
Submitted:
21 June 2024
Posted:
21 June 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Various symptoms have been reported to persist beyond acute phase of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, which is referred as long coronavirus disease 19 (long COVID-19). Over 65 million individuals suffer from long COVID-19. However, the causes of long COVID-19 have been largely unknown. Since long COVID-19 symptoms are observed throughout the body, vascular endothelial dysfunction may be a strong candidate to induce long COVID-19. The angioten-sin-converting enzyme 2 (ACE2), the entry receptor of SARS-CoV-2, is ubiquitously expressed in endothelial cells. We previously found that the risk factors for athero-sclerotic cardiovascular disease (ASCVD) and the history of ASCVD can be the risk for severe COVID-19, suggesting a contribution of pre-existing endothelial dysfunction to severe COVID-19. Here, we show a significant association of endothelial dysfunction with development of long COVID-19 and show that biomarkers for endothelial dysfunction in patients with long COVID-19 are also crucial players in development of ASCVD. We consider the influence of long COVID-19 on development of chronic kidney disease (CKD) and ASCVD. Furthermore, we suggest the therapeutic interventions for long COVID-19 by considering endothelial dysfunction as the treatment tar-gets for long COVID-19. Such interventions may prevent pandemic of CKD and ASCVD in post COVID-19 era.
Keywords:
atherosclerotic cardiovascular disease
; chronic kidney disease
; endothelial dysfunction
; long coronavirus disease 19.
1. Introduction
A broad range of symptoms have been reported to persist beyond the acute phase of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, which is referred as “long coronavirus disease 19 (long COVID-19)” [1,2,3]. Preliminary reports have shown that at least 65 million individuals are estimated to suffer from long COVID-19, with cases increasing daily [4,5] The SARS-CoV-2 invades human cells via angiotensin-converting enzyme 2 (ACE2), and injures multiple organs [2]. Typical symptoms of long COVID-19 include fatigue/malaise, joint pain, muscle pain, cough, sputum, shortness of breath, chest pain, hair loss, memory impairment, decreased concentration, headache, depression, olfactory dysfunction, taste disturbance, palpitations, diarrhea, abdominal pain, sleep disturbances, and muscle weakness [6,7]. The causes of long COVID-19 have been largely unknown. Since the symptoms of long COVID-19 are observed throughout the body, it can be said that disorder of blood vessels distributed throughout the body, in short, vascular endothelial dysfunction may be a strong candidate for the cause of long COVID-19. The ACE2, the entry receptor of SARS-CoV-2, is ubiquitously expressed in endothelial cells [8,9], supporting a possible contribution of endothelial dysfunction to the development of long COVID-19. We previously reported that the risk factors for atherosclerotic cardiovascular disease (ASCVD) such as diabetes and hypertension and the history of ASCVD can be the risk factor for severe COVID-19 [10], suggesting that pre-existing endothelial dysfunction which is an initial lesion of atherosclerosis may be associated with the development of severe COVID-19.
Here, we will show a significant association of endothelial dysfunction with the development of long COVID-19, and show that the biomarkers for endothelial dysfunction in patients with long COVID-19 are crucial players in the development of ASCVD. We will consider the influence of long COVID-19 on atherosclerotic risk factors such as diabetes and hypertension, and on the development of chronic kidney disease (CKD) and ASCVD. Furthermore, we suggest the therapeutic interventions for long COVID-19 when considering vascular endothelial dysfunction as the treatment targets for long COVID-19.
2. The Association of Pre-Existing Endothelial Dysfunction with the Development of Severe COVID-19
Diabetes and hypertension are important risk factors for ASCVD and induce endothelial dysfunction which is an early lesion of atherosclerosis. To understand the association of ASCVD risk factors inducing endothelial dysfunction with the development of severe COVID-19, we previously performed the meta-analysis by using PubMed. In the meta-analysis, the prevalence of diabetes in severe patients was significantly higher than that in non-severe patients (odds ratio [OR], 3.52; 95% confidence interval (CI), 2.65 to 4.67), and the prevalence of hypertension in severe patients was significantly higher than that in non-severe patients (OR, 2.69; 95% CI, 2.16 to 3.34) [10]. The prevalence of ASCVD in severe COVID-19 patients was significantly higher than that in non-severe patients (OR, 5.37; 95% CI, 3.73 to 7.74), supporting a significant association between vascular damage and severity of COVID-19.
Obesity is also one of important ASCVD risk factors. Patients with overweight and obesity admitted in a medical ward for COVID-19, despite their younger age, required more frequently assisted ventilation and access to intensive care unit (ICU) than normal-weight patients [11]. ACE2 is the cellular entry receptor of SARS-CoV-2 [2], and increased ACE2 expression in the bronchial epithelium of overweight patients compared to non-overweight patients was observed [12], indicating that SARS-CoV-2 is more likely to enter the human body in obese people as compared with non-obese people. It has been proposed that the increase of secretion of interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α) by adipose tissue in obesity-induced insulin resistance, could underlie the associations of insulin resistance with endothelial dysfunction and coagulopathy [13]. Insulin resistance is associated with increased expression and secretion of plasminogen activator inhibitor-1 (PAI-1) by endothelial cells [13]. Von Willebrand factor (VWF) is elevated in insulin-resistant states, suggesting insulin-resistance induces endothelial dysfunction [13]. Elevation of inflammatory cytokines, endothelial dysfunction and procoagulant state already exist in obese people even before SARS-CoV-2 infection [14]. SARS-CoV-2 infection may enhance elevation of inflammatory cytokines which leads to cytokine storm and induces further endothelial injury. Elevated PAI-1 and endothelial injury induce thrombosis. Multivariable regression showed remarkably increasing odds of in-hospital death associated with d-dimer (the marker for thrombosis) greater than 1 µg/mL (18.42, 2.64 - 128.55; p = 0.0033) on admission [15]. Elevated VWF (the marker for endothelial injury) level was also observed in COVID-19 patients in ICU [16]. Fibrinogen and d-dimer also increased in patients in ICU, such patients showed hypercoagulability together with a severe inflammatory state [16], which I would call as “systemic severe coagulopathic vasculitis (SSCV)” which is the most crucial cause for severe COVID-19 [14].
The underlying mechanisms for the development of severe COVID-19 in patients with metabolic syndrome were shown in Figure 1. Metabolic syndrome and its components such as obesity, diabetes and hypertension are significantly associated with the susceptibility to SARS-CoV-2 infection and severity of COVID-19 [17]. Enhanced ACE2 expression, pre-existing endothelial dysfunction and procoagulant state induced by adipocytokines dysregulation in metabolic syndrome may play a crucial role for the development of severe COVID-19 [17]. Secretion of inflammatory cytokines such IL-6 and TNF-α is increased in patients with obesity. Increased inflammatory cytokines induce insulin resistance, diabetes, dyslipidemia and hypertension, which cause endothelial dysfunction. Dysfunctional endothelial cells release more IL-6, TNF-α, and PAI-1 and VWF, which induce highly inflammatory and procoagulant state in patients with obesity. The entry of SARS-CoV-2 via overexpressed ACE2 induce cytokine storm and thrombosis, which develop severe COVID-19. A systematic review and meta-analysis showed that IL-6 and d-dimer were significantly elevated in patients with severe COVID-19 [18].
3. A significant Association of Endothelial Dysfunction with the Development of Long COVID-19
The generation of abnormal levels of oxidants under a COVID-19-induced cytokine storm causes the irreversible oxidation of a wide range of macromolecules and subsequent damage to cells, tissues, and organs [19]. Clinical studies have shown that oxidative stress initiates endothelial damage, which increases the risk of complications in COVID-19 and post-COVID-19 or long-COVID-19 cases [19].
Lasting effects and long-term sequelae could persist after the infection and may be due to persistent endothelial dysfunction. The endothelial quality index in a large cohort of long COVID-19 patients was evaluated [20]. In multivariate analysis, endothelial dysfunction was one of independent risk factors of long COVID-19 [20]. Long COVID-19 symptoms, specifically non-respiratory symptoms, are due to persistent endothelial dysfunction.
The endothelial function of patients hospitalized for COVID-19 was assessed by brachial artery flow-mediated dilation (FMD) [21]. FMD was significantly impaired in the COVID-19 group compared to the control. ICU-treated subjects presented significantly impaired FMD compared to those treated in the medical ward. Although, a significant improvement in FMD was noted during the follow-up, FMD remained impaired compared to control at 1 month and 6 months after hospital discharge.
The meta-analysis showed that a total of 644 convalescent COVID-19 patients showed significantly lower FMD values as compared to 662 controls (MD [mean difference], -2.31%; 95% CI, -3.19 to -1.44; p < 0.0001) [22]. Similar results were obtained in the sensitivity analysis of the studies that involved participants in either group with no cardiovascular risk factors or history of coronary artery disease (CAD). Meta-regression models showed that an increasing prevalence of post-acute sequelae of COVID-19 was linked to a higher difference in FMD between cases and controls.
To understand whether endothelial cell activation may be sustained in convalescent COVID-19 patients and contribute to long COVID-19 pathogenesis, endothelial cell activation was assessed in COVID-19 patients [23]. Endogenous thrombin potential and thrombin were increased in convalescent COVID-19 patients. The biomarkers for endothelial dysfunction including VWF, factor VIII and soluble thrombomodulin levels were significantly elevated in convalescent COVID-19 compared with controls, suggesting that endothelial dysfunction may contribute to long COVID-19 pathogenesis. In another study, compared to controls, long COVID-19 patients had increased levels of endothelial dysfunction indices L-selectin and P-selectin [24].
Several studies have found persistence of vascular damage with increased circulating markers of endothelial dysfunction, coagulation abnormalities with heightened thrombin generation capacity, and abnormalities in platelet counts in long COVID-19 [25]. Platelets contribute to elevated levels of thrombo-inflammatory mediators and pro-coagulant extracellular vesicles in individuals with long COVID-19 [26].
4. The Biomarkers for Endothelial Dysfunction in Patients with Long COVID-19 as Risk Factors for ASCVD
Various atherogenic factors induced by vascular endothelial dysfunction caused by infection with SARS-CoV-2 were shown in Figure 2.
