
Submitted:
27 June 2024
Posted:
29 June 2024
You are already at the latest version
Abstract
Nonsense mutations affect about 11% of patients with Cystic Fibrosis and produce a premature termination codon in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) mRNA causing early termination of the polypeptide translation.
A potential therapy for nonsense mutations allows small molecules to overcome the premature termination codon (PTC) by a readthrough mechanism leading to the synthesis of a complete CFTR protein. However, the efficiency of the readthrough molecules is limited by a quality control cellular pathway known as Nonsense-mediated mRNA Decay (NMD). This pathway provides the degradation of the mRNA harboring a premature termination codon, to prevent the production of altered polypeptides. In contrast, the activity of this pathway interferes with the effectiveness of the readthrough drugs, limiting the CFTR mRNA amount to the rescue.
In this work, we reduced the activity of the NMD pathway, silencing the Upf1 protein, one of the main components of the pathway.
Our strategy was to increase the amount of the mRNA target to improve the efficiency of readthrough of TRIDs (Translational Readthrough Inducing Drugs) molecules in cells that ectopically express the W1282X-CFTR mRNA. The reduction of the Upf1 expression/activity was correlated with the increase of the efficiency of readthrough and so the increase of CFTR protein rescue.
Keywords:
premature termination codon
; nonsense mutation
; Nonsense-mediated decay
; translational readthrough inducing drugs
; oxadiazoles.
1. Introduction
With 70.000 affected people and a mortality rate of 90%, Cystic Fibrosis (CF) is one of the most widespread lethal genetic diseases [Rafeeq M.M., 2017]. The clinical manifestations of this monogenic disorder are caused by mutations of a single gene, encoding for a channel protein named CFTR (Cystic Fibrosis Transmembrane Conductance Regulator). This protein is expressed on the apical surface of epithelial cells of secretory organs, where it regulates the chloride ion transport and subsequently the osmotic water passage, needful to fluidify the mucus layer of these tissues [Farinha C.M., 2017].
Almost two thousand different mutations of the CFTR gene have been identified as the cause of structural and/or functional alterations of the channel. They are associated to a more or less severe dysfunction of the chloride flux, inducing duct obstruction and tissue damage [Quon B.S., 2016].
Much progress has been made in recent years in the treatment of CF with correctors or potentiators of the CFTR channel. However, these molecules act respectively by correcting the folding, so driving the channel to the plasmatic membrane, and increasing the activity of the channel; therefore correctors and potentiators, in order to be able to act, require the presence of the protein. Nevertheless, 11% of the population affected by CF presents a premature termination codon (PTC) that causes the synthesis of an incomplete and non-functional CFTR protein, on which therefore, correctors or potentiators have no effects [Quon B.S., 2016].
A potential therapy for nonsense mutations aims to reduce the fidelity of the translation termination process using small molecules named Translational readthrough-inducing drugs (TRIDs) [Campofelice A., 2019; Samanta A., 2019]. TRIDs molecules allow the insertion of a near cognate tRNA (nc-tRNA), that, even if is not perfectly complementary to the PTC in the A site, allows the ribosome to skip the PTC and thus leads to the formation of a full-length protein [Dabrowski M. 2015].
In this context, a promising molecule is a synthetic aminoglycoside ELX02; it showed the rescue of the CFTR channel activity in human organoids from CF patients [Kerem E., 2020], but respect these promising results it did not achieve statistical significance for efficacy endpoints in Phase 2 study in Class 1 CF mutations [NCT04135495]. Currently, Ataluren or PTC124 is the only TRID approved by the FDA, and it has been tested in phase II and III trials both for CF and Duchenne Muscular Dystrophy (DMD, another genetic disorder caused by nonsense mutations) [Ardicli D., 2019]. However, the conflicting results in Cystic Fibrosis led to the stop of the trials for this disease and the approval of Ataluren only for the treatment of DMD [Richardson R., 2017]. For this reason, it is important to identify new readthrough molecules with low toxic effects and high efficiency of action.
Many efforts have been devoted to the search for new TRIDs [Pibiri I., 2015; Pibiri I., 2016, Pibiri I., 2018] and the investigation of their mechanism of action [Tutone M., 2019; Tutone M, 2020]. In our recent work [Pibiri I., 2020] we identified three molecules with interesting readthrough activity (NV848, NV914, and NV930) on the two most frequent Cystic Fibrosis stop mutations: G542X and W1282X mutations. Despite the demonstrated efficacy of these molecules, the efficiency of the readthrough mechanism depends on different features, including of course the genetic contest of the mutation but also inter-individual genetic variability [Loughran G., 2014]. Indeed, different patients, characterized by the same mutation, show a different response to the therapy, proving the involvement of mutation-independent factors affecting CFTR mRNA expression [Pranke I., 2019; Bongiorno R., 2021].
Among these factors, a significant role is performed by the Nonsense-Mediated decay (NMD) pathway. It is a pathway with an extremely efficient variability in population and responsible for the decay of mRNA with a PTC, to reduce the production of truncated proteins that could be toxic for the cell, due to their dominant negative function [Chang Y., 2004; Keeling K., 2013; Karousis E.D., 2016].
A prominent role in the NMD pathway is played by the up-frameshift factor 1 (Upf1), the protein that, in a phosphorylated state and the presence of a PTC, can interact with the termination factors eRF1 and eRF3, causing the recruitment of endonucleases inducing mRNA decay [Popp M.W., 2018]. The mechanisms by which Upf1 discriminates its target are mainly two: exon junction complex (EJC)-dependent and EJC-independent.
The EJC is a large protein complex recruited upstream of the splicing site during mRNA maturation and normally removed by the ribosome during translation.
According to the EJC-dependent NMD mechanism, an EJC present after a stop codon will not be removed, representing so a signal for NMD activation [Neu-Yilik G., 2017].
The EJC-dependent mechanism has been considered the only way for NMD activation for a long time. However, NMD has been reported to have a role not only in targeting PTC-containing mRNA but also in fine quality control of the transcriptome, implying a mechanism independent of EJC presence [Lou C.H., 2016]. Indeed, mRNAs without introns can similarly be recognized by NMD [Hogg L., 2010; Bongiorno R., 2021]. An alternative model, “faux 3’UTR”, proposes that the distance from the termination codon to the poly (A) tail provides for the targeting of NMD due to the competitive binding of the major NMD regulator, Upf1, and cytoplasmic poly (A) binding protein 1 (PABPC1) with eukaryotic polypeptide chain release factor 3 (eRF3), which interacts with eRF1 and recognizes termination codons including the PTC [Sato H., 2021].
Several pieces of evidence demonstrate that NMD can also act on transcripts not containing an EJC [Hogg L., 2010, Metze S., 2013].
In the EJC-independent mechanism, the recognition of a PTC by NMD is possible thanks to a competition mechanism depending on the PTC position. Indeed, eRF3 can either interact with Upf1, so activating the NMD pathway or cooperate with the polyA binding protein (PABP), then inhibiting Upf1 recruiting [Ivanov I. P. 2008, Peixeiro I., 2012; Fritz S., 2020, Sato H., 2021] (Figure 1).
In general, the NMD pathway is a limiting factor for TRIDs action, thus a combined use of readthrough agents and factors that minimize the EJC-dependent and the EJC-independent nonsense mRNA decay can be considered a promising strategy for nonsense mutation treatment. In this regard, post-transcriptional silencing of Upf1 or inhibition of Upf1 phosphorylation, by using caffeine, could be possible strategies to stabilize the CFTR mRNA and so increase the readthrough effects of TRIDs [Roy B., 2016; Lentini L., 2019].
