Introduction
The industrial production of recombinant proteins secreted by E. coli is essential in the biotechnology industry (Yoon et al., 2010). E. coli has advantageous properties, such as rapid growth to high cell densities on a low-cost carbon source, and serves as a platform for recombinant protein production (Baneyx & Mujacic, 2004). Due to its extensive use as a model system, E. coli genetics have been extensively characterized, resulting in the development of numerous tools that aid in chromosome engineering, gene replication, and gene expression (Mergulhao et al., 2005). Recombinant proteins can be targeted to different compartments within E. coli, including the cytoplasm, periplasm, and extracellular space. However, cytoplasmic expression of recombinant proteins is challenging. Proteins expressed in the cytoplasm may be susceptible to degradation by cellular proteases or accumulate as insoluble inclusion bodies, rendering them inactive (Mergulhao & Monteiro, 2007). In addition, cytoplasmic expression of certain proteins can have toxic effects on host cells, leading to reduced protein yields or even cell death (Mierendorf et al., 1998). In contrast, extracellular expression in E. coli offers several advantages for the production of recombinant proteins, including simplified downstream processing, reduced host cell contamination, high expression levels, reduced protein degradation, and increased solubility (Choi & Lee, 2004). E. coli has two major secretory pathways, the Sec and Tat pathways (Costa et al., 2015). The Sec pathway, which facilitates the transport of proteins from the cytoplasmic membrane, was the first secretory pathway identified in bacteria (Beckwith, 2013). This pathway has demonstrated the ability to translocate proteins in an unfolded state, thereby facilitating the efficient export of target proteins (Crane & Randall, 2017). In contrast, the Tat pathway has the distinct ability to transport fully folded proteins across the cytoplasmic membrane (Choi & Lee, 2004).
Chitosanase (EC 3.2.1.132) catalyzes the hydrolysis of β-1,4-glucosidic linkages in the polymeric chitosan, a type of polysaccharide. Chitosan is a high molecular weight polymer that requires enzymatic degradation for human consumption. It is broken down into chitosan oligosaccharides by extracellular chitosanases found in various bacteria. Chitosan oligosaccharides offer more significant biological functions compared to chitosan, including higher levels of antibacterial, antioxidant, blood cholesterol reduction, blood pressure improvement, infection prevention, arthritis control, and antitumor effects (Yabuki et al., 1988). The GH-46 type chitosanases, especially those derived from Bacillus and Streptomyces, have been extensively studied for their catalytic functions, enzymatic mechanisms, and protein structures (Wang et al., 2008).
Previously, our studies have shown that chitosanase derived from B. subtilis CH1 and B. subtilis CH2 are secreted into the extracellular space of B. subtilis (Oh & Lee, 2006; Oh et al., 2011). In this study, we compared the efficiency of two chitosanase signal peptides from B. subtilis in mediating the extracellular secretion of chitosanase when introduced into E. coli.
Materials and Methods
Molecular Characterization of Chitosanases from B. subtilis Strains
Gene Cloning of Chitosanases from B. subtilis CH1 and CH2
Primers were designed to amplify the coding region, including the signal sequences, from the genomic DNA of B. subtilis strains CH1 and CH2. The forward primer, CH-F (5′-GAGACATATGAAAATCAGTATGCAAAAAGC-3′), contained a NdeI site, and the reverse primer, CH-R (5′-GAGAGGATCCTTATTTGATTACAAAATTACCGTA-3′), contained a BamHI site. The PCR mixture consisted of 5 μL of 10X Ex Taq DNA polymerase buffer, 8 μL of 2.5 mM dNTP mixture, 5 μL of 25 mM MgCl2, 100 pmol of each primer, 1 μg of genomic DNA, and 3 units of Ex Taq DNA polymerase (Takara Bio Inc., Shiga, Japan) in a final volume of 50 μL. PCR cycling conditions were set as follows: initial denaturation at 94 °C for 5 minutes, followed by 30 cycles of 94 °C for 1 minute, 55 °C for 30 seconds, and 72 °C for 1 minute, with a final extension at 72 °C for 5 minutes. PCR products were verified by 1% agarose gel electrophoresis and purified using a gel purification kit (Bioneer Co., Daejeon, South Korea). The verified PCR products and the pET-11a vector (Novagen, Madison, USA) were digested with NdeI and BamHI restriction enzymes (Takara Bio Inc., Shiga, Japan) according to the manufacturer's instructions. The digested fragments were then ligated into the vector using T4 DNA ligase (Takara Bio Inc., Shiga, Japan). The ligation mixture was transformed into E. coli DH5alpha cells by the heat shock method. Transformed colonies were selected on LB agar plates containing 100 μg/mL ampicillin (Merck, Darmstadt, Germany) and incubated in 4 mL LB broth at 37°C overnight with shaking at 180 rpm. Plasmid DNA (pET11a-CH1 and pET11a-CH2) was extracted from the cultured cells using an Accuprep Nano-plus Plasmid Mini Extraction Kit (Bioneer Co., Daejeon, South Korea). The nucleotide sequences of the plasmids were confirmed by sequencing at Macrogen (Seoul, South Korea).
