
Submitted:
27 June 2024
Posted:
01 July 2024
You are already at the latest version
Abstract
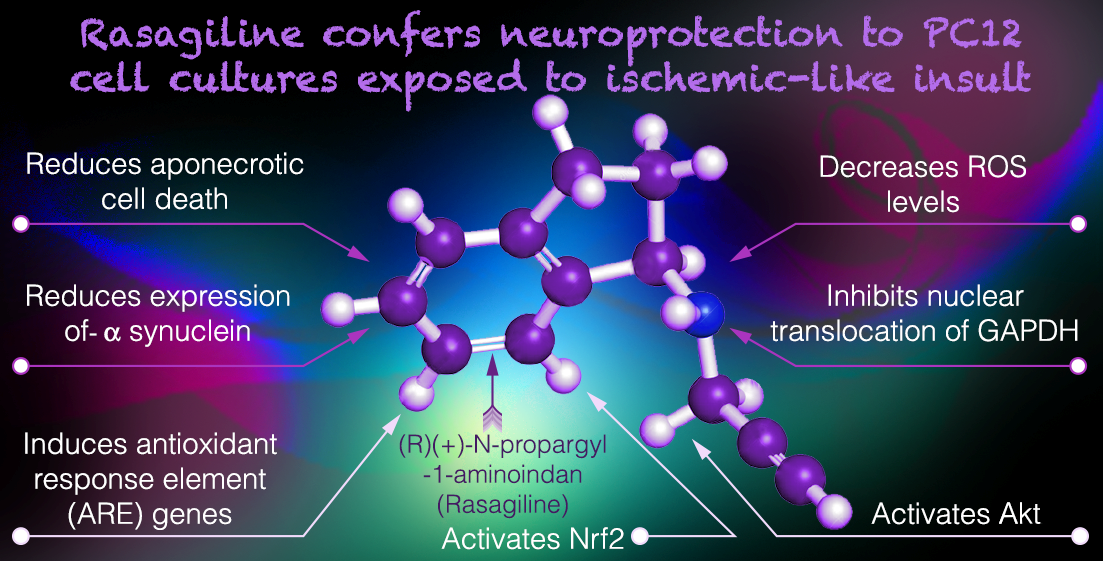
Rasagiline (Azilect®) is a selective monoamine oxidase B (MAO-B) inhibitor that provides symp-tomatic benefit in Parkinson’s disease (PD) treatment and found to exert preclinical neuroprotec-tive effects. Here, we investigated the neuroprotective signaling pathways of rasagiline in the PC12 neuronal cultures exposed to an ischemic-like insult. Exposure of neurons to oxy-gen-glucose deprivation for 4 h followed by 18 h of reoxygenation caused 40% aponecrotic cell death. Rasagiline induced dose-dependent neuroprotection by decreasing cell death and produc-tion of the neurotoxic reactive oxygen species, and reducing the nuclear translocation of glycer-aldehyde-3-phosphate dehydrogenase (GAPDH). Rasagiline increased protein kinase B (Akt) phosphorylation and decreased protein expression of the ischemia-induced α-synuclein protein, in correlation to the neuroprotective effect. Treatment with rasagiline induced nuclear shuttling of transcription factor Nrf2 and increased the mRNA levels of the antioxidant enzymes heme oxy-genase-1, (NAD (P) H- quinone dehydrogenase, and catalase. These results indicate that rasagiline provides neuroprotection in the ischemic neuronal cultures with inhibition of α-synuclein and GAPDH-mediated aponecrotic cell death, and via mitochondrial protection, as by increasing mi-tochondria-specific antioxidant enzymes by a mechanism involving the Akt/Nrf2 redox-signaling pathway. These findings may be exploited for neuroprotective drug development in PD and stroke therapy.
Keywords:
Akt
; cell death
; GAPDH
; ischemia-like insult
; neuroprotection
; Nrf2
; phosphorylation
; Rasagiline
; ROS
; PC 12 neuronal model
; α-synuclein
; Parkinson’s disease
; stroke
1. Introduction
Parkinson's disease (PD) is a complex neurodegenerative disease of the dopaminergic neurons located in the brain substantia nigra pars compacta and characterized by progressive dopaminergic neuron degeneration and Lewy body formation [1]. This degeneration and the decline in the functional presynaptic dopaminergic activity in the PD brain leads to a dramatic decrease in dopamine (DA) level [2], triggering the typical clinical symptoms of bradykinesia, rigidity, resting tremors, postural instability, etc., as well as anatomical changes in brain tissue as shown by imaging techniques [3]. The etiology of PD is likely multifactorial, and involves interplay among aging, genetic susceptibility, and environmental factors [4]. Although the pathophysiologic mechanism of PD remains unknown, many studies have shown that oxidative stress plays an important role in the cell death of the dopaminergic neurons [5,6,7]. Dopamine can undergo oxidation to induce the production of reactive oxygen species (ROS) and electrophilic quinone molecules [8], explaining the susceptibility of dopaminergic neurons to oxidative and electrophilic stress [9]. Moreover, targeting oxidative stress received a widespread recognition, considering that reactive oxygen species (ROS) play key roles in normal brain function and pathology in the context of many neurological diseases including stroke, traumatic brain injury, etc [10].
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) besides its function in converting glyceraldehyde 3-phosphate to 1, 3-biphosphoglycerate during glycolysis, is implicated in several non-metabolic processes, including transcriptional activation and cell apoptosis [11]. GAPDH is overexpressed and accumulates in the nucleus during apoptosis in response to oxidative stress, triggered by its nuclear translocation induced by a variety of insults in diverse cell types including PC12 cells [12,13]. Phosphatidylinositol 3-kinase-protein kinase B (PI3K-Akt) signaling pathway regulates signal transduction and biological processes such as cell proliferation, apoptosis, and metabolism, and regulates neurotoxicity and mediates the survival of neurons [14]. Many findings indicate that Akt phosphorylates several targets suggesting that it modulates neuronal cell death by both impinging on the cytoplasmic cell death machinery and by regulating nuclear proteins [15]. One mechanism by which Akt kinase suppresses glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-mediated apoptosis is via phosphorylating GAPDH at threonine 237 and decreasing its nuclear translocation [16].
The neuron maintains a robust antioxidant defense mechanism consisting of several neuroprotective genes and enzymes whose expression is controlled by antioxidant response element (ARE) which further depends on activation of nuclear factor erythroid 2-related factor 2 (Nrf2). In response to oxidative or electrophilic stress, this redox-sensitive transcription factor Nrf2 binds to ARE and rescues the cells from oxidative stress by the induction of ARE-mediated expression of phase II detoxifying antioxidant enzymes, including NAD(P)H-quinone oxidoreductase1 (NQO1), heme oxygenase-1 (HO-1), catalase, etc., [17]. The nuclear factor erythroid 2-related factor 2 (Nrf2)-Keap1 orchestrates the antioxidant response in the brain by promoting the expression of several antioxidant enzymes that exert neuroprotective effects against oxidative damage and mitochondrial impairment [18,19]. Activations of Nrf2 by drugs or genetic manipulations are demonstrated to alleviate PD induced by agents, such as MPP+, Rotenone, and H2O2, as well as genetic factors that can protect in vitro and in vivo neurons against DA-related neurotoxicity [20]. Hence, drugs that can induce the upregulation of ARE-mediated expression of phase II detoxifying antioxidant enzymes via the Nrf2/ARE pathway would be a promising approach for neuroprotection in PD and stroke [21,22].
α-synuclein is a neuronal synaptic protein that is a major component of Lewy bodies, neuronal cytoplasmic inclusions of aggregated proteins, that are typical biomarkers of idiopathic and familial forms of PD [23]. Extensive evidence shows that α-synuclein is neurotoxic and that it is implicated in the pathophysiology of PD [24] and stroke [25]. Recent studies also provided evidence of its relation to neuroprotection, as it can inhibits apoptosis in response to various pro-apoptotic signals [26]. α-synuclein is expressed and selectively up-regulated in response to nerve growth factor treatment in PC12 cell cultures [27]. Despite conflicting data that still exist, understanding the homeostatic balance of α-synuclein expression and activity is important and calls for further investigations regarding the physiological role of this neuronal protein during PD and stroke and upon treatment with anti-Parkinsonian drugs.
Rasagiline (Azilect®, Teva Co.) is a selective, irreversible B-type monoamine oxidase (MAO-B) second generation inhibitor that is clinically used in PD patients [28]. Rasagiline has been demonstrated to be neuroprotective in PD and oxidative stress model systems by preventing the formation of reactive oxygen species derived from the oxidation of dopamine by MAO-B and via an anti-apoptotic action, which appears to be independent of MAO-B inhibition, and related to its pharmacophore N-propargyl moiety[29,30,31]. Furthermore, chronic treatment with rasagiline induces indirect, antioxidant activity by enhancing the expression of anti-oxidative enzymes such as SOD1, SOD2, and catalase [32].
To investigate the cellular and molecular neuroprotective mechanisms of rasagiline under ischemia, we used pheochromocytoma PC12 dopaminergic neuronal cell cultures that express MAO-B, and exposed to an oxygen-glucose-deprivation (OGD) and reoxygenation protocol, representing a well-established in vitro model of the ischemic insult [33,34]. We hypothesized that rasagiline conferred neuroprotection to PC12 cell cultures exposed to ischemic-like oxidative stress insult by decreasing the nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase and α-synuclein protein levels, and by activating the Akt/Nrf2 redox-signaling pathway. Therefore, we investigated whether rasagiline induced the Nrf2-ARE signaling pathway leading to an increase in ARE-dependent antioxidant enzyme mRNA expression, in relation to its neuroprotective effect in PC12 cell cultures exposed to ischemic-like insult. We also explored the temporal effects of Akt activation and GAPDH nuclear localization in relation to rasagiline-induced neuroprotection. The findings provide a mechanistic proposal for rasagiline’s neuroprotective effects towards ischemic-like insults that may add to its future drug development and repurposing for some aspects of PD and stroke therapy.
2. Materials and Methods
2.1. Materials
Rasagiline mesylate was supplied by Teva pharmaceutics company, Israel Ltd., and stored as a stock solution of 10 mM ( 5 mg dissolved in 1.87 ml DMSO). Tempol, (4-hydroxy-2, 2, 6, 6-tetramethylpiperidine-d17-1-oxyl) a membrane-permeable radical scavenger and LY294002, a cell-permeable, potent and reversible specific inhibitor of PI3-kinase were purchased from Sigma-Aldrich Chemical Co. (Israel), and used as received. The collagen type Ι was purchased from BD Biosciences (Bedford, MA, USA). MitoSOX™ Red mitochondrial superoxide probe (M36008) was purchased from Invitrogen Molecular Probes (Eugene, OR, USA). Polyvinylidene difluoride (PVDF) membranes were from Invitrogen (Invitrogen Corporation Carlsbad, CA, USA). mNGF HPLC grade was purchased from Alomone Labs (Jerusalem, Israel). Dulbecco’s modified Eagle’s medium (DMEM), fetal calf serum, donor horse serum, penicillin, and streptomycin were all purchased from Biological Industries (Beit Haemek, Afula, Israel)
2.2. PC12 Cell Cultures
Rat pheochromocytoma, PC12 cells, were propagated in 25 cm2 flasks in growth medium composed of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 7 % fetal calf serum (FCS), 7 % horse serum (HS), 10,000 U/ml penicillin and 100 μg/mL streptomycin. The medium was replaced every second day, and cells were grown at 37 °C, in a humidified atmosphere of 6% CO2 [35]. To differentiate the cells, PC12 cell cultures grown on 6 well plates ( 0.5 x 105 cells/well), and glass coverslips (1 x 104 cells/coverslip), were treated with 50 ng/ml mouse β-NGF (Alomone Labs, Jerusalem, Israel) in DMEM with serum and antibiotics, every 48 h, for a period of 8-10 days, before submission to the ischemic insult.
