
Submitted:
02 July 2024
Posted:
03 July 2024
You are already at the latest version
Abstract
GABAA receptors are channel proteins crucial to mediating neuronal balance in the CNS. The structure of GABAA receptors allows for multiple binding sites and is key to drug development. Yet, the formation mechanism of the receptor’s distinctive pentameric structure is still unknown. This study aims to investigate the role of three predominant subunits of the human GABAA receptor in the formation of protein pentamers. Through purifying and refolding the protein fragments of the GABAA receptor α1, β2, and γ2 subunits, homopentamers were visualised with negative staining and Cryo-EM. To aid the analysis, AlphaFold2 was used to compare the structures. Results show that α1 and β2 subunit fragments successfully formed homo-oligomers, particularly homopentameric structures, while the predominant heteropentameric GABAA receptor was also replicated through the combination of the three subunits. However, it is still unclear whether γ2 subunits can form homopentamers or their main purpose is to bring together the α1 and β2 subunits to form GABAA receptors. Nevertheless, this study will lead to a deeper understanding of the atomic structure of GABAA receptors, especially in the formation mechanism of complex protein structures, and hopefully pave the way to the development of novel therapeutics for neuropsychiatric diseases.
Keywords:
α1β2γ2 GABAA receptor
; pentameric structure
; homopentamers
1. Introduction
Neurotransmitters function to transmit signals from nerve cells to other target cells through the synapse and are important for maintaining neuronal balance. Inhibitory neurotransmitters reduce the chances of an action potential and interact with corresponding receptors to prevent over-excitation. γ-aminobutyric acid (GABA) functions as the major inhibitory neurotransmitter in the human central nervous system, and its main corresponding receptor is the GABAA receptor [1,2,3]. When GABA binds to its receptor, GABAA receptors undergo conformational changes, leading to an influx of negatively charged chloride ions, and terminating an action potential by causing hyperpolarisation [3,4].
GABAA receptors are part of the superfamily of Cys-loop pentameric ligand-gated ion channels (pLGICs), which include other neurotransmitter receptors, such as the glycine receptor (GlyR) [5]. GlyRs are closely related to GABAA receptors [6,7]. Both receptors possess the ability to form heteropentamers with different subunits, while several specific subunits can also form homopentamers [8]. Currently, 19 subunits of the GABAA receptor have been discovered in humans, namely α1-6, β1-3, γ1-3, δ, ε, θ, π and ρ1-3 subunits [9,10]. The structure of each subunit usually includes a hydrophilic extracellular domain with the Cys-loop, and four hydrophobic transmembrane domains [10,11]. Various combinations of subunits may be assembled to construct the receptors. Still, the most common form consists of two α, two β and one γ subunits, with the most abundant subtype being α1β2γ2 in a 2:2:1 ratio, accounting for 43% of all GABAA receptors in the brain [1,12,13]. In this predominant subtype of GABAA receptors, alternating α1 and β2 subunits are connected by a single γ2 subunit. Apart from the GABA binding site, the GABAA receptors also have a benzodiazepine (BZ) drug binding site. The GABA binding site is located between the α and β subunits, while the BZ binding site is located between the α and γ subunits [14,15]. Dysfunction of GABAA receptors can lead to neuropsychiatric disorders [16], which cause the GABAA receptors to play an important role in the study of receptor-based drugs, such as BZs. To design specific drugs and study the ligand binding mechanism of the receptors, there is an urgent need to identify the atomic structure of the receptor.
Ligand-binding sites in the GABAA receptor are widely studied. The α1 subunit is the most highly expressed GABAA receptor subunit [17,18,19], and some of its residues have been identified to interact with GABA agonists [20,21,22,23]. It is well known that the aromatic amino acids – phenylalanine (Phe), tyrosine (Tyr), and tryptophan (Trp), are frequently involved in π interactions, which often play an important role in protein structural formation and ligand binding [24,25]. Indeed, GABA agonist binding sites are lined with aromatic residues, including Phe200 and Tyr205 on the β subunit, and Phe46 and Phe65 on the α subunit [26]. Interestingly, Phe65 (Phe64 on bovine homolog [27]) is conserved among the six α subunits as well as the γ2 subunit and is one of the residues that have been indicated to interact with the GABA agonist muscimol [20,28]. Amongst the 20 amino acids, Trp is the largest and contains two aromatic rings. Perhaps due to its complexity, Trp is the least abundant amino acid and the most energy-consuming [29]. It has been reported to play a role in stabilizing the structures of membrane proteins [30,31,32]. In fact, the invariant residues Trp-69 and Trp-94 in the α1 subunit have been identified as crucial residues for the GABAA receptor pentameric assembly [33]. Previous studies have suggested various roles in these two residues, associating with channel opening [34] and folding of loops [35], leading to potential altering conformational characteristics. One study performed point mutation on these residues, expressing the α1 peptide with β2 and γ2 subunits. The results demonstrated that the mutant α1 peptide did not form pentamers with β2 and γ2 subunits and could not bind to the BZ ligand, [3H]flunitrazepam [33]. Thus, aromatic residues in the α1 subunit may play a role in the GABAA receptor pentamer formation and ligand binding mechanisms.
Incorporating an additional β2 subunit in the GABAA receptor has been proposed to increase the binding sensitivity of GABA [36], suggesting an intriguing role of the β2 subunit in GABA binding and structural formation. Again, mutation at aromatic residues Tyr97, Tyr157, and Tyr205 on the β2 subunit greatly reduced the binding rate of GABA [37]. Similarly, important domains have been investigated within the γ2 subunit, for example, a region for flunitrazepam binding that consisted of Tyr-58 as an essential residue for high-affinity binding [38]. Hence, evidence highlights the significance of aromatic residues in ligand binding in the most predominant form of α1β2γ2 GABAA receptors. These studies surely aid in developing pharmacological drugs targeting the GABAA receptor and give insight into the binding mechanism of ligands. Still, structural studies are needed to explore potential conformational changes and intermolecular interactions with ligands.
Previously, researchers have overexpressed the major subunits of the GABAA receptor in E.coli to extract structural information and identify key binding residues [39,40,41]. Subsequently, the C139–L269 fragment of the α1 subunit has been successfully expressed, resulting in the formation of rosette-like structures [40,41,42]. Methods such as ala-scanning and base substitution revealed several important residues. In particular, five conserved Cys and Trp residues were identified in the membrane-proximal β-rich (MPB) domain and associated with the structural stability of an “immunoglobulin-like” (Ig-like) fold of the α1 subunit [42]. For instance, in some Ig-like proteins with two packed β-sheets, an additional Trp residue is packed against the cysteine disulphide bridge between the β-sheets [43]. Similarly, Trp residues located in the proximity of the Cys-loop were shown to affect the structural stability of Cys-loop receptors [42,44]. This provides invaluable insights into the secondary structure of the GABAA receptor subunits and the potential motifs in identifying Ig-like domains in Cys-loop receptors.
More recently, high-resolution atomic structures of the GABAA receptor have been determined with the aid of technological advancements and methods such as 3D classification, negative staining microscopy, and Cryo-EM. The earliest high-resolution Cryo-EM structures of the α1β2γ2 GABAA receptor were successfully determined in 2018 [45,46]., providing an additional foundation for investigating the receptor’s architecture and ligand binding. Yet, some structures described in the studies were unusual [47], particularly in one conformation where the γ2 subunit collapsed into the pore of the receptor [45]. Another Cryo-EM structure was solved using full-length subunits of the α1β3γ2 receptor subtype, but the membrane-spanning segments of M3-M4 loops were reported to be disordered [15]. The majority of studies focus on the hetero-pentameric receptor, with some including ligands in complex with the receptor [48,49]. Interestingly, one particular study presented the first 3D crystal structure of a truncated β3 subunit homo-pentamer [50], encouraging more researchers to explore the possibility of the homo-pentameric structures of the GABAA receptor subunits.
Constant improvement of GABAA receptor models is needed for identifying the binding mechanisms of ligands, understanding the interactions between subunits, and explaining the assembly pathway of the receptor. However, exploring eukaryotic membrane proteins is challenging due to their functions depending on their native environment. Therefore, using previous studies as a reference [39,40], the present study utilises truncated fragments of the α1, β2 and γ2 subunit protein samples, aiming for a higher-resolution structure of the key extracellular residues of GABAA receptors. Although GABAA receptors may consist of various subunits, the most abundant form consists of α1, β2 and γ2 only. Yet, the formation mechanism of the predominant GABAA receptor is still unknown, such as intermediate structures and how different subunits interact with each other freely within their native environment. This study aims to utilize state-of-the-art tools, such as Cryo-EM and neural network-based model AlphaFold2 [51] for elucidating the structures of GABAA receptors. By investigating the pentameric formation mechanism of the three major subunits of the GABAA receptor, this study will hopefully increase our understanding of the mechanism of complex protein structures and potentially pave the way to the development of novel therapeutics for neuropsychiatric diseases.
2. Results
2.1. Amino Acid Sequence Alignment
The amino acid sequence and predicted molecular structure of the α1, β2 and γ2 subunits have been previously identified by multiple studies. Each subunit consists of a long extracellular N-terminus containing a Cys-loop, four transmembrane domains, an intracellular loop, and a short C-terminus [2]. The ligand binding sites of GABA and BZ are located in the extracellular domain of the receptor [45,52,53]. Due to the complication of expressing and purifying whole protein subunits, extracellular domains involving most of the binding sites on the GABAA receptors were expressed and purified. The selected domains were Gln28-Arg248 in the α1 subunit (25 kDa), Gln25-Gly243 in the β2 subunit (25.2kDa), and Gln40-Gly273 in the γ2 subunit (27.5 kDa).
To gain initial insights into the possible structural similarities and differences between the α1, β2 and γ2 subunits of the GABAA receptor, amino acid sequence alignment was performed, as shown in Figure 1. The three subunit fragments of key residues have sequence homology of >26% with multiple conserved sequences. The percent identity matrix on each subunit (Table 1) indicates that α1 and γ2 subunits have higher sequence homology compared with β2. Genetic analysis indicated the subunit genes are clustered on chromosome 5, suggesting a possibility of gene duplication of clusters during evolution [2,9,11].
2.2. Expression and Purification of α1, β2, and γ2 Subunit Fragments
After the three fragments were expressed in a prokaryotic vector and purified by washing and precipitation. Solubilized protein pellet was loaded for fast protein liquid chromatography (FPLC) gel filtration and collected into eluents corresponding to consecutive fraction numbers. Measurement of optical density at 280nm (OD280) shows protein elution at the absorbing peaks and indicates the concentration of protein obtained. When the purified proteins were applied to the Superdex 200 column, bound proteins eluted between fraction numbers 20-35, as observed in Figure 2. Higher OD280 suggests a higher protein concentration [54], thus allowing dialysis for further structural visualization and negative staining. SDS-PAGE was used to confirm the molecular weight of the purified protein fragments. As shown in Figure 2, the peak of each protein contains a polypeptide band with a molecular mass corresponding to each fragment size. In an attempt to recreate the human heteropentameric α1β2γ2 GABAA receptor, purified proteins were combined in a 2:2:1 ratio of α1, β2 and γ2 respectively. However, the concentration of the complex obtained was relatively low.
2.3. Visualisation of Protein Structures Using Cryo-EM
In pursuit of a high-resolution molecular structure, negative staining was performed following the confirmation of molecular weights. Figure 3a depicts α1 subunit negative staining, showing clear protein structure without unusual aggregation. 2D classification in Figure 3b shows top views indicating a pentameric shape and side views suggesting a central cavity. This verifies that α1 subunit fragments successfully formed homopentamers with rosette-like structures and a central cavity as previously described in Xue et al [40]. Although negative staining and 2D classification of β2 subunit fragments also suggest a homopentameric structure similar to the α1 subunit homopentamer, there seemed to be unusual aggregation, as seen in Figure 3c. Also, the concentration of the β2 subunit fragments was relatively lower than the other fragments obtained (Figure 2b), which could be a reason why the Cryo-EM images obtained were not of high resolution. On the other hand, the concentration of γ2 subunit fragments obtained was much higher (Figure 2c), but negative staining (Figure 3e) did not identify any pentameric structures for further imaging. In addition to the subunit fragments, the predominant GABAA receptor structure was also analysed.