Overexpression of Chitosanases from B. subtilis CH1 and CH2 in E. coli
The recombinant plasmids pET11a-CH1 and pET11a-CH2 were transformed into E. coli BL21(DE3) (Novagen, Madison, USA) cells. Transformed cells were plated on LB agar (BD, New jersey, USA) containing 100 μg/mL ampicillin and incubated at 37°C for 16 hours. Single colonies were inoculated into 20 mL LB broth supplemented with 100 μg/mL ampicillin and cultured at 37°C with shaking at 180 rpm. When the optical density at 600 nm reached 0.4, recombinant protein expression was induced by the addition of IPTG (Merck, Darmstadt, Germany) at concentrations of 0.1 mM and 1 mM. Cultures were then incubated at 30°C with shaking at 180 rpm for three days. Samples were collected on day 1 and day 3 post-induction, with 1 mL aliquots taken for each time point. Samples were centrifuged at 7,000 × g for 5 minutes to separate the pellet and supernatant. The collected pellets were resuspended in 1 mL cold distilled water and disrupted using a sonicator (Qsonica, Connecticut, USA) to release intracellular contents. The cell lysates and supernatants were analyzed by 10% SDS-polyacrylamide gel electrophoresis (PAGE) using MOPS buffer (Thermo Fisher Scientific, Massachusetts, USA). The gels were then stained with Coomassie Brilliant Blue (Merck, Darmstadt, Germany) to visualize proteins.
Protein Measurement
Protein quantification was performed using ImageJ software (NIH, Maryland, USA). Bovine serum albumin (BSA) protein (Thermo Fisher Scientific, Massachusetts, USA) was used as the standard for protein quantification.
Chitosanase Assay
Chitosanase activity was measured using soluble chitosan as the substrate. To prepare soluble chitosan, 1 mL of acetic acid was added to 80 mL of distilled water, and 1 g of chitosan (Merck, Darmstadt, Germany) was dissolved in this mixture. The pH of the solution was then adjusted to 5.5 using 10 N NaOH. Subsequently, the volume was augmented to 100 mL through the addition of distilled water. Subsequently, the enzyme solution was combined with 1% soluble chitosan, thereby initiating the reaction. The reaction was conducted at 50 °C for 10 minutes, in accordance with the methodology described by (Rondle & Morgan, 1955). One unit (U) of enzyme activity was defined as the amount of enzyme required to produce 1 μmol of reducing sugar per minute. Glucosamine was employed as the standard for quantification.
Application of mCsn2-SP to hSOD
The hSOD nucleotide sequence linked with mCsn2-SP was genetically modified through the implementation of codon optimization using the codon optimization tool (
https://sg.idtdna.com/CodonOpt), which was submitted to GenBank under the accession numbers OR661283. The gene sequence was synthesized and cloned into the pTOP Blunt V2 vector by Macrogen (Seoul, South Korea). Primers were designed for the purpose of amplifying the synthesized gene and inserting it into the pET-11a vector via the In-Fusion method. The forward primer was designed as 5'-TAGCATAATATA
CATATGAAAATTTCTATGCAG-3', while the reverse primer was designed as 5'-TTGTTAGCAGCC
GGATCCTTACTGTGCGATCC-3'. The polymerase chain reaction (PCR) mixture consisted of 5 μL of 10X Ex
Taq DNA polymerase buffer, 8 μL of a 2.5 mM dNTP mixture, 20 pmol of each primer, 10 ng of template, and 3 units of Ex
Taq DNA polymerase, resulting in a final volume of 50 μL. The PCR cycling conditions were set as follows: an initial denaturation at 94 °C for 5 minutes was performed, followed by 2 cycles of 94 °C for 30 seconds, 50 °C for 30 seconds, and 72 °C for 40 seconds. Subsequently, 15 cycles of 94 °C for 30 seconds, 60 °C for 30 seconds, and 72 °C for 40 seconds were conducted, followed by a final extension at 72 °C for 5 minutes. The PCR product was purified using a gel purification kit. The PCR product and the pET-11a vector digested with
NdeI and
BamHI were fused using the EZ-Fusion
TM Cloning Kit (Enzynomics, Daejeon, Korea) according to manual. It was then subcloned into
E. coli DH5alpha as described above and finally transformed into
E. coli BL21(DE3) cells. The cells were cultured in 100 mL of LB broth containing 100 μg/mL ampicillin at 37 °C with shaking at 180 rpm. At an optical density of 0.7 at 600 nm, protein expression was induced by the addition of IPTG to a final concentration of 1 mM. The induction was conducted at 20 °C with agitation at 180 rpm for 24 hours. The inducted sample 1mL was centrifuged at 7,000 × g for 5 minutes to separate the pellet and supernatant. The pellets were resuspended in 1 mL of cold distilled water and disrupted using a sonicator. The lysed cells were subjected to centrifugation at 15,000 × g for one hour with the objective of separating the soluble and insoluble proteins. The supernatant was then transferred to a new tube, while the remaining pellet was diluted with 1 ml of distilled water. Additionally, periplasmic protein isolation was conducted using 50 ml of induced cells, in accordance with the established protocol for the osmotic shock method (French et al., 1996). The proteins were analyzed by 10% SDS- PAGE using MES buffer (Thermo Fisher Scientific, Massachusetts, USA).