Fisher rat thyroid (FRT) cell model system, transfected with a vector harboring the CFTR cDNA with the W1282X stop mutation was used to demonstrate the potentiality of these combined treatments and in particular to contrast the effect of the EJC-independent nonsense mRNA decay and to promote TRIDs activity.
2. Materials and Methods
TRIDs used in this study have been synthesized as reported in the literature [Pibiri I.,2020; Lentini L., 2014], and all reagents and solvents were purchased from commercial sources. All synthesized compounds were first purified by silica flash chromatography and analyzed (see supplementary material) by IR, NMR, and HPLC/MS, assessing their purity grade (>95%).
2.1. Cell Cultures
FRT YFP+/+CFTRWT: rat epithelial thyroid cells transfected with both pcDNA3.1 and pTRACER-CFTR-WT(pTCF-wt) plasmid vectors. So these cells express CFTR wt protein.
FRT YFP+/+CFTRW1282X-opal: rat epithelial thyroid cells expressing the YFP protein and transfected with pTRACER-CFTR-W1282X plasmid vector, that we obtained by replacing the tryptophan UGG codon, in 1282 position, with the UGA opal stop codon, through site-specific mutagenesis of pTCF-wt.
2.2. Condition of Culture and TRIDs Treatment
The culture medium is prepared by dissolving the Nutrient Mixture Coon’s F-12 Ham and sodium bicarbonate (2,68 g) in sterile water for cell culture (1l). The solution is filtered and then added with fetal bovine serum (10%, Corning, USA) and glutamine, penicillin and streptomycin (1%) (removed before TRIDs treatment). Cells were cultured in a humidified atmosphere of 5% CO2 in air at 37°C. TRIDs were used at 12μM for 24 hours.
2.3. Real-Time RT-PCR
Total RNA was extracted from cells by using the RNAeasy Mini kit according to the manufacturer's instructions (Qiagen, Milano, Italy). RNA was reverse-transcribed in a final volume of 20 μl using the High Capacity cDNA Archive kit (Applied Biosystems, Monza, Italy) for 10 min at 25 °C and 2 h at 37 °C. For each sample 2 μl of cDNA, corresponding to 100 ng of reverse transcribed RNA, was analyzed by real-time RT-PCR (95 °C for 15 s, 60 °C for 60 s repeated for 40 cycles), in triplicate, using the ABI PRISM 7300 instrument (Applied Biosystems, Monza, Italy). Real-Time RT-PCR was done in a final volume of 25μl comprising 1x Master Mix SYBR Green (Applied Biosystems, Monza, Italy) and 2μM of forward and reverse primers for CFTR (Fwd: 5′-GCACCATTAAGAAAATATCATCTT-3'; Rev:5′-TTGTCTTTCTCTGCAAACTTGG-3′) and Upf1 (Fwd:5′-CAGACTACCCTTCCCAACAG-3′; Rev:5′AGGGCTGGCTCATGGACACA-3′). RNA level was determined by using the SDS software version (Applied Biosystems, Monza, Italy) according to the 2−ΔΔCt method (Fernandes P., 2021) and Ct values was normalized to the internal control actin.
2.4. Western Blotting
Proteins (30 μg) were separated by 3–8% SDS-PAGE (Bolt, Life Technologies) containing 0.1% SDS and transferred to PVDF membranes (Thermo Scientific, Rockford, USA) by electroblotting. For CFTR detection the membrane was incubated with an anti–CFTR antibody (Abcam 1:1000), and HRP-conjugated anti-mouse (Thermo Scientific Rockford, USA, 1:5000). The target protein was detected by the use of SuperSignalWest Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, USA). We used an anti-β-tubulin antibody (mouse; Sigma-Aldrich 1:10.000) to confirm equal protein loading. Gel bands were quantified by ImageJ software. WB assays were repeated at least two times.
2.5. Immunofluorescence Microscopy
FRT pTRACER-CFTR-WT and FRT pTRACER-CFTR-W1282X-opal cells have been plated on rounded glass coverslips and suitably treated. Cells were then fixed with methanol and blocked with 0.1% BSA for one hour at room temperature. The coverslips were incubated with a mouse monoclonal antibody against the first extracellular loop of human CFTR (Abcam, 1:500) overnight at 4°C, followed by a goat polyclonal anti-mouse Alexa-Fluor-488 (Abcam, 1:1000) secondary antibodies for 1 h at 37 °C. The cell membrane and Golgi apparatus were stained by the wheat germ agglutinin Alexa 594 conjugated Alexa 594 antibody (Thermo Scientific, USA 1:1000) while nuclei were visualized with DAPI. Cells were observed under a fluorescence microscope (Carl Zeiss) at 63X magnification.
2.6. Functionality Assay: Quenching of Fluorescence of YFP Protein
The functionality of the CFTR channel has been evaluated, on pTRACER-CFTR-WT e FRT pTRACER-CFTR-W1282X cells, through both quantitative (50.000) and qualitative (250.000) assays. After the respective times of treatment with caffeine, siRNA, and TRIDs, the fluorescence was evaluated after the 60s and 120s from the addition of PBS NaI solution added with Forskolin (5μM). The fluorescence of YFP protein has been quantitative measured through the use of a fluorometer and qualitative evaluated thanks to a fluorescence microscope (Zeiss Observer.D1).
2.7. Reduction of NMD Pathway Activity
The reduction of NMD pathway activity has been done with two different approaches:
caffeine treatment for 24h at a concentration of 0,75 mM to inhibit the kinase of Upf1.
post-transcriptional silencing of Upf1 by use of a specific siRNA against Upf1 (Silencer Select Pre-designed siRNA product, Ambion) (5’ – CUACCUCGGUGAUGAGUUUtt – 3’), for 72h at a concentration of 100nM. For this treatment, pTRACER-CFTR-W1282X cells (250.000) have been transfected with siRNA by using lipofectamine 2000 (5μl) and Optimem (24μl).
Non-specific siRNA, consisting in the experimental control, siGFP (Eurofins, 5’-GGCUACGUCCAGGAGCGCAdTdT – 3’) was used with the same condition of siUPF1.
3. Results
3.1. NMD Pathway Inhibition Increases CFTR mRNA Level Favoring TRIDs Readthrough Action
Recently we identified three potential translational readthrough-inducing drugs named NV848, NV914, and NV930 [Pibiri I., 2020]. To increase the nonsense CFTR mRNA levels and to optimize the TRIDs readthrough effect, we used two different approaches aimed at NMD pathway inhibition. The first approach was based on the use of caffeine, an inhibitor of the Upf1 phosphorylation. The second one, relying on post-transcriptional silencing of Upf1 mRNA, was designed to reduce Upf1 activity and to evaluate the possible increase of the readthrough effect by our TRIDs.
FRT cells stably transfected with the pTRACER-CFTRW1282X vector were treated with 0.75 mM caffeine for 24 hours, in parallel FRT pTRACER-CFTRW1282X were transfected with siUpf1 and siGFP (used as not specific siRNA) for 72 hours to reduce the Upf1 expression, thus weakening the nonsense-mediated decay pathway. The use of 0.75mM caffeine was justified by previous results, showing the increase in CFTR mRNA expression without toxic effects [Lentini L., 2019].
