
Submitted:
24 June 2024
Posted:
24 June 2024
You are already at the latest version
Abstract
Background: Colorectal adenoma undergoes neoplastic progression via the normal epithelium-adenoma-adenocarcinoma sequence as reported in the Vogelgram. The risk of developing cancer is strongly associated with the number and size of adenoma and with the subtype. Currently, adenomatous polyps can be distinguished on the basis of histology: the prevalence are tubular, 5-15% are villous and tubular/villous. Considering the increased risk for malignant transformation described for tubular/villous adenomas, patients diagnosed with adenomatous polyposis are at increased risk of developing CRC. The Wnt/β-catenin pathway plays a key role in the onset of colorectal adenoma, in particular intestinal cells first acquire loss-of-function mutations in APC gene that induce the formation of adenomas.
Methods: Wnt/β-catenin pathway APC, Wnt3a, Wnt5a, LEF1, BCL9 genes and protein expression analyses were conducted by qRT-PCR and western blot in 68 colonic samples (polyps and adjacent mucosa) from 41 patients, of which 17 affected by FAP. Ten normal colonic mucosal samples were collected from 10 healthy donors.
Results: In this study both APC gene and protein resulted less expressed in colon tumor compared to the adjacent colonic mucosa. Conversely, activated β-catenin was more expressed in polyps than in the adjacent mucosa. All results confirmed literature data on carcinomas. A statistically significant correlation between Wnt3a and BCL9 both in polyps and in the adjacent mucosa underlies that the canonical Wnt pathway is activated in early colon carcinogenesis and that the adjacent mucosa is already altered.
Conclusion: This is the first study analyzing the difference in expression of Wnt/β-catenin pathway in human colorectal adenomas. Understanding the progression from adenomas to colorectal carcinomas is essential for the development of new therapeutic strategies and improving clinical outcomes.
Keywords:
CRA
; colorectal adenoma
; APC
; Wnt3a
; Wnt5a
; LEF1
; BCL9
; polyps
; early carcinogenesis
; Wnt/β-catenin
1. Introduction
The prevalence of colorectal adenomas (CRA) increases with age, mainly in Western populations, and it is of 30-40% in the people over 50 years, predominantly in men [1,2]. The annual rate of adenoma progression to colorectal cancer (CRC) is ~0.25% [3].
CRA is associated with colorectal cancer (CRC), and at least 80% of CRC undergo neoplastic progression via the normal epithelium-adenoma-adenocarcinoma sequence as reported in the Vogelgram model [4,5]. The incidence of cancer after a negative colonoscopy is significant because adenomas may be missed during colonoscopy or biological changes in tumor growth rate may occur [6]. Screening and surveillance programs can help identify precursor lesions and prevent death from CRC [7]. Thus, it is important to understand the progression from CRA to carcinomas to facilitate the development of novel treatment strategies and improve clinical outcomes.
The malignancy of adenomas is highly correlated with the occurrence of colon cancer and its depending on the subtype [8]. In addition, the risk of developing cancer is strongly associated with the number and size of (previously) encountered polyps [9]. Therefore, the development of multiple colonic polyps with malignant potential will result in an increased lifetime risk of developing CRC. At least three subtypes of polyps can be distinguished on the basis of histology and the underlying molecular pathway: adenomatous, serrated or hyperplastic (non-neoplastic) [10,11,12]. The first are characterized by an adenomatous histotype, whereas both sessile/traditional serrated adenomas and hyperplastic polyps have a serrated histotype [13]. The adenomatous can be tubular (more than 80%), villous (5-15%) and tubular/villous (5-15%). Hyperplastic polyps are very common and have a very low malignant potential [8]. Even though the different types of polyps may be disseminated in all the large bowel, adenomatous and hyperplastic polyps are prevalently located in distal colon [14,15,16] and sessile serrated polyps are often found in proximal colon [17,18,19,20]. Owing to the malignant potential of tubular/villous adenomas, patients diagnosed with adenomatous polyposis, i.e. the constitutive development of multiple colorectal adenomas, are at increased risk of developing CRC.
Most studies investigating the carcinogenesis of CRA have focused on villous and familial adenomatous polyps, which have the highest rates of carcinogenesis [21,22]. Whereas, few studies have investigated sporadic tubular adenoma, which has the highest clinical incidence [23,24].
Adenomas are the main precursors of colorectal cancer in high-risk family groups with a history of polyposis syndrome and in the general population. Indeed, genetic events such as gain or loss of function of molecules required for intestinal cell functional homeostasis can lead to the development of polyps [25].
The Wnt/β-catenin signaling deregulation is an early event in the onset of colorectal adenoma [26]. Its up-regulation is mainly due to the altered functions of the adenomatous polyposis coli (APC) protein, which reduces differentiation of intestinal epithelial cells (IEC), leading to the onset of adenoma and CRC progression. Furthermore, mutations and LOH of APC alter the quantitative regulation of the β-catenin protein, which accumulates in the nucleus, favoring the activity of transcription factors for cell proliferation genes expression and reducing differentiation [27]. Less than 1% of CRC cases belong to Familial adenomatous polyposis (FAP), an autosomal dominant inherited CRC syndrome as result from a germline mutation in the APC gene. Most patients (~70%) have a family history of colorectal polyps and cancer. FAP is characterized by the development of many tens to thousands of adenomas in the rectum and colon during the second and third decade of life. APC is essential for IEC homeostasis and its inactivation facilitates tumorigenesis. Indeed, APC somatic truncation mutations are observed in more than 90% of human colon cancers [28,29]. Wnt ligands may activate the canonical (β-catenin dependent) and the non-canonical (β-catenin independent) pathways. They are in concert to maintain renewal, defense and metabolic homeostasis of colon epithelia [30,31].
Most of the cellular β-catenin is confined to the adherens junctions on the plasma membrane. Cytosolic β-catenin associates in a complex with APC and AXIN1 proteins, which mediate the N-terminal phosphorylation of β-catenin. This event conducts to the ubiquitination of β-catenin by the E3 ubiquitin ligase β-transducin repeat-containing protein (β-TRCP) following proteasomal degradation. When Wnt ligands bind to the Frizzled receptors, Dvl/Dsh is phosphorylated and, in turn, recruits AXIN1 and GSK3β adjacent to the plasma membrane, therefore preventing the building of the degradation complex. As a consequence, unphosphorylated β-catenin eludes recognition by β-TRCP and translocates into the nucleus, where it binds to the T cell factor (TCF) and lymphoid enhancer-binding protein family (LEF) transcription factors.
The activated β-catenin/TCF/LEF complex induces the transcription of genes controlling cell proliferation and survival. In normal cells two LEF1 isoforms regulate Wnt-dependent pathways as apoptosis, motility and gene transcription and its expression in human colon tissue gradually increased from normal colon, low-grade adenoma, high-grade adenoma to adenocarcinoma [32]. Then, β-catenin accumulates in the cytoplasm and in the nucleus [33].
B-cell CLL/lymphoma 9 (BCL9) protein is a novel co-factor of canonical Wnt/β-catenin signaling [34,35,36]. It forms a complex with β-catenin-LEF/TCF to activate transcription of Wnt target genes, after hyper-activation of canonical Wnt signaling [37]. In CRC tissues Wnt3 is highly expressed to sustain autocrine Wnt activity and CRC progression by EMT and is indicative of advanced stages with poor prognoses [38]. Inhibiting Wnt3 secretion inhibits colon cancer cells proliferation [39]. Wnt3a expression was also increased and associated with EMT, which is indicative of advanced stages with poor prognoses [40]. Also, Wnt3a was overexpressed in CRC primary tissues than in metastatic areas, suggesting that Wnt3a was expressed early in cancer rather than appearing as it progressed. A more recent study discovered that Wnt3a inhibits the ability of human colon myofibroblasts to proliferate and migrate [41]. Thus, various CRC subgroups have distinct molecular and cellular properties contributing to Wnt3a’s context-dependent nature.
Wnt5a is one of the most effective non-canonical Wnt ligands which can significantly antagonize and eventually suppress canonical Wnt ligand functions [42]. Recent findings demonstrate that Wnt5a non-canonical ligand, as tumor suppressor and oncogenic agent, can promote and inhibit tumor processes through canonical and non-canonical Wnt signaling [43,44]. The exact role of Wnt5A in CRC is contradictory [45].
As described above, Wnt/β-catenin signaling is well known in human CRC, but less studied in the early stages of colon tumorigenesis. The main aim of this study was to investigate the relationships of some key elements of Wnt/β-catenin signaling in the early stage of colorectal adenoma carcinogenesis. Based on what is reported in the current literature, there are no studies analyzing these aspects on human adenomas. In this study, the analysis of the gene and protein expression of some components of the Wnt/β-catenin pathway was conducted on pathological samples (polyps and related adjacent mucosa) derived from patients both with sporadic adenomas and suffering from FAP.
2. Materials and Methods
2.1. Tissues
Pathological and control tissues were recruited from patients with sporadic adenomas undergoing endoscopic biopsy at the Digestive Endoscopy and Gastroenterology Unit, SS. Annunziata Hospital of Chieti, while those from patients belonging to families with FAP were recruited at the Surgery Unit, Careggi University Hospital in Florence. The collection of each pathological sample was accompanied by the normal-appearing tissues sample obtained from areas that were at least 5 centimeters away from the margins of the primary lesion. Finally, normal colonic mucosal samples were collected from healthy donors without inflammatory bowel disease and family history of cancer, who had undergone follow-up colonoscopy at the Digestive Endoscopy and Gastroenterology Unit, “SS. Annunziata” of Chieti (UOD of Digestive Endoscopy and Gastroenterology). After the surgical removal, the tissue fragments were stored in RNAlater™ solution (Thermo Fisher Scientific, Waltham, MA, USA) to stabilize the RNA and to preserve proteins; subsequently stored at - 80˚C. The study protocol was approved by the local Ethics Committee and each participant tissue donor provided written informed consent. The study was conducted according to the declaration of Helsinki and approval was granted from the Institutional Review Board (Prot. Id RICH1KHE). Adenoma tissues were classified according to conventional histopathological criteria, as defined by WHO (World Health Organization). Patient characteristics and polyps histotype are shown in Table 1 [46].
