
Submitted:
19 June 2024
Posted:
20 June 2024
You are already at the latest version
Abstract
(max 200 words): The contamination of groundwater is a serious concern for ecosystems and human health. In this regard, biochar represents a promising low-cost and sustainable material for in-situ groundwater remediation. Herein, biochar waste of biomass energy production process derived from pine wood gasification at 950°C was used as active carbonaceous material in combination with different biopolymers: chitosan (CS), alginate (ALG), potato starch (PST), and carboxymethylcellulose (CMC) to produce BC@biopolymer composite materials. Different concentrations of biochar (0.3–1.0 g·L-1) and biopolymers (0.2–20.0 g·L-1) were tested and blending mode was used as simple and straightforward strategy to obtain BC composites. Scanning electron microscopy (FE-SEM), UV-Visible, infrared (FT-IR) spectroscopy, specific surface area (BET method), pore volume, and pore size distribution (textural parameters), dynamic light scattering (DLS), and ζ-potential were used to investigate the composite chemical structure, stability, and to assess the interaction among counterparts. On optimized formulations, breakthrough tests and batch adsorption isotherms on trichloroethylene (TCE) as a model contaminant were performed to evaluate the capability of composites to be delivered through a porous medium determining the transport properties, (hydrodynamic dispersion retention percentage, and adsorption capacity). This biopolymer-modified biochar holds significant promise as a sustainable and effective solution to address various groundwater pollution challenges.
Keywords:
colloidal biochar
; biopolymers
; chitosan
; alginate
; potato starch
; carboxymethylcellulose
; composite material
; trichloroethylene (TCE)
; in-situ remediation
; groundwater remediation
1. Introduction
In recent years, water pollution has been a pervasive issue worldwide and has posed a significant threat to both human health and the ecosystem [1,2]. Among different waterbodies and natural water resources, groundwater is one of the most important freshwater sources and is seriously threatened by anthropogenic activities, such as industrial and agricultural practices [3]. Concerning groundwater pollution, inorganic (mainly heavy metals denoted as potentially toxic elements (PTEs)), and organic (e.g., chlorinated hydrocarbons, (CHCs), polycyclic aromatic hydrocarbons (PAHs) and poly- and perfluoroalkyl substances (PFAS), dyes, nitro compounds, pesticides, etc.) contaminants can be recalcitrant under natural conditions [4,5]. Due to their chemical characteristics, these substances do not easily biodegrade. Hence, they have high persistence in the environment and are difficult to remove after subsurface penetration [5]. Notably, according to their chemical nature, upon release, dense hydrophobic organic compounds tend to migrate downward and penetrate the water table (the so-called dense non-aqueous phase liquids (DNAPL)), causing extensive subsurface contamination [6]. Therefore, it is necessary to develop effective groundwater remediation technologies which enable the cleanup of the contaminated site reducing pollution to a desirable and safe level via sustainable, cost-effective, and eco-friendly methods [7].
To date, lots of groundwater remediation technologies have been developed both in-situ (treatment of the contaminated matrix in its location) and ex-situ (removal of the contaminated matrix and subsequent treatment in a different location) [8]. Depending on the mechanism underlying the contaminant removal or degradation, they can be classified into three main categories: chemical (e.g., chemical reduction or oxidation), physical (e.g., soil washing, stabilization/solidification, adsorption), and biological (e.g., microbial remediation, biostimulation, bioaugmentation) [8,9]. Besides common ex-situ treatments, such as pump-and-treat methods, thermal desorption, oxidation, and solvent extraction, in-situ treatment technologies are potentially more sustainable from an economic and an environmental standpoint [10]. Thus, in-situ treatments represent preferable options, and, in this regard, absorption methods (physical capture) are gaining attention due to their operational simplicity, recyclability, high efficiency, and environmental friendliness [11]. The most traditional in-situ adsorption systems are Permeable Reactive Barriers (PRBs), constituted by a trench perpendicular to groundwater flow filled with inert and reactive material (e.g., activated carbon) allowing the contaminant plume to pass through it while adsorbing contaminants [12,13]. Recently, new in-situ adsorption processes have been developed based on injectable colloidal activated carbon (CAC) stabilized by humic acids and carboxymethylcellulose (CMC) [14,15]. This approach allows remediation at greater depths in that digging a trench at great depths (over 30 m) is significantly more complex than drilling [16]. However, implementing CAC in in-situ remediation approaches remains niche due to challenges in distributing the colloid effectively without clogging porous media. Proper suspension retention is by no means a simple task: too little retention may cause failure in achieving plume break, while too high retention may cause clogging issues that results groundwater bypass [15]. Thus, besides activated carbons (ACs), a wide variety of sorbents have been used for groundwater remediation purposes, such as zeolites [17,18], organoclays [19,20], organo-hydrotalcites [21], and graphene oxides [22]. As a cheap and eco-friendly material, biochar (BC) is one of the most interesting alternatives to conventional absorbers. As with ACs, BC is a biomass derived C-rich material obtained via pyrolysis or gasification of different feedstock, such as wood, agricultural waste, manure, and wastewater sludge. It is highly hydrophobic material characterized by reduced stability in water due to its particle size [23]. Although, there is usually a fraction of the particle size distribution of BC that is fine enough to be considered as a colloid (particles with diameters smaller than a few tens of micrometers) [24], and thus characterized by greater stability in water. However, this fraction is a minority: 1-20 mg/g [25]. Biochar itself possesses a large specific surface area, high porosity, and thermal resistance, although its properties and sorption behavior depend not only on biomass feedstock but also on production temperature [26,27]. Several studies demonstrated that biochar could sorb a wide range of contaminants, ranging from organic compounds to metal and metalloids [28,29,30,31]. Nevertheless, the hydrophobic nature and reduced stability of colloidal biochar in the aqueous environment influence its retaining and adsorption capacity [29]. Indeed, colloid retention by porous media is primarily controlled by straining processes [32], which occurs when colloids are trapped by bottlenecks formed by collector particles [33]. Despite extensive research over the past years, it remains a complex mechanism. A critical ratio between colloid diameter (dp) and collector diameter (dc) determines straining (these ratios vary between 0.027 [34] and 0.0017 [35]), although straining is also controlled by physicochemical factors such as ionic strength [34] and fluid dynamics: increasing velocity reduces straining intensity by destabilizing colloids attached to secondary minimum straining sites [36].
To improve stability, adsorption, and retention properties (e.g., carbon structure, surface area, pore size) and widen potential applications for environmental remediation, several physical, mechanical, and chemical modification processes of biochar have been carried out in recent years [37]. In this framework, BC shows intriguing properties as a superior filler in polymer and biopolymer-based composites [29,38,39]. Concerning polymer matrices, BC has been used in several composite formulations in combination with polyolefins, such as polypropylene (PP), polyethylene (PE), polyethylene terephthalate (PET), polyamide, polyester, and thermosetting matrices (epoxy resin, unsaturated polyester resin, rubber), with significant enhancement of either mechanical or electrical properties even at low concentrations [38]. Despite the improved properties, synthetic and petroleum-based polymers suffer from a lack of biodegradability. Thus, biopolymers such as polylactic acid (PLA), poly(3-hydroxybutyrate) (PHB), starch-based polymers, and biopolymers from algae biomass have the potential to be used as valuable flexible matrices to produce high-quality biochar composites and to establish a sustainable circular economy globally [40,41]. Nevertheless, biopolymer-based BC composites still require optimization to reach performances comparable to traditional active carbon materials and take advantage over the synergistic effects of biochar dispersed in polymers.
In this work, biochar powder (BC) derived from pine wood gasification at 950° were used as active carbonaceous material filler of polymer matrices. Specifically, four different BC samples were studied, based on different sieving and grinding processes, as described in Table 1. On selected BC sample (AP sample), water-based BC@biopolymer composite materials were obtained combining raw BC with commercially available biopolymers, such as chitosan (CS), alginate (ALG), potato starch (PST), and carboxymethylcellulose (CMC) as host matrices. The aim was to (i) modify and enhance the surface and physical characteristics of the biochar, (ii) increase its stability in the aqueous environment via dispersion in a hydrophilic matrix, and (iii) enhance diffusion properties in porous media, thus, broadening the contaminants which can be removed from the water. Different concentrations of biochar (0.3–1 g·L−1) and biopolymers (0.2–20 g·L−1) were tested, and a one-pot blending mode at different temperatures (from room temperature up to 80 °C, depending on polymer solubility) was used as a simple and straightforward strategy to obtain BC@biopolymer composites. To improve physicochemical properties of potato starch-based composite, an alkylpolyglucoside-based surfactant (APG2, a non-ionic surfactant) was added in a 0.1–5.0% v/v range. APG-based surfactant was chosen according to its eco-friendliness, nontoxicity, and biodegradability [42]. Raw pine wood biochar (BC) and the as-prepared composite materials were characterized via field-emission scanning electron microscopy (FE-SEM), N2 adsorption/desorption measurements, and UV-Visible spectroscopy, whereas infrared (FT-IR) spectroscopy was used further to confirm the composite chemical structure and interaction among counterparts. BC@biopolymer materials were compared with pristine biochar in terms of stability and hydrodynamic size in water at their native pH.
The functional properties of optimized formulations were tested in breakthrough tests in a 140 × 25 mm sediment packed PMMA column to assess the capability of the composite to be delivered through a porous medium. Herein, different granulometry glass beads were used (600–800 µm) to determine transport properties, such as hydrodynamic dispersion and retention percentage in various aquifer configurations. Batch adsorption isotherms on trichloroethylene (TCE) as a model contaminant were carried out to confirm the adsorptive capacity of the BC-based composites, which has already been validated in the literature [43]. TCE was selected as the contaminant of choice due to its importance in contamination scenarios [44] and because it has been demonstrated (in previous studies) how the adsorption of TCE on BCs can promote the establishment of a beneficial biological reductive dechlorination process [45]. The preparation of herein presented stable biopolymer-based biochar composites represents a promising low-cost, green, and effective sorbent in groundwater remediation strategies.

Table 1.
Biochar (BC) samples notation studied in this work. All samples were obtained at 950 °C.
| Sieving at 0.5 mm | Sieving at 64 µm and grinding |
|---|---|
| A | AP |
| B | BP |
* P denotes additional manual grinding on sample.
2. Materials and Methods
2.1. Pine Wood Biochar Production
Pine wood biochar (BC) was used to produce the composite material. The BC used is a waste product of a biomass energy production process implemented by Burkhardt Energy and Building Technology. The energy production involves the use of two machines in series: the first (V3.90) carries out the gasification of pine wood pellets at 950 °C, while the second (CHP ECO 220) produces thermal and electrical energy from the combustion of the previously produced syngas. The BC in question is, therefore, a by-product of the heat and power generation process that is usually disposed of as waste. Two replicated batches were used (A and B) and compared with the two samples obtained after sieving and grinding procedure (AP and BP), respectively (see Table 1).
2.2. Preparation of BC@biopolymer Composites
For the synthesis of BC@biopolymer composites, biochar from pine wood and commercially available bio-based polymers (all Merck Sigma-Aldrich, Milan) were used: chitosan (CS), sodium alginate (ALG), potato starch (PST), and sodium carboxymethylcellulose (CMC). To optimize the composite formulation, biochar in the 0.3–1.0 g·L−1 range were mixed with different concentration of biopolymers: 0.2, 5.0, 7.5, 10.0, 15.0, 20.0 g·L−1. Pilot tests were conducted in a total volume of 10 mL, whereas the final formulations were scaled-up to 1.5 L and g·L−1. of BC. In a typical procedure, the selected amount of polymer powder was mixed with BC and the solid mixture homogenized. 10 mL of ultra-pure water (H2Oup, 18.3 MΩ·cm, produced with a Zeneer Power I Scholar-UV instrument, Full Tech Instruments, Italy) were added, and the mixture sonicated for 30 minutes. Then, to obtain the final composite the aqueous suspension was vigorously stirred at different experimental conditions depending on polymer solubility, as reported in Table 2.
At the end, BC@PST composite was allowed to cool down to room temperature and 1.0% v/v of APG2 surfactant (Chimec, Italy) was added, whereas BC@CS, BC@ALG, and BC@CMC were synthesized in absence of further additives. The as-prepared final composites were used without further modification or purification. As a blank sample, pristine BC aqueous suspension (denoted as BC) and BC + 1.0% v/v APG2 aqueous solution (denoted as BC/APG2) in the 0.2–5.0% v/v range were prepared.
2.3. Sedimentation Tests
To investigate the stability over time of BC composites, sedimentation tests were performed as follows: the as-prepared BC@polymer composites (10 mL) were introduced in a glass vial and the suspensions were kept at quiescent state for 7 days without stirring. Then, a sample aliquot (1 mL) was taken from 1/3 of the total volume and diluted 1:3 v/v with H2Oup prior to UV-Vis analysis. For the test, the biochar concentration was fixed at 0.3 g/L and the absorbance value at 650 nm at 0 h, 24 h, 7 days (Abst) was measured. The sedimentation percentage of BC inside the polymer matrix at different times was calculated using equation (1).
Sedimentation (%) = (1 − (Abst/Abst0))·100
2.4. Adsorption Isotherms
Batch adsorption tests were carried out with TCE to verify the efficiency of the raw BC in immobilizing the chosen contaminant in an aquifer medium. The BC used in these tests was subjected to the same treatment as that used in the distribution tests (paragraph 2.5), i.e., sieving at 64 µm and manual grinding. For TCE adsorption tests, the 25 mg·L−1 solution was prepared in a 1 L Tedlar bag (Supelco, Bellefonte, PA, USA) to avoid headspace formation; a volume of 17 µL of pure TCE (ACS ≥ 99.5%, Sigma-Aldrich, St. Louis, MO, USA) was spiked to achieve the decided concentration. The solution was horizontally shaken for three days to reach complete solubilization of TCE. To verify the initial effective TCE concentration, the contaminated solution was sampled and analyzed before setting up the tests. The batch reactors (VWR International glass vials, Milan, Italy) were prepared by weighing a known amount of BC and fully filling with approximately 0.02 L of the contaminated solution. The reactor was sealed by a Teflon butyl stopper (Wheaton, Millville, NJ, USA) and an aluminum cap and mechanically shaken for 24 h. Five different loads of BC were placed in the borosilicate glass reactors: 10, 20, 30, 40, and 50 mg, then, the 25 mg·L−1 solution of TCE was added. Each load of BC was performed in triplicate to strengthen the data.
The experimental data obtained were subsequently analyzed using SigmaPlot 12.0 software and fitted with a Freundlich-type isotherm (Qe, equation (2)) as it gave an optimal representation of the trend.
Qe = KFCeqn
The Freundlich constants KF and n were determined using SigmaPlot 12.0, while the equilibrium concentration Ceq in the liquid was carried out via gas chromatographic analysis sampling the contaminated solution. The determination of the concentration of TCE adsorbed on the BC (defined as S, mgTCE·gBC−1) is calculated using equation (3):
where C0 is the initial TCE concentration in the contaminated solution, V and m are the volume of solution and the mass of BC loaded in each reactor, respectively.
S = (C0V − CeqV)/m
2.5. Continuous Flow Column Distribution Tests
Column transport tests were performed to verify the deliverability of the produced composites and their retention in the simulated aquifer. The columns used in these tests are made of PMMA and have dimensions of 14 × 2.5 cm. The columns were packed with 600–800 µm diameter glass beads to represent an aquifer consisting of medium sand. The columns were equipped with two sampling points: one for the inlet and one for the outlet.
