Submitted:
21 June 2024
Posted:
24 June 2024
You are already at the latest version
Abstract
Long noncoding RNAs (lncRNAs) are known to play key roles in breast cancers; however, detailed mechanistic studies of lncRNA function have not been conducted in large cohorts of breast cancer tumors, nor have inter-donor and inter-subtype variabilities been taken into consideration. Here we provide the first identification and annotation of the human BORG lncRNA gene. Using multiple tumor cohorts of human breast cancers, we show that while BORG expression is strongly induced in breast tumors as compared to normal breast tissues, the extent of BORG induction varies widely between breast cancer subtypes and even between different tumors within the same subtype. Elevated levels of BORG in breast tumors are associated with the acquisition of core cancer aggression pathways, including those associated with basal tumor and pluripotency phenotypes, and with epithelial-mesenchymal transition (EMT) programs. While a subset of BORG-associated pathways was present in high BORG-expressing tumors across all breast cancer subtypes, many were specific to triple-negative breast cancer tumors. Finally, we show that genes induced by heterologous expression of BORG in murine models of TNBC both in vitro and in vivo strongly overlap with those associated with high BORG levels in human TNBC tumors, indicating that human BORG is a novel driver of the highly aggressive basal TNBC tumor phenotype.
Keywords:
long non-coding RNAs
; lncRNAs
; TNBC
; Cancer stem cells
; basal breast cancer
; breast cancer
Introduction
Despite significant improvements in the early detection and treatment of breast cancers, this disease persists as a global health concern that ranks among the leading causes of mortality in women [1,2]. Breast cancers are highly heterogeneous at both the cellular and molecular levels and are classified clinically by immunohistochemistry and/or in situ hybridization-based detection of estrogen receptor-a (ER-a), progesterone receptor (PR), and HER2/ErbB-2 expression [3,4,5]. Importantly, the presence of these receptors continues to serve as some of the most effective therapeutic targets in all of oncology, particularly for patients harboring breast tumors belonging to the luminal A (ER-a- and PR-positive), luminal B (ER-a and HER2-positive), and HER2 subtypes [6]. In stark contrast, patients harboring triple-negative breast cancers (TNBCs), which lack notable expression of ER-a, PR, and HER2, exhibit the highest disease grades and proliferation indices that culminate in (i) the worst progression-free and overall survival rates of all breast cancer subtypes, and (ii) rapid relapse and early death within 5 years of initial diagnosis and treatment [6,7,8]. Despite the similarities needed to classify breast cancers into subtypes, there remains significant variability amongst individual human breast tumors, even those from the same subtype, which further complicates the choice of therapy and significantly affects patient response to treatment and prognosis.
Transcriptomic profiling of TNBCs using multiple methods such as PAM50 [9] and TNBCtype-4 [10] finds that these tumors predominantly fall into the basal-like category, which are highly metastatic and robustly express genes typically found in basal/myoepithelial cells, such as cytokeratins 14, and 16 [11]. Moreover, TNBCs are also enriched in the expression of transcriptional programs coupled to breast cancer stem cells (e.g., CD44, ITGA6/Integrinα6, ALDH1A1, and CD133/PROM1 [12,13,14,15,16]) and epithelial-mesenchymal transition (EMT) programs [6]. They are also characterized by (i) elevated expression of the receptors for epidermal growth factor (EGFR) and stem cell growth factor (KIT) [17,18,19]; and (ii) mutational inactivation of BRCA and p53, resulting in impaired DNA damage response and high mutational burden [20,21]. Finally, recurrent TNBCs frequently acquire resistance to standard-of-care chemotherapeutic agents (e.g., doxorubicin, cyclophosphamides, and taxanes) through mechanisms that remain incompletely understood. More recently, immunotherapy approaches (e.g., monoclonal antibodies against programmed death-ligand (PD-L1)) have made inroads into the breast oncology space, particularly for the treatment of TNBCs due to their higher mutational burden, higher infiltration of tumor lymphocytes, and higher expression of PD-L1 [7]. Unfortunately, this strategy has failed to significantly improve overall patient survival and outcomes in monotherapy settings [22]. This failure stems from our lack of understanding of the variability and complex nature of the molecular networks that drive the behavior of breast cancer tumors, even those from the same subtype, in different patients. Clearly, a new paradigm in cancer research that takes tumor variability into consideration is urgently needed for identification of new therapeutic strategies that can achieve curative outcomes for TNBC patients.
Long noncoding RNAs (lncRNAs) are a heterogeneous group of cellular RNAs that are transcribed by RNA polymerases I, II or III and thus, may or may not be spliced and polyadenylated [23,24,25]. Although most lncRNAs are less evolutionarily conserved, expressed at lower copy numbers, and less efficiently spliced compared to protein-coding genes [26,27], these noncoding transcripts constitute the dominant output of the human genome in terms of the fraction of the genome comprising their loci [23,24,25]. LncRNAs range from around two hundred to tens of thousands of nucleotides in length, with many containing short open reading frames that may even code for short peptides. Nonetheless, they perform an RNA-mediated cellular function [23,24,25,28]. A large fraction of lncRNAs are predominantly nuclear localized, where they play critical roles in regulation of transcription and epigenetic regulation [24,28,29,30]. Recent studies have pointed to the involvement of this highly understudied class of RNAs in all aspects of tumor development and metastatic progression that transpires in essentially all human tumors [31,32,33,34,35,36]. However, nearly all mechanistic studies of lncRNA function have been performed using animal models or cultured cells employing knockdown and forced overexpression studies, which may not reflect their function in the context of the complex tumor milieu. Importantly, these studies are inherently unable to address the donor-to-donor variability inherent in human cancers.
We previously characterized and described the role of the murine lncRNA BORG (BMP2/OP1 Responsive Gene [37]) in breast cancer. Indeed, we found BORG expression to be aberrantly upregulated in human and murine TNBC cell lines and tumors, particularly those exhibiting aggressive metastatic and chemoresistant behaviors [38]. In the mouse, BORG (annotated as GM45924) is a Pol II-transcribed, nuclear-localized, spliced and polyadenylated intergenic RNA [39]. Mechanistically, we showed that BORG binds to the E3 SUMO ligase TRIM28/KAP1, and that the resulting complex induces latent breast cancer cells to resume proliferative programs coupled to metastatic outgrowth and recurrence [38]. Moreover, BORG:TRIM28 complexes also govern the self-renewal and expansion of breast cancer stem cells, doing so by inducing the expression of Nanog, Aldh1a3, and Itgs6/a6 integrins [40,41]. Finally, we observed BORG to interact physically with RPA1, an event coupled to the acquisition of resistance to doxorubicin via activation of the prosurvival NF-B axis [42]. Despite these intriguing findings, a human gene corresponding to the murine BORG has not been annotated nor identified and thus, the existence of a human ortholog for the mouse BORG gene was uncertain. In addressing this knowledge gap, the overall objectives of the current study were to (i) determine whether a human ortholog for BORG existed, and if successful, (ii) characterize the gene and define its genomic architecture, and (iii) define the function of human BORG in developing and progressing human breast cancers, particularly TNBCs, at molecular level in the context of large numbers of human tumor samples that account for donor-to-donor and tumor subtype variability.
Results
Identification and Architecture of Human BORG Genomic Locus
We identified the human BORG lncRNA through conservation of a small domain located close to the 3' end of mouse BORG, which we have previously characterized [38,39,40,42], additional conservation patterns between vertebrate species, syntenic preservation of its locus, genomic direction of transcription and its 3’ processing region (Figure 1A, Figure S1 and S2). In humans, BORG genic region is located on chromosome 8 (hg38 chr8: 103095156-103119540) and is transcribed in the antisense genomic direction. The human BORG gene is 24.3 kb long, and as observed in other lncRNAs, is poorly spliced. It also contains a conserved 3’ end that lies in close vicinity to a canonical polyadenylation signal (Figure 1A and Figure S1). Its transcription start site overlaps a genomic region with H3K4Me1 and H3K27AC marks, consistent with an active/poised enhancer. Interestingly, this enhancer also gives rise to another lncRNA (LINC01181) through a bidirectional promoter, with the two transcription start sites less than 2000 nucleotides apart (Figure 1A). Finally, we confirmed that human BORG RNA lacks protein coding capacity in a manner analogous to its murine ortholog (GM45924; Figure 1A, phyloCSF tracks).
Human BORG rna Is Strongly Upregulated in a Subset of Human TNBC Tumors
To determine if human BORG RNA plays a role in breast cancer development or progression, we compared its expression pattern in normal versus cancerous breast tissue from human donors (Figure 1B and Figure S3). BORG expression in breast tumors was significantly elevated compared to breast organoids derived from non-cancerous primary breast cells (Figure 1B), especially in a subset of TNBC tumors. To confirm these results in a larger cohort of human patients, we took advantage of the breast cancer dataset of The Cancer Genome Atlas (TCGA BRCA) (Figure S4A). Comparison of the BORG expression levels between distinct breast cancer histological and immunohistochemical subtypes (Figure S4B–E) indicated that BORG expression was indeed dramatically elevated in a subset of human basal TNBC tumors (Figure 1A,C). Interestingly, despite the overall increased expression of BORG in basal TNBC tumors, we observed that within each histological group, there is a range of BORG expression levels (Figure 1C).