Real-time RT PCR analysis performed after 72 hours from siRNA transfection revealed that Upf1 mRNA was reduced by 0.4 folds in siUpf1 transfected cells compared to siGFP-transfected cells (Figure 2A).
To evaluate the CFTR mRNA expression levels, real-time RT PCR was performed on total mRNA extracts of siUpf1 silenced FRT pTRACER-CFTRW1282X cells and of FRT pTRACER-CFTRW1282X cells treated for 24 hours with caffeine (Figure 2-B). We observed that the reduction of Upf1 expression by siRNA induced a significant effect on CFTR expression. Indeed, FRT pTRACER-CFTRW1282X cells, where Upf1 mRNA expression was partially silenced, showed an increase of 38% in CFTR transcript respect to siGFP transfected cells (Figure 2-B). On the other hand, caffeine treatment showed a 1.87-fold change (87%) in CFTR mRNA level (Figure 2-B).
3.2. Combined Treatment of the NMD pathway Inhibition and Readthrough Induction with NV914 or NV848 TRID Molecules Increases CFTR Expression
To increase the expression of the CFTR protein in FRT pTRACER-CFTRW1282X cells, we combined the two described approaches, by treating the cells with TRIDs (NV848 and NV914) [Pibiri I., 2020] and the NMD pathway inhibitors (siUpf1 and caffeine). The choice of these molecules was made based on our previous results, showing that both NV914 and NV848 molecules were able to rescue CFTR expression and functionality in FRT pTRACER-CFTRW1282X cells [Pibiri I., 2020].
FRT pTRACER-CFTRW1282X cells were treated with 0.75 mM caffeine and 12 μM of NV914 or NV848 for 24 hours. In the alternative, FRT pTRACER-CFTRW1282X cells were transfected with Upf1 siRNA for 72 hours with the addition of the NV914 or NV848 in the last 24 hours. CFTR expression was evaluated by immunofluorescence and Western blot analyses (Figure 3 and Figure 4).
We detected that FRT pTRACER-CFTRW1282X cells showed increased CFTR expression in comparison with the untreated sample after 24 hours of NV914 and NV848 treatment. Moreover, the combined treatment with TRIDs/siUpf1 for 72 hours induced a further increase in CFTR expression (Figure 3 and Figure 4).
The caffeine treatment did not induce CFTR protein expression. Combined TRIDs/caffeine treatment, although it seems to increase CFTR rescue, failed to achieve significance compared to samples treated only with TRIDs.
Western Blot analysis of the UPF1 protein was made to verify UPF1 silencing efficiency (Figure 4).
3.3. Functional Analysis of CFTR Channel after TRIDs Treatment
Aiming to verify if TRID administration associated with NMD pathway inhibition can improve rescuing the CFTR channel functionality, in addition to the protein expression recovery, an indirect CFTR functionality assay has been performed.
Taking advantage of the expression of the YFP fluorescent protein in FRT cells, [Galietta L., 2001], this assay is based on the YFP characteristic of being sensitive to halides (as I-), due to the presence of a binding pocket closely approached to the YFP chromophore. When these ions enter cells through the CFTR channel, they bind to YFP protein so inducing fluorescence quenching.
Consequently, a progressive quenching of green fluorescence is indicative of channel functionality.
To evaluate CFTR functionality, FRT pTRACER-CFTRW1282X cells were treated with caffeine and NV914 or NV848 for 24 hours. In the alternative, FRT pTRACER-CFTRW1282X cells were transfected with siUpf1 for 72 hours with the addition of the NV914 or NV848 in the last 24 hours.
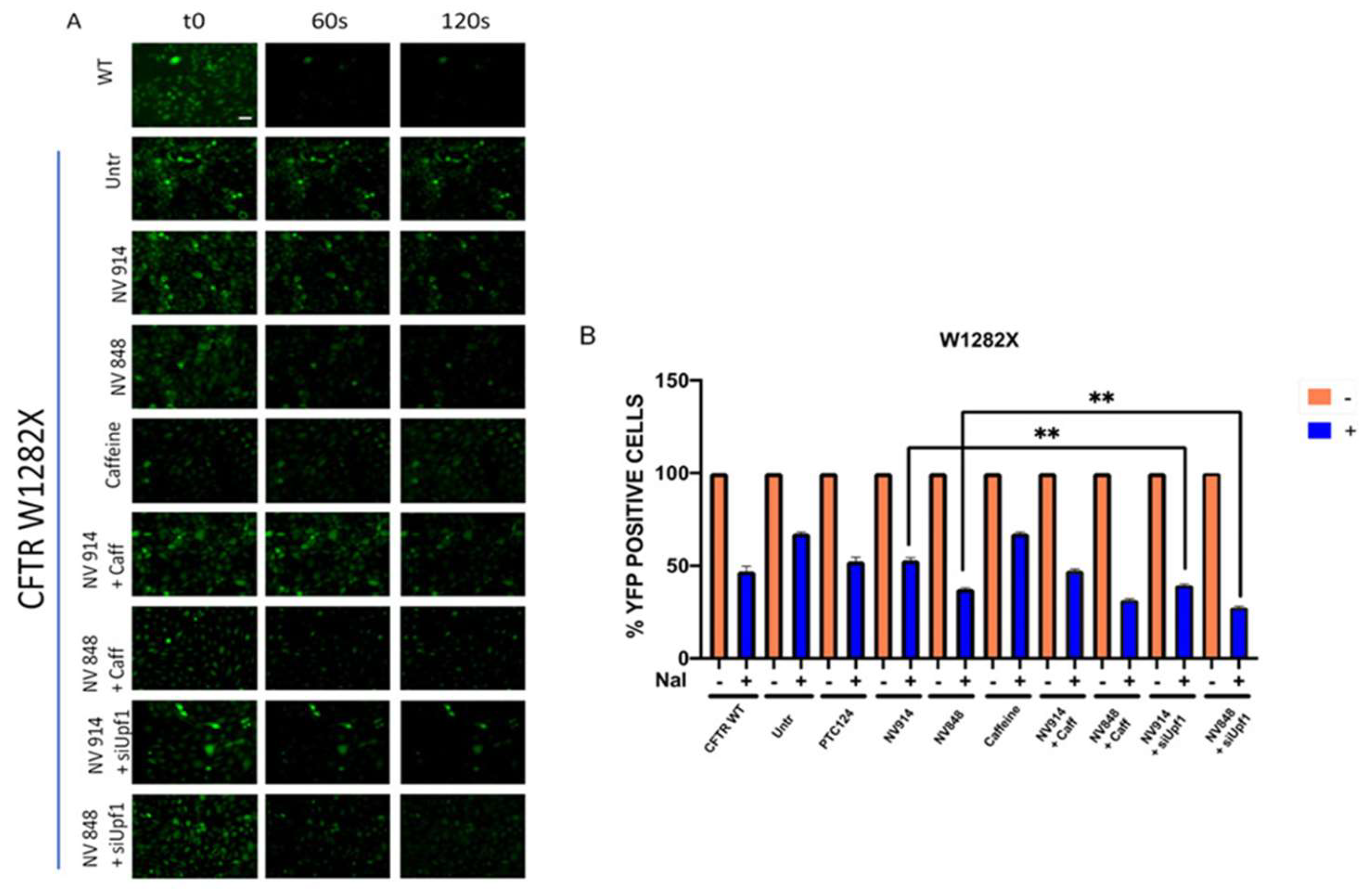
After 24h, cells were pre-stimulated with Forskolin, a CFTR channel activator, and observed by microscopy before and after 60 seconds from the addition of PBS NaI (Figure 5-A). YFP fluorescence quenching was quantitatively measured after 60 seconds from the addition of PBS NaI through the use of a fluorimeter (Figure 5-B). The observation of the YFP fluorescence was further extended to 120 seconds.