The experimental setup involved the continuous feeding of the solution taken from a magnetically stirred beaker via a Gilson miniplus evolution peristaltic pump and then injected into the column in an up-flow configuration to avoid the formation of air pockets. The solution exiting the top of the column discharged in a Gilson Fraction Control 201-202 that was used to monitor the effluent leaving the column. The concentration of BC and BC-based composites in the effluent was monitored by measuring the turbidity of the suspension with a Shimadzu UV-1800 photometer at 650 nm. A calibration curve was performed for each test with a different batch of BC composite to ensure accurate quantification. The tests were carried out in two phases marked by a change of feed: (i) distribution of the BC suspension within the column and (ii) washing with water to verify the amount of BC retained by the column and thus, close the mass balance.
The amount of BC retained in the column was determined by the mass balance in equation 4.
where Qi is the flow rate at i-th time (although designed as constant flow tests some fluctuated significantly), CIN is the concentration of BC entering the column, CiOUT is the concentration of BC leaving the column at i-th time, and m is the BC mass retained by the column. Prior to the BC distribution tests, a tracer test with 150 mg·L−1 of Cl− (supplied as NaCl) feeding solution was carried out to determine pore volume (PV), effective porosity (ε), and hydraulic retention time (θ). The effluent samples obtained by the aforementioned fraction collector were subsequently analyzed with a Dionex ICS-1000 IC ion chromatograph in the case of tracer tests and with a Shimadzu UV-1800 photometer at 650 nm in the case of BC distribution tests. Both tracer and distribution tests were carried out with flow rates of approximately 0.6 mL·min−1 and apparent velocity of 0.3 cm·min−1.
m = Σin(QiCIN − QiCiOUT)
2.6. Characterization Techniques and Analytical Methods
Absorption spectra were recorded using a UV–Visible spectrophotometer Varian Cary100 instrument in a 200–800 nm wavelength region. Quartz cuvettes having a path length of 1 cm were used in all experiments.
Turbidity measurements on column effluent for BC composites distribution tests were carried out with a UV-Vis-NIR Shimadzu UV1800 photometer at 650 nm, with 1 cm cuvettes in polystyrene.
pH measurements were carried out using pH600 Eutech Instruments pHmeter calibrated with standard solutions (pH 4–10) before measurement.
Fourier transform infrared (FT-IR) spectroscopy in Attenuated Total Reflectance (ATR) mode was used to analyze the composition of the materials and assess potential interaction between counterparts. Measurements were done using a Bruker Vertex70 instrument over the wavenumber range of 4000–600 cm−1 with a resolution of 4 cm−1 and 32 scans. Samples were deposited as a solid directly onto the diamond-coated ATR crystal.
The specific surface area (Brunauer–Emmett–Teller, BET method in the 0-0.1 p/p° interval) [46], total pore volume, micro-pore volume, and pore size distribution were determined by adsorption/desorption of N2 at −196 °C (77 K) using a 3Flex 3500 MIcromeritics analyzer. 0.250 g of powder samples were pretreated at 250 °C for 24 h in an oven (ambient pressure) to remove excess of absorbed water (calculated as weight loss %), at 300 °C for 2 h, and at 350 °C for 1 h, under vacuum via thermally-controlled heating mantles, up to a residual pressure lower than 0.5 Pa. Pore size distribution was determined by the Barrett–Joyner–Halenda (BJH) method [47] from the adsorption isotherm. Total pore volume was obtained by the rule of Gurvitsch [48]. Micro-pore volume was obtained by the t-plot. The uncertainty was ± 5 m2·g−1 for the specific surface area values, ±0.005 cm3·g−1 for the total pore volume values.
The size as hydrodynamic diameter (<2RH>), size distribution (PDI) and ζ-potential were evaluated by dynamic light scattering (DLS) using a Malvern Nano-ZetaSizer apparatus, operating in backscattering mode (173°) and equipped with a 5 mW HeNe laser (λ = 632.8 nm). Both size and ζ-potential were measured at 25 °C using a minimum of ten replicates.
Surface morphology of the materials was investigated by Field-Emission Scanning Electron Microscopy (FESEM) on an Auriga Zeiss instrument. Samples were drop-casted onto a silicon stub from their aqueous suspension without any conductive coating and air-dried. The acceleration voltage was set at 1.5 kV. FESEM images were analyzed using ImageJ software.
Chloride analysis for the tracer tests was performed with a Dionex ICS-1000 IC ion chromatograph equipped with an electrical conductivity detector and Dionex AS-40 autosampler. The instrument is equipped with a Dionex IonPac AG14 pre-column (4 × 50 mm), a Dionex IonPac AS14 IC column and a Thermo Fisher Scientific AESR 500 4 mm suppressor. The eluent phase was prepared with 3.5 mM Na2CO3 and 1.0 mM NaHCO3 with 1.2 mL·min−1 as flow rate. The chloride calibration curve was realized from 5 to 160 mg·L−1 of Cl− (supplied as NaCl).
TCE concentration for isothermal curve determination was carried out with a DANI MASTER GC (DANI Instruments, Contone, Switzerland) gas chromatograph, equipped with DANI 86.50 headspace auto-sampler, TRB624 capillary column (30 m × 0.53 mm ID × 3 µm,) and a Flame Ionization Detector (FID) was used. The DANI 86.50 was set-up as follows: oven temperature 80 °C, manifold temperature 120 °C, transfer line temperature 180 °C, shaking softly for 1 min. The GC conditions were: N2 carrier gas (flow 10 mL·min−1), 180 °C injector temperature split injection 1:2; 200 °C detector temperature with air, N2 and H2 for the FID (flows 240, 25, 60 mL min−1). The oven temperature was programmed as follows: 60 °C for 3 min, 30 min to 120 °C, then 6 min at 120 °C. For the quantitative determination of TCE, a calibration curve was obtained by dilution of a TCE/ethanol stock solution in standards with concentration range 0.1–25 mg·L−1.
3. Results and Discussion
Herein, pine wood biochars obtained via gasification at 950 °C were used as starting material to obtain composites with different biopolymers. The aim is to synthesize a cheap, colloidally stable and environmentally friendly material with application as adsorbent towards chlorinated hydrocarbons. In this regard, biochar produced at high temperature is reported to be more effective than that those produced at lower temperatures (200–600 °C), mainly due to its improved capacity to act as organic compound sequester, its higher surface area, and nanoporosity [49]. In the following, extensive characterization of both raw biochar and composite materials is presented, together with preliminary transport and adsorption tests on optimized formulations using TCE as a model contaminant.
3.1. Raw Pine Wood Biochar Characterization
Prior to application in composites, pine wood biochars (BCs) were characterized as raw materials. Four different BC samples (replicates of two different batches) obtained at 950 °C gasification temperature were studied, based on different sieving and grinding processes: A, B (sieving at 0.5 mm), and AP, BP (sieving 64 µm and manual grinding), see Table 1.
3.1.1. Fourier Transform Infrared Spectroscopy (FTIR) and Morphological Analysis
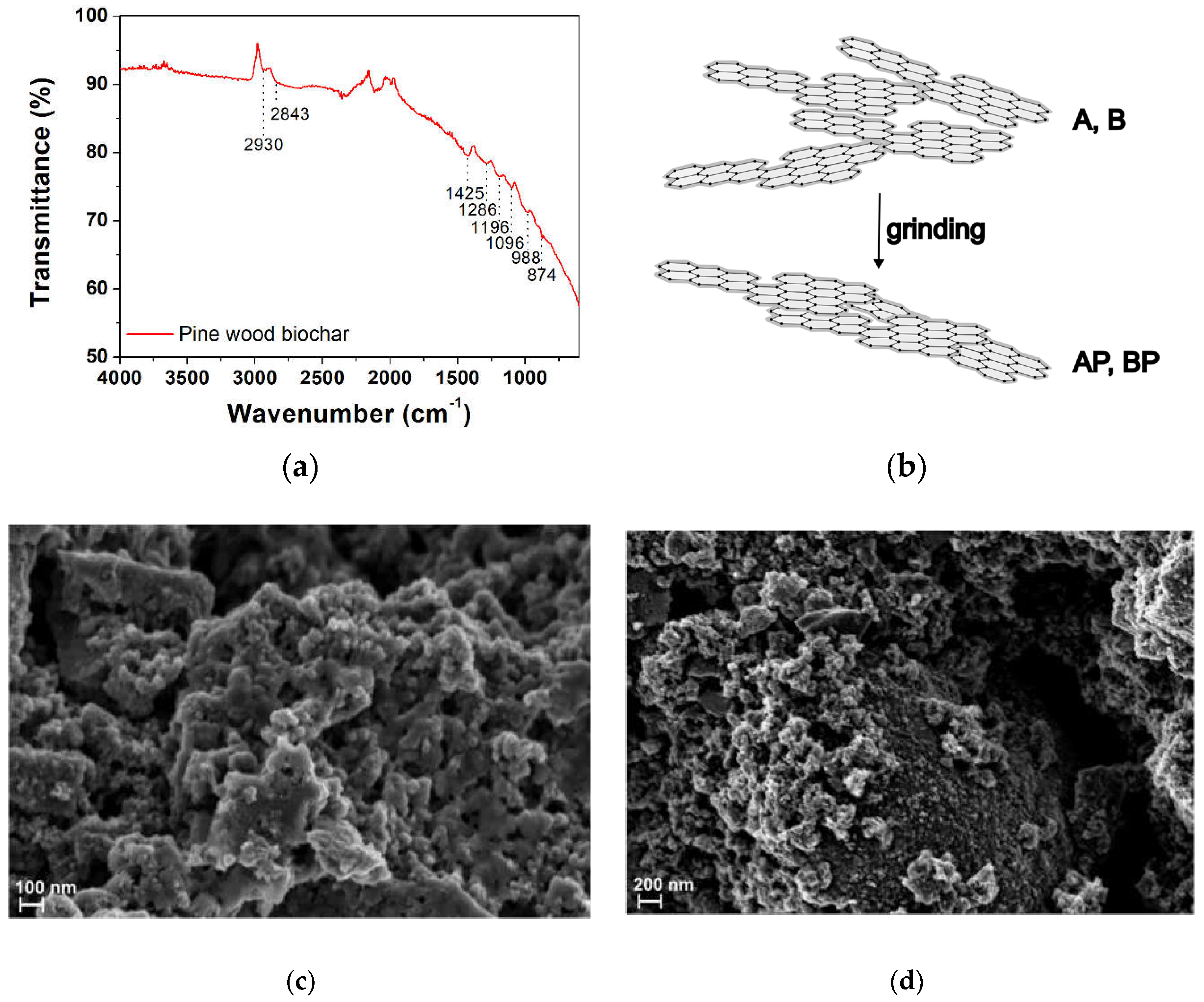
Infrared spectroscopy was used for functional groups characterization. Since gasification temperature (950 °C for all samples) determines the content of surface functionalities, a representative FTIR spectrum in ATR mode of AP sample is presented (Figure 1a). The bands between 2950 cm−1 and 2850 cm−1 were associated with the asymmetric and symmetric C–H stretching vibrations (νas, νs) of aliphatic carbons νas(–CH2) = 2930 cm−1 and νs(–CH2) = 2843 cm−1 which was also confirmed by the –CH2 scissoring vibration (partially overlapped with C–C stretching vibrations of aromatic ring) at 1425 cm−1 and 2890 cm−1 –CH– stretching. Bands at 1286 cm−1, 1096 cm−1 and 1196 cm−1 correspond to C–O and C–O–C stretching, attributable to the presence cellulose, hemicellulose, and lignin residual fractions in the biochar structure [50]. Weak bands between 988 and 874 cm−1 can be attributed to aromatic C–H bending vibrations, suggesting the presence of aromatic hydrogens in the biochar structure, as expected for lignin-based BC. No presence of typical hydroxyl group stretching vibration (ca. 3200 cm−1), as well as carboxyl C=O stretching (ca. 1730 cm−1) was detected due to dehydration reactions, although a small quantity of oxygen-containing organic groups was still retained even during pyrolysis or gasification at elevated temperature [51,52,53]. The limited presence of functional groups and aromatic portion in the spectrum implied that BCs are essentially graphitic in nature (Figure 1b). Indeed, at the used gasification temperature, the aromatic structures condense, forming highly disordered graphite-like domains with less residual functional groups. The absence of specific absorption arose from the UV-Vis spectra of all biochars (Figure S1). Due to their structural features, these materials are commonly classified as hard carbon due to their high mechanical hardness [38].
Morphologically, the raw AP biochar was examined via scanning electron microscopy. Images presented in Figure 1c,d showed a sponge-like topography of the surface with a grain size of (37 ± 8) nm, see also Figure S2. Pores were quasi-spherical with an average pore size (taken from a total of ten SEM images) from (19 ± 6) nm to (123 ± 59) nm, depending on pore and measurement orientation. The fluffier and inhomogeneous structure of the biochar agree with a high pyrolysis/gasification temperature (above 650 °C), which reduce the high percentage of cellulose, hemicellulose, and lignin of the biomass [54].
Figure 1.
(a) FTIR-ATR spectrum of raw pine wood biochar deposited as a solid powder. Spectrum of AP sample is reported as a representative sample; (b) graphitic structure of BC according to its structural features; (c,d) representative field-emission scanning electron microscopy (FE-SEM) images of raw pine wood biochar (AP sample) at different magnification. Sample was drop-casted onto a silicon stub from its aqueous suspension. Accelerating voltage was 1.50 kV.
Figure 1.
(a) FTIR-ATR spectrum of raw pine wood biochar deposited as a solid powder. Spectrum of AP sample is reported as a representative sample; (b) graphitic structure of BC according to its structural features; (c,d) representative field-emission scanning electron microscopy (FE-SEM) images of raw pine wood biochar (AP sample) at different magnification. Sample was drop-casted onto a silicon stub from its aqueous suspension. Accelerating voltage was 1.50 kV.
3.1.2. Brunauer–Emmett–Teller (BET) Surface Area and TEXTURAL PARAmeters
The specific surface area and textural parameters (total pore volume and micro-pore volume) influence adsorption performances of biochar and play a role considering BC as a filler in polymer matrices. High surface areas and pore volumes are reported to improve mechanical properties of BC@polymer materials, due to infiltration of polymer matrix during the mixing process [55]. Prior to analysis, samples were treated at 250 °C for 24 h in a conventional over to remove excess of absorbed water, and results calculated as weight loss % (compared with untreated sample) are reported in Table 3.
The N2 adsorption/desorption isotherms of different biochar samples (A, AP, B, BP) subjected to different sieving and manual grinding procedures are reported in Figure 2. The curves are consistent with a type IV isotherm and type H4 hysteresis loop [56]. Results of N2 adsorption/desorption measurements are given in Table 3. All raw BC samples resulted to be micro/mesoporous materials with a continuous pore size distribution in the 0-40 nm range (with maximum of the distribution at pore size < 1.5 nm). It is worthy to note that a reduction in the specific surface area (m2·g−1) occurs in samples after manual grinding (AP and BP samples). The grinding process (e.g., impact or shear) has been reported to alter the pristine molecular structure of graphite-based samples, and due to weak intralayer bonds, additional structural distortions (besides pre-existing dislocations and stacking faults), such as rotation, translation, curvature, and fluctuation of interlayer spacing of graphene layers can be easily induced [57]. Particularly, an increase in the specific surface area is accompanied by an increase in the interplanar distance of graphite layers. However, a suppression of surface area (i.e., reduction of interplanar distances) can be induced through mild manual grinding (with mortar and pestle), in which shock (fracturing) forces predominates over shear (abrasive) forces, forming aggregates [58,59].
Table 3.
Weight loss, BET specific surface area, total pore volume, and micro-pore volume.
| BC sample * | Weight loss (%) | BET Specific surface area (m2·g−1) | Total pore Volume (cm3·g−1) | Micro-pore volume (cm3·g−1) |
|---|---|---|---|---|
| A | 6.59% | 457 ± 5 | 0.511 | 0.098 |
| AP | 3.31% | 293 ± 5 | 0.421 | 0.046 |
| B | 12.97% | 523 ± 5 | 0.578 | 0.140 |
| BP | 3.37% | 378 ± 5 | 0.474 | 0.087 |
* P denotes additional manual grinding on sample.
