
Submitted:
19 June 2024
Posted:
19 June 2024
You are already at the latest version
Abstract
The MAPK signaling pathway with BRAF mutations has been shown to drive the pathogenesis of 40-60% of melanomas. Inhibitors of this pathway's BRAF and MEK components are currently used to treat these malignancies. However, responses to these treatments are not always successful. Therefore, identifying noninvasive biomarkers to predict treatment responses is essential for personalized medicine of melanoma patients. Using noninvasive 1H magnetic resonance spectroscopy (1H MRS), we previously showed that BRAF inhibition reduces lactate and alanine tumor levels at early stages of effective therapy and could be construed as metabolic imaging biomarkers for drug response. The present work aimed to demonstrate if these metabolic changes and those assessed by 31P MRS are also found in preclinical human melanoma models treated with MEK inhibitors. Apart from 1H and 31P MRS, additional supporting in vitro biochemical analyses are described. Our results indicate significant early metabolic correlations with response levels to MEK inhibition in the melanoma models and are consistent with our previous study of BRAF inhibition. Given these results, our study supports the potential clinical utility of noninvasive MRS to objectively image metabolic biomarkers for early predicting melanoma response to MEK inhibition.
Keywords:
melanoma
; trametinib
; 1H/31P magnetic resonance spectroscopy
; oxygen consumption rate
; extracellular acidification rate
; glucose uptake
; lactate production.
1. Introduction
Melanoma, the deadliest form of human skin cancer, originates from melanocytes and exhibits a high tendency to metastasize, accounting for approximately 90% of skin cancer-related mortality [1]. Melanoma represents 1.7% of all global cancer diagnoses and ranks as the fifth most common cancer in the United States [2]. While melanoma is highly treatable when confined to its primary site, metastatic melanoma presents a grim outlook, with a median survival of only approximately six months [3,4,5]. Furthermore, systemic therapies currently employed in patients with metastatic melanoma exhibit varying response rates, with rapid development of tumor resistance observed in the majority of patients [3,6,7]. While surgical intervention remains the primary treatment for melanoma, recent breakthroughs in immunotherapy and targeted molecular therapies for metastatic melanoma offer significant potential [8].
Melanoma treatment, incorporating surgery, chemotherapy, immunotherapy, and radiotherapy, faces challenges due to melanoma resistance, primarily attributed to melanin production. Recent focus has been on targeting the mitogen-activated protein kinases (MAPK) pathway to overcome resistance and improve therapeutic outcomes [9,10]. Inhibiting the BRAF or MEK portions of the MAPK pathway exerts primarily cytostatic effects compared to chemotherapy. MEK inhibition in BRAF mutant cells may induce apoptotic and cytostatic impact [11]. Early noninvasive biomarkers are crucial to assess target modulation and treatment efficacy due to the potential absence of tumor shrinkage. BRAF, activated by RAS, is highly expressed in melanocytes, neural tissue, testes, and hematopoietic cells. Phosphorylated BRAF activates MEK (a kinase component of the MAPK pathway), which in turn activates ERK (MAPK) by phosphorylation and thus stimulates growth and transformation [12]. Unfortunately, melanoma cells have a hypermutable genome leading to tumor resistance by responding to the blockage of the MAPK pathway by rerouting to an alternate path. Therefore, most patients develop resistance to targeted therapy within weeks to months of initiation of treatment.
Melanoma often exhibits a glycolytic phenotype driven by the constitutive activation of the BRAF gene mutation (Figure 1) [13]. This activation increases glucose uptake and glycolysis, mediated by the MAPK pathway and its induction of hypoxia-inducible factor 1α (HIF-1α), a key regulator of glycolytic activity [14,15]. In response to varying energy demands and environmental cues, melanomas demonstrate metabolic plasticity, adjusting between glycolysis and oxidative phosphorylation (OXPHOS) [16]. Glutamine could also become a primary energy source, facilitated by heightened glutaminolysis and upregulation of glutamine transporters. Furthermore, resistance to BRAF and MEK inhibition primarily arises from metabolic adaptations within the MAPK signaling pathway [16,17].
Published research, including ours, shows that magnetic resonance spectroscopy (MRS) is a noninvasive avenue for monitoring metabolic changes in melanoma models following diverse anticancer treatments [10,11]. We demonstrated that MRS discerns significant changes in metabolic signatures in successfully treated melanoma models with dabrafenib, a BRAF inhibitor, showcasing early therapy-related changes in metabolomics, pH, and bioenergetics [10]. This elucidation underscores the potential of MRS in delineating early-treatment metabolic responses to targeted therapies. Notably, the MRS availability in clinical MRI systems guarantees the translation of the metabolic signatures as possible biomarkers of treatment efficacy, supporting personalized therapeutic approaches in clinical melanoma management.
This report examines four human melanoma cell lines characterized for their BRAF sensitivity: a wild-type (WM3918), a BRAF resistance mutant-type (WM983BR), and two BRAF-sensitive mutant-types (WM983B and DB-1). These studies tested the cell lines’ response to trametinib, a MEK kinase inhibitor targeting the MAPK signaling pathway (Figure 1). Inhibition of the MEK kinase is known to elicit treatment responses in melanoma models, as the hyperactive BRAF precedes MEK in the MAPK pathway. To discern trametinib-induced metabolic changes, we grew the melanoma cell lines in athymic nude mice xenografts and cell cultures. We employed in vivo 1H and 31P MRS noninvasively in the xenografts alongside other in vitro analytical methods in the cell cultures to assess the metabolic response of human melanoma.
2. Materials and Methods
2.1. Reagents and MEK Inhibitor
The reagents used for cell cultures and biochemical analyses, including 3-aminopropylphosphonate (3-APP) and their sources, are listed in our previous study [10]. Trametinib was obtained from LC Laboratories (Woburn, MA, USA). Trametinib was dissolved in 0.5% hydroxypropyl methylcellulose and 0.2% Tween 80 buffer solution in Milli-Q filtered water as the vehicle for mouse injection.
2.2. Cell Lines and Cell Culture Conditions
Briefly, WM3918 (Accession No: CVCL_C279) and WM983BR (Accession No: CVCL_AP81) melanoma cells were cultured in MCDB153/L-15 media supplemented with antibiotics, calcium chloride, and 2% FBS. WM983BR media also contained 100 nM dabrafenib. DB-1 (Accession No: CVCL_D902) and WM983B (Accession No: CVCL_6809) cells were cultured in MEM-α media supplemented with glucose, glutamine, antibiotics, MEM NEAA, and FBS. More detailed culturing conditions are described elsewhere [10].
2.3. Extracellular Glucose and Lactate Measurements
Melanoma cell lines were seeded in T-75 tissue culture flasks and incubated at 37 °C with 5% CO2 for 24 hours to allow adhesion. Afterward, either vehicle (0.1% DMSO) or 7 nmol/L trametinib was added to create control and treated groups. After 48 hours of treatment, media samples were collected, and extracellular glucose and lactate concentrations were measured using a YSI 2300 STAT PLUS Biochemistry Analyzer (YSI Incorporated - Yellow Springs – Xylem, Yellow Springs, Ohio 45387-0279 USA).
2.4. Mitochondrial Stress and Cell Energy Phenotype Assays
Immediately before the assays, the cell lines were incubated in the presence of either 0.1% DMSO (control) or 7 nmol/L trametinib in 0.1% DMSO for 48 hours. Cell mitochondrial stress and energy phenotype assays were carried out using the Seahorse XFe96 Extracellular Flux Analyzer (Agilent Technologies; Santa Clara, CA, USA). Melanoma cells were seeded in the Seahorse 96-well cell culture microplates at 15-20 × 103 cells per well and incubated for 24 hours before the assay was started. Fresh stock solutions of oligomycin (100 µM), FCCP (100 µM), and rotenone/antimycin-A (50 µM) were prepared on the assay day. Using these stocks, we prepared working oligomycin, FCCP, and rotenone/antimycin-A solutions at 15 µM, 5 µM, and 5 µM, respectively. The final concentration of these reagents in the microplate wells was 1/10 of the working solutions. Additionally, 30 µL of 20 mM Hoechst 33342 solution (Thermo Scientific, Waltham, MA, USA) was added to the rotenone/antimycin-A solution to stain the cells for cell counting. These experiments followed the user guide of the flux analyzer. Oxygen Consumption Rates (OCR) and Extracellular Acidification Rates (ECAR) were determined for each of the four melanoma lines and used to create a cell energy phenotype map.
After the assays, the microplates were transferred to a BioTek Cytation 5 cell imaging multimode reader (Agilent Technologies) using fluorescent scanning for cell count and viability. The OCR and ECAR data were normalized to viable cell count for comparisons.
2.5. In Vitro Measurement of Intracellular Metabolites by High-Resolution 1H-MRS
We conducted 1H-MRS on extracted melanoma cells grown in T-182 tissue culture flasks. The cells were grown in either vehicle (0.1% DMSO) or 7 nmol/L trametinib in 0.1% DMSO for 48 hours. After treatment, 10-15 × 106 cells were harvested, centrifuged, and washed with cold phosphate-buffered saline (PBS), then stored at -80 °C. Frozen cell pellets were resuspended in an 80% methanol-water solution, homogenized, sonicated, and centrifuged to extract metabolites. The supernatant containing the metabolites was lyophilized and resuspended in deuterium oxide (D2O) with trimethylsilyl propanoic acid (TSP) and transferred to a 5 mm NMR tube. The method is described in detail elsewhere [10]. High-resolution 1H-MRS spectra were acquired using a PRESAT pulse sequence (water suppression with presaturation pulses) on a 9.4T/8.9 cm vertical bore Varian NMR spectrometer, with a flip angle of 45°, repetition time (TR) of 8.8 sec, spectral width (SW) of 6756.8 Hz, 16384 points, and 128 transients. The MestRec software (Mestrelab Research, Santiago de Compostela, Spain) was used for data processing. Spectra were filtered with a 1 Hz exponential function to improve the apparent signal-to-noise ratio (SNR). Peak areas were integrated and normalized by the TSP signal and the number of protons in the signal to obtain molar concentrations.
2.6. Mouse Preparation for Proton (1H) and Phosphorus (31P) MRS Studies
The MRS study cohort comprised 48 human melanoma-bearing male athymic nude mice obtained from Charles River Laboratories (https://www.criver.com/). The mice were divided into four groups, each xenografted with a different human melanoma cell line. Before injection, cells were cultured in a monolayer at 37 °C in a controlled environment with 5% CO2 using Hank’s Balanced Salt Solution (HBSS). This controlled culture setup allowed the cells to grow and multiply until they reached the desired density for injection. Animals were injected subcutaneously into their right thigh with 0.1 mL of a 109/mL suspension of the melanoma cells in HBSS. After injection, the xenografted tumors were allowed to grow until their volume reached approximately 250 mm3. The tumors were permitted to establish themselves in the host mouse to develop characteristics like those in human melanoma during their initial growth period, providing a relevant preclinical model for studying the disease and potential treatments.