4.1. NADPH Oxidase (NOX) 2 (NOX2), IL-6 and Monocyte Chemoattractant Protein-1 (MCP-1)
4.1.1. The Association of NOX2, IL-6 and MCP-1 with Long COVID-19
Activation of NADPH oxidase (NOX) family oxidases induce endothelial dysfunction, while augmenting inflammation to further deteriorate endothelial cell barrier dysfunction/injury. Upon binding of spike protein (S protein) to its membrane receptor ACE2, S protein stimulates NOX2 dependent reactive oxygen species (ROS) production [27]. Excessive ROS production by S protein induces ROS dependent cellular signaling including induction of cytokines such as IL-6 and MCP-1 [27]. IL-6 also induces ROS production in a NOX2 dependent manner, aggravating endothelial oxidative stress, which in turn sustains endothelial dysfunction. The meta-analysis showed that increased IL-6 correlates with long COVID-19, suggesting IL-6 as a basic determinant to predict long COVID-19 [28]. MCP-1 was higher in long COVID-19 patients with the most frequent symptoms [29].
4.1.2. The Association of NOX2, IL-6 and MCP-1 with ASCVD
Activation of vascular NOX and the production of ROS by these enzyme systems are common in ASCVD. Animal studies showed that ROS produced by NOX2 contributes to ASCVD including atherosclerosis and hypertension [30]. Recently, efforts have been devoted to developing inhibitors of NOX that will provide useful experimental tools and might have therapeutic potential in the treatment of human diseases. Inflammatory processes are deeply implicated in the pathogenesis of ASCVD. The meta-analysis including 17 studies showed that baseline IL-6 levels were significantly higher in ASCVD cases than in non-ASCVD controls (MD, 0.36 pg/ml; 95%CI, 0.28 to 0.44 pg/ml) [31], suggesting that higher IL-6 levels in healthy individuals are associated with ASCVD risk. MCP-1 is expressed by mainly inflammatory cells and endothelial cells [32]. The expression level is upregulated after proinflammatory stimuli and tissue injury which are associated with atherosclerotic lesion [32]. MCP-1 has been reported to play an important role in the pathogenesis of atherosclerosis. Genetic instruments for 41 cytokines and growth factors were obtained from a genome-wide association study (GWAS) of 8293 healthy adults [33]. Genetic predisposition to higher MCP-1 levels was associated with higher risk of any stroke (OR per 1 SD increase, 1.06; 95% CI, 1.02-1.09; p = 0.0009), any ischemic stroke (OR, 1.06; 95% CI, 1.02-1.10; p = 0.002), large-artery stroke (OR, 1.19; 95% CI, 1.09-1.30; p = 0.0002), and cardioembolic stroke (OR, 1.14; 95% CI, 1.06-1.23; p = 0.0004). Genetically determined higher MCP-1 levels were further associated with CAD (OR, 1.04; 95% CI, 1.00-1.08; p = 0.04) and myocardial infarction (OR, 1.05; 95% CI, 1.01-1.09; p = 0.02).
4.2. VWF, Factor VIII and ADAMST-13
4.2.1. The Association of VWF, Factor VIII and ADAMST-13 with Long COVID-19
The biomarkers for endothelial dysfunction such as VWF, factor VIII and soluble thrombomodulin levels were significantly elevated in convalescent COVID-19 compared with controls [23]. Plasma VWF facilitates platelet aggregation and adhesion to the sites of vascular injury reinforcing the pro-coagulation effect of primary and secondary hemostasis. Vascular injury leads to generation of a platelet plug at the site of injury, which is subsequently stabilized through activation of the coagulation cascade and formation of a cross-linked fibrin network. Platelet adhesion, activation, and aggregation, together with concurrent thrombin generation, are central events in this response. The VWF has been elevated in COVID-19 patients, acting as a marker of acute and sustained endothelial cell activation and predictor of poor outcomes [34,35]. The FVIII–VWF complex plays critical roles in regulating both platelet responses and the normal coagulation cascade [36]. The VWF activity is largely regulated by ADAMST-13, which is generated from endothelial cells. ADAMST-13 cleaves VWF multimers reducing their pro-adhesive and pro-coagulant activity [37]. A higher VWF:ADAMST-13 ratio together with endothelial injury, coagulopathy, and poor prognosis has been found in acute and long-COVID-19 [38,39,40,41].
4.2.2. The Association of VWF, Factor VIII and ADAMST-13 with ASCVD
VWF is involved in pathogenesis of ASCVD. The meta-analysis using 9 eligible studies including 576 cases and 632 controls showed that plasma VWF levels was significantly higher in type 2 diabetic patients with ASCVD than type 2 diabetic patients without ASCVD (MD, 0.61; 95% CI, 0.32 to 0.90; p < 0.00001) [42]. Another meta-analysis showed that men in the top third of baseline VWF values (tertile cutoff >126 IU/dl) had an OR for CAD of 1.83 (95% CI, 1.43 to 2.35; p < 0.0001) compared with those in the bottom third (tertile cutoff < 90 IU/dl), after adjustments for age [43].
Factor VIII and its carrier protein VWF are associated with risk of arterial and venous thrombosis. Sabater-Lleal, M.; et al. meta-analyzed genome-wide association results from 46354 individuals of European, African, East Asian, and Hispanic ancestry [44]. Mendelian randomization suggested causal effects of plasma FVIII activity levels on venous thrombosis and CAD risk and plasma VWF levels on ischemic stroke risk.
The meta-analysis based on published results confirmed a significant association of ADAMTS-13 levels with ischemic stroke (relative risk [RR], 2.72; 95% CI, 1.52 to 4.85, for low versus high ADAMTS-13 levels) [45]. The individual patient data meta-analysis from observational studies investigating the association between ADAMTS-13 levels and myocardial infarction showed that low ADAMTS-13 levels were associated with myocardial infarction risk, with an OR of 1.89 (95% CI, 1.15 to 3.12) for values below the 5th percentile versus above, suggesting that low ADAMTS-13 levels are associated with an increased risk of myocardial infarction [46].
4.3. Tissue Plasminogen Activator (tPA) and PAI-1
4.3.1. The Association of tPA and PAI-1 with Long COVID-19
The fibrinolytic activity is essential to dissolve the fibrin clot in tertiary hemostasis and it is mainly determined by the balance between the endothelial tPA and PAI-1 [47]. Endothelial cells are crucially involved in maintaining blood fluidity and providing controlled vascular hemostasis at sites of injury [48]. Under physiological conditions, endothelial cells constitute a non-adhesive surface preventing activation of platelets and the coagulation cascade [48]. Multiple fibrinolytic and antithrombotic properties act on their cell surface contributing to the maintenance of blood fluidity [48]. SARS-CoV-2 infection-induced endothelial dysfunction may imbalance fibrinolysis. Endothelial damage triggers the release of anti-fibrinolytic mediators such as PAI-1, which, in turn, inhibits fibrinolysis. In a study conducted by Yu Zuo et al., markedly elevated tPA and PAI-1 levels were observed in patients hospitalized with COVID-19. Both factors demonstrated strong correlations with neutrophil counts and markers of neutrophil activation. High levels of tPA and PAI-1 were associated with worse respiratory status [49]. A hypo-fibrinolysis mainly associated with increased PAI-1 levels was observed in COVID-19 patients admitted to ICU [50]. COVID-19 patients showed significantly elevated levels of tPA and PAI-1 compared to the control group [51]. Higher levels of t-PA at the time of admission were associated with lower survival rates [51]. Persistent hypo-fibrinolysis has been reported to contribute to long COVID-19 manifestations [52].
4.3.2. The Association of tPA and PAI-1 with ASCVD
The meta-analysis result suggested that PAI-1 gene polymorphism was associated with an increased risk of ischemic stroke, myocardial infarction and CAD [53,54,55]. In another meta-analysis, patients with major adverse cardiovascular events (MACE) had higher PAI-1 antigen levels with a MD of 6.11 ng/mL (95% CI, 3.27 to 8.96), suggesting that elevated plasma PAI-1 antigen levels are associated with MACE [56].
4.4. Neutrophil Extracellular Traps (NETs)
4.4.1. The Association of NETs with Long COVID-19
It was reported that neutrophils undergo a form of cell death distinct from apoptosis or necrosis termed neutrophil extracellular traps (NETs) formation [57]. Neutrophil extracellular traps (NETs), composed of DNA, histones, and antimicrobial protein such as myeloperoxidase (MPO), neutrophil elastase, cathelicidin, calprotectin, are released by neutrophils in response to pathogens [58]. NETs are important mediators of tissue damage in inflammatory diseases. SARS-CoV-2 virus induces the formation of NETs, which is dependent on virus binding to ACE2 on neutrophil [59]. Neutrophil elastase included in NETs can cleave S protein, resulting in an easier SARS-CoV-2 entry into the cell through ACE2, potentially increasing virus infectivity and its ability to stimulate immune response [60]. Neutrophils and NETs are implicated in thrombosis formation during severe SARS-CoV-2 infection [61]. The production of NETs is increased in COVID-19 and their concentration is associated with severity of the disease and thrombosis [62]. During COVID-19, SARS–CoV-2 triggers complement activation [63]. Subsequently, C3a might activate platelets, while C5a and platelets-derived thrombin induce both neutrophil tissue factor (TF) expression and NETs carrying active TF [63]. These thrombogenic NETs may induce endothelial cell activation toward TF expression, thus increasing their procoagulant activity. Increased NETs formation during SARS-CoV-2 infection has been linked to ischemic stroke [64].
NETs formation levels strongly correlated with illness severity/duration, platelet activation markers, and coagulation factors were significantly reduced upon dexamethasone treatment and recovery [65]. Patients with long COVID maintained higher NETs formation compared to recovered convalescent patients [65]. The persistence of NETs is associated with pulmonary fibrosis, cardiovascular abnormalities, and neurological dysfunction in long COVID-19 [66].
4.4.2. The Association of NETs with ASCVD
NETs have been found to be present in atherosclerotic lesions [67]. NETs can promote endothelial dysfunction and vascular inflammation contributing to the initiation and progression of atherosclerotic plaques both by activation and damage of endothelial cells via type I interferon response [68], and through recruitment of other immune cells, mainly macrophages [69]. MPO accumulating in NETs drives further ROS release and the modification of low-density lipoprotein (LDL) to oxidized LDL, thus promoting the development of foam cells [70].
4.5. Selectins
4.5.1. The Association of Selectins with Long COVID-19
Compared to controls, long COVID-19 patients had increased levels of L-selectin and P-selectin [24]. Another study showed that P-selectin levels were elevated in long COVID-19 patients [71]. L-selectin binds multiple ligands expressed on endothelial cells, while P-selectin interacts exclusively with P-selectin glycoprotein ligand-1 (PSGL-1) on leukocytes [72]. L-selectin binding to PSGL-1 expressed by leukocytes may mediate neutrophil rolling on stationary leukocytes bound to cytokine-induced endothelial cells [72]
4.5.2. The Association of Selectins with ASCVD
The meta-analysis showed that E-Selectin gene polymorphism was associated with an increased risk of ischemic stroke [73]. Another meta-analysis suggested there is an increase in the risk of CAD conferred by the Ser128Arg polymorphism of selectin gene (OR, 1.33; 95% CI, 1.04 to 1.69; p = 0.02) and the thr715Pro polymorphism may be a protective factor of myocardial infarction (OR, 0.81; p = 0.04) [74]. Selectin gene polymorphisms (A561C, G98T) were also reported to be significantly associated with increased risk of CAD [75].
