
Submitted:
24 June 2024
Posted:
26 June 2024
You are already at the latest version
Abstract
High systolic blood pressure and increased blood pressure variability after the onset of ischemic stroke are associated with poor clinical outcomes. One of the key determinants of blood pressure is arteriolar size, determined by vascular smooth muscle tone and vasodilatory and vasoconstrictor substances that are released by the endothelium. The aim of this study is to outline alterations in vasomotor function in isolated peripheral arteries following ischemic stroke. The reactivity of thoracic aortic segments from male C57BL/6 mice to dilators and constrictors was quantified using wire myography. Acetylcholine induced endothelium-dependent vasodilation was impaired after ischemic stroke (LogIC50 Sham=-7.499, LogIC50 Stroke=-7.350, P=0.0132, n= 19, 31 respectively). Vasodilatory responses to SNP were identical in isolated aortas in sham and stroke groups. Phenylephrine induced vasoconstriction was impaired in aortas isolated from stroke animals in comparison to sham counterparts (Sham LogEC50= -6.652 vs Stroke LogEC50=-6.475, P< 0.001). Our study demonstrates that 24 hours post-ischemic stroke, peripheral vascular responses are impaired in remote arteries. Aortas from stroke animals exhibited reduced vasoconstrictor and endothelium-dependent vasodilator responses, while endothelium-independent vasodilatory responses were preserved. Since both vasodilatory and vasoconstrictor responses of peripheral arteries are impaired following ischemic stroke, our findings might explain increased blood pressure variability following ischemic stroke.
Keywords:
Ischemic Stroke
; Blood Pressure
; Hypertension
; Hypotension
; Vasodilation
; Vasoconstriction
; Peripheral Vessels
; Myography
; alpha-adrenergic receptors
; middle cerebral artery occlusion
1. Introduction
Ischemic stroke is a devastating disease and the fifth leading cause of death in the United States. It is also one of the main causes of long-term disability with an estimated 7.6 million annual stroke survivors. Stroke-related disability places a great burden on society. For example, the estimated cost of stroke treatment and related morbidity cost in 2018 was $52.8 billion. [1] Despite the high impact of ischemic stroke on general public health, current therapies are limited to maintenance of normal homeostasis and prevention and prompt treatment of secondary insults. Despite numerous pharmacological therapies tested to prevent the sequelae of ischemic stroke, the experimental therapies have not been successfully translated to clinical settings.[2] While many therapeutic approaches target the neuronal death resulting from ischemia, a critical gap exists in the literature regarding the co-existing morbidities and their underlying mechanisms in stroke patients. These co-morbidities include dysregulation of blood pressure, cardiac dysfunction, metabolic and immune alterations that are related to stroke.[3,4] These co-morbidities responsible for dysregulation of blood pressure triggered by cerebral ischemia and reperfusion are significant and require development of novel treatment strategies.[3,5,6]
It has been established that high systolic blood pressure[7] and increased blood pressure variability after the onset of ischemic stroke are associated with poor outcomes [5,8,9] and increased mortality in patients.[10] Systemic mean arterial blood pressure is determined by cardiac output and systemic vascular resistance. One of the key determinants of systemic vascular resistance is arteriolar size. Arteriolar size is further determined by the contractility of the vascular smooth muscle and the vasodilatory and vasoconstrictor substances that are released by the endothelial cells. Any change in the responses of either endothelial cells and/or vascular smooth muscle would alter the mean arterial blood pressure. Although clinical studies suggest that endothelium-dependent and independent vasodilation are impaired in stroke patients[11,12], the mechanisms that underlie impaired peripheral vascular responses in ischemic stroke remain unclear. Local alterations in the peripheral vascular responses such as endothelium-dependent and independent vasodilation and vascular smooth muscle contraction following ischemic stroke might be the underlying causes of the mean arterial blood pressure variability following ischemic stroke. Since blood pressure variability is a risk factor following ischemic stroke, it is of paramount importance to address the underlying mechanisms of impaired vascular responses in ischemic stroke.
The main underlying concept of current study is that ischemic stroke is accompanied by one or more features of vascular dysfunction at remote sites of the body other than the brain itself. This contention is supported by three major lines of evidence: a) It has been established that high systolic blood pressure[7] and increased blood pressure variability after the onset of ischemic stroke are associated with poor outcomes[5,8] and increased mortality in patients.[10] b) Following ischemic stroke patients exhibit impaired peripheral vasodilation due to endothelial dysfunction compared to hypertension-matched controls and healthy control groups.[11] c) There is a correlation between the expression of proinflammatory cytokines and endothelial dysfunction observed in the patients following ischemic stroke.[13]
Although clinical studies document an association between ischemic stroke and endothelial-dependent vascular dysfunction at peripheral circulation, there are limited studies that address the mechanism behind vascular dysfunction following ischemic stroke. Additionally, there is a lack of established studies that identify and measure both vasoconstriction and endothelium-dependent as well as endothelium-independent vasodilation following an ischemic stroke. Consequently, the aim of this study is to outline the changes in vasomotor function in isolated peripheral arteries following ischemic stroke.
2. Materials and Methods
Animals: All experimental procedures were performed on male wildtype C57Bl/6 mice. The animal experiments will be performed according to the criteria outlined by the National Institutes of Health and approved by the City College, City University of New York, IACUC. A sham group (wild-type C57Bl/6J [WT] exposed to sham operation; (n=31) and the sham group exposed to 1 hour of middle cerebral artery occlusion (MCAO) n=19 and exposed to 23 hours reperfusion were used.
Ischemic Stroke Model: The mice were anesthetized by inhaled isoflurane. Transient (60 minutes) focal cerebral ischemia and reperfusion induced by occlusion and reperfusion of the left middle cerebral artery (MCAo/R). A blunted tip of a 6-0 nylon monofilament is advanced to the level of the carotid bifurcation via the internal carotid artery until light resistance is felt. The monofilament removed after 60 minutes of occlusion. In the sham group, these arteries are visualized but not disturbed. At the end of experiments, mice were euthanized, and the brains were removed, and then stained with 2% 2, 3, 5-triphenyl tetrazolium chloride (TTC) to confirm the production of an infarct.[14] NIH Image J is used for image processing and quantification of the percent of infarct area.
Wire Myography: The reactivity of mouse thoracic aortic segments to different dilators and constrictors were quantified using wire myography.[15] 2-mm long segments of the aorta were isolated 24 hours after the onset of either will be mounted in a small vessel wire myograph (DMT, Denmark). Vessel rings will be maintained in 5-ml organ baths with oxygenated physiological saline solution (95% O2 and 5% CO2) at 37.1°C. Aortic rings will be rinsed with a 60mM KCl solution for vascular smooth muscle activation and to determine the maximal contractile response. The aortic rings will be then precontracted with 10-6 M phenylephrine (PE) to obtain submaximal contraction, and after obtaining a stable plateau phase of contraction, the endothelium-dependent responses of the endothelium wire assessed by increasing cumulative concentrations of acetylcholine (ACh: 10-9 –10-4 M). Endothelium-dependent dilations were expressed as the percent dilation from the precontraction response to 10-6 M PE. Endothelium-independent vasorelaxation are determined by application of increasing cumulative concentrations of sodium nitroprusside (SNP; 10-9-10-6 M), while vascular smooth muscle contraction is determined by increasing cumulative concentrations of PE (10-9-10-4 M).
Statistical Analysis: All values are reported as means ± SE. Comparisons between two groups of animals or treatments will be made by t-test, and comparisons among groups of more than 2 will be analyzed by one-way ANOVA. Statistical analysis, nonlinear analysis of the dose response curves, curve fitting and half maximal effective concentration (EC50) and the half-maximal inhibitory concentration (IC50) values were performed using GraphPad Prism version 10.2.3 (347), for MacOS, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com. A null hypothesis of EC50 and IC50 are same for data sets are tested. At P<0.05 null hypothesis rejected and is reported as significant.
3. Results
3.1.1. Endothelium-Dependent Vasodilation Is Impaired Following Ischemic Stroke
Endothelium-dependent nitric oxide mediated vasodilation was assessed by increasing concentration of acetylcholine. Acetylcholine induced endothelium-dependent vasodilation was impaired after ischemic stroke in thoracic aorta (Figure 1). The greatest difference in acetylcholine induced vasodilatation were observed in the concentration range of 10-8 M to 10-6 M of acetylcholine between stroke and sham groups. A 1.42-fold increase in concentration of acetylcholine was required to achieve 50% of vasodilation in aortas isolated form stroke mice in comparison to sham mice. LogIC50s for ACh were different in sham vs stroke vessels (LogIC50 Sham=-7.499 vs LogIC50 Stroke=-7.350, P=0.0132, n= 19, 31 respectively) (Figure 2).
