
Submitted:
03 July 2024
Posted:
04 July 2024
You are already at the latest version
Abstract
The work presents the results of studying dilute aqueous solutions of commercial Ln(NO3)3 · xH2O salts with Ln=Ce-Lu using X-ray diffraction (XRD), IR spectroscopy, X-ray absorption spectroscopy (XAS: EXAFS/XANES) and pH measurements. As a reference point, XRD and XAS measurements for characterized Ln(NO3)3 · xH2O microcrystalline powder samples were performed. The local structure of Ln-nitrate complexes in 20 mM Ln(NO3)3 · xH2O aqueous solution was studied under total external reflection conditions and EXAFS geometry was applied to obtain high-quality EXAFS data for solutions with low concentrations of Ln3+ ions. Results obtained by EXAFS spectroscopy showed significant contraction of the first coordination sphere during the dissolution process for metal ions located in the middle of the lanthanide series. It was established that in Ln(NO3)3 · xH2O solutions with Ln=Ce, Sm, Gd, Yb (c=134, 100, 50 and 20 mM) there are coordinated and, to a greater extent, non-coordinated nitrate groups with bidentate and predominantly monodentate bonds with Ln ions, the number of which increases upon transition from cerium to ytterbium. Cross-relationships between the concentration of solutions, pH value and antimicrobial activity with the type of Ln = Ce, Sm, Gd, Yb were established, as well as the absence of biocidal properties of solutions with a concentration of 20 mM, except for Ln = Yb. The important role of experimental conditions in obtaining and interpreting the results was noted.
Keywords:
Ln nitrate solutions
; X-ray diffraction
; X-ray absorption spectroscopy
; IR spectroscopy
; antimicrobial activity
1. Introduction
Traditionally, the coordination chemistry of lanthanides has been a matter of exceptional interest for wide ranging industrial applications, first of all for development of effective methods for separation and purification of rare earth elements. The efficiency of extraction process is greatly dependent on solvation properties of lanthanide ions, such as complexation with extractants, solubility of salts, dissociation/association of ion pairs, etc. It is clear that optimization of industrial technologies for extraction of rare-earth elements, as well as elaboration of novel “green” methods for their recycling and recovering, requires deeper insight into molecular mechanisms regulating metal-ligand interaction in aqueous/organic media. Ln(NO3)3 · xH2O salts are excellent precursors for production of ultra-high purity compounds and certain catalyst and nanoscale (nanoparticles and nanopowders) materials. In addition, Ln(NO3)3 · xH2O are used in the growth of large-sized polyfunctional crystals doped with Ln3+ ions from aqueous solutions.
Recently much interest to the aqueous chemistry of lanthanides has been stimulated by growing implications of these elements in medicine and biotechnologies. Cerium, one of the most abundant lanthanides, has been successfully used in clinical practice since the end of the 19th century. Cerium nitrate solution is commonly recognized as the topical agent of choice for the treatment of burn wounds [1,2]. Additionally, cerium ions exhibit superior antibacterial activity. In particular, Ce(NO3)3 · 6H2O salt solutions (~ 50 mM) are used as antiseptic agent for the treatment of indwelling medical devices (implants, catheters) [2]. In general, lanthanide compounds are now considered to be potential alternative to antibiotics in antimicrobial and antifungal therapy [3,4].
Investigations of hydration behavior of lanthanides in diluted solutions are of special interest for biological sciences, when low Ln3+ concentrations are used to reduce possible cytotoxic effects. It is also important to bear in mind that coordination environment of Ln3+ ion is strongly dependent on lanthanide salt concentration and can differ significantly in diluted compared with more concentrated aqueous solutions [5]. Thus, increasing attention in lanthanide biochemistry has been paid to experimental techniques that provide atomic-level information on the solution structure of Ln3+ ions under diluted conditions. On the other hand, while maintaining the functional properties of diluted solutions, a decrease in the content of Ln3+ ions is ensured when drugs are taken orally.
The techniques for studying the nature (size and/or structure) of the Ln shell in solutions can be classified as direct or indirect methods [6]. The direct methods include X-ray [7,8] and neutron diffraction [9], X-ray absorption spectroscopy (EXAFS) [10,11] FT-IR [12,13] and Raman spectroscopy [13,14], and nuclear magnetic resonance (NMR) [15]. The indirect methods involve compressibility, NMR exchange and adsorption spectroscopy measurements. In addition, ab initio quantum mechanical calculations [16] and molecular dynamics (MD) are applied using the classical or mixed quantum/classical interaction potential, which are combined with UV-visible spectroscopy [17] or X-ray absorption spectroscopy (XAS), the combination of MD simulations and XAS spectroscopy [18]. Experimental highly sensitive microcalorimetry is used to calculate the thermodynamic characteristics of weak interactions in solutions [19] together with luminescence emission spectra and the lifetime of Ln [20]. X-ray absorption spectroscopy has been proven to be exceptionally informative to investigate coordination environment of Ln3+ ions in aqueous solutions [10,18,21,22,23,24,25,26]. Most X-ray absorption spectroscopy studies reported in the literature are focused on the solutions of Ln3+ chloride salts. Their results cannot be transferred to solutions of other salts, as well as the approaches used.
The purpose of this work is to identify the structural features of dilute aqueous solutions of lanthanide nitrates and to establish the role of concentration and pH of the medium in the implementation of antimicrobial properties.
To characterize aqueous solutions of Ln(NO3)3 · xH2O (Ln=Ce-Lu), modern informative methods of X-ray diffraction (XRD), infrared spectroscopy (FT-IR), and X-ray absorption spectroscopy (XAS) were chosen. In the present study we used the main advantage of X-ray absorption spectroscopy under total external reflection (TER), namely the extremely low intensity of background scattering and accordingly high signal-to-noise ratio that allows for detection of week spectroscopic signal from samples with small amounts of absorbing atoms. X-ray absorption spectroscopy measurements under TER have been performed for 20 mM Ln(NO3)3 · xH2O aqueous solutions across the lanthanide series from Ce to Lu. In addition, X-ray standing wave (XRSW) technique was applied to examine the adsorption behavior of Ln3+ ions at the air/liquid interface.
2. Results
The local structure of Ln3+ nitrate complexes in salt and dilute aqueous solution has been systematically studied across the lanthanide series from Ce to Lu.
2.1. X-ray Standing Wave Studies
Additional XRSW measurements were carried out to examine the adsorption behavior of Ln3+ ions at solution surface, in particular possible enrichment of Ln3+ ions at the air/liquid interface. In XRSW experiments the intensity of characteristic fluorescence exited by the incident X-ray beam is recorded as a function of the incident angle θ. The key idea of XRSW method is as follows: the angular dependence of fluorescence yield is highly sensitive to the position of atoms in the direction normal to the sample surface. Thus, XRSW measurements offer an opportunity to locate the atoms directly from the analysis of the corresponding fluorescence curve [27].
Experimental XRSW data, obtained for studied Ln3+ aqueous solutions, exhibited essentially similar behavior, as a typical example Figure 1 shows angular dependence of Ce L3-fluorescence from Се(NO3)3 · 6H2O salt solution. This section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
Generally, two factors should be kept in mind, when analyzing XRSW data collected under TER. The first factor is the dramatic changes in electric field intensity above the reflecting surface as the incidence angle is scanned within the TER region. As a direct result of these changes large modulations arise in angular dependence of fluorescence yield from atoms located in the near-surface region; calculated angular dependence, presented in Figure 1, is a perfect illustration of this characteristic feature. The calculations have been performed using the recursion formalism developed by Parratt [28]. As can be seen in Figure 1 angular dependence of fluorescence yield for near-surface distribution of atoms drastically increases from zero at θ = 0° and reaches the maximum value in the vicinity of the critical angle θC.
In marked contrast, angular dependence of fluorescence yield from the atoms, which are present in the bulk liquid subphase, exhibits absolutely different behavior. In this case the most important factor is the changes in the penetration depth of the electric field into the subphase. As the incident angle increases above the critical angle θС the penetration depth abruptly rises from several nanometers (for θ<θС) to several hundreds of microns (for θ > θС), that in turn results in the sharp increase in fluorescence signal. From Figure 1 it is clear that Ce-fluorescence curve, recorded in our experiments, corresponds to this type of angular dependence. These observations evidence, that no enhancement of Ln3+ concentration in the near-surface region occurs in the dilute aqueous solutions of Ln(NO3)3 · xH2O salts and the distribution of Ln3+ ions can be considered as homogeneous.
2.2. X-ray Absorption Spectroscopy Studies
As a reference point in studying Ln(NO3)3 · xH2O aqueous solutions (c=20mM) we performed X-ray absorption spectroscopy measurements also for Ln(NO3)3 · xH2O microcrystalline powder samples (Table S1). For appropriate fitting of the first intense peak in Fourier-transformed spectrum one should take into account two coordination shells around metal ion: oxygen atoms (in crystalline Ln(NO3)3 · xH2O: СNLn=4OH2O+6ONO3 except CNCe=11 - 5OH2O+6ONO3, CNYb(Lu)=9 - 3OH2O+6ONO3; Ln bond is monodentate with water oxygen and bidentate with oxygen of nitrate groups), nitrogen atoms (in crystalline Ln(NO3)3 · LH2O coordination number is 3: CNLn=3NNO3). Results of such fitting presented in Table S1 and in Figure S1.
These fits performed with fixed coordination numbers. Such fitting allows to estimate the possibilities of EXAFS technique to determine structural parameters. Observation results presented in Table S1 demonstrate that only first coordination shell radius could be determined this appropriate precision.
Further, such analysis applied for experimental EXAFS data for solutions. But in case of solutions coordination numbers for oxygen and nitrogen coordination shells were treated like free parameters, but Debay-Waller parameter for nitrogen coordination shell was fixed to decrease the number of independent parameters during the fitting. Results also presented in Table S1 and in Figure S2. In case of solutions the only parameter which could be determined from EXAFS fitting with appropriate precision is an interatomic distance Ln-O for the first coordination shell. Figure 2 contains the dependence of the Ln–O bond distances over the Ln series.
Also plotted error bars associated with each data point are based on EXAFS fitting uncertainty. It’s clearly seen from presented Ln-O distances that Ln-O for Ln = Tb, Dy, Ho, Er, Tm ions demonstrate significant contraction of oxygen shell in solutions. Such observation is also in agreement with measured XANES spectra, which are plotted in Figure S3. Vertical lines show the position of the second maximum in XANES spectra, shifting these lines towards higher energies indicates shrinking of the first coordination shell.
Concluding the results obtained by examination of XANES spectra and fitting of the EXAFS ones, the contraction of the first coordination shell for metals Ln = Tb, Dy, Ho, Er, Tm is observed. Stability of the nitrate complexes couldn’t be determined by simple EXAFS fitting procedure.
It should be noted that nitrogen coordination number in aquatic solutions has been a subject of disagreement. Questions such as whether an eventual coordination change occurs within each series or whether inner or outer sphere complexation occurs for specific ligands have not been completely resolved. Thus, according to [19] the «light» lanthanides in aqueous solutions are nine-coordinated, whereas the “heavy” ones are eight-coordinated. According to [29] La-Nd has larger coordination numbers than Tb-Lu, and in the region between them (Ln=Sm-Gd) there are transitional structures or a mixture of structures. The authors of [30] believe that the coordination numbers change in the Nd-Dy region. It is interesting to note that the authors of the review [31] do not exclude fractional coordination (between 8 and 9) Ln=Gd-Ho in Ln(NO3) solutions. According to [32] the exact coordination numbers of rare-earth ions in the nitrate complexes of solutions are unknown. The latter statement seems to be correct, since CNLn in aquatic solutions depends on the concentration: in highly concentrated solutions, coordination with fewer solvent molecules can be realized than in diluted ones, and even more so in significantly diluted ones, due to a shortage of water molecules.
Analysis of Figure 2 and Table S1 data allows distinguishing two Ln(NO3)3 · xH2O regions: Ln=Ce-Eu (region 1) and Ln=Eu-Lu (region 2). Moreover, it is not excluded that in region 1 of Ln(NO3)3 · xH2O solutions both bidentate and monodentate bonds of Ln with nitrate groups are realized, and in region 2 only monodentate bonds occur.
