
Submitted:
24 June 2024
Posted:
25 June 2024
You are already at the latest version
Abstract
Dinoflagellates are one of the largest groups of marine microalgae and exhibit diverse trophic strategies. Within these niches, dinoflagellates can produce secondary metabolites that are known to be toxic, which can lead to ecologically harmful blooms. Amphidinium carterae is one species of dinoflagellate that produces toxic compounds and is used as a model for dinoflagellate studies. The extent to which the microbiome surrounding A. carterae affects its growth and secondary metabolite synthesis is still being assessed, as well as the impact of bacterial data on sequencing and assembly. An antibiotic cocktail was previously shown to eliminate 16S amplification from the dinoflagellate culture. Even with drastically reduced bacterial numbers, bacterial populations are likely to be present. In this experiment we used novel Nanopore long-read sequencing techniques on A. carterae cultures to assemble 15 full bacterial genomes ranging from 2.9-6.0 Mb and found that the use of antibiotics decreased the percentage of reads mapping back to bacteria. We also identified shifts in the microbiome composition and identified a potentially deleterious bacterial species arising in the absence of the antibiotic-treatment. Multiple antibiotic-resistance genes were identified, as well as evidence that the bacterial population do not contribute to toxic secondary metabolite synthesis.
Keywords:
dinoflagellate
; microbiome
; long-read sequencing
1. Introduction
Dinoflagellates are unicellular organisms, which occupy multiple trophic levels in marine ecosystems, ranging from photo-autotrophy, predation, mixotrophy, to even parasitism [1,2,3]. They are a significant component of phytoplankton communities, responsible for producing a large portion of the world’s oxygen and serve as the primary food source for a wide array of marine organisms, including zooplankton, small fish, and filter feeders [4,5,6]. Their ability to photosynthesize and sometimes feed enables them to thrive in various environmental conditions, making them vital for maintaining and disturbing ecological balance and supporting biodiversity in aquatic habitats.
Some species of dinoflagellates are also known to cause harmful algal blooms (HABs), which can have devastating ecological and economic impacts [7,8,9]. These blooms occur when dinoflagellate populations grow explosively under favorable conditions, producing toxins that can kill fish, shellfish, and other marine life, and pose serious health risks to humans through seafood consumption or direct exposure. The toxins can also lead to significant economic losses in fisheries, tourism, and public health sectors. On a broader level the single-species dominance of a bloom influences the microbial community both directly and indirectly.
Many dinoflagellate cultures contain abundant bacterial populations, likely originating from the original sample isolated from the environment [10,11,12]. The role of bacterial populations on dinoflagellate toxin production is one that remains hotly debated, with prior research showing evidence of dinoflagellate dependence on bacteria for toxin-production and others showing the opposite [13,14,15,16,17,18]. Some of these studies involve the filtering of bacteria from dinoflagellate cultures, followed by null amplification of 16S regions as verification to prove a culture is axenic, and subsequent high-performance liquid chromatography (HPLC) analysis to show if toxin is still produced in the axenic culture [19,20,21]. Results of these studies have been contradictory.
Currently, consistent culturing of dinoflagellate cultures with antibiotics is a common practice to select for organisms of interest and maintain monoclonal conditions. Eukaryotic cultures can be maintained with antibiotics to decrease the effects of bacterial populations on downstream analysis such as sequencing efforts, toxin analysis, and translation rate studies [22]. Although antibiotic use in dinoflagellate culturing can be useful, there is concern about bacterial antibiotic resistance allowing for re-growth of bacterial populations, as well as the impact antibiotics can have on the growth of the dinoflagellate culture.
We propose a new approach to investigate bacterial contributions to dinoflagellate cultures, by way of full bacterial genome assembly from long-read sequencing to identify pathways present in those bacterial populations, determine microbial taxonomic composition, and estimate relative abundance [23,24]. Here, we use this approach in a toxin-producing Amphidinium carterae CCMP1314 culture with a long history of laboratory growth in the presence of carbenicillin, kanamycin A, and spectinomycin antibiotics. This approach provides a potentially more complete, unbiased measure of bacterial diversity and metabolism within the A. carterae culture with and without ongoing antibiotic treatment.
Amphidinium carterae was chosen for this study firstly because of its cosmopolitan appearance in nature: it is one of the most common species found in sediments in multiple ecosystems [25,26,27]. Cultures of A. carterae are also easy to maintain and are known to grow to relatively high density compared to other dinoflagellate species [28,29]. This species is also often used as a model athecate photosynthetic peridinin-pigmented dinoflagellate due to a smaller genome compared to other free-living dinoflagellate species, and because it is a relatively early diverging toxic species [30].
In this study we have found populations of bacteria in an A. carterae culture that have thrived in the presence of antibiotics, identified antimicrobial genes within these populations, and observed how the dinoflagellate culture responds when reverted back to antibiotic-free growth conditions.
2. Results
2.1. Bacterial Growth
The growth of A. carterae cultures grown both with and without antibiotics was found to be similar during both lag and log phases, with daily maximum growth rate being about 63 ± 9% and 65 ± 9% for both treatments, respectively. Maximum cell densities were similar, with the antibiotic-free culture reaching 142,166 ± 15,382 cells mL-1 and the antibiotic-treated culture reaching 126,000 ± 1311 cells mL-1. Growth differed between antibiotic and control treatments once the stationary phase began. In the case of the antibiotic-treated culture, cell growth became stagnant and cell density remained relatively the same, while the cultures grown without antibiotics began to show significant decreases in cell number and signs of lysing and death under microscopy (Figure 1A). Cells within the antibiotic-free culture appeared to form aggregates, and dinoflagellate cells imaged at the beginning of stationary phase in the antibiotic-free cultures displayed mass lysing and the appearance of rod-shaped bacteria overwhelming the culture (Figure 1B), alluding to algicidal properties. These bacteria were not observed by microscopy in the antibiotic-treated cultures.
2.2. Bacterial Identification
From the A. carterae cultures, metagenome assembly of 15 bacterial contigs from 2.9 to 6.0 Mb were identified based on 16S rDNA identity (Table 1). All 15 were single contig genomes and the Flye assembly reported 14 of the 15 being circular (all but O. alexandrii). Nearest neighbors of the 16S sequences found in these genomes gave consensus to their identification from 98 to 100% identity (Figure 1A). The bacteria present included 10 Alphaproteobacteria, 1 Gammaproteobacteria, 3 Bacteroidia, and 1 Planctomycetota. Based on the very high (>98%) full-length 16S rDNA identity to previously described species the names of these high identity 16S rDNA matches are used as an operational taxonomy for the remainder of the text.
Within the 15 genomes between 2,890 and 5,758 genes were annotated; of these 43-70% had functional assignments according to BV-BRC (Supplementary Table 2A). Genome quality was qualified as good for all of the genomes, with CheckM Completeness ranging from 77.5% to 100%, and a majority of the genomes showing above 90% completeness [31]. CheckM Contamination scores were below 5% for all the genomes as well.

Table 1.
Bacterial genome characteristics.
| NCBI Sample Title | length | AT% |
|---|---|---|
| Labrenzia sp. strain ac12 | 6,053,707 | 40.9 |
| Hoeflea alexandrii strain ac4 | 4,829,646 | 38.4 |
| Algihabitans albus strain ac2 | 4,739,774 | 34.3 |
| Ahrensia marina strain ac1 | 4,424,055 | 42.8 |
| Seohaeicola saemankumensis strain ac14 | 4,275,009 | 36.6 |
| Roseovarius mucosus strain ac13 | 3,969,289 | 38.9 |
| Rhodophyticola porphyridii strain ac11 | 3,902,216 | 35.8 |
| Oceaniradius stylonematis strain ac10 | 3,664,187 | 35.3 |
| Oceanicaulis alexandrii strain ac9 | 2,992,841 | 37.7 |
| Nitratireductor sp. strain ac15 | 2,917,504 | 39.8 |
| Marinobacter adhaerens strain ac5 | 4,424,055 | 42.9 |
| Cyclobacterium xiamenense strain ac3 | 5,806,256 | 51.6 |
| Marivirga tractuosa strain ac6 | 4,787,102 | 65.3 |
| Muricauda sp. strain ac8 | 4,366,883 | 58.0 |
| Phycisphaeraceae SM1A02 strain ac7 | 3,415,114 | 34.9 |
1 16S sequence identification found using both the SILVA ACT service and NCBI BLASTn feature [32]. ⥉ Contig 2139 was ‘Unclassified’ according to SILVA but identified by NCBI as an Alphaproteobacteria.
2.2. Bacterial Abundance
In the antibiotic-free cultures 52.2 % of the total bases and 49.9 % of reads mapped to the 15 bacterial genomes while in antibiotic-treated treated cultures 39.8% of total bases and 33.5% of reads mapped to bacteria (Figure 2a). Ten of the 15 bacterial genomes identified account for 90% of the total bacterial reads mapped in both conditions (Figure 2b) and the remaining five taxa accounted for <10% of total bacterial reads.
All 15 bacteria were detectable at some level in all the batches of sequencing data, but the relative abundance differed substantially for 7 bacterial genomes. Under antibiotic treatment R. mucosus was the most abundant single bacterium (at 53%) and then decreased to 6.16% without antibiotics. Without antibiotics, Muricauda sp., Marivirga tractuosa, Ahrensia marina, and Seohaeicola saemankumensis rose to the top 90% of the bacterial reads. Finally, when antibiotics were in use, populations of O. stylonematis and C. xiamenense were found in the top 90% of bacterial reads but were not abundant under antibiotic treatment.
