
Submitted:
18 June 2024
Posted:
25 June 2024
You are already at the latest version
Abstract
Parkinson’s disease (PD) is a common multisystem neurodegenerative disorder affecting 1% of the population above 60 years. The main neuropathological features of PD are the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the presence of alpha synuclein (Syn)-rich Lewy bodies both manifesting with classical motor signs. Syn has emerged as a key protein in PD pathology as it can spread through synaptic networks to reach several anatomical regions of the body contributing to the appearance of non-motor symptoms (NMS) considerate prevalent among individuals before PD diagnosis and persisting throughout the patient’s life. NMS mainly include loss of taste and smell, constipation, psychiatric disorders, dementia, rapid eye movement (REM) sleep behavior impairment, urogenital dysfunction, and cardiovascular impairment. This review summarizes the more recent findings showing the impact of Syn deposits on several prodromal NMS and emphasizes the importance of early detection of Syn toxic species in biofluids and peripheral biopsies as prospective biomarkers in PD.
Keywords:
alpha synuclein
; Parkinson’s disease
; non-motor symptoms
; early diagnosis
1. Introduction
Parkinson’ disease (PD) is one of the most common neurodegenerative disorders, second only to Alzheimer’s disease, that refers to the group of α-synucleinopathies characterized by loss of the dopaminergic and monoaminergic neurons in the substantia nigra pars compacta (SNpc) and ventral tegmental area, as well as by the accumulation, aggregation and spread of α-Synuclein (αSyn) within neurons and non-neuronal cells including microglia, pericytes, astrocytes, and oligodendrocytes [1]. PD is the most common form of movement disorder clinically manifested by bradykinesia, dyskinesia, rest tremor, rigidity, slowness of movement, freezing, dizziness and postural instability as a result of the loss of 70-80% of the dopaminergic neurons in the substantia nigra pars compacta (SNpc) [2]. Aside from motor manifestations, PD impacts a plethora of non-motor symptoms (NMS) which may antedate the onset of movement disorders by several years [3,4]. They include smell-related alterations [5,6], dysfunction of the circadian rhythms [7], autonomic disorders such as orthostatic hypotension constipation, urinary difficulties, gastrointestinal disorders, anxiety, depression, and impairment or loss of sensory perception [4,8,9]. Non-motor manifestations have been recognized as significant contributors to the overall impact of the disease on the individual’s quality of life [10,11,12].
According to Braak stage 1, early NMS such as smell disorders experienced by PD patients are caused by neuronal degeneration in the olfactory bulb and in anterior olfactory nucleus, while Braak stage 2 involves the lower brainstem i.e., raphe nucleus, locus coeruleus, pedunculopontine nucleus and thalamocortical system, which are associated with sleep disturbance, autonomic dysfunctions, visual hallucinations, and rapid eye movements [13].
Dysfunction of the medullary nuclei located in the brainstem, namely nucleus tractus solitarius, dorsal motor nucleus of the vagus, and nucleus ambigus, regulate various involuntary functions including arterial blood pressure, heart rate, respiratory activity, and renal function. These conditions are associated with orthostatic hypotension, cardiovascular abnormalities, and other autonomic disorders in PD [14]. The emergence of the classic motor symptoms of PD typically occurs at Braak stages 3 and 4. These clinical signs coincide with the involvement of key regions such as the SNpc and other deep nuclei of the midbrain and the forebrain [15]. In the final stages Braak 5 and 6, Lewy bodies are found in the limbic structures and mature neocortex. At these stages, patients with PD can experience a variety of neuropsychiatric symptoms, including depression, cognitive impairment, and visual hallucinations [15]. The subthalamic nucleus plays a crucial role in PD. It receives inputs from sensorimotor, associative, and limbic brain regions which serve distinct functions within the basal ganglia circuitry. Specifically, pallidosubthalamic projection is thought to underlie cognitive, emotional, and motivational NMS. This intricate organization underscores the diverse roles of the subthalamic nucleus in both motor and non-motor aspects of PD.
Several studies have reported effective role for the subthalamic nucleus deep brain stimulation (STN-DBS) on motor and NMS, although PD patients with longer disease duration had limited motor benefits from STN-DBS [16,17,18,19]. Case reports and data from a meta-analysis conducted on dozens of studies indicate that STN-DBS improve NMS and PD patient quality of life [20,21,22,23]. Due to the complexity of PD, a more comprehensive understanding of the underlying mechanisms beyond the scope of the Braak staging model is required. Certainly, the recognition of non-motor signs can contribute to early diagnosis of PD and help to adopt strategies aimed at improving the management of the disease [24]. In this review, we highlight recent advance in αSyn neurotoxicity that have contributed to our understanding of NMS onset and could aid in the development of future treatment strategies and/or early diagnosis.
2. αSyn Toxicity
From pathological point of view, NMS are the consequence of the formation of intracytoplasmic Lewy bodies and neurites rich in αSyn aggregates in nigral and extranigral areas.
αSyn is a small, 140 amino acid presynaptic acidic protein encoded by the SNCA gene whose main function seems to be the control of neurotransmitter release [25]. It is a vesicle-bound multimer consisting of three distinct regions: 1) the N-terminus (residues 1-60) crucial for its interaction with lipid membranes [26,27]; 2) the central hydrophobic region (residues 61-95) prone to aggregation named non-amyloid-ß component (NAC region) [28] 3) the unstructured C-terminus (residues 96-140) highly negatively charged with Ca2+ binding and chaperone-like activity [29].
In the CNS, it exists in a soluble cytosolic fraction (for as much as 1% of the total protein) and in two membrane- and vesicle-binding forms: via C-terminal domain it interacts with vesicle-associated membrane protein 2 leading to the formation of vesicle clusters [30] and affecting vesicle docking as well as the inhibition of vesicle fusion, while via N-terminal region αSyn was found to bind membrane lipids. This membrane-binding pool seems to prevent the pathological aggregation [31,32,33]. Mutations in this domain are associated with PD pathology [34,35]. On the contrary, the cytosolic fraction is intrinsically disordered and behaves like a natively unfolded protein, contributing to the formation of aggregate species [36].
Different factors, such as genetic mutations [37], elevated levels of αSyn, mitochondrial dysfunction [38], oxidative stress (OS) [39], endoplasmic reticulum (ER) stress [40] dysregulation of synaptic vesicle recycling [41], and autophagy-lysosomal system [42], are well-known to concur to the αSyn misfolding, formation of β-sheet rich oligomers and fibrils in Lewy bodies or Lewy neurites accompanying NMS [43,44,45,46]. Studies also support the contribution of neuroinflammation in NMS [47,48,49], pointing to the essential components of the innate immune response such as the toll-like receptors (TLRs) [50,51,52,53]. In particular, pathogen associated molecular patterns and damage-associated molecular patterns (DAMPs) can prime and activate the TLRs creating a neuroinflammatory status which may culminate with neuronal death in specific brain areas [54]. Human studies suggest a role for TLR2 and TLR4 in the recognition of toxic species of αSyn as a DAMP which precede the αSyn aggregation [55,56]. Furthermore, polymorphisms in the SNCA gene are also well recognized factors concurring in the non-motor signs development [57]. NMS also are the consequence of the diffusion of the pathological species of αSyn. In fact, αSyn aggregates spread between interconnected brain areas in a cell-to-cell and prion-like fashion which involve neurons and non-neuronal cells [58,59]. Transsynaptical transmission of αSyn may be triggered by the oligomeric αSyn-mediate microglia activation in the early phases of the disease [60] or by interactions between endogenous αSyn and mitochondria [61,62]. However, cells differentially mediate the uptake of the αSyn fibrils involving processes such as receptor-mediated endocytosis, extracellular vesicles and tunneling nanotubes. Fibrils can be sequestered and degraded into lysosomes or be trafficked into the cytosol via endocytic pathways, where they can interact with and recruit monomeric αSyn into mature pathological inclusions [63]. In addition, fibrils can be released by damaged or dead cells following mitochondrial dysfunction [38,51,64], disruption of redox balance [39], nitrosative stress [65], impaired autophagic flux [42], and prolonged ER stress [40]. Oligomeric αSyn may alter the voltage-gate receptors resulting in impairment of calcium efflux [66,67].
A schematic representation of the relation between αSyn toxic species and cellular dysfunctions is reported in Figure 1.
2.1. Oxidative/Nitrosative Stress
A bidirectional relationship between oxidative/nitrosative stress and αSyn toxicity exists [68]. High levels of reactive oxidative species (ROS) within the neurons have been suggested to induce αSyn toxicity [69,70,71]. Excessive OS, cause peroxidation of the membrane lipids followed by the production of the highly toxic 4-hydroxy-2-nonenal. This compound induces the formation of beta sheets and toxic soluble oligomers of αSyn which are believed to promote neuronal damage [72,73]. Over, hydroxyl radical species can react with tyrosine residues on αSyn contributing to the formation of intramolecular oxidative covalent cross-linkage between two tyrosine residues known as the dityrosine bond [74]. Two opposing effects have been described for dityrosine: (i) formation and stabilization of αSyn insoluble fibrils or aggregates [75], and (ii) inhibition of αSyn fibrillation by dityrosine-modified monomer and dimers [74]. As aggregation proceeds, dityrosine formation shifts from an aggregation-inhibiting to an aggregation-promoting element [76]. The higher-order assembly as well as increased stability of growing fibrils appear significantly to reduce their capacity to seed further aggregation because to the minor available surface area for serving as effectives nucleation site and the conversion to less dynamic structures. Again, changes in hydrophobicity or charge distribution aside from changing the equilibrium between monomers, oligomers, and fibrils could influence the incorporation of additional monomers and aggregation kinetics [77].
Hypochlorite-oxidized cysteinyl-dopamine obtained by exposing cysteinyl-dopamine to hypochlorite is considered a potent redox cycler able to accelerate OS and contribute to an excessive demand for autophagy and ultimately to cell death [78]. Additionally, post-translationally aberrant S-nitrosylation by reactive nitrogen species and nitric oxide in neurons is known to induce to aSyn misfolding and toxicity, axo-dendritic-, and dopamine dysfunction [79]. Moreover, Kumar et al. [80] showed that S-nitrosylation of the ubiquitin C-terminal hydrolase-1 provides a nucleation to the native aSyn accelerating the protein aggregation.
2.2. Mitochondrial Dysfunction
Mitochondrial dysfunction plays a central role in the pathogenesis of PD [81]. Several neurotoxins have been found to provoke high levels of ROS, complex I inhibition and neuronal damage [70]. Additionally, abnormal levels or misfolded forms of αSyn can disrupt the balance of mitochondrial fission and fusion and transport, leading to fragmentate or aggregate mitochondria, impairment of mitochondrial trafficking, electron transport chain, and calcium signaling [81,82]. αSyn can interact with complex I, resulting in decreased ATP production and increased ROS [83]. The selective autophagic degradation of damaged mitochondria can overwhelm the cellular proteostasis, leading to the accumulation of misfolded αSyn and mitochondrial dysfunction [84,85,86]. αSyn can trigger the opening of the mitochondrial permeability transition pore, causing mitochondrial swelling and loss of membrane potential [87] and can interact with other proteins involved in mitochondrial quality control, such as Parkin and PINK [88]. Dysregulation of these interactions can impair mitochondrial function and dynamics, contributing to the pathogenesis of PD.
2.3. ER Stress
The accumulation of newly synthesized or improperly folded proteins in the ER can saturate the folding machinery, leading to ER stress which promptly actives the unfolded protein response (UPR) aims to restore normal ER function. In response to prolonged stress, UPR loses the capacity to assist in protein folding and its dysregulation culminates with neuronal death as a protective measure to prevent further damage [40]. In this condition, the activation of C/EBP Homologous Protein by Protein Kinase R-like ER Kinase, Activating Transcription Factor 4 (ATF4) or ATF6 or X-box Binding Protein 1 appears to be central for the induction of ER stress-driven apoptotic signal, ensuring that cells with irreparable damage are eliminated to maintain tissue homeostasis. ER-phagy, a selective form of autophagy targeting the ER for degradation, plays a crucial role in the clearance of misfolded aSyn. The interaction with ER-resident autophagy receptor FAM134B and the involvement of calnexin are key components for the recruitment of unfolded aSyn, encapsulation of ER fragments into autophagosomes, transport to- and fusion with lysosomes, and degradation and clearance of misfolded synuclein [89].
2.4. Lipids
There is increasing evidence that the oligomeric high levels of polyunsaturated fatty acids (PUFA’s) play an important role in neuronal toxicity. Notably, lipid peroxidation of PUFAs represents a feature of PD [90]. Specifically, there is a substantial increase in reactive aldehyde species such as malondialdehyde, 4-hydroxy-2-nonenal, cholesterol lipid hydroperoxide, and F2-isoprostanes in the SNpc of PD brain as well as in the anterior cingulate cortex of PD subjects [90,91].
Many studies also showed physical interaction between αSyn and PUFAs which results in a higher propensity to pathological aggregation [92,93,94,95]. The mutual electrostatic interaction between αSyn and membrane containing negatively charged lipids is found to affect the αSyn characteristics and membrane composition. In particular, changes in the levels of fatty acids, sphingolipids, and cholesterol have been reported [96].
2.5. Autophagic/Lysosomal Disruption
Autophagic/lysosomal disruption is closely linked to αSyn toxicity, particularly in the context of PD. Autophagy-lysosome pathway is a crucial mechanism deputy to degradation and recycling of damaged misfolded proteins, which include autophagosome formation, fusion with lysosomes and degradation by lysosomal hydrolases. Overexpression or aggregation of αSyn can disrupt the autophagy-lysosome pathway leading to the accumulation of misfolded αSyn in the brain, while, inefficient clearance of αSyn aggregates facilitates their spread to neighboring cells, propagating the proteotoxicity. The translocation of cytosolic αSyn to the lysosome lumen is mediated by binding of heat shock cognate 70 chaperone to the KFERQ sequence which, with other co-chaperones, directs the αSyn to the chaperone-mediated autophagy (CMA) adaptor Lysosomal-Associated Membrane Protein 2 (LAMP2A) located on the lysosomal membrane. Under stress conditions, exposure of the KFERQ motif can trigger CMA to remove harmful αSyn. Dysregulation of CMA is related to impaired degradation of αSyn and may contribute to the pathogenesis of PD [97].
