
Submitted:
04 July 2024
Posted:
04 July 2024
You are already at the latest version
Abstract
Hypoxia contributes to tumor progression and metastasis, and hypoxically dysregulated RNA molecules may thus be implicated in poor outcomes. Canine oral melanoma (COM) has a par-ticularly poor prognosis, and some hypoxia-mediated miRNAs are known to exist in this cancer; however, equivalent data on hypoxically dysregulated other non-coding RNAs (ncRNAs) are lacking. Accordingly, we aimed to elucidate non-miRNA ncRNAs that may be mediated by hy-poxia, targeting primary-site and metastatic COM cell lines and clinical COM tissue samples in next generation sequencing (NGS), with subsequent qPCR validation and quantification in COM primary and metastatic cells, plasma and extracellular vesicles (EVs) for any identified ncRNA of interest. Findings suggested that a number of non-miRNA ncRNA species are hypoxically up- or downregulated in COM. We identified one ncRNA, the long ncRNA fragment EN-SCAFT00000084705.1, as a molecule of interest due to its consistent downregulation in COM tissues, hypoxically and normoxically cultured primary and metastatic cell lines, when compared to the oral tissues from healthy dogs. However, this molecule was undetectable in plasma and plasma EVs, suggesting that its expression may be tumor tissue-specific and it has little potential as a biomarker. Here, we provide evidence of hypoxic transcriptional dysregulation for ncRNAs other than miRNA in COM for the first time, and suggest that ncRNA ENSCAFT00000084705.1 is a molecule of interest for future research on the role of the transcriptome in hypoxia-mediated progression of this aggressive cancer.
Keywords:
Hypoxia
; melanoma
; ncRNA
; NGS
; dog
; metastasis
1. Introduction
Canine oral melanoma is a major killer of dogs, and a hypoxic tumor microenvironment may be implicated in its highly aggressive nature and high propensity to metastasize [1]. As a solid tumor, COM sees a fall in oxygen pressure (to levels of 1% to 2%), with the oxygen supply proving unequal to the tumor growth [2,3] Crucially, hypoxia can cause upregulation of tumorigenic factors and downregulation of anti-tumorigenic factors, in addition to mediating tumor angiogenesis and cancer stem cell quiescence [4,5,6]. Thus, in a number of ways, hypoxia may act as a driver of tumor aggression and promote the progression to metastasis, and is particularly relevant for highly aggressive and frequently metastasizing malignant tumors such as COM.
COM research commands a high priority in veterinary medicine. Melanomas represent around seven per cent of all malignant tumors in dogs, and most frequently occur in the oral cavity [7,8,9]. COM has the poorest prognosis of any melanoma in dogs, with a median survival time for stage II and stage III disease of around six to eight months from diagnosis [9]. Moreover, the survival time reduced to two months if pulmonary metastasis develops at the time of diagnosis of COM [9]. COM research may have wider application, since these canine tumors resemble some human mucosal melanomas macroscopically [10] and human non-UV-induced cutaneous melanomas clinically and histopathologically [11]. Advances in the diagnosis and treatment of this disease in dogs may this also ultimately benefit human patients [8,12].
One promising line of COM research involves identifying elements of the transcriptome that may be dysregulated hypoxically. Changes in long non-coding RNA (lncRNA) profiles [13] and genomic alteration [14] are reportedly involved in metastatic COM. Hypoxia is known to alter gene expression and RNA profiles within the tumor microenvironment [15], and dysregulated molecules thus may have utility as therapeutic targets or diagnostic biomarkers. We previously identified a number of molecules of interest based on dysregulated expression. These reports encompass microRNAs (miRNAs) [16], and snoRNA, snRNA, piRNA, and tRNA fragments (tRFs) [17] in tumor tissue from COM patients. In further reports, we have highlighted dysregulated miRNAs [18], and lncRNA and tRFs [19] as exosomal biomarkers in plasma from COM patients. However, to the best of our knowledge, previous molecular investigations in COM have involved hypoxically dysregulated miRNAs [18] and Y-RNA [1], and a full understanding of hypoxic mechanism require evidence on the entire RNA transcriptome. Thus, further investigations of non-coding RNAs (ncRNAs) other than miRNAs are needed. Recent advances in molecular analysis technologies have added identification of transcriptome elements, with next generation sequencing (NGS) playing a particularly important role for disease diagnosis and progression.
Against this background, we set out in this study to investigate hypoxic mediation of non-miRNA ncRNAs in COM, by identifying hypoxically deregulated ncRNAs through comparative NGS analysis of primary-site and metastatic COM cell lines, COM tissue samples, and oral tissue from healthy dogs. Any ncRNA identified as a target of interest was then to have its expression levels validated in qPCR assays. Furthermore, we evaluated the target of interest as a potential biomarker by measuring its levels in plasma and plasma extracellular vesicles (EVs).