The antibiotic-free and antibiotic-treated cultures both included 3 taxa at roughly similar proportions: SM1A02, O. alexandrii, and A. albus. When cultures were grown without antibiotics, the abundance of SM1A02 and O. alexandrii slightly increased by 2.6 and 1.3%, while A. albus slightly decreased by 2.8%. The remaining 5 bacterial genomes were present at low levels in both culture conditions.
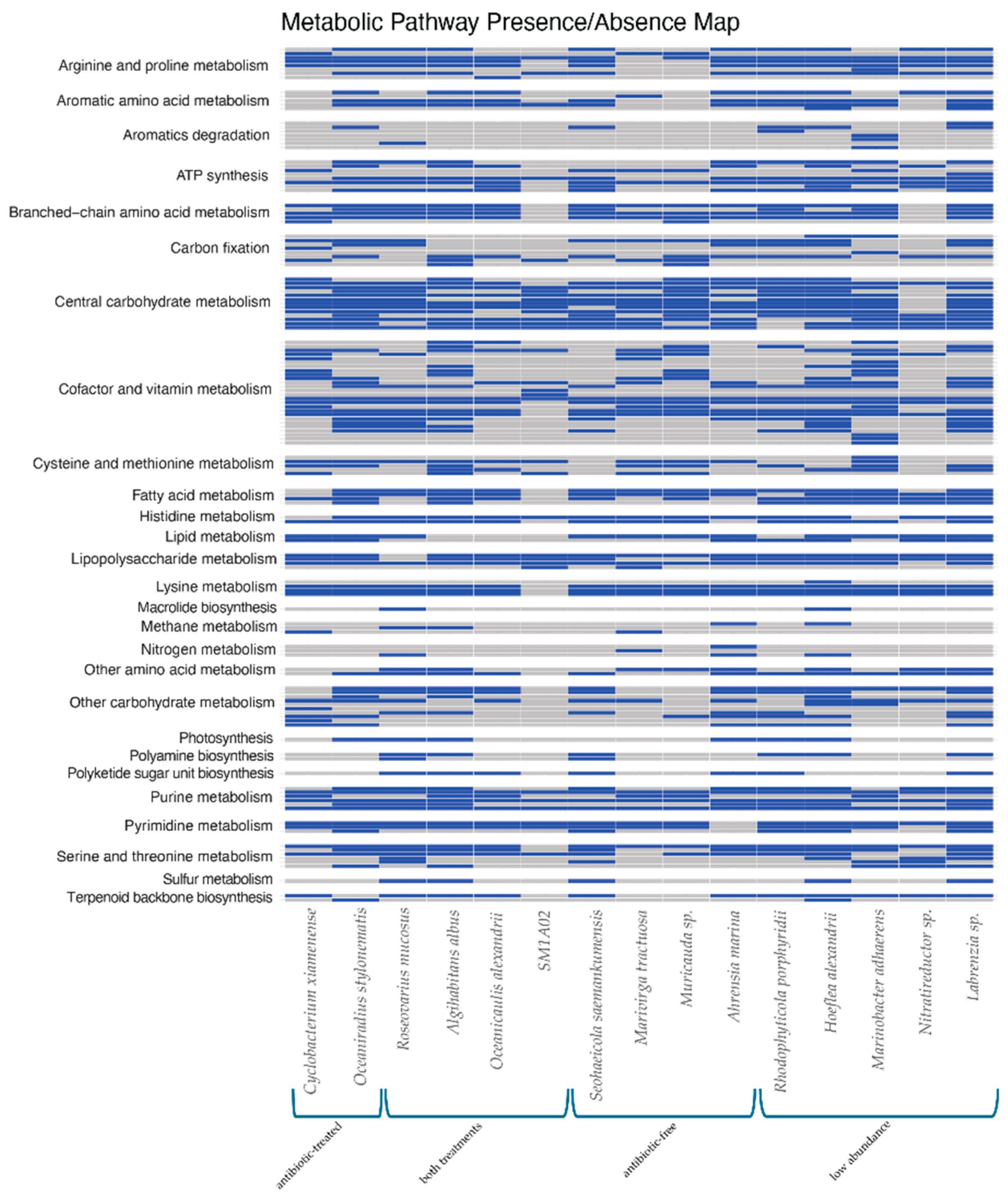
2.3. Bacterial Metabolic Pathways
The metabolic abilities of the bacterial genomes were assessed using the KEGG database, showing a wide variety of potential pathways (Figure 3, Supplementary Table 3A). Many of the bacterial genomes have potential pathways for the production of amino acids, such as arginine, proline, serine, and threonine. Basic metabolic pathways such as carbohydrate metabolism and ATP synthesis were identified. Some of the bacterial genomes, including R. mucosus, R. porphyridii, H. alexandrii, O. stylonematis, A. marina, and A. albus were found to potentially contain photosynthesis machinery for photosystem II, as well as some carbon fixation pathways. The Labrenzia sp. genome, although the largest bacterial genome assembled, was also in low abundance in the antibiotic-treated culture and contained part of the reductive phosphate pentose cycle and the Calvin cycle, but not PSII.
Nitrate reduction pathways were found in 3 of the bacterial genomes, and denitrification machinery was identified only in the M. tractuosa genome. The Nitratireductor sp. found in low abundance the antibiotic-treated culture, was negative for nitrate-reduction pathways despite the genera name, which aligns with prior research [33].
Figure 3.
Functional traits identified via CARD based on the KEGG database [34]. Specific pathway names for each row are listed in Supplementary Table 3A.
Figure 3.
Functional traits identified via CARD based on the KEGG database [34]. Specific pathway names for each row are listed in Supplementary Table 3A.
2.4. Secondary Metabolite Synthesis
The assembled bacterial genomes were found to contain regions associated with the production of a variety of secondary metabolite compound classes, including building-block lactone structures such as butyrolactones, betalactones, and hserlactones (Figure 4). Terpenes and aromatic compound synthase genes were the major natural product occurring amongst the genomes. Some polyketide synthases (PKSs) and non-ribosomal peptide synthases (NRPSs) were identified, but none were predicted to participate in the biosynthesis of larger chemical structures according to antiSMASH.
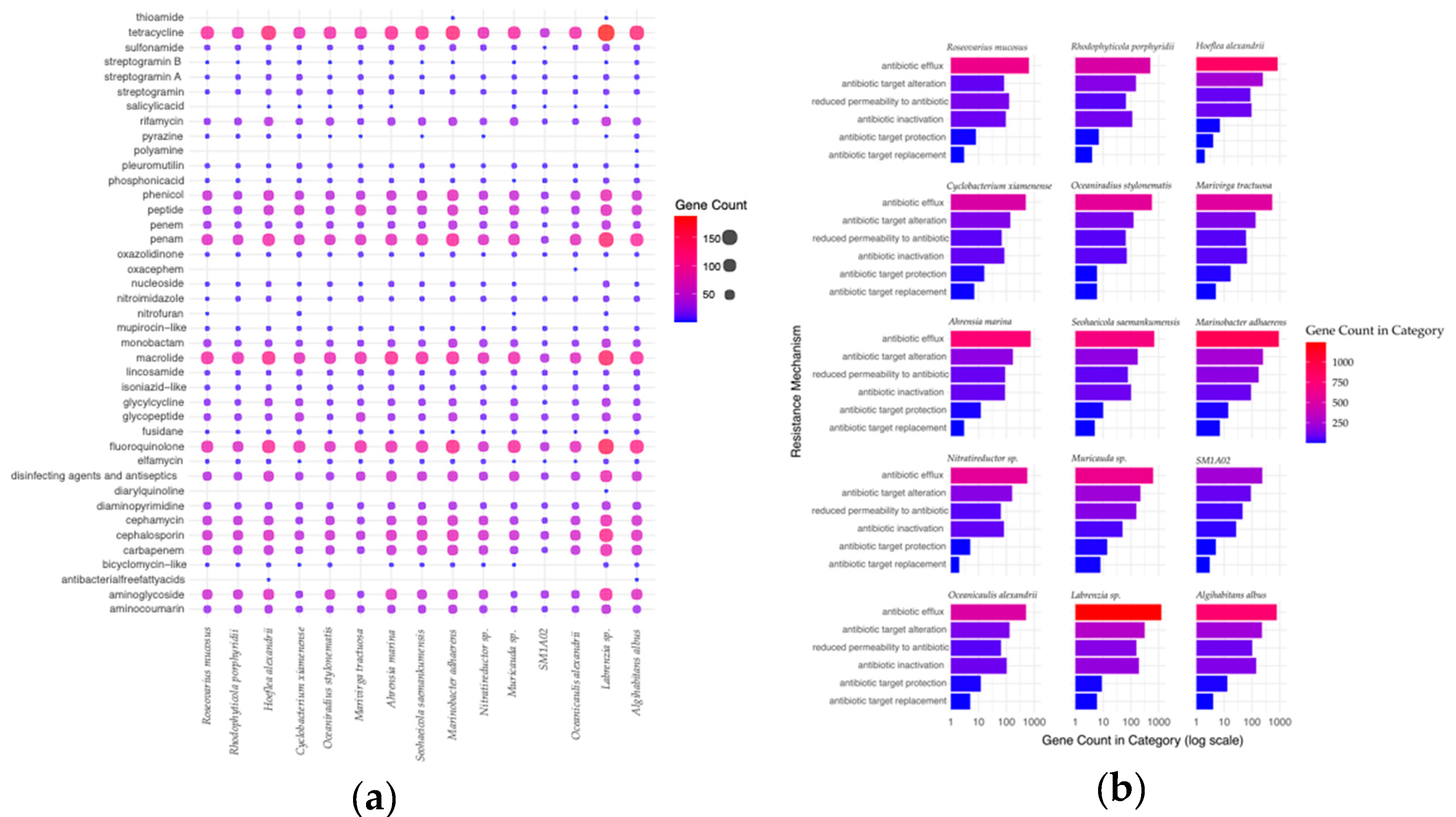
2.5. Antibiotic-Resistance
Generally, all of the bacterial genomes had antibiotic resistance genes in the CARD database search (Figure 5A). Interestingly, all of the genomes have a large number of resistance genes to tetracycline. The antibiotic resistance genes in the bacterial genomes found in the antibiotic-treated cultures do not appear to indicate a strong pattern of drug class resistance to the antibiotic cocktail used. Most of the antibiotic resistance genes were part of antibiotic efflux systems, especially in the assembled Labrenzia sp. genome (Figure 5B). This was generally followed by mechanisms including drug target alterations, reduction of drug permeability, drug inactivation, drug target protection, and drug target replacement.