Tissues were selected based on RNA availability. Samples with insufficient quantity of target in the cDNA template or protein were not included in the gene and protein expression analyses. The study included 68 colonic samples. 58 biopsies (33 polyps and 25 adjacent mucosa) belonged to 41 patients, of which 17 affected by FAP. Ten normal colonic mucosal samples were collected from 10 healthy donors.
2.2. Gene Expression by Real-Time Quantitative PCR Analysis (qRT-PCR)
Total RNA was isolated from colon tissues homogenized in liquid nitrogen with a mortar and pestle, using TRIzol® Reagent (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions at RNase free environment. The synthesis of complementary DNA (cDNA) were performed as previously described [47]. The mRNA levels were evaluated by SYBR Green and TaqMan assay by quantitative real-time PCR (qRT-PCR) analysis using StepOne™ 2.0 (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA). Data were analyzed by the comparative Ct method and were graphically indicated as 2−ΔCt ± SD. In accordance with the method, the mRNA amounts of the target genes were normalized by the ratio on the median value of the endogenous housekeeping gene (GUSB). Primers sequences are available in Catalano et al 2021 [47] The cycling conditions were performed as follow, 10 min at 95 °C and 40 cycles of 15 s at 95 °C followed by 1 min at 60 °C and final elongation of 15 s at 95°C. All data were validated in a second analysis.
2.3. Western Blotting
Total proteins were isolated from pathological and control tissues using RIPA lysis buffer (Cell Signaling Technology, Beverly, MA, USA). Protein were quantified by Bradford Assay (Thermo Fisher Scientific, Waltham, CA, USA) and analyzed by western blot, using the primary antibodies: Active-β-Catenin and APC (Merck-Millipore, Burlington, MA, USA). The β-actin primary antibody (Sigma-Aldrich, St. Louis, MI, USA) was used as a protein loading control. Secondary antibodies were HRP-conjugated anti-rabbit or anti-mouse (Bethyl Laboratories, Montgomery, TX, USA). The immune complexes were visualized using the ECL Western blot detection system (EuroClone) by using AllianceLD2 hardware (UVItec Limited, Cambridge, UK).
2.4. Statistical Analysis
All measurements were made after three independent experiments, and for each data is shown a representative value of all experiments plus standard deviation (SD). Statistical analyses were performed using the t-test or one-way analysis of variance (ANOVA) as appropriate. All p-values are two-sided and a p-value ≤ 0.05 was considered as significant. All analyses were performed using SPSS software. Kruskal-Wallis non parametric test for independent samples was used to compare the expression of genes in polyps, adjacent mucosa and normal tissue. To assess for a possible correlation between the five genes, Spearman's ρ correlation coefficient was evaluated. All p-value are twotailes and a p-value of <0.05 was considered as significant. Statistical analyses and descriptive statistics were carried out using GraphPad Prism version 9.0 (GraphPad Software Inc., La Jolla, CA, USA).
3. Results
We analyzed the expression of five genes of Wnt/β-catenin family in all available tissues, grouping them into three categories: colorectal adenomas, adjacent mucosa and normal tissues. To evaluate any alterations in the protein expression of APC and β-catenin in the mucosa adjacent to the adenoma transition, we performed protein expression in the cases in which matched adenoma and adjacent mucosal tissues were available.
3.1. Gene Expression of APC, Wnt3A, Wnt5A, BCL9 and LEF1 in FAP and Tubular-Villous Adenomas
Gene expression of five Wnt/β-catenin signaling genes (APC, BCL9, LEF1, Wnt3a, Wnt5a) was conducted on 52 colonic samples. 42 biopsies (25 polyps and 17 adjacent mucosa) belonged to 33 patients, of which 15 affected by FAP, and 10 normal colonic mucosal samples from 10 healthy donors.
The expressions of APC, Wnt3A, Wnt5A, BCL9 and LEF1 in polyps, adjacent mucosa and normal tissue were compared by Kruskal-Wallis non parametric test for independent samples (Figure 1). A statistically significance difference was detected in APC expression in adjacent mucosa compared to normal tissue (P=0.01). Statistically significance differences were also detected between Wnt5a expression in polyps compared to normal tissue (P=0.004) and in adjacent mucosa compared to normal tissue (P=0.01).
Furthermore, the Spearman's ρ analysis revealed a statistically significance correlation between Wnt3a and BCL9 (P= 0.0294) when polyps and adjacent mucosa were considered in the same category (Table 2). A statistically significance correlation between Wnt5a and LEF1 (P= 0.025097) was detected in healthy tissues (Table 2).
Gene expression results were graphically represented using a heat map. APC, Wnt3a, Wnt5a, BCL9, LEF1, tend to reduce from healthy to pathological tissue (Figure 2). APC, Wnt5a, LEF1 are the most expressed genes in healthy mucosa (Figure 2).
3.2.1. Protein Expression Assay by Western Blotting of APC and β-Catenin
A 50% reduction of APC gene expression in the adjacent mucosa compared to the healthy mucosa, and an more accentuated reduction in polyps, have been detected (Figure 1). Therefore we wanted to investigate the levels of APC and β-catenin proteins, in still available tissues. Then a Western blot analysis of matched polyp-adjacent mucosal tissues was performed. Matched adenoma and adjacent mucosal tissues were available for 6 FAP patients and for 7 patients with sporadic adenomas. The analyses revealed the expression of the full-length APC protein (300 kDa band) in all matched adenoma-adjacent mucosa samples (Figure 3a and Figure 4a). A decrease in APC expression is visible in all the adenomas, both familial than sporadic, compared to adjacent mucosa. As we expected, as regards β-catenin expression, its active form appeared increased in all the familial and sporadic adenomas compared to adjacent mucosa (Figure 3a and Figure 4a). Sample 42FI-P does not show a decrease in APC expression and intriguingly it shows a reduced active β-catenin expression, also visible in matched adjacent mucosa. Also for sample 43FI, a reduced expression of active β-catenin was noted, apparently not associated with APC modulation. Relative expression quantification of the selected proteins (APC, active β-catenin) detected by WB in available matched adenoma and adjacent mucosal tissues are shown in Figure 3b,c and Figure 4b,c.
4. Discussion
The current literature lacks significant Wnt/β-catenin/APC gene expression studies on adenomas. Our study is the first to analyze the modulation of Wnt/β-catenin pathway in early carcinogenesis. Understanding the significance of benign pre-cancerous lesions is widely recognized, yet studying them is complicated in terms of sampling, mainly due to the difficulty of obtaining biopsies and their small size. The long period required for polyps and cancer to develop, as well as the tendency of early-stage cancers to be asymptomatic in many individuals, allows a window of opportunity for polypectomy and cancer prevention, as well as for early diagnosis and highly effective drug administration for early-stage cancers. Blood-based tests could detect genetic changes associated with polyps released into circulation. Promoting the uptake and completion of follow-up testing and treatment holds significant potential to save lives. Therefore, understanding the progression from colorectal adenomas to colorectal carcinomas is essential for the development of new therapeutic strategies and improving clinical outcomes.
Tumor and adjacent mucosa biopsies are the definitive diagnostic procedures for histological evaluation. The adjacent mucosa, also known as "apparently healthy" mucosa, being closely associated with polyps in the tumor microenvironment (TME), may have already molecularly been impacted. Our data supports this hypothesis.
It is already known that APC is inactivated in FAP polyps. This study focused on evaluating APC gene and protein expression not only on pathological FAP tissue and tubular-villous adenomas, but also on apparently normal endoscopic mucosal tissue sampled at the 5-cm proximal. Very interestingly, lower APC expression was detected in the adjacent mucosa compared to the normal one.
APC has a functional role in the canonical (β-catenin-dependent) Wnt signaling pathways [48]. Most cases of CRCs are initiated by APC inactivating mutations, leading to constitutive activation of the Wnt/β-catenin signaling pathway. Most of the literature has extensively assessed the role of APC somatic and germline mutations in familial as well as sporadic forms of CRC.
It is known that APC is inactivated in FAP polyps. Our results showed a reduction in APC expression both in FAP and sporadic polyps. Additionally, a lower APC expression was detected in adjacent mucosa.
Moreover, APC can inhibit the initiation and development of colorectal tumor, independently of canonical Wnt signaling. APC assists in chromosome segregation, establishes cellular polarity and migration, and represses DNA replication [49]. APC mutations contribute in early adenoma creation leading to chromosomal instability by triggering spindle abnormalities and deregulation of microtubules/actin cytoskeleton. Moreover, APC mutations increase cell migration by reducing cell adhesion via deregulation of β-catenin and E-cadherin distributions among the cytoplasm and the cell membrane [50]. Xu et al. demonstrated that over-expression of erythropoietin-producing hepatocyte (EphB6), a member of the tyrosine kinase family, along with APC gene mutations, increases proliferation, migration, and invasion in colon epithelial cell line, IMCE, supporting the role of APC mutations in promoting tumorigenesis in CRC [51]. In our study APC gene was less expressed in colon tumor compared to the adjacent colonic mucosa and to the mucosa of healthy controls. Also APC protein resulted less expressed in colon tumor compared to the adjacent colonic mucosa. All results confirmed literature data in carcinomas.