5. The Association of Long COVID-19 with Atherosclerotic Risk Factors
5.1. Diabetes
In the systematic review and meta-analysis including 10 articles involving 11 retrospective cohorts with a total of 47.1 million participants showed a 64 % greater risk (RR, 1.64, 95% CI, 1.51 to 1.79) of diabetes in patients with COVID-19 compared with non-COVID-19 controls. COVID-19 is strongly associated with the risk of incident diabetes, including both type 1 and type 2 diabetes [76]. It remains undetermined whether burden of diabetes newly detected during acute COVID-19 persist in post-acute COVID-19 phase. Pooled analysis of 5787,027 subjects from four observational studies showed 59 % higher risk of developing incident diabetes in post-acute COVID-19 phase versus healthy controls (hazard ratio (HR), 1.59; 95 % CI, 1.40 to 1.81, p < 0.001) [77].
5.2. Hypertension
The risk of new-onset hypertension in COVID-19 survivors within one year from the index infection by a systematic review and meta-analysis of the available data was assessed. Overall, 19,293,346 patients were included in this analysis [78]. Pooled analysis revealed that recovered COVID-19 patients presented an increased risk of new-onset hypertension (HR, 1.70; 95% CI, 1.46 to 1.97; p < 0.0001) within seven months [78].
5.3. Dyslipidemia
In the post-acute phase of the SARS-CoV-2 infection, compared with the non-infected contemporary control group, those in the COVID-19 group had higher risks of incident dyslipidemia, including total cholesterol greater than 200 mg/dL (HR, 1.26; 95% CI, 1.22 to 1.29), triglycerides greater than 150 mg/dL (HR, 1.27; 95% CI, 1.23 to 1.31), LDL cholesterol greater than 130 mg/dL (HR, 1.24; 95% CI, 1.20 to 1.29), and HDL cholesterol lower than 40 mg/dL (HR, 1.20; 95% CI, 1.16 to 1.25) [79]. There was also increased risk of incident lipid-lowering medications use (HR, 1.54; 95% CI,1.48 to 1·61) [79]. Post-acute care for those with COVID-19 should involve attention to dyslipidemia as a potential post-acute sequela of SARS-CoV-2 infection.
6. The Association of Long COVID-19 with Chronic Kidney Disease (CKD) and Cardiovascular Diseases
In CKD patients, systemic vascular endothelial damage is observed from an early stage, which can explain the frequent development of CVD in CKD patients [80,81]. Cardiomyocyte is the main player in cardiac function and the development of heart failure (HF); however, its function is underpinned by non-cardiomyocytes such as vascular endothelial cells [81]. Vascular endothelial cells are important cells for maintaining blood perfusion to myocardial cells. Endothelial dysfunction is a very early event in atherosclerosis.
Therefore, endothelial dysfunction is a crucial determinant for the development and progression of CKD, HF and ASCVD such as CAD and ischemic stroke [81]. Considering that vascular endothelial dysfunction is the underlying pathological condition of long COVID-19, it is thought that long COVID-19 has a negative impact on CKD, ischemic stroke and ASCVD.
6.1. CKD
Severe COVID-19 is often complicated by acute kidney injury (AKI), which may transition to CKD. SARS-CoV-2-induced endothelial injury initiates platelet activation, platelet-neutrophil interaction, inducing apoptosis of renal tubular cells, and enhancing renal fibrosis, and 15-30% have protracted renal injury, raising the specter of transition from AKI to CKD [82]. The changes of renal function were longitudinally investigated in patients with Omicron COVID-19 for 6 months [83]. Compared with renal function in the hospital, serum creatinine levels at 6 months increased remarkably; meanwhile, eGFR decreased significantly in all patients [83]. The tendency of CKD is one of the manifestations of long COVID-19 and deserves attention. The effect of long COVID-19 on kidney function was investigated among patients followed in post-COVID-19 recovery clinics in British Columbia, Canada [84]. There was an estimated 2.96 mL/min/1.73 m2 decrease in eGFR within 1 year after COVID-19 that was equivalent to 3.39% reduction from the baseline [84]. More than 40% of patients were at risk of CKD. People with long COVID-19 experienced a substantial decline in eGFR within 1 year from the infection date. The prevalence of proteinuria appeared to be high. The retrospective, multi-database cohort study of patients with COVID-19 from the Hong Kong Hospital Authority the UK Biobank databases showed that patients with COVID-19 incurred greater risk of end-stage renal disease (HR, 1.76; 95% CI, 1.31 to 2.38) and AKI (HR, 2.14; 95% CI, 1.69 to 2.71) [85].
6.2. HF
From the onset of the pandemic, evidence of cardiac involvement in acute COVID-19 abounded. Cardiac presentations ranged from arrhythmias to ischemia, myopericarditis/myocarditis, ventricular dysfunction to acute HF, and cardiogenic shock. SARS-CoV-2 was reportedly detected in endothelial cells and cardiac myocytes. SARS-CoV-2 myocarditis and SARS-CoV-2 infection-induced endothelial injury may lead to cardiac pathophysiology of long COVID-19 [86]. Hospitalized patients recovering from COVID-19 were characterized by a high prevalence of left ventricular concentric remodeling, predominantly Grade I diastolic dysfunction, and a mild decrease in the longitudinal systolic function. These changes were largely persisted throughout the 1-month follow-up [87]. The Hong Kong Hospital Authority and the UK Biobank databases showed that patients with COVID-19 incurred greater risk of HF (HR 1.82; 95% CI 1.65, 2.01) during their post-acute phase of infection [85]. The meta-analysis showed that recovered COVID-19 patients showed an increased risk of incident HF (HR, 1.90; 95% CI, 1.54 to 3.24, p < 0.0001) [88]. COVID-19 survivors had an additional 90% risk of developing HF after COVID-19 in the early post-acute phase of COVID-19. The Mendelian randomization study which investigated a causal association between the genetic predisposition to COVID-19 and long COVID-19 revealed that a genetic predisposition to COVID-19 was significantly causally linked to an increased risk of developing HF [89].
6.3. CAD
The Hong Kong Hospital Authority the UK Biobank databases showed that patients with COVID-19 incurred greater risk of CAD (HR, 1.32; 95% CI, 1.07 to 1.63) and cardiovascular mortality (HR, 2.86; 95% CI, 1.25 to 6.51) during their post-acute phase of infection [85]. Over a mean follow-up of 8.5 months, among COVID-19 recovered patients acute myocardial infarction occurred in 3.5 cases per 1.000 individuals compared to 2.02 cases per 1.000 individuals in the control cohort. COVID-19 patients showed an increased risk of incident acute myocardial infarction (HR, 1.93; 95% CI, 1.65 to 2.26, p < 0.0001) [90].
6.4. Ischemic Stroke
In a meta-analysis to assess the risk of ischemic stroke within 1 year after the post-acute phase of COVID-19, over a mean follow-up of 9.2 months, recovered COVID-19 patients presented a higher risk of ischemic stroke (HR, 2.06; 95% CI, 1.75 to 2.41; p < 0.0001) compared to people who did not have COVID-19 [91].
7. Possible Therapeutic Interventions for Long COVID-19 Considering Endothelial Dysfunction as the Therapeutic Target
Endothelial nitric oxide (NO) synthase (eNOS) produces NO in endothelial cells, and eNOS is closely associated with the regulation of anti-atherogenic processes such as vasorelaxation, an inhibition of the adhesion between leukocytes and endothelial cells, the suppression of migration and proliferation of vascular smooth muscle cells, and inhibition of platelet aggregation [92,93,94]. Endothelial dysfunction reduces NO bioavailability and increases oxidative stress [81]. Therefore, NO and anti-oxidants can be possible therapeutic options for long COVID-19.
In addition to hypolipidemic effects, statins exert pleiotropic effects without a clear relationship LDL-C levels [95]. The pleiotropic properties of statins, such as an improvement of endothelial function and increase in the bioavailability of NO [96], anti-inflammatory [97], immunomodulatory and anti-oxidant effects [98], which may contribute to an improvement of long COVID-19. A recent meta-analysis showed that n-3 polyunsaturated fatty acids (PUFAs) supplementation has been reported to improve inflammation and endothelial function as estimated by FMD [99]. Statins and n-3 PUFAs can be also possible candidates for the treatment of long COVID-19.
Beyond lowering plasma glucose levels, sodium-glucose cotransporter 2 inhibitors (SGLT2is) significantly reduce hospitalization for HF and retard the progression of CKD [100,101,102,103,104,105,106]. SGLT2is have been shown to improve endothelial dysfunction, as assessed by FMD, in individuals at high risk of ASCVD [107,108,109]. The suppression of the development of HF and progression of CKD achieved by SGLT2is might have been largely induced by their capacity to improve vascular endothelial function [81]. SGLT2is may contribute to treat long COVID-19.
7.1. NO
The safety and efficacy of the combined inhalation of NO and molecular hydrogen (H2) in long COVID-19 patients with respiratory manifestations was evaluated [110]. By the combined inhalation of NO and H2, the symptoms severity, such as dyspnea, cough, fatigue and palpitations significantly decreased (p < 0.005), and other makers for quality of life improved. The distance walked (p = 0.01) and the saturation of percutaneous oxygen (p = 0.04) in 6-minute walk test, the volumetric blood flow velocity in venules (p < 0.001), and the oxidative damage (p < 0.001) and anti-oxidant activity (p = 0.03) were all improved by the combined inhalation of NO and H2.
7.2. Anti-Oxidants
7.2.1. Hydrogen-Rich Water
Hydrogen may be a potential therapeutic agent for managing long COVID-19 due to its antioxidant and anti-inflammatory properties. In the randomized, single-blind, placebo-controlled study (RCT), hydrogen-rich water was effective to alleviate fatigue and improve cardiorespiratory endurance, musculoskeletal function, and sleep quality [111].
7.2.2. L-arginine with Vitamin C
L-arginine is an amino acid that acts as a substrate for eNOS [112], and it has been previously shown to significantly improve endothelial function in COVID-19 patients [113]. Additionally, beneficial effects of l-arginine on the regulation of immune responses have been reported [114]. Vitamin C potentiates NO synthesis in cultured human endothelial cells by enhancing eNOS enzymatic activity [115,116,117,118].