Statistical Analysis
All experiments were performed in triplicate, and the data were analyzed using GraphPad Prism version 8 (GraphPad Software, Califonia, USA). Results are presented as the mean ± standard deviation. Significant differences between the groups were determined using analysis of variance (ANOVA), with a p-value of less than 0.05 considered statistically significant.
Results
Molecular Characterization of Chitosanase
The nucleotide (nt) sequence of the CH1CSN gene contains an 831-base pair (bp) open reading frame (ORF) encoding a chitosanase protein of 277 amino acid (aa) residues (GenBank accession number GU001718). Conversely, the nt sequence of the CH2CSN gene has an 813-bp ORF encoding a chitosanase protein of 271 aa residues (GenBank accession number GU001716).
Figure 1 show the nt and aa sequences in comparison with the chitosanase from
B. subtilis strain 168 (168CSN). The signal peptides of CH1CSN and CH2CSN, predicted to be located in the N-terminal region, consist of 35 aa and 29 aa, respectively, and were identified as Sec pathway signal peptides using the SignalP program (Supplementary Figure 1). Despite an 11 bp difference in the mature chitosanase N-terminal sequences of CH1CSN and CH2CSN, their amino acid sequences remained identical. Notwithstanding, the signal sequence exhibited 20 bp mismatches in the nt sequence, 18 nt sequences were deleted, resulting in the removal of six amino acids, and two nt sequences were replaced, though this did not affect the amino acids. Of interest, a repeated sequence (AAAAAGCAG) was identified in the signal sequences of 168CSN and CH1CSN, and based on these sequences, we were able to confirm that some sequences were deleted in the signal sequence of CH2CSN (