3.1.3. Hydrodynamic Particle Size Distribution
The mean particle size, evaluated as hydrodynamic diameter (<2RH>), of the raw biochars were evaluated via dynamic light scattering technique, in water. Results of freshly prepared biochar suspension are presented in Figure 3. Biochar A (Figure 3a, red line) showed a population (90% of the particles) centered at (425 ± 98) nm, whereas a less intense population appears at (138 ± 34) nm. Manual grinding (sample AP, Figure 3a, blue line) slightly improves the intensity percentage of the smaller population centered at (247 ± 7) nm, although aggregates were found above 1000 nm. Biochar B resulted in a broad distribution centered at (455 ± 174) nm (Figure 3b, red line), whereas in the BP sample two population can be distinguished, the first appearing at (271 ± 46) nm and a second one at ca. 1000 nm (Figure 3b, blue line). The presence of a large population in the AP and BP samples further confirms the formation of larger aggregates after the manual grinding process (fracturing-type process) with a decrease in the interplanar distance of graphite layers constituting the biochar samples, as per specific surface area results (paragraph 3.1.2). Despite their hydrophobic nature, BCs showed a ζ-potential in the -19 to -30 mV range, at their native pH of 9 (determined in a 2:1 w/v of solid biochar to water). Results were as follows: A = (-31.9 ± 0.9) mV, AP = (-24.7 ± 0.8) mV, B = (-25.3 ± 0.9) mV, BP = (-19.1 ± 0.9) mV. After two hours of static aging, all BCs water suspensions showed an increase in the hydrodynamic diameter with a single population above 1500 nm (Figure S3) and a decrease in the ζ-potential value (A = (-13.9 ± 0.7) mV, AP = (-15.9 ± 0.4) mV, B = (-17.2 ± 02) mV, BP = (-5 ± 1) mV, Figure S3), highlighting the poor stability in aqueous environment of high-temperature biochar samples.
Based on the above characterization results, it can be stated that gasification at 950 °C produce biochars with reproducible features, for sample A and B. The manual grinding process increase the intensity distribution of the smaller population (although showing an higher amount of aggregates compared with untreated samples), with AP sample showing the higher percentage of colloidal fraction, i.e., it showed the most intense population below 200 nm, respect to other BC samples. Thus, the AP biochar sample was selected as a reference material for further studies, as reported in the following.
3.2. Adsorption Isotherm Curve of Trichloroethylene (TCE) of Raw Biochar
In Figure 4, the isotherm of TCE adsorption obtained from AP biochar sample is reported. As can be seen, experimental data follows a classic adsorption pattern of a solute on a solid adsorbent. In this case, one is working at relatively low ratios of mass of contaminant to the mass of adsorbent, thus staying well below the saturation threshold of the raw BC, which is confirmed by the curve pattern well described by a Freundlich isotherm rather than a Langmuir-type one. Freundlich’s characteristic parameters obtained from the fit were K = 9.2 ± 0.3 and n = 0.61 ± 0.02, indicating very good adsorption capacities for TCE compared to literature values [43]. This preliminary evaluation validates the use of AP biochar (sieving at 64 µm and grinding) as a inorganic filler for subsequent polymeric composites and related column distribution tests.
3.3. BC@biopolymer Composites Characterization
Biopolymer-based biochar composite materials (obtained with the AP biochar sample) were prepared via a simple and straightforward blending approach at different temperature, according to the chemical nature of polymers (see paragraph 2.2), using biochar derived from pine wood (950 °C gasification temperature). Indeed, raw biochar suffers from a lack of stability in water and to overcome these limitations, emphasis was put on the modification of biochar with different polymers in order to enhance its surface functionality (with positive impact on water stability) and pore structures. The polymers used in this study are reported in Figure 5.
3.3.1. Sedimentation Tests
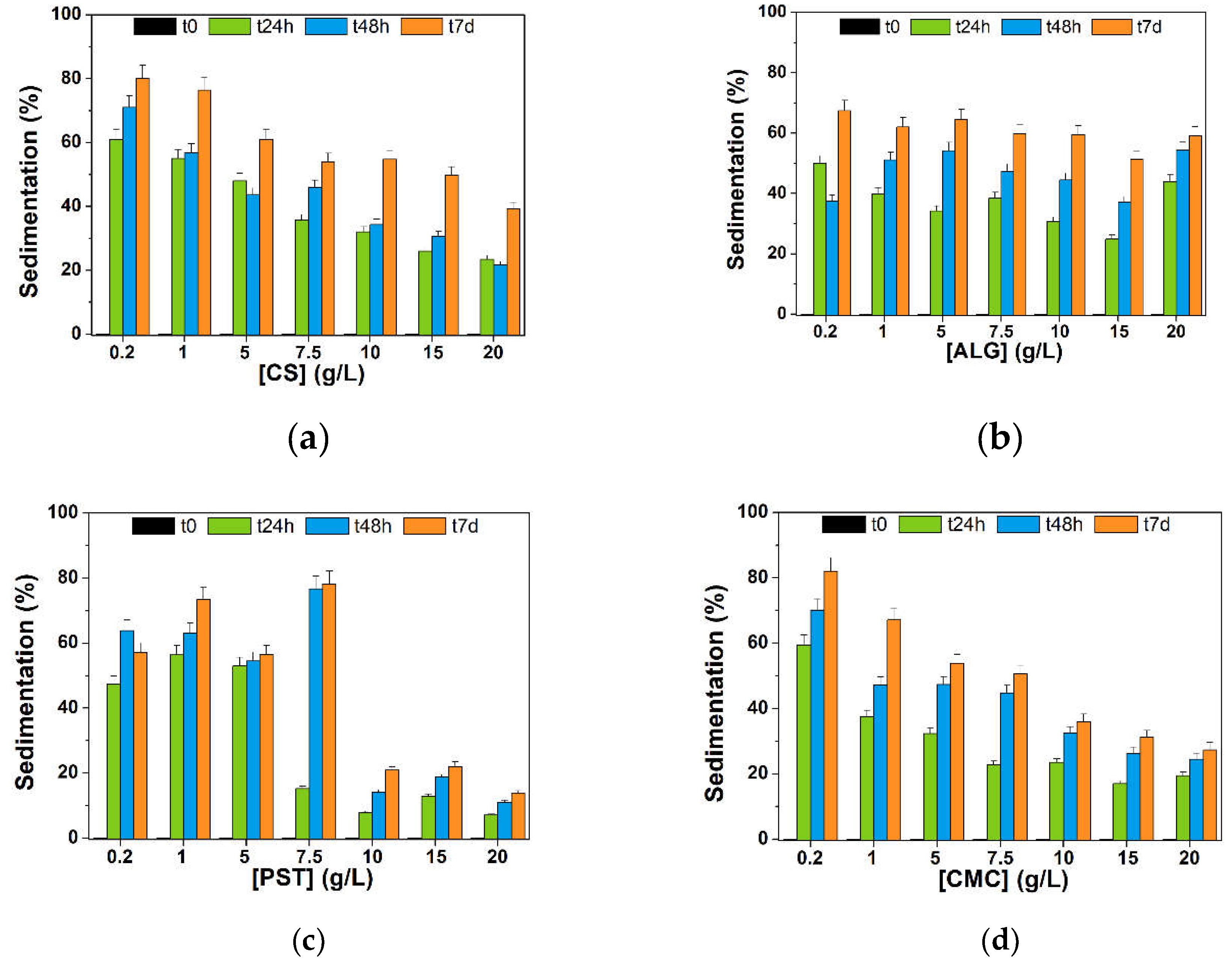
To evaluate the stability in water over time compared with pristine biochars (stability < 2 h in water, see paragraph 3.1.3), preliminary sedimentation tests were conducted on different BC@polymer formulations fixing the concentration of biochar at 0.3 g/L and varying the polymer concentration in the 0.2-20 g/L range (details can be found in paragraph 2.2). The sedimentation was calculated monitoring the absorbance value at 650 nm for each formulation (after 1:3 v/v with H2O) at different time points (0, 24 h, 7 days) and their absorption intensity compared. As can be seen in Figure 6, a polymer concentration below 10 g/L resulted in an unstable dispersion (≥50% sedimentation) over a week in all formulations herein studied.
In the case of chitosan (Figure 6a) and sodium alginate (Figure 6b), formulations at higher polymer concentration (10.0, 15.0, 20.0 g/L) sedimented rapidly after 24 hours, reaching (27 ± 5)% (on average) and (32 ± 10)% (on average), respectively. In this regard, sedimentation in BC@ALG composites settled at around 60% after 7 days, demonstrating the feeble steadiness of BC@ALG composites. Compared with sodium alginate, BC@CS composites showed an improved stability within a week (39 ± 2 %) although chitosan showed the best solubility in acidic environments via primary amine protonation [60]. Indeed, the less stable formulation, i.e., BC@CS 0.2 g/, showed a pH value of 6.63, whereas a higher CS concentration in the composite resulted in an acidic pH in the 4-5 range (see Table S1 for complete pH values). The poor solubility of CS matrix at neutral and basic pHs strongly limits its applicability in environmental applications.
Potato starch-based composites (Figure 6c) showed a higher stability in the 10-20 g/L concentration range, in the presence of APG2 surfactant as additive in the mixture. Some important points should be highlighted when considering BC@PST composites. First, potato starch is a densely packed biopolymer which consists of a mixture of two polysaccharides: amylose (20 to 35%) and amylopectin. Heating starch (below 100 °C) in excess water cause swelling of PST granules and loss of crystallinity in a semi-cooperative process. The subsequent cooling results in a transition from a liquid system with dispersed granules to an elastic gel (referred to as sol-gel transition) and for longer storage periods at room temperature a reordering of amylopectin chains also occurs with an increase in the elastic modulus of starch [61]. As a first attempt, the viscous gel-like structure formed after retrogradation can be responsible for a physical encapsulation of BC granules. Indeed, potato starch-based systems have been used over the years to physically entrap hydrophobic molecules (e.g., food ingredients, aroma compounds etc.) [62]. However, the major drawback of PST-based composites is the high intrinsic viscosity, ca. 103 times higher than the viscosity of water (expressed in cP) [63]. Thus, a slight improvement in the viscosity and stability of BC@PST composites was achieved via addition of APG2 biodegradable surfactant (compared with composites without APG2 addition, data not shown). APG2 allowed to improve wetting properties and water tolerance of the composite in a wide range of pHs [64]. The appropriate amount of surfactant was visually chosen upon stability tests within 7 days in the presence of 0.3 g/L of raw biochar, and results presented in Figure S4. Evaluating ten different APG2 concentrations (0.1, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 2.0, 5.0 %v/v) above its critical micelle concentration (0.0071 wt% for APG2 [42]), the highest stability was shown by 1% v/v dispersion, thus being selected for BC@PST composites preparation. The properties of PST and APG2 combination allowed to obtained stable composites, in which sedimentation is far below 30% (14 ± 1%) within 7 days of storage.
Considering the use of sodium carboxymethylcellulose as a polymer matrix for BC dispersion, results in Figure 6d showed that sedimentation percentage settled around 30% after a week: (36 ± 2)%, (31 ± 2)% and (27 ± 2)% for 10.0, 15.0, and 20.0 g/L, respectively. Being an anionic polysaccharide, CMC show coagulation characteristic and tends to form micellar-like structure absorbing a considerable amount of water [65], thus being suitable as a support matrix for biochar. Moreover, CMC shows some advantages compared with other polymers, (i) it does not require heating for complete dissolution, (ii) it is soluble in water in a wide range of pHs, and (iii) led to a slightly basic suspension (ca. pH 9, see Table S1 for complete pH values) with long-term stability. The most stable and versatile composites resulted to be BC@PST + APG2 1.0% v/v (Figure 6c) and BC@CMC (Figure 6d), although in the latter case ca. 50% of dispersed biochar sedimented after 7 days.
Figure 6.
Dispersion stability over time evaluated as sedimentation percentage for 0.3 g/L of BC blended with: (a) chitosan (CS); (b) sodium alginate (ALG); (c) potato starch (PST) + APG2 1.0% v/v; (d) sodium carboxymethylcellulose (CMC) at different concentrations.
Figure 6.
Dispersion stability over time evaluated as sedimentation percentage for 0.3 g/L of BC blended with: (a) chitosan (CS); (b) sodium alginate (ALG); (c) potato starch (PST) + APG2 1.0% v/v; (d) sodium carboxymethylcellulose (CMC) at different concentrations.
3.3.2. Hydrodynamic Parameters of BC@PST and BC@CMC Composites
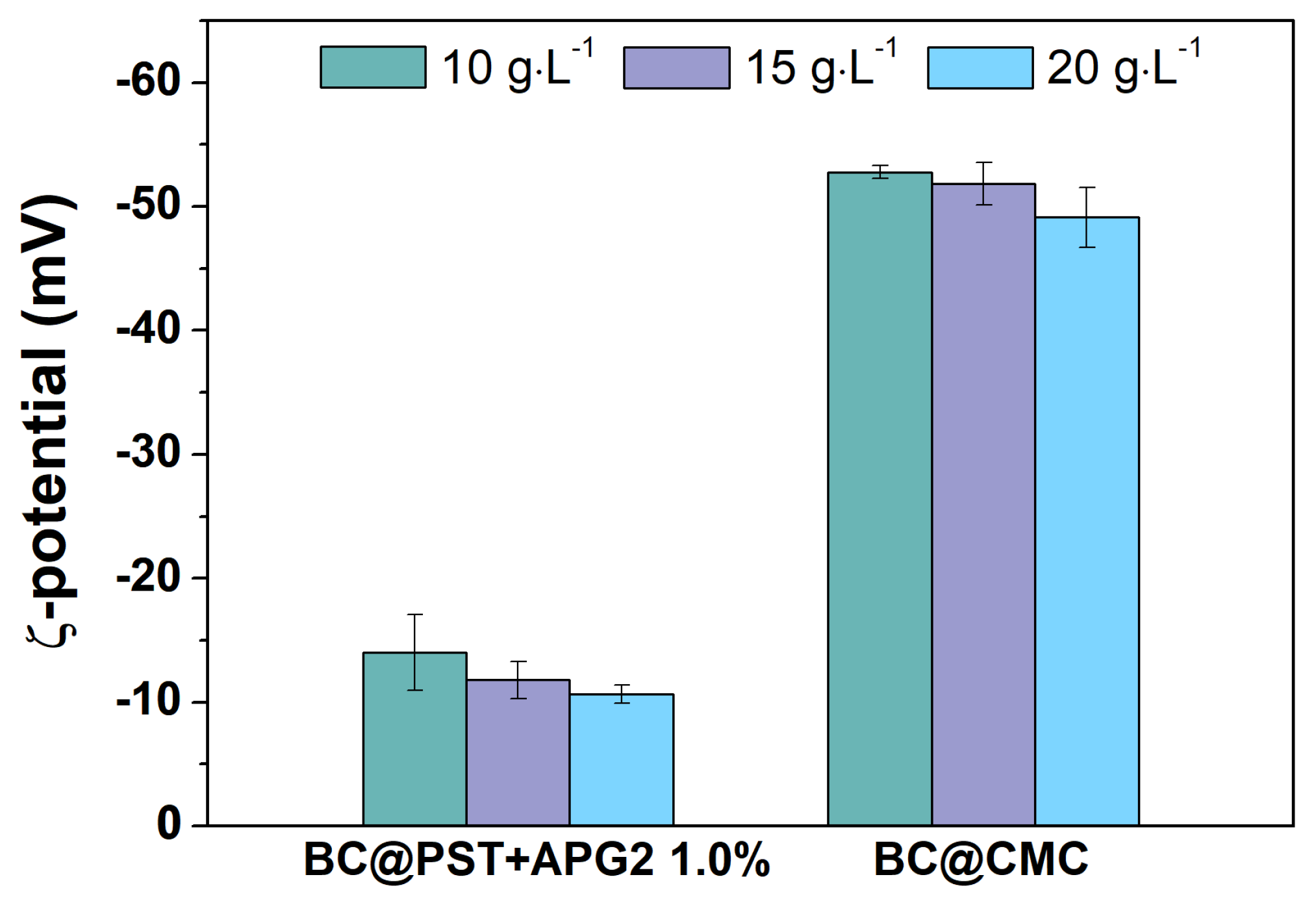
Hydrodynamic diameter (<2RH>) and ζ-potential were recorded at different time points to further evaluate the stability and hydrodynamic behavior of BC@polymer formulations. According to sedimentation tests, the ζ-potential values of BC@PST + APG2 1.0% and BC@CS selected composites after 7 days aging are reported in Figure 7 and discussed in the following, whereas full data (0 h, 24 h, 7 days for all composites) can be found in Table S1.