As a first step toward understanding the impact of BORG expression in human breast tumors, we compared tumors whose expression of BORG fell within the top and bottom quartiles of all breast cancers. Tumors from all four PAM50 subtypes (luminal A, luminal B, HER2+ and basal/TNBCs) were represented in both groups (Figure S5A). We identified 2180 and 798 protein-coding genes that showed increased and decreased expression, respectively, in high BORG-expressing tumors compared to those with lower BORG expression levels (>1.5-fold change and FDR<0.05, Figure S5B). Pathway analysis using the Hallmark gene lists of the Molecular Signatures Dabatase (mSigDB) pointed to enrichment of pathways related to cellular proliferation and TGFβ signaling in high BORG-expressing tumors (Figure 2A and Figure S6A,B). Analysis of the expression pattern of genes known to be involved in aggressive cancer phenotype indicated that most of these genes were indeed upregulated in high BORG-expressing tumors (Figure 2B and Figure S6C), including several genes associated with the basal phenotypes, EMT programs, pluripotency, and tumor invasion and metastasis [18,20,43,44,45] (Figure S6C). Similarly, genes coding for multiple cytokines and surface protein markers such as CD44, KIT, EGFR and PROM1 that are known to be associated with the basal phenotype, pluripotency and cancer aggression [14,15,20], were also significantly upregulated in the high BORG-expressing tumors (Figure S7A,B). Collectively, these findings pointed to the intriguing possibility that increased levels of BORG may be associated with key carcinogenic pathways in human breast cancers of all subtypes.
Increased Expression of BORG Is Associated with the Induction of Basal Transcriptomic Signatures
To compare the observed differences in high versus low BORG-expressing tumors to the transcriptomic patterns observed in cancers, we performed an enrichment analysis using the C2 gene lists from the mSigDB. Intriguingly, the top positively enriched gene list contained genes upregulated in basal breast cancers, while the most negatively enriched gene list comprises genes downregulated in basal breast cancers (Figure 2C). Multiple additional breast cancer-related gene lists were also included among the top 15 positively- and negatively-enriched gene lists. These results strongly pointed to the association of higher BORG expression levels with the gene expression program found in basal tumors, including enrichment of proliferation-related pathways and a reduction in oxidative phosphorylation (Figure 3A, see also Figure S8B). Interestingly, even after removing the basal tumors from the comparison groups, the pattern of changes in gene expression between the high and low BORG-expressing groups remained largely unchanged (Figure S8A,B and Figure 3A). Likewise, genes found in breast cancer progenitor cells were enriched in high BORG-expressing tumors in every PAM50 tumor subtype (Figure 3B,C,D), confirming our previous contention that BORG plays a significant role in governing the self-renewal and expansion of breast cancer stem cells [40].
Taken together, these findings point to the presence of a shared transcriptomic signature associated with higher expression levels of BORG in all breast tumor subtypes, leading to increased proliferation, altered TGF-β signaling, and induction of progenitor phenotypes. However, it should also be noted that higher BORG expression was also associated with differential enrichment of many critical genes and pathways, such as those resulting in cancer aggression, upon inclusion of the basal/TNBC tumors in comparison groups (Figure 2B and Figure S6C), but not when only the non-basal breast cancer subtypes were analyzed (Figure S8C). Thus, in addition to the effects that BORG elicits in all breast cancer subtypes, aberrant BORG expression may also drive additional aspects of tumor gene expression program specifically in the basal/TNBC breast cancer subtype.
Induction of Invasive Signatures in High BORG-Expressing TNBC Tumors
We next focused on defining the transcriptomic patterns specifically associated with elevated BORG expression within the basal/TNBC subset of breast cancers. Among the top 25 genes differentially expressed between TNBC tumors with the highest and lowest levels of BORG were TCF20, ARID1B and UVSSA, which are involved in neoplastic processes such as pluripotency and DNA repair (Figure S9A). We also identified several differentially expressed genes known to induce tumor invasiveness in breast cancers, the majority of which were upregulated in high BORG-expressing TNBC tumors (Figure S9B). The levels of several mRNAs that code for surface proteins with roles in cancer were similarly upregulated, including ALK, PTPRM, PTPRF, PTPRK and SVEP1 (Figure S9C). Interestingly, pathway analysis using the curated Hallmark genelists indicated a strong induction of the EMT pathways, known to be involved in breast cancer invasiveness and metastasis; they also identified pathways involved in cellular proliferation and the TGFβ signaling (Figure 4A). As with pathway analyses in other breast cancer groups, oxidative phosphorylation was the top downregulated pathway (Figure 4A).
To obtain information on BORG-associated changes in a wider range of pathways and gene lists, we again used the C2 gene list database of mSigDB. Interestingly, multi-cancer invasive signature was the top enriched genelist in high BORG-expressing tumors (Figure S9D,E), and four additional breast cancer-related pathways were among the top 15 enriched gene lists (Figure S9D). Among the subset of C2 pathways related to cancer, enriched pathways pointed to association of high BORG expression levels with high grade cancer phenotypes (Figure 4B,C). We showed that targets of a number of key carcinogenic transcription factors were enriched amongst genes upregulated in high BORG-expressing cells (Figure 4D), including those associated with metastasis and invasive phenotype (FOXD1, EVI1/MECOM, MEF2C), EMT (EVI1/MECOM, OCT1/POU2F1, MEF2A), brain metastasis (MEF2C), poor prognosis (FOXD1), and multi-drug resistance (NKX-2.5) [46,47,48,49,50,51,52,53] (Figure 4D,E,F). Interestingly, our previous work in murine models of breast cancer development and metastatic progression demonstrated robust acquisition of pro-metastatic and drug-resistance phenotypes following BORG overexpression [38,40,42]. Collectively, these findings indicate that higher BORG expression levels in basal/TNBC tumors are associated with pathways involved in induction of highly invasive phenotypes, including the EMT, metastasis and drug-resistance pathways.
Aberrant BORG Expression Leads to Induction of Genes Associated with Aggressive Basal Phenotypes
To determine if increased BORG expression has a causative effect on induction of basal/TNBC phenotypes, we leveraged RNA-seq analyses of the D2.HAN breast cancer model that consists of indolent D2.OR cells that express little-to-no BORG and their aggressive D2.A1 counterparts that endogenously express robust quantities of BORG [38,40,41,42]. Importantly, heterologous expression of murine BORG in D2.OR cells confers aggressive and metastatic phenotypes reminiscent of their D2.A1 counterparts (Figure S10A) [38,42]. RNA-seq analyses of parental and BORG-expressing D2.OR cells identified 387 protein-coding genes that were consistently differentially expressed as a result of aberrant BORG expression. Mapping these differentially expressed genes to pathways and gene lists involved in breast cancer identified multiple positively- and negatively-enriched pathways. The top enriched pathways activated by Borg included those we observed to be enriched in high BORG-expressing human breast cancer tumors, including the basal breast cancer and breast cancer progenitor gene lists (Figure S10B). Taken together, these findings suggest that increased BORG expression plays a causative role in eliciting the formation of aggressive basal breast cancers.
