitale, F.; Giliberto, L.; Ruiz, S.; Steslow, K.; Marambaud, P.; D’abramo, C. Anti-tau conformational scFv MC1 antibody efficiently reduces pathological tau species in adult JNPL3 mice. Acta Neuropathol. Commun. 2018, 6, 82. [CrossRef]
- Smolek, T.; Jadhav, S.; Brezovakova, V.; Cubinkova, V.; Valachova, B.; Novak, P.; Zilka, N. First-in-Rat Study of Human Alzheimer’s Disease Tau Propagation. Mol. Neurobiol. 2018, 56, 621–631. [CrossRef]
- Ferrer, I.; Zelaya, M.V.; García, M.A.; Carmona, M.; López-González, I.; Andrés-Benito, P.; Lidón, L.; Gavín, R.; Garcia-Esparcia, P.; del Rio, J.A. Relevance of host tau in tau seeding and spreading in tauopathies. Brain Pathol. 2020, 30, 298–318. [CrossRef]
- Ferrer, I.; García, M.A.; Carmona, M.; Andrés-Benito, P.; Torrejón-Escribano, B.; Garcia-Esparcia, P.; del Rio, J.A. Involvement of Oligodendrocytes in Tau Seeding and Spreading in Tauopathies. Front. Aging Neurosci. 2019, 11, 112. [CrossRef]
- Masuda-Suzukake, M.; Suzuki, G.; Hosokawa, M.; Nonaka, T.; Goedert, M.; Hasegawa, M. Dextran sulphate-induced tau assemblies cause endogenous tau aggregation and propagation in wild-type mice. Brain Commun. 2020, 2. [CrossRef]
- Mezias, C.; Rey, N.; Brundin, P.; Raj, A. Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: Involvement of retrograde and anterograde axonal propagation. Neurobiol. Dis. 2019, 134, 104623–104623. [CrossRef]
- Veys, L.; Van Houcke, J.; Aerts, J.; Van Pottelberge, S.; Mahieu, M.; Coens, A.; Melki, R.; Moechars, D.; De Muynck, L.; De Groef, L. Absence of Uptake and Prion-Like Spreading of Alpha-Synuclein and Tau After Intravitreal Injection of Preformed Fibrils. Front. Aging Neurosci. 2021, 12. [CrossRef]
- Courte, J.; Bousset, L.; Von Boxberg, Y.; Villard, C.; Melki, R.; Peyrin, J.-M. The expression level of alpha-synuclein in different neuronal populations is the primary determinant of its prion-like seeding. Sci. Rep. 2020, 10, 1–13. [CrossRef]
- Jimenez-Ferrer, I.; Bäckström, F.; Dueñas-Rey, A.; Jewett, M.; Boza-Serrano, A.; Luk, K.C.; Deierborg, T.; Swanberg, M. The MHC class II transactivator modulates seeded alpha-synuclein pathology and dopaminergic neurodegeneration in an in vivo rat model of Parkinson’s disease. Brain, Behav. Immun. 2020, 91, 369–382. [CrossRef]
- Thomsen, M.B.; Ferreira, S.A.; Schacht, A.C.; Jacobsen, J.; Simonsen, M.; Betzer, C.; Jensen, P.H.; Brooks, D.J.; Landau, A.M.; Romero-Ramos, M. PET imaging reveals early and progressive dopaminergic deficits after intra-striatal injection of preformed alpha-synuclein fibrils in rats. Neurobiol. Dis. 2020, 149, 105229. [CrossRef]
- Dutta D, Jana M, Majumder M, Mondal S, Roy A, Pahan K. Selective targeting of the TLR2/MyD88/NF-κB pathway reduces α-synuclein spreading in vitro and in vivo. Nat Commun. 2021;12(1):5382. [CrossRef]
- Bassil, F.; Meymand, E.S.; Brown, H.J.; Xu, H.; Cox, T.O.; Pattabhiraman, S.; Maghames, C.M.; Wu, Q.; Zhang, B.; Trojanowski, J.Q.; et al. α-Synuclein modulates tau spreading in mouse brains. J. Exp. Med. 2020, 218. [CrossRef]
- Matsuo, K.; Kawahata, I.; Melki, R.; Bousset, L.; Owada, Y.; Fukunaga, K. Suppression of α-synuclein propagation after intrastriatal injection in FABP3 null mice. Brain Res. 2021, 1760, 147383. [CrossRef]
- Pan L, Li C, Meng L, Tian Y, He M, Yuan X, et al. Tau accelerates α-synuclein aggregation and spreading in Parkinson’s disease. Brain. 2022;145(10):3454-71. [CrossRef]
- Dutta D, Paidi RK, Raha S, Roy A, Chandra S, Pahan K. Treadmill exercise reduces α-synuclein spreading via PPARα. Cell Rep. 2022;40(2):111058. [CrossRef]
- Garcia P, Jürgens-Wemheuer W, Uriarte Huarte O, Michelucci A, Masuch A, Brioschi S, et al. Neurodegeneration and neuroinflammation are linked, but independent of alpha-synuclein inclusions, in a seeding/spreading mouse model of Parkinson’s disease. Glia. 2022;70(5):935-60. [CrossRef]
- Vasili, E.; Dominguez-Meijide, A.; Flores-León, M.; Al-Azzani, M.; Kanellidi, A.; Melki, R.; Stefanis, L.; Outeiro, T.F. Endogenous Levels of Alpha-Synuclein Modulate Seeding and Aggregation in Cultured Cells. Mol. Neurobiol. 2022, 59, 1273–1284. [CrossRef]
- Sun, F.; Salinas, A.G.; Filser, S.; Blumenstock, S.; Medina-Luque, J.; Herms, J.; Sgobio, C. Impact of α-synuclein spreading on the nigrostriatal dopaminergic pathway depends on the onset of the pathology. Brain Pathol. 2021, 32, e13036. [CrossRef]
- Rahayel, S.; Mišić, B.; Zheng, Y.-Q.; Liu, Z.-Q.; Abdelgawad, A.; Abbasi, N.; Caputo, A.; Zhang, B.; Lo, A.; Kehm, V.; et al. Differentially targeted seeding reveals unique pathological alpha-synuclein propagation patterns. Brain 2021, 145, 1743–1756. [CrossRef]
- Lloyd, G.M.; Sorrentino, Z.A.; Quintin, S.; Gorion, K.-M.M.; Bell, B.M.; Paterno, G.; Long, B.; Prokop, S.; Giasson, B.I. Unique seeding profiles and prion-like propagation of synucleinopathies are highly dependent on the host in human α-synuclein transgenic mice. Acta Neuropathol. 2022, 143, 663–685. [CrossRef]
- Jain, N.; Lewis, C.A.; Ulrich, J.D.; Holtzman, D.M. Chronic TREM2 activation exacerbates Aβ-associated tau seeding and spreading. J. Exp. Med. 2022, 220. [CrossRef]
- De Gerando, A.M.; Welikovitch, L.A.; Khasnavis, A.; Commins, C.; Glynn, C.; Chun, J.E.; Perbet, R.; Hyman, B.T. Tau seeding and spreading in vivo is supported by both AD-derived fibrillar and oligomeric tau. Acta Neuropathol. 2023, 146, 191–210. [CrossRef]
- Deng H, Wang P, Jankovic J. The genetics of Parkinson disease. Ageing Res Rev. 2018;42:72-85. [CrossRef]
- Schulte C, Gasser T. Genetic basis of Parkinson’s disease: inheritance, penetrance, and expression. Appl Clin Genet. 2011;4:67-80. [CrossRef]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [CrossRef]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [CrossRef]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [CrossRef]
- Song, H.; Chen, J.; Huang, J.; Sun, P.; Liu, Y.; Xu, L.; Wei, C.; Mu, X.; Lu, X.; Wang, W.; et al. Epigenetic modification in Parkinson’s disease. Front. Cell Dev. Biol. 2023, 11, 1123621. [CrossRef]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear Localization of alpha-Synuclein and Its Interaction with Histones. Biochemistry 2003, 42, 8465–8471. [CrossRef]
- Mouradian, M.M. MicroRNAs in Parkinson’s disease. Neurobiol. Dis. 2012, 46, 279–284. [CrossRef]
- McMillan, K.J.; Murray, T.K.; Bengoa-Vergniory, N.; Cordero-Llana, O.; Cooper, J.; Buckley, A.; Wade-Martins, R.; Uney, J.B.; O’neill, M.J.; Wong, L.F.; et al. Loss of MicroRNA-7 Regulation Leads to α-Synuclein Accumulation and Dopaminergic Neuronal Loss In Vivo. Mol. Ther. 2017, 25, 2404–2414. [CrossRef]
- Doxakis, E. Post-transcriptional Regulation of alpha-Synuclein Expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [CrossRef]