These analyses showed that combined treatment of siUpf1 with NV914 or NV848 determines a higher recovery of CFTR channel functionality than the sample treated with only NV914 or NV848 (Figure 5). In contrast, TRIDs/Caffeine treatment, although increasing CFTR rescue, failed to achieve significance compared to the samples treated only with TRIDs.
4. Discussion
In order to overcome the presence of PTC, in the last years, different molecules allowing ribosomes to readthrough the premature stop codon have been identified.
Despite the good results obtained from the readthrough approach, TRIDs efficiency is considerably reduced by the poor amount of target transcript, that is the mRNA containing the PTC. Indeed, the readthrough does not occur on the totality of the target transcript because of its degradation mediated by the NMD.
Being the NMD pathway a limiting factor, the identification and the use of approaches that induce nonsense mRNA stabilization could be a strategy to improve the readthrough efficiency of TRIDs.
In this work, we alternatively reduce Upf1 expression, through post-transcriptional silencing, and limit Upf1 activity, by using caffeine treatment in a Cystic Fibrosis cell model. In both cases, the association of the inhibition of NMD and treatment with NV914 and NV848 causes an increase of CFTRW1282X mRNA level followed also by the rescue of CFTR expression and functionality in FRT pTRACER-CFTRW1282X cells. However, unexpectedly, despite the higher CFTRW1282X mRNA level in caffeine-treated samples, both expression and functionality of CFTR rescue obtained by this treatment are slightly lower than the recovery achieved by Upf1 silencing.
These observations led us to hypothesize the existence of a positive feedback mechanism in which, the increase of CFTRW1282X mRNA level by caffeine treatment, could determine the transcriptional activation of the Upf1 gene and induce Upf1 protein expression (Figure 4). This could be an attempt of the cell to reduce the level of nonsense transcripts, which could potentially have deleterious dominant-negative effects. However, this high level of Upf1 cannot activate the NMD pathway because of the presence of caffeine. In this vision, in which both Upf1 and his transcript target (CFTRW1282X) result high, a competition between Upf1 and our TRIDs for the interaction with nonsense mRNA could be present, thus resulting in a reduction of readthrough efficiency. Furthermore, the presence of this positive feedback mechanism could also explain the usual difficulties that we encountered in Upf1 silencing and the high concentrations of siRNA used in different works to reduce UPF1 expression [Salvatori F. 2018; Sharma N., 2018].
Conclusions
Although the sole NMD pathway inhibition cannot be considered as a solution to nonsense pathologies, our results indicate that modulation of the NMD pathway, although still to be optimized, could be a promising approach in combination with TRIDs, to rescue expression and functionality of CFTR protein. Indeed, the synergy between these two approaches, NMD modulation, and readthrough, allows greater response to treatment, increasing the quantity of produced protein and boosting its activity.
5. Patents
L.L., I.P., R.M., and A.P. are co-inventors of the patent WO2019101709 “Oxadiazole derivatives for treating genetic diseases due to nonsense mutations”.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
The manuscript was written through the contributions of all authors. Conceptualization, L.L., and I.P.; methodology, L.L., R.B., R.M., E.V., and D.R.; validation, R.B., and L.L; formal analysis, A.P. P.S.C.; investigation, R.B., L.L.; resources, L.L. and I.P.; data curation, R.B., L.L., and I.P.; writing—original draft preparation, R.B. and L. L.; writing—review and editing, R.B., L.L., R.M., P.S.C., A.P and I.P.; project administration, L.L.; funding acquisition, L.L., and I.P. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: The research leading to these results has received funding from the European Union - NextGenerationEU through the Italian Ministry of University and Research under PNRR - M4C2-I1.3 Project PE_00000019 "HEAL ITALIA" to Ivana Pibiri and Laura Lentini CUP B73C22001250006 (Dip. STEBICEF-University of Palermo), and by Italian Cystic Fibrosis Research Foundation with FFC#06/2020 grant to Laura Lentini and Ivana Pibiri. The views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or the European Commission. Neither the European Union nor the European Commission can be held responsible for them.
Data Availability Statement
Data supporting the findings of this study are available from the corresponding author upon request.
Acknowledgments
We thank Prof. Louis Galietta (TIGEM, Pozzuoli, Italy) for kindly providing us with the FRT cells and the pTracer CFTRWT vector.
Conflicts of Interest
L.L., I.P., R.M., and A.P. are co-inventors of the patent WO2019101709 “Oxadiazole derivatives for treating genetic diseases due to nonsense mutations”. All other authors declare no conflict of interest.
References
- Ardicli, D.; Haliloglu, G.; Alikasifoglu, M.; Topaloglu, H. (2019). Diagnostic Pathway to Nonsense Mutation Dystrophinopathy: A Tertiary-Center, Retrospective Experience. Neuropediatrics 2019, 50, 41–45. [CrossRef]
- Bongiorno R., Colombo M.P., Lecis D. (2021). Deciphering the nonsense-mediated mRNA decay pathway to identify cancer cell vulnerabilities for effective cancer therapy. J Exp Clin Cancer Res. 2021 Dec 1;40(1):376. PMID: 34852841; PMCID: PMC8638473. [CrossRef]
- Campofelice A., Lentini L., Di Leonardo A., Melfi R., Tutone M., Pace A. and Pibiri I. (2019). Strategies against Nonsense: Oxadiazoles as Translational Readthrough-Inducing Drugs (TRIDs). Int. J. Mol. Sci. 2019, 20, 3329;. [CrossRef]
- Chang, Y.F.; Imam, J. S.; Wilkinson, M. F. (2007). The Nonsense- Mediated Decay RNA Surveillance Pathway. Annu. Rev. Biochem. 2007, 76, 51−74. [CrossRef]
- Dabrowski M., Bukowy-Bieryllo Z., Zietkiewicz E. (2015). Translational readthrough potential of natural termination codons in eucaryotes--The impact of RNA sequence. RNA Biol; 12(9):950-8. PMID: 26176195; PMCID: PMC4615788. [CrossRef]
- Farinha, C.M.; Canato, S. (2017). From the endoplasmic reticulum to the plasma membrane: Mechanisms of CFTR folding and trafficking Cell. Mol. Life Sci. 2017, 74, 39–55. [CrossRef]
- Fernandes P., Miotto B., Saint-Ruf C., Said M., Barra V., Nähse V., Ravera S., Cappelli E., Naim V. FANCD2 modulates the mitochondrial stress response to prevent common fragile site instability. Commun Biol.2021 Jan 29;4(1):127.
- Fritz S. E., Ranganathan S., Wang C. D. , Hogg R. J. (2020). The RNA-binding protein PTBP1 promotes ATPase-dependent dissociation of the RNA helicase UPF1 to protect transcripts from nonsense-mediated mRNA decay, Journal of biological chemistry Volume 295, Issue 33, 14 August 2020, Pages 11613-11625. ISSN 0021-9258.