2.4. AlphaFold2 Prediction of α1, β2, and γ2 Homopentameric Structures
The sequence of the fragments obtained from UniProt [55] was applied to AlphaFold2 and tested as homopentamers. For each pentameric structure, a total of 5 models with different structures and confidence levels were created by AlphaFold2, as shown in Supplementary Figure S1. The best model with the highest per residue confidence (pLDDT) was chosen for further analysis in Chimera software. For α1 homopentamer, the confidence score from AlphaFold2 is 86.3 with the predicted template modelling score (pTM) of 0.864, above the 0.75 confidence cut-off (Supplementary Figure S2e). The high concentration and resolution from negative staining, as well as the high structural prediction confidence in AlphaFold2, indicates that the α1 subunit fragment can successfully form homopentameric structures, as expected. Furthermore, the β2 subunit fragment homopentamer structural prediction has a high pLDDT of 91.2 and pTM of 0.9 (Supplementary Figure S2f). The confidence of prediction is even higher than the α1 homopentamer, suggesting a high potential of the β2 subunit forming homopentamers. This is also evident in the negative staining and Cryo-EM results. Similarly, the pentameric GABAA receptor structural prediction had a high pLDDT of 87.8 and pTM of 0.889, supporting its pentameric formation seen in negative staining. Although AlphaFold2 successfully predicted the γ2 subunit pentamer formation with pLDDT=82.5 and pTM=0.833, no pentameric structure was seen in negative staining results Figure 4e. To further investigate the possible reasons, the secondary structure of the highest-ranked prediction model was analyzed in Chimera.
The α1 homopentamer and β2 homopentamer AlphaFold2 models have similar structures (Figure 4a and 4b), with a smooth pentameric shape and a clear central cavity. In particular, the central cavity of the β2 seems to be slightly larger than the central cavity in α1 homopentamer. In contrast, γ2 homopentamer has protruding residues that are not tucked into the pentameric structure, which may be affecting the ability of the fragments to form homopentamers. Interestingly, AlphaFold2 prediction of γ2 homopentamer consisted of subunits with slightly different secondary structures. Four of the five γ2 subunits in the homopentamer have a short 3-residue helical structure near the beginning of the fragment (Figure 4c). This demonstrates two possible folding methods of the γ2 subunit fragment, which may be difficult to control during protein refolding, thus again, affecting their ability to form pentamers in vitro. Another captivating point from the AlphaFold2 predictions is that the γ2 subunit in the predicted GABAA receptor heteropentamer shows no helical structure in the first 20 residues (Figure 4d). This suggests that the γ2 folding without the 3-residue helix is preferred and crucial for forming the predominant human GABAA receptor.
2.5. Cryo-EM Images and AlphaFold2 Prediction Comparison
To validate the prediction of AlphaFold2 models, the Cryo-EM images obtained for α1 homopentamer, β2 homopentamer, and GABAA receptor heteropentamer were compared with the AlphaFold2 protein structures. The highest-ranked models were inputted into Chimera software to generate a surface model that could match the shapes of the Cryo-EM images. Since the resolution of the α1 homopentamer is the highest, four Cryo-EM images were chosen for comparison, while two images of the β2 homopentamer, and GABAA receptor heteropentamer were used. α1 homopentamer models show high similarities with the Cryo-EM images, as seen in Figure 5a-d with different points of view of the protein structure. β2 homopentamer has a rough structure of a pentamer and the side view shows a similar shape, but due to the low resolution of the Cryo-EM images obtained, it was difficult to model and match with AlphaFold2 models. GABAA receptor heteropentamer only had top and bottom views of the pentamer and not enough images of the side view. Nevertheless, the two images chosen successfully show the pentameric structure and a central cavity in the protein structure, which is also modelled by AlphaFold2. Interestingly, the protruding residues of the γ2 subunit also seem to be shown on the Cryo-EM images (red arrow in Figure 5g-h), suggesting that the γ2 subunit fragment obtained in the experiment can indeed bind and form heteropentamers.
2.6. Conserved Trp Residues in the GABAA Receptor α1, β2, and γ2 Subunits
Previous studies show that Trp residues located in the Cys-loop’s proximity may affect the structural stability of the receptors [42,44]. Hence, the AlphaFold2 structures were examined for any Trp residues packed in or near the Cys-loop, but none was found. Since Trp is one of the least common amino acids, further exploration was performed on all the Trp residues in the subunit fragments. Observation identified two Trp residues in close proximity to each other in all three subunit fragments. Subsequent analysis showed that these two residues are an identified pair of Trp residues conserved in the ECD of Cys-loop receptors within the WxD and WxPD motifs [44], as shown in Figure 10. All three pairs of Trp residues are less than 6Å apart, and some may say that they are within structural contact [83], perhaps for establishing pi-pi stacking of aromatic rings. However, other studies mentioned that inter-residue proximity requires a minimum atom distance of less than 5Å [84], thus the actual form of molecular interaction between these two residues cannot be firmly established with the currently available information. Yet, it can be certain that these two conserved residues play a role in the formation of GABAA receptors, and may function to stabilize the ECD [44].
Figure 6.
The conserved tryptophan pair in the α1, β2, and γ2 subunits. (a-c) conserved Trp residues are closely related to each other, with distances of less than 6Å apart, as depicted with Chimera. (d) Sequence comparison of the α1, β2, and γ2 subunits show the WxD and WxPD motifs, highlighted in red. The pair of conserved Trp residues are outlined with a black box. The Trp residues are all 25 residues apart.
Figure 6.
The conserved tryptophan pair in the α1, β2, and γ2 subunits. (a-c) conserved Trp residues are closely related to each other, with distances of less than 6Å apart, as depicted with Chimera. (d) Sequence comparison of the α1, β2, and γ2 subunits show the WxD and WxPD motifs, highlighted in red. The pair of conserved Trp residues are outlined with a black box. The Trp residues are all 25 residues apart.

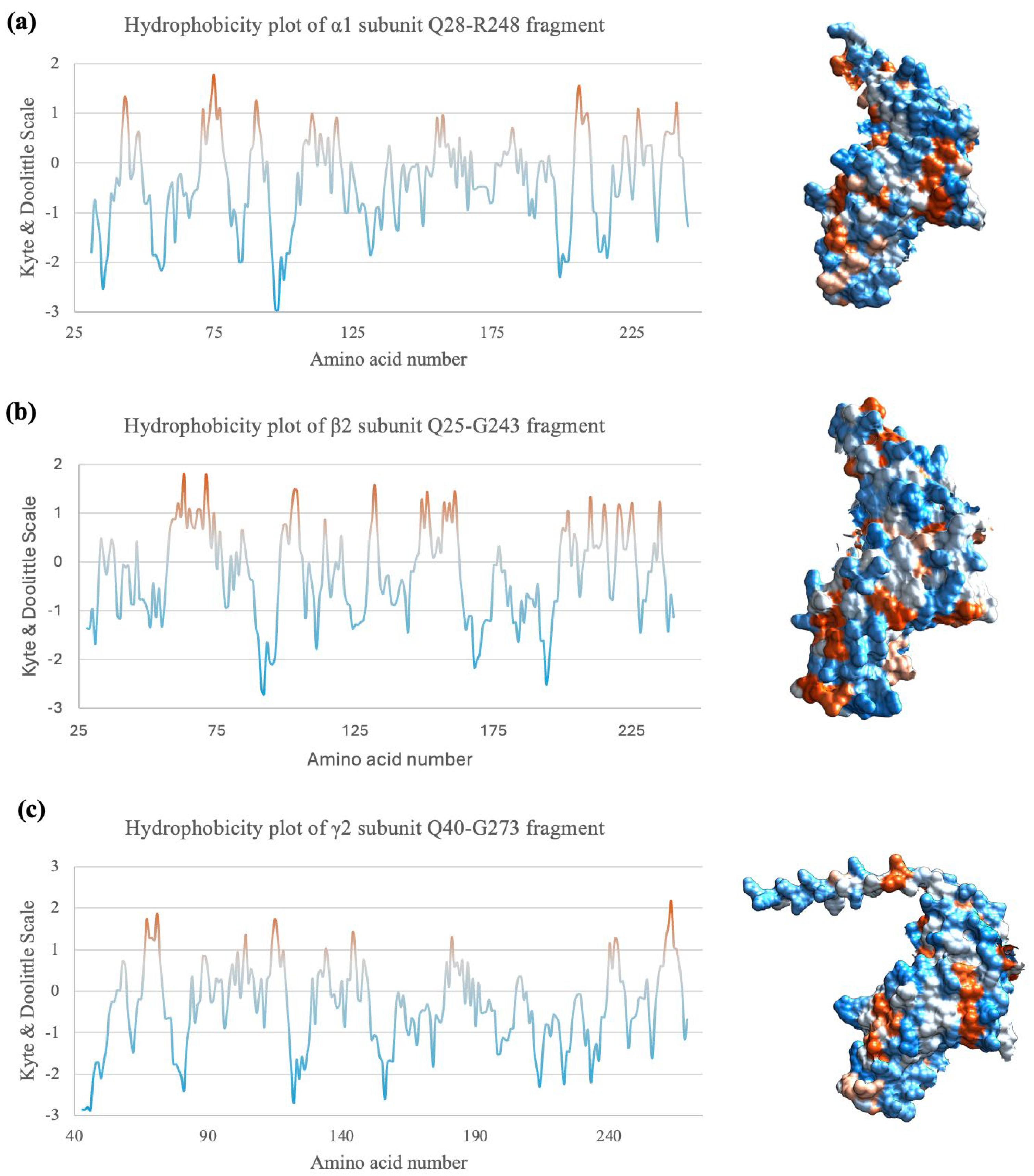
2.7. Hydrophobicity Plot Indicates Subunit Fragments Are Relatively Hydrophilic
The GABAA receptor subunits contain a hydrophilic extracellular domain (ECD) and four transmembrane domains. The fragments expressed in this study are all part of the ECD, thus it should be more hydrophilic on average [10,11]. Indeed, the hydrophobicity plots computed with ProtScale using Kyte-Doolittle analysis [56,57] in Figure 6 indicate that all three subunit fragments are relatively hydrophilic. In addition, according to the Kyte-Doolittle scale, a given segment with an average hydrophobicity score of +1.6 or above may suggest a possible transmembrane domain [58]. To confirm this on the AlphaFold2 model and test the accuracy of the models, the Q28-L296 fragment of the α1 subunit [40,59] was computed with AlphaFold2 and ProtScale for an additional homopentamer prediction. AlphaFold2 prediction of secondary structure is more than 97% identical with the shorter Q28-R248 fragment, with one residue difference in secondary structure at two positions, and a missing 3-residue alpha helix structure (Supplementary Figure S3). A previous study indicated that the transmembrane domains TM1 and TM2 of the α1 subunit sequence correspond to the residues F254-L274 and P280-R301 [45]. The full TM1 segment and part of the TM2 are included in the additionally computed Q28-R248 fragment. The hydrophobicity plot indeed had a much higher hydrophobicity in the transmembrane domain sequences, but only with an average of around 0.95 for TM1 and 0.45 for part of TM2 (Supplementary Figure S4). However, only a partial segment of the TM2 was computed, thus it may not be very reliable. Nevertheless, surface hydrophobicity may affect protein stability and assembly of protein structures [60,61]. Hence, hydrophobicity studies may help determine the biochemistry of protein complex assembly.
Figure 7.
Hydrophobicity of the three subunit fragments. (a-c) Hydrophobicity plots according to the amino acid sequence of each subunit fragment. A negative score on the Kyle-Doolittle scale represents hydrophilicity and a positive score means the residue is relatively hydrophobic. Hydrophobicity surface depiction on the right, as portrayed by Chimera. Orange = hydrophobic residues, white = neutral residues, and blue = hydrophilic residues.
Figure 7.
Hydrophobicity of the three subunit fragments. (a-c) Hydrophobicity plots according to the amino acid sequence of each subunit fragment. A negative score on the Kyle-Doolittle scale represents hydrophilicity and a positive score means the residue is relatively hydrophobic. Hydrophobicity surface depiction on the right, as portrayed by Chimera. Orange = hydrophobic residues, white = neutral residues, and blue = hydrophilic residues.