- Tan, Y.-Y.; Wu, L.; Zhao, Z.-B.; Wang, Y.; Xiao, Q.; Liu, J.; Wang, G.; Ma, J.-F.; Chen, S.-D. Methylation of α-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Park. Relat. Disord. 2014, 20, 308–313. [CrossRef]
- de Boni, L.; Riedel, L.; Schmitt, I.; Kraus, T.F.; Kaut, O.; Piston, D.; Akbarian, S.; Wüllner, U. DNA methylation levels of α-synuclein intron 1 in the aging brain. Neurobiol. Aging 2015, 36, 3334.e7–3334.e11. [CrossRef]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [CrossRef]
- Masliah E, Dumaop W, Galasko D, Desplats P. Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics. 2013;8(10):1030-8. [CrossRef]
- Wang, Y.; Liu, H.; Zhang, B.-S.; Soares, J.C.; Zhang, X.Y. Low BDNF is associated with cognitive impairments in patients with Parkinson’s disease. Park. Relat. Disord. 2016, 29, 66–71. [CrossRef]
- Habibi, E.; Masoudi-Nejad, A.; Abdolmaleky, H.M.; Haggarty, S.J. Emerging roles of epigenetic mechanisms in Parkinson’s disease. Funct. Integr. Genom. 2011, 11, 523–537. [CrossRef]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, et al. DNA Methylation-Related Chromatin Remodeling in Activity-Dependent Bdnf Gene Regulation. Science. 2003;302(5646):890-3. [CrossRef]
- Howells, D.; Porritt, M.; Wong, J.; Batchelor, P.; Kalnins, R.; Hughes, A.; Donnan, G. Reduced BDNF mRNA Expression in the Parkinson’s Disease Substantia Nigra. Exp. Neurol. 2000, 166, 127–135. [CrossRef]
- Muñoz, P.C.; A Aspé, M.; Contreras, L.S.; Palacios, A.G. Correlations of recognition memory performance with expression and methylation of brain-derived neurotrophic factor in rats. Biol. Res. 2010, 43, 251–258. [CrossRef]
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28(42):10576-86. [CrossRef]
- Yang, X.; Xu, S.; Qian, Y.; He, X.; Chen, S.; Xiao, Q. Hypermethylation of the Gene Coding for PGC-1α in Peripheral Blood Leukocytes of Patients With Parkinson’s Disease. Front. Neurosci. 2020, 14, 97. [CrossRef]
- Su X, Chu Y, Kordower JH, Li B, Cao H, Huang L, et al. PGC-1α Promoter Methylation in Parkinson’s Disease. PLoS One. 2015;10(8):e0134087. [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [CrossRef]
- Zhao Y, Zhang J, Zheng Y, Zhang Y, Zhang XJ, Wang H, et al. NAD(+) improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1α pathway. J Neuroinflammation. 2021;18(1):207. [CrossRef]
- Han, B.; Jiang, W.; Liu, H.; Wang, J.; Zheng, K.; Cui, P.; Feng, Y.; Dang, C.; Bu, Y.; Wang, Q.M.; et al. Upregulation of neuronal PGC-1 α ameliorates cognitive impairment induced by chronic cerebral hypoperfusion. Theranostics 2020, 10, 2832–2848. [CrossRef]
- Coupland, K.G.; Mellick, G.D.; Silburn, P.A.; Mather, K.; Armstrong, N.J.; Sachdev, P.S.; Brodaty, H.; Huang, Y.; Halliday, G.M.; Hallupp, M.; et al. DNA methylation of the MAPT gene in Parkinson’s disease cohorts and modulation by vitamin E In Vitro. Mov. Disord. 2013, 29, 1606–1614. [CrossRef]
- Setó-Salvia N, Clarimón J, Pagonabarraga J, Pascual-Sedano B, Campolongo A, Combarros O, et al. Dementia risk in Parkinson disease: disentangling the role of MAPT haplotypes. Arch Neurol. 2011;68(3):359-64. [CrossRef]
- Winder-Rhodes, S.E.; Hampshire, A.; Rowe, J.B.; Peelle, J.E.; Robbins, T.W.; Owen, A.M.; Barker, R.A. Association between MAPT haplotype and memory function in patients with Parkinson’s disease and healthy aging individuals. Neurobiol. Aging 2014, 36, 1519–1528. [CrossRef]
- Tunold, J.-A.; Geut, H.; Rozemuller, J.M.A.; Henriksen, S.P.; Toft, M.; van de Berg, W.D.J.; Pihlstrøm, L. APOE and MAPT Are Associated With Dementia in Neuropathologically Confirmed Parkinson’s Disease. Front. Neurol. 2021, 12. [CrossRef]
- Jiskoot, L.C.; Panman, J.L.; van Asseldonk, L.; Franzen, S.; Meeter, L.H.H.; Kaat, L.D.; van der Ende, E.L.; Dopper, E.G.P.; Timman, R.; van Minkelen, R.; et al. Longitudinal cognitive biomarkers predicting symptom onset in presymptomatic frontotemporal dementia. J. Neurol. 2018, 265, 1381–1392. [CrossRef]
- Pal S, Tyler JK. Epigenetics and aging. Sci Adv. 2016;2(7):e1600584. [CrossRef]
- Levenson, J.M.; Sweatt, J.D. Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 2005, 6, 108–118. [CrossRef]
- Hwang, J.-Y.; Aromolaran, K.A.; Zukin, R.S. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [CrossRef]
- Federman N, de la Fuente V, Zalcman G, Corbi N, Onori A, Passananti C, et al. Nuclear factor κB-dependent histone acetylation is specifically involved in persistent forms of memory. J Neurosci. 2013;33(17):7603-14. [CrossRef]
- de la Fuente V, Federman N, Zalcman G, Salles A, Freudenthal R, Romano A. NF-κB transcription factor role in consolidation and reconsolidation of persistent memories. Front Mol Neurosci. 2015;8:50. [CrossRef]
- Basavarajappa, B.S.; Subbanna, S. Histone Methylation Regulation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2021, 22, 4654. [CrossRef]
- Chatterjee, P.; Roy, D.; Bhattacharyya, M.; Bandyopadhyay, S. Biological networks in Parkinson’s disease: an insight into the epigenetic mechanisms associated with this disease. BMC Genom. 2017, 18, 721. [CrossRef]
- Morse, S.J.; Butler, A.A.; Davis, R.L.; Soller, I.J.; Lubin, F.D. Environmental Enrichment Reverses Histone Methylation Changes in the Aged Hippocampus and Restores Age-Related Memory Deficits. Biology 2015, 4, 298–313. [CrossRef]
- Gupta-Agarwal, S.; Jarome, T.J.; Fernandez, J.; Lubin, F.D. NMDA receptor- and ERK-dependent histone methylation changes in the lateral amygdala bidirectionally regulate fear memory formation. Learn. Mem. 2014, 21, 351–362. [CrossRef]
- Gupta-Agarwal, S.; Franklin, A.V.; DeRamus, T.; Wheelock, M.; Davis, R.L.; McMahon, L.L.; Lubin, F.D. G9a/GLP Histone Lysine Dimethyltransferase Complex Activity in the Hippocampus and the Entorhinal Cortex Is Required for Gene Activation and Silencing during Memory Consolidation. J. Neurosci. 2012, 32, 5440–5453. [CrossRef]
- Riddle, N.C.; Minoda, A.; Kharchenko, P.V.; Alekseyenko, A.A.; Schwartz, Y.B.; Tolstorukov, M.Y.; Gorchakov, A.A.; Jaffe, J.D.; Kennedy, C.; Linder-Basso, D.; et al. Plasticity in patterns of histone modifications and chromosomal proteins in Drosophila heterochromatin. Genome Res. 2010, 21, 147–163. [CrossRef]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [CrossRef]
- Gupta, S.; Kim, S.Y.; Artis, S.; Molfese, D.L.; Schumacher, A.; Sweatt, J.D.; Paylor, R.E.; Lubin, F.D. Histone Methylation Regulates Memory Formation. J. Neurosci. 2010, 30, 3589–3599. [CrossRef]
- Södersten, E.; Feyder, M.; Lerdrup, M.; Gomes, A.-L.; Kryh, H.; Spigolon, G.; Caboche, J.; Fisone, G.; Hansen, K. Dopamine Signaling Leads to Loss of Polycomb Repression and Aberrant Gene Activation in Experimental Parkinsonism. PLOS Genet. 2014, 10, e1004574. [CrossRef]
- Gibney E, Nolan C. Epigenetics and gene expression. Heredity. 2010;105(1):4-13.