- Galietta L.J., Haggie P.M., Verkman A.S. (2001). Green fluorescent protein-based halide indicator s with improved chloride and iodide affinities. FEBS Letters, pp 220-24. [CrossRef]
- Hogg L. Goff. S., (2010). Upf1 senses 3′UTR length to potentiate mRNA decay. Cell 2010 Oct 29; 143 (3): 379-389.
- Ivanov, P. V.; Gehring, N. H.; Kunz, J. B.; Hentze, M. W.; Kulozik, A. E. (2008). Interactions between Upf1, ERFs, PABP and the Exon Junction Complex Suggest an Integrated Model for Mammalian NMD Pathways. EMBO J. 2008, 27 (5), 736−747. [CrossRef]
- Karousis, E. D.; Nasif, S.; Mühlemann, O. (2016). Nonsense-Mediated MRNA Decay: Novel Mechanistic Insights and Biological Impact. Wiley Interdiscip Rev RNA 2016, 7 (5), 661−682. doi: 10.1002/wrna.1357.
- Keeling K., Wang D., Dai Y., Murugesan S., Chenna B., Clark J., Belakhov V, Kandasamy J., Velu S., Baasov T., Bedwell DM. (2013). Attenuation of Nonsense- Mediated mRNA Decay Enhances In Vivo Nonsense Suppression, Plos One; 8(4):e60478. [CrossRef]
- Kerem E. (2020). ELX-02: an investigational read-through agent for the treatment of nonsense mutation-related genetic disease. Expert Opin Investig Drugs. Dec;29(12):1347-1354. Epub. 2020 Oct 12. PMID: 32972261. [CrossRef]
- Lentini L., Melfi R., Cancemi P., Pibiri I., Di Leonardo A. (2019). Caffeine boosts Ataluren's readthrough activity. Molecular Biology, Biochemistry, Volume 5, Issue 6 . [CrossRef]
- Lentini L., Melfi R., Di Leonardo A., Spinello A., Barone G., Pace A., Palumbo Piccionello A., Pibiri I. (2014). Toward a rationale for the PTC124 (Ataluren) promoted readthrough of premature stop codons: a computational approach and GFP-reporter cell-based assay. Mol Pharm. 2014 Mar 3;11(3):653-64. Epub 2014 Feb 7. PMID: 24483936; PMCID: PMC4167060. [CrossRef]
- Lou C.H., Dumdie J., Nonsense-Mediated RNA Decay Influences Human Embryonic Stem Cell Fate Stem Cell Reports 2016 Jun 14;6(6):844-857. [CrossRef]
- Loughran G, Chou M-Y, Ivanov IP, Jungreis I, Kellis M, Kiran AM, Baranov PV, Atkins JF. (2014). Evidence of efficient stop codon readthrough in four mammalian genes. Nucleic Acids Res, 42:8928-38. [CrossRef]
- Metze S., Herzog V.A, Ruepp M., and Mühlemann O. Comparison of EJC-enhanced and EJC-independent NMD in human cells reveals two partially redundant degradation pathways. RNA. 2013 Oct; 19(10): 1432–1448. [CrossRef]
- Neu-Yilik, Raimondeau E., et al.Dual function of UPF3B in early and late translation termination EMBO J, 2017 Oct 16;36(20):2968-2986. Epub 2017 Sep 12. [CrossRef]
- Peixeiro I, Inácio Â, Barbosa C, Silva AL, Liebhaber SA, Romão L. (2012). Interaction of PABPC1 with the translation initiation complex is critical to the NMD resistance of AUG-proximal nonsense mutations. Nucleic Acids Res. Feb;40(3):1160-73. Epub 2011 Oct 11. PMID: 21989405; PMCID: PMC3273812. [CrossRef]
- Pibiri I., Lentini L., Melfi R., Gallucci G., Pace A., Spinello A., Barone G., Di Leonardo A. (2015). Enhancement of premature stop codon readthrough in the CFTR gene by Ataluren (PTC124) derivatives. Eur J Med Chem. 2015 Aug 28;101:236-44. Epub 2015 Jun 21. PMID: 26142488. [CrossRef]
- Pibiri I., Lentini L., Melfi R., Tutone M., Baldassano S., Ricco Galluzzo P., Di Leonardo A., Pace A. (2018). Rescuing the CFTR protein function: Introducing 1,3,4-oxadiazoles as translational readthrough inducing drugs. Eur J Med Chem. 2018 Nov 5;159:126-142. Epub 2018 Sep 26. PMID: 30278331. [CrossRef]
- Pibiri I., Lentini L., Tutone M., Melfi R., Pace A., Di Leonardo A. (2016). Exploring the readthrough of nonsense mutations by non-acidic Ataluren analogues selected by ligand-based virtual screening. European Journal of Medicinal Chemistry. [CrossRef]
- Pibiri I., Melfi R., Tutone M., Di Leonardo A., Pace A. and Lentini L. (2020). Targeting Nonsense: Optimization of 1,2,4-Oxadiazole TRIDs to Rescue CFTR Expression and Functionality in Cystic Fibrosis Cell Model Systems Int. J. Mol. Sci. 2020, 21, 6420;. [CrossRef]
- Popp M.W., Maquat L.E. (2018). Nonsense-mediated mRNA Decay and Cancer. Curr Opin Genet Dev. 2018 Feb;48:44-50. Epub 2017 Nov 7. PMID: 29121514; PMCID: PMC5869107. [CrossRef]
- Pranke I., Golec A., Hinzpeter A., Edelman A., Sermet-Gaudelus I. (2019). Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front Pharmacol. 2019 Feb 27;10:121. PMID: 30873022; PMCID: PMC6400831. [CrossRef]
- Quon B.S., Rowe S.M. (2016). New and emerging targeted therapies for cystic. BMJ. Mar 30;352:i859. PMID: 27030675; PMCID: PMC4817245. [CrossRef]
- Rafeeq M.M., Murad H.A.S. (2017). Cystic fibrosis: current therapeutic targets and future approaches. J Transl Med. 2017 Apr 27;15(1):84. PMID: 28449677; PMCID: PMC5408469. [CrossRef]
- Richardson R., Smart M., Tracey-White D., Webster A., Moosajee M. (2017) Mechanism and evidence of nonsense suppression therapy for genetic eye disorders, Experimental Eye Research. [CrossRef]
- Roy B., Friesen W.J., Tomizawa Y., Leszyk J.D., Zhuo J., Johnson B., Dakka J., Trotta C.R., Xue X., Mutyam V., Keeling K.M., Mobley J.A., Rowe S.M., Bedwell D.M., Welch E.M., Jacobson A. (2016). Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc Natl Acad Sci U S A. Nov 1;113(44):12508-12513. Epub 2016 Oct 4. PMID: 27702906; PMCID: PMC5098639. [CrossRef]
- Salvatori F., Pappadà M., Breveglieri G., D'Aversa E., Finotti A., Lampronti I., Gambari R. and Borgatti M. (2018). UPF1 silenced cellular model systems for screening of read-through agents active on ß039 thalassemia point mutation. BMC biotechnology, 18(1), 28.
- Samanta A., Stingl K., Kohl S., Ries J., Linnert J., Nagel-Wolfrum K. (2019). Ataluren for the Treatment of Usher Syndrome 2A Caused by Nonsense Mutations. Int J Mol Sci.Dec 12;20(24). pii: E6274. [CrossRef]
- Sato H., Singer R.(2021) Cellular variability of nonsense-mediated mRNA decay Nat Commun 2021 Dec 10;12(1):7203.