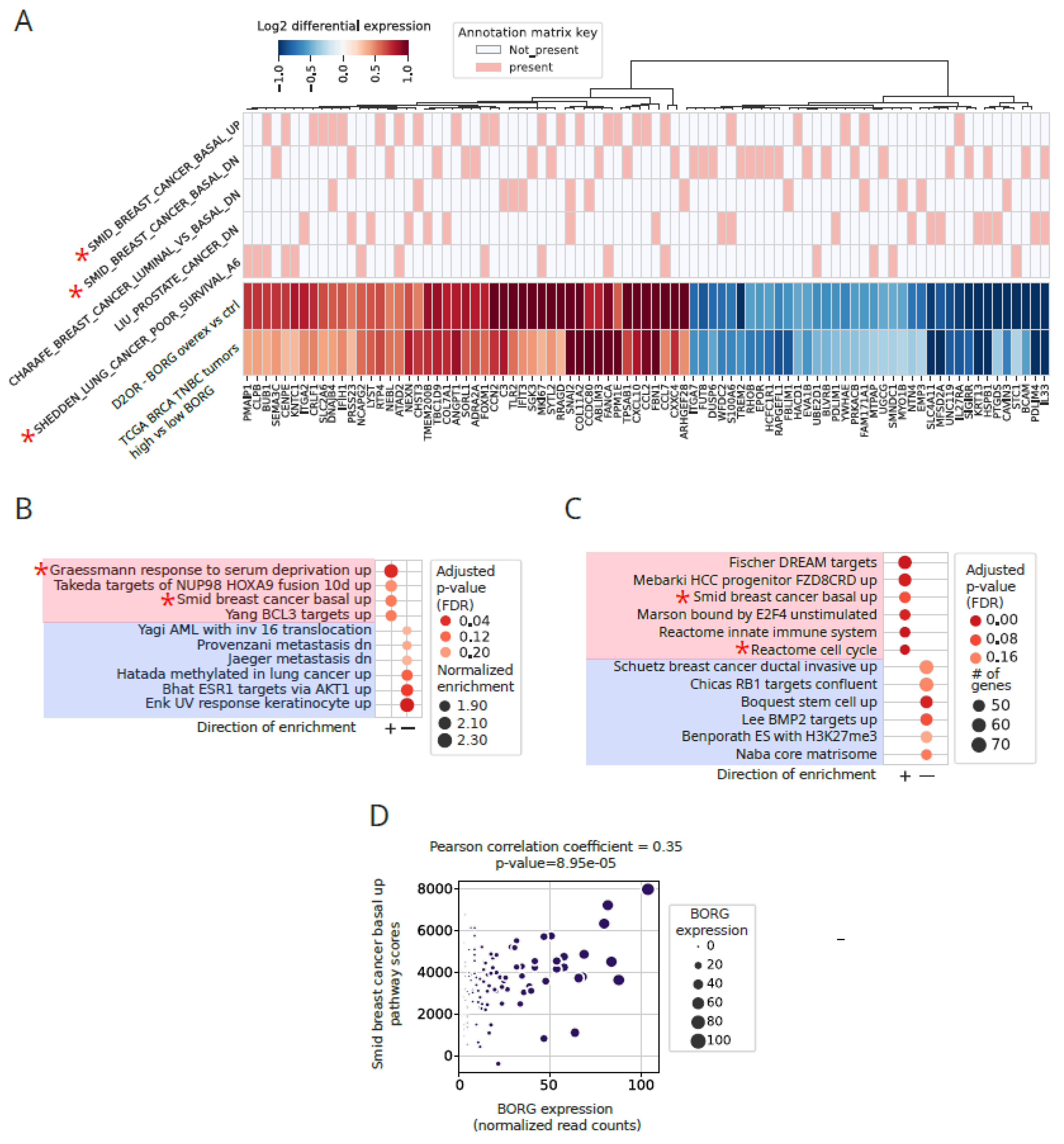
To further investigate this intriguing possibility, we compared the genes that were differentially expressed following heterologous BORG expression in D2.OR cells to those upregulated in high BORG-expressing human breast tumors. Interestingly, over 270 of the 387 genes that were differentially expressed in BORG-overexpressing D2.OR cells were also differentially expressed in the same direction in high BORG-expressing human TNBC tumors as compared to their low BORG-expressing counterparts (Figure 5A). These results suggest that at least part of the gene expression difference between high and low BORG-expressing TNBC tumors is likely the direct result of higher BORG expression levels in these tumors. Interestingly, these BORG-associated genes corresponded to those upregulated in basal breast cancers (e.g., Smid breast cancer basal up genelist) and key cancer-related processes and pathways, such as oncogenesis, metastasis, and stress response (Figure 5A,B and Figure S11A), many of which were also enriched in high BORG-expressing breast tumors (Figure S10B, Figure 2C and Figure 3A). These intriguing results indicate that the BORG-induced gene expression pattern plays a key role in induction of the aggressive, pro-metastatic basal phenotype, which is strongly associated with the TNBC tumors.
Finally, we also studied the pattern of expression of BORG-dependent genes in the CMI/MBC cohort of the TCGA, which includes paired primary and metastatic breast cancers from multiple donors (n=6 pairs). Genes differentially expressed in paired metastatic versus primary tumors included many of the BORG-dependent genes and mapped to pathways and gene lists which overlapped with those upregulated in high BORG-expressing TNBCs, and in D2.OR cells engineered to express BORG (e.g., Smid breast cancer basal up gene list, Figure 5C). These findings further point to the relevance of this group of genes in cancer aggression and the metastatic process. To directly assess the impact of BORG on the expression of this group of genes, we compared BORG expression levels versus the aggregate expression levels of genes associated with the basal breast cancer phenotype (Smid breast cancer basal up gene list) in the TCGA cohort of breast tumors. Importantly, in basal TNBC tumors, higher levels of BORG showed direct correlation with the aggregate expression score of the Smid breast cancer basal up genes, many of which are BORG-dependent genes (Figure 5A and Figure S10B), with a Pearson correlation coefficient of 0.35 for the entire basal TNBC cohort (n=118) and 0.49 for tumors in the top three quartiles in terms of BORG expression (Figure 5D and Figure S11B,C). Collectively, these results indicate that BORG expression is likely to be a major driving force for induction of the basal gene expression pattern in human breast cancer.
Discussion
We provide the first identification of the human ortholog of the murine BORG lncRNA, and in addition to characterizing its locus, we further define the impact of changes in the expression of human BORG observed in large cohorts of human breast tumors. These analyses show that BORG is aberrantly elevated in primary human breast tumors compared to normal human mammary tissue, and importantly, shows significant variability in expression level in all breast cancer subtypes. As we had described for the murine BORG RNA, higher expression levels of human BORG in tumors are also associated with the induction of invasive, metastatic, and “stemness” gene signatures commonly observed in metastatic tumors [38,40,41,42]. Accordingly, a significant fraction of genes differentially expressed in BORG-overexpressing murine TNBCs show concordant expression changes when high BORG-expressing human tumors were compared to their low BORG-expressing counterparts. It is noteworthy that BORG, like many other studied lncRNAs, is poorly conserved in its primary sequence [23,24,25], having only two relatively short (300-400 nucleotides long) stretches of evolutionarily conserved sequences in its genic region. However, as observed for many other poorly conserved lncRNAs [26,27], the function of BORG in the human and mouse TNBC tumors is strongly conserved.
Our study of the protein interactome of the mouse BORG indicates that it binds TRIM28/KAP1, an E3 Sumo ligase known to be involved in regulation of pluripotency-related pathways [38,54,55]. Interestingly, in both mice and humans, increased expression of BORG is associated with increased breast cancer stem cell characteristics in the tumors. Indeed, aberrant expression of BORG in mouse and human breast cancers was associated with acquisition of breast cancer stem cell-like properties and upregulation of known breast cancer stem cell markers, such as NANOG, ALDH1 and ITGA6/α6Integrin/CD49f. Collectively, these findings indicate that induction of stemness properties in breast cancers is a conserved function of BORG, thereby explaining the observed association of BORG with metastatic properties in tumors.
Our analysis comparing high versus low BORG-expressing tumors indicate that across all breast cancer subtypes, higher BORG levels were associated with enhanced stem cell/progenitor phenotypes and increased expression of genes that are typically associated with basal tumors. This finding points to the presence of a core set of pathways which are induced in all breast cancer subtypes in association with the elevated level of BORG. On the other hand, several key oncogenic pathways associated with aggressive breast cancer behavior, such as EMT and invasive signatures, were either uniquely or much more prominently induced in high BORG-expressing basal/TNBC tumors compared to other breast cancer subtypes analyzed in this study. Taken together, these findings point to a subtype-specific impact for increased BORG expression, thereby creating a nuanced picture of BORG function in breast cancer with potential clinical and therapeutic relevance.
Another consequence of the strong similarity of the transcriptomic signature of enforced BORG expression in murine TNBC models and the gene expression pattern observed when high and low BORG-expressing TNBC tumors are compared is proving a causative role for BORG in inducing at least a fraction of the gene expression patterns observed in high BORG tumors. Over 70% of genes differentially expressed in mouse TNBC cells engineered to overexpress BORG were concordantly differentially expressed in high BORG-expressing human TNBC tumors. Further, BORG-expressing mouse TNBCs induced an overlapping set of cellular pathways when compared to high BORG-expressing tumors. We showed that a significant fraction of the transcriptomic signature associated with aberrant BORG expression is mediated through induction of expression and/or activation of a handful of transcription factors, including EMT- and pluripotency-regulating factors such as OCT1, MEF2A and MEF2C. It is plausible that BORG, in association with its complement of RNA-binding proteins, may directly affect chromatin modification or transcriptional activity at MEF2C and OCT1 loci, thus setting a pro-metastatic, pro-invasiveness program into motion. Taken together, the above results point to the existence of a human ortholog for BORG RNA, and a key pro-metastatic function for the human BORG. By establishing the overall importance of BORG in activating pro-metastatic cellular pathways and defining the inter-donor and subtype variability of BORG’s effect, these results indicate its unique suitability not only as a target for development of therapeutic agents, but also as a biomarker to guide the choice of therapeutic strategy.
Methods
BORG Overexpression Studies in Mouse Cell Lines
Murine D2.HAN (D2.OR, RRID:CVCL_0I88; and D2.A1, RRID:CVCL_0I90) cells used in this study were obtained from Dr. Fred Miller (Wayne State University, Detroit, MI). The cell lines were propagated in DMEM media (Sigma-Aldrich) that were supplemented with 10% FBS in addition to 1% Pen/Strep. D2.OR cells were routinely authenticated using short tandem repeat analysis (ATCC), and were regularly tested for mycoplasma infection using MycoAlert Mycoplasma Detection Kit (Lonza, catalog #LT07 218). D2OR cells used in the described studies were passaged fewer than 20 times before data acquisition.