According to Raman spectroscopy data [33], in a wide range of Се(NO3)3 concentrations in aqueous solution, nitrate ions are bound to Ce both monodentately and bidentately, which is consistent with our data (Table S1, Figure 2). Complexation of Nd3+ and Eu3+ ions with nitrate group was studied by spectrophotometry and microcalorimetry [20]. The authors are of the opinion that both inner-sphere and outer-sphere nitrate complexes of Ln3+ ions exist in solutions. Based on thermodynamic and spectroscopic data, it is assumed that the weak complex of Nd3+ with nitrate in solution forms an inner sphere, and the nature of complex formation increases with an increase in temperature. In addition, it is possible that nitrate binds Eu3+ and possibly also Nd3+ bidentately in aqueous solutions.
Bonal S. et al. [19] used microcalorimetry to determine the stability constant, Gibbs energy, enthalpies and entropies to analyze the very weak complexation of Ln=La-Lu nitrate anion in dilute aqueous solutions of Ln(NO3)3 · xH2O at room temperature (298.15 K) with analysis of changes in the thermodynamic properties of (LnNO3)2+ across the lanthanide series. With a decrease in the ionic radius of Ln3+, it is more difficult for the nitrate anion with a slightly larger size of the NO3 group than a water molecule to “penetrate” into the inner sphere of the Ln3+ cation. Therefore, for Tm-Lu the inner sphere consists exclusively of water molecules, which was confirmed by studying aqueous solutions of rare earth metal nitrates in a glassy state by Raman spectroscopy [32]. Moreover, as the cation becomes smaller, the preference for monodentate nitrate binding increases, due to avoided repulsions in the first coordination sphere. These literature data do not contradict our analysis of EXAFS results (Table S1, Figure 2).
Dobler et al. [16] showed by quantum chemical investigation that an increase in the number of water molecules in the first coordination sphere of Ln3+ promotes monodentate coordination of nitrate. The change from bidentate to monodentate coordination is also observed before the salt dissociation, i.e. in an aqueous solution, nitrates on the way to dissociation pass from bidentate to monodentate coordination. The thermodynamic explanation for this process is interesting. In solution, the enthalpy (ΔH) and entropy (ΔS) energy components are antagonists. Bidentate binding may be promoted by entropy, which reduces the number of “frozen” water molecules coordinated with Ln3+ ions. On the other hand, monodentate bonding may be preferable from an enthalpy point of view, since up to six hydrogen bonds can be formed with the oxygens of the three monodentate nitrates (ONO3) instead of three with the bidentate nitrate groups. If we compare the course of interatomic distances (Table S1, Figure 2) according to EXAFS data with the curves ΔS of complexes (LnNO3)2+ and ΔH from Ln [16], then we can detect similarities for Ln=Ce-Eu with ΔS curve. This may be indirect confirmation of the presence of a certain amount of nitrate groups bidentately bound to Ln=Ce-Eu.
The authors of [17] used MD with explicit polarization and UV-visible spectroscopy to study solutions of Nd3+ and Dy3+ nitrates from a “highly diluted” solution to experimental saturation. It has been established that the bidentate mode is somewhat more stable for Nd3+ than for Dy3+; at the end of the lanthanide series, the ratio between the bidentate and monodentate conformations decreases, while only the monodentate mode is present in Lu.
Moreover, in solution, hydrated nitrate complexes can exhibit an equilibrium between several polyhydrate forms involving different types of nitrate binding modes [34].
Analysis of the limited literature data on the study of dilute solutions of Ln(NO3)3 · xH2O salts confirms the different structural behavior of Ln3+ ions in them, and different results are observed depending on the calculation methods and experimental conditions. It should be noted that experimental concentrations are typically far from standard and cannot be correctly extrapolated to infinite dilution to obtain a thermodynamic equilibrium constant that is valid only for a particular medium and concentration range (ionic strength).
2.3. X-ray Diffraction
According to the structural analysis of Ln(NO3)3 · xH2O salts [35,36,37,38,39,40,41,42,43,44,45], with an increase in the Ln atomic number, the total content of water molecules decreases: x=6 for Ce - Sm, x=6 and 5 for Eu - Tb, x=5 for Dy-Yb (except for Tm with x=6 and 5), x=4 and 3 for Lu. In the inner sphere (in square brackets) of coordination compounds of the form [Ln(O2NO3)3(OH2)n] · (x-n)H2O, the number of water molecules also decreases along the Ln series (n=5 for Ce, n=4 for Pr-Yb, n=3 for Lu) connected monodentantly to Ln. In the outer sphere (outside square brackets), the number of water molecules (x-n) for all Ln except Ce, and the content of (NO3)1- groups bidentantly coordinated with Ln ions remains constant (3 NO3).
It should be noted that in the Ln(NO3)3 · 6H2O structures, starting from Gd, the length of one Ln-ONO3 bond is greatly increased compared to others (bond asymmetry: Δ=202Å). The bond asymmetry increases in the structure with Tb (Δ=220Å) and is maximum (Δ=523Å) in the structure with Tm [36]. This gave reason to the authors of [36] to assume one monodentate and two bidentate bonds of NO3 with Tm3+ ions (there is no asymmetry of bonds in the Tm(NO3)3 · 5H2O structure). This asymmetric bonding seems to be associated with a steric effect of the coordinating water molecules. The increasing asymmetry in the binding mode of one nitrate group is related to the decrease in the ionic radius of the Ln.
An analysis of the interatomic distances given in the literature showed that for the same Ln, for which x=5 and x=6 in the Ln(NO3)3 · xH2O composition, with an increase in molecules of crystallization water, the average Ln-OH2O distance decreases (ΔLn-O(H2O) ~0.032Å), and Ln-ONO3, on the contrary, increases, reaching a maximum value (ΔLn-O(NO3) =0.058Å) for Tm [36]. In this case, the average Ln-O interatomic distance in the first coordination sphere increases upon transition from x=5 to x=6. However, for Lu, in the transition from x=3 to x=4, the value of ΔLn-O(NO3) =0.004Å, which contributes to an increase in Ln-O (R,Å) in the Lu(NO3)3 · 3H2O structure compared to Lu(NO3)3 · 4H2O Lu(NO3)3 · 3H2O (Table S3, Figure 3).
The X-ray diffraction study of commercial Ln(NO3)3 · xH2O samples [46] indicates single-phase Ln(NO3)3 · 6H2O with Ce (structure 1; x=6), Pr-Tb (structure 2; x=6), Tm (structure 3; x=5), two-phase with Dy-Er (structure 2 + structure 3), Yb (structure 3 + unknown structure 4) and uncertainty with Lu (unknown structure 5 or non-single-phase sample). Although the crystal structures of 1-3 differ from each other (the closest are structures 2 and 3), and the structures of 4 and 5 are unknown, their main structural fragments are the same.
Table S3 and Figure 3a-c show the average interatomic distances Ln-O (R, Å) without separation into Ln-OH2O and Ln-ONO3, calculated based on the Ln coordination in the inner sphere, taking into account the quantitative analysis of non-single-phase Ln(NO3)3 · xH2O with Ln = Dy, Ho, Er (Figure 3a, c, d; red dots). The structural parameters are consistent with the data given in [16] for Ln(NO3)3 · xH2O with Ln=Ce-Sm (x=6; CN=11 for Ce, CN=10 for Nd-Sm), but differ for Eu (x=6 according to our data, x=5 according to [16]) and interatomic Lu-O distances (Figure 3a, blue double dots), which we either calculated or took from structural data [43].
X-ray diffraction patterns of Ln(NO3)3 · xH2O solutions (Figure 4) are represented by two pronounced halos with interplanar distances in the intervals d = ~7.16- ~6.34 Å (1st peak) and d = 3.423-3.119 Å (2nd peak) and diffuse peak with a maximum at d ~ 2.15Å (3rd peak).
A comparison of diffuse reflections of Ln(NO3)3 · xH2O solutions with Ln=Ce, Sm, Gd, Yb (Figure 4) with similar reflections of a dilute aqueous solution of lanthanum nitrate [47] shows their similarity. The main diffuse peaks for water (Figure 4, 1st and 2nd peaks) occur at 14.17 and 27.92°(2θ) (d = 6.24 and 3.193 Å) and a very weak and diffuse peaks at ~39.85°(2θ) (d ~ 2.26 Å) (Figure 4, 3rd peak) are present in the diffraction patterns of all solutions.
If we take the intensities of the 1st (I(1)rel) and 2nd (I(2)rel) diffuse reflection for water as reference, then the intensity I(1)rel>I(2)rel for all concentrations of Ln(NO3)3 · xH2O solutions (Figure 4). Moreover, I(1)rel>I(2)rel with c=134 Mm for Ln=Ce, Sm (for Sm to a greater extent), for Yb with c=50 and 134 mM the values of I(1)rel and I(2)rel are almost identical, and for Gd the 1st and 2nd diffuse reflections with c = 50 mM are most pronounced (Figure 4). This pattern of changes in the reflection intensity of Ln(NO3)3 · xH2O (Ln = Ce, Sm, Gd, Yb) solutions corresponds to the characteristic structural features of commercial samples of these compositions.
When comparing diffraction patterns of aqueous solutions [31,47,48] and Ln(NO3)3 · xH2O salts [35,37,38,43], the position of the 1st diffuse peak generally coincides with the region of the most intense Bragg reflections in the range of 2θ ~12÷~15°, in particular, 10 with d~6.7 Å, caused by Ln ions. These values are in the regions of increased electron density observed in the radial distribution functions of aqueous solutions.
It is necessary to pay attention to the background level in the diffraction patterns of solutions and water (Figure 4), in particular, in the low-angle region adjacent to 2θ=2°, which logically increases with an increase in the Ln(NO3)3 · xH2O concentration from 0 (water) to c = 134 mM. This may be an indirect indication of the presence of clusters in solutions, the size of which increases with an increase in the solution concentration, since it is known that a system consisting of weakly bound atoms, ions or molecules forms a so-called cluster ion. It occupies an intermediate position between van der Waals molecules and molecular formations with a covalent chemical bond, and the components of the cluster retain their specific individuality.
2.4. FT-IR Spectroscopy Data
Figure 5a shows the IR spectra of solutions of commercial Ln(NO3)3 · xH2O salts (c=20 mM and 50 mM) with Ln = Ce, Sm, Gd, Yb, and the bandwidth correspondence is presented in Table S4.
The band shift ~1044 cm-1 is associated with the bond covalency degree of nitrate ions with Ln ones, which increases along the Ln series. Based upon this shift in the 1050 cm-1 region, the following series may be set up for increasing covalency of metal-nitrate bond: Ce<La<Gd=Y [49] This should lead to a general tendency for interatomic distances and coordination numbers to decrease due to a decrease in the size of Ln, which is confirmed by the data in Figure 2. A very weak band at 1146 cm-1 (Figure 5b) may correspond to bending vibrations of the hydronium ion δ (H3O+), which can be present in a solution with pH<7 [50]. The intensity of this band increases with an increase in Ln concentration in Ln(NO3)3 · xH2O solutions with Ln=Gd and Yb (Figure 5b).
The splitting of the ~1400 cm-1 band into two (~1345 and ~1400 cm-1) (Figure 5c) indicates an asymmetric vibration of the uncoordinated hydrated nitrate ion. The difference (Δν~125 cm-1) between the bands ~1345 and ~1470 cm-1 indicates partial coordination of the nitrate ions to Ln [51]. With an increase in the concentration of the Ln(NO3)3 · xH2O solutions (from 20 mM to 100 mM), the ~1470 cm-1 band becomes more clearly defined (Figure 5c). This means an increase in the number of bound nitrate ions and their gradual dominance over water molecules in competition to enter the inner coordination sphere. The band at 1470 cm-1 is quite wide and, in accordance with [12], apparently results from the superposition of bands of monodentate and bidentate coordinated nitrate ions [52,53].
Strong band at ~1635 см-1 is the result of superposition of water scissoring bending and N=O stretching: the vibrational peak for N=O stretching of bidentate coordinated nitrate ions usually appears at 1630-1788 cm-1 (Figure 5d). The band at 1640 cm–1 is related to the intermolecular bending oscillation ν2 of the water molecule, which is due to the change in the angle between OH bands [54]. From the above results it can be concluded that all bands in the 1625-1524 cm-1 region characterize isolated N=O bonds (consistent with bidentate structure of nitrates) and the bands below 1520 cm-1 are coupled vibrations, consistent with monodentate (but not only) nitrate structure [55].