- Sharma N., Evans T. A., Pellicore M. J., Davis E., Aksit, M. A., McCague A. F., Joynt A. T., Lu Z., Han S. T., Anzmann A. F., Lam A. N., Thaxton A., West N., Merlo C., Gottschalk L. B., Raraigh K. S., Sosnay P. R., Cotton C. U. and Cutting G. R. (2018). Capitalizing on the heterogeneous effects of CFTR nonsense and frameshift variants to inform therapeutic strategy for cystic fibrosis. PLoS genetics, 14(11), e1007723.
- Tutone M., Pibiri I., Lentini L., Pace A., Almerico A.M. (2019). Deciphering the Nonsense Readthrough Mechanism of Action of Ataluren: An in Silico Compared Study. ACS Med Chem Lett. 2019 Feb 7;10(4):522-527. PMID: 30996790; PMCID: PMC6466511. [CrossRef]
- Tutone M., Pibiri I., Perriera R., Campofelice A., Culletta G., Melfi R., Pace A., Almerico A.M., Lentini L. (2020). Pharmacophore-Based Design of New Chemical Scaffolds as Translational Readthrough-Inducing Drugs (TRIDs). ACS Med Chem Lett. 2020 Feb 18;11(5):747-753. PMID: 32435380; PMCID: PMC7236267. [CrossRef]
Figure 1.
Schematic representation of the competition mechanism that regulates premature stop codon recognition by the NMD pathway.
Figure 1.
Schematic representation of the competition mechanism that regulates premature stop codon recognition by the NMD pathway.
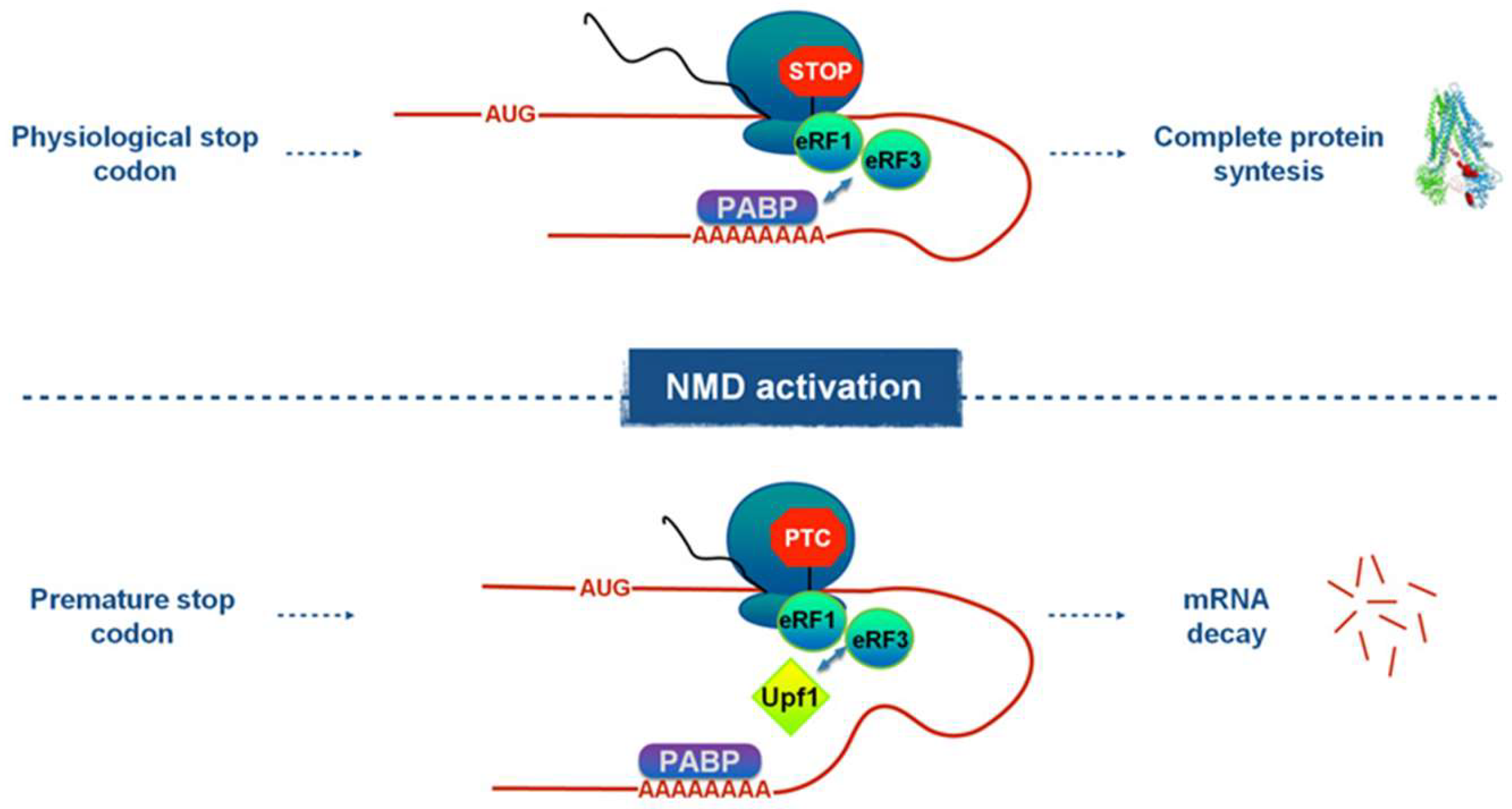
Figure 2.
A) Real-time RT PCR analysis of Upf1 transcripts on FRT-CFTRW1282X cell transfected with siRNA against Upf1 and GFP (non-specific siRNA). B) Real-time RT PCR analysis of CFTR transcripts on FRT CFTRW1282X cells treated with caffeine and siUpf1. Images are representative of two different independent experiments done in triplicate. Data were analyzed by GraphPad Prism 6 software, expressed as mean values ± standard error of the mean (SEM). Symbol (*) represents the statistical significance of treatment with siUpf1 versus treatment with siGFP (p< 0.05); Three symbols (***) represent the statistical significance of caffeine treatment versus siGFP treated sample (p < 0.005) (Unpaired t-test).
Figure 2.
A) Real-time RT PCR analysis of Upf1 transcripts on FRT-CFTRW1282X cell transfected with siRNA against Upf1 and GFP (non-specific siRNA). B) Real-time RT PCR analysis of CFTR transcripts on FRT CFTRW1282X cells treated with caffeine and siUpf1. Images are representative of two different independent experiments done in triplicate. Data were analyzed by GraphPad Prism 6 software, expressed as mean values ± standard error of the mean (SEM). Symbol (*) represents the statistical significance of treatment with siUpf1 versus treatment with siGFP (p< 0.05); Three symbols (***) represent the statistical significance of caffeine treatment versus siGFP treated sample (p < 0.005) (Unpaired t-test).