Autophagy of mitochondria, or mitophagy, is intricately regulated by specific posttranslational modifications that “tag” the cargo for degradation, and by the Tank-binding kinase 1-mediated phosphorylation of optineurin that enhances its binding to ubiquitin and light chain 3 (LC3). Once bound to the ubiquitinated mitochondria, adaptors recruit LC3 so that the mitochondria are encapsulated by a double membrane structure called the autophagosome. Then, the autophagosome fuses with a lysosome forming an autolysosome, where the mitochondria are degraded and recycled [98]. Elevated αSyn levels inhibit macrophagic flux. Moreover, mutant forms of αSyn, such as A53T and A30P, exhibit a stronger binding affinity for LAMP2A which impairs the efficiency of αSyn clearance [99]. A53T and E46K αSyn variants also engage functional LC3B monomers into insoluble microaggregates on the surface of late endosomes favoring αSyn exosome excretion and seeding [79].
2.6. Metals
Although the precise role of metals in the pathogenesis of PD is still debated, multiple evidence suggest their involvement in conformational effects related to the binding of metal ions to αSyn and subsequent aggregation and accumulation. Elevated levels of metals such as iron, zinc, aluminum, lead, and copper have been found in the brain and in the cerebrospinal fluid (CSF) of PD patients [100]. Albeit with different affinities and stoichiometries, metals, especially copper, can establish electrostatic interactions with C-terminus of αSyn, more strongly with Tyr-125 and Ser-129 phosphorylate αSyn (p-αSyn), increasing their propensity to fibrillation. The aggregation speed of the acetylated A53T variant is higher with respect to wild type protein, suggesting an intrinsic self-assembly of the αSyn mutants into aggregates [101]. The toxicity of iron has also been investigated. Abeyawardhane et al. [102] suggest that the impact of Fe(II) on aSyn structure is higher than Fe(III) because its more elevated reactivity with O2 which resulting in the production of H2O2 and triggering b-sheet generation; it act either as initiator and as a potential allosteric cofactor of protein misfolding [103]. αSyn interact with both oxidation states of copper ions (Cu(I) and (Cu(II)) generating ROS, and contributing to OS and potentially leading to aggregation into pathological fibrils [104]. Aluminum is a metal that can cross the blood-brain barrier (BBB) and accumulate in the brain [105]. It has been found to co-localize with Biondi ring tangles in PD brains [106]. There is evidence suggesting that aluminum can induce OS, or directly interact with αSyn, or disrupt the balance of essential ions in the brain, thus accelerating the fibrillation process of αSyn aggregation and contributing to the development and progression of PD.
3. Distribution of αSyn in Tissues and Fluids in Parkinson’s Disease
The presence of different proteoforms of αSyn has been observed not only in the central nervous system (CNS) but also in peripheral tissues and biofluids such as cerebrospinal fluid, plasma, saliva, olfactory mucosa, skin, salivary glands, retina, adrenal medulla, heart, and gastrointestinal tract in PD cases, even if conflicting reports exist and regard the consistency and significance of this occurrence [107,108,109]. αSyn aggregates have been detected in various peripheral biopsies such as the gastrointestinal tract, skin, and salivary glands, even in the early stages of PD [110] supporting the hypothesis that PD might start in the peripheral nervous system before affecting the brain. For instance, αSyn pathology has been found in the enteric nervous system (ENS), leading to the theory that the disease may originate in the gut and then spread to the brain via the vagus nerve [111]. Similarly, skin biopsies have shown the presence of αSyn deposits, suggesting that peripheral tissues could serve as accessible sites for early diagnosis [110]. Peng et al. [112] showed that the presence of dermal phosphorylated α-syn serve the identification of the lesions in early stage of disease and predict clinical phenotypes. Figure 2 describes the different organs and tissues in which αSyn can be detected and used as biomarker for the development of diagnostic assays.-
However, studies have failed to consistently detect pathological αSyn in peripheral tissues of PD patients, raising questions about the reliability and specificity of these findings. For example, CSF α-synuclein inversely correlates with non-motor symptoms in a cohort of PD patients [41,113,114]. Differences in detection methods, sample handling, and the stage of the disease at which samples are collected could account for these discrepancies. Therefore, the presence of opposing data requires the development of standardized methodologies and more comprehensive studies for allowing peripheral αSyn become a robust biomarker for early diagnosis of PD and monitoring of disease progression [113].
4. Non-Motor Symptoms Associated with αSyn Pathology
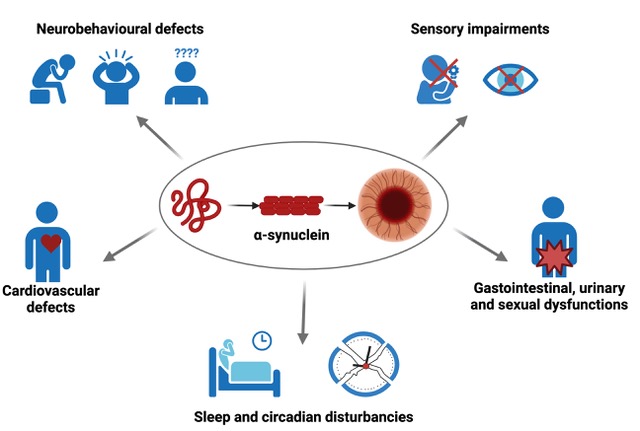
Non-motor symptoms in PD include both sympathetic and parasympathetic dysfunctions and may concern, among others, neurobehavioral changes, pain, olfaction impairment, sleep and circadian dysfunctions, gastrointestinal symptoms, uro-genital disturbances and cardiovascular problems.
Figure 3 describes the cardinal non-motor symptoms in the prodromal stage of PD.
4.1. Depression
The wide spectrum of NMS implicates the molecular interaction between the dopaminergic, glutamatergic, noradrenergic, and serotoninergic systems in which a significant number of neurons are loss [115]. Depression is a psychiatric condition estimated to affect approximately half of all PD patients [116]. Around 17% recapitulate symptoms consistent with major depression [117,118]. Research indicates that αSyn pathology begins its early accumulation in the olfactory system, particularly in the anterior olfactory nucleus and then spreads to the limbic system. Moreover, neuroimaging studies revealed alterations in limbic system including cingulate gyrus, hippocampus, amygdala, hypothalamus, nucleus accumbens, ventral striatum, orbitofrontal cortex [119]. Unfortunately, L-DOPA, the gold standard for the motor symptoms treatment do not alleviate depressive symptoms, indicating that depression is associated with deficits in serotoninergic neurotransmission in brainstem, raphe nuclei, and limbic system circuitry [120,121]. Serotonin and its metabolite, 5-HIAA, were decreased in the plasma of PD patients with depression [122]. Although the precise mechanisms for depression in PD are not fully elucidated, growing data reported the presence of aggregates of αSyn in monoaminergic brainstem, specifically in dendritic and axonal processes that, associated with loss of neurons in the above-mentioned brain areas, give rise to disturbances of neuroplasticity and depressive signs occurring in PD [123,124,125]. A recent study conducted in a mouse model of synucleinopathy showed that the overexpression of human αSyn in the raphe nuclei triggers progressive accumulation, phosphorylation, and aggregation of αSyn altering the 5-HT neurotransmission which results in a depressive-like phenotype [126].
4.2. Anxiety
Anxiety has a relative high prevalence (22.2%-66.7%) in PD patients with female being at higher risk than male patients [127,128,129]. Several kinds of anxiety are described in PD, including generalized anxiety, panic attacks, social phobia, agoraphobia, and obsessive-compulsive disorder. Stoyka et al. [130] showed that intrastriatal injections of fibrils of αSyn causes abnormal accumulation of αSyn inclusion in the cortex and amygdala. Likewise, injection of αSyn preformed fibrils into the bilateral olfactory bulb of A53T transgenic mice spreads into connected regions provoking severe pathology in the hippocampus, bed nucleus of the stria terminalis and central nucleus of the amygdala. Animals exhibited hyposmia, anxiety-like behavior, and memory impairment, consequence of atrophy, neuronal loss and gliosis [131]Increase in hippocampal αSyn expression has been detected in rats with high levels of innate anxiety, suggesting that anxiety can cause αSyn accumulation, besides being a contributor of synuclein progression [129].
4.3. Psychosis
Psychosis is a premotor neuropsychiatric condition affecting PD patients, characterized by a spectrum of mental symptoms including illusions, delusions, visual hallucinations, schizophrenia (SCZ), and rapid eye movement sleep behavior disorder that are the result of dopamine hyperactivity in the mesolimbic pathway [132,133] and dysfunction of neurotransmitter systems [134]. In the past years, psychotic individuals such as schizophrenic treated with antidopaminergic and neuroleptic drugs manifested parkinsonism but not was actual PD patients. The use of atypical newer antipsycotic drugs has significantly reduced the incidence of the psychiatric symptoms [135]. On the other hand, PD psychosis (PDP) is a common side effect derived from dopamine use to treat motor symptoms. However, several data indicate that psychosis may predate motor signs even in absence of therapy and in PD patients and when manifested is associated with high mortality and morbidity and heavily impacts on the quality of life of many patients. There is controversial evidence about the role of αSyn in the pathophysiology of PDP. For example, studies reported downregulation of the αSyn expression [136] and lower levels of serum of αSyn in SCZ patients than healthy controls [137] while another study did not reveal significant differences between SCZ patients and healthy controls [138] In one case study was found that duplication of the SNCA gene was associated with progression of PD [139]. Also, αSyn aggregates could contribute to neuroinflammation, OS, synaptic dysfunction and downstream neurotransmitter imbalance, the main feature of schizophrenia[140]. Furthermore, a pilot study revealed the association between rs356219 polymorphism in SNCA gene and psychiatric disorders [141] compared to healthy controls. Data from a human study described αSyn deposition in specific regions involved in directing attention toward visual target eliciting hallucination in dementia with Lewy bodies [142]. Furthermore, transgenic rats overexpressing human wild-type αSyn l (αSyn-BAC, harboring the entire human full-length SNCA locus) showed elevated levels of dopamine as a compensatory response, also accompanied by aberrant αSyn formation such as hyperphosphorylation, monomeric C-terminal truncation followed by aggregation pathology. These alterations produced a psychosis-like phenotype [133].
4.4. Cognitive Impairment
Cognitive decline is a prominent feature of PD and dementia with Lewy bodies (DLB), which comprises deficits that ranging from mild cognitive impairment to severe PD dementia (PDD).
It is noteworthy that αSyn directly impacts cognition and cognitive progression in PD. Many microRNA including miR-7, miR-129, miR-135a, miR-153 have been recognized to post-transcriptionally regulate αSyn levels involved in cognitive decline [143]. High CSF and plasma αSyn levels have recently been associated with higher prevalence of cognitive decline and faster progression of clinical dementia. Likewise, LB pathology evaluated by seed amplification assay [144,145,146,147] is associated with increased progression of DLB and rapid early alterations in cognitive performance [148]. These findings suggest a transport of CNS αSyn to the periphery which could help clinicians to detect by non-invasive practices αSyn levels as early as possible serving as a potential surrogate biomarker of risk of cognitive impairment. A recent transcriptomic study in an αSyn-based brain model of PD shows region-specific gene expression changes associated with cognitive deficit [149].
4.5. Pain
Pain is a common uncomfortable NMS in PD which can develop many years before the onset of classic motor signs and significantly impacts on depression and patient’s quality of life. It has been described in about 60-70% of patients and can be of different nature and prevalence. According to Ford’s classification, five types of pain are proposed: musculoskeletal pain, radicular-neuropathic pain, dystonic pain, central neuropathic pain, and akathisia [150]. Central pathways involved in PD pain comprise the lateral pain pathways with projections to thalamus and primary sensory cortex, and the medial spinoreticulothalamic pathways intimately associated with autonomic nervous system, containing fibers that project to the medullary core and mesencephalon. Neuronal loss and Lewy body formation occur in medial pain pathway, specifically in parabrachial nucleus coeruleus and periaqueductal grey [151]. αSyn misfolding and aggregation may start in peripheral nerves fibers and spread alongside neuroanatomical sensory connections causing a multitude of painful perceptions [152].
In a spared nerve injury model of neuropathic pain, the expression of αSyn has been detected in peptidergic and non-peptidergic nociceptive neurons in the dorsal horn of the spinal cord, signifying an involvement in pain transmission in the CNS. In the same model, the inhibition of αSyn was associated with suppression of pronociceptive MAP kinase signalling [153]. In addition, Chen et al. [154] showed that δ-Opioid receptor activation attenuated the MPP(+) and hypoxia induced αSyn overexpression/aggregation by enhancing CREB phosphorylation and TORC1/SIK1/CREB pathways.
4.6. Olfaction Dysfunction
Olfactory dysfunction (OD) is an early NMS symptom of PD affecting more than 90% of patients whose underlying mechanisms is partly defined. Post-mortem studies of PD indicate that αSyn pathology starts in the OB and lower brainstem and developments in limbic system and connected brain regions leading to hyposmia [155].
Animal models created by injecting recombinant αSyn preformed fibrils in the bilateral OB exhibit Lewy Body-like pathology and OD [156,157]. Additionally, human A30P mutant αSyn-expressing mouse model causes OD along with reduction in OB neurogenesis and alterations in synaptic vesicular transport [158]. The induction of αSyn aggregates via overexpression of double mutant human αSyn (A53T and A30P) in OB, negatively impacts on neural activity and odor-evoked response in the OB [112]. A recent study using A53T mutant mice, revealed hyperactivity of mitral/tufted cells and disruption in excitation/inhibition balance in OB by impairing GABAergic transmission and abnormal expression of GABA transporters [159].
4.7. Visual Impairment
A variety of visual alterations, including ocular, visuoperceptive, and visuospatial impairments have been associated with PD. Therefore, eye movement, visual acuity, tritan axis of color perception, recognition of visual stimuli and spatial relationships between objects result strongly compromised [160]. αSyn aggregates and p-Syn positive Lewy body-like and neurites have been described in the retina of PD patients and correlates with motor scores at the stage of the disease [161]. Phosphorylated form of αSyn has also been detected in wholemount human retinal nerve fiber layer and ganglion cell layer from PD and PDD subjects. Importantly, αSyn accumulation in these structures leads to detrimental consequences on dopaminergic neuron functions [162]. Overall, these findings suggest that detecting αSyn in the retina could represent a promising approach for searching early marker of PD.
4.8. Sleep and Circadian Dysfunctions
Sleep and circadian dysfunctions are recurrent non-motor symptoms of synucleinopathies, and significantly correlate with poorer quality of life [163].