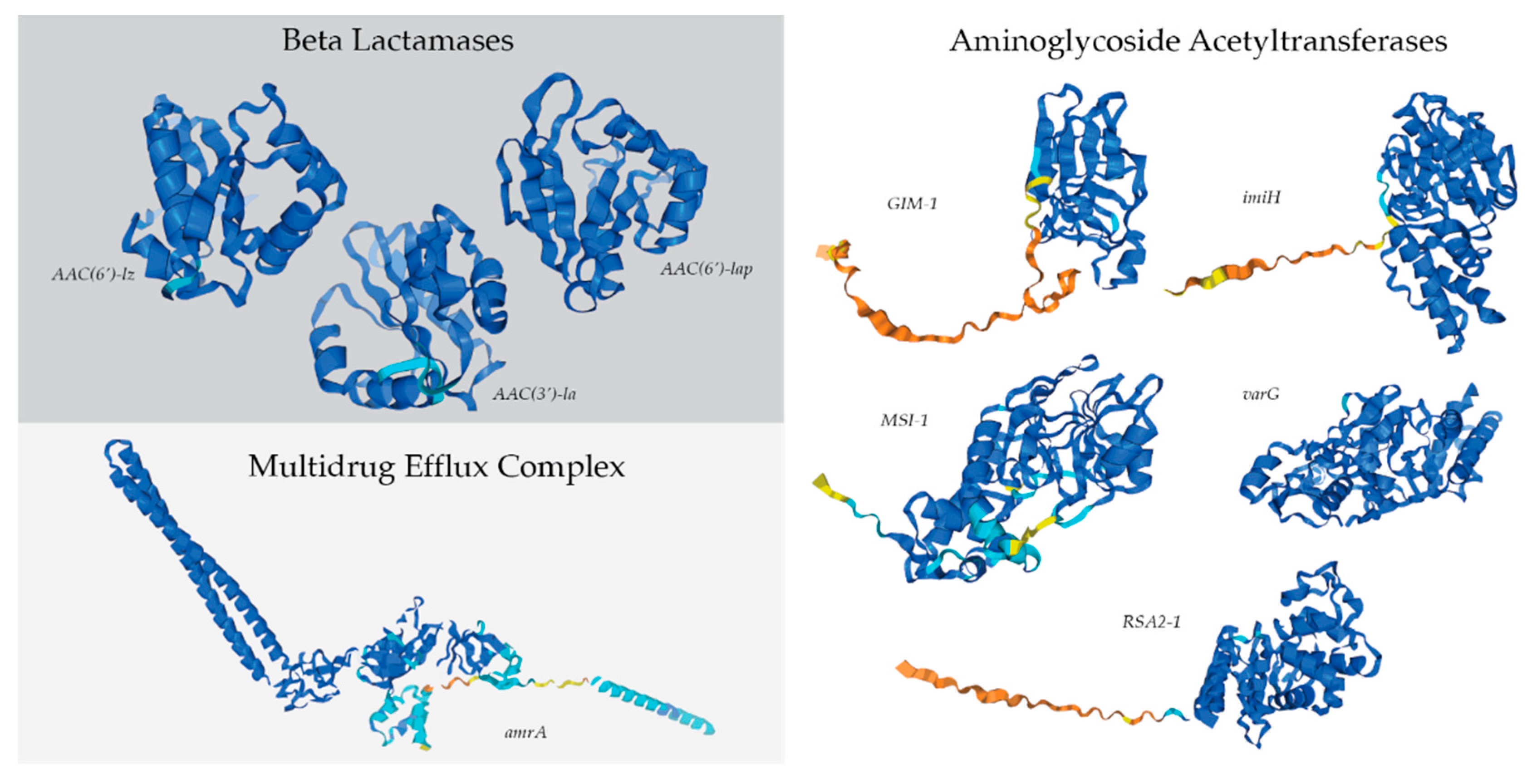
The dominant bacterial genome in the antibiotic-treated culture, R. mucosus, contained 13 resistance genes that were either completely unique to the taxa, or only shared with other bacterial genomes found in the treated culture (Suppementary Table 2A). Of those 13, nine had the potential to confer resistance to carbenicillin, kanamycin A, and/or spectinomycin (Figure 6). Three of these genes encoded potential beta-lactamases, which inactivate beta-lactam antibiotics by hydrolyzing the peptide bond of the beta-lactam ring and may confer resistance to carbenicillin. One of the genes is similar to the efflux subunit of the AmrAB-OprM multidrug efflux complex, which impairs aminoglycoside accumulation. Lastly, 5 genes in the R. mucosus genome were identified as potential aminoglycoside acyltransferases, which may catalyze the AcCoA-dependent N-acetylation of amino groups on aminoglycoside molecule (such as kanamycin A and spectinomycin).
3. Discussion
Of the 15 genomes assembled every genome has a high (>95%) identity 16S rDNA match to an existing annotated sequence suggesting that the culture does not contain any entirely novel bacteria. Many of the assembled genomes were identified as bacterial species that have previously been observed as co-existing with dinoflagellates. Interestingly, the Planctomycetota bacteria assembled was identified as SM1A02, an uncultured strain associated with many dinoflagellate cultures [39]. This species’ genome has previously been reconstructed using metagenomic assembly and binning. Research by Baker et al. found two of these SM1A02 genomes to be 2.6 and 2.9Mb, while we have assembled a 3.4Mb genome here. Most recently, an assembly of SM1A02 was produced from a Karlodinium culture which agrees with our genome length of 3.4Mb [40]. SM1A02 is thought to likely be an anammox bacteria – efficient at nutrient removal, specifically through anaerobic ammonium oxidation [41]. This ability to oxidize ammonium to nitrogen gas may have an impact on the close association with dinoflagellate species.
Roseovarius mucosus was also identified, similar to a species found with the dinoflagellate Alexandrium ostenfeldii. In prior research, genes in R. mucosus were found that may play crucial roles in the interrelationship of the bacterium and dinoflagellate, such as genes for dimethyl sulfoniopropionate (DMSP) utilization. Research on the close interactions of DMSP-degrading Roseobacter species with DMSP-producing dinoflagellates are well-established [42]. Our metabolic results show R. mucosus likely has pathways for thiosulfate oxidation, supporting these past findings [43]. The ability for thiosulfate oxidation may also have a connection to the common sulfation of toxic amphidinol products produced by A. carterae or more broadly for sulfur cycling within the cultures [44,45]. The pathway for assimilatory nitrate reduction found in the R. mucosus genome has been observed as a potential nitrogen source by some [46,47]. The pathway for polyamine biosynthesis for putrescine and spermidine was also found in the R. mucosus genome, which may play an essential role in dinoflagellate growth [48,49,50]. The presence of PSII and other carbon-fixing pathways also aligns well with prior research of this species [51].
Hoeflea alexandrii and Oceanicaulis alexandrii were discovered with a dinoflagellate species of Alexandrium as well as bacterial species Labrenzia alexandrii and Nitratireductor alexandrii, which may be closely related to the Labrenzia and Nitratireductor species assembled here [52,53,54,55]. Marinobacter adhaerens has been found in close association with Pyrodinium, another toxin-producing dinoflagellate [56]. Muricauda species have been previously associated with Amphidinium as well [57]. A Seohaeicola species genome was recently assembled from a culture of Karlodinium, another toxin-producing dinoflagellate species [40]. Some species such as O. stylonematis, A. s albus, C. xiamenense and R. porphyridii were discovered in association with other kinds of microalgae such as diatoms and red algae [58,59,60,61,62].
Some species found only in the antibiotic-free cultures may be opportunistic due to the nutrient availability caused by dinoflagellate senescence. Both the assembled R. porphyridii genome and the H. alexandrii genome were found in very low abundances in the antibiotic-free culture. The R. porphyridii species in a genus of purple non-sulphur bacteria known to be halophilic and have the ability to perform photosynthesis [58]. Thiosulfate oxidation pathway genes were also found in the H. alexandrii genome.
The transition from antibiotic treatment to untreated is unlikely to have resulted in the introduction of new bacteria to the culture as all taxa were seen and could be at least partially assembled in sequencing of either antibiotic treated or untreated culture. Thus, the microbiome shift is more likely due to stronger growth of some species over others when antibiotics were present or absent rather than recruitment of novel species during culture changes, which were performed under sterile conditions.