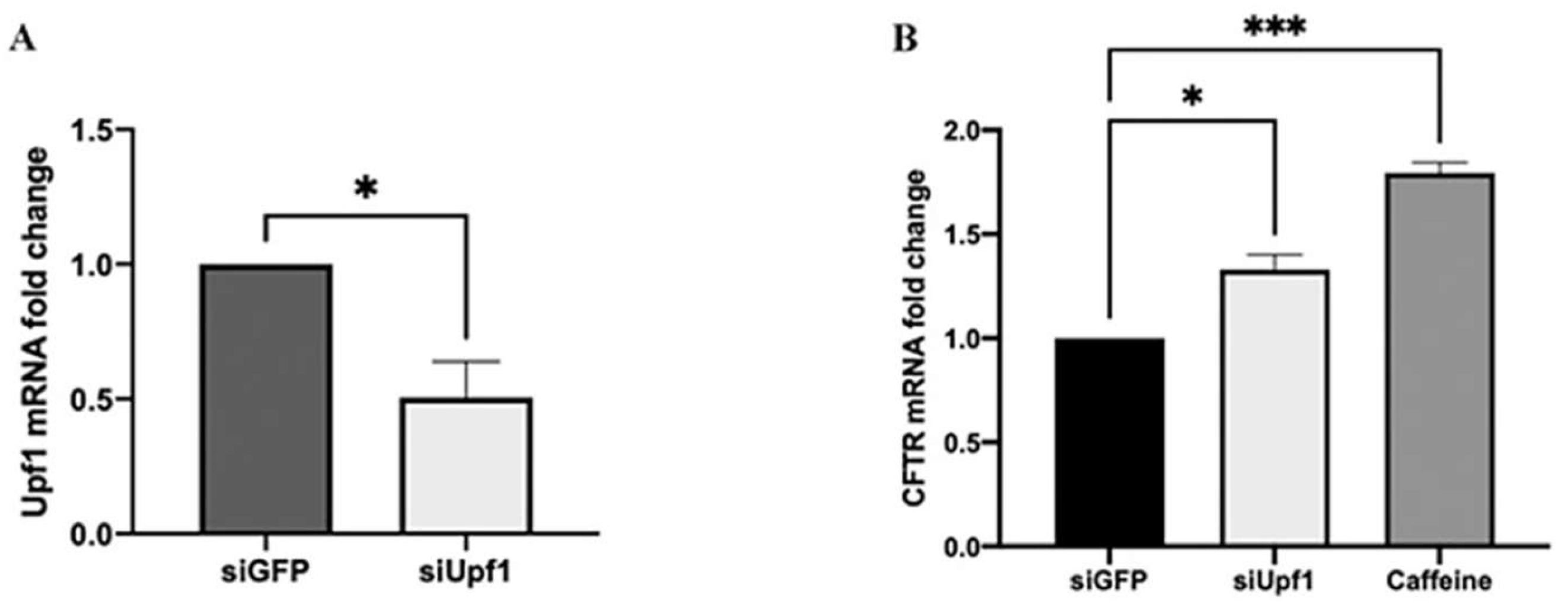
Figure 3.
Immunofluorescence analysis of CFTR protein on FRT pTRACER-CFTRWT (positive control) FRT CFTRW1282X untreated (negative control) or treated as indicated. Images are representative of three independent experiments and Quantification of CFTR expression was done manually by using ImageJ software. The background was subtracted and the integrated signal intensity in the selected area (one cell) was measured. Data were analyzed by GraphPad Prism 6 software, expressed as mean values ± standard error of the mean (SEM). Two symbols (**) represent the statistical significance of treatment with siUpf1 and NV914 versus treatment with only NV914 (p< 0.005); Three symbols (***) represent the statistical significance of treatment with siUpf1 and NV848 versus treatment with only NV848 (p< 0.0005)(Unpaired t-test).
Figure 3.
Immunofluorescence analysis of CFTR protein on FRT pTRACER-CFTRWT (positive control) FRT CFTRW1282X untreated (negative control) or treated as indicated. Images are representative of three independent experiments and Quantification of CFTR expression was done manually by using ImageJ software. The background was subtracted and the integrated signal intensity in the selected area (one cell) was measured. Data were analyzed by GraphPad Prism 6 software, expressed as mean values ± standard error of the mean (SEM). Two symbols (**) represent the statistical significance of treatment with siUpf1 and NV914 versus treatment with only NV914 (p< 0.005); Three symbols (***) represent the statistical significance of treatment with siUpf1 and NV848 versus treatment with only NV848 (p< 0.0005)(Unpaired t-test).
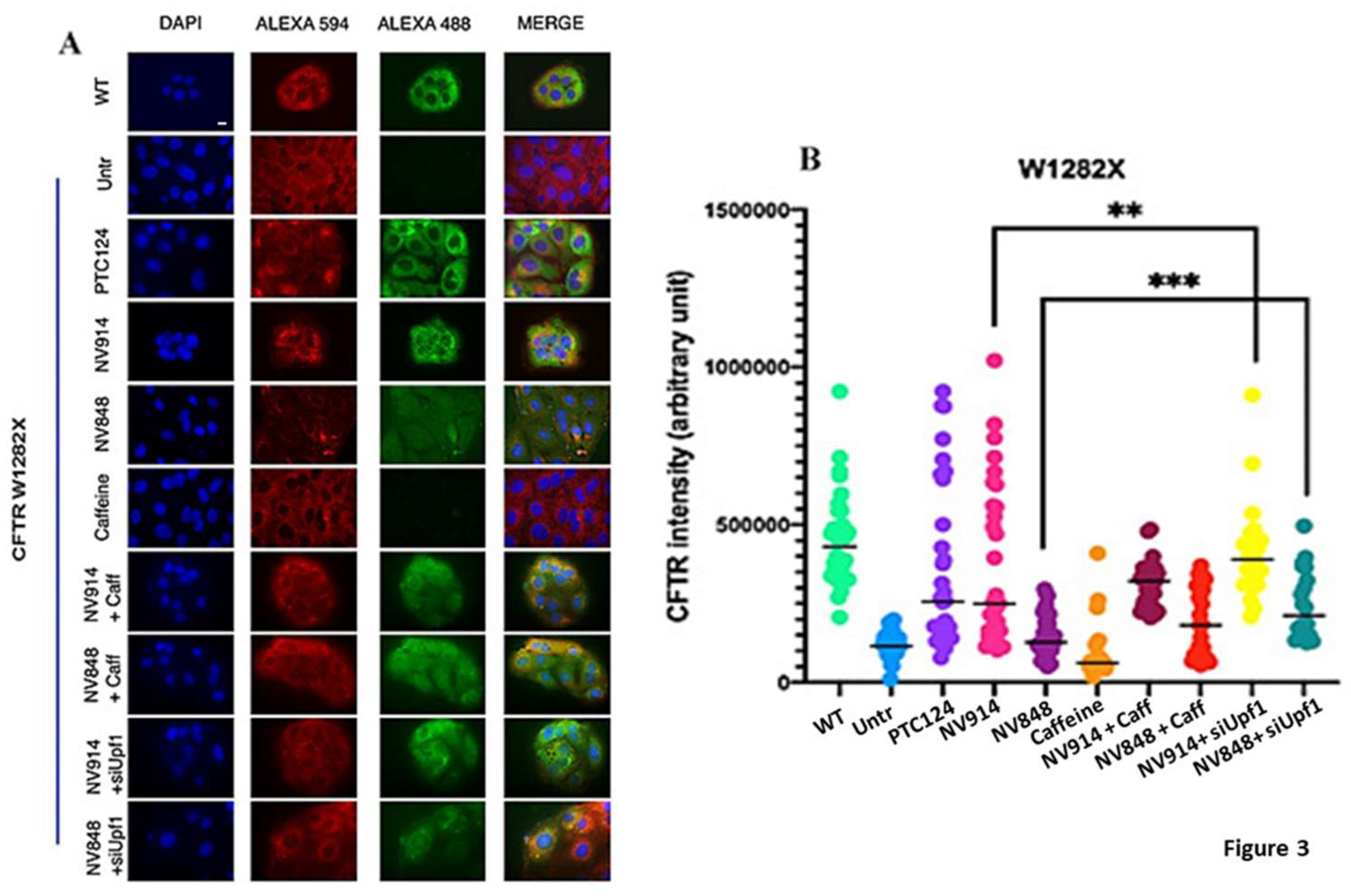
Figure 4.