Among the most common sleep-wake disturbances there is rapid eye movement (REM) behavior disorder (RBD), a non-familial sleep disorder characterized by the loss of muscle atonia during REM sleep (REM sleep without atonia or RSWA) and dream-enactment behaviors [164]. RBD is strictly associate to synucleinopaties [165] to the point that idiopathic/isolated RBD (iRBD), that may precede the onset of motor features by decades, is considered a prodromal form of synucleinopathy and therefore a highly specific marker for future development of a synucleinopathy. Furthermore, αSyn aggregates are considered as a biomarker for the development of diagnostic assays. As example, αSyn aggregates in stool samples of iRBD patients can be detected and measured, supporting the diagnosis of prodromal synucleinopathies [166].
Transgenic mice overexpressing human A53T α-syn (SNCAA53T/-) in its natively expressed regions exhibited REM sleep without atonia, similar to RBD patients, already at 5 months of age [167]. Compared to wild-type mice, in which REM sleep is characterized by sinusoidal theta-frequency electroencephalogram activity and absent or minimal electromyogram activity, SNCAA53T/- mice displayed excessive muscle twitches in body and limb during REM sleep and transient increases in muscle activity [165,168,169].
Restless legs syndrome (RLS), a very common movement disorder manifested by disturbing symptoms in lower limbs, more severe in the evening or night, has a high prevalence in PD patients [170] and some studies report the onset of PD at younger age in subjects with RLS [171].
Insomnia and excessive daytime sleepiness (EDS) are also common in PD patients. Insomnia has been associated with depressed mood, autonomic symptoms fatigue and age [172]. Subjective EDS has been reported in up to 50% of patients with PD [169]; it has been associated to the neurodegenerative process itself, that is extended to the dopaminergic and non-dopaminergic neurons in the lower brainstem and midbrain [173] involved in sleep-wake regulation [174]. These observations of poorly consolidated rest/activity patterns in humans are paralleled by animal models. Rodent models for PD display impairment in the sleep-wake parameters, such as deficit in REM sleep, overwhelming episodes of sleep, similar to “sleep attacks”, increased sleepiness [175,176,177]. In Drosophila, the pan-neuronal overexpression of pre-fibrillar human αSyn oligomers impacts on the sleep-like rest behavior (defined as absence of movement for 5 consecutive minutes), in terms of number and length of sleep episodes and total sleep [178,179,180,181]. Flies exhibit decreased sleep (especially in the night), increased number of sleep bouts (indicating sleep fragmentation) and decreased wake activity [178,180,181]. These alterations precede the onset of motor symptoms and can be related to the presence of pre-fibrillar αSyn oligomers that prevent protein aggregation [178,179,181].
Sleep is coordinated by the interaction of homeostatic and circadian mechanisms, regulating the sleep debt and the periodicity of sleep/wake propensity, respectively [182]. Circadian rhythms are controlled by an endogenous mechanism that comprises: a pacemaker, orchestrated by interconnected transcriptional/translational feedback loops, input pathways for light and other stimuli that synchronize the pacemaker to the environment and output pathways that convert the molecular oscillation of clock components in overt rhythms [183]. In the primary loop, the molecular oscillation involves the transcription factors CLOCK and BMAL1, which, as heterodimer, regulate the rhythmic expression of output genes. The CLOCK/BMAL1complex also activates the expression of the negative elements of the loop, Period and Cryptochrome genes, whose products inhibit their own expression by inactivating the activity of CLOCK/BMAL1 [183]. The circadian physiology is based on a hierarchical network of central and peripheral oscillators; the central pacemaker (master clock) is located in the suprachiasmatic nuclei (SCN) of the hypothalamus: it receives signals from the environment and transmits temporal information to downstream peripheral clocks, located in organs such as heart, lungs, liver and adrenal glands, through neurotransmitters and neuromodulators [183]. Melatonin secretion is a rhythmic output generated by the SCN: melatonin and circadian clock gene expression can be easily measured in serum and peripheral blood mononuclear cells, respectively, serving as reliable markers of circadian rhythmicity [183].
Several studies have reported lower levels and dampened melatonin oscillation and a lack of time-dependent variation in Bmal1 expression in PD patients, compared to healthy controls, indicating that the peripheral molecular clock is affected in PD [94,169,184].
Also, in Drosophila, the sleep defects reported in flies overexpressing pre-fibrillar human αSyn oligomers are associated with a severe impairment of two key circadian feature, the anticipation of the dark/light transition and the circadian periodicity [178].
4.9. Gastrointestinal Symptoms
For a long time αSyn has been identified as an important player in the complex process leading to GI symptoms in PD. Several large cohort and case-control studies show correlations between gastrointestinal (GI) dysfunctions and PD, DLB, and PDD during the disease course and overlap with autonomic symptoms [185,186].
GI disorders can cause PD on one hand and increase the risk of developing PD on the other two distinct subtypes of PD have been initially proposed based on the origin and progression of αSyn pathology: a “brain-first” subtype, in which αSyn pathology originates in the OB or other brainstem regions and then spreads to the peripheral autonomic nervous system, and a “body-first” subtype in which the pathological process starts in the ESN and then ascends via the vagus nerve or other autonomic pathways to reach the brain [187]. However, recent post-mortem studies showed that Lewy pathology is triggered in the GI tract or autonomic nervous system without concomitant involvement of the OB, therefore supporting a single-hit brain-first hypothesis [188,189].
Loss of enteric dopamine cells and degeneration of vague nuclei represent the main cause of GI symptoms such as dysphagia, sialorrhea, bloating, nausea, vomiting, gastroparesis, and constipation.
p-αSyn deposited in vagus nerve Schwann cells resulting in inflammatory response induced by interaction and activation with a family of pattern recognition receptor, Toll-like receptor 2 (TLR2), implicated in regulation of αSyn release [190]. On the other hand, the interaction between p-αSyn and TLR2/4 has been previously described in murine models of PD as well as in post-mortem human PD brain and associated with pro-inflammatory response and PD pathology [191,192].
GI symptoms affect nearly 80% of subjects before or after onset of motor manifestations and some of them are life-threatening symptoms, as an example dysphagia, due to the risk of pneumonia. Furthermore, PD patients experience a more widespread onset of lifelong GI symptoms and constipation is widely recognized as one of the most prevalent NMS of PD. Intestinal microbiome also plays a role in the bidirectional communication between the brain and the gut that has emerged as a significant aspect of PD pathology [193,194,195].
Aberrant aggregation of αSyn is responsible for vagus nerve and ENS degeneration, slow down intestinal peristalsis, and constipation [196,197]. A study revealed that human A53T αSyn transgenic mice exhibit severe signs of GI dysfunction together with αSyn aggregation in the ESN [198]. Interestingly, it has also been shown that pathological species of αSyn can be carried through red blood cells extravesicles to the GI tract, contributing to the onset and/or progression of the PD pathology.
In recent years, some authors speculate about the role of short-chain fatty acids (SCFAs) produced by the gut microbiome, especially butyrate and propionate, on development of synculeinopathies. Paradoxal and contracting effects of SCFAs were obtained, probably depending on the injection rout ways, the mixture composition and concentration [199,200,201,202]. For example, lower SCFA levels and higher calprotectin quantity detected in stool of PD patients correlate with GI symptoms [203]. An interesting preclinical study conducted in αSyn pre-formed fibrils (αSyn PFFs)-induced rat model of PD, documented the protective effects of sodium butyrate by reducing inflammatory markers such as TNFα, IL-1, IL-6, and increasing DA content [204]. Moreover, fecal transplant of gut microbiota from PD patients to αSyn overexpressing mice seems to promote αSyn-dependent activation of microglia and motor deficit worsening [205].
4.10. Sexual Dysfunction and Urinary Dysfunction
Sexual and urinary dysfunctions are common NMS in PD patients that involve neuronal cytoplasmic αSyn inclusions in the peripheral autonomic small nerve fibers. In about 70% of subjects, nocturia accompanied by urge urinary incontinence and detrusor hyperactivity often followed by the appearance of motor symptoms [206,207]. Sexual performance and relationship dissatisfaction have evaluated by employing multidimensional self-administered questionnaires. Data analysis suggest that there is a high prevalence of sexual dysfunction among young-onset male PD and female compared to healthy subjects [208], which is related to severity of depressive symptoms especially in female [209]. A number of studies have shown a range of symptoms ranging from hyposexuality to hypersexuality behaviors. For example, erectile dysfunction has been reported in diagnosed male PD and DLB cases, and loss of libido and orgasmic dysfunction were found in both women and men [210]while hypersexuality behaviors are often associated with PD treatment [201]. Impairment in courtship rituals and in copulation were observed also in a Drosophila A30P-mediated PD model [211].
4.11. Cardiovascular Symptoms
Epidemiological studies have suggested a possible relationship between the incidence of cardio-cerebrovascular disease and PD, either as risk factors or as manifestations of PD itself [212,213]. Studies using Mendelian randomization method reveal that PD is correlated to high risk of coronary artery disease, stroke, ischemic stroke, and cardioembolic stroke compared with age- and gender-matched general population [214,215]. On the other hand, the deposition of αSyn in the brain of PD may be a potential pathogenic factor for cardiovascular disease and stroke [216]. The involvement of the cardiovascular system in PD reflects αSyn deposition in sympathetic noradrenergic nerves, defects of the autonomous cardiac innervation and abnormal function of residual noradrenergic endings [217,218].
The loss of myocardial noradrenergic innervation together with the loss of myocardial norepinephrine caused by Lewy bodies deposition, are responsible for orthostatic hypotension with substantial fall in systolic and diastolic blood pressures of at least 20 mmHg and 10 mmHg, respectively, during the change from supine to standing position [219]. In PD patients a significant alteration in the electrophysiological activity and remodeling of the myocardium has also been described [220]. PD entails αSyn multi-organ deposition including myocardial tissue signifying that αSyn fibrils dispersed in the myocardium may be pathogenic. Myocardial tissues from PD autopsies display p-αSyn deposits and Lewy body pathology which correlate with cardiac sympathetic denervation [221]. Accumulation of αSyn aggregate in the paravertebral sympathetic ganglia seems to be chronologically preceded by those in the distal axons of the cardiac sympathetic nervous system, suggesting centripetal degeneration [222]. To note, cardiac dysfunction can be present even when the CNS αSyn deposition is limited to the brainstem only, with doubtless no clinical symptoms [223,224].
5. Conclusions
Given the growing incidence and the long list of NMS in the prodromic stage of PD, that negatively impact on health-related quality of life, it is imperative for clinicians to achieve an early diagnosis aimed at prompt intervention to alleviate the symptoms and hopefully to slow the progression of the disease. The correlation between αSyn deposit and the appearance of NMS in the 2- to 20-year pre-motor period is well-documented, although the exact mechanisms have to be fully elucidated. Interestingly, the possibility to detect and quantify αSyn conformers in biofluids and in tissue biopsies reflects the multisystem nature of the disorder and represents a promising diagnostic approach at preclinical PD stage, when neurodegeneration starts. Randomized controlled trials, cohort studies/cross-sectional studies and the validation of PD-specific screening questionnaire together with non-invasive and painless withdrawal procures and sophisticated technology for αSyn measurement could contribute to achievement of timely diagnosis, monitoring and management of PD.
Author Contributions
Conceptualization, C.C. and G.M.M.; Writing—original draft, CC and GMM; Writing-review and editing, C.C. and G.M.M; Visualization C.C and G.M.M.; Supervision C.C. and GMM.
Funding
This work was supported by University of Perugia, Fondo Ricerca di Base di Ateneo, grant number 6RICBASE22 to CC; University of Padova, Budget Integrato per la Ricerca dei Dipartimenti (BIRD), BIRD213814 and BIRD234310 to GMM.
Institutional Review Board Statement
Not applicable.
Acknowledgments
Figures have been created with Biorender.com.
Conflicts of Interest
Both authors declare no conflicts of interest with respect to the authorship and/or publication of this review.