- Mu, M.-D.; Qian, Z.-M.; Yang, S.-X.; Rong, K.-L.; Yung, W.-H.; Ke, Y. Therapeutic effect of a histone demethylase inhibitor in Parkinson’s disease. Cell Death Dis. 2020, 11, 1–16. [CrossRef]
- Mullen, R.J.; Buck, C.R.; Smith, A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Development 1992, 116, 201–211. [CrossRef]
- Kim, K.K.; Adelstein, R.S.; Kawamoto, S. Identification of Neuronal Nuclei (NeuN) as Fox-3, a New Member of the Fox-1 Gene Family of Splicing Factors. J. Biol. Chem. 2009, 284, 31052–31061. [CrossRef]
- Guhathakurta S, Kim J, Adams L, Basu S, Song MK, Adler E, et al. Targeted attenuation of elevated histone marks at SNCA alleviates α-synuclein in Parkinson’s disease. EMBO Molecular Medicine. 2021;13(2):e12188. [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [CrossRef]
- Srivastav, S.; Walitza, S.; Grünblatt, E. Emerging role of miRNA in attention deficit hyperactivity disorder: a systematic review. Adhd Atten. Deficit Hyperact. Disord. 2017, 10, 49–63. [CrossRef]
- Tang, G.-B.; Zeng, Y.-Q.; Liu, P.-P.; Mi, T.-W.; Zhang, S.-F.; Dai, S.-K.; Tang, Q.-Y.; Yang, L.; Xu, Y.-J.; Yan, H.-L.; et al. The Histone H3K27 Demethylase UTX Regulates Synaptic Plasticity and Cognitive Behaviors in Mice. Front. Mol. Neurosci. 2017, 10, 267–267. [CrossRef]
- Harrison, I.F.; Smith, A.D.; Dexter, D.T. Pathological histone acetylation in Parkinson’s disease: Neuroprotection and inhibition of microglial activation through SIRT 2 inhibition. Neurosci. Lett. 2017, 666, 48–57. [CrossRef]
- Hait, N.C.; E Wise, L.; Allegood, J.C.; O’Brien, M.; Avni, D.; Reeves, T.M.; E Knapp, P.; Lu, J.; Luo, C.; Miles, M.F.; et al. Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory. Nat. Neurosci. 2014, 17, 971–980. [CrossRef]
- Peixoto, L.; Abel, T. The Role of Histone Acetylation in Memory Formation and Cognitive Impairments. Neuropsychopharmacology 2012, 38, 62–76. [CrossRef]
- Huang, M.; Malovic, E.; Ealy, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Microglial immune regulation by epigenetic reprogramming through histone H3K27 acetylation in neuroinflammation. Front. Immunol. 2023, 14. [CrossRef]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(7):522-31. [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [CrossRef]
- Duffy, C.P.; McCoy, C.E. The Role of MicroRNAs in Repair Processes in Multiple Sclerosis. Cells 2020, 9, 1711. [CrossRef]
- Zetterberg, H.; Burnham, S.C. Blood-based molecular biomarkers for Alzheimer’s disease. Mol. Brain 2019, 12, 1–7. [CrossRef]
- Prabhakar P, Chandra SR, Christopher R. Circulating microRNAs as potential biomarkers for the identification of vascular dementia due to cerebral small vessel disease. Age Ageing. 2017;46(5):861-4. [CrossRef]
- Nadim WD, Simion V, Benedetti H, Pichon C, Baril P, Morisset-Lopez S. MicroRNAs in Neurocognitive Dysfunctions: New Molecular Targets for Pharmacological Treatments? Curr Neuropharmacol. 2017;15(2):260-75.
- Miquel, S.; Champ, C.; Day, J.; Aarts, E.; Bahr, B.A.; Bakker, M.; Bánáti, D.; Calabrese, V.; Cederholm, T.; Cryan, J.; et al. Poor cognitive ageing: Vulnerabilities, mechanisms and the impact of nutritional interventions. Ageing Res. Rev. 2018, 42, 40–55. [CrossRef]
- Aarsland D, Creese B, Politis M, Chaudhuri KR, Ffytche DH, Weintraub D, et al. Cognitive decline in Parkinson disease. Nat Rev Neurol. 2017;13(4):217-31. [CrossRef]
- Junn, E.; Lee, K.-W.; Jeong, B.S.; Chan, T.W.; Im, J.-Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [CrossRef]
- Zhou, Y.; Lu, M.; Du, R.-H.; Qiao, C.; Jiang, C.-Y.; Zhang, K.-Z.; Ding, J.-H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 1–15. [CrossRef]
- Yao, G.-Y.; Zhu, Q.; Xia, J.; Chen, F.-J.; Huang, M.; Liu, J.; Zhou, T.-T.; Wei, J.-F.; Cui, G.-Y.; Zheng, K.-Y.; et al. Ischemic postconditioning confers cerebroprotection by stabilizing VDACs after brain ischemia. Cell Death Dis. 2018, 9, 1–15. [CrossRef]
- Li, S.; Lv, X.; Zhai, K.; Xu, R.; Zhang, Y.; Zhao, S.; Qin, X.; Yin, L.; Lou, J. MicroRNA-7 inhibits neuronal apoptosis in a cellular Parkinson’s disease model by targeting Bax and Sirt2. Am J Transl Res. 2016;8(2):993-1004.
- Geng, L.; Liu, W.; Chen, Y. miR-124-3p attenuates MPP+-induced neuronal injury by targeting STAT3 in SH-SY5Y cells. Exp. Biol. Med. 2017, 242, 1757–1764. [CrossRef]
- Han, J.; Pu, C.-X.; Xiao, Q.-X.; Tang, L.-J.; Liu, T.; He, L.; Ren, Y.-B.; Liu, Q.; Zhang, Y. miRNA-124-3p targeting of LPIN1 attenuates inflammation and apoptosis in aged male rats cardiopulmonary bypass model of perioperative neurocognitive disorders. Exp. Gerontol. 2021, 155, 111578. [CrossRef]
- Gan, L.; Li, Z.; Lv, Q.; Huang, W. Rabies virus glycoprotein (RVG29)-linked microRNA-124-loaded polymeric nanoparticles inhibit neuroinflammation in a Parkinson’s disease model. Int. J. Pharm. 2019, 567, 118449. [CrossRef]
- Xing RX, Li LG, Liu XW, Tian BX, Cheng Y. Down regulation of miR-218, miR-124, and miR-144 relates to Parkinson’s disease via activating NF-κB signaling. Kaohsiung J Med Sci. 2020;36(10):786-92. [CrossRef]
- Pantano, L.; Friedländer, M.R.; Escaramís, G.; Lizano, E.; Pallarès-Albanell, J.; Ferrer, I.; Estivill, X.; Martí, E. Specific small-RNA signatures in the amygdala at premotor and motor stages of Parkinson’s disease revealed by deep sequencing analysis. Bioinformatics 2015, 32, 673–681. [CrossRef]
- Rajasethupathy, P.; Fiumara, F.; Sheridan, R.; Betel, D.; Puthanveettil, S.V.; Russo, J.J.; Sander, C.; Tuschl, T.; Kandel, E. Characterization of Small RNAs in Aplysia Reveals a Role for miR-124 in Constraining Synaptic Plasticity through CREB. Neuron 2009, 63, 803–817. [CrossRef]
- Makeyev EV, Zhang J, Carrasco MA, Maniatis T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell. 2007;27(3):435-48.
- Konopka, W.; Kiryk, A.; Novak, M.; Herwerth, M.; Parkitna, J.R.; Wawrzyniak, M.; Kowarsch, A.; Michaluk, P.; Dzwonek, J.; Arnsperger, T.; et al. MicroRNA Loss Enhances Learning and Memory in Mice. J. Neurosci. 2010, 30, 14835–14842. [CrossRef]
- Kang, Q.; Xiang, Y.; Li, D.; Liang, J.; Zhang, X.; Zhou, F.; Qiao, M.; Nie, Y.; He, Y.; Cheng, J.; et al. MiR-124-3p attenuates hyperphosphorylation of tau protein-induced apoptosis via caveolin-1-PI3K/Akt/GSK3β pathway in N2a/APP695swe cells. Oncotarget 2017, 8, 24314–24326. [CrossRef]
- Bartsch, D.; Casadio, A.; A Karl, K.; Serodio, P.; Kandel, E.R. CREB1 Encodes a Nuclear Activator, a Repressor, and a Cytoplasmic Modulator that Form a Regulatory Unit Critical for Long-Term Facilitation. Cell 1998, 95, 211–223. [CrossRef]
- Yang, Y.; Shu, X.; Liu, D.; Shang, Y.; Wu, Y.; Pei, L.; Xu, X.; Tian, Q.; Zhang, J.; Qian, K.; et al. EPAC Null Mutation Impairs Learning and Social Interactions via Aberrant Regulation of miR-124 and Zif268 Translation. Neuron 2012, 73, 774–788. [CrossRef]
- Blount, G.S.; Coursey, L.; Kocerha, J. MicroRNA Networks in Cognition and Dementia. Cells 2022, 11, 1882. [CrossRef]
- Ren Y, Li H, Xie W, Wei N, Liu M. MicroRNA-195 triggers neuroinflammation in Parkinson’s disease in a Rho-associated kinase 1-dependent manner. Mol Med Rep. 2019;19(6):5153-61. [CrossRef]
- Ai, J.; Sun, L.-H.; Che, H.; Zhang, R.; Zhang, T.-Z.; Wu, W.-C.; Su, X.-L.; Chen, X.; Yang, G.; Li, K.; et al. MicroRNA-195Protects Against Dementia Induced by Chronic Brain Hypoperfusion via Its Anti-Amyloidogenic Effect in Rats. J. Neurosci. 2013, 33, 3989–4001. [CrossRef]
- Sun LH, Ban T, Liu CD, Chen QX, Wang X, Yan ML, et al. Activation of Cdk5/p25 and Tau phosphorylation following chronic brain hypoperfusion in rats involves microRNA-195 down-regulation. J Neurochem. 2015;134(6):1139-51. [CrossRef]
- Fischer, A.; Sananbenesi, F.; Schrick, C.; Spiess, J.; Radulovic, J. Cyclin-Dependent Kinase 5 Is Required for Associative Learning. J. Neurosci. 2002, 22, 3700–3707. [CrossRef]
- Junker, A.; Krumbholz, M.; Eisele, S.; Mohan, H.; Augstein, F.; Bittner, R.; Lassmann, H.; Wekerle, H.; Hohlfeld, R.; Meinl, E. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain 2009, 132, 3342–3352. [CrossRef]
- Sun, Q.; Wang, S.; Chen, J.; Cai, H.; Huang, W.; Zhang, Y.; Wang, L.; Xing, Y. MicroRNA-190 alleviates neuronal damage and inhibits neuroinflammation via Nlrp3 in MPTP-induced Parkinson’s disease mouse model. J. Cell. Physiol. 2019, 234, 23379–23387. [CrossRef]
- Liu, Q.; Hou, A.; Zhang, Y.; Guo, Y.; Li, J.; Yao, Y.; Niu, K.; Li, H.; Ma, Y.; Cao, J. MiR-190a potentially ameliorates postoperative cognitive dysfunction by regulating Tiam1. BMC Genom. 2019, 20, 1–12. [CrossRef]
- Jiang, C.; Dong, N.; Feng, J.; Hao, M. MiRNA-190 exerts neuroprotective effects against ischemic stroke through Rho/Rho-kinase pathway. Pfl?gers Arch. Eur. J. Physiol. 2020, 473, 121–130. [CrossRef]
- Iyer, A.; Zurolo, E.; Prabowo, A.; Fluiter, K.; Spliet, W.G.M.; van Rijen, P.C.; Gorter, J.A.; Aronica, E. MicroRNA-146a: A Key Regulator of Astrocyte-Mediated Inflammatory Response. PLOS ONE 2012, 7, e44789. [CrossRef]
- Lukiw WJ, Dua P, Pogue AI, Eicken C, Hill JM. Upregulation of micro RNA-146a (miRNA-146a), a marker for inflammatory neurodegeneration, in sporadic Creutzfeldt-Jakob disease (sCJD) and Gerstmann-Straussler-Scheinker (GSS) syndrome. J Toxicol Environ Health A. 2011;74(22-24):1460-8. [CrossRef]
- Alexandrov, P.N.; Dua, P.; Hill, J.M.; Bhattacharjee, S.; Zhao, Y.; Lukiw, W.J. microRNA (miRNA) speciation in Alzheimer’s disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int. J. Biochem. Mol. Biol. 2012, 3, 365–373.