3.1.2. Endothelium-Independent Vasodilation Is Preserved Following Ischemic Stroke
Endothelium-independent vasodilation was assessed by administration of increasing SNP, a non-enzymatic nitric oxide donor. Vasodilatory responses to SNP were identical in isolated aortas in sham and stroke groups (Figure 3). (Log IC50 Sham = -7.924 vs Log IC50 Stroke=-7.875, P=0.3637). There were no differences between the curves (Figure 4).
3.1.3. Vascular Smooth Muscle Contraction Is Impaired Following Ischemic Stroke
Vascular smooth muscle contraction in aorta was assessed by administration of increasing concentrations of alpha-1 receptor agonist phenylephrine. Phenylephrine induced vasoconstriction was impaired in aortas isolated from stroke animals in comparison to sham counterparts (Figure 5). Sham and stroke dose response curves have significantly different LogEC50s (Sham LogEC50= -6.652 vs Stroke LogEC50=-6.475, P<0.001) (Figure 6). This impairment was prominent in the concentration range of 10-7 M-10-5 M of phenylephrine concentration. A 1.5-fold increase in phenylephrine concentration was required to achieve 50% of maximum vasoconstriction were required in the aortas isolated form stroke animals in comparison to sham counterparts (Figure 5).
3.1.4. Infarct Size
Infarct size was calculated as the percent of largest cross-sectional area of infarct to the whole brain area for each brain sample. In sham group no infract was observed. In stroke group, 24 hours after the induction of MCAO/R, average infarct size of 23.2±1.5 % (n=19) is observed. A representative of infarct vs sham brain samples with mean infarct percentage of the stroke group shown in Figure 7.
4. Discussion
Our study demonstrates that 24 hours following ischemic stroke peripheral vascular responses are impaired in remote arteries (aortas). The aortas isolated from stroke animals exhibited both reduced vasoconstrictor and reduced endothelium-dependent vasodilator responses while endothelium-independent vasodilatory responses were preserved. Our findings indicate local dysregulation of peripheral vascular responses following ischemic stroke.
Endothelium-dependent vasodilator responses depend on a healthy endothelial cell layer, and healthy nitric oxide production in endothelial cells. Our results indicate an impaired vasodilatory response to acetylcholine. Acetylcholine binds to its muscarinic receptor and activates eNOS IP3/Ca++ dependent manner. In this reaction arginine is converted to citrulline and nitric oxide is produced by activated eNOS Nitric oxide then diffuses to underlying smooth muscle activating guanylyl cylase which leads to vasodilation.[16] An impairment of endothelium-dependent vasodilation in the presence of preserved endothelium-independent vasodilation in response to nitric oxide releasing agent SNP suggests that nitric oxide dependent vascular smooth muscle relaxation is preserved and impairment in vasodilatory responses is due to endothelial dysfunction following ischemic stroke. It is possible that direct endothelial cell damage, a decreased expression or activity of eNOS, low substrate availability, endogenous inhibition of eNOS or reduced bioavailability of NO would be the culprits of decreased vasodilatory responses[16] that is observed following ischemic stroke. Clinical studies[17] addressed an impaired vasodilatory response in stroke patients our results suggest local damage to endothelial layer which excludes the contribution of nervous system to the observed impairment of endothelial dependent vasodilation following stroke.
Vascular smooth muscle tone is locally mediated by nitric oxide production in the endothelial cells. Impairment of endothelium derived nitric oxide dependent vasodilation leads to increased vascular resistance and increased blood pressure. [18,19] Our results suggest a relation between ischemic stroke induced endothelial damage and increased blood pressure. Ischemic stroke may induce high blood pressure[20]. Clinical significance of impaired endothelial vasodilation following ischemic stroke has not been well-defined. While increased blood pressure following ischemic stroke has been reported as a poor prognostic factor [21,22] and associated with all-cause mortality, in different patient populations lower systolic blood pressure is associated with favorable outcome.[23] or a U-shaped correlation with the adverse outcomes and blood pressure is reported.[21] Nevertheless it is possible that endothelial dysfunction contributes to increased blood pressure observed in ischemic stroke due to increased systemic vascular resistance.
Isolated blood vessels are devoid of the acute influence of humoral and nervous system modifications that might be activated following ischemic stroke. Since endothelial cells are exposed to proinflammatory cytokines it is possible that proinflammatory cytokines that are increased due to ischemic stroke might link the brain ischemia to endothelial dysfunction. Potential candidates that would impact distant endothelial cell layer following ischemic stroke are IFN-gamma, IL-6, IL-10, TGF-beta [24,25,26] . Cerebral ischemia accompanies an increase in the expression of endothelial adhesion molecules in the vasculature [27]. Although studies are mostly limited to modifications of cerebral adhesion molecules it is possible that cerebral ischemia may induce endothelial cell adhesion molecules at distant vasculature. For example, increase in adhesion molecule VCAM-1 was associated with lower flow mediated vasodilation in stroke patients. [13] Increased expression of endothelial adhesion molecules such as β2-integrins (CD11/CD18), ICAM-1, and P-selectin might play a role in the recruitment of leukocytes as well as platelets in the peripheral vessels following ischemic stroke and potentially leading to cell mediated dysfunction of endothelial cells.
Our results implicate a humoral factor that is induced by ischemic stroke with a potential to induce endothelial dysfunction in peripheral vessels. In support of our findings, in previous studies serum collected from ischemic stroke patients in the first 24 hours of onset of ischemic stroke were shown to induce an impairment of endothelial dependent vascular responses in cerebral and mesenteric arteries [28]. In addition, impairment of endothelium dependent vascular responses were also reported with serum isolated from stroke patients with atherosclerosis 7 days to a year after the onset of ischemic event. [29] Although our study and previous studies implicate cerebral ischemia as a triggering factor for subsequent endothelial dysfunction whether brain being the source of humoral factor or factors that exerts this dysfunction remains unexplored. In one study atherosclerotic plaque associated proinflammatory factors such as IL-1β and subsequent activation of cyclooxygenase are implicated[29] as triggering factors for remote endothelial dysfunction in stroke patients, albeit in our study, in the absence of atherosclerosis we still observed endothelial dysfunction. The nature of substances that might trigger peripheral endothelial dysfunction following ischemic stroke has been partially addressed in the current literature. Remote endothelial dysfunction triggered by cerebral ischemia has been associated with proinflammatory milieu and increased oxidative stress[28], increased IL-1β and cyclooxygenase products[29], and with circulating white blood cells[30]
Ischemic stroke is accompanied by profound changes in humoral and cellular immune responses.[24,26] while it is possible that endothelial impairment might be due to activation of circulating white blood cells[30], in first 24 hours a suppression of peripheral immune responses rather than an activation has been reported [24].
Our study discovered a diminished vasoconstrictor response to the alpha-1 agonist phenylephrine in aortas isolated from stroke animals, compared to sham animals. This represents a novel finding.Alpha-1 receptors are prominent at mice aorta[31] and activation of these receptors by selective apha-1 agonist phenylephrine induce vascular smooth muscle contraction and vasoconstriction.[32] Our study shows that a 1.5 fold increase in phenylephrine concentration is need to achieve the same level of vasoconstriction in aortas isolated from stroke animals in comparison to sham aortas. These results indicate a blunted vasoconstrictor response to alpha-1 adrenergic agonist following ischemic stroke. A reduction of in response to alpha-1 receptor agonist induced vasoconstriction might be due to decreased alpha-1 receptor activity in response to proinflammatory cytokines. In support of this contention phenylephrine mediated vasoconstrictor responses were reported to be blunted with the treatment of isolated aortas with IL-6.[33]
Sympathetic nervous system induces vasoconstriction via activating alpha-1 receptors by releasing norepinephrine and epinephrine. Minute-to minute regulation of blood pressure is achieved by baroreceptor reflex mechanism. In case of low blood pressure, increase in sympathetic outflow and subsequent activation of alpha-1 receptors on vascular smooth muscle results in vasoconstriction which increases total peripheral resistance and reverts the blood pressure back to normal values. Our results show that vascular smooth muscle vasoconstriction in response to alpha-1 adrenergic receptor activation is blunted following ischemic stroke. Impaired vasoconstrictor response to alpha-1 agonists after ischemic stroke might be one of the mechanisms that would impair blood pressure regulation in ischemic stroke patients. We have observed an approximate 10% reduction in the vasoconstrictor response to the same concentration of phenylephrine in aortas isolated from stroke animals. This 10% reduction in vessel size could result in around a 30% decrease in blood pressure, provided that flow is constant to the vascular beds. The implications of this blood pressure reduction following ischemic stroke may include impaired cerebral perfusion and potential hemodynamic instability. This instability poses a significant risk for stroke patients who are already vulnerable to immobility, secondary infections, and increased deep vein thrombosis. In ischemic stroke patients, hypotension is accompanied with ischemic stroke and is an independent prognostic factor for poor outcomes.[21,34] Our results suggest that impaired alpha-1 adrenergic receptor mediated vasoconstriction might be one of the factors that lead to hypotension following ischemic stroke. Although larger patient groups needed for conclusive results[35], pharmacological elevation of blood pressure in acute phases of ischemic stroke using phenylephrine appears to be associated with a better prognosis in patients.[36,37]
In our study, vasodilatory responses to nitric oxide releasing SNP [38] were identical in vessels isolated from both sham and stroke animals. This finding indicates that nitric oxide dependent vasorelaxation is preserved after ischemic stroke in first 24 hours after the onset of cerebral ischemia. NO activates soluble guanylyl cyclase (sGC) and increases cGMP levels that leads to vascular smooth muscle relaxation.[32] Preservation of vasodilatory responses to NO suggests that downstream events to NO production such as guanylyl cyclase activity, cGMP production and cGMP coupled vascular smooth muscle relaxation dynamics are conserved after ischemic stroke.