3. Discussion
3.1. α1. and β2 Subunits of GABAA Receptors Form Homopentamers
The current study successfully expressed and purified the Gln28-Arg248 fragment of the α1 subunit, Gln25-Gly243 of the β2 subunit, and Gln40-Gly273 of the γ2 subunit of the GABAA receptor. Fast protein liquid chromatography (FPLC), SDS-PAGE and negative staining were carried out for each sample to confirm the sizes and purity of the proteins, as well as observe the aggregation levels. Results showed low protein aggregation and high homogeneity for α1 subunit, but some aggregation were present for β2 subunit, possibly due to issues related to the low concentration and strength of the detergent used, which will need further refinement. The concentration of the γ2 subunit was high but seems to be unable to form homopentamers. Yet, it may also be due to possible detergent issues or complications during the protein refolding stage, hence more trials would be needed to purify both the β2 and γ2 proteins to confirm the results obtained in this study. Nevertheless, results from negative staining and 2D classification of α1 and β2 protein samples demonstrate that they can form rosette-like homo-oligomers, mostly homopentamers.
3.2. Gln28-Arg248 α1 Subunit Fragments Form Stable Homopentamers
Previously, homopentamer formation from the Cys 166-Leu 248 segment of the rat mature α1 subunit protein was demonstrated [40]. Essential protein residues contributing to the benzodiazepine (BZ) binding pocket on the α1 subunit were indicated in the study, including His101, Tyr159, and Tyr209, as numbered in rat mature protein [41]. Converting to GABRA1 amino acid sequence numbering would assign the residues as His102, Tyr 160, and Try 209 (with Gln28 as Gln1). These residues have also been shown to be essential to the binding of BZ with the GABAA receptor α1 subunit in various studies [46,62,63,64]. The α1 fragment designed in the current study also consists of these residues. These residues correlate to another study indicating the BZ binding pocket is composed of a series of residues forming five binding-site loop regions, with three loops on the α1 subunit and two loops on the γ2 subunit [65]. If we hypothesise that the formation of homopentamers acts as intermediates before the assembly of α1β2γ2 GABAA receptors [66], it is natural to investigate whether the folding of α1 homopentamers correlates with the BZ binding pocket structure. In Figure 6, the binding-site loops are shown with the hydrogen bonds (H-bonds) identified by AlphaFold2 between the α1 fragments, giving insights into the assembly of α1 homopentamers. Interestingly, the majority of the H-bonds were within the BZ binding pocket loops, which may suggest the importance of correct refolding of the BZ binding pocket during the α1 homopentamer assembly mechanism. Supplementary Figure S5 shows the other H-bonds identified by AlphaFold2 and only Tyr160 is involved in the H-bonds and considered as one of the main residues affecting BZ binding. This may suggest that not only Try160 plays a role in BZ binding, but also the initial formation of the BZ binding site, further indicating the importance of this residue.
Figure 8.
AlphaFold2 structure of α1 subunit homopentamer with BZ binding-site loops and hydrogen bonds. Two chains of the α1 fragments are shown to visualise the interface, other chains are hidden for clarity. The three BZ binding-site loops are coloured as Loop A (Trp95 – Lys105), Loop B (Leu155 – Val167), and Loop C (Thr197 – Thr215) in orange, green and pink respectively, numbered with Gln28 as Gln1. All images are visualised from Chimera. (a) the α1- α1 interface of the homopentameric model in AlphaFold2 prediction. (b) BZ binding pocket loops in between the two fragments, expanded. (c) Inter-residue hydrogen bonds identified in AlphaFold2 prediction of α1 subunit homopentamer. Residues involved are labelled with name, and residue number on both fragment chains. Residues are numbered based on Gln28 as Gln1. Each chain is linked to its neighbour by hydrogen bonds depicted as red lines between residues.
Figure 8.
AlphaFold2 structure of α1 subunit homopentamer with BZ binding-site loops and hydrogen bonds. Two chains of the α1 fragments are shown to visualise the interface, other chains are hidden for clarity. The three BZ binding-site loops are coloured as Loop A (Trp95 – Lys105), Loop B (Leu155 – Val167), and Loop C (Thr197 – Thr215) in orange, green and pink respectively, numbered with Gln28 as Gln1. All images are visualised from Chimera. (a) the α1- α1 interface of the homopentameric model in AlphaFold2 prediction. (b) BZ binding pocket loops in between the two fragments, expanded. (c) Inter-residue hydrogen bonds identified in AlphaFold2 prediction of α1 subunit homopentamer. Residues involved are labelled with name, and residue number on both fragment chains. Residues are numbered based on Gln28 as Gln1. Each chain is linked to its neighbour by hydrogen bonds depicted as red lines between residues.

The most abundant form of the GABAA receptor consists of two α1 subunits, with the BZ binding site between the α1 and γ2 subunits, and the GABA binding site between the α1 and β2 subunits. This suggests that the α1 subunit is vital for GABAergic transmissions. However, one study showed that α subunits in GABAA receptors are not essential for the binding of GABA. The study also suggested that when α subunit expression is low, GABA receptors with only the β2 and γ2 subunits can be formed to replace the predominant α1β2γ2 form GABAA receptors [67], signifying equal importance of β2 and γ2 subunits. Besides, this may suggest the notion of formation of α1 homopentamers as an intermediate before the assembly of α1β2γ2 GABAA receptors [66]. A possibility can be considered that the formation of subunit homo-oligomers may control the availability of subunits to the formation of the receptor, or key residues in the subunits may signal the assembly of the receptor [68]. This undoubtedly creates invaluable insights into the flexibility and complexity of GABAA receptor formation to allow the critical function of GABAergic transmission.
3.3. β2. Subunit Fragments Homopentamers Possible Assembly Mechanism
A previous study has shown that the GABAA receptor β3 subunit can form homopentamers [50]. Miller and Aricescu have indicated several attributes in the assembly of the crystal structure of a β3 homopentamer. The homopentamer was analysed from the extracellular domain interface, which is also the case for the β2 fragment expressed in this study. Figure 7 shows the amino acid sequence of the β3 subunit, which is mostly conserved in the β2 subunit fragment used in this study. Coincidentally, Gln26 is considered as Gln1 in Miller and Aricescu’s β3 subunit crystal structure, thus making the model easily comparable to the Gln25-Gly243 β2 subunit fragment in the current study, as the sequence inputted to AlphaFold2 begins with β2 Gln25 as Gln1.
Figure 9.
Sequence alignment of the β2 Gln25-Gly243 fragment and the β3 subunit sequence. Alignment shows that the two sequences are 89.95% identical.
Figure 9.
Sequence alignment of the β2 Gln25-Gly243 fragment and the β3 subunit sequence. Alignment shows that the two sequences are 89.95% identical.

Specifically, Miller and Aricescu demonstrated a series of hydrogen bonds between the extracellular domains at the side chains Arg26 and Asp17 to Asp24 and Lys13, along with hydrogen bonds surrounding Arg86. Since the subunit fragments are both extracellular domains involving most of the binding sites, the AlphaFold2 prediction of β2 subunit homopentamer can be compared with the model of the β3 subunit crystal structure. As shown in Figure 8a, there are hydrogen bonds formed between residues Arg26 and Asp17, as well as between Asp23 and Lys13, which correlates with Miller and Aricescu’s study. However, AlphaFold2 presents a bond at Asp84 instead of Arg86, which may suggest slight structural differences in the interaction of residues. Yet, it is important to note that AlphaFold2 only provides a prediction of protein structures based on existing models, hence it is still necessary to perform experimental studies to confirm the prediction on complex structures such as the pentamers in this study.
Figure 10.
Inter-residue hydrogen bonds identified in AlphaFold2 prediction of β2 subunit homopentamer. (a) β2 homopentamer with chains A and B shown, the other three chains are hidden for clarity. Black and red squares correspond to the (b-c) two detailed architecture of hydrogen bonds between two chains of the β2 subunit homopentamer. Residues involved are highlighted in orange and labelled with name, residue number and different chains (.A = chain A, .B = chain B). Each chain is linked to its neighbour by hydrogen bonds depicted as red lines between residues. Residues are numbered based on β2 subunit Gln25 as Gln1.
Figure 10.
Inter-residue hydrogen bonds identified in AlphaFold2 prediction of β2 subunit homopentamer. (a) β2 homopentamer with chains A and B shown, the other three chains are hidden for clarity. Black and red squares correspond to the (b-c) two detailed architecture of hydrogen bonds between two chains of the β2 subunit homopentamer. Residues involved are highlighted in orange and labelled with name, residue number and different chains (.A = chain A, .B = chain B). Each chain is linked to its neighbour by hydrogen bonds depicted as red lines between residues. Residues are numbered based on β2 subunit Gln25 as Gln1.

3.4. Complexity of γ2 Subunit of the GABAA Receptor
In this study, negative staining of the γ2 subunit fragments after protein refolding did not show a formation of homo-oligomers. As seen in Figure 4c, the two different secondary structures of the γ2 fragment may indicate that possible conformational changes necessary for the assembly of homopentamers are not induced [66]. This aligns with previous studies that showed the expression of the γ2 subunit alone could not produce functional GABA receptors [69,70,71,72]. However, one study revealed that the human γ2L subunit was able to form functional receptors in Xenopus oocytes [73]. Thus, current evidence is inconsistent and further investigations are required to confirm the ability of the γ2 subunit to form functional homopentamers.
It has also been suggested that homo-oligomeric formation may be a preferred intermediary mechanism when other types of subunits are absent [66]. This is suggested by the evidence that no two of the same subunits are adjacent to each other in the predominant GABAA receptors. If it is eventually confirmed that γ2 does not form homo-oligomers, perhaps it can be further hypothesized that γ2 is the last subunit to be incorporated into the receptor as there are no necessary intermediary structures. Although it is undetermined whether γ2 simply cannot form homo-oligomers, or whether they merely do not prefer to form homo-oligomers due to their structure, their function in the GABAA receptor is undeniably crucial. In fact, more than 50% of the GABAA receptor isoforms in the rodent brain consist of the γ2 subunit [74].
Interestingly, one study demonstrated that α1 subunits can reach the cell surface only in the presence of β2 and γ2 subunits [30]. Multiple studies have elaborated on the relationship between γ2 and receptor trafficking. In addition, Golgi-specific zinc finger protein (GODZ) was presented as an important binding partner of intracellular γ2 subunits, suggesting an association with vesicular membrane receptor coupling [75]. Nonetheless, the main evidence of γ2’s association with receptor trafficking is its relationship with gephyrin – a submembrane scaffolding protein. Gephyrin is a multifunctional protein known for its role in postsynaptic clustering and anchoring of GABAA receptors [76,77,78,79,80]. In particular, one study indicated that the fourth transmembrane domain of the γ2 subunit is essential for the postsynaptic clustering of GABAA receptors [79]. Some studies also indicate a role of the α2 subunit in binding gephyrin [81,82], but only the γ2 subunit was shown to rescue postsynaptic clustering of GABAA receptors in γ2-/- neurons [79].
These studies all highlight the potential importance of γ2 subunits in the trafficking of α1β2γ2 GABAA receptors. Perhaps it can be further hypothesized that the γ2 subunit is the limiting factor of postsynaptic clustering of GABAA receptors, thus supporting the initial hypothesis of the γ2 subunit being the last subunit to be assembled, as once the γ2 subunit is present, postsynaptic clustering of the predominant receptor will occur. Regardless, many more studies are needed to investigate these hypotheses and thoroughly explore the assembly mechanisms of GABAA receptors. Understanding these mechanisms may also provide invaluable insights into the trafficking mechanism of similar receptors, as well as their ligand-binding abilities.
3.5. Constructed α1β2γ2 Heteropentameric GABAA Receptors
Apart from homopentamer construction, expressed fragments of α1, β2, and γ2 subunits were combined in the predominant 2:2:1 ratio to study the oligomeric structure. Results indicated that subunits combined in this ratio were able to form heteropentamers in a stable manner (Figure 3g). This confirms that the three subunit fragments expressed in the study can form heteromeric structures with each other. Although the 2:2:1 pentamers had stable protein structure, negative staining results showed some aggregation and the sample concentration was relatively low. Therefore, further investigations are needed to obtain samples with higher concentration and purity. Overall, the protein samples still provide a useful model for studying the homopentameric nature of the receptors. In our future studies, higher resolution of the GABAA receptors’ structure will be needed to obtain the atomic structure of the receptors and identify the key residues of drug binding.
