
Submitted:
28 June 2024
Posted:
01 July 2024
You are already at the latest version
Abstract
Glucocorticoids (GCs) are widely used in blood cancer treatment despite their multiple adverse effects. Biological response to GCs is mediated by glucocorticoid receptor (GR) by transactivation (TA), associated with side effects, and transrepression (TR), mediating anticancer effects. Selective GR agonists (SEGRAs) preferentially activating GR TR could be better option for cancer treatment. One of well-characterized SEGRAs is 2-(4-acetoxyphenyl)-2-chloro-N-methylethylammonium-chloride (CpdA), demonstrated anticancer activity in vitro and in vivo, however its translational potential is limited due to chemical instability. CpdA derivatives CpdA-01-08 were designed by two synthetic strategies. Derivative CpdA-03 demonstrated superior GR affinity and stability compared to CpdA. Analysis of CpdA-03 ligand properties in lymphoma Granta and leukemia CEM cell lines revealed its SEGRA profile: it induced GR TR without triggering GR TA, as evidenced by changes in expression of GR target genes. Effects of CpdA-03 on cell viability, growth, and apoptosis were similar to reference GR ligand, dexamethasone (Dex), and the proto-type compound CpdA. In vivo testing of CpdA-03 anti-lymphoma activity in transplantable P388 murine lymphoma model showed that CpdA-03 reduced tumor volume threefold, outperforming Dex and CpdA. In conclusion, this work introduces CpdA-03 as promising new SEGRA with potential for further development as anti-lymphoma drug with fewer side effects.
Keywords:
selective glucocorticoid receptor agonists
; transrepression
; transactivation
; CpdA-03
; anti-lymphoma activity
; mice
# These authors contributed equally to this work.
1. Introduction
Glucocorticoids (GCs) are essential regulators of many physiological processes including homeostasis, embryonic and post-embryonic development as well as stress response and modulation of immunity [1]. Specific cytotoxic effects of GCs on a number of immune cells are responsible for their usage in the treatment of hematological malignancies [2]. Combined GC-based therapies remain the mainstay treatment of blood cancers for the last decades, however, their long-term therapeutic administration is limited by major debilitating side effects: skin and muscle atrophy, osteoporosis, Cushing syndrome and steroid-induced diabetes [3,4,5,6,7,8].
Pleiotropic effects of GCs are realized through the glucocorticoid receptor (GR), a well-known transcription factor. In the absence of ligand, GR is located in cytoplasm in multi-chaperone complex. Upon GC binding, GR can regulate the transcription of target genes through: (i) GC-responsive element (GRE)-mediated direct transactivation (TA), or (ii) transrepression (TR) of transcriptional activators, such as NFκB, AP1, and STAT3 [9,10,11,12]. Therapeutic anti-cancer and anti-inflammatory activity of GCs is mostly associated with GR TR, whereas GC-induced metabolic complications are mediated via GR TA. Selective GR agonists (SEGRAs) that shift GR function towards TR appear to be a safer option for treatment of hematological malignancies. Several SEGRAs were discovered, synthesized and characterized during the past two decades: mapracorat [13], CpdA [14], CpdX [12,15,16], ZK 216348 [17], AL-438 [18], Org 214007–0 [19], and AZD9567 [20]. Some of them entered the clinical trials as anti-inflammatory agents with reduced side effects [21,22], however, none of them reached clinical practice, in particular, cancer treatment.
2-(4-acetoxyphenyl)-2-chloro-N-methylethylammonium-chloride also called Compound A (CpdA) is a synthetic analogue of hydroxyphenyl aziridine precursor isolated from the Namibian shrub Salsola tuberculatiformis Botschantzev [23]. It was demonstrated that CpdA competes with GCs for GR binding, does not induce GR TA but activates GR TR, reveals anti-cancer and anti-inflammatory effects in different models in vitro and in vivo [8,14,24] and, in contrast to GCs, has fewer side effects related to maintenance of hypothalamic-pituitary-adrenal (HPA) axis, and bone metabolism [8,24]. However, it was also shown that in water solution CpdA decomposes to azidirine derivative [25] referred as 2B carcinogen according to the classification of International Agency for Research on Cancer (IARC) [26]. Stable CpdA analogues have not been described.
Here we report the synthesis of eight CpdA chemical derivatives and evaluation of their and putative SEGRA cytotoxic effects. Two alternative synthetic strategies were applied to obtain the test compounds. One of the new compounds, Cpda-03, revealed the most prominent cytotoxic effects in vitro, demonstrating SEGRA properties and anti-lymphoma activity in vitro and in vivo.
2. Results
2.1. Synthesis of CpdA Chemical Derivatives
CpdA derivatives synthesized and tested here as well as template molecule, CpdA, are the analogues of synephrine, natural alkaloid from citrus fruits, widely used for weight loss/weight management, sports performance, appetite control, energy, and mental focus and cognition [27,28]. An efficient method based on the use of commercially available compounds has been proposed for the synthesis of analogs of CpdA (CpdA-01-04, Figure 1). To minimize the content of impurities, first, heavy metal impurities, all stages of this synthesis were optimized. The second synthetic protocol (the synthesis of CpdA-05-08, Figure 2) is based on an approach where the key step is the preparation of amino alcohols by the reaction of azomethine ylide with aromatic aldehydes [29]. However, in order to improve the yields of the target compounds and eliminate additional purification at the last stage, each step of this synthetic protocol was optimized.
2.2. Screening of Cytotoxic Effects of Newly Synthesized CpdA Derivatives
Screening for cytotoxicity was performed for eight CpdA derivatives in three cell lines originated from hematological malignancies: acute lymphoblastic leukemia CCRF-CEM (CEM) cells, chronic myeloid leukemia K562 cells and mantle cell lymphoma Granta-519 (Granta). All three cell lines differ in sensitivity to tested compound including active comparators Dex and CpdA. Specifically, cytotoxicity of CpdA derivatives was higher in CEM and Granta cells characterized earlier by the high GR expression level (Table 1). K562 cells were less sensitive and, therefore, were used further only in ligand binding assay to keep the maximal amount of GR for competitive binding. Among the novel CpdA derivatives, CpdA-03 was the most cytotoxic. Due to the multiplicity of tests in our study, we performed the further experiments with CpdA-03.
2.3. Affinity of CpdA-03 to GR
CpdA-03 was tested in radioligand binding assay for the affinity to GR. For this purpose, CpdA-03 was tested at concentration range of 0.001 - 10 uM. We have demonstrated that binding IC50 for reference GR ligands Dex and CpdA were 0.23 uM and 2.5 uM, respectively, which is in accordance with literature data [30,31,32] and proved the accuracy of the assay used. CpdA-03 showed a relative binding affinity to the GR with the IC50 values of 0.15 uM (Figure 3). These data demonstrated that CpdA-03 affinity was higher than original compound and, therefore, worth to be further investigated.
2.4. Anti-Cancer Effect of CpdA-03 In Vitro and In Vivo
To evaluate anti-cancer potential of CpdA-03, we studied CpdA-03 effect on the proliferation of CEM and Granta cells in comparison with template compound CpdA and reference GC Dex. CpdA and Dex were used in the concentration of 1 uM, which we tested in the previous work [24,33]. Growth inhibitory effect of CpdA-03 first was studied at wide concentration range of 10 nM-10 µM in three cell lines (Table 1). We have chosen the concentration of 1 uM for CpdA-03 as close to IC50 and proved that CpdA-03 exerted significant cytotoxic effects at similar 1 uM concentrations (Figure 4B). Importantly, GR blockage by shGR-expressing lentivirus (Figure 4A), resulted in a drastic loss of sensitivity to CpdA-03 as well as CpdA and Dex in both cell lines (Figure 4B). These data provide an additional proof that CpdA-03 is GR ligand and its anti-lymphoma effects are realized via GR activation.
Next, we examined CpdA-03 effect on cell survival and demonstrated that this CpdA derivative induced apoptosis in both cell lines. More specific, flow cytometry analysis revealed that the number of apoptotic cells (sub-G1-phase cell population) reached 15-17% after 48 h incubation with CpdA-03 compared to 2-4% in control (Figure 4C). These results were comparable with the level of apoptosis induction caused by CpdA and Dex (Figure 4C).
For the experiment in vivo we used P388 murine lymphoma model, which could grow both subcutaneously and intraperitoneally, and was described in previous studies for the evaluation of GC anti-cancer effect in comparison with different cytostatic drugs [34,35]. We have chosen subcutaneous grafting of P388 cells to make possible the comparison of tumor growth in dynamics in groups treated with Dex (1 mg/kg), CpdA (7,5 mg/kg), CpdA-03 (7,5 mg/kg) and vehicle (5% DMSO in PBS). Dex and CpdA doses were selected previously in the studies of our and some other research groups [8,36]. I.p. injections of CpdA, Dex and CpdA-03 were started on the 11th day after P388 implantation (after the formation of the first tumor nodules). Comparative analysis of tumor volume on the 12th day of injection (the 23rd day of experiment) demonstrated the statistically significant tumor growth inhibition by 57% in the group treated with Dex, by 66% in the group treated with CpdA and by 78% in the group treated with CpdA-03 (Figure 4D).
2.5. Effects of CpdA-03 on GR Functions
To assess the effect of CpdA-03 on GR TA function, we used CEM and Granta cells transduced with GRE.Luc reporter, Dex and CpdA were used for comparative analysis. CpdA-03 as well as CpdA and in contrast with Dex did not induced GRE.Luciferase activity in both cell lines (Figure 5A). Attenuation of GR TA was further confirmed by Q-PCR analysis of known GR-target genes including GR chaperone FKBP51 [24,37] and mediator of GC anti-inflammatory effects GILZ [38,39] (Figure 5B) as well as by Western blot analysis of the amount of phosphorylated GR (Ser211) in cells (Figure 5C).
GR TR function was analyzed using NF-kB.Luc reporter assay. NF-kB activity was strongly reduced by CpdA-03 according to NF-kB.Luciferase test. Dex and CpdA also inhibited the Luciferase activity (Figure 6A), which is in accordance with our previous papers [8,24,33]. This finding was further corroborated by Q-PCR analysis of negative regulation of several endogenous genes such as cell cycle regulators cyclins CCND1/D2 [40,41,42] in CEM cells, and interleukins IL1 and IL6 involved in lymphoid cell survival for Granta cell line (Figure 6B).