The weak band in the region of ~1750 cm-1 (Figures 5a, 5e) corresponds to the combination band of the symmetrical stretch and in-plane bending of the nitrate-ion. With an increase in the Ln atomic number, the position of this band shifts (Figures 5a, 5e) to the long-wavelength region (1745 cm-1 for Ce, 1749.6 cm-1 for Sm, 1752.3 cm-1 for Yb; it was not possible to detect it for Gd), due to a weakening of the O–N bond in the nitrate ligand owing to an increase in electron transfer to the Ln3+ metal ion with an increase in the charge density.
Bands above 1600 cm-1 are usually attributed to N=O bond and suggest bidentate coordination. In contrast, when the highest frequency band is below ca. 1500 cm-1, this is an indication of monodentate coordination [55].
The position and intensity of the transmission bands corresponding to vibrations of nitrate ions in these spectra are very close for all samples, and the intensity of the bands increases with an increase in the concentration of solutions. It should be noted that from the IR spectra of Ln(NO3)3 · xH2O solutions with low-intensity, overlapping or even partially absent transmission bands, it is quite difficult to identify nitrate ions coordinated monodentately by Ln3+ ions. However, judging by the spectra, part (from ~10% to ~50%) of nitrate ions is coordinated by Ln3+ ions mono- or bidentately (mainly monodentately), and the remaining nitrate groups are uncoordinated.
A wide weak band at ~2114 cm-1 (Figure 5a) corresponds to the composite vibration of water molecules: bending vibration together with stretching, intermolecular, due to the rotation of the water molecule. The broad strong band ~3314 cm-1 corresponds to the stretching of free, hydrogen-bonded and coordinated water molecules. The band is greatly broadened, which indicates the implementation of strong hydrogen bonding in Ln(NO3)3 · xH2O solutions (Figure 5a).
The wide intensive band at 2700–3700 cm–1 corresponds to three oscillations of the water molecule: asymmetric stretching oscillation (3490 cm–1) symmetric stretching oscillation (3280 cm–1, and the overtone of the bending oscillation (3250 cm–1). The intensity of the maximum of the bending band, on the contrary, decreases with a reduction of the temperature in the same range [54].
A wide intense band below ~800 cm-1, responsible for stretching vibrations of water molecules, overlaps the bands of bending vibrations of nitrate ions lying in this spectrum region (Figure 5a).
The IR spectra of Ln(NO3)3 · xH2O salts contain transmission bands both observed for solutions (marked with * in Table S4 and in the text) and those belonging only to the salts. Thus, bidentately coordinated nitrate ions correspond to intense bands at ~1460 cm-1* and 1280 cm-1, medium-intensity bands at ~1660 cm-1* (in solutions, this band is present for Gd and Yb) (Figure 5d), and 1044 cm-1*. The difference between the band values at 1280 and 1044 cm-1 (Δ=236 cm-1) indicates bidentate chelating of nitrate ions [55].
The ν(H2O) band splits into ~3560 cm-1, 3480 cm-1, ~3200 cm-1, with the exception of the salt with Ce, for which a broad band is observed at ~3500 cm-1 due to hydrolysis in air, characteristic of salts with Ln=La-Sm (mostly for La). The intense band at 1635 cm-1* refers to bending vibrations of water molecules.
Weak transmission bands at ~810 and 750 cm-1 are attributed to stretching and bending vibrations of (NO3); very weak bands at 675 and 610 cm-1 belong to Ln-О (νLn-O) vibrations.
Thus, the results of IR spectroscopy of commercial Ln(NO3)3 · xH2O salts with Ln=Ce, Sm, Gd, Yb confirm the structural analysis data known from the literature on the implementation of only bidentately coordinated nitrate groups in them. As for Ln(NO3)3 · xH2O solutions, according to IR spectroscopy, in dilute solutions of 20 mM there are Ln3+ ions coordinated by nitrate groups, but in very small quantities, bidentantly and monodentantly (mainly) linked with Ln3+ ions. The number of monodentate nitrate groups increases from Ce to Yb: the ratio of the intensity of the band (shoulder) at 1750 cm-1 to the intensity of the water bending vibration band at 1635 cm-1 increases from Ce to Yb (Figure 5e).
3. Discussion
In aqueous solutions of Ln(NO3)3 · xH2O salts, hydrolysis occurs - a chemical reaction between salt ions with H+ and OH- ions of water. Ln(NO3)3 · xH2O salts are formed by the weak base of the multivalent Ln3+ ion and the strong HNO3 acid, therefore hydrolysis occurs through cations (pH<7), which bind to the water anion in a stepwise manner:
Stage I:
Ln(NO3)3 + HOH ↔ LnOH(NO3)2 + HNO3
Stage II:
LnOH(NO3)2 + HOH ↔ Ln(OH)2(NO3) + HNO3
Stage III:
Ln(OH)2(NO3) + HOH ↔ Ln(OH)3 + HNO3
Hydrolysis at stage II and, in particular, at stage III practically does not occur at room temperature, at which we conducted the experiment. This is also facilitated by ions that are formed during hydrolysis at the stage I, which suppress hydrolysis at the stage II, shifting the equilibrium of reactions to the left. In accordance with Le Chatelier's principle, when diluting solutions (decreasing the concentration of hydrogen ions), the degree of hydrolysis increases. The tendency to hydrolysis increases with an increase in atomic number and a decrease in ionic radius of Ln [56]: when moving from “light” to “heavy” Ln, hydrolysis increases. The degree of hydrolysis can be influenced by the composition of the substances involved in hydrolysis and the concentration of hydrolysis products, as well as the process temperature. The structure of Ln(NO3)3 in solutions depends on the degree of hydrolysis of solutions: the composition of the outer and inner spheres (the presence and content of nitrate ions and water molecules) and the density of ligands (the same groups).
For solutions of Ln(NO3)3 · xH2O salts (t=25°C; Ln = Ce, Sm, Tb, Gd, Yb) pH<7, but the pH values are different (Figure 6).
For Ln=Ce and Gd, with increasing solution concentration, the pH value decreases (Figure 6); for Ln=Sm pH values first decrease from pH=5.52 (c=134mM) to 5.35 (c=100mM), and then increase from pH=5.39 (c=50mM) to 5.56 (c=5.56mM); at c > 20 mM, turbidity of the solution was observed; the pH value of a solution with Yb increases with an increase in its concentration with a maximum value of pH=6.20 at c=50mM. As can be seen from Figure 6, the Tb(NO3)3 · xH2O salts solution is distinguished from other solutions by lower pH values, which smoothly decrease with increasing solution concentration. This behavior of the Ln(NO3)3 · xH2O solution with Ln=Tb is consistent with the EXAFS data (Figure 3) for the Tb(NO3)3 · xH2O solution.
All these experimental results indicate different hydrolysis processes in the systems under study depending on the type of Ln and the phase purity of the salt, as well as possible intermediate complexes formed in the system. It is necessary to pay attention to the obvious increase in pH with increasing temperature from 21°C (the temperature at which the XAS solution experiments were performed) to 25°C (the temperature at which IR spectra were obtained and the pH of the solutions was determined), which should be accompanied by an increase in hydrolysis. In the case of a salt formed by a weak base and a strong acid (our case), the Ln3+ cation undergoes hydrolysis and the reaction is accompanied by the formation of Н3О+ ions (the presence was not excluded when analyzing the IR spectra of solutions) (Table S4, Figure 5). An increase in the Н3О+ content in the solution leads to a decrease in the concentration of OH- ions.
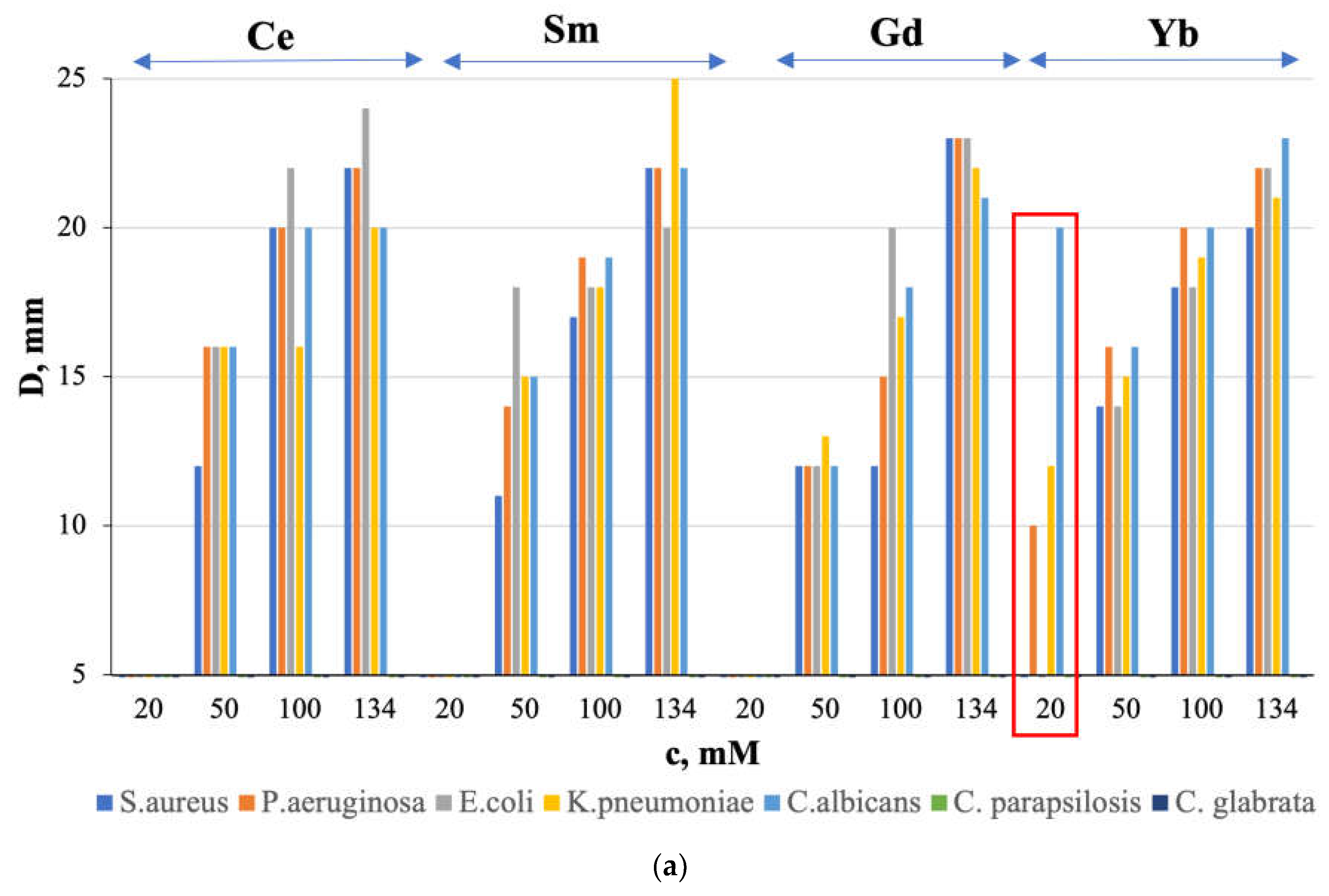
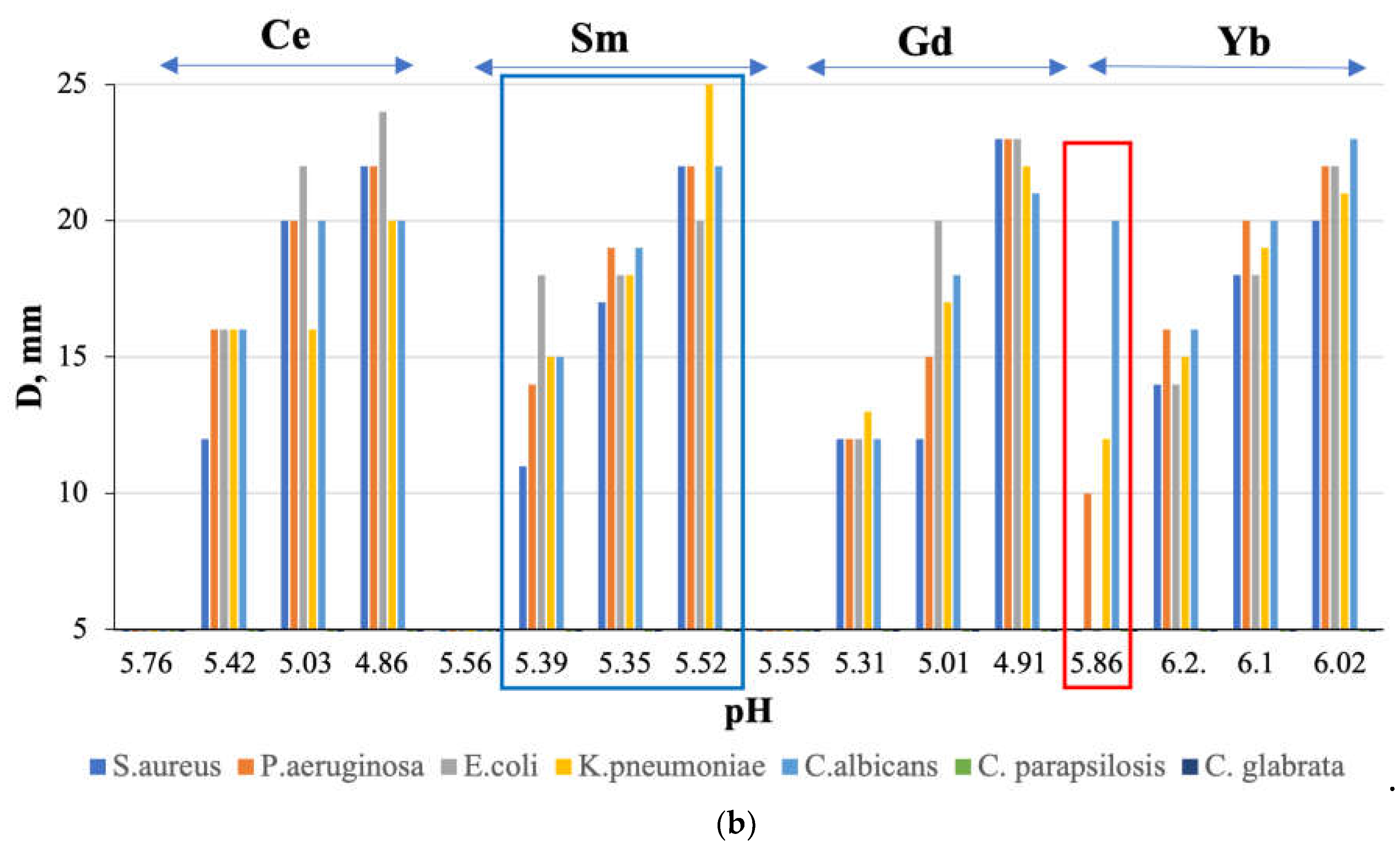
The obtained characteristics of Ln(NO3)3 · xH2O solutions with Ln=Ce,Sm,Gd,Yb do not contradict their antimicrobial activity (Figure 7): microorganism growth inhibition zone (D,mm value) has a general tendency to decrease with increasing solution concentration (Figure 7a) and increasing pH value (Figure 7b), with the exception of Ln = Sm, for which higher values of D,mm were obtained at higher pH (Figure 7b, blue rectangle).