2. Materials and Methods
2.1. Clinical Samples
Tumor tissue and blood samples were collected from dogs with COM undergoing treatment in the Kagoshima University Veterinary Teaching Hospital (KUVTH) or an affiliated veterinary clinic between 2014 and 2022. Oral tissue samples were collected from COM-free dogs for use as healthy controls. All procedures involving animals received approval from the animal ethics committee at KUVTH (Approval No. KVH220001). Individual sample information has been described previously [20]. Briefly, the sample donors included 22 males and 10 females, with a median age of 12 years (range: 7-16 years), and were drawn mainly from small breeds. The healthy controls were beagles that had been purpose bred for research at Shin Nippon Biomedical Laboratories, Ltd., Drug Safety Laboratories (Kagoshima, Japan).
Consent for sample collection was obtained from the owner of each dog. Certified pathologists employed by KUVTH diagnosed each sample as COM tumor tissue, or as a tumor-free healthy oral tissue, based on histopathological examination. The tissue samples had been transferred to RNAlater immediately after collection for storage. Blood samples were centrifuged to obtain plasma, in accordance with our standard protocol stated earlier [21]. All samples were stored at -80°C until analysis.
2.2. Cell Lines and Cell Culture
KMeC and LMeC are canine oral melanoma cell lines originating from primary and metastatic tumor sites, respectively, and were preserved in a freezing medium (039-23511, CultureSure, Fujifilm Wako Pure Chemical Corporation, Osaka, Japan). The cell lines were cultured under previously described conditions [22]. In brief, cells were cultured in a medium containing Roswell Park Memorial Institute (RPMI) media-1640 (Gibco), 10% fetal bovine serum (BI, Biological Industries), L-glutamine solution (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan) and antibiotics (penicillin-streptomycin; Sigma). Cells were cultured (biological replicates/line:6) in parallel under normoxic (5% CO2, 21% O2, 37°C) or hypoxic (5% CO2, 2% O2, 37°C) conditions for the specified duration. A hypoxic environment was achieved using a hypoxic incubator (HeracellTM 150i, Thermo Fisher Scientific, Waltham, MA, USA). The culture medium was changed at 48-h intervals. Cells were grown until confluency. Cells were counted using an automated cell counter (LUNA II, Logos Biosystems by Aligned Genetics, Inc., USA). Cell lysates were obtained at the relevant time intervals, and RNA was subsequently extracted from each lysate for ncRNA expression analysis. To explore the impact of hypoxia on melanoma, cells were cultured for up to 96 hours. We set the cell lysate collection times at 12, 48, and 96 h, so as to mitigate any immediate effect of medium change.
2.3. Isolation of EVs
EVs were isolated using the Total Exosome RNA and Protein Isolation Kit (Invitrogen, Waltham, MA, USA, Thermo Fisher Scientific) from plasma, in accordance with the manufacturer’s instructions and the procedure described earlier [23]. In summary, a 300-µL aliquot from the plasma sample was thoroughly mixed with a half-volume of 1X PBS, followed by the addition of 90 µL of exosome precipitation reagent. The resultant mixture was thoroughly vortexed, and then centrifuged at 10,000×g for 5 min. Supernatant was discarded to isolate the pellet containing the EVs. Subsequently, 150 µL of 1X PBS was added to resuspend the pellet, which was then stored at −80°C until analysis.
2.4. RNA Isolation from Cells, Plasma, and Plasma EVs
Total RNA was extracted from the COM cells and tissues, using a mirVana™ RNA Isolation Kit (Thermo Fisher Scientific), and from plasma and plasma EVs using a mirVana PARIS Kit (Thermo Fisher Scientific, Waltham, MA, USA), following the instructions provided by the manufacturer. Prior to the RNA extraction, 5μl of synthetic cel-miR-39 was mixed with each plasma or plasma-derived EV sample in order to normalize the differences in expression. Briefly, each cell lysates or plasma sample was mixed with an equal amount of 2× denaturation solution. miRNA homogenate was added at a ratio of 10:1, and the mixture was then incubated at 4°C for 10 min, after which acid-phenol:chloroform (Ambion®) was added at a volume equal to that of the initial cell lysate. The resultant mixture was vortexed thoroughly, and then centrifuged at 15,000×g for 5 min at 25°C. After centrifugation, the supernatant was carefully removed and mixed with 1.25 volumes of ethanol (99.9% purity) in an Eppendorf tube. The tube was then centrifuged using the spin column provided with the kit to trap the RNA particles on the filter paper. Finally, RNA was eluted from the filter membrane by the elution solution pre-heated to 95°C. The concentration of total RNA was measured using the NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific). RNA quality and integrity were assessed using an Agilent 2100 Bioanalyzer (G2939BA, Agilent Technologies, Santa Clara, CA, USA). The RNA Integrity Numbers (RINs) for the cell samples ranged from 8.5 to 9.5.