3. Discussion
It should be mentioned that identification of novel methods that decrease GC-induced side effects is a critically important clinical problem. There are two major approaches to attenuate GR TA induction and decrease the development of metabolic and atrophic complications: (i) combination of GCs with the compounds protecting tissues against steroid adverse effects [37,42,43], (ii) development of SEGRAs that shift GR activity towards TR [12,13,14,15,16,18,19,20,44].
Previous studies of our research group was focused on the anti-cancer effects of Compound A (CpdA), a unique non-steroidal SEGRA, which is a stable synthetic analogue of a compound found in the Namibian shrub, Salsola tuberculatiformis Botschantzev [23]. However, CpdA is very labile and decomposes in aqueous medium to mutagen aziridine and then to polyphenol synephrine [25]. In spite of this fact that CpdA can be stabilized by binding to plasma proteins and remain biologically active in vivo [25], decomposition can occur during storage and preparation of medication, and, therefore, affect the activity of the compound.
Here we proposed to modify CpdA molecule in such a manner that allowed to increase the stability of the compound but reserve the relative negative electron charges facilitating the formation of H-bonds with several amino acids lining these binding cavities of GR (Asn564 and Arg611) [31]. An advantage of the developed approach to the synthesis of CpdA analogues (CpdA-01-04) is the use of commercially available compounds and the exclusion of the use of metal-containing catalysts and toxic solvents.
The most prospective compound from the CpdA modification list, CpdA-03, is more inert and stable analogue of alkaloid synephrine, unable to form the aziridine derivative because of the absence of proton at the nitrogen atom. CpdA-04, the chemical derivative of CpdA with less affinity to GR, is more hydrophilic synephrine analogue. CpdA-01, CpdA-02, and CpdA-05-08 are characterized by a large number of substituting groups, which could impede the binding of the molecule with Asn564 and Arg611 in GR active center.
CpdA-03 revealed anti-cancer activity in vitro and in vivo comparable with Dex and CpdA (Figure 4, [8,24]). In addition, CpdA-03 was less likely to induce therapy resistance, as it did not activate FKBP51, the direct GR target gene [45] involved in feedback control of GR signaling via retention of GR in the cytoplasm and associated with GC resistance in patients with brain and prostate cancer, melanomas and lymphomas [46,47].
In conclusion, novel SEGRA, 4-(1-hydroxy-2-(piperidin-1-yl)ethyl)phenol or CpdA-03, retains the therapeutic anti-cancer potential of GCs and, more important, of template compound CpdA, along with abrogation of GR TA and, therefore, potential decrease in the induction of side effects and therapy resistance. Taken together with the data of molecule stability, this makes CpdA-03 a very attractive candidate for further investigations of future therapeutic applications in the treatment of hematological malignancies.
4. Materials and Methods
4.1. Chemistry
4.1.1. General Information
The structures of compounds were established using 1D NMR (1H, 13C) spectroscopy (for spectra see Supplementary Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7 and Figure 8). NMR spectra were acquired on Bruker 300 spectrometers at 293; the chemical shifts were measured in ppm relative to the solvent (1H: DMSO-d6, d 2.50 ppm; 13C: DMSO-d6, d 39.50 ppm). Splitting patterns are designated as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, double doublet; ddd, double double doublet; dt, doublet triplet. The coupling constants (J) are in Hertz. High-resolution mass spectra (HRMS) were measured using electrospray ionization (ESI) in positive ion mode (interface capillary voltage 4,500V); the mass range was from m/z 50 to 3,000 Da; external/internal calibration was performed using an electrospray calibrant solution. A syringe injection was used for solutions in CH3CN (flow rate 3 ml/min). Melting points were measured on a Boetius capillary melting point apparatus and are uncorrected. Analytical thin-layer chromatography (TLC) was carried out on silica gel plates (silica gel 60 F254 aluminum supported plates); the visualization was accomplished with an UV lamp (254/365 nm). All chemicals and anhydrous solvents were purchased from commercial sources and used without further purification. Silica column chromatography was performed using silica gel 60 (70-230 mesh); TLC analysis was conducted on silica gel 60 F254 plates.
4.1.2. Synthesis and Characterization of CpdA-01 – CpdA-04
The amino alcohol 2 was obtained by reduction ketone 1 with sodium borohydride (Figure 1). The target compound 3 was obtained by reflux previously synthesized amino alcohol in a stream of dry hydrogen chloride in absolute diethyl ether. The characteristics of piperidine analogues of CpdA are summarized in Table 2.
Stage 1. To a solution of aminoketone 1 (3.0 mmol) cooled to 10°С in abs. methanol (30 ml) was added portionwise sodium borohydride (30 mmol) and the solution was stirred at room temperature for 5 hours. The reaction mixture was poured into water (150 ml), extracted with ethyl acetate (3 × 30 ml), the extract was washed with water (50 ml) and evaporated. The residue was purified by chromatography (petroleum ether/EtOAc/Et3N 1:1:0.05), the diastereomers were not separated. The residue was recrystallized from methylene chloride-heptane mixture.
Stage 2. Through a solution of amino alcohol 2 (3.0 mmol) in abs. diethyl ether (60 ml), dry hydrogen chloride was passed with stirring, after which the reaction mixture was refluxed for 20 hours. After cooling the reaction mixture, the precipitated white precipitate was filtered off and thoroughly washed with absolute diethyl ether. The residue was dried under vacuum at room temperature.
4.1.3. Synthesis and Characterization of CpdA-05 – CpdA-08
These analogues of the CpdA were prepared according previously reported method [48] from corresponding aromatic aldehydes (Figure 2). The characteristics of CpdA-05 – CpdA-08 are summarized in Table 2.
A mixture of the corresponding aromatic aldehyde (1.0 mmol), finely ground sarcosine (0.13 g, 1.5 mmol), and paraformaldehyde (0.09 g, 3.0 mmol) was refluxed in dry benzene (3.3 mL), with magnetic stirring and removal of formed water by means of a Dean-Stark trap, for 6-8 h. The resulting solution was evaporated in vacuo to give the oily 5-aryl-3-methyloxazolidines. The oxazolidine 5 was dissolved in MeOH (1 mL) and treated with concentrated HCl (0.10 mL, 1.2 mmol). The resulting mixture was refluxed in a fume hood with partial evaporation of the solvent for 1.5 h (for the removing of dimethoxymethane). The MeOH was evaporated in vacuo and H2O (0.5 mL) was added. The mixture was extracted with Et2O (2 × 1 mL) followed by basification with an excess of a cold concentrated solution of NaOH. Extraction with CH2Cl2 (2 × 2 mL), drying over Na2SO4, and evaporation gave the crude 1-aryl-2-(methylamino)ethanol, which was recrystallized from CH2Cl2-heptane mixture. Ammonium salts 7a,b were obtained as described above.
4.2. Biology
4.2.1. Cell Cultures and Treatments
Acute lymphoblastic leukemia cell line CCRF-CEM, chronic myeloid leukemia cell line K562 and mantle cell lymphoma cell line Granta-519 (hereinafter – CEM, K562, and Granta) were obtained from ATCC and cultured as described [24]. Cells were treated with Dexamethasone (Dex), CpdA, CpdA-01-08 or vehicle (0.01% DMSO) for 24 h.
4.2.2. MTT Cytotoxicity Assay
Cell cultures were seeded into 96-well plates and incubated overnight under 5% CO2 at 37°C for 24 h. Then cells were treated with Dex, CpdA, CpdA-01-08 (0.1–100 μM) or vehicle for 24 h. Then, 20 μL MTT solution was added to each well and mixed. After 4 h, the supernatants were removed and 100 μL DMSO was added to each well to dissolve the precipitate. The cells viability was estimated by measuring absorbance at 570 nm using the MultiScan MCC 340 spectrophotometer (Thermo Fisher, Waltham, MA, USA). The IC50 values of on various cell lines were determined using GraphPad Prizm software, Ver.7.0. Results are presented as the mean ± standard deviation (SD).
4.2.3. Western Blot Analysis
Western blot analysis was performed as described previously [24,49]. Proteins were resolved by SDS-PAGE. Membranes were blocked with 5% nonfat milk solution and then incubated with primary antibodies against GR (H-300) and phosphor-GR (pGR) (Santa Cruz Biotechnology) overnight at 4°C. To verify equal protein loading and adequate transfer, the membranes were probed with GAPDH antibodies (Cell Signaling). Goat anti-rabbit IgGs (Jackson Immuno Research) were used as secondary antibodies. Signals were detected using the ECL reagent and an ImageQuant LAS4000 system (GE HealthCare). ImageJ software (NIH) was used for densitometry.
4.2.4. Cell Viability
Cell viability was measured by direct cell counting. Cells were plated at 104 cells/well onto 24-well plates and cultured in complete medium in the presence of CpdA, CpdA-03, Dex or vehicle for 24 h.
4.2.5. Distribution of Cells by Cell Cycle Phases
Distribution of cells by cell cycle phases was evaluated using propidium iodide (PI) staining as described in [24]. Cells were resuspended in 70% ethanol, fixed for 2h at 4°C, placed in PBS containing 50 mg/mL PI, 0.1% sodium citrate and 0.3% NP-40 and incubated for 30 min at room temperature. Analysis by FACScan flow cytometer (Becton Dickinson) was carried out to discriminate between live and apoptotic cells.
4.2.6. RNA Extraction and Q-PCR
4.2.7. Lentiviral Technology
Lentiviral stocks were generated using lentiviral expression vectors obtained from Northwestern University SDRC DNA/RNA delivery Core as described previously [24,49]: pGIPZ encoding short hairpin RNA (shRNA) against GR or empty vector as control (Open Biosystems); NF-kB.Luc and GRE.Luc encoding Firefly Luciferase reporters or Firefly Luciferase under minimal CMV promoter as control (System Biosciences). Lymphoma cells lines stably infected with lentiviruses were selected 0.5 ug/ml puromycin.
4.2.8. Luciferase Assay
Cells (104 cells/well in 24well plate), stably expressing Firefly Luciferase under NF-kB or GRE promoters, were treated for 24h with CpdA, CpdA-03, Dex or vehicle. Luciferase activity was measured as described [42,43] using commercial Luciferase Assay (Promega) and Luminometer TD 20/20 (Turner Design Instruments). Cells stably expressing Firefly Luciferase under minimal CMV promoter were used as a control to adjust for non-specific toxicity. The results were normalized by total protein amount in each sample.