References
- Stevenson, T.J.; Murray, H.C.; Turner, C.; Faull, R.L.M.; Dieriks, B. V.; Curtis, M.A. α-Synuclein Inclusions Are Abundant in Non-Neuronal Cells in the Anterior Olfactory Nucleus of the Parkinson’s Disease Olfactory Bulb. Sci Rep 2020, 10, 6682. [Google Scholar] [CrossRef] [PubMed]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s Disease: Biomarkers, Treatment, and Risk Factors. Front Neurosci 2018, 12. [Google Scholar] [CrossRef]
- Durcan, R.; Wiblin, L.; Lawson, R.A.; Khoo, T.K.; Yarnall, A.J.; Duncan, G.W.; Brooks, D.J.; Pavese, N.; Burn, D.J. Prevalence and Duration of Non-motor Symptoms in Prodromal Parkinson’s Disease. Eur J Neurol 2019, 26, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-Motor Features of Parkinson Disease. Nat Rev Neurosci 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.C.G.; Silva, F.G.; Costa, L.O.P.; Freitas, S.M.S.F. Smell Tests Can Discriminate Parkinson’s Disease Patients from Healthy Individuals: A Meta-Analysis. Clin Neurol Neurosurg 2021, 211, 107024. [Google Scholar] [CrossRef] [PubMed]
- Iravani, B.; Arshamian, A.; Schaefer, M.; Svenningsson, P.; Lundström, J.N. A Non-Invasive Olfactory Bulb Measure Dissociates Parkinson’s Patients from Healthy Controls and Discloses Disease Duration. NPJ Parkinsons Dis 2021, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.; Coulson, E.J.; Rajnarayanan, R.; Oster, H.; Videnovic, A.; Rawashdeh, O. Sleep and Circadian Rhythms in Parkinson’s Disease and Preclinical Models. Mol Neurodegener 2022, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.-C.; Shi, R.; Gao, C.; Yuan, X.-L.; Wu, Y.; Liu, Z.-G.; Wang, C.-D.; Zhao, S.-R.; Chen, X.; Yuan, C.-X.; et al. Autonomic Function and Motor Subtypes in Parkinson’s Disease: A Multicentre Cross-Sectional Study. Sci Rep 2023, 13, 14548. [Google Scholar] [CrossRef]
- Fanciulli, A.; Wenning, G.K. Autonomic Failure: A Neglected Presentation of Parkinson’s Disease. Lancet Neurol 2021, 20, 781–782. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of Oligomeric Forms of A-synuclein Protein in Human Plasma as a Potential Biomarker for Parkinson’s Disease. The FASEB Journal 2006, 20, 419–425. [Google Scholar] [CrossRef]
- Kosaka, K. Latest Concept of Lewy Body Disease. Psychiatry Clin Neurosci 2014, 68, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Pritzkow, S. Protein Misfolding, Aggregation, and Conformational Strains in Neurodegenerative Diseases. Nat Neurosci 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; Dauer, W.T.; Vonsattel, J.P.G. A Critical Evaluation of the Braak Staging Scheme for Parkinson’s Disease. Ann Neurol 2008, 64, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Pyatigorskaya, N.; Mongin, M.; Valabregue, R.; Yahia-Cherif, L.; Ewenczyk, C.; Poupon, C.; Debellemaniere, E.; Vidailhet, M.; Arnulf, I.; Lehéricy, S. Medulla Oblongata Damage and Cardiac Autonomic Dysfunction in Parkinson Disease. Neurology 2016, 87, 2540–2545. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Tredici, K. Del; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Dams, J.; Siebert, U.; Bornschein, B.; Volkmann, J.; Deuschl, G.; Oertel, W.H.; Dodel, R.; Reese, J. Cost-effectiveness of Deep Brain Stimulation in Patients with Parkinson’s Disease. Movement Disorders 2013, 28, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Geng, X.; Huang, B.; Chen, Y.; Jiang, H.; Zou, Y.; Wang, W.; Li, Y.; Li, Y.; Yin, L.; et al. Outcomes of STN-DBS in PD Patients With Different Rates of Disease Progression Over One Year of Follow-Up. Front Neurol 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Fundament, T.; Eldridge, P.R.; Green, A.L.; Whone, A.L.; Taylor, R.S.; Williams, A.C.; Schuepbach, W.M.M. Deep Brain Stimulation for Parkinson’s Disease with Early Motor Complications: A UK Cost-Effectiveness Analysis. PLoS ONE 2016, 11, e0159340. [Google Scholar] [CrossRef] [PubMed]
- Lilleeng, B.; Gjerstad, M.; Baardsen, R.; Dalen, I.; Larsen, J.P. Motor Symptoms after Deep Brain Stimulation of the Subthalamic Nucleus. Acta Neurol Scand 2015, 131, 298–304. [Google Scholar] [CrossRef]
- Listik, C.; Cury, R.G.; Casagrande, S.C.B.; Listik, E.; Arnaut, D.; Santiago, N.; Da Silva, V.A.; Galhardoni, R.; Machado, J. de L.A.; Almeida, J.C. de; et al. Improvement of Non-Motor Symptoms and Quality of Life After Deep Brain Stimulation for Refractory Dystonia: A 1-Year Follow-Up. Front Neurol 2021, 12. [Google Scholar] [CrossRef]
- Eghlidos, Z.; Rahimian, Z.; Vadiee, G.; Jahangiri, S. Effects of Subthalamic Deep Brain Stimulation on Non-motor Symptoms of Parkinson’s Disease: A Meta-analysis. Acta Neurol Scand 2022, 146, 115–125. [Google Scholar] [CrossRef]
- Alonso-Frech, F.; Fernandez-Garcia, C.; Gómez-Mayordomo, V.; Monje, M.H.G.; Delgado-Suarez, C.; Villanueva-Iza, C.; Catalan-Alonso, M.J. Non-Motor Adverse Effects Avoided by Directional Stimulation in Parkinson’s Disease: A Case Report. Front Neurol 2022, 12. [Google Scholar] [CrossRef]
- El Ghazal, N.; Nakanishi, H.; Martinez-Nunez, A.E.; Al Sabbakh, N.K.; Segun-Omosehin, O.A.; Bourdakos, N.E.; Nasser, M.; Matar, R.H.; Than, C.; Danoun, O.A.; et al. The Effects of Deep Brain Stimulation on Mood and Quality of Life in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Cureus 2023. [Google Scholar] [CrossRef]
- Gupta, S.; Shukla, S. Non-Motor Symptoms in Parkinson’s Disease: Opening New Avenues in Treatment. Current Research in Behavioral Sciences 2021, 2, 100049. [Google Scholar] [CrossRef]
- Maroteaux, L.; Campanelli, J.; Scheller, R. Synuclein: A Neuron-Specific Protein Localized to the Nucleus and Presynaptic Nerve Terminal. The Journal of Neuroscience 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of Native Brain α-Synuclein. Nature 2013, 498, E4–E6. [Google Scholar] [CrossRef]
- Cholak, E.; Bugge, K.; Khondker, A.; Gauger, K.; Pedraz-Cuesta, E.; Pedersen, M.E.; Bucciarelli, S.; Vestergaard, B.; Pedersen, S.F.; Rheinstädter, M.C.; et al. Avidity within the N-terminal Anchor Drives A-synuclein Membrane Interaction and Insertion. The FASEB Journal 2020, 34, 7462–7482. [Google Scholar] [CrossRef]
- Hashimoto, M.; Takenouchi, T.; Mallory, M.; Masliah, E.; Takeda, A.; Culvenor, J.G.; McLean, C.A.; Campbell, B.C.V.; Masters, C.L.; Li, Q.-X.; et al. The Role of NAC in Amyloidogenesis in Alzheimer’s Disease. Am J Pathol 2000, 156, 734–735. [Google Scholar] [CrossRef]
- Gao, V.; Briano, J.A.; Komer, L.E.; Burré, J. Functional and Pathological Effects of α-Synuclein on Synaptic SNARE Complexes. J Mol Biol 2023, 435, 167714. [Google Scholar] [CrossRef]
- Diao, J.; Burré, J.; Vivona, S.; Cipriano, D.J.; Sharma, M.; Kyoung, M.; Südhof, T.C.; Brunger, A.T. Native α-Synuclein Induces Clustering of Synaptic-Vesicle Mimics via Binding to Phospholipids and Synaptobrevin-2/VAMP2. Elife 2013, 2. [Google Scholar] [CrossRef]
- Bell, R.; Thrush, R.J.; Castellana-Cruz, M.; Oeller, M.; Staats, R.; Nene, A.; Flagmeier, P.; Xu, C.K.; Satapathy, S.; Galvagnion, C.; et al. N-Terminal Acetylation of α-Synuclein Slows down Its Aggregation Process and Alters the Morphology of the Resulting Aggregates. Biochemistry 2022, 61, 1743–1756. [Google Scholar] [CrossRef]
- Horsley, J.R.; Jovcevski, B.; Pukala, T.L.; Abell, A.D. Designer D-Peptides Targeting the N-Terminal Region of α-Synuclein to Prevent Parkinsonian-Associated Fibrilization and Cytotoxicity. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2022, 1870, 140826. [Google Scholar] [CrossRef] [PubMed]
- Hmila, I.; Vaikath, N.N.; Majbour, N.K.; Erskine, D.; Sudhakaran, I.P.; Gupta, V.; Ghanem, S.S.; Islam, Z.; Emara, M.M.; Abdesselem, H.B.; et al. Novel Engineered Nanobodies Specific for N-terminal Region of Alpha-synuclein Recognize Lewy-body Pathology and Inhibit In-vitro Seeded Aggregation and Toxicity. FEBS J 2022, 289, 4657–4673. [Google Scholar] [CrossRef] [PubMed]
- Daida, K.; Shimonaka, S.; Shiba-Fukushima, K.; Ogata, J.; Yoshino, H.; Okuzumi, A.; Hatano, T.; Motoi, Y.; Hirunagi, T.; Katsuno, M.; et al. A-Synuclein V15A Variant in Familial Parkinson’s Disease Exhibits a Weaker Lipid-Binding Property. Movement Disorders 2022, 37, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Diaw, S.H.; Borsche, M.; Streubel-Gallasch, L.; Dulovic-Mahlow, M.; Hermes, J.; Lenz, I.; Seibler, P.; Klein, C.; Brüggemann, N.; Vos, M.; et al. Characterization of the Pathogenic α-Synuclein Variant V15A in Parkinson´s Disease. NPJ Parkinsons Dis 2023, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Emin, D.; Zhang, Y.P.; Lobanova, E.; Miller, A.; Li, X.; Xia, Z.; Dakin, H.; Sideris, D.I.; Lam, J.Y.L.; Ranasinghe, R.T.; et al. Small Soluble α-Synuclein Aggregates Are the Toxic Species in Parkinson’s Disease. Nat Commun 2022, 13, 5512. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, F.S.; Flagmeier, P.; Kumita, J.R.; Meisl, G.; Chirgadze, D.Y.; Bongiovanni, M.N.; Knowles, T.P.J.; Dobson, C.M. The Influence of Pathogenic Mutations in α-Synuclein on Biophysical and Structural Characteristics of Amyloid Fibrils. ACS Nano 2020, 14, 5213–5222. [Google Scholar] [CrossRef]
- Krzystek, T.J.; Banerjee, R.; Thurston, L.; Huang, J.; Swinter, K.; Rahman, S.N.; Falzone, T.L.; Gunawardena, S. Differential Mitochondrial Roles for α-Synuclein in DRP1-Dependent Fission and PINK1/Parkin-Mediated Oxidation. Cell Death Dis 2021, 12, 796. [Google Scholar] [CrossRef] [PubMed]
- Helwig, M.; Ulusoy, A.; Rollar, A.; O’Sullivan, S.A.; Lee, S.S.L.; Aboutalebi, H.; Pinto-Costa, R.; Jevans, B.; Klinkenberg, M.; Di Monte, D.A. Neuronal Hyperactivity–Induced Oxidant Stress Promotes in Vivo α-Synuclein Brain Spreading. Sci Adv 2022, 8. [Google Scholar] [CrossRef]
- Stojkovska, I.; Wani, W.Y.; Zunke, F.; Belur, N.R.; Pavlenko, E.A.; Mwenda, N.; Sharma, K.; Francelle, L.; Mazzulli, J.R. Rescue of α-Synuclein Aggregation in Parkinson’s Patient Neurons by Synergistic Enhancement of ER Proteostasis and Protein Trafficking. Neuron 2022, 110, 436–451. [Google Scholar] [CrossRef]
- Kang, N.; Han, X.; Li, Z.; Liu, T.; Mi, X.; Li, Y.; Guo, X.; Han, D.; Yang, N. Rapamycin Affects the Hippocampal SNARE Complex to Alleviate Cognitive Dysfunction Induced by Surgery in Aged Rats. Brain Sci 2023, 13, 598. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, D.; Cisternas-Olmedo, M.; Arcos, J.; Nassif, M.; Vidal, R.L. Contribution of Autophagy-Lysosomal Pathway in the Exosomal Secretion of Alpha-Synuclein and Its Impact in the Progression of Parkinson’s Disease. Front Mol Neurosci 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.; Homma, D.; Bloem, B.; Gibb, L.G.; Amemori, K.; Hu, D.; Delcasso, S.; Truong, T.F.; Yang, J.; Hood, A.S.; et al. Chronic Stress Alters Striosome-Circuit Dynamics, Leading to Aberrant Decision-Making. Cell 2017, 171, 1191–1205. [Google Scholar] [CrossRef] [PubMed]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front Mol Neurosci 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-Synuclein Oligomers: A New Hope. Acta Neuropathol 2017, 134, 819–838. [Google Scholar] [CrossRef] [PubMed]
- Alegre-Abarrategui, J.; Brimblecombe, K.R.; Roberts, R.F.; Velentza-Almpani, E.; Tilley, B.S.; Bengoa-Vergniory, N.; Proukakis, C. Selective Vulnerability in α-Synucleinopathies. Acta Neuropathol 2019, 138, 681–704. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, D.; Kaufman, E.; Brundin, L.; Hall, S.; Surova, Y.; Hansson, O. Non-Motor Symptoms in Patients with Parkinson’s Disease – Correlations with Inflammatory Cytokines in Serum. PLoS ONE 2012, 7, e47387. [Google Scholar] [CrossRef]
- Kim, R.; Kim, H.; Shin, J.H.; Lee, C.Y.; Jeon, S.H.; Jeon, B. Serum Inflammatory Markers and Progression of Nonmotor Symptoms in Early Parkinson’s Disease. Movement Disorders 2022, 37, 1535–1541. [Google Scholar] [CrossRef]
- Chavan, S.S.; Pavlov, V.A.; Tracey, K.J. Mechanisms and Therapeutic Relevance of Neuro-Immune Communication. Immunity 2017, 46, 927–942. [Google Scholar] [CrossRef]
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.D.; Lucassen, P.J.; van Dam, A.-M. Microglial Phenotypes and Toll-like Receptor 2 in the Substantia Nigra and Hippocampus of Incidental Lewy Body Disease Cases and Parkinson’s Disease Patients. Acta Neuropathol Commun 2014, 2, 90. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J. Mitochondrial Complex I Deficiency and Parkinson Disease. Nat Rev Neurosci 2023, 24, 193–193. [Google Scholar] [CrossRef]
- Li, X.; Xue, L.; Sun, J.; Sun, Y.; Xie, A. Single Nucleotide Polymorphisms in the Toll-like Receptor 2 (TLR2) Gene Are Associated with Sporadic Parkinson’s Disease in the North-Eastern Han Chinese Population. Neurosci Lett 2017, 656, 72–76. [Google Scholar] [CrossRef]