After 28 days of l-arginine plus vitamin C supplementation, serum l-arginine concentrations increased significantly compared with placebo. This supplement may therefore be proposed as a remedy to increase NO bioavailability in people with long COVID-19 [119].
In the LINCOLN (L-Arginine and Vitamin C improves Long-COVID) survey, patients were divided into two groups, with a 2:1 ratio: the first group included patients that received l-arginine + vitamin C, whereas the second group received a multivitamin combination (alternative treatment) [120]. The survey included effort perception, measured using the Borg scale. The l-arginine + vitamin C treatment arm had significantly lower scores compared to patients who had received the multivitamin combination. When examining effort perception, a significantly lower value (p < 0.0001) was observed in patients receiving l-arginine + vitamin C compared to the alternative-treatment arm.
In another RCT, participants were randomized 1:1 to receive twice-daily orally either a combination of 1.66 g l-arginine plus 500 mg liposomal vitamin C or a placebo for 28 days [121]. At 28 days, l-arginine plus vitamin C increased the 6-minute walk distance (median (interquartile range) +30 (40.5) m; placebo: +0 (75) m, p = 0.001) and induced a greater improvement in handgrip strength (+3.4 (7.5) kg) compared with the placebo (+1 (6.6) kg, p = 0.03). The FMD was greater in the active group than in the placebo (14.3% (7.3) vs. 9.4% (5.8), p = 0.03). At 28 days, fatigue was reported by two participants in the active group (8.7%) and 21 in the placebo group (80.1%; p < 0.0001). L-arginine plus vitamin C supplementation improved walking performance, muscle strength, endothelial function, and fatigue in adults with long COVID-19.
7.2.3. Coenzyme Q10 and Alpha Lipoic Acid
Implementations of mitochondrial nutrients with diet are important for the clinical anti-oxidant effects. An association of supplementation of coenzyme Q10 and alpha lipoic acid with long COVID-19 symptoms was examined [122]. A Fatigue Severity Scale (FSS) complete response was reached in 53.5% of patients in treatment group and in 3.5% of patients in control group. A reduction in FSS core < 20% from baseline (non-response) was observed in 9.5% of patients in the treatment group and in 25.9% of patients in the control group (p < 0.0001).
7.3. Statins
The prospective cohort study was performed to explore the effect of statins on long-term respiratory symptoms and pulmonary fibrosis in COVID-19 patients with diabetes [123]. Non-statin patients with > 5 years of diabetes were more likely to exhibit a significantly higher pulmonary fibrosis score during the follow-up period as compared to the Statin group. The use of statins was associated with a lower risk of developing chronic cough and dyspnea in diabetic patients with COVID-19, and may reduce pulmonary fibrosis associated with COVID-19 in patients with long-term (> 5 years) diabetes. Another study showed that a 6-week treatment with statins plus angiotensin II type 1 receptor blockers improved clinical symptoms in patients with long COVID-19 [124].
7.4. N-3 PUFAs
Two relatively large RCTs are currently underway to test the hypothesis that treatment of severe forms of COVID-19 with n-3 PUFAs is beneficial [125]. However, clinical interventional studies investigating the role of n-3 PUFAs in individuals with long COVID-19 are currently not available. Nonetheless, due to their generally favorable profiles from various standpoints (psychiatric, cardiovascular benefits), n-3 PUFAs can be considered a potential health supplement to help maintain physical and mental health in patients with long COVID-19.
7.5. SGLT2is
In patients with COVID-19, proinflammatory cytokines induced a redox-sensitive upregulation of SGLT2 expression in endothelial cells, which in turn promoted endothelial injury, senescence, platelet adhesion, aggregation, and thrombin generation [126]. SGLT2 inhibition appeared as an attractive strategy to restore vascular homeostasis in COVID-19.
Furthermore, SGLT2is suppress progression of CKD and the development of HF and ASCVD, which is beneficial to prevent future pandemic of such diseases induced by long COVID-19.
8. Conclusion
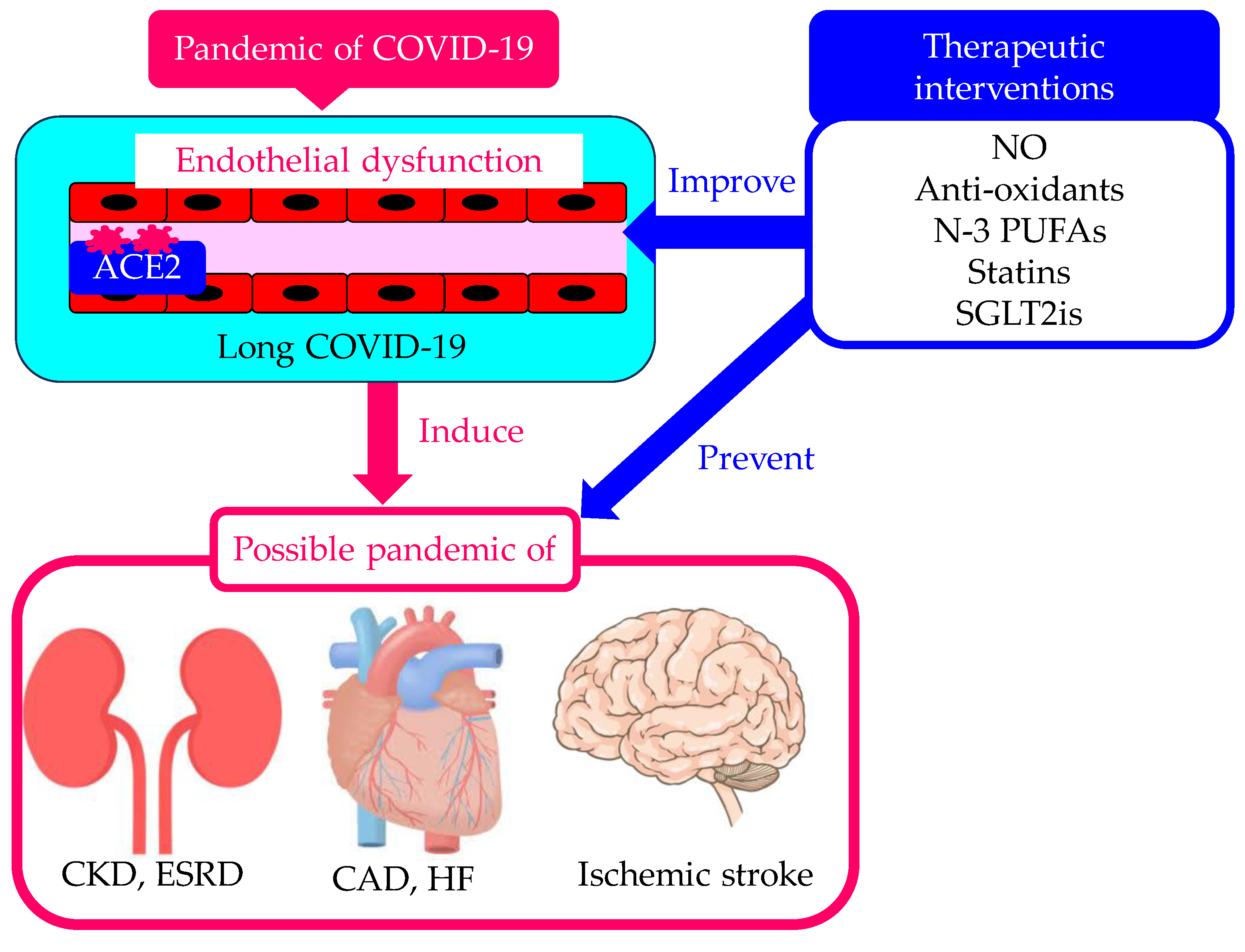
The pandemic of COVID-19 created a great number of long COVID-19 patients. Endothelial dysfunction in long COVID-19 can induce pandemic of CKD, CAD, HF and ischemic stroke (Figure 3). Therapeutic interventions for long COVID-19 considering endothelial dysfunction as the therapeutic target are needed to prevent pandemics of such diseases, which is the worst-case scenario.
Author Contributions
H.Y., M.H., H.A., H.K., and A.S. conceived the review; H. Y. wrote the paper; A.S. edited the paper and provided critical guidance. All authors read and approved the final version of this paper.
Funding
This review research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest in relation to the present review paper.