Figure 1). In the presence of the signal peptide, the predicted molecular weight of CH1CSN is 31.5 kDa, whereas in its absence, it is 27.4 kDa. Similarly, the predicted molecular weight for CH2CSN is 30.7 kDa with the signal peptide and 27.4 kDa without it. All chitosanase sequences lacked cysteine residues and predicted the GH46 domain including two catalytic residues using the protein BLAST tool (
Figure 1b).
Expression and Extracellular Secretion Analysis of Chitosanase in E. coli
To express CH1CSN and CH2CSN from
B. subtilis in
E. coli and evaluate their secretion efficiency, the chitosanase genes including the signal sequences were amplified by PCR and cloned into the pET11a vector. The constructs were then transformed into
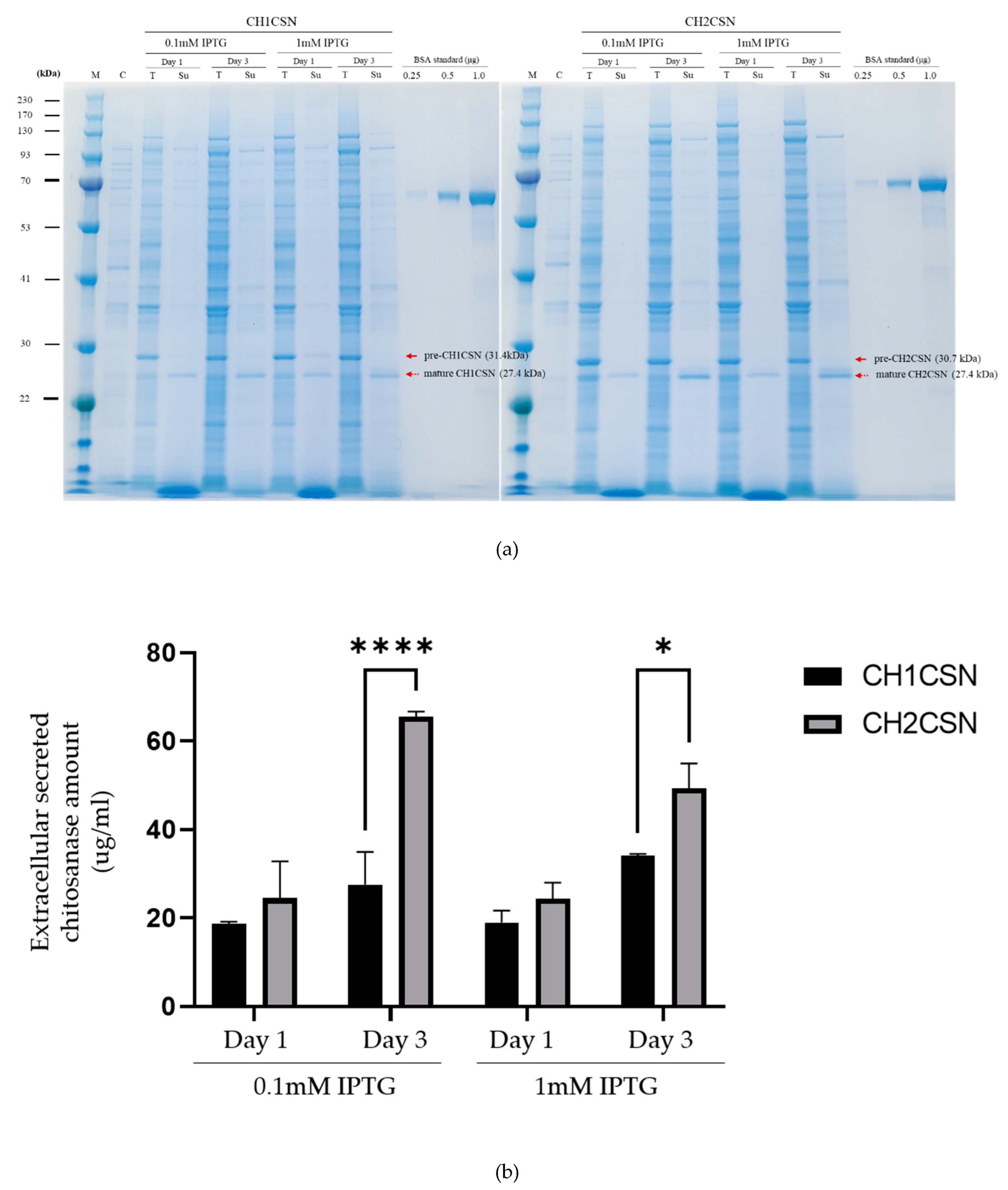
E. coli BL21(DE3) cells. The chitosanase proteins were induced by different IPTG concentrations (0.1 and 1 mM) and different times (1 and 3 days) at 30°C. SDS-PAGE was employed to differentiate chitosanase proteins based on the cleavage status of their signal peptides. Proteins with intact signal peptides remained in the cytoplasm, while those with cleaved signal peptides were located in the periplasm and secreted extracellularly (
Figure 2a). Quantitative analysis of these proteins was conducted using image analysis software, and the detailed results are presented in
Table 1. The data confirm that the signal peptides not only facilitated the export of chitosanase from the cytoplasm to the periplasm but also enabled its subsequent secretion outside the cell. The induction of CH1CSN expression with 0.1 mM and 1 mM IPTG resulted in an increase in protein secretion on day 3 compared to day 1. In the case of CH2CSN, secretion levels were found to be comparable at both IPTG concentrations on day 1. However, on day 3, a higher level was observed at 0.1 mM than at 1 mM IPTG (
Figure 2). It is noteworthy that the concentration of chitosanase was significantly elevated in the CH2CSN group relative to the CH1CSN group on day 3 with 0.1 mM IPTG. Similarly, chitosanase secretion levels were elevated in the CH2CSN group on day 3 with 1 mM IPTG (
