
Submitted:
17 June 2024
Posted:
18 June 2024
You are already at the latest version
Abstract
The key factor that enables pathogenic bacteria to establish successful infections lies largely in their ability to escape the host's immune response and adhere to host surfaces. Vitronectin (Vn) is a multi-domain glycoprotein ubiquitously present in blood and extracellular matrix of several tissues, where it plays important roles as regulator of membrane attack complex (MAC) formation and as mediator of cell adhesion. Vn has emerged as intriguing target for several microorganisms. Vn-binding by bacterial receptors confers protection from lysis resulting from MAC deposition. Furthermore, through its Arg-Gly-Asp (RGD) motif, Vn can bind several host cell integrins. Therefore, Vn recruited to the bacterial cell functions as a molecular bridge between bacteria and host surfaces, where it triggers several host signalling events that could promote bacterial internalisation. Each bacterium uses different receptors that recognise specific Vn-domains. In this review, we update the current knowledge of Vn receptors of major bacterial pathogens, emphasising the role they may play in the host upon Vn-binding. Focusing on the structural properties of bacterial proteins, we provide details on the residues involved in their interaction with Vn. Furthermore, we discuss the possible involvement of Vn adsorption on biomaterials in promoting bacterial adhesion on abiotic surfaces.
Keywords:
Vitronectin
; bacterial surface proteins
; bacterial-host interaction
; bacterial adhesion
; bacterial immune evasion
; human ligands
1. Introduction
1.1. Vitronectin Structure and Physio/Patho-Logical Role in the Host
Vitronectin (Vn) is a multidomain glycoprotein of 75 kDa synthetized mostly in the liver and secreted in the plasma, produced to a lesser extent at the level of platelets and macrophages. After an initial synthesis as a single-chain protein, Vn undergoes a proteolytic cleavage at Arg379-Ala380 due to a Vn polymorphism (substitution of methionine to threonine at position 381 in the C-terminal domain), resulting in a disulfide-bonded two-chain form [1].
Vn is highly present in plasma (200-400 μg/mL), extracellular matrix, and bone matrix, where it binds several structurally different molecules [2]. To do so, this protein is organized into a variety of distinct domains. The N-terminal domain contains a 44 residues region, identical to somatomedin B (SMB), therefore referred to as the SMB domain. The SMB domain is crucial to wound healing as it is also involved in Vn binding by the plasminogen activator inhibitor-1 (PAI-1) and the urokinase/plasminogen activator receptor (uPAR) [3,4,5]. This region is followed by the RGD domain that mediates Vn binding to integrins [6]. Following the RGD domain, there is the collagen binding region followed by the first out of three heparin-binding domains, the HBD-1 [7]. Near the N-terminal domain, the central region comprises 3 hemopexin-like domains, including a highly charged sequence involved in heparin binding, that constitutes the second heparin-binding domain (HBD-2) [8,9]. The C-terminal region is the multi-functional portion that contains the third heparin-binding domain (HBD-3) [10] and the hemopexin domain 4. Moreover, this region constitutes a binding site for plasminogen [11], thrombin [12], elastase, [13] and a second binding site for PAI-1 and uPAR [4,5,12] (Figure 1).
Vn is a conformationally labile molecule, and this characteristic plays an important role in the regulation of protein function. Vn is normally present in blood in a native, inactive form. In the native conformation, different sensitive epitopes of the protein, such as the RGD integrin binding domain, are not exposed. For this reason, blood Vn is commonly not an adhesive glycoprotein [6]. The in vivo activation of Vn occurs either in the presence of heparin or through Vn complexes formation, such as Vn-thrombin-antithrombin III, Vn-terminal complement proteins, or Vn-PAI-1. Formation of these complexes is likely to occur in areas of tissue injury and thrombosis [14]. Upon these complexes’ formation, Vn undergoes conformational changes that expose the cell adhesion domain of Vn, which can then avidly bind to integrins. Active Vn is also present on the extracellular matrix and in platelets, organized in active multimeric tissue forms [15]. Changes in Vn conformation and multimerizations are fundamental to regulate Vn functions. Nonetheless, each motif of Vn has a specific structure and organization that allows this multivalent protein to express its function and interaction with a specific biological ligand. Interestingly, in the wound healing area Vn is cleaved by thrombin, elastase and plasmin, so that the protein is no more able to bind the PAI-1. The protein switches from antifibrinolytic protein impeding the conversion of plasminogen to plasmin, to a profibrinolytic protein, which dissociates PAI-1 and converts it into a non-inhibitory protein [13]. Binding activity of Vn can be triggered in vitro upon urea treatment or heating. These processes induce a conformational change that triggers a transition of the inactive, native form to active form of Vn. Lastly, residues Y56 and Y59 of Vn have been determined as tyrosine sulphation sites of this protein. Sulphated tyrosines play a role in conformation stability of Vn, as their acidic nature promotes intramolecular binding to the basic HBD3 located at the C-terminal region of Vn, stabilizing the protein in its inactive native form [16].
Vn plays several physiological functions in the host. At the level of the extracellular matrix (ECM), Vn binds all the major ECM components through its several domains, integrins, and several receptors such as proteoglycans or collagen, mediating important roles in cell adhesion and migration, as well as cell spreading and wound healing [17]. Blood Vn functions as biological “superglue”: through binding to complement, heparin, and thrombin-antithrombin III complexes, this protein is in fact involved in regulation of clot formation, playing a fundamental role as a key controller of mammalian tissue repair [17]. Importantly, this aspect spills over in case of sepsis, a condition associated to highest mortality rates for patients in intensive care units [18]. At a pathological level, the main event that triggers microvascular thrombosis and multi-organ dysfunction till death during sepsis is the uncontrolled activation of the coagulation cascade [19]. In particular, several bacterial toxins, such as the lipopolysaccharide (LPS) from Gram-negative bacteria, target the haemostasis system, altering the coagulation cascade and fibrinolytic systems, thus playing a detrimental role in the development of sepsis. Interestingly, sepsis caused by Gram-negative bacteria is also associated to acute kidney injury (AKI) due to an increased renal PAI-1 activity observed in patients affected by sepsis. PAI-1 at the level of kidneys increases renal fibrin deposition leading to AKI. Vn is among the causes of AKI during sepsis, as through binding to PAI-1, it traps it in its active form in the ECM [18].
From a pathological aspect, altered levels of Vn have been reported in different types of cancer, such as breast cancer or neuroblastoma, as malignant cells exploit Vn to migrate to different body districts [20,21]. Indeed, serum concentration of Vn is considered a good biomarker for tumour progression evaluation.
1.2. Vitronectin as Regulator of Complement System
Also known as complement cascade, the complement system is a part of the human immune system that complements the phagocytic cells in defending the host against invading microorganisms to prevent bacterial infections. The complement system is made up of several distinct plasmatic proteins, that react with each other to clear bacteria from the system. Once activated, the complement system leads to the release of antimicrobial compounds, complement peptides (anaphylatoxins), opsonization through C3b deposition on bacterial surfaces and finally formation of the terminal complement complex (TCC) or soluble membrane attack complex (MAC) [22]. As a matter of fact, plasma from patients suffering from bacterial infections often display increased levels of MAC biomarkers [23].
The complement system can be activated through three different pathways, alternative, classical and lectin pathway, depending on the activation induced by spontaneous or specific recognition molecules. Activation of any pathway leads to the assembly of the C5b, C6, C7, C8 and C9 proteins, that form a pore in the cell membrane, leading to cell lysis. However, not all cells are vulnerable to lysis by MAC deposition. The thick cell-wall of Gram-positive bacteria for example naturally protects them from MAC-mediated cell lysis, even though a sub-lytic activity of the MAC is still able to trigger signal transduction pathways [24]. Complement activation promotes activation of an inflammatory process as well, leading to opsonization, increased phagocytosis and recruitment of leucocytes [25]. Upon triggering, the complement system must be tightly regulated by soluble and membrane-bound proteins in order to avoid an excessive response and subsequent self-damage to host tissues. Proteins that inhibit the complement system are the C4b-binding protein (C4BP) for the factor-I, factor H, clusterin and Vn. The latter contributes to complement system regulation by binding the C5b-7 peptide at the metastable membrane-binding site, therefore inhibiting MAC insertion in the terminal pathway. Moreover, Vn can also act as a direct inhibitor of C9 polymerization [26].
1.3. Vitronectin as Mediator of Cell Migration and Adhesion
Beyond the role as regulator of complement system, Vn mediates cell adhesion as well. Vn is anchored to the ECM via its collagen binding- or heparin binding-domains and it mediates cell adhesion, spreading and migration via interaction with several classes of integrins (α3β1, αvβ1, αvβ3, αvβ5 and αIIbβ3) [17]. Integrins in the various host cell types are usually found in clusters at cell attachment sites, named focal adhesions. Integrins binding by receptors outside of the cell promotes internally the attachment and remodeling of the intracellular actin cytoskeleton, stimulating the activity of different signaling molecules. Notably, the Arg-Gly-Asp (RGD) cell-binding site at the level of the N-terminal region of Vn promotes cell adhesion via Vn docking and activating of integrin receptors on the cell membrane [27]. Integrins bound by Vn trigger signalling pathways that activate different processes, such as cytoskeletal reorganization, intracellular ion transport or gene expression [28].
2. Bacterial Engagement of Vitronectin as a Weapon to Escape the Immune System
The complement system promotes either direct bacterial lysis by MAC deposition or tags the pathogens to be killed by phagocytic cells. Interestingly, pathogens use related evasion strategies to counteract the complement system. Among the several strategies evolved, they all bind the human complement regulators Factor H, Factor H like protein 1, C4BP as well as plasminogen to their surface. Moreover, many of them acquire human Vn as a very common approach to inhibit the TCC deposition and subsequent cell lysis [29].