In the case of potato starch-based composites, the ζ-potential value settled around (-14 ± 3) mV, (-12 ± 2) mV and (-10.7 ± 0.8) mV, for 10.0, 15.0, and 20.0 g/L, respectively, with a decrease of about 15% over a week. Values for BC@CMC fluctuates around -55 mV at all time points. Although BC@PST + APG2 formulations showed the less sedimentation percentage compared with BC@CMC, the latter showed the highest colloidal stability over time. It is worthy to note that the ζ-potential value in polymers depends on surface properties and size of granules, thus being a strong pH-dependent parameter [66]. Specifically, starch, both in its native and pasted form, is a very complex dispersion that cannot be clearly classified as a solution, colloid or suspension, and its hydrodynamic properties are affected by many factors, e.g., pH and presence of dispersed phases [66].
Figure 7.
ζ-potential values for BC@PST + APG2 1.0% v/v and BC@CMC after 7 days at 10.0 g/L (green bar), 15.0 g/L (violet bar), and 20.0 g/L (light blue bar). Complete values can be found in Table S1.
Figure 7.
ζ-potential values for BC@PST + APG2 1.0% v/v and BC@CMC after 7 days at 10.0 g/L (green bar), 15.0 g/L (violet bar), and 20.0 g/L (light blue bar). Complete values can be found in Table S1.
3.3.3. FT-IR Characterization of BC@PST and BC@CMC Composites
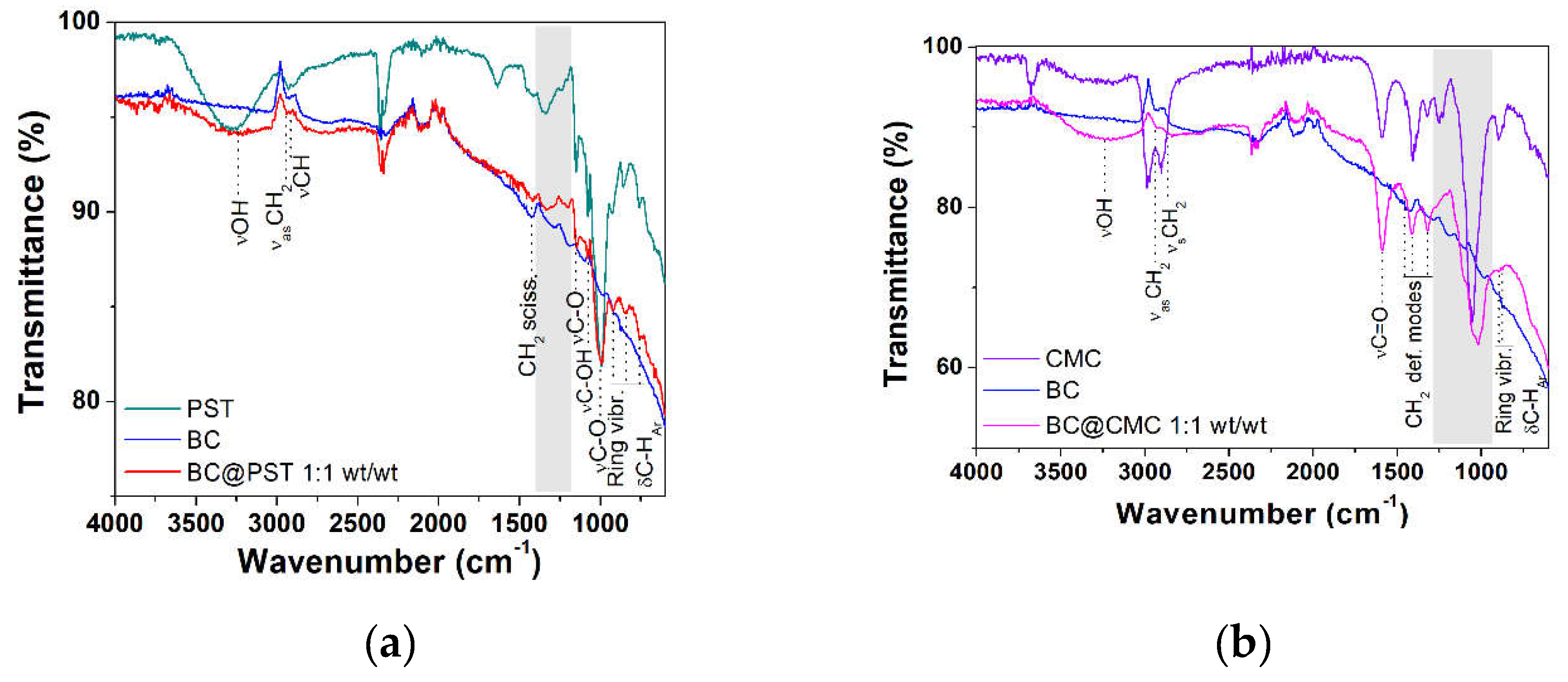
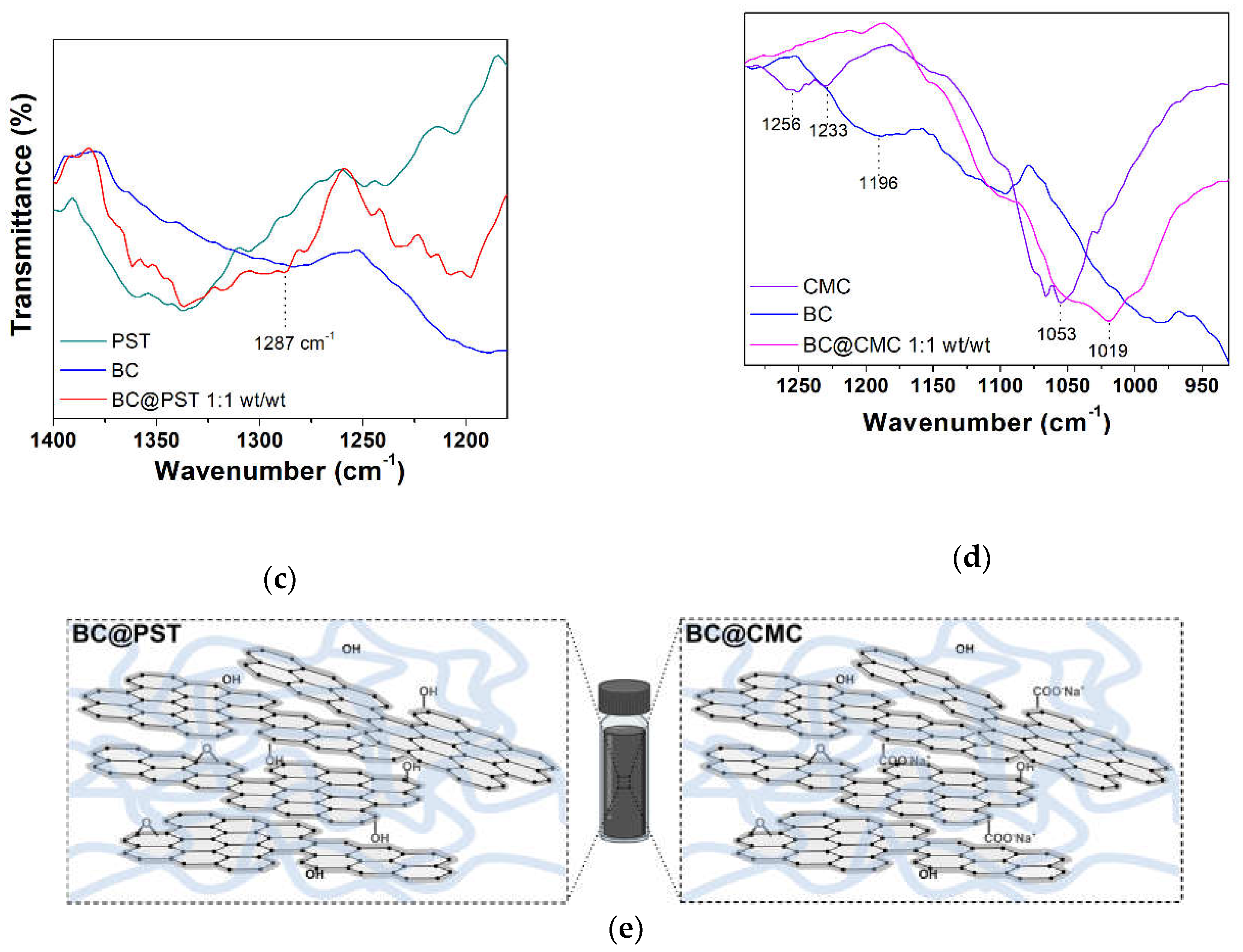
To further examine the structural properties of PST- and CMC-based composites, FT-IR spectra in ATR mode were recorded on dried composites. The FT-IR spectrum of pristine potato starch and CMC taken as reference can be found in Figures S5 and S6, respectively, with complete band assignment. To reduce sampling bias and improve the poor signal-to-noise ratio in reflectance FTIR measurements, an ad-hoc sample was prepared for each composite at 1:1 wt/wt ratio. BC@PST and BC@CMC composites full FT-IR spectra (4000-600 cm−1) are presented in Figure 8. In both spectra (red and magenta lines in Figure 8a,b), a broad stretching (ν) vibration band is observed in the 3030-3650 cm−1 region centered at 3238 cm−1 for BC@PST and at 3255 cm−1 for BC@CMC, which indicates the presence hydroxyl (−OH) functional groups involved in hydrogen bonds. Absorption bands at ca. 2930 and 2840 cm−1 labeled as νas(CH2) and ν(CH) are due to methylene −CH2 and −CH stretching vibrations, respectively, arising from both BC and polymers aliphatic chains (see also paragraph 3.1.1), together with the −CH2 scissoring deformation modes in the 1420-1310 cm−1 wavenumber region coupled with alcoholic O−H in-plane bending [67]. In the case of CMC polymer matrix, the strong band at 1590 cm−1 pertain to hydrated carboxylic C=O stretching in the anion form of CMC functional group. In the latter, bands at 898 and 874 cm−1 are assigned to C−O−C vibrations of cellulose ring coupled with aromatic C−H bending of biochar) [69]. In Bc@PST stretching of C−O−C and C−OH groups can be found at 1150, 1078, and 995 cm−1. In addition, the bands appeared at 926 (band type I, asymmetric deformation of the ring), 843 (band type II, deformation vibration of the −CH bond) and 753 cm−1 (band type III, characteristic for α-glycosidic bond) correspond to the vibration of carbohydrate ring [68,69], partially overlapped with stretching vibration of aromatic C−H of BC. A noticeable difference among pristine polymers, raw BC, and composite spectra can be seen in the fingerprint region (highlighted in grey in Figure 8a,b) and reported in Figure 8c,d. To establish a reasonable interaction occurring between biochar and polymer matrices, a calculated FT-IR spectrum of BC@PST and BC@CMC composite (obtained via 1:1 weighted sum of raw BC and pristine polymer spectra) were compared with the experimental one in Figure S7.
In the BC@PST spectrum (1400-1250 cm−1, Figure 8c), a broadening of the band centered at 1287 cm−1 which involve C−O stretching of ethers vibrations occurs, with no significant shift compared with the calculated one. However, in the experimental spectrum band due to ν(−OH) at 3238 cm−1 is much less intense compared with a calculated physical mix of BC and PST (Figure S7), whereas that calculated profile linearly follows the experimental one in the 1400-1180 cm−1 region, making possible to hypothesize that the above-mentioned broadening at 1287 cm−1 was due to BC and PST absorption combination. Then, the wavenumber of hydroxyl stretching vibration shifted from 3290 cm−1 (pristine PST) to a lower wavenumber 3238 cm−1 in the composite, while the characteristic band is absent in raw BC (see Figure 1 for a better comparison). Blueshift in ν(−OH) vibration is due to formation of intermolecular hydrogen bonds [70]. Furthermore, the heat treatment (applied in the preparation of the composite) is known to reduce the intramolecular hydrogen bonds among starch molecules [71]. Heating starch at temperature ≥ 70 °C, cause hydrogen bonds break, with water molecules that combine with −OH groups of starch and tend to swell, beginning an irreversible thermal transition called gelatinization and condensation (due to amylose fractions) which reflects in a sol formation [71,72]. Upon cooling, the amylose molecules re-associate to form a network during retrogradation [73]. Thus, based on (1) decrease in the ν(−OH) band intensity, (2) blueshift in the wavenumber of ν(−OH) in the FT-IR spectrum, and (3) PST gel-like network formation upon retrogradation, it is possible to ascribe the BC stabilization in the BC@PST composite to a combination of both hydrogen bonds and physical entrapment. Considering the sedimentation tests (paragraph 3.2.1) and ζ-potential results (paragraph 3.2.2), physical entrapment plays a major role in the stabilization, since a higher concentration of PST reduce BC sedimentation over time.
Conversely, in the BC@CMC composite spectrum (1290-930 cm−1 range, Figure 8d) the bands centered at 1256 and 1233 cm−1 related to C−O stretching of carboxylic group [69], typical of pristine CMC, are absent as well as the band at 1196 cm−1 of raw BC. Moreover, a shift of 34 cm−1 occurs in the C−OH stretching vibration (from 1053 cm−1 in CMC to 1019 cm−1 in BC@CMC). The observed shift refers to the changes occurred in the molecular structure of CMC [74]. The consistent broadening of this band may result in an interaction-driven combination of both C−O stretching vibrations of CMC and BC. Compared with the mere sum of absorptions, the experimental spectrum maintains its peculiar features: (a) the bandwidth of ν(−OH) centered at 3255 cm−1, and (b) the 34 cm−1 blueshift of ν(C−O) at 1019 cm−1, making possible to hypothesize a strong hydrogen bond interaction involving C−O−C groups as H-acceptors and aliphatic C−O/C−OH as both H-donors/acceptors, thus stabilizing the composite structure to some extent. These results indicated no covalent bonds formation between polymers and BC but rather a combination of hydrogen bonds, hydrophobic forces, and physical entrapment as roughly represented in Figure 8e.
Figure 8.
FT-IR ATR spectra: (a) full spectrum of potato starch (PST, dark cyan line), raw biochar (BC, blue line), and potato starch-based composite in a 1:1 wt/wt ratio (BC@PST, red line); (b) full spectrum of sodium carboxymethylcellulose (CMC, violet line), raw biochar (BC, blue line), and CMC-based composite in a 1:1 wt/wt ratio (BC@CMC, magenta line); (c) 1400-1250 cm−1 wavenumber region taken from (a); (d) 1290-930 cm−1 wavenumber region taken from (b); (e) schematic representation of BC@polymer composite structure.
Figure 8.