D2.OR cells were stably transfected with a plasmid carrying the full-length mouse BORG cDNA as a transgene and the expression of the transgene was validated by analysis of the level of BORG expression compared to empty vector-transfected controls. Total cellular RNA was obtained from BORG overexpressing and empty vector-transfected D2.OR cells for bulk RNA-seq analysis. For each experimental condition three technical replicates were included.
Bulk RNA-Seq Analyses
RNA-seq studies were performed on parental (transfected with empty vector) and BORG-overexpressing D2.OR derivatives (transfected with a plasmid carrying the mouse BORG cDNA transgene) that were propagated in 3D-culture for 7 days, followed by harvesting total cellular RNA from the resulting organoids. Sample preparation was performed as described [42]. RNA sequencing reactions were performed on a HiSeq 2500 Illumina platform instrument yielding an average of ~66 million paired end, 100 nucleotide long reads for each sample. The raw sequencing data has been deposited in the NCBI SRA (accession number below).
Quality of the resulting sequenced reads was assessed using FastQC (Babraham Bioinformatics), followed by pre-processing using Trim Galore (Babraham Bioinformatics), which is a wrapper based on Cutadapt and FastQC, to remove any leftover adaptor-derived sequences, and sequences with Phred score less than 30. Any reads shorter than 40 nucleotides after the trimming were not used in alignment. Subsequently the trimmed reads were aligned to the mouse genome (Mus musculus, GRCm38/mm10) using kallisto [56] followed by normalization using Sleuth [57]. Pairwise differential expression tests were performed using generalized linear models as implemented in edgeR (QL) [58], and false discovery rate (FDR) values were calculated for each differential expression value. Protein-coding genes that were expressed at a minimum abundance of 5 transcripts per million (TPM) were used for pathway analysis with fold-change values as the ranking parameter while controlling false discovery rate at 0.05. Gene Set Enrichment Analysis (GSEA) package was used to identify the enriched pathways and promoter elements using mSigDB and KEGG databases. Pathways that showed an FDR q-value <= 0.25 were considered significantly enriched, per the GSEA package guidelines. The number of genes contributing to the enrichment score was calculated using the leading-edge output of GSEA (tag multiplied by size).
The Human bulk RNA-seq data used in this study were downloaded from SRA (bioproject accession PRJNA227137) [59] or The Cancer Genome Atlas [4] (TCGA-BRCA and CMI-MBC). The quality control and pre-processing steps were performed as above. The pre-processed reads were aligned to the human genome (hg38/GRCh38) with the Gencode release 28 as the reference annotations using STAR version 2.7.2b [60], followed by gene-level quantitation using htseq-count [61]. In parallel, the pre-processed reads were pseudo-aligned using Kallisto (v0.43.1 [56]), with 100 rounds of bootstrapping to the Gencode release 28 of the human transcriptome. The resulting values were normalized using Sleuth. The two pipelines yielded concordant results. The genomic alignments were used for defining the transcriptional architecture of the BORG locus.
Identification of the Human BORG Locus
For defining the human BORG locus, de novo transcriptome assembly packages RefShannon [62] and Cufflinks [63] were used. Expression patterns of nearby genes were evaluated to ensure that the reads assigned to the BORG locus were not resulting from run-through transcription activity from nearby genes. Samples that showed such a run-through pattern were eliminated from analysis. Direction of transcriptional activity at the human BORG locus was determined using strand-specific RNA-seq studies. Representative TNBC and non-TNBC tumors for use in read histogram representations were identified as the samples in which the expression of BORG was closest to mean BORG expression value within each group.
Filtering and Preparation of the Study Cohort
TCGA BRCA dataset was filtered to eliminate all samples that had a high percentage of normal, non-cancerous cells. Only tumor samples that were unequivocally assigned to one of the basal TNBC, luminal A, luminal B or HER2+ subtypes were used in the study. Tumors that did not have a histological type (PAM50 subtype) assigned were not included in the analysis. Similarly, due to their small number, non-basal TNBC tumors were not included in the study. PAM50 subtype assignments were further verified using gene expression analysis including ERBB2, ESR1, and basal and luminal cytokeratin expression patterns. Tumors labeled as metastatic or secondary, which formed a very small group of tumors in this study, were eliminated from the analysis. The triple-negative status of TNBC cells were verified using both RNA expression patterns and protein expression data included in the TCGA data repository.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
F.N.S., W.P.S. and S.V. designed the study. S.S. contributed key datasets. K.A.P. performed the studies in mouse cell line models. S.J.M. made gene expression figures and pathway analysis data for the mouse cell line studies. F.N.S. and S.V. performed the computational analyses. All authors participated in the interpretation of the data. S.V and W.P.S. wrote the manuscript. All authors reviewed the manuscript, provided comments, and approved the manuscript prior to submission.
Data Availability Statement
The raw sequencing files for the mouse data presented in this study are available in NCBI Sequence Read Archive (SRA) at https://www.omicsdi.org/dataset/geo/GSE99234.
Acknowledgments
Members of the Valadkhan and Schiemann laboratories are thanked for critical comments and reading of the manuscript. Research support was provided in part by the National Institutes of Health to W.P. Schiemann and S. Valadkhan (NCI CA236273). Additional support was graciously provided by pilot funding from the Case Comprehensive Cancer (W.P. Schiemann and S. Valadkhan). The results shown in this manuscript are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. This work made use of the High Performance Computing Resource in the Core Facility for Advanced Research Computing at Case Western Reserve University.
Conflicts of Interest
The authors declare no competing interests.
Materials and Correspondence
Correspondence should be addressed to Drs. Saba Valadkhan and William P. Schiemann.
References
- Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J Clin 72, 7–33 (2022).
- Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J Clin 73, 17–48 (2023).
- Sorlie, T. et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A 100, 8418–8423 (2003). [CrossRef]
- Koboldt, D. C. et al. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
- Sørlie, T. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 98, 10869–10874 (2001). [CrossRef]
- Gooding, A. J. & Schiemann, W. P. Epithelial-Mesenchymal Transition Programs and Cancer Stem Cell Phenotypes: Mediators of Breast Cancer Therapy Resistance. Mol Cancer Res 18, 1257–1270 (2020). [CrossRef]
- Loizides, S. & Constantinidou, A. Triple negative breast cancer: Immunogenicity, tumor microenvironment, and immunotherapy. Front Genet 13, 1095839 (2022). [CrossRef]
- Anders, C. & Carey, L. A. Understanding and treating triple-negative breast cancer. Oncology (Williston Park) 22, 1233–1239; discussion 1239-1240, 1243 (2008).
- Parker, J. S. et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J Clin Oncol 27, 1160–1167 (2009). [CrossRef]
- Lehmann, B. D. et al. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS One 11, e0157368 (2016).
- Castaneda, M., den Hollander, P., Kuburich, N. A., Rosen, J. M. & Mani, S. A. Mechanisms of cancer metastasis. Semin Cancer Biol 87, 17–31 (2022). [CrossRef]
- Morrison, B. J., Schmidt, C. W., Lakhani, S. R., Reynolds, B. A. & Lopez, J. A. Breast cancer stem cells: implications for therapy of breast cancer. Breast Cancer Research 10, 210 (2008). [CrossRef]
- Hu, T., Zhou, R., Zhao, Y. & Wu, G. Integrin α6/Akt/Erk signaling is essential for human breast cancer resistance to radiotherapy. Sci Rep 6, 33376 (2016). [CrossRef]
- Abdoli Shadbad, M. et al. A Systematic Review to Clarify the Prognostic Values of CD44 and CD44+CD24- Phenotype in Triple-Negative Breast Cancer Patients: Lessons Learned and The Road Ahead. Frontiers in Oncology 11, (2021). [CrossRef]
- Honeth, G. et al. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res 10, R53 (2008).
- Olsson, M., Larsson, P., Johansson, J., Sah, V. R. & Parris, T. Z. Cancer stem cells are prevalent in the basal-like 2 and mesenchymal triple-negative breast cancer subtypes in vitro. Frontiers in Cell and Developmental Biology 11, (2023). [CrossRef]
- Shan, N. L., Shin, Y., Yang, G., Furmanski, P. & Suh, N. Breast Cancer Stem Cells: A Review of Their Characteristics and The Agents That Affect Them. Mol Carcinog 60, 73–100 (2021). [CrossRef]
- Bobbitt, J. R., Seachrist, D. D. & Keri, R. A. Chromatin Organization and Transcriptional Programming of Breast Cancer Cell Identity. Endocrinology 164, bqad100 (2023). [CrossRef]
- Prat, A. et al. Molecular Characterization of Basal-Like and Non-Basal-Like Triple-Negative Breast Cancer. Oncologist 18, 123–133 (2013). [CrossRef]
- MacDonald, I., Nixon, N. A. & Khan, O. F. Triple-Negative Breast Cancer: A Review of Current Curative Intent Therapies. Curr Oncol 29, 4768–4778 (2022). [CrossRef]
- Aysola, K. et al. Triple Negative Breast Cancer – An Overview. Hereditary Genet 2013, 001 (2013).