There are two major bacterial binding regions along Vn. The great majority of bacterial proteins described to date for their Vn binding interact with the C-terminus of the glycoprotein. In particular, many microbial proteins bind Vn at the same 23 Residues (Region 352–374) [30]. Even though to a lesser extent, different bacteria recruit Vn at the level of the N-terminal or central domains.
Haemophilus influenzae is a Gram-negative occasional pathogen commonly carried in the upper respiratory tract of around 80% of healthy children. The Nontypeable H. influenzae (NTHi) can give rise to conjunctivitis, otitis media, sinusitis, and eventually pneumonia [31]. The human host is usually able to counteract NTHi infections through complement system activation. On the other hand, this bacterium is able to recruit Vn to its surface through binding by the surface adhesins Protein E (PE) [32,33] and Protein F (PF) [34]. PE is a lipoptrotein present as a dimer in solution and each monomer comprises 6-stranded antiparallel β-sheets linked by loops and a rigid α-helix at the C-terminus, which is tethered to the concave side of the sheet by a disulfide bridge (Figure 2 A) [33]. Hallström et al. have identified three different Vn-binding sites on PE protein: the major Vn-binding region is in the central part of the protein (amino acid residues 84–108), spanning the 4 and 5 antiparallel β-sheets, with Lys 85 and Arg 86, localized at the level of the loop 4, being the most important amino acid residues involved in the interaction (Figure 2 A and Table 1). Two additional Vn- binding regions are localised at the level of residues 41–68 and residues 64–88, albeit with a lower binding affinity (Figure 2 A and Table 1) [32,33,34,35]. The Vn domain involved in PE-interaction is the C-terminal heparin-binding domain (HBD-3) corresponding to residues 353–363 (Figure 1 and Table 1) [33]. PF is constituted by distinct N- and C-terminal globular domains, interlinked by a long helix backbone (Figure 2B) [36]. Its Vn-binding region is located at the N-terminus of PF, more precisely within the Lys23-Glu48 residues (Figure 2 B and Table 1) [34]. PF recruits Vn at the level of the HBD-3 (residues 348-361) and the C-terminal PAI-1 binding site (residues 348-370) of the Vn molecule (Figure 1 and Table 1) [34].
Interaction of PE and PF with Vn promotes resistance to complement-mediated killing and is heparin-dependent. Yu-Ching Su et al. have observed that bacterial binding to Vn does not interfere with Vn functions, therefore the human protein retains its ability to delay MAC formation on bacterial membrane. As this is a successful strategy for bacterial survival in the host, the PF protein appears to be highly conserved and detected in all clinical NTHi isolates under investigation. PF promotes Vn-dependent bacterial adhesion and complement-mediated killing resistance by delaying MAC formation on the bacterial surface [34]. Among the typeable H. influenzae strains, the serotype f strain associated to the development of increasing invasive diseases exploits the Factor H-binding Protein (PH) to bind the C-terminal region of Vn by interacting with Vn residues 352–362, and resist against complement-mediated killing (Figure 1 and Table 1) [37]. H. influenzae type B (Hib) express on its surface the Haemophilus surface fibril (Hsf), a major trimeric autotransporter adhesin (TAA), which is a family of adhesins that generally enable many Gram-negative pathogens to adhere to/interact with the host. The bacterium adopts Hsf to bind Vn and therefore inhibit the terminal pathway and MAC deposition. TAAs are modular, highly repetitive proteins commonly present in the outer membrane of many Gram-negative bacterial species, that mediate adhesion to external surfaces [38]. Hsf is a multidomain molecule, that displays three epithelial cell-binding domains (BDs) and three putative domains (PDs) of unknown function (Figure 2C). Vn-interaction occurs at the level of the BD2 domain, and with a lower affinity also at the level of the PD2, BD1, and 1047–1751 fragment (Figure 2C and Table 1). Hsf recruits Vn at the level of the Vn residues 352 to 374 on its HBD3 on the C-terminal region, indeed this interaction is heparin-dependent (Figure 1 and Table 1) [39].
Moraxella catarrhalis is another nasopharyngeal Gram-negative pathogen spread among children, that commonly causes otitis media. M. catarrhalis is able to subvert complement-mediated killing through C4BP and Vn recruitment through the trimeric autotransporter adhesins Ubiquitous surface proteins A2 and A2H (UspA2, UspA2H, respectively), with UspA2 being the major Vn ligand [40]. The UspA protein can be divided into a distant head, followed by a stalk and membrane anchoring region (MA) (Figure 2D). UspA2 exploits the N-terminal head domain (residues 30-177) to target Vn at the level of the C-terminal located HBD3 (Figure 1, 2D and Table 1). Notably, only the trimeric UspA230-177 can bind Vn, whereas monomeric UspA230–177 does not [41]. Interestingly, the N-terminal head domain is highly diverse among bacterial strains, nonetheless all the various variants bind Vn at the level of the C-terminal located HBD3 domain (residues 312-396) (Figure 1 and Table 1) [42].
Rickettsia is a family of Gram-negative, obligate intracellular bacteria transmitted by arthropod vectors to mammals, that cause fever and other diseases. The Rickettsia adhesin Adr1 is a surface associated protein expressed by the species R. conorii, that promotes bacterial resistance to host immune system through Vn binding. Adr1 is composed of 8 trans-membrane beta sheets, constituting the membrane spanning barrel, and four connecting beta strands termed “loops” that protrude into the extracellular environment (Figure 2E). It has been reported that loops 3 (residues 190-202) or 4 (residues 238-252) are sufficient to bind Vn and escape the host-immune system (Figure 2E and Table 1) [43]. For this bacterial species as well, interaction with Vn resides at the level of the C-terminal domain of the human protein, precisely between amino acid residues 363 and 373 (Figure 1 and Table 1) [44]. Interestingly, addition of increasing concentrations of NaCl can inhibit the Vn-Adr1 interaction, suggesting an electrostatic type of interaction. On the other hand, the presence of heparin does not affect the interaction between the two proteins. For this reason, the Adr1/Vn interaction differs from other Gram-negative bacterial/Vn interactions, as they usually are heparin dependent as well documented in literature [30].
A similar interaction mechanism as per the Rickettsia family is adopted by Neisseria meningitidis (Nm), cause of meningitis and septicaemia worldwide. Once in the blood stream, Nm survival relies on the ability to avoid killing by host immune system. Among the several surface proteins already characterised for their interaction with the various complement system components, this Gram-negative bacterium is able to recruit Vn on its surface through the Outer membrane protein C OpC and avoid complement-mediated killing by inhibition of the deposition of the MAC [45]. N. meningitidis is able to bind Vn through another molecule as well, the Meningococcal surface fibril (Msf), in a heparin-independent manner. Both OpC and Msf interaction with Vn occurs at the level of the N-terminal region, precisely at amino acids 43-68 of Vn (Figure 1 and Table 1). Msf is a trimeric autotransporter adhesin sharing common structural architecture with Hsf from H. influenzae [46]. The Vn-binding region of Msf protein is located between amino acids 39–82 of the mature protein (Table 1). Interestingly, this region can elicit and antibody response that could reduce pathogen survival within the host and therefore could be used as a potential vaccine antigen candidate [47].
The enterobacterial species of Yersiniae and Salmonellae are Gram-negative bacteria that infect humans upon ingestion of contaminated food or water, causing gastrointestinal diseases associated with several symptoms, such as diarrhea or enterocolitis. Both these species are able to bind Vn on their bacterial surface to escape complement-mediated killing. Yersinia pestis, the agent of plague, binds Vn through the Attachment invasion locus outer membrane protein Ail [48] Vn recruitment by the Ail protein occurs at the level of the hemopexin domains of Vn [49]. The outer membrane Protease E PgtE from Salmonella instead directly cleaves Vn and PAI-1 [50]. Y. enterocolitica adopts the Yersinia adhesin A (YadA) to interact with Vn via Its C-Terminal Heparin-binding domain HBD-3 and is able to evade the host complement system [51]. YadA is a trimeric autotransporter adhesin structurally homologous to UspA2 from M. catarrhalis [52]. As for UspA2, Vn-binding by YadA occurs at the level of its head domain. Interestingly, this region is able to recognize glycan moyeties, therefore YadA is able to bind glycosylated Vn (Table 1) [53].
Helicobacter pylori is a spiral-shaped Gram-negative coloniser of the human stomach of over half of the global population. H. pylori binds Vn through the Katalase A, KatA, acquiring complement resistance and ability to evade the innate host immune response. The primary binding site for KatA binding to Vn is located within amino acids 229 and 339 of Vn, corresponding to the hemopexin-like domains 2 and 3 (Figure 1 and Table 1). Generally, H. pylori does not enter the bloodstream, nonetheless it is exposed to complement system, since both complement factors and regulators, Vn included, are present at the level of the gastric epithelium during bacterial infection [54]. KatA structure is similar to other katalases, that are organized in tetramers. Each monomer is made of an N-terminal protruding arm, a central β-barrel domain and a C-terminal helical domain, linked to the β-barrel one by an extended ‘wrapping’ loop (Figure 2F). KatA binding to Vn occurs at the level of the central region of the extended wrapping loop (residues 316-428) (Figure 2F and Table 1) [54].
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen bacterium of humans, that can cause several chronic diseases. It is also a common cause of acute lung infections in cystic fibrosis patients. The Dihydrolipoamide dehydrogenase Lpd is a surface exposed moonlighting protein, present also in the cytoplasm of P. aeruginosa. Lpd binds several human plasma proteins and complement regulators, included Vn. Like for many other bacterial proteins, Vn-Lpd interaction is inhibited dose-dependently by increasing concentrations of heparin, as it occurs via the C-terminal heparin-binding domain (HBD) of Vn. More precisely, Vn is bound by Lpd at the level of a ten amino acid long region (residues 354–363) within the HBD3 domain (Figure 1 and Table 1). Moreover, Lpd has a second binding site on Vn, at the level of the middle region of the protein as well (residues 161–287) (Figure 1 and Table 1), suggesting the importance of this interaction for MAC inhibition [55]. Interestingly, Paullson et al. demonstrated that both H. influenzae and P. aeruginosa are able to trigger Vn production through outer membrane vescicles release in mice lungs and in human cell cultures. Higher levels of Vn in the bronchoalveolar space are then used by bacteria to gain protection from complement-mediated killing [56].