FT-IR ATR spectra: (a) full spectrum of potato starch (PST, dark cyan line), raw biochar (BC, blue line), and potato starch-based composite in a 1:1 wt/wt ratio (BC@PST, red line); (b) full spectrum of sodium carboxymethylcellulose (CMC, violet line), raw biochar (BC, blue line), and CMC-based composite in a 1:1 wt/wt ratio (BC@CMC, magenta line); (c) 1400-1250 cm−1 wavenumber region taken from (a); (d) 1290-930 cm−1 wavenumber region taken from (b); (e) schematic representation of BC@polymer composite structure.
3.4. Continuous Flow Column Distribution Tests on BC@polymer Composites
Column distribution tests were carried out on the most promising BC composites in terms of stability over time to assess their fitness as an injectable adsorption material. Therefore, the tests aimed to verify the actual possibility of distributing the suspension in a porous medium and to evaluate possible modifications to the composition of the suspension to optimize its fluid-dynamic behavior. The main qualities of BC composites evaluated were (i) their ability to be transported throughout the whole column without generating clogging (ii) while also being retained by the porous media during subsequent water flushing tests. In the following, continuous flow column distribution tests on BC@PST, BC@CMC with CMC at 20 g/L, and BC@CMC with CMC 10 g/L are presented.
3.4.1. Continuous Flow Column Distribution Tests on BC@PST Composite
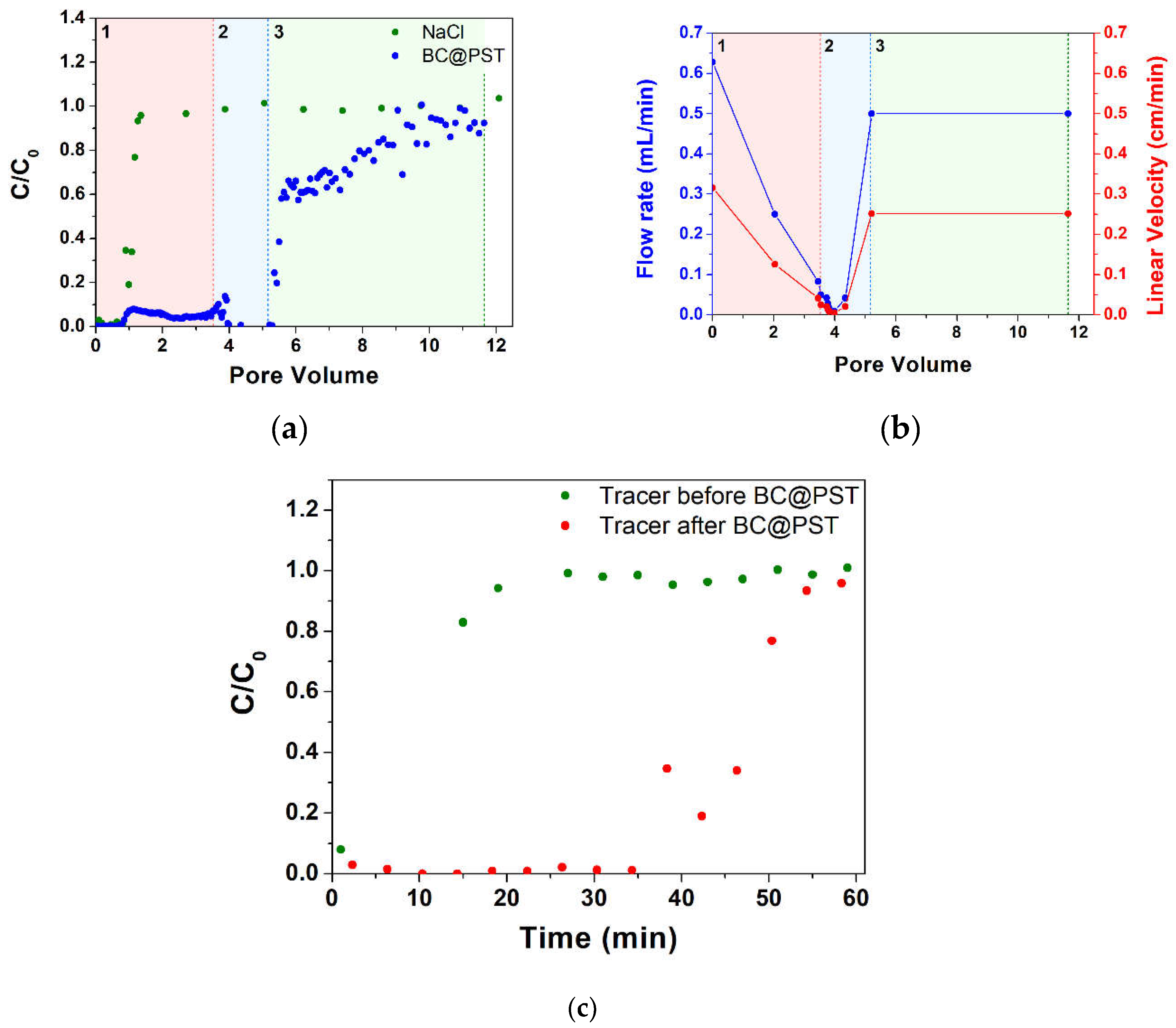
The first formulation that was deemed suitable for column distribution tests was BC@PST. On the column, packed as per paragraph 2.5, a tracer test was performed (Figure 9a) showing a pore volume (PV) of 27.8 mL corresponding to an effective porosity (ε) of 0.41 with an initial hydraulic retention time (θ) of 44.3 min. Flow rate and linear velocity values in the 0-12 pore volume range are reported in Figure 9b. Observing the trend of the breakthrough curves, the test can be divided into three sections: (1) in PV 0-3.46, (2) in PV 3.46-5.21 and (3) in PV 5.21-11.64 range. Section (1) highlights, in accordance with the tracer test, that the C/C0 of BC at the output is 0 up to 0.78 PV, after which there is an increase in the latter up to PV 1.13, where it stands at a value of 0.08 with a very stable trend. At this point, again, with a very stable trend, there is a halving of C/C0 to 0.04-0.05 up to PV 3.46. At the same time (between PV 0 and 3.46), a progressive decrease in the flow rate from 0.63 to 0.083 mL/min is registered. In the first part of section (2), between PV 3.46 and PV 3.99, there is a destabilization of the C/C0 value, varying between 0.01 and 0.14, which has been very stable up to now, while at the same time, there is a sharp decrease in the flow rate from 0.083 down to 0.0083 mL/min. The next part of section (2) is characterized by very high retention with C/C0 stable at 0.01 and a flow rate around 0.0083 to 0.042 mL/min. In section (3), a rapid increase in C/C0 from 0.01 to 0.58 in the range PV 5.28-5.57 can be observe. Coherently, in the range PV 4.35-5.21 a flow rate increase from 0.042 to 0.5 mL/min was registered, which managed to remain at more or less constant values for the rest of the test. The unclogging was achieved by increasing the rpm of the pump for a couple of seconds. The rapid increase in C/C0 has a sharp drop in slope in the final section from PV 5.57 onwards, finally reaching an average value of 0.92 C/C0 at PV ca. 11. The residual mass in the column at the end of the test calculated using equation (4) was 31.3 mg. Looking at the above results, a correlation between BC retention and work flow rate is clear, which is in agreement with various authors who find higher quantity of strained colloid for lower pore water velocity [36,75,76].
In order to assess the residual PV when loaded with BC@PST, a second test was performed with NaCl tracer at the same flow rate as the first (Figure 9c). Due to the absence of points at the inflection of the test performed after loading with BC@PST, it is not possible to determine the exact θ value, which is, however, approximately a quarter of the initial one, thus giving us an ε of roughly 0.1 that justifies clogging issues. Due to the low mass retained in the column, the low maximum working concentration, and the difficulties in distribution due to clogging, it was subsequently decided to proceed with the study of other composites. In this case, the clogging is not due to mechanical filtration as there is no evidence of cake formation on the feed side of the column and is, therefore, inevitably attributable to physical straining.
Figure 9.
Tracer test on BC@PST: (a) step input tracer test with NaCl curve and BC@PST distribution test curve. C0 is the feed concentration while C is the discharge concentration; (b) flow rate during the distribution test. Section A (red), section B (blue), section C (green) are highlighted; (c) comparison between step input tracer test before and after BC@PST distribution test.
Figure 9.
Tracer test on BC@PST: (a) step input tracer test with NaCl curve and BC@PST distribution test curve. C0 is the feed concentration while C is the discharge concentration; (b) flow rate during the distribution test. Section A (red), section B (blue), section C (green) are highlighted; (c) comparison between step input tracer test before and after BC@PST distribution test.
3.4.2. Continuous Flow Column Distribution Tests on BC@CMC 20 g·L−1 Composite
The tracer test conducted on BC@CMC with CMC 20 g/L (Figure 10) showed a PV of 27.5 mL corresponding to an ε of 0.40 with a θ of 44.3 min. Similarly to the previous test, the BC in the effluent remains at 0 throughout the first PV, then rises sharply to about 0.75 at PV 1.1, and then remains about constant until PV 6.4. At this point, a slow growth can be observed until it stabilizes around PV 30 at the end of the elution test, where an average C/C0 value of 0.98 is obtained between PV 30 and 37. At the end of the distribution test, a water rinse test was performed to remove the BC in the column not retained by the bed. However, after about 0.3 PV the column became completely clogged and it was impossible to continue the test. One hypothesis on clogging could be due to a sudden decrease in ionic strength following flushing. The retention of a colloid in a porous medium by straining is, in fact, strongly controlled by the ionic strength [36,77], whereas the latter increases, the retention of the colloid increases. Elution with deionized water causes an immediate zeroing of the ionic strength in the column, which had previously been at 81 mM (given by the CMC), generating an immediate release of a fraction of BC retained by attachment processes to clog the column by physical straining. In this case, it was possible to calculate the dp/dc ratio, which stands at a value of 0.00361–0.00481, in line with theoretical values that guarantee physical straining [35,78], confirming the aforementioned filtration mechanism. In this case, due to the higher concentration achievable (C0), the residual mass in the column at the end of the test calculated was much higher than 128 mg previously obtained. From this test, it can certainly be deduced that CMC provides better properties than PST with regard to the transport of BC (as the distribution test was performed), without any variation in the flow rate and therefore did not give any clogging problems. However, it is evident that further optimization of the suspension composition is required to avoid blocking the column, which then prevents future adsorption tests.
3.4.3. Continuous Flow Column Distribution Tests on BC@CMC 10 g·L−1 Composite
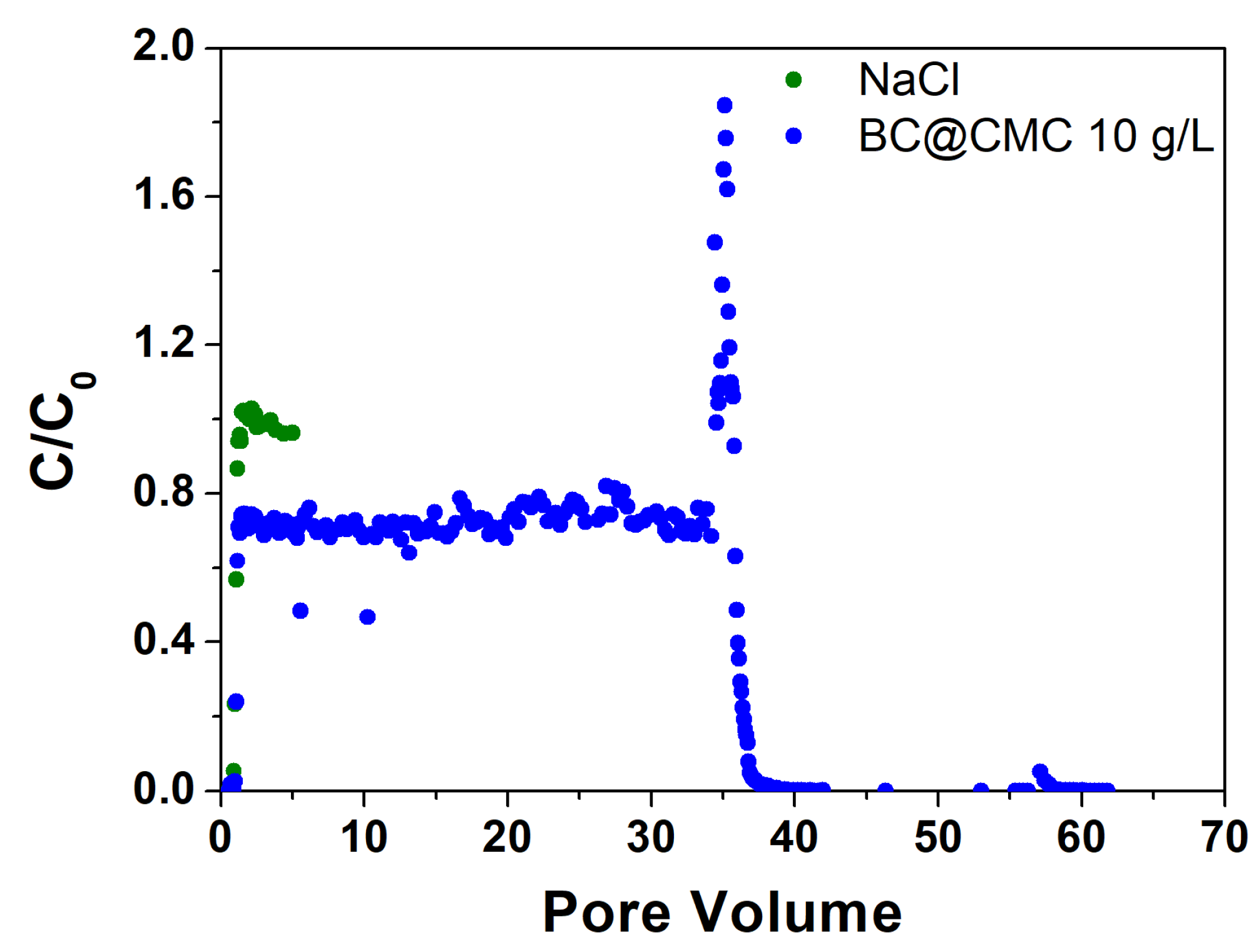
Based on the results obtained with BC@PST, BC@CMC with CMC 20 g·L−1, the composite formulation was optimized by reducing the CMC concentration at 10 g·L−1. The related column distribution tests are presented in Figure 11. The reduction of CMC concentration slightly reduces the stability of the BC, as can be observed from the sedimentation tests (Figure 6d in Section 3.3.1). However, it reduces the hydrodynamic diameter of the particles that remain suspended (see Table S1), reducing the probability of clogging by physical straining following a massive release caused by ionic strength collapse. For this trial, the tracer test showed a PV of 29.8 mL, corresponding to an ε of 0.43 with a θ of 48.0 min. In agreement with data collected in previous tests, the concentration of BC begins to increase rapidly in the effluent shortly after the tracer breakthrough, although in this case it reaches a C/C0 plateau of about 0.72 between PV 1.6 and 34.1 (where the flushing test is initiated), mobilizing significant amounts of BC with a C/C0 peak of 1.85. Thereafter, the concentration drops very rapidly and remains near zero for the duration of the flushing test of another 25 PV. Again, it is evident how the sudden lowering of the ionic strength leads to the mobilization of BC withheld by attachment only due to a higher ionic strength (see paragraph 3.4.2) [77]. Instead of obtaining a clogging of the column, a strong momentary release is registered, evidenced by the C/C0 peak at 1.85. This result indicates that, in this case, the hydrodynamic diameter is small enough (1122 ± 300) nm vs (2887 ± 950) nm to not to clog the porous medium following massive release. A further interesting aspect of this test is the obtaining of a plateau C/C0 of 0.72, significantly lower than the values of the previous tests of 0.92 and 0.98 for BC@PST and BC@CMC 20 g·L, respectively. This indicates a significantly more effective retention than in previous trials, which does not lead to clogging. This could be due to a physical straining of a specific particle size fraction that is systematically retained by the porous medium, generating the steady state between PV 1.6 and 34.1 [34]. In this case, the residual mass in the column at the end of the test was higher than the one previously achieved, which was around 365 mg. It was possible to calculate the dp/dc ratio to be 0.0014 and 0.0019, partially in line with literature values that guarantee physical straining [35,78]. However, the aforementioned mechanism is dominant over attachment, as rinsing with deionized water removed only a fraction of the retained BC [77].