- Keenan, T. E. & Tolaney, S. M. Role of Immunotherapy in Triple-Negative Breast Cancer. J Natl Compr Canc Netw 18, 479–489 (2020). [CrossRef]
- Mattick, J. S. et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol 24, 430–447 (2023). [CrossRef]
- Rinn, J. L. & Chang, H. Y. Long Noncoding RNAs: Molecular Modalities to Organismal Functions. Annu. Rev. Biochem. 89, 283–308 (2020). [CrossRef]
- Amaral, P. et al. The status of the human gene catalogue. Nature 622, 41–47 (2023). [CrossRef]
- Pang, K. C., Frith, M. C. & Mattick, J. S. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet 22, 1–5 (2006). [CrossRef]
- Ulitsky, I., Shkumatava, A., Jan, C. H., Sive, H. & Bartel, D. P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147, 1537–1550 (2011). [CrossRef]
- Gil, N. & Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 21, 102–117 (2020). [CrossRef]
- McDonel, P. & Guttman, M. Approaches for Understanding the Mechanisms of Long Noncoding RNA Regulation of Gene Expression. Cold Spring Harb Perspect Biol 11, (2019). [CrossRef]
- Sun, Q., Hao, Q. & Prasanth, K. V. Nuclear Long Noncoding RNAs: Key Regulators of Gene Expression. Trends Genet 34, 142–157 (2018). [CrossRef]
- Richard, J. L. C. & Eichhorn, P. J. A. Deciphering the roles of lncRNAs in breast development and disease. Oncotarget 9, 20179–20212 (2018). [CrossRef]
- Su, J., Deng, L. & Wang, Y.-D. Roles and Mechanisms of Long Non-Coding RNAs in Breast Cancer. Int J Mol Sci 24, 89 (2022). [CrossRef]
- Amelio, I., Bernassola, F. & Candi, E. Emerging roles of long non-coding RNAs in breast cancer biology and management. Semin Cancer Biol 72, 36–45 (2021). [CrossRef]
- Ferrer, J. & Dimitrova, N. Transcription regulation by long non-coding RNAs: mechanisms and disease relevance. Nat Rev Mol Cell Biol (2024). [CrossRef]
- Olivero, C. E. & Dimitrova, N. Identification and characterization of functional long noncoding RNAs in cancer. FASEB J 34, 15630–15646 (2020). [CrossRef]
- Winkler, L. & Dimitrova, N. A mechanistic view of long noncoding RNAs in cancer. Wiley Interdiscip Rev RNA 13, e1699 (2022). [CrossRef]
- Takeda, K. et al. Identification of a novel bone morphogenetic protein-responsive gene that may function as a noncoding RNA. J. Biol. Chem 273, 17079–17085 (1998). [CrossRef]
- Gooding, A. J. et al. The lncRNA BORG Drives Breast Cancer Metastasis and Disease Recurrence. Sci Rep 7, 12698 (2017). [CrossRef]
- Zhang, B. et al. A Novel RNA Motif Mediates the Strict Nuclear Localization of a Long Noncoding RNA. Mol. Cell. Biol. 34, 2318–2329 (2014). [CrossRef]
- Parker, K. A., Gooding, A. J., Valadkhan, S. & Schiemann, W. P. lncRNA BORG:TRIM28 Complexes Drive Metastatic Progression by Inducing α6 Integrin/CD49f Expression in Breast Cancer Stem Cells. Mol Cancer Res 19, 2068–2080 (2021). [CrossRef]
- Aj, G., Ka, P., S, V. & Wp, S. The IncRNA BORG: A novel inducer of TNBC metastasis, chemoresistance, and disease recurrence. Journal of cancer metastasis and treatment 5, (2019).
- Gooding, A. J. et al. The lncRNA BORG facilitates the survival and chemoresistance of triple-negative breast cancers. Oncogene 1 (2018). [CrossRef]
- Joseph, C. et al. Elevated MMP9 expression in breast cancer is a predictor of shorter patient survival. Breast Cancer Res Treat 182, 267–282 (2020). [CrossRef]
- Radisky, E. S. & Radisky, D. C. Matrix metalloproteinases as breast cancer drivers and therapeutic targets. Front Biosci (Landmark Ed) 20, 1144–1163 (2015).
- Liu, H. et al. The role of MMP-1 in breast cancer growth and metastasis to the brain in a xenograft model. BMC Cancer 12, 583 (2012). [CrossRef]
- Ogura, T. et al. OCT1 Is a Poor Prognostic Factor for Breast Cancer Patients and Promotes Cell Proliferation via Inducing NCAPH. Int J Mol Sci 22, 11505 (2021). [CrossRef]
- Galego, S., Kauppila, L. A., Malhó, R., Pimentel, J. & Brito, M. A. Myocyte Enhancer Factor 2C as a New Player in Human Breast Cancer Brain Metastases. Cells 10, 378 (2021). [CrossRef]
- Kumegawa, K., Yang, L., Miyata, K. & Maruyama, R. FOXD1 is associated with poor outcome and maintains tumor-promoting enhancer–gene programs in basal-like breast cancer. Frontiers in Oncology 13, (2023). [CrossRef]
- Long, Y. et al. FOXD1-dependent RalA-ANXA2-Src complex promotes CTC formation in breast cancer. Journal of Experimental & Clinical Cancer Research 41, 301 (2022).
- Hwang-Verslues, W. W. et al. Loss of corepressor PER2 under hypoxia up-regulates OCT1-mediated EMT gene expression and enhances tumor malignancy. Proceedings of the National Academy of Sciences 110, 12331–12336 (2013). [CrossRef]
- Lim, J.-S., Jung, G. Y. & Park, S.-Y. Nkx-2.5 Regulates MDR1 Expression via Its Upstream Promoter in Breast Cancer Cells. J Korean Med Sci 34, e100 (2019). [CrossRef]
- Pradeepa, null et al. EVI1 promotes metastasis by downregulating TIMP2 in metastatic colon and breast cancer cells. Int J Biochem Cell Biol 142, 106118 (2022). [CrossRef]
- Wang, H. et al. Prominent Oncogenic Roles of EVI1 in Breast Carcinoma. Cancer Res 77, 2148–2160 (2017). [CrossRef]
- Asimi, V. et al. Hijacking of transcriptional condensates by endogenous retroviruses. Nat Genet 54, 1238–1247 (2022). [CrossRef]
- Oleksiewicz, U. et al. TRIM28 and Interacting KRAB-ZNFs Control Self-Renewal of Human Pluripotent Stem Cells through Epigenetic Repression of Pro-differentiation Genes. Stem Cell Reports 9, 2065–2080 (2017). [CrossRef]
- Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527 (2016). [CrossRef]
- Pimentel, H., Bray, N. L., Puente, S., Melsted, P. & Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Meth 14, 687–690 (2017). [CrossRef]
- Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [CrossRef]
- Eswaran, J. et al. Transcriptomic landscape of breast cancers through mRNA sequencing. Sci Rep 2, 264 (2012). [CrossRef]
- Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [CrossRef]
- Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [CrossRef]
- Mao, S., Pachter, L., Tse, D. & Kannan, S. RefShannon: A genome-guided transcriptome assembler using sparse flow decomposition. PLOS ONE 15, e0232946 (2020). [CrossRef]
- Roberts, A., Pimentel, H., Trapnell, C. & Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 27, 2325–2329 (2011). [CrossRef]
Figure 1.
The genomic locus, architecture and expression pattern of human BORG RNA. The genomic locus of the human BORG RNA and its exon-intron architecture is shown. The position of the transcription start site of a divergent lncRNA which originates from a shared promoter with BORG is marked. The short red lines above the gene model indicate the regions conserved between the mouse and human BORG. The blue line marks a conserved sequence close to the 3’ end of the human BORG that likely contributes to its 3’ end processing and is in close vicinity of a canonical polyadenylation signal sequence located 30 nucleotides upstream of the 3’ end of BORG transcript, which is shown in Figure S1. Read histograms for representative tumors from TNBC and non-TNBC groups are shown. PhyloCSF tracks, shown in the three reading frames corresponding to the direction of transcription of BORG, indicate the lack of protein-coding capacity in human BORG RNA. Tracks showing markers of enhancer elements from the UCSC genomes indicate the presence of an enhancer element close to the 5’ end of BORG. Conservation tracks point to the conserved regulatory regions close to the 3’ end of BORG and the area conserved between mouse and human. The position of repeat elements is shown at the bottom. B. BORG expression is strongly induced in breast cancers, especially in a subset of the TNBC tumors, compared to normal mammary organoids. Dots within the box plots mark the expression level of individual samples. C. BORG is highly expressed in a subset of human breast cancers of basal TNBC subtype. Red and black horizontal lines indicate the mean and median, respectively. Gray dots within the box plots mark the expression level of individual tumors.
Figure 1.