2.3. Ischemic Insult Protocol
PC12 cells (1 x105 cells/well) were applied to 12-well plates or glass coverslips in 6-well plates, pre-coated with 200 μg/mL collagen type-I, and grown for 2 days. On the day of the experiment, the cell medium was replaced to glucose-free DMEM (hypoglycemic insult), and the cultures were introduced into an ischemic chamber with an oxygen level below 1% (hypoxic insult), for 4 h at 37 °C under oxygen and glucose deprivation (OGD), as previously described [35]. To mimic in vivo reperfusion conditions, at the end of the OGD insult, 4.5 mg/mL glucose was added and cultures were incubated for an additional 18 h under normoxic conditions (reoxygenation) to complete the ischemic insult. Operationally, ischemic insult represents therefore a combination of both OGD and reperfusion phases. Control cultures were maintained under regular atmospheric conditions (normoxia) in the presence of 6% CO2 with high glucose DMEM. The addition of 10 μM Rasagiline was performed before OGD, and maintained during the reperfusion. At the end of the reperfusion phase, cell death was measured using the LDH release method [36] at 340 nm using a spectrofluorometer (TECAN, SPECTRA Fluor PLUS, Salzburg, Austria). LDH release was expressed as percent of total LDH released into the medium upon subtracting the basal values of LDH release. The neuroprotective effect, defined as the percent decrease in LDH release in the presence of Rasagiline was normalized to untreated ischemic cultures.
2.4. Measurement of Mitochondrial Reactive Oxygen Species (ROS) Levels
Mitochondrial superoxide in live cells was detected using the MitoSOX™ Red mitochondrial superoxide probe. PC12 cells were cultured in 6-well plates (5x105 cells/well) and exposed to the OGD ischemic insult (4 h hypoxia followed by 18 h reperfusion) in the presence or absence of 10 µM Rasagiline or 1 mM Tempol, used as a positive control. The plates were then centrifuged at 1000 rpm for 5 min and the medium was removed. The cell cultures were incubated with 5 mM MitoSOX™ reagent dissolved in dimethyl sulfoxide working solution in Hanks' Balanced Salt Solution (HBSS/Ca2+/Mg2+) (Bet Haemek, Afula, Israel) at 37ºC for 10 min, in the dark. The cells were washed gently three times with HBSS/Ca2+/Mg2+, and the fluorescence intensity of the cells was detected by flow cytometry. For each sample, 105–106 events were acquired with a BD LSRII flow cytometer (BD Biosciences). ROS levels were expressed as the mean fluorescence intensity of 105 cells/sample, calculated by the FCS Express 6 Plus analysis software (De Novo Software) [37].
2.5. Nuclear Protein Extraction
PC12 cells (2 X 106) naïve or knocked-down for Nrf2, were grown for 24 h in a 10-cm dish under normoxia, or exposed to the OGD insult, and then Rasagiline was added into the medium of each dish. At the end of the ischemic insult the cell cultures were harvested by scraping. The cell pellets were washed by PBS and then re-suspended in 50 μl of the extraction buffer I (10 mM KCl, 10 mM HEPES, 1.5 mM MgCl2; with 0.5 mM DTT and 0.1% NP-40 freshly added before use) and incubated on ice for 15 min, and centrifuged at 6000 rpm, 4°C for 10 min. The pellets containing the nuclei were re-suspended in 50 μl of nuclear extraction buffer II (20 mM HEPES (pH 7.9), 25% glycerol, 420 mM NaCl, 0.2 mM EDTA, 1.5 mM MgCl2, with 0.5 mM DTT, and 0.5 mM phenylmethylsulfonyl fluoride added freshly), and incubated on ice for 15 min, and then centrifuged at 3000 rpm for 10 min at 4°C. The supernatant containing nuclear proteins was diluted with 50 μl of buffer III (20 mM HEPES (pH 7.9), 20 % glycerol, 50 mM KCl, 0.2 mM EDTA with 0.5 mM DTT and 0.5 mM phenylmethylsulfonyl fluoride added freshly) and stored at −80°C [13]. A BCA protein assay kit (PIERCE) was used to determine the protein concentration.
2.6. Western Blotting
At the end of the ischemic insult, PC12 cell cultures were collected by centrifugation at 2000 rpm for 5 min and washed with 1 mL fresh PBS. Thereafter, the cells were suspended in lysis buffer (20 mM HEPES pH 7.4 containing 2 mM EGTA, 50 mM glycerol phosphate, 1% Triton X-100,10 % glycerol, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 10 mg/mL leupeptin, 10 mg/mL aprotinin, 1 mM Na3VO4, and 5 mM NaF) and incubated for 10 min on ice. The lysed samples were sonicated for 5 sec, and centrifuged at 15,000 rpm, for 15 min, to separate the protein extract from the pellet. The protein concentration was determined according to by BCA protein assay kit (PIERCE). Samples containing 30 μg of cell protein were boiled for 5 min in SDS sample buffer and separated by 10 %, 12 %, or 15 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).The protein bands were subsequently transferred to PVDF membranes. The membranes were blocked at room temperature for 1 h using TBST (20 mM Tris-HCl, 137 mM NaCl, pH 7.5, and 0.1% Tween-20) containing 5% milk. After washing with TBST, the membranes were incubated overnight at 4º C with the primary antibodies and then washed with TBST. The primary antibodies employed were directed towards: cleaved caspase-3 (Santa Cruz Biotechnology, 1:100); cleaved PARP (Santa Cruz Biotechnology, 1:500); Bcl-2 (Santa Cruz Biotechnology, 1:200); phospho-Akt (Ser 473, Cell Signaling Technology, 1:2000); pan-Akt (Cell Signaling Technology, 1:1000); α-synuclein (BD Transduction Laboratories, 1 : 500); nuclear factor erythroid 2-related factor 2 (Cell Signaling Technology, 1:500); NAD(P)H: quinone oxidoreductase 1 (Calbiochem, NQO1; 1:1000); β-actin (Santa Cruz Biotechnology, 1:3000); tubulin (anti-βIII tubulin, Chemicon –Millipore, 1:200) and lamin B (Santa Cruz Biotechnology, 1:500). In the next step, the membranes were washed three times with TBST for 10 min, before the treatment for 2 h with the secondary antibody−HRP conjugates of either sheep anti-mouse (1:3000), donkey anti-rabbit (1:3000) (Jackson Immunoresearch, PA, USA) or rabbit antibodies to goat IgG (1:3000, Santa Cruz Biotechnology). For Akt bands, the densitometric values were obtained for the phosphorylated and the pan antibodies. The background of each film was subtracted and the relative density of the band of phosphorylated Akt was divided by the density of the respective band of the pan-protein. The data are presented as the percentage of phosphorylated Akt from the total pan-Akt. Specific antibody binding was measured by enhanced chemiluminescence (ECL), visualized by Image Lab Software 5.1 (Bio-Rad Laboratories, Hercules, CA), and quantified [38].
2.7. Knockdown of Nrf 2 Gene Expression Using siRNA
The commercial Amine Transfection Agent protocol was followed (Ambion, Austin, TX, USA). SiRNASiPORT™ Amine–Polyamine–Based Transfection Agent is a propriety blend of polyamines formulated for transfection of small RNAs. The reagent functions by complexing with RNA and facilitating its transfer into the cells. In brief, 10 nM of Silencer® Select Predesigned mixture of siRNAs (Ambion) directed against Nrf-2α and Nrf-2β. The sense sequences of Nrf-2α and Nrf-2β siRNAs were 5′-GAAUUCAGCAUGACCGAUAtt-3′ and 5′-GGUGGAACUUUUAAUCAAAtt-3′, respectively [39]. Nrf2 siRNAs or Silencer® Select Negative Control (scrambled) siRNA (Ambion), diluted in Gibco™ OPTI-MEM®I (Invitrogen, NY, USA), was combined with siPORT™ Amine Transfection Agent diluted in Gibco™ OPTI-MEM®I. This mixture was added to a 6-well NUNC™ TC plate containing 2 × 105 PC12 cells per well. The cell cultures were subjected to daily treatment with Rasagiline, starting 12 h after treatment with the siRNAs, and up to 6 days post-transfection, followed by RNA extraction and RT-PCR analyses. Following 72 h of incubation, a new transfection reagent was added, containing Nrf2 or scrambled siRNA in the appropriate groups, an approach previously used by us to knock-down the tuftelin gene [40].
2.8. RNA Extraction and RT-PCR
Total RNA from PC12 cell cultures was extracted using Trizol (Invitrogen Corp., NY, USA) and total RNA concentration was determined using the NanoDrop ND-1000 spectrophotometer (Nano-Drop Technologies, Wilmington, DE, USA). Thereafter, 1µg of total RNA of each sample was subjected to reverse transcription according to the manufacturer’s protocol using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems®, Foster City, CA, USA) and real-time quantitative PCR was performed using Applied Biosystems 7300 Real-Time-PCR System with Applied Biosystems TaqMan® AOD (Assays-On-Demand) and Master Mixes (TaqMan®, Gene Expression Assays). Briefly, 2 µL cDNA was mixed with GoTaq Green Master Mix (Promega) and with primers for NQO1, heme oxygenase 1 (HO-1), glutathione peroxidase 2 (GPx2), catalase and GAPDH (internal control). The primers for these genes were: NQO1, 50-TTC TGT GGC TTCCAG GTC TT-30 (forward), and 50-AGG CTG CTT GGA GCA AAA TA-30 (reverse); HO-1, 50-CTT TCA GAA GGG TCA GGT GTC-30 (forward), and 50-TGC TTG TTT CGC TCT ATC TCC-30 (reverse); Catalase, 50-AAA TGC TTC AGG GCC GCC TT -30 (forward), and 50-GTAGGG ACA GTT CAC AGG TA-30 (reverse); and GAPDH, 50-GTA TTGGGC GCC TGG TCA CC-30 (forward), and 50-CGC TCC TGG AAG ATGGTG ATG G-30 (reverse). The PCR products were also electrophoresed in 2% agarose gel and visualized by ethidium bromide. The PCR parameters were as follows: initial denaturation at 95ºC for 10 min, followed by 40 cycles of denaturation at 95ºC for 10 sec, annealing at 60ºC for 15 sec, and elongation at 72ºC for 20 sec. The mRNA levels were expressed as the relative copy number of each target mRNA to GAPDH for each sample, and the cycle threshold of the control group was normalized to 1 [40].
2.9. Immunofluorescence and Confocal Microscopy
PC12 cell cultures grown on microscope glass coverslips under normoxia, or exposed to the OGD insult, with and without Rasagiline treatment, were permeabilized with Fix & Perm Cell Permeabilization Kit (Life Technologies), for 30 min at 4º C. Thereafter, the cells were immunostained with a mouse anti-GAPDH antibody (1:200-1:1000; Santa Cruz Biotechnology), or mouse monoclonal antibody anti α-synuclein-1 (1:50; Transduction Laboratories), or rabbit polyclonal anti α-synuclein (1:100; Chemicon), or mouse monoclonal anti-Nrf2 (1:100; Santa Cruz Biotechnology Inc). The secondary antibodies used for immunofluorescence (1:1000-1:5000) were goat anti-mouse or anti-rabbit conjugated to Alexa Fluor 594 (red, Abcam) or Alexa-Fluor-488 (green, Abcam). Nuclear staining was performed with DAPI diluted 1:1000 in PBS for 3 min (blue staining). The cells were examined by a fluorescent microscope (Nikon 50i), at magnifications of 100x or 200x. In other experiments, the coverslips were observed at a magnification of 600x by a confocal microscope (Zeiss LSM 710). Photographs of different random fields (4–5 per slide) were acquired and 60-80 cells were analyzed for the fluorescence intensity (arbitrary units) using the Image J software. Representative fields were quantified and displayed [40].
2.10. Statistical Analysis
The results were statistically analyzed using SPSS 19.0 software (SPSS Inc., Chicago, IL, USA). The results are presented as mean ± SEM. Each experiment was performed four to six times in sixplicate wells. Comparisons between experimental groups were performed by using the analysis of variance program (ANOVA) followed by Dunnett’s multiple comparison test. A p-value of 0.05 or less was considered significant for all comparisons.
3. Results
3.1. Rasagiline Conferred Neuroprotection towards Ischemic Insult
3.1.1. Rasagiline Inhibited Ischemic Insult-Induced Necrotic Cell Death
The ability of rasagiline to exert its neuroprotective effect was tested in a PC12 cells model exposed to oxygen and glucose deprivation (OGD) followed by reoxygenation. Briefly, monolayer cultures of PC12 cells were exposed to 4 h OGD followed by 18 h reoxygenation, in the presence of different micromolar concentrations of rasagiline [34], or 1500 μM tempol antioxidant, a concentration previously found most effective as neuroprotective [35] (Figure 1A). It is evident that the addition of 10 μM rasagiline significantly (p ≤ 0.01%) decreased by 33% necrotic cell death as measured by LDH release, compared to control OGD-exposed, but untreated cells. This cytoprotective effect indicates that 10 μM rasagiline conferred 80% neuroprotection was twofold higher, compared to the effect of tempol, respectively (Figure 1A-insert).