Among the animal colonizers, the Riemerella anatipestifer is a Gram-negative bacterium that elicits infections in poultry, especially in ducklings and gees, causing deep losses in industries. The outer membrane protein 76, OMP76, from this bacterium has been recently described as a key escape-associated virulence factor in this important pathogen, as it is able to bind Vn (Table 1) and enhance sero-sensitivity of the bacterium to complement-mediated escape [57].
Leptospira is a genus of bacteria able colonize the kidneys of reservoir animals and cause leptospirosis, that can also infect humans as occasional hosts. Leptospira interrogans displays on its surface the leptospiral complement regulator-acquiring protein A (Lcpa) protein, able to bind several human complement molecules simultaneously. Da Silva et al. demonstrated that this protein is also able to bind Vn at the level of the heparin binding domains (Table 1) [58]. Given the simultaneous ability to bind different complement system proteins, LcpA might exploit Vn to evade the immune system.
Borrelia miyamotoi is a relapsing fever microorganism that infects humans causing fever, headache, myalgia, arthralgia, and eventually meningitis. This bacterium displays on its surface the Borrelia miyamotoi protein BOM1093, a Vn-binding protein that contributes to serum resistance in vitro. Vn-binding occurs through the C-terminal region of BOM1093 (residues 209–308) (Table 1) [59].
As Gram-positive bacteria are known to be intrinsically resistant to MAC-dependent killing, Vn recruitment to escape the TCC deposition on their surface is a strategy pursued and described mostly by Gram-negative bacteria [60].
Nonetheless, the Gram-positive Streptococcus pneumoniae serotype 3 is also able to adopt Vn binding, together with factor H binding, to evade the immune system. This bacterium is often a harmless colonizer of the human nasopharynx, that can cause several diseases of the mucosa and respiratory tract, such as sinusitis or pneumonia. S. pneumoniae possess several approaches to fight and escape the host immune system. Among the several strategies adopted, strains expressing class I pneumococcal surface protein C proteins (PspC) are able to recruit Vn to their surface to escape the host defence [61]. In particular, binding of the PspC-like protein factor H-binding inhibitor of complement Hic to Vn at the level of the C-terminal heparin-binding domain of Vn allows the bacterium to prevent the TCC formation during pneumococcal infection of the host. Hic is composed of a long stretch containing regions with predicted α-helical conformation, followed by a proline-rich repeats domain and a cell-wall spanning domain (W) (Figure 2G) [62]. Binding of Hic to Vn involves the central α-helical region of Hic encompassing amino acid residues 151 to 201 (Figure 2 G and Table 1) [63]. Even though the Hic protein and the classical PspC proteins from the same bacterial specie share only a slight sequence homology, they share a similar binding behaviour, as they all evolved to bind Vn at its C-terminal HBD3 (Figure 1 and Table 1), suggesting the importance of Vn binding for this bacterial specie [64]. Interestingly, increasing salt concentrations inhibit progressively the ability of the Hic to bind Vn, suggesting an electrostatic interaction between the negatively charged amino acids in the bacterial protein and Vn. This mechanism is very similar to the R. conorii Adr1/Vn interaction [44].
Many different respiratory pathogens recruit Vn via the third heparin binding region at the level of the C-terminal region of the protein (Table 1). This segment represents the major binding region of the human TCC inhibitor for several microbes. This interaction, together with the benefits that the bacterium gains, identify a common evolutionary pathway for bacterial evasion of the host immune system, that highlights its importance for their survival.
3. Bacterial Targeting of Vitronectin for Host Colonization
Bacterial adhesion to host cells constitutes the first fundamental step to colonize and establish an infection. Bacterial pathogens display several factors on their surface to adhere to host surfaces, such as pili and different surface-exposed membrane or cell-wall anchored proteins, called adhesins [65,66], which recognize the different receptors present on the host cell surface. In case of tissue damages, bacteria might gain access to the ECM and therefore bind their receptors, colonizing and eventually invading the host. Being both an ECM and plasma component, Vn is a common route adopted by both Gram-positive and Gram-negative bacteria to adhere to host cells through different adhesins/systems (Table 1). Moreover, Vn displays distinct binding sites for pathogens and epithelial cells, therefore it can constitute a molecular bridge between several pathogenic microorganisms and integrins, whose integrin-dependent signalling can promote bacterial entry into host cells [67].
3.1. Gram-Positive
Streptococci are Gram-positive bacteria ubiquitously found in the human body, mostly in the urogenital and gastrointestinal tract, that can act either as harmless commensal or deadly pathogens in humans. Streptococcal interaction with Vn for host cell adhesion purposes was firstly described in 1988, when the authors described how different strains of Streptococci were able to adhere to endothelial cells from blood vessels via Vn-binding [68]. Thereafter, different streptococcal adhesins have been described for their binding to Vn. In 1990, different S. dysgalactiae isolates from cattle with mastitis were described for their ability to adhere to bovine epithelial cells through Vn-binding (Table 1) [69]. Vn promotes S. pneumoniae adhesion to A549 alveolar epithelial and human brain-derived microvascular endothelial cells (HBMEC). Moreover, S. pneumoniae is able to invade nasopharyngeal epithelial cells through the Vn-mediated interaction with αvβ3 integrins. Vn binding of S. pneumoniae occurs at the level of the C-terminal heparin binding site, as increasing concentrations of heparin are able to inhibit S. pneumoniae interaction with Vn. Interestingly, Bergmann et al. showed that bacterial interaction with Vn promotes cellular invasion through a dynamic actin cytoskeleton rearrangement mediated by an integrin-linked kinase activated by Vn-binding to integrin αvβ3. Moreover, the authors showed that the bacterium binds preferentially multimeric forms of Vn rather than monomeric forms, indicating that this bacterium has cell-bound multimeric Vn as a favourite host target [70].
Lastly, S. agalactiae adopts the cell wall-anchored Plasminogen binding surface Protein PbsP to adhere to and invade the pulmonary and intestinal epithelial cells, by binding to Vn and exploiting the host Vn/αv integrin axis. PbsP is a well characterized GBS virulence factor containing two repeated Streptococcal Surface Repeat (SSURE) domains (residues 123-422) following the N-terminal region, and a methionine and lysine-rich region, defined as the MK-rich region in the C-terminal region, involved in plasminogen binding (Figure 2H) [71]. The SSURE 1 and 2 domains are homologous to domains found in other streptococcal species and are the two domains involved in interaction with Vn (Figure 2 H and Table 1) [72].
Staphylococci are colonizers of the human skin and opportunistic pathogens that can cause severe diseases, such as osteomyelitis, endocarditis, skin infections, infections associated with indwelling medical devices [73]. Staphylococcal binding to Vn has been firstly reported in 1987 [74,75]. According to the work of Dai-Qing et al., S. epidermidis displays different proteins on its surface able to bind Vn, highlighting the importance of multiple recognition sites on bacterial surface to promote bacterial interaction with Vn and subsequent colonization of the host. Interestingly, the various adhesins identified are expressed at different levels on the basis of the growth conditions tested (e.g., growth on blood agar or in Todd-Hewitt broth) [76]. The autolysin E (AtlE) from S. epidermidis is a surface-associated protein that mediates attachment to polystyrene surfaces. AtlE exhibits Vn-binding activity, suggesting a function in bacterial adhesion that might involve specific interactions with plasma proteins present on polymer surfaces [77]. The full length AtlE is processed proteolytically in vivo after removal of the N-terminal pro-peptide into an N-terminal amidase domain, containing the amidase catalytic site (amidase cat) and two repeating regions furtherly subdivided into a- and b-type subunits (R1ab and R2ab), and a C-terminal glucosaminidase domain, containing a third repeat region (R3ab) at its N-terminal region and the glucosaminidase catalytic site (glucosaminidase cat) (Figure 2I). Both glucosaminidase and amidase are important bacterial enzymes, in which the repeating structures interact with teichoic acids and peptidoglycan to guide the enzymes toward the site of cell separation [78]. AtlE recruitment of Vn occurs at the level of the R1ab and R2ab repeating structures (amino acid residues 598-839) (Figure 2 I and Table 1) [78,79].
S. aureus cell-wall anchored adhesin Iron-regulated surface determinant B (IsdB) is a receptor for host Vn. This adhesin is not constantly displayed on bacterial surface, but it is expressed under iron starvation conditions, as it is involved in iron uptake from the environment. IsdB displays in its central region two structurally conserved near iron transporter (NEAT) motifs that bind haemoglobin and heme, flanked by two disordered regions at the N-terminal and C-terminal regions (Figure 2 J). IsdB recruits Vn through both its subdomains NEAT1 and NEAT2 independently, at the binding occurs at the level of the heparin binding site(s) (Figure 2 J and Table 1). Interestingly, the bacterium is able to sequester Vn from plasma through IsdB, and to promote bacterial adhesion and invasion of HeLa cervicovaginal epithelial cell monolayers and HUVEC endothelial cell monolayers through Vn-interaction [80]. It is worth noting that recent single-molecule analyses conducted through Single-molecule force spectroscopy experiments have recently revealed that IsdB binding to Vn and integrins is strongly enhanced by mechanical stress conditions, that promote staphylococcal adhesion to the host [81].