LEF1 is a downstream mediator and nuclear effector of the Wnt/β-catenin signaling pathway [52]. In normal cells two LEF1 isoforms are in regulation of Wnt-dependent pathways as apoptosis, motility and gene transcription. The role of LEF1 is controversial, usually is detected and upregulated in most colonic carcinomas enhancing the progression [53]. Nevertheless, little is known about the expression of LEF1 in early carcinogenesis. LEF1 is generally excessively expressed in malignant tumors and may play a role in tumor growth and metastasis [54]. LEF1 knockdown in glioblastoma multiforme cells inhibits invasion, migration, proliferation, and the self-renewal potential of stem-like cells [55]. Myc induces the expression of LEF1 to activate the Wnt pathway in colon cancer [56]. LSD1 (lysine-specific demethylase 1) promotes bladder cancer progression by upregulating LEF1 and enhancing EMT [57]. LEF1 expression may contribute to cancer development [58,59,60], but there is a lack of evidence to support malignant phenotype changes. The underlying molecular mechanisms of LEF1 regulation for colonic adenocarcinoma progression remain unknown. Xiao et al. 2021 study findings, support LEF1 as a potentiator and potential therapeutic target for colonic adenocarcinoma. LEF1 enhances the motility of colonic adenocarcinoma cells via remodeling of lamellipodia/filopodia and the polymerization of F-actin/β-tubulin. LEF1 maintains the viability and growth of colonic adenocarcinoma cells through increasing proliferation, Lamin B1 expression, and decreasing apoptosis. In addition, LEF1 is closely related to EMT. However, an in vivo study published on Science [61] showed that LEF1 restricts ectopic crypt formation and tumor cell growth in colon adenomas from APC-deficient mice. Loss of Lef1 markedly increased tumor initiation and tumor cell proliferation, reduced the expression of several Wnt antagonists, and increased Myc proto-oncogene expression and formation of ectopic crypts in Apc-mutant adenomas. These results uncover a previously unknown negative feedback mechanism in CRC, in which ectopic Lef1 expression suppresses intestinal tumorigenesis by restricting adenoma cell dedifferentiation to a crypt-progenitor phenotype and by reducing the formation of cancer stem cell niches. Recent literature data therefore demonstrate how the controversy on the function of LEF1 in colorectal tumorigenesis is still open.
In our study, the expression of LEF1 in healthy tissues decreased in the adjacent mucosa and the polyp, supporting the recent hypothesis of LEF1's role as a tumour suppressor, already in adenomas.
BCL9 is considered a key developmental regulator and a well-established oncogenic driver in multiple cancer types, mainly through potentiating the Wnt/β-catenin signaling. BCL9 has been identified as an adapter and essential core component of nuclear β-catenin complexes [62] and functions as an adapter protein providing binding sites for the transcriptional machinery of Wnt signaling [63]. As an oncoprotein, BCL9 promotes cancer development mainly through sustaining cancer cell division [64], promoting proliferation and migration, inhibiting apoptosis [65], remodeling TME and immune system, and regulating chromosomal instability and karyotype for tumor evolution [66].
Wnt3a is a ligand that activates the canonical Wnt pathway. Wnt3a stimulates tumor progression in glioblastoma [67], breast and prostate cancers [68,69], and malignant mesothelioma [70]. Other studies have shown that Wnt3a serves as a tumor suppressor based on two main findings. One is that bones engrafted with Wnt3a-expressing multiple myeloma H929 cells are preserved; the other is that treatment of myelomatous SCID mice carrying the primary disease with recombinant Wnt3a stimulates bone formation and attenuates multiple myeloma growth [71]. Marit et al. reported that Wnt3a inhibits the proliferation of several B-acute lymphoblastic leukemia cell lines [72]. However, Qi L. et al., analyzed Wnt3a expression in a large array of colon cancer tissue samples to determine its role in colon-cancer progression and observed a significant correlation between Wnt3a expression and histological differentiation, clinical stages, metastasis, and recurrence. The resulting data indicates that the upper stream factor of the Wnt signaling pathway may play an important role in colon cancer progression and agree with a recent study on colorectal cancer, in which results reveal that Wnt3a is highly expressed in the primary and metastatic sites and is significantly associated with expression of the metastasis-related protein matrix metalloproteinase (MMP)-9 [73]. In our study the correlation between BCL9 and Wnt3a both in polyps and in the adjacent mucosa confirms two aspects: the first is that the canonical Wnt pathway is already activated in the adjacent mucosa; the second that it is also activated in early carcinogenesis. Finally our results confirm Wnt3a as tumor suppressor.
There is no evidence to prove the exact function of Wnt5a. Some studies suggest that Wnt5a is a tumor suppressor, while others suggest the opposite. During the development of colorectal cancer, Wnt5a showed different functions in different signal transduction pathways [74]
Wnt5a mRNA is expressed in most normal tissues including the colon, but is highly up-regulated in the progression from normal tissue to carcinomas [75]. The expression of Wnt5a protein, however, seems to be downregulated as it is frequently inactivated in CRC by tumour-specific methylation and, thus, could be a potential biomarker [76]. Wnt5a is suggested to act as a tumor suppressor for CRC by antagonizing the Wnt/β-catenin signaling [76].
Traditionally, Wnt5a is believed to be the non-canonical Wnt ligand and activates Ca2+-dependent effectors and other non-canonical pathways through small Rho-GTPases and c-Jun-NH2-kinase [77]. However, its role in the progression of CRC is complicated and seems to be contradictory. Several studies proved that Wnt5a was silenced in most CRC cell lines and specimens due to frequent methylation in its promoter region [78,79]. The expression of Wnt5a was negatively correlated with the degree of tumor differentiation and the aggressive behavior [78,80]. Meanwhile, promoter methylation of Wnt5a was strongly associated with the microsatellite instability status of patients with CRC, and multiple histone modifications of Wnt5a were involved in its silencing and might promote colon cancer metastasis, providing evidence that epigenetic events may promote Wnt5a-mediated signaling in CRC [81,82]. On the contrary, other studies demonstrated that Wnt5a was upregulated consistently in intestinal polyps and tumor samples, and increased Wnt5a expression predicted the early recurrence or metastasis in colon cancer patients [83,84]. Wnt5a could also promote the migration of CRC cells by activating Fzd7-driven non-canonical Wnt signaling and enhance the cell stemness of CRC through activating the canonical Wnt signaling [85,86]
In our study the correlation between Wnt5a and LEF1 detected in healthy tissues could indicate that the Wnt non-canonical pathway is active in normal colonic tissue. Furthermore, these results also could denote a possible inflammatory state of donors undergoing screening colonoscopy, as both Wnt5a and LEF-1 are linked to the inflammatory state. Aberrant Wnt signaling is linked to defects in the chronic inflammatory response. Indeed, in the still normal colonic mucosa, various inflammatory mediators can actively contribute to the creation of a TME favorable to cell transformation, survival and proliferation [87]. The mutual interaction of epithelial cells within the TME influences the stages of tumorigenesis driven to a large extent by inflammation This may be an attractive therapeutic target to control inflammation in the colonic mucosa [88] Furthermore, IEC can drive the plasticity of stromal cells in the TME under the influence of extrinsic factors, such as diet, and microbiota composition contributing to inflammation and tumorigenesis in CRC [87,89] However, understanding the interactions between Wnt signaling and inflammatory/immune responses in tumor onset remains a necessary goal for both prevention and therapy, given that the majority of CRCs appear to be immunologically “cold” tumors unresponsive to therapies with immune checkpoint inhibitors [90].
5. Conclusions
This is the first study analyzing the gene expression of APC, Wnt3A, Wnt5A, BCL9 and LEF1 in colon polyps vs. adjacent mucosa and vs. normal mucosa from control individuals. These findings of altered expression levels of Wnt genes in apparently normal adjacent mucosa from patients with familial and sporadic colon polyps underline an interplay between tumor and surrounding colonic epithelium. This may aid in identifying patients at risk of developing cancer.
In conclusion, our study will improve the understanding of the pathogenesis in CRA. Finding essential genes, exploring their potential pathogenesis of colorectal adenoma, and developing gene-targeted drugs are urgent clinical and scientific problems to be solved.
Author Contributions
Conceptualization, M.C.C. and G.M.A.; Methodology, D.L.D., F.F., C.M., A.F.; Recruitment of patients: R.V., F.F., S.S., K.E., M.N.; Writing – Review & Editing, M.C.C, D.L.D., G.M.A.; Supervision, M.C.C.
Funding
This research received no external funding.
Institutional Review Board Statement
.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bonnington SN, Rutter MD. Surveillance of colonic polyps: Are we getting it right? World J Gastroenterol. 2016 Feb 14;22(6):1925-34. [CrossRef] [PubMed]
- Click B, Pinsky PF, Hickey T, Doroudi M, Schoen RE. Association of Colonoscopy Adenoma Findings With Long-term Colorectal Cancer Incidence. JAMA. 2018 May 15;319(19):2021-2031.
- Eide TJ. Risk of colorectal cancer in adenoma-bearing individuals within a defined population. Int J Cancer. 1986 Aug 15;38(2):173-6. [CrossRef] [PubMed]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990 Jun 1;61(5):759-67. [CrossRef] [PubMed]
- Yang B, Mao L, Li Y, Li Q, Li X, Zhang Y, Zhai Z. β-catenin, leucine-rich repeat-containing G protein-coupled receptor 5 and GATA-binding factor 6 are associated with the normal mucosa-adenoma-adenocarcinoma sequence of colorectal tumorigenesis. Oncol Lett. 2018;15:2287–2295.