Peripheral vasomotor responses following a stroke may also be potently influenced by the interference with normal angiotensin converting enzyme-2 (ACE-2) activity. ACE-2 plays a crucial role in regulating the balance between Angiotensin-1 -7 and Angiotensin-2, with Angiotensin-1-7 generally promoting vasodilation and Angiotensin-2 supporting vasoconstriction. After a stroke, the impairment of ACE-2 function could skew this balance towards a vasoconstrictor-dominant environment. This shift results in heightened vascular resistance and reduced vasodilation capacity, contributing to the compromised vasomotor responses observed in peripheral arteries post-stroke.
Additionally, the aberrant ratio of Angiotensin-1 to Angiotensin-2 not only favors vasoconstriction but also promotes hypercoagulation and inflammation, further exacerbating vascular dysfunction. [42] The increase in Angiotensin-2 can lead to the activation of pro-inflammatory pathways and the stimulation of pro-thrombotic mechanisms, thereby heightening the risk of clot formation and inflammatory damage within the vasculature. These combined effects of impaired ACE-2 function create a hostile vascular environment that may contribute to the increased blood pressure variability and the overall vascular instability seen in patients after an ischemic stroke. Additionally, disturbances in the receptor for Ang-1-7, the Mas receptor may also be a suppressed target after stroke which is dysregulated to provoke these types of responses possibly in response to inflammatory cytokines which may govern expression of ACE-2, Ang-1-7, Mas receptor or other components of this signaling module. [42–46]
Our study is limited by the use of thoracic aorta as the target of vascular alterations following ischemic stroke. Given that the aorta functions as a transportation vessel rather than a resistance artery, it is possible that the vascular responses we elicited may exhibit a different profile compared to resistance arteries. It is documented that arteries with vascular resistance elements such as mesenteric, splenic, hepatic and distal omental arteries have higher density of alpha-adrenergic receptors than the aorta [39] It is plausible that these arteries would exhibit pronounced responses to adrenergic receptor dysfunction following ischemic stroke. Similar studies in in resistance arteries remain warranted. Under this study, we have not included potential humoral and neuronal signaling mechanisms that could lead to peripheral vascular changes associated with ischemic stroke. The vascular dysfunction associated with ischemic stroke might be related to an alteration in neuronal input to the peripheral tissues such as dysfunction of autonomic nervous system. Furthermore, since ischemic stroke is associated with profound modification of immune system responses[26], it is possible that endothelium or vascular smooth muscle dysfunction could be a by-product of ongoing inflammatory changes during a stroke. In addition, possible changes of cardiac function[40] that might lead to blood pressure variability associated with ischemic stroke has not been addressed in this study.
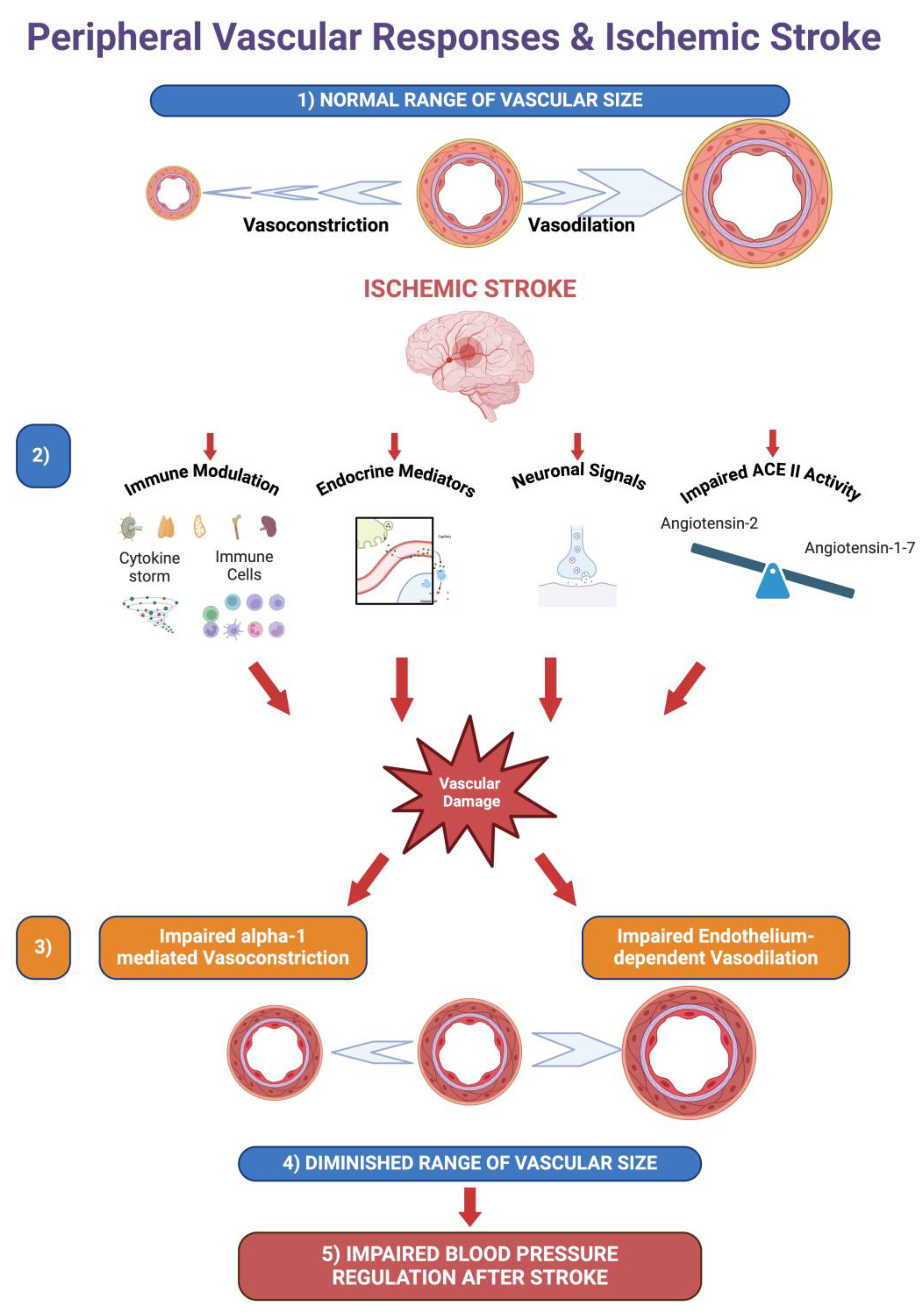
Our results implicate both vasodilatory and vasoconstrictor responses are impaired following ischemic stroke. Blood pressure variability following ischemic stroke is associated with poor clinical outcomes and higher mortality [5,8,9]. Our findings might explain increased blood pressure variability following ischemic stroke. Local vascular changes in arteries would explain blood pressure changes and increased blood pressure variability following an ischemic stroke. A reduced capacity in alpha-1 receptor mediated vasoconstriction would partially impair the ability of vascular system to increase the peripheral resistance and reset of the blood pressure to normal physiological range when needed, whereas a reduced capacity of vasodilation would impair the reduction of vascular resistance in response to increased blood pressure. Since both vasoconstrictor and vasorelaxant properties of the arteries are impaired, blood pressure dysregulation following ischemic stroke might be partially explained by impairment of local vascular responses in stroke patients (Figure 8). For reduced morbidity it is vital to keep the blood pressure controlled not only following ischemic stroke but also after endovascular therapies for stroke treatment.[41] Our findings might shed light to the mechanisms of blood pressure control associated with ischemic stroke.
Author Contributions
Conceptualization, G.Y. and J.S.A.; methodology, G.Y.; software, G.Y.; validation, G.Y.; formal analysis, G.Y.; investigation, G.Y.; resources, G.Y.; data curation, G.Y.; writing—original draft preparation, G.Y. and J.S.A.; writing—review and editing, G.Y. and J.S.A; visualization, G.Y. and J.S.A; supervision, G.Y. and J.S.A; project administration, G.Y.; funding acquisition, G.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by PSC CUNY Cycle 54 Award, 66608-00 54 and CUNY Start-up funds.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board (or Ethics Committee) of the City College, City University of New York, IACUC (Protocol No.: 1121.A1 and March 23, 2023).