4.2.9. GR Binding Affinity Assay
The K562 cells (2.5 million cells/ml) were cultured in a serum-free culture medium in the presence of 0.5 uM [3H]Dex and unlabeled Dex, CpdA or CpdA-03 in various concentrations. After 90 minutes of incubation, cells were washed twice by centrifugation in a cold serum-free medium (1500 rpm, 5 min). Then cells were lysed with RIPA buffer (50 mM Tris (pH 8.0) 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1% triton-X100, 10% glycerin, protease inhibitor) for 20 min. Lysates were transferred to a scintillation liquid, and radioactivity was measured by a RackBeta 1215 LC spectrometer for 4 minutes. The relative binding percentages of Dex, CpdA, and CpdA-03 were measured. IC50 was determined by using GraphPad Prizm software, Ver.7.0. Measured values represented in dpm (decay per minute) of different concentrations of Dex were calculated using a method of non-linear regression (curve fit) with equation - [Inhibitor] vs response (three parameters: «top», «bottom», IC50). «Top» meant the highest value in dpm and was equal to 100%, while the «bottom» was considered as the lowest value (0%). Dpm for CpdA and CpdA-03 compounds were calculated in the same way using the «bottom» of Dex in the individual biological repeat. These calculations led to plotting curves, the most precisely fit the pattern of competitive inhibition by a ligand of binding [3H]Dex to GR. The IC50 values for each ligand were determined based on the plotting of these curves.
4.2.10. Anti-cancer Study In Vivo
The protocols for the experiments involving mice were approved by the Animal Care and Use Committee of the N.N. Blokhin National Research Center of Oncology and corresponded to the guidelines for the welfare and use of animals in cancer research adopted by The United Kingdom Coordinating Committee on Cancer Prevention Research [50]. P388 cells were injected i.p. in 5 weeks old female DBA/2 mice (Stolbovaya Farm, Moscow region, Russia, http://www.scbmt.ru). After 15 days, cells were collected from the peritoneum, washed and resuspended in PBS. For experiments, 0.1 ml containing 2 million cells obtained from the ascites was inoculated s.c. in BDF1 mice (Stolbovaya Farm, Moscow region, Russia, http://www.scbmt.ru). Mice were randomly divided into four groups, 10 animals per group, and treated i.p. 3 times/week with Dex (1 mg/kg), CpdA (7.5 mg/kg), CpdA-03 (7.5 mg/kg) of vehicle (1% DMSO in PBS). Tumor size was measured twice a week by digital calipers.
4.2.11. Statistical Analysis
Mean and standard deviation values were calculated using Microsoft Excel and GraphPad Prizm (Ver.7.0) software. The treatment effects in each experiment were compared by one-way ANOVA or t-test. Differences between groups were considered significant at p<0.05. All experiments were repeated three times. In animal experiments, we used 10 animals/experimental group.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1,. Figure S1: Copy of 1H NMR spectra of 1-(2-chloro-2-(4-methoxyphenyl) ethyl)piperidin-1-ium chloride (CpdA-01); Figure S2: Copy of 1H NMR spectra of 1-(4-methoxyphenyl)-2-(piperidin-1-yl)ethanol (CpdA-02); Figure S3: Copy of 1H NMR spectra of 4-(1-hydroxy-2-(piperidin-1-yl)ethyl)phenol (CpdA-03); Figure S4: Copy of 1H NMR spectra of 1-(2-chloro-2-(4-hydroxyphenyl)ethyl)piperidin-1-ium chloride (CpdA-04); Figure S5: Copy of 1H NMR spectra of 1-(4-methoxyphenyl)-2-(methylamino)ethanol (CpdA-05); Figure S6: Copy of 1H NMR spectra of 1-(4-methoxyphenyl)-2-(methylamino)ethanol (CpdA-06); Figure S7: Copy of 1H NMR spectra of 1-(3,4-dimethoxyphenyl)-2-(methylamino)ethanol (CpdA-07); Figure S8: Copy of 1H NMR spectra of 2-chloro-2-(3,4-dimethoxyphenyl)-N-methylethanaminium chloride (CpdA-08); Table S1: Primer sets for Q-PCR analysis.
Author Contributions
Conceptualization, E.M.Z., I.V.B., V.Z.S., E.P.K., M.G.Y., E.A.L.; investigation, E.M.Z., T.I.F., L.R.T., K.I.K., G.A.B., M.G.C., V.Z.S., A.D.E.; writing—original draft preparation, E.M.Z., K.I.K., V.Z.S., M.G.Y., E.A.L.; writing—review and editing, E.M.Z., I.V.B., E.P.K., M.G.Y., E.A.L.; visualization, E.M.Z., L.R.T., A.D.E., E.A.L.; supervision, E.A.L.; project administration, E.M.Z., E.A.L.; funding acquisition, E.A.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation, grant number 23-15-00321 (to E.L.).
Institutional Review Board Statement
The animal study protocol was approved by Local N.N. Blokhin National Medical Research Center Ethical committee on 19.11.2018, approval number 2018-37.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sacta, M.A.; Chinenov, Y.; Rogatsky, I. Glucocorticoid Signaling: An Update from a Genomic Perspective. Annu. Rev. Physiol. 2016, 78, 155–180. [Google Scholar] [CrossRef] [PubMed]
- Gulliver, L.S.M. Xenobiotics and the Glucocorticoid Receptor. Toxicol. Appl. Pharmacol. 2017, 319, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Zhidkova, E.M.; Lylova, E.S.; Grigoreva, D.D.; Kirsanov, K.I.; Osipova, A.V.; Kulikov, E.P.; Mertsalov, S.A.; Belitsky, G.A.; Budunova, I.; Yakubovskaya, M.G.; et al. Nutritional Sensor REDD1 in Cancer and Inflammation: Friend or Foe? Int. J. Mol. Sci. 2022, 23, 9686. [Google Scholar] [CrossRef] [PubMed]
- Lesovaya, E.A.; Chudakova, D.; Baida, G.; Zhidkova, E.M.; Kirsanov, K.I.; Yakubovskaya, M.G.; Budunova, I.V. The Long Winding Road to the Safer Glucocorticoid Receptor (GR) Targeting Therapies. Oncotarget 2022, 13, 408–424. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, M.C.; Sauders, R.H.; Schwartz, L.I.; Zannos, L.; Perez Santiago, E.; Dameshek, W. The Use of Adrenocorticotropic Hormone and Cortisone in the Treatment of Leukemia and Leukosarcoma. Blood 1951, 6, 804–823. [Google Scholar] [CrossRef] [PubMed]
- Hachemi, Y.; Rapp, A.E.; Picke, A.-K.; Weidinger, G.; Ignatius, A.; Tuckermann, J. Molecular Mechanisms of Glucocorticoids on Skeleton and Bone Regeneration after Fracture. J. Mol. Endocrinol. 2018, 61, R75–R90. [Google Scholar] [CrossRef] [PubMed]
- Conaway, H.H.; Henning, P.; Lie, A.; Tuckermann, J.; Lerner, U.H. Activation of Dimeric Glucocorticoid Receptors in Osteoclast Progenitors Potentiates RANKL Induced Mature Osteoclast Bone Resorbing Activity. Bone 2016, 93, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Lesovaya, E.; Yemelyanov, A.; Swart, A.C.; Swart, P.; Haegeman, G.; Budunova, I. Discovery of Compound A--a Selective Activator of the Glucocorticoid Receptor with Anti-Inflammatory and Anti-Cancer Activity. Oncotarget 2015, 6, 30730–30744. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Wahli, W. A Trilogy of Glucocorticoid Receptor Actions. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 1115–1117. [Google Scholar] [CrossRef] [PubMed]
- Ratman, D.; Vanden Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; De Bosscher, K. How Glucocorticoid Receptors Modulate the Activity of Other Transcription Factors: A Scope beyond Tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Langlais, D.; Couture, C.; Balsalobre, A.; Drouin, J. The Stat3/GR Interaction Code: Predictive Value of Direct/Indirect DNA Recruitment for Transcription Outcome. Mol. Cell 2012. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Ganti, K.P.; Chambon, P. Glucocorticoid-Induced Tethered Transrepression Requires SUMOylation of GR and Formation of a SUMO-SMRT/NCoR1-HDAC3 Repressing Complex. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E635–43. [Google Scholar] [CrossRef] [PubMed]
- Baiula, M.; Bedini, A.; Baldi, J.; Cavet, M.E.; Govoni, P.; Spampinato, S. Mapracorat, a Selective Glucocorticoid Receptor Agonist, Causes Apoptosis of Eosinophils Infiltrating the Conjunctiva in Late-Phase Experimental Ocular Allergy. Drug Des. Devel. Ther. 2014, 8, 745–757. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Vanden Berghe, W.; Beck, I.M.E.; Van Molle, W.; Hennuyer, N.; Hapgood, J.; Libert, C.; Staels, B.; Louw, A.; Haegeman, G. A Fully Dissociated Compound of Plant Origin for Inflammatory Gene Repression. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15827–15832. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Zein, N.; Daubeuf, F.; Chambon, P. Glucocorticoid Receptor Modulators CpdX and CpdX-D3 Exhibit the Same in Vivo Antiinflammatory Activities as Synthetic Glucocorticoids. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 14191–14199. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Zein, N.; Paulen, L.; Chambon, P. The Glucocorticoid Receptor Agonistic Modulators CpdX and CpdX-D3 Do Not Generate the Debilitating Effects of Synthetic Glucocorticoids. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 14200–14209. [Google Scholar] [CrossRef] [PubMed]
- Schäcke, H.; Schottelius, A.; Döcke, W.-D.; Strehlke, P.; Jaroch, S.; Schmees, N.; Rehwinkel, H.; Hennekes, H.; Asadullah, K. Dissociation of Transactivation from Transrepression by a Selective Glucocorticoid Receptor Agonist Leads to Separation of Therapeutic Effects from Side Effects. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 227–232. [Google Scholar] [CrossRef]
- Coghlan, M.J.; Jacobson, P.B.; Lane, B.; Nakane, M.; Lin, C.W.; Elmore, S.W.; Kym, P.R.; Luly, J.R.; Carter, G.W.; Turner, R.; et al. A Novel Antiinflammatory Maintains Glucocorticoid Efficacy with Reduced Side Effects. Mol. Endocrinol. Baltim. Md 2003, 17, 860–869. [Google Scholar] [CrossRef] [PubMed]
- van Lierop, M.-J.C.; Alkema, W.; Laskewitz, A.J.; Dijkema, R.; van der Maaden, H.M.; Smit, M.J.; Plate, R.; Conti, P.G.M.; Jans, C.G.J.M.; Timmers, C.M.; et al. Org 214007-0: A Novel Non-Steroidal Selective Glucocorticoid Receptor Modulator with Full Anti-Inflammatory Properties and Improved Therapeutic Index. PloS One 2012, 7, e48385. [Google Scholar] [CrossRef]
- Ripa, L.; Edman, K.; Dearman, M.; Edenro, G.; Hendrickx, R.; Ullah, V.; Chang, H.-F.; Lepistö, M.; Chapman, D.; Geschwindner, S.; et al. Discovery of a Novel Oral Glucocorticoid Receptor Modulator (AZD9567) with Improved Side Effect Profile. J. Med. Chem. 2018, 61, 1785–1799. [Google Scholar] [CrossRef]
- Schäcke, H.; Zollner, T.M.; Döcke, W.D.; Rehwinkel, H.; Jaroch, S.; Skuballa, W.; Neuhaus, R.; May, E.; Zügel, U.; Asadullah, K. Characterization of ZK 245186, a Novel, Selective Glucocorticoid Receptor Agonist for the Topical Treatment of Inflammatory Skin Diseases. Br. J. Pharmacol. 2009, 158, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Du, S.; Tunca, C.; Braden, T.; Long, K.R.; Lee, J.; Webb, E.G.; Dietz, J.D.; Hummert, S.; Rouw, S.; et al. The Antagonists but Not Partial Agonists of Glucocorticoid Receptor Ligands Show Substantial Side Effect Dissociation. Endocrinology 2011, 152, 3123–3134. [Google Scholar] [CrossRef] [PubMed]
- Swart, P.; Swart, A.C.; Louw, A.; van der Merwe, K.J. Biological Activities of the Shrub Salsola Tuberculatiformis Botsch.: Contraceptive or Stress Alleviator? BioEssays News Rev. Mol. Cell. Dev. Biol. 2003, 25, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Lesovaya, E.; Yemelyanov, A.; Kirsanov, K.; Popa, A.; Belitsky, G.; Yakubovskaya, M.; Gordon, L.I.; Rosen, S.T.; Budunova, I. Combination of a Selective Activator of the Glucocorticoid Receptor Compound A with a Proteasome Inhibitor as a Novel Strategy for Chemotherapy of Hematologic Malignancies. Cell Cycle Georget. Tex 2013, 12, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Louw, A.; Swart, P.; de Kock, S.S.; van der Merwe, K.J. Mechanism for the Stabilization in Vivo of the Aziridine Precursor --(4-Acetoxyphenyl)-2-Chloro-N-Methyl-Ethylammonium Chloride by Serum Proteins. Biochem. Pharmacol. 1997, 53, 189–197. [Google Scholar] [CrossRef] [PubMed]
- IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Man: Some Aziridines, N-, S- & O-Mustards and Selenium. IARC Monogr. Eval. Carcinog. Risk Chem. Man 1975, 9, 1–268.