- Mazzotta, G.M.; Ceccato, N.; Conte, C. Synucleinopathies Take Their Toll: Are TLRs a Way to Go? Cells 2023, 12, 1231. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Toll-like Receptors in the Pathogenesis of Neuroinflammation. J Neuroimmunol 2019, 332, 16–30. [Google Scholar] [CrossRef]
- Kouli, A.; Horne, C.B.; Williams-Gray, C.H. Toll-like Receptors and Their Therapeutic Potential in Parkinson’s Disease and α-Synucleinopathies. Brain Behav Immun 2019, 81, 41–51. [Google Scholar] [CrossRef]
- Heidari, A.; Yazdanpanah, N.; Rezaei, N. The Role of Toll-like Receptors and Neuroinflammation in Parkinson’s Disease. J Neuroinflammation 2022, 19, 135. [Google Scholar] [CrossRef]
- Yoo, J.M.; Lin, Y.; Heo, Y.; Lee, Y.-H. Polymorphism in Alpha-Synuclein Oligomers and Its Implications in Toxicity under Disease Conditions. Front Mol Biosci 2022, 9. [Google Scholar] [CrossRef]
- Ma, J.; Gao, J.; Wang, J.; Xie, A. Prion-Like Mechanisms in Parkinson’s Disease. Front Neurosci 2019, 13. [Google Scholar] [CrossRef]
- Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int J Mol Sci 2021, 22, 8338. [Google Scholar] [CrossRef]
- Garcia, P.; Jürgens-Wemheuer, W.; Uriarte Huarte, O.; Michelucci, A.; Masuch, A.; Brioschi, S.; Weihofen, A.; Koncina, E.; Coowar, D.; Heurtaux, T.; et al. Neurodegeneration and Neuroinflammation Are Linked, but Independent of Alpha-synuclein Inclusions, in a Seeding/Spreading Mouse Model of Parkinson’s Disease. Glia 2022, 70, 935–960. [Google Scholar] [CrossRef]
- Dening, Y.; Straßl, T.; Ruf, V.; Dirscherl, P.; Chovsepian, A.; Stievenard, A.; Khairnar, A.; Schmidt, F.; Giesert, F.; Herms, J.; et al. Toxicity of Extracellular Alpha-Synuclein Is Independent of Intracellular Alpha-Synuclein. Sci Rep 2022, 12, 21951. [Google Scholar] [CrossRef] [PubMed]
- Vasili, E.; Dominguez-Meijide, A.; Flores-León, M.; Al-Azzani, M.; Kanellidi, A.; Melki, R.; Stefanis, L.; Outeiro, T.F. Endogenous Levels of Alpha-Synuclein Modulate Seeding and Aggregation in Cultured Cells. Mol Neurobiol 2022, 59, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Karpowicz, R.J.; Trojanowski, J.Q.; Lee, V.M.-Y. Transmission of α-Synuclein Seeds in Neurodegenerative Disease: Recent Developments. Laboratory Investigation 2019, 99, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Holper, L.; Ben-Shachar, D.; Mann, J. Multivariate Meta-Analyses of Mitochondrial Complex I and IV in Major Depressive Disorder, Bipolar Disorder, Schizophrenia, Alzheimer Disease, and Parkinson Disease. Neuropsychopharmacology 2019, 44, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Tapias, V.; Hu, X.; Luk, K.C.; Sanders, L.H.; Lee, V.M.; Greenamyre, J.T. Synthetic Alpha-Synuclein Fibrils Cause Mitochondrial Impairment and Selective Dopamine Neurodegeneration in Part via INOS-Mediated Nitric Oxide Production. Cellular and Molecular Life Sciences 2017, 74, 2851–2874. [Google Scholar] [CrossRef]
- Yamamoto, K.; Izumi, Y.; Arifuku, M.; Kume, T.; Sawada, H. α-Synuclein Oligomers Mediate the Aberrant Form of Spike-Induced Calcium Release from IP3 Receptor. Sci Rep 2019, 9, 15977. [Google Scholar] [CrossRef] [PubMed]
- Leandrou, E.; Chalatsa, I.; Anagnostou, D.; Machalia, C.; Semitekolou, M.; Filippa, V.; Makridakis, M.; Vlahou, A.; Anastasiadou, E.; Vekrellis, K.; et al. α-Synuclein Oligomers Potentiate Neuroinflammatory NF-ΚB Activity and Induce Cav3.2 Calcium Signaling in Astrocytes. Transl Neurodegener 2024, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Shvachiy, L.; Geraldes, V.; Outeiro, T.F. Uncovering the Molecular Link Between Lead Toxicity and Parkinson’s Disease. Antioxid Redox Signal 2023, 39, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein Binds to TOM20 and Inhibits Mitochondrial Protein Import in Parkinson’s Disease. Sci Transl Med 2016, 8. [Google Scholar] [CrossRef]
- Shan, L.; Heusinkveld, H.J.; Paul, K.C.; Hughes, S.; Darweesh, S.K.L.; Bloem, B.R.; Homberg, J.R. Towards Improved Screening of Toxins for Parkinson’s Risk. NPJ Parkinsons Dis 2023, 9, 169. [Google Scholar] [CrossRef]
- Won, S.J.; Fong, R.; Butler, N.; Sanchez, J.; Zhang, Y.; Wong, C.; Tambou Nzoutchoum, O.; Huynh, A.; Pan, J.; Swanson, R.A. Neuronal Oxidative Stress Promotes α-Synuclein Aggregation In Vivo. Antioxidants 2022, 11, 2466. [Google Scholar] [CrossRef]
- Almandoz-Gil, L.; Welander, H.; Ihse, E.; Khoonsari, P.E.; Musunuri, S.; Lendel, C.; Sigvardson, J.; Karlsson, M.; Ingelsson, M.; Kultima, K.; et al. Low Molar Excess of 4-Oxo-2-Nonenal and 4-Hydroxy-2-Nonenal Promote Oligomerization of Alpha-Synuclein through Different Pathways. Free Radic Biol Med 2017, 110, 421–431. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Villamil Giraldo, A.M.; Öllinger, K.; Hallbeck, M.; Civitelli, L. Lipid Vesicles Affect the Aggregation of 4-Hydroxy-2-Nonenal-Modified α-Synuclein Oligomers. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2018, 1864, 3060–3068. [Google Scholar] [CrossRef]
- Sahin, C.; Østerlund, E.C.; Österlund, N.; Costeira-Paulo, J.; Pedersen, J.N.; Christiansen, G.; Nielsen, J.; Grønnemose, A.L.; Amstrup, S.K.; Tiwari, M.K.; et al. Structural Basis for Dityrosine-Mediated Inhibition of α-Synuclein Fibrillization. J Am Chem Soc 2022, 144, 11949–11954. [Google Scholar] [CrossRef]
- Al-Hilaly, Y.K.; Biasetti, L.; Blakeman, B.J.F.; Pollack, S.J.; Zibaee, S.; Abdul-Sada, A.; Thorpe, J.R.; Xue, W.-F.; Serpell, L.C. The Involvement of Dityrosine Crosslinking in α-Synuclein Assembly and Deposition in Lewy Bodies in Parkinson’s Disease. Sci Rep 2016, 6, 39171. [Google Scholar] [CrossRef]
- Wördehoff, M.M.; Shaykhalishahi, H.; Groß, L.; Gremer, L.; Stoldt, M.; Buell, A.K.; Willbold, D.; Hoyer, W. Opposed Effects of Dityrosine Formation in Soluble and Aggregated α-Synuclein on Fibril Growth. J Mol Biol 2017, 429, 3018–3030. [Google Scholar] [CrossRef]
- Buell, A.K.; Galvagnion, C.; Gaspar, R.; Sparr, E.; Vendruscolo, M.; Knowles, T.P.J.; Linse, S.; Dobson, C.M. Solution Conditions Determine the Relative Importance of Nucleation and Growth Processes in α-Synuclein Aggregation. Proceedings of the National Academy of Sciences 2014, 111, 7671–7676. [Google Scholar] [CrossRef]
- Mehta, N.; Marwah, P.; Njus, D. Are Proteinopathy and Oxidative Stress Two Sides of the Same Coin? Cells 2019, 8, 59. [Google Scholar] [CrossRef]
- Stykel, M.G.; Humphries, K.M.; Kamski-Hennekam, E.; Buchner-Duby, B.; Porte-Trachsel, N.; Ryan, T.; Coackley, C.L.; Bamm, V. V.; Harauz, G.; Ryan, S.D. α-Synuclein Mutation Impairs Processing of Endomembrane Compartments and Promotes Exocytosis and Seeding of α-Synuclein Pathology. Cell Rep 2021, 35, 109099. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Jangir, D.K.; Verma, G.; Shekhar, S.; Hanpude, P.; Kumar, S.; Kumari, R.; Singh, N.; Sarovar Bhavesh, N.; Ranjan Jana, N.; et al. S-Nitrosylation of UCHL1 Induces Its Structural Instability and Promotes α-Synuclein Aggregation. Sci Rep 2017, 7, 44558. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Eckert, A. Brain Aging and Neurodegeneration: From a Mitochondrial Point of View. J Neurochem 2017, 143, 418–431. [Google Scholar] [CrossRef]
- Park, J.-S.; Koentjoro, B.; Davis, R.L.; Sue, C.M. Loss of ATP13A2 Impairs Glycolytic Function in Kufor-Rakeb Syndrome Patient-Derived Cell Models. Parkinsonism Relat Disord 2016, 27, 67–73. [Google Scholar] [CrossRef]
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.M.; Höllerhage, M.; Höglinger, G.U.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial Damage by α-Synuclein Causes Cell Death in Human Dopaminergic Neurons. Cell Death Dis 2019, 10, 865. [Google Scholar] [CrossRef]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef]
- Lehtonen, Š.; Sonninen, T.-M.; Wojciechowski, S.; Goldsteins, G.; Koistinaho, J. Dysfunction of Cellular Proteostasis in Parkinson’s Disease. Front Neurosci 2019, 13. [Google Scholar] [CrossRef]
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease With the Alpha-Synuclein Protein. Front Pharmacol 2020, 11. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A. V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson’s Disease. Nat Commun 2018, 9, 2293. [Google Scholar] [CrossRef]
- Wang, X.-L.; Feng, S.-T.; Wang, Y.-T.; Yuan, Y.-H.; Li, Z.-P.; Chen, N.-H.; Wang, Z.-Z.; Zhang, Y. Mitophagy, a Form of Selective Autophagy, Plays an Essential Role in Mitochondrial Dynamics of Parkinson’s Disease. Cell Mol Neurobiol 2022, 42, 1321–1339. [Google Scholar] [CrossRef]
- Kim, D.Y.; Shin, J.Y.; Lee, J.E.; Kim, H.N.; Chung, S.J.; Yoo, H.S.; Kim, S.J.; Cho, H.J.; Lee, E.-J.; Nam, S.J.; et al. A Selective ER-Phagy Exerts Neuroprotective Effects via Modulation of α-Synuclein Clearance in Parkinsonian Models. Proceedings of the National Academy of Sciences 2023, 120. [Google Scholar] [CrossRef]
- Fu, Y.; He, Y.; Phan, K.; Bhatia, S.; Pickford, R.; Wu, P.; Dzamko, N.; Halliday, G.M.; Kim, W.S. Increased Unsaturated Lipids Underlie Lipid Peroxidation in Synucleinopathy Brain. Acta Neuropathol Commun 2022, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, T.; Li, J.; Xia, M.; Li, Y.; Wang, X.; Liu, C.; Zheng, T.; Chen, R.; Kan, D.; et al. Oxidative Stress and 4-Hydroxy-2-Nonenal (4-HNE): Implications in the Pathogenesis and Treatment of Aging-Related Diseases. J Immunol Res 2022, 2022, 1–12. [Google Scholar] [CrossRef]
- De Franceschi, G.; Frare, E.; Bubacco, L.; Mammi, S.; Fontana, A.; de Laureto, P.P. Molecular Insights into the Interaction between α-Synuclein and Docosahexaenoic Acid. J Mol Biol 2009, 394, 94–107. [Google Scholar] [CrossRef]
- De Franceschi, G.; Frare, E.; Pivato, M.; Relini, A.; Penco, A.; Greggio, E.; Bubacco, L.; Fontana, A.; de Laureto, P.P. Structural and Morphological Characterization of Aggregated Species of α-Synuclein Induced by Docosahexaenoic Acid. Journal of Biological Chemistry 2011, 286, 22262–22274. [Google Scholar] [CrossRef]
- Cai, Y.; Lendel, C.; Österlund, L.; Kasrayan, A.; Lannfelt, L.; Ingelsson, M.; Nikolajeff, F.; Karlsson, M.; Bergström, J. Changes in Secondary Structure of α-Synuclein during Oligomerization Induced by Reactive Aldehydes. Biochem Biophys Res Commun 2015, 464, 336–341. [Google Scholar] [CrossRef]
- Beal, M.F.; Chiluwal, J.; Calingasan, N.Y.; Milne, G.L.; Shchepinov, M.S.; Tapias, V. Isotope-Reinforced Polyunsaturated Fatty Acids Improve Parkinson’s Disease-like Phenotype in Rats Overexpressing α-Synuclein. Acta Neuropathol Commun 2020, 8, 220. [Google Scholar] [CrossRef]
- Galvagnion, C. The Role of Lipids Interacting with α-Synuclein in the Pathogenesis of Parkinson’s Disease. J Parkinsons Dis 2017, 7, 433–450. [Google Scholar] [CrossRef]
- Bonam, S.R.; Tranchant, C.; Muller, S. Autophagy-Lysosomal Pathway as Potential Therapeutic Target in Parkinson’s Disease. Cells 2021, 10, 3547. [Google Scholar] [CrossRef]
- Kinnart, I.; Manders, L.; Heyninck, T.; Imberechts, D.; Praschberger, R.; Schoovaerts, N.; Verfaillie, C.; Verstreken, P.; Vandenberghe, W. Elevated α-Synuclein Levels Inhibit Mitophagic Flux. NPJ Parkinsons Dis 2024, 10, 80. [Google Scholar] [CrossRef]
- Stykel, M.G.; Ryan, S.D. Nitrosative Stress in Parkinson’s Disease. NPJ Parkinsons Dis 2022, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Bisi, N.; Feni, L.; Peqini, K.; Pérez-Peña, H.; Ongeri, S.; Pieraccini, S.; Pellegrino, S. α-Synuclein: An All-Inclusive Trip Around Its Structure, Influencing Factors and Applied Techniques. Front Chem 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, N.; Lemminger, L.; Pedersen, J.N.; Nielsen, S.B.; Otzen, D.E. The N-terminus of A-synuclein Is Essential for Both Monomeric and Oligomeric Interactions with Membranes. FEBS Lett 2014, 588, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Abeyawardhane, D.L.; Fernández, R.D.; Murgas, C.J.; Heitger, D.R.; Forney, A.K.; Crozier, M.K.; Lucas, H.R. Iron Redox Chemistry Promotes Antiparallel Oligomerization of α-Synuclein. J Am Chem Soc 2018, 140, 5028–5032. [Google Scholar] [CrossRef]
- Chang, C.J. Bioinorganic Life and Neural Activity: Toward a Chemistry of Consciousness? Acc Chem Res 2017, 50, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Bubacco, L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules 2020, 10, 195. [Google Scholar] [CrossRef]
- Exley, C.; Mold, M.J. Aluminium in Human Brain Tissue: How Much Is Too Much? JBIC Journal of Biological Inorganic Chemistry 2019, 24, 1279–1282. [Google Scholar] [CrossRef]