Data Availability Statement
Dataset available on request from the authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- O'Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; van der Worp, B.H.; Howells, D.W. 1,026 experimental treatments in acute stroke. Ann Neurol 2006, 59, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, M.J.; Liebeskind, D.S.; Chan, S.L. The importance of comorbidities in ischemic stroke: Impact of hypertension on the cerebral circulation. J Cereb Blood Flow Metab 2018, 38, 2129–2149. [Google Scholar] [CrossRef]
- Gallacher, K.I.; Jani, B.D.; Hanlon, P.; Nicholl, B.I.; Mair, F.S. Multimorbidity in Stroke. Stroke 2019, 50, 1919–1926. [Google Scholar] [CrossRef]
- Manning, L.S.; Rothwell, P.M.; Potter, J.F.; Robinson, T.G. Prognostic Significance of Short-Term Blood Pressure Variability in Acute Stroke: Systematic Review. Stroke 2015, 46, 2482–2490. [Google Scholar] [CrossRef]
- Hawkes, M.A.; Anderson, C.S.; Rabinstein, A.A. Blood Pressure Variability After Cerebrovascular Events: A Possible New Therapeutic Target: A Narrative Review. Neurology 2022, 99, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, S.; Silvestrini, M.; Provinciali, L. Elevated blood pressure in the acute phase of stroke and the role of Angiotensin receptor blockers. Int J Hypertens 2013, 2013, 941783. [Google Scholar] [CrossRef]
- de Havenon, A.; Bennett, A.; Stoddard, G.J.; Smith, G.; Chung, L.; O'Donnell, S.; McNally, J.S.; Tirschwell, D.; Majersik, J.J. Determinants of the impact of blood pressure variability on neurological outcome after acute ischaemic stroke. Stroke Vasc Neurol 2017, 2, 1–6. [Google Scholar] [CrossRef]
- Chung, J.-W.; Kim, N.; Kang, J.; Park, S.H.; Kim, W.-J.; Ko, Y.; Park, J.H.; Lee, J.S.; Lee, J.; Yang, M.H.; et al. Blood pressure variability and the development of early neurological deterioration following acute ischemic stroke. Journal of Hypertension 2015, 33, 2099–2106. [Google Scholar] [CrossRef]
- de Havenon, A.; Stoddard, G.; Saini, M.; Wong, K.H.; Tirschwell, D.; Bath, P.; Collaborators, V.I.-A. Increased blood pressure variability after acute ischemic stroke increases the risk of death: A secondary analysis of the Virtual International Stroke Trial Archive. JRSM Cardiovasc Dis 2019, 8, 2048004019856496. [Google Scholar] [CrossRef]
- Stenborg, A.; Terent, A.; Lind, L. Endothelium-dependent vasodilatation in forearm is impaired in stroke patients. J Intern Med 2006, 259, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Wang, P.Y.; Sheu, W.H.; Chen, Y.T.; Ho, Y.P.; Hu, H.H.; Hsu, H.Y. Changes of brachial flow-mediated vasodilation in different ischemic stroke subtypes. Neurology 2006, 67, 1056–1058. [Google Scholar] [CrossRef]
- Billinger, S.A.; Sisante, J.V.; Mattlage, A.E.; Alqahtani, A.S.; Abraham, M.G.; Rymer, M.M.; Camarata, P.J. The relationship of pro-inflammatory markers to vascular endothelial function after acute stroke. Int J Neurosci 2017, 127, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Alexander, J.S.; Erkuran Yilmaz, C.; Granger, D.N. Induction of neuro-protective/regenerative genes in stem cells infiltrating post-ischemic brain tissue. Exp Transl Stroke Med 2010, 2, 11. [Google Scholar] [CrossRef]
- del Campo, L.; Ferrer, M. Wire Myography to Study Vascular Tone and Vascular Structure of Isolated Mouse Arteries. Methods Mol Biol 2015, 1339, 255–276. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ Res 2016, 119, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Pretnar-Oblak, J.; Sabovic, M.; Pogacnik, T.; Sebestjen, M.; Zaletel, M. Flow-mediated dilatation and intima-media thickness in patients with lacunar infarctions. Acta Neurol Scand 2006, 113, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N Engl J Med 1990, 323, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Panza, J.A.; Garcia, C.E.; Kilcoyne, C.M.; Quyyumi, A.A.; Cannon, R.O. , 3rd. Impaired endothelium-dependent vasodilation in patients with essential hypertension. Evidence that nitric oxide abnormality is not localized to a single signal transduction pathway. Circulation 1995, 91, 1732–1738. [Google Scholar] [CrossRef]
- Bath, P.; Chalmers, J.; Powers, W.; Beilin, L.; Davis, S.; Lenfant, C.; Mancia, G.; Neal, B.; Whitworth, J.; Zanchetti, A.; et al. International Society of Hypertension (ISH): Statement on the management of blood pressure in acute stroke. J Hypertens 2003, 21, 665–672. [Google Scholar] [CrossRef]
- Leonardi-Bee, J.; Bath, P.M.; Phillips, S.J.; Sandercock, P.A.; Group, I.S.T.C. Blood pressure and clinical outcomes in the International Stroke Trial. Stroke 2002, 33, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Jillella, D.V.; Calder, C.S.; Uchino, K.; Qeadan, F.; Ikram, A.; Casul, Y.R.; Tran, H.Q. Blood Pressure and Hospital Discharge Outcomes in Acute Ischemic Stroke Patients Undergoing Reperfusion Therapy. J Stroke Cerebrovasc Dis 2020, 29, 105211. [Google Scholar] [CrossRef] [PubMed]
- Maier, B.; Gory, B.; Taylor, G.; Labreuche, J.; Blanc, R.; Obadia, M.; Abrivard, M.; Smajda, S.; Desilles, J.P.; Redjem, H.; et al. Mortality and Disability According to Baseline Blood Pressure in Acute Ischemic Stroke Patients Treated by Thrombectomy: A Collaborative Pooled Analysis. J Am Heart Assoc 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Hagmann, S.; Zschoche, C.; Adamek, J.; Zhou, W.; Sun, L.; Hug, A.; Zorn, M.; Dalpke, A.; Nawroth, P.; et al. The spectrum of systemic immune alterations after murine focal ischemia: Immunodepression versus immunomodulation. Stroke 2009, 40, 2849–2858. [Google Scholar] [CrossRef] [PubMed]