- Kim, J.-H.; Jou, I.; Joe, I.J.A.E.-H. Suppression of miR-155 Expression in IFN-γ-Treated Astrocytes and Microglia by DJ-1: A Possible Mechanism for Maintaining SOCS1 Expression. Exp. Neurobiol. 2014, 23, 148–154. [CrossRef]
- Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology. 2012;135(1):73-88. [CrossRef]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA Feedback Circuit in Midbrain Dopamine Neurons. Science 2007, 317, 1220–1224. [CrossRef]
- Zhang, Y.; Liu, J.; Xie, C.; Wu, P. Overexpression of miR-133b protects against isoflurane-induced learning and memory impairment. Exp. Ther. Med. 2021, 22, 1–7. [CrossRef]
- Yang, Q.; Zhao, Q.; Yin, Y. miR-133b is a potential diagnostic biomarker for Alzheimer’s disease and has a neuroprotective role. Exp. Ther. Med. 2019, 18, 2711–2718. [CrossRef]
- Schulz J, Takousis P, Wohlers I, Itua IOG, Dobricic V, Rücker G, et al. Meta-analyses identify differentially expressed micrornas in Parkinson’s disease. Ann Neurol. 2019;85(6):835-51. [CrossRef]
- Schlaudraff, F.; Gründemann, J.; Fauler, M.; Dragicevic, E.; Hardy, J.; Liss, B. Orchestrated increase of dopamine and PARK mRNAs but not miR-133b in dopamine neurons in Parkinson’s disease. Neurobiol. Aging 2014, 35, 2302–2315. [CrossRef]
- Villar-Menéndez, I.; Porta, S.; Buira, S.P.; Pereira-Veiga, T.; Díaz-Sánchez, S.; Albasanz, J.L.; Ferrer, I.; Martín, M.; Barrachina, M. Increased striatal adenosine A2A receptor levels is an early event in Parkinson’s disease-related pathology and it is potentially regulated by miR-34b. Neurobiol. Dis. 2014, 69, 206–214. [CrossRef]
- Kim, W.; Lee, Y.; McKenna, N.D.; Yi, M.; Simunovic, F.; Wang, Y.; Kong, B.; Rooney, R.J.; Seo, H.; Stephens, R.M.; et al. miR-126 contributes to Parkinson’s disease by dysregulating the insulin-like growth factor/phosphoinositide 3-kinase signaling. Neurobiol. Aging 2014, 35, 1712–1721. [CrossRef]
- Briggs, C.E.; Wang, Y.; Kong, B.; Woo, T.-U.W.; Iyer, L.K.; Sonntag, K.C. Midbrain dopamine neurons in Parkinson׳s disease exhibit a dysregulated miRNA and target-gene network. Brain Res. 2015, 1618, 111–121. [CrossRef]
- Kong, F.; Zhou, J.; Zhou, W.; Guo, Y.; Li, G.; Yang, L. Protective role of microRNA-126 in intracerebral hemorrhage. Mol. Med. Rep. 2017, 15, 1419–1425. [CrossRef]
- Yu, P.; Venkat, P.; Chopp, M.; Zacharek, A.; Shen, Y.; Ning, R.; Liang, L.; Li, W.; Zhang, L.; Landschoot-Ward, J.; et al. Role of microRNA-126 in vascular cognitive impairment in mice. J. Cereb. Blood Flow Metab. 2018, 39, 2497–2511. [CrossRef]
- Hoss, A.G.; Labadorf, A.; Beach, T.G.; Latourelle, J.C.; Myers, R.H. microRNA Profiles in Parkinson’s Disease Prefrontal Cortex. Front. Aging Neurosci. 2016, 8, 36. [CrossRef]
- Tatura, R.; Kraus, T.; Giese, A.; Arzberger, T.; Buchholz, M.; Höglinger, G.; Müller, U. Parkinson’s disease: SNCA-, PARK2-, and LRRK2- targeting microRNAs elevated in cingulate gyrus. Park. Relat. Disord. 2016, 33, 115–121. [CrossRef]
- Sun, L.; Zhao, M.; Zhang, J.; Liu, A.; Ji, W.; Li, Y.; Yang, X.; Wu, Z. MiR-144 promotes β-amyloid accumulation-induced cognitive impairments by targeting ADAM10 following traumatic brain injury. Oncotarget 2017, 8, 59181–59203. [CrossRef]
- Wang, J.; Chen, T.; Shan, G. miR-148b Regulates Proliferation and Differentiation of Neural Stem Cells via Wnt/β-Catenin Signaling in Rat Ischemic Stroke Model. Front. Cell. Neurosci. 2017, 11, 329–329. [CrossRef]
- Yang, H.; Zhang, Y.; Chen, H.; Zhu, Y.; Li, Y.; Ouyang, F.; Chu, L.; Liu, D. Mir-184 Contributes to Brain Injury Through Targeting PPAP2B Following Ischemic Stroke in Male Rats. Front. Mol. Neurosci. 2021, 14. [CrossRef]
- Mendes-Silva, A.P.; Fujimura, P.T.; Silva, J.R.d.C.; Teixeira, A.L.; Vieira, E.M.; Guedes, P.H.G.; Barroso, L.S.S.; Nicolau, M.d.S.; Ferreira, J.D.R.; Bertola, L.; et al. Brain-enriched MicroRNA-184 is downregulated in older adults with major depressive disorder: A translational study. J. Psychiatr. Res. 2019, 111, 110–120. [CrossRef]
- McKiernan, R.C.; Jimenez-Mateos, E.M.; Sano, T.; Bray, I.; Stallings, R.L.; Simon, R.P.; Henshall, D.C. Expression profiling the microRNA response to epileptic preconditioning identifies miR-184 as a modulator of seizure-induced neuronal death. Exp. Neurol. 2012, 237, 346–354. [CrossRef]
- Shan, Y.; Hu, J.; Lv, H.; Cui, X.; Di, W. miR-221 Exerts Neuroprotective Effects in Ischemic Stroke by Inhibiting the Proinflammatory Response. J. Stroke Cerebrovasc. Dis. 2020, 30, 105489. [CrossRef]
- Song, D.; Li, G.; Hong, Y.; Zhang, P.; Zhu, J.; Yang, L.; Huang, J. miR-199a decreases Neuritin expression involved in the development of Alzheimer’s disease in APP/PS1 mice. Int. J. Mol. Med. 2020, 46, 384–396. [CrossRef]
- Tolosa E, Botta-Orfila T, Morató X, Calatayud C, Ferrer-Lorente R, Martí MJ, et al. MicroRNA alterations in iPSC-derived dopaminergic neurons from Parkinson disease patients. Neurobiol Aging. 2018;69:283-91. [CrossRef]
- Nakahama T, Hanieh H, Nguyen NT, Chinen I, Ripley B, Millrine D, et al. Aryl hydrocarbon receptor-mediated induction of the microRNA-132/212 cluster promotes interleukin-17–producing T-helper cell differentiation. Proceedings of the National Academy of Sciences. 2013;110(29):11964-9. [CrossRef]
- Wayman, G.A.; Davare, M.; Ando, H.; Fortin, D.; Varlamova, O.; Cheng, H.-Y.M.; Marks, D.; Obrietan, K.; Soderling, T.R.; Goodman, R.H.; et al. An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc. Natl. Acad. Sci. USA 2008, 105, 9093–9098. [CrossRef]
- Cong, L.; Cong, Y.; Feng, N.; Liang, W.; Wu, Y. Up-regulated microRNA-132 reduces the cognition-damaging effect of sevoflurane on Alzheimer’s disease rats by inhibiting FOXA1. Genomics 2021, 113, 3644–3652. [CrossRef]
- Deng, Y.; Zhang, J.; Sun, X.; Ma, G.; Luo, G.; Miao, Z.; Song, L. miR-132 improves the cognitive function of rats with Alzheimer’s disease by inhibiting the MAPK1 signal pathway. Exp. Ther. Med. 2020, 20, 1–1. [CrossRef]
- Hansen, K.F.; Karelina, K.; Sakamoto, K.; Wayman, G.A.; Impey, S.; Obrietan, K. miRNA-132: a dynamic regulator of cognitive capacity. Brain Structure and Function. 2013;218(3):817-31. [CrossRef]
- Wang RY, Phang RZ, Hsu PH, Wang WH, Huang HT, Liu IY. In vivo knockdown of hippocampal miR-132 expression impairs memory acquisition of trace fear conditioning. Hippocampus. 2013;23(7):625-33.