Western blot analysis of CFTR and Upf1 proteins on FRT pTRACER-CFTRWT (positive control) FRT CFTRW1282X untreated (negative control) or treated as indicated. Images are representative of two independent experiments; β-tubulin was used as a control for the protein loading. The experiment was performed in duplicate and data was normalized by β-tubulin expression, and analyzed by GraphPad Prism 6 software. Data are expressed as mean values ± standard error of the mean (SEM). Symbols (*) represent the statistical significance of treatment with siUpf1 and NV914/NV848 versus treatment with only NV914/NV848 (p< 0.05). Two symbols (**), p < 0.005 (Unpaired t-test).
Figure 4.
Western blot analysis of CFTR and Upf1 proteins on FRT pTRACER-CFTRWT (positive control) FRT CFTRW1282X untreated (negative control) or treated as indicated. Images are representative of two independent experiments; β-tubulin was used as a control for the protein loading. The experiment was performed in duplicate and data was normalized by β-tubulin expression, and analyzed by GraphPad Prism 6 software. Data are expressed as mean values ± standard error of the mean (SEM). Symbols (*) represent the statistical significance of treatment with siUpf1 and NV914/NV848 versus treatment with only NV914/NV848 (p< 0.05). Two symbols (**), p < 0.005 (Unpaired t-test).
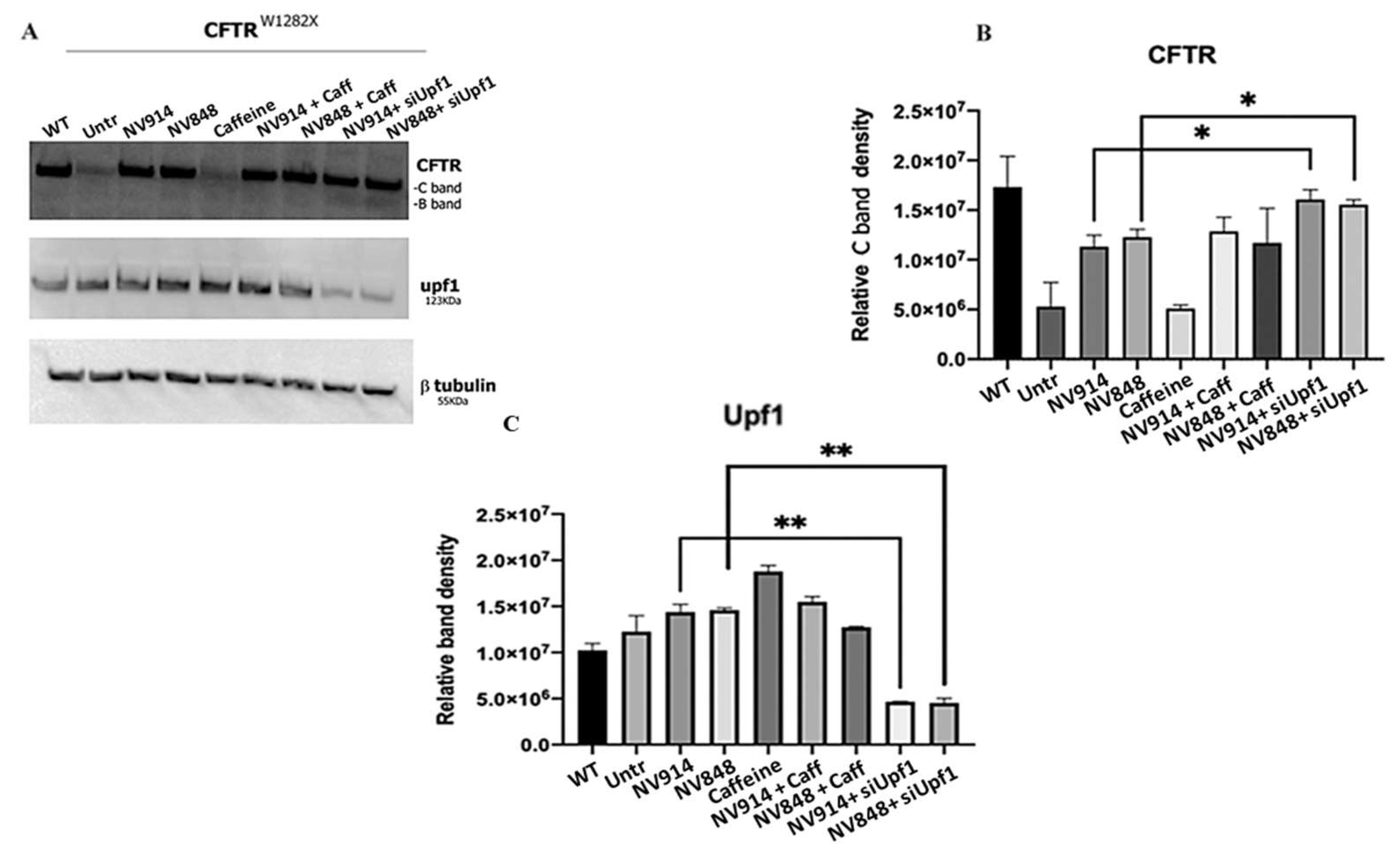
Figure 5.
CFTR functionality in FRT cells measured by the yellow fluorescent protein (YFP) assay on FRT pTRACER-CFTRWT (positive control) FRT CFTRW1282X untreated (negative control) or treated as indicated. Images were taken before (t0) and after (t60, t120) the addition of NaI (A). Percentages of YFP-positive cells before (− dark green bars) and after NaI addition (+ blue bars); data were analyzed by GraphPad (n = 3) and expressed as mean values ± standard error of the mean (SEM) (B Data are expressed as mean values ± standard error of the mean (SEM) and were analyzed by GraphPad Prism 6 software (n = 2) comparing treatment with siUpf1 and NV914/NV848 versus treatment with only NV914/NV848. Two symbols (**) represent statistical significance (p < 0.005) (Unpaired t-test).
Figure 5.
CFTR functionality in FRT cells measured by the yellow fluorescent protein (YFP) assay on FRT pTRACER-CFTRWT (positive control) FRT CFTRW1282X untreated (negative control) or treated as indicated. Images were taken before (t0) and after (t60, t120) the addition of NaI (A). Percentages of YFP-positive cells before (− dark green bars) and after NaI addition (+ blue bars); data were analyzed by GraphPad (n = 3) and expressed as mean values ± standard error of the mean (SEM) (B Data are expressed as mean values ± standard error of the mean (SEM) and were analyzed by GraphPad Prism 6 software (n = 2) comparing treatment with siUpf1 and NV914/NV848 versus treatment with only NV914/NV848. Two symbols (**) represent statistical significance (p < 0.005) (Unpaired t-test).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