References
- Lancet The. Facing up to long COVID. Lancet. 2020, 396, 1861. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler G, Wu, N-H. ; Nitsche, A.; Müller, M.A.; Drosten, C.; Pöhlmann, S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Wadman, M.; Couzin-Frankel, J.; Kaiser, J.; Matacic, C. A rampage through the body. Science. 2020, 368, 356–360. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Morrone, M.C.; Patrono, C.; Santoro, M.G.; Schiaffino, S.; Remuzzi, G.; Bussolati, G. Long Covid: where we stand and challenges ahead. Cell. Death. Differ. 2022, 29, 1891–1900. [Google Scholar] [CrossRef]
- Yelin, D.; Wirtheim, E.; Vetter, P.; Kalil, A.C.; Bruchfeld, J.; Runold, M.; Guaraldi, G.; Mussini, C.; Gudiol, C.; Pujol, M.; et al. Long-term consequences of COVID-19: research needs. Lancet. Infect. Dis. 2020, 20, 1115–1117. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: What do we know about “long covid”? BMJ. 2020, 370, m2815. [Google Scholar] [CrossRef]
- Su, S.; Cui, H.; Wang, T.; Shen, X.; Ma, C. Pain: A potential new label of COVID-19. Brain. Behav. Immun. 2020, 87, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Hoshijima, H.; Mihara, T.; Seki, H.; Hyuga, S.; Kuratani, N.; Shiga, T. Incidence of long-term post-acute sequelae of SARS-CoV-2 infection related to pain and other symptoms: A systematic review and meta-analysis. PLoS. One. 2023, 18, e0250909. [Google Scholar] [CrossRef]
- Yanai, H. A Significance of High Prevalence of Diabetes and Hypertension in Severe COVID-19 Patients. J. Clin. Med. Res. 2020, 12, 389–392. [Google Scholar] [CrossRef]
- Busetto, L.; Bettini, S.; Fabris, R.; Serra, R.; Dal Pra, C.; Maffei, P.; Rossato, M.; Fioretto, P.; Vettor, R. Obesity and COVID-19: An Italian Snapshot. Obesity 2020, 28, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Higham, A.; Singh, D. Increased ACE2 Expression in Bronchial Epithelium of COPD Patients who are Overweight. Obesity 2020, 28, 1586–1589. [Google Scholar] [CrossRef] [PubMed]
- Yudkin, J.S. Abnormalities of coagulation and fibrinolysis in insulin resistance. Evidence for a common antecedent? Diabetes. Care. 1999, 22 (Suppl 3), C25–30. [Google Scholar] [PubMed]
- Yanai, H. Adiposity is the Crucial Enhancer of COVID-19. Cardiol. Res. 2020, 11, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020, 395, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in intensive care unit: A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1742. [Google Scholar] [CrossRef]
- Yanai, H. Metabolic Syndrome and COVID-19. Cardiol. Res. 2020, 11, 360–365. [Google Scholar] [CrossRef]
- Moutchia, J.; Pokharel, P.; Kerri, A.; McGaw, K.; Uchai, S.; Nji, M.; Goodman, M. Clinical laboratory parameters associated with severe or critical novel coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. PLoS. One. 2020, 15, e0239802. [Google Scholar]
- Georgieva, E.; Ananiev, J.; Yovchev, Y.; Arabadzhiev, G.; Abrashev, H.; Abrasheva, D.; Atanasov, V.; Kostandieva, R.; Mitev, M.; Petkova-Parlapanska, K.; et al. COVID-19 Complications: Oxidative Stress, Inflammation, and Mitochondrial and Endothelial Dysfunction. Int. J. Mol. Sci. 2023, 24, 14876. [Google Scholar] [CrossRef]
- Charfeddine, S.; Ibn Hadj Amor, H.; Jdidi, J.; Torjmen, S.; Kraiem, S.; Hammami, R.; Bahloul, A.; Kallel, N.; Moussa, N.; Touil, I.; et al. Long COVID 19 Syndrome: Is It Related to Microcirculation and Endothelial Dysfunction? Insights From TUN-EndCOV Study. Front. Cardiovasc. Med. 2021, 8, 745758. [Google Scholar]
- Oikonomou, E.; Souvaliotis, N.; Lampsas, S.; Siasos, G.; Poulakou, G.; Theofilis, P.; Papaioannou, T.G.; Haidich, A.B.; Tsaousi, G.; Ntousopoulos, V.; et al. Endothelial dysfunction in acute and long standing COVID-19: A prospective cohort study. Vascul Pharmacol. 2022, 144, 106975. [Google Scholar] [PubMed]
- Nicolai, L.; Kaiser, R.; Stark, K. Thromboinflammation in long COVID-the elusive key to postinfection sequelae? J. Thromb. Haemost. 2023, 21, 2020–2031. [Google Scholar]
- Fogarty, H.; Townsend, L.; Morrin, H.; Ahmad, A.; Comerford, C.; Karampini, E.; Englert, H.; Byrne, M.; Bergin, C.; O’Sullivan, J.M.; et al. Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J. Thromb. Haemost. 2021, 19, 2546–2553. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Liberale, L.; Portincasa, P.; Khalil, M.; Galerati, I.; Farella, I.; Noto, A.; JohnBritto, S.; Moriero, M.; Michelauz, C.; et al. Neutrophil degranulation, endothelial and metabolic dysfunction in unvaccinated long COVID patients. Eur. J. Clin. Invest. 2024, 54, e14155. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Sanduzzi Zamparelli, S.; Mosella, M.; Formisano, R.; Molino, A.; Spedicato, G.A.; Papa, A.; Motta, A.; Di Minno, M.N.D.; Maniscalco, M. Clinical assessment of endothelial function in convalescent COVID-19 patients: a meta-analysis with meta-regressions. Ann. Med. 2022, 54, 3234–3249. [Google Scholar]
- Martins-Gonçalves, R.; Hottz, E.D.; Bozza, P.T. Acute to post-acute COVID-19 thromboinflammation persistence: Mechanisms and potential consequences. Curr. Res. Immunol. 2023, 4, 100058. [Google Scholar] [CrossRef]
- Youn, J.Y.; Zhang, Y.; Wu, Y.; Cannesson, M.; Cai, H. Therapeutic application of estrogen for COVID-19: Attenuation of SARS-CoV-2 spike protein and IL-6 stimulated, ACE2-dependent NOX2 activation, ROS production and MCP-1 upregulation in endothelial cells. Redox. Biol. 2021, 46, 102099. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.X.; Agbana, Y.L.; Sun, Z.S.; Fei, S.W.; Zhao, H.Q.; Zhou, X.N.; Chen, J.H.; Kassegne, K. Increased interleukin-6 is associated with long COVID-19: a systematic review and meta-analysis. Infect. Dis. Poverty. 2023, 12, 43. [Google Scholar] [PubMed]
- Alfadda, A.A.; Rafiullah, M.; Alkhowaiter, M.; Alotaibi, N.; Alzahrani, M.; Binkhamis, K.; Siddiqui, K.; Youssef, A.; Altalhi, H.; Almaghlouth, I.; et al. Clinical and biochemical characteristics of people experiencing post-coronavirus disease 2019-related symptoms: A prospective follow-up investigation. Front. Med (Lausanne). 2022, 9, 1067082. [Google Scholar]
- Cai, H.; Griendlingm, K.K.; Harrison, D.G. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends. Pharmacol. Sci. 2003, 24, 471–478. [Google Scholar]
- Zhang, B.; Li, X.L.; Zhao, C.R.; Pan, C.L.; Zhang, Z. Interleukin-6 as a Predictor of the Risk of Cardiovascular Disease: A Meta-Analysis of Prospective Epidemiological Studies. Immunol. Invest. 2018, 47, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Kakkar, V.; Lu, X. Impact of MCP-1 in atherosclerosis. Curr. Pharm. Des. 2014, 20, 4580–4588. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; Gill, D.; Rannikmäe, K.; Traylor, M.; Anderson, C.D.; Lee, J.M.; Kamatani, Y.; Hopewell, J.C.; Worrall, B.B.; Bernhagen, J.; et al. Genetically Determined Levels of Circulating Cytokines and Risk of Stroke. Impact of MCP-1 in atherosclerosis. Circulation 2019, 139, 256–268. [Google Scholar] [CrossRef]
- Ward, S.E.; Curley, G.F.; Lavin, M.; Fogarty, H.; Karampini, E.; McEvoy, N.L.; Clarke, J.; Boylan, M.; Alalqam, R.; Worrall, A.P.; et al. Von Willebrand factor propeptide in severe coronavirus disease 2019 (COVID-19): evidence of acute and sustained endothelial cell activation. Br. J. Haematol. 2021, 192, 714–719. [Google Scholar] [CrossRef]
- Wibowo, A.; Pranata, R.; Lim, M.A.; Akbara, M.R.; Martha, J.W. Endotheliopathy marked by high von Willebrand factor (vWF) antigen in COVID-19 is associated with poor outcome: a systematic review and meta-analysis. Int. J. Infect. Dis. 2022, 117, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Terraube, V.; O’Donnell, J.S.; Jenkins, P.V. Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia. 2010, 16, 3–13. [Google Scholar] [CrossRef]
- Zheng, X.L. ADAMTS13 and von willebrand factor in thrombotic thrombocytopenic purpura. Annu. Rev. Med. 2015, 66, 211–225. [Google Scholar] [CrossRef]
- Prasannan, N.; Heightman, M.; Hillman, T.; Wall, E.; Bell, R.; Kessler, A.; Neave, L.; Doyle, A.; Devaraj, A.; Singh, D.; et al. Impaired exercise capacity in post-COVID-19 syndrome: the role of VWF-ADAMTS13 axis. Blood. Adv. 2022, 6, 4041–4048. [Google Scholar] [CrossRef]
- Mancini, I.; Baronciani, L.; Artoni, A.; Colpani, P.; Biganzoli, M.; Cozzi, G.; Novembrino, C.; Boscolo Anzoletti, M.; De Zan, V.; Pagliari, M.T.; et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J. Thromb. Haemost. 2021, 19, 513–521. [Google Scholar] [CrossRef]
- Xu, X.; Feng, Y.; Jia, Y.; Zhang, X.; Li, L.; Bai, X.; Jiao, L. Prognostic value of von willebrand factor and ADAMTS13 in patients with COVID-19: a systematic review and meta-analysis. Thromb. Res. 2022, 218, 83–98. [Google Scholar] [CrossRef]
- Mei, Z.W.; van Wijk Xander, M.R.; Pham, H.P.; Marin, M.J. Role of von willebrand factor in COVID-19 associated coagulopathy. J. Appl. Lab. Med. 2021, 6, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wang, X.; Fan, M.; Zhao, J.; Lin, L.; Liu, J. Plasma levels of von Willebrand factor in type 2 diabetes patients with and without cardiovascular diseases: A meta-analysis. Diabetes. Metab. Res. Rev. 2020, 36, e3193. [Google Scholar] [CrossRef] [PubMed]
- Whincup, P.H.; Danesh, J.; Walker, M.; Lennon, L.; Thomson, A.; Appleby, P.; Rumley, A.; Lowe, G.D. von Willebrand factor and coronary heart disease: prospective study and meta-analysis. Eur. Heart. J. 2002, 23, 1764–1770. [Google Scholar] [CrossRef] [PubMed]