Heteropentameric GlyR has been suggested to be formed in 2:3 or 3:2 ratios of α and β subunits respectively [80,85], thus this study also aimed to find out whether GABAA receptors α and β subunits can form heteropentamers in similar 2:3 and 3:2 ratios. However, no protein complexes were seen in combining the two subunits. Since these combinations may not be naturally present in the human body, perhaps AlphaFold2 can first be used to model the structures and predict the possibility of them forming pentamers. This not only aids in the understanding of subunit binding mechanisms of the GABAA receptor but may also indicate the essential role of γ2 subunits in receptor formation – if α and β subunits truly do not prefer to form pentamers without a third subunit type.
3.6. Potential of Using AlphaFold2 for Aided Experimental Design and Evaluation
Since the launch of the AlphaFold2 system and the release of the open-source software, its potential for various applications in biology and medicine has allowed it to gain a reputation. It has been suggested that AlphaFold2 can be harnessed in processes such as drug discovery [86] and modelling protein-protein interaction [87,88]. Still, the majority of studies apply AlphaFold2 to aid the determination of protein structures. This prediction algorithm can be used for homology modelling, de-novo modelling and ML-based modelling [89], most importantly, the predicted structures can be used along with other structural protein techniques to accelerate the process of protein structure identification. There have been numerous successful studies utilizing AlphaFold2 with X-ray crystallography [90,91] and cryo-EM [92,93,94,95]. Although the system has proved to be a useful tool in the structural biology field, it is still crucial to perform experiments to confirm the predictions. Accordingly, the conclusive structure of the GABAA receptor and its subunit homopentamers can only be confirmed by more structural studies such as X-ray crystallography, cryo-EM, or mass spectrometry, especially when studies are needed to mimic the native environment of the receptor.
An increasing number of studies have revealed the association of GABAA receptors with various neuropsychiatric disorders, such as schizophrenia, epilepsy, and Alzheimer’s disease [96,97,98,99]. Thus, understanding the mechanisms of the GABAA receptor is crucial for combatting these CNS disorders. Ultimately, a high-resolution structure for an important family of neurotransmitter receptors at the atomic level will pave the way to new drug design and therapeutics for various neuropsychiatric diseases.
4. Materials and Methods
Methods are adapted from Xue et al. [59]
4.1. Buffers
The truncated protein samples of each subunit were obtained as inclusion bodies and refolded into their active forms using buffers. Then the proteins were further purified in soluble forms by using different detergents, aiding the proper refolding and improving the homogeneity of the samples for further, high-resolution studies. The buffers used were:
- Wash Buffer A: 50mM Tris-Cl, pH 8.0 and 10mM EDTA.
- Lysis buffer: 100mM Tris-Cl, pH 8.0, 10mM EDTA 5mM DTT, 100mM NaCl, 10% Glycerol and 200µg/ml lysozyme.
- Wash Buffer B: 100mM Tris-Cl, pH 8.0, 10mM EDTA 5mM DTT, 100mM NaCl, 10% Glycerol 2M urea and 2% deoxycholic acid.
- Elution Buffer: 10 mM glycine pH 10.3 and 2% sodium dodecyl sulfate (SDS).
4.2. Cloning and Protein Expression
Prokaryotic expression vector, specifically the pTrcHis vector, was used to introduce the bovine gene of interest into E. coli strain – NovaBlue, to express the proteins. The culture was prepared using 1ml stock of the competent cells, 100ml of LB broth and 100μg/ml of ampicillin. The culture was incubated overnight at 37ºC and then transferred into 1L baffled bottles containing LB broth and 100μg/ml ampicillin. Optical density (OD) at 600nm was observed until reached between 0.4-0.5 and then induced with IPTG with a final concentration of 0.8mM. Cells were collected after 3-4 hours of growth at 37ºC shaker. Cells were harvested by centrifugation at 4200rpm, 4ºC for 20 minutes. The pellet containing the bacterial cells was washed with 200ml of wash buffer A and centrifuged again at 5500rpm, 4ºC for 20 minutes. The cell pellet was kept at -20ºC before purification.
4.3. Cell Purification
The bacterial cell pellet was resuspended in lysis buffer and shaken for 1 hour at 37°C. Cells were centrifuged at 12000 rpm at 4°C for 20 minutes and the supernatant was discarded. The pellet was resuspended in wash buffer B and sonicated for 30s then centrifuged at 12000 rpm at 4°C for 20 minutes and the supernatant was discarded. The cell lysate was washed with the same experimental steps with wash buffer B for 4 times. The pellet was washed with 4M urea, followed by Milli-Q water, and centrifuged at 12000 rpm at 4°C for 20 minutes with each wash respectively. Then, 7M guanidine hydrochloride (GnCl) was added to the lysate and shaken at room temperature for an hour. Centrifuge again at 18000 rpm and 4°C for 20 minutes. The function of GnCl was to solubilise the inclusion bodies and allow the protein to bind with water. After washing with GnCl, the supernatant was kept instead of the pellet. To form precipitation of the protein, 5.3g of (NH4)2SO4 was gradually added into the supernatant, making sure it dissolved properly. The solution was left at room temperature for a minimum of 4 hours or even overnight, then the precipitate was washed with ice-cold distilled water 3 times and centrifuged at 18000 rpm, 4°C for 15 minutes. The protein pellet was collected and solubilized in 4% SDS and 100μl β-mercaptoethanol followed by shaking overnight.
4.4. Protein Refolding
After solubilization, a 0.22µm syringe filter was used to remove large particles from the pellet. The filtered pellet was loaded into a 200 Superdex 200 HR 60/60 column for Fast Protein Liquid Chromatography gel filtration (FPLC). The samples were collected by adding elution buffer into the column at a rate of 1ml/min. Eluents were collected in numbered tubes and a NanoDrop Spectrophotometer (at A280) was used to measure the concentration of proteins in each tube. The samples with higher protein concentrations were selected and used for 12% SDS-PAGE with commissive brilliant blue stain. After SDS-PAGE, samples that were confirmed to have the correct size and higher purity were dialyzed for the negative staining.
4.5. Constructing Heteropentamers
Other than the individual subunits, the samples were also combined into a GABAA receptor pentamer with a 2:2:1 ratio before column elution to investigate their ability to form heteropentamers. 2ml of the α1 subunit, 2ml of the β2 subunit, and 1ml of the γ2 subunit of the same were combined and put into the shaker for one hour to allow them to interact and form oligomers. 10% SDS-PAGE was done for the complex.
Previously, studies showed that α1 and β3 subunits formed pentamers in vitro [100] α1 and β2 subunits combinations produced functional surface expression [33,101,102], hence in this study, the α1 and β2 subunit fragments were also combined in 2:3 and 3:2 ratios to attempt pentameric formation. Unfortunately, the pentameric formation was not achieved and no structures were seen.
4.6. Proteins Dialysis
Protein samples were transferred into semipermeable membranes submerged in different concentrations of SDS and Glycine solutions. Firstly, the samples were submerged in 2 litres of 1.5% SDS with 10mM Glycine (pH 10.3), then transferred to 2 litres of 1.0% SDS with 10mM Glycine. Then gradual dilutions from 0.8% SDS → 0.6% SDS → 0.4% SDS → 0.2% SDS, each with 10mM Glycine. Lastly, dialysis was done in a 4*4 litre pure 10mM Glycine solution (pH10.3) for 4 days. Then, protein samples were filtered, and OD and molecular weight were checked before negative staining.
4.7. Negative Staining of the Protein
The copper mesh used for negative staining was hydrophilized using a plasma cleaner and vacuumed for 2 minutes. After that, both hydrophilization and vacuum pump were turned off until the balance was completed, and the copper mesh was removed. 2.5 µl of the protein samples were loaded into the copper mesh and incubated for 1 minute. 4µl of 2% uranyl acetate was added and incubated for 1 minute. Excess liquid was absorbed using filter paper. Only a thin layer of uranium acetate was retained, dried, and examined by electron microscopy.
4.8. Computational Prediction of Protein Structure Using AlphaFold2
Novel computational approaches have emerged in recent years due to the increasing focus on bioinformatics and the advancement of technology. It is not surprising that artificial intelligence and machine learning have been used to develop a model for the prediction of 3D protein structures. AlphaFold [51] is an open-source neural network-based method for predicting protein structures with atomic accuracy. By using multi-sequence alignment and available knowledge about the target protein, AlphaFold can create a visual model of the structure [103].
First, the sequence of the fragments is obtained from UniProt [55] and applied to the open-source AlphaFold2 interactive Python Notebook in Google Colab. Sampling options were edited based on a few parameters. Recycling uses the previous output for the next iteration’s input. The number of recycling stops after a certain tolerance level (tol), which was set to RMSD of 0.5Å. When subsequent predictions reach the threshold of 0.5Å difference from the previous prediction, recycling will terminate, and a predicted model will be generated. The predicted local-distance difference test (pLDDT) score shows the confidence level of the prediction. The estimate of template modelling score (pTM) assesses the topological similarity of protein structures. It is suggested that the multimer interface pTM score (ipTM) score can have a confidence cut-off of 0.75 [104]. AlphaFold2 Multimer_v3 Model was used at the time of performing this study. The highest-ranked prediction model was then analysed in Chimera software for secondary structure identification.
4.9. Hydrophobicity Plots
Hydrophobicity plots were generated with Protein Identification and Analysis Tools on the ExPASy Server – ProtScale. The UniProt/SwissProt accession number is used to retrieve the amino acid sequence of each subunit, and the amino acid range is selected according to each fragment. The window size of 7 is used for the subunit fragments, as it is aimed at finding hydrophilic regions exposed on the surface and may potentially be antigenic [57]. In this case, the ECD regions exposed on the cell surface are related to ligand binding. A higher window size of 19 was recommended to identify more hydrophobic residues and transmembrane domains [57]. In this case, the Q28-L296 fragment included two transmembrane domains of the α1 subunit, hence the window size 19 was used for the additional hydrophobicity plot (Supplementary Figure 3).
5. Conclusions
The α1 and β2 subunit fragments expressed in this study successfully formed homo-oligomers, mostly homopentamers, as seen from negative staining and Cryo-EM results. However, it is still undetermined whether the γ2 subunit can form homo-oligomers like its counterparts. Efforts were also made to form the α1β2γ2 GABAA receptors, and results show a pentameric structure with a central cavity. AlphaFold2 was used to model each pentameric structure to predict the structural characteristics and assembly mechanism of the subunits and the receptor. Results were compared with previous studies to gain more insights into the possible assembly mechanism of an important neurotransmitter receptor that may affect ligand-binding processes and drug development for neuropsychiatric diseases associated with the GABAA receptor.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Figure S1. Prediction quality and error estimates by AlphaFold2; Supplementary Figure S2. AlphaFold2 multiple sequence alignment (MSA) and predicted pentameric structures coloured by pLDDT. Supplementary Figure S3. α1 subunit Q28-L296 sequence and structure prediction from AlphaFold2. Supplementary Figure S4. Hydrophobicity of the α1 subunit Q28-L296 fragment. Supplementary Figure S5. AlphaFold2 prediction of hydrogen-bonds (H-bond) between two α1 subunit fragments in the homopentamer.
Author Contributions
Conceptualization, A.U., C.K., and H.X.; methodology, A.U., C.K., and H.X.; software, C.K.; validation, A.U. and C.K.; formal analysis, C.K.; investigation, A.U. and C.K.; resources, H.X.; data curation, A.U. and C.K.; writing—original draft preparation, A.U. and C.K.; writing—review and editing, C.K. and H.X.; visualization, C.K.; supervision, H.X.; project administration, H.X.; funding acquisition, C.K. and H.X. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the University Grants Council (UROP20SC15). We thank the HKUST UROP Support Grant for funding and supporting our study.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We acknowledge the support of HKUST and would like to thank Dr. Flora Mat for her support. We would also like to thank Professor Shangyu Dang and his team for the support of Cryo-EM imaging.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sieghart, W. Structure, Pharmacology, and Function of GABAA Receptor Subtypes. In Advances in Pharmacology; GABA; Academic Press, 2006; Vol. 54, pp. 231–263.