Antimicrobial activity is absent for solutions with a concentration of c = 20 mM, except for a solution with Ln = Yb (Figure 7, red rectangle). At the same time, the sensitivity of the fungus C. albicans to a solution with c = 20 mM is higher than to a solution with c = 50 mM. We detected the observed “homeopathic” effect for the first time for these objects.
4. Materials and Methods
The nitrate salts Ln(NO3)3 · xH2O with 99.9 % Ln= Ce(x=6), Pr, Nd(x=6), Sm(x=6), Eu, Gd, Tb(x=6), Dy(x=5), Ho(x=5), Er, Tm, Yb, Lu were purchased from LANHIT Ltd (Russia) and used as received. We studied 20 (in most measurements), 50, 100, 134 mM Ln(NO3)3 · xH2O aqueous solutions (separate measurements were performed for solutions with Ln=Ce, Sm, Gd, Yb) with water pH 6.2. All solutions were prepared using ultrapure water (Milli-Q Advantage A10 Water Purification System, Millipore, France).
4.1. X-ray Studies
X-ray experiments have been carried out at the LANGMUIR beamline (Kurchatov Center for Synchrotron Radiation, Russia) [57]. The Langmuir trough was filed with water vapor saturated helium to decrease X-ray scattering. Aqueous solutions of Ln(NO3)3 · xH2O salts at a concentration of 20 mM were filled in the Langmuir trough mounted on the diffractometer. All measurements were performed at room temperature t = 21°C.
4.1.1. X-ray Absorption Spectroscopy Measurements at Liquid Surface
Ln(NO3)3 aqueous solutions were measured in fluorescence mode under TER geometry. The channel-cut monochromator Si (111) with spectral width of ~2 eV has been used to perform energy scan over the range of 400 eV. Ln3+ L3-edge absorption spectra were collected at the fixed incidence angle of 0.9×θC (θC is the critical angle of TER for water). The energy dispersive Vortex EX detector was mounted above the water subphase at the angle of 90°. To obtain high-quality EXAFS spectra we averaged several energy scans recorded in the multipass mode of data acquisition. The reproducibility of the energy position of monochromator was determined to be within 0.18 eV.
4.1.2. X-ray Absorption Spectroscopy Measurements of Powder Samples
Ln(NO3)3 · xH2O microcrystalline samples have been studied at the Structural Materials Science beamline (Kurchatov Center for Synchrotron Radiation, Russia) [58]. EXAFS spectra at the lanthanide L3-edge were collected in transmission mode using two ionization chambers filled with N2/Ar mixtures.
4.1.3. XRSW Measurements
The fluorescence intensity was recorded by energy dispersive Vortex EX detector mounted above the water subphase at an angle of 90°. The characteristic fluorescence spectra were collected for each angle of incidence in the angular range corresponding to TER region.
4.2. EXAFS Data Analysis
The extraction of the experimental fine-structure χ(k) from the atomic background function was performed using conventional procedures described elsewhere [59,60]. EXAFS data analysis was carried out in the single-scattering approximation based on the equation, which describes EXAFS as a sum of the contributions from different coordination shells with radii Ri and coordination numbers Ni
where is a many-body reduction factor, that accounts for amplitude damping due multielectron effect; and are the backscattering amplitude and phase shift for photoelectron scattered by neighbor atoms; λ(k) is the mean-free-path of the photoelectron; is Debye-Waller factor that represents the mean-square displacement of atom from equilibrium position. Backscattering amplitude and phase shift were calculated using the ab initio code FEFF8.5L [61,62]. The photoelectron inelastic losses were accounted for within the one-plasmon approximation using the complex exchange-correlation Hedin−Lundqvist potential [63]. FEEF calculations were performed for atomic clusters, which include nearest environment of lanthanide ions: 4 H2O molecules and 3 NO3 ligands. Such calculations allow performing EXAFS spectra fitting using paths associated with H2O and NO3 molecules. Fourier transformation is calculated for χ(k)×k2 using k-range = 2 – 9 Å-1; k2-weighted EXAFS data were fitted in space of their Fourier transforms |FT(χ(k)×k2)|. The value of factor was fixed at 0.9. All data processing and quantitative analysis of EXAFS spectra were performed using Larch software package [64].
EXAFS spectra obtained in our experiments for Ce and Pr exhibited distinct sing of multi-electron excitation, which is a well-known feature of electronic structure of these ions [65]. Thus, in analysis of the EXAFS spectra for Ce and Pr we used ATHENA program from the IFFEFIT package [66] to exclude the influence of multi-electron excitation effect.
4.3. X-ray diffraction experiments with Ln(NO3)3 · xH2O aqueous solutions of the beginning (Ce and Sm), middle (Gd) and end (Yb) ions of the Ln series with concentrations c=50 and 134 mM were carried out using Al container and difractometer PowDiX 600 (ADANI, Belarussia) equipped with a MYTHEN2 R 1D (Dectris) detector at room temperature (t=21°C) using monochromatic CuKα1 radiation (λ=1.5406Å) with Ni-filter 0.02 mm thick, on a diffracted beam; θ-2θ, 2θ ± 0.01°), continuous shooting in the range of 2 - 60° (30 kV, 10 mA).
4.4. FT-IR Spectroscopy
Fourier transform infrared spectra of Ln(NO3)3 · xH2O (Ln=Ce, Sm, Gd, Yb) solutions (с=20 and 50 for Ln=Ce, Sm, Gd, Yb; c=100 for Ln=Yb) were collected in a FSM-2201 (Infraspec) spectrometer in the 500–4000 cm-1 range. An average of 15 scans per sample with a resolution of 1 cm-1 formed each spectrum. Aqueous solutions were analyzed using an ZnSe Attenuated Total Reflectance (ATR) accessory. A Fourier transform infrared spectrometer WQF-530A (China) was used to study Ln(NO3)3 · xH2O (Ln=Ce, Sm, Gd, Yb) salts: wavenumber range 500-4000 cm-1, spectral resolution 0.85 cm-1, number of scans 256.
4.5. The Antimicrobial Activity
The study of antimicrobial activity (AMA) of Ln(NO3)3 · xH2O (Ln=Ce, Sm, Gd, Yb) solutions (c=20 and 50) was carried out by the disk diffusion method antimicrobial activity against bacteria (S.aureus, E.coli, P.aeruginosa, K.Pneumoniae) and fungi (C.albicans, C.glabrata, C.parapsilosis). The test bacterium was seeded as a «lawn» in Petri dishes on Mueller–Hinton agar. A thin-walled cylinder (6–8 mm in diameter) was used to make holes on the agar surface, into which test sample s were added in the powder form with the addition of 2–3 drops of NaCl physiological solution (0.9 wt.%). Next, Petri dishes were placed in a thermostat at 37°C for 24 hours. The sensitivity of microorganisms to the objects under study was assessed by the bacterial growth inhibition zone (D, mm) around the hole using a ruler. According to the classification [67] there are four groups of growth inhibition zone diameters of microorganisms in the presence of antimicrobial agents: D < 10 mm indicates a lack of sensitivity, D = 11–15 mm indicates low sensitivity, D = 15–25 mm denotes an average sensitivity, and D > 25 mm represents high sensitivity.
5. Conclusions
Based on the analysis of the results of studying commercial Ln(NO3)3 · xH2O salts (X-ray diffraction, IR spectroscopy, XAS) and their diluted solutions (XAS), as well as solutions with Ln=Ce, Sm, Gd, Yb (X-ray diffraction, pH measurement, IR spectroscopy) together with literature data, it was shown that in in solution, a few nitrate anions can be coordinated bidentantly and monodentantly depending on the type of Ln, and also be uncoordinated. Water molecules are chemically bonded to Ln ions via oxygen and, in addition, form multiple hydrogen bonds with water molecules and nitrate groups.
For the first time X-ray absorption spectroscopy under total external reflection geometry has been used to study the local environment of lanthanide ions in dilute aqueous solutions (20 mM Ln(NO3)3 · xH2O). The applied experimental technique allowed for high-quality EXAFS data to be collected for all elements from Ce to Lu (except for Pm). Essentially, all of the solutions were measured under the identical experimental conditions, EXAFS spectra were all treated by the same software package and modeling procedures that provided essential benefits in comparative studies of lanthanide coordination. A comparison of Ln-O interatomic distances for salts and their solutions, determined by the EXAFS method, showed that for a number of lanthanides from the middle of the lanthanide series, the oxygen coordination sphere contracts, which may be due to partial or complete dissociation of the nitrate complex in water. The absence of biocidal properties to a solution with c = 20 mM was found, excluding the Yb(NO3)3·xH2O one.
The understanding of molecular interactions through physicochemical properties is of utmost importance because they provide a potentially valuable insight into the forces that persist in liquid mixtures.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Structural parameters derived from EXAFS fitting procedure; Table S2: The interatomic distances (R,Å) and the Debye-Waller factors (σ), determined from analysis of EXAFS data for 20 mM aqueous solution of Ln(NO3)3 · xH2O salts with water molecules in the Ln local environment; Table S3: Average Ln-O interatomic distances in the first coordination sphere in the structures of Ln(NO3)3 · xH2O salts; Table S4: Assignment of the bands in the IR spectra of the Ln(NO3)3 · xH2O aqueous solutions; Figure S1: Fourier-transformed EXAFS spectra for Ln(NO3)3 · xH2O salts. Experimental spectra are shown using black color, and red color is used to plot fitted curved; Figure S2: Fourier-transformed EXAFS spectra for Ln(NO3)3 · xH2O aqueous solutions. Experimental spectra are shown using black color, and red color is used to plot fitted curved; Figure S3: Ln LIII XANES spectra for Ln(NO3)3 · xH2O salts (black color) and aqueous solutions (red color). Vertical lines are used to mark the position of the second maximum.