- Kim N. H., Jung Y. S., Jeong W. S., et al. Miss rate of colorectal neoplastic polyps and risk factors for missed polyps in consecutive colonoscopies. Intestinal Research. 2017;15(3):411–418. [CrossRef]
- International Agency for Research on Cancer. Colorectal cancer screening. In IARC Handbooks of Cancer Prevention; International Agency for Research on Cancer: Lyon, France, 2019; Volume 17, p. 300.
- Lieberman D. A., Rex D. K., Winawer S. J., et al. Guidelines for colonoscopy surveillance after screening and polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2012;143(3):844–857. [CrossRef]
- Aceto GM, Catalano T, Curia MC. Molecular Aspects of Colorectal Adenomas: The Interplay among Microenvironment, Oxidative Stress, and Predisposition. Biomed Res Int. 2020 Mar 16;2020:1726309. [CrossRef] [PubMed]
- Dornblaser D, Young S, Shaukat A. Colon polyps: updates in classification and management. Curr Opin Gastroenterol. 2024 Jan 1;40(1):14-20. Epub 2023 Nov 1. [CrossRef] [PubMed]
- Galuppini F, Fassan M, Mastracci L, Gafà R, Lo Mele M, Lazzi S, Remo A, Parente P, D'Amuri A, Mescoli C, Tatangelo F, Lanza G. The histomorphological and molecular landscape of colorectal adenomas and serrated lesions. Pathologica. 2021 Jun;113(3):218-229. [CrossRef] [PubMed]
- Dubé C, Yakubu M, McCurdy BR, et al.. Risk of advanced adenoma, colorectal cancer, and colorectal cancer cortality in people with low-risk adenomas at baseline colonoscopy: a systematic review and meta-analysis. Am J Gastroenterol 2017;112:1790-1801. [CrossRef]
- Lucci-Cordisco E., Risio M., Venesio T., Genuardi M. The growing complexity of the intestinal polyposis syndromes. American Journal of Medical Genetics. Part A. 2013;161(11):2777–2787. [CrossRef]
- Corley D. A., Jensen C. D., Marks A. R., et al. Variation of adenoma prevalence by age, sex, race, and colon location in a large population: implications for screening and quality programs. Clinical Gastroenterology and Hepatology. 2013;11(2):172–180. [CrossRef]
- Pommergaard H. C., Burcharth J., Rosenberg J., Raskov H. The association between location, age and advanced colorectal adenoma characteristics: a propensity-matched analysis. Scandinavian Journal of Gastroenterology. 2017;52(1):1–4. [CrossRef]
- Klein J. L., Okcu M., Preisegger K. H., Hammer H. F. Distribution, size and shape of colorectal adenomas as determined by a colonoscopist with a high lesion detection rate: influence of age, sex and colonoscopy indication. United European Gastroenterology Journal. 2016;4(3):438–448.
- IJspeert J. E., van Doorn S. C., van der Brug Y. M., et al. The proximal serrated polyp detection rate is an easy-to-measure proxy for the detection rate of clinically relevant serrated polyps. Gastrointestinal Endoscopy. 2015;82(5):870–877. [CrossRef]
- Kim K. H., Kim K. O., Jung Y., et al. Clinical and endoscopic characteristics of sessile serrated adenomas/polyps with dysplasia/adenocarcinoma in a Korean population: a Korean Association for the Study of Intestinal Diseases (KASID) multicenter study. Scientific Reports. 2019;9(1):p. 3946. [CrossRef]
- Kahi C. J., Hewett D. G., Norton D. L., Eckert G. J., Rex D. K. Prevalence and variable detection of proximal colon serrated polyps during screening colonoscopy. Clinical Gastroenterology and Hepatology. 2011;9(1):42–46. [CrossRef]
- Leggett B., Whitehall V. Role of serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138(6):2088–2100. [CrossRef]
- Lochhead P, Chan AT, Giovannucci E, Fuchs CS, Wu K, Nishihara R, O'Brien M and Ogino S: Progress and opportunities in molecular pathological epidemiology of colorectal premalignant lesions. Am J Gastroenterol. 109:1205–1214. 2014.
- Dinarvand P, Davaro EP, Doan JV, Ising ME, Evans NR, Phillips NJ, Lai J and Guzman MA: Familial adenomatous polyposis syndrome: An update and review of extraintestinal manifestations. Arch Pathol Lab Med. 143:1382–1398. 2019.
- Taherian M, Lotfollahzadeh S, Daneshpajouhnejad P, Arora K. Tubular Adenoma. 2023 Jun 3. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan–. [PubMed]
- Tischoff I, Tannapfel A. Präkanzerosen im Kolon [Precancerous colorectal tumors]. Internist (Berl). 2013 Jun;54(6):691-8. German. [CrossRef] [PubMed]
- Zhang L, Shay JW. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J Natl Cancer Inst. 2017 Aug 1;109(8):djw332. [CrossRef] [PubMed]
- Schatoff EM, Leach BI, Dow LE. Wnt Signaling and Colorectal Cancer. Curr Colorectal Cancer Rep. 2017 Apr;13(2):101-110. Epub 2017 Feb 28. [CrossRef] [PubMed]
- Hankey W, Frankel WL, Groden J. Functions of the APC tumor suppressor protein dependent and independent of canonical WNT signaling: implications for therapeutic targeting. Cancer Metastasis Rev. 2018 Mar;37(1):159-172. [CrossRef] [PubMed]
- Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001 Oct;1(1):55-67. [CrossRef] [PubMed]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997 Mar 21;275(5307):1787-90. [CrossRef] [PubMed]
- Steinhart Z, Angers S. Wnt signaling in development and tissue homeostasis. Development. 2018 Jun 8;145(11):dev146589. [CrossRef] [PubMed]
- Han, W., He, L., Cao, B., Zhao, X., Zhang, K., Li, Y., … Wang, H. (2017). Differential expression of LEF1/TCFs family members in colonic carcinogenesis. Molecular Carcinogenesis, 56(11), 2372–2381. [CrossRef]
- F.H. Brembeck, Maria Wiese, Nathalie Zatula, Tamara Grigoryan, Yiyang Dai, Johannes Fritzmann, Walter Birchmeier, BCL9-2 Promotes Early Stages of Intestinal Tumor Progression,Gastroenterology,Volume 141, Issue 4,2011,Pages 1359-1370.e3,ISSN 0016-5085. [CrossRef]
- Brembeck FH, Schwarz-Romond T, Bakkers J, Wilhelm S, Hammerschmidt M, Birchmeier W. Essential role of BCL9-2 in the switch between beta-catenin's adhesive and transcriptional functions. Genes Dev. 2004 Sep 15;18(18):2225-30. Epub 2004 Sep 1. [CrossRef] [PubMed]
- Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S, Murone M, Züllig S, Basler K. Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell. 2002 Apr 5;109(1):47-60. [CrossRef] [PubMed]
- Thompson B, Townsley F, Rosin-Arbesfeld R, Musisi H, Bienz M. A new nuclear component of the Wnt signalling pathway. Nat Cell Biol. 2002 May;4(5):367-73. [CrossRef] [PubMed]
- Wu M, Dong H, Xu C, Sun M, Gao H, Bu F, Chen J. The Wnt-dependent and Wnt-independent functions of BCL9 in development, tumorigenesis, and immunity: Implications in therapeutic opportunities. Genes Dis. 2023 Apr 11;11(2):701-710. [CrossRef] [PubMed]
- Tufail, M., Wu, C. Wnt3a is a promising target in colorectal cancer. Med Oncol 40, 86 (2023). [CrossRef]
- Voloshanenko O, Erdmann G, Dubash TD, Augustin I, Metzig M, Moffa G, Hundsrucker C, Kerr G, Sandmann T, Anchang B, Demir K, Boehm C, Leible S, Ball CR, Glimm H, Spang R, Boutros M. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun. 2013;4:2610. [CrossRef] [PubMed]
- Qi L, Sun B, Liu Z, Cheng R, Li Y, Zhao X. Wnt3a expression is associated with epithelial-mesenchymal transition and promotes colon cancer progression. J Exp Clin Cancer Res. 2014 Dec 11;33(1):107. [CrossRef] [PubMed]
- Ferrer-Mayorga G, Niell N, Cantero R, González-Sancho JM, Del Peso L, Muñoz A, Larriba MJ. Vitamin D and Wnt3A have additive and partially overlapping modulatory effects on gene expression and phenotype in human colon fibroblasts. Sci Rep. 2019 May 30;9(1):8085. [CrossRef] [PubMed]
- Torres M. A., Yang-Snyder J. A., Purcell S. M., DeMarais A. A., McGrew L. L., Moon R. T. Activities of the Wnt-1 class of secreted signaling factors are antagonized by the Wnt-5A class and by a dominant negative cadherin in early Xenopus development. Journal of Cell Biology . 1996;133(5):1123–1137. [CrossRef]
- Bauer M., Bénard J., Gaasterland T., Willert K., Cappellen D. WNT5A encodes two isoforms with distinct functions in cancers. PLoS One . 2013;8(11). [CrossRef]
- Asem M., Buechler S., Wates R., Miller D., Stack M. Wnt5a signaling in cancer. Cancers . 2016;8(9):p. 79. [CrossRef]
- Muhammad Tufail, Changxin Wu,WNT5A: a double-edged sword in colorectal cancer progression,Mutation Research/Reviews in Mutation Research,Volume 792,2023,108465,ISSN 1383-5742. [CrossRef]
- Ficari F, Cama A, Valanzano R, Curia MC, Palmirotta R, Aceto G, Esposito DL, Crognale S, Lombardi A, Messerini L, Mariani-Costantini R, Tonelli F, Battista P. APC gene mutations and colorectal adenomatosis in familial adenomatous polyposis. Br J Cancer. 2000 Jan;82(2):348-53. [CrossRef] [PubMed]
- Catalano T, D'Amico E, Moscatello C, Di Marcantonio MC, Ferrone A, Bologna G, Selvaggi F, Lanuti P, Cotellese R, Curia MC, Lattanzio R, Aceto GM. Oxidative Distress Induces Wnt/β-Catenin Pathway Modulation in Colorectal Cancer Cells: Perspectives on APC Retained Functions. Cancers (Basel). 2021 Nov 30;13(23):6045. [CrossRef] [PubMed]
- Silviera ML, Smith BP, Powell J, Sapienza C. Epigenetic differences in normal colon mucosa of cancer patients suggest altered dietary metabolic pathways. Cancer Prev Res (Phila). 2012 Mar;5(3):374-84. Epub 2012 Feb 1. [CrossRef] [PubMed]
- Zhan, T., Rindtorff, N. & Boutros, M. Wnt signaling in cancer. Oncogene 36, 1461–1473 (2017). [CrossRef]
- Hankey, W., Frankel, W.L. & Groden, J. Functions of the APC tumor suppressor protein dependent and independent of canonical WNT signaling: implications for therapeutic targeting. Cancer Metastasis Rev 37, 159–172 (2018). [CrossRef]
- Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007 Oct 1;120(Pt 19):3327-35. [CrossRef] [PubMed]
- Xu D., Yuan L., Liu X., Li M., Zhang F., Gu X., Zhang D., Yang Y., Cui B., Tong J., Zhou J., Yu Z. EphB6 overexpression and Apc mutation together promote colorectal cancer. Oncotarget. 2016; 7: 31111-31121. Retrieved from https://www.oncotarget.com/article/9080/text/.