- Stohs, S.J. Safety, Efficacy, and Mechanistic Studies Regarding Citrus Aurantium (Bitter Orange) Extract and p-Synephrine. Phytother. Res. PTR 2017, 31, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Dodonova, S.A.; Zhidkova, E.M.; Kryukov, A.A.; Valiev, T.T.; Kirsanov, K.I.; Kulikov, E.P.; Budunova, I.V.; Yakubovskaya, M.G.; Lesovaya, E.A. Synephrine and Its Derivative Compound A: Common and Specific Biological Effects. Int. J. Mol. Sci. 2023, 24, 17537. [Google Scholar] [CrossRef] [PubMed]
- Moshkin, V.S.; Sosnovskikh, V.Ya. A Simple Two-Step Synthesis of 2-(Alkylamino)-1-Arylethanols, Including Racemic Adrenaline, from Aromatic Aldehydes via 5-Aryloxazolidines. Tetrahedron Lett. 2013, 54, 5869–5872. [Google Scholar] [CrossRef]
- Brandon, D.D.; Kendall, J.W.; Alman, K.; Tower, P.; Loriaux, D.L. Inhibition of Dexamethasone Binding to Human Glucocorticoid Receptor by New World Primate Cell Extracts. Steroids 1995, 60, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Yemelyanov, A.; Czwornog, J.; Gera, L.; Joshi, S.; Chatterton, R.T.; Budunova, I. Novel Steroid Receptor Phyto-Modulator Compound a Inhibits Growth and Survival of Prostate Cancer Cells. Cancer Res. 2008, 68, 4763–4773. [Google Scholar] [CrossRef] [PubMed]
- Ronacher, K.; Hadley, K.; Avenant, C.; Stubsrud, E.; Simons, S.S.; Louw, A.; Hapgood, J.P. Ligand-Selective Transactivation and Transrepression via the Glucocorticoid Receptor: Role of Cofactor Interaction. Mol. Cell. Endocrinol. 2009, 299, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Lesovaya, E.A.; Yemelyanov, A.Y.; Kirsanov, K.I.; Yakubovskaya, M.G.; Budunova, I. V Antitumor Effect of Non-Steroid Glucocorticoid Receptor Ligand CpdA on Leukemia Cell Lines CEM and K562. Biochem. Biokhimiia 2011, 76, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Robak, T.; Szmigielska, A. Dexamethasone Does Not Enhance Antileukemic Activity of Cladribine in Mice with Leukemias L1210 and P388. Neoplasma 2000, 47, 168–171. [Google Scholar] [PubMed]
- Trafalis, D.T.P.; Chrysogelou, E.; Dalezis, P.; Geromichalos, G.; Kontos, M.; Andreadis, C.; Ziras, N.; Koutsilieris, M.; Athanassiou, A.E.; Pangalis, G.A.; et al. Octreotide Neutralizes Dexamethasone Antitumor Actions on P388 Murine Lymphocytic Leukemia in Vivo. J. BUON Off. J. Balk. Union Oncol. 10, 89–94.
- Saksida, T.; Vujicic, M.; Nikolic, I.; Stojanovic, I.; Haegeman, G.; Stosic-Grujicic, S. Compound A, a Selective Glucocorticoid Receptor Agonist, Inhibits Immunoinflammatory Diabetes, Induced by Multiple Low Doses of Streptozotocin in Mice. Br. J. Pharmacol. 2014, 171, 5898–5909. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Mirzoeva, S.; Readhead, B.; Dudley, J.T.; Budunova, I. PI3K Inhibitors Protect against Glucocorticoid-Induced Skin Atrophy. EBioMedicine 2019, 41, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Baban, B.; Isales, C.M.; Shi, X.-M. Role of Glucocorticoid-Induced Leucine Zipper (GILZ) in Inflammatory Bone Loss. PloS One 2017, 12, e0181133. [Google Scholar] [CrossRef]
- Fan, H.; Kao, W.; Yang, Y.H.; Gu, R.; Harris, J.; Fingerle-Rowson, G.; Bucala, R.; Ngo, D.; Beaulieu, E.; Morand, E.F. Macrophage Migration Inhibitory Factor Inhibits the Antiinflammatory Effects of Glucocorticoids via Glucocorticoid-Induced Leucine Zipper. Arthritis Rheumatol. Hoboken NJ 2014, 66, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Tonsing-Carter, E.; Hernandez, K.M.; Kim, C.R.; Harkless, R. V; Oh, A.; Bowie, K.R.; West-Szymanski, D.C.; Betancourt-Ponce, M.A.; Green, B.D.; Lastra, R.R.; et al. Glucocorticoid Receptor Modulation Decreases ER-Positive Breast Cancer Cell Proliferation and Suppresses Wild-Type and Mutant ER Chromatin Association. Breast Cancer Res. BCR 2019, 21, 82. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Hua, C.; Geng, Y.; Chen, Q.; Niu, L.; Tao, S.; Ni, Y.; Zhao, R. Chronic Dexamethasone Exposure Activates the TLR4-Mediated Inflammation Pathway and Induces Epithelial Apoptosis in the Goat Colon. Biochem. Biophys. Res. Commun. 2019, 518, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lesovaya, E.; Agarwal, S.; Readhead, B.; Vinokour, E.; Baida, G.; Bhalla, P.; Kirsanov, K.; Yakubovskaya, M.; Platanias, L.C.; Dudley, J.T.; et al. Rapamycin Modulates Glucocorticoid Receptor Function, Blocks Atrophogene REDD1, and Protects Skin from Steroid Atrophy. J. Invest. Dermatol. 2018, 138, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Baida, G.; Bhalla, P.; Kirsanov, K.; Lesovaya, E.; Yakubovskaya, M.; Yuen, K.; Guo, S.; Lavker, R.M.; Readhead, B.; Dudley, J.T.; et al. REDD1 Functions at the Crossroads between the Therapeutic and Adverse Effects of Topical Glucocorticoids. EMBO Mol. Med. 2015, 7, 42–58. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Beck, I.M.; Haegeman, G. Classic Glucocorticoids versus Non-Steroidal Glucocorticoid Receptor Modulators: Survival of the Fittest Regulator of the Immune System? Brain. Behav. Immun. 2010, 24, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- U, M.; Shen, L.; Oshida, T.; Miyauchi, J.; Yamada, M.; Miyashita, T. Identification of Novel Direct Transcriptional Targets of Glucocorticoid Receptor. Leukemia 2004, 18, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lou, Z.; Wang, L. The Role of FKBP5 in Cancer Aetiology and Chemoresistance. Br. J. Cancer 2011, 104, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Li, L.; Fridley, B.L.; Jenkins, G.D.; Kalari, K.R.; Lingle, W.; Petersen, G.; Lou, Z.; Wang, L. FKBP51 Affects Cancer Cell Response to Chemotherapy by Negatively Regulating Akt. Cancer Cell 2009, 16, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Bähn, S.; Tillack, A.; Imm, S.; Mevius, K.; Michalik, D.; Hollmann, D.; Neubert, L.; Beller, M. Ruthenium--catalyzed Selective Monoamination of Vicinal Diols. ChemSusChem 2009, 2, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Yemelyanov, A.; Czwornog, J.; Chebotaev, D.; Karseladze, A.; Kulevitch, E.; Yang, X.; Budunova, I. Tumor Suppressor Activity of Glucocorticoid Receptor in the Prostate. Oncogene 2007, 26, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.H.; Glennie, M.J.; et al. Guidelines for the Welfare and Use of Animals in Cancer Research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Synthesis of piperidine analogues of CpdA.