2.5. Next Generation Sequencing
NGS targeting total RNA was commissioned to a specialist genetics analysis laboratory (Hokkaido System Science, Hokkaido, Japan), and conducted as described previously [3]. Briefly, small RNA libraries were prepared using the TruSeq Small RNA Library preparation kit from 1 µg of total RNA, following the manufacturer’s instructions (Illumina, San Diego, CA). Adapters (5′ and 3′) were then ligated to the small RNAs, and cDNA was synthesized using reverse transcription, followed by substantial amplification. The amplified cDNA was subjected to gel electrophoresis to check its purity and then sequenced using an Illumina/Hiseq2500 system by Hokkaido System Science (Hokkaido, Japan). The sequencing yielded high quality reads with Phred scores >35. Finally, the sequences were stored in the NCBI Sequence Read Archive (SRA) database with an accession number of PRJNA629070.
NGS data was obtained from analyses of canine oral healthy tissue (control tissues, n = 3), clinical COM tissue (n = 8), and KMeC and LMeC cell lines (n = 3 replicates for each cells in either normoxic or hypoxic condition), under normoxic and hypoxic conditions. The sequence reads were stored in the Sequence Read Archive (SRA) (www.ncbi.nlm.nih.gov/sra) with the accession number PRJNA629070. Summary of the NGS data analysis has already been documented in previous studies [3,24]. NGS data were trimmed, quality checked, and underwent subsequent analysis as described in earlier studies [1,3,24]. miRBase and Ensembl databases were utilized to annotate the reads. All cell line replicates clustered together under normoxic and hypoxic conditions, suggesting they are appropriate for further differential expression analysis [3].
2.6. Bioinformatic Analysis
NGS reads were examined using the CLC Genomics Workbench, V10.0 and 12.0, in accordance with the developer’s instructions (https://digital insights.qiagen.com, CLC Bio, Qiagen, Germany). Final analysis preceded adapter trimming, QC checking, and sorting of the ambiguous reads. Parameters were set as the recommended defaults for all analysis runs. Initially, adapters and low-quality, ambiguous reads were removed. Small RNA sequences were extracted and counted from the clean reads. Subsequently, annotation of the extracted reads was obtained using miRBase and canine and human non-coding RNA databases from ENSEMBL (Canis_familiris/canfam3.1.ncrna and Homo_sapiens/GRCh37.ncrna). A previous study from our laboratory had reported miRNAs annotated from miRbase [3]. Non-miRNA ncRNAs were investigated in this study. Sequence counts were regarded as the expression values for ncRNAs. Empirical analysis of differential gene expression (EDGE) was used to identify any significantly dysregulated ncRNAs, based on the criteria: FDR p-value < 0.05, |FC| > 2, and minimum expression > 10 [mature count] per replicate.
2.7. qPCR
The expression level of the target ncRNA was measured using TaqMan gene expression assays (Thermo Fisher Scientific). qPCR was performed following previously described procedures [24]. First, cDNA was prepared from 2 ng of RNA sample using a TaqMan MicroRNA Reverse Transcription kit following the manufacturer’s protocol (4366597, Thermo Fisher Scientific). In the next step, qPCR was performed with a StepOnePlus real-time PCR system (Thermo Fisher Scientific) using a TaqMan First Advanced Master Mix kit. The thermocycling conditions for qPCR were: 50°C for 2 min, 95°C for 20s; followed by 40 cycles of 1s denaturation at 95°C and 20s annealing/extension at 60°C. All experimental data were reproduced at least twice. RNU6B, miR-16, and miR-186 were used as internal controls for the evaluation of the relative expression of tumor tissue and cell lines, plasma, and plasma EVs, respectively [18]. The final expression level was calculated following the 2− ΔΔCT method [25]. A qPCR test providing cycle threshold (Ct) value greater than 35 was regarded as undetected for that specified sample. Expression values of the control miRNAs (miR-16 for plasma and miR-186 for plasma EVs) were evaluated using TaqMan microRNA assays. The primer IDs for the internal controls were: RNU6B: 001093, miR-16: 000391, and miR-186: 002285. Information on each primer can be found at: https://www.thermofisher.com/order/genome-database/. Primers for the selected ncRNA (Ensembl ID; ENSCAFT00000084705.1; sequence: 5′- ATTCCTGGACTCACGGATACT-3′) was custom-designed.
2.8. Statistical Analysis
Differences between groups were assessed using Kruskal–Wallis test and Mann–Whitney U test to evaluate the relative expression of the target ncRNA. A statistical test with a p-value of less than 0.05 (p < 0.05) was considered significant. All statistical analyses and graph visualizations were conducted using GraphPad Prism 9 (https://www.graphpad.com/).