- Terao, S.; Yilmaz, G.; Stokes, K.Y.; Russell, J.; Ishikawa, M.; Kawase, T.; Granger, D.N. Blood cell-derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia-reperfusion. Stroke 2008, 39, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 2006, 113, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Granger, D.N. Cell adhesion molecules and ischemic stroke. Neurol Res 2008, 30, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Canavero, I.; Sherburne, H.A.; Tremble, S.M.; Clark, W.M.; Cipolla, M.J. Effects of Acute Stroke Serum on Non-Ischemic Cerebral and Mesenteric Vascular Function. Translational Stroke Research 2016, 7, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Puertas-Umbert, L.; Puig, N.; Camacho, M.; Dantas, A.P.; Marín, R.; Martí-Fàbregas, J.; Jiménez-Xarrié, E.; Benitez, S.; Camps-Renom, P.; Jiménez-Altayó, F. Serum from Stroke Patients with High-Grade Carotid Stenosis Promotes Cyclooxygenase-Dependent Endothelial Dysfunction in Non-ischemic Mice Carotid Arteries. Translational Stroke Research 2024, 15, 140–152. [Google Scholar] [CrossRef]
- Asano, S.; O'Connell, G.C.; Lemaster, K.C.; DeVallance, E.R.; Branyan, K.W.; Simpkins, J.W.; Frisbee, J.C.; Barr, T.L.; Chantler, P.D. Circulating leucocytes perpetuate stroke-induced aortic dysfunction. Exp Physiol 2017, 102, 1321–1331. [Google Scholar] [CrossRef]
- van Langen, J.T.; Van Hove, C.E.; Schrijvers, D.M.; Martinet, W.; De Meyer, G.R.; Fransen, P.; Bult, H. Contribution of alpha-adrenoceptor stimulation by phenylephrine to basal nitric oxide production in the isolated mouse aorta. J Cardiovasc Pharmacol 2013, 61, 318–323. [Google Scholar] [CrossRef]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. alpha1 -Adrenoceptor activation of PKC-epsilon causes heterologous desensitization of thromboxane receptors in the aorta of spontaneously hypertensive rats. Br J Pharmacol 2015, 172, 3687–3701. [Google Scholar] [CrossRef]
- Ohkawa, F.; Ikeda, U.; Kanbe, T.; Kawasaki, K.; Shimada, K. Effects of inflammatory cytokines on vascular tone. Cardiovascular Research 1995, 30, 711–715. [Google Scholar] [CrossRef]
- Zhu, Y.; Wu, M.; Wang, H.; Zheng, Y.; Zhang, S.; Wang, X.; Wang, S.; Fang, Z. Daily blood pressure variability in relation to neurological functional outcomes after acute ischemic stroke. Front Neurol 2022, 13, 958166. [Google Scholar] [CrossRef]
- Stromsnes, T.A.; Kaugerud Hagen, T.J.; Ouyang, M.; Wang, X.; Chen, C.; Rygg, S.E.; Hewson, D.; Lenthall, R.; McConachie, N.; Izzath, W.; et al. Pressor therapy in acute ischaemic stroke: An updated systematic review. Eur Stroke J 2022, 7, 99–116. [Google Scholar] [CrossRef]
- Bang, O.Y.; Chung, J.W.; Kim, S.K.; Kim, S.J.; Lee, M.J.; Hwang, J.; Seo, W.K.; Ha, Y.S.; Sung, S.M.; Kim, E.G.; et al. Therapeutic-induced hypertension in patients with noncardioembolic acute stroke. Neurology 2019, 93, e1955–e1963. [Google Scholar] [CrossRef]
- Kim, H.J.; Kang, D.W. Induced hypertensive therapy in an acute ischemic stroke patient with early neurological deterioration. J Clin Neurol 2007, 3, 187–191. [Google Scholar] [CrossRef]
- Bates, J.N.; Baker, M.T.; Guerra, R., Jr.; Harrison, D.G. Nitric oxide generation from nitroprusside by vascular tissue. Evidence that reduction of the nitroprusside anion and cyanide loss are required. Biochem Pharmacol 1991, 42 Suppl, S157–165. [Google Scholar] [CrossRef]
- Rudner, X.L.; Berkowitz, D.E.; Booth, J.V.; Funk, B.L.; Cozart, K.L.; D'Amico, E.B.; El-Moalem, H.; Page, S.O.; Richardson, C.D.; Winters, B.; et al. Subtype specific regulation of human vascular alpha(1)-adrenergic receptors by vessel bed and age. Circulation 1999, 100, 2336–2343. [Google Scholar] [CrossRef]
- Chen, J.; Cui, C.; Yang, X.; Xu, J.; Venkat, P.; Zacharek, A.; Yu, P.; Chopp, M. MiR-126 Affects Brain-Heart Interaction after Cerebral Ischemic Stroke. Transl Stroke Res 2017, 8, 374–385. [Google Scholar] [CrossRef]
- Rasmussen, M.; Schonenberger, S.; Henden, P.L.; Valentin, J.B.; Espelund, U.S.; Sorensen, L.H.; Juul, N.; Uhlmann, L.; Johnsen, S.P.; Rentzos, A.; et al. Blood Pressure Thresholds and Neurologic Outcomes After Endovascular Therapy for Acute Ischemic Stroke: An Analysis of Individual Patient Data From 3 Randomized Clinical Trials. JAMA Neurol 2020, 77, 622–631. [Google Scholar] [CrossRef]
Figure 1.
Endothelium- dependent vasodilation. 1 hour brain ischemia followed by 23 hours reperfusion resulted in impaired vasodilation in isolated mice thoracic aorta in response to increasing concentrations of acetylcholine (ACh) in organ bath (P=0.0132, IC50 of ACh sham vs stroke, n=31, 19 respectively).
Figure 1.
Endothelium- dependent vasodilation. 1 hour brain ischemia followed by 23 hours reperfusion resulted in impaired vasodilation in isolated mice thoracic aorta in response to increasing concentrations of acetylcholine (ACh) in organ bath (P=0.0132, IC50 of ACh sham vs stroke, n=31, 19 respectively).
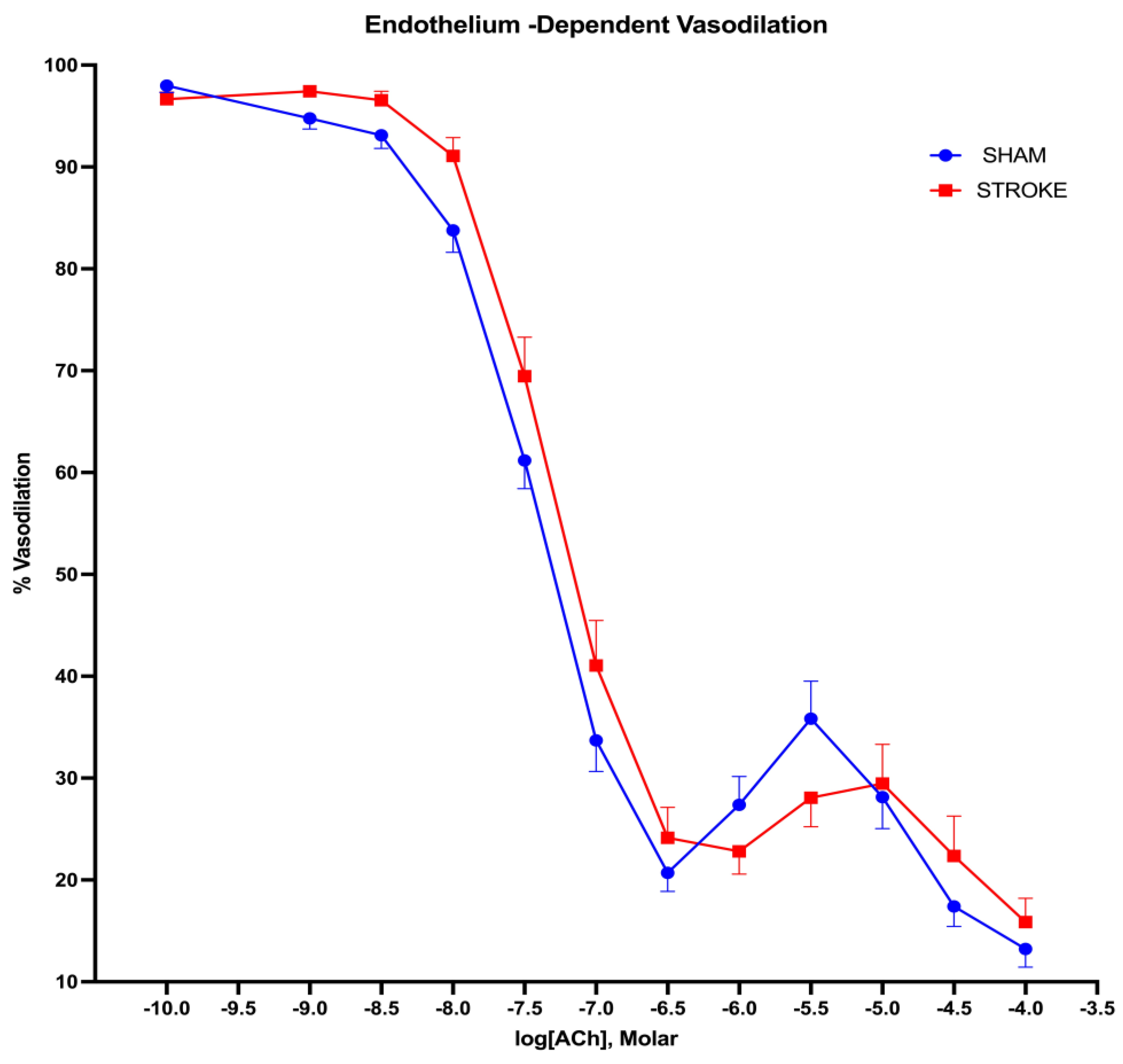
Figure 2.
Non-linear fitting of acetylcholine dose-response curve. IC50 is different for sham and stroke groups (P=0.0132, IC50 of ACh sham vs stroke groups, whereas top, bottom and Hill Slope (P=0.8569) are shared with the curves and are not significantly different for the sham and stroke curves.
Figure 2.
Non-linear fitting of acetylcholine dose-response curve. IC50 is different for sham and stroke groups (P=0.0132, IC50 of ACh sham vs stroke groups, whereas top, bottom and Hill Slope (P=0.8569) are shared with the curves and are not significantly different for the sham and stroke curves.