The genomic locus, architecture and expression pattern of human BORG RNA. The genomic locus of the human BORG RNA and its exon-intron architecture is shown. The position of the transcription start site of a divergent lncRNA which originates from a shared promoter with BORG is marked. The short red lines above the gene model indicate the regions conserved between the mouse and human BORG. The blue line marks a conserved sequence close to the 3’ end of the human BORG that likely contributes to its 3’ end processing and is in close vicinity of a canonical polyadenylation signal sequence located 30 nucleotides upstream of the 3’ end of BORG transcript, which is shown in Figure S1. Read histograms for representative tumors from TNBC and non-TNBC groups are shown. PhyloCSF tracks, shown in the three reading frames corresponding to the direction of transcription of BORG, indicate the lack of protein-coding capacity in human BORG RNA. Tracks showing markers of enhancer elements from the UCSC genomes indicate the presence of an enhancer element close to the 5’ end of BORG. Conservation tracks point to the conserved regulatory regions close to the 3’ end of BORG and the area conserved between mouse and human. The position of repeat elements is shown at the bottom. B. BORG expression is strongly induced in breast cancers, especially in a subset of the TNBC tumors, compared to normal mammary organoids. Dots within the box plots mark the expression level of individual samples. C. BORG is highly expressed in a subset of human breast cancers of basal TNBC subtype. Red and black horizontal lines indicate the mean and median, respectively. Gray dots within the box plots mark the expression level of individual tumors.
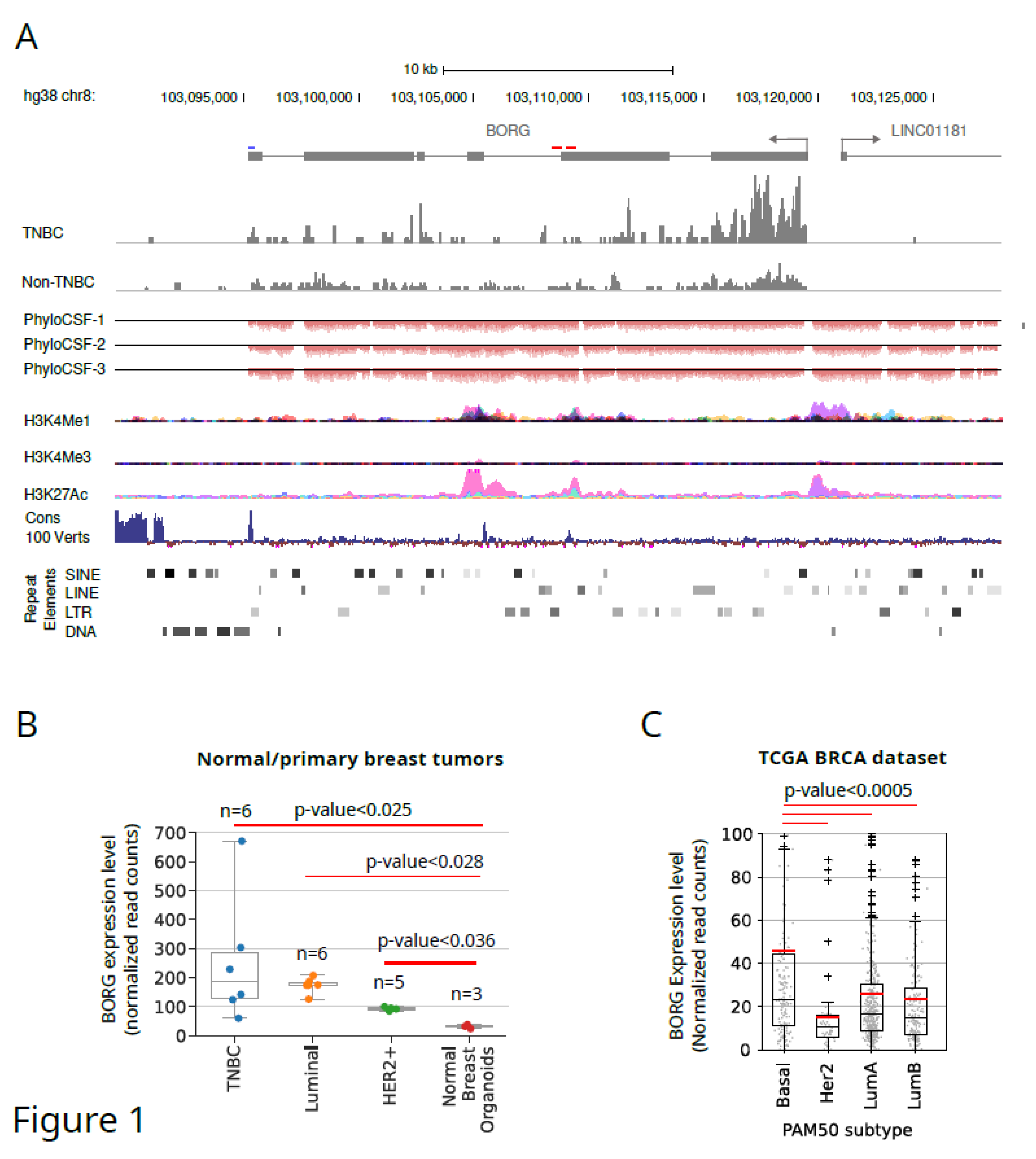
Figure 2.
Elevated expression of BORG is associated with increased cancer aggression markers and induction of a basal phenotype. A. Comparison of differential pathway enrichment patterns between breast cancer samples falling in the upper versus lower quartiles with respect to the expression level of BORG among breast cancers of all histological subtypes. Molecular Signatures Database (mSigDB) Hallmark gene lists are used. Significantly enriched pathways in the positive (pink rectangle) versus negative (blue rectangle) directions are shown. Proliferation-related terms and TGFβ signaling, known to be involved in cancer aggressiveness, are significantly induced. B. Several genes known to associate with the aggressive breast cancer phenotype are upregulated in tumors with a high level of expression of BORG (high BORG) compared to those with low BORG expression levels (low BORG). The rotated bar plot to the right of the dotplot indicates the number of tumors in each group. C. Pathway analysis as described in A, using C2 gene lists of mSigDB, indicates a strong induction of basal phenotype among tumors showing higher BORG expression levels. breast cancer-related pathways are marked by an asterisk.
Figure 2.
Elevated expression of BORG is associated with increased cancer aggression markers and induction of a basal phenotype. A. Comparison of differential pathway enrichment patterns between breast cancer samples falling in the upper versus lower quartiles with respect to the expression level of BORG among breast cancers of all histological subtypes. Molecular Signatures Database (mSigDB) Hallmark gene lists are used. Significantly enriched pathways in the positive (pink rectangle) versus negative (blue rectangle) directions are shown. Proliferation-related terms and TGFβ signaling, known to be involved in cancer aggressiveness, are significantly induced. B. Several genes known to associate with the aggressive breast cancer phenotype are upregulated in tumors with a high level of expression of BORG (high BORG) compared to those with low BORG expression levels (low BORG). The rotated bar plot to the right of the dotplot indicates the number of tumors in each group. C. Pathway analysis as described in A, using C2 gene lists of mSigDB, indicates a strong induction of basal phenotype among tumors showing higher BORG expression levels. breast cancer-related pathways are marked by an asterisk.