Figure 2,
Table 1). The total expression of chitosanase, which includes cytoplasmic, and extracellular components, was highest at 117.28 μg/mL when CH2CSN was treated with 0.1 mM IPTG for 3 days to induce expression. These results confirmed that a naturally mutated signal peptide from
B. subtilis affected the efficiency of recombinant protein secretion in
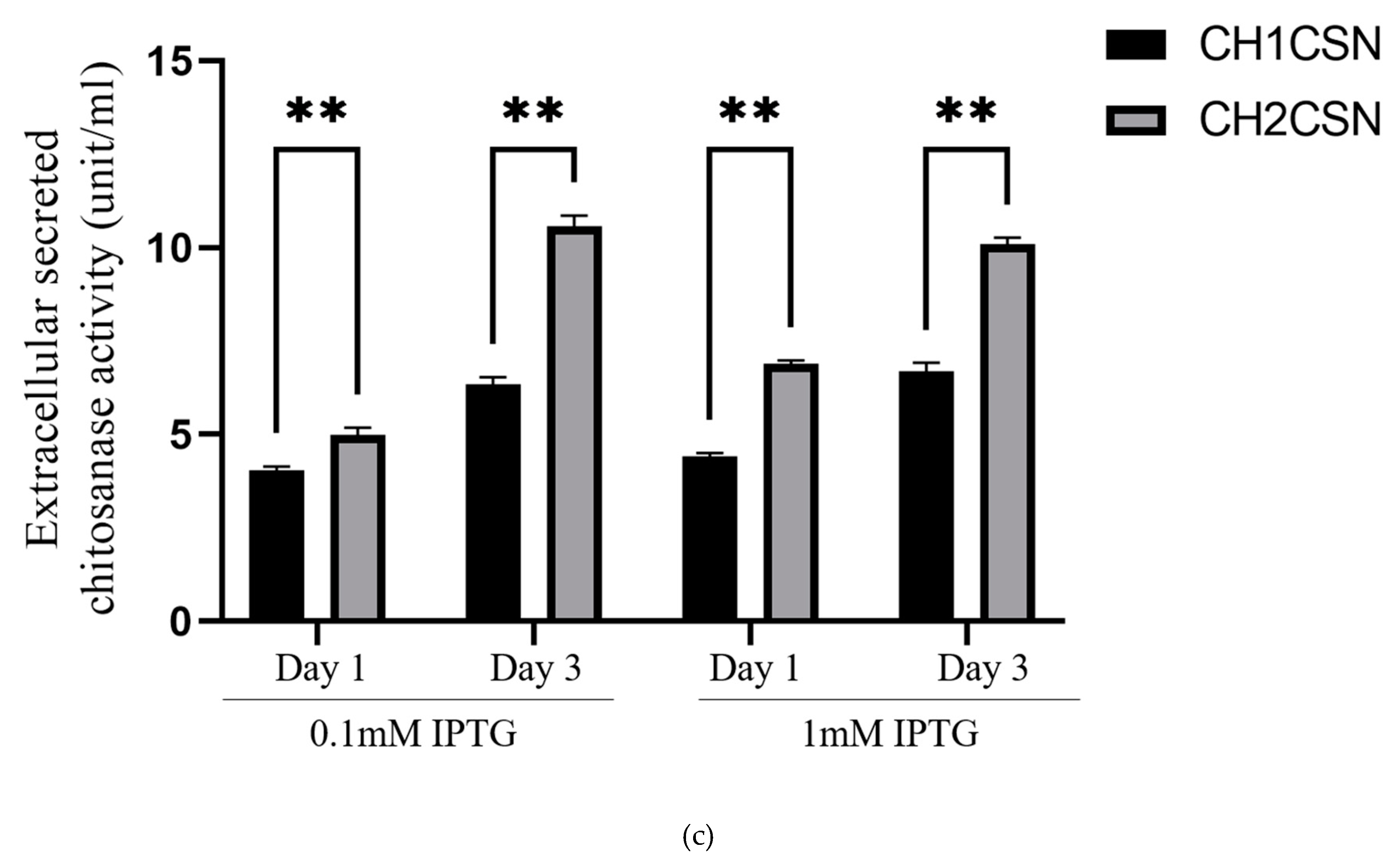
E. coli. The activity of CH1CSN was observed to be 4.04 U/ml and 6.35 U/ml on day 1 and day 3, respectively, at a 0.1 mM IPTG concentration. At a 1 mM IPTG concentration, the activity was observed to be 4.41 U/ml on day 1 and 6.69 U/ml on day 3. The activity of CH2CSN at a 0.1 mM IPTG concentration was recorded as 4.99 U/ml and 10.56 U/ml on day 1 and day 3, respectively. At a 1 mM IPTG concentration, the activity was 6.88 U/ml on day 1 and 10.09 U/ml on day 3 (
Figure 2c).
Expression of mCsn2-SP Linked hSOD in E. coli
The gene sequence of mCsn2-SP-linked hSOD was synthesized and subsequently inserted into the pET-11a vector, which was then transformed into
E. coli BL21(DE3) cells. An attempt was also made to induce expression at 30°C, but no hSOD expression was detected (data not shown). Therefore, the experiment was performed at 20°C only. The results of inducing protein expression at 20°C for 24 hours are shown in
Figure 3, where two distinct bands were observed, corresponding to pre-hSOD and mature hSOD forms, respectively. The predicted molecular weight of the pre-hSOD protein was 19.1 kDa. That of the mature hSOD protein was 15.8 kDa. To confirm that mature hSOD migrated to the periplasm, the osmotic shock method was used. The majority of mature hSOD that migrated to the periplasmic region was soluble, while the pre-hSOD that remained in the cytoplasmic region was insoluble. No protein was secreted into the supernatant.