3. Results
3.1. NGS Profiling of Hypoxia-mediated ncRNAs
To initially determine ncRNAs that are differentially expressed in clinical COM tissue, and primary-site (KMeC) and metastatic (LMeC) COM cells versus healthy oral tissue, and in hypoxic versus normoxic KMeC and LMeC cells, we applied stringent filtering criteria to NGS reads (FDR p-value < 0.05, |FC| > 2, and minimum expression > 10 [mature count] per replicate), and listed the ncRNAs thus classified as upregulated or downregulated. The numbers of differentially expressed ncRNAs for all comparisons are shown in Figure 1.
Focusing on hypoxia versus normoxia comparisons in COM cells, we identified 928 and 1367 hypoxically dysregulated ncRNAs for KMeC and LMeC cells, respectively. For the KMeC cells, 242 of these ncRNAs were upregulated and 686 were downregulated, and for LMeC cells, 266 were upregulated and 1101 were downregulated (Figure 1, Table S1-S4). These findings suggest that hypoxia mediates ncRNAs expression in COM.
3.2. Identification of Target ncRNAs
To identify ncRNAs of interest, we then subjected data on differentially expressed ncRNAs to Venn diagramming to find any molecules showing a consistent pattern across clinical COM tissue and the COM cell lines. Comparing expression between hypoxia and normoxia in cell lines may not lead to sufficiently robust identification of target molecules because of the possibility to develop unintended hypoxia in the cultured cells. Accordingly, we set out to identify hypoxically dysregulated, melanoma-specific target ncRNAs which were commonly up- or down-regulated in clinical COM tissue, and hypoxic KMeC and LMeC cells versus healthy oral tissue, and in hypoxic KMeC and LMeC cells versus normoxically cultured equivalents. Target ncRNAs were to be identified as those in the intersection of three comparisons in the relevant Venn diagram. The results are visually presented, separating upregulated and downregulated ncRNAs in Figure 2, with a full breakdown of numbers in Supplementary Table S1-S10.
Although 16 ncRNAs were commonly upregulated in clinical COM tissue and hypoxic KMeC or LMeC versus healthy oral tissue or the relevant normoxic cells, none of them satisfied the criterion for selection as a target ncRNA because they were not upregulated in LMeC (metastatic) vs. KMeC (primary-site) COM cells.
By contrast, one ncRNA (ENSCAFT00000084705.1) was commonly downregulated in COM tumor tissue and hypoxic KMeC or LMeC versus healthy oral tissue or the relevant normoxic cells in NGS (Figure 2), and thus satisfied the criteria for selection as a target ncRNA based on downregulation in LMeC vs. KMeC cells. The relative expression levels of the target ncRNA (ENSCAFT00000084705.1) across all types of NGS samples are depicted in Figure 3.
3.3. qRT-PCR Validation of the Downregulated ncRNA (ENSCAFT00000084705.1)
3.3.1. Relative expression in hypoxic KMeC and LMeC cells
To validate our NGS findings, we investigated the expression of ENSCAFT00000084705.1 at three different time intervals (12, 48, and 96 h) in hypoxic KMeC and LMeC cells. The expression level decreased significantly (p < 0.05) in both cells at 48 and 96h under hypoxic conditions compared to 12h (Figure 4). Moreover, expression significantly differed (p < 0.01) between LMeC and KMeC cells at 96 h (Figure 5), having shown a non-significant trend to a similar at 48h (p > 0.05). Accordingly, ENSCAFT00000084705.1 expression appears to decrease progressively with hypoxia, and the magnitude of this decrease is greater in metastatic (LMeC) cells than primary-site (KMeC) cells.