Previous studies have demonstrated that use of the KAS-antibiotic treatment (with kanamycin A, ampicillin, and streptomycin) can be used to favor pigmented bacterial species [63]. Similar mechanisms may be why we saw such a shift towards bacterial populations with PSII and carbon fixation systems with the use of our antibiotic cocktail, from 12.5% to 50% of the highly abundant bacterial species having PSII pathways with the antibiotic-treatment. The R. mucosus identified in past dinoflagellate cultures, which dominated the antibiotic-treated microbiome in this experiment, has been shown to contain bacteriochlorophyll a [51]. There is also evidence of dinoflagellates protecting certain pigmented bacterial populations from antibiotics as the pigmented bacteria may be protecting the microalgal cells from light stress via carotenoid production, which was previously shown to be produced by multiple assembled bacterial species (Figure 4) [63]. In prior research regarding coral symbiont dinoflagellate Symbiodinium, the bacterial microbiome was observed to support the dinoflagellate’s PSII yield and decrease the production or reactive oxygen species (ROSs) [64]
Antimicrobial resistance appears to be generally common amongst bacterial populations found in dinoflagellate cultures. In the case of the assembled genomes here, all of them had hundreds of potential antibiotic-resistance genes that likely allow their broad prevalence (Figure 5). The reasons for why some bacteria were found to survive better in antibiotic-treated or antibiotic-free conditions could be due to multiple causes. One may be that the minimum inhibitory concentration of the antibiotic used may differ from the actual concentration tested [65]. This may be due to the mechanism of resistance, or the antibiotic’s resistance to degradation (such as in the case of carbenicillin compared to ampicillin) [66]. The specific genes found within each of these resistance groups may have varying efficacy against the antibiotics as well. In the case of R. mucosus, which best-endured the antibiotic-treatment used here, it may be that one of the nine antibiotic-resistant genes has greater efficacy over one or more of the antibiotics used compared to the machinery found in the other genomes (Figure 6).
The extent to which the bacteria from the assembled genomes are mutual, commensal, or deleterious to the A. carterae population is still obscure. The fact that the dinoflagellate population significantly decreased and showed signs of mass lysis without antibiotics leads us to believe that at least one abundant bacterial species in the antibiotic-free culture is likely the cause. Prior research on antibiotic effects on dinoflagellate growth have shown various results. In some cases, dinoflagellates appear to require their associated microbiomes to survive [11,67]. In the case of the antibiotic cocktail used here, the growth results align with previous observations of the antibiotic-treated Amphidinium cultures having a slightly extended growth phase and the ability to maintain higher densities of dinoflagellate cells [22]. Based on our microscopy analysis, we suspect the 10µm length rod-shaped bacteria that began to accumulate around the start of mass-cell-lysis is likely harmful to the dinoflagellates and may possess some algicidal properties. We could deduce the culprit may be Marivirga tractuosa or Seohaeicola saemankumensis due to the increased abundance found in the antibiotic-free culture (Figure 2B), as well as prior descriptions of this species being rod-shaped. Cells of M. tractuosa can be between 10-50µm length, while S. saemankumensis has been shown to be up to 5µm in length [68,69,70]. The M. tractuosa genome lacks many main metabolic pathways, such as amino acid biosynthesis suggesting this species requires resources gained from the lysed dinoflagellate cells. The M. tractuosa genome was also found to have complete denitrification pathways, which may contribute to a loss of bioavailable nitrogen in the culture [71]. The Coenzyme M pathway alludes to methanogenic abilities and production of methane, and potentially the use of dinoflagellate-released DMSP as a precursor [72,73]. Harmful algal blooms have been observed to precede methane increases in aquatic environments, which may be in part due to the shift in microbial community [73].
Prior research has been contentious over the secondary metabolite synthesis potential of dinoflagellate microbiomes, and contrasting results have identified toxin production as a product either of the bacterial community or the dinoflagellate cells themselves [13,14,15,16,17,74,75,76,77]. The fifteen apparently complete genomes assembled from this culture likely represent the bulk of prokaryotic diversity due to the extent of our sequencing and the production of full, well-covered genomes. Any missing diversity would likely be in very low abundance to evade detection and are not likely to be present at a level to contribute to toxin biosynthesis. Similarly, the gene annotation provides a potentially complete picture of the culture metabolic potential. However, a large fraction of predicted genes was unannotated, likely due to imperfect prediction of protein coding genes as well as knowledge gaps of every possible bacterial pathway. Our genome analysis into secondary metabolite synthesis has shown no evidence of potential bacterial origin for a processive multidomain PKS gene responsible for the amphidinol toxins associated with our A. carterae culture. Since multidomain, processive PKS genes are very large open reading frames rich in easily defined conserved domains these genes would be unlikely to have been missed in the genomes described here. Several multidomain PKS / NRPS genes derived from bacteria are present broadly across core dinoflagellate transcriptomes which generally express a surfeit of domains associated with toxin production and lipid synthesis whether or not they are document toxin-producing species [18]. More research could be done to see what effect metabolic pathways, such as thiosulfate oxidation, may have on toxin production. Although it seems most likely that A. carterae independently synthesizes amphidinols, the bacterial populations may contribute resources for the task, such as acetate [45,78,79]. Of the bacterial genomes assembled, only M. adhaerens was shown to have a complete pathway for a phosphate acetyltransferase−acetate kinase pathway which produces acetate from acetyl-CoA (Figure 3). Oceaniradius stylonematis, S. saemankumensis, R. porphyridii, H. alexandrii, and the Labrenzia sp. all had complete phenylacetate degradation pathways to produce acetyl−CoA which may serve as a precursor to acetate synthesis, and O. stylonematis, S. saemankumensis, R. porphyridii, R. mucosus, A. albus, O. alexandrii, M. adhaerens, and the Labrenzia sp. had complete pathways for leucine degradation to acetyl−CoA as well.
The microbiome of algal species has been shown to contribute necessary vitamins and products to dinoflagellate species, the most recognized being cyanocobalamin (B12) [80]. Based on our metabolic findings, the introduction of B vitamins into dinoflagellate growth media does not appear to be redundant with the biosynthetic abilities of the microbiome. The vitamins added to our ESAW media preparation include biotin (H), B12, and thiamine (B1) to increase growth rate and final yield [81]. Of the bacterial genomes assembled, only the M. adhaerens genome had a pathway identified to synthesize biotin, and this species was in very low abundance. The only highly abundant species with aerobic and anaerobic pathways for the synthesis of B12 were R. mucosus and O. stylonematis, which were significantly more abundant in the antibiotic-treated cultures, and their decline without antibiotics may have been a factor in the cell mortality of Amphidinium as the nutrients in the culture diminished over the log phase. Only A. albus within the highly abundant bacterial species had a pathway for vitamin B1 synthesis, and yet this was only through a salvage pathway which utilizes precursors or similar compounds in the surrounding media for B1 biosynthesis [82].
The Planctomycetota bacteria SM1A02 is the only assembled genome in the Amphidinium culture to have full pathways for menaquinone (vitamin K2) biosynthesis. Although vitamin K1 is conventionally known as a redox cofactor in plants and green algae, vitamin K2 can also be a secondary electron acceptor of PSI in some algal and archaeal species [83]. Vitamin K2 can also shuttle electrons between different respiratory complexes in anaerobic respiration or aerobic respiration in a microaerophilic environment [84]. The effect of vitamin K2 bioavailability for dinoflagellate species remains to be seen.
4. Materials and Methods
4.1. Cell Culturing
Cultures of Amphidinium carterae were grown in ESAW artificial marine media with a salinity of 20 ppt supplemented with f/2 nutrients without silicates at a starting concentration of 10K cells mL-1 and allowed to acclimate for 14 days in 14:10 h light:dark period in 100 μmol photons m-2 sec-1 at 25°C [81]. Cultures have been continuously grown with an antibiotic cocktail of 100 μg/mL carbenicillin, 50 μg/mL kanamycin A and 50 μg mL-1 spectinomycin (“antibiotic-treated”). For analysis, some cultures were weaned off growth in the presence of antibiotics over a month before subsequent DNA extraction (“antibiotic-free”). Cell counts were done using a Scepter 3.0 Cell Counter (Figure 1).
4.2. DNA Extraction and Long-Read Sequencing
For each dinoflagellate culture, the cells were spun down at 10,000 x g for 10 minutes to form cell pellets. Cell pellets were then resuspended in 0.1M EDTA and 0.5% SDS with 200 μg proteinase K and allowed to incubate for 10 minutes at 50°C. Following this incubation, equal 16.6% volume of 2% CTAB and 5M NaCl were added and once again the sample was allowed to incubate at 50°C for 10 minutes. After this, the solution was mixed 1:1 with chloroform and allowed to sit at room temperature for 10 minutes. The sample was then spun for 10 minutes at 10,000 x g and the aqueous layer was transferred to a new tube and mixed with binding buffer from the DNA Clean & Concentrator kit (Zymo Research). Aliquots of 750ul were spun through the columns, followed by washing and finally elution in 20-40 μL of water. Small fragments were filtered out using the PacBio SRE/SRE-XL kit according to manufacturer’s directions. A high molecular weight DNA library was then prepped using the Nanopore whole genome sequencing kit (SQK-LS114), followed by sequencing on a MinION, GridION, or PromethION device (Supplementary Table 1A).
4.3. Genome Assembly
Following sequencing, DNA data in pod5 files were basecalled by Oxford Nanopore’s Dorado basecaller (7.2.13) using the dna_r10.4.1_e8.2_400bps_sup@v4.3.0 model. Sequences from the antibiotic-free culture runs or antibiotic-treated runs were pooled and assembled using the Flye de novo assembler (2.9.1-b1780), filtering for sequences over 200 bases [85]. Bacterial contigs were identified using BLASTn for 16S sequences from NCBI’s 16S RefSeq or 16S microbial ribosomal databases. Bacterial matches from each assembly were identified, and duplicate matches between the antibiotic-free and antibiotic-treated runs were compared, and the most complete bacterial genomes were chosen for further analysis.