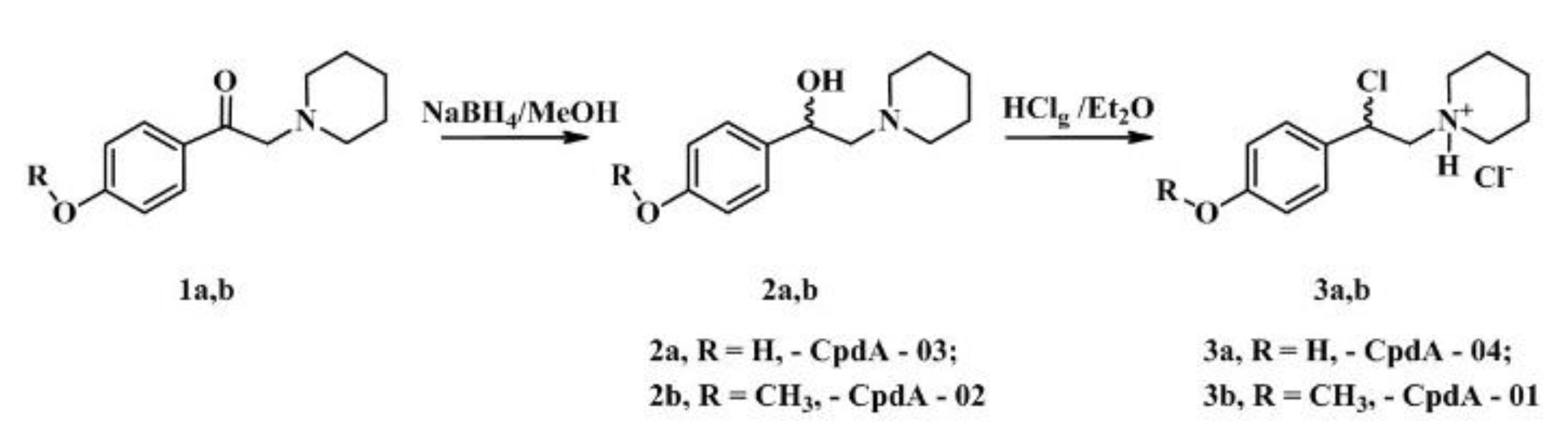
Figure 2.
Synthesis of CpdA analogues CpdA-05 - CpdA-08.
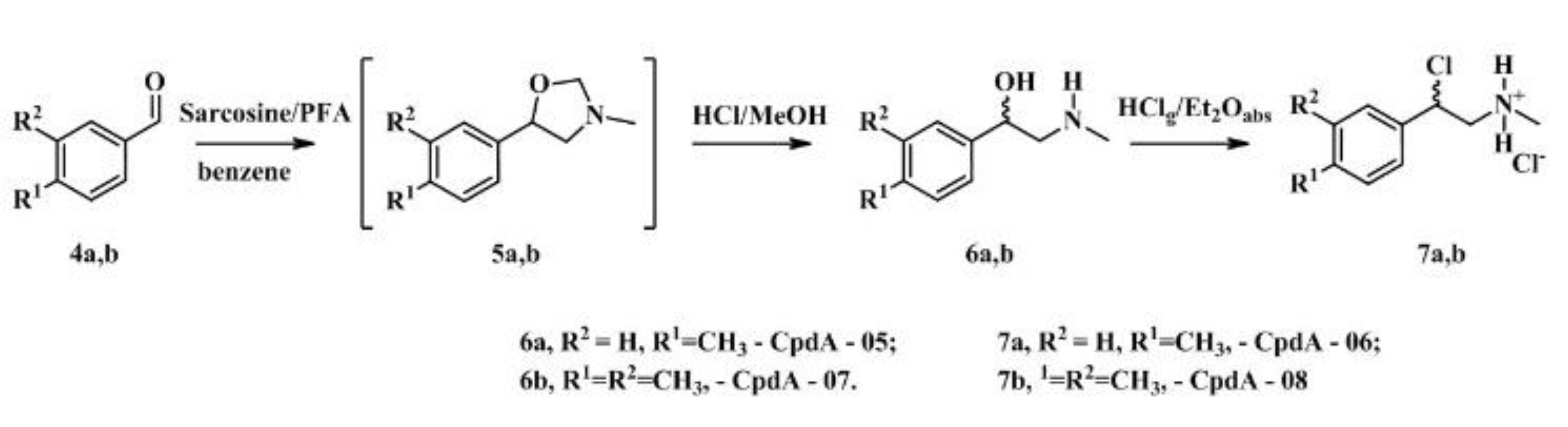
Figure 3.
Evaluation of CpdA-03 affinity to GR. GR binding affinity was evaluated using radioligand binding assay. Dex, CpdA and CpdA-03 in various concentration were added to each reaction tube and incubated for 90 min. The cell lysate radioactivity was measured by a RackBeta 1215 LC spectrometer for 4 minutes. The experimental results were presented as percentage to [3Н]Dex alone signal.
Figure 3.
Evaluation of CpdA-03 affinity to GR. GR binding affinity was evaluated using radioligand binding assay. Dex, CpdA and CpdA-03 in various concentration were added to each reaction tube and incubated for 90 min. The cell lysate radioactivity was measured by a RackBeta 1215 LC spectrometer for 4 minutes. The experimental results were presented as percentage to [3Н]Dex alone signal.
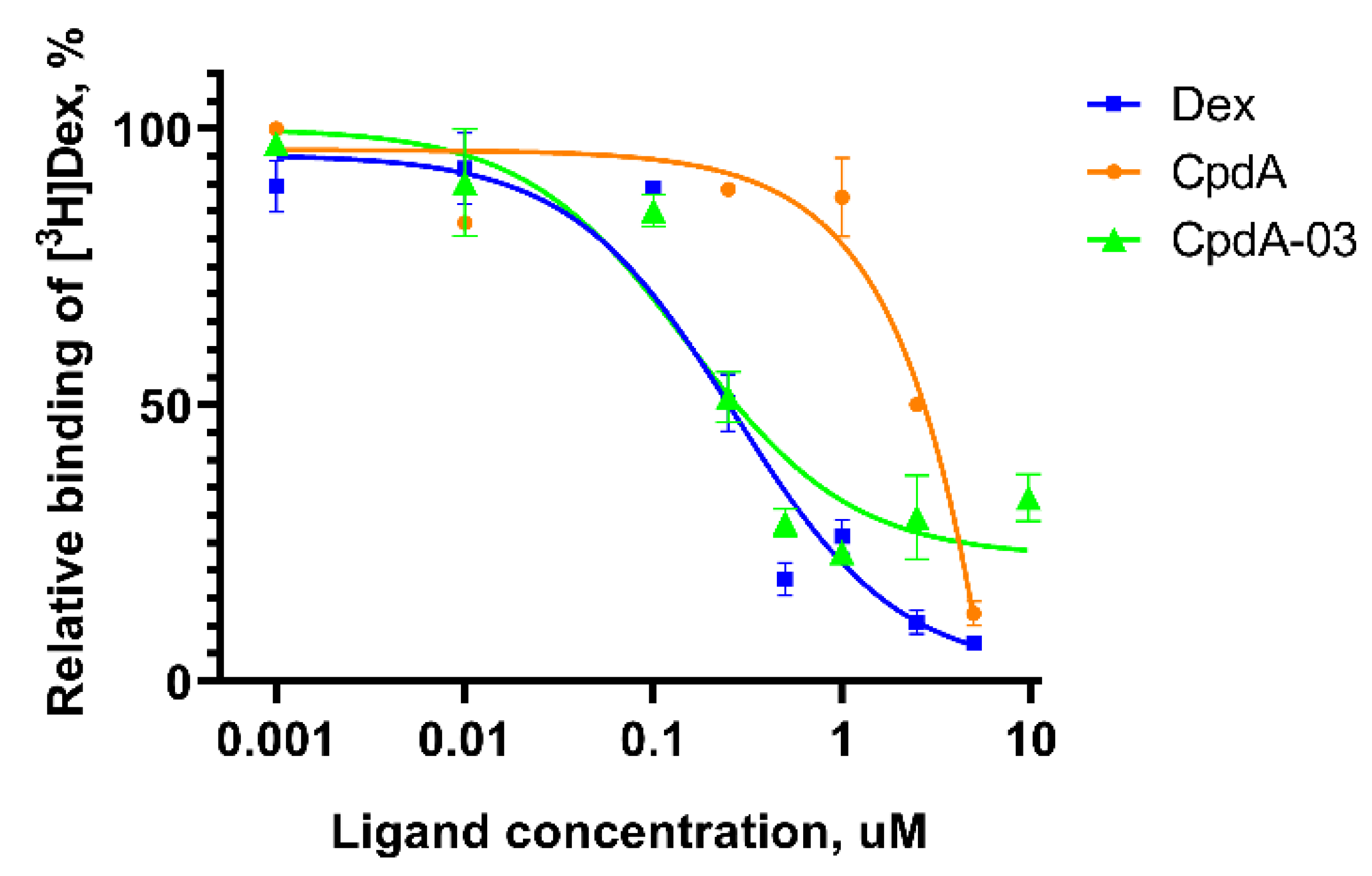
Figure 4.
Anti-cancer activity of CpdA-03 in vitro and in vivo. The knock-down of GR in CEM and Granta cells was confirmed by Western blot analysis (A). CEM, CEM-shGR, Granta, and Granta-shGR were treated with solvent (control), Dex, CpdA, CpdA-03 (all compounds in 1 uM concentration) for 24 h, and the effect on cell growth (B) was estimated by cell counting. The effect on apoptosis (C) was determined by flow cytometry using propidium iodide staining (the amount of cells in sub-G1-phase was calculated as percentage to all cells in sample). (D) Anti-cancer study in vivo was evaluated using transplantable P388 murine lymphoma. Animals were treated i.p. 3 times/week with Dex (1 mg/kg), CpdA (7,5 mg/kg), CpdA-03 (7,5 mg/kg) of vehicle (5% DMSO in PBS). Tumor nodule size was measured twice a week by digital calipers. * - statistically significant difference (p<0.05) compared to control, # - statistically significant difference (p<0.05) between corresponding samples in control cells and GR-knockdown cells.
Figure 4.