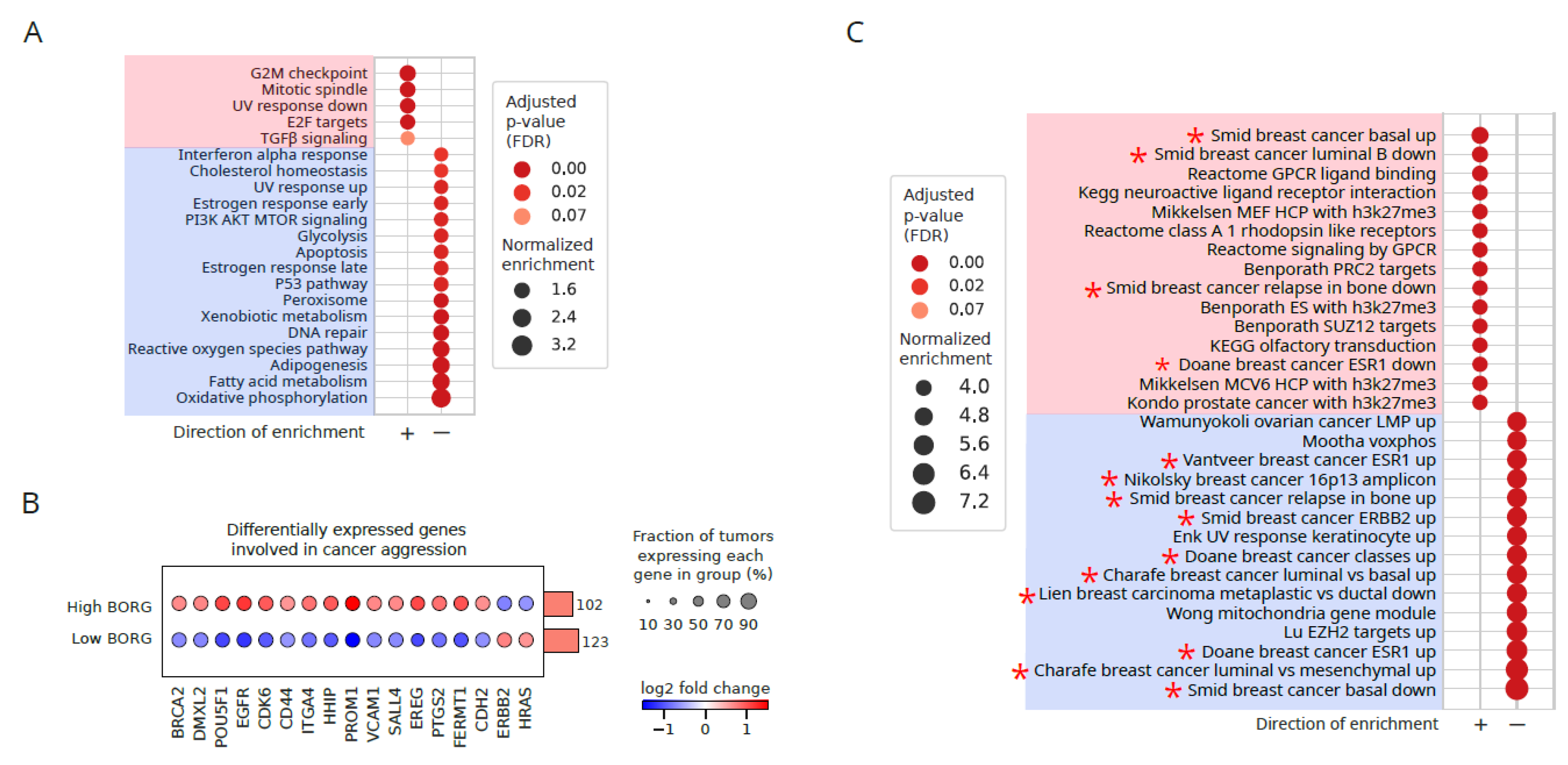
Figure 3.
Many gene expression patterns associated with higher BORG expression levels are shared between all TNBC subtypes. A. Pathway analysis on genes differentially expressed in high BORG-expressing cells in non-basal breast cancer subtypes as a group points to the association of basal transcriptomic signatures with high BORG expression, even in non-basal breast cancer subtypes. Interestingly, genes found in breast cancer progenitor cells were strongly enriched in non-basal tumors with high BORG expression levels. Key pathways and gene lists are marked with asterisks. B and C. Heatmap of genes constituting those associated with breast cancer progenitors indicates their upregulation in high BORG-expressing tumors in all breast cancer subtypes, including basal, shown as a group (B) and separately for each subtype (C). D. Aggregate scores for genes upregulated in breast cancer progenitor cells indicate their overall increase in high BORG-expressing tumors in all breast cancer subtypes. Each dot represent a tumor. P-values are shown at the top. For HER2+ tumors, the number of tumors was too small to derive a reliable p-value.
Figure 3.
Many gene expression patterns associated with higher BORG expression levels are shared between all TNBC subtypes. A. Pathway analysis on genes differentially expressed in high BORG-expressing cells in non-basal breast cancer subtypes as a group points to the association of basal transcriptomic signatures with high BORG expression, even in non-basal breast cancer subtypes. Interestingly, genes found in breast cancer progenitor cells were strongly enriched in non-basal tumors with high BORG expression levels. Key pathways and gene lists are marked with asterisks. B and C. Heatmap of genes constituting those associated with breast cancer progenitors indicates their upregulation in high BORG-expressing tumors in all breast cancer subtypes, including basal, shown as a group (B) and separately for each subtype (C). D. Aggregate scores for genes upregulated in breast cancer progenitor cells indicate their overall increase in high BORG-expressing tumors in all breast cancer subtypes. Each dot represent a tumor. P-values are shown at the top. For HER2+ tumors, the number of tumors was too small to derive a reliable p-value.
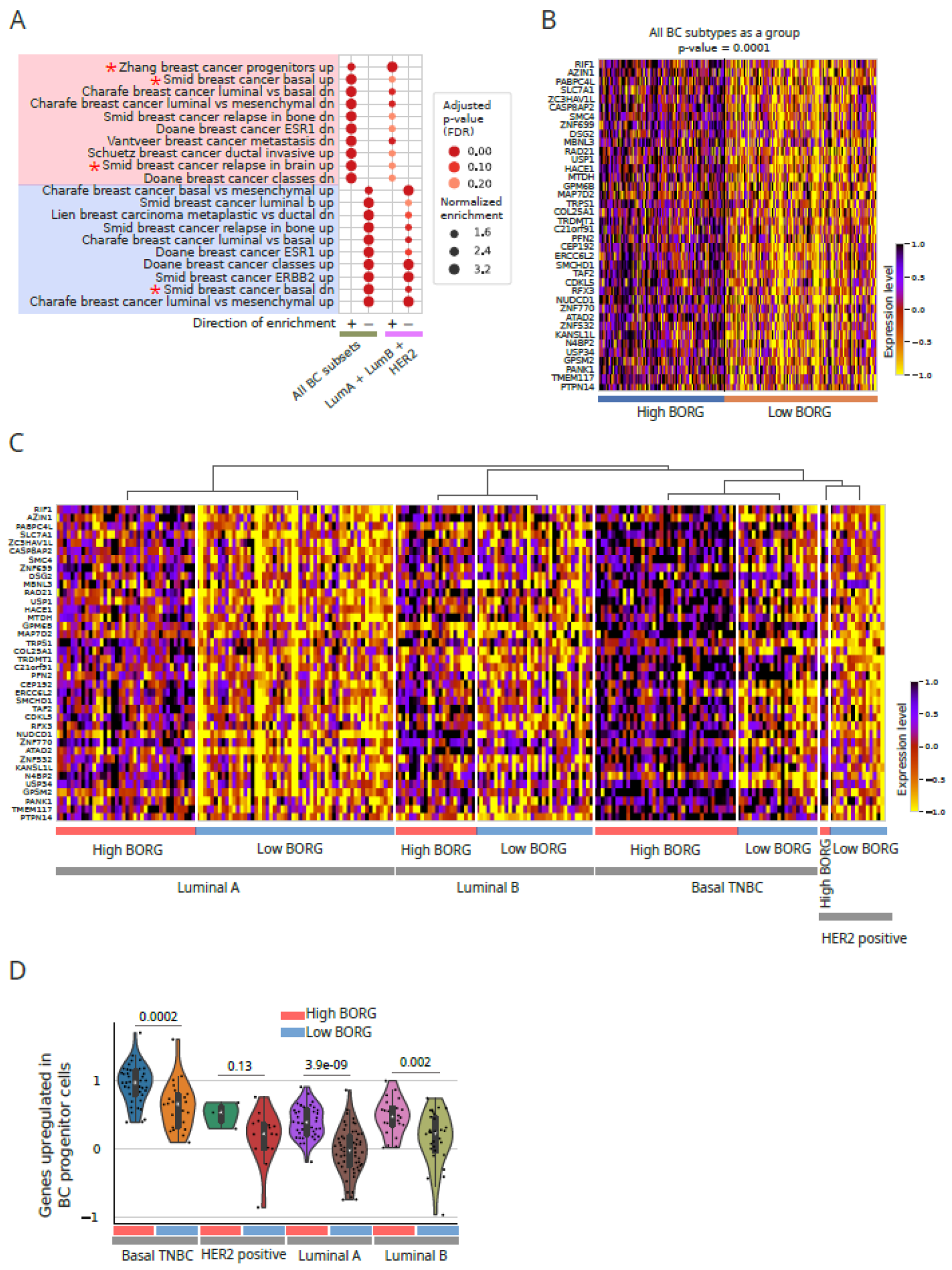
Figure 4.
Higher expression of BORG in basal TNBC tumors is associated with induction of invasiveness and pro-metastatic gene expression programs. A. Pathway analysis comparing high versus low BORG-expressing basal TNBC tumors points to the induction of the EMT and TGFβ pathways. B. Using the cancer-related subset of pathways and gene lists from the mSigDB C2 database of genes points to a set of genes comprising a multi-cancer invasiveness signature as the top enriched genelist in basal TNBCs (marked by an asterisk). C. Multi-cancer invasiveness signature genes (shown on the left of the heatmap) are strongly upregulated in high BORG-expressing TNBC tumors compared to low BORG-expressing ones. Donors IDs are shown at the bottom. D. Transcription factor binding motif analysis indicates that genes induced in high BORG-expressing basal TNBC tumors are enriched for targets of multiple pro-neoplastic, pro-metastatic transcription factors. E. The pro-metastatic MEF2 and FOXD1 transcription factors are transcriptionally upregulated in high BORG-expressing TNBCs. F. Genes regulated by the five transcription factors in Figure 3D are upregulated in high BORG-expressing TNBC tumors, indicative of a global induction of a pro-metastatic gene expression program.
Figure 4.