Author Contributions
Conceptualization, Galina Kuz’micheva; methodology, Galina Kuz’micheva; validation, Galina Kuz’micheva, Alexander Trigub; formal analysis, Galina Kuz’micheva, Elena Domoroshchina; investigation, Galina Kuz’micheva, Alexander Trigub, Alexander Rogachev and Andrey Dorokhov; data curation, Galina Kuz’micheva, Elena Domoroshchina; writing—original draft preparation, Galina Kuz’micheva and Alexander Trigub; writing—review and editing, Galina Kuz’micheva; visualization, Elena Domoroshchina; supervision, Galina Kuz’micheva; funding acquisition, Galina Kuz’micheva. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Higher Education of the Russian Federation (grant no. FSFZ-2024-0026).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available on request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Garner, J.P.; Heppell, P.S.J. Cerium nitrate in the management of burns. Burns 2005, 31, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Silva-Dias, A.; Miranda, I.M.; Branco, J.; Cobrado, L.; Monteiro-Soares, M.; Pina-Vaz, C.; Rodrigues, A.G. In vitro antifungal activity and in vivo antibiofilm activity of cerium nitrate against Candida species. J. Antimicrob Chemother. 2015, 70, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Cota, I.; Marturano, V.; Tylkowski, B. Ln complexes as double faced agents: Study of antibacterial and antifungal activity. Coordin. Chem. Rev. 2019, 396, 49–71. [Google Scholar] [CrossRef]
- Kuz’micheva, G. M.; Timaeva, O. I.; Novikova, N. N.; Yakunin, S. N.; Rogachev, A. V.; Svetogorov, R. D.; Pashkin, I. I.; Terekhovа, R. P. Antimicrobial Activity of Composite Hydrogels in the Poly(N-vinylpyrrolidone)–RE(NO3)3 · xH2O (RE Are Rare-Earth Ions) System. Crystallography Reports 2020, 65, 922–932. [Google Scholar] [CrossRef]
- Beuchat, С.; Hagberg, D.; Spezia, R.; Gagliardi, L. Hydration of Lanthanide Chloride Salts: A Quantum Chemical and Classical Molecular Dynamics Simulation Study. J. Phys. Chem. B 2010, 114, 15590–15597. [Google Scholar] [CrossRef]
- Rizkalla, E. N.; Choppin, G. R. Lanthanides and actinides hydration and hydrolysis. In Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K. A. Jr., Eying, L., Choppin, G. R., Lander, G. H., Eds.; Elsevier Science: Amsterdam, 1994; Volume 18, pp. 529–558. [Google Scholar]
- Yokoyama, H; Johansson G. Structures of Nitrate Complexes of Erbium in Aqueous Solutions. Acta Chemica Scandinavica 1990, 4, 567–573. [Google Scholar]
- Smirnov, P.R.; Grechin, O.V.; Trostin, V.N. Concentration Dependence of the Structure of Aqueous Solutions of Lutetium Nitrate According to X-ray Diffraction. Russian Journal of Physical Chemistry A 2014, 88, 250–253. [Google Scholar] [CrossRef]
- Butcher, T.A.; Formon, G.J.M.; Dunne, P.; Hermans, T.M.; Ott, F.; Noirez, L.; Coey, J.M.D. Neutron imaging of liquid-liquid systems containing paramagnetic salt solutions. Applied Physics Letters 2020, 116, 022405. [Google Scholar] [CrossRef]
- Yaita, T.; Narita, H.; Suzuki, S.; Tachimori, Sh. Structural study of lanthanides(III) in aqueous nitrate and chloride solutions by EXAFS. J Radioanal Nucl Chem. 1999, 239, 371–375. [Google Scholar] [CrossRef]
- Ohta, A.; Kagi, H.; Tsuno, H.; Nomura, M.; Kawabe, I. Influence of multi-electron excitation on EXAFS spectroscopy of trivalent rare-earth ions and elucidation of change in hydration number through the series. American Mineralogist, 2008; 93, 1384–1392. [Google Scholar]
- Nelson, D.L.; Irish, D.E. Interactions in Lanthanide Systems. I. A Raman and Infrared Study of Aqueous Gadolinium Nitrate. The journal of chemical physics. 1971, 54, 4479–4489. [Google Scholar] [CrossRef]
- Onghena, B.; Papagni, E.; Rezende Souza, E.; Banerjee, D.; Binnemans, K.; Hoogerstraete, T. Speciation of lanthanide ions in the organic phase after extraction from nitrate media by basic extractants. RSC Adv. 2018, 8, 32044–32054. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, W.W.; Irmer, G. ; On the Hydration of the Rare Earth Ions in Aqueous Solution. J Solution Chem. 2020, 49, 316–331. [Google Scholar] [CrossRef]
- Fratiello, A.; Kubo-Anderson, V.; Azimi, S.; Flores, T.; Martiinez, E.; Matejka, D.; Perrigan, R.; Vigil, M. A hydrogen-1, nitrogen-15, and chlorine-35 NMR coordination study of Lu(ClO4)3 and Lu(NO3)3 in aqueous solvent mixtures. J Solution Chem. 1990, 19, 811–829. [Google Scholar] [CrossRef]
- Dobler, M.; Guilbaud, P.; Dedieub, A.; Wipff, G. Interaction of trivalent lanthanide cations with nitrate anions: a quantum chemical investigation of monodentate/bidentate binding modes. New J. Chem. 2001, 25, 1458–1465. [Google Scholar] [CrossRef]
- Duvail, M.; Ruas, A.; Venault, L.; Moisy, P.; Guilbaud, P. Molecular Dynamics Studies of Concentrated Binary Aqueous Solutions of Lanthanide Salts: Structures and Exchange Dynamics. Inorg. Chem. 2010, 49, 519–530. [Google Scholar] [CrossRef]
- Migliorati, V.; Serva, A.; Sessa, F.; Lapi, A.; D’Angelo, P. Influence of Counterions on the Hydration Structure of Lanthanide Ions in Dilute Aqueous Solutions. J. Phys. Chem. B. 2018, 122, 2779–2791. [Google Scholar] [CrossRef] [PubMed]
- Bonal, C. ; Morel, J-P.; Morel-Desrosiers, N. Interactions between lanthanide cations and nitrate anions in water Part 2. Microcalorimetric determination of the Gibbs energies, enthalpies and entropies of complexation of Y3‘ and trivalent lanthanide cations. J. Chem. Soc., Faraday Trans, 1998; 94, 1431–1436. [Google Scholar]
- Rao, L.; Tian, G. Complexation of Lanthanides with Nitrate at Variable Temperatures: Thermodynamics and Coordination Modes. Inorg. Chem. 2009, 48, 964–970. [Google Scholar] [CrossRef]
- Allen, P.G.; Bucher, J.J.; Shuh, D.K.; Edelstein, N.M.; Craig, I. Coordination Chemistry of Trivalent Lanthanide and Actinide Ions in Dilute and Concentrated Chloride Solutions. Inorg. Chem. 2000, 39, 595–601. [Google Scholar] [CrossRef]
- Bera, M.K.; Luo, G.; Schlossman, M.L.; Soderholm, L.; Lee, S.; Antonio, M.R. Erbium(III) Coordination at the Surface of an Aqueous Electrolyte. J. Phys. Chem. B. 2015, 119, 8734–8745. [Google Scholar] [CrossRef]
- Shiery, R.C.; Fulton, J.L.; Balasubramanian, M, Nguyen, M. -T., Lu, J.-B., Li, J., Rousseau, R., Glezakou, V.-A., Cantu, D.C. Coordination Sphere of Lanthanide Aqua Ions Resolved with Ab Initio Molecular Dynamics and X-ray Absorption Spectroscopy. Inorg. Chem. 2021, 60, 3117–3130. [Google Scholar] [CrossRef]
- Persson, I.; D'Angelo, P.; De Panfilis, S.; Sandström, M.; Erikssonet, L. Hydration of Lanthanoid (III) Ions in Aqueous Solution and Crystalline Hydrates Studied by EXAFS Spectroscopy and Crystallography: The Myth of the “Gadolinium Break”. Chem.-Eur. J. 2008, 14, 3056–3066. [Google Scholar] [CrossRef] [PubMed]
- Solera, J. A.; García, J.; Proietti, M. G. Multielectron excitations at the L edges in rare-earth ionic aqueous solutions. Phys. Rev. B. 1995, 51, 2678. [Google Scholar] [CrossRef] [PubMed]
- Plakhova, T.; Romanchuk, A.; Yakunin, S.; Dumas, T.; Demir, S.; Wang, S.; Minasian, S.; Shuh, D.; Tyliszczak, T.; Shiryaev, A.; Egorov, A.; Ivanov, V.; Kalmykov, S. Solubility of Nanocrystalline Cerium Dioxide: Experimental Data and Thermodynamic Modeling. J. Phys. Chem. C. 2016, 120, 22615–22626. [Google Scholar] [CrossRef]
- Zegenhagen, J.; Kazimirov, A. X-ray Standing Wave Technique, Principles and Applications, World Scientific Publishing, Singapore, 2013; 556 p.
- Parratt, L.G. Surface Studies of Solids by Total Reflection of X-Rays. Phys. Rev. 1954, 95, 359–369. [Google Scholar] [CrossRef]
- Rizkalla, E.N.; Choppin, G.R. Hydration and hydrolysis of lanthanides. In Handbook on the Physics and Chemistry of Rare; Gschneidner, K.A.; Eyring, L., Eds.; Elsevier, Amsterdam, 1991, Volume 15, pp. 393-443.
- Rizkalla, E.N.; Choppin, G.R. Hydration of lanthanides and actinides in solution. Journal of Alloys and Compounds 1992, 180, 325–336. [Google Scholar] [CrossRef]
- Smirnov, P.R.; Trostin, V.N. Structural parameters of the immediate environment of ions in aqueous solutions of inorganic electrolytes. Ivanovo Publishing house, Russia, 2011; 400 p. (RU).
- Kanno, Н.; Hiraishi, J. Raman study of aqueous rare earth nitrate solutions in liquid and glassy states. J. Phys. Chem. 1984, 88, 2787–2792. [Google Scholar] [CrossRef]
- Choppin, G. R.; Strazik, W. F. Complexes of Trivalent Lanthanide and Actinide Ions. I. Outer-Sphere Ion Pairs. Inorg. Chem. 1965, 4(9), 1250–1254.
- Klimov, V.D.; Chudinov, E.G. Application of infrared spectroscopy to study bonds in complexes of rare earth nitrates with alkylammonium nitrates. Moscow: Order-of-Lenin I. V. Kurchatov Institute of Atomic Energy, Russia, 1974. (RU).