Anti-cancer activity of CpdA-03 in vitro and in vivo. The knock-down of GR in CEM and Granta cells was confirmed by Western blot analysis (A). CEM, CEM-shGR, Granta, and Granta-shGR were treated with solvent (control), Dex, CpdA, CpdA-03 (all compounds in 1 uM concentration) for 24 h, and the effect on cell growth (B) was estimated by cell counting. The effect on apoptosis (C) was determined by flow cytometry using propidium iodide staining (the amount of cells in sub-G1-phase was calculated as percentage to all cells in sample). (D) Anti-cancer study in vivo was evaluated using transplantable P388 murine lymphoma. Animals were treated i.p. 3 times/week with Dex (1 mg/kg), CpdA (7,5 mg/kg), CpdA-03 (7,5 mg/kg) of vehicle (5% DMSO in PBS). Tumor nodule size was measured twice a week by digital calipers. * - statistically significant difference (p<0.05) compared to control, # - statistically significant difference (p<0.05) between corresponding samples in control cells and GR-knockdown cells.
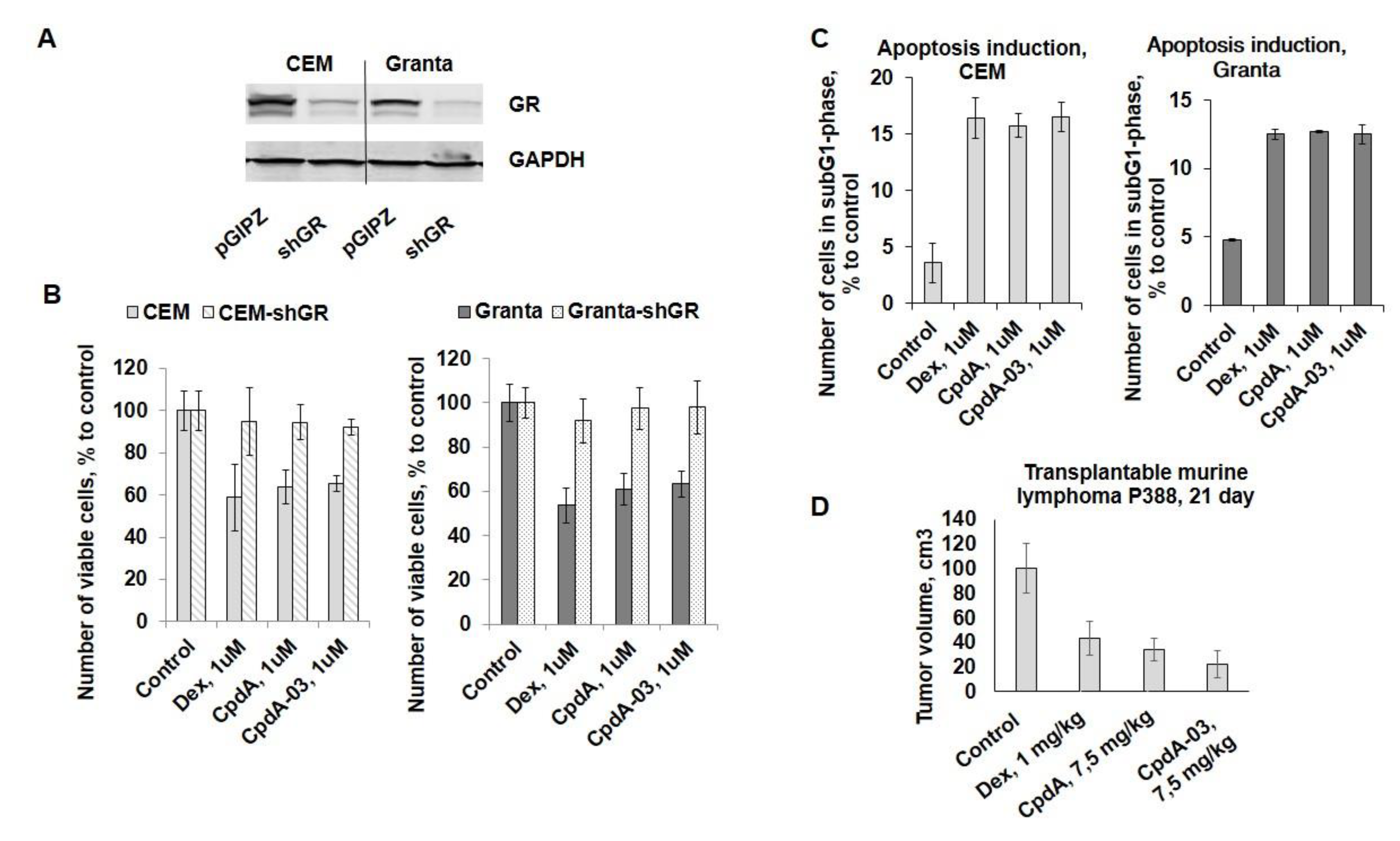
Figure 5.
CpdA-03 does not affect GR transactivation but induces GR transrepression in lymphoma and leukemia cells. (A) CEM and Granta cells stably infected with lentiviruses expressing Luciferase reporters: GRE.Luc. Cells were incubated for 24 hrs with solvent (Control), Dex or CpdA. Luciferase activity was determined as described in “Materials and Methods”. (B) Q-PCR analysis of FKBP51 and GILZ mRNA expression in CEM and Granta cells. The Q-PCR results were normalized to the expression of housekeeping gene RPL27, and presented as fold change compared to control. The mean +/- SD was calculated for three individual samples/condition. *- statistically significant difference (p<0.05) compared to control. (C) The levels of phospho-GR (Ser211) and GR were determined by Western blot analysis. GAPDH was used as loading control.
Figure 5.
CpdA-03 does not affect GR transactivation but induces GR transrepression in lymphoma and leukemia cells. (A) CEM and Granta cells stably infected with lentiviruses expressing Luciferase reporters: GRE.Luc. Cells were incubated for 24 hrs with solvent (Control), Dex or CpdA. Luciferase activity was determined as described in “Materials and Methods”. (B) Q-PCR analysis of FKBP51 and GILZ mRNA expression in CEM and Granta cells. The Q-PCR results were normalized to the expression of housekeeping gene RPL27, and presented as fold change compared to control. The mean +/- SD was calculated for three individual samples/condition. *- statistically significant difference (p<0.05) compared to control. (C) The levels of phospho-GR (Ser211) and GR were determined by Western blot analysis. GAPDH was used as loading control.
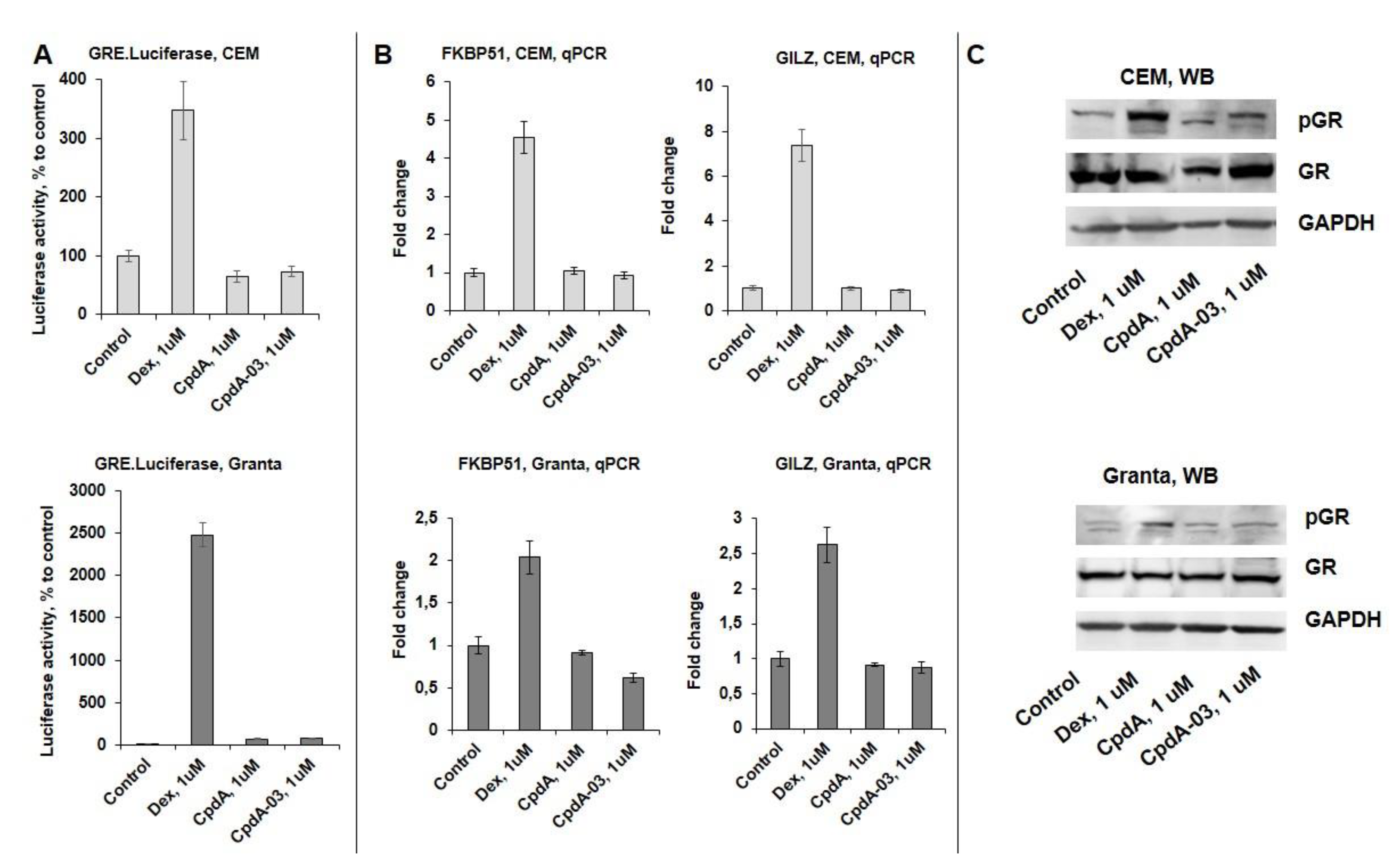
Figure 6.
CpdA-03 induces GR transrepression in lymphoma and leukemia cells. (A) CEM and Granta cells stably infected with lentiviruses expressing NF-kB.Luciferase reporter. Cells were incubated for 24 hrs with solvent (Control), Dex or CpdA. Luciferase activity was determined as described in “Materials and Methods”. (B) СCND1 and CCND2 mRNA expression in CEM cells, IL1 and IL6 mRNA expression in Granta cells. The Q-PCR results were normalized to the expression of housekeeping gene RPL27, and presented as fold change compared to control. The mean +/- SD was calculated for three individual samples/condition. * - statistically significant difference (p<0.05) compared to control.