- Gong, X.; Huang, M.; Chen, L. Mechanism of miR-132-3p Promoting Neuroinflammation and Dopaminergic Neurodegeneration in Parkinson’s Disease. eneuro 2021, 9, 1–17. [CrossRef]
- Clovis, Y.M.; Enard, W.; Marinaro, F.; Huttner, W.B.; De Pietri Tonelli, D. Convergent repression of Foxp2 3′UTR by miR-9 and miR-132 in embryonic mouse neocortex: implications for radial migration of neurons. Development 2012, 139, 3332–3342. [CrossRef]
- Gascon, E.; Lynch, K.; Ruan, H.; Almeida, S.; Verheyden, J.M.; Seeley, W.W.; Dickson, D.W.; Petrucelli, L.; Sun, D.; Jiao, J.; et al. Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med. 2014, 20, 1444–1451. [CrossRef]
- Pan, S.; Feng, W.; Li, Y.; Huang, J.; Chen, S.; Cui, Y.; Tian, B.; Tan, S.; Wang, Z.; Yao, S.; et al. The microRNA-195 - BDNF pathway and cognitive deficits in schizophrenia patients with minimal antipsychotic medication exposure. Transl. Psychiatry 2021, 11, 1–8. [CrossRef]
- Kondratyev, A.; Gale, K. Intracerebral injection of caspase-3 inhibitor prevents neuronal apoptosis after kainic acid-evoked status epilepticus. Mol. Brain Res. 2000, 75, 216–224. [CrossRef]
- Caggiu, E.; Paulus, K.; Mameli, G.; Arru, G.; Sechi, G.P.; Sechi, L.A. Differential expression of miRNA 155 and miRNA 146a in Parkinson’s disease patients. eNeurologicalSci 2018, 13, 1–4. [CrossRef]
- Chen, L.; Dong, R.; Lu, Y.; Zhou, Y.; Li, K.; Zhang, Z.; Peng, M. MicroRNA-146a protects against cognitive decline induced by surgical trauma by suppressing hippocampal neuroinflammation in mice. Brain, Behav. Immun. 2019, 78, 188–201. [CrossRef]
- Zhan-Qiang H, Hai-Hua Q, Chi Z, Miao W, Cui Z, Zi-Yin L, et al. miR-146a aggravates cognitive impairment and Alzheimer disease-like pathology by triggering oxidative stress through MAPK signaling. Neurologia (Engl Ed). 2021. [CrossRef]
- Liu, D.; Zhao, D.; Zhao, Y.; Wang, Y.; Zhao, Y.; Wen, C. Inhibition of microRNA-155 Alleviates Cognitive Impairment in Alzheimer’s Disease and Involvement of Neuroinflammation. Curr. Alzheimer Res. 2019, 16, 473–482. [CrossRef]
- Zago E, Dal Molin A, Dimitri G, Xumerle L, Pirazzini C, Bacalini MG, et al. Early downregulation of hsa-miR-144-3p in serum from drug-naïve Parkinson’s disease patients. Scientific reports. 2022;12:1330. [CrossRef]
- Li K, Zhang J, Ji C, Wang L. MiR-144-3p and Its Target Gene β-Amyloid Precursor Protein Regulate 1-Methyl-4-Phenyl-1,2-3,6-Tetrahydropyridine-Induced Mitochondrial Dysfunction. Mol Cells. 2016;39(7):543-9. [CrossRef]
- Murphy CP, Li X, Maurer V, Oberhauser M, Gstir R, Wearick-Silva LE, et al. MicroRNA-Mediated Rescue of Fear Extinction Memory by miR-144-3p in Extinction-Impaired Mice. Biological Psychiatry. 2017;81(12):979-89. [CrossRef]
- da Silva FC, Iop RdR, Vietta GG, Kair DA, Gutierres Filho PJB, de Alvarenga JGS, et al. microRNAs involved in Parkinson’s disease: A systematic review. Mol Med Rep. 2016;14(5):4015-22. [CrossRef]
- Zamanian, M.Y.; Ivraghi, M.S.; Gupta, R.; Prasad, K.D.V.; Alsaab, H.O.; Hussien, B.M.; Ahmed, H.; Ramadan, M.F.; Golmohammadi, M.; Nikbakht, N.; et al. miR-221 and Parkinson’s disease: A biomarker with therapeutic potential. Eur. J. Neurosci. 2023, 59, 283–297. [CrossRef]
- Ma, W.; Li, Y.; Wang, C.; Xu, F.; Wang, M.; Liu, Y. Serum miR-221 serves as a biomarker for Parkinson’s disease. Cell Biochem. Funct. 2016, 34, 511–515. [CrossRef]
- Li, L.; Xu, J.; Wu, M.; Hu, J.M. Protective role of microRNA-221 in Parkinson’s disease. Bratisl. Med J. 2018, 119, 22–27. [CrossRef]
- Ishihara, Y.; Yamada, H.; Munetuna, E.; Hagiwara, C.; Fujii, R.; Yamazaki, M.; Hatta, T.; Iwahara, A.; Ohashi, K.; Ishikawa, H.; et al. 486Serum levels of miRNA-221 and -222 may be predictive biomarkers for cognitive decline. International Journal of Epidemiology. 2021;50. [CrossRef]
- Gullett, J.M.; Chen, Z.; O’shea, A.; Akbar, M.; Bian, J.; Rani, A.; Porges, E.C.; Foster, T.C.; Woods, A.J.; Modave, F.; et al. MicroRNA predicts cognitive performance in healthy older adults. Neurobiol. Aging 2020, 95, 186–194. [CrossRef]
- Ba R-Q, Liu J, Fan X-J, Jin G-L, Huang B-G, Liu M-W, et al. Effects of miR-199a on autophagy by targeting glycogen synthase kinase 3β to activate PTEN/AKT/mTOR signaling in an MPP+ in vitro model of Parkinson’s disease. Neurological Research. 2020;42(4):308-18. [CrossRef]
- Nakashima, H.; Tsujimura, K.; Irie, K.; Imamura, T.; Trujillo, C.A.; Ishizu, M.; Uesaka, M.; Pan, M.; Noguchi, H.; Okada, K.; et al. MeCP2 controls neural stem cell fate specification through miR-199a-mediated inhibition of BMP-Smad signaling. Cell Rep. 2021, 35, 109124. [CrossRef]
- Wang RY, Phang RZ, Hsu PH, Wang WH, Huang HT, Liu IY. In vivo knockdown of hippocampal miR-132 expression impairs memory acquisition of trace fear conditioning. Hippocampus. 2013;23(7):625-33. [CrossRef]
- Harvey J, Smith AR, Weymouth LS, Smith RG, Castanho I, Hubbard L, et al. Epigenetic insights into neuropsychiatric and cognitive symptoms in Parkinson’s disease: A DNA co-methylation network analysis. Res Sq. 2023. [CrossRef]
- Chuang, Y.-H.; Lu, A.T.; Paul, K.C.; Folle, A.D.; Bronstein, J.M.; Bordelon, Y.; Horvath, S.; Ritz, B. Longitudinal Epigenome-Wide Methylation Study of Cognitive Decline and Motor Progression in Parkinson’s Disease. J. Park. Dis. 2019, 9, 389–400. [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.-C.; McLean, P.J.; et al. Sirtuin 2 Inhibitors Rescue α-Synuclein-Mediated Toxicity in Models of Parkinson’s Disease. Science 2007, 317, 516–519. [CrossRef]
- Rane, P.; Shields, J.; Heffernan, M.; Guo, Y.; Akbarian, S.; King, J.A. The histone deacetylase inhibitor, sodium butyrate, alleviates cognitive deficits in pre-motor stage PD. Neuropharmacology 2012, 62, 2409–2412. [CrossRef]
- Song, C.; Anantharam, V.; Sun, F.; Kanthasamy, A.G. Environmental Neurotoxic Pesticide Increases Histone Acetylation to Promote Apoptosis in Dopaminergic Neuronal Cells: Relevance to Epigenetic Mechanisms of Neurodegeneration. Mol. Pharmacol. 2010, 77, 621–632. [CrossRef]
Figure 1.