- Sabater-Lleal, M.; Huffman, J.E.; de Vries, P.S.; Marten, J.; Mastrangelo, M.A.; Song, C.; Pankratz, N.; Ward-Caviness, C.K.; Yanek, L.R.; Trompet, S.; et al. Genome-Wide Association Transethnic Meta-Analyses Identifies Novel Associations Regulating Coagulation Factor VIII and von Willebrand Factor Plasma Levels. Circulation. 2019, 139, 620–635. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, M.A.; de Maat, M.P.; Leebeek, F.W. von Willebrand factor and ADAMTS13 in arterial thrombosis: a systematic review and meta-analysis. Blood. Rev. 2014, 28, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Maino, A.; Siegerink, B.; Lotta, L.A.; Crawley, J.T.; le Cessie, S.; Leebeek, F.W.; Lane, D.A.; Lowe, G.D.; Peyvandi, F.; Rosendaal, F.R. Plasma ADAMTS-13 levels and the risk of myocardial infarction: an individual patient data meta-analysis. J. Thromb. Haemost. 2015, 13, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Medcalf, R.L.; Keragala, C.B. The fibrinolytic system: mysteries and opportunities. Hemasphere. 2021, 5, e570. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, K.; Zieger, B. Endothelial cells and coagulation. Cell. Tissue. Res. 2022, 387, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Warnock, M.; Harbaugh, A.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Knight, J.S.; Kanthi, Y.; Lawrence, D.A. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci. Rep. 2021, 11, 1580. [Google Scholar] [CrossRef]
- Nougier, C.; Benoit, R.; Simon, M.; Desmurs-Clavel, H.; Marcotte, G.; Argaud, L.; David, J.S.; Bonnet, A.; Negrier, C.; Dargaud, Y. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J. Thromb. Haemost. 2020, 18, 2215–2219. [Google Scholar] [CrossRef]
- Cabrera-Garcia, D.; Miltiades, A.; Yim, P.; Parsons, S.; Elisman, K.; Mansouri, M.T.; Wagener, G.; Harrison, N.L. Plasma biomarkers associated with survival and thrombosis in hospitalized COVID-19 patients. Int. J. Hematol. 2022, 116, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, E.; Barion, B.G.; da Rocha, T.R.F.; Di Giacomo, G.; Ho, Y.L.; Rothschild, C.; Fatobene, G.; de Carvalho Moraes, B.D.G.; Stefanello, B.; Villaça, P.R.; et al. Persistent hypofibrinolysis in severe COVID-19 associated with elevated fibrinolysis inhibitors activity. J. Thromb. Thrombolysis. 2024, 57, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Jarahzadeh, M.H.; Dastgheib, S.A.; Seifi-Shalamzari, N.; Raee-Ezzabadi, A.; Sadeghizadeh-Yazdi, J.; Akbarian, E.; Neamatzadeh, H. Association of PAI-1 rs1799889 Polymorphism with Susceptibility to Ischemic Stroke: a Huge Meta-Analysis based on 44 Studies. ACTA MEDICA 2020, 63, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulos, G.K.; Bagos, P.G.; Tsangaris, I.; Tsiara, C.G.; Kopterides, P.; Vaiopoulos, A.; Kapsimali, V.; Bonovas, S.; Tsantes, A.E. The association between plasminogen activator inhibitor type 1 (PAI-1) levels, PAI-1 4G/5G polymorphism, and myocardial infarction: a Mendelian randomization meta-analysis. Clin. Chem. Lab. Med. 2014, 52, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y. Plasminogen activator inhibitor-1 4G/5G gene polymorphism and coronary artery disease in the Chinese Han population: a meta-analysis. PLoS. One. 2012, 7, e33511. [Google Scholar] [CrossRef] [PubMed]
- Jung, R.G.; Motazedian, P.; Ramirez, F.D.; Simard, T.; Di Santo, P.; Visintini, S.; Faraz, M.A.; Labinaz, A.; Jung, Y.; Hibbert, B. Association between plasminogen activator inhibitor-1 and cardiovascular events: a systematic review and meta-analysis. Thromb. J. 2018, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science. 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS. Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef] [PubMed]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Watanabe, R.; Matsuyama, S.; Shirato, K.; Maejima, M.; Fukushi, S.; Morikawa, S.; Taguchi, F. Entry from the cell surface of severe acute respiratory syndrome coronavirus with cleaved S protein as revealed by pseudotype virus bearing cleaved S protein. J. Virol. 2008, 82, 11985–11991. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Invest. 2020, 130, 6151–6157. [Google Scholar] [CrossRef] [PubMed]
- Pramitasuri, T.I.; Laksmidewi, A.A.A.P.; Putra, I.B.K.; Dalimartha, F.A. Neutrophil Extracellular Traps in Coronavirus Disease-19-Associated Ischemic Stroke: A Novel Avenue in Neuroscience. Exp. Neurobiol. 2021, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Krinsky, N.; Sizikov, S.; Nissim, S.; Dror, A.; Sas, A.; Prinz, H.; Pri-Or, E.; Perek, S.; Raz-Pasteur, A.; Lejbkowicz, I.; et al. NETosis induction reflects COVID-19 severity and long COVID: insights from a 2-center patient cohort study in Israel. J. Thromb. Haemost. 2023, 21, 2569–2584. [Google Scholar] [CrossRef] [PubMed]
- Shafqat, A.; Omer, M.H.; Albalkhi, I.; Alabdul Razzak, G.; Abdulkader, H.; Abdul Rab, S.; Sabbah, B.N.; Alkattan, K.; Yaqinuddin, A. Neutrophil extracellular traps and long COVID. Front. Immunol. 2023, 14, 1254310. [Google Scholar] [CrossRef] [PubMed]
- Megens, R.T.; Vijayan, S.; Lievens, D.; Doring, Y.; van Zandvoort, M.A.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T. , et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Zernecke, A.; Eriksson, E.E.; Rothfuchs, A.G.; Pham, C.T.; Herwald, H.; Bidzhekov, K.; Rottenberg, M.E.; Weber, C. : Lindbom, L. Neutrophil secretion products pave the way for inflammatory monocytes. Blood. 2008, 112, 1461–1471. [Google Scholar] [CrossRef]
- Tangeten, C.; Zouaoui Boudjeltia, K.; Delporte, C.; Van Antwerpen, P.; Korpak, K. Unexpected Role of MPO-Oxidized LDLs in Atherosclerosis: In between Inflammation and Its Resolution. Antioxidants. 2022, 11, 874. [Google Scholar] [CrossRef]
- Garcia-Larragoiti, N.; Cano-Mendez, A.; Jimenez-Vega, Y.; Trujillo, M.; Guzman-Cancino, P.; Ambriz-Murillo, Y.; Viveros-Sandoval, M.E. Inflammatory and Prothrombotic Biomarkers Contribute to the Persistence of Sequelae in Recovered COVID-19 Patients. Int. J. Mol. Sci. 2023, 24, 17468. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Chen, A.; Delahunty, M.D.; Moore, K.L.; Watson, S.R.; McEver, R.P.; Tedder, T.F. L-selectin binds to P-selectin glycoprotein ligand-1 on leukocytes: interactions between the lectin, epidermal growth factor, and consensus repeat domains of the selectins determine ligand binding specificity. J. Immunol. 1996, 157, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Wang, J.; Liu, K.; Huang, B.; Deng, W.; He, T. Association of E-Selectin gene rs5361 polymorphism with ischemic stroke susceptibility: a systematic review and Meta-analysis. Int. J. Neurosci. 2021, 131, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lou, Y.; Lu, L.; Liu, Y.; Chen, Q.; Chen, X.; Jin, W. Heterogeneous effect of two selectin gene polymorphisms on coronary artery disease risk: a meta-analysis. PLoS. One. 2014, 9, e88152. [Google Scholar] [CrossRef] [PubMed]
- Wang X, Zhang J, Du X, Song M, Jia C, Liu H. Association of A561C and G98T polymorphisms in E-selectin gene with coronary artery disease: a meta-analysis. PLoS ONE 2013, 8, e79301.
- Lai, H.; Yang, M.; Sun, M.; Pan, B.; Wang, Q.; Wang, J.; Tian, J.; Ding, G.; Yang, K.; Song, X.; et al. Risk of incident diabetes after COVID-19 infection: A systematic review and meta-analysis. Metabolism. 2022, 137, 155330. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Pal, R.; Dutta, S. Risk of incident diabetes post-COVID-19: A systematic review and meta-analysis. Prim. Care. Diabetes. 2022, 16, 591–593. [Google Scholar] [CrossRef]
- Zuin, M.; Rigatelli, G.; Bilato, C.; Pasquetto, G.; Mazza, A. Risk of Incident New-Onset Arterial Hypertension After COVID-19 Recovery: A Systematic Review and Meta-analysis. High. Blood. Press. Cardiovasc. Prev. 2023, 30, 227–233. [Google Scholar] [CrossRef]
- Xu, E.; Xie, Y.; Al-Aly, Z. Risks and burdens of incident dyslipidaemia in long COVID: a cohort study. Lancet. Diabetes. Endocrinol. 2023, 11, 120–128. [Google Scholar] [CrossRef]
- Yilmaz, M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; Ozcan, O.; Acikel, C.; et al. The determinants of endothelial dysfunction in CKD: oxidative stress and asymmetric dimethylarginine. Am. J. Kidney. Dis. 2006, 47, 42–50. [Google Scholar] [CrossRef]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Significance of Endothelial Dysfunction Amelioration for Sodium-Glucose Cotransporter 2 Inhibitor-Induced Improvements in Heart Failure and Chronic Kidney Disease in Diabetic Patients. Metabolites. 2023, 13, 736. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.C.; Imig, J.D.; Kalantar-Zadeh, K.; Gupta, A. Kidney in the net of acute and long-haul coronavirus disease 2019: a potential role for lipid mediators in causing renal injury and fibrosis. Curr. Opin. Nephrol. Hypertens. 2022, 31, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.; Song, X.; Zhang, M.; Han, Y.; Chang, G.; Chang, W.; Shen, Z. The pattern of cytokines expression and dynamic changes of renal function at 6 months in patients with Omicron COVID-19. J. Med. Virol. 2023, 95, e28477. [Google Scholar] [CrossRef] [PubMed]
- Atiquzzaman, M.; Thompson, J.R.; Shao, S.; Djurdjev, O.; Bevilacqua, M.; Wong, M.M.Y.; Levin, A.; Birks, P.C. Long-term effect of COVID-19 infection on kidney function among COVID-19 patients followed in post-COVID-19 recovery clinics in British Columbia, Canada. Nephrol. Dial. Transplant. 2023, 38, 2816–2825. [Google Scholar] [CrossRef] [PubMed]
- Lam, I.C.H.; Wong, C.K.H.; Zhang, R.; Chui, C.S.L.; Lai, F.T.T.; Li, X.; Chan, E.W.Y.; Luo, H.; Zhang, Q.; Man, K.K.C.; et al. Long-term post-acute sequelae of COVID-19 infection: a retrospective, multi-database cohort study in Hong Kong and the UK. EClinicalMedicine. 2023, 60, 102000. [Google Scholar] [CrossRef]