3.1.2. Rasagiline Inhibited Ischemic Insult-Induced ROS Production
To examine whether rasagiline treatment suppressed the ischemic insult-induced oxidative stress, the levels of mitochondrial ROS was measured in PC12 cell cultures exposed to ischemic insult (OGD) in the presence or absence of 10 μM rasagiline, by comparison to normoxic cultures, with and without rasagiline treatment (Figure 1B,C). The levels of ROS measured in the PC12 cell cultures exposed to ischemic insult in the absence or presence of 10 μM rasagiline, were 185 ± 5%, and 162 ± 4 %, respectively, as compared to 100 ± 15% in the control normoxic cells, with and without rasagiline treatment. Therefore, the ischemic insult-induced elevation of ROS in PC12 cell cultures was significantly decreased by about 15% upon rasagiline treatment (p ≤ 0.05, Figure 1B, C), as also reported upon Tempol treatment [35].
3.1.3. Rasagiline Inhibited Ischemic Insult-Induced Apoptotic Cell Death
PC12 cell cultures exposed to ischemic insult showed increased apoptosis, which significantly decreased in cell cultures treated with 10 μM rasagiline (Figure 1D). Ischemic insult exposure ) OGD (+ increased the activation of apoptotic effectors, including the cleavage of caspase-3 and PARP, while pretreatment with 10 μM rasagiline during the ischemic insult (OGD+/ Rasagiline+) significantly reduced by 60-80% the levels of these proteins, and increased by 70% the expression of the anti-apoptotic protein, Bcl-2 (upon comparing the protein level of expression relative to beta-actin levels). These results indicate that rasagiline inhibited ischemic insult-mediated apoptosis in the PC12 cell cultures.
3.2. Rasagiline Potentiated PI3K/Akt Signaling in PC12 Cell Cultures Exposed to Ischemic Insult
Activation of the PI3K/Akt signaling pathway by ischemic insult in the PC12 cell cultures has been reported in the past [41]. To elucidate weather this intracellular signaling pathway could be involved in the rasagiline-induced neuroprotection towards the ischemic insult, we investigated the degree of activation of Akt by the ischemic insult in the presence or absence of rasagiline (Figure 2).
The phosphorylation experiment indicated strong phosphorylation of Akt in cultures exposed to the ischemic insult (OGD) in the presence of 10 µM rasagiline, but lower by 50% in the untreated, ischemic cultures (Figure 2; p < 0.01). Based on the results described above, we examined whether a PI3K inhibitor could inhibit rasagiline-induced PI3K phosphorylation. PI3K inhibitor LY29004 inhibited ischemic insult-induced, as well as rasagiline-ischemic insult-induced phosphorylation of Akt (Figure 2). In normoxic cultures, the effects of rasagiline either alone or in the presence of LY294002 on the phosphorylation of Akt were non-significant. In parallel experiments, the effect of the PI3K inhibitor LY29004 was investigated on the neuroprotective effect of rasagiline towards the ischemic insult. It was found that LY29004 treatment reduced significantly by 36% rasagiline-induced neuroprotective effect (Table 1, p < 0.05), indicating a relationship between the partial reversal of rasagiline-induced Akt activation and the rasagiline-induced neuroprotective effect during the ischemic insult, further proposing that additional mechanisms may contribute to neuroprotection.
3.3. Rasagiline Inhibited the Ischemic Insult-Induced GAPDH Nuclear Translocation
GAPDH’s nuclear translocation participates in neuronal and non-neuronal cell death [42]. Therefore, we sought to investigate whether the ischemic insult could cause the translocation of GAPDH from the cytosol into the nucleus in PC12 cell cultures, either undifferentiated, or differentiated with NGF, and if rasagiline could modulate this effect (Figure 3). In the control group (normoxia), immunostaining indicated that the GAPDH level was low and was found predominantly in the cytosol (Figure 3A insert, red staining) in both undifferentiated and NGF-differentiated cell cultures. However, the GAPDH signal increased 3-4 fold compared to the control, upon exposure to the ischemic insult (Figure 3A; *p < 0.01 compared to control). The treatment with 10 μM rasagiline, inhibited by 2.6 fold GAPDH immunostaining induced by the ischemic insult, to a higher intensity in the NGF-differentiated, compared to the undifferentiated cell cultures (Figure 3A; *p < 0.01 compared to OGD). To confirm the quantitative results of confocal microscopy, GAPDH levels were determined by Western blotting, to investigate the effect of OGD and rasagiline on the nuclear localization of GAPDH (Figure 3B). The results indicated that OGD induced an increased in the translocation from the cytosol to the nucleus of GAPDH, an effect attenuated by 75-90 % by 10 µM Rasagiline treatment in both undifferentiated, and NGF-differentiated cells (Figure 3B). These results may suggest that rasagiline inhibition of the ischemic insult-induced cell death may be due to the attenuation of the translocation and transcriptional activity of GAPDH in the PC12 cell nuclei.
3.4. Rasagiline Increased the Translocation of Nrf2 into the Nucleus and Transcription of ARE Phase II Antioxidant Enzyme Genes during The Ischemic Insult
Considering that the transcription factor Nrf2 is a master regulator of the acute antioxidant defense [43], we hypothesized that rasagiline could modulate its expression and subcellular distribution during the ischemic oxidative insult. Immunostaining and western blotting analyses were performed to determine the extent of nuclear translocation of Nrf2 in response to the ischemic insult with, or without treatment with different concentrations of rasagiline (Figure 4). The fluorescence microscopy staining indicates that under normoxic conditions, Nrf2 has a low cytoplasmic distribution, which was increased by the ischemic insult, and upon treatment with 10 µM rasagiline localized in part with the DAPI-blue stained nuclei (Figure 4A). Western blotting results showed that the pretreatment with 1 and 5 µM rasagiline significantly increased by 40 % and 90 %, respectively the nuclear levels of Nrf2 (Figure 4B,C; p < 0.01).To further determine the role of Nrf2 in the mechanism of action of rasagiline, we used siRNA treatment to significantly reduce the expression level of Nrf2 in PC12 cells. The reduced level of Nrf2 was confirmed by Western blotting analysis (Figure 4B, C). In PC12 cells transfected with non-targeting scramble siRNA, no evidence of silencing was found because the Nrf2 levels were most probably similar to the control. By contrast, in the Nrf2 siRNA-transfected PC12 cells, the rasagiline increase in the nuclear level of Nrf2 during the ischemic insult was reduced by about 60 % (Figure 4B, C; p < 0.01).
In parallel experiments, the effect of the Nrf2- siRNA was investigated on the neuroprotective effect of rasagiline towards the ischemic insult. It was found that Nrf2- siRNA, but not scramble RNA, treatment reduced by 70% rasagiline-induced neuroprotective effect (Table 1; p < 0.01), suggesting an apparent direct correlation during the ischemic insult between the rasagiline-induced increased translocation of Nrf2 into the nucleus and the rasagiline-induced neuroprotective effect.
Increased levels of antioxidant enzymes such as NQO1, HO-1, catalase, etc., by the Nrf2 pathway, provide a major defense against oxidative stress in Parkinson’s [44] and stroke [45]. To investigate whether rasagiline affected the expression of these antioxidant genes in PC12 cell cultures exposed to the ischemic insult, the mRNA expression of NQO1, HO-1, and catalase was determined (Figure 5). The ischemic insult increased by about twofold the mRNA levels of NQO1, HO-1, and catalase, as compared to the normoxic cultures (Figure 5; p < 0.05). The increase in the mRNA levels of NQO1, HO-1, and catalase was amplified to 6.4 and 3.5 fold, respectively upon treatment with rasagiline during the ischemic insult (Figure 5; p < 0.01), indicative of an increased transcription of the ARE-mediated phase II detoxifying antioxidant enzyme genes.
3.5. Rasagiline Decreased the Expression Levels of α-Synuclein during Ischemic Insult
Since α-synuclein protein is involved in cell death and oxidative stress in PC12 cells [46], and preventing its expression is neuroprotective after stroke [25], we sought to investigate the effect of Rasagiline on both undifferentiated and NGF-differentiated PC12 cell cultures exposed to the ischemic insult (Figure 6). Undifferentiated and 50 ng/ml NGF-differentiated PC12 cell cultures grown under normoxic conditions, or exposed to ischemic insult, in the presence or absence of 10 µM Rasagiline, were fixed, permeabilized, and immunostained. The ischemic insult increased by about twofold the level of α-synuclein expression in the ischemic cultures as compared to the normoxic cultures (Figure 6; p < 0.05). This increased effect was attenuated in the ischemic cultures treated with Rasagiline (p < 0.01, Figure 6A, B). The representative western immunoblots and densitometry data for the α-synuclein protein levels of the undifferentiated, and NGF-differentiated PC12 cell cultures protein lysates, indicated that both the monomeric (about 14-19 kDa), and tetrameric (about 56-76 kDa) forms of α-synuclein were significantly increased in the ischemic cultures, compared to the normoxic cultures, and reduced by about 50% in the ischemic cultures treated with 10 µM Rasagiline (Figure 6 B).
4. Discussion
Current major treatment strategies for PD and stroke are directed to improve and/or reduce the symptoms of the disease without modifying its underlying multifactorial pathology [47,48]. Thus, the quest for efficient disease-modifying treatments (attempts to delay/slow progression by addressing the underlying pathology of the disease) with chronic therapeutic effects on disease progression has been envisioned as a novel approach, with many drugs in active clinical trials [49,50]. Type-B monoamine oxidase inhibitors, such as rasagiline (Azilect) and selegiline (Deprenyl), treat Parkinson’s patients by ameliorating motor symptoms and improving motor fluctuations and their evaluation in preclinical studies indicated that they hold a strong neuroprotective potential in Parkinson’s and other neurodegenerative diseases for reducing oxidative stress [51]. However, MAO-B inhibitors were poorly characterized in vitro and in vivo for neuroprotection in ischemic stroke [52].
In the past, we investigated in vitro neuroprotective features of chemicals, drugs, natural products, and stem cells on PC12 clonal cell lines temporarily deprived of oxygen and glucose (OGD) followed by reoxygenation. These catecholaminergic neurons have been used previously to mimic some of the properties of in vivo brain ischemia-reperfusion-injury (IRI) and have been instrumental in identifying common mechanisms such as calcium overload, redox potential, lipid peroxidation, MAPKs modulation, etc. [33,53,54]. The present study characterize in vitro, using the PC12 cell culture model, several signaling pathways beneficial in the neuroprotective effects of rasagiline towards ischemia-like-induced neuronal injury as schematically presented in Figure 7.
First, rasagiline, a selective MAO-B inhibitor, attenuated the ischemia-induced aponecrotic PC12 cell death, by decreasing the generation of free radicals (Figure 7, pathways 1 and 3). Similar findings were observed in the brains of rodent models, in which rasagiline reduced dopamine oxidative metabolism by both MAO B inhibition, and by a direct, MAO-B-independent, antioxidant effect [55,56,57].
Secondly, rasagiline decreased the nuclear translocation of GAPDH thereby reducing cell death (Figure 7, pathway 2), as expected from accumulated evidence demonstrating that GAPDH nuclear translocation plays a critical role in ischemic cell death [58]. In support of the present findings, there are studies indicating in different cell systems that GAPDH-MAO-B-mediated cell death, induced by different insults was prevented by rasagiline [13,30,59] and selegiline [60,61,62]. A plausible mechanism by which rasagiline decreased the nuclear translocation of GAPDH, is the activation of Akt during the ischemic insult (Figure 7, pathway 4), which in turn phosphorylated Thr-237 of GAPDH and decreased its nuclear translocation, an essential step for GAPDH-mediated apoptosis.
Thirdly, rasagiline decreased ROS levels /oxidative stress and mitochondrial dysfunction (Figure 7, pathway 3), most probably by a direct antioxidant effect, since its propargylamine pharmacophore can directly scavenge free radicals [57]. Propargylamine residue is composed of an amine group in β-position to an alkyne moiety and compounds with a carbon-carbon triple bond can behave as electrophilic substrates and as electron sources in nucleophilic reactions, providing a direct antioxidant effect. Other studies supporting Figure 7 pathway 3, indicate that selegiline inhibited NOS in the brain’ mitochondria, potentiated mitochondrial cytochrome oxidase activity, and reduced ROS/NOS production [63,64].