Discussion
In the field of biotechnology, it has become increasingly apparent that there is no universal signal peptide that can universally enhance the optimal secretion of desired target proteins across different bacterial expression hosts (Freudl, 2018). The first systematic study to explore the effects of signal peptide variations on the secretion production of heterologous proteins was reported by Brockmeier et al. (Brockmeier et al., 2006). One surprising outcome of this study was that signal peptides optimal for one protein were often ineffective for another, underscoring the necessity for a specific combination between the signal peptide and the target protein. Therefore, the discovery of new signal peptides provides fresh options for the production of recombinant proteins, highlighting the tailored approach needed in optimizing secretion systems for diverse proteins.
In this study, we investigated the genetic characterization of chitosanase in two
B. subtilis strains and produced it as a recombinant protein in
E. coli to determine the secretion properties of the signal peptide. We compared the genetic profile of the known 168CSN with that of CH1CSN and CH2CSN, as shown in
Figure 1a. The results show that there is a small difference of only 1 bp between 168CSN and CH1CSN, while there is a larger difference of 30 bp in CH2CSN. There is also a difference of 31 bp between CH1CSN and CH2CSN. Looking at the signal sequence alone, there is no difference between Csn168-SP and Csn1-SP, while a 20 bp difference was observed in mCsn2-SP. Interestingly, the sequences of Csn168-SP and Csn1-SP contain a repeat sequence (AAAAAGCAG) that appears to be central to the 18 bp deletion mutation identified in mCsn2-SP. This finding is consistent with the work of Bzymek and Lovett, who showed that genetic diversity in bacteria often results from genetic rearrangements in which simple sequence repeats result in a variety of insertions or deletions (Bzymek & Lovett, 2001). As shown in
Figure 1b, a comparison of the amino acid sequences of the chitosanases revealed that the mature chitosanase sequences were all identical. However, mCsn2-SP showed a deletion of six consecutive amino acids (ADFWKK) compared to Csn168-SP and Csn1-SP. Signal peptides are typically divided into three main regions: the N-region, which carries a positive charge and assists in the translocation process; the H-region, which is hydrophobic and binds to the membrane in an alpha-helix structure; and the C-region, which is polar and contains the cleavage site that releases the mature protein (Rusch & Kendall, 2007). Specially, the N-region plays a crucial role in facilitating protein translocation by interacting with the negatively charged phosphate groups in the lipid bilayer, which is vital for the effective movement of proteins. Despite the H-region and C-region of mCsn2-SP being identical to those of Csn168-SP and Csn1-SP, the N-region of mCsn2-SP exhibited a net charge decrease from +2 to +1 due to the deletion of six amino acids. Although the net positive charge in the N-region of a signal peptide is crucial for the efficient translocation of target proteins, an increase in net positive charge does not always result in beneficial outcomes. In fact, higher positive charges can lead to negative consequences such as protein aggregation, jamming of the secretion translocase, and activation of stress-related genes (Owji et al., 2018).
We determined the chitosanase secretion efficiency of Csn1-SP and mCsn2-SP as a function of concentration and time of IPTG treatment, a protein expression inducer in
E. coli. SDS-PAGE showed that both forms of pre-chitosanase, in which the signal peptide is not cleaved and is located in the cytoplasm, and mature chitosanase, in which the signal peptide has been cleaved and translocated to the periplasmic region, were observed in the total protein of the cells, and some of the protein was released into the culture supernatant (
Figure 2a). The difference in length of the signal peptides resulted in the size of pre-CH1CSN (31.5 kDa) being larger in molecular weight than pre-CH2CSN (30.7 kDa), while the mature forms of CH1CSN and CH2CSN were the same size (27.4 kDa). The results show that the signal peptide of the naturally mutated CH2CSN significantly improves the efficiency of protein secretion compared to CH1CSN under all conditions used in the experiment (
Figure 2b,
Table 1). This result suggests that minor alterations in the signal peptide sequence can have substantial impacts on the protein export process in bacterial systems. Consequently, these results may imply that such mutations contribute to the enhancement of genomic diversity and highlight the potential of adaptive evolutionary processes in microbial genetics (Vale et al., 2022). To determine the potential for the use of this mCsn2-SP in the production of other heterologous recombinant proteins, we ligated human-derived hSOD. Previous studies have employed the OmpA signal peptide to facilitate the translocation of hSOD to the periplasmic region in
E. coli (Mao et al., 2010; Takahara et al., 1988). Similarly, our results demonstrated that mCsn2-SP was also effective in translocating hSOD to the periplasmic region of
E. coli, thereby confirming its usefulness (
Figure 3).