3.3.2. Relative expression in plasma and plasma-derived EVs
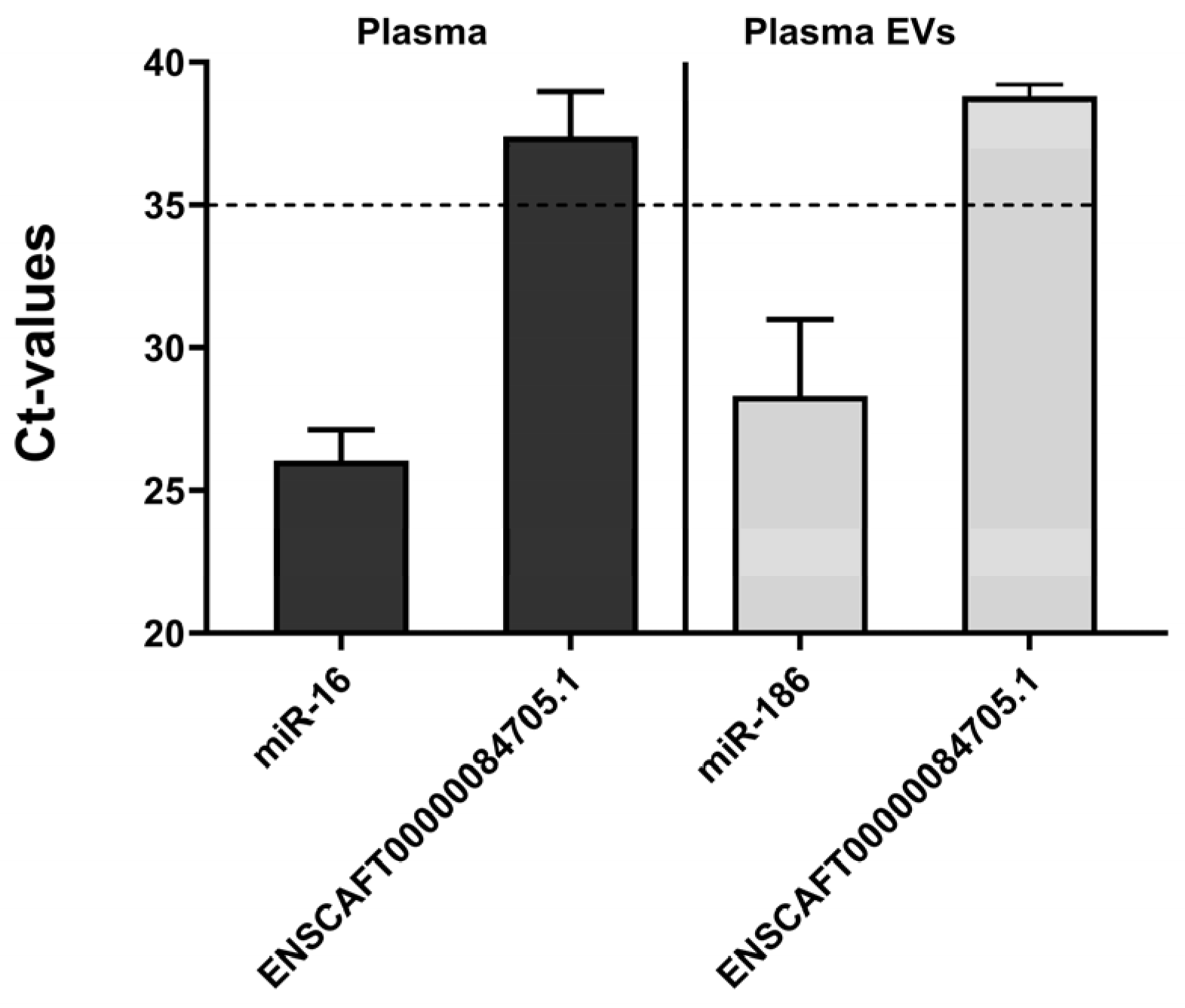
We further examined the expression of ncRNA in plasma and plasma-derived EVs from clinical COM patients. Although the internal control (miR-186) was detectable in plasma EVs, ENSCAFT00000084705.1 was undetectable in both plasma and plasma EVs (Figure 6). The cycle threshold (Ct) values during qPCR validation surpassed 35, failing to meet the criteria for our experimental conditions. Thus, our results imply that ENSCAFT00000084705.1 was either absent or minimally expressed in plasma and plasma EVs.
4. Discussion
In this study, we aimed to build on previous research on RNA molecules, and evaluate expression of ncRNAs other than miRNAs in COM and investigate their links to hypoxia, which may be heavily implicated in the progression and metastasis of this cancer. To the best or our knowledge, this is the first report on such hypoxic dysregulation of non-miRNA ncRNAs.
In the initial findings in our study, we demonstrated transcriptional dysregulation for non-miRNA ncRNAs, with 928 (242 up and 686 downregulated) and 1367 (266 up and 1101 downregulated) relevant molecules in hypoxically cultured KMeC and LMeC cells, versus their normoxic counterparts respectively, based on NGS results. Thus, ncRNAs expression appears to be linked with hypoxic dysregulation. Moreover, the number of dysregulated ncRNAs was greater in metastatic than primary-site COM cells, suggesting these species might exhibit greater efficacy in coping with hypoxic cellular stress in the metastatic stage. Our findings for non-miRNA ncRNAs broadly reflect the extent of dysregulation we previously identified for miRNAs in comparable screening experiments [3]. The functional relationships between miRNAs and other ncRNAs have yet to be elucidated; however, considering that hypoxia is known to alter gene expression in multiple ways, our findings may indicate that RNA molecules across the whole transcriptome are affected by or may interact in some way with a hypoxic tumor microenvironment. We consider that non-miRNA ncRNAs are thus also of potential interest to veterinary oncologists seeking to better understand why COM is such an aggressive cancer.