- Goetz, T.; Arslan, A.; Wisden, W.; Wulff, P. GABA(A) Receptors: Structure and Function in the Basal Ganglia. Prog. Brain Res. 2007, 160, 21–41. [Google Scholar] [CrossRef]
- Olsen, R.W.; Li, G.-D. Chapter 18 - GABA. In Basic Neurochemistry (Eighth Edition); Brady, S.T., Siegel, G.J., Albers, R.W., Price, D.L., Eds.; Academic Press: New York, 2012; pp. 367–376; ISBN 978-0-12-374947-5. [Google Scholar]
- Elgarf, A.A.; Siebert, D.C.B.; Steudle, F.; Draxler, A.; Li, G.; Huang, S.; Cook, J.M.; Ernst, M.; Scholze, P. Different Benzodiazepines Bind with Distinct Binding Modes to GABAA Receptors. ACS Chem. Biol. 2018, 13, 2033–2039. [Google Scholar] [CrossRef] [PubMed]
- Salari, R.; Murlidaran, S.; Brannigan, G. Pentameric Ligand-Gated Ion Channels : Insights from Computation. Mol. Simul. 2014, 40, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Becker, C.-M.; Betz, H. Inhibitory Glycine Receptors: An Update. J. Biol. Chem. 2012, 287, 40216–40223. [Google Scholar] [CrossRef]
- Waxham, M.N. Chapter 10 - Neurotransmitter Receptors. In From Molecules to Networks (Third Edition); Byrne, J.H., Heidelberger, R., Waxham, M.N., Eds.; Academic Press: Boston, 2014; pp. 285–321; ISBN 978-0-12-397179-1. [Google Scholar]
- Nemecz, Á.; Prevost, M.S.; Menny, A.; Corringer, P.-J. Emerging Molecular Mechanisms of Signal Transduction in Pentameric Ligand-Gated Ion Channels. Neuron 2016, 90, 452–470. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.; Wakimoto, H.; Fujita, N.; Lalande, M.; Barnard, E.A. Analysis of the Set of GABAA Receptor Genes in the Human Genome*. J. Biol. Chem. 2004, 279, 41422–41435. [Google Scholar] [CrossRef]
- Ghit, A.; Assal, D.; Al-Shami, A.S.; Hussein, D.E.E. GABAA Receptors: Structure, Function, Pharmacology, and Related Disorders. J. Genet. Eng. Biotechnol. 2021, 19, 123. [Google Scholar] [CrossRef]
- Sigel, E.; Steinmann, M.E. Structure, Function, and Modulation of GABAA Receptors. J. Biol. Chem. 2012, 287, 40224–40231. [Google Scholar] [CrossRef]
- Baumann, S.W.; Baur, R.; Sigel, E. Forced Subunit Assembly in Alpha1beta2gamma2 GABAA Receptors. Insight into the Absolute Arrangement. J. Biol. Chem. 2002, 277, 46020–46025. [Google Scholar] [CrossRef]
- Barki, M.; Xue, H. GABRB2, a Key Player in Neuropsychiatric Disorders and Beyond. Gene 2022, 809, 146021. [Google Scholar] [CrossRef]
- Puthenkalam, R.; Hieckel, M.; Simeone, X.; Suwattanasophon, C.; Feldbauer, R.V.; Ecker, G.F.; Ernst, M. Structural Studies of GABAA Receptor Binding Sites: Which Experimental Structure Tells Us What? Front. Mol. Neurosci. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Renard, S.; Olivier, A.; Granger, P.; Avenet, P.; Graham, D.; Sevrin, M.; George, P.; Besnard, F. Structural Elements of the γ-Aminobutyric Acid Type A Receptor Conferring Subtype Selectivity for Benzodiazepine Site Ligands*. J. Biol. Chem. 1999, 274, 13370–13374. [Google Scholar] [CrossRef] [PubMed]
- Claxton, D.P.; Gouaux, E. Expression and Purification of a Functional Heteromeric GABAA Receptor for Structural Studies. PLOS ONE 2018, 13, e0201210. [Google Scholar] [CrossRef]
- Barker, J.S.; Hines, R.M. Regulation of GABAA Receptor Subunit Expression in Substance Use Disorders. Int. J. Mol. Sci. 2020, 21, 4445. [Google Scholar] [CrossRef] [PubMed]
- Sieghart, W.; Fuchs, K.; Tretter, V.; Ebert, V.; Jechlinger, M.; Höger, H.; Adamiker, D. Structure and Subunit Composition of GABAA Receptors. Neurochem. Int. 1999, 34, 379–385. [Google Scholar] [CrossRef]
- Laurie, D.J.; Wisden, W.; Seeburg, P.H. The Distribution of Thirteen GABAA Receptor Subunit mRNAs in the Rat Brain. III. Embryonic and Postnatal Development. J. Neurosci. 1992, 12, 4151–4172. [Google Scholar] [CrossRef]
- Smith, G.B.; Olsen, R.W. Identification of a [3H]Muscimol Photoaffinity Substrate in the Bovine Gamma-Aminobutyric acidA Receptor Alpha Subunit. J. Biol. Chem. 1994, 269, 20380–20387. [Google Scholar] [CrossRef] [PubMed]
- Westh-Hansen, S.E.; Rasmussen, P.B.; Hastrup, S.; Nabekura, J.; Noguchi, K.; Akaike, N.; Witt, M.R.; Nielsen, M. Decreased Agonist Sensitivity of Human GABA(A) Receptors by an Amino Acid Variant, Isoleucine to Valine, in the Alpha1 Subunit. Eur. J. Pharmacol. 1997, 329, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Westh-Hansen, S.E.; Witt, M.R.; Dekermendjian, K.; Liljefors, T.; Rasmussen, P.B.; Nielsen, M. Arginine Residue 120 of the Human GABAA Receptor Alpha 1, Subunit Is Essential for GABA Binding and Chloride Ion Current Gating. Neuroreport 1999, 10, 2417–2421. [Google Scholar] [CrossRef]
- Nors, J.W.; Gupta, S.; Goldschen-Ohm, M.P. A Critical Residue in the α1M2–M3 Linker Regulating Mammalian GABAA Receptor Pore Gating by Diazepam. eLife 10, e64400. [CrossRef]
- Shao, J.; Kuiper, B.P.; Thunnissen, A.-M.W.H.; Cool, R.H.; Zhou, L.; Huang, C.; Dijkstra, B.W.; Broos, J. The Role of Tryptophan in π Interactions in Proteins: An Experimental Approach. J. Am. Chem. Soc. 2022, 144, 13815–13822. [Google Scholar] [CrossRef]
- Dougherty, D.A. Cation-Pi Interactions Involving Aromatic Amino Acids. J. Nutr. 2007, 137, 1504S–1508S; discussion 1516S-1517S. [Google Scholar] [CrossRef] [PubMed]
- Laverty, D.; Desai, R.; Uchański, T.; Masiulis, S.; Stec, W.J.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Steyaert, J.; Miller, K.W.; et al. Cryo-EM Structure of the Human A1β3γ2 GABAA Receptor in a Lipid Bilayer. Nature 2019, 565, 516–520. [Google Scholar] [CrossRef]
- Sigel, E.; Baur, R.; Kellenberger, S.; Malherbe, P. Point Mutations Affecting Antagonist Affinity and Agonist Dependent Gating of GABAA Receptor Channels. EMBO J. 1992, 11, 2017–2023. [Google Scholar] [CrossRef]
- O’Shea, S.M.; Harrison, N.L. Arg-274 and Leu-277 of the γ-Aminobutyric Acid Type A Receptor A2 Subunit Define Agonist Efficacy and Potency *. J. Biol. Chem. 2000, 275, 22764–22768. [Google Scholar] [CrossRef] [PubMed]
- Barik, S. The Uniqueness of Tryptophan in Biology: Properties, Metabolism, Interactions and Localization in Proteins. Int. J. Mol. Sci. 2020, 21, 8776. [Google Scholar] [CrossRef]
- Khemaissa, S.; Sagan, S.; Walrant, A. Tryptophan, an Amino-Acid Endowed with Unique Properties and Its Many Roles in Membrane Proteins. Crystals 2021, 11, 1032. [Google Scholar] [CrossRef]
- Perraut, C.; Clottes, E.; Leydier, C.; Vial, C.; Marcillat, O. Role of Quaternary Structure in Muscle Creatine Kinase Stability: Tryptophan 210 Is Important for Dimer Cohesion. Proteins Struct. Funct. Bioinforma. 1998, 32, 43–51. [Google Scholar] [CrossRef]
- Skoging, U.; Liljeström, P. Role of the C-Terminal Tryptophan Residue for the Structure-Function of the Alphavirus Capsid Protein1. J. Mol. Biol. 1998, 279, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Nichols, C.J.; Lawless, G.M.; Olsen, R.W.; Tobin, A.J. Two Invariant Tryptophans on the A1 Subunit Define Domains Necessary for GABAA Receptor Assembly*. J. Biol. Chem. 1999, 274, 26633–26638. [Google Scholar] [CrossRef]
- Smith, G.B.; Olsen, R.W. Functional Domains of GABAA Receptors. Trends Pharmacol. Sci. 1995, 16, 162–168. [Google Scholar] [CrossRef]
- Galzi, J.-L.; Changeux, J.-P. Neurotransmitter-Gated Ion Channels as Unconventional Allosteric Proteins. Curr. Opin. Struct. Biol. 1994, 4, 554–565. [Google Scholar] [CrossRef]
- Hartiadi, L.Y.; Ahring, P.K.; Chebib, M.; Absalom, N.L. High and Low GABA Sensitivity A4β2δ GABAA Receptors Are Expressed in Xenopus Laevis Oocytes with Divergent Stoichiometries. Biochem. Pharmacol. 2016, 103, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Laha, K.T.; Tran, P.N. Multiple Tyrosine Residues at the GABA Binding Pocket Influence Surface Expression and Mediate Kinetics of the GABAA Receptor. J. Neurochem. 2013, 124, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Kucken, A.M.; Wagner, D.A.; Ward, P.R.; Teissére, J.A.; Boileau, A.J.; Czajkowski, C. Identification of Benzodiazepine Binding Site Residues in the Gamma2 Subunit of the Gamma-Aminobutyric Acid(A) Receptor. Mol. Pharmacol. 2000, 57, 932–939. [Google Scholar] [PubMed]
- Shi, H.; Tsang, S.Y.; Tse, M.K.; Xu, Z.; Xue, H. Recombinant Extracellular Domain of the Three Major Subunits of GABAA Receptor Show Comparable Secondary Structure and Benzodiazepine Binding Properties. Protein Sci. Publ. Protein Soc. 2003, 12, 2642–2646. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Zheng, H.; Li, H.M.; Kitmitto, A.; Zhu, H.; Lee, P.; Holzenburg, A. A Fragment of Recombinant GABA(A) Receptor Alpha1 Subunit Forming Rosette-like Homo-Oligomers. J. Mol. Biol. 2000, 296, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Hang, J.; Shi, H.; Li, D.; Liao, Y.; Lian, D.; Xiao, Y.; Xue, H. Ligand Binding and Structural Properties of Segments of GABAA Receptor A1 Subunit Overexpressed in Escherichia Coli *. J. Biol. Chem. 2000, 275, 18818–18823. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Fang, S.; Shi, H.; Li, H.; Deng, Y.; Liao, Y.; Wu, J.-M.; Zheng, H.; Zhu, H.; Chen, H.-M.; et al. Topology Characterization of a Benzodiazepine-Binding β-Rich Domain of the GABAA Receptor A1 Subunit. Protein Sci. 2005, 14, 2622–2637. [Google Scholar] [CrossRef]
- Lesk, A.M.; Chothia, C. Evolution of Proteins Formed by β-Sheets: II. The Core of the Immunoglobulin Domains. J. Mol. Biol. 1982, 160, 325–342. [Google Scholar] [CrossRef]
- Braun, N.; Lynagh, T.; Yu, R.; Biggin, P.C.; Pless, S.A. Role of an Absolutely Conserved Tryptophan Pair in the Extracellular Domain of Cys-Loop Receptors. ACS Chem. Neurosci. 2016, 7, 339–348. [Google Scholar] [CrossRef]
- Zhu, S.; Noviello, C.M.; Teng, J.; Walsh, R.M.; Kim, J.J.; Hibbs, R.E. Structure of a Human Synaptic GABAA Receptor. Nature 2018, 559, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Phulera, S.; Zhu, H.; Yu, J.; Claxton, D.P.; Yoder, N.; Yoshioka, C.; Gouaux, E. Cryo-EM Structure of the Benzodiazepine-Sensitive α1β1γ2S Tri-Heteromeric GABAA Receptor in Complex with GABA. eLife 2018, 7, e39383. [Google Scholar] [CrossRef] [PubMed]
- Kasaragod, V.B.; Schindelin, H. Structure of Heteropentameric GABAA Receptors and Receptor-Anchoring Properties of Gephyrin. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Gharpure, A.; Teng, J.; Zhuang, Y.; Howard, R.J.; Zhu, S.; Noviello, C.M.; Walsh, R.M.; Lindahl, E.; Hibbs, R.E. Shared Structural Mechanisms of General Anaesthetics and Benzodiazepines. Nature 2020, 585, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Masiulis, S.; Desai, R.; Uchański, T.; Serna Martin, I.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABAA Receptor Signalling Mechanisms Revealed by Structural Pharmacology. Nature 2019, 565, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.S.; Aricescu, A.R. Crystal Structure of a Human GABAA Receptor. Nature 2014, 512, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Kaczor, P.T.; Wolska, A.D.; Mozrzymas, J.W. A1 Subunit Histidine 55 at the Interface between Extracellular and Transmembrane Domains Affects Preactivation and Desensitization of the GABAA Receptor. ACS Chem. Neurosci. 2021, 12, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.S.; Smart, T.G. Binding, Activation and Modulation of Cys-Loop Receptors. Trends Pharmacol. Sci. 2010, 31, 161–174. [Google Scholar] [CrossRef]