Fourth, rasagiline activated the PI3K-Akt-Nrf2 pathway in the PC12 cell cultures exposed to the ischemic insult with the induction of the transcription of the antioxidant response element (ARE) genes (Figure 7, pathway 4). Selegiline and rasagiline induced in different oxidative stress cell types models the nuclear translation of Nrf2, increased binding to the antioxidant response element (ARE), enhanced expression of the antioxidant thioredoxin, and increased activities of glutathione-dependent antioxidant enzymes, and anti-peroxidative enzymes, catalase, and superoxide dismutase [65,66,67]. Cerebral ischemic stroke involves many pathological processes such as oxidative stress, inflammation, and mitochondrial dysfunction. Nrf2, as one of the most critical antioxidant transcription factors in cells, can coordinate various cytoprotective antioxidant enzymes and factors to inhibit oxidative stress. Targeting Nrf2 is considered as a potential strategy to prevent and treat cerebral ischemic injury [45] and therefore, rasagiline drug may be repurposed for ischemic stroke therapy.
Together, these events ultimately lowered the expression of the monomeric and tetramer neurotoxic α-synuclein protein level (Figure 7, pathway 5), minimizing cell death. α-synuclein is a small, soluble, disordered protein that is widely expressed in the nervous system. Although its physiological functions are not yet fully understood, it is mainly involved in synaptic vesicle transport, neurotransmitter synthesis and release, cell membrane homeostasis, mitochondrial and lysosomal activities, etc. The complex pathological manifestations of α-synuclein are attributed to its structural complexity, misfolding, and different posttranslational modifications. These properties cause mitochondrial dysfunction, oxidative stress, and neuroinflammation, resulting in neuronal cell death and neurodegeneration. Several recent studies have discovered the pathogenic roles of α-synuclein in traumatic and vascular central nervous system diseases, such as traumatic spinal cord injury, brain injury, stroke, and in aggravating the processes of neurodegeneration [68]. The beneficial effect of rasagiline in lowering the expression of α-synuclein protein levels further stresses its potential clinical use in the therapy of synucleinopathic diseases [57].
Rasagiline monotherapy is established in early Parkinson's disease (PD) for motor benefit in patients from Eastern and Western countries, as evident from recent meta-analysis of randomized controlled clinical trials [69], and longer duration of MAO-B inhibitor exposure is associated with less clinical decline in Parkinson's disease [70]. In a phase II, randomized, double blind, and placebo-controlled study, selegiline treatment facilitated recovery after stroke [71]. However, additional preclinical studies as reported here, combined with necessary clinical studies are required to unambigously proof the concept of a disease-modifying-neuroprotective effect of MAO-B inhibitors in PD and stroke.
5. Conclusions
Rasagiline (Azilect) is a selective MAO-B inhibitor drug that provides symptomatic benefits in PD treatment and found to exert neuroprotective effects in preclinical cellular and animal models of ischemic stroke. However, slowing or halting the neurodegenerative process have not yet been accomplished in PD and stroke patients using these drugs and therefore, neuroprotection is still considered an unmet clinical need. We investigated in the PC12 dopaminergic neuronal cell culture model, exposed to oxygen-glucose deprivation (OGD) followed by reoxygenation (ischemic-like insult), the neuroprotective signaling pathways of rasagiline. Rasagiline decreased the production of neurotoxic reactive oxygen species and aponecrotic cell death, activated protein kinase B (Akt) phosphorylation activity, decreased the nuclear translocation of GAPDH, induced the nuclear shuttling of transcription factor Nrf2, and increased the mRNA expression of antioxidant enzymes, in temporal relation to the neuroprotective effect. These results indicate that rasagiline conferred neuroprotection via improving mitochondrial integrity, as well as increasing mitochondria-specific antioxidant enzymes by a mechanism involving the Akt/Nrf2 redox-signaling pathway. These findings may be exploited to develop a third generation of MAO-B inhibitors with improved neuroprotection in PD and stroke disease-modifying therapy but, further investigations are required to proof this concept in neuronal stroke models in vitro and in vivo and in clinical trials.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, western blots of Figure S1-S4,S6 with molecular weight markers.
Author Contributions
SL: AL, MK, MSN and JD performed experiments, analyzed results, reviewed references, and wrote the manuscript; LR discussed and provided input on the manuscript; WZ and PL conceived and supervised the study, discussed data, wrote and reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by internal funds of the Hebrew University (PL) and the National Natural Science Foundation of China (32070969), The Science and Technology Development Fund, Macau SAR (File No. 0104/2022/A2, 0038/2020/AMJ) (WZ).
Informed Consent Statement
Not applicable.
Data Availability Statement
All data associated with this study are available in the main text or are available through the corresponding author upon request.
Acknowledgments
The authors are grateful to Ms. Zehava Cohen for the help with the artwork. Philip Lazarovici holds the Jacob Gitlin Chair in Physiology and is affiliated with, and acknowledge the support by the Grass Center for Drug Design and Synthesis of Novel Therapeutics, David R. Bloom Center of Pharmacy, and the Adolph and Klara Brettler Medical Research Center at the Hebrew University of Jerusalem, Israel.
Conflicts of Interest
SL is a director at CBX Therapeutics, which develops novel therapies for neurodegenerative disorders; all other authors declare no conflict of interest.
References
- Kalia, L. V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired Dopamine Metabolism in Parkinson’s Disease Pathogenesis. Mol. Neurodegener. 2019, 14, 1–21. [Google Scholar] [CrossRef]
- Maetzler, W.; Liepelt, I.; Berg, D. Progression of Parkinson’s Disease in the Clinical Phase: Potential Markers. Lancet Neurol. 2009, 8, 1158–1171. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.Y.; Ho, P.W.L.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The Interplay of Aging, Genetics and Environmental Factors in the Pathogenesis of Parkinson’s Disease. Transl. Neurodegener. 2019, 8, 1–11. [Google Scholar] [CrossRef]
- Dionísio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative Stress and Regulated Cell Death in Parkinson’s Disease. Ageing Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Moreno, N.; Lane, J.D. Autophagy and Redox Homeostasis in Parkinson’s: A Crucial Balancing Act. Oxid. Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative Stress in the Aging Substantia Nigra and the Etiology of Parkinson’s Disease. Aging Cell 2019, 18, 1–23. [Google Scholar] [CrossRef]
- Smythies, J. Redox Aspects of Signaling by Catecholamines and Their Metabolites. Antioxid Redox Signal. 2000, 2, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Kim, S.W.; Lee, S.Y.; Hwang, O. Dopamine-Dependent Cytotoxicity of Tetrahydrobiopterin: A Possible Mechanism for Selective Neurodegeneration in Parkinson’s Disease. J. Neurochem. 2003, 86, 143–152. [Google Scholar] [CrossRef]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef]
- Nicholls, C.; Li, H.; Liu, J.P. GAPDH: A Common Enzyme with Uncommon Functions. Clin. Exp. Pharmacol. Physiol. 2012, 39, 674–679. [Google Scholar] [CrossRef]
- Huang, J.; Xiong, N.; Chen, C.; Xiong, J.; Jia, M.; Zhang, Z.; Cao, X.; Liang, Z.; Sun, S.; Lin, Z.; et al. Glyceraldehyde-3-Phosphate Dehydrogenase: Activity Inhibition and Protein Overexpression in Rotenone Models for Parkinson’s Disease. Neuroscience 2011, 192, 598–608. [Google Scholar] [CrossRef]
- Ou, X.M.; Lu, D.; Johnson, C.; Chen, K.; Youdim, M.B.H.; Rajkowska, G.; Shih, J.C. Glyceraldehyde-3-Phosphate Dehydrogenase-Monoamine Oxidase B-Mediated Cell Death-Induced by Ethanol Is Prevented by Rasagiline and 1-R-Aminoindan. Neurotox. Res. 2009, 16, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Agrawal, A.; Verma, A.; Dubey, N. The PI3K-AKT Pathway: A Plausible Therapeutic Target in Parkinson’s Disease. Exp. Mol. Pathol. 2022, 129, 104846. [Google Scholar] [CrossRef]
- Ahn, J.-Y. Neuroprotection Signaling of Nuclear Akt in Neuronal Cells. Exp. Neurobiol. 2014, 23, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Lan, F.; Zheng, Z.; Xie, F.; Han, J.; Dong, L.; Xie, Y.; Zheng, F. Akt2 Kinase Suppresses Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH)-Mediated Apoptosis in Ovarian Cancer Cells via Phosphorylating Gapdh at Threonine 237 and Decreasing Its Nuclear Translocation. J. Biol. Chem. 2011, 286, 42211–42220. [Google Scholar] [CrossRef]
- Boas, S.M.; Joyce, K.L.; Cowell, R.M. The Nrf2-Dependent Transcriptional Regulation of Antioxidant Defense Pathways: Relevance for Cell Type-Specific Vulnerability to Neurodegeneration and Therapeutic Intervention. Antioxidants 2022, 11. [Google Scholar] [CrossRef]
- Villavicencio Tejo, F.; Quintanilla, R.A. Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease. Antioxidants 2021, 10. [Google Scholar] [CrossRef]
- Cho, M.-R.; Han, J.-H.; Lee, H.-J.; Park, Y.K.; Kang, M. Emerging Functional Cross-Talk between the Keap1 Nrf2 System and Mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kumar H, Koppula S, Kim IS, More SV, Kim BW, Choi DK. Nuclear Factor Erythroid 2-Related Factor 2 Signaling in Parkinson Disease: A Promising Multi Therapeutic Target Against Oxidative Stress, Neuroinflammation and Cell Death. CNS Neurol Disord Drug Targets. 2012, 11, 1015–1029. [Google Scholar] [CrossRef]
- Niu, Y.; Zhang, J.; Dong, M. Nrf2 as a Potential Target for Parkinson’s Disease Therapy. J. Mol. Med. 2021, 99, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Farina, M.; Vieira, L.E.; Buttari, B.; Profumo, E.; Saso, L. The Nrf2 Pathway in Ischemic Stroke: A Review. Molecules 2021, 26, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Ma ZL, Wang ZL, Zhang FY, Liu HX, Mao LH, Yuan L. Biomarkers of Parkinson's Disease: From Basic Research to Clinical Practice. Aging Dis. 2023. [CrossRef]
- Maries, E.; Dass, B.; Collier, T.J.; Kordower, J.H.; Steece-Collier, K. The Role of α-Synuclein in Parkinson’s Disease: Insights from Animal Models. Nat. Rev. Neurosci. 2003, 4, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Mehta, S.L.; Kaimal, B.; Lyons, K.; Dempsey, R.J.; Vemuganti, R. Poststroke Induction of α-Synuclein Mediates Ischemic Brain Damage. J. Neurosci. 2016, 36, 7055–7065. [Google Scholar] [CrossRef] [PubMed]
- Sidhu A, Wersinger C, Moussa CE, Vernier P. The Role of Alpha-Synuclein in Both Neuroprotection and Neurodegeneration. Ann N Y Acad Sci. 2004, 1035, 250–270. [Google Scholar] [CrossRef]
- Stefanis, L.; Kholodilov, N.; Rideout, H.J.; Burke, R.E.; Greene, L.A. Synuclein-1 Is Selectively up-Regulated in Response to Nerve Growth Factor Treatment in PC12 Cells. J. Neurochem. 2001, 76, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Lecht, S.; Haroutiunian, S.; Hoffman, A.; Lazarovici, P. Rasagiline - A Novel MAO B Inhibitor in Parkinson’s Disease Therapy. Ther. Clin. Risk Manag. 2007, 3, 467–474. [Google Scholar] [PubMed]
- Weinreb, O.; Amit, T.; Riederer, P.; Youdim, M.B.H.; Mandel, S.A. Neuroprotective Profile of the Multitarget Drug Rasagiline in Parkinson’s Disease; 1st ed.; Elsevier Inc., 2011; Vol. 100; ISBN 9780123864673.