- Tsai, E.J.; Cˇiháková, D.; Tucker, N.R. Cell-Specific Mechanisms in the Heart of COVID-19 Patients. Circ. Res. 2023, 132, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Honchar, O.; Ashcheulova, T. Short-term echocardiographic follow-up after hospitalization for COVID-19: a focus on early post-acute changes. Front. Cardiovasc. Med. 2023, 10, 1250656. [Google Scholar] [CrossRef]
- Zuin, M.; Rigatelli, G.; Roncon, L.; Pasquetto, G.; Bilato, C. Risk of incident heart failure after COVID-19 recovery: a systematic review and meta-analysis. Heart. Fail. Rev. 2023, 28, 859–864. [Google Scholar] [CrossRef]
- Shenoy, P.U.; Udupa, H.; Ks, J.; Babu, S. K. N.; Jain, N.; Das, R.; Upadhyai, P. The impact of COVID-19 on pulmonary, neurological, and cardiac outcomes: evidence from a Mendelian randomization study. Front. Public. Health. 2023, 11, 1303183. [Google Scholar] [CrossRef]
- Zuin, M.; Rigatelli, G.; Battisti, V.; Costola, G.; Roncon, L.; Bilato, C. Increased risk of acute myocardial infarction after COVID-19 recovery: A systematic review and meta-analysis. Int. J. Cardiol. 2023, 372, 138–143. [Google Scholar] [CrossRef]
- Zuin, M.; Mazzitelli, M.; Rigatelli, G.; Bilato, C.; Cattelan, A.M. Risk of ischemic stroke in patients recovered from COVID-19 infection: A systematic review and meta-analysis. Eur. Stroke. J. 2023, 8, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ming, X.F. Recent advances in understanding endothelial dysfunction in atherosclerosis. Clin. Med. Res. 2006, 4, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Bauer, V.; Sotnikova, R. Nitric oxide-the endothelium-derived relaxing factor and its role in endothelial functions. Gen. Physiol. Biophys. 2010, 29, 319–340. [Google Scholar] [CrossRef] [PubMed]
- Stankevicius, E.; Kevelaitis, E.; Vainorius, E.; Simonsen, U. Role of nitric oxide and other endothelium-derived factors. Medicina. 2003, 39, 333–341. [Google Scholar]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory Action of Statins in Cardiovascular Disease: The Role of Inflammasome and Toll-Like Receptor Pathways. Clin. Rev. Allergy. Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Bakogiannis, C.; Leeson, P.; Guzik, T.J.; Zhang, M.H.; Tousoulis, D.; Antonopoulos, A.S.; Demosthenous, M.; Marinou, K.; Hale, A.; et al. Rapid, direct effects of statin treatment on arterial redox state and nitric oxide bioavailability in human atherosclerosis via tetrahydrobiopterin-mediated endothelial nitric oxide synthase coupling. Circulation. 2011, 124, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Sola, S.; Mir, M.Q.; Lerakis, S.; Tandon, N.; Khan, B.V. Atorvastatin improves left ventricular systolic function and serum markers of inflammation in nonischemic heart failure. J. Am. Coll. Cardiol. 2006, 47, 332–337. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Jiang, H.; Sun, A.; Wang, Y.; Zou, Y.; Ge, J.; Chen, H. Effects of statin therapy on inflammatory markers in chronic heart failure: A meta-analysis of randomized controlled trials. Arch. Med. Res. 2010, 41, 464–471. [Google Scholar] [CrossRef]
- Ibrahim Mohialdeen Gubari, M. Effect of omega-3 fatty acid supplementation on markers of inflammation and endothelial function in patients with chronic heart disease: A systematic review and meta-analysis. Cell. Mol. Biol. 2024, 70, 171–177. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Yanai, H. Sodium-glucose cotransporter 2 inhibitors and death and heart failure in type 2 diabetes. Ann. Transl. Med. 2017, 5, 470. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Cherney, D.Z.I.; Zinman, B.; Inzucchi, S.E.; Koitka-Weber, A.; Mattheus, M.; von Eynatten, M.; Wanner, C. Effects of empagliflozin on the urinary albumin-to-creatinine ratio in patients with type 2 diabetes and established cardiovascular disease: An exploratory analysis from the EMPA-REG OUTCOME randomised, placebo-controlled trial. Lancet. Diabetes. Endocrinol. 2017, 5, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Ikehara, K.; Kanda, E.; Uchino, H.; Hirose, T. Effectiveness of dapagliflozin on vascular endothelial function and glycemic control in patients with early-stage type 2 diabetes mellitus: DEFENCE study. Cardiovasc. Diabetol. 2017, 16, 84. [Google Scholar] [CrossRef]
- Ramirez, A.J.; Sanchez, M.J.; Sanchez, R.A. Diabetic patients with essential hypertension treated with amlodipine: Blood pressure and arterial stiffness effects of canagliflozin or perindopril. J. Hypertens. 2019, 37, 636–642. [Google Scholar] [CrossRef]
- Sezai, A.; Sekino, H.; Unosawa, S.; Taoka, M.; Osaka, S.; Tanaka, M. Canagliflozin for Japanese patients with chronic heart failure and type II diabetes. Cardiovasc. Diabetol. 2019, 18, 76. [Google Scholar] [CrossRef]
- Pozdnyakova, D.D.; Bakhareva, T.А.; Baranova, I.A.; Selemir, V.D.; Chuchalin, A.G. Rehabilitation program of post-COVID-19 syndrome with the use of nitric oxide and molecular hydrogen. Ter. Arkh. 2024, 96, 260–265. [Google Scholar] [CrossRef]
- Tan, Y.; Xie, Y.; Dong, G.; Yin, M.; Shang, Z.; Zhou, K.; Bao, D.; Zhou, J. The Effect of 14-Day Consumption of Hydrogen-Rich Water Alleviates Fatigue but Does Not Ameliorate Dyspnea in Long-COVID Patients: A Pilot, Single-Blind, and Randomized, Controlled Trial. Nutrients. 2024, 16, 1529. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Meininger, C.J.; McNeal, C.J.; Bazer, F.W.; Rhoads, J.M. Role of l-Arginine in nitric oxide synthesis and health in humans. Adv. Exp. Med. Biol. 2021, 1332, 167–187. [Google Scholar] [PubMed]
- Adebayo, A.; Varzideh, F.; Wilson, S.; Gambardella, J.; Eacobacci, M.; Jankauskas, S.S.; Donkor, K.; Kansakar, U.; Trimarco, V.; Mone, P.; et al. l-Arginine and COVID-19: An Update. Nutrients. 2021, 13, 3951. [Google Scholar] [CrossRef]
- Bronte, V.; Zanovello, P. Regulation of immune responses by l-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef]
- Morelli, M.B.; Gambardella, J.; Castellanos, V.; Trimarco, V.; Santulli, G. Vitamin C and Cardiovascular Disease: An Update. Antioxidants (Basel). 2020, 9, 1227. [Google Scholar] [CrossRef] [PubMed]
- Heller, R.; Munscher-Paulig, F.; Grabner, R.; Till, U. L-Ascorbic acid potentiates nitric oxide synthesis in endothelial cells. J. Biol. Chem. 1999, 274, 8254–8260. [Google Scholar] [CrossRef]
- Huang, A.; Vita, J.A.; Venema, R.C.; Keaney, J.F. Jr. Ascorbic acid enhances endothelial nitric-oxide synthase activity by increasing intracellular tetrahydrobiopterin. J. Biol. Chem. 2000, 275, 17399–17406. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.A.; Milstien, S.; Katusic, Z.S. Effect of vitamin C on the availability of tetrahydrobiopterin in human endothelial cells. J. Cardiovasc. Pharmacol. 2001, 37, 333–338. [Google Scholar] [CrossRef]
- Calvani, R.; Gervasoni, J.; Picca, A.; Ciciarello, F.; Galluzzo, V.; Coelho-Júnior, H.J.; Di Mario, C.; Gremese, E.; Lomuscio, S.; Paglionico, A.M.; et al. Effects of l-Arginine Plus Vitamin C Supplementation on l-Arginine Metabolism in Adults with Long COVID: Secondary Analysis of a Randomized Clinical Trial. Int. J. Mol. Sci. 2023, 24, 5078. [Google Scholar] [CrossRef] [PubMed]
- Izzo, R.; Trimarco, V.; Mone, P.; Aloè, T.; Capra Marzani, M.; Diana, A.; Fazio, G.; Mallardo, M.; Maniscalco, M.; Marazzi, G.; et al. Combining L-Arginine with vitamin C improves long-COVID symptoms: The LINCOLN Survey. Pharmacol. Res. 2022, 183, 106360. [Google Scholar] [CrossRef]
- Tosato, M.; Calvanim, R.; Picca, A.; Ciciarello, F.; Galluzzo, V.; Coelho-Júnior, H.J.; Di Giorgio, A.; Di Mario, C.; Gervasoni, J.; Gremese, E.; Leone, P.M.; et al. Effects of l-Arginine Plus Vitamin C Supplementation on Physical Performance, Endothelial Function, and Persistent Fatigue in Adults with Long COVID: A Single-Blind Randomized Controlled Trial. Nutrients. 2022, 14, 4984. [Google Scholar] [CrossRef]
- Barletta, M.A.; Marino, G.; Spagnolo, B.; Bianchi, F.P.; Falappone, P.C.F.; Spagnolo, L.; Gatti, P. Coenzyme Q10 + alpha lipoic acid for chronic COVID syndrome. Clin. Exp. Med. 2023, 23, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Sadeghdoust, M.; Aligolighasemabadi, F.; Dehesh, T.; Taefehshokr, N.; Sadeghdoust, A.; Kotfis, K.; Hashemiattar, A.; Ravandi, A.; Aligolighasemabadi, N.; Vakili, O.; et al. The Effects of Statins on Respiratory Symptoms and Pulmonary Fibrosis in COVID-19 Patients with Diabetes Mellitus: A Longitudinal Multicenter Study. Arch. Immunol. Ther. Exp (Warsz). 2023, 71, 8. [Google Scholar] [CrossRef] [PubMed]
- Grote, K.; Schaefer, A.C.; Soufi, M.; Ruppert, V.; Linne, U.; Mukund Bhagwat, A.; Szymanski, W.; Graumann, J.; Gercke, Y.; Aldudak, S.; et al. Targeting the High-Density Lipoprotein Proteome for the Treatment of Post-Acute Sequelae of SARS-CoV-2. Int. J. Mol. Sci. 2024, 25, 4522. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.P.; Chang, C.M.; Yang, C.C.; Pariante, C.M.; Su, K.P. Long COVID and long chain fatty acids (LCFAs): Psychoneuroimmunity implication of omega-3 LCFAs in delayed consequences of COVID-19. Brain. Behav. Immun. 2022, 103, 19–27. [Google Scholar] [CrossRef]
- Mroueh, A.; Fakih, W.; Carmona, A.; Trimaille, A.; Matsushita, K.; Marchandot, B.; Qureshi, A.W.; Gong, D.S.; Auger, C.; Sattler, L.; et al. COVID-19 promotes endothelial dysfunction and thrombogenicity: role of proinflammatory cytokines/SGLT2 prooxidant pathway. J. Thromb. Haemost. 2024, 22, 286–299. [Google Scholar] [CrossRef]
Figure 1.
The underlying mechanisms for the development of severe COVID-19 in patients with metabolic syndrome. Up arrows indicate increase in expression of molecules, respectively ACE2, angiotensin-converting enzyme 2; IL-6, interleukin-6; PAI-1, plasminogen activator inhibitor-1; TNF-α, tumor necrosis factor-alpha; VWF, von Willebrand factor.