Overall, the secretory capability of E. coli provides valuable advantages in terms of increased yield, simplified purification, reduced cell toxicity, improved post-translational modifications, and scalability, making it a popular choice for recombinant protein expression and production (Baldi et al., 2007). The Sec pathway is a well-known secretory pathway in E. coli and is involved in the secretion of various proteins. Representative proteins secreted via the Sec pathway in E. coli include MalE (Beena et al., 2004), OmpA (Pechsrichuang et al., 2016), OmpC (Wang et al., 2016), OmpF (Forster et al., 2013), PelB (Shi et al., 2021), and PhoA (Miksch et al., 2008) signal peptides. As signal peptides in the Sec pathway, Csn1-SP and mCsn2-SP were also identified. We also identified a mutant chitosanase signal peptide, mCsn2-SP, and successfully demonstrated secretion of recombinant proteins in E. coli. This study confirmed whether mCsn2-SP has a higher secretion efficiency than Csn1-SP in E. coli. A previous study by another group showed that the E. coli OmpA signal sequence was more effective in both expression and secretion than the native Bacillus signal peptide (Pechsrichuang et al., 2016). In Gram-negative bacteria, such as E. coli, the Sec pathway plays a key role in protein secretion. It is also known to recognize cleavage sites and selectively target the Sec translocase. Nevertheless, these results confirm that the signal peptide, originally from Gram-positive bacteria, operates effectively even when interacting with distinct receptors within the secretion system of Gram-negative bacteria.
Collectively, we compared the nucleotide and amino acid sequences of CH1CSN and CH2CSN from B. subtilis and found that a repetitive sequence in the Csn1-SP resulted in an 18 bp base deletion in the mCsn2-SP. This was manifested as a deletion of 6 aa at the end of the N-region of mCsn2-SP. When CH1CSN and CH2CSN were produced as recombinant proteins in E. coli, the secretion rate of CH2CSN was found to be superior to that of CH1CSN under all expression induction conditions, demonstrating that the deletion of the N-region that affects translocation within mCsn2-SP is an evolutionary consequence of its evolution relative to Csn1-SP. Also, we have confirmed that mCsn2-SP can also be applied to the production of heterologous proteins. The findings presented herein offer a potential avenue for the utilization of naturally mutated mCsn2-SP for the secretion of heterologous proteins for academic or industrial production purposes.
Acknowledgements
This research was supported by the Development of Technology for Biomaterialization of Marine Fishery Byproducts of the Korea Institute of Marine Science & Technology Promotion (KIMST), funded by the Ministry of Oceans and Fisheries (KIMST-20220128) and the Korea Institute of Ocean Science and Technology (PEA0124).
Conflicts of Interest
The authors have no financial conflicts of interest to declare.
Ethical Statements
Not applicable.
References
- Baldi, L., Hacker. Recombinant protein production by large-scale transient gene expression in mammalian cells: state of the art and future perspectives. Biotechnology letters 2007, 29, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, F., & Mujacic, M. (2004). Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol, 22(11), 1399-1408. [CrossRef]
- Beckwith, J. (2013). The Sec-dependent pathway. Res Microbiol, 164(6), 497-504. [CrossRef]
- Beena, K., Udgaonkar. Effect of signal peptide on the stability and folding kinetics of maltose binding protein. Biochemistry 2004, 43, 3608–3619. [Google Scholar] [CrossRef] [PubMed]
- Brockmeier, U., Caspers, M., Freudl, R., Jockwer, A., Noll, T., & Eggert, T. (2006). Systematic screening of all signal peptides from B. subtilis: A powerful strategy in optimizing heterologous protein secretion in gram-positive bacteria. Journal of Molecular Biology, 362(3), 393-402. [CrossRef]
- Bzymek, M., & Lovett, S. T. (2001). Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc Natl Acad Sci U S A, 98(15), 8319-8325. [CrossRef]
- Choi, J. H., & Lee, S. Y. (2004). Secretory and extracellular production of recombinant proteins using Escherichia coli. Appl Microbiol Biotechnol, 64(5), 625-635. [CrossRef]
- Costa, T. R., Felisberto-Rodrigues, C., Meir, A., Prevost, M. S., Redzej, A., Trokter, M., & Waksman, G. (2015). Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nature Reviews Microbiology, 13(6), 343-359. [CrossRef]
- Crane, J. M., & Randall, L. L. (2017). The Sec System: Protein Export in Escherichia coli. EcoSal Plus, 7(2). [CrossRef]