Subsequently, each group of mice was divided into two subgroups. One received vehicle (placebo), and the other trametinib (10 mg/kg) once daily through oral gavage for five days. The number of mice for groups WM3918 and WM983BR was n = 5 for the placebo and trametinib subgroups, while for groups WM983B and DB-1 was n = 6 for the placebo and n = 8 for the trametinib subgroups.
2.7. Experiments Involving In Vivo 1H and 31P MRS
In vivo MRS exams were conducted noninvasively in each mouse on Day 0, Day 2, and Day 5 regarding the start of placebo or trametinib administration. For these exams, mice were anesthetized with 1% isoflurane in O2/CO2 (95%/5%) at a rate of 1 L/min. To measure extracellular pH (pHe) during 31P MRS, 3-APP (300 mg/mL solution in water) was injected into the mouse peritoneum before placing it in the spectrometer. The animal’s core temperature (37 ± 1 °C) and breathing were monitored during the experiment.
In vivo, MRS exams were performed on a 9.4 T Bruker horizontal bore spectrometer (Bruker Instruments, Billerica, MA, USA). 1H MRS was carried out using a slice-selective, double-frequency, Hadamard-selective, multiple quantum coherence (HDMD-Sel-MQC) transfer pulse sequence to acquire the methyl 1H signals of lactate and alanine selectively [18]. For these studies, the tumor was positioned in a homemade single frequency (1H) slotted-tube resonator (15 mm outer diameter, 13 mm inner diameter, 16.5 mm depth). The following acquisition parameters were used: TR = 4 sec, 1000 points, and 32 transients. A localized water spectrum was also acquired using the same sequence without water suppression (TR = 4 sec, 4 transients).
31P MRS exams were performed as described in detail previously [19]. In summary, the tumor was placed in a homemade dual-frequency (1H/31P) slotted-tube resonator with a diameter of 10 mm. The Image Selected In vivo Spectroscopy (ISIS) pulse sequence was employed with the following parameters: Acquisition time (AT) of 64 ms, flip angle = 90°, TR = 2 sec, SW = 7979 Hz, offset frequency = 430, and 512 points. 31P MRS spectra were obtained using a decoupling duration of 64.16 ms, decoupling power of 3.12 W, and CPD sequence element of 0.5 ms. A scout image was initially acquired to set the acquisition voxel to minimize contamination of tumor metabolite signals with exogenous signals from lipids and muscle.
All spectroscopic data were processed using the NUTS (Acorn NMR Inc., Livermore, CA, USA) and MestRec programs. Ten and 40 Hz exponential filters were applied to enhance the apparent SNR of 1H and 31P MRS, respectively. Baseline correction was performed before plotting and calculating peak areas. In the 1H spectra acquired, the 1H methyl signals from lactate and alanine were integrated. In addition, the unsuppressed water spectrum was used to integrate the 1H signal from water (H2O) to obtain the in vivo lactate/H2O and alanine/H2O values in the tumors. Similarly, the 31P signals from the center phosphate of NTP (i.e., b-NTP) and inorganic phosphate (Pi) were integrated to obtain the b-NTP/Pi ratio. Furthermore, the centroid of the Pi, 3-APP, and a-NTP signals was measured, and the pH values of the intracellular (chemical shift difference between Pi and a-NTP) and extracellular compartments (chemical shift difference between 3-APP and a-NTP) were calculated using adaptations of the Henderson-Hasselback equation [20].
2.8. Measurement of Tumor Volume
Tumor dimensions in the mouse xenografts were assessed using calipers. Measurements were taken in three orthogonal directions, and tumor volume was calculated using the formula V = π (l × w × d)/6, where l = length, w = width, and d = depth of the tumor.
2.9. Statistical Analysis
The data are presented graphically and summarized using either bar graphs denoting mean ± standard deviation (s ± SD – Figure 2, Figure 3 and Figure 4 and Table 1) or box-and-whiskers plots showing the median, mean, quartiles, data points, and outliers (Figure 5, Figure 6 and Figure 7). Two-tailed Student’s t-tests assuming variance homogeneity were conducted to compare untreated (control) vs. trametinib-treated pairs, setting the significance level at α = 0.05. In Figure 5, Figure 6 and Figure 7, the data is presented as time-related changes. Although an exponential behavior is expected for biological data like those presented in Figure 5, Figure 6 and Figure 7 (e.g., tumor growth), due to the limited timespan of the studies and the fact that linear and exponential regressions reported R2 values within 95% of each other, we opted to use linear regressions. We assumed linearity and variance homogeneity for these graphs, and outliers were included in the analyses presented. The adjusted R2 and p-values of each time course were determined (Figure 5, Figure 6 and Figure 7), but we only show the adjusted R2 values with a substantial effect size (adjusted R2 ≥ 0.26). Similarly, non-significant p-values (> a) have been omitted. Tumor volume was normalized for individual tumors by the mean volumes for Day 0 in all groups to obtain the relative tumor volume (Figure 7). These mean values were then averaged across tumors to obtain mean values for individual days.
3. Results
3.1. Trametinib’s Impact on Glucose Consumption and Lactate Production in Melanoma In Vitro
Extracellular glucose and lactate concentrations were assessed using the YSI 2300 STAT PLUS Glucose Lactate Analyzer, unveiling significant variations in response between untreated cells and those subjected to trametinib treatment (7 nM for 48 hours), as depicted in Figure 2. Specifically, a significant decrease in glucose consumption was observed in all cell lines in the presence of trametinib. Furthermore, lactate production was increased in the wild-type WM3918 and BRAF-resistant WM983BR melanoma cell lines, while the sensitive human melanoma cell lines (WM983B and DB-1) exhibited a decrease in extracellular lactate production. This divergence in lactate production highlights the distinct metabolic phenotype of these cell lines affecting their response to trametinib treatment.
3.2. Oxygen Consumption and Extracellular Acidification Rates In Vitro
As shown in Figure 3 and Table 1, WM3918 cells treated with trametinib showed no significant differences in OCR or the OCR/ECAR ratio but a statistically significant decrease in ECAR relative to their control values. Conversely, trametinib-treated WM983BR cells showed significant shifts in OCR and ECAR vs. control cells. Still, the resulting shifts in this cell line’s OCR/ECAR ratio were insignificant, probably due to the high dispersion of the measurements.
In comparison, trametinib-treated WM983B and DB-1 cells showed substantial shifts in the OCR/ECAR ratio compared to control cells. Trametinib-treated WM983B cells showed decreased ECAR and increased OCR, while treated DB-1 cells showed an overall decrease in energy (i.e., reduced OCR and ECAR). Significant shifts have occurred in the OCR/ECAR ratio of the DB-1 and WM983B cells with trametinib (Figure 3 and Table 1). However, while DB-1 cells were significantly de-energized by trametinib (i.e., drop in OCR and ECAR, blue arrow), WM983B cells showed a more respiratory phenotype after trametinib (i.e., decreased ECAR and increased OCR, red arrow).
3.3. In Vitro 1H MRS to Test the Metabolic Response to Trametinib in Isolated Melanoma Cells
We conducted 1H MRS at 9.4T in suspensions of the four melanoma cell lines to determine the metabolic effect of trametinib. For these experiments, untreated and trametinib-treated cells (7 nM for 48 h) were studied. As shown in Figure 4, we focused on lactate and alanine as these metabolites relate to the Warburg effect, an abnormal metabolic reprogramming in cancer that affects glycolysis [21,22,23,24].
Figure 4 shows that the lactate levels in the untreated (control) cultured melanoma cells vary substantially. Student t-tests indicated that the lactate values of untreated cells were significantly different when the WM3918 cell line was compared with WM983B and DB-1 but not with WM983BR (asterisks on top of red bars in Figure 4). Conversely, when comparing WM3918 with the rest of the cell lines, only the WM983BR cell line showed a significantly reduced alanine value. Furthermore, trametinib-treated WM3918 and WM983BR cells did not show statistically significant changes in lactate vs. their untreated counterparts. However, this comparison was significant for the lactate reductions on WM983B and DB-1 cell lines. Moreover, trametinib-related changes in alanine were significant for all cell lines except WM983BR (p-values on top of yellow bars in Figure 4).
3.4. In vivo 1H- and 31P- MRS of Melanoma Xenografts in Mice
The trametinib treatment effect on the metabolomics of human melanoma xenografts in mice was noninvasively tested using MRS. The melanoma implants on the mice’s flank underwent in vivo 1H and 31P MRS evaluation before (Day 0) and during trametinib therapy (Day 2 and Day 5).
Figure 5 shows the trametinib’s impact on intracellular lactate and alanine levels tracked noninvasively in melanoma xenografts. The HDMD-Sel-MQC transfer pulse sequence facilitated the selective detection and integration of the lactate and alanine methyl resonances. The water signal from the acquired unsuppressed spectrum was also integrated and used to normalize the lactate and alanine signals. The Lactate/H2O [25] and Alanine/H2O ratios were considered representative of the intracellular metabolites’ molar concentrations. Figure 5 shows that untreated mice have a significant, time-related lactate and alanine ratio increase regardless of cell line (panels B and C, control row in red), with a substantial effect size (i.e., R2 ≥ 0.26) and significant regression in all cell lines. In comparison, the tumor lactate and alanine values in treated mice (panels A and B, trametinib row in yellow) show striking differences. While the WM3918 melanoma cell line exhibited similar lactate and alanine time-related increases to those in its control group (with substantial effect size and p-value), the rest of the cell lines did not show metabolite increases during therapy. Instead, the WM983BR cell line showed no time dependency for the lactate and alanine tumor content (i.e., near-zero slope, low adjusted R2, and no significance on the linear regression analysis), while the WM983B and DB-1 showed significant time-related decreases in the metabolites’ tumor content. Furthermore, the WM3918 cell line did not show statistical differences in lactate or alanine between the control and trametinib-treated groups by comparing slopes or group means of each day. Regarding lactate, the results in WM3918 xenografts match the studies in isolated cells. Still, we did not reproduce the significant trametinib-related reduction of alanine in isolated cells of this cell line (Figure 4). In addition, the variability of lactate and alanine found in untreated cultured cells was not reproduced in any of the three studies of the untreated (control) xenografts except for an increase in lactate in the untreated DB-1 xenografts.