The probable role of DNA methylation in PD-related cognition impairment. The Figure was created with BioRender.com.
Figure 1.
The probable role of DNA methylation in PD-related cognition impairment. The Figure was created with BioRender.com.
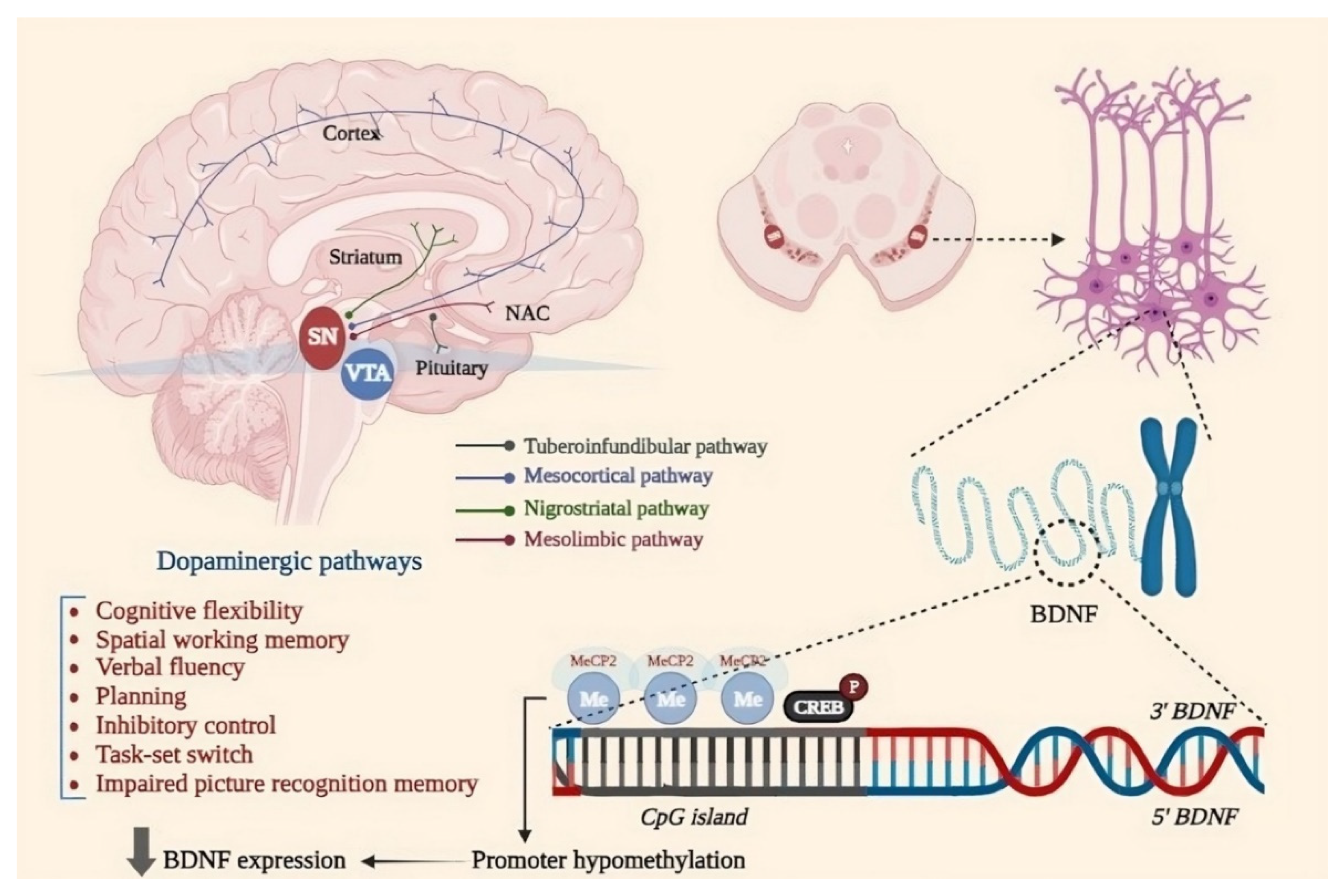
Figure 2.
The organization of nucleosomes and potential epigenetic pathways implicated in cognition processes in PD. Created with BioRender.com.
Figure 2.
The organization of nucleosomes and potential epigenetic pathways implicated in cognition processes in PD. Created with BioRender.com.
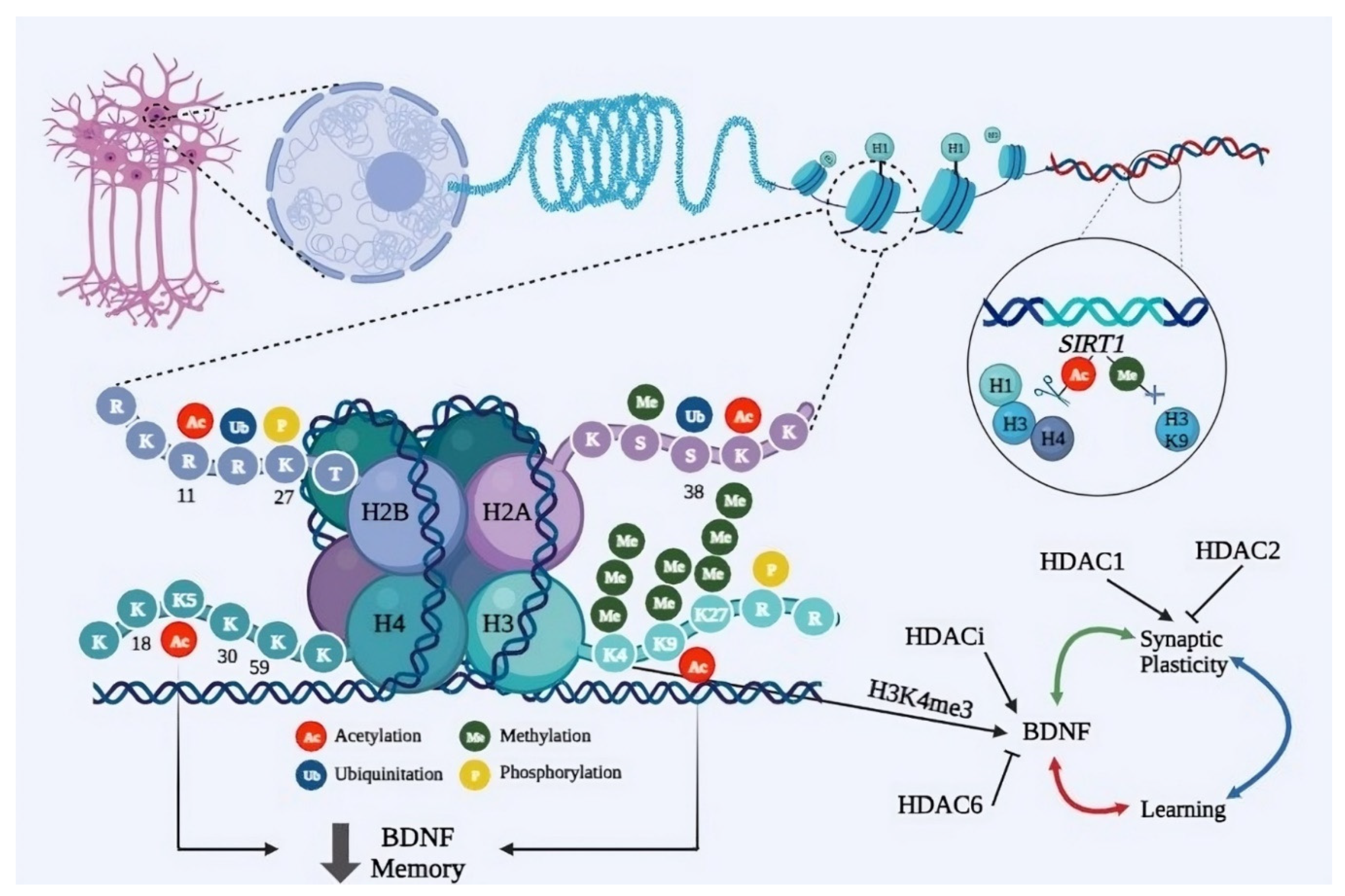
Figure 3.
Role of mir-7 in the pathogenesis of PD. Created with BioRender.com.