- Milinski, N.; Ribar, B.; Sataric, M. Pentaaquatrinitratocerium(III) monohydrate, Ce(H2O)5(NO3)3 · H2O. Crystal Structure Communication 1980, 9, 473–477. [Google Scholar]
- Klein, W. Crystal structures of the penta- and hexahydrate of thulium nitrate. Acta Cryst. 2020, E76, 1863–1867. [Google Scholar] [CrossRef] [PubMed]
- Taha, Z.A.; Ajlouni, A.; Hijazi, A.K.; Kühn, F.E.; Herdtweck, E. Redetermination of [Gd(NO3)3(H2O)4]·2H2O. Acta Cryst. 2012, E68, i56–i57. [Google Scholar] [CrossRef]
- Kawashima, R.; Sasaki, M.; Satoh, S.; Isoda, H.; Kino, Y.; Shiozaki, Y. Report on Temperature Dependence of Crystal Structure for Samarium Nitrate Having Metastable Phenomena. J. Phys. Soc. Jpn. 2000, 69, 3297–3303. [Google Scholar] [CrossRef]
- Decadt, R.; Van Der Voort, P.; Van Driessche, I.; Van Deun, R.; Van Hecke, K. Redetermination of [Pr(NO3)3(H2O)4]_2H2O. Acta Cryst. 2012, E68, i59–i60. [Google Scholar]
- Stumpf, T.; Bolte, M. Tetraaquatrinitratoeuropium (III) dehydrate. Acta Cryst. 2001, E57, i10–i11. [Google Scholar]
- Moret, E.; Bunzli, J-C. G.; Schenk, K.J. Structural and luminescence study of europium and terbium nitrate hexahydrates. Inorg. Chim. Acta 1990, 178, 83–88. [Google Scholar] [CrossRef]
- Rogers, D.J.; Taylor, N.J.; Toogood, G.E. Tetraaquatrinitratoneodymium(III) dihydrate, [Nd(NO3)3(H2O)4].2H2O. Acta Cryst. 1983, C39, 939–941. [Google Scholar] [CrossRef]
- Junk, P.C.; Kepert, D.L.; Skelton, B.W.; White, A.H. Structural Systematics of Rare Earth Complexes. XIII. ("Maximally") Hydrated (Heavy) Rare Earth Nitrates. Aust. J. of Chem. 1999, 52, 497–505. [Google Scholar]
- Rincke, C.; Schmidt, H.; Voigt, W. Rebuttal of the Existence of Solid Rare Earth Bicarbonates and the Crystal Structure of Holmium Nitrate Pentahydrate. Zeitschrift Für Anorganische Und Allgemeine Chemie, 2017; 643, 437–442. [Google Scholar]
- Klein, W. Crystal structure of tetraaqua-tris(nitrato-κ2O,O′) erbium(III) monohydrate, Er(NO3)3·5H2O, H10ErN3O14. Zeitschrift fur Kristallographie - New Crystal Structures, 2022; 237, 265–266. [Google Scholar]
- Setyaeva, E.A.; Glushko, A.A.; Kuzmicheva, G.M.; Neznanov, A.A.; Terekhova, R.P.; Svetogorov, R.D. Application of information technologies for the selection of salts and solutions RE(NO3)3·xH2O (RE=La-Lu,Y, Sc) with optimal structural characteristics and biocidal and neutron studies. In Book of reports of the Kurchatov Forum of Synchrotron and Neutron Research, Moscow, Russia, 24-27 October 2023. (RU).
- Grechin, O.V.; Smirnov, P.R.; Trostin, V.N. X-Ray Diffraction Study of Aqueous solutions of Lanthanum Chloride and Nitrate. Chemistry and Chemical Technology 2013, 56, 15–20 (RU). [Google Scholar]
- Smirnov, P.R.; Grechin, O.V.; Vashurin, A.S. Ion Coordination in Aqueous Lanthanum Chloride and Lanthanum Nitrate Solutions as Probed by X-Ray Diffraction. Russian Journal of Inorganic Chemistry 2022, 67, 382–387. [Google Scholar] [CrossRef]
- Rabadanov, K.Sh.; Gafurov, M.M.; Aliev, A.R. Study of vitrified homogeneous and heterophase nitrate systems using vibrational spectroscopy methods. In Proceedings of the XVII All-Russian Conference “Optics and Spectroscopy of Condensed Matter”, Krasnodar, Russia, 12-14 September 2011. (RU). [Google Scholar]
- Khakhalin, A.V.; Koroleva, A.V. Investigation of the temperature dependence for the spectra of supercooled water in the middle infrared. Moscow University Physics Bulletin. 2014, 1, 66–69. [Google Scholar] [CrossRef]
- Vratny, F. Infrared Spectra of Metal Nitrates. Applied Spectroscopy. 1959, 13(3), 59–70. [Google Scholar] [CrossRef]
- Egorov, N.B.; Shagalov, V.V. Infrared spectroscopy of rare and trace elements. Guidelines for conducting laboratory work in the course “Physico-chemical methods of analysis”; Tomsk: TPU Publishing House, Russia, 2012; 20 p. (RU). [Google Scholar]
- Sidyakin, P.V.; Karpov, V.L.; Egorov, B.N.; Egorova, Z.S. Radiation transformations in epoxy oligomers based on epichlorohydrin and n,n’-dioxydiphenylpropane. Polymer Science, Series A. 1971; 8, 2195–2206. [Google Scholar]
- Trivedi, M.K.; Branton, A.; Trivedi, D.; Jana, S. Spectroscopic Characterization of Disodium Hydrogen Orthophosphate and Sodium Nitrate after Biofield Treatment. Journal of Chromatography & Separation Techniques, 2015; 6, 2–5. [Google Scholar]
- Mihaylov, M.Y.; Zdravkova, V.R.; Ivanova, E.Z.; Aleksandrov, H.A.; Petko, P.S.; Vayssilov, G.N.; Hadjiivanov, K.I. Infrared Spectra of Surface Nitrates: Revision of the Current Opinions Based on the Case Study of Ceria. Journal of Catalysis. 2021, 394, 245–258. [Google Scholar] [CrossRef]
- Bentouhami, E.; Bouet, G.M.; Meullemeestre, J.; Verling, F.; Khan, M.A. Physicochemical study of the hydrolysis of Rare-Earth elements (III) and thorium (IV). Comptes Rendus Chimie. 2004, 7(5), 537–545. [Google Scholar] [CrossRef]
- Yakunin, S.N.; Novikova, N.N.; Rogachev, А.V.; Trigub, А.L.; Kuzmicheva, G.M.; Stepina, N.D.; Rozenberg, O.A.; Yurieva, E.A.; Kovalchuk, М. V. Spectral-Selective X-Ray Studies at the “Langmuir” Beamline of the Kurchatov Synchrotron Radiation Source. Crystallogr. Rep. 2022, 67, 799–812. [Google Scholar] [CrossRef]
- Chernyshov, A.A.; Veligzhanin, A.A.; Zubavichus, Y.V. Structural materials science end-station at the Kurchatov synchrotron radiation source: recent instrumentation up-grades and experimental results. Nucl. Instr. Methods Phys. Res. 2009, A603, 95–98. [Google Scholar] [CrossRef]
- Bunker, G. Introduction to XAFS: A Practical Guide to X-ray Absorption Fine Structure Spectroscopy; Cambridge University Press, UK, 2010.
- Aksenov, V.L.; Koval’chuk, M.V.; Kuz’min, A.Y. Purans, Yu.; Tyutyunnikov S.I. Development of methods of EXAFS spectroscopy on synchrotron radiation beams: Review. Crystallogr. Rep. 2006, 51, 908–935. [Google Scholar] [CrossRef]
- Ankudinov, A. L.; Ravel, B.; Rehr, J. J.; Conradson, S. D. RealSpace Multiple-Scattering Calculation and Interpretation of X-RayAbsorption Near-Edge Structure. Phys. Rev. B. 1998, 58, 7565. [Google Scholar] [CrossRef]
- Rehr, J.J.; Albers, R.C. Theoretical Approaches to X-Ray Absorption Fine Structure. Rev. Mod. Phys. 2000, 72, 621. [Google Scholar] [CrossRef]
- Hedin, L.; Lundqvist, B. I. Explicit Local Exchange-Correlation Potentials. J. Phys. C Solid State Phys. 1971, 4, 2064. [Google Scholar] [CrossRef]
- Newville, M.L. An Analysis Package for XAFS and Related Spectroscopies. J. Phys.: Conf. Ser. 2013, 430, 012007. [Google Scholar] [CrossRef]
- Fonda, E.; Andreatta, D.; Colavita, P.E.; Vlaica, G. EXAFS analysis of the L3 edge of Ce in CeO2: effects of multi-electron excitations and final-state mixed valence. J. Synchrotron Rad. 1999, 6, 34–42. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Blatun, L. A. Wounds and wound infections. The prof. B.M. Kostyuchenok journal 2015, 2, 36–44. [Google Scholar]
Figure 1.
Experimental angular dependence of Ce- fluorescence yield from 20 mM aqueous solution of Се(NO3)3 · 6H2O salts (squares). The dashed line represents the calculated angular dependence from the film, in which metal ions are distributed in the layer of 10 Å thick at the air/liquid interface. The energy of the incident beam was 13.6 keV.
Figure 1.
Experimental angular dependence of Ce- fluorescence yield from 20 mM aqueous solution of Се(NO3)3 · 6H2O salts (squares). The dashed line represents the calculated angular dependence from the film, in which metal ions are distributed in the layer of 10 Å thick at the air/liquid interface. The energy of the incident beam was 13.6 keV.
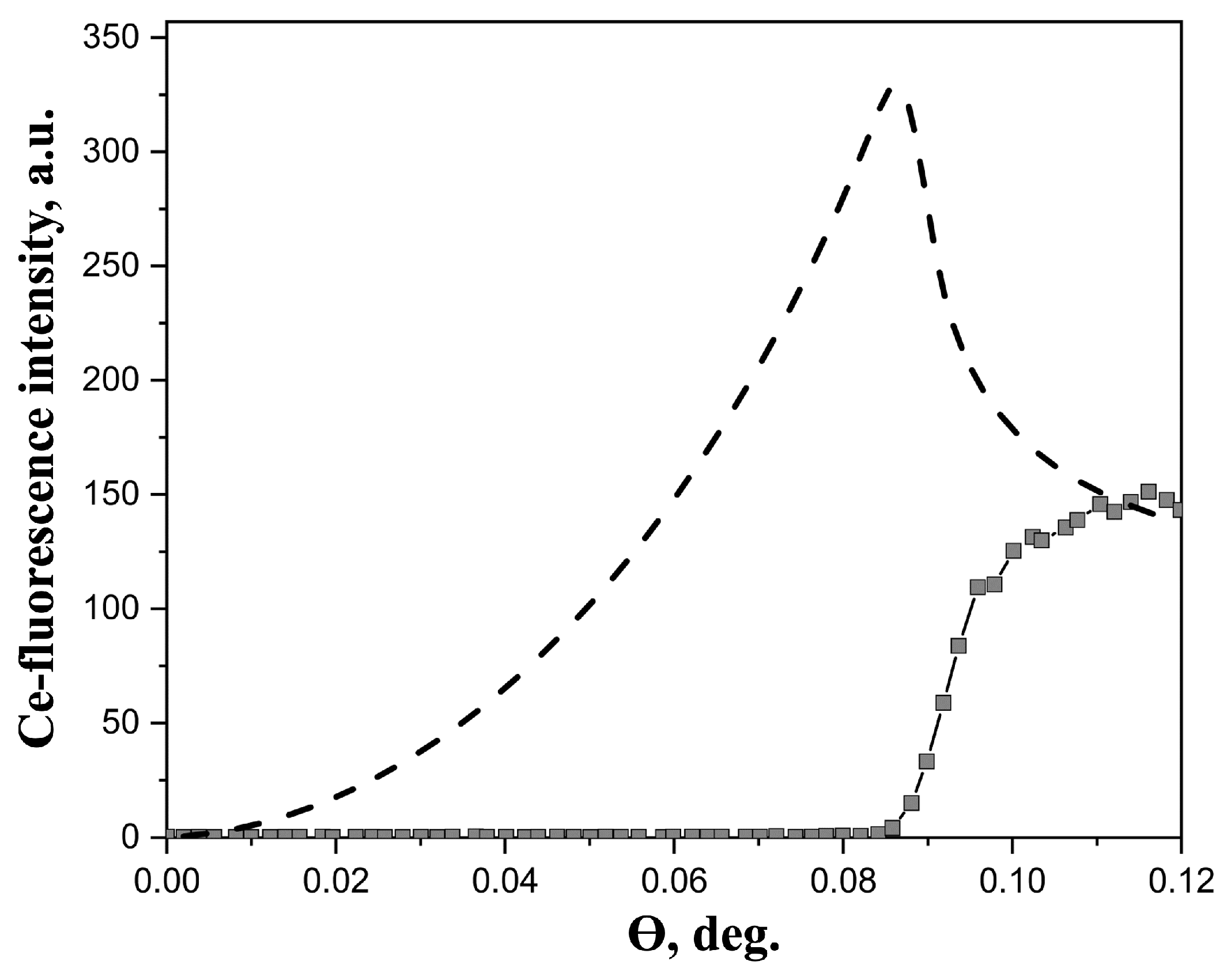
Figure 2.
Dependence of Ln-O distance from central atom type. Red and green curves are using to plot data for solutions and solids, respectively.
Figure 2.
Dependence of Ln-O distance from central atom type. Red and green curves are using to plot data for solutions and solids, respectively.