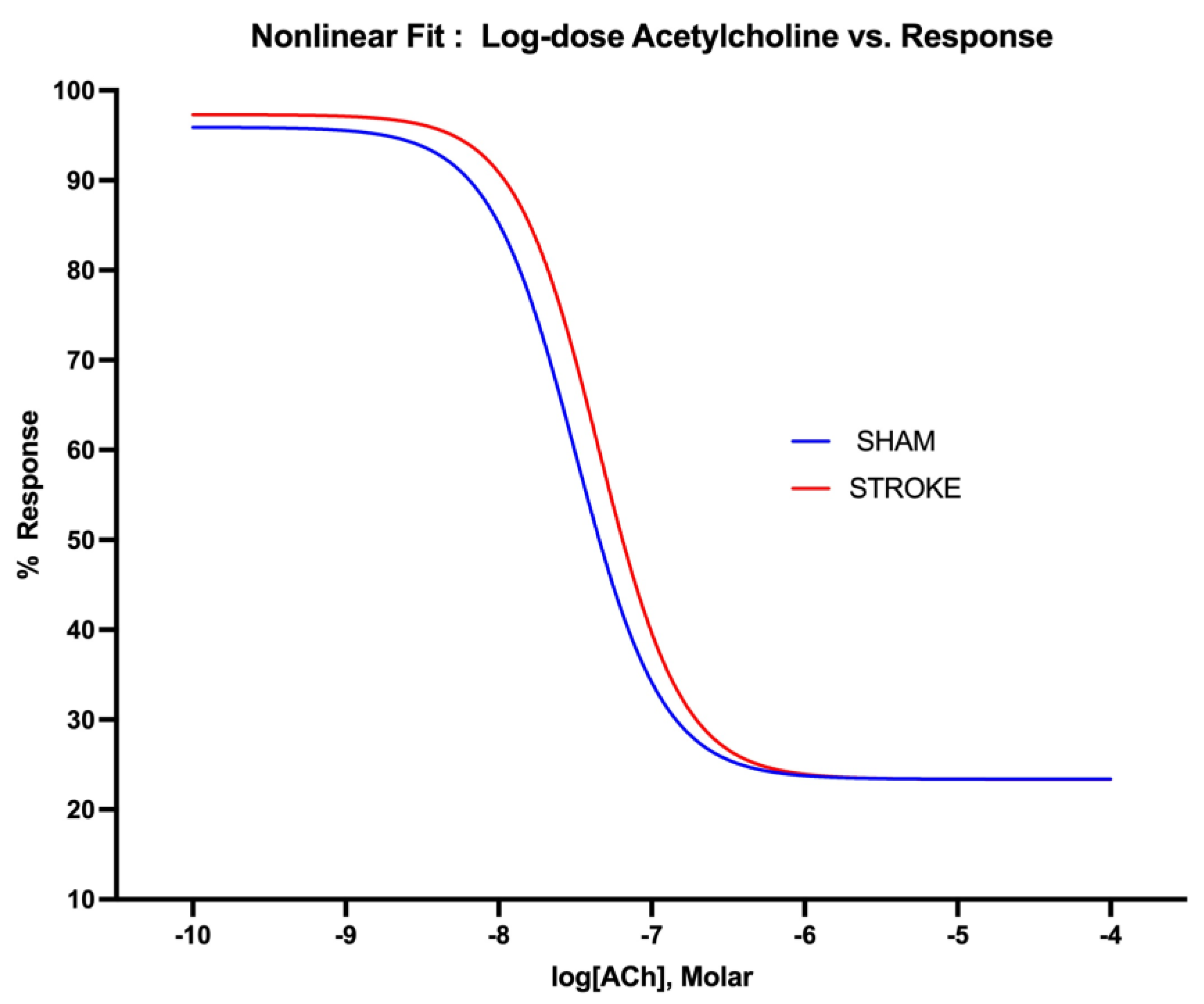
Figure 3.
Endothelium- independent vasodilation. There were no differences in endothelium-independent vasodilation responses in aortas isolated from sham and stroke animals. 1 hour brain ischemia followed by 23 hours reperfusion did not change endothelium-independent vasodilation in isolated mice thoracic aorta in response to increasing concentrations of sodium nitroprusside (SNP), a nitric oxide donor, in organ baths. (P= 0.3637, IC50 of SNP sham vs stroke, n=31, 19 respectively).
Figure 3.
Endothelium- independent vasodilation. There were no differences in endothelium-independent vasodilation responses in aortas isolated from sham and stroke animals. 1 hour brain ischemia followed by 23 hours reperfusion did not change endothelium-independent vasodilation in isolated mice thoracic aorta in response to increasing concentrations of sodium nitroprusside (SNP), a nitric oxide donor, in organ baths. (P= 0.3637, IC50 of SNP sham vs stroke, n=31, 19 respectively).
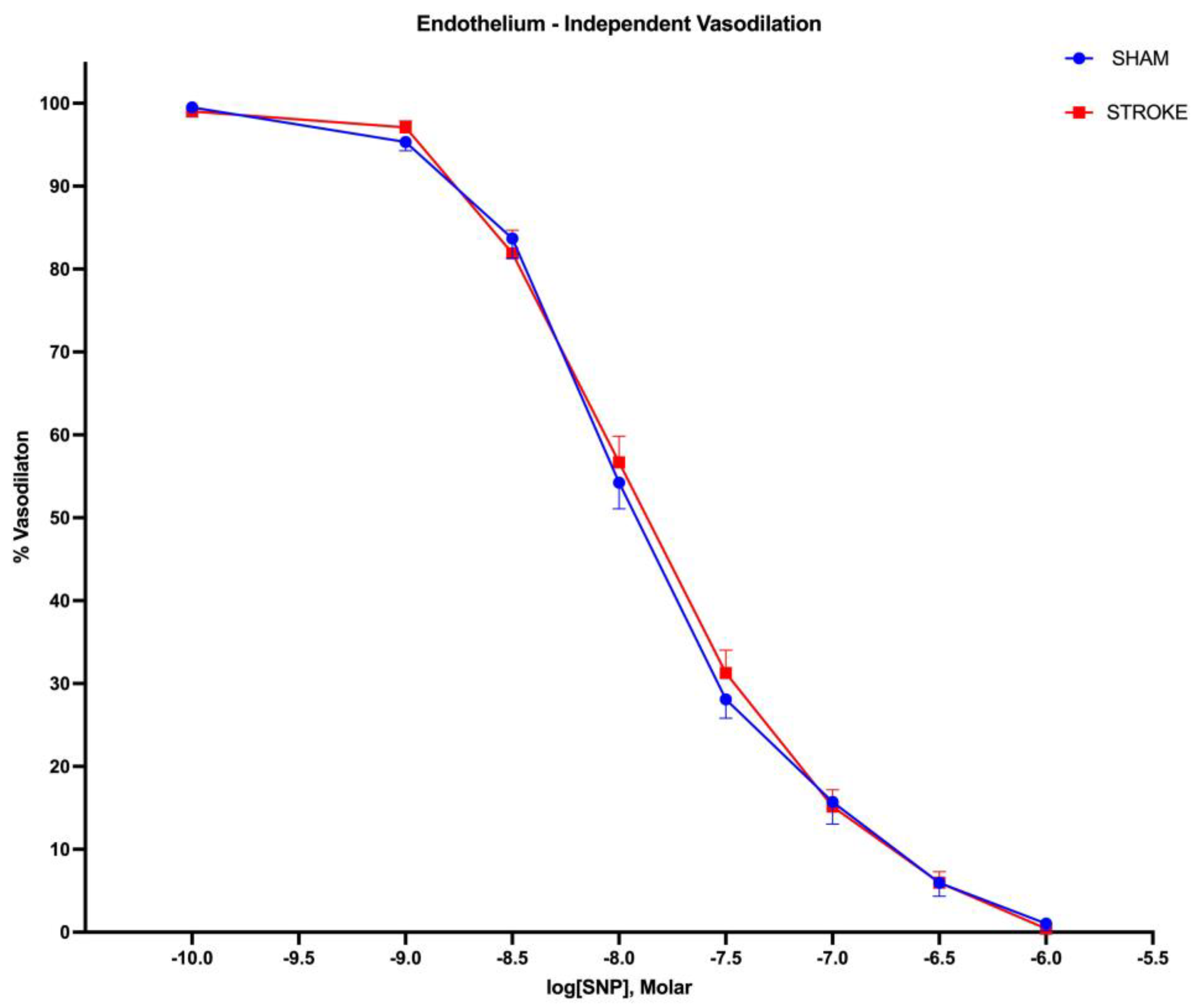
Figure 4.
Non-linear fitting of sodium nitroprusside dose-response curve. IC50, top, bottom and Hill Slope values are not different for sham and stroke groups P> 0.05.
Figure 4.
Non-linear fitting of sodium nitroprusside dose-response curve. IC50, top, bottom and Hill Slope values are not different for sham and stroke groups P> 0.05.