- Tabakman, R.; Lecht, S.; Lazarovici, P. Neuroprotection by Monoamine Oxidase B Inhibitors: A Therapeutic Strategy for Parkinson’s Disease? BioEssays 2004, 26, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, Y.; Shohami, E.; Weinstock, M. Neuroprotective Effect of Rasagiline, a Selective Monoamine Oxidase-B Inhibitor, against Closed Head Injury in the Mouse. Eur. J. Pharmacol. 1999, 366, 127–135. [Google Scholar] [CrossRef]
- Carrillo MC, Kanai S, Nokubo M, Kitani K. (-) Deprenyl Induces Activities of Both Superoxide Dismutase and Catalase but Not of Glutathione Peroxidase in the Striatum of Young Male Rats. Life Sci. 1991, 48, 517–521. [Google Scholar] [CrossRef]
- Tabakman R, Jiang H, Shahar I, Arien-Zakay H, Levine RA, Lazarovici P. Neuroprotection by NGF in the PC12 In Vitro OGD Model: Involvement of Mitogen-Activated Protein Kinases and Gene Expression. Ann N Y Acad Sci. 2005, 1053, 84–96. [Google Scholar] [CrossRef]
- Abu-Raya, S.; Tabakman, R.; Blaugrund, E.; Trembovler, V.; Lazarovici, P. Neuroprotective and Neurotoxic Effects of Monoamine Oxidase-B Inhibitors and Derived Metabolites under Ischemia in PC12 Cells. Eur. J. Pharmacol. 2002, 434, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Lahiani, A.; Hidmi, A.; Katzhendler, J.; Yavin, E.; Lazarovici, P. Novel Synthetic PEGylated Conjugate of α-Lipoic Acid and Tempol Reduces Cell Death in a Neuronal PC12 Clonal Line Subjected to Ischemia. ACS Chem. Neurosci. 2016, 7, 1452–1462. [Google Scholar] [CrossRef]
- Maatuf Y, Priel A, Lazarovici P. Measurements of Cell Death Induced by Snake and Spider's Venoms and Derived Toxins. Methods Mol Biol. 2020, 2068, 239–268. [Google Scholar] [CrossRef]
- Gincberg, G.; Shohami, E.; Trembovler, V.; Alexandrovich, A.G.; Lazarovici, P.; Elchalal, U. Nerve Growth Factor Plays a Role in the Neurotherapeutic Effect of a CD45 + Pan-Hematopoietic Subpopulation Derived from Human Umbilical Cord Blood in a Traumatic Brain Injury Model. Cytotherapy 2018, 20, 245–261. [Google Scholar] [CrossRef]
- Ettinger, K.; Lecht, S.; Arien-Zakay, H.; Cohen, G.; Aga-Mizrachi, S.; Yanay, N.; Saragovi, H.U.; Nedev, H.; Marcinkiewicz, C.; Nevo, Y.; et al. Nerve Growth Factor Stimulation of ERK1/2 Phosphorylation Requires Both P75NTR and A9β1 Integrin and Confers Myoprotection towards Ischemia in C2C12 Skeletal Muscle Cell Model. Cell. Signal. 2012, 24, 2378–2388. [Google Scholar] [CrossRef]
- Bruni, F.; Polosa, P.L.; Gadaleta, M.N.; Cantatore, P.; Roberti, M. Nuclear Respiratory Factor 2 Induces the Expression of Many but Not All Human Proteins Acting in Mitochondrial DNA Transcription and Replication. J. Biol. Chem. 2010, 285, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Shilo, D.; Cohen, G.; Blumenfeld, A.; Goren, K.; Hanhan, S.; Sharon, S.; Haze, A.; Deutsch, D.; Lazarovici, P. Tuftelin Is Required for NGF-Induced Differentiation of PC12 Cells. J. Mol. Neurosci. 2019, 68, 135–143. [Google Scholar] [CrossRef]
- Qi, H.; Han, Y.; Rong, J. Potential Roles of PI3K/Akt and Nrf2-Keap1 Pathways in Regulating Hormesis of Z-Ligustilide in PC12 Cells against Oxygen and Glucose Deprivation. Neuropharmacology 2012, 62, 1659–1670. [Google Scholar] [CrossRef]
- Sawa, A.; Khan, A.A.; Hester, L.D.; Snyder, S.H. Glyceraldehyde-3-Phosphate Dehydrogenase: Nuclear Translocation Participates in Neuronal and Nonneuronal Cell Death. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 11669–11674. [Google Scholar] [CrossRef]
- Zamanian, M.Y.; Parra, R.M.R.; Soltani, A.; Kujawska, M.; Mustafa, Y.F.; Raheem, G.; Al-Awsi, L.; Lafta, H.A.; Taheri, N.; Heidari, M.; et al. Targeting Nrf2 Signaling Pathway and Oxidative Stress by Resveratrol for Parkinson’s Disease: An Overview and Update on New Developments. Mol. Biol. Rep. 2023, 50, 5455–5464. [Google Scholar] [CrossRef] [PubMed]
- Tahavvori, A.; Gargari, M.K.; Yazdani, Y.; Mamalo, A.S.; Beilankouhi, E.A.V.; Valilo, M. Involvement of Antioxidant Enzymes in Parkinson’s Disease. Pathol. Res. Pract. 2023, 249. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, X.; Xiong, X.; Zhu, H.; Chen, R.; Zhang, S.; Chen, G.; Jian, Z. Nrf2 Regulates Oxidative Stress and Its Role in Cerebral Ischemic Stroke. Antioxidants 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Saberzadeh, J.; Arabsolghar, R.; Takhshid, M.A. Alpha Synuclein Protein Is Involved in Aluminum-Induced Cell Death and Oxidative Stress in PC12 Cells. Brain Res. 2016, 1635, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Regensburger, M.; Ip, C.W.; Kohl, Z.; Schrader, C.; Urban, P.P.; Kassubek, J.; Jost, W.H. Clinical Benefit of MAO-B and COMT Inhibition in Parkinson’s Disease: Practical Considerations. J. Neural Transm. 2023, 130, 847–861. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, B.; Testai, F.D. Navigating Antiplatelet Treatment Options for Stroke: Evidence-Based and Pragmatic Strategies. Curr. Neurol. Neurosci. Rep. 2022, 22, 789–802. [Google Scholar] [CrossRef] [PubMed]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Fiske, B.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2023 Update. J. Parkinsons. Dis. 2023, 13, 427–439. [Google Scholar] [CrossRef]
- Safouris A, Magoufis G, Tsivgoulis G. Emerging Agents for the Treatment and Prevention of Stroke: Progress in Clinical Trials. Expert Opin Investig Drugs. 2021, 30, 1025–1035. [Google Scholar] [CrossRef]
- Alborghetti, M.; Bianchini, E.; De Carolis, L.; Galli, S.; Pontieri, F.E.; Rinaldi, D. Type-B Monoamine Oxidase Inhibitors in Neurological Diseases. Neural Regen. Res. 2024, 19, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E.; Binder, H.; Birle, C.; Bornstein, N.; Diserens, K.; Groppa, S.; Homberg, V.; Lisnic, V.; Pugliatti, M.; Randall, G.; et al. European Academy of Neurology and European Federation of Neurorehabilitation Societies Guideline on Pharmacological Support in Early Motor Rehabilitation after Acute Ischaemic Stroke. Eur. J. Neurol. 2021, 28, 2831–2845. [Google Scholar] [CrossRef]
- Lahiani, A.; Brand-Yavin, A.; Yavin, E.; Lazarovici, P. Neuroprotective Effects of Bioactive Compounds and Mapk Pathway Modulation in “Ischemia”—Stressed PC12 Pheochromocytoma Cells. Brain Sci. 2018, 8. [Google Scholar] [CrossRef]
- Peng, T.; Jiang, Y.; Farhan, M.; Lazarovici, P.; Chen, L.; Zheng, W. Anti-Inflammatory Effects of Traditional Chinese Medicines on Preclinical in Vivo Models of Brain Ischemia-Reperfusion-Injury: Prospects for Neuroprotective Drug Discovery and Therapy. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Aluf, Y.; Vaya, J.; Khatib, S.; Loboda, Y.; Finberg, J.P.M. Selective Inhibition of Monoamine Oxidase A or B Reduces Striatal Oxidative Stress in Rats with Partial Depletion of the Nigro-Striatal Dopaminergic Pathway. Neuropharmacology 2013, 65, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Seif-El-Nasr M, Atia AS, Abdelsalam RM. Effect of MAO-B Inhibition Against Ischemia-Induced Oxidative Stress in the Rat Nrain. Comparison with a Rational Antioxidant. Arzneimittelforschung. 2008, 58, 160–167. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M. Neuroprotective Function of Rasagiline and Selegiline, Inhibitors of Type B Monoamine Oxidase, and Role of Monoamine Oxidases in Synucleinopathies. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhai, D.; Chin, K.; Wang, M.; Liu, F. Disruption of the Nuclear P53-GAPDH Complex Protects against Ischemia-Induced Neuronal Damage. Mol. Brain 2014, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, W.; Akao, Y,; Youdim, M. B.; Davis, B.A.; Naoi, M. Transfection-enforced Bcl-2 Overexpression and an Anti-Parkinson Drug, Rasagiline, Prevent Nuclear Accumulation of Glyceraldehyde-3-phosphate Dehydrogenase Induced by an Endogenous Dopaminergic Neurotoxin, N-methyl(R)salsolinol. J Neurochem. 2001, 78, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Thomas, B.; Cascio, M.B.; Bae, B. Il; Hester, L.D.; Dawson, V.L.; Dawson, T.M.; Sawa, A.; Snyder, S.H. Neuroprotection by Pharmacologic Blockade of the GAPDH Death Cascade. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 3887–3889. [Google Scholar] [CrossRef]
- Tatton, W.; Chalmers-Redman, R.; Tatton, N. Neuroprotection by Deprenyl and Other Propargylamines: Glyceraldehyde-3-Phosphate Dehydrogenase Rather than Monoamine Oxidase B. J. Neural Transm. 2003, 110, 509–515. [Google Scholar] [CrossRef]
- Sharma, S.K.; Carlson, E.C.; Ebadi, M. Neuroprotective Actions of Selegiline in Inhibiting 1-Methyl, 4-Phenyl, Pyridinium Ion (MPP+)-Induced Apoptosis in SK-N-SH Neurons. J. Neurocytol. 2003, 32, 329–343. [Google Scholar] [CrossRef]
- Czerniczyniec, A.; Bustamante, J.; Lores-Arnaiz, S. Modulation of Brain Mitochondrial Function by Deprenyl. Neurochem. Int. 2006, 48, 235–241. [Google Scholar] [CrossRef]
- Irer, S. V.; Alper, G.E.; Sezer, E.D.; Duman, E.; Saatcioglu, F.; Yilmaz, C. The Effect of L-Deprenyl on Tissue mRNA Expressions of NOS Isoforms and NO Levels in an Experimental Diabetes Mellitus Model. J. Neural Transm. 2007, 114, 811–815. [Google Scholar] [CrossRef]
- Nakaso, K.; Nakamura, C.; Sato, H.; Imamura, K.; Takeshima, T.; Nakashima, K. Novel Cytoprotective Mechanism of Anti-Parkinsonian Drug Deprenyl: PI3K and Nrf2-Derived Induction of Antioxidative Proteins. Biochem. Biophys. Res. Commun. 2006, 339, 915–922. [Google Scholar] [CrossRef]
- Andoh, T.; Boon Chock, P.; Murphy, D.L.; Chiueh, C.C. Role of the Redox Protein Thioredoxin in Cytoprotective Mechanism Evoked by (-)-Deprenyl. Mol. Pharmacol. 2005, 68, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Liu, K.W.K.; Liang, Y.; Ip, M.S.M.; Mak, J.C.W. Inhibition of Monoamine Oxidase-B by Selegiline Reduces Cigarette Smoke-Induced Oxidative Stress and Inflammation in Airway Epithelial Cells. Toxicol. Lett. 2017, 268, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Liu, N.; Liu, X.X.; Yang, Y.Y.; Zhou, M.W. α-Synuclein in Traumatic and Vascular Diseases of the Central Nervous System. Aging (Albany. NY). 2020, 12, 22313–22334. [Google Scholar] [CrossRef] [PubMed]
- Chang,H. Y.;Li,Y.Y.;Hong,C.T.;Kuan,Y.C.Efficacy of rasagiline monotherapy for early Parkinson disease: A systematic review and meta-analysis of randomized controlled trials. J Psychopharmacol. 2022, 36, 704–714. [Google Scholar] [CrossRef]
- Hauser,R. A.;Li,R.;Perez,A.;Ren,X.;Weintraub,D.;Elm,J.;Goudreau,J.L.;Morgan,J.C.Fang,J.Y.;Aminoff,M.J. et. all. Longer duration of MAO-B inhibitor exposure is associated with less clinical decline in Parkinson's disease: An Analysis of NET-PD LS1. J Parkinsons Dis. 2017, 7, 117–127. [Google Scholar] [CrossRef]
- Sivenius, J.; Sarasoja,T. ; Aaltonen,H.; Heinonen,E.; Kilkku,O.; Reinikainen,K. Selegiline treatment facilitates recovery after stroke. Neurorehabil Neural Repair. 2001, 15, 183–190. [Google Scholar] [CrossRef]
Figure 1.