- Mold, M.J.; Exley, C. Aluminium Co-Localises with Biondi Ring Tangles in Parkinson’s Disease and Epilepsy. Sci Rep 2022, 12, 1465. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.G.; Andrews, H.; Amara, A.; Naito, A.; Alcalay, R.N.; Shaw, L.M.; Taylor, P.; Xie, T.; Tuite, P.; Henchcliffe, C.; et al. Cerebrospinal Fluid, Plasma, and Saliva in the BioFIND Study: Relationships among Biomarkers and Parkinson’s Disease Features. Movement Disorders 2018, 33, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Chahine, L.M.; Beach, T.G.; Brumm, M.C.; Adler, C.H.; Coffey, C.S.; Mosovsky, S.; Caspell-Garcia, C.; Serrano, G.E.; Munoz, D.G.; White, C.L.; et al. In Vivo Distribution of α-Synuclein in Multiple Tissues and Biofluids in Parkinson Disease. Neurology 2020, 95. [Google Scholar] [CrossRef]
- Ma, L.-Y.; Liu, G.-L.; Wang, D.-X.; Zhang, M.-M.; Kou, W.-Y.; Feng, T. Alpha-Synuclein in Peripheral Tissues in Parkinson’s Disease. ACS Chem Neurosci 2019, 10, 812–823. [Google Scholar] [CrossRef]
- Lee, J.M.; Derkinderen, P.; Kordower, J.H.; Freeman, R.; Munoz, D.G.; Kremer, T.; Zago, W.; Hutten, S.J.; Adler, C.H.; Serrano, G.E.; et al. The Search for a Peripheral Biopsy Indicator of α-Synuclein Pathology for Parkinson Disease. J Neuropathol Exp Neurol 2017, nlw103. [Google Scholar] [CrossRef]
- Chen, M.; Mor, D.E. Gut-to-Brain α-Synuclein Transmission in Parkinson’s Disease: Evidence for Prion-like Mechanisms. Int J Mol Sci 2023, 24, 7205. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, W.; Liu, P.; Wang, Z.; Zhou, Y.; Liu, X.; Li, A. α-Synuclein Aggregation in the Olfactory Bulb Induces Olfactory Deficits by Perturbing Granule Cells and Granular–Mitral Synaptic Transmission. NPJ Parkinsons Dis 2021, 7, 114. [Google Scholar] [CrossRef]
- Oliveira, L.M.A.; Gasser, T.; Edwards, R.; Zweckstetter, M.; Melki, R.; Stefanis, L.; Lashuel, H.A.; Sulzer, D.; Vekrellis, K.; Halliday, G.M.; et al. Alpha-Synuclein Research: Defining Strategic Moves in the Battle against Parkinson’s Disease. NPJ Parkinsons Dis 2021, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, T.; Sancesario, G.M.; Di Lazzaro, G.; Biticchi, B.; Colona, V.L.; Mercuri, N.B.; Bernardini, S.; Pisani, A. CSF α-Synuclein Inversely Correlates with Non-Motor Symptoms in a Cohort of PD Patients. Parkinsonism Relat Disord 2019, 61, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Maillet, A.; Météreau, E.; Tremblay, L.; Favre, E.; Klinger, H.; Lhommée, E.; Le Bars, D.; Castrioto, A.; Prange, S.; Sgambato, V.; et al. Serotonergic and Dopaminergic Lesions Underlying Parkinsonian Neuropsychiatric Signs. Movement Disorders 2021, 36, 2888–2900. [Google Scholar] [CrossRef] [PubMed]
- Hayley, S.; Vahid-Ansari, F.; Sun, H.; Albert, P.R. Mood Disturbances in Parkinson’s Disease: From Prodromal Origins to Application of Animal Models. Neurobiol Dis 2023, 181, 106115. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.; Lim, J.; Choi, H.J. Recent Advances in the Pathology of Prodromal Non-Motor Symptoms Olfactory Deficit and Depression in Parkinson’s Disease: Clues to Early Diagnosis and Effective Treatment. Arch Pharm Res 2021, 44, 588–604. [Google Scholar] [CrossRef]
- Stocchi, F.; Angelo Antonini; Barone, P. ; Bellelli, G.; Fagiolini, A.; Ferini Strambi, L.; Sorbi, S.; Padovani, A. Exploring Depression in Parkinson’s Disease: An Italian Delphi Consensus on Phenomenology, Diagnosis, and Management. Neurological Sciences 2023, 44, 3123–3131. [Google Scholar] [CrossRef] [PubMed]
- Banwinkler, M.; Theis, H.; Prange, S.; van Eimeren, T. Imaging the Limbic System in Parkinson’s Disease—A Review of Limbic Pathology and Clinical Symptoms. Brain Sci 2022, 12, 1248. [Google Scholar] [CrossRef]
- Prange, S.; Metereau, E.; Maillet, A.; Lhommée, E.; Klinger, H.; Pelissier, P.; Ibarrola, D.; Heckemann, R.A.; Castrioto, A.; Tremblay, L.; et al. Early Limbic Microstructural Alterations in Apathy and Depression in de Novo Parkinson’s Disease. Movement Disorders 2019, 34, 1644–1654. [Google Scholar] [CrossRef]
- Buchanan, A.M.; Mena, S.; Choukari, I.; Vasa, A.; Crawford, J.N.; Fadel, J.; Maxwell, N.; Reagan, L.; Cruikshank, A.; Best, J.; et al. Serotonin as a Biomarker of Toxin-Induced Parkinsonism. Molecular Medicine 2024, 30, 33. [Google Scholar] [CrossRef]
- Tong, Q.; Zhang, L.; Yuan, Y.; Jiang, S.; Zhang, R.; Xu, Q.; Ding, J.; Li, D.; Zhou, X.; Zhang, K. Reduced Plasma Serotonin and 5-Hydroxyindoleacetic Acid Levels in Parkinson’s Disease Are Associated with Nonmotor Symptoms. Parkinsonism Relat Disord 2015, 21, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Kamagata, K.; Nakatsuka, T.; Sakakibara, R.; Tsuyusaki, Y.; Takamura, T.; Sato, K.; Suzuki, M.; Hori, M.; Kumamaru, K.K.; Inaoka, T.; et al. Diagnostic Imaging of Dementia with Lewy Bodies by Susceptibility-Weighted Imaging of Nigrosomes versus Striatal Dopamine Transporter Single-Photon Emission Computed Tomography: A Retrospective Observational Study. Neuroradiology 2017, 59, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Butkovich, L.M.; Houser, M.C.; Chalermpalanupap, T.; Porter-Stransky, K.A.; Iannitelli, A.F.; Boles, J.S.; Lloyd, G.M.; Coomes, A.S.; Eidson, L.N.; De Sousa Rodrigues, M.E.; et al. Transgenic Mice Expressing Human α-Synuclein in Noradrenergic Neurons Develop Locus Ceruleus Pathology and Nonmotor Features of Parkinson’s Disease. The Journal of Neuroscience 2020, 40, 7559–7576. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Li, G.; Luo, H.; Pan, Y.; Xu, Q.; Ma, K. Hippocampal Alpha-Synuclein Mediates Depressive-like Behaviors. Brain Behav Immun 2021, 95, 226–237. [Google Scholar] [CrossRef]
- Miquel-Rio, L.; Alarcón-Arís, D.; Torres-López, M.; Cóppola-Segovia, V.; Pavia-Collado, R.; Paz, V.; Ruiz-Bronchal, E.; Campa, L.; Casal, C.; Montefeltro, A.; et al. Human α-Synuclein Overexpression in Mouse Serotonin Neurons Triggers a Depressive-like Phenotype. Rescue by Oligonucleotide Therapy. Transl Psychiatry 2022, 12, 79. [Google Scholar] [CrossRef]
- Dissanayaka, N.N.W.; White, E.; O’Sullivan, J.D.; Marsh, R.; Silburn, P.A.; Copland, D.A.; Mellick, G.D.; Byrne, G.J. Characteristics and Treatment of Anxiety Disorders in Parkinson’s Disease. Mov Disord Clin Pract 2015, 2, 155–162. [Google Scholar] [CrossRef]
- Khedr, E.M.; Abdelrahman, A.A.; Elserogy, Y.; Zaki, A.F.; Gamea, A. Depression and Anxiety among Patients with Parkinson’s Disease: Frequency, Risk Factors, and Impact on Quality of Life. Egypt J Neurol Psychiatr Neurosurg 2020, 56, 116. [Google Scholar] [CrossRef]
- Lai, T.T.; Gericke, B.; Feja, M.; Conoscenti, M.; Zelikowsky, M.; Richter, F. Anxiety in Synucleinopathies: Neuronal Circuitry, Underlying Pathomechanisms and Current Therapeutic Strategies. NPJ Parkinsons Dis 2023, 9, 97. [Google Scholar] [CrossRef]
- Stoyka, L.E.; Arrant, A.E.; Thrasher, D.R.; Russell, D.L.; Freire, J.; Mahoney, C.L.; Narayanan, A.; Dib, A.G.; Standaert, D.G.; Volpicelli-Daley, L.A. Behavioral Defects Associated with Amygdala and Cortical Dysfunction in Mice with Seeded α-Synuclein Inclusions. Neurobiol Dis 2020, 134, 104708. [Google Scholar] [CrossRef]
- Uemura, N.; Ueda, J.; Yoshihara, T.; Ikuno, M.; Uemura, M.T.; Yamakado, H.; Asano, M.; Trojanowski, J.Q.; Takahashi, R. A-Synuclein Spread from Olfactory Bulb Causes Hyposmia, Anxiety, and Memory Loss in BAC-SNCA Mice. Movement Disorders 2021, 36, 2036–2047. [Google Scholar] [CrossRef] [PubMed]
- Taddei, R.N.; Cankaya, S.; Dhaliwal, S.; Chaudhuri, K.R. Management of Psychosis in Parkinson’s Disease: Emphasizing Clinical Subtypes and Pathophysiological Mechanisms of the Condition. Parkinsons Dis 2017, 2017, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Polissidis, A.; Koronaiou, M.; Kollia, V.; Koronaiou, E.; Nakos-Bimpos, M.; Bogiongko, M.; Vrettou, S.; Karali, K.; Casadei, N.; Riess, O.; et al. Psychosis-Like Behavior and Hyperdopaminergic Dysregulation in Human α -Synuclein <scp>BAC</Scp> Transgenic Rats. Movement Disorders 2021, 36, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Pagonabarraga, J.; Bejr-Kasem, H.; Martinez-Horta, S.; Kulisevsky, J. Parkinson Disease Psychosis: From Phenomenology to Neurobiological Mechanisms. Nat Rev Neurol 2024, 20, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Savica, R.; Grossardt, B.R.; Bower, J.H.; Ahlskog, J.E.; Mielke, M.M.; Rocca, W.A. Incidence and Time Trends of Drug-Induced Parkinsonism: A 30-Year Population-Based Study. Movement Disorders 2017, 32, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Göverti, D.; Büyüklüoğlu, N.; Nazik Yüksel, R.; Kaya, H.; Yücel, Ç.; Göka, E. Decreased Serum Levels of A-synuclein in Patients with Schizophrenia and Their Unaffected Siblings. Early Interv Psychiatry 2023, 17, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Demirel, Ö.F.; Cetin, İ.; Turan, Ş.; Sağlam, T.; Yıldız, N.; Duran, A. Decreased Expression of α-Synuclein, Nogo-A and UCH-L1 in Patients with Schizophrenia: A Preliminary Serum Study. Psychiatry Investig 2017, 14, 344. [Google Scholar] [CrossRef] [PubMed]
- Noori-Daloii, M.R.; Kheirollahi, M.; Mahbod, P.; Mohammadi, F.; Astaneh, A.N.; Zarindast, M.R.; Azimi, C.; Mohammadi, M.R. Alpha- and Beta-Synucleins MRNA Expression in Lymphocytes of Schizophrenia Patients. Genet Test Mol Biomarkers 2010, 14, 725–729. [Google Scholar] [CrossRef]
- Takamura, S.; Ikeda, A.; Nishioka, K.; Furuya, H.; Tashiro, M.; Matsushima, T.; Li, Y.; Yoshino, H.; Funayama, M.; Morinobu, S.; et al. Schizophrenia as a Prodromal Symptom in a Patient Harboring SNCA Duplication. Parkinsonism Relat Disord 2016, 25, 108–109. [Google Scholar] [CrossRef]
- Mosquera, F.E.C.; Guevara-Montoya, M.C.; Serna-Ramirez, V.; Liscano, Y. Neuroinflammation and Schizophrenia: New Therapeutic Strategies through Psychobiotics, Nanotechnology, and Artificial Intelligence (AI). J Pers Med 2024, 14, 391. [Google Scholar] [CrossRef]
- Gerlach, M.; Sharma, M.; Romanos, M.; Lesch, K.-P.; Walitza, S.; Conzelmann, H.A.; Krüger, R.; Renner, T.J. Family-Based Association Study on Functional α-Synuclein Polymorphisms in Attention-Deficit/Hyperactivity Disorder. ADHD Attention Deficit and Hyperactivity Disorders 2019, 11, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Erskine, D.; Thomas, A.J.; Taylor, J.-P.; Savage, M.A.; Attems, J.; McKeith, I.G.; Morris, C.M.; Khundakar, A.A. Neuronal Loss and A-Synuclein Pathology in the Superior Colliculus and Its Relationship to Visual Hallucinations in Dementia with Lewy Bodies. The American Journal of Geriatric Psychiatry 2017, 25, 595–604. [Google Scholar] [CrossRef]
- Doxakis, E. Post-Transcriptional Regulation of α-Synuclein Expression by Mir-7 and Mir-153. Journal of Biological Chemistry 2010, 285, 12726–12734. [Google Scholar] [CrossRef]
- Yang TT, L.C.G.S.Z.Y.W.P. The Serum Exosome Derived MicroRNA-135a, -193b, and -384 Were Potential Alzheimer’s Disease Biomarkers. Biomed Environ Sci 2018. [Google Scholar]
- Han, L.; Tang, Y.; Bai, X.; Liang, X.; Fan, Y.; Shen, Y.; Huang, F.; Wang, J. Association of the Serum MicroRNA-29 Family with Cognitive Impairment in Parkinson’s Disease. Aging 2020, 12, 13518–13528. [Google Scholar] [CrossRef]
- Li, S.; Lei, Z.; Sun, T. The Role of MicroRNAs in Neurodegenerative Diseases: A Review. Cell Biol Toxicol 2023, 39, 53–83. [Google Scholar] [CrossRef] [PubMed]
- Stabile, F.; Torromino, G.; Rajendran, S.; Del Vecchio, G.; Presutti, C.; Mannironi, C.; De Leonibus, E.; Mele, A.; Rinaldi, A. Short-Term Memory Deficit Associates with MiR-153-3p Upregulation in the Hippocampus of Middle-Aged Mice. Mol Neurobiol 2024, 61, 3031–3041. [Google Scholar] [CrossRef] [PubMed]
- Palmqvist, S.; Rossi, M.; Hall, S.; Quadalti, C.; Mattsson-Carlgren, N.; Dellavalle, S.; Tideman, P.; Pereira, J.B.; Nilsson, M.H.; Mammana, A.; et al. Cognitive Effects of Lewy Body Pathology in Clinically Unimpaired Individuals. Nat Med 2023, 29, 1971–1978. [Google Scholar] [CrossRef]
- Manchinu, M.F.; Pala, M.; Palmas, M.F.; Diana, M.A.; Maschio, A.; Etzi, M.; Pisanu, A.; Diana, F.I.; Marongiu, J.; Mansueto, S.; et al. Region-Specific Changes in Gene Expression Are Associated with Cognitive Deficits in the Alpha-Synuclein-Induced Model of Parkinson’s Disease: A Transcriptomic Profiling Study. Exp Neurol 2024, 372, 114651. [Google Scholar] [CrossRef]
- Ford, B. Pain in Parkinson’s Disease. Movement Disorders 2010, 25. [Google Scholar] [CrossRef]