- McKechnie, W.S.; Tugcu, N.; Kandula, S. Accurate and Rapid Protein Concentration Measurement of In-Process, High Concentration Protein Pools. Biotechnol. Prog. 2018, 34, 1234–1241. [Google Scholar] [CrossRef]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, 2005; pp. 571–607; ISBN 978-1-58829-343-5. [Google Scholar]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Chu, R.; Hang, J.; Lee, P.; Zheng, H. Fragment of GABA(A) Receptor Containing Key Ligand-Binding Residues Overexpressed in Escherichia Coli. Protein Sci. Publ. Protein Soc. 1998, 7, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, D.; Xia, G.; Shen, W.; Macdonald, R.L.; Xu, D.; Kang, J.-Q. Differential Protein Structural Disturbances and Suppression of Assembly Partners Produced by Nonsense GABRG2 Epilepsy Mutations: Implications for Disease Phenotypic Heterogeneity. Sci. Rep. 2016, 6, 35294. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Lin, S.L.; Nussinov, R. Protein Binding versus Protein Folding: The Role of Hydrophilic Bridges in Protein Associations. J. Mol. Biol. 1997, 265, 68–84. [Google Scholar] [CrossRef]
- Klausberger, T.; Fuchs, K.; Mayer, B.; Ehya, N.; Sieghart, W. GABAA Receptor Assembly. J. Biol. Chem. 2000, 275, 8921–8928. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, D.; Gibbs, T.T.; Farb, D.H. Docking of 1,4-Benzodiazepines in the A1/Γ2 GABAA Receptor Modulator Site. Mol. Pharmacol. 2009, 76, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Richter, L.; de Graaf, C.; Sieghart, W.; Varagic, Z.; Mörzinger, M.; de Esch, I.J.P.; Ecker, G.F.; Ernst, M. Diazepam-Bound GABAA Receptor Models Identify New Benzodiazepine Binding-Site Ligands. Nat. Chem. Biol. 2012, 8, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Morlock, E.V.; Czajkowski, C. Different Residues in the GABAA Receptor Benzodiazepine Binding Pocket Mediate Benzodiazepine Efficacy and Binding. Mol. Pharmacol. 2011, 80, 14–22. [Google Scholar] [CrossRef]
- Sarto-Jackson, I.; Sieghart, W. Assembly of GABAA Receptors (Review). Mol. Membr. Biol. 2008, 25, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Wongsamitkul, N.; Maldifassi, M.C.; Simeone, X.; Baur, R.; Ernst, M.; Sigel, E. α Subunits in GABAA Receptors Are Dispensable for GABA and Diazepam Action. Sci. Rep. 2017, 7, 15498. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.M.; Thomas, P.; Gorrie, G.H.; Connolly, C.N.; Smart, T.G.; Moss, S.J. Identification of Amino Acid Residues within GABAAReceptor β Subunits That Mediate Both Homomeric and Heteromeric Receptor Expression. J. Neurosci. 1999, 19, 6360–6371. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, D.B.; Sontheimer, H.; Shivers, B.D.; Ymer, S.; Kettenmann, H.; Schofield, P.R.; Seeburg, P.H. Importance of a Novel GABAA Receptor Subunit for Benzodiazepine Pharmacology. Nature 1989, 338, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Boileau, A.J.; Li, T.; Benkwitz, C.; Czajkowski, C.; Pearce, R.A. Effects of γ2S Subunit Incorporation on GABAA Receptor Macroscopic Kinetics. Neuropharmacology 2003, 44, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Kittler, J.T.; Wang, J.; Connolly, C.N.; Vicini, S.; Smart, T.G.; Moss, S.J. Analysis of GABAA Receptor Assembly in Mammalian Cell Lines and Hippocampal Neurons Using Γ2 Subunit Green Fluorescent Protein Chimeras. Mol. Cell. Neurosci. 2000, 16, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Connolly, C.N.; Uren, J.M.; Thomas, P.; Gorrie, G.H.; Gibson, A.; Smart, T.G.; Moss, S.J. Subcellular Localization and Endocytosis of Homomeric Gamma2 Subunit Splice Variants of Gamma-Aminobutyric Acid Type A Receptors. Mol. Cell. Neurosci. 1999, 13, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Torres, A.; Miledi, R. Expression of Functional Receptors by the Human γ-Aminobutyric Acid A Γ2 Subunit. Proc. Natl. Acad. Sci. 2004, 101, 3220–3223. [Google Scholar] [CrossRef]
- Stephens, D.N.; King, S.L.; Lambert, J.J.; Belelli, D.; Duka, T. GABAA Receptor Subtype Involvement in Addictive Behaviour. Genes Brain Behav. 2017, 16, 149–184. [Google Scholar] [CrossRef]
- Keller, C.A.; Yuan, X.; Panzanelli, P.; Martin, M.L.; Alldred, M.; Sassoè-Pognetto, M.; Lüscher, B. The Gamma2 Subunit of GABA(A) Receptors Is a Substrate for Palmitoylation by GODZ. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 5881–5891. [Google Scholar] [CrossRef]
- Tyagarajan, S.K.; Fritschy, J.-M. Gephyrin: A Master Regulator of Neuronal Function? Nat. Rev. Neurosci. 2014, 15, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Choii, G.; Ko, J. Gephyrin: A Central GABAergic Synapse Organizer. Exp. Mol. Med. 2015, 47, e158–e158. [Google Scholar] [CrossRef] [PubMed]
- Essrich, C.; Lorez, M.; Benson, J.A.; Fritschy, J.-M.; Lüscher, B. Postsynaptic Clustering of Major GABAA Receptor Subtypes Requires the Γ2 Subunit and Gephyrin. Nat. Neurosci. 1998, 1, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Alldred, M.J.; Mulder-Rosi, J.; Lingenfelter, S.E.; Chen, G.; Lüscher, B. Distinct Γ2 Subunit Domains Mediate Clustering and Synaptic Function of Postsynaptic GABAA Receptors and Gephyrin. J. Neurosci. 2005, 25, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Kasaragod, V.B.; Schindelin, H. Structure–Function Relationships of Glycine and GABAA Receptors and Their Interplay With the Scaffolding Protein Gephyrin. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Saiepour, L.; Fuchs, C.; Patrizi, A.; Sassoè-Pognetto, M.; Harvey, R.J.; Harvey, K. Complex Role of Collybistin and Gephyrin in GABAA Receptor Clustering. J. Biol. Chem. 2010, 285, 29623–29631. [Google Scholar] [CrossRef]
- Tretter, V.; Jacob, T.C.; Mukherjee, J.; Fritschy, J.-M.; Pangalos, M.N.; Moss, S.J. The Clustering of GABA(A) Receptor Subtypes at Inhibitory Synapses Is Facilitated via the Direct Binding of Receptor Alpha 2 Subunits to Gephyrin. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Hockenberry, A.J.; Wilke, C.O. Evolutionary Couplings Detect Side-Chain Interactions. PeerJ 2019, 7, e7280. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.S.; Colwell, L.J.; Sheridan, R.; Hopf, T.A.; Pagnani, A.; Zecchina, R.; Sander, C. Protein 3D Structure Computed from Evolutionary Sequence Variation. PLOS ONE 2011, 6, e28766. [Google Scholar] [CrossRef] [PubMed]
- Patrizio, A.; Renner, M.; Pizzarelli, R.; Triller, A.; Specht, C.G. Alpha Subunit-Dependent Glycine Receptor Clustering and Regulation of Synaptic Receptor Numbers. Sci. Rep. 2017, 7, 10899. [Google Scholar] [CrossRef]
- Nussinov, R.; Zhang, M.; Liu, Y.; Jang, H. AlphaFold, Allosteric, and Orthosteric Drug Discovery: Ways Forward. Drug Discov. Today 2023, 28, 103551. [Google Scholar] [CrossRef]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved Prediction of Protein-Protein Interactions Using AlphaFold2. Nat. Commun. 2022, 13, 1265. [Google Scholar] [CrossRef]
- Bartolec, T.K.; Vázquez-Campos, X.; Norman, A.; Luong, C.; Johnson, M.; Payne, R.J.; Wilkins, M.R.; Mackay, J.P.; Low, J.K.K. Cross-Linking Mass Spectrometry Discovers, Evaluates, and Corroborates Structures and Protein–Protein Interactions in the Human Cell. Proc. Natl. Acad. Sci. 2023, 120, e2219418120. [Google Scholar] [CrossRef]
- Yang, Z.; Zeng, X.; Zhao, Y.; Chen, R. AlphaFold2 and Its Applications in the Fields of Biology and Medicine. Signal Transduct. Target. Ther. 2023, 8, 1–14. [Google Scholar] [CrossRef]
- Nagaratnam, N.; Martin-Garcia, J.M.; Yang, J.-H.; Goode, M.R.; Ketawala, G.; Craciunescu, F.M.; Zook, J.D.; Sonowal, M.; Williams, D.; Grant, T.D.; et al. Structural and Biophysical Properties of FopA, a Major Outer Membrane Protein of Francisella Tularensis. PLoS ONE 2022, 17, e0267370. [Google Scholar] [CrossRef]
- Paul, B.; Weeratunga, S.; Tillu, V.A.; Hariri, H.; Henne, W.M.; Collins, B.M. Structural Predictions of the SNX-RGS Proteins Suggest They Belong to a New Class of Lipid Transfer Proteins. Front. Cell Dev. Biol. 2022, 10, 826688. [Google Scholar] [CrossRef]
- Jin, Y.; Fyfe, P.K.; Gardner, S.; Wilmes, S.; Bubeck, D.; Moraga, I. Structural Insights into the Assembly and Activation of the IL-27 Signaling Complex. EMBO Rep. 2022, 23, e55450. [Google Scholar] [CrossRef]
- Skalidis, I.; Kyrilis, F.L.; Tüting, C.; Hamdi, F.; Chojnowski, G.; Kastritis, P.L. Cryo-EM and Artificial Intelligence Visualize Endogenous Protein Community Members. Struct. Lond. Engl. 1993 2022, 30, 575–589e6. [Google Scholar] [CrossRef]
- Liu, H.; Hong, X.; Xi, J.; Menne, S.; Hu, J.; Wang, J.C.-Y. Cryo-EM Structures of Human Hepatitis B and Woodchuck Hepatitis Virus Small Spherical Subviral Particles. Sci. Adv. 2022, 8, eabo4184. [Google Scholar] [CrossRef]
- Tai, L.; Zhu, Y.; Ren, H.; Huang, X.; Zhang, C.; Sun, F. 8 Å Structure of the Outer Rings of the Xenopus Laevis Nuclear Pore Complex Obtained by Cryo-EM and AI. Protein Cell 2022, 13, 760–777. [Google Scholar] [CrossRef]
- Yeung, R.K.; Xiang, Z.-H.; Tsang, S.-Y.; Li, R.; Ho, T.Y.C.; Li, Q.; Hui, C.-K.; Sham, P.-C.; Qiao, M.-Q.; Xue, H. Gabrb2-Knockout Mice Displayed Schizophrenia-like and Comorbid Phenotypes with Interneuron-Astrocyte-Microglia Dysregulation. Transl. Psychiatry 2018, 8, 128. [Google Scholar] [CrossRef]
- Lo, W.-S.; Harano, M.; Gawlik, M.; Yu, Z.; Chen, J.; Pun, F.W.; Tong, K.-L.; Zhao, C.; Ng, S.-K.; Tsang, S.-Y.; et al. GABRB2 Association with Schizophrenia: Commonalities and Differences Between Ethnic Groups and Clinical Subtypes. Biol. Psychiatry 2007, 61, 653–660. [Google Scholar] [CrossRef]
- Sperk, G.; Furtinger, S.; Schwarzer, C.; Pirker, S. GABA and Its Receptors in Epilepsy. Adv. Exp. Med. Biol. 2004, 548, 92–103. [Google Scholar] [CrossRef]
- Limon, A.; Reyes-Ruiz, J.M.; Miledi, R. Loss of Functional GABAA Receptors in the Alzheimer Diseased Brain. Proc. Natl. Acad. Sci. 2012, 109, 10071–10076. [Google Scholar] [CrossRef] [PubMed]
- Tretter, V.; Ehya, N.; Fuchs, K.; Sieghart, W. Stoichiometry and Assembly of a Recombinant GABAA Receptor Subtype. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- Connolly, C.N.; Krishek, B.J.; McDonald, B.J.; Smart, T.G.; Moss, S.J. Assembly and Cell Surface Expression of Heteromeric and Homomeric γ-Aminobutyric Acid Type A Receptors (∗). J. Biol. Chem. 1996, 271, 89–96. [Google Scholar] [CrossRef]
- Taylor, P.M.; Connolly, C.N.; Kittler, J.T.; Gorrie, G.H.; Hosie, A.; Smart, T.G.; Moss, S.J. Identification of Residues within GABAA Receptor α Subunits That Mediate Specific Assembly with Receptor β Subunits. J. Neurosci. 2000, 20, 1297–1306. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Yin, R.; Feng, B.Y.; Varshney, A.; Pierce, B.G. Benchmarking AlphaFold for Protein Complex Modeling Reveals Accuracy Determinants. Protein Sci. 2022, 31, e4379. [Google Scholar] [CrossRef]
Figure 1.