Figure 6.
CpdA-03 induces GR transrepression in lymphoma and leukemia cells. (A) CEM and Granta cells stably infected with lentiviruses expressing NF-kB.Luciferase reporter. Cells were incubated for 24 hrs with solvent (Control), Dex or CpdA. Luciferase activity was determined as described in “Materials and Methods”. (B) СCND1 and CCND2 mRNA expression in CEM cells, IL1 and IL6 mRNA expression in Granta cells. The Q-PCR results were normalized to the expression of housekeeping gene RPL27, and presented as fold change compared to control. The mean +/- SD was calculated for three individual samples/condition. * - statistically significant difference (p<0.05) compared to control.
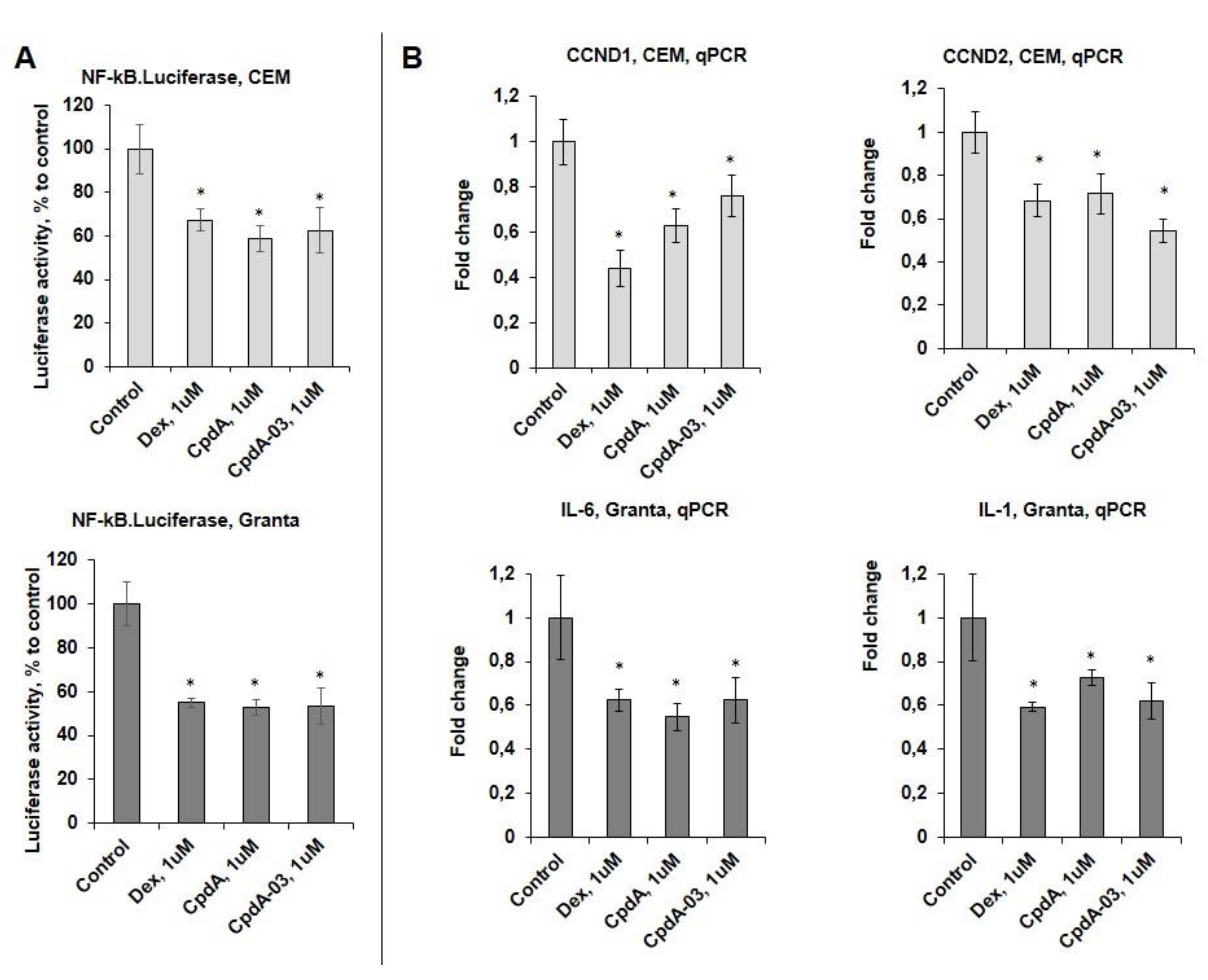

Table 1.
Cytotoxicity of novel CpdA derivatives in MTT-test.
| Compound | IC50, uМ, CEM | IC50, uМ, Granta | IC50, uМ, K562 |
|---|---|---|---|
| Dex | 1,35±0,48 | 3,55±0,69 | 8,87±0,91 |
| CpdA | 1,36±0,27 | 5,56±0,27 | 14,61±1,27 |
| CpdA-01 | 15,33±2,44 | 19,55±2,85 | 25,04±3,49 |
| CpdA-02 | 18,34±2,71 | 26,54±1,84 | 38,74±5,92 |
| CpdA-03 | 3,26±0,22 | 5,78±0,63 | 18,81±1,62 |
| CpdA-04 | 24,33±3,34 | 28,65±3,50 | 34,41±3,63 |
| CpdA-05 | 9,67±0,51 | 11,71±2,01 | 25,78±3,17 |
| CpdA-06 | 9,13±1,78 | 19,25±1,79 | 33,91±5,17 |
| CpdA-07 | 18,61±1,62 | 20,61±2,02 | 39,87±3,68 |
| CpdA-08 | 27,93±3,94 | 19,95±2,56 | 47,38±5,47 |
Table 2.
Structure and characterization of CpdA-01-08.
| Compound | Structure | Characterization |
|---|---|---|
| 1-(2-chloro-2-(4-methoxyphenyl)ethyl)piperidin-1-ium chloride (CpdA-01) |
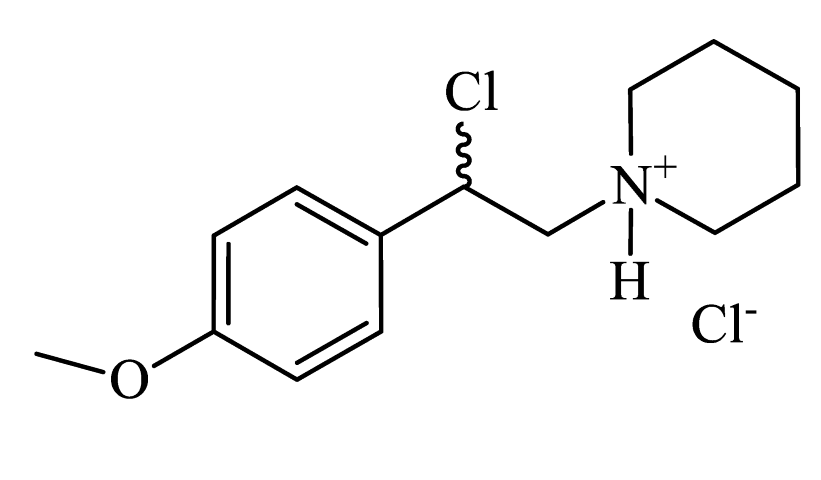 |
Colorless powder, 99% yield (858 mg). 1H NMR (300 MHz, DMSO-d6): δ = 1.35-1.42 (m, 1H, CH2); 1.65-1.79 (m, 5H, CH2); 2.99-3.01 (m, 2H, CH2); 3.37-3.60 (m, 4H, CH2); 3.74 (s, 3H, OCH3); 6.28-6.30 (m, 1H, CH); 6.95-6.98 (m, 2H, Harom); 7.36-7.40 (m, 2H, Harom); 10.61 (s, 1H, NH). MS (EI): m/z (%) = 253 [M]+. HRMS (ESI-TOF) m/z: [M + H]+ Calculated for C14H20ClNO: 254.1306; found: 254.1317. |
| 1-(4-methoxyphenyl)-2-(piperidin-1-yl)ethanol (CpdA-02) |
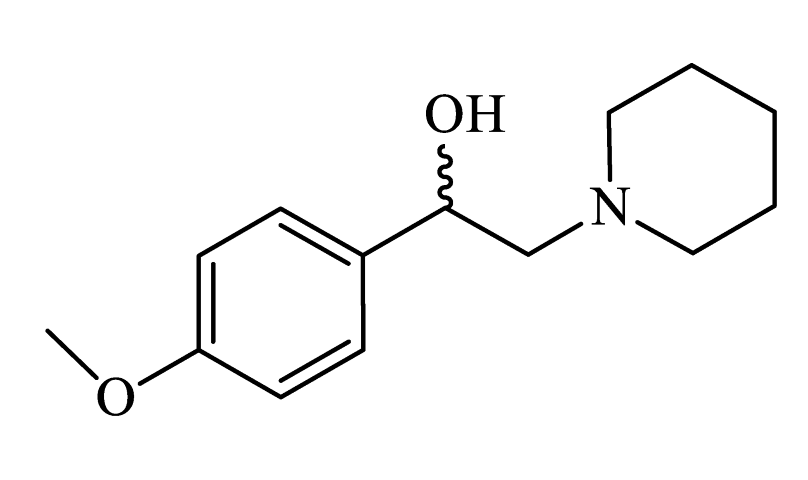 |
Colorless powder, 71% yield (500 mg). 1H NMR (300 MHz, CDCl3) δ = 1.49 (dd, J = 11.3, 5.6 Hz, 2H, CH2); 1.61-1.67 (m, 4H, CH2); 2.37-2.45 (m, 4H, CH2); 2.70-2.76 (m, 2H, CH2); 3.81 (s, 3H, OCH3); 4.69 (dd, J = 9.8, 4.3 Hz, 1H, CH); 6.89 (d, J = 8.6 Hz, 2H, Harom); 7.26-7.32 (m, 2H, Harom). Anal. Calculated for C14H21NO2: C, 71.38; H, 9.00; N, 5.85. Found: C, 71.46; H, 8.99; N, 5.95. Lit. [S1]. |
| 4-(1-hydroxy-2-(piperidin-1-yl)ethyl)phenol (CpdA-03) |