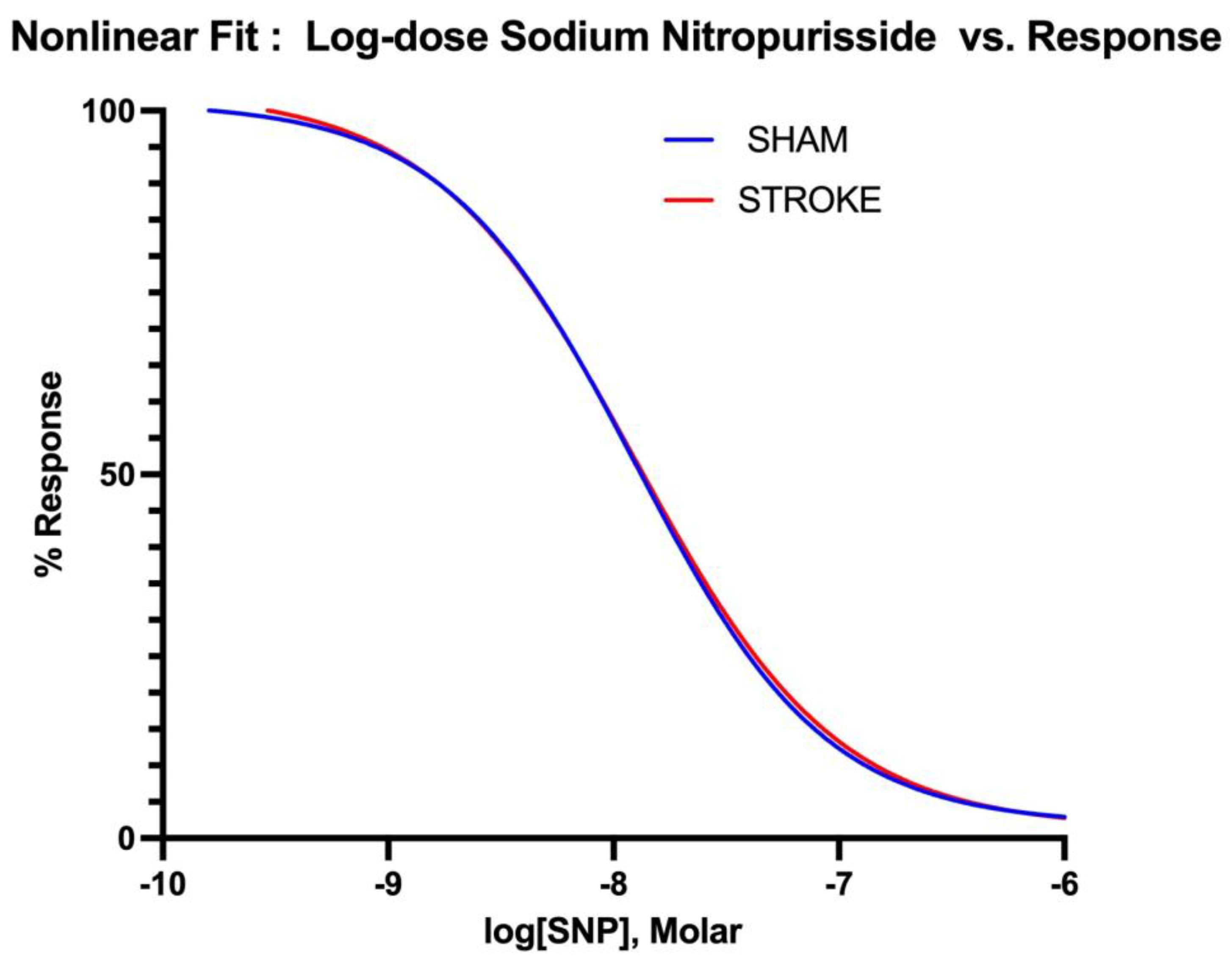
Figure 5.
Vascular smooth muscle contraction. 1 hour brain ischemia followed by 23 hours reperfusion resulted in impaired vasoconstriction in isolated mice thoracic aorta in response to increasing concentrations of phenylephrine (PE) in organ bath (P <0.0001, EC50 of PE sham vs stroke, n=31, 19 respectively).
Figure 5.
Vascular smooth muscle contraction. 1 hour brain ischemia followed by 23 hours reperfusion resulted in impaired vasoconstriction in isolated mice thoracic aorta in response to increasing concentrations of phenylephrine (PE) in organ bath (P <0.0001, EC50 of PE sham vs stroke, n=31, 19 respectively).
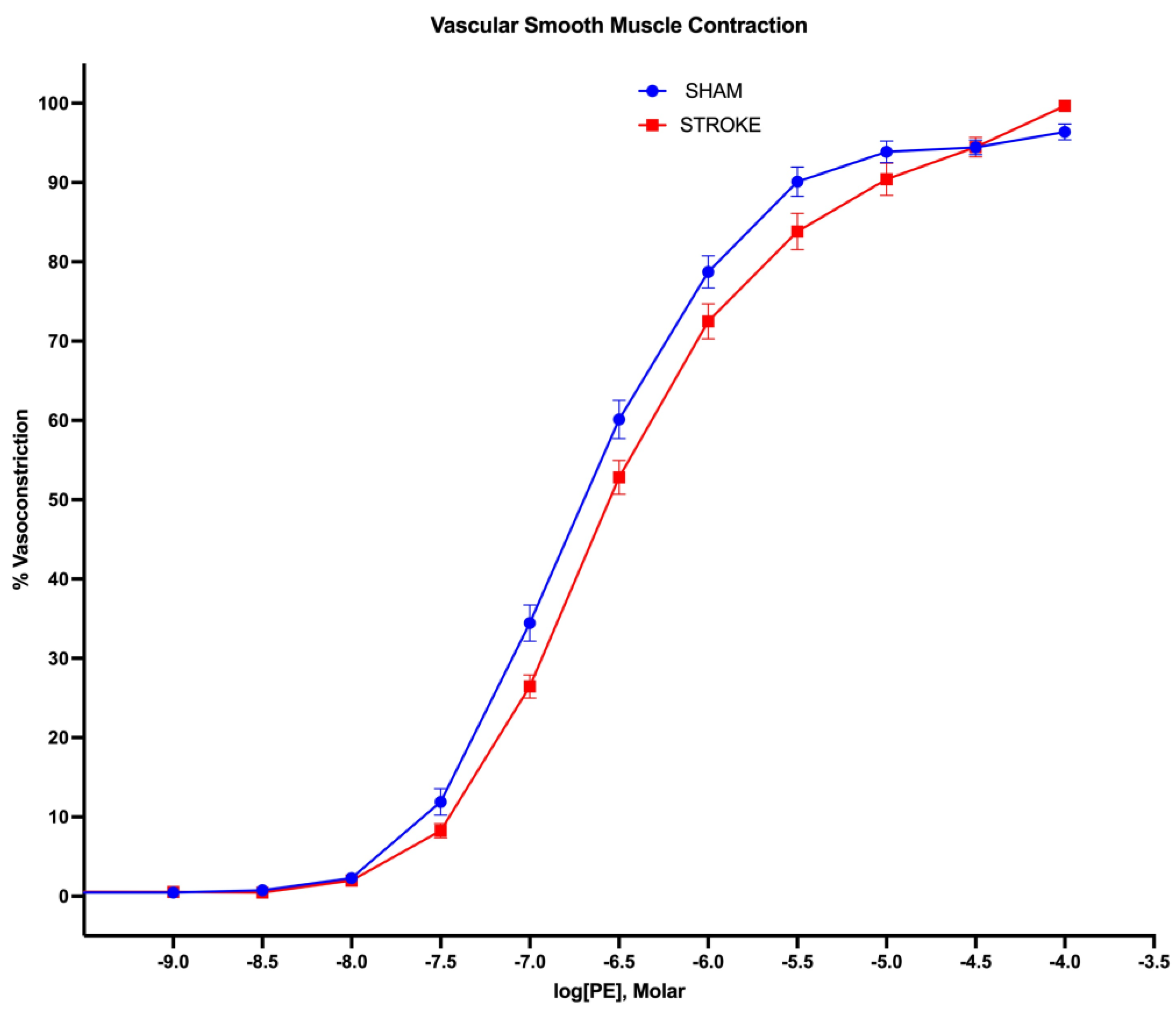
Figure 6.
Non-linear fitting of phenylephrine dose-response curve. EC50s is different for sham and stroke groups (Sham LogEC50= -6.652 vs Stroke LogEC50=-6.475, P< 0.05), whereas top, bottom and Hill Slope are shared and not significantly different for the sham and stroke PE curves P> 0.05.
Figure 6.
Non-linear fitting of phenylephrine dose-response curve. EC50s is different for sham and stroke groups (Sham LogEC50= -6.652 vs Stroke LogEC50=-6.475, P< 0.05), whereas top, bottom and Hill Slope are shared and not significantly different for the sham and stroke PE curves P> 0.05.