Figure 1.
The underlying mechanisms for the development of severe COVID-19 in patients with metabolic syndrome. Up arrows indicate increase in expression of molecules, respectively ACE2, angiotensin-converting enzyme 2; IL-6, interleukin-6; PAI-1, plasminogen activator inhibitor-1; TNF-α, tumor necrosis factor-alpha; VWF, von Willebrand factor.
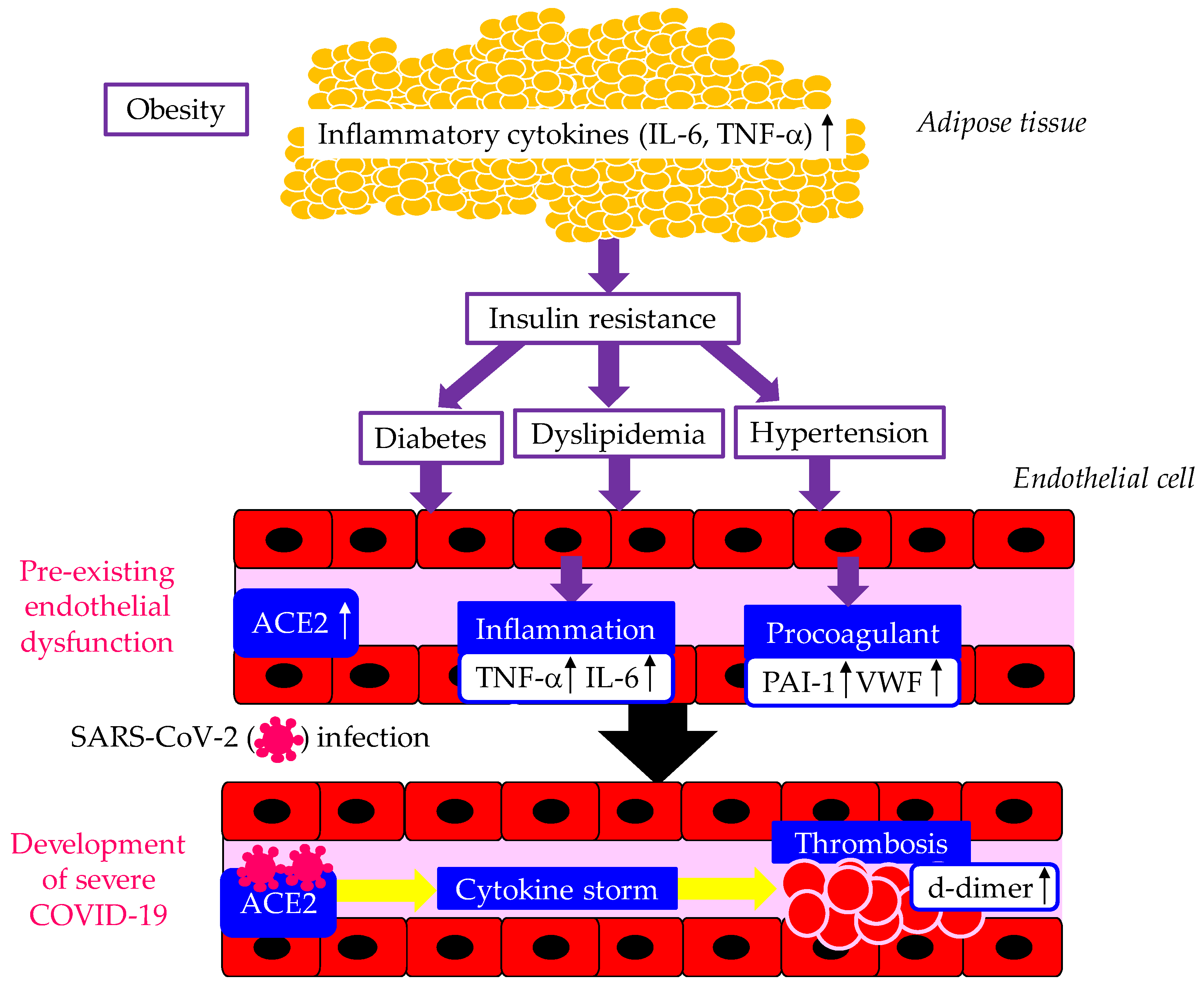
Figure 2.
Various atherogenic factors induced by vascular endothelial dysfunction caused by infection with SARS-CoV-2. Up arrows indicate an increase in degree of atherogenic and thrombogenic factors. ACE2, angiotensin-converting enzyme 2; IL-6, interleukin-6; LDL, low-density lipoprotein; MCP-1, monocyte chemoattractant protein-1; NETs, Neutrophil extracellular traps NOX2, NADPH oxidase 2; PAI-1, plasminogen activator inhibitor-1; ROS, reactive oxygen species; SR, scavenger receptor; TF, tissue factor; tPA, tissue plasminogen activator; VWF, von Willebrand factor.
Figure 2.
Various atherogenic factors induced by vascular endothelial dysfunction caused by infection with SARS-CoV-2. Up arrows indicate an increase in degree of atherogenic and thrombogenic factors. ACE2, angiotensin-converting enzyme 2; IL-6, interleukin-6; LDL, low-density lipoprotein; MCP-1, monocyte chemoattractant protein-1; NETs, Neutrophil extracellular traps NOX2, NADPH oxidase 2; PAI-1, plasminogen activator inhibitor-1; ROS, reactive oxygen species; SR, scavenger receptor; TF, tissue factor; tPA, tissue plasminogen activator; VWF, von Willebrand factor.
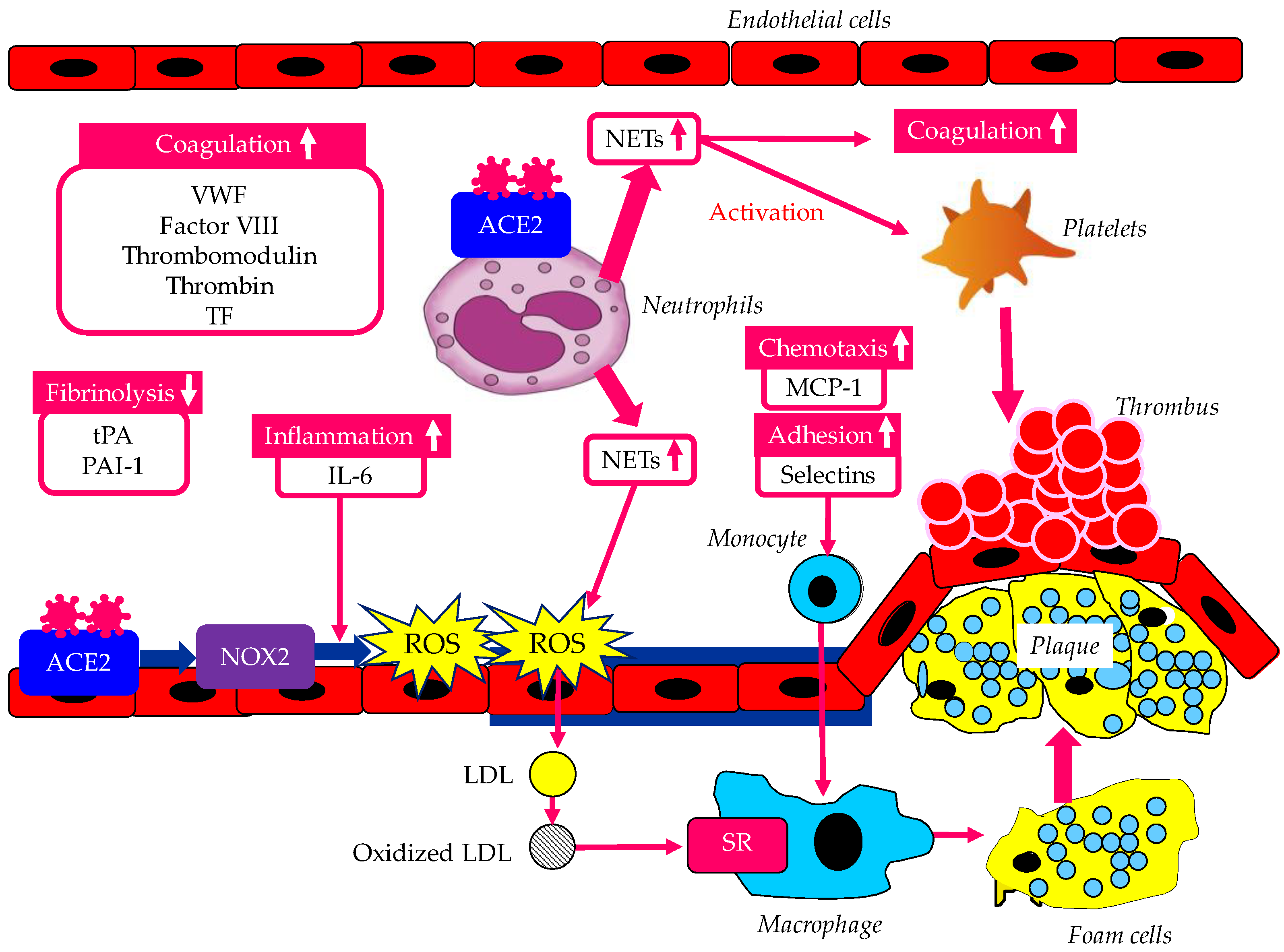
Figure 3.
Possible induction of pandemic of CKD, cardiovascular disease and ischemic stroke due to endothelial dysfunction in long COVID-19 and prevention of such pandemic by therapeutic interventions for long COVID-19 considering endothelial dysfunction as the therapeutic target. ACE2, angiotensin-converting enzyme 2; CAD, coronary artery disease; CKD, chronic kidney disease; ESRD, end stage renal disease; HF, heart failure; NO, nitric oxide; PUFAs, polyunsaturated fatty acids; SGLT2is, sodium-glucose cotransporter 2 inhibitors.
Figure 3.
Possible induction of pandemic of CKD, cardiovascular disease and ischemic stroke due to endothelial dysfunction in long COVID-19 and prevention of such pandemic by therapeutic interventions for long COVID-19 considering endothelial dysfunction as the therapeutic target. ACE2, angiotensin-converting enzyme 2; CAD, coronary artery disease; CKD, chronic kidney disease; ESRD, end stage renal disease; HF, heart failure; NO, nitric oxide; PUFAs, polyunsaturated fatty acids; SGLT2is, sodium-glucose cotransporter 2 inhibitors.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