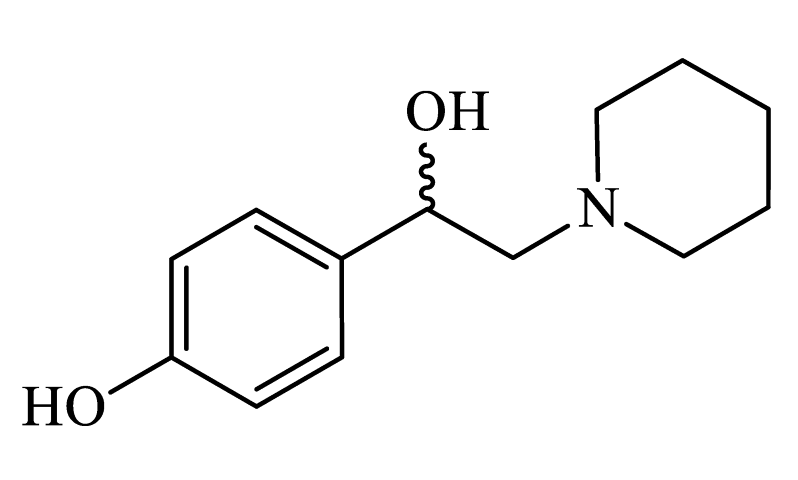 |
Colorless powder, 33% yield (219 mg). 1H NMR (300 MHz, DMSO-d6): δ = 1.32-1.39 (m, 2H, CH2); 1.46-1.47 (m, 4H, CH2); 2.24-2.44 (m, 6H, CH2); 4.54-4.56 (m, 1H, CH); 6.66-6.69 (m, 2H, Harom); 7.09-7.12 (m, 2H, Harom); 9.18 (s, 1H, OH). Anal. Calculated for C13H19NO2: C, 70.45; H, 8.65; N 6.20, Found: C, 70.56; H, 8.65; N, 6.33. Lit. [S2]. |
| 1-(2-chloro-2-(4-hydroxyphenyl)ethyl)piperidin-1-ium chloride (CpdA-04) |
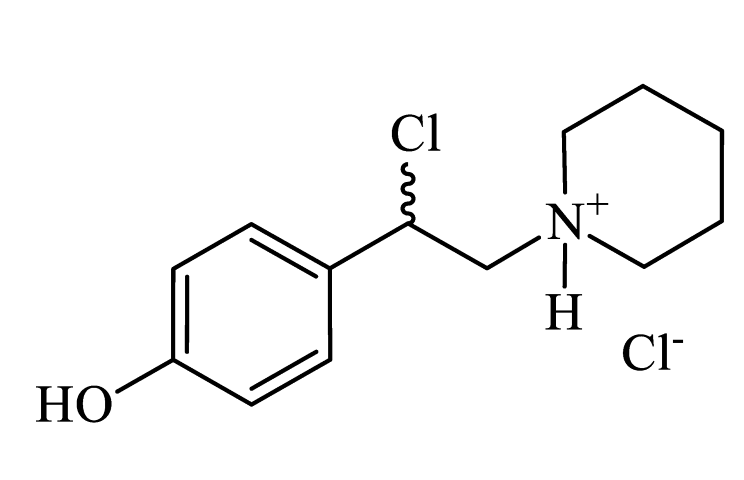 |
Colorless powder, 99% yield (817 mg). 1H NMR (300 MHz, DMSO-d6): δ = 1.33-1.49 (m, 1H, CH2); 1.65-1.81 (m, 5H, CH2); 2.94-3.11 (m, 2H, CH2); 3.36-3.839 (m, 4H, CH2); 6.37-6.41 (m, 1H, CH); 7.31-7.35 (m, 2H, Harom); 7.55-7.68 (m, 2H, Harom); 10.75 (s, 1H, NH). MS (EI): m/z (%) = 203 [M-HCl]+. |
| 1-(4-methoxyphenyl)-2-(methylamino)ethanol (CpdA-05) |
 |
Colorless powder, 33% yield (60 mg). 1H NMR (300 MHz, DMSO-d6): δ = 2.29 (s, 3H, CH3); 2.50-2.56 (m, 2H, CH2); 3.73 (s, 3H, OCH3); 4.53-4.59 (m, 1H, CH); 6.87 (d, J = 8.40 Hz, 2H, Harom); 7.24 (d, J = 8.40 Hz, 2H, Harom). MS (EI): m/z (%) = 181 [M]+. Anal. Calculated for C10H15NO2: C, 66.13; H, 8.42; N, 7.35. Found: C, 66.27; H, 8.34; N, 7.73. Lit. [S3]. |
| 2-chloro-2-(4-methoxyphenyl)-N-methylethanaminium chloride (CpdA-06) |
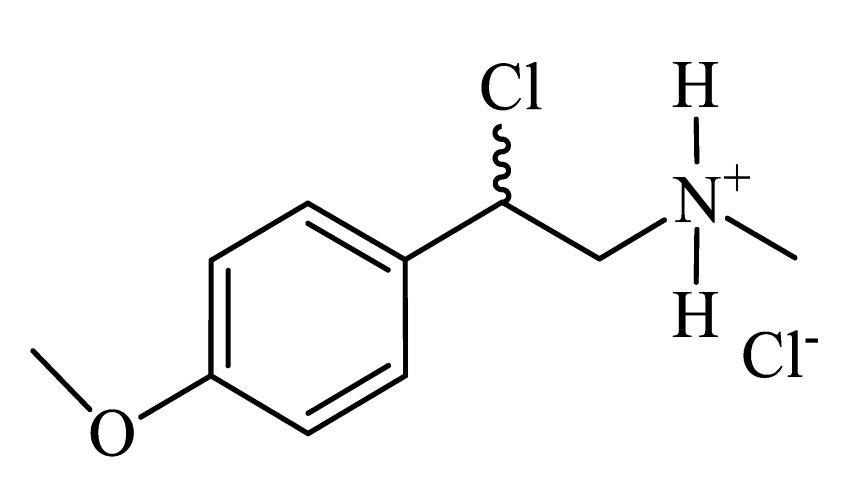 |
Colorless powder, 33% yield (233 mg). 1H NMR (300 MHz, DMSO-d6): δ = 2.63 (s, 3H, CH3); 3.76 (s, 5H, OCH3+CH2); 6.13-6.19 (m, 1H, CH); 7.00 (d, J = 8.06 Hz, 2H, Harom); 7.41 (d, J = 8.06 Hz, 2H, Harom); 9.29 (s, 2H, NH). MS (EI): m/z (%) = 199 [M]+. |
| 1-(3,4-dimethoxyphenyl)-2-(methylamino)ethanol (CpdA-07) |
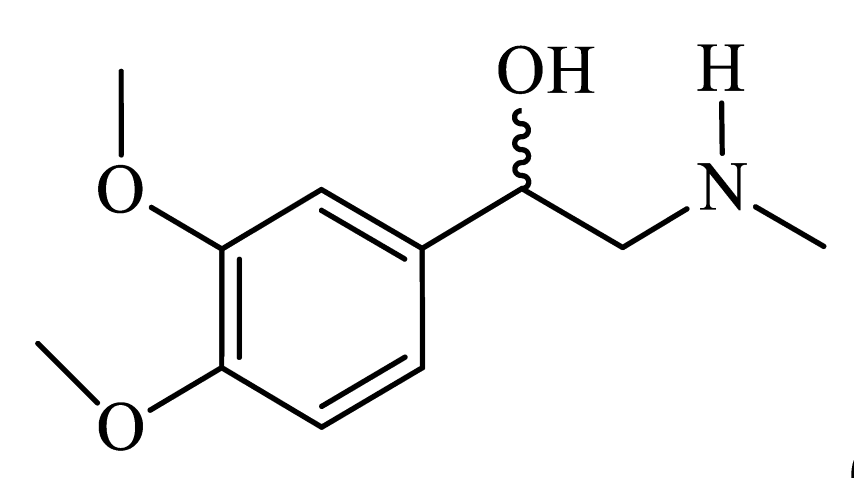 |
Colorless powder, 63% yield (133 mg). 1H NMR (300 MHz, CDCl3) δ = 2.44 (s, 3H, CH3); 2.66-2.80 (m, 2H, CH2); 2.88 (s, 2H, OH+NH); 3.86 (s, 3H, OCH3); 3.88 (s, 3H, OCH3);4.67-4.72 (dd, J = 8.4, 4.3 Hz, 1H, CH); 6.84 (s, 1H, Harom); 6.86-6.87 (m, 1H, Harom); 6.93-6.94 (m, 1H, Harom). MS (EI): m/z (%) 211 [M]+. HRMS (ESI-TOF) m/z: [M + H]+. Calculated for C11H17NO3: 212.1281; found: 212.1275. Lit. [S3]. |
| 2-chloro-2-(3,4-dimethoxyphenyl)-N-methylethanaminium chloride (CpdA-08) |
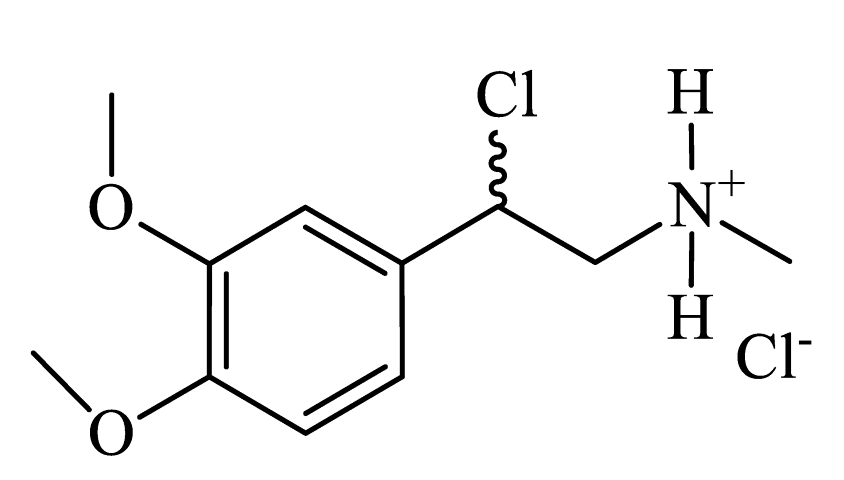 |
Colorless powder, 33% yield (262 mg). 1H NMR (300 MHz, DMSO-d6) δ = 2.62 (s, 3H, CH3); 3.74 (s, 4H, CH+OCH3); 3.76 (s, 4H, CH+OCH3); 6.12-6.16 (m, 1H, CH); 6.98 (s, 2H, Harom); 7.06 (s, 1H, Harom); 9.21 (s, 2H, NH). MS (EI): m/z (%) = 198 [M - MeO]+. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