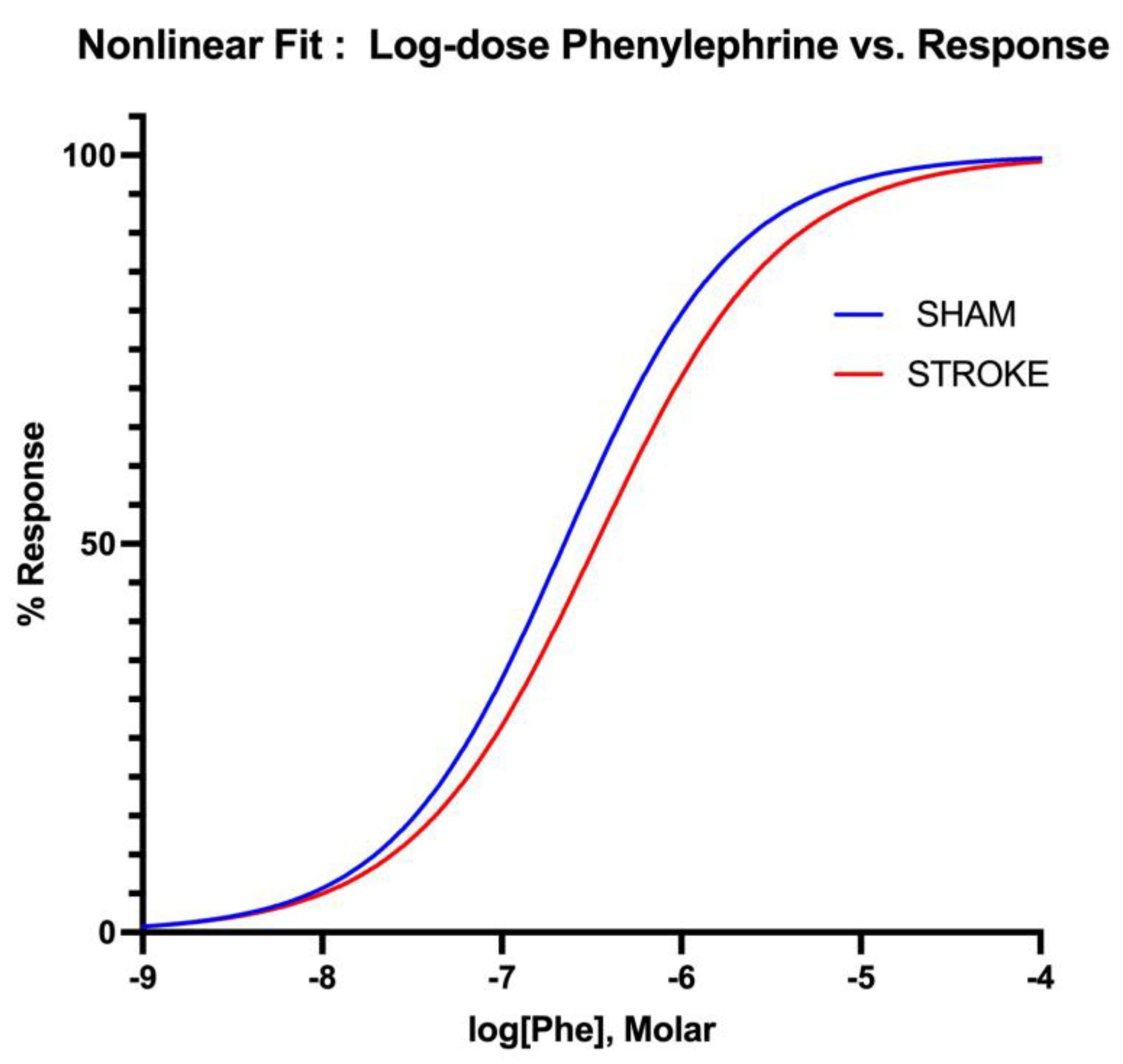
Figure 7.
Infarct percentage after 1 hour of middle cerebral artery occlusion and 23 hours of reperfusion. Infarct percentage was 23.2±1.5 % (n=19) assessed by TTC staining of brain sections. No infarct detected at sham group. Representative photographs of TTC staining of sham and stroke brains are shown.
Figure 7.
Infarct percentage after 1 hour of middle cerebral artery occlusion and 23 hours of reperfusion. Infarct percentage was 23.2±1.5 % (n=19) assessed by TTC staining of brain sections. No infarct detected at sham group. Representative photographs of TTC staining of sham and stroke brains are shown.
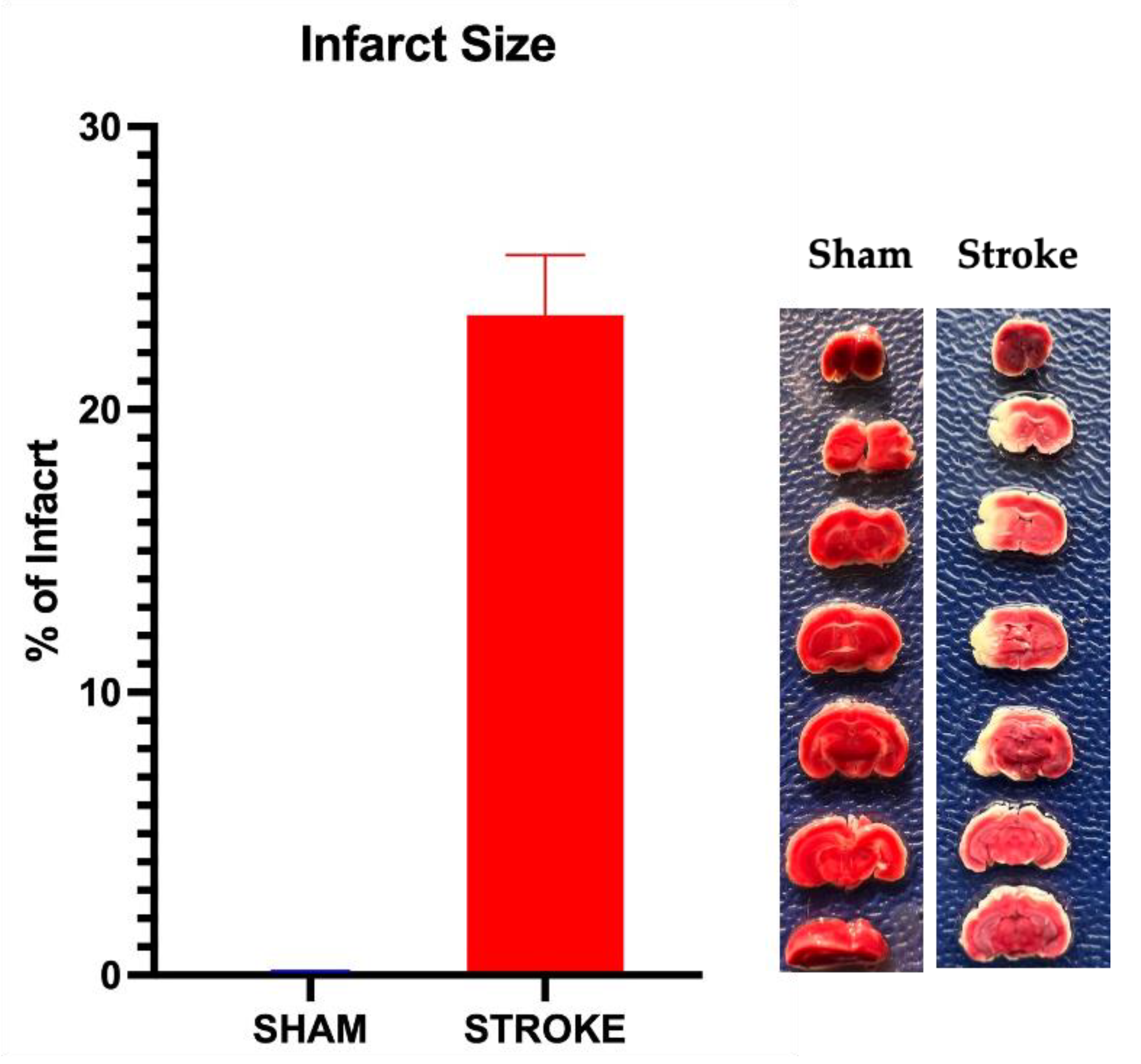
Figure 8.
A Notional Summary of Vascular Alterations following Ischemic Stroke. 1) Arteries and arterioles act within a broad range of sizes to regulate vascular resistance. 2) Ischemic stroke causes immunomodulation, the release of endocrine factors, modulation of neuronal signals and an imbalance of Angiotensin-1 -7 and Angiotensin-2 due to impairment ofACE II that potentially damages peripheral vessels. 3) Ischemic stroke impairs alpha-1 mediated vasoconstriction and endothelium-dependent vasodilation. 4) The impairment of vasoconstriction and vasodilation diminishes the range of vascular sizes and, as a result, impairs the regulation of vascular resistance in response to blood pressure changes. 5) Due to the reduced range of vascular reactivity, blood pressure regulation is impaired following ischemic stroke. Created with BioRender.com .
Figure 8.
A Notional Summary of Vascular Alterations following Ischemic Stroke. 1) Arteries and arterioles act within a broad range of sizes to regulate vascular resistance. 2) Ischemic stroke causes immunomodulation, the release of endocrine factors, modulation of neuronal signals and an imbalance of Angiotensin-1 -7 and Angiotensin-2 due to impairment ofACE II that potentially damages peripheral vessels. 3) Ischemic stroke impairs alpha-1 mediated vasoconstriction and endothelium-dependent vasodilation. 4) The impairment of vasoconstriction and vasodilation diminishes the range of vascular sizes and, as a result, impairs the regulation of vascular resistance in response to blood pressure changes. 5) Due to the reduced range of vascular reactivity, blood pressure regulation is impaired following ischemic stroke. Created with BioRender.com .
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