- Behrens J, von Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996 Aug 15;382(6592):638-42. [CrossRef] [PubMed]
- Xiao, L.; Zhang, C.; Li, X.; Jia, C.; Chen, L.; Yuan, Y.; Gao, Q.; Lu, Z.; Feng, Y.; Zhao, R.; et al. LEF1 Enhances the Progression of Colonic Adenocarcinoma via Remodeling the Cell Motility Associated Structures. Int. J. Mol. Sci. 2021, 22, 10870. [Google Scholar] [CrossRef] [PubMed]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. [CrossRef]
- Gao X., Mi Y., Ma Y., Jin W. LEF1 regulates glioblastoma cell proliferation, migration, invasion, and cancer stem-like cell self-renewal. Tumour Biol. 2014;35:11505–11511. [CrossRef]
- Hao Y.H., Lafita-Navarro M.C., Zacharias L., Borenstein-Auerbach N., Kim M., Barnes S., Kim J., Shay J., DeBerardinis R.J., Conacci-Sorrell M. Induction of LEF1 by MYC activates the WNT pathway and maintains cell proliferation. Cell Commun. Signal. 2019;17:129. [CrossRef]
- Xie Q, Tang T, Pang J, Xu J, Yang X, Wang L, Huang Y, Huang Z, Liu G, Tong D, Zhang Y, Wang L, Zhang D, Lan W, Liu Q, Jiang J. LSD1 Promotes Bladder Cancer Progression by Upregulating LEF1 and Enhancing EMT. Front Oncol. 2020 Jul 28;10:1234. [CrossRef] [PubMed]
- Yuan M., Zhang X., Zhang J., Wang K., Zhang Y., Shang W., Zhang Y., Cui J., Shi X., Na H., et al. DC-SIGN-LEF1/TCF1-miR-185 feedback loop promotes colorectal cancer invasion and metastasis. Cell Death Differ. 2020;27:379–395. [CrossRef]
- Kim G.H., Fang X.Q., Lim W.J., Park J., Kang T.B., Kim J.H., Lim J.H. Cinobufagin Suppresses Melanoma Cell Growth by Inhibiting LEF1. Int. J. Mol. Sci. 2020;21:6706. [CrossRef]
- Blazquez R., Rietkötter E., Wenske B., Wlochowitz D., Sparrer D., Vollmer E., Müller G., Seegerer J., Sun X., Dettmer K., et al. LEF1 supports metastatic brain colonization by regulating glutathione metabolism and increasing ROS resistance in breast cancer. Int. J. Cancer. 2020;146:3170–3183. [CrossRef]
- Heino S, Fang S, Lähde M, Högström J, Nassiri S, Campbell A, Flanagan D, Raven A, Hodder M, Nasreddin N, Xue HH, Delorenzi M, Leedham S, Petrova TV, Sansom O, Alitalo K. Lef1 restricts ectopic crypt formation and tumor cell growth in intestinal adenomas. Sci Adv. 2021 Nov 19;7(47):eabj0512. Epub 2021 Nov 17. [CrossRef] [PubMed]
- Sampietro J, Dahlberg CL, Cho US, Hinds TR, Kimelman D, Xu W. Crystal structure of a beta-catenin/BCL9/Tcf4 complex. Mol Cell. 2006 Oct 20;24(2):293-300. [CrossRef] [PubMed]
- Habib SJ, Acebrón SP. Wnt signalling in cell division: from mechanisms to tissue engineering. Trends Cell Biol. 2022 Dec;32(12):1035-1048. Epub 2022 Jun 15. [CrossRef] [PubMed]
- Chen J, Rajasekaran M, Xia H, Kong SN, Deivasigamani A, Sekar K, Gao H, Swa HL, Gunaratne J, Ooi LL, Xie T, Hong W, Hui KM. CDK1-mediated BCL9 phosphorylation inhibits clathrin to promote mitotic Wnt signalling. EMBO J. 2018 Oct 15;37(20):e99395.Epub 2018 Sep 14. [CrossRef] [PubMed]
- Suzuki K, Okuno Y, Kawashima N, Muramatsu H, Okuno T, Wang X, Kataoka S, Sekiya Y, Hamada M, Murakami N, Kojima D, Narita K, Narita A, Sakaguchi H, Sakaguchi K, Yoshida N, Nishio N, Hama A, Takahashi Y, Kudo K, Kato K, Kojima S. MEF2D-BCL9 Fusion Gene Is Associated With High-Risk Acute B-Cell Precursor Lymphoblastic Leukemia in Adolescents. J Clin Oncol. 2016 Oct 1;34(28):3451-9. Epub 2016 Aug 9. [CrossRef] [PubMed]
- Feng M, Wu Z, Zhou Y, Wei Z, Tian E, Mei S, Zhu Y, Liu C, He F, Li H, Xie C, Jin J, Dong J, Yang D, Yu K, Qian J, Lambrechts D, Wang MW, Zhu D. BCL9 regulates CD226 and CD96 checkpoints in CD8+ T cells to improve PD-1 response in cancer. Signal Transduct Target Ther. 2021 Aug 20;6(1):313. [CrossRef] [PubMed]
- Kaur N, Chettiar S, Rathod S, Rath P, Muzumdar D, Shaikh ML, Shiras A. Wnt3a mediated activation of Wnt/β-catenin signaling promotes tumor progression in glioblastoma. Mol Cell Neurosci. 2013 May;54:44-57. Epub 2013 Jan 19. [CrossRef] [PubMed]
- Lamb R, Ablett MP, Spence K, Landberg G, Sims AH, Clarke RB. Wnt pathway activity in breast cancer sub-types and stem-like cells. PLoS One. 2013 Jul 4;8(7):e67811. [CrossRef] [PubMed]
- Verras M, Brown J, Li X, Nusse R, Sun Z. Wnt3a growth factor induces androgen receptor-mediated transcription and enhances cell growth in human prostate cancer cells. Cancer Res. 2004 Dec 15;64(24):8860-6. [CrossRef] [PubMed]
- Fox SA, Richards AK, Kusumah I, Perumal V, Bolitho EM, Mutsaers SE, Dharmarajan AM. Expression profile and function of Wnt signaling mechanisms in malignant mesothelioma cells. Biochem Biophys Res Commun. 2013 Oct 11;440(1):82-7. Epub 2013 Sep 13. [CrossRef] [PubMed]
- Qiang YW, Shaughnessy JD Jr, Yaccoby S. Wnt3a signaling within bone inhibits multiple myeloma bone disease and tumor growth. Blood. 2008 Jul 15;112(2):374-82. Epub 2008 Mar 14. [CrossRef] [PubMed]
- Nygren MK, Døsen G, Hystad ME, Stubberud H, Funderud S, Rian E. Wnt3A activates canonical Wnt signalling in acute lymphoblastic leukaemia (ALL) cells and inhibits the proliferation of B-ALL cell lines. Br J Haematol. 2007 Feb;136(3):400-13. Epub 2006 Dec 8. [CrossRef] [PubMed]
- Lee MA, Park JH, Rhyu SY, Oh ST, Kang WK, Kim HN. Wnt3a expression is associated with MMP-9 expression in primary tumor and metastatic site in recurrent or stage IV colorectal cancer. BMC Cancer. 2014 Feb 24;14:125. [CrossRef] [PubMed]