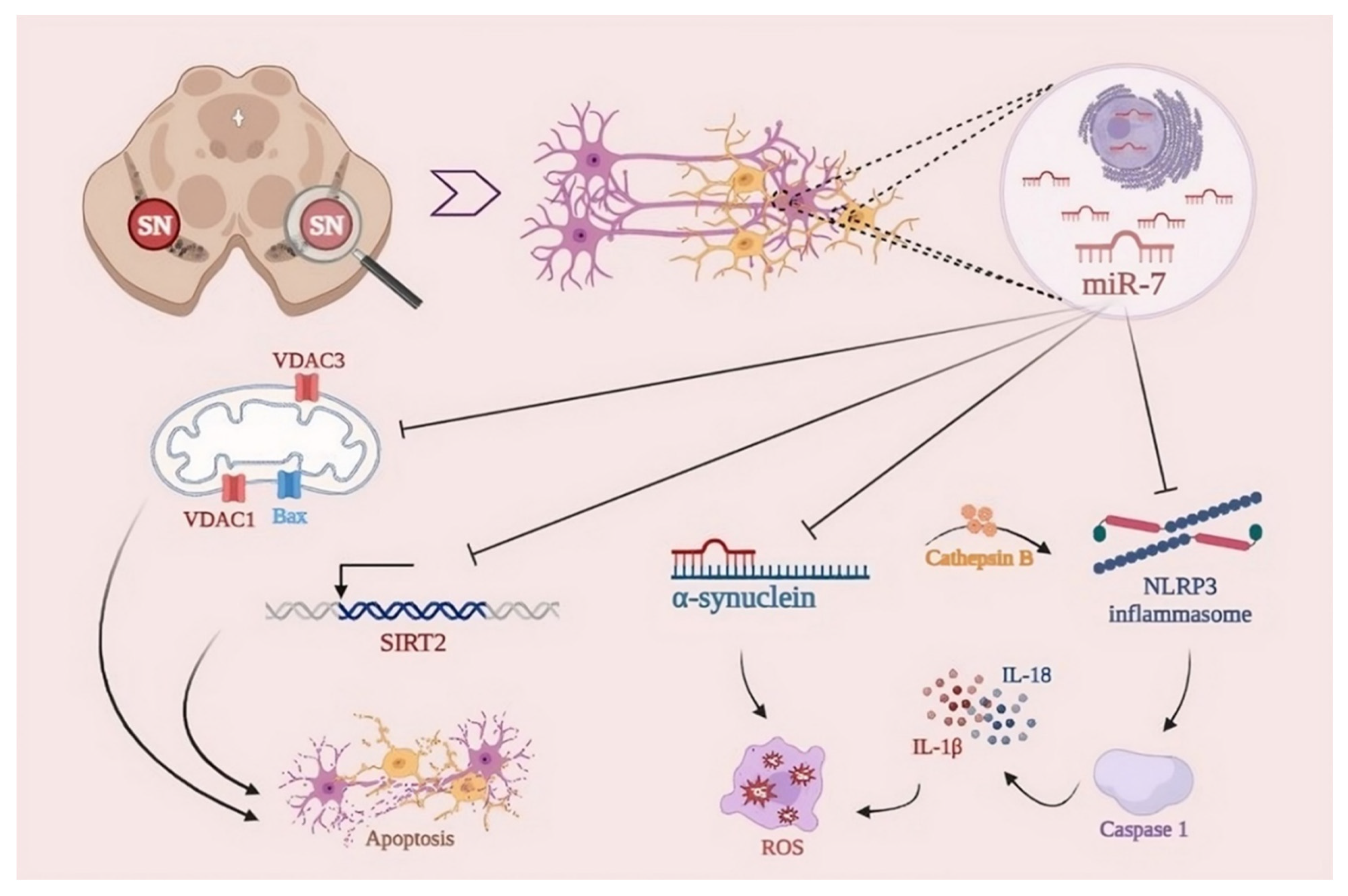

Table 1.
Synuclein and Tau ‘seeding and spreading’ experiments over the past decade.
| Study | Protein | Findings | Reference |
|---|---|---|---|
| Caillierez et al. 2013 | α-Syn | AT8 immunoreactivity spans from the CA1 to the cortex in rats receiving LV-hTau46WT injections, while, in rats injected with LV-hTau46P301L, it is restricted to the hippocampal formation. | [53] |
| Clavaguera et al. 2013 | Tau | Self-spreading of Tau inclusions in a manner like prions, not influenced by other pathological mechanisms | [54] |
| Holmes et al. 2013 | Tau and α-Syn | Tau fibrils enter cells via macropinocytosis; HSPGs act as receptors for binding and uptake of Tau, inhibition of HSPG blocks aggregate propagations, HSPGs mediate α-Syn uptake | [55] |
| Levarska et al. 2013 | Tau | Different spreading capacities of disease-modified Tau strains as Spreading only of the SHR72 4R Tau variant | [56] |
| Masuda-Suzukake et al. 2013 | α-Syn | Intracerebral injection of insoluble α-Syn induces aggregation of endogenous mouse α-Syn through a prion-like spreading. | [57] |
| Oliveras-Salvá et al. 2013 | α-Syn | Following viral vector-mediated α-Syn overexpression, dose-dependent dopaminergic neuron loss in SNpc 8 months post-injection occurred, also motor issues, and protein aggregates in remaining surviving neurons. | [58] |
| Pooler et al. 2013 | Tau | AMPA-stimulated Tau release is dependent on pre-synaptic vesicle secretion instead of exosome extrusion | [59] |
| Sacino et al. 2013 | α-Syn | Injection of amyloidogenic and non-amyloidogenic human α-Syn induced limited α-Syn inclusions. Delayed Strong induction of neuroinflammation with or without α-Syn inclusions | [60] |
| Ulusoy et al. 2013 | α-Syn | Propagation from medulla oblongata to rostral brain regions | [61] |
| Watts et al. 2013 | α-Syn | Lethality upon transmission to animals resembles how kuru, CJD, and similar diseases spread in nonhuman primates | [62] |
| Wu et al. 2013 | Tau | Transferring through the cell via anterograde and retrograde endocytosis of Small Misfolded Tau species | [63] |
| Ahmed et al. 2014 | Tau | Spreading relied on synaptic connectivity | [64] |
| Clavaguera et al. 2014 | Tau | Seeded Tau aggregates can spread to the CNS, peripherally | [65] |
| Dujardin et al. 2014 | Tau | Indication of trans-synaptic protein transfer due to Wild-type human Tau protein could transferred via the axons from ventral hippocampus neurons to connected secondary neurons e.g., olfactory and limbic systems | [66] |
| Holmes et al. 2014 | Tau | The seeding activity of mutant mice rises with age, interaction with synuclein | [67] |
| Holmqvist et al. 2014 | α-Syn | Spreading of α-Syn from intestine to the brain via the vagal nerve | [68] |
| Recasens and Dehay 2014 | α-Syn | Regardless of whether it was injected into the striatum or the SNpc. LB-induced degeneration manifested earlier and more extensively in the striatal dopaminergic axon terminals rather than the cell bodies in the SNpc, | [69] |
| Reyes et al. 2014 | α-Syn | Reuptake of α-Syn monomers, oligomers, and fibrils by oligodendrocytes, internalization of α-Syn by in vivo Oligodendrocyte, Transfer of α-Syn from host brain to grafted oligodendroglial cells | [70] |
| Rotermund et al. 2014 | α-Syn | Nutritional factors can have a significant impact on α-Synucleinopathy. Possibility of diet-induced obesity as a risk factor for α-Synucleinopathy | [71] |
| Sacino et al. 2014 | α-Syn | Fibrillar α-Syn intramuscular injection led to CNS inclusion pathology | [72] |
| Sacino et al. 2014 | α-Syn | Spreading of inclusion pathology only in M83 mice after 4 months | [73] |
| Sanders et al. 2014 | Tau | Spreading to the ipsilateral and contralateral entorhinal cortex, retrosplenial cortex, and contralateral hippocampus from the left hippocampus after 5 weeks | [74] |
| Asai et al. 2015 | Tau | Microglia’s significant role in Tau protein spreading and neurotoxicity | [75] |
| Bernis et al. 2015 | α-Syn | Spreading of α-Syn after intrastriatal injection to the contralateral side | [76] |
| Bourdenx et al. 2015 | α-Syn | Spreading into the striatal area and throughout the whole mesencephalon in rats 16 weeks after surgery. Absence of phosphorylation levels of α-Syn and neurodegeneration in rats and marmosets | [77] |
| Daher et al. 2015 | α-Syn | Neurodegeneration in the contralateral side | [78] |
| Abounit et al. 2016 | Tau | Tau fibrils capability to enter cells and induce TNT (tunneling nano tubules) | [79] |
| d’Abramo et al. 2016 | Tau | Tau detection in serum | [80] |
| Domert et al. 2016 | α-Syn | Cell-to-cell transfer in of α-Syn between neuron-like cell model |
[81] |
| Helwig et al. 2016 | α-Syn | Spreading from the medulla oblongata to rostral brain regions | [82] |
| Koprich et al. 2016 | α-Syn | Accumulation of A53T shows an age-related progressive rise, higher levels, and a broader extent of degeneration with A53T | [83] |
| Aulić et al. 2017 | α-Syn | Spreading and the entrance of α-Syn amyloid in N2a cells is facilitated by cells expressing PrPc | [84] |
| Cavaliere et al. 2017 | α-Syn | Quantification of α-Syn uptake in neurons and astrocytes | [85] |
| DeVos et al. 2017 | Tau | Reduction and prevention of Tau seeding capability by human Tau | [86] |
| Loria et al. 2017 | α-Syn | Transfer of α-Syn between primary cortical astrocytes and neurons | [87] |
| Ngolab et al. 2017 | α-Syn | Intracellular aggregation of α-Syn is correlated with internalization of exosomes via endocytosis | [88] |
| Shimozawa et al. 2017 | α-Syn | Retrograde spreading of abnormal α-Syn and neurotoxicity following intracerebral injection of synthetic α-Syn fibrils, Clearance of α-Syn inclusions by microglial cells | [89] |
| Ulusoy et al. 2017 | α-Syn | The Vagus nerve acts as a conduit for α-Syn to reach from the brain to peripheral tissues e.g., stomach | [90] |
| Wang et al. 2017 | Tau | Exomes from CSF have the capability of stimulating Tau aggregation within cultured cells | [91] |
| Polanco et al. 2018 | Tau | When interconnected axons extend in proximity, they exchange exomes | [92] |
| Rusconi et al. 2018 | α-Syn | Sustained overexpression of α-Syn is crucial for continuous propagation. Increased spreading of α-Syn through the brain following increased level α-Syn in the medulla oblongata | [93] |
| Vitale et al. 2018 | Tau | Diffusion to distant areas | [94] |
| Smolek et al. 2019 | Tau | Tau pathology induction following bilateral hippocampus injection | [95] |
| Ferrer et al. 2020 | Tau | Potential role of oligodendropathy and neuronopathy in Tauopathies progression. 3RTau and 4RTau production and deposition and activation specific Tau kinases following injection of human Tau inoculations leads to modification of Tau metabolism | [96] |
| Ferrer et al. 2019 | Tau | Role of oligodendrocytes in seeding and spreading of Tauopathies in white matter | [97] |
| Masuda-Suzukake et al. 2020 | Tau | Tau spreading after dextran sulphate-induced Tau assemblies, propagation of Tau-assemblies does not depend on Tau to be either mutated or overexpressed. | [98] |