- Forster, S., Brandt, M., Mottok, D. S., Zschuttig, A., Zimmermann, K., Blattner, F. R., Gunzer, F., & Pohlmann, C. (2013). Secretory expression of biologically active human Herpes virus interleukin-10 analogues in Escherichia coli via a modified Sec-dependent transporter construct. BMC Biotechnol, 13, 8. [CrossRef]
- French, C., Keshavarz-Moore, E., & Ward, J. M. (1996). Development of a simple method for the recovery of recombinant proteins from the Escherichia coli periplasm. Enzyme and Microbial Technology, 19(5), 332-338. [CrossRef]
- Freudl, R. (2018). Signal peptides for recombinant protein secretion in bacterial expression systems. Microbial Cell Factories, 17. [CrossRef]
- Mao, L., Stathopulos, P. B., Ikura, M., & Inouye, M. (2010). Secretion of human superoxide dismutase in Escherichia coli using the condensed single-protein-production system. Protein Sci, 19(12), 2330-2335. [CrossRef]
- Mergulhao, F. J., & Monteiro, G. A. (2007). Periplasmic targeting of recombinant proteins in Escherichia coli. Methods Mol Biol, 390, 47-61. [CrossRef]
- Mergulhao, F. J., Summers, D. K., & Monteiro, G. A. (2005). Recombinant protein secretion in Escherichia coli. Biotechnol Adv, 23(3), 177-202. [CrossRef]
- Mierendorf, R. C., Morris, B. B., Hammer, B., & Novy, R. E. (1998). Expression and Purification of Recombinant Proteins Using the pET System. Methods Mol Med, 13, 257-292. [CrossRef]
- Miksch, G., Ryu. Factors that influence the extracellular expression of streptavidin in Escherichia coli using a bacteriocin release protein. Applied Microbiology and Biotechnolog 2008, 81, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.-H., & Lee. Isolation, purification and characterization of chitosanase from Bacillus subtilis CH1. Journal of Aquaculture 2006, 19(1), 40–46. [Google Scholar]
- Oh, C., De Zoysa, M., Kang, D. H., Lee, Y., Whang, I., Nikapitiya, C., Heo, S. J., Yoon, K. T., Affan, A., & Lee, J. (2011). Isolation, purification, and enzymatic characterization of extracellular chitosanase from marine bacterium Bacillus subtilis CH2. J Microbiol Biotechnol, 21(10), 1021-1025. [CrossRef]
- Owji, H., Nezafat, N., Negandaripour, M., Hajiebrahimi, A., & Ghasemi, Y. (2018). A comprehensive review of signal peptides: Structure, roles, and applications. European Journal of Cell Biology, 97(6), 422-441. [CrossRef]
- Pechsrichuang, P., Songsiriritthigul, C., Haltrich, D., Roytrakul, S., Namvijtr, P., Bonaparte, N., & Yamabhai, M. (2016). OmpA signal peptide leads to heterogenous secretion of B. subtilis chitosanase enzyme from E. coli expression system. Springerplus, 5, 1-10.
- Rondle, C. J., & Morgan, W. T. (1955). The determination of glucosamine and galactosamine. Biochem J, 61(4), 586-589. [CrossRef]
- Rusch, S. L., & Kendall, D. A. (2007). Interactions that drive Sec-dependent bacterial protein transport. Biochemistry, 46(34), 9665-9673. [CrossRef]
- Shi, L., Liu, H., Gao, S., Weng, Y., & Zhu, L. (2021). Enhanced Extracellular Production of IsPETase in Escherichia coli via Engineering of the pelB Signal Peptide. J Agric Food Chem, 69(7), 2245-2252. [CrossRef]
- Takahara, M., Sagai, H., Inouye, S., & Inouye, M. (1988). Secretion of Human Superoxide Dismutase in Escherichia Coli. Bio/Technology, 6(2), 195-198. [CrossRef]
- Teufel, F., Almagro Armenteros, J. J., Johansen, A. R., Gíslason, M. H., Pihl, S. I., Tsirigos, K. D., Winther, O., Brunak, S., von Heijne, G., & Nielsen, H. (2022). SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature biotechnology, 40(7), 1023-1025.
- Vale, F. F., Lehours, P., & Yamaoka, Y. (2022). Editorial: The Role of Mobile Genetic Elements in Bacterial Evolution and Their Adaptability [Editorial]. Frontiers in Microbiology, 13. [CrossRef]
- Wang, J., Zhou, W., Yuan, H., & Wang, Y. (2008). Characterization of a novel fungal chitosanase Csn2 from Gongronella sp. JG. Carbohydr Res, 343(15), 2583-2588. [CrossRef]
- Wang, P., Ma, J., Zhang, Y., Zhang, M., Wu, M., Dai, Z., & Jiang, M. (2016). Efficient Secretory Overexpression of Endoinulinase in Escherichia coli and the Production of Inulooligosaccharides. Appl Biochem Biotechnol, 179(5), 880-894. 5). [CrossRef]
- Yabuki, M., Uchiyama, A., Suzuki, K., Ando, A., & Fujii, T. (1988). Purification and Properties of Chitosanase from Bacillus-Circulans Mh-K1. Journal of General and Applied Microbiology, 34(3), 255-270. [CrossRef]
- Yoon, S. H., Kim, S. K., & Kim, J. F. (2010). Secretory production of recombinant proteins in Escherichia coli. Recent Pat Biotechnol, 4(1), 23-29. [CrossRef]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).