Clostridioides difficile is a strict anaerobic Gram-positive pathogen transmitted through the fecal-oral route, that produces highly resistant spores persistent in the environment. Castro-Còrdova et al. have recently demonstrated that the spore produced by this bacterium are able to entry into the intestinal barrier via pathways dependent on host fibronectin-α5β1 and Vn-αvβ1. In particular, Vn- binding occurs at the level of the (Bacillus ortholog) exosporium collagen-like protein BclA3, expressed on the spore surface (Table 1) [82].
3.2. Gram-Negative
Gram-negative bacteria exploit Vn binding not only to escape the host complement system, but to adhere to host surfaces as well. Singh et al. observed that bacteria lacking Haemophilus surface fibril Hsf displayed a dramatically reduced adherence to and invasion of monolayers of the alveolar epithelial cell line A549 compared to wild-type (wt) strains in presence of Vn. The results suggest that the Hsf fibrils may contribute to bacterial adhesion and invasion through Vn binding (Table 1) [39]. Among H- influenzae serotypes, the NTHi non-typeable one as well was reported for its ability to penetrate into bronchial epithelial cells through binding of Vn, expressed on the surface of BEAS-2B epithelial cells. Initially characterized as an extracellular pathogen, according to diverse studies this bacterium is actually able to break into bronchial epithelial cells, probably to evade the host immune system [83].
The Ail outer membrane protein in Y. pestis previously characterized for its role in complement evasion [48] has been recently reported ad involved in invasion of Chinese Hamster Ovary (CHO) cells as well [84]. Whether this newly identified role is mediated by Ail interaction with Vn remains to be still elucidated.
Adhesion of the Gram-negative bacillus P. aeruginosa to the human airways is a key initial step in the establishment of infection in cystic fibrosis patients [85]. P. aeruginosa adopts the αvβ5-Vn pathway to adhere to human A549 pulmonary epithelial cells, underlying the importance of Vn-interaction for this bacterium not only to evade the host immune system, but for a successful colonization of the host as well [86].
Neisseria meningitidis Opc protein does not simply mediate bacterium evasion of host immune system. This protein acts as an invasin as well, as it promotes bacterial adhesion and invasion of Human Brain Endothelial Cells through binding of Vn at the level of residues 43–68, more precisely at the sulphated tyrosines (Table 1). Moreover, N. meningitidis is able to interact with Vn at the level of a second sites, the C-terminal heparin binding HBD-3 of Vn (Figure 1 and Table 1) [45].
Mycoplasma hyorhinis is a common pathogen of swine, that is also associated with different human tumors. There is still lack of knowledge concerning the pathogenic mechanism of this species. M. hyorhinis is able to adhere to swine PK-15 cells and human NCI-H292 cells through the DnaK protein. DnaK is a highly conserved protein belonging to the heat-shock protein family of molecular chaperones. Recently, the DnaK protein has been characterized for its ability to interact with different ECM proteins, Vn included (Table 1) [87]. Vn-DnaK interaction still needs to be further elucidated.
4. Vitronectin-Binding by Bacteria with a Yet to Be Defined Activity
The most diverse bacterial species recruit Vn on their surface to subvert the host immune system or adhere to the host surfaces. Despite various research, many interactions are still unknown and need to be studied more intensively. Other than IsdB protein, already characterized for its involvement in bacterial adhesion and invasion of human cells through Vn binding [80], S. aureus displays the autolysin A (AtlA) protein, that displays a high similarity both in sequence and domain organization with AtlE from S. epidermidis [88] and is able to bind caprine Vn. Patak et al. have recently showed that AtlA interacts with caprine Vn through a binding site which differs from the domains usually involved in bacterial adhesins recruitment (heparin binding domain and the second RGD motif of goat Vn), but the exact binding domain, has well as the role in AtlA interaction with Vn, has not been identified yet (Table 1) [89].
Besides AtlE from S. epidermidis, this bacterium as well displays another a surface-exposed protein, the autolysin/adhesin Aae, able to bind Vn. The biological significance of this interaction has not been identified yet (Table 1) [90].
Brucellosis is a highly common bacterial zoonotic disease. Brucella display on their surface several adhesion factors, necessary to adhere to and invade different cell types and tissues. Among these, the Brucella protein Bp26 is able to bind several ECM ligands, including Vn [91]. The protein binds Vn through different N-terminal domains (amino acids 46–65, 96–115 and 146–160) and through two regions located at the C-terminal domain (amino acids 176–190 and 231–250), suggesting the importance of this interaction for the bacterium (Table 1). Nonetheless, the advantages for this interaction have not been described yet.
Haemophilus ducreyi is a Gram-negative pathogen causative of sexually transmitted genital ulcer disease. Among the several proteins that confer serum resistance to this bacterium, Ducreyi serum resistance A DsrA is an outer membrane protein belonging to the trimeric autotransporter adhesins family, structurally similar to UspA2 [40]. DsrA mediates bacterial protection from host complement activity and is involved in Vn binding as well through the C-terminal region of the passenger domain of DsrA (Table 1). Despite Vn role as regulator of complement system, Leduc et al. have observed that Vn binding by DsrA is not required for H. ducreyi serum resistance, therefore the role of DsrA binding to Vn needs to be still clarified [92].
Streptococcus pyogenes is a human specific pathogen causing from mild to severe infections, like necrotizing fasciitis and toxic shock syndrome. This bacterium is able to bind Vn at the level of the Hemopexin-Type Repeats, however the adhesin involved in this interaction, as well as the role of this interaction are still unknown (Figure 1 and Table 1) [93].
Streptococcus suis is an emergent zoonotic etiologic agent of septicemia, pneumonia, endocarditis, arthritis, and meningitis both in pigs and humans. During a screening regarding S. suis serotype 2 ability to bind different extracellular matrix components, bacterium ability to Vn has been reported, but the adhesin involved in this interaction has not been identified yet (Table 1) [94].
5. Vitronectin Adsorption on Biomaterial Surfaces: A Double-Edged Weapon
The ability of newly implanted biomaterials to adhere to cells is a critical aspect to consider during evaluation of the biocompatibility of implants. It is fundamental to create an implant with proper physicochemical characteristics of the surface, that will promote cell affinity on the basis of the amount of adsorbed proteins from the extracellular matrix [95]. As major component of the extracellular matrix, Vn is a crucial mediator protein to be adsorbed on the surface of biomaterials. Interestingly, a recent study from Li et al. has provided new insight into the structural evolution of Vn on material surfaces [96]. The authors observed that Vn seems to be preferably adsorbed by negatively charged surfaces rather than positively charged ones, as Vn itself is negatively charged. In particular, a negatively charged surface promotes layering of Vn molecules, accumulating them to form a high-density protein layer. On the other hand, non-charged hydrophobic surfaces crushed the Vn molecules, stacking them into a high-density multilayer by tracking adsorption [96]. Collectively, according to each substrate characteristic, the patterns for Vn adsorption can change. Charged substrates promote cell adhesion to biomaterials due to the orientation of the RGD domains on the N-terminal region of Vn. More precisely, upon interaction with the biomaterial, the SMB domain unfolds into an RGD-flexible orientation, conferring Vn a higher cell-binding capability. Noteworthy, Vn attachment on biomaterial surfaces may represent a double-edged sword: if on one hand it can promote biomaterial vascularization and cell adhesion on the implant, increasing the biocompatibility of the implant for the host, on the other hand it could constitute a route for bacterial dissemination on the biomaterial, where the bacterium could potentially create biofilms and cause infections in the patient difficult to eradicate. Several adhesins involved in Vn binding are exploited by bacterial species to form biofilms as well. Biofilms are bacterial communities that can help microorganisms to adhere to the surfaces of medical devices, such as joint protheses, mechanical heart valves or catheter, causing biofilm-associated infections of the bioimplants. Bacteria embedded in biofilms are difficult to eradicate and become naturally protected against antimicrobial agents [97,98]. Moreover, Hessenauer et al. recently demonstrated that Vn is able to promote the vascularization of porous polyethylene biomaterial, frequently used in reconstructive surgery [99]. As a consequence, the Vn-mediated intensified implant vascularization could favour the biomaterial implementation at usually unfavourable sites for implantation. Concomitantly, bacterial pathogens might use the newly created vascularization of the implant to further disseminate in the host. Therefore, Vn adsorption on biomaterial surfaces needs to be further investigated in order to modulate its functionality on advanced biomaterials and to design implants safe from risks of bacterial contamination.
6. Conclusions
Microorganisms have evolved several strategies to escape the host immune system and disseminate in the host. Vn is a multidomain glycoprotein that plays the most varied functions in the host. As a regulator of the host complement system, Vn recruitment allows many bacterial pathogens to inhibit the MAC deposition on their cell wall, and therefore escape the complement system attack. In this review, we have updated the list of the several surface proteins from a wide variety of microorganisms involved in this strategy. Up to date, the great majority of bacteria involved in Vn recruitment to escape the innate host immunity belongs to Gram-negative bacteria. The reason behind this resides in the biological structure of Gram-positive bacterial cells. Gram-positive cell-wall is very thick compared to the peptidoglycan layer of Gram-negative bacteria, therefore Gram-positive cells are naturally protected from MAC-mediated cell lysis. For this reason, Vn recruitment by Gram-positive bacterial pathogens is not deeply investigated. However, Vn recruitment by the Gram-positive S. pneumoniae for prevention of MAC deposition and subsequent escape of the host immune system during pneumococcal infection of the host has been described. Moreover, a sub-lytic activity of the MAC is still able to trigger signal transduction pathways to induce cell-lysis [24]. Therefore, in view of the fact that Vn recruitment as a route to evade the complement system is commonly used by Gram-negatives, but it has also been reported for a Gram-positive species, Vn recruitment by Gram-positive bacteria for the latter strategy needs further studies.