Rasagiline reduced aponecrotic cell death and production of mitochondrial reactive oxygen species in PC12 cell cultures exposed to ischemic insult. A. PC12 cells (1.2 × 106 cells/well) were applied to 12-well plates and grown for 3 days. At the start of the experiment, the normoxic (white bar) and ischemic insult exposed cell cultures (black bar) were treated with different concentrations of rasagiline or tempol (1500 μM). The OGD insult was carried out for 4 h followed by 18 h reperfusion. Aliquots from the culture media were taken for LDH release measurements indicating that rasagiline decreased necrotic cell death. *p ≤ 0.01 vs normoxia; **p ≤ 0.01 vs OGD, control; Insert: Neuroprotection; *p ≤ 0.01 vs control; B. Mitochondrial reactive oxygen species (ROS) production was examined by flow cytometry and typical traces are presented. C. Quantitation of ROS indicating that the elevation of ROS level by the ischemic insult was significantly attenuated by treatment with rasagiline; *p ≤ 0.01 vs control, normoxia; **p ≤ 0.05 vs OGD. D. Western blots of cell culture extracts indicate that rasagiline treatment during the ischemic insult (OGD) inhibited the expression levels of apoptotic effectors (cleaved caspase-3 and cleaved PARP) and increased the levels of the anti-apoptotic protein, Bcl-2.
Figure 1.
Rasagiline reduced aponecrotic cell death and production of mitochondrial reactive oxygen species in PC12 cell cultures exposed to ischemic insult. A. PC12 cells (1.2 × 106 cells/well) were applied to 12-well plates and grown for 3 days. At the start of the experiment, the normoxic (white bar) and ischemic insult exposed cell cultures (black bar) were treated with different concentrations of rasagiline or tempol (1500 μM). The OGD insult was carried out for 4 h followed by 18 h reperfusion. Aliquots from the culture media were taken for LDH release measurements indicating that rasagiline decreased necrotic cell death. *p ≤ 0.01 vs normoxia; **p ≤ 0.01 vs OGD, control; Insert: Neuroprotection; *p ≤ 0.01 vs control; B. Mitochondrial reactive oxygen species (ROS) production was examined by flow cytometry and typical traces are presented. C. Quantitation of ROS indicating that the elevation of ROS level by the ischemic insult was significantly attenuated by treatment with rasagiline; *p ≤ 0.01 vs control, normoxia; **p ≤ 0.05 vs OGD. D. Western blots of cell culture extracts indicate that rasagiline treatment during the ischemic insult (OGD) inhibited the expression levels of apoptotic effectors (cleaved caspase-3 and cleaved PARP) and increased the levels of the anti-apoptotic protein, Bcl-2.

Figure 2.
Rasagiline potentiates Akt signaling in PC12 cell cultures exposed to ischemic insult. PC12 cell cultures were subjected to 4 h OGD followed by 30 min reperfusion in the absence (lane 4) or presence of 10 μM rasagiline (lane 3), or with (lane 6) and without 50 μM LY29004 (lane 3). Normoxic cultures were subjected to drugs (lanes 2, 7), or left untreated (lane 1). Cell extracts were prepared for Western blot analyses of Akt phosphorylation (top blots) and for total enzyme level using pan antibodies (bottom blots). The relative phosphorylation of the kinase was calculated by the ratio between the phosphorylated and total (pan) levels. Data is expressed as a percentage above control cells and represents mean ± SEM (n = 4); *p < 0.01 vs normoxia (lane 1); **p < 0.05 vs OGD alone (lane 4) or OGD and Rasagiline (lane 3).
Figure 2.
Rasagiline potentiates Akt signaling in PC12 cell cultures exposed to ischemic insult. PC12 cell cultures were subjected to 4 h OGD followed by 30 min reperfusion in the absence (lane 4) or presence of 10 μM rasagiline (lane 3), or with (lane 6) and without 50 μM LY29004 (lane 3). Normoxic cultures were subjected to drugs (lanes 2, 7), or left untreated (lane 1). Cell extracts were prepared for Western blot analyses of Akt phosphorylation (top blots) and for total enzyme level using pan antibodies (bottom blots). The relative phosphorylation of the kinase was calculated by the ratio between the phosphorylated and total (pan) levels. Data is expressed as a percentage above control cells and represents mean ± SEM (n = 4); *p < 0.01 vs normoxia (lane 1); **p < 0.05 vs OGD alone (lane 4) or OGD and Rasagiline (lane 3).
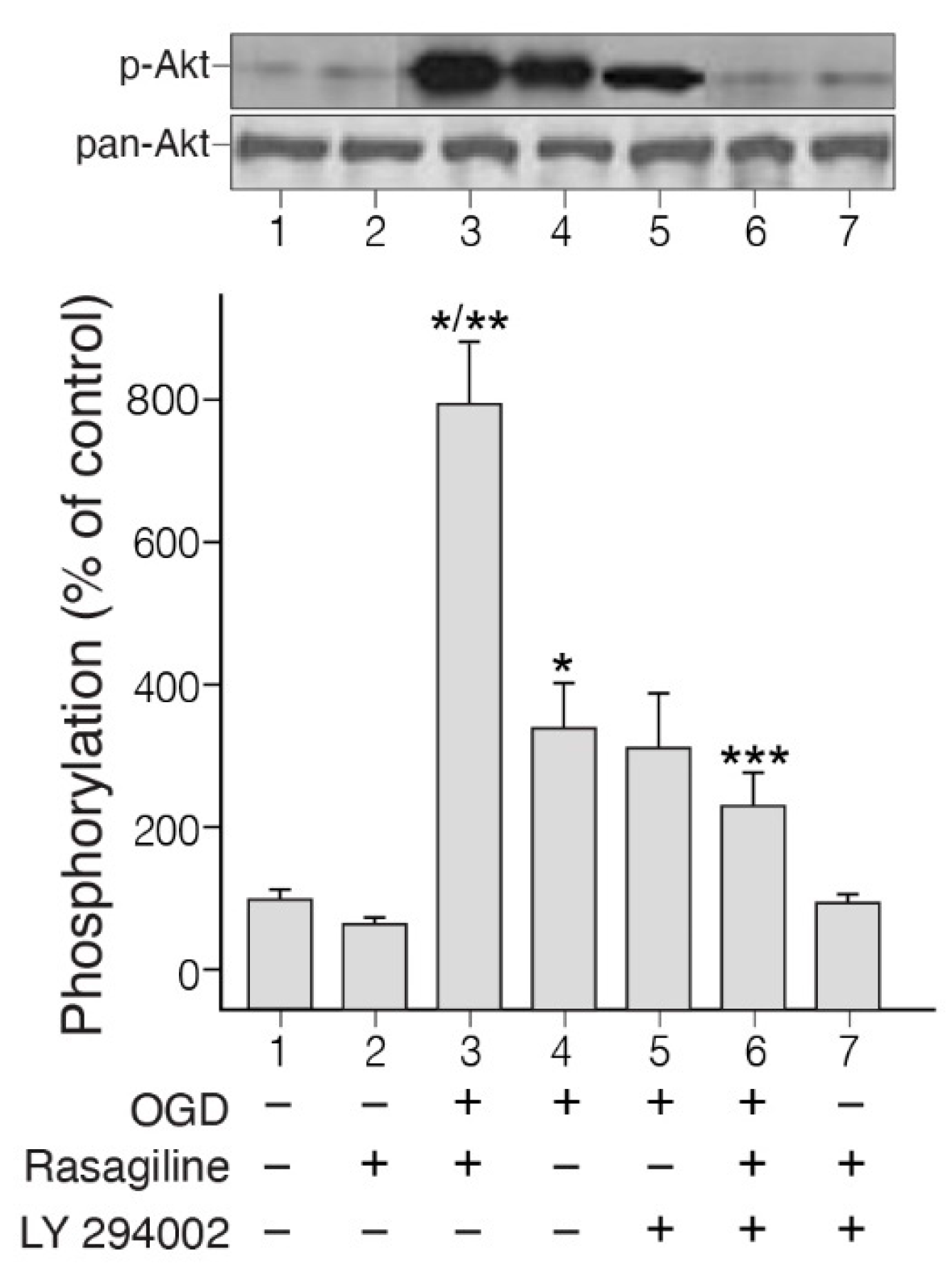
Figure 3.
Figure 3. Rasagiline decreased ischemic insult-induced, nuclear translocation of GAPDH. Undifferentiated (black bar) and NGF-differentiated (white bar). PC12 cell cultures were subjected to normoxia (control), or ischemic insult (OGD), in the absence (-) or presence (+) of 10 μM rasagiline (OGD + Rasagiline). A. Quantitative analysis of the fluorescence intensity of cultures grown on coverslips, fixed and immunostained with anti-GAPDH antibody (red) and analyzed by a confocal microscope (insert), indicating that was mostly located in the cytosol and the level increased by the ischemic insult and reduced by rasagiline treatment. *p < 0.01 vs control; **p < 0.01 vs OGD; B. Western blotting analysis of the 36 kDa nuclear GAPDH protein; the nuclear protein of 66 kDa Lamin-B was used as control; GAPDH level was increased by the ischemic insult and decreased in rasagiline- treated cultures.
Figure 3.
Figure 3. Rasagiline decreased ischemic insult-induced, nuclear translocation of GAPDH. Undifferentiated (black bar) and NGF-differentiated (white bar). PC12 cell cultures were subjected to normoxia (control), or ischemic insult (OGD), in the absence (-) or presence (+) of 10 μM rasagiline (OGD + Rasagiline). A. Quantitative analysis of the fluorescence intensity of cultures grown on coverslips, fixed and immunostained with anti-GAPDH antibody (red) and analyzed by a confocal microscope (insert), indicating that was mostly located in the cytosol and the level increased by the ischemic insult and reduced by rasagiline treatment. *p < 0.01 vs control; **p < 0.01 vs OGD; B. Western blotting analysis of the 36 kDa nuclear GAPDH protein; the nuclear protein of 66 kDa Lamin-B was used as control; GAPDH level was increased by the ischemic insult and decreased in rasagiline- treated cultures.
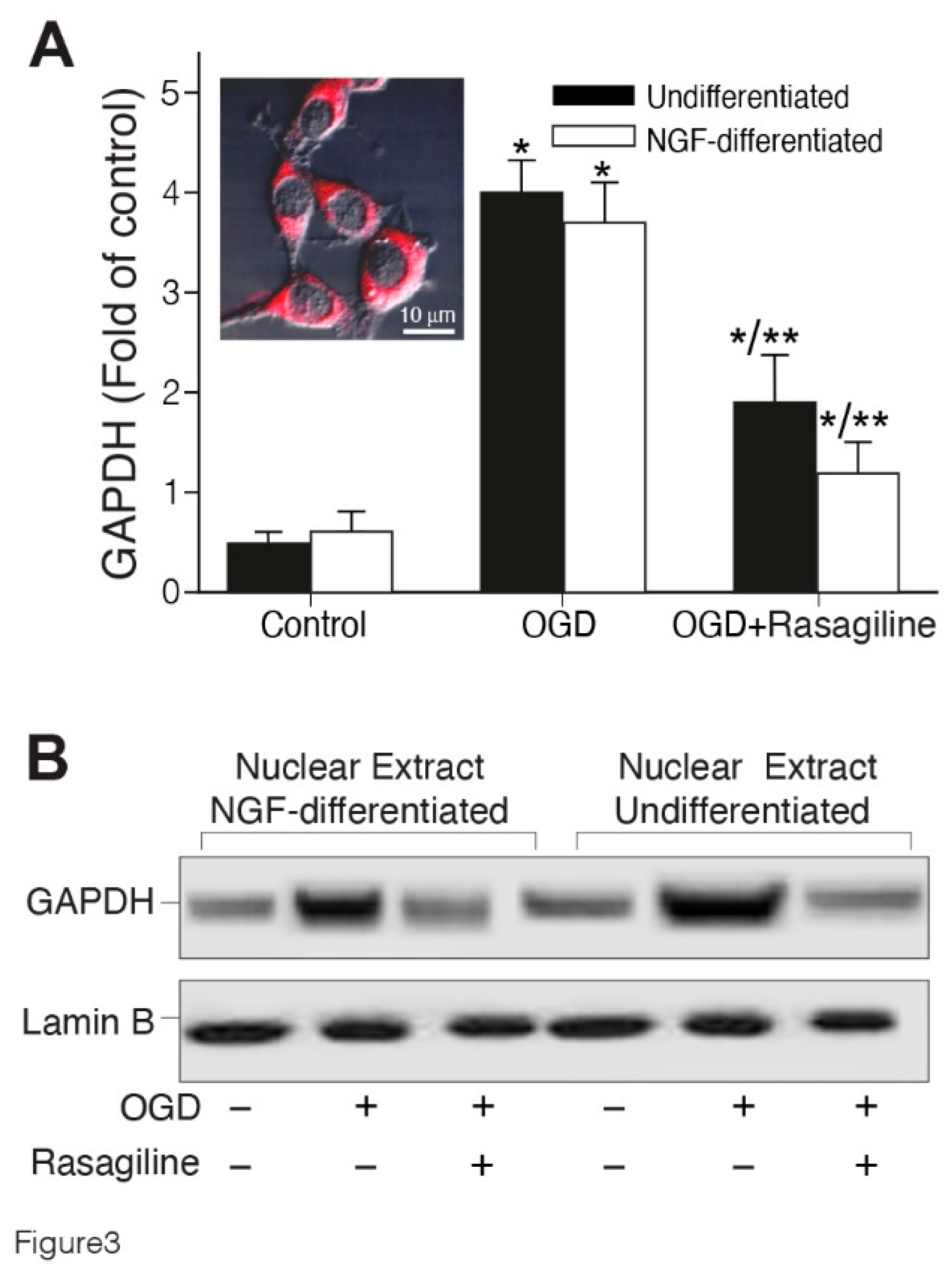
Figure 4.