This test showed positive results concerning: (1) high amount of BC retained in the column, (2) efficient distribution of the suspension in the porous medium, and (3) constant flow rate throughout the transport and flushing test.
Figure 11.
Step input tracer test with NaCl curve and BC@CMC 10 g·L−1 distribution test curve.
4. Conclusions
In this work, biochars (BCs) waste obtained from gasification at 950 °C of pine wood pellets for energy production were used as fillers in polymer matrices, to obtain BC@biopolymer composites for in-situ groundwater remediation. Four different biochar samples were studied, based on different sieving and grinding processes: A, B (sieving at 0.5 mm), and AP, BP (sieving 64 µm and manual grinding). Extensive morphostructural characterizations via UV-Visible and FT-IR spectroscopy, dynamic light scattering (DLS), and scanning electron microscopy (FESEM) were carried out on both BC and BC@polymer composites. Raw BCs showed typical graphite-based structure with little to no residual functional groups, as evidenced by FTIR and UV-Visible spectroscopy. In water suspension, the grinding process resulted in a more intense population centered at around 200 nm with the formation of larger aggregates above 1000 nm (AP, BP samples), with a sponge-like morphology of the surface and quasi-spherical pores. The micro-/mesoporous nature of BCs arose from BET and textural parameters analysis, with a reduction of the specific surface area of about 30% after grinding process. Based on the percentage of colloidal fraction, detected by DLS, AP sample was used to obtain water-based polymer composites with chitosan (CS), alginate (ALG), potato starch (PST), and sodium carboxymethylcellulose (CMC) as polymer matrices (0.2-20.0 g/L concentration range). According to pilot sedimentation tests, after 7 days BC@PST (BC 0.3 g/L, PST 20.0 g/L + APG2 surfactant 1.0% v/v) and BC@CMC (BC 0.3 g/L, CMC 10.0 and 20.0 g/L) resulted as the most stable suspensions with a sedimentation percentage of (14 ± 1)%, (31 ± 2)%, (27 ± 2)% for BC@PST, BC@CMC 10 g/L, and BC@CMC 20.0 g/L, respectively. Enhanced stability of the mentioned composites was ascribed to a combination of both hydrogen bonds and physical entrapment, as studied by FTIR. Final composites formulations were optimized increasing the BC concentration up to 1.0 g/L.
Before distribution tests on BC@PST and BC@CMC, adsorption performances of raw BCs were validated via adsorption isotherm using trichloroethylene (TCE) as model contaminant. Continuous flow column distribution tests on composites resulted in a clogging of porous media in the case of BC@PST and BC@CMC 20.0 g/L, probably due to a higher hydrodynamic diameter of composite particles in relation to the pore size of the media. Conversely, BC@CMC 10.0 g/L composite showed an optimized distribution behavior, with high retention (ca. 365 mg) and without column clogging. As a future perspective, continuous flow adsorption tests of TCE will be carried out. This study showed that biopolymer-based biochar composites can be used as a green and sustainable alternative solution to address various groundwater pollution challenges.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: UV-Vis spectra of raw biochars; Figure S2: FESEM images of AP biochar sample; Figure S3: DLS of biochars after two hours aging; Figure S4: pictures of selected sedimentation tests; Figure S5: FTIR-ATR spectrum of commercial potato starch and its assignment; Figure S6: FTIR-ATR spectrum of commercial sodium carboxymethylcellulose and its assignment; Figure S7: calculated FTIR ATR spectra of composites; Table S1: Hydrodynamic parameters of BC composites at different time points.
Author Contributions
Conceptualization, M.P.P. and I.F.; methodology, S.C. and D.F.; validation, S.C. and D.F.; formal analysis, S.C., D.F. and I.P.; investigation, S.C., D.F., and I.P.; resources, M.P.P. and I.F.; data curation, S.C. and D.F.; writing—original draft preparation, S.C. and D.F.; writing—review and editing, M.P.P., S.C., D.F. and I.F.; visualization, L.L.; supervision, M.P.P. and I.F.; project administration, M.P.P. and I.F.; funding acquisition, M.P.P. All authors have read and agreed to the published version of the manuscript.
Funding
I.F. gratefully acknowledge for the financial support Sapienza University of Rome funding Grant Ateneo Ricerca 2022 (RM1221867C322C1). S.C. gratefully acknowledge for the financial support Sapienza University of Rome funding Grant Progetti per Avvio alla Ricerca-Tipo 2 2023 (AR223188AFDCFD42).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
M.P.P. and D.F. carefully acknowledge Cecilia Riccioni for the collaborative work and support in conducting the experimental studies on biochar outlined in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, H.-M.; Wu, M.-T. Residential exposure to chlorinated hydrocarbons from groundwater contamination and the impairent of renal function-An ecological study. Sci. Rep. 2019, 7, 40283. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yuan, J.; Lu, X.; Su, C.; Zhang, Y.; Wang, C.; Cao, X.; Li, Q.; Su, J.; Ittekkot, V.; Garbutt, R.A.; Bush, S.; Fletcher, S.; Wagey, T.; Kachur, A.; Sweijd, N. Major threats of pollution and climate change to global coastal ecosystems and enhanced management for sustainability. Environ. Pollut. 2018, 236, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Kurwadkar, S. Occurrence and distribution of organic and inorganic pollutants in groundwater. Water Environ. Res. 2019, 91, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Lyu, T.; Guo, J.; Zhao, C.; Yang, Y. Simultaneous removal of organic micropollutants and inorganic heavy metals by nano-calcium peroxide induced Fenton-like treatment. Sep. Purif. Technol. 2023, 305, 122474. [Google Scholar] [CrossRef]
- Padhye, L.P.; Srivastava, P.; Jasemizad, T.; Bolan, S.; Hou, D.; Shaheen, S.M.; Rinklebe, J.; O’Connor, D.; Lamb, D.; Wang, H.; Siddique, K.H.M.; Bolan, N. Contaminant containment for sustainable remediation of persistent contaminants in soil and groundwater. J. Hazard. Mat. 2023, 455, 131575. [Google Scholar] [CrossRef] [PubMed]
- Ciampi, P.; Esposito, C.; Bartsch, E.; Alesi, E.J.; Rehner, G.; Petrangeli Papini, M. Remediation of chlorinated aliphatic hydrocarbons (CAHs) contaminated site coupling groundwater recirculation well (IEG-GCW®) with a peripheral injection of soluble nutrient supplement (IEG-C-MIX) via multilevel-injection wells (IEG-MIW). Heliyon 2022, 8, e11402. [Google Scholar] [CrossRef] [PubMed]
- Cerra, S.; Fratoddi, I. Nanoscale Solutions: The Transformative Applications of Functionalized Nanomaterials in Environmental Remediation. Appl. Nano 2024, 5, 14–19. [Google Scholar] [CrossRef]
- Aparicio, J.D.; Raimondo, E.E.; Saez, J.M.; Costa-Gutierrez, S.B.; Álvarez, A.; Benimeli, C.S.; Polti, M.A. The current approach to soil remediation: A review of physicochemical and biological technologies, and the potential of their strategic combination. J. Environ. Chem. Eng. 2022, 10, 107141. [Google Scholar] [CrossRef]
- Lyu, H.; Hu, K.; Wu, Z.; Shen, B.; Tang, J. Functional materials contributing to the removal of chlorinated hydrocarbons from soil and groundwater: Classification and intrinsic chemical-biological removal mechanisms. Sci. Total Environ. 2023, 879, 163011. [Google Scholar] [CrossRef]
- Hussain, A.; Rehman, F.; Rafeeq, H.; Waqas, M.; Asghar, A.; Afsheen, N.; Rahdar, A.; Bilal, M.; Iqbal, H.M.M. In-situ, Ex-situ, and nano-remediation strategies to treat polluted soil, water, and air—A review. Chemosphere 2022, 289, 133252. [Google Scholar] [CrossRef]
- Pochampally, S.V.; Krishnaswamy, P.; Obra, C.; Mortazavian, S.; Marti, E.; Moon, J. Adsorption of chlorinated hydrocarbons onto non-activated biochars: Biochar physicochemical characteristics and governing factors. Bioresour. Technol. 2023, 22, 101465. [Google Scholar] [CrossRef]
- Obiri-Nyarko, F.; Grajales-Mesa, S.J.; Malina, G. An overview of permeable reactive barriers for in situ sustainable groundwater remediation. Chemosphere 2014, 111, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Powell, R.M.; Blowes, D.W.; Gillham, R.W.; Schultz, D.; Sivavec, T.; Puls, R.W.; Vogan, J.L.; Powell, P.D.; Landis, R. Permeable reactive barrier technologies for contaminant remediation. US EPA 1998, 600, 1–94. [Google Scholar]
- Mackenzie, K.; Schierz, A.; Georgi, A.; Kopinke, F.D. Colloidal activated carbon and carbo-iron–Novel materials for in-situ groundwater treatment. Global NEST Journal 2008, 10, 54–61. [Google Scholar]
- Georgi, A.; Schierz, A.; Mackenzie, K.; Kopinke, F.D. Colloidal activated carbon for in-situ groundwater remediation—Transport characteristics and adsorption of organic compounds in water-saturated sediment columns. J. Contam. Hydrol. 2015, 179, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Sethi, R.; Day, S.; Di Molfetta, A. Clamshell vs. backhoe excavation of permeable reactive barriers. Am. J. Environ. Sci. 2011, 7, 463–467. [Google Scholar] [CrossRef]
- Ma, J.; Stevens, G.W.; Mumford, K.A. The performance of diphenyldichlorosilane coated ammonium exchange zeolite and its application in the combination of adsorption and biodegradation of hydrocarbon contaminated ground water. Chem. Eng. J. 2018, 347, 415–423. [Google Scholar] [CrossRef]
- Meiramkulova, K.; Kydyrbekova, A.; Devrishov, D.; Nurbala, U.; Tuyakbayeva, A.; Zhangazin, S.; Ualiyeva, R.; Kolpakova, V.; Yeremeyeva, Y.; Mkilima, T. Comparative Analysis of Natural and Synthetic Zeolite Filter Performance in the Purification of Groundwater. Water 2023, 15, 588. [Google Scholar] [CrossRef]
- Javadi, S.; Ghavami, M.; Zhao, Q.; Bate, B. Advection and retardation of non-polar contaminants in compacted clay barrier material with organoclay amendment. Appl. Clay Sci 2017, 142, 30–39. [Google Scholar] [CrossRef]
- Ni, H.; Fan, R.-D.; Reddy, K.R.; Du, Y.-J. Containment of phenol-impacted groundwater by vertical cutoff wall with backfill consisting of sand and bentonite modified with hydrophobic and hydrophilic polymers. J. Hazard. Mater. 2024, 461, 132627. [Google Scholar] [CrossRef]
- Alonso-de-Linaje, V.; Mangayayam, M.C.; Tobler, D.J.; Dietmann, K.M.; Espinosa, R.; Rives, V.; Dalby, K.N. Sorption of chlorinated hydrocarbons from synthetic and natural groundwater by organo-hydrotalcites: Towards their applications as remediation nanoparticles. Chemosphere 2019, 236, 124369. [Google Scholar] [CrossRef]
- Song, T.; Tian, W.; Qiao, K.; Zhao, J.; Chu, M.; Du, Z.; Wang, L.; Xie, W. Adsorption Behaviors of Polycyclic Aromatic Hydrocarbons and Oxygen Derivatives in Wastewater on N-Doped Reduced Graphene Oxide. Sep. Purif. Technol. 2021, 254, 117565. [Google Scholar] [CrossRef]
- Kinney, T.J.; Masiello, C.A.; Dugan, B.; Hockaday, W.C.; Dean, M.R.; Zygourakis, K.; Barnes, R.T. Hydrologic properties of biochars produced at different temperatures. Biomass Bioenergy 2012, 41, 34–43. [Google Scholar] [CrossRef]
- Yang, W.; Shang, J.; Li, B.; Flury, M. Surface and Colloid Properties of Biochar and Implications for Transport in Porous Media. Crit. Rev. Environ. Sci. Technol. 2020, 50, 2484–2522. [Google Scholar] [CrossRef]
- Meng, Q.; Jin, L.; Cheng, L.; Fang, J.; Lin, D. Release and sedimentation behaviors of biochar colloids in soil solutions. J. Environ. Sci. 2021, 100, 269–278. [Google Scholar] [CrossRef]
- Janu, R.; Mrlik, V.; Ribitsch, D.; Hofman, J.; Sedláček, P.; Bielská, L.; Soja, G. Biochar surface functional groups as affected by biomass feedstock, biochar composition and pyrolysis temperature. Carbon Resour. Convers. 2021, 4, 36–46. [Google Scholar] [CrossRef]
- Al-Wabel, M.I.; Al-Omran, A.; El-Naggar, A.H.; Nadeem, M.; Usman, A.R. A Pyrolysis temperature induced changes in characteristics and chemical composition of biochar produced from conocarpus wastes. Bioresour. Technol 2013, 131, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Silvani, L.; Di Palma, P.R.; Riccardi, C.; Eek, E.; Hale, S.E.; Viotti, P.; Petrangeli Papini, M. Use of biochar as alternative sorbent for the active capping of oil contaminated sediments. J. Environ. Chem. Eng. 2017, 5, 5241–5249. [Google Scholar] [CrossRef]