Higher expression of BORG in basal TNBC tumors is associated with induction of invasiveness and pro-metastatic gene expression programs. A. Pathway analysis comparing high versus low BORG-expressing basal TNBC tumors points to the induction of the EMT and TGFβ pathways. B. Using the cancer-related subset of pathways and gene lists from the mSigDB C2 database of genes points to a set of genes comprising a multi-cancer invasiveness signature as the top enriched genelist in basal TNBCs (marked by an asterisk). C. Multi-cancer invasiveness signature genes (shown on the left of the heatmap) are strongly upregulated in high BORG-expressing TNBC tumors compared to low BORG-expressing ones. Donors IDs are shown at the bottom. D. Transcription factor binding motif analysis indicates that genes induced in high BORG-expressing basal TNBC tumors are enriched for targets of multiple pro-neoplastic, pro-metastatic transcription factors. E. The pro-metastatic MEF2 and FOXD1 transcription factors are transcriptionally upregulated in high BORG-expressing TNBCs. F. Genes regulated by the five transcription factors in Figure 3D are upregulated in high BORG-expressing TNBC tumors, indicative of a global induction of a pro-metastatic gene expression program.
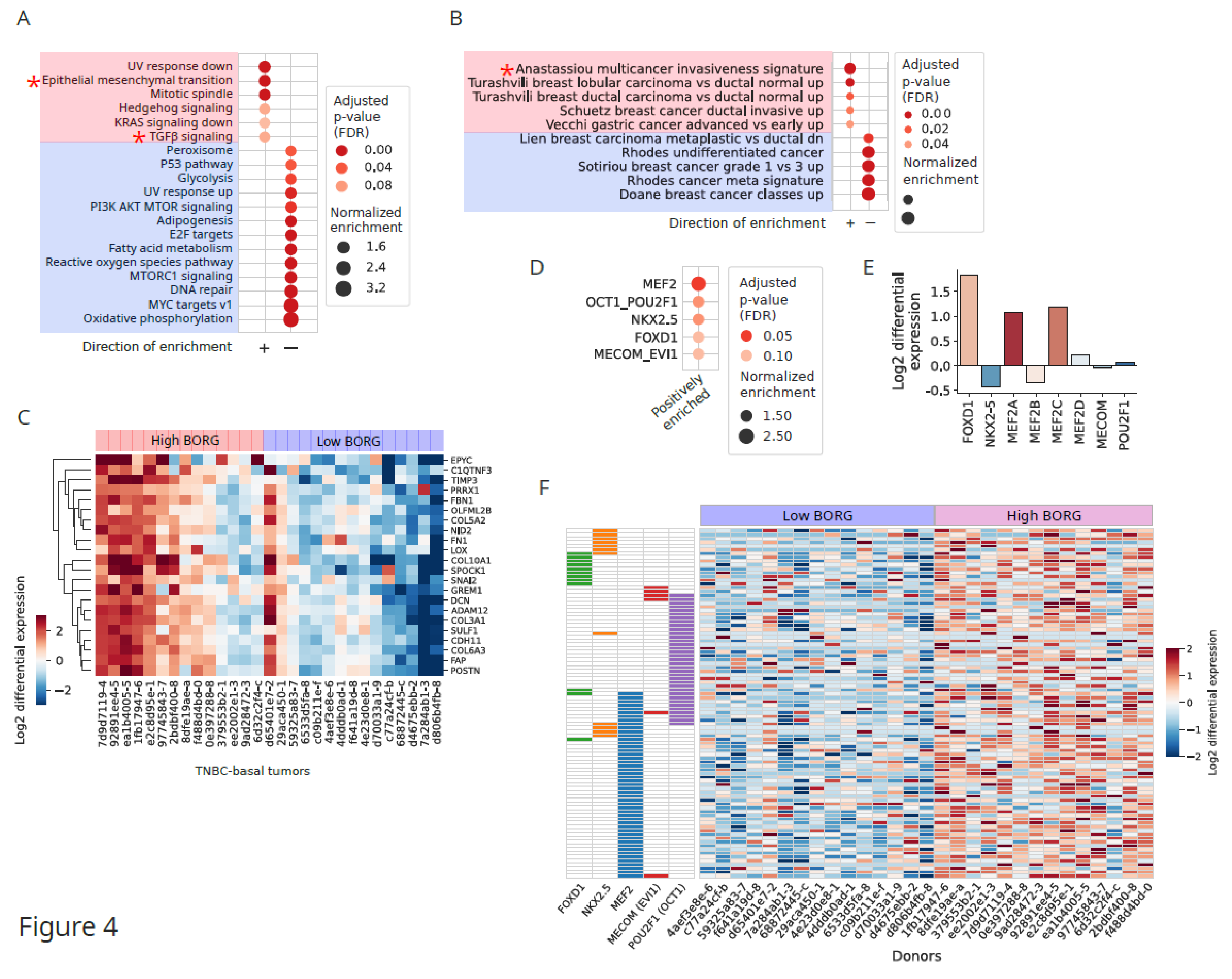
Figure 5.
Increased expression of BORG has a causative role in induction of an aggressive, basal like transcriptomic signature in breast tumors. A. Forced BORG overexpression from a transgene in D2OR cells and high BORG-expressing TNBC tumors share differentially expressed genes that include those upregulated in basal breast cancer. Heatmap at the bottom shows shared differentially expressed genes that map to the top five most enriched pathways. Rows contain the differential expression pattern of genes when BORG overexpressing cells are compared to empty vector-transfected cells (D2OR-BORG Overex vs ctrl) and those that are differentially expressed in high versus low BORG-expressing TNBCs (TCGA BRCA TNBC tumors high vs low BORG). The annotation matrix at the top maps each gene to the pathways it’s a member of. Identity of the pathways/gene lists are shown to the left. Asterisks mark gene lists associated with poor outcome and aggressive basal breast cancer signature. B. Pathway analysis using the largest mSigDB pathway list on genes that show concordant differential expression between D2OR BORG overexpressing cells and high BORG-expressing basal TNBC tumors. The result shows the most positively enriched pathways among these shared genes include Smid breast cancer basal up gene list, consistent with BORG-mediated induction of expression of basal-specific gene expression patterns. C. Comparison of paired primary and metastatic breast cancers from the same donor (n=6 pairs) indicates the upregulation of multiple gene lists and pathways in metastatic tumors, including genes upregulated in basal tumors (Smid breast cancer basal up). D. The extent of upregulation of genes induced in basal breast cancer (Smid breast cancer basal up genes) is positively correlated with the level of expression of BORG in TNBC tumors. Pearson’s correlation coefficient for the entire group of basal TNBC tumors (n=118) is shown. In tumors with BORG expression in the top three quartiles, the correlation coefficient was 0.49 (p-value 7.32e-06) indicating a stronger correlation between BORG level and the basal phenotype at higher BORG expression levels. The aggregate expression score of the Smid breast cancer basal up genes was calculated as the average expression of the set of genes subtracted with the average expression of a reference set of genes. The reference set is randomly sampled from the gene expression table for each binned expression value.
Figure 5.
Increased expression of BORG has a causative role in induction of an aggressive, basal like transcriptomic signature in breast tumors. A. Forced BORG overexpression from a transgene in D2OR cells and high BORG-expressing TNBC tumors share differentially expressed genes that include those upregulated in basal breast cancer. Heatmap at the bottom shows shared differentially expressed genes that map to the top five most enriched pathways. Rows contain the differential expression pattern of genes when BORG overexpressing cells are compared to empty vector-transfected cells (D2OR-BORG Overex vs ctrl) and those that are differentially expressed in high versus low BORG-expressing TNBCs (TCGA BRCA TNBC tumors high vs low BORG). The annotation matrix at the top maps each gene to the pathways it’s a member of. Identity of the pathways/gene lists are shown to the left. Asterisks mark gene lists associated with poor outcome and aggressive basal breast cancer signature. B. Pathway analysis using the largest mSigDB pathway list on genes that show concordant differential expression between D2OR BORG overexpressing cells and high BORG-expressing basal TNBC tumors. The result shows the most positively enriched pathways among these shared genes include Smid breast cancer basal up gene list, consistent with BORG-mediated induction of expression of basal-specific gene expression patterns. C. Comparison of paired primary and metastatic breast cancers from the same donor (n=6 pairs) indicates the upregulation of multiple gene lists and pathways in metastatic tumors, including genes upregulated in basal tumors (Smid breast cancer basal up). D. The extent of upregulation of genes induced in basal breast cancer (Smid breast cancer basal up genes) is positively correlated with the level of expression of BORG in TNBC tumors. Pearson’s correlation coefficient for the entire group of basal TNBC tumors (n=118) is shown. In tumors with BORG expression in the top three quartiles, the correlation coefficient was 0.49 (p-value 7.32e-06) indicating a stronger correlation between BORG level and the basal phenotype at higher BORG expression levels. The aggregate expression score of the Smid breast cancer basal up genes was calculated as the average expression of the set of genes subtracted with the average expression of a reference set of genes. The reference set is randomly sampled from the gene expression table for each binned expression value.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