Furthermore, the time-related differences in the tumor lactate and alanine content between untreated and trametinib-treated groups in the WM983BR, WM983B, and DB-1 xenograft were highly significant (slope comparison p-value in Figure 5). Figure 5 shows statistical differences between the control vs. trametinib pairs on these cell lines on Day 2 and Day 5, except for Day 2 of WM983BR. Given that this late appearance of a significant lactate reduction in the trametinib-treated WM983BR xenografts (until Day 5) could be the reason for the non-significant lactate reduction found in the isolated cells of treated WM983BR, we can conclude that the trametinib effect on qualitative metabolic changes (or lack thereof) in cultured cells and xenografts of the four melanoma cell lines match.
Figure 6 (A) displays the in vivo 31P MRS of the melanoma models demonstrating distinct low-field resonances, including the NTP’s three signals, Pi, and the exogenously added 3-APP. Given that the most concentrated NTP in biological tissues is adenosine triphosphate (ATP) and that there is a rapid exchange of the terminal phosphates of all the NTPs, we considered the NTP/Pi ratio as an acceptable measure of the cellular ATP hydrolytic state, a reliable indicator of the bioenergetic status. We calculated the NTP/Pi ratio by integrating the b-signal of NTP because it does not overlap with other signals compared to the a- and g-NTP signals.
Figure 6 (B) shows that time changes of β-NTP/Pi were not demonstrated in control mice regardless of cell line (near-zero slope, low adjusted R2, and p > 0.05). In comparison, except for the trametinib-treated WM3918, the remaining three xenografts showed significant time-related increases in the β-NTP/Pi ratio with substantial effect sizes (adjusted R2 ≥ 0.026). Again, excluding the WM3918 data, the comparison of the control vs. trametinib-treated time courses of β-NTP/Pi in the remaining xenografts is significant. In addition, comparing the control vs. trametinib pairs at each timepoint shows significance for Day 2 and Day 5, except for Day 2 of the WM983BR.
Figure 6 (C) shows the pHi and (D) the pHe values determined by 31P MRS. Only DB-1 xenografts exhibited a significant time-dependent decrease of pHi under trametinib treatment. In addition, the untreated xenografts of the same DB-1 xenografts showed significant pHe reductions but no time change during treatment. Therefore, the time course comparison of the pHe data in the control vs. trametinib groups of the DB-1 xenografts is statistically significant.
3.5. Assessment of Tumor Growth Following Trametinib Treatment
Figure 7 illustrates the changes in tumor volume over time in the different mouse xenografts. Untreated mice (control) showed the expected timely exponential and fast tumor size increase of cancer. Trametinib did not change this behavior in the WM3981 xenografts, but the remaining cell lines (WM983BR, WM983B, and DB-1) displayed a significantly reduced time-related tumor volume during therapy. However, while W983BR showed a time-related increase in volume (although not as fast as its untreated counterpart), WM983B and DB-1 showed a significant time-related decrease in tumor volume. As expected, the comparisons of control vs. trametinib at each treatment day show differences, but they were significant only on Day 5 of WM983B and Day 2 and Day 5 of DB-1 xenografts.
4. Discussion
BRAF/MEK inhibition has become a standard-of-care option for BRAFV600-mutated melanoma. Dabrafenib, a BRAF inhibitor, and trametinib, a MEK inhibitor, have received approval from the U.S. Food and Drug Administration for treating BRAF-driven melanoma [21]. BRAF and MEK are crucial components of the MAPK signaling pathway, which regulates various essential cell functions, including cell growth and apoptosis [26,27,28]. We previously reported significant metabolic changes in preclinical melanoma models with mutated BRAF dependency sensitive to dabrafenib [10]. In the present work, we expanded these observations, demonstrating again the significant metabolomic phenotype variability in melanomas with high dependency on mutated BRAF [10,29,30]. However, different from our previous work inhibiting the altered BRAF protein directly using dabrafenib [10], in the present work, we use trametinib to inhibit MEK, which is a subsequent step after BRAF in the MAPK pathway (Figure 1). Thus, we reduced MAPK signaling by indirectly affecting the hyperactive BRAF protein, inhibiting MEK. Our findings strongly suggest a tight relationship between altered tumor metabolism and MAPK inhibition. Molecularly targeted agents inducing selective MEK inhibition play a crucial role in melanoma by inhibiting the abnormal MAPK signaling pathway and restoring apoptosis [31,32,33]. Hence, maximizing the benefits of inhibiting the MAPK pathway is crucial to achieving a high objective therapy response. Our noninvasive metabolic measurements (i.e., 1H and 31P MRS), which could be translatable to the clinical arena, may be valuable in determining the metabolic effect of BRAF and MEK inhibitors and, thus, help as clinical biomarkers of response to these therapies.
We evaluated glucose uptake by isolated tumor cells, considering the correlation of this uptake with lactate production in cancer cells due to the Warburg effect [34]. Our results in Figure 2 demonstrate that trametinib reduces glucose uptake in all melanoma cell lines. However, the two sensitive cell lines showed reduced lactate production with trametinib, while the resistant ones significantly increased it. The decreased glucose uptake with increased lactate production may be due to a different energy source the resistant cell lines utilize besides glucose (e.g., glutaminolysis, lipolysis, etc.). As an aside, these results could explain, at least in part, the difficulties of predicting response by positron emission tomography (PET) [35,36,37]. PET is the modality for staging patients with solid malignancies, including melanoma [37,38,39]. For these purposes, the tumor uptake of the radiotracer 18F-2-fluoro-2-deoxy glucose (FDG) is followed by PET. FDG is an analog of glucose that cannot be metabolized further than the first step of glycolysis. As trametinib reduces the glucose uptake in all melanoma cell lines tested (Figure 2), an FDG-PET exam might not be able to demonstrate differences to predict response. In contrast, 1H MRS shows lactate and alanine differences (Figure 5) associated with response and may be assessed noninvasively in melanoma patients.
Our results with the OCR and ECAR measurements (Figure 3 and Table 1) demonstrate that the wild-type cell line WM3918 under trametinib treatment showed no significant differences in OCR and the smallest, although statistically significant, decrease in ECAR of all cell lines compared to its control group. As expected, the OCR/ECAR did not change with trametinib therapy in WM3918. In comparison, the remaining cell lines showed significant but variable changes in OCR and ECAR. Trametinib proportionally increased both parameters in the resistant mutant WM983BR cell line, maintaining the OCR/ECAR constant at the lowest level of all cell lines. Both mutant cell lines, resistant (WM983BR) and sensitive (WM983B), had an increased OCAR but differed in ECAR. These changes demonstrate an upregulation of respiration in both cell lines, with an increase in glycolytic activity for WM983BR and a decrease in WM983B. Finally, the three parameters decreased in the remaining sensitive mutant cell line, DB-1. The trametinib-related changes in DB-1 demonstrate a decrease in respiratory and glycolytic activities, with a reduction in overall energy production.
Furthermore, WM983B and DB-1 cell lines are BRAF-positive mutants, showing significant shifts in cell energy phenotype. These shifts reveal that MEK inhibition via trametinib impacts glycolytic activity in WM983B and DB-1. The trametinib-related decrease in ECAR in the sensitive mutant cell lines WM983B and DB-1 established them as less glycolytic (Figure 3). Figure 3 also shows that WM983B cells relied more on respiration with trametinib treatment and had the second-largest change in energy phenotype. DB-1, the most energetic melanoma line, was the most significantly affected and had the most prominent energy phenotype shift with treatment (Figure 3 and Table 1). These facts were corroborated by the decreased significantly glucose uptake and lactate production shown in Figure 2 and the reduced tumor lactate values in vitro and in vivo depicted in Figure 4 and Figure 5, respectively. However, given that WM983B and WM983BR have a higher OCR with trametinib, they differ from DB-1, which has a lower OCR. Although we expected to see a reflection of these results in the bioenergetic status of the cell lines, the xenografts of these three cell lines had a trametinib-related increase of β-NTP/Pi (Figure 6). We theorize that despite the decreased cellular respiration found in DB-1 in the controlled in vitro OCR studies (Figure 3), its increased bioenergetic status in vivo could be due to the inhibition of the hyperactive MAPK pathway and the concomitant reduction of the Warburg effect. Under these in vivo conditions, instead of synthesizing lactate, glycolytic intermediates, mainly pyruvate, could be available for cellular respiration, albeit the fact that respiration seems to be deficient in DB-1 in the in vitro studies (Figure 3). The inhibition of the Warburg effect is supported by the substantial reduction in lactate production shown in Figure 2 and the reduction of tumor lactate content depicted in Figure 4 and Figure 5 for DB-1 and WM983B. However, it is also possible that the DB-1 inconsistencies may be due to the different experimental conditions of cultured tumor cells vs. in vivo xenografts.
The present results complement the ones we obtained on the effect of BRAF inhibition on the same melanoma models [10]. WM3918, the wild-type melanoma cell line, and WM983BR, the BRAF inhibitor-resistant mutant line, showed no significant shifts in OCR/ECAR ratio with trametinib treatment, indicating that the effect of trametinib was minimal. Other studies on MEK signaling inhibition in wild-type melanoma have shown similar findings [40]. However, WM3918 did show a significant drop in ECAR, which suggests that trametinib had some effect on glycolytic activity in the wild-type line. WM983BR showed small but significant increases in OCR and ECAR with trametinib treatment. Still, the OCR/ECAR ratio shift was not significant, probably due to high measurement deviations. Other studies on BRAF/MEK inhibition found that some BRAF mutants rapidly resisted BRAF and MEK inhibitors through genetic or epigenetic alterations [41]. The in vitro resistance can also be seen in our results in the dabrafenib-resistant WM983BR. However, more work is needed to elucidate the discrepancies in the response of WM983BR to trametinib.
Our present results show that the cellular effects of a positive response to MEK inhibition were accompanied by a reduction of lactate production (Figure 2) and a time-dependent reduction in tumor lactate levels (Figure 4 and Figure 5). These results corroborate the report by Falck Miniotis et al. [11]. However, our methodology also allowed us to demonstrate changes in alanine, bioenergetics, and pH. As shown in Figure 5, the lactate changes matched alanine’s in WM983B and DB-1. However, reduced tumor lactate levels were found on Day 5, but alanine was not significantly reduced on that day in the WM983BR cell line. These data match the fact that WM983BR has a decreased but not significant tumor volume with trametinib treatment during our observation period, showing only a statistical trend (p = 0.06, Figure 7). This suboptimal response matches with a significant tumor lactate reduction but not with one for alanine, suggesting that WM983BR has a unique metabolic phenotype responding differently to trametinib than the two fully responding cell lines. Therefore, finding ways to coax an alanine reduction in WM983BR (e.g., using different therapies or combining other drugs with trametinib) may help improve its response. In comparison, WM983B and DB-1, the responding cell lines, showed the expected decrease in tumor growth while on trametinib therapy (Figure 7), correlating with significant lactate and alanine changes (Figure 5).