| Mezias et al. 2020 | α-Syn | After injection of protein fibrils, the α-Syn spreading follows neural networks | [99] |
| Veys et al. 2020 | Tau, α-Syn | α-Syn PFFs similar to the Tau K18 PFFs fail to enter the retina after IVT injection | [100] |
| Courte et al. 2020 | α-Syn | The ability of exogenous α-Syn seeding is dependent on the level of α-Syn expression within the regional neurons | [101] |
| Jimenez-Ferrer et al. 2021 | α-Syn | The significant role of Mhc2a as the key regulator of MHCII expression for regulation of seeding, spreading, and toxicity of α-Syn in vivo | [102] |
| Thomsen et al. 2021) | α-Syn | Striatal injection of α-Syn fibrils causes gradual impairment of synaptic function before cell death, detectable by PET scan, α-Syn striatal injection creates progressive α-Syn pathology as found in human PD | [103] |
| Dutta et al. 2021 | α-Syn | Decrease in α-Syn spreading by Intranasal administration of wtTIDM peptide, NEMO-binding domain (wtNBD) peptide, or genetic deletion of TLR2 | [104] |
| Bassil et al. 2021 | Tau, α-Syn | Tau spreading is reduced by endogenous α-Syn in mice while α-Syn seeding and spreading are not affected by endogenous Tau | [105] |
| Matsuo et al. 2021 | α -Syn | Deletion of Fabp3 results in the prevention of exogenous α-Syn spreading into SNpc | [106] |
| Pan et al. 2022 | α-Syn | Striatal injection of Tau-modified α-Syn fibrils results in more severe α-Syn pathology and motor and cognitive symptoms comparing pure α-Syn injection | [107] |
| Dutta et al. 2022 | α-Syn | Treadmill exercise reduces α-Syn spreading | [108] |
| Garcia et al. 2022 | α-Syn | Microgliosis occurs as a part of response independent from α-Syn inclusion formation, neurodegeneration could occur even if α-Syn inclusion is not present. | [109] |
| Vasili et al. 2022 | α-Syn | Seeding and accumulation of pS129A-Syn is induced by adding α-Syn PFFs, endogenous α-Syn aggregates are dependent on α-Syn levels | [110] |
| Sun et al. 2022 | α-Syn | Pathological mechanisms induced by the spreading of misfolded α-Syn throughout the nigrostriatal pathway vary based on the age of the dopamine network, resulting in a striatal dopamine release decline specifically observed in adult mice. | [111] |
| Rahayel et al. 2022 | α-Syn | Differentially targeted seeding of pathological α-Syn led to distinct spreading patterns observed over 24 months indicating most brain regions were susceptible to this pathology | [112] |
| Lloyd et al. 2022 | α-Syn | The phenotypic and pathological progression of the disease.is significantly influenced by initial inoculation | [113] |
| Jain et al. 2023 | Tau | Chronic administration of activatingTREM2 antibody enhances the activation of microglia around plaques. This amplification correlates with an increase in peri-plaque NP-Tau pathology and neuritic dystrophy, while the presence of Aβ plaques remains unaffected. | [114] |
| Mate De Gerando et al. 2023 | Tau | Soluble HMW Tau has similar seeding potential comparing fibrillar sarkosyl-insoluble Tau but could be more bioactive in terms of spreading across neural systems | [115] |
Table 2.
Commonly involved microRNAs in both cognitive impairment and PD.
| miRNA | miR Expression |
Studied Population | Cognition Impaired | Studied Species/Specimen | References |
|---|---|---|---|---|---|
| miR-7 | ↑ | PD patients/ MPTP mice model/ MPP+-SH-SY5Y cell model |
Acute ischemic stroke | Human/blood | [180]/ [182] |
| ↑ | MPTP mouse model | Neuroprotection | Human/brain | [181]/ [183] | |
| miR-124-3p | ↓ | MPP+-SH-SY5Y cell model | [184] | ||
| ↑ | Ease cognitive function, inflammation and apoptosis in PND |
Rat/ brain | [185] | ||
| miR-124 | ↓ | LPS-BV-2 cell models MPTP mice model, MPP+-SH-SY5Y cell model |
AD/suppress memory and learning; Suppress CREB and GluA2 |
Mouse /brain /hepatopancreas, muscle, heart, ovotestis, and central nervous system | [186]/ [189,190,191,192,193] |
| ↓ | prefrontal cortex of the left cerebral hemisphere | Impaired LTP and learning and memory, spatial learning, and social interactions |
Mouse/brain | [187]/ [194] | |
| ↑ | amygdala | FTD | Mouse/ | [188]/ [195] | |
| miR-195 | ↓ | LPS-BV-2 cell model | Impaired spatial memory | Rat/brain | [196]/ [197] |
| ↓ | AD | Rat/brain | [198,199,200] | ||
| miR-190 | ↓ | LPS-BV-2 cell model, MPTP mice model | Upreg.ameliorates POCD | Mice/brain | [201] /[202] |
| ↑ | Neuroprotective | Rat | [203] | ||
| miR-146a | ↑ | human glial cell lines | AD | human, mouse/archived tissue, or total RNA extract sources | [204]/ [205] |
| ↑ | AD | human/CSF & ECF | [206] | ||
| ↓ | AD | human/plasma and CSF | [175] | ||
| miR-155 | ↑ | microglia and astrocytes cultured from DJ-1-knockout mouse brain | neuroprotective effect | mouse/brain | [207]/ [208] |
| miR-133b | ↓ | aphakia mice 6OHDA-treated mice |
[209] | ||
| ↑ | protects against isoflurane-induced learning and memory impairment | rat/brain | [210] | ||
| ↑ | neuroprotective in AD | AD vs. HC /serum | [211] | ||
| ↓ | midbrain | [209,212,213] | |||
| miR-34b | ↓ | putamen / FC / amygdala / SN / cerebellum | [214] | ||
| miR-126 | ↑ | DA neurons / amygdala | improved hemorrhagic lesion and the number of apoptotic cortical neurons | rat/brain | [188,215,216]/ [217] |
| ↓ | induces cognitive impairment and neuroinflammation | mice/serum | [218] | ||
| miR-204 | ↑ | putamen | [7] | ||
| ↓ | amygdala | [188] | |||
| miR-144 | ↑ | the prefrontal cortex anterior cingulate gyrus |
promoted cognitive impairments induced by β-amyloid accumulation post-TBI via suppres- sing ADAM10 expression (spatial learning and memory). | human, rat/brain | [219,220] / [221] |
| ↓ | the prefrontal cortex of the left cerebral hemisphere | [187] | |||
| miR-148b | ↓ | the prefrontal cortex/amygdala | [188,219] | ||
| ↑ | attenuated neuroprotection by inhibiting Wnt/ β-catenin signaling | rat | [222] | ||
| miR-184 | ↑ | DA neurons and amygdala | increased viability and reduced apoptosis | human, rat/brain | [188,216] / [223] |
| ↓ | worse learning and memory capacity | MDD vs. HC/blood | [224] | ||
| ↓ | Neuroprotective | mice/brain | [225] | ||
| miR-221 | ↑ | putamen / anterior cingulate gyrus, and amygdala | [7,188,220] | ||
| ↓ | neuroprotection | human/plasma | [226] | ||
| miR-199a | ↑ | amygdala | spatial memory ability began to decrease at 4 months and was significantly decreased at 7 months | mice/brain | [188]/ [227] |
| ↓ | iPSC-derived DNs from PD patients | [228] | |||
| miR-132 | ↓ | prefrontal cortex, and in a meta-analysis of different PD brain specimens | [212,219] | ||
| ↑ | MS | human/ | [229] | ||
| regulates the process of cognitive impairment after stress | rat/ | [230] | |||
| reduces the cognition-damaging effect of sevoflurane | rat/brain | [231,232] | |||
| spatial memory mission | /brain | [233] | |||
| significantly damage the cognition of spatial memory | mice/brain | [234] | |||
| midbrain | axon growth, neural migration, and plasticity | mice/dorsal root ganglion | [235]/ [236] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