As a component of the extracellular matrix, the second main function of Vn consists in mediating cell adhesion through its RGD integrin-binding domain. Because of this very reason, Vn is often exploited by bacterial pathogens as a link to adhere to and eventually invade host epithelial cells. Here we provide a comprehensive overview of most of the bacterial molecules that behave as adhesins interacting with Vn, and some of them are also involved in bacterial internalization in host cells. The vast majority of bacterial proteins bind Vn at the level of the C-terminal region. By binding different classes of integrins present on the host cell surfaces through its free the N-terminal RGD motif, Vn can thus act like a physical molecular bridge for bacterial cells, promoting their adhesion and colonization of the host. Moreover, Vn interaction with host-cell integrins can initiate signal transduction pathways, that would trigger cytoskeleton remodelling and reorganization. This feature can in some cases favour bacterial internalization in the host cells, that could then disseminate in the host, gaining an increased survival.
Recent studies indicate that Vn is well adsorbed on biomaterials used to create auxiliary aids for hospitals, or protheses implants, moreover its adsorption can promote vascularization of the implants. This peculiar behaviour of Vn could confer a successful implantation in the patient, but at the same time it could represent a novel route for bacterial dissemination on abiotic surfaces. As many bacterial pathogens tend to form biofilm on abiotic surfaces, further work is needed in this direction.
The exact mechanism of bacterial proteins/factors interactions with Vn has been widely studied but has not been clarified completely yet. Here, we report the structure of the bacterial proteins involved in Vn binding characterized up to date and indicate their regions involved in interaction with Vn. However, the structure of many factors that bind Vn is still missing. High-resolution crystal structure of these proteins in their apo-form and/or in complex with full length Vn or Vn domains may help to provide a deeper understanding of this interaction, and hopefully give new insights for the design of novel bacterial therapeutic agents. Many of the here described bacterial proteins involved in Vn-interaction are highly conserved among bacterial strains. Conserved protein antigens expressed on the surface of bacteria constitute critical factors for the formulation of new therapies / preventive medicines against bacteria. It is also important to identify the bacterial surface proteins that interact with Vn in clinically relevant bacterial species, but that have not been assessed yet. In light of these considerations, it is deemed essential to investigate the molecular mechanisms underlying the interaction between bacteria and Vn and to conduct further investigations to identify new bacterial adhesins for Vn. These results will be of great support in the development of new antibacterial strategies, especially in view of the increasing number of antibiotic resistances in all Gram-positive and -negative strains.
Author Contributions
A.P. designed and constructed the study, wrote the original draft, figures and table; G.P. conceptualized the topic, designed and constructed the study, analyzed the bibliography, and carried out the final editing of the text and figures and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by EU funding within the NextGeneration EU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (project no. PE00000007, INF-ACT) to G.P
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Gibson AD, Peterson CB. Full-length and truncated forms of vitronectin provide insight into effects of proteolytic processing on function. Biochim Biophys Acta. 2001. 1545:289-304. [CrossRef]
- Hayman EG, Pierschbacher MD, Ohgren Y, Ruoslahti E. Serum spreading factor (vitronectin) is present at the cell surface and in tissues. Proc Natl Acad Sci U S A. 1983. 80:4003-4007. [CrossRef]
- Zhou, A. Functional structure of the somatomedin B domain of vitronectin. Protein Sci. 2007. 16:1502-1508. [CrossRef]
- Zhou A, Huntington JA, Pannu NS, Carrell RW, Read RJ. How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration. Nat Struct Biol. 2003. 10:541-544. [CrossRef]
- Wei Y, Waltz DA, Rao N, Drummond RJ, Rosenberg S, Chapman HA. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J Biol Chem. 1994. 269:32380-32388.
- Seiffert D, Smith JW. The cell adhesion domain in plasma vitronectin is cryptic. J Biol Chem. 1997. 272:13705-13710. [CrossRef]
- Gebb C, Hayman EG, Engvall E, Ruoslahti E. Interaction of vitronectin with collagen. J Biol Chem. 1986. 261:16698-16703.
- Chillakuri CR, Jones C, Mardon HJ. Heparin binding domain in vitronectin is required for oligomerization and thus enhances integrin mediated cell adhesion and spreading. FEBS Lett. 2010. 584:3287-3291. [CrossRef]
- Hunt LT, Barker WC, Chen HR. A domain structure common to hemopexin, vitronectin, interstitial collagenase, and a collagenase homolog. Protein Seq Data Anal. 1987. 1:21-26.
- Tschopp J, Masson D, Schäfer S, Peitsch M, Preissner KT. The heparin binding domain of S-protein/vitronectin binds to complement components C7, C8, and C9 and perforin from cytolytic T-cells and inhibits their lytic activities. Biochemistry. 1988. 27:4103-4109. [CrossRef]
- Preissner, KT. Specific binding of plasminogen to vitronectin. Evidence for a modulatory role of vitronectin on fibrin(ogen)-induced plasmin formation by tissue plasminogen activator. Biochem Biophys Res Commun. 1990. 168:966-971. [CrossRef]
- de Boer HC, Preissner KT, Bouma BN, de Groot PG. Binding of vitronectin-thrombin-antithrombin III complex to human endothelial cells is mediated by the heparin binding site of vitronectin. J Biol Chem. 1992. 267:2264-2268.
- Gechtman Z, Belleli A, Lechpammer S, Shaltiel S. The cluster of basic amino acids in vitronectin contributes to its binding of plasminogen activator inhibitor-1: evidence from thrombin-, elastase- and plasmin-cleaved vitronectins and anti-peptide antibodies. Biochem J. 1997. 325:339-349. [CrossRef]
- Stoop AA, Lupu F, Pannekoek H. Colocalization of thrombin, PAI-1, and vitronectin in the atherosclerotic vessel wall: A potential regulatory mechanism of thrombin activity by PAI-1/vitronectin complexes. Arterioscler Thromb Vasc Biol. 2000. 20:1143-1149. [CrossRef]
- Yoneda A, Ogawa H, Kojima K, Matsumoto I. Characterization of the ligand binding activities of vitronectin: interaction of vitronectin with lipids and identification of the binding domains for various ligands using recombinant domains. Biochemistry. 1998. 37:6351-6360. [CrossRef]
- Jenne D, Hille A, Stanley KK, Huttner WB. Sulfation of two tyrosine-residues in human complement S-protein (vitronectin). Eur J Biochem. 1989. 185:391-395. [CrossRef]
- Schvartz I, Seger D, Shaltiel S. Vitronectin. Int J Biochem Cell Biol. 1999. 31:539-544. [CrossRef]
- Wang C, Cui Y, Miao H, Sun T, Lu Y, Zhang Y. Circulating Vitronectin Predicts Liver Injury and Mortality in Children With Sepsis: A Prospective Observational Study. Clin Appl Thromb Hemost. 2020. 26:1076029620935201. [CrossRef]
- Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis, thrombosis and organ dysfunction. Thromb Res. 2012. 129:290-295. [CrossRef]
- Bera A, Subramanian M, Karaian J, et al. Functional role of vitronectin in breast cancer. PLoS One. 2020. 15:e0242141. Published 2020 Nov 19. [CrossRef]
- Burgos-Panadero R, Noguera I, Cañete A, Navarro S, Noguera R. Vitronectin as a molecular player of the tumor microenvironment in neuroblastoma. BMC Cancer. 2019. 19:479. Published 2019. 22 May. [CrossRef]
- Sarma JV, Ward PA. The complement system. Cell Tissue Res. 2011. 343:227-235. [CrossRef]
- Westra D, Volokhina EB, van der Molen RG, et al. Serological and genetic complement alterations in infection-induced and complement-mediated hemolytic uremic syndrome. Pediatr Nephrol. 2017. 32:297-309. [CrossRef]
- Laarman A, Milder F, van Strijp J, Rooijakkers S. Complement inhibition by gram-positive pathogens: molecular mechanisms and therapeutic implications. J Mol Med (Berl). 2010. 88:115-120. [CrossRef]
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010. 11:785-797. [CrossRef]
- Podack ER, Preissner KT, Müller-Eberhard HJ. Inhibition of C9 polymerization within the SC5b-9 complex of complement by S-protein. Acta Pathol Microbiol Immunol Scand Suppl. 1984. 284:89-96.