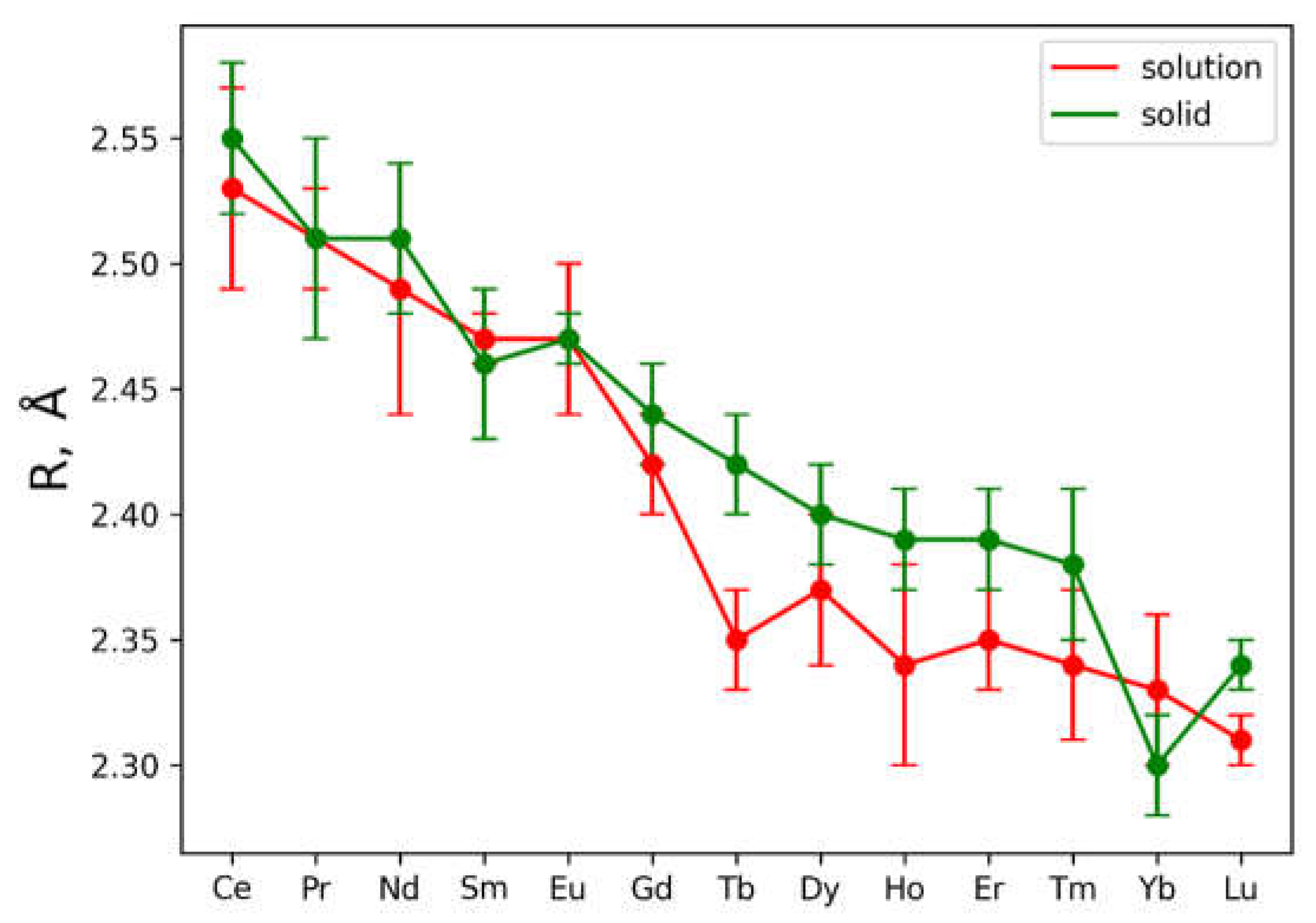
Figure 3.
(a) Average Ln-O interatomic distances (R, Å) in the structures of Ln(NO3)3 · xH2O salts according to literature data highlighting the (b) Pr-Sm, (c) Sm-Dy, (d) Dy-Yb regions. Red dots: interatomic distances calculated from phase analysis of the commercial samples we studied.
Figure 3.
(a) Average Ln-O interatomic distances (R, Å) in the structures of Ln(NO3)3 · xH2O salts according to literature data highlighting the (b) Pr-Sm, (c) Sm-Dy, (d) Dy-Yb regions. Red dots: interatomic distances calculated from phase analysis of the commercial samples we studied.
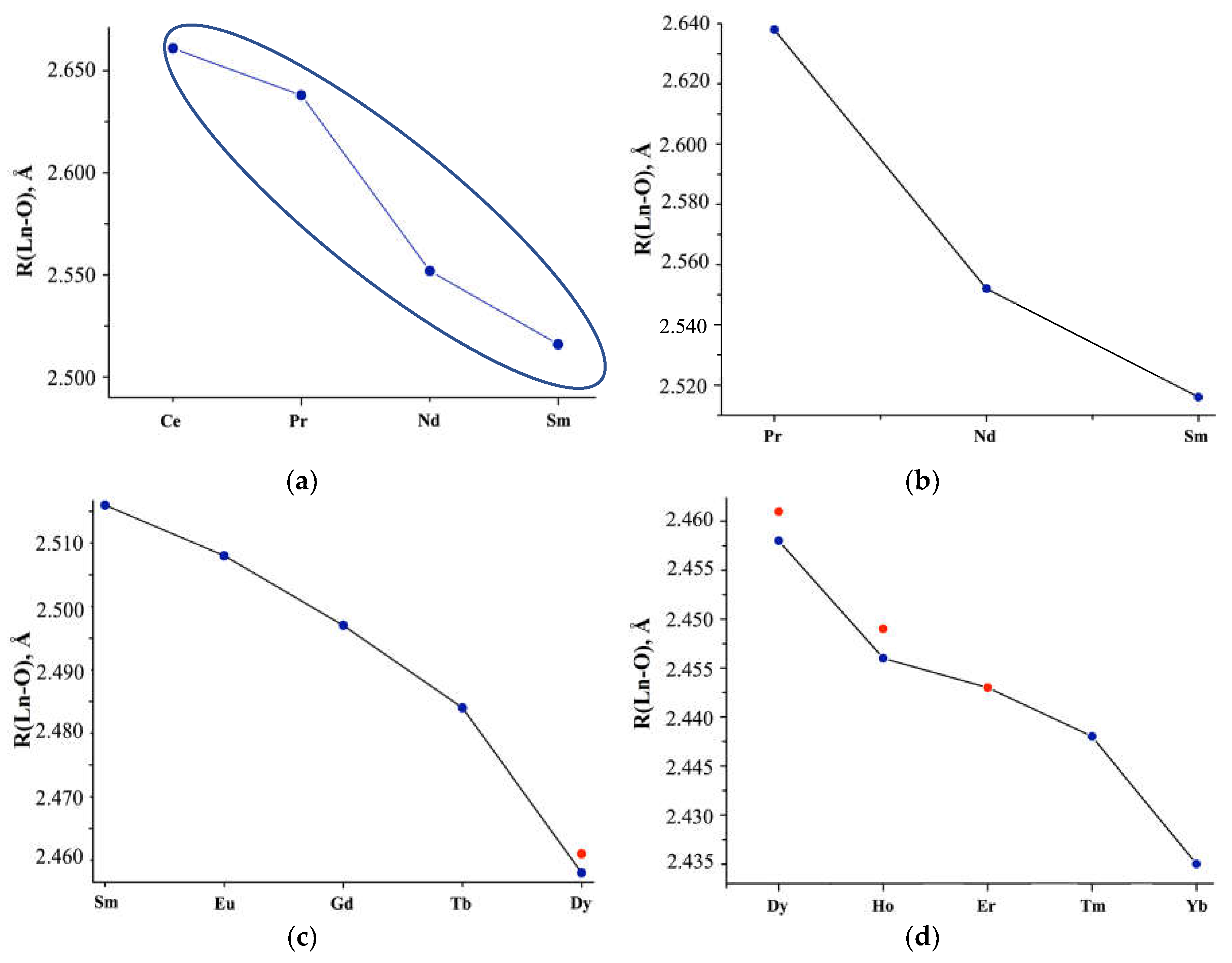
Figure 4.
Diffraction patterns of water and solutions with c=50 mM and c=134 mM: Ln(NO3)3 · xH2O with (a) Ln=Ce, (b) Sm, (c) Gd, (d) Yb.
Figure 4.
Diffraction patterns of water and solutions with c=50 mM and c=134 mM: Ln(NO3)3 · xH2O with (a) Ln=Ce, (b) Sm, (c) Gd, (d) Yb.
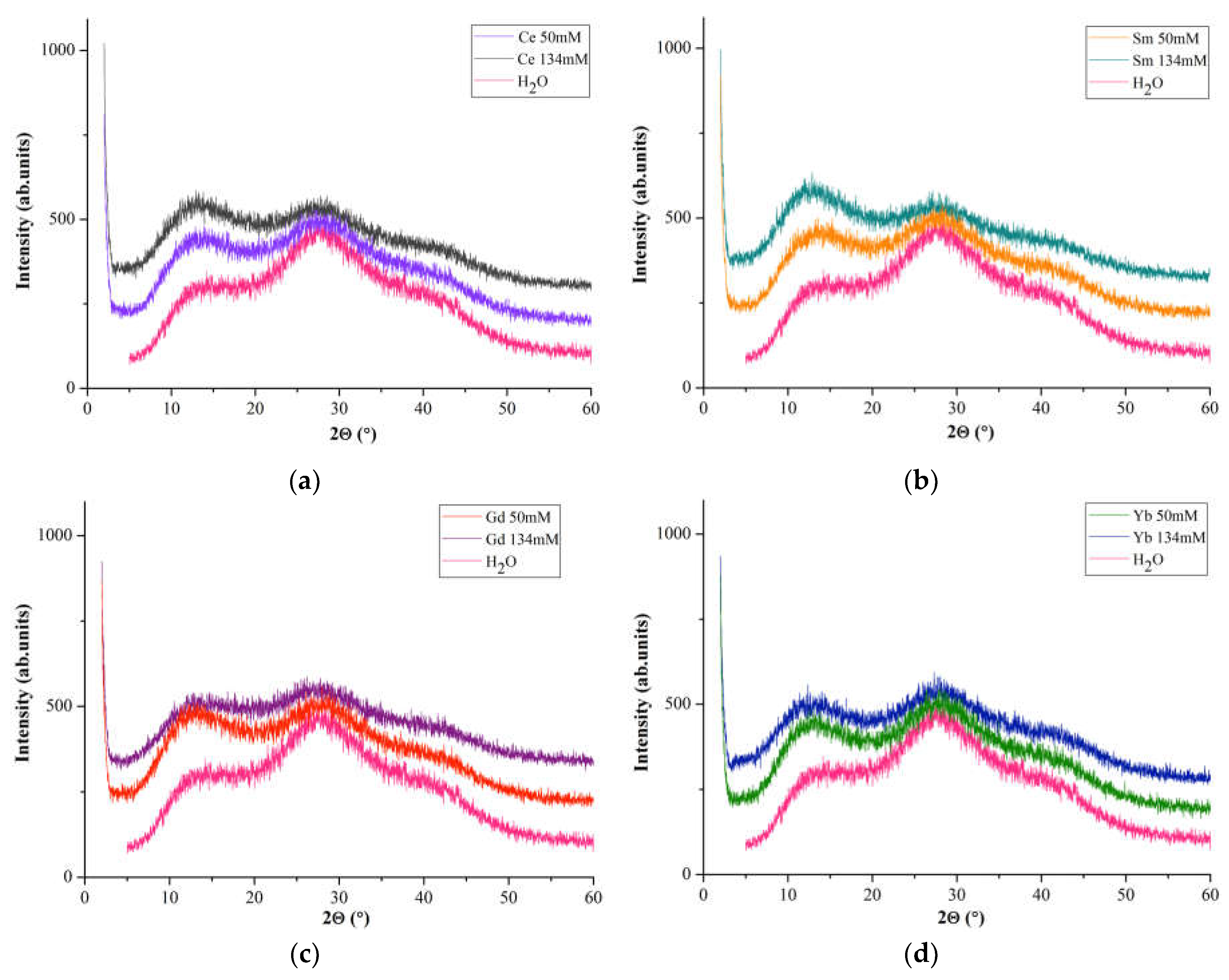
Figure 5.
(a) FT-IR spectra of Ln(NO3)3 · xH2O solutions (Ln = Ce, Sm, Gd, Yb); (c) part of the spectrum (1200-1500 cm-1) of Yb(NO3)3 · xH2O solution (c=50 mM and 100 mM); (b, d and e for c=20 mM) spectrum sections.
Figure 5.