4.4. Phylogenetic Analysis
Once the bacterial genomes were identified, 16S sequences were extracted and identified using SILVA’s Alignment, Classification, and Tree (ACT) service according to the global SILVA alignment for rRNA genes, as well as through NCBI’s BLASTn feature [32]. A consensus of the two was chosen based on match percentage. A phylogenetic tree was constructed using RaxML including 10 nearest neighbors identified by SILVA ACT from the database (Supplementary Figure 1A).
4.5. Read Abundance Mapping
To quantify the relative abundance of bacterial genomes within the antibiotic-free and antibiotic-treated cultures, reads from each condition were aligned to the single unified set of bacterial contigs using minimap2, using ordinary minimizers as seeds [86]. Alignment SAM files were converted to BAM using Samtools, then sorted, indexed, and finally coverage and stats files were created [87]. Genomes with greater than 90% coverage were considered present in the cultures, and abundance was calculated by the percent of reads mapping to that genome out of the total bacterial reads mapped in each condition.
4.6. Metabolic and Secondary Metabolites Analysis
4.7. Antimicrobial Resistance Analysis
The Comprehensive Antibiotic Resistance Database (CARD) Resistance Gene Identifier (RGI) software was used for the prediction of antibiotic resistance genes in the bacterial genomes [36]. Perfect, Strict and Loose hits for genes were allowed but partial genes were excluded. Protein predictions of antimicrobial resistance genes of interest were created using the AlphaFold Google DeepMind AI system [37,38]
5. Conclusions
Although A. carterae may be grown without antibiotics, the viability of the culture and subsequent sequencing analyses may benefit from their use. We observed die off of cells in the A. carterae cultures during the end of the log phase without antibiotics present, likely due to proliferation of one of the more antibiotic-sensitive bacteria identified. We also found a decrease in the proportion of bacterial reads sequenced with the use of antibiotics. As novel sequencing technologies, such as efficient long-read sequencing, becomes more readily available, the use of techniques to target sequences of interest will allow for more productive sequencing efforts. The data suggest both comprehensive and quantitative microbial genome sequencing can be accomplished from this culture, which could in future be expanded to work with in situ sequencing of dinoflagellate blooms.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: 16S Phylogenetic Tree; Table S1: Sequencing Runs; Table S2: Basic Stats on Bacterial Genomes; Table S3: METABOLIC Pathway Identifiers; Table S4: Roseovarius Unique AMR Genes;.
Author Contributions
Conceptualization, M. Judd, J. Wira, A.R. Place, and T. Bachvaroff; methodology, M. Judd, J. Wira, and T. Bachvaroff; software, M. Judd, J. Wira, and T. Bachvaroff; validation, M. Judd, J. Wira, and T. Bachvaroff; formal analysis, M. Judd and T. Bachvaroff; investigation, M. Judd, J. Wira, and T. Bachvaroff; resources, M. Judd, J. Wira, A.R. Place, and T. Bachvaroff; data curation, M. Judd and T. Bachvaroff; writing—original draft preparation, M. Judd; writing—review and editing, M. Judd, J. Wira, A.R. Place, and T. Bachvaroff; visualization, M. Judd and T. Bachvaroff; supervision, A.R. Place and T. Bachvaroff; project administration, A.R. Place and T. Bachvaroff; funding acquisition, A.R. Place and T. Bachvaroff. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Vetlesen foundation and the IMET Angel Investor Program.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
All of the raw sequence and assembled data has been deposited under the NCBI project number SUB14439053 and SRA numbers SRR28996185, SRR28996184, SRR28996183, SRR28996182.
Acknowledgments
The hardware used in the computational studies is part of the UMBC High Performance Computing Facility (HPCF). The facility is supported by the U.S. National Science Foundation through the MRI program (grant nos. CNS–0821258, CNS–1228778, OAC–1726023, and CNS–1920079) and the SCREMS program (grant no. DMS–0821311), with additional substantial support from the University of Maryland, Baltimore County (UMBC). See hpcf.umbc.edu for more information on HPCF and the projects using its resources.The BioAnalytical Services Laboratory (BASLab) at the Institute of Marine and Environmental Technology (IMET) was used for Nanopore GridION and MinION long-read sequencing. See https://www.umces.edu/baslab for more information on the BASLab capabilities.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix A
Table A1.
Nanopore sequencing runs.
| Antibiotic-Free | Antibiotic-Treated | |||||
|---|---|---|---|---|---|---|
| Date | Reads | Device used | Date | Reads | Device used | |
| 2/1/2024 | 86,499 | GridION | 5/10/2023 | 122,303 | MinION | |
| 2/2/2024 | 62,172 | GridION | 5/12/2023 | 59,113 | MinION | |
| 2/3/2024 | 1,615 | GridION | 5/13/2023 | 180,995 | MinION | |
| 3/15/2024 | 295,191 | GridION | 5/16/2023 | 222,574 | MinION | |
| 3/17/2024 | 188,915 | GridION | 5/17/2023 | 152,303 | MinION | |
| 3/18/2024 | 152,194 | GridION | 10/30/2023 | 3,345,955 | PromethION | |
| Total Reads | 786,585 | 4,083,243 | ||||
| Total Bases | 23,622,147,065 | 151,572,689,672 | ||||
Figure A1.
Phylogenetic tree including bacterial 16S sequences found in the A. carterae microbiome. Tree was generated using SILVA ACT Service using RaxML, the GTR model, and the Gamma rate model, with a minimum sequencing identity of 0.95 and including 10 neighbors.
Figure A1.
Phylogenetic tree including bacterial 16S sequences found in the A. carterae microbiome. Tree was generated using SILVA ACT Service using RaxML, the GTR model, and the Gamma rate model, with a minimum sequencing identity of 0.95 and including 10 neighbors.

References
- Adolf, J.E.; Stoecker, D.K.; Harding, L.W. The Balance of Autotrophy and Heterotrophy during Mixotrophic Growth of Karlodinium micrum (Dinophyceae). J Plankton Res 2006, 28, 737–751. [Google Scholar] [CrossRef]
- Place, A.R.; Bowers, H.A.; Bachvaroff, T.R.; Adolf, J.E.; Deeds, J.R.; Sheng, J. Karlodinium veneficum-The Little Dinoflagellate with a Big Bite. Harmful Algae 2012, 14, 179–195. [Google Scholar] [CrossRef]
- Not, F.; Siano, R.; Kooistra, W.H.C.F.; Simon, N.; Vaulot, D.; Probert, I. Diversity and Ecology of Eukaryotic Marine Phytoplankton; Elsevier, 2012; Vol. 64, ISBN 9780123914996. [Google Scholar]
- Hansen, P.J. The Role of Photosynthesis and Food Uptake for the Growth of Marine Mixotrophic Dinoflagellates1. Journal of Eukaryotic Microbiology 2011, 58, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Prézelin, B.B.; Alberte, R.S. Photosynthetic Characteristics and Organization of Chlorophyll in Marine Dinoflagellates. Proceedings of the National Academy of Sciences 1978, 75, 1801–1804. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.Y.B.; Oliveira, C.D.L.; Müller, M.N.; Santos, E.P.; Dantas, D.M.M.; Gálvez, A.O. A Scientometric Overview of Global Dinoflagellate Research. Publications 2020, 8, 50. [Google Scholar] [CrossRef]
- Botana, L.M. Seafood and Freshwater Toxins: Pharmacology, Physiology, and Detection, Second Edition. In Seafood and Freshwater Toxins: Pharmacology, Physiology, and Detection, Second Edition; 2008; pp. 1–943. [Google Scholar]
- Kuhlisch, C.; Shemi, A.; Barak-Gavish, N.; Schatz, D.; Vardi, A. Algal Blooms in the Ocean: Hot Spots for Chemically Mediated Microbial Interactions. Nat Rev Microbiol 2024, 22, 138–154. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M. Red Tides, 2nd ed. Scientific American, 1994; Vol. 271.