- Mayasundari A, Whittemore NA, Serpersu EH, Peterson CB. The solution structure of the N-terminal domain of human vitronectin: proximal sites that regulate fibrinolysis and cell migration. J Biol Chem. 2004. 279:29359-29366. [CrossRef]
- Meredith JE Jr, Winitz S, Lewis JM, et al. The regulation of growth and intracellular signaling by integrins. Endocr Rev. 1996. 17:207-220. [CrossRef]
- Singh B, Su YC, Riesbeck K. Vitronectin in bacterial pathogenesis: a host protein used in complement escape and cellular invasion. Mol Microbiol. 2010. 78:545-560. [CrossRef]
- Hallström T, Singh B, Kraiczy P, et al. Conserved Patterns of Microbial Immune Escape: Pathogenic Microbes of Diverse Origin Target the Human Terminal Complement Inhibitor Vitronectin via a Single Common Motif. PLoS One. 2016. 11:e0147709. Published 2016 Jan 25. [CrossRef]
- Foxwell AR, Kyd JM, Cripps AW. Nontypeable Haemophilus influenzae: pathogenesis and prevention. Microbiol Mol Biol Rev. 1998. 62:294-308. [CrossRef]
- Hallström T, Blom AM, Zipfel PF, Riesbeck K. Nontypeable Haemophilus influenzae protein E binds vitronectin and is important for serum resistance. J Immunol. 2009. 183:2593-2601. [CrossRef]
- 33. Singh B, Jalalvand F, Mörgelin M, Zipfel P, Blom AM, Riesbeck K. Haemophilus influenzae, 8: the C-terminal domain of vitronectin and modulates the membrane attack complex. Mol Microbiol. 2011. 81, 2011. [CrossRef]
- 34. Su YC, Jalalvand F, Mörgelin M, Blom AM, Singh B, Riesbeck K. Haemophilus influenzae, 1: via the ubiquitous Protein F to subvert host innate immunity. Mol Microbiol. 2013. 87, 2013. [CrossRef]
- Singh B, Al-Jubair T, Mörgelin M, Thunnissen MM, Riesbeck K. The unique structure of Haemophilus influenzae protein E reveals multiple binding sites for host factors. Infect Immun. 2013. 81:801-814. [CrossRef]
- 36. Jalalvand F, Su YC, Mörgelin M, et al. Haemophilus influenzae, 8: binding to laminin and human pulmonary epithelial cells. J Infect Dis. 2013. 207, 2013. [CrossRef]
- 37. Al-Jubair T, Mukherjee O, Oosterhuis S, et al. Haemophilus influenzae, 5: Vitronectin Using Protein H To Resist Host Innate Immunity and Adhere to Pulmonary Epithelial Cells. J Immunol. 2015. 195, 2015. [CrossRef]
- Linke D, Riess T, Autenrieth IB, Lupas A, Kempf VA. Trimeric autotransporter adhesins: variable structure, common function. Trends Microbiol. 2006. 14:264-270. [CrossRef]
- Singh B, Su YC, Al-Jubair T, et al. A fine-tuned interaction between trimeric autotransporter haemophilus surface fibrils and vitronectin leads to serum resistance and adherence to respiratory epithelial cells. Infect Immun. 2014. 82:2378-2389. [CrossRef]
- Attia AS, Ram S, Rice PA, Hansen EJ. Binding of vitronectin by the Moraxella catarrhalis UspA2 protein interferes with late stages of the complement cascade. Infect Immun. 2006. 74:1597-1611. [CrossRef]
- Singh B, Blom AM, Unal C, Nilson B, Mörgelin M, Riesbeck K. Vitronectin binds to the head region of Moraxella catarrhalis ubiquitous surface protein A2 and confers complement-inhibitory activity. Mol Microbiol. 2010. 75:1426-1444. [CrossRef]
- Su YC, Hallström BM, Bernhard S, Singh B, Riesbeck K. Impact of sequence diversity in the Moraxella catarrhalis UspA2/UspA2H head domain on vitronectin binding and antigenic variation. Microbes Infect. 2013. 15:375-387. [CrossRef]
- Riley SP, Patterson JL, Nava S, Martinez JJ. Pathogenic Rickettsia species acquire vitronectin from human serum to promote resistance to complement-mediated killing. Cell Microbiol. 2014. 16:849-861. [CrossRef]
- Fish AI, Riley SP, Singh B, Riesbeck K, Martinez JJ. The Rickettsia conorii Adr1 Interacts with the C-Terminus of Human Vitronectin in a Salt-Sensitive Manner. Front Cell Infect Microbiol. 2017. 7:61. [CrossRef]
- 45. Sa E Cunha C, Griffiths NJ, Virji M. Neisseria meningitidis, e: to the sulphated tyrosines of activated vitronectin to attach to and invade human brain endothelial cells. PLoS Pathog. 2010. 6, 20 May 2010. [CrossRef]
- Andreae CA, Sessions RB, Virji M, Hill DJ. Bioinformatic analysis of meningococcal Msf and Opc to inform vaccine antigen design. PLoS One. 2018. 13:e0193940. Published 2018 Mar 16. [CrossRef]
- Hill DJ, Griffiths NJ, Borodina E, Andreae CA, Sessions RB, Virji M. Identification and therapeutic potential of a vitronectin binding region of meningococcal msf. PLoS One. 2015. 10:e0124133. [CrossRef]
- 48. Bartra SS, Ding Y, Miya Fujimoto L, et al. Yersinia pestis, 2: membrane protein to recruit vitronectin. Microbiology (Reading). 2015. 161, 2015. [CrossRef]
- Shin K, Lechtenberg BC, Fujimoto LM, et al. Structure of human Vitronectin C-terminal domain and interaction with Yersinia pestis outer membrane protein Ail. Sci Adv. 2019. 5:eaax5068. [CrossRef]
- Krukonis ES, Thomson JJ. Complement evasion mechanisms of the systemic pathogens Yersiniae and Salmonellae. FEBS Lett. 2020. 594:2598-2620. [CrossRef]
- Mühlenkamp MC, Hallström T, Autenrieth IB, et al. Vitronectin Binds to a Specific Stretch within the Head Region of Yersinia Adhesin A and Thereby Modulates Yersinia enterocolitica Host Interaction. J Innate Immun. 2017. 9:33-51. [CrossRef]
- Brooks MJ, Sedillo JL, Wagner N, et al. Modular arrangement of allelic variants explains the divergence in Moraxella catarrhalis UspA protein function. Infect Immun. 2008. 76:5330-5340. [CrossRef]
- Meuskens I, Leva-Bueno J, Millner P, Schütz M, Peyman SA, Linke D. The Trimeric Autotransporter Adhesin YadA of Yersinia enterocolitica Serotype O:9 Binds Glycan Moieties. Front Microbiol. 2022. 12:738818. [CrossRef]
- Richter C, Mukherjee O, Ermert D, et al. Moonlighting of Helicobacter pylori catalase protects against complement-mediated killing by utilising the host molecule vitronectin. Sci Rep. 2016. 6:24391. [CrossRef]
- 55. Hallström T, Uhde M, Singh B, Skerka C, Riesbeck K, Zipfel PF. Pseudomonas aeruginosa, e: Dehydrogenase (Lpd) to Bind to the Human Terminal Pathway Regulators Vitronectin and Clusterin to Inhibit Terminal Pathway Complement Attack. PLoS One. 2015. 10, 2015. [CrossRef]
- Paulsson M, Che KF, Ahl J, et al. Bacterial Outer Membrane Vesicles Induce Vitronectin Release Into the Bronchoalveolar Space Conferring Protection From Complement-Mediated Killing. Front Microbiol. 2018. 9:1559. [CrossRef]
- Li S, Wang Y, Yang R, et al. Outer membrane protein OMP76 of Riemerella anatipestifer contributes to complement evasion and virulence by binding to duck complement factor vitronectin. Virulence. 2023. 14:2223060. [CrossRef]
- da Silva LB, Miragaia Ldos S, Breda LC, et al. Pathogenic Leptospira species acquire factor H and vitronectin via the surface protein LcpA. Infect Immun. 2015. 83:888-897. [CrossRef]
- Sato K, Kumagai Y, Sekizuka T, et al. Vitronectin binding protein, BOM1093, confers serum resistance on Borrelia miyamotoi. Sci Rep. 2021. 11:5462. Published 2021 Mar 9. [CrossRef]
- Brown, EJ. Interaction of gram-positive microorganisms with complement. Curr Top Microbiol Immunol. 1985. 121:159-187. [CrossRef]
- Voss S, Hallström T, Saleh M, et al. The choline-binding protein PspC of Streptococcus pneumoniae interacts with the C-terminal heparin-binding domain of vitronectin. J Biol Chem. 2013. 288:15614-15627. [CrossRef]
- Janulczyk R, Iannelli F, Sjoholm AG, Pozzi G, Bjorck L. Hic, a novel surface protein of Streptococcus pneumoniae that interferes with complement function. J Biol Chem. 2000. 275:37257-37263. [CrossRef]
- Kohler S, Hallström T, Singh B, et al. Binding of vitronectin and Factor H to Hic contributes to immune evasion of Streptococcus pneumoniae serotype 3. Thromb Haemost. 2015. 113:125-142. [CrossRef]
- Voss S, Hallström T, Saleh M, et al. The choline-binding protein PspC of Streptococcus pneumoniae interacts with the C-terminal heparin-binding domain of vitronectin. J Biol Chem. 2013. 288:15614-15627. [CrossRef]
- Adegbola, RA. Bacterial adhesion and pathogenicity. Afr J Med Med Sci. 1988. 17:63-69.
- Speziale P, Pietrocola G, Rindi S, et al. Structural and functional role of Staphylococcus aureus surface components recognizing adhesive matrix molecules of the host. Future Microbiol. 2009. 4:1337-1352. [CrossRef]
- Scibelli A, Roperto S, Manna L, et al. Engagement of integrins as a cellular route of invasion by bacterial pathogens. Vet J. 2007. 173:482-491. [CrossRef]
- Valentin-Weigand P, Grulich-Henn J, Chhatwal GS, Müller-Berghaus G, Blobel H, Preissner KT. Mediation of adherence of streptococci to human endothelial cells by complement S protein (vitronectin). Infect Immun. 1988. 56:2851-2855. [CrossRef]
- Filippsen LF, Valentin-Weigand P, Blobel H, Preissner KT, Chhatwal GS. Role of complement S protein (vitronectin) in adherence of Streptococcus dysgalactiae to bovine epithelial cells. Am J Vet Res. 1990. 51:861-865.