Rasagiline increased ischemic insult-induced, nuclear translocation of Nrf2. PC12 cell cultures were subjected to normoxia, or ischemic insult (OGD), in the absence (control) or presence of 10 μM (A) or different concentrations of rasagiline (B). A. Representative immunofluorescence images of cultures grown on coverslips, fixed and stained with anti-Nrf2 antibody (green), nuclei stained with DAPI, and analyzed by a fluorescent microscope. Inserts: a few cells at higher magnification. B. Representative images of the Western blotting analysis of cytosol (left), nuclear extracts treated during the ischemic insult (OGD) with different concentrations of rasagiline (middle), and nuclear extracts of untreated (control), scramble RNA (SC) and Nrf2-siRNA (siRNA) cultures (right). β-tubulin and Lamin B were used as controls for the cytosol and nucleus, respectively. C. Quantitative analysis of the nuclear extracts presented in part B. Rasagiline increased dose-dependently the nuclear Nrf2 protein level during the ischemic insult in naïve cells (white bar) compared to normoxia (black bar) (top), an effect inhibited in Nrf2-siRNA treated cultures (bottom); *p < 0.01; vs control or respective normoxic level; **p < 0.01 vs Rasagiline (R).
Figure 4.
Rasagiline increased ischemic insult-induced, nuclear translocation of Nrf2. PC12 cell cultures were subjected to normoxia, or ischemic insult (OGD), in the absence (control) or presence of 10 μM (A) or different concentrations of rasagiline (B). A. Representative immunofluorescence images of cultures grown on coverslips, fixed and stained with anti-Nrf2 antibody (green), nuclei stained with DAPI, and analyzed by a fluorescent microscope. Inserts: a few cells at higher magnification. B. Representative images of the Western blotting analysis of cytosol (left), nuclear extracts treated during the ischemic insult (OGD) with different concentrations of rasagiline (middle), and nuclear extracts of untreated (control), scramble RNA (SC) and Nrf2-siRNA (siRNA) cultures (right). β-tubulin and Lamin B were used as controls for the cytosol and nucleus, respectively. C. Quantitative analysis of the nuclear extracts presented in part B. Rasagiline increased dose-dependently the nuclear Nrf2 protein level during the ischemic insult in naïve cells (white bar) compared to normoxia (black bar) (top), an effect inhibited in Nrf2-siRNA treated cultures (bottom); *p < 0.01; vs control or respective normoxic level; **p < 0.01 vs Rasagiline (R).
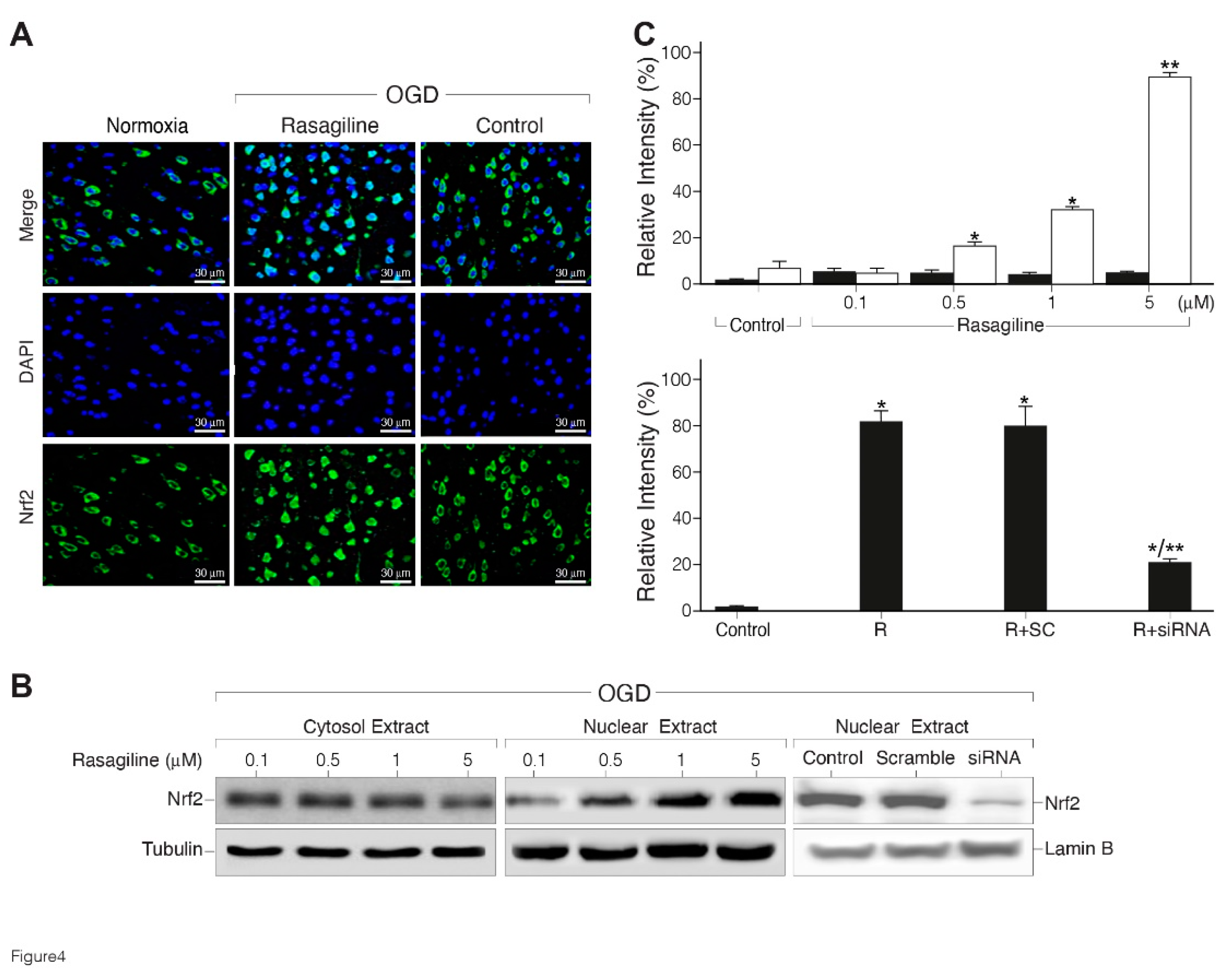
Figure 5.
Rasagiline increased the mRNA levels of ARE-mediated expression of phase II antioxidant enzymes. PC12 cell cultures were subjected to normoxia or ischemic insult in the absence or presence of 10 μM Rasagiline that significantly increased the mRNA levels of NQO1, HO-1, and catalase, as compared to normoxic untreated or treated cultures; *p < 0.01 vs normoxia; **p < 0.01 vs OGD.
Figure 5.
Rasagiline increased the mRNA levels of ARE-mediated expression of phase II antioxidant enzymes. PC12 cell cultures were subjected to normoxia or ischemic insult in the absence or presence of 10 μM Rasagiline that significantly increased the mRNA levels of NQO1, HO-1, and catalase, as compared to normoxic untreated or treated cultures; *p < 0.01 vs normoxia; **p < 0.01 vs OGD.
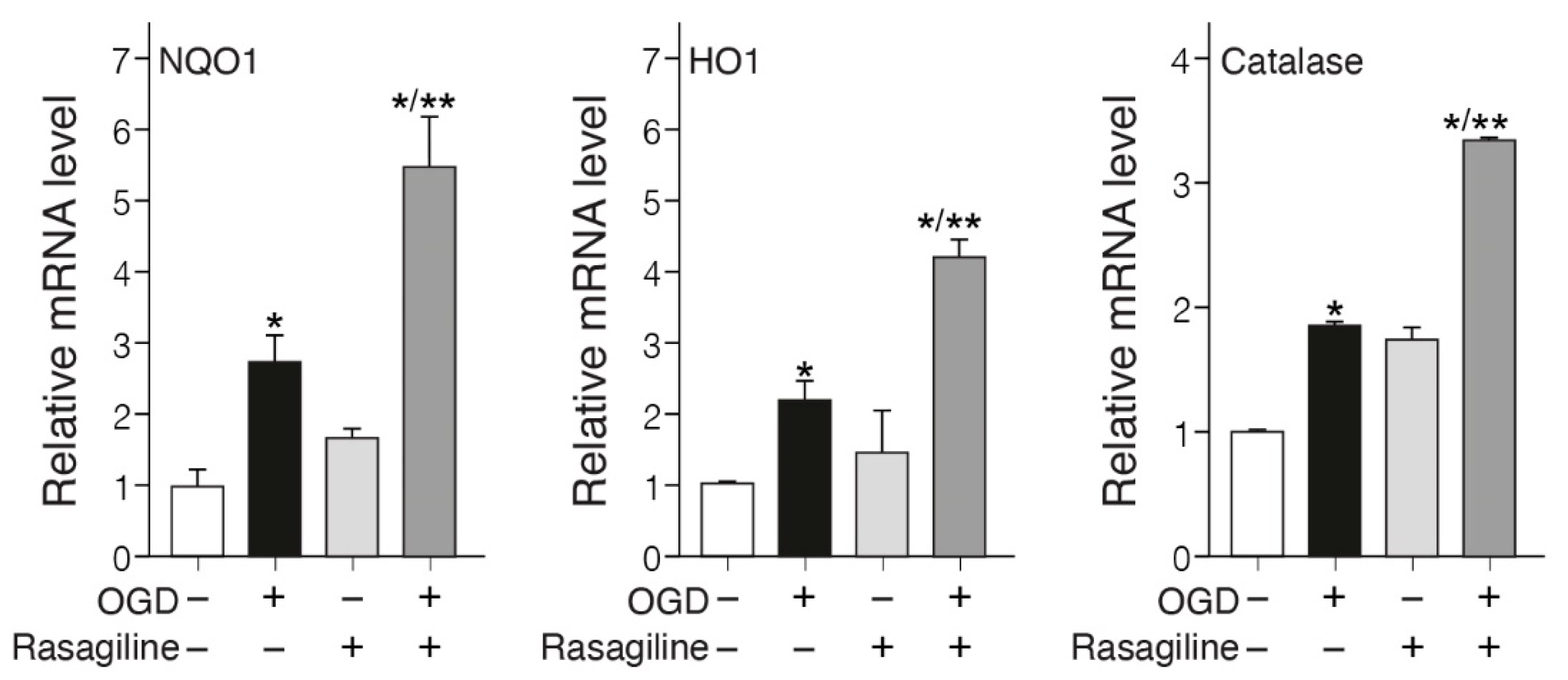
Figure 6.
Rasagiline decreased the ischemic insult-induced α-synuclein protein level. Undifferentiated (black bar) or NGF-differentiated (white bar) PC12 cell cultures were subjected to normoxia (control), or ischemic insult (OGD), in the absence or presence of 10 μM rasagiline (OGD + Rasagiline). A. Quantitative analysis of the fluorescence intensity of cultures grown on coverslips, fixed and immunostained with anti- α-synuclein antibody (green), and analyzed by a confocal microscope; its level was increased by the ischemic insult and reduced by rasagiline treatment. *p < 0.05 vs control; **p < 0.05 vs OGD; LM-light microscopy photos of undifferentiated cells B. Western blotting analysis of the 19 and 65 kDa - α-synuclein proteins; their level was increased by the ischemic insult and significantly decreased in the rasagiline-treated cultures.