- Guangshun Sun, Liangliang Wu, Guoqiang Sun, Xuesong Shi, Hongyong Cao & Weiwei Tang (2021) WNT5a in Colorectal Cancer: Research Progress and Challenges, Cancer Management and Research,, 2483-2498. [CrossRef]
- Smith K, Bui TD, Poulsom R, Kaklamanis L, Williams G, Harris AL. Up-regulation of macrophage wnt gene expression in adenoma-carcinoma progression of human colorectal cancer. Br J Cancer. 1999 Oct;81(3):496-502. [CrossRef] [PubMed]
- Ying J, Li H, Yu J, Ng KM, Poon FF, Wong SC, Chan AT, Sung JJ, Tao Q. WNT5A exhibits tumor-suppressive activity through antagonizing the Wnt/beta-catenin signaling, and is frequently methylated in colorectal cancer. Clin Cancer Res. 2008 Jan 1;14(1):55-61. [CrossRef] [PubMed]
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005 Apr 14;434(7035):843-50. [CrossRef] [PubMed]
- Abdelmaksoud-Dammak R, Miladi-Abdennadher I, Saadallah-Kallel A, Khabir A, Sellami-Boudawara T, Frikha M, Daoud J, Mokdad-Gargouri R. Downregulation of WIF-1 and Wnt5a in patients with colorectal carcinoma: clinical significance. Tumour Biol. 2014 Aug;35(8):7975-82. Epub 2014 May 16. [CrossRef] [PubMed]
- Hibi K, Mizukami H, Goto T, Kitamura Y, Sakata M, Saito M, Ishibashi K, Kigawa G, Nemoto H, Sanada Y. WNT5A gene is aberrantly methylated from the early stages of colorectal cancers. Hepatogastroenterology. 2009 Jul-Aug;56(93):1007-9. [PubMed]
- Cao YC, Yang F, Liu XH, Xin X, Wang CC, Geng M. [Expression of Wnt5a, APC, β-catenin and their clinical significance in human colorectal adenocarcinoma]. Zhonghua Zhong Liu Za Zhi. 2012 Sep;34(9):674-8. Chinese. [CrossRef] [PubMed]
- Rawson JB, Mrkonjic M, Daftary D, Dicks E, Buchanan DD, Younghusband HB, Parfrey PS, Young JP, Pollett A, Green RC, Gallinger S, McLaughlin JR, Knight JA, Bapat B. Promoter methylation of Wnt5a is associated with microsatellite instability and BRAF V600E mutation in two large populations of colorectal cancer patients. Br J Cancer. 2011 Jun 7;104(12):1906-12. Epub 2011 May 17. [CrossRef] [PubMed]
- Li Q, Chen H. Silencing of Wnt5a during colon cancer metastasis involves histone modifications. Epigenetics. 2012 Jun 1;7(6):551-8. Epub 2012 Jun 1. [CrossRef] [PubMed]
- Lai C, Robinson J, Clark S, Stamp G, Poulsom R, Silver A. Elevation of WNT5A expression in polyp formation in Lkb1+/- mice and Peutz-Jeghers syndrome. J Pathol. 2011 Apr;223(5):584-92. Epub 2011 Feb 21. [CrossRef] [PubMed]
- Bakker ER, Das AM, Helvensteijn W, Franken PF, Swagemakers S, van der Valk MA, ten Hagen TL, Kuipers EJ, van Veelen W, Smits R. Wnt5a promotes human colon cancer cell migration and invasion but does not augment intestinal tumorigenesis in Apc1638N mice. Carcinogenesis. 2013 Nov;34(11):2629-38. Epub 2013 Jun 12. [CrossRef] [PubMed]
- Dong X, Liao W, Zhang L, Tu X, Hu J, Chen T, Dai X, Xiong Y, Liang W, Ding C, Liu R, Dai J, Wang O, Lu L, Lu X. RSPO2 suppresses colorectal cancer metastasis by counteracting the Wnt5a/Fzd7-driven noncanonical Wnt pathway. Cancer Lett. 2017 Aug 28;402:153-165. Epub 2017 Jun 6. [CrossRef] [PubMed]
- Chen Z, Tang C, Zhu Y, Xie M, He D, Pan Q, Zhang P, Hua D, Wang T, Jin L, Qi X, Zhu Y, Yao X, Jin J, Ma X. TrpC5 regulates differentiation through the Ca2+/Wnt5a signalling pathway in colorectal cancer. Clin Sci (Lond). 2017 Feb 1;131(3):227-237. Epub 2016 Nov 28. [CrossRef] [PubMed]
- Schmitt M, Greten FR. The inflammatory pathogenesis of colorectal cancer. Nat Rev Immunol. 2021 Oct;21(10):653-667. [CrossRef]
- Jridi I, Canté-Barrett K, Pike-Overzet K, Staal FJT. Inflammation and Wnt Signaling: Target for Immunomodulatory Therapy? Front Cell Dev Biol. 2021 Feb 4;8:615131. [CrossRef]
- Cui G. TH9, TH17, and TH22 Cell Subsets and Their Main Cytokine Products in the Pathogenesis of Colorectal Cancer. Front Oncol. 2019 Oct 4;9:1002. [CrossRef]
- Liu JL, Yang M, Bai JG, Liu Z, Wang XS. "Cold" colorectal cancer faces a bottleneck in immunotherapy. World J Gastrointest Oncol. 2023 Feb 15;15(2):240-250. [CrossRef]
Figure 1.
The figure shows the five Wnt/β-catenin pathway genes (APC, Wnt3a, Wnt5a, BCL9, LEF1) expression means in healthy colorectal mucosa, adjacent mucosa and polyps, compared by Kruskal-Wallis non parametric test for independent samples. Statistically significance differences were detected in APC and Wnt5a expressions in adjacent mucosa compared to normal tissue (P=0.01), and, for Wnt5a expression, also in polyps compared to normal tissue (P=0.004).
Figure 1.
The figure shows the five Wnt/β-catenin pathway genes (APC, Wnt3a, Wnt5a, BCL9, LEF1) expression means in healthy colorectal mucosa, adjacent mucosa and polyps, compared by Kruskal-Wallis non parametric test for independent samples. Statistically significance differences were detected in APC and Wnt5a expressions in adjacent mucosa compared to normal tissue (P=0.01), and, for Wnt5a expression, also in polyps compared to normal tissue (P=0.004).
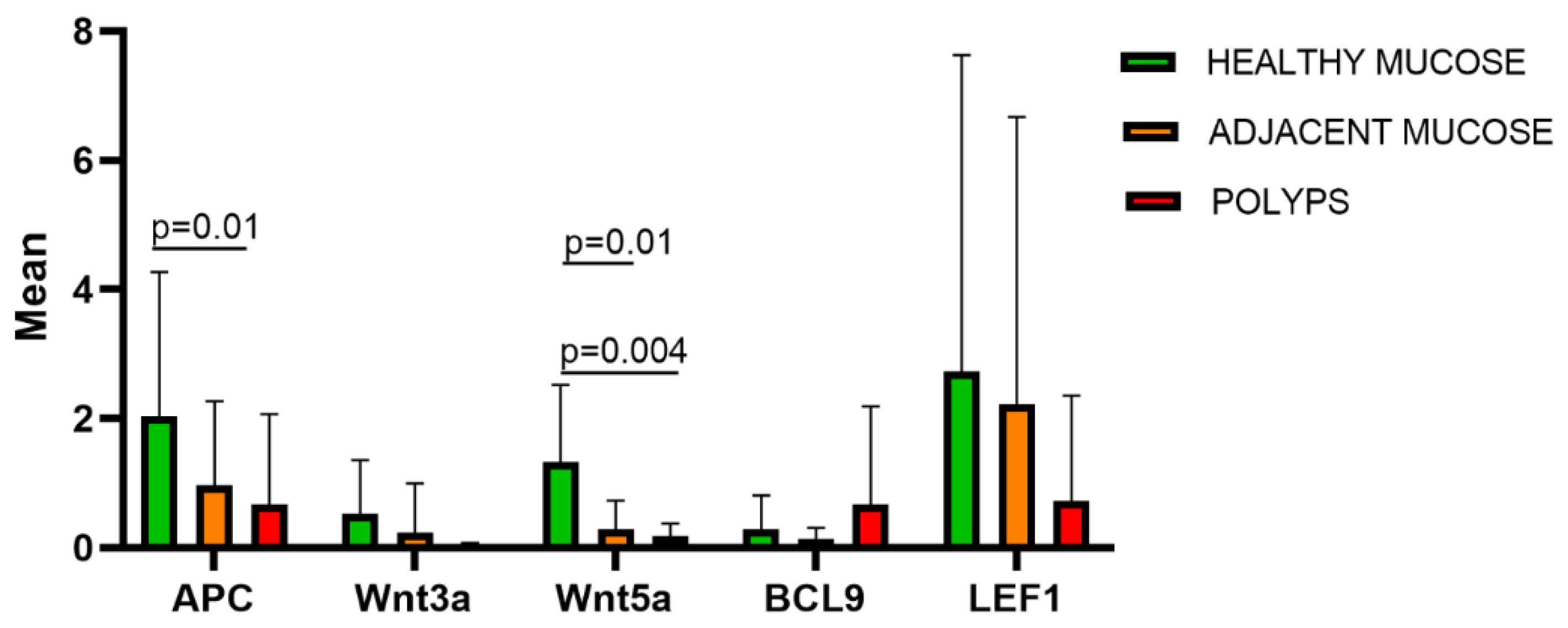
Figure 2.
The figure represents an heat map of the genes expression means of APC, Wnt3a, Wnt5a, BCL9, LEF1, in healthy mucosa (a), adjacent mucosa (b) and polyps (c). It is shown how gene expression tends to reduce from healthy to pathological tissue. APC, Wnt5a, LEF1 are the most expressed genes in healthy mucosa.
Figure 2.