- Bergmann S, Lang A, Rohde M, et al. Integrin-linked kinase is required for vitronectin-mediated internalization of Streptococcus pneumoniae by host cells. J Cell Sci. 2009. 122:256-267. [CrossRef]
- Buscetta M, Firon A, Pietrocola G, et al. PbsP, a cell wall-anchored protein that binds plasminogen to promote hematogenous dissemination of group B Streptococcus. Mol Microbiol. 2016. 101:27-41. [CrossRef]
- De Gaetano GV, Pietrocola G, Romeo L, et al. The Streptococcus agalactiae cell wall-anchored protein PbsP mediates adhesion to and invasion of epithelial cells by exploiting the host vitronectin/αv integrin axis. Mol Microbiol. 2018. 110:82-94. [CrossRef]
- Coates R, Moran J, Horsburgh MJ. Staphylococci: colonizers and pathogens of human skin. Future Microbiol. 2014. 9:75-91. [CrossRef]
- Chhatwal GS, Preissner KT, Müller-Berghaus G, Blobel H. Specific binding of the human S protein (vitronectin) to streptococci, Staphylococcus aureus, and Escherichia coli. Infect Immun. 1987. 55:1878-1883. [CrossRef]
- Paulsson M, Wadström T. Vitronectin and type-I collagen binding by Staphylococcus aureus and coagulase-negative staphylococci. FEMS Microbiol Immunol. 1990. 2:55-62. [CrossRef]
- Li DQ, Lundberg F, Ljungh A. Characterization of vitronectin-binding proteins of Staphylococcus epidermidis. Curr Microbiol. 2001. 42:361-367. [CrossRef]
- Heilmann C, Hussain M, Peters G, Götz F. Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol Microbiol. 1997. 24:1013-1024. [CrossRef]
- Zoll S, Schlag M, Shkumatov AV, et al. Ligand-binding properties and conformational dynamics of autolysin repeat domains in staphylococcal cell wall recognition. J Bacteriol. 2012. 194:3789-3802. [CrossRef]
- Kohler TP, Gisch N, Binsker U, et al. Repeating structures of the major staphylococcal autolysin are essential for the interaction with human thrombospondin 1 and vitronectin. J Biol Chem. 2014. 289:4070-4082. [CrossRef]
- Pietrocola G, Pellegrini A, Alfeo MJ, Marchese L, Foster TJ, Speziale P. The iron-regulated surface determinant B (IsdB) protein from Staphylococcus aureus acts as a receptor for the host protein vitronectin. J Biol Chem. 2020. 295:10008-10022. [CrossRef]
- Mathelié-Guinlet M, Viela F, Pietrocola G, Speziale P, Dufrêne YF. Nanonewton forces between Staphylococcus aureus surface protein IsdB and vitronectin. Nanoscale Adv. 2020. 2:5728-5736. [CrossRef]
- Mora-Uribe P, Miranda-Cárdenas C, Castro-Córdova P, et al. Characterization of the Adherence of Clostridium difficile Spores: The Integrity of the Outermost Layer Affects Adherence Properties of Spores of the Epidemic Strain R20291 to Components of the Intestinal Mucosa. Front Cell Infect Microbiol. 2016. 6:99. [CrossRef]
- Ikeda M, Enomoto N, Hashimoto D, et al. Nontypeable Haemophilus influenzae exploits the interaction between protein-E and vitronectin for the adherence and invasion to bronchial epithelial cells. BMC Microbiol. 2015. 15:263. [CrossRef]
- Zhang Y, Ying X, He Y, et al. Invasiveness of the Yersinia pestis ail protein contributes to host dissemination in pneumonic and oral plague. Microb Pathog. 2020. 141:103993. [CrossRef]
- Davies, JC. Pseudomonas aeruginosa in cystic fibrosis: pathogenesis and persistence. Paediatr Respir Rev. 2002. 3:128-134. [CrossRef]
- Leroy-Dudal J, Gagnière H, Cossard E, Carreiras F, Di Martino P. Role of alphavbeta5 integrins and vitronectin in Pseudomonas aeruginosa PAK interaction with A549 respiratory cells. Microbes Infect. 2004. 6:875-881. [CrossRef]
- Li Y, Wang J, Liu B, et al. DnaK Functions as a Moonlighting Protein on the Surface of Mycoplasma hyorhinis Cells. Front Microbiol. 2022. 13:842058. [CrossRef]
- Biswas R, Voggu L, Simon UK, Hentschel P, Thumm G, Götz F. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol Lett. 2006. 259:260-268. [CrossRef]
- 89. Pathak H, Sokkalingam M, Prasanth L, Devi K, Joshi P. Staphylococcus aureus, 6: with caprine vitronectin without involving the heparin binding domain and the second arginine-glycine-aspartic acid motif of the host protein. Arch Microbiol. 2019. 201, 2019. [CrossRef]
- Heilmann C, Thumm G, Chhatwal GS, Hartleib J, Uekötter A, Peters G. Identification and characterization of a novel autolysin (Aae) with adhesive properties from Staphylococcus epidermidis. Microbiology (Reading). 2003. 149:2769-2778. [CrossRef]
- ElTahir Y, Al-Araimi A, Nair RR, et al. Correction to: Binding of Brucella protein, Bp26, to select extracellular matrix molecules. BMC Mol Cell Biol. 2020. 21:16. [CrossRef]
- Leduc I, Olsen B, Elkins C. Localization of the domains of the Haemophilus ducreyi trimeric autotransporter DsrA involved in serum resistance and binding to the extracellular matrix proteins fibronectin and vitronectin. Infect Immun. 2009. 77:657-666. [CrossRef]
- Liang OD, Preissner KT, Chhatwal GS. The hemopexin-type repeats of human vitronectin are recognized by Streptococcus pyogenes. Biochem Biophys Res Commun. 1997. 234:445-449. [CrossRef]
- 94. Esgleas M, Lacouture S, Gottschalk M. Streptococcus suis, 3: to extracellular matrix proteins. FEMS Microbiol Lett. 2005. 244, 2005. [CrossRef]
- Abdallah MN, Tran SD, Abughanam G, et al. Biomaterial surface proteomic signature determines interaction with epithelial cells. Acta Biomater. 2017. 54:150-163. [CrossRef]
- Li T, Hao L, Li J, Du C, Wang Y. Insight into vitronectin structural evolution on material surface chemistries: The mediation for cell adhesion. Bioact Mater. 2020. 5:1044-1052. [CrossRef]
- Li P, Yin R, Cheng J, Lin J. Bacterial Biofilm Formation on Biomaterials and Approaches to Its Treatment and Prevention. Int J Mol Sci. 2023. 24:11680. [CrossRef]
- Kreve S, Reis ACD. Bacterial adhesion to biomaterials: What regulates this attachment? A review. Jpn Dent Sci Rev. 2021. 57:85-96. [CrossRef]
- Hessenauer MET, Lauber K, Zuchtriegel G, et al. Vitronectin promotes the vascularization of porous polyethylene biomaterials. Acta Biomater. 2018. 82:24-33. [CrossRef]
Figure 1.
Structure of Vitronectin and ligand binding sites. Schematic representation of linear Vn structure showing the positions of Vn domains. Vn natural ligands are reported on the right (in black). Bacterial ligands are indicated on the left (in red). The N-terminal region of Vn contains a region homologous to somatomedin B (SMB), indicated in pink. The cell-binding motif RGD constitutes the integrin binding domain, indicated in red. The three heparin-binding domains (HBD) are reported in purple (HBD-1, HBD-2, HBD-3). The central and the C-terminal domains contain 4 regions homologous to hemopexin, the hemopexin-like domains, reported in orange. The C-terminal region is the one involved in binding by most ligands. The big majority of bacterial pathogens binds to C-terminal HBD-3. N. meningitidis is the only bacterial species that has been shown to bind to the N-terminal region of Vn. References of the various ligands are reported in the main text.
Figure 1.
Structure of Vitronectin and ligand binding sites. Schematic representation of linear Vn structure showing the positions of Vn domains. Vn natural ligands are reported on the right (in black). Bacterial ligands are indicated on the left (in red). The N-terminal region of Vn contains a region homologous to somatomedin B (SMB), indicated in pink. The cell-binding motif RGD constitutes the integrin binding domain, indicated in red. The three heparin-binding domains (HBD) are reported in purple (HBD-1, HBD-2, HBD-3). The central and the C-terminal domains contain 4 regions homologous to hemopexin, the hemopexin-like domains, reported in orange. The C-terminal region is the one involved in binding by most ligands. The big majority of bacterial pathogens binds to C-terminal HBD-3. N. meningitidis is the only bacterial species that has been shown to bind to the N-terminal region of Vn. References of the various ligands are reported in the main text.
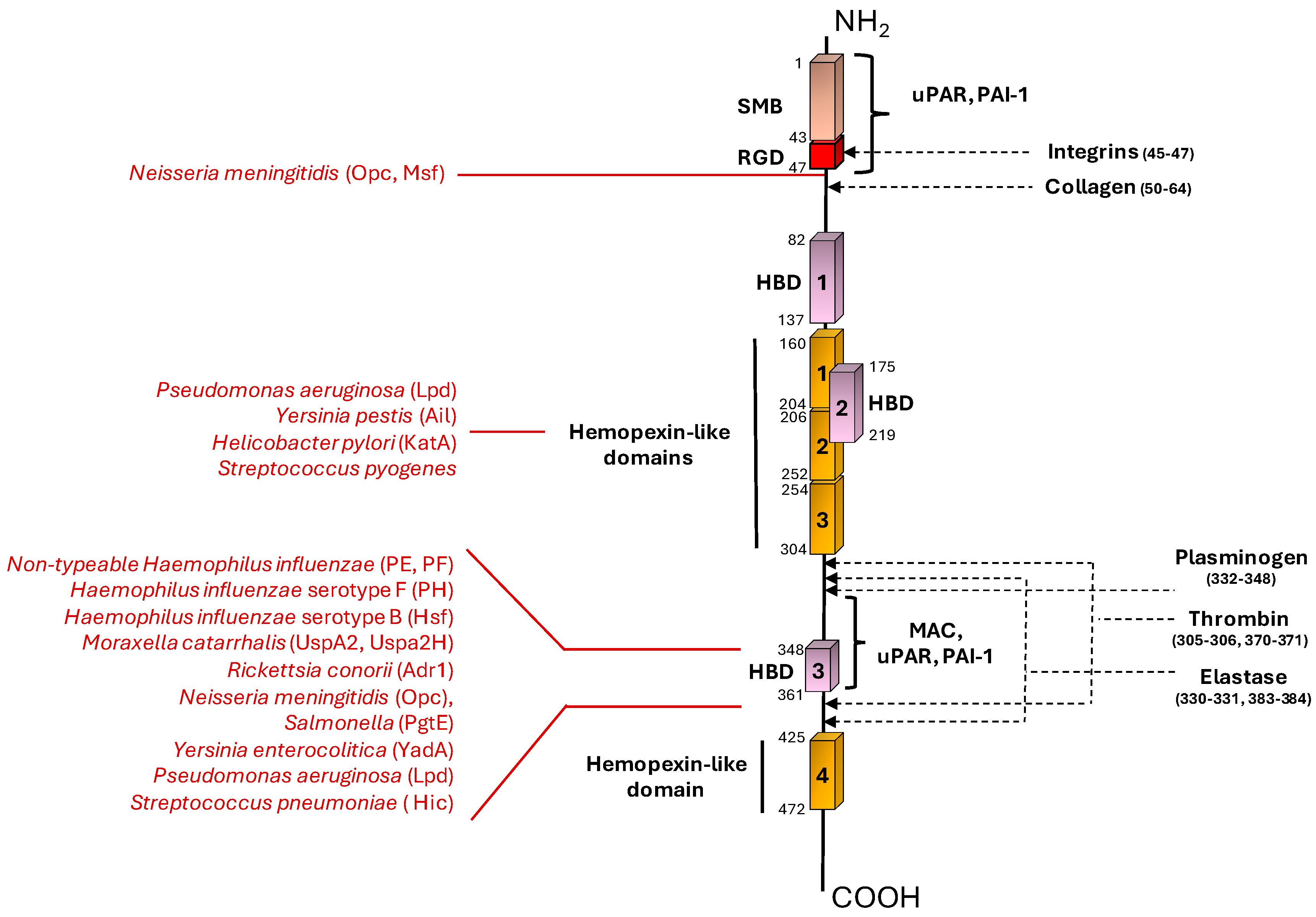
Figure 2.