An essential objective of this work was to explore the clinical translatability of our standardized noninvasive tumor metabolic assessment. Experimental measurements using 1H MRS have revealed that the reduction in lactate observed after BRAF/MEK inhibition was more pronounced in cells containing the oncogenic BRAFV600E mutants than non–BRAF-driven cells [11]. These metabolic differences suggest that BRAF-independent cells do not experience a robust metabolic response to treatment, as lactate levels remained unchanged. However, a reduction in alanine was observed following MEK inhibition in the cell lines containing the oncogenic BRAFV600E mutation and in non–BRAF-driven cells (Figure 5). Alanine, a product of pyruvate amination and an essential component of the glutaminolysis pathway, plays a crucial role in cellular metabolism. Our findings in Figure 4 and Figure 5 demonstrate decreased lactate and alanine concentration with MEK inhibition, as observed by in vitro and in vivo 1H MRS of preclinical melanoma models.
To demonstrate prediction by lactate and alanine, we adapted the clinically used RECIST (response evaluation criteria in solid tumors) [42,43] to determine response using the mean group value of tumor volume on Day 5 of the trametinib treatment xenografts depicted in Figure 7. On Day 5, WM3918 had a 50% increase in tumor volume; thus, RECIST classifies it as a progressive disease (PD). In comparison, WM983BR had a tumor increase below 20%, and WM983B had a decrease of less than 30% in tumor volume, classifying both as stable diseases (SD). Finally, DB-1 shows a 40% decrease, so RECIST considers it a partial response (PR). Following this convention, the only xenograft with a positive response on Day 5 is DB-1 (PR). However, continued trametinib therapy could have brought some of these tumors to achieve a complete response (CR). The linear regressions for WM983B and DB-1 in Figure 7 predict that CR (i.e., a complete absence of tumor) could have been achieved by Day 24 and Day 14, respectively. Notably, these two xenografts have significantly and sustainably reduced lactate and alanine since Day 2. In comparison, the time course of the tumor volume of WM983BR predicts that CR can never be reached. WM983BR had a tumor lactate value significantly reduced by Day 5, but the alanine value did not change (Figure 5). These analyses suggest that early and sustained reductions of tumor lactate and alanine values predict a subsequent CR response in melanoma. They also suggest that a delayed reduction of lactate without changes in alanine (e.g., WM983BR) or no changes in both metabolites (e.g., WM3918) predict a negative response.
Using 31P MRS, we found that higher bioenergetic levels (i.e., β-NTP/Pi) correlate with an increased response to trametinib in responsive melanoma lines (Figure 6). However, only the DB-1 sensitive line showed a significant increase in extracellular pH (pHe). Tumors with higher glycolytic or energetic profiles, such as WM983B and DB-1, show better responsiveness to trametinib than wild-type (WM3918) and BRAF-resistant (WM983BR) melanoma cell lines. The reason for this correlation between cellular energy state and glycolytic capacity remains unknown. However, this selective tumor de-energization could enhance the tumor’s response to therapeutic agents like trametinib.
Although we expected metabolic responses similar to trametinib compared to dabrafenib, as both affect the same regulatory pathway, we found subtle differences amongst the cell lines. For example, dabrafenib increased OCR significantly in WM983B, while the OCR change was insignificant with trametinib. Furthermore, when comparing control with trametinib therapy, WM983BR has reduced (or inverted) time-related slopes for lactate and alanine in the xenograft studies. In comparison, both slopes are positive and not significantly different when using dabrafenib. The same WM983BR has a much steeper slope for the b-NTP/Pi increase in response to trametinib than dabrafenib [10].
Finally, the differences between the lactate levels before therapy could also be important as potential biomarkers of response in patients with melanoma. The in vivo value of lactate in the untreated (control) DB-1 xenografts (the best responder to trametinib) is significantly larger in comparison to the rest of the xenografts (Figure 5) We hypothesize that finding a large tumor lactate value in melanoma patients before therapy could be predictive of a positive response with trametinib.
5. Conclusions
Reducing the hyperactive MAPK signaling using trametinib, we found diverse metabolomic changes in phenotypically diverse models of melanoma. Notably, these metabolic changes are correlated with the response to the inhibitor (i.e., reduction in tumor growth) and thus have the potential to be used as biomarkers of prediction or follow-up in melanoma patients. Our results suggest that the noninvasive 1H/31P MRS assessment of lactate, alanine, bioenergetics levels (β-NTP/Pi), and pH (pHi and pHe), especially when complemented with multiparametric MR imaging of the tumor microenvironment (e.g., vascularity and cellularity) and FDG-PET, will offer valuable information for the personalized medicine approach of the patient with melanoma. Our current study provides proof of principle for investigating lactate and alanine metabolism, bioenergetics levels (β-NTP/Pi), and pH using MRS in BRAF/MEK inhibitor-treated patients with metastatic melanoma and possibly other solid tumors.
This study evaluated the metabolic effects of trametinib, an MEK inhibitor, in preclinical human melanoma models. Using 1H and 31P MRS and complementary biochemical assays, we observed significant changes in tumor metabolomics, including modifications in lactate and alanine levels and bioenergetics. These changes were more pronounced in cells with the BRAFV600E mutation, indicating a more significant response to trametinib. Additionally, trametinib treatment decreased tumor volume in both wild-type and mutant melanoma xenografts, with the most considerable effects observed in sensitive cell lines. Biochemical measurements of glycolysis and bioenergetics could provide valuable predictive estimates of melanoma patients’ response to treatment, particularly in assessing the energetic demands of therapy. This evidence may also apply to monitoring and predicting responses in other cancers and treatment modalities, including BRAF, MEK inhibitors, and immunotherapy. The study highlights the potential of metabolic imaging techniques like MRS in assessing treatment response and guiding therapy in melanoma patients.
Author Contributions
Conceptualization, K.N., P.K.G., and D.S.N.; methodology, K.N. P.K.G., and D.S.N.; software, P.K.G., K.N., and D.S.N., ; validation, P.K.G., K.N., F.A.M., D.S.N., and S.O.; formal analysis, P.K.G., F.A.M., K.N., D.S.N., and S.O.; investigation, P.K.G., K.N., D.S.N., and S.O.; resources, K.N., D.S.N., P.K.G., and S.O.; data curation, P.K.G., F.A.M., K.N., D.S.N., and S.O.; writing—original draft preparation, P.K.G., K.N., D.S.N., and S.O.; writing—review and editing, P.K.G., F.A.M., K.N., D.S.N. and S.O.; visualization, P.K.G., F.A.M., K.N., D.S.N., and S.O.; supervision, K.N. P.K.G and D.S.N.; project administration, K.N., and D.S.N.; funding acquisition, K.N., and D.S.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NIH, grant numbers: R01CA250102, R01CA228457, R01CA268601, and R21CA280523.
Institutional Review Board Statement
Mouse studies were performed under the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC; Protocol code#: 806891; Approval Date: 24 February 2023) for studies involving animals.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are available upon request.
Acknowledgments
We thank Meenhard Herlyn of the Wistar Institute in Philadelphia for providing WM983B, WM983BR, and WM3918 melanoma cells and for consultations regarding the research conducted. In addition, we thank Anatoliy V. Popov and Stephen Pickup, Department of Radiology, University of Pennsylvania, Philadelphia, for assistance with processing and analyzing samples for high-resolution 1H MRS. We want to thank Skyler Nova, Meklit Tedla, Aria Osborne, and Mikal Banks for their help with cell culture on this project. We want this publication to recognize Jerry D Glickson and Dennis B Leeper. Drs. Glickson and Leeper were fundamental in inspiring this project. We honor their legacy in melanoma research by continuing the work that they initiated.