Figure 6.
Rasagiline decreased the ischemic insult-induced α-synuclein protein level. Undifferentiated (black bar) or NGF-differentiated (white bar) PC12 cell cultures were subjected to normoxia (control), or ischemic insult (OGD), in the absence or presence of 10 μM rasagiline (OGD + Rasagiline). A. Quantitative analysis of the fluorescence intensity of cultures grown on coverslips, fixed and immunostained with anti- α-synuclein antibody (green), and analyzed by a confocal microscope; its level was increased by the ischemic insult and reduced by rasagiline treatment. *p < 0.05 vs control; **p < 0.05 vs OGD; LM-light microscopy photos of undifferentiated cells B. Western blotting analysis of the 19 and 65 kDa - α-synuclein proteins; their level was increased by the ischemic insult and significantly decreased in the rasagiline-treated cultures.
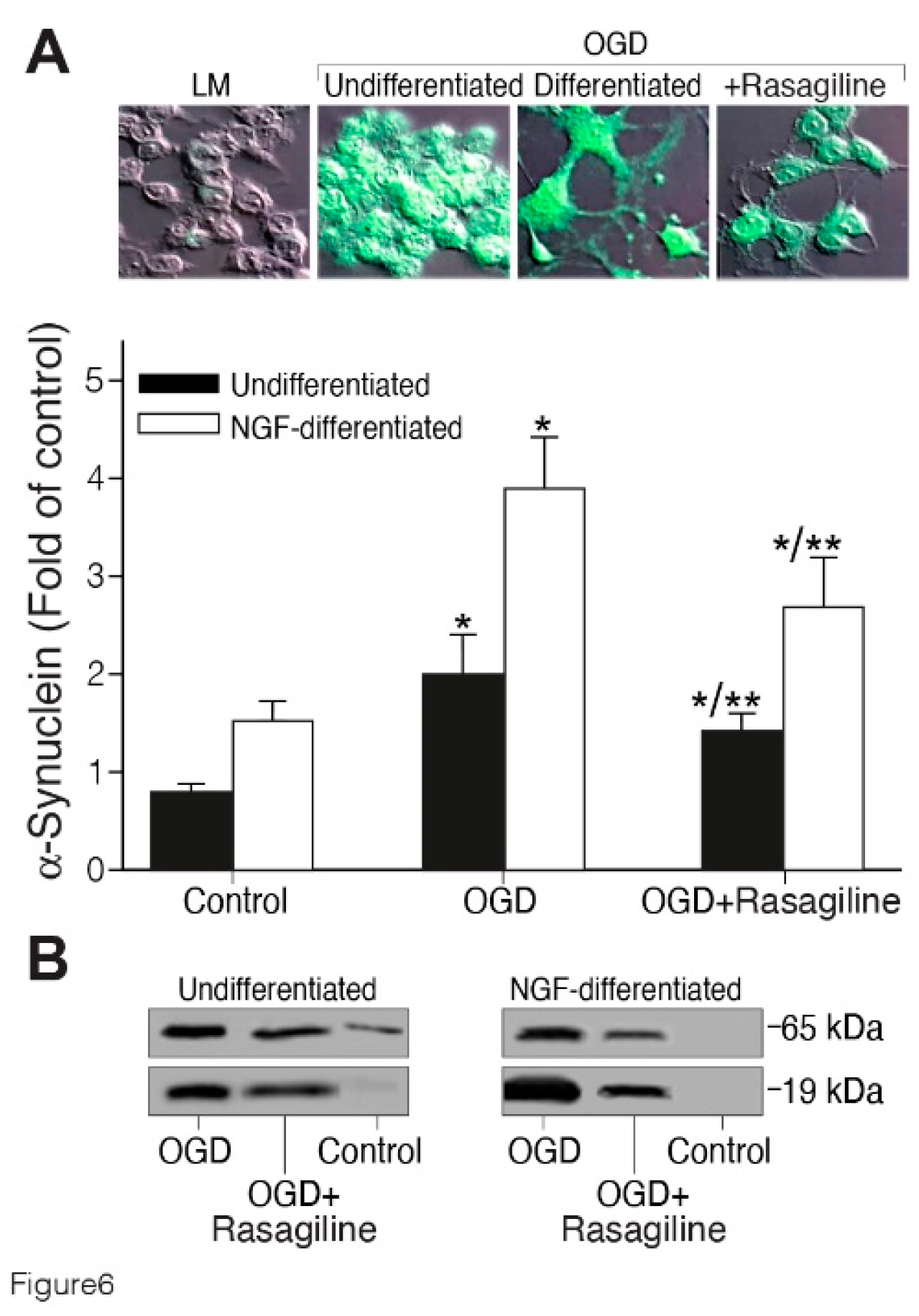
Figure 7.
Summary of the underlying mechanisms by which rasagiline conferred neuroprotection towards ischemic-like insults of PC12 cell cultures. Rasagiline, a selective MAO-B inhibitor, attenuated the ischemia-induced neuronal injury through the following actions: 1. Inhibition of MAO B activity of metabolizing dopamine and therefore, reducing the generation of free radicals during the ischemia/reperfusion; 2. Decrease of the nuclear translocation of GAPDH and thereby reducing cell death; 3. Decrease of the ROS levels /oxidative stress and mitochondrial dysfunction by a direct antioxidant effect since its propargylamine pharmacophore can directly scavenge free radicals; 4. Activation of the PI3K-Akt-Nrf2 pathway and induction of the antioxidant response element (ARE) genes; 5. Together, these events and others ultimately lowered the expression of the monomeric and/or tetramer neurotoxic α-synuclein protein level, minimizing cell death. Black sharp arrow, activate; red blunt arrow, inhibit; Abbreviations: MAO-B, monoamine oxidase B; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Akt, protein kinase B; Bcl2, B-cell lymphoma 2 protein; Nrf2, nuclear factor erythroid 2-related factor 2; HO-1, heme oxygenase 1; NQQ1, NAD(P)H quinone oxidoreductase 1; ROS, reactive oxygen species.
Figure 7.
Summary of the underlying mechanisms by which rasagiline conferred neuroprotection towards ischemic-like insults of PC12 cell cultures. Rasagiline, a selective MAO-B inhibitor, attenuated the ischemia-induced neuronal injury through the following actions: 1. Inhibition of MAO B activity of metabolizing dopamine and therefore, reducing the generation of free radicals during the ischemia/reperfusion; 2. Decrease of the nuclear translocation of GAPDH and thereby reducing cell death; 3. Decrease of the ROS levels /oxidative stress and mitochondrial dysfunction by a direct antioxidant effect since its propargylamine pharmacophore can directly scavenge free radicals; 4. Activation of the PI3K-Akt-Nrf2 pathway and induction of the antioxidant response element (ARE) genes; 5. Together, these events and others ultimately lowered the expression of the monomeric and/or tetramer neurotoxic α-synuclein protein level, minimizing cell death. Black sharp arrow, activate; red blunt arrow, inhibit; Abbreviations: MAO-B, monoamine oxidase B; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Akt, protein kinase B; Bcl2, B-cell lymphoma 2 protein; Nrf2, nuclear factor erythroid 2-related factor 2; HO-1, heme oxygenase 1; NQQ1, NAD(P)H quinone oxidoreductase 1; ROS, reactive oxygen species.
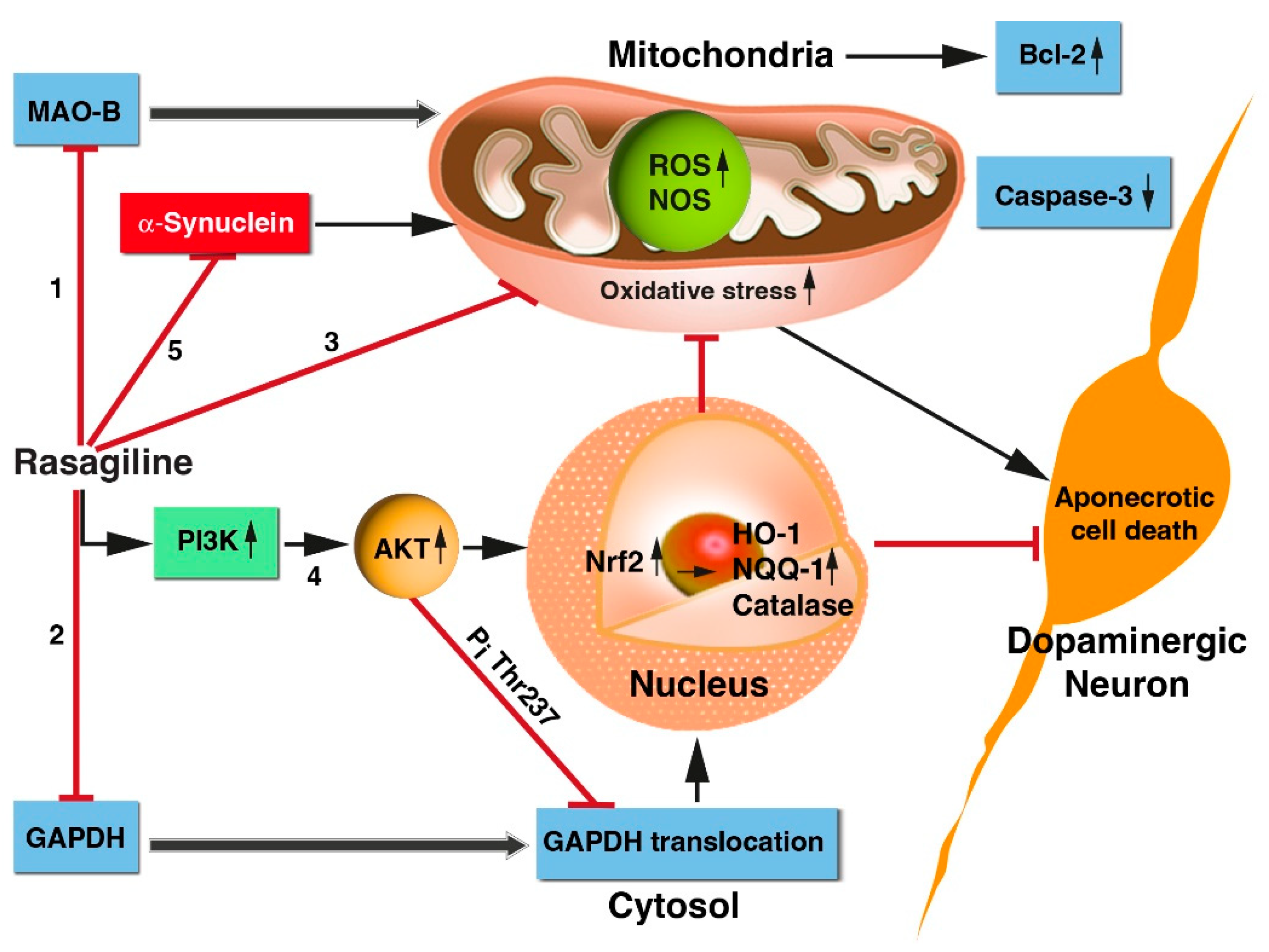

Table 1.
The effect of different inhibitory chemicals on the Rasagiline-induced neuroprotection towards ischemic like insult in PC12 cell cultures.
Table 1.
The effect of different inhibitory chemicals on the Rasagiline-induced neuroprotection towards ischemic like insult in PC12 cell cultures.
| Treatment | LDH release (% of total) | Neuroprotection (% of Rasagiline) |
|
|---|---|---|---|
| OGD | OGD + Rasagiline | ||
| Control | 38±9 | 10±3 | 74±7 |
| LY294002 | 42±7 | 22±61* | 48±9 |
| Scramble RNA | 47±5 | 15±52 | 68±6 |
| Nrf2 siRNA | 36±10 | 28±62* | 20±4 |
1 PC12 cell cultures were subjected to 4 h OGD followed by 30 min reperfusion in the absence (OGD) or presence (OGD + Rasagiline) of 10 μM rasagiline with and without 50 μM LY29004. *p < 0.01 compared to control. Under normoxia, LDH release was 2% of total. 2 PC12 cell cultures treated for 5 days with scramble RNA or Nrf2-siRNA were subjected to the ischemic insult, in the absence (OGD) or presence (OGD + Rasagiline) of 10 μM rasagiline. The OGD insult was carried out for 4 h followed by 18h reperfusion. Aliquots from the culture media were taken for LDH release measurements. *p < 0.01 compared to control or scramble RNA.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