- Loc, N.X.; Tuyen, P.T.T.; Mai, L.C.; Phuong, D.T.M. Chitosan-Modified Biochar and Unmodified Biochar for Methyl Orange: Adsorption Characteristics and Mechanism Exploration. Toxics 2022, 10, 500. [Google Scholar] [CrossRef]
- Li, N.; Yin, M.; Tsang, D.C.W.; Yang, S.; Liu, J.; Li, X.; Song, G.; Wang, J. Mechanisms of U(VI) removal by biochar derived from Ficus microcarpa aerial root: A comparison between raw and modified biochar. Sci. Total Environ. 2019, 697, 134115. [Google Scholar] [CrossRef]
- Zhong, Y.; Igalavithana, A.D.; Zhang, M.; Li, X.; Rinklebe, J.; Hou, D.; Tack, F.M.G.; Alessi, D.S.; Tsang, D.C.W.; Ok, Y.S. Effects of aging and weathering on immobilization of trace metals/metalloids in soils amended with biochar. Environ. Sci. Processes Impacts 2020, 22, 1790–1808. [Google Scholar] [CrossRef] [PubMed]
- Herzig, J.P.; Leclerc, D.M.; Goff, P.L. Flow of suspensions through porous media—application to deep filtration. Ind. Eng. Chem. 1970, 62, 8–35. [Google Scholar] [CrossRef]
- McDowell-Boyer, L.M. Chemical mobilization of micron-sized particles in saturated porous media under steady flow conditions. Environ. Sci. Technol. 1992, 26, 586–593. [Google Scholar] [CrossRef]
- Shen, C.; Huang, Y.; Li, B.; Jin, Y. Effects of solution chemistry on straining of colloids in porous media under unfavorable conditions. Water Resour. Res. 2008, 44, W05419. [Google Scholar] [CrossRef]
- Bradford, S.A.; Yates, S.R.; Bettahar, M.; Simunek, J. Physical factors affecting the transport and fate of colloids in saturated porous media. Water Resour. Res. 2002, 38, 1327. [Google Scholar] [CrossRef]
- Du, Y.; Shen, C.; Zhang, H.; Huang, Y. Effects of flow velocity and nonionic surfactant on colloid straining in saturated porous media under unfavorable conditions. Transp. Porous Med. 2013, 98, 193–208. [Google Scholar] [CrossRef]
- Rajapaksha, A.U.; Chen, S.S.; Tsang, D.C.W.; Zhang, M.; Vithanage, M.; Mandal, S.; Gao, B.; Bolan, N.S.; Ok, Y.S. Engineered/designer biochar for contaminant removal/immobilization from soil and water: Potential and implication of biochar modification. Chemosphere 2016, 148, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.; Arrigo, R.; Malucelli, G.; Tagliaferro, A.; Duraccio, D. Recent Advances in Biochar Polymer Composites. Polymers 2022, 14, 2506. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.M.; Alfano, S.; Amanat, N.; Andreini, F.; Lorini, L.; Martinelli, A.; Petrangeli Papini, M. A Polyhydroxybutyrate (PHB)-Biochar Reactor for the Adsorption and Biodegradation of Trichloroethylene: Design and Startup Phase. Bioengineering 2022, 9, 192. [Google Scholar] [CrossRef]
- Infurna, G.; Caruso, G.; Dintcheva, N.T. Sustainable Materials Containing Biochar Particles: A Review. Polymers 2023, 15, 343. [Google Scholar] [CrossRef]
- Alazaiza, M.Y.D.; Albahnasawi, A.; Eyvaz, M.; Al Maskari, T.; Nassani, D.E.; Amr, S.S.A.; Abujazar, M.S.S.; Bashir, M.J.K. An Overview of Green Bioprocessing of Algae-Derived Biochar and Biopolymers: Synthesis, Preparation, and Potential Applications. Energies 2023, 16, 791. [Google Scholar] [CrossRef]
- Amanat, N.; Barbati, B.; Rossi, M.M.; Bellagamba, M.; Buccolini, M.; Galantini, L.; Petrangeli Papini, M. Synthetic and Natural Surfactants for Potential Application in Mobilization of Organic Contaminants: Characterization and Batch Study. Water 2022, 14, 1182. [Google Scholar] [CrossRef]
- Rossi, M.M.; Silvani, L.; Amanat, N.; Petrangeli Papini, M. Biochar from pine wood, rice husks and iron-eupatorium shrubs for remediation applications: Surface characterization and experimental tests for trichloroethylene removal. Materials 2021, 14, 1776. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.J.; Zogorski, J.S.; Squillace, P.J. Chlorinated solvents in groundwater of the United States. Environ. Sci. Technol. 2007, 41, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.M.; Matturro, B.; Amanat, N.; Rossetti, S.; Petrangeli Papini, M. Coupled adsorption and biodegradation of trichloroethylene on biochar from pine wood wastes: A combined approach for a sustainable bioremediation strategy. Microorganisms 2022, 10, 101. [Google Scholar] [CrossRef]
- Rouquerol, J.; Llewellyn, P.; Rouquerol, F. Is the Bet Equation Applicable to Microporous Adsorbents? In Studies in Surface Science and Catalysis; Elsevier, 2007; Volume 160, pp. 49–56. ISBN 978-0-444-52022-7. [Google Scholar]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Gurvitsch, L.G. Physicochemical attractive force. J. Russ. Phys. Chem. Soc. 1915, 47, 805–827. [Google Scholar]
- Yu, X.-Y.; Ying, G.-G.; Kookana, R.S. Reduced plant uptake of pesticides with biochar additions to soil. Chemosphere 2009, 76, 665–671. [Google Scholar] [CrossRef]
- Kim, P.; Johnson, A.; Edmunds, C.W.; Radosevich, M.; Vogt, F.; Rials, T.G.; Labbé, N. Surface Functionality and Carbon Structures in Lignocellulosic-Derived Biochars Produced by Fast Pyrolysis. Energy Fuels 2011, 25, 4693–4703. [Google Scholar] [CrossRef]
- Wang, X.; Sotoudehniakarani, F.; Yu, Z.; Morrell, J.J.; Cappellazzi, J.; McDonald, A.G. Evaluation of corrugated cardboard biochar as reinforcing fiber on properties, biodegradability and weatherability of wood-plastic composites. Polym. Degrad. Stab. 2019, 168, 108955. [Google Scholar] [CrossRef]
- Liu, Y.; He, Z.; Uchimiya, M. Comparison of Biochar Formation from Various Agricultural By-Products Using FTIR Spectroscopy. Mod. Appl. Sci. 2015, 9, 246–253. [Google Scholar] [CrossRef]
- Fang, Q.; Chen, B.; Lin, Y.; Guan, Y. Aromatic and Hydrophobic Surfaces of Wood-derived Biochar Enhance Perchlorate Adsorption via Hydrogen Bonding to Oxygen-containing Organic Groups. Environ. Sci. Technol. 2014, 48, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Zhang, M.; Liu, Y.; Gou, J.; Wei, Q.; Shen, B. Physico-chemical characteristics and the adsorption of ammonium of biochar pyrolyzed from distilled spirit lees, tobacco fine and Chinese medicine residues. J. Anal. Appl. Pyrolysis 2021, 156, 105148. [Google Scholar] [CrossRef]
- Elnour, A.Y.; Alghyamah, A.; Shaikh, H.M.; Poulose, A.M.; Al-Zahrani, S.M.; Anis, A.; Al-Wabel, M. Effect of Pyrolysis Temperature on Biochar Microstructural Evolution, Physicochemical Characteristics, and Its Influence on Biochar/Polypropylene Composites. Appl. Sci. 2019, 9, 1149. [Google Scholar] [CrossRef]
- Sing, K.S.W. Reporting Physisorption Data for Gas/Solid Systems with Special Reference to the Determination of Surface Area and Porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Li, Z.Q.; Lu, C.J.; Xia, Z.P.; Zhou, Y.; Luo, Z. X-ray diffraction patterns of graphite and turbostratic carbon. Carbon 2007, 45, 1686–1695. [Google Scholar] [CrossRef]
- Disma, F. Effect of Mechanical Grinding on the Lithium Intercalation Process in Graphites and Soft Carbons. J. Electrochem. Soc. 1996, 143, 3959. [Google Scholar] [CrossRef]
- Abulikemu, G.; Wahman, D.G.; Sorial, G.A.; Nadagouda, M.; Stebel, E.K.; Womack, E.A.; Smith, S.J.; Kleiner, E.J.; Gray, B.N.; Taylor, R.D.; et al. Role of Grinding Method on Granular Activated Carbon Characteristics. Carbon Trends 2023, 11, 100261. [Google Scholar] [CrossRef] [PubMed]
- Aranaz, I.; Alcántara, A.R.; Civera, M.C.; Arias, C.; Elorza, B.; Caballero, A.H.; Acosta, N. Chitosan: An Overview of Its Properties and Applications. Polymers 2021, 13, 3256. [Google Scholar] [CrossRef]
- Keetelsl, C.J.A.M.; van Vliet, T.; Walstra, P. Gelation and retrogradation of concentrated starch systems: 1. Gelation. Food Hydrocoll. 1996, 10, 343–353. [Google Scholar] [CrossRef]
- Lafarge, C.; Cayot, N. Potential Use of Mixed Gels from Konjac Glucomannan and Native Starch for Encapsulation and Delivery of Aroma Compounds: A Review. Starch 2018, 70, 1700159. [Google Scholar] [CrossRef]
- Yassaroh, Y.; Woortman, A.J.J.; Loos, K. A new way to improve physicochemical properties of potato starch. Carbohydr. Polym. 2019, 204, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Geetha, D.; Tyagi, R. Alkyl Poly Glucosides (APGs) Surfactants and Their Properties: A Review. Tenside Surfactants Deterg. 2012, 49, 417–427. [Google Scholar] [CrossRef]
- Han, F.; Zhao, Y.; Liu, M.; Hu, F.; Peng, Y.; Ma, L. Effect of Sodium Carboxymethyl Cellulose on the Dynamic Wetting Characteristics of the Dust Suppression Droplet Impacting the Coal Surface. ACS Omega 2023, 8, 18414–18424. [Google Scholar] [CrossRef] [PubMed]
- Makowska, A.; Dwiecki, K.; Kubiak, P.; Baranowska, H.M.; Lewandowicz, G. Polymer-Solvent Interactions in Modified Starches Pastes–Electrokinetic, Dynamic Light Scattering, Rheological and Low Field Nuclear Magnetic Resonance Approach. Polymers 2022, 14, 2977. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.; Webster, M.F.X.; Kiemle, D.J. Chapter 2: Infrared Spectrometry. In Spectrometric identification of organic compounds, 7th ed.; John Wiley & Sons, Inc.: Hoboken, New Jersey, 2005; pp. 88–92. [Google Scholar]
- Dankar, I.; Haddarah, A.; Omar, F.E.L.; Pujolà, M.; Sepulcre, F. Characterization of food additive-potato starch complexes by FTIR and X-ray diffraction. Food Chem. 2018, 160, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Avram, M.; Mateescu, GH.D. Carbohydrates. In Infrared Spectroscopy: Applications in Organic Chemistry, 2nd ed.; R. E. Krieger Publishing Company: Huntington, New York, 1978; pp. 474–478. [Google Scholar]
- Lu, Z.-H.; Donner, E.; Yada, R.Y.; Liu, Q. Physicochemical properties and in vitro starch digestibility of potato starch/protein blends. Carbohydr. Polym. 2016, 154, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Gao, W.; Kang, X.; Dong, Y.; Liu, P.; Yan, S.; Yu, B.; Guo, L.; Cui, B.; El-Aty, A.M.A. Structural changes in corn starch granules treated at different temperatures. Food Hydrocoll. 2021, 118, 106770. [Google Scholar] [CrossRef]
- da Silva, A.M.; Gräbin Scherer, L.; Moschini Daudt, R.; Corralo Spada, J.; Medeiros Cardozo, N.S.; Damasceno Ferreira Marczak, L. Effects of starch source and treatment type—Conventional and ohmic heating—On stability and rheological properties of gels. LWT 2019, 109, 7–12. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wang, Y.; Yang, H. Characterizing digestibility of potato starch with cations by SEM, X-ray, LF-NMR, FTIR. Food Chem. 2023, 424, 136396. [Google Scholar] [CrossRef]
- Badry, R.; Ezzat, H.A.; El-Khodary, S.; Morsy, M.; Elhaes, H.; Nada, N.; Ibrahim, M. Spectroscopic and thermal analyses for the effect of acetic acid on the plasticized sodium carboxymethyl cellulose. J. Mol. Struct. 2021, 1224, 129013. [Google Scholar] [CrossRef]
- Bradford, S.A.; Torkzaban, S.; Walker, S.L. Coupling of physical and chemical mechanisms of colloid straining in saturated porous media. Water Res. 2007, 41, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.P.; Li, X.; Yal, G. Colloid retention in porous media: mechanistic confirmation of wedging and retention in zones of flow stagnation. Environ. Sci. Technol. 2007, 41, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Mitropoulou, P.N.; Syngouna, V.I.; Chrysikopoulos, C.V. Transport of colloids in unsaturated packed columns: Role of ionic strength and sand grain size. Chem. Eng. J. 2013, 232, 237–248. [Google Scholar] [CrossRef]
- Xu, S.; Gao, B.; Saiers, J.E. Straining of colloidal particles in saturated porous media. Water Resour. Res. 2006, 42, W12S16. [Google Scholar] [CrossRef]
Figure 2.
N2 adsorption/desorption isotherm for different raw biochar samples obtained at 950 °C: (a,c) sample A, B (sieving at 0.5 mm); (b,d) sample AP, BP (sieving at 64 µm and grinding).
Figure 2.
N2 adsorption/desorption isotherm for different raw biochar samples obtained at 950 °C: (a,c) sample A, B (sieving at 0.5 mm); (b,d) sample AP, BP (sieving at 64 µm and grinding).
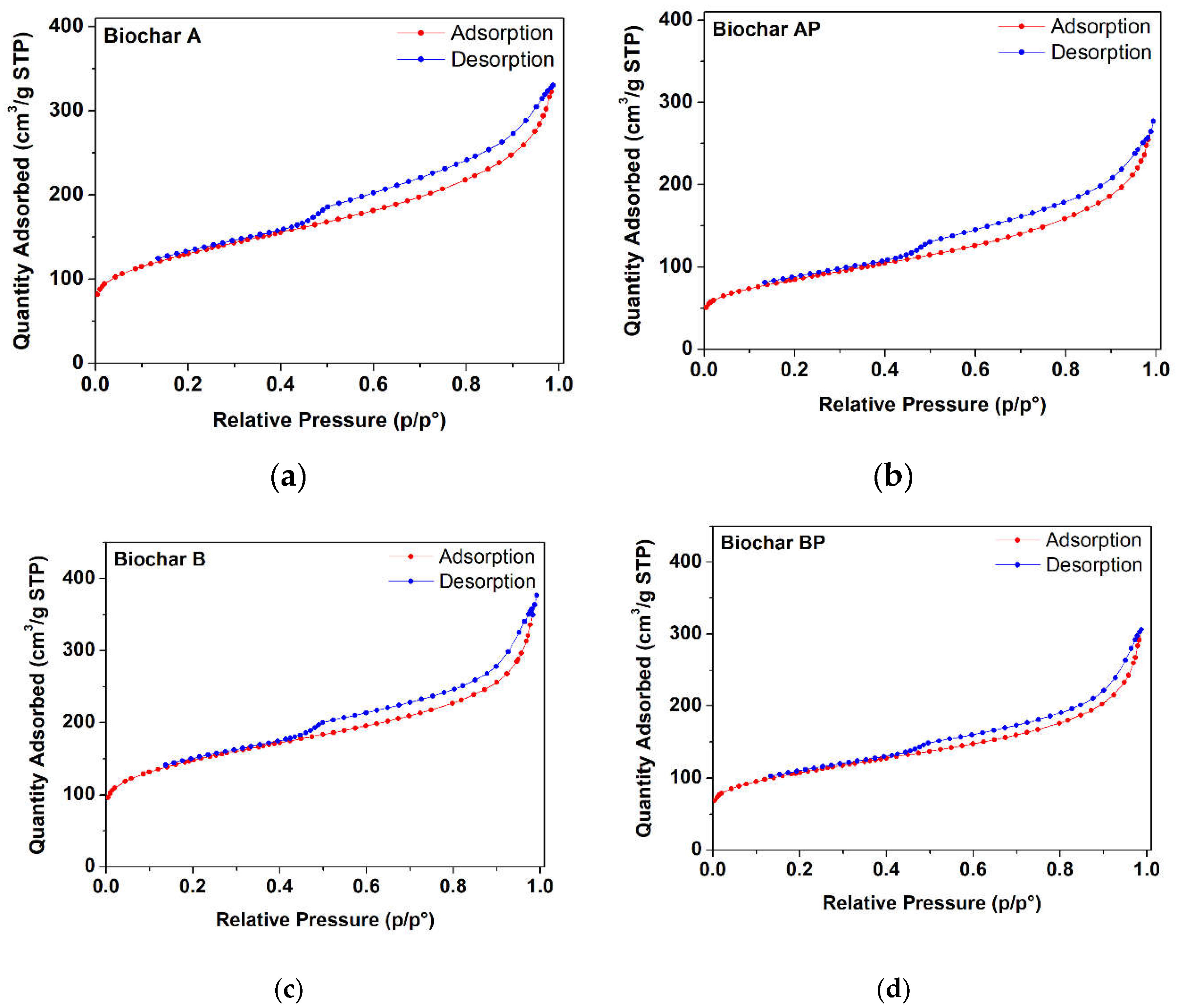
Figure 3.
DLS size distributions in H2O of freshly prepared BC samples: (a) overlap A, AP; (b) overlap B, BP.
Figure 3.
DLS size distributions in H2O of freshly prepared BC samples: (a) overlap A, AP; (b) overlap B, BP.