Conflicts of Interest
Author Fernando Arias-Mendoza was employed by Advanced Imaging Research, Inc. The remaining authors declare that the research was conducted without commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Roesch, A.; Berking, C. Melanoma. In Braun-Falco’s Dermatology, Plewig, G., French, L., Ruzicka, T., Kaufmann, R., Hertl, M., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2022; pp. 1855–1871. [Google Scholar]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med Sci (Basel) 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol Ther 2019, 20, 1366–1379. [Google Scholar] [CrossRef]
- Sandru, A.; Voinea, S.; Panaitescu, E.; Blidaru, A. Survival rates of patients with metastatic malignant melanoma. J Med Life 2014, 7, 572–576. [Google Scholar] [PubMed]
- Quaglino, P.; Fava, P.; Tonella, L.; Rubatto, M.; Ribero, S.; Fierro, M.T. Treatment of Advanced Metastatic Melanoma. Dermatol Pract Concept 2021, 11, e2021164S. [Google Scholar] [CrossRef]
- Polkowska, M.; Ekk-Cierniakowski, P.; Czepielewska, E.; Kozłowska-Wojciechowska, M. Efficacy and safety of BRAF inhibitors and anti-CTLA4 antibody in melanoma patients-real-world data. Eur J Clin Pharmacol 2019, 75, 329–334. [Google Scholar] [CrossRef]
- Wang, X.; Luo, Z.; Chen, J.; Chen, Y.; Ji, D.; Fan, L.; Chen, L.; Zhao, Q.; Hu, P.; Sun, P.; et al. First-in-human phase I dose-escalation and dose-expansion trial of the selective MEK inhibitor HL-085 in patients with advanced melanoma harboring NRAS mutations. BMC Med 2023, 21, 2. [Google Scholar] [CrossRef]
- Pavri, S.N.; Clune, J.; Ariyan, S.; Narayan, D. Malignant Melanoma: Beyond the Basics. Plast Reconstr Surg 2016, 138, 330e–340e. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.; Rodrigues, C.M.P.; Gaspar, M.M.; Reis, C.P. Melanoma Management: From Epidemiology to Treatment and Latest Advances. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Gupta, P.K.; Orlovskiy, S.; Arias-Mendoza, F.; Nelson, D.S.; Osborne, A.; Pickup, S.; Glickson, J.D.; Nath, K. Metabolic Imaging Biomarkers of Response to Signaling Inhibition Therapy in Melanoma. Cancers (Basel) 2024, 16. [Google Scholar] [CrossRef]
- Falck Miniotis, M.; Arunan, V.; Eykyn, T.R.; Marais, R.; Workman, P.; Leach, M.O.; Beloueche-Babari, M. MEK1/2 inhibition decreases lactate in BRAF-driven human cancer cells. Cancer Res 2013, 73, 4039–4049. [Google Scholar] [CrossRef]
- Strashilov, S.; Yordanov, A. Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Meyle, K.D.; Lange, M.K.; Klima, M.; Sanderhoff, M.; Dahl, C.; Abildgaard, C.; Thorup, K.; Moghimi, S.M.; Jensen, P.B.; et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget 2013, 4, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Kasraian, Z.; Rezvani, H.R. Energy metabolism in skin cancers: A therapeutic perspective. Biochim Biophys Acta Bioenerg 2017, 1858, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.; Lok, H.C.; Sahni, S.; Lane, D.J.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim Biophys Acta 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Radi, R.H.; Arbiser, J.L. Mitochondrial Metabolism in Melanoma. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Baenke, F.; Chaneton, B.; Smith, M.; Van Den Broek, N.; Hogan, K.; Tang, H.; Viros, A.; Martin, M.; Galbraith, L.; Girotti, M.R.; et al. Resistance to BRAF inhibitors induces glutamine dependency in melanoma cells. Mol Oncol 2016, 10, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Pickup, S.; Lee, S.C.; Mancuso, A.; Glickson, J.D. Lactate imaging with Hadamard-encoded slice-selective multiple quantum coherence chemical-shift imaging. Magn Reson Med 2008, 60, 299–305. [Google Scholar] [CrossRef]
- Nath, K.; Nelson, D.S.; Ho, A.M.; Lee, S.C.; Darpolor, M.M.; Pickup, S.; Zhou, R.; Heitjan, D.F.; Leeper, D.B.; Glickson, J.D. (31) P and (1) H MRS of DB-1 melanoma xenografts: lonidamine selectively decreases tumor intracellular pH and energy status and sensitizes tumors to melphalan. NMR Biomed 2013, 26, 98–105. [Google Scholar] [CrossRef]
- Rata, M.; Giles, S.L.; deSouza, N.M.; Leach, M.O.; Payne, G.S. Comparison of three reference methods for the measurement of intracellular pH using 31P MRS in healthy volunteers and patients with lymphoma. NMR Biomed 2014, 27, 158–162. [Google Scholar] [CrossRef]
- Goodwin, M.L.; Gladden, L.B.; Nijsten, M.W.; Jones, K.B. Lactate and cancer: revisiting the warburg effect in an era of lactate shuttling. Front Nutr 2014, 1, 27. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Orlovskiy, S.; Gupta, P.K.; Roman, J.; Arias-Mendoza, F.; Nelson, D.S.; Koch, C.J.; Narayan, V.; Putt, M.E.; Nath, K. Lonidamine Induced Selective Acidification and De-Energization of Prostate Cancer Xenografts: Enhanced Tumor Response to Radiation Therapy. Cancers (Basel) 2024, 16. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Dummer, R.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Contribution of MEK Inhibition to BRAF/MEK Inhibitor Combination Treatment of BRAF-Mutant Melanoma: Part 2 of the Randomized, Open-Label, Phase III COLUMBUS Trial. J Clin Oncol 2023, 41, 4621–4631. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.K.S.; Li, S.; Macrae, E.R.; Park, J.I.; Mitchell, E.P.; Zwiebel, J.A.; Chen, H.X.; Gray, R.J.; McShane, L.M.; Rubinstein, L.V.; et al. Dabrafenib and Trametinib in Patients With Tumors With BRAF(V600E) Mutations: Results of the NCI-MATCH Trial Subprotocol H. J Clin Oncol 2020, 38, 3895–3904. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.; Lopez, A.; Lin, E.; Sales, D.; Perets, R.; Jain, P. Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Guan, H.; Yuan, S.; Cheng, H.; Zheng, J.; Zhang, Z.; Liu, Y.; Yu, Y.; Meng, Z.; Zheng, X.; et al. Targeting myeloid derived suppressor cells reverts immune suppression and sensitizes BRAF-mutant papillary thyroid cancer to MAPK inhibitors. Nat Commun 2022, 13, 1588. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012, 367, 107–114. [Google Scholar] [CrossRef]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Signal Transduct Target Ther 2023, 8, 455. [Google Scholar] [CrossRef]
- Li, L.; Zhao, G.D.; Shi, Z.; Qi, L.L.; Zhou, L.Y.; Fu, Z.X. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol Lett 2016, 12, 3045–3050. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Xiao, Z.; Chen, T.; Liang, S.H.; Guo, H. Glucose Metabolism on Tumor Plasticity, Diagnosis, and Treatment. Front Oncol 2020, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Beasley, G.M.; Parsons, C.; Broadwater, G.; Selim, M.A.; Marzban, S.; Abernethy, A.P.; Salama, A.K.; Eikman, E.A.; Wong, T.; Zager, J.S.; et al. A multicenter prospective evaluation of the clinical utility of F-18 FDG-PET/CT in patients with AJCC stage IIIB or IIIC extremity melanoma. Ann Surg 2012, 256, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Strobel, K.; Skalsky, J.; Steinert, H.C.; Dummer, R.; Hany, T.F.; Bhure, U.; Seifert, B.; Perez Lago, M.; Joller-Jemelka, H.; Kalff, V. S-100B and FDG-PET/CT in therapy response assessment of melanoma patients. Dermatology 2007, 215, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Van de Wiele, C.; Juanito, G.; Vander, B.K.; Lawal, I.; Sathekge, M.; Maes, A.; De Spiegeleer, B. Practical Considerations When Interpreting FDG PET/CT Imaging for Staging and Treatment Response Assessment in Melanoma Patients. Semin Nucl Med 2021, 51, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Perng, P.; Marcus, C.; Subramaniam, R.M. (18)F-FDG PET/CT and Melanoma: Staging, Immune Modulation and Mutation-Targeted Therapy Assessment, and Prognosis. AJR Am J Roentgenol 2015, 205, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Weber, P.; Arnold, A.; Hohmann, J. Comparison of 18F-FDG PET/CT and ultrasound in staging of patients with malignant melanoma. Medicine (Baltimore) 2022, 101, e31092. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Purwin, T.J.; Heilman, S.A.; Chervoneva, I.; McCue, P.A.; Berger, A.C.; Davies, M.A.; Gershenwald, J.E.; Krepler, C.; Aplin, A.E. ErbB3 Targeting Enhances the Effects of MEK Inhibitor in Wild-Type BRAF/NRAS Melanoma. Cancer Res 2018, 78, 5680–5693. [Google Scholar] [CrossRef]
- Chen, W.; Park, J.I. Tumor Cell Resistance to the Inhibition of BRAF and MEK1/2. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Schwartz, L.H.; Seymour, L.; Litiere, S.; Ford, R.; Gwyther, S.; Mandrekar, S.; Shankar, L.; Bogaerts, J.; Chen, A.; Dancey, J.; et al. RECIST 1.1 - Standardisation and disease-specific adaptations: Perspectives from the RECIST Working Group. Eur J Cancer 2016, 62, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Litiere, S.; Isaac, G.; De Vries, E.G.E.; Bogaerts, J.; Chen, A.; Dancey, J.; Ford, R.; Gwyther, S.; Hoekstra, O.; Huang, E.; et al. RECIST 1.1 for Response Evaluation Apply Not Only to Chemotherapy-Treated Patients But Also to Targeted Cancer Agents: A Pooled Database Analysis. J Clin Oncol 2019, 37, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of the metabolomics research in human melanoma models treated with trametinib. The red arrows highlight the effects of the mutated BRAF protein (i.e., increased growth and cell proliferation, increased glycolysis, and reduced pyruvate oxidation). Trametinib (structure in A; from istrockphoto.com) exerts its effect, inhibiting MEK and, thus, blocking the mutated BRAF effects (white block arrow). Mutated BRAF enhances the metabolic pathways highlighted with orange arrows, while those inhibited are shown with blue arrows. The green arrows indicate the procedures for determining the highlighted metabolites or cellular processes. Summarizing, we measured extracellular glucose and lactate levels (B; see Figure 2 for a detailed explanation), intracellular lactate and alanine levels by noninvasive 1H MRS (C; see Figure 4 and Figure 5), the effect of trametinib on the extracellular acidification rate (ECAR) and oxygen consumption rate (OCR; D; see Figure 3), bioenergetic parameters and intra- and extracellular pH by noninvasive 31P MRS (E; see Figure 6), and tumor growth (F; see Figure 7).
Figure 1.
Schematic representation of the metabolomics research in human melanoma models treated with trametinib. The red arrows highlight the effects of the mutated BRAF protein (i.e., increased growth and cell proliferation, increased glycolysis, and reduced pyruvate oxidation). Trametinib (structure in A; from istrockphoto.com) exerts its effect, inhibiting MEK and, thus, blocking the mutated BRAF effects (white block arrow). Mutated BRAF enhances the metabolic pathways highlighted with orange arrows, while those inhibited are shown with blue arrows. The green arrows indicate the procedures for determining the highlighted metabolites or cellular processes. Summarizing, we measured extracellular glucose and lactate levels (B; see Figure 2 for a detailed explanation), intracellular lactate and alanine levels by noninvasive 1H MRS (C; see Figure 4 and Figure 5), the effect of trametinib on the extracellular acidification rate (ECAR) and oxygen consumption rate (OCR; D; see Figure 3), bioenergetic parameters and intra- and extracellular pH by noninvasive 31P MRS (E; see Figure 6), and tumor growth (F; see Figure 7).

Figure 2.
The extracellular glucose consumption (A) and lactate production (B) were measured in the four cultured melanoma cell lines in the absence (control) or presence of trametinib (7 nmol/L) for 48 hours. Relative values (y-axes) were obtained by dividing the extracellular metabolite content by the number of cells. Error bars denote the standard deviation (SD). An asterisk (*) indicates a statistically significant difference (p < 0.05) between control (n = 3) and trametinib-treated (n = 3) groups.
Figure 2.
The extracellular glucose consumption (A) and lactate production (B) were measured in the four cultured melanoma cell lines in the absence (control) or presence of trametinib (7 nmol/L) for 48 hours. Relative values (y-axes) were obtained by dividing the extracellular metabolite content by the number of cells. Error bars denote the standard deviation (SD). An asterisk (*) indicates a statistically significant difference (p < 0.05) between control (n = 3) and trametinib-treated (n = 3) groups.