Amino acid sequence alignment for the extracellular domain fragments of human GABAA receptor α1, β2, and γ2 subunits. Alignment by MAFFT in CLUSTAL format. Percent identity is >26%. The Cys-loop sequence is highlighted in red. Asterisks (*) show conserved and identical sequences, two dots (:) show conservative mutation, and one dot (.) shows semi-conservative mutations. Blue highlighted regions are identical and aligned sequences between two fragments. There are 47 amino acids aligned between α1 and γ2 fragments, 22 aligned between β2 and γ2, and 16 aligned between α1 and β2 fragments.
Figure 1.
Amino acid sequence alignment for the extracellular domain fragments of human GABAA receptor α1, β2, and γ2 subunits. Alignment by MAFFT in CLUSTAL format. Percent identity is >26%. The Cys-loop sequence is highlighted in red. Asterisks (*) show conserved and identical sequences, two dots (:) show conservative mutation, and one dot (.) shows semi-conservative mutations. Blue highlighted regions are identical and aligned sequences between two fragments. There are 47 amino acids aligned between α1 and γ2 fragments, 22 aligned between β2 and γ2, and 16 aligned between α1 and β2 fragments.
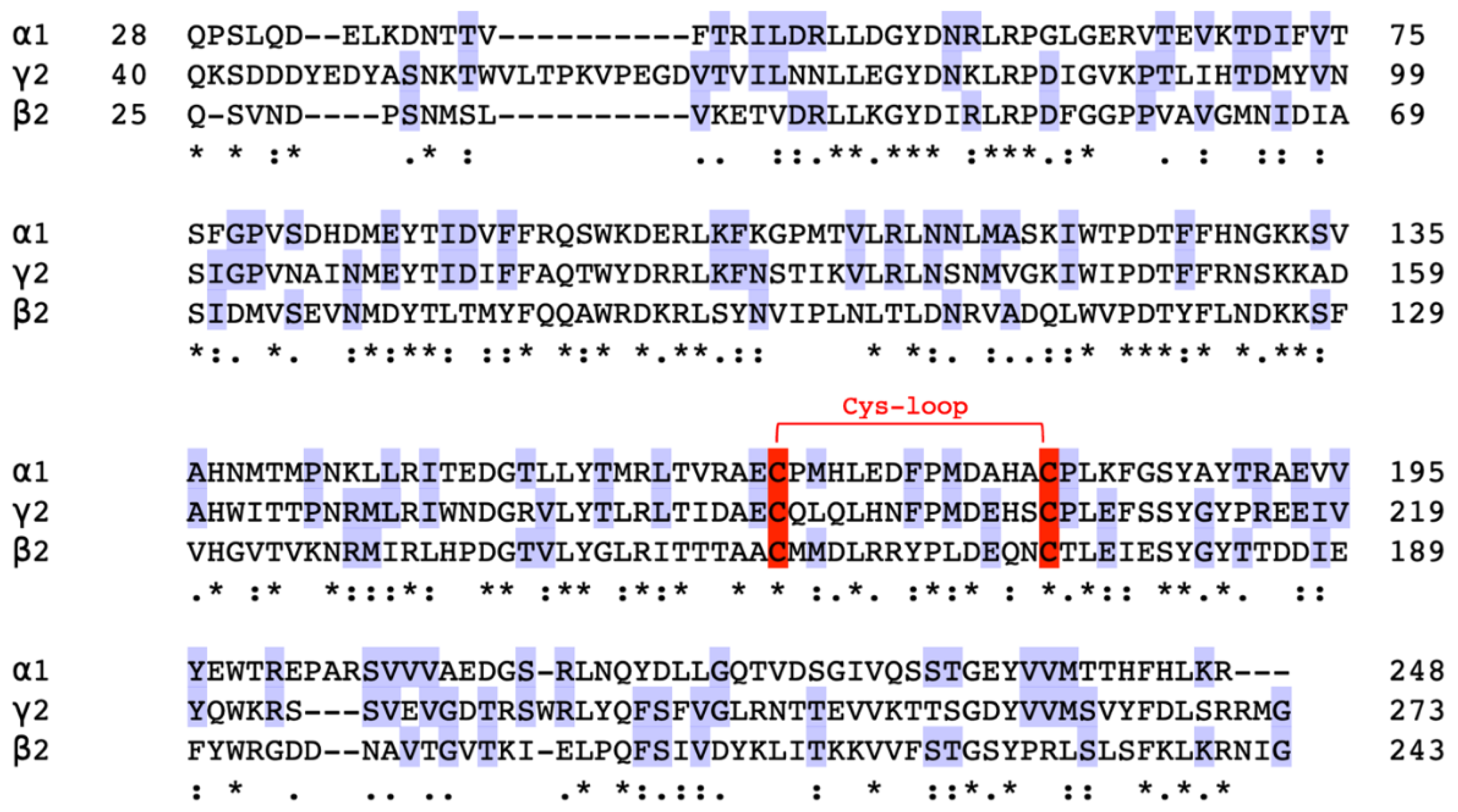
Figure 2.
Purification of the protein fragments by fast protein liquid chromatography (FPLC). Eluents were collected in numbered Falcon tubes corresponding to the fraction number. The concentration of protein in fraction numbers 21-35 indicated by OD280 measured by NanoDrop. M = marker from Bio-Rad SDS-PAGE Broad Range Standards. Prestained standard was used in (a, b, d), and unstained standard in (c) was visualised with Coomassie brilliant blue R-250. 12% SDS-PAGE was performed for fraction numbers with the highest OD280, representing the highest concentration. (a) concentrations of α1 subunit (Gln28-Arg248 fragment) in consecutive fractions, with eluent numbers 25 and 26 having the highest concentrations. SDS-PAGE visualises the molecular weight to be between 20.6kDa and 28.9kDa, corresponding to the 25kDa size of the fragment. (b) concentration of β2 subunit (Gln25-Gly243 fragment) in consecutive fractions, with eluent numbers 29 and 30 having the highest concentrations. SDS-PAGE visualises the molecular weight to be between 20.6kDa and 28.9kDa, corresponding to the 25.2kDa size of the fragment. (c) concentration of γ2 subunit (Gln40-Gly273 fragment) in consecutive fractions, with eluent numbers 30 and 31 having the highest concentrations. SDS-PAGE visualises the molecular weight to lie slightly below the 28.9kDa band of the marker, which corresponds to the 27.5kDa of the fragment. (d) expression and purification analysis of 2α:2β:1γ GABAA receptors complex. Eluent number 23 showed the highest concentration of oligomers (red arrow). Fractions after 25 were considered as concentrations of individual subunits. SDS-PAGE shows the concentration and size of the fragments from eluents number 23 and 24.
Figure 2.
Purification of the protein fragments by fast protein liquid chromatography (FPLC). Eluents were collected in numbered Falcon tubes corresponding to the fraction number. The concentration of protein in fraction numbers 21-35 indicated by OD280 measured by NanoDrop. M = marker from Bio-Rad SDS-PAGE Broad Range Standards. Prestained standard was used in (a, b, d), and unstained standard in (c) was visualised with Coomassie brilliant blue R-250. 12% SDS-PAGE was performed for fraction numbers with the highest OD280, representing the highest concentration. (a) concentrations of α1 subunit (Gln28-Arg248 fragment) in consecutive fractions, with eluent numbers 25 and 26 having the highest concentrations. SDS-PAGE visualises the molecular weight to be between 20.6kDa and 28.9kDa, corresponding to the 25kDa size of the fragment. (b) concentration of β2 subunit (Gln25-Gly243 fragment) in consecutive fractions, with eluent numbers 29 and 30 having the highest concentrations. SDS-PAGE visualises the molecular weight to be between 20.6kDa and 28.9kDa, corresponding to the 25.2kDa size of the fragment. (c) concentration of γ2 subunit (Gln40-Gly273 fragment) in consecutive fractions, with eluent numbers 30 and 31 having the highest concentrations. SDS-PAGE visualises the molecular weight to lie slightly below the 28.9kDa band of the marker, which corresponds to the 27.5kDa of the fragment. (d) expression and purification analysis of 2α:2β:1γ GABAA receptors complex. Eluent number 23 showed the highest concentration of oligomers (red arrow). Fractions after 25 were considered as concentrations of individual subunits. SDS-PAGE shows the concentration and size of the fragments from eluents number 23 and 24.

Figure 3.
Negative staining and visualization by cryo-EM. Results on carbon-coated mica sheets were visualised under EM. 910 particles selected from 1030 particles were manually picked from 20 cryo-EM images (micrographs) and used to generate 2D averages. (a) negative staining of α1 subunit (Gln28-Arg248) and (b) 2D classification of α1 homopentamer structures from 2D averages. (c) negative staining of β2 subunit (Gln25-Gly243) and (d) 2D classification of Gln25-Gly243 protein fragments from 2D averages suggest a similar pentameric structure. (e) negative staining of γ2 subunit (Gln40-Gly273). (f) Negative staining of the 2α:2β:1γ GABAA receptor complex. The protein structures circled red are considered as stable particles, which were subsequently picked and used for (g) 2D classification, indicating pentameric structures.
Figure 3.