The figure represents an heat map of the genes expression means of APC, Wnt3a, Wnt5a, BCL9, LEF1, in healthy mucosa (a), adjacent mucosa (b) and polyps (c). It is shown how gene expression tends to reduce from healthy to pathological tissue. APC, Wnt5a, LEF1 are the most expressed genes in healthy mucosa.
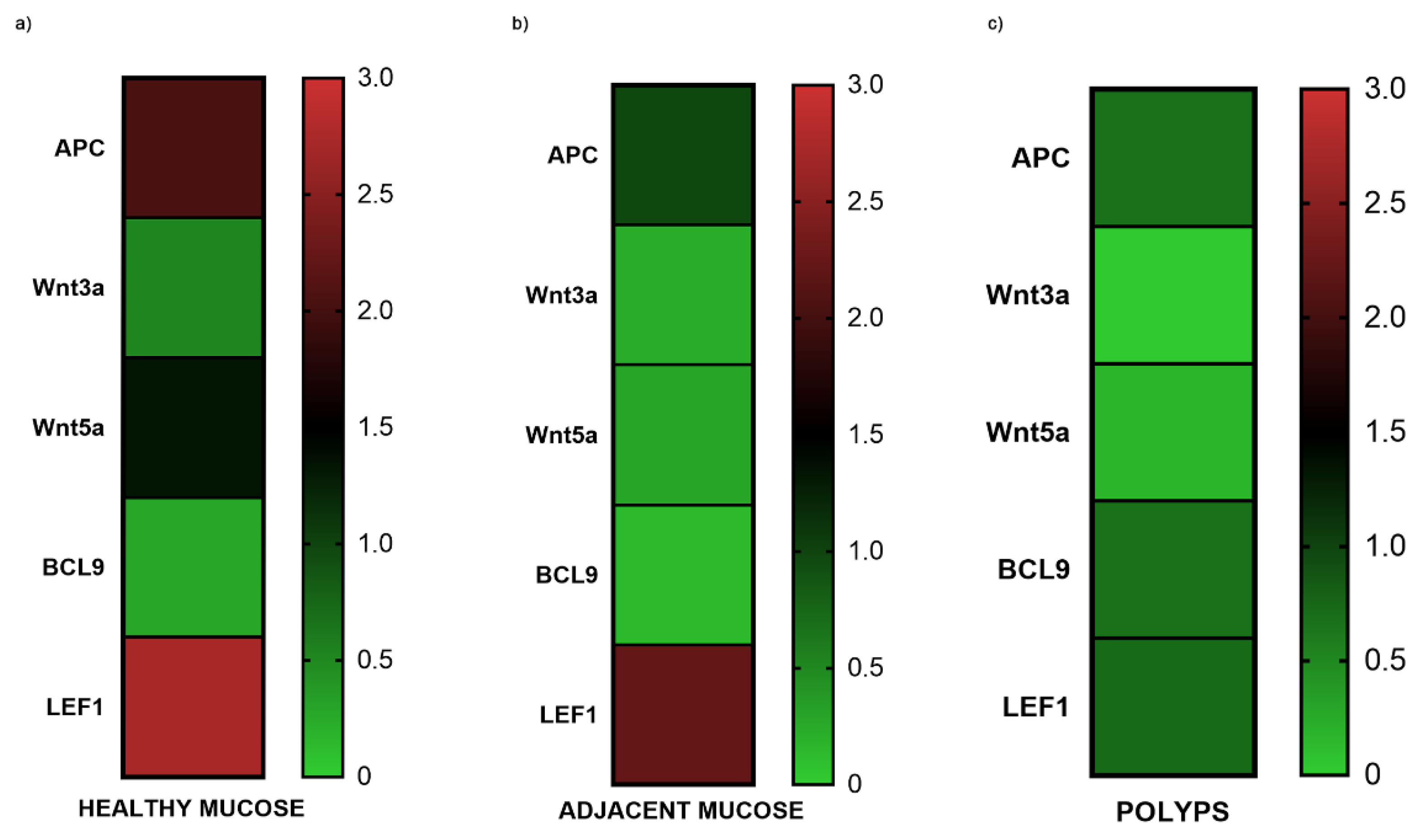
Figure 3.
Western blotting analysis in familial adenomas determining the protein expression levels of APC and β-Catenin in polyp (P) vs. adjacent mucosa (M). Data shows are representative of three independent experiments. The average expression levels of panel “a” were determined by densitometric analysis (panel b and c) and calculated in relation to the β-Actin level. kD: Kilodalton as protein molecular weight unit.
Figure 3.
Western blotting analysis in familial adenomas determining the protein expression levels of APC and β-Catenin in polyp (P) vs. adjacent mucosa (M). Data shows are representative of three independent experiments. The average expression levels of panel “a” were determined by densitometric analysis (panel b and c) and calculated in relation to the β-Actin level. kD: Kilodalton as protein molecular weight unit.
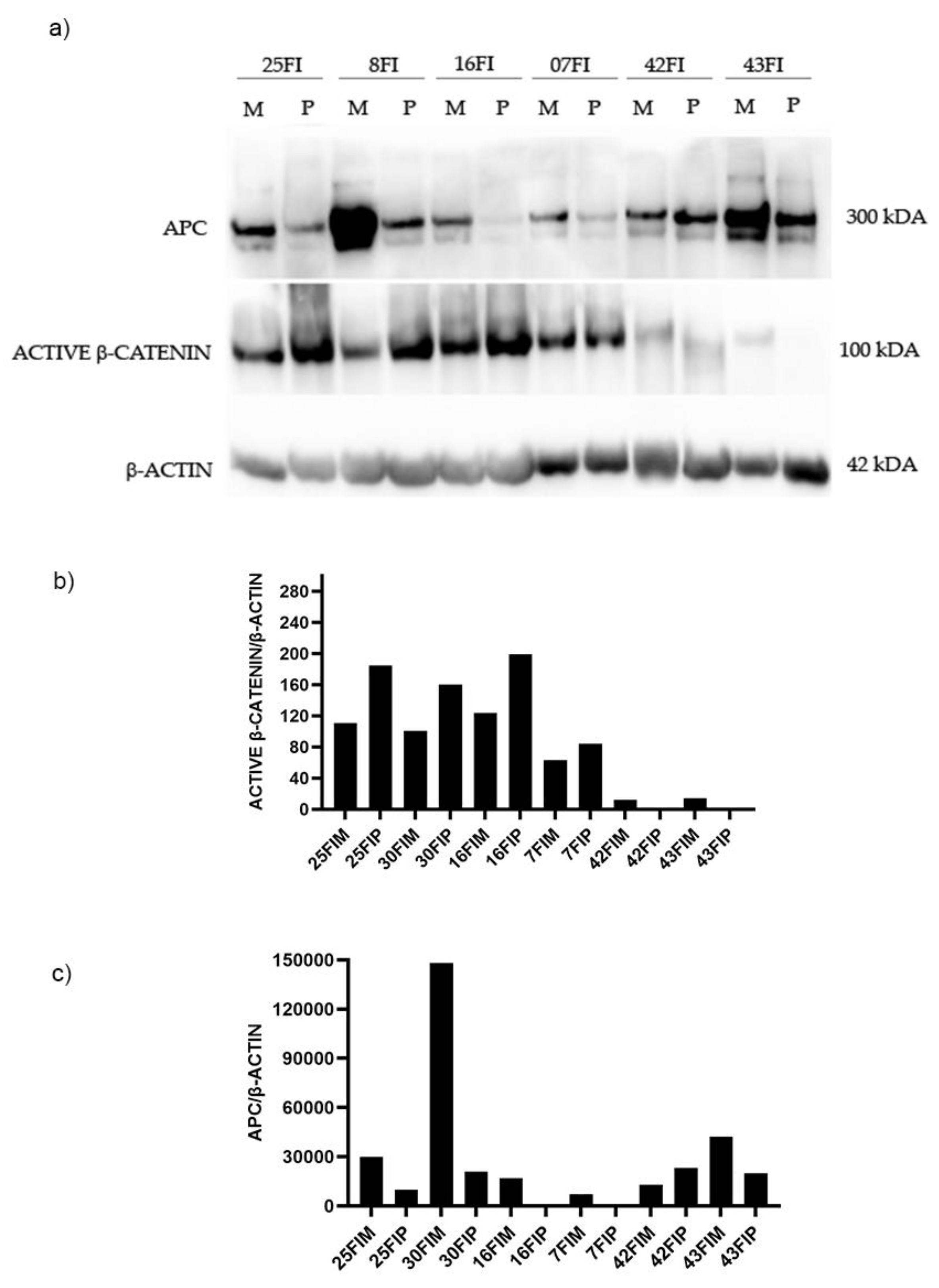
Figure 4.
Western blotting analysis in sporadic adenomas determining the protein expression levels of APC and β-Catenin in polyp (P) vs. adjacent mucosa (M). Data shows are representative of three independent experiments. The average expression levels of panel “a” were determined by densitometric analysis (panel b and c) and calculated in relation to the β-Actin level. kD: Kilodalton as protein molecular weight unit.
Figure 4.
Western blotting analysis in sporadic adenomas determining the protein expression levels of APC and β-Catenin in polyp (P) vs. adjacent mucosa (M). Data shows are representative of three independent experiments. The average expression levels of panel “a” were determined by densitometric analysis (panel b and c) and calculated in relation to the β-Actin level. kD: Kilodalton as protein molecular weight unit.