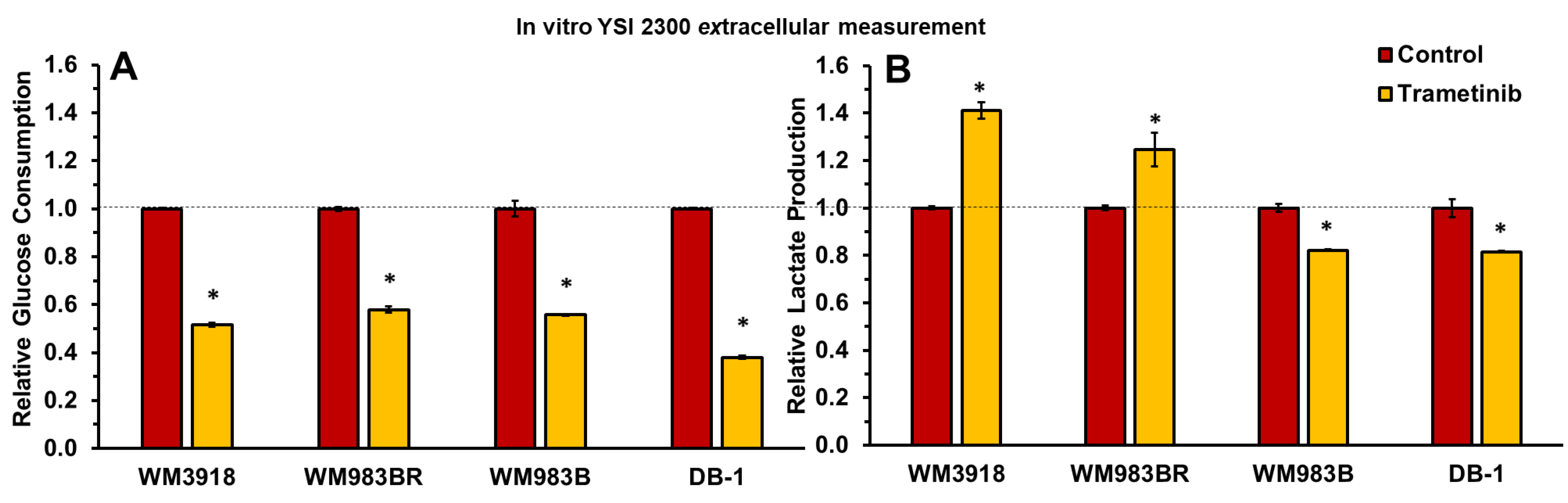
Figure 3.
Effect of trametinib treatment on OCR and ECAR of cultured melanoma cells. The graph shows the four melanoma cell lines’ OCR vs. ECAR under basal conditions. Open symbols represent untreated cells, while solid symbols represent trametinib-treated cells. The dashed arrows designate cell lines with statistically significant shifts in the OCR/ECAR ratio (Table 1), which indicates different cell energy phenotypes and shows shifts in the preference of energy pathways for each cell line under trametinib.
Figure 3.
Effect of trametinib treatment on OCR and ECAR of cultured melanoma cells. The graph shows the four melanoma cell lines’ OCR vs. ECAR under basal conditions. Open symbols represent untreated cells, while solid symbols represent trametinib-treated cells. The dashed arrows designate cell lines with statistically significant shifts in the OCR/ECAR ratio (Table 1), which indicates different cell energy phenotypes and shows shifts in the preference of energy pathways for each cell line under trametinib.
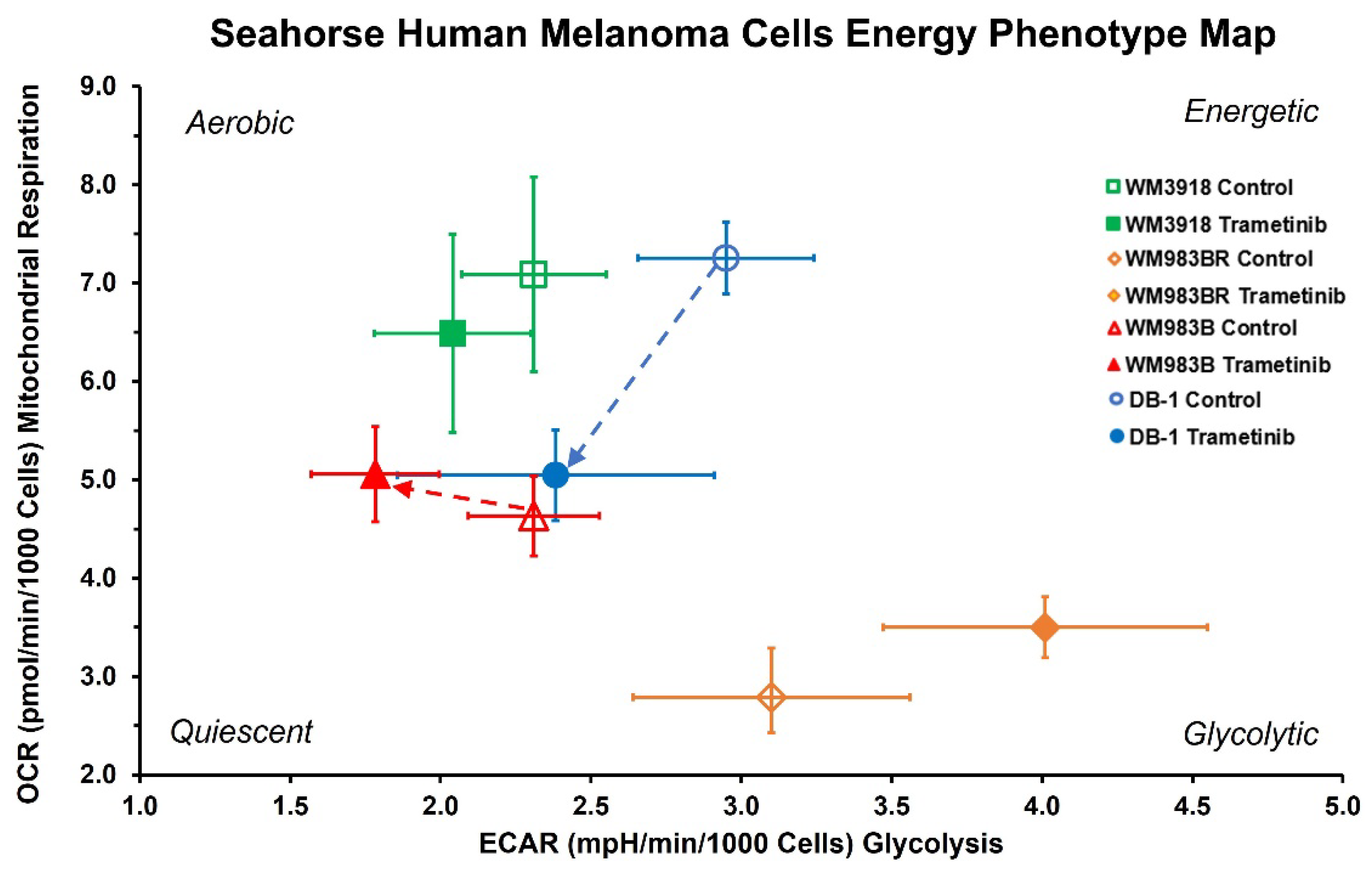
Figure 4.
In vitro 1H MRS of human melanoma cell lines. (A) Representative high-resolution 1H MRS spectra depicting alanine and lactate in the four cell lines are illustrated. Control spectra are displayed in red, while trametinib-treated spectra are shown in green. (B) Mean values ± SD (n=3) of the intracellular lactate and alanine concentrations were determined by integrating their 1H MRS signals in (A) and normalized by the trimethylsilyl propanoic acid (TSP) content protons to obtain mM/cell concentrations. The asterisks (*) denote a statistically significant difference (p < 0.05) between the control values of lactate or alanine in the WM3918 cell line (in red) vs. the control values in the other three cell lines. The p-values on top of the trametinib-treated data (in yellow) demonstrate the statistical significance of the trametinib-related reduction vs. its control value.
Figure 4.
In vitro 1H MRS of human melanoma cell lines. (A) Representative high-resolution 1H MRS spectra depicting alanine and lactate in the four cell lines are illustrated. Control spectra are displayed in red, while trametinib-treated spectra are shown in green. (B) Mean values ± SD (n=3) of the intracellular lactate and alanine concentrations were determined by integrating their 1H MRS signals in (A) and normalized by the trimethylsilyl propanoic acid (TSP) content protons to obtain mM/cell concentrations. The asterisks (*) denote a statistically significant difference (p < 0.05) between the control values of lactate or alanine in the WM3918 cell line (in red) vs. the control values in the other three cell lines. The p-values on top of the trametinib-treated data (in yellow) demonstrate the statistical significance of the trametinib-related reduction vs. its control value.
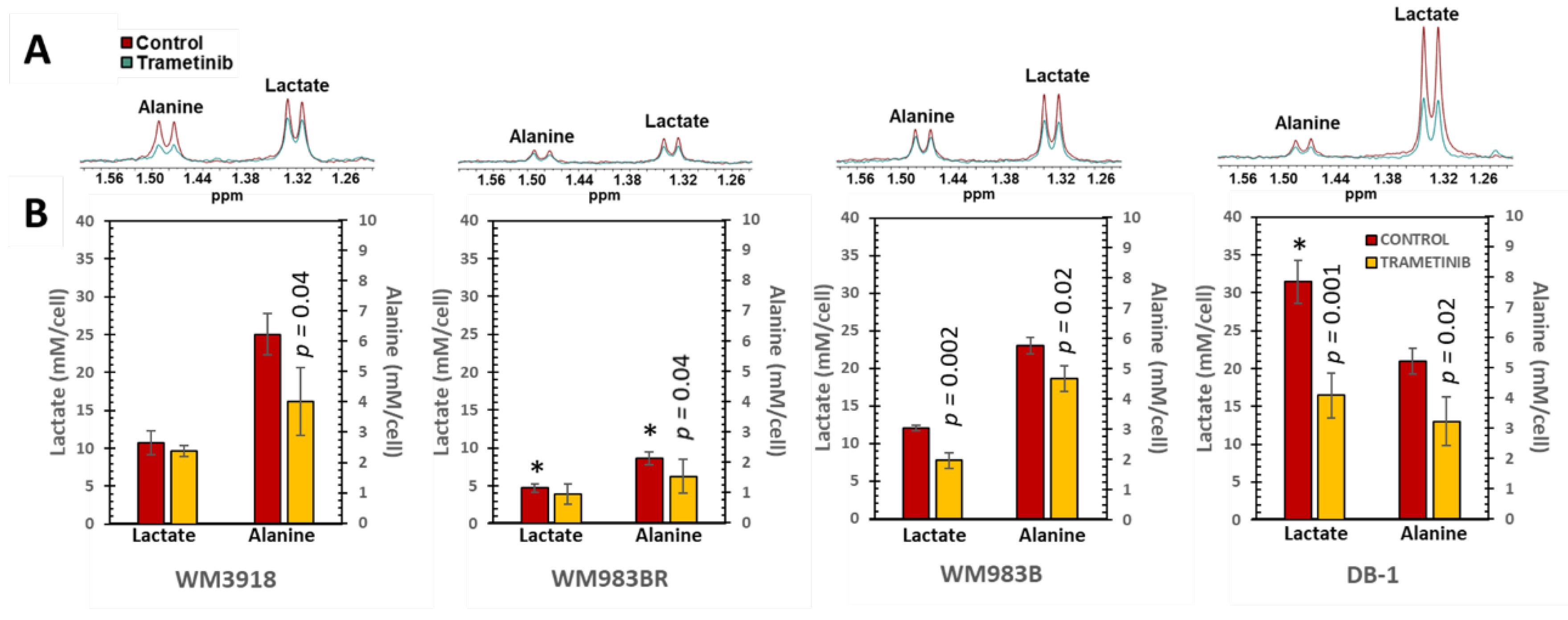
Figure 5.