- Buhidma, Y.; Hobbs, C.; Malcangio, M.; Duty, S. Periaqueductal Grey and Spinal Cord Pathology Contribute to Pain in Parkinson’s Disease. NPJ Parkinsons Dis 2023, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.; Gonçalves, N.P.; Jan, A.; Jensen, N.M.; van der Laan, A.; Mohseni, S.; Vægter, C.B.; Jensen, P.H. Trans-Synaptic Spreading of Alpha-Synuclein Pathology through Sensory Afferents Leads to Sensory Nerve Degeneration and Neuropathic Pain. Acta Neuropathol Commun 2021, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Möller, M.; Möser, C. V.; Weiß, U.; Niederberger, E. The Role of AlphαSynuclein in Mouse Models of Acute, Inflammatory and Neuropathic Pain. Cells 2022, 11, 1967. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, J.; Chao, D.; Sandhu, H.K.; Liao, X.; Zhao, J.; Wen, G.; Xia, Y. δ-Opioid Receptor Activation Reduces α-Synuclein Overexpression and Oligomer Formation Induced by MPP+ and/or Hypoxia. Exp Neurol 2014, 255, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; White, C.L.; Hladik, C.L.; Sabbagh, M.N.; Connor, D.J.; Shill, H.A.; Sue, L.I.; Sasse, J.; Bachalakuri, J.; Henry-Watson, J.; et al. Olfactory Bulb α-Synucleinopathy Has High Specificity and Sensitivity for Lewy Body Disorders. Acta Neuropathol 2009, 117, 169. [Google Scholar] [CrossRef] [PubMed]
- Mason, D.M.; Wang, Y.; Bhatia, T.N.; Miner, K.M.; Trbojevic, S.A.; Stolz, J.F.; Luk, K.C.; Leak, R.K. The Center of Olfactory Bulb-seeded A-synucleinopathy Is the Limbic System and the Ensuing Pathology Is Higher in Male than in Female Mice. Brain Pathology 2019, 29, 741–770. [Google Scholar] [CrossRef]
- Johnson, M.E.; Bergkvist, L.; Mercado, G.; Stetzik, L.; Meyerdirk, L.; Wolfrum, E.; Madaj, Z.; Brundin, P.; Wesson, D.W. Deficits in Olfactory Sensitivity in a Mouse Model of Parkinson’s Disease Revealed by Plethysmography of Odor-Evoked Sniffing. Sci Rep 2020, 10, 9242. [Google Scholar] [CrossRef] [PubMed]
- Martin-Lopez, E.; Vidyadhara, D.J.; Liberia, T.; Meller, S.J.; Harmon, L.E.; Hsu, R.M.; Spence, N.; Brennan, B.; Han, K.; Yücel, B.; et al. α-Synuclein Pathology and Reduced Neurogenesis in the Olfactory System Affect Olfaction in a Mouse Model of Parkinson’s Disease. The Journal of Neuroscience 2023, 43, 1051–1071. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Wang, K.; Deng, X.-H.; Wei, Y.-H.; Guo, R.; Liu, S.-F.; Zhu, Y.-F.; Zhong, J.-J.; Zheng, J.-Y.; Wang, M.-D.; et al. Amelioration of Olfactory Dysfunction in a Mouse Model of Parkinson’s Disease via Enhancing GABAergic Signaling. Cell Biosci 2023, 13, 101. [Google Scholar] [CrossRef]
- Nieto-Escamez, F.; Obrero-Gaitán, E.; Cortés-Pérez, I. Visual Dysfunction in Parkinson’s Disease. Brain Sci 2023, 13, 1173. [Google Scholar] [CrossRef]
- Ortuño-Lizarán, I.; Esquiva, G.; Beach, T.G.; Serrano, G.E.; Adler, C.H.; Lax, P.; Cuenca, N. Degeneration of Human Photosensitive Retinal Ganglion Cells May Explain Sleep and Circadian Rhythms Disorders in Parkinson’s Disease. Acta Neuropathol Commun 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, E.; Indrieri, A.; Esposito, F.; Tarallo, V.; Carboncino, A.; Alvino, F.G.; De Falco, S.; Franco, B.; De Risi, M.; De Leonibus, E. α-Synuclein Overexpression in the Retina Leads to Vision Impairment and Degeneration of Dopaminergic Amacrine Cells. Sci Rep 2020, 10, 9619. [Google Scholar] [CrossRef] [PubMed]
- Samizadeh, M.-A.; Fallah, H.; Toomarisahzabi, M.; Rezaei, F.; Rahimi-Danesh, M.; Akhondzadeh, S.; Vaseghi, S. Parkinson’s Disease: A Narrative Review on Potential Molecular Mechanisms of Sleep Disturbances, REM Behavior Disorder, and Melatonin. Brain Sci 2023, 13, 914. [Google Scholar] [CrossRef] [PubMed]
- Roguski, A.; Rayment, D.; Whone, A.L.; Jones, M.W.; Rolinski, M. A Neurologist’s Guide to REM Sleep Behavior Disorder. Front Neurol 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.-C.; Lee, H.-H.; Hong, C.-T.; Hu, C.-J.; Wu, D. REM Sleep Behavior Disorder (RBD) in Dementia with Lewy Bodies (DLB). Behavioural Neurology 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Schaffrath, A.; Schleyken, S.; Seger, A.; Jergas, H.; Özdüzenciler, P.; Pils, M.; Blömeke, L.; Cousin, A.; Willbold, J.; Bujnicki, T.; et al. Patients with Isolated REM-Sleep Behavior Disorder Have Elevated Levels of Alpha-Synuclein Aggregates in Stool. NPJ Parkinsons Dis 2023, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Ikuno, M.; Hondo, M.; Parajuli, L.K.; Taguchi, K.; Ueda, J.; Sawamura, M.; Okuda, S.; Nakanishi, E.; Hara, J.; et al. α-Synuclein BAC Transgenic Mice Exhibit RBD-like Behaviour and Hyposmia: A Prodromal Parkinson’s Disease Model. Brain 2020, 143, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, A.; Fernández-Arcos, A.; Tolosa, E.; Serradell, M.; Molinuevo, J.L.; Valldeoriola, F.; Gelpi, E.; Vilaseca, I.; Sánchez-Valle, R.; Lladó, A.; et al. Neurodegenerative Disorder Risk in Idiopathic REM Sleep Behavior Disorder: Study in 174 Patients. PLoS ONE 2014, 9, e89741. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.P.; Vuono, R.; Nawarathna, U.; Fisher, K.; Shneerson, J.M.; Reddy, A.B.; Barker, R.A. Sleep and Circadian Rhythm Regulation in Early Parkinson Disease. JAMA Neurol 2014, 71, 589. [Google Scholar] [CrossRef]
- Diaconu, Ş.; Irincu, L.; Ungureanu, L.; Ciopleiaș, B.; Țînț, D.; Falup-Pecurariu, C. Restless Legs Syndrome in Parkinson’s Disease. J Pers Med 2023, 13, 915. [Google Scholar] [CrossRef]
- Azmin, S.; Khairul Anuar, A.M.; Nafisah, W.Y.; Tan, H.J.; Raymond, A.A.; Hanita, O.; Shah, S.A.; Norlinah, M.I. Restless Legs Syndrome and Its Associated Risk Factors in Parkinson’s Disease. Parkinsons Dis 2013, 2013, 1–5. [Google Scholar] [CrossRef]
- Chung, S.; Bohnen, N.I.; Albin, R.L.; Frey, K.A.; Müller, M.L.T.M.; Chervin, R.D. Insomnia and Sleepiness in Parkinson Disease: Associations with Symptoms and Comorbidities. Journal of Clinical Sleep Medicine 2013, 09, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the Development of Parkinson’s Disease-Related Pathology. Cell Tissue Res 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Lu, J.; Sherman, D.; Devor, M.; Saper, C.B. A Putative Flip–Flop Switch for Control of REM Sleep. Nature 2006, 441, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.M.S.; Andersen, M.L.; Reksidler, A.B.; Vital, M.A.B.F.; Tufik, S. The Role of the Substantia Nigra Pars Compacta in Regulating Sleep Patterns in Rats. PLoS ONE 2007, 2, e513. [Google Scholar] [CrossRef] [PubMed]
- Willison, L.D.; Kudo, T.; Loh, D.H.; Kuljis, D.; Colwell, C.S. Circadian Dysfunction May Be a Key Component of the Non-Motor Symptoms of Parkinson’s Disease: Insights from a Transgenic Mouse Model. Exp Neurol 2013, 243, 57–66. [Google Scholar] [CrossRef]
- McDowell, K.A.; Shin, D.; Roos, K.P.; Chesselet, M.-F. Sleep Dysfunction and EEG Alterations in Mice Overexpressing Alpha-Synuclein. J Parkinsons Dis 2018, 4, 531–539. [Google Scholar] [CrossRef]
- Gajula Balija, M.B.; Griesinger, C.; Herzig, A.; Zweckstetter, M.; Jäckle, H. Pre-Fibrillar α-Synuclein Mutants Cause Parkinson’s Disease-Like Non-Motor Symptoms in Drosophila. PLoS ONE 2011, 6, e24701. [Google Scholar] [CrossRef]
- Dhanushkodi, N.R.; Abul Khair, S.B.; Ardah, M.T.; Haque, M.E. ATP13A2 Gene Silencing in Drosophila Affects Autophagic Degradation of A53T Mutant α-Synuclein. Int J Mol Sci 2023, 24, 1775. [Google Scholar] [CrossRef]
- Abul Khair, S.B.; Dhanushkodi, N.R.; Ardah, M.T.; Chen, W.; Yang, Y.; Haque, M.E. Silencing of Glucocerebrosidase Gene in Drosophila Enhances the Aggregation of Parkinson’s Disease Associated α-Synuclein Mutant A53T and Affects Locomotor Activity. Front Neurosci 2018, 12. [Google Scholar] [CrossRef]
- Ito, K.; Kawasaki, H.; Suzuki, T.; Takahara, T.; Ishida, N. Effects of Kamikihito and Unkei-to on Sleep Behavior of Wild Type and Parkinson Model in Drosophila. Front Psychiatry 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Borbély, A. The Two-process Model of Sleep Regulation: Beginnings and Outlook. J Sleep Res 2022, 31. [Google Scholar] [CrossRef]
- Patke, A.; Young, M.W.; Axelrod, S. Molecular Mechanisms and Physiological Importance of Circadian Rhythms. Nat Rev Mol Cell Biol 2020, 21, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Videnovic, A.; Noble, C.; Reid, K.J.; Peng, J.; Turek, F.W.; Marconi, A.; Rademaker, A.W.; Simuni, T.; Zadikoff, C.; Zee, P.C. Circadian Melatonin Rhythm and Excessive Daytime Sleepiness in Parkinson Disease. JAMA Neurol 2014, 71, 463. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, T.; Schäfer, K.-H.; Claus, I.; Del Tredici, K.; Jost, W.H. Gastrointestinal Involvement in Parkinson’s Disease: Pathophysiology, Diagnosis, and Management. NPJ Parkinsons Dis 2022, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Camerucci, E.; Mullan, A.F.; Bower, J.H.; Bharucha, A.E.; Turcano, P.; Stang, C.D.; Benarroch, E.E.; Boeve, B.F.; Ahlskog, J.E.; Savica, R. Lifelong Constipation in Parkinson’s Disease and Other Clinically Defined Alpha-Synucleinopathies: A Population-Based Study in Southeast Minnesota. Parkinsonism Relat Disord 2023, 107, 105244. [Google Scholar] [CrossRef] [PubMed]
- Borghammer, P.; Horsager, J.; Andersen, K.; Van Den Berge, N.; Raunio, A.; Murayama, S.; Parkkinen, L.; Myllykangas, L. Neuropathological Evidence of Body-First vs. Brain-First Lewy Body Disease. Neurobiol Dis 2021, 161, 105557. [Google Scholar] [CrossRef] [PubMed]
- Horsager, J.; Andersen, K.B.; Knudsen, K.; Skjærbæk, C.; Fedorova, T.D.; Okkels, N.; Schaeffer, E.; Bonkat, S.K.; Geday, J.; Otto, M.; et al. Brain-First versus Body-First Parkinson’s Disease: A Multimodal Imaging Case-Control Study. Brain 2020, 143, 3077–3088. [Google Scholar] [CrossRef] [PubMed]
- Borghammer, P.; Just, M.K.; Horsager, J.; Skjærbæk, C.; Raunio, A.; Kok, E.H.; Savola, S.; Murayama, S.; Saito, Y.; Myllykangas, L.; et al. A Postmortem Study Suggests a Revision of the Dual-Hit Hypothesis of Parkinson’s Disease. NPJ Parkinsons Dis 2022, 8, 166. [Google Scholar] [CrossRef]
- Li, Y.; Tong, Q.; Wang, Y.; Cheng, Y.; Geng, Y.; Tian, T.; Yuan, Y.; Fan, Y.; Lu, M.; Zhang, K. Phosphorylated α-Synuclein Deposited in Schwann Cells Interacting with TLR2 Mediates Cell Damage and Induces Parkinson’s Disease Autonomic Dysfunction. Cell Death Discov 2024, 10, 52. [Google Scholar] [CrossRef]
- Dutta, D.; Jana, M.; Majumder, M.; Mondal, S.; Roy, A.; Pahan, K. Selective Targeting of the TLR2/MyD88/NF-ΚB Pathway Reduces α-Synuclein Spreading in Vitro and in Vivo. Nat Commun 2021, 12, 5382. [Google Scholar] [CrossRef]
- Conte, C.; Ingrassia, A.; Breve, J.; Bol, J.J.; Timmermans-Huisman, E.; van Dam, A.-M.; Beccari, T.; van de Berg, W.D.J. Toll-like Receptor 4 Is Upregulated in Parkinson’s Disease Patients and Co-Localizes with PSer129αSyn: A Possible Link with the Pathology. Cells 2023, 12, 1368. [Google Scholar] [CrossRef] [PubMed]
- Conte, C.; Sichetti, M.; Traina, G. Gut–Brain Axis: Focus on Neurodegeneration and Mast Cells. Applied Sciences 2020, 10, 1828. [Google Scholar] [CrossRef]
- Ryman, S.; Vakhtin, A.A.; Richardson, S.P.; Lin, H.C. Microbiome–Gut–Brain Dysfunction in Prodromal and Symptomatic Lewy Body Diseases. J Neurol 2023, 270, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.H.; Chuah, K.H.; Beh, Y.Y.; Schee, J.P.; Mahadeva, S.; Lim, S.-Y. Gastrointestinal Dysfunction in Parkinson’s Disease: Neuro-Gastroenterology Perspectives on a Multifaceted Problem. J Mov Disord 2023, 16, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Liddle, R.A. Parkinson’s Disease from the Gut. Brain Res 2018, 1693, 201–206. [Google Scholar] [CrossRef]
- Xu, J.; Wang, L.; Chen, X.; Le, W. New Understanding on the Pathophysiology and Treatment of Constipation in Parkinson’s Disease. Front Aging Neurosci 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Rota, L.; Pellegrini, C.; Benvenuti, L.; Antonioli, L.; Fornai, M.; Blandizzi, C.; Cattaneo, A.; Colla, E. Constipation, Deficit in Colon Contractions and Alpha-Synuclein Inclusions within the Colon Precede Motor Abnormalities and Neurodegeneration in the Central Nervous System in a Mouse Model of Alpha-Synucleinopathy. Transl Neurodegener 2019, 8, 5. [Google Scholar] [CrossRef]
- Qiao, C.-M.; Sun, M.-F.; Jia, X.-B.; Li, Y.; Zhang, B.-P.; Zhao, L.-P.; Shi, Y.; Zhou, Z.-L.; Zhu, Y.-L.; Cui, C.; et al. Sodium Butyrate Exacerbates Parkinson’s Disease by Aggravating Neuroinflammation and Colonic Inflammation in MPTP-Induced Mice Model. Neurochem Res 2020, 45, 2128–2142. [Google Scholar] [CrossRef]
- Mahbub, N.U.; Islam, M.M.; Hong, S.-T.; Chung, H.-J. Dysbiosis of the Gut Microbiota and Its Effect on α-Synuclein and Prion Protein Misfolding: Consequences for Neurodegeneration. Front Cell Infect Microbiol 2024, 14. [Google Scholar] [CrossRef]
- Bhattacharyya, K.B.; Rosa-Grilo, M. Sexual Dysfunctions in Parkinson’s Disease: An Underrated Problem in a Much Discussed Disorder. In; 2017; pp. 859–876.