From among the hypoxically dysregulated ncRNAs, we then set out to find molecules of interest that showed a broadly consistent pattern, either upregulation or downregulation, across the COM cell lines and tumor tissue samples, in comparison with healthy oral tissue. COM tumor tissue samples were included in these comparisons to safeguard against mis-identifying a transcriptome element as dysregulated, due to any unintended hypoxia that might occur during cell culture. As a very aggressive, solid cancer, COM develops hypoxic microenvironments quickly, which play a role in metastasis [26,27]. Thus, the final part of the consistent pattern we were seeking involved the trend toward upregulation or downregulation being evident in the metastatic cell line versus the primary-site cell line (LMeC vs. KMeC cells). Eventually, we were only able to identify one candidate ncRNA, (ENSCAFT00000084705.1), which was commonly downregulated in every relevant comparison (no candidate satisfied the criteria for the upregulation pattern). These findings differ somewhat from our previously reported results with miRNAs, where we found 15 (14 upregulated and one downregulated) commonly hypoxically dysregulated miRNAs in COM [3]. as evaluated with the same criteria used in this study. Taken together, these results may tentatively indicate miRNA species show sensitivity to a hypoxic microenvironment in greater numbers than other ncRNA species, but considerable further research is needed.
Although ENSCAFT00000084705.1 was the sole target ncRNA we identified, its association with hypoxia appears robust. This ncRNA was downregulated in COM cells and tumor tissue versus healthy oral tissue, and in hypoxic cells versus normoxic cells. Furthermore, the downregulation in metastatic cells was consistent with a progressive trend to downregulation as COM, an aggressive cancer with a high propensity to metastasize, progresses. We were able to validate our NGS findings for this ncRNA in PCR assays with KMeC and LMeC cells, and demonstrated chronologically progressive downregulation of greater magnitude in metastatic cells. We thus speculate that ENSCAFT00000084705.1 plays a distinct and important role in the hypoxic COM microenvironment that is linked to tumor progression and metastasis. Hypoxia is known to cause downregulation of anti-tumorigenic factors like p53, dicer, e-cadherin and so on [28,29,30], indicating that ENSCAFT00000084705.1 may have a role in the development of future therapeutic strategies. Searches with the Ensembl genome browser (https://asia.ensembl.org/index.html) revealed that the ENSCAFT00000084705.1 sequence is a lncRNA fragment in both humans and dogs; however, at this moment in time, we are unable to determine what roles it may play in the two species. Nevertheless, this finding offers the interesting possibility of links between canine and human melanomas.
Having established ENSCAFT00000084705.1 as an ncRNA of interest, we then set out to evaluate its level in plasma and plasma EVs. As EV may carry a cargo from tumor cells to other regions in the body [31], their target molecule concentrations may reveal any potential role in the spread of a tumor beyond it primary site [32]. However, ENSCAFT00000084705.1 proved almost undetectable in plasma and plasma EVs, in qPCR assays, and we deduced that it is either absent or minimally expressed in these matrices. Its expression would thus appear to be tissue specific, and we regard ENSCAFT00000084705.1 as having little promise as a biomarker for COM generally, or for metastatic versus non-metastatic COM. This contrasts with our findings in a previous study, on another lncRNA (ENSCAFT00000069599.1), which we suggested is a potential exosomal biomarker for differentiating COM cases from healthy dogs [19].
Our findings add to the understanding of the transcriptional factors in COM. Transcriptome analysis is typically further advanced in human, then canine medicine. It is now well established that non-coding RNAs are implicated in the initiation and progression of a number of human cancers, with hypoxic tumor microenvironments suggested to play a role in many cases, with modulation of the hypoxia/HIF pathway being one part of a possible mechanism [33]. Aberrant lncRNA expression has also been found in human melanoma [34], for which it has been implicated in tumor formation and progression [35]. Even though thousands of lncRNAs have been identified in dogs [13], their role in canine cancers remains poorly understood and few have been pinpointed for roles in COM. Our identification of ENSCAFT00000084705.1 as a hypoxia-mediated ncRNA is thus a useful addition to a still relatively small base of evidence.
The current study has a number of limitations. Firstly, the hypoxic levels of the tissue samples used for NGS analysis were not investigated. However, we believed that the tumor mass was large enough to develop hypoxic condition inside it. Secondly, clinical validation was conducted with a relatively small population, and our findings on the target ncRNA thus require further validation with a larger sample size to draw more robust conclusions. Finally, there is a need to explore the genes targeted by this lncRNA and their associated signaling pathways. Additionally, investigating the underlying molecular mechanisms and functional role of ENSCAFT00000084705.1 in melanoma development and progression is essential.