(previous page). In vivo localized 1H MRS results. (A) Noninvasive 1H MR spectra were acquired using the Hadamard-selective MQC transfer pulse sequence to measure lactate and alanine. The spectra at Day 0 (red), Day 2 (green), and Day 5 (blue) of trametinib therapy in each xenograft model are shown from bottom to top. Box-and-whiskers plots of time-related in vivo changes are shown in (B) for tumor lactate and (C) for alanine in mice xenografts of each cell line. At the top of B and C, in red, the box plots show the lactate and alanine changes in untreated (control) groups, while at the bottom, in yellow, the lactate and alanine changes in the trametinib-treated mice. The p-values in (B) and (C) denote the statistical significance of the mean difference of lactate (B) and alanine (C) for the control vs. trametinib groups at each time point. The tables below (B) and (C) summarize the time-related analysis of each metabolite using linear regressions. The equation that describes the time course, the adjusted R2, and the significance of the regression are shown for the control (red) and trametinib time curves (yellow). The last row in both tables shows the statistical significance when the slopes of control vs. trametinib are determined. Empty spaces denote and adjusted R2 > 0.26 or p-values that were non-significant.
Figure 5.
(previous page). In vivo localized 1H MRS results. (A) Noninvasive 1H MR spectra were acquired using the Hadamard-selective MQC transfer pulse sequence to measure lactate and alanine. The spectra at Day 0 (red), Day 2 (green), and Day 5 (blue) of trametinib therapy in each xenograft model are shown from bottom to top. Box-and-whiskers plots of time-related in vivo changes are shown in (B) for tumor lactate and (C) for alanine in mice xenografts of each cell line. At the top of B and C, in red, the box plots show the lactate and alanine changes in untreated (control) groups, while at the bottom, in yellow, the lactate and alanine changes in the trametinib-treated mice. The p-values in (B) and (C) denote the statistical significance of the mean difference of lactate (B) and alanine (C) for the control vs. trametinib groups at each time point. The tables below (B) and (C) summarize the time-related analysis of each metabolite using linear regressions. The equation that describes the time course, the adjusted R2, and the significance of the regression are shown for the control (red) and trametinib time curves (yellow). The last row in both tables shows the statistical significance when the slopes of control vs. trametinib are determined. Empty spaces denote and adjusted R2 > 0.26 or p-values that were non-significant.
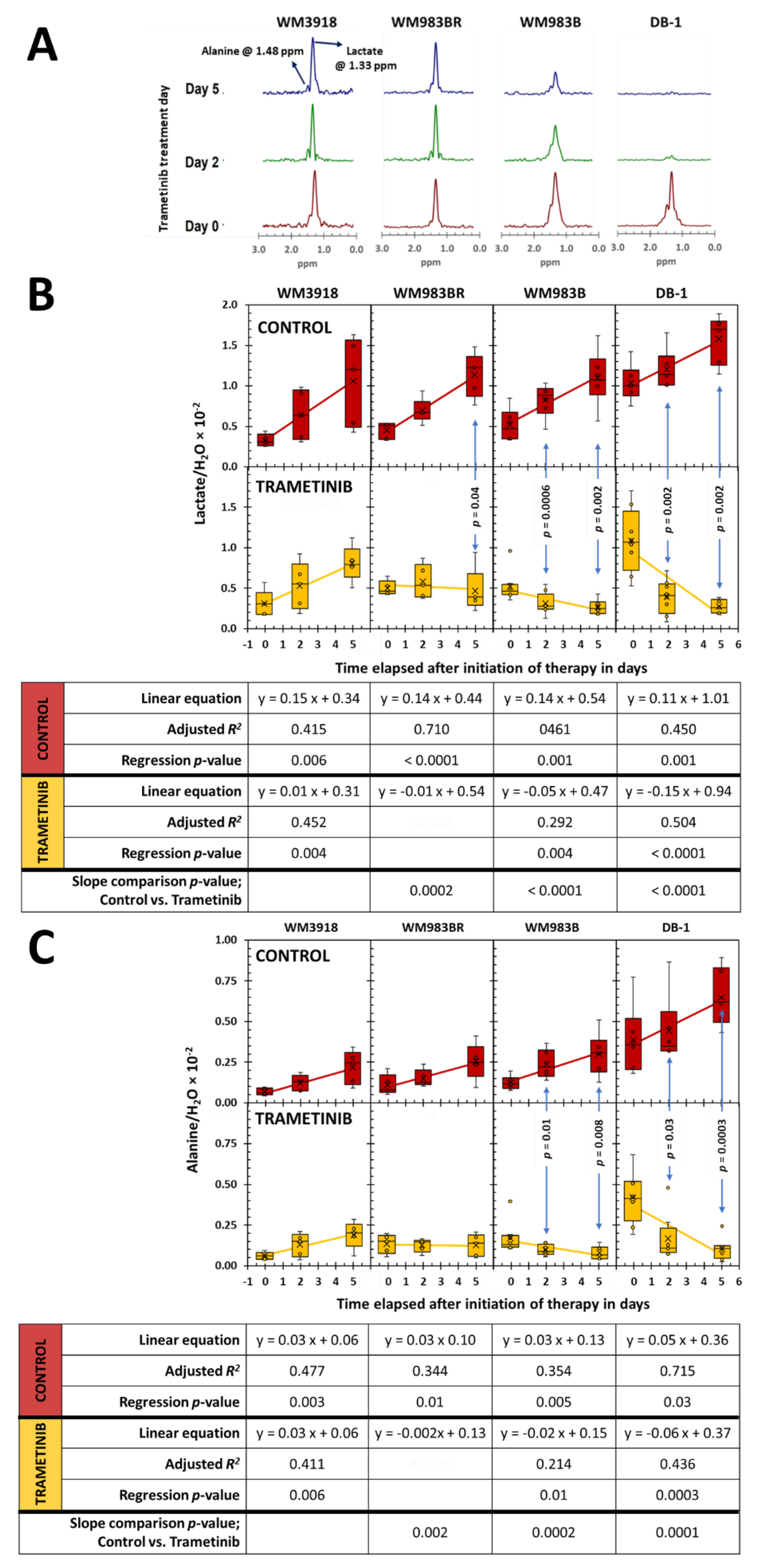
Figure 6.
(previous page). In vivo localized 31P MRS results. (A) Noninvasive MR spectra were acquired using the ISIS pulse sequence of DB-1 human melanoma xenografts at Day 0 (red), Day 2 (green), and Day 5 (blue) of trametinib treatment. Peak assignments: 3-APP, 3-amino propylphosphonic acid; PME, phosphomonoesters; Pi, inorganic phosphate; PDE, phosphodiesters; NTP, the a, b, and g 31P signals of nucleoside triphosphates. We determined the chemical shift differences between Pi and α-NTP and those of 3-APP and α-NTP to obtain the pH values of the intracellular (pHi) and extracellular compartments (pHe), respectively. The intensity variability of 3-APP shown is due to factors like animal weight and administration method but does not interfere with the pHe measurements. (B) Box-and-whiskers plots of the time-related changes of the b-NTP/Pi ratio measured in 31P MR spectra. In red, the top row of box plots depicts the data of untreated mice set apart by the xenografted cell line, while on the bottom row of box plots, in yellow, the trametinib-treated results are shown. The p-value of the mean comparison of the control vs. treated pair at each time point is shown in the graphs. Like in Figure 5, the bottom table in (B) summarizes the fitting analysis of the data to time-related linear regressions and the slope comparisons between control vs. trametinib-treated in each cell line. The pH values in the intracellular (C) and extracellular compartments (D) determined by 31P MRS in the four melanoma xenografts are also shown. Only valuable time-dependent data was obtained for pHi and pHe in the DB-1 xenografts: pHi during trametinib treatment (i.e., y = -0.021x +6.9, adjusted R2 = 0.240, and regression’s p-value = 0.01), and pHe in the DB-1 controls (i.e., y = -0.020 x + 7.0, adjusted R2 = 0.235, and regression’s p-value = 0.02). In addition, the comparison of slopes for pHe of DB-1 between untreated (m = -0.020) and trametinib treated (m = 0.010) was statistically significant (p = 0.02).
Figure 6.
(previous page). In vivo localized 31P MRS results. (A) Noninvasive MR spectra were acquired using the ISIS pulse sequence of DB-1 human melanoma xenografts at Day 0 (red), Day 2 (green), and Day 5 (blue) of trametinib treatment. Peak assignments: 3-APP, 3-amino propylphosphonic acid; PME, phosphomonoesters; Pi, inorganic phosphate; PDE, phosphodiesters; NTP, the a, b, and g 31P signals of nucleoside triphosphates. We determined the chemical shift differences between Pi and α-NTP and those of 3-APP and α-NTP to obtain the pH values of the intracellular (pHi) and extracellular compartments (pHe), respectively. The intensity variability of 3-APP shown is due to factors like animal weight and administration method but does not interfere with the pHe measurements. (B) Box-and-whiskers plots of the time-related changes of the b-NTP/Pi ratio measured in 31P MR spectra. In red, the top row of box plots depicts the data of untreated mice set apart by the xenografted cell line, while on the bottom row of box plots, in yellow, the trametinib-treated results are shown. The p-value of the mean comparison of the control vs. treated pair at each time point is shown in the graphs. Like in Figure 5, the bottom table in (B) summarizes the fitting analysis of the data to time-related linear regressions and the slope comparisons between control vs. trametinib-treated in each cell line. The pH values in the intracellular (C) and extracellular compartments (D) determined by 31P MRS in the four melanoma xenografts are also shown. Only valuable time-dependent data was obtained for pHi and pHe in the DB-1 xenografts: pHi during trametinib treatment (i.e., y = -0.021x +6.9, adjusted R2 = 0.240, and regression’s p-value = 0.01), and pHe in the DB-1 controls (i.e., y = -0.020 x + 7.0, adjusted R2 = 0.235, and regression’s p-value = 0.02). In addition, the comparison of slopes for pHe of DB-1 between untreated (m = -0.020) and trametinib treated (m = 0.010) was statistically significant (p = 0.02).