- Alavi, M.; Miller, T.; Erlandson, K.; Schneider, R.; Belas, R. Bacterial Community Associated with Pfiesteria-like Dinoflagellate Cultures. Environ Microbiol 2001, 3, 380–396. [Google Scholar] [CrossRef] [PubMed]
- Bolch, C.J.S.; Bejoy, T.A.; Green, D.H. Bacterial Associates Modify Growth Dynamics of the Dinoflagellate Gymnodinium catenatum. Front Microbiol 2017, 8. [Google Scholar] [CrossRef]
- Green, D.H.; Llewellyn, L.E.; Negri, A.P.; Blackburn, S.I.; Bolch, C.J.S. Phylogenetic and Functional Diversity of the Cultivable Bacterial Community Associated with the Paralytic Shellfish Poisoning Dinoflagellate Gymnodinium catenatum. FEMS Microbiol Ecol 2004, 47, 345–357. [Google Scholar] [CrossRef]
- Albinsson, M.E.; Negri, A.P.; Blackburn, S.I.; Bolch, C.J.S. Bacterial Community Affects Toxin Production by Gymnodinium catenatum. PLoS One 2014, 9, e104623. [Google Scholar] [CrossRef]
- Tarazona-Janampa, U.I.; Cembella, A.D.; Pelayo-Zárate, M.C.; Pajares, S.; Márquez-Valdelamar, L.M.; Okolodkov, Y.B.; Tebben, J.; Krock, B.; Durán-Riveroll, L.M. Associated Bacteria and Their Effects on Growth and Toxigenicity of the Dinoflagellate Prorocentrum Lima Species Complex From Epibenthic Substrates Along Mexican Coasts. Front Mar Sci 2020, 7. [Google Scholar] [CrossRef]
- Bui, Q.T.N.; Pradhan, B.; Kim, H.-S.; Ki, J.-S. Environmental Factors Modulate Saxitoxins (STXs) Production in Toxic Dinoflagellate Alexandrium: An Updated Review of STXs and Synthesis Gene Aspects. Toxins (Basel) 2024, 16, 210. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Evans, A.N.; Kulis, D.M.; Hackett, J.D.; Erdner, D.L.; Anderson, D.M.; Bhattacharya, D. Transcriptome Profiling of a Toxic Dinoflagellate Reveals a Gene-Rich Protist and a Potential Impact on Gene Expression Due to Bacterial Presence. PLoS One 2010, 5, e9688. [Google Scholar] [CrossRef] [PubMed]
- Stüken, A.; Orr, R.J.S.; Kellmann, R.; Murray, S.A.; Neilan, B.A.; Jakobsen, K.S. Discovery of Nuclear-Encoded Genes for the Neurotoxin Saxitoxin in Dinoflagellates. PLoS One 2011, 6, e20096. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.; Bachvaroff, T.; Place, A. Dinoflagellate Phosphopantetheinyl Transferase (PPTase) and Thiolation Domain Interactions Characterized Using a Modified Indigoidine Synthesizing Reporter. Microorganisms 2022, 10, 687. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Yang, X.; Zheng, T.; Hong, H. An Efficient Method to Obtain Axenic Cultures of Alexandrium tamarense—a PSP-Producing Dinoflagellate. J Microbiol Methods 2007, 69, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.C.-H.; Chan, P.-L.; Tam, N.F.-Y.; Xu, S.J.-L.; Lee, F.W.-F. Establish Axenic Cultures of Armored and Unarmored Marine Dinoflagellate Species Using Density Separation, Antibacterial Treatments and Stepwise Dilution Selection. Sci Rep 2021, 11, 202. [Google Scholar] [CrossRef] [PubMed]
- Hold, G.L.; Smith, E.A.; Harry Birkbeck, T.; Gallacher, S. Comparison of Paralytic Shellfish Toxin (PST) Production by the Dinoflagellates Alexandrium lusitanicum NEPCC 253 and Alexandrium tamarense NEPCC 407 in the Presence and Absence of Bacteria. FEMS Microbiol Ecol 2001, 36, 223–234. [Google Scholar] [CrossRef]
- Liu, C.L.; Place, A.R.; Jagus, R. Use of Antibiotics for Maintenance of Axenic Cultures of Amphidinium carterae for the Analysis of Translation. Mar Drugs 2017, 15. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Assembling the Perfect Bacterial Genome Using Oxford Nanopore and Illumina Sequencing. PLoS Comput Biol 2023, 19, e1010905. [Google Scholar] [CrossRef]
- Sereika, M.; Kirkegaard, R.H.; Karst, S.M.; Michaelsen, T.Y.; Sørensen, E.A.; Wollenberg, R.D.; Albertsen, M. Oxford Nanopore R10.4 Long-Read Sequencing Enables the Generation of near-Finished Bacterial Genomes from Pure Cultures and Metagenomes without Short-Read or Reference Polishing. Nat Methods 2022, 19, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.; Patterson, D.J. The Benthic Dinoflagellate Genus Amphidinium in South-Eastern Australian Waters, Including Three New Species. Eur J Phycol 2002, 37, S0967026202003591. [Google Scholar] [CrossRef]
- Karafas, S.; Teng, S.T.; Leaw, C.P.; Alves-de-Souza, C. An Evaluation of the Genus Amphidinium (Dinophyceae) Combining Evidence from Morphology, Phylogenetics, and Toxin Production, with the Introduction of Six Novel Species. Harmful Algae 2017, 68, 128–151. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yan, Z.; Chen, J.; Wang, D.; Li, K. Acute Toxicity of the Dinoflagellate Amphidinium carterae on Early Life Stages of Zebrafish (Danio Rerio). Toxics 2023, 11, 370. [Google Scholar] [CrossRef]
- DIXON, G.K.; SYRETT, P.J. The Growth of Dinoflagellates in Laboratory Cultures. New Phytologist 1988, 109, 297–302. [Google Scholar] [CrossRef]
- Baig, H.S.; Saifullah, S.M.; Dar, A. Occurrence and Toxicity of Amphidinium carterae Hulburt in the North Arabian Sea. Harmful Algae 2006, 5, 133–140. [Google Scholar] [CrossRef]
- Murray, S.; Flø Jørgensen, M.; Daugbjerg, N.; Rhodes, L. Amphidinium Revisited. II. Resolving Species Boundaries in the Amphidinium operculatum Species Complec (Dinophyceae). J Phycol 2004, 40, 366–382. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Marasco, R.; Michoud, G.; Sefrji, F.O.; Fusi, M.; Antony, C.P.; Seferji, K.A.; Barozzi, A.; Merlino, G.; Daffonchio, D. The Identification of the New Species Nitratireductor thuwali sp. Nov. Reveals the Untapped Diversity of Hydrocarbon-Degrading Culturable Bacteria from the Arid Mangrove Sediments of the Red Sea. Front Microbiol 2023, 14. [Google Scholar] [CrossRef]
- Zhou, Z.; Tran, P.Q.; Breister, A.M.; Liu, Y.; Kieft, K.; Cowley, E.S.; Karaoz, U.; Anantharaman, K. METABOLIC: High-Throughput Profiling of Microbial Genomes for Functional Traits, Metabolism, Biogeochemistry, and Community-Scale Functional Networks. Microbiome 2022, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. AntiSMASH: Rapid Identification, Annotation and Analysis of Secondary Metabolite Biosynthesis Gene Clusters in Bacterial and Fungal Genome Sequences. Nucleic Acids Res 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The Comprehensive Antibiotic Resistance Database. Antimicrob Agents Chemother 2013, 57, 3348–3357. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing Structure Coverage for over 214 Million Protein Sequences. Nucleic Acids Res 2024, 52, D368–D375. [Google Scholar] [CrossRef] [PubMed]
- Rambo, I.M.; Dombrowski, N.; Constant, L.; Erdner, D.; Baker, B.J. Metabolic Relationships of Uncultured Bacteria Associated with the Microalgae Gambierdiscus. Environ Microbiol 2020, 22, 1764–1783. [Google Scholar] [CrossRef] [PubMed]
- Tizabi, D.; Bachvaroff, T. Nanopore Sequencing of Amoebophrya sp. Reveals Novel Collection of Bacteria Putatively Associated with Karlodinium veneficum. Genome Biol Evol, 2024. (In Review) [Google Scholar]
- Vico, P.; Iriarte, A.; Bonilla, S.; Piccini, C. Metagenomic Analysis of Raphidiopsis raciborskii Microbiome: Beyond the Individual. Biodivers Data J 2021, 9. [Google Scholar] [CrossRef]
- Miller, T.R.; Belas, R. Dimethylsulfoniopropionate Metabolism by Pfiesteria-Associated Roseobacter spp. Appl Environ Microbiol 2004, 70, 3383–3391. [Google Scholar] [CrossRef]
- Ding, W.; Wang, S.; Qin, P.; Fan, S.; Su, X.; Cai, P.; Lu, J.; Cui, H.; Wang, M.; Shu, Y.; et al. Anaerobic Thiosulfate Oxidation by the Roseobacter Group Is Prevalent in Marine Biofilms. Nat Commun 2023, 14, 2033. [Google Scholar] [CrossRef]
- Nuzzo, G.; Cutignano, A.; Sardo, A.; Fontana, A. Antifungal Amphidinol 18 and Its 7-Sulfate Derivative from the Marine Dinoflagellate Amphidinium carterae. J Nat Prod 2014, 77, 1524–1527. [Google Scholar] [CrossRef]
- Haq, S.; Oyler, B.L.; Williams, E.; Khan, M.M.; Goodlett, D.R.; Bachvaroff, T.; Place, A.R. Investigating A Multi-Domain Polyketide Synthase in Amphidinium carterae. Mar Drugs 2023, 21, 425. [Google Scholar] [CrossRef] [PubMed]
- LeKieffre, C.; Spero, H.J.; Fehrenbacher, J.S.; Russell, A.D.; Ren, H.; Geslin, E.; Meibom, A. Ammonium Is the Preferred Source of Nitrogen for Planktonic Foraminifer and Their Dinoflagellate Symbionts. Proceedings of the Royal Society B: Biological Sciences 2020, 287, 20200620. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, M.; Robertson, E.K.; Edler, L.; Arneborg, L.; Whitehouse, M.J.; Ploug, H. Nitrate and Ammonium Fluxes to Diatoms and Dinoflagellates at a Single Cell Level in Mixed Field Communities in the Sea. Sci Rep 2019, 9, 1424. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Coyne, K.J. Molecular Insights into the Synergistic Effects of Putrescine and Ammonium on Dinoflagellates. Int J Mol Sci 2024, 25, 1306. [Google Scholar] [CrossRef]
- Nishibori, N.; Imai, I. Polyamines Control the Growth of the Fish-Killing Dinoflagellate Karenia mikimotoi in Culture. Harmful Algae 2013, 29, 10–13. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, W.H.; Dong, B.; Li, B.; Yang, X.H. Changes in Intracellular and Extracellular Free Polyamines during the Growth Cycle of Prorocentrum donghaiense. IOP Conf Ser Earth Environ Sci 2019, 344, 012060. [Google Scholar] [CrossRef]