5. Conclusions
Present study investigated the hypoxically dysregulated non-miRNA ncRNAs from COM for the first time and eventually, our findings revealed that hypoxia may contribute to the dysregulation of ncRNA expression in COM. Our results here allow us to identify one ncRNA, ENSCAFT00000084705.1 as a hypoxia-mediated factor in COM. Although it may lack potential as a diagnostic biomarker, we consider that ENSCAFT00000084705.1 is a molecule of interest for future study on this aggressive cancer in dogs.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1. Table S1: Differentially expressed up-regulated ncRNAs (except miRNA) between KMeC normoxic and hypoxic cell lines; Table S2: Differentially expressed down-regulated ncRNAs (except miRNA) between KMeC normoxic and hypoxic cell lines; Table S3: Differentially expressed up-regulated ncRNAs (except miRNA) between LMeC normoxic and hypoxic cell lines; Table S4: Differentially expressed down-regulated ncRNAs (except miRNA) between LMeC normoxic and hypoxic cell lines; Table S5: Differentially expressed up-regulated ncRNAs (except miRNA) between normal tissue (healthy oral tissue) and KMeC hypoxic cell lines; Table S6: Differentially expressed down-regulated ncRNAs (except miRNA) between normal tissue (healthy oral tissue) and KMeC hypoxic cell lines; Table S7: Differentially expressed up-regulated ncRNAs (except miRNA) between normal tissue (healthy oral tissue) and LMeC hypoxic cell lines; Table S8: Differentially expressed down-regulated ncRNAs (except miRNA) between normal tissue (healthy oral tissue) and LMeC hypoxic cell lines; Table S9: Differentially expressed up-regulated ncRNAs (except miRNA) between normal tissue (healthy oral tissue) and melanoma (COM) tissue; Table S10: Differentially expressed down-regulated ncRNAs (except miRNA) between normal tissue (healthy oral tissue) and melanoma (COM) tissue.
Author Contributions
Conceptualization, Yasunori Hino and Naoki Miura; Formal analysis, Yasunori Hino, Md Mahfuzur Rahman and MD Nazmul Hasan; Funding acquisition, Naoki Miura; Investigation, Yasunori Hino, Md Mahfuzur Rahman, Al Asmaul Husna and MD Nazmul Hasan; Methodology, Yasunori Hino, Al Asmaul Husna and Naoki Miura; Project administration, Naoki Miura; Resources, Naoki Miura; Validation, Yasunori Hino, Mohammad Arif, Md Mahfuzur Rahman, Al Asmaul Husna and MD Nazmul Hasan; Visualization, Yasunori Hino, Mohammad Arif, Md Mahfuzur Rahman, Al Asmaul Husna and MD Nazmul Hasan; Writing – original draft, Yasunori Hino and Mohammad Arif; Writing – review & editing, Yasunori Hino, Mohammad Arif and Naoki Miura.
Funding
This work was supported by JSPS KAKENHI (Grant nos. 21H02366, 20K21375).
Institutional Review Board Statement
The animal study protocol was approved by the Ethics Committee of Kagoshima University (Approval No. KVH220001).
Informed Consent Statement
Informed consent was obtained from the dog owner prior to the sample collection. Furthermore, the study does not include any human participants.
Data Availability Statement
Sequence reads of this study were stored in the SRA (www.ncbi.nlm.nih.gov/sra) database (Accession number; PRJNA629070). Other data are included within this article and Supplementary Materials.
Acknowledgments
The authors express their gratitude to Professor Dr. Henry Smith for editing a draft of this manuscript.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Hasan, M.N.; Rahman, M.M.; Husna, A.A.; Kato, D.; Nakagawa, T.; Arif, M.; Miura, N. Hypoxia-Related Y RNA Fragments as a Novel Potential Biomarker for Distinguishing Metastatic Oral Melanoma from Non-Metastatic Oral Melanoma in Dogs. Vet. Q. 2024, 44, 1–8. [CrossRef]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The Role of Hypoxia in the Tumor Microenvironment and Development of Cancer Stem Cell: A Novel Approach to Developing Treatment. Cancer Cell Int. 2021, 21, 62. [CrossRef]