Schematic representation of bacterial vitronectin-binding proteins from Gram-positive and Gram-negative bacteria. Each protein is identified with a capital letter (A-J) and by its acronym. N- and C-terminal sites of each protein are indicated with their respective residues reported in black. Vitronectin binding sites are indicated for each protein by red curly brackets, and respective amino acid residues are reported in round brackets. Protein loops (A, E) are indicated by red numbers. For further details, see the text. The structures reported here represent those for which the structural organization and Vn-binding sites have been determined.
Figure 2.
Schematic representation of bacterial vitronectin-binding proteins from Gram-positive and Gram-negative bacteria. Each protein is identified with a capital letter (A-J) and by its acronym. N- and C-terminal sites of each protein are indicated with their respective residues reported in black. Vitronectin binding sites are indicated for each protein by red curly brackets, and respective amino acid residues are reported in round brackets. Protein loops (A, E) are indicated by red numbers. For further details, see the text. The structures reported here represent those for which the structural organization and Vn-binding sites have been determined.
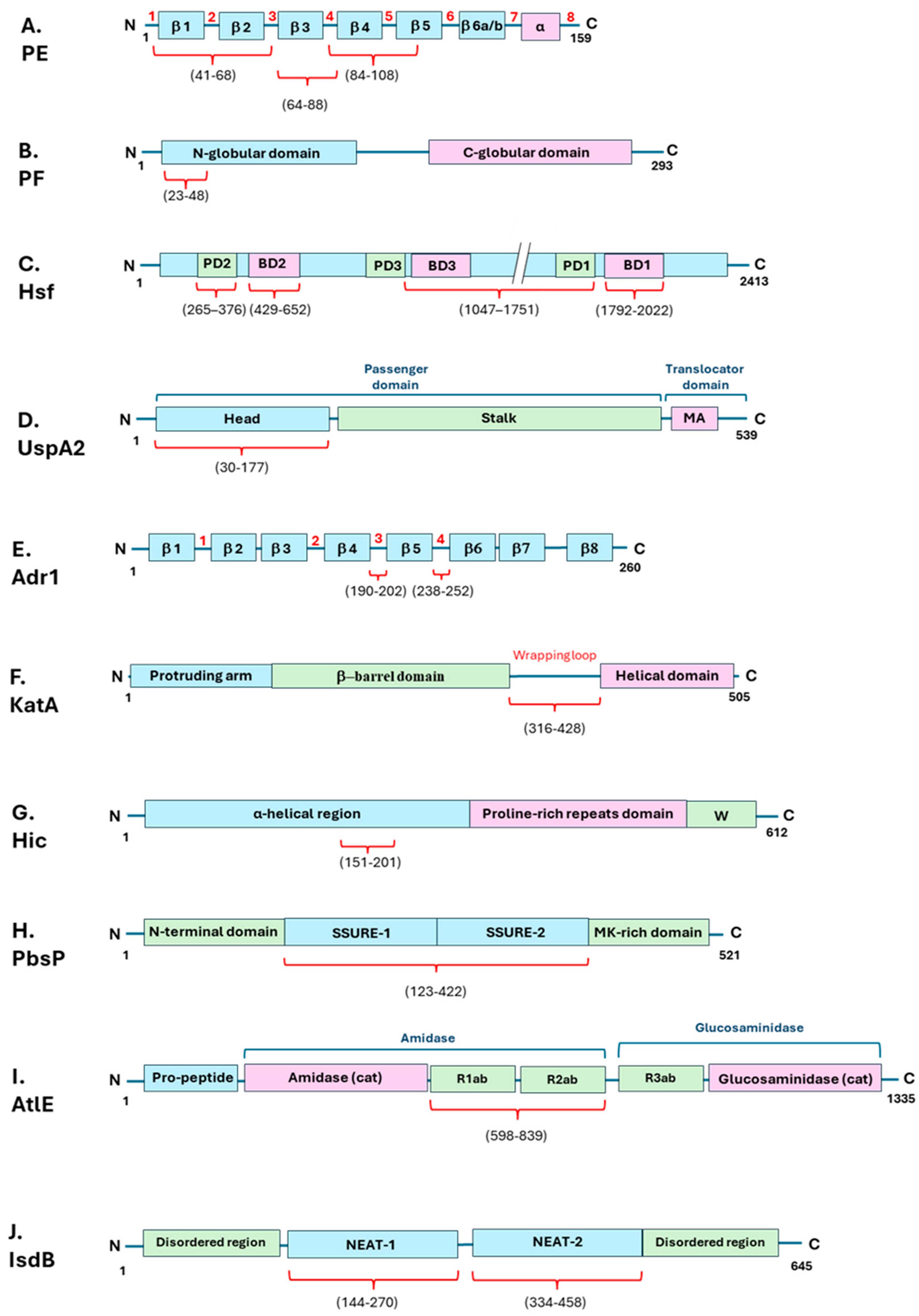

Table 1.
Bacterial factors involved in Vn-interaction.
| Bacterial species | Bacterial Protein interacting with Vn | Bacterial protein region involved in Vn binding (amino acid residues) | Vn region bound (amino acid residues) | Role in Complement evasion | Role in Cell Adhesion / invasion | Ref. |
|---|---|---|---|---|---|---|
| Gram-negative | ||||||
| Nontypeable Haemophilus influenzae | PE | 84–108, 41–68, 64–88, |
HBD-3 (353-363) |
+ | U | [33,35] |
| “ | PF | 23-48 | HBD-3 (348-361), PAI-1 binding site (348-370) |
+ | U | [34,36] |
| Haemophilus influenzae serotype F | PH | U | C-terminal (352–362) |
+ | U | [37] |
| Haemophilus influenzae serotype B | Hsf | 265-376, 429-652, 1047-1751, 1792-2022 |
C-terminal (352–374) |
+ | + | [38,39] |
| Moraxella catarrhalis | UspA2 | 30-177 | HBD-3 (312-396) |
+ | U | [40,41,42] |
| “ | UspA2H | 30-177 | HBD-3 (312-396) |
+ | U | [42] |
| Rickettsia conorii | Adr1 | 190-202, 238-252 |
C-terminal (363-373) |
+ | U | [43,44] |
| Neisseria meningititis | Opc | U | N-terminal (43-68), HBD-3 |
+ | + | [45] |
| “ | Msf | 39-82 | N-terminal (43-68) |
+ | U | [46,47] |
| Salmonella | Pgte | U | C-terminal | + | U | [50] |
| Yersinia enterocolitica | YadA | Head domain | HBD-3 | + | U | [51,53] |
| Yersinia pestis | Ail | U | Hemopexin domain | + | U | [48,49] |
| Helicobacter pylori | KatA | 316-428 | C-terminal (229-339) |
+ | U | [54] |
| Pseudomonas aeruginosa | LpD | U | C-terminal (354-363) Hemopexin-like repeats (161-287) |
+ | U | [55] |
| Riemerella anatipestifer | OMP76 | U | U | + | U | [57] |
| Leptospira interrogans | Lcpa | U | HBD (s) | + | U | [58] |
| Brucella | Bp26 | 46–65, 96–115, 146–160, 176–190, 231–250) |
U | U | U | [91] |
| Borrelia miyamotoi | BOM1093 | 209-308 | U | + | U | [59] |
| Mycoplasma hyorhinis | DnaK | U | U | U | + | [87] |
| Haemophilus ducreyi | DsrA | C-terminal passenger domain | U | U | U | [92] |
| Gram-positive | ||||||
| Streptococcus pneumoniae | Hic | 151-201 | C-terminal (HBD-3) | + | U | [63,64] |
| Streptococcus dysagalactiae | U | U | U | U | + | [69] |
| Streptococcus pyogenes | U | U | Hemopexin-Type Repeats | U | U | [92] |
| Streptococcus suis | U | U | U | U | U | [93] |
| Streptococcus agalactiae | PbsP | 123-422 | U | U | + | [72] |
| Staphylococcus epidermidis | AtlE | 598-839 | U | U | + | [77,79] |
| “ | Aae | U | U | U | U | [90] |
| Staphylococcus aureus | AtlA | U | U | U | U | [88,89] |
| “ | IsdB | 144-270, 334-458 | HBD (s) | U | + | [80] |
| Clostridioides difficile | BclA3 | U | U | U | + | [82] |
U = unknown.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