- Biebl, H.; Allgaier, M.; Lünsdorf, H.; Pukall, R.; Tindall, B.J.; Wagner-Döbler, I. Roseovarius mucosus sp. Nov., a Member of the Roseobacter Clade with Trace Amounts of Bacteriochlorophyll a. Int J Syst Evol Microbiol 2005, 55, 2377–2383. [Google Scholar] [CrossRef]
- Biebl, H.; Pukall, R.; Lünsdorf, H.; Schulz, S.; Allgaier, M.; Tindall, B.J.; Wagner-Döbler, I. Description of Labrenzia alexandrii Gen. Nov., Sp. Nov., a Novel Alphaproteobacterium Containing Bacteriochlorophyll a, and a Proposal for Reclassification of Stappia aggregata as Labrenzia aggregata Comb. Nov., of Stappia marina as Labrenzia marina Comb. Nov. and of Stappia alba as Labrenzia alba Comb. Nov., and Emended Descriptions of the Genera Pannonibacter, Stappia and Roseibium, and of the Species Roseibium denhamense and Roseibium hamelinense. Int J Syst Evol Microbiol 2007, 57, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Palacios, L.; Arahal, D.R.; Reguera, B.; Marín, I. Hoeflea alexandrii sp. Nov., Isolated from the Toxic Dinoflagellate Alexandrium minutum AL1V. Int J Syst Evol Microbiol 2006, 56, 1991–1995. [Google Scholar] [CrossRef]
- Strompl, C. Oceanicaulis alexandrii Gen. Nov., Sp. Nov., a Novel Stalked Bacterium Isolated from a Culture of the Dinoflagellate Alexandrium tamarense (Lebour) Balech. Int J Syst Evol Microbiol 2003, 53, 1901–1906. [Google Scholar] [CrossRef]
- Jiang, Z.; Duan, Y.; Yang, X.; Yao, B.; Zeng, T.; Wang, X.; Feng, Q.; Qi, M.; Yang, Q.; Zhang, X. Nitratireductor alexandrii Sp. Nov., from Phycosphere Microbiota of Toxic Marine Dinoflagellate Alexandrium tamarense. Int J Syst Evol Microbiol 2020, 70, 4390–4397. [Google Scholar] [CrossRef] [PubMed]
- Chin, G.J.W.L.; Jani, J.; Law, S.V.; Rodrigues, K.F. Whole Genome Sequence Data of a Marine Bacterium, Marinobacter adhaerens PBVC038, Associated with Toxic Harmful Algal Bloom. Data Brief 2023, 46, 108768. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Z.; Wang, H. Muricauda Amphidinii Sp. Nov., a Novel Marine Bacterium Isolated from the Phycosphere of Dinoflagellate Amphidinium carterae. Int J Syst Evol Microbiol 2019, 71. [Google Scholar] [CrossRef]
- Jung, H.S.; Jeong, S.E.; Chun, B.H.; Quan, Z.-X.; Jeon, C.O. Rhodophyticola porphyridii Gen. Nov., Sp. Nov., Isolated from a Red Alga, Porphyridium marinum. Int J Syst Evol Microbiol 2019, 69, 1656–1661. [Google Scholar] [CrossRef]
- Chernikova, T.N.; Bargiela, R.; Toshchakov, S.V.; Shivaraman, V.; Lunev, E.A.; Yakimov, M.M.; Thomas, D.N.; Golyshin, P.N. Hydrocarbon-Degrading Bacteria Alcanivorax and Marinobacter Associated With Microalgae Pavlova lutheri and Nannochloropsis oculata. Front Microbiol 2020, 11. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, L.; Li, Y.; Lai, Q.; Zhang, H.; Wei, J.; Zhou, Y.; Lei, X.; Zheng, W.; Tian, Y.; et al. Cyclobacterium xiamenense Sp. Nov., Isolated from Aggregates of Chlorella autotrophica, and Emended Description of the Genus Cyclobacterium. Int J Syst Evol Microbiol 2014, 64, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Z.; Li, C.; Zhang, M.; Zhao, D.; Li, J.; Zhang, Y. Algihabitans albus Gen. Nov., Sp. Nov., Isolated from a Culture of the Green Alga Ulva prolifera. Int J Syst Evol Microbiol 2019, 69, 828–832. [Google Scholar] [CrossRef]
- Jeong, S.E.; Kim, K.H.; Lhee, D.; Yoon, H.S.; Quan, Z.-X.; Lee, E.-Y.; Jeon, C.O. Oceaniradius stylonematis Gen. Nov., Sp. Nov., Isolated from a Red Alga, Stylonema cornu-cervi. Int J Syst Evol Microbiol 2019, 69, 1967–1973. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Aoyama, K.; Motone, K.; Aburaya, S.; Yamashiro, H.; Miura, N.; Inoue, K. Mutualistic Interactions between Dinoflagellates and Pigmented Bacteria Mitigate Environmental Stress. Microbiol Spectr 2023, 11. [Google Scholar] [CrossRef]
- Motone, K.; Takagi, T.; Aburaya, S.; Miura, N.; Aoki, W.; Ueda, M. A Zeaxanthin-Producing Bacterium Isolated from the Algal Phycosphere Protects Coral Endosymbionts from Environmental Stress. mBio 2020, 11. [Google Scholar] [CrossRef]
- Kowalska-Krochmal, B.; Dudek-Wicher, R. The Minimum Inhibitory Concentration of Antibiotics: Methods, Interpretation, Clinical Relevance. Pathogens 2021, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Rolinson, G. Forty Years of Beta-Lactam Research. Journal of Antimicrobial Chemotherapy 1998, 41, 589–603. [Google Scholar] [CrossRef]
- Bolch, C.J.S.; Subramanian, T.A.; Green, D.H. The Toxic Dinoflagellate Gymnodinium catenatum Require Marine Bacteria for Growth. J Phycol 2011, 47, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-H.; Kang, S.-J.; Lee, S.-Y.; Oh, K.-H.; Oh, T.-K. Seohaeicola saemankumensis Gen. Nov., Sp. Nov., Isolated from a Tidal Flat. Int J Syst Evol Microbiol 2009, 59, 2675–2679. [Google Scholar] [CrossRef]
- Nedashkovskaya, O.I.; Vancanneyt, M.; Kim, S.B.; Bae, K.S. Reclassification of Flexibacter tractuosus (Lewin 1969) Leadbetter 1974 and ‘Microscilla sericea’ Lewin 1969 in the Genus Marivirga Gen. Nov. as Marivirga tractuosa Comb. Nov. and Marivirga Sericea Nom. Rev., Comb. Nov. Int J Syst Evol Microbiol 2010, 60, 1858–1863. [Google Scholar] [CrossRef]
- Pagani, I.; Chertkov, O.; Lapidus, A.; Lucas, S.; Del Rio, T.G.; Tice, H.; Copeland, A.; Cheng, J.-F.; Nolan, M.; Saunders, E.; et al. Complete Genome Sequence of Marivirga tractuosa Type Strain (H-43T). Stand Genomic Sci 2011, 4, 154–162. [Google Scholar] [CrossRef]
- Dagenais-Bellefeuille, S.; Morse, D. Putting the N in Dinoflagellates. Front Microbiol 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Shang, L.; Wang, X.; Hu, Z.; Du, H.; Wang, H. Different Dimethylsulphoniopropionate-Producing Ability of Dinoflagellates Could Affect the Structure of Their Associated Bacterial Community. Algal Res 2021, 57, 102359. [Google Scholar] [CrossRef]
- Damm, E.; Kiene, R.P.; Schwarz, J.; Falck, E.; Dieckmann, G. Methane Cycling in Arctic Shelf Water and Its Relationship with Phytoplankton Biomass and DMSP. Mar Chem 2008, 109, 45–59. [Google Scholar] [CrossRef]
- Tse, S.P.K.; Lee, F.W.F.; Mak, D.Y.L.; Kong, H.K.; Chan, K.K.Y.; Lo, P.Y.; Lo, S.C.L. Production of Paralytic Shellfish Toxins (PSTs) in Toxic Alexandrium catenella Is Intertwined with Photosynthesis and Energy Production. Toxins (Basel) 2020, 12, 1–25. [Google Scholar] [CrossRef]
- Uribe, P.; Espejo, R.T. Effect of Associated Bacteria on the Growth and Toxicity of Alexandrium catenella. Appl Environ Microbiol 2003, 69, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yao, M.; Zhou, J.; Tan, S.; Jin, H.; Zhang, F.; Mak, Y.L.; Wu, J.; Lai Chan, L.; Cai, Z. Growth and Toxin Production of Gambierdiscus spp. Can Be Regulated by Quorum-Sensing Bacteria. Toxins (Basel) 2018, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Monroe, E.A.; Johnson, J.G.; Wang, Z.; Pierce, R.K.; Van Dolah, F.M. Characterization and Expression of Nuclear-Encoded Polyketide Synthases in the Brevetoxin-Producing Dinoflagellate Karenia brevis. J Phycol 2010, 46, 541–552. [Google Scholar] [CrossRef]
- Rein, K.S.; Borrone, J. Polyketides from Dinoflagellates: Origins, Pharmacology and Biosynthesis. Comp Biochem Physiol B Biochem Mol Biol 1999, 124, 117–131. [Google Scholar] [CrossRef]
- Houdai, T.; Matsuoka, S.; Murata, M.; Satake, M.; Ota, S.; Oshima, Y.; Rhodes, L.L. Acetate Labeling Patterns of Dinoflagellate Polyketides, Amphidinols 2, 3 and 4. Tetrahedron 2001, 57, 5551–5555. [Google Scholar] [CrossRef]
- Cruz-López, R.; Maske, H. The Vitamin B1 and B12 Required by the Marine Dinoflagellate Lingulodinium Polyedrum Can Be Provided by Its Associated Bacterial Community in Culture. Front Microbiol 2016, 7. [Google Scholar] [CrossRef]
- Berges, J.A.; Franklin, D.J.; Harrison, P.J. Evolution of an Artificial Seawater Medium: Improvements in Enriched Seawater, Artificial Water Over the Last Two Decades. J Phycol 2001, 37, 1138–1145. [Google Scholar] [CrossRef]
- Gonçalves, C.; Gonçalves, P. Multilayered Horizontal Operon Transfers from Bacteria Reconstruct a Thiamine Salvage Pathway in Yeasts. Proceedings of the National Academy of Sciences 2019, 116, 22219–22228. [Google Scholar] [CrossRef] [PubMed]
- Del Mondo, A.; Smerilli, A.; Sané, E.; Sansone, C.; Brunet, C. Challenging Microalgal Vitamins for Human Health. Microb Cell Fact 2020, 19, 201. [Google Scholar] [CrossRef]
- Cenci, U.; Qiu, H.; Pillonel, T.; Cardol, P.; Remacle, C.; Colleoni, C.; Kadouche, D.; Chabi, M.; Greub, G.; Bhattacharya, D.; et al. Host-Pathogen Biotic Interactions Shaped Vitamin K Metabolism in Archaeplastida. Sci Rep 2018, 8, 15243. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat Biotechnol 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Amphidinium carterae growth with and without antibiotics (a) growth curves of A. carterae cultures; (b) Image of lysed A. carterae (red arrow) taken at 100X, and presence of rod-shaped bacteria (blue arrow).