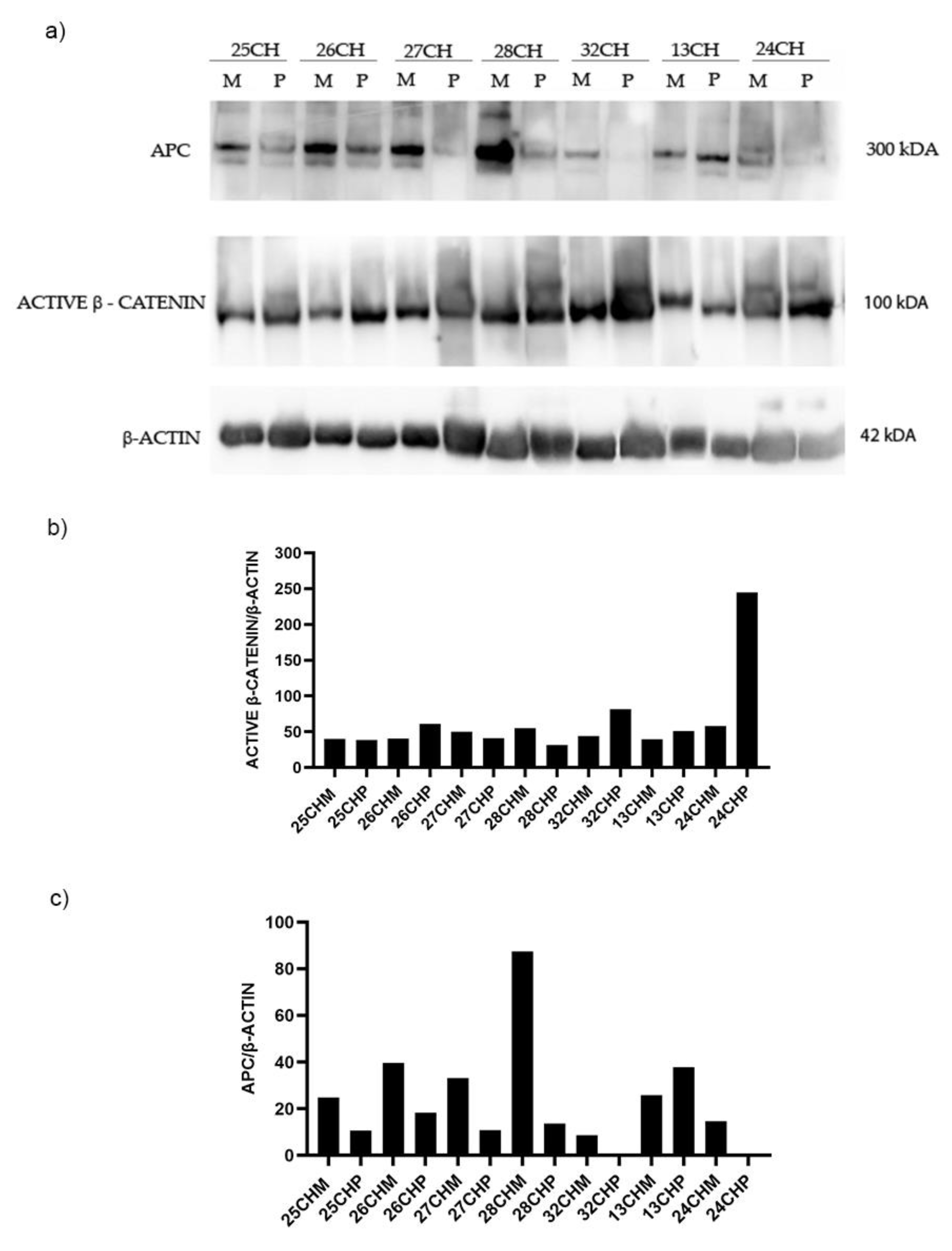

Table 1.
Clinical and histopathological characteristics of 41 patients with FAP and sporadic adenomas analyzed.
Table 1.
Clinical and histopathological characteristics of 41 patients with FAP and sporadic adenomas analyzed.
| Patients with FAP polyps | ||||||
|---|---|---|---|---|---|---|
| Case | age | sex | Phenotype | Site and size of polyps | Dysplasia (L or H) |
n.of polyps |
| 5FI | 25 | F | Adenomatous | Diffuse or “carpet”, <1cm | HGD | 1060 |
| 6FI a,e | 58 | M | Adenomatous | Diffuse | HGD | 25 |
| 7FI a,e | 28 | F | Adenomatous | Diffuse | HGD | 375 |
| 8FI b,e | 18 | F | Adenomatous (Tubular-villous) |
Diffuse | HGD | 415 |
| 9FI c,e | 15 | F | Adenomatous | Diffuse | LGD | 375 |
| 16FI | n.a | F | Adenomatous | Diffuse | LGD | n.a |
| 25FI | n.a. | M | Adenomatous | Diffuse | LGD | n.a. |
| 26FI | 46 | F | Adenomatous | Diffuse | LGD | 835 |
| 31FI | M | Adenomatous | Diffuse | |||
| 33FI | 49 | F | Adenomatous | Diffuse | LGD | 97 |
| 35FI c,e | 31 | M | Adenomatous | Diffuse | LGD | 550 |
| 36FI | 42 | F | Adenomatous | Diffuse | LGD | 250 |
| 39FI | 42 | M | Adenomatous | Diffuse | LGD | 430 |
| 40FI a,d,e | 61 | F | Adenomatous | Diffuse | HGD | 730 |
| 41FI | 49 | M | Adenomatous | Diffuse | LGD | 1025 |
| 42FI | 42 | F | Adenomatous and amartomatous | Diffuse | LGD | 210 |
| 43FI | 36 | M | Adenomatous | Diffuse | LGD? | n.a |
| Patients with sporadic polyps | ||||||
| case | age | sex | Phenotype | Site and size of polyps |
Dysplasia (L or H) |
Morphology |
| 1CH | 50 | M | Hyperplastic | Sigma, 6mm | LGD | Spl |
| 2CH | 67 | M | Tubular | Sigma, 10mm | LGD | Spl |
| 3CH | 49 | M | Hyperplastic | Sigma, 4mm | LGD | Spl |
| 9CH | 47 | M | Tubular-villous | Retto, 15mm | LGD | Ppl |
| 11CH | 57 | F | Hyperplastic-adenomatous | Descending, 4mm |
LGD | Spl |
| 13CH | 83 | F | Tubular | Descending, 15mm | LGD | Spl |
| 15CH | 37 | F | Villous | Sigma, 50mm |
HGD | Ppl |
| 16CH | 60 | M | Tubular | Cecum, 15mm |
LGD | Ppl |
| 17CH | 66 | M | Tubular-villous | Sigma, 15mm | LGD | Ppl |
| 18CH | 64 | M | Tubular-villous | Descending, 8mm | LGD | Spl |
| 21CH | 78 | M | Tubular-villous | Sigma, 10mm |
LGD | Spl |
| 22CH | 67 | M | Tubular-villous | Rectum, 10mm |
LGD | Spl |
| 23CH | 68 | F | Tubular-villous | Sigma, 10mm |
LGD | Ppl |
| 24CH | 59 | M | Tubular-villous | Ascending | LGD | Ppl |
| 25CH | 77 | M | Tubular-villous | Descending | HGD | Ppl |
| 26CH | 69 | M | Tubular | Splenic flexure, 10mm | LGD | Spl |
| 27CH | 61 | F | Tubular-villous | Sigma, 15mm | LGD | Ppl |
| 28CH | 77 | M | Tubular-villous | Hepatic flexure, 5mm | LGD | Spl |
| 29CH | 47 | M | Hyperplastic-adenomatous | Descending, 20mm | Not atypical | Ppl |
| 30CH | 53 | M | Hyperplastic-adenomatous | Retto-sigma, 7mm |
Not atypical | Spl |
| 31CH | 76 | M | Tubular | Ascending, 5mm | LGD | Spl |
| 32CH | 51 | M | Tubular-villous | Ascending, 45mm | LGD | Spl |
| 33CH | 68 | F | Tubular-villous | Colon, 40mm |
LGD | LST-G |
| 34CH | 67 | M | Tubular | Colon sx, 7mm |
LGD | Ppl |
a Presence of rectal cancer; b APC mutation: c.4666_4665ins(p.Thr1556fs);cAPC mutation: c.2805 C>(p.Tyr935X); d APCmutation: c.3927_3931del(p.Glu1309_Asp.fsx1312); eFicari F, Cama A, et all., Br J Cancer. 2000 Jan;82(2):348-53. Ppl: Pedunculated polypoid lesion; Spl: Sessile polypoid lesion; LST-G: Laterally Spreading Tumor (LST)-granular shape (G); HGD: High Grade Dysplasia; LGD: Low grade Dysplasia.
Table 2.
Correlation among APC, Wnt3a, Wnt5a, BCL9, LEF1 gene expression in 52 colon tissue samples. Correlation is significant at the 0.05 level (2-tailed).
Table 2.
Correlation among APC, Wnt3a, Wnt5a, BCL9, LEF1 gene expression in 52 colon tissue samples. Correlation is significant at the 0.05 level (2-tailed).
| Wnt3a | BCL9 | |||||
|---|---|---|---|---|---|---|
| Group |
Spearman’s rho (2-tailed) |
P-value |
Spearman’s rho (2-tailed) |
P-value | ||
| Polyps and adjacent mucosa | n. 42 | |||||
| Wnt3a | 0,336376 | 0,0294 | ||||
| BCL9 | 0,336376 | 0,0294 | ||||
| Wnt5a | LEF1 | |||||
| Group |
Spearman’s rho (2-tailed) |
P-value |
Spearman’s rho (2-tailed) |
P-value | ||
| Normal colonic mucosa | n. 10 | |||||
| Wnt5a | 0,69697 | 0,025097 | ||||
| LEF1 | 0,69697 | 0,025097 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