Negative staining and visualization by cryo-EM. Results on carbon-coated mica sheets were visualised under EM. 910 particles selected from 1030 particles were manually picked from 20 cryo-EM images (micrographs) and used to generate 2D averages. (a) negative staining of α1 subunit (Gln28-Arg248) and (b) 2D classification of α1 homopentamer structures from 2D averages. (c) negative staining of β2 subunit (Gln25-Gly243) and (d) 2D classification of Gln25-Gly243 protein fragments from 2D averages suggest a similar pentameric structure. (e) negative staining of γ2 subunit (Gln40-Gly273). (f) Negative staining of the 2α:2β:1γ GABAA receptor complex. The protein structures circled red are considered as stable particles, which were subsequently picked and used for (g) 2D classification, indicating pentameric structures.
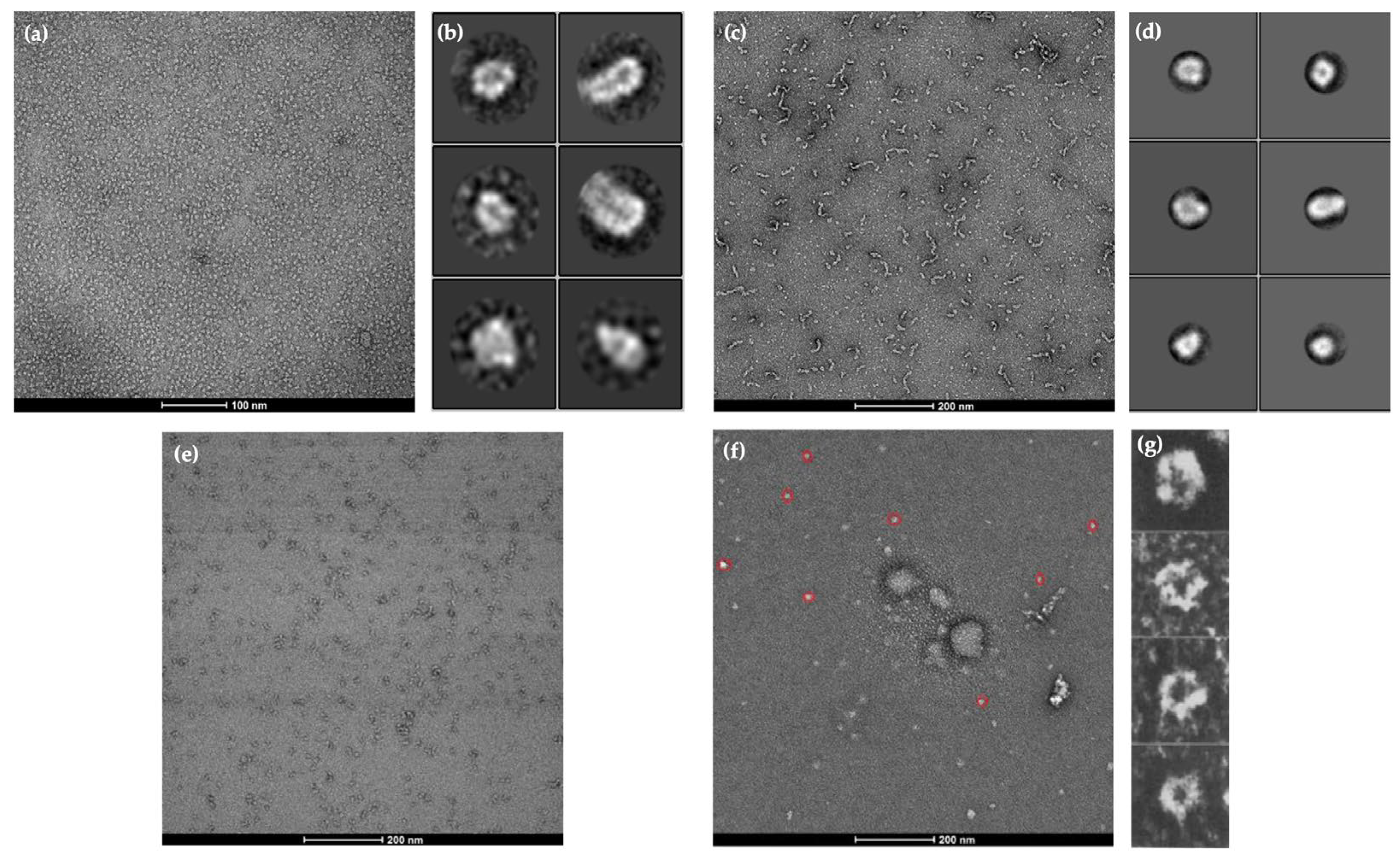
Figure 4.
AlphaFold2 predicted structures. Alpha helices are depicted as green “pipes”, beta sheets as cyan “planks” and red strands as coils. The secondary structure is aligned under the amino acid sequence of a single subunit fragment of the corresponding homopentameric structure. The scale bar denotes 10Å (1nm). (a) α1 subunit fragment, (b) β2 subunit fragment and (c) γ2 subunit fragment all show the potential of forming homopentamers based on AlphaFold2 prediction. Black arrows show the two different secondary structures of the individual γ2 subunit fragment, one without the 3-residue helix (left) and one with (right).
Figure 4.
AlphaFold2 predicted structures. Alpha helices are depicted as green “pipes”, beta sheets as cyan “planks” and red strands as coils. The secondary structure is aligned under the amino acid sequence of a single subunit fragment of the corresponding homopentameric structure. The scale bar denotes 10Å (1nm). (a) α1 subunit fragment, (b) β2 subunit fragment and (c) γ2 subunit fragment all show the potential of forming homopentamers based on AlphaFold2 prediction. Black arrows show the two different secondary structures of the individual γ2 subunit fragment, one without the 3-residue helix (left) and one with (right).

Figure 5.
Comparison of Cryo-EM images and AlphaFold2 predictions of α1 homopentamer, β2 homopentamer, and GABAA receptor heteropentamer. (a-d) Cryo-EM images and AlphaFold2 structure prediction of α1 homopentamer have a similar shape, including (a) bottom view of whole pentamer, (b) side view with one subunit hidden to mimic the exposed central cavity, (c) side view of whole pentamer, and (d) side view with two subunits hidden for clearer comparison. (e-f) Cryo-EM images and AlphaFold2 structure prediction of β2 homopentamer with (e) top view of pentamer and (f) side view with two subunits hidden to mimic the exposed central cavity. (g-h) Cryo-EM images and AlphaFold2 structure prediction of α1β2γ2 GABAA receptor. The α1, β2, and γ2 subunits are shown in blue, yellow and magenta colours respectively. The AlphaFold2 model aims to match the shape of the structure obtained by Cryo-EM, with (g) bottom view of the pentamer, and (h) top view of the whole pentamer. The red arrow identifies the structure resembling the protruding residues of the γ2 subunit.
Figure 5.
Comparison of Cryo-EM images and AlphaFold2 predictions of α1 homopentamer, β2 homopentamer, and GABAA receptor heteropentamer. (a-d) Cryo-EM images and AlphaFold2 structure prediction of α1 homopentamer have a similar shape, including (a) bottom view of whole pentamer, (b) side view with one subunit hidden to mimic the exposed central cavity, (c) side view of whole pentamer, and (d) side view with two subunits hidden for clearer comparison. (e-f) Cryo-EM images and AlphaFold2 structure prediction of β2 homopentamer with (e) top view of pentamer and (f) side view with two subunits hidden to mimic the exposed central cavity. (g-h) Cryo-EM images and AlphaFold2 structure prediction of α1β2γ2 GABAA receptor. The α1, β2, and γ2 subunits are shown in blue, yellow and magenta colours respectively. The AlphaFold2 model aims to match the shape of the structure obtained by Cryo-EM, with (g) bottom view of the pentamer, and (h) top view of the whole pentamer. The red arrow identifies the structure resembling the protruding residues of the γ2 subunit.
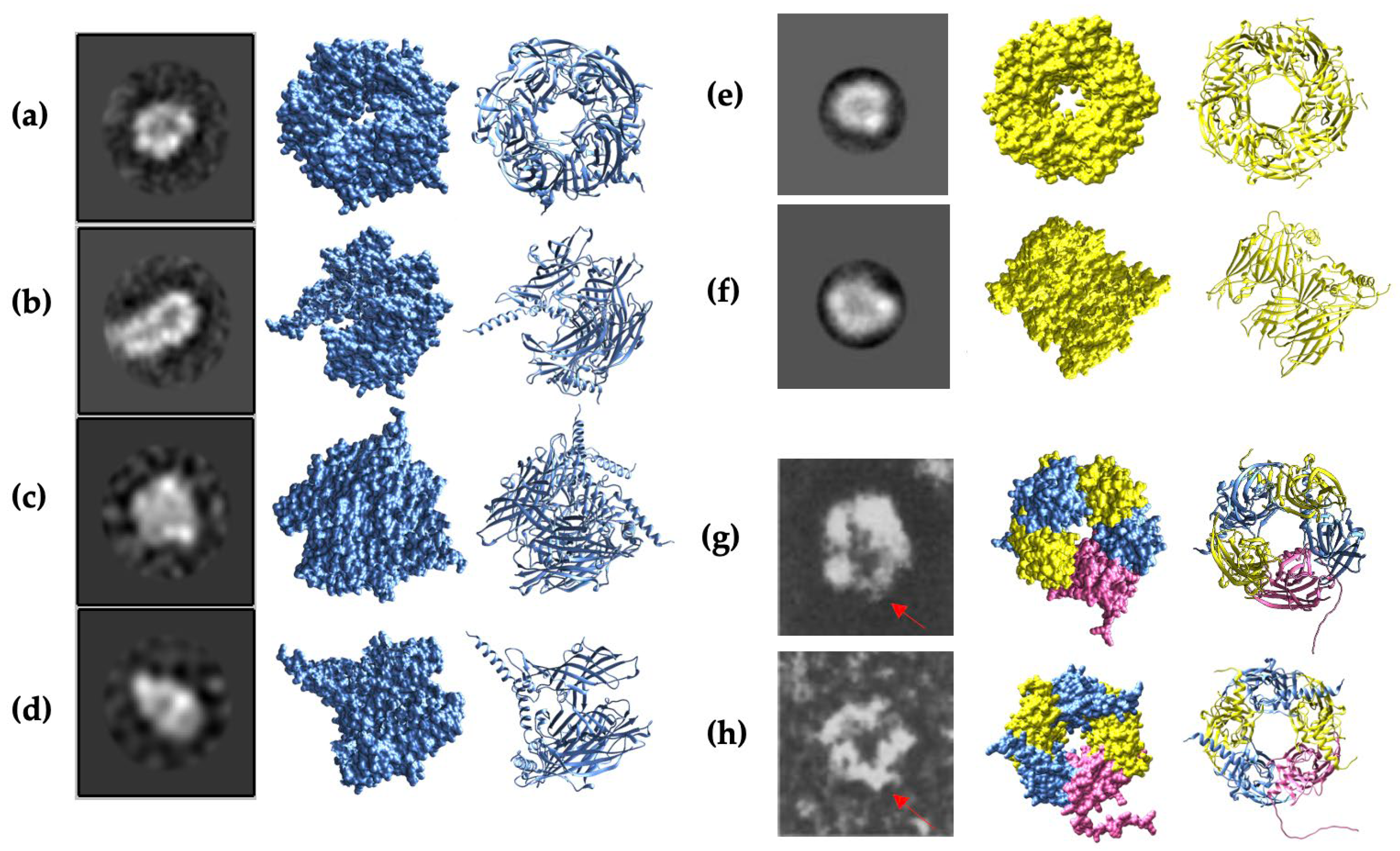

Table 1.
Percent identity matrix for sequence alignment of GABAA receptor α1, β2 and γ2 subunit protein fragments generated by MAFFT. Sequence homology is highest between α1 and γ2 fragments, lowest between α1 and β2.
Table 1.
Percent identity matrix for sequence alignment of GABAA receptor α1, β2 and γ2 subunit protein fragments generated by MAFFT. Sequence homology is highest between α1 and γ2 fragments, lowest between α1 and β2.
| α1 | β2 | γ2 | |
| α1 | 100% | 36.57% | 51.83% |
| β2 | 36.57% | 100% | 39.45% |
| γ2 | 51.83% | 39.45% | 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