Figure 1.
Amphidinium carterae growth with and without antibiotics (a) growth curves of A. carterae cultures; (b) Image of lysed A. carterae (red arrow) taken at 100X, and presence of rod-shaped bacteria (blue arrow).
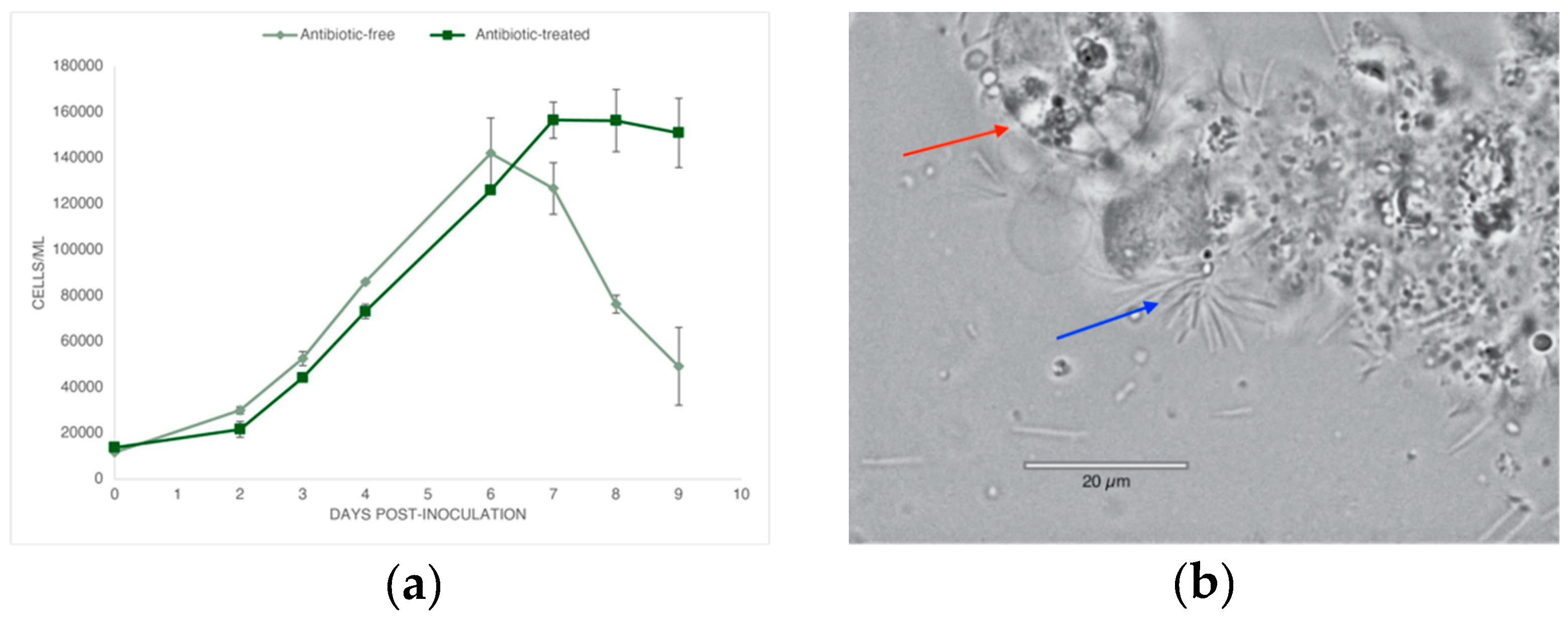
Figure 2.
Bacterial populations with and without antibiotics (a) Percent of reads and bases mapping back to bacterial genomes from both the antibiotic-free and antibiotic-treated cultures, aligned using minimap2; (b) Bacterial abundance based on read mapping in the antibiotic-free and antibiotic-treated cultures.
Figure 2.
Bacterial populations with and without antibiotics (a) Percent of reads and bases mapping back to bacterial genomes from both the antibiotic-free and antibiotic-treated cultures, aligned using minimap2; (b) Bacterial abundance based on read mapping in the antibiotic-free and antibiotic-treated cultures.
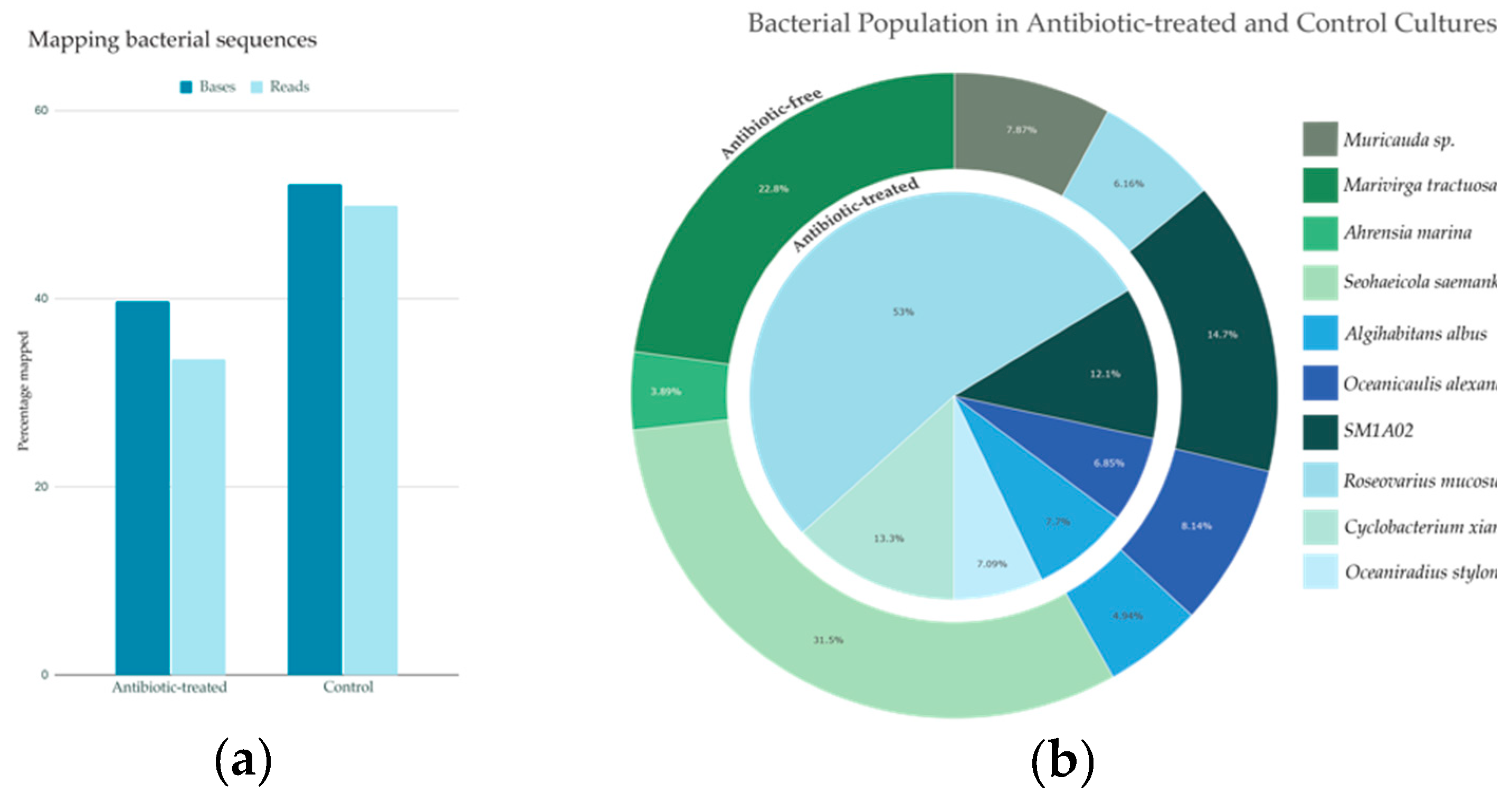
Figure 4.
Secondary metabolites present in the bacterial genomes, identified by antiSMASH [35].
Figure 4.
Secondary metabolites present in the bacterial genomes, identified by antiSMASH [35].

Figure 5.
Antimicrobial resistance in assembled microbiome (a) Antimicrobial resistance genes within each assembled bacterial genome (b) Specialty genes identified from the CARD database [36].
Figure 5.
Antimicrobial resistance in assembled microbiome (a) Antimicrobial resistance genes within each assembled bacterial genome (b) Specialty genes identified from the CARD database [36].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