- Duan, W.-X.; Wang, F.; Liu, J.-Y.; Liu, C.-F. Relationship Between Short-Chain Fatty Acids and Parkinson’s Disease: A Review from Pathology to Clinic. Neurosci Bull 2024, 40, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Aho, V.T.E.; Houser, M.C.; Pereira, P.A.B.; Chang, J.; Rudi, K.; Paulin, L.; Hertzberg, V.; Auvinen, P.; Tansey, M.G.; Scheperjans, F. Relationships of Gut Microbiota, Short-Chain Fatty Acids, Inflammation, and the Gut Barrier in Parkinson’s Disease. Mol Neurodegener 2021, 16, 6. [Google Scholar] [CrossRef] [PubMed]
- Kakoty, V.; K C, S.; Dubey, S.K.; Yang, C.-H.; Taliyan, R. Neuroprotective Effects of Trehalose and Sodium Butyrate on Preformed Fibrillar Form of α-Synuclein-Induced Rat Model of Parkinson’s Disease. ACS Chem Neurosci 2021, 12, 2643–2660. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, R.; Tateno, F.; Yamamoto, T.; Uchiyama, T.; Yamanishi, T. Urological Dysfunction in Synucleinopathies: Epidemiology, Pathophysiology and Management. Clinical Autonomic Research 2018, 28, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, G.; Liu, J. Autonomic Dysfunction in Parkinson’s Disease: Implications for Pathophysiology, Diagnosis, and Treatment. Neurobiol Dis 2020, 134, 104700. [Google Scholar] [CrossRef]
- Shalash, A.; Hamid, E.; Elrassas, H.; Abushouk, A.I.; Salem, H.H. Sexual Dysfunction in Male Patients with Parkinson’s Disease: Related Factors and Impact on Quality of Life. Neurological Sciences 2020, 41, 2201–2206. [Google Scholar] [CrossRef] [PubMed]
- Varanda, S.; Ribeiro da Silva, J.; Costa, A.S.; Amorim de Carvalho, C.; Alves, J.N.; Rodrigues, M.; Carneiro, G. Sexual Dysfunction in Women with Parkinson’s Disease. Movement Disorders 2016, 31, 1685–1693. [Google Scholar] [CrossRef]
- Hasan, S.; Mielke, M.M.; Ahlskog, J.E.; Bower, J.; Turcano, P.; Savica, R. Erectile Dysfunction Preceding Clinically Diagnosed Synucleinopathies: A Case-Control Study in Olmsted County. Parkinsons Dis 2019, 2019, 1–6. [Google Scholar] [CrossRef]
- Shaltiel-Karyo, R.; Davidi, D.; Menuchin, Y.; Frenkel-Pinter, M.; Marcus-Kalish, M.; Ringo, J.; Gazit, E.; Segal, D. A Novel, Sensitive Assay for Behavioral Defects in Parkinson’s Disease Model Drosophila. Parkinsons Dis 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Hatate, J.; Miwa, K.; Matsumoto, M.; Sasaki, T.; Yagita, Y.; Sakaguchi, M.; Kitagawa, K.; Mochizuki, H. Association between Cerebral Small Vessel Diseases and Mild Parkinsonian Signs in the Elderly with Vascular Risk Factors. Parkinsonism Relat Disord 2016, 26, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Potashkin, J.; Huang, X.; Becker, C.; Chen, H.; Foltynie, T.; Marras, C. Understanding the Links Between Cardiovascular Disease and Parkinson’s Disease. Movement Disorders 2020, 35, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhang, M.; Fang, Q.; Huang, J. Relationship between Parkinson’s Disease and Cardio-Cerebrovascular Diseases: A Mendelian Randomized Study. Sci Rep 2023, 13, 20428. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xu, S. Association between Parkinson’s Disease and the Risk of Adverse Cardiovascular Events: A Systematic Review and Meta-Analysis. Front Cardiovasc Med 2023, 10. [Google Scholar] [CrossRef] [PubMed]
- Lamotte, G.; Holmes, C.; Wu, T.; Goldstein, D.S. Long-Term Trends in Myocardial Sympathetic Innervation and Function in Synucleinopathies. Parkinsonism Relat Disord 2019, 67, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Goldstein, D.S. Cardiovascular Dysautonomia in Parkinson Disease: From Pathophysiology to Pathogenesis. Neurobiol Dis 2012, 46, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Isonaka, R.; Sullivan, P.; Goldstein, D.S. Pathophysiological Significance of Increased α-Synuclein Deposition in Sympathetic Nerves in Parkinson’s Disease: A Post-Mortem Observational Study. Transl Neurodegener 2022, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Goldstein, D.S. Cardiovascular Dysautonomia in Parkinson Disease: From Pathophysiology to Pathogenesis. Neurobiol Dis 2012, 46, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Scorza, F.A.; Fiorini, A.C.; Scorza, C.A.; Finsterer, J. Cardiac Abnormalities in Parkinson’s Disease and Parkinsonism. Journal of Clinical Neuroscience 2018, 53, 1–5. [Google Scholar] [CrossRef]
- Park, D.G.; Kang, J.; An, Y.-S.; Chang, J.; Yoon, J.H. Association of Plasma α-Synuclein with Cardiac 123I-MIBG Scintigraphy in Early Parkinson’s Disease. Neurosci Lett 2022, 770, 136399. [Google Scholar] [CrossRef]
- Orimo, S.; Uchihara, T.; Nakamura, A.; Mori, F.; Kakita, A.; Wakabayashi, K.; Takahashi, H. Axonal -Synuclein Aggregates Herald Centripetal Degeneration of Cardiac Sympathetic Nerve in Parkinson’s Disease. Brain 2008, 131, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Javanshiri, K.; Drakenberg, T.; Haglund, M.; Englund, E. Sudden Cardiac Death in Synucleinopathies. J Neuropathol Exp Neurol 2023, 82, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Grosu, L.; Grosu, A.; Crisan, D.; Zlibut, A.; Perju-Dumbrava, L. Parkinson’s Disease and Cardiovascular Involvement: Edifying Insights (Review). Biomed Rep 2023, 18, 25. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Bi-directional relation between αSyn toxic species and cellular dysfunctions. Different factors are well-known to concur to the generation of misfolded αSyn toxic species. On the other hand, abnormal levels or misfolded forms of αSyn impact on several aspects of neuronal function, thus contributing to the pathogenesis of PD. Created with Biorender.com.
Figure 1.
Bi-directional relation between αSyn toxic species and cellular dysfunctions. Different factors are well-known to concur to the generation of misfolded αSyn toxic species. On the other hand, abnormal levels or misfolded forms of αSyn impact on several aspects of neuronal function, thus contributing to the pathogenesis of PD. Created with Biorender.com.
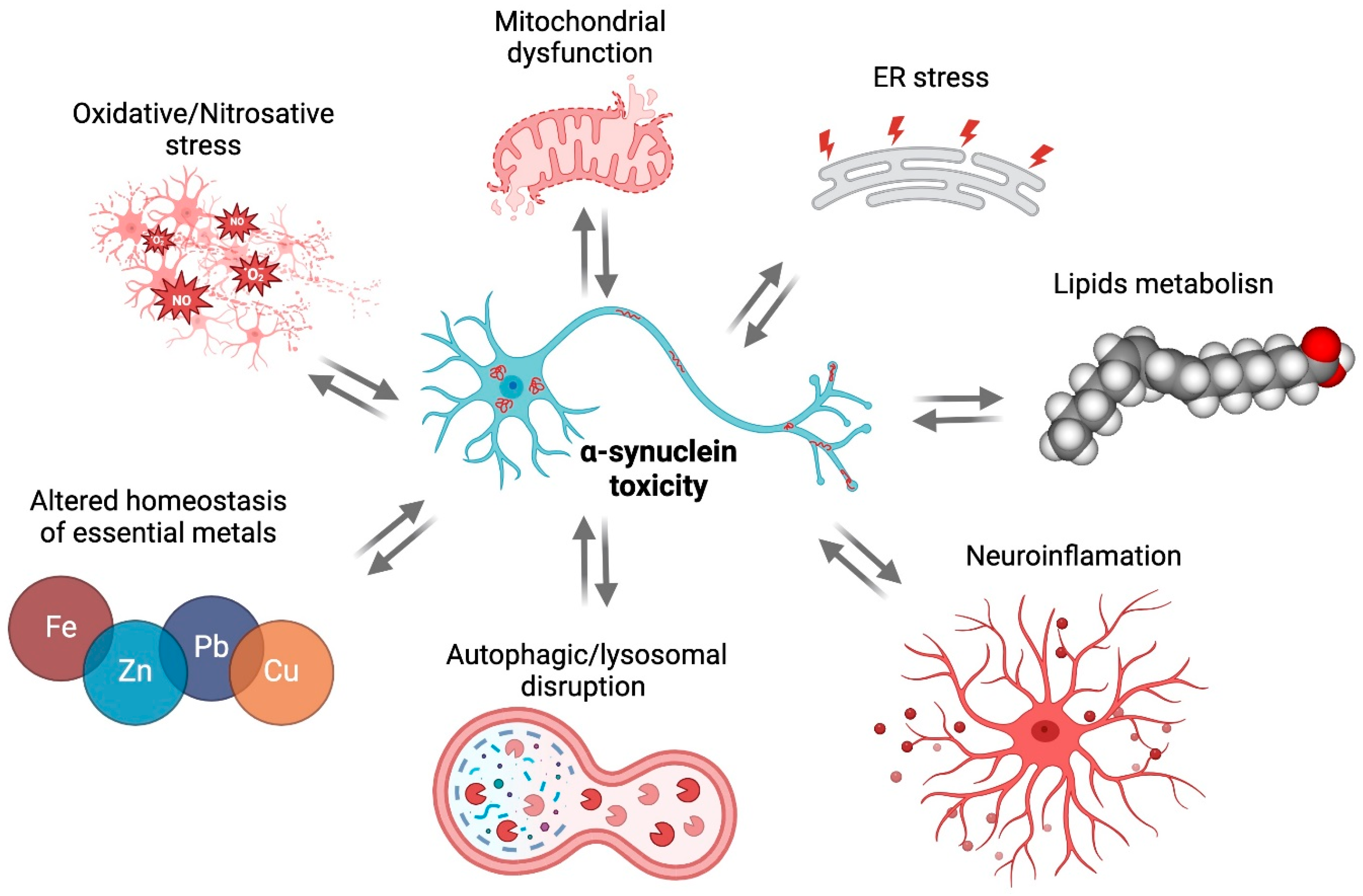
Figure 2.
αSyn is distributed in different organs and tissues. Besides the central nervous system, αSyn aggregates can be observed in peripheral organs, tissues and biofluids. The detection in peripheral tissues (skin), biofluids (plasma, tears, saliva urine, stool) has made αSyn aggregates a biomarker for the development of non-invasive diagnostic assays. Created with Biorender.com.
Figure 2.
αSyn is distributed in different organs and tissues. Besides the central nervous system, αSyn aggregates can be observed in peripheral organs, tissues and biofluids. The detection in peripheral tissues (skin), biofluids (plasma, tears, saliva urine, stool) has made αSyn aggregates a biomarker for the development of non-invasive diagnostic assays. Created with Biorender.com.

Figure 3.
Prodromal non-motor symptoms in synucleopathies. In the prodromal stage of synucleinopathies, several non-motor symptoms, can appear up to 20 years before cardinal motor symptoms manifest. Created with Biorender.com.
Figure 3.
Prodromal non-motor symptoms in synucleopathies. In the prodromal stage of synucleinopathies, several non-motor symptoms, can appear up to 20 years before cardinal motor symptoms manifest. Created with Biorender.com.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