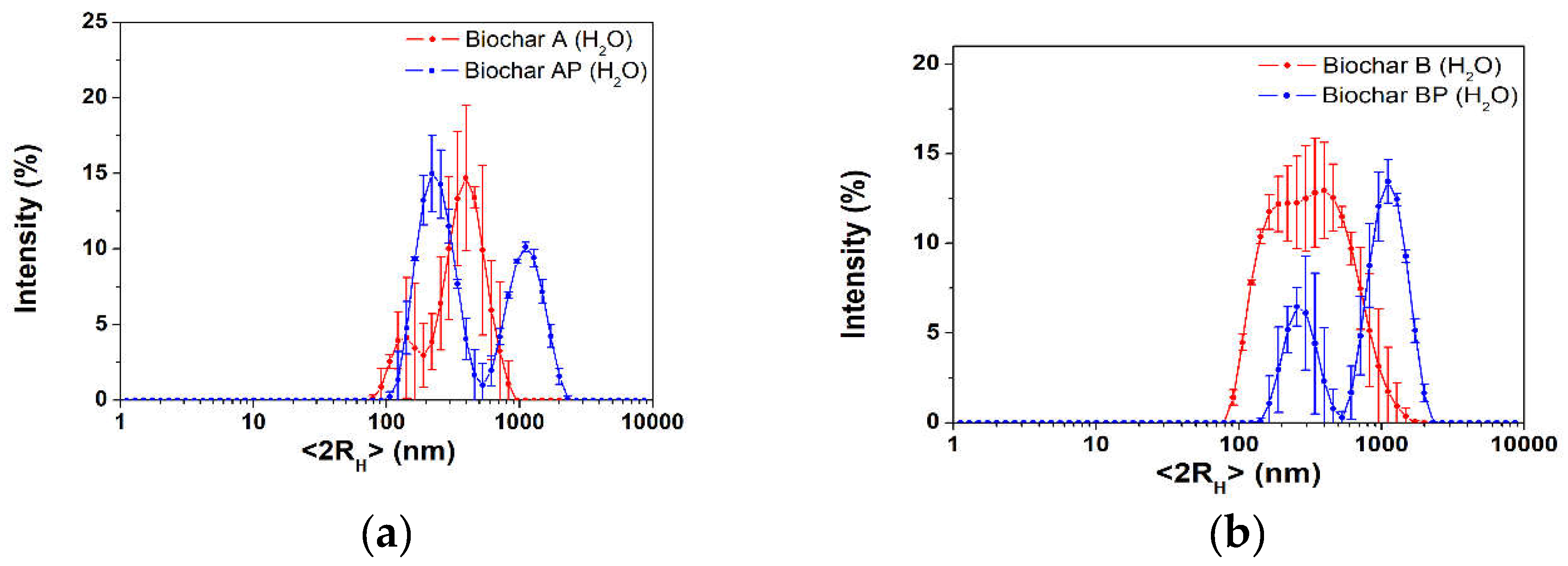
Figure 4.
Isotherm of TCE adsorption on BC with Freundlich fitting equation. Sample AP was taken as representative for this study.
Figure 4.
Isotherm of TCE adsorption on BC with Freundlich fitting equation. Sample AP was taken as representative for this study.
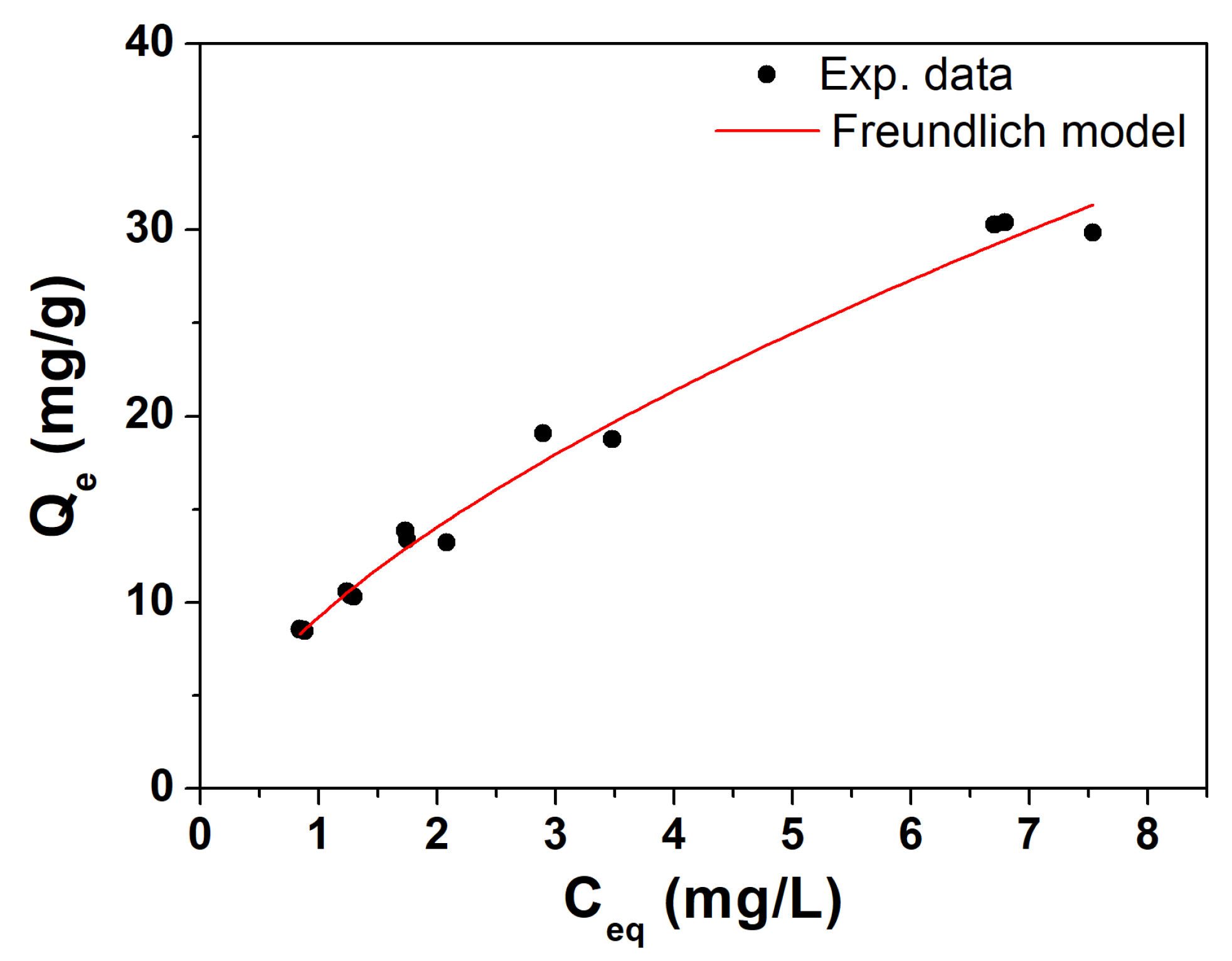
Figure 5.
Molecular structure of naturally available polymers used for BC composites: (a) chitosan (CS); (b) sodium alginate (ALG); (c) potato starch (PST); (d) sodium carboxymethylcellulose (CMC).
Figure 5.
Molecular structure of naturally available polymers used for BC composites: (a) chitosan (CS); (b) sodium alginate (ALG); (c) potato starch (PST); (d) sodium carboxymethylcellulose (CMC).
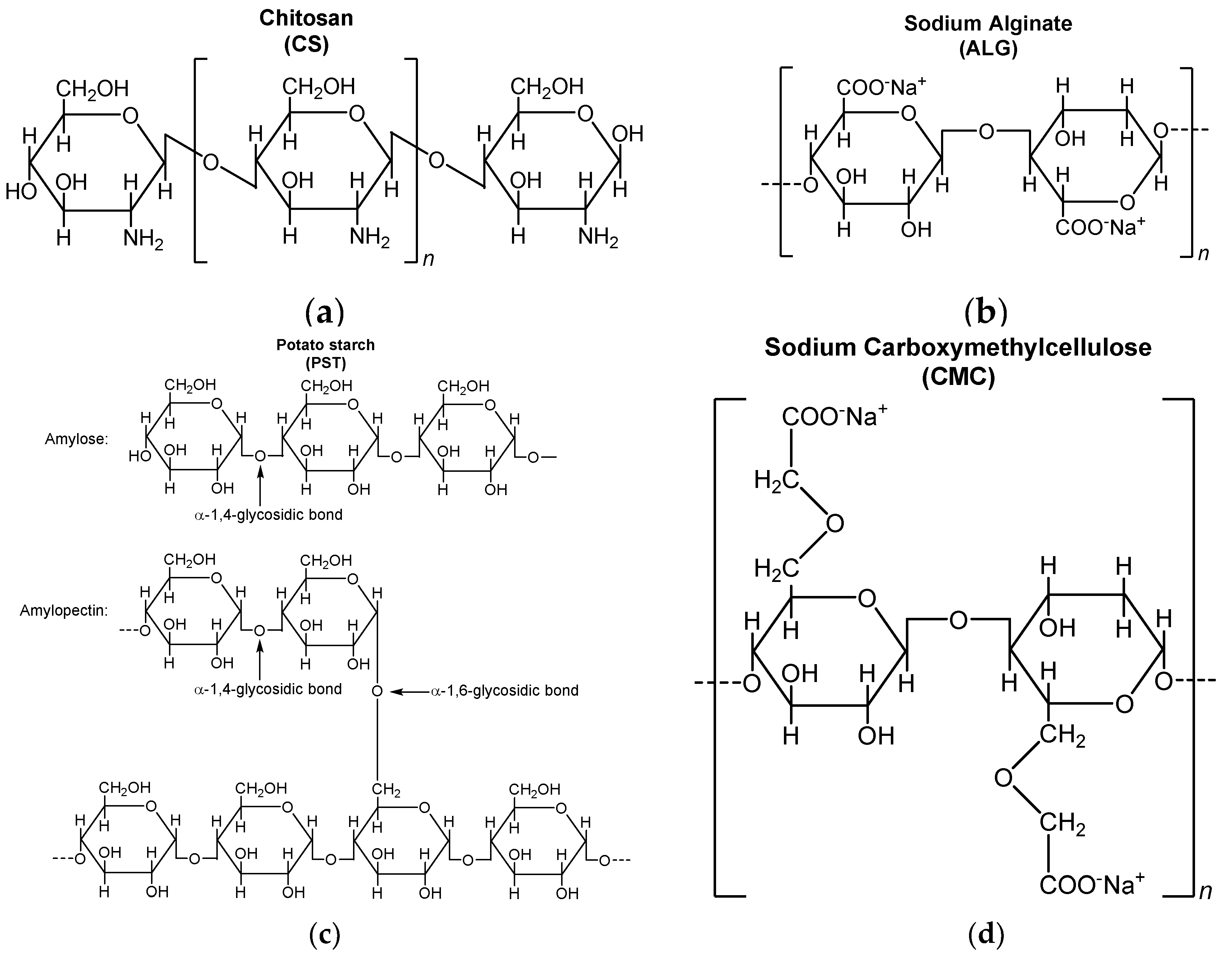
Figure 10.
Step input tracer test with NaCl curve and BC@CMC with 20 g·L−1 distribution test curve.
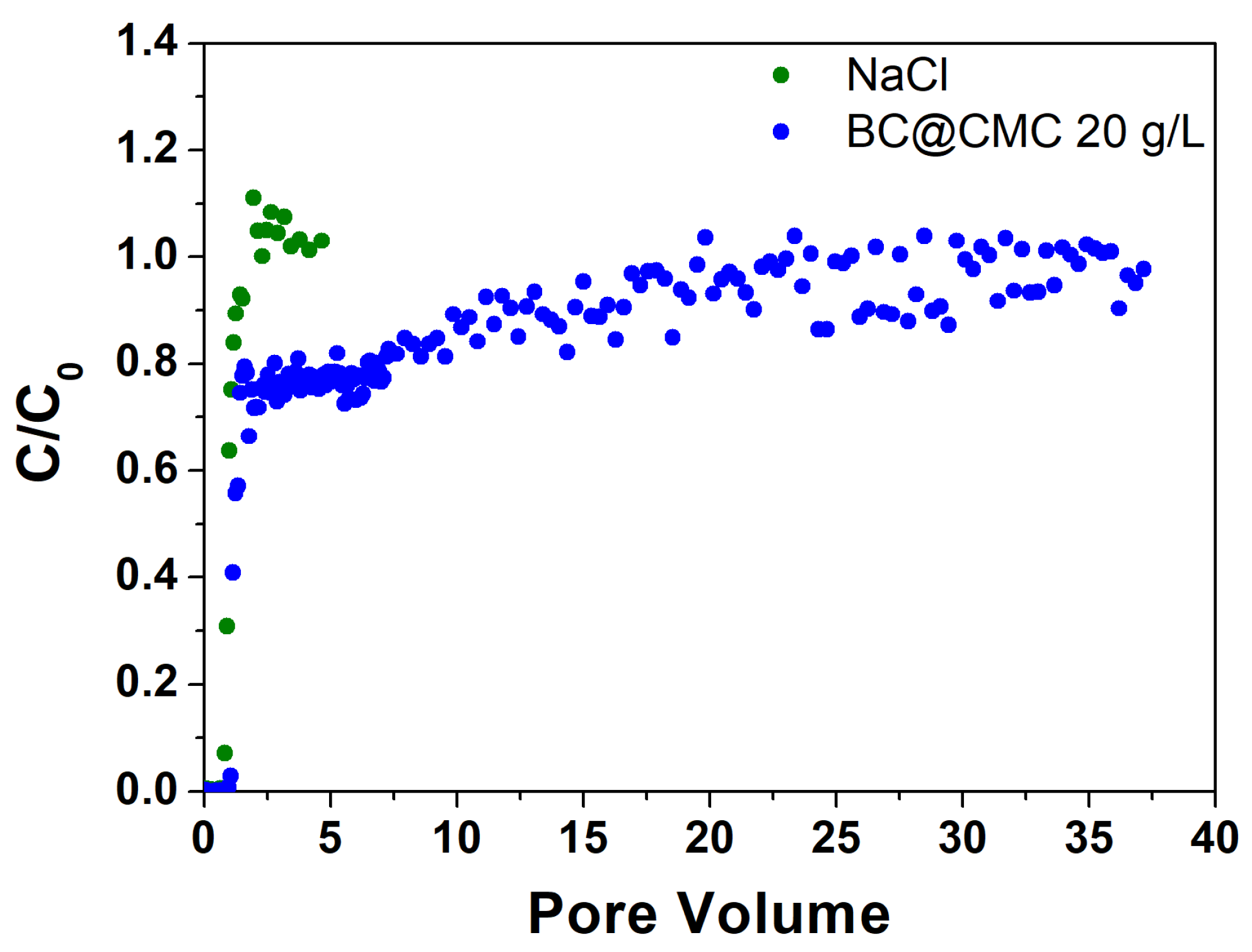
Table 2.
Conditions applied to obtain different BC@biopolymer composites in H2Oup depending on the polymer nature. These conditions were applied regardless of the BC (0.3–1.0 g·L−1) and polymer (0.2–20.0 g·L−1) concentrations used.
Table 2.
Conditions applied to obtain different BC@biopolymer composites in H2Oup depending on the polymer nature. These conditions were applied regardless of the BC (0.3–1.0 g·L−1) and polymer (0.2–20.0 g·L−1) concentrations used.
| Type of biopolymer | Temperature* | Reaction time | Additive |
|---|---|---|---|
| Chitosan | Room temperature | 2 h | - |
| Sodium alginate | Room temperature | 2 h | - |
| Potato starch | 70 °C | 1 h | APG2 1.0% v/v |
| Sodium carboxymethylcellulose | Room temperature | 2 h | - |
* Temperature is supplied via immersion in an oil bath.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