(a) FT-IR spectra of Ln(NO3)3 · xH2O solutions (Ln = Ce, Sm, Gd, Yb); (c) part of the spectrum (1200-1500 cm-1) of Yb(NO3)3 · xH2O solution (c=50 mM and 100 mM); (b, d and e for c=20 mM) spectrum sections.
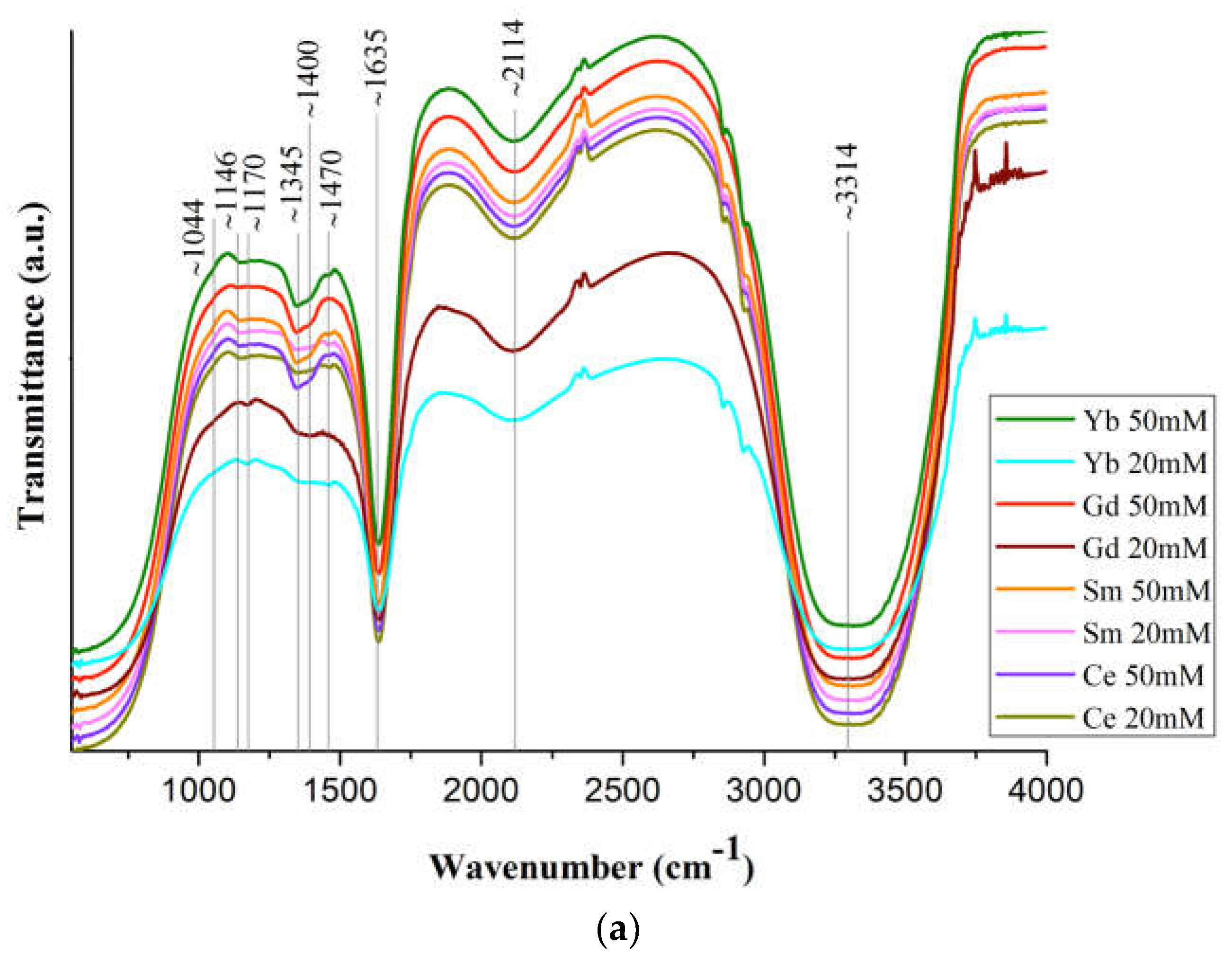
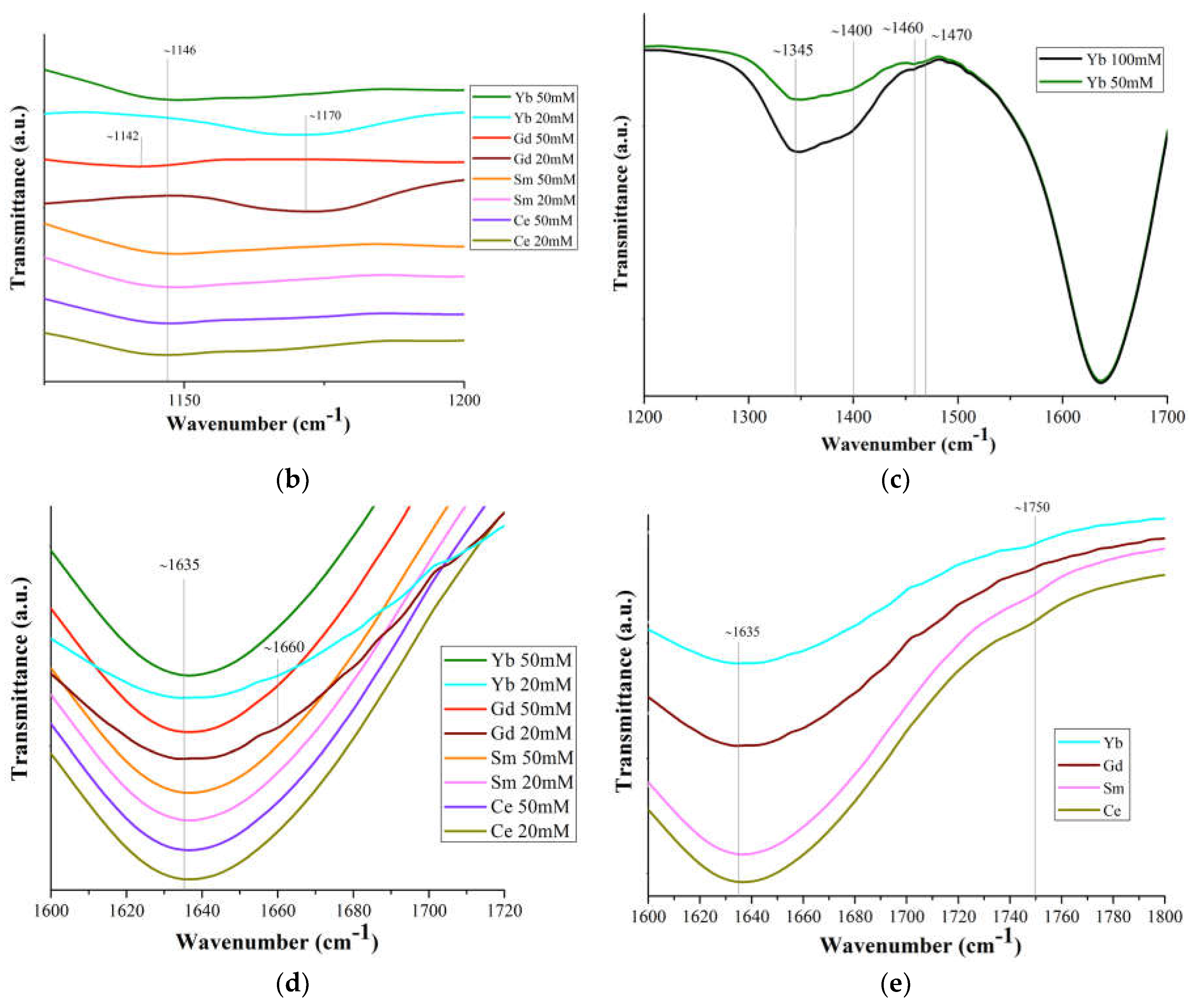
Figure 6.
Relationship between the pH value and the concentration of Ln(NO3)3 · xH2O solution with x=6 for Ln=Ce, Sm, Tb, Gd and x is unknown for Yb.
Figure 6.
Relationship between the pH value and the concentration of Ln(NO3)3 · xH2O solution with x=6 for Ln=Ce, Sm, Tb, Gd and x is unknown for Yb.
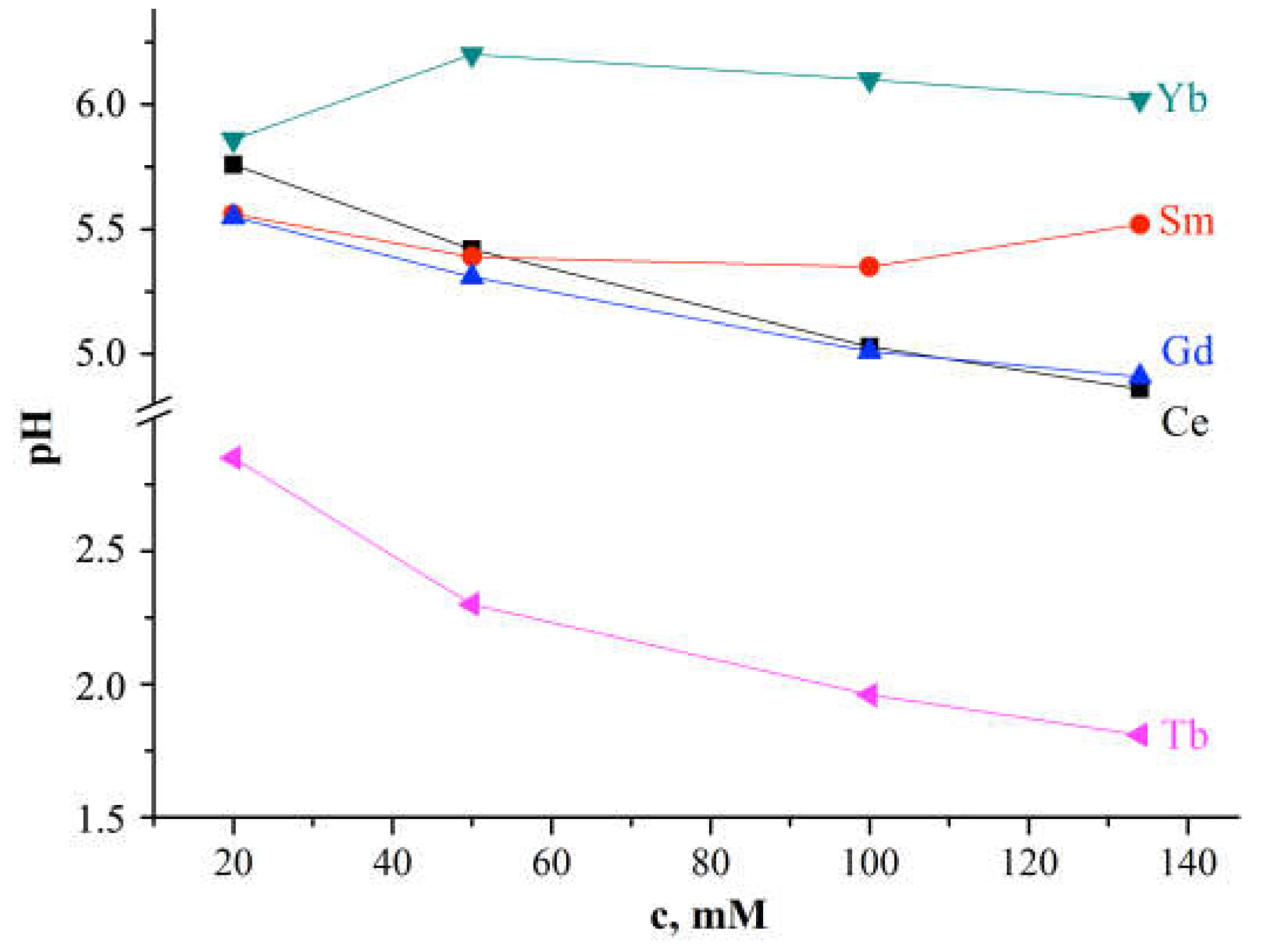
Figure 7.
Relationship between the growth inhibition zone (D, mm) and (a) concentration (c, mM) and (b) pH of Ln(NO3)3 · xH2O solutions (Ln=Ce, Sm, Gd, Yb).
Figure 7.
Relationship between the growth inhibition zone (D, mm) and (a) concentration (c, mM) and (b) pH of Ln(NO3)3 · xH2O solutions (Ln=Ce, Sm, Gd, Yb).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