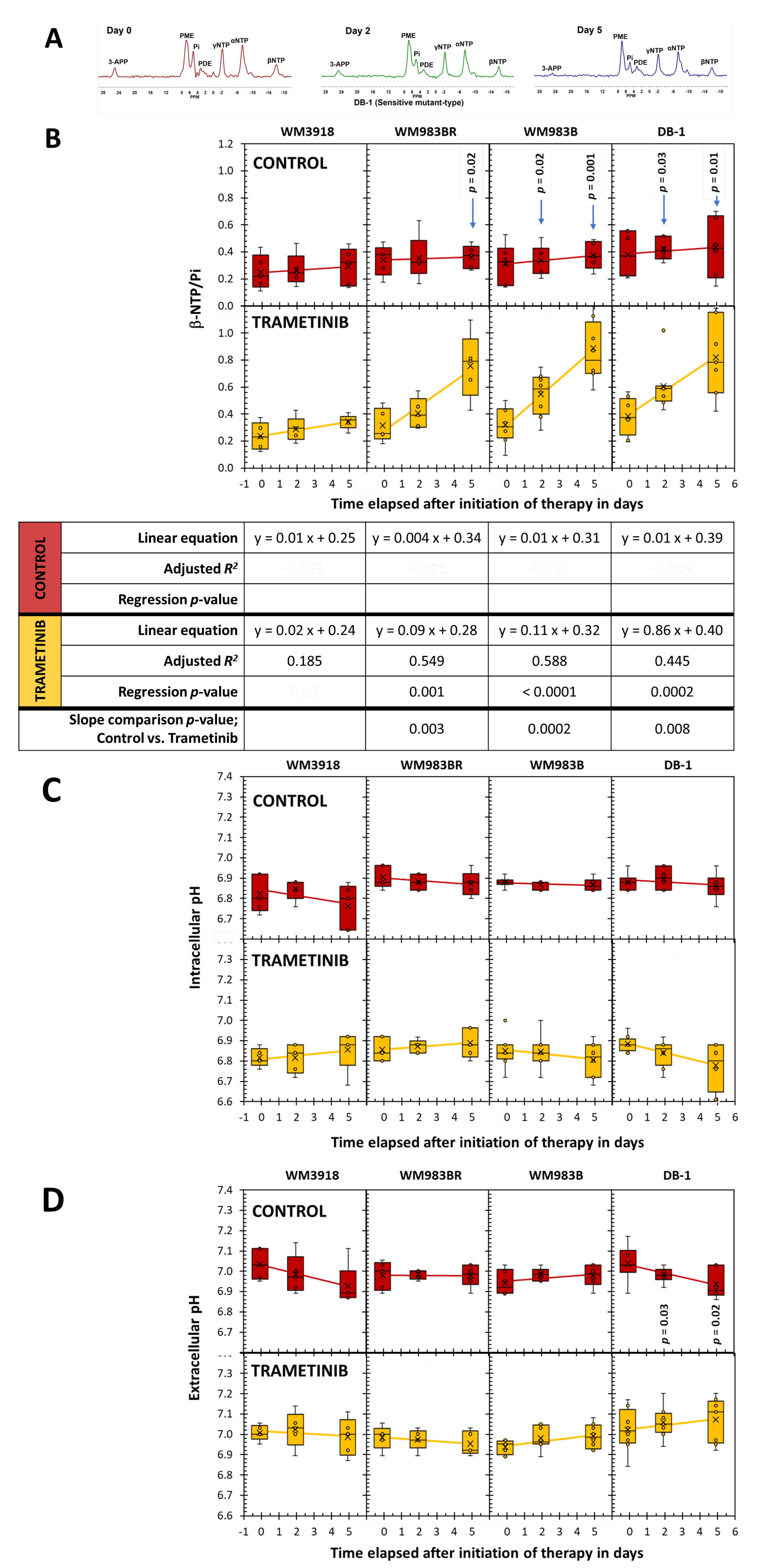
Figure 7.
Comparison of tumor growth on Day 0, Day 2, and Day 5 between untreated controls (red) and trametinib-treated xenografts (yellow). The p-values in the graphs denote the significance of comparing the untreated (control) vs. trametinib-treated pair at specific times. The table shows the parameters related to the statistical analysis of the data as time-dependent changes.
Figure 7.
Comparison of tumor growth on Day 0, Day 2, and Day 5 between untreated controls (red) and trametinib-treated xenografts (yellow). The p-values in the graphs denote the significance of comparing the untreated (control) vs. trametinib-treated pair at specific times. The table shows the parameters related to the statistical analysis of the data as time-dependent changes.
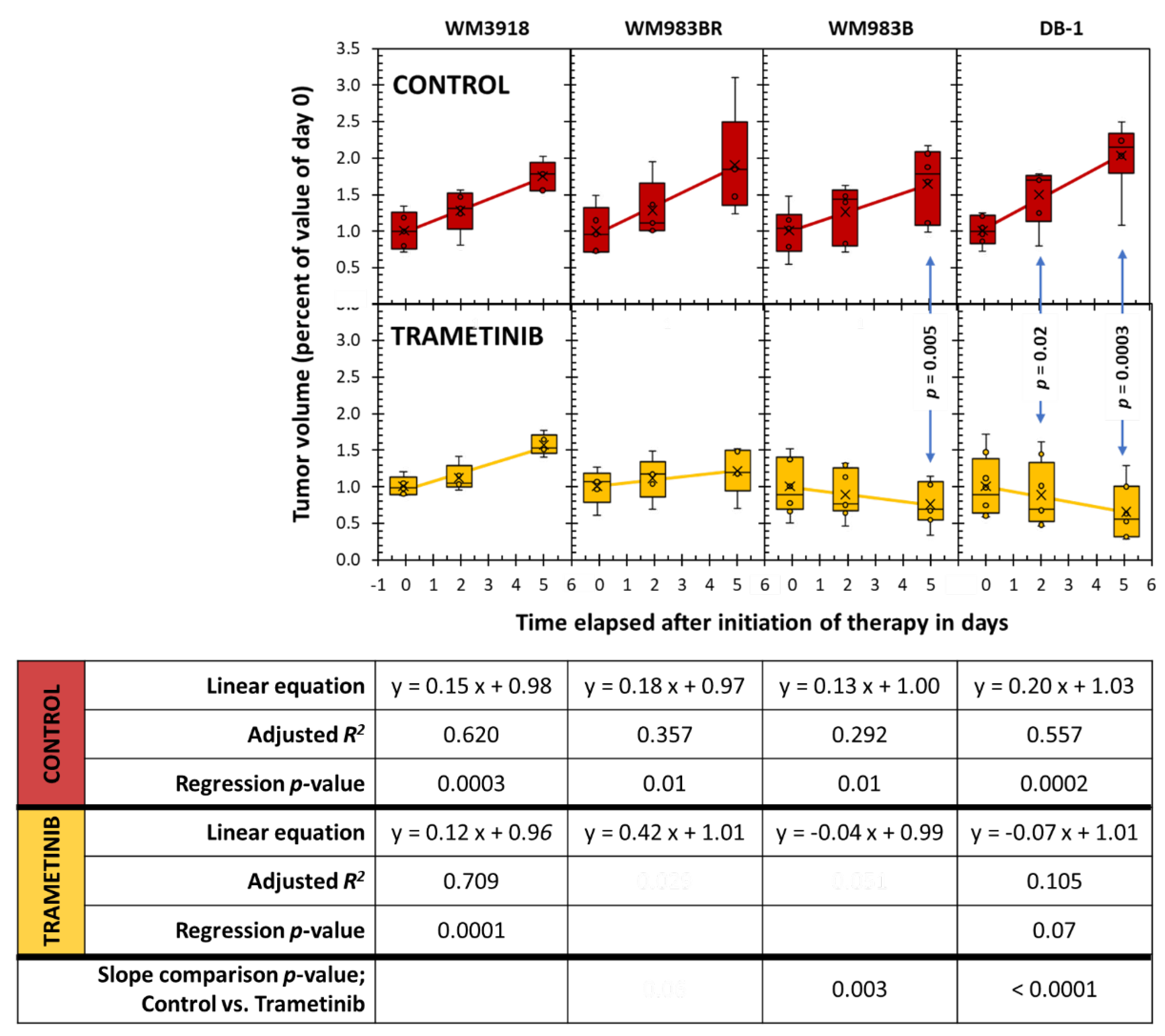

Table 1.
Effect of trametinib in OCR, ECAR, and the OCR/ECAR value in melanoma cell lines. Data are shown as mean value ± standard deviation with n = 8 per group. We used the Student-independent t-test to determine statistical significance.
Table 1.
Effect of trametinib in OCR, ECAR, and the OCR/ECAR value in melanoma cell lines. Data are shown as mean value ± standard deviation with n = 8 per group. We used the Student-independent t-test to determine statistical significance.
| Group | Mean ± SD (n = 8) | p-value | |
|---|---|---|---|
| Control | Trametinib | ||
| Oxygen Consumption Rate (pmol/min/1000cells) | |||
| WM3918 | 7.09±0.99 | 6.49±1.01 | 0.13 |
| WM983BR | 2.79±0.50 | 3.50±0.31 | 0.007 |
| WM983B | 4.63±0.41 | 5.06±0.48 | 0.014 |
| DB-1 | 7.25±0.36 | 5.05±0.46 | <0.001 |
| Extracellular Acidification Rate (pmol/min/1000cells) | |||
| WM3918 | 2.31±0.24 | 2.04±0.26 | 0.011 |
| WM983BR | 3.10±0.46 | 4.01±0.54 | 0.015 |
| WM983B | 2.31±0.22 | 1.78±0.21 | 0.001 |
| DB-1 | 2.95±0.29 | 2.38±0.53 | 0.001 |
| OCR/ECAR Ratio – Cell Energy Phenotype | |||
| WM3918 | 3.08±0.37 | 3.18±0.29 | 0.46 |
| WM983BR | 0.90±0.12 | 0.88±0.08 | 0.71 |
| WM983B | 2.02±0.22 | 2.89±0.55 | <0.001 |
| DB-1 | 2.48±0.28 | 2.17±0.33 | 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