- Hino, Y.; Rahman, M.M.; Lai, Y.C.; Husna, A.A.; Chen, H. wen; Hasan, M.N.; Nakagawa, T.; Miura, N. Hypoxic miRNAs Expression are Different Between Primary and Metastatic Melanoma Cells. Gene 2021, 782, 145552 . [CrossRef]
- Barreca, M.M.; Zichittella, C.; Alessandro, R.; Conigliaro, A. Hypoxia-induced Non-coding RNAs Controlling Cell Viability in Cancer. Int. J. Mol. Sci. 2021, 22, 1857. [CrossRef]
- Singh, D.; Arora, R.; Kaur, P.; Singh, B.; Mannan, R.; Arora, S. Overexpression of Hypoxia-Inducible Factor and Metabolic Pathways: Possible Targets of Cancer. Cell Biosci. 2017, 7, 62. [CrossRef]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia Inducible Factors in Cancer Stem Cells. Br. J. Cancer 2010, 102, 789–795. [CrossRef]
- Gillard, M.; Cadieu, E.; De Brito, C.; Abadie, J.; Vergier, B.; Devauchelle, P.; Degorce, F.; Dréano, S.; Primot, A.; Dorso, L.; et al. Naturally Occurring Melanomas in Dogs as Models for Non-UV Pathways of Human Melanomas. Pigment Cell Melanoma Res. 2014, 27, 90–102. [CrossRef]
- Prouteau, A.; André, C. Canine Melanomas as Models for Human Melanomas: Clinical, Histological, and Genetic Comparison. Genes 2019, 10, 501. [CrossRef]
- Hardwick, L. A Comparative View on Molecular Alterations and Potential Therapeutic Strategies for Canine Oral Melanoma. Vet. Sci. 2021, 8, 286. [CrossRef]
- Hernandez, B.; Adissu, H.A.; Wei, B.R.; Michael, H.T.; Merlino, G.; Mark Simpson, R. Naturally Occurring Canine Melanoma as a Predictive Comparative Oncology Model for Human Mucosal and Other Triple Wild-Type Melanomas. Int. J. Mol. Sci. 2018, 19, 394. [CrossRef]
- Gardner, H.L.; Fenger, J.M.; London, C.A. Dogs as a Model for Cancer. Annu. Rev. Anim. Biosci. 2016, 4, 199–222. [CrossRef]
- Van Der Weyden, L.; Patton, E.E.; Wood, G.A.; Foote, A.K.; Brenn, T.; Arends, M.J.; Adams, D.J. Cross-Species Models of Human Melanoma. J. Pathol. 2016, 238, 152–165. [CrossRef]
- Hitte, C.; Le Béguec, C.; Cadieu, E.; Wucher, V.; Primot, A.; Prouteau, A.; Botherel, N.; Hédan, B.; Lindblad-Toh, K.; André, C.; et al. Genome-Wide Analysis of Long Non-Coding RNA Profiles in Canine Oral Melanomas. Genes 2019, 10, 477. [CrossRef]
- Rahman, M.M.; Lai, Y.C.; Husna, A.A.; Chen, H.W.; Tanaka, Y.; Kawaguchi, H.; Hatai, H.; Miyoshi, N.; Nakagawa, T.; Fukushima, R.; et al. Transcriptome Analysis of Dog Oral Melanoma and Its Oncogenic Analogy with Human Melanoma. Oncol. Rep. 2020, 43, 16–30. [CrossRef]
- Sutherland, R.M. Tumor Hypoxia and Gene Expression - Implications for Malignant Progression and Therapy. Acta Oncol. 1998, 37, 567–574. [CrossRef]
- Ushio, N.; Rahman, M.M.; Maemura, T.; Lai, Y.C.; Iwanaga, T.; Kawaguchi, H.; Miyoshi, N.; Momoi, Y.; Miura, N. Identification of Dysregulated MicroRNAs in Canine Malignant Melanoma. Oncol. Lett. 2019, 17, 1080–1088. [CrossRef]
- Rahman, M.M.; Lai, Y.C.; Husna, A.A.; Chen, H.W.; Tanaka, Y.; Kawaguchi, H.; Hatai, H.; Miyoshi, N.; Nakagawa, T.; Fukushima, R.; et al. Aberrantly Expressed snoRNA, snRNA, piRNA and tRFs in Canine Melanoma. Vet. Comp. Oncol. 2020, 18, 353–361. [CrossRef]
- Husna, A.A.; Rahman, M.M.; Lai, Y.C.; Chen, H.W.; Hasan, M.N.; Nakagawa, T.; Miura, N. Identification of Melanoma-Specific Exosomal miRNAs as the Potential Biomarker for Canine Oral Melanoma. Pigment Cell Melanoma Res. 2021, 34, 1062–1073. [CrossRef]
- Husna, A.A.; Rahman, M.M.; Chen, H.W.; Hasan, M.N.; Nakagawa, T.; Miura, N. Long Non-Coding RNA and Transfer RNA-Derived Small Fragments in Exosomes are Potential Biomarkers for Canine Oral Melanoma. Vet. Comp. Oncol. 2022, 20, 653–663. [CrossRef]
- Hasan, M.N.; Rahman, M.M.; Husna, A.A.; Arif, M.; Jasineviciute, I.; Kato, D.; Nakagawa, T.; Miura, N. Upregulation and Functional Roles of miR-450b in Canine Oral Melanoma. Non-coding RNA Res. 2024, 9, 376–387. [CrossRef]
- Hasan, M.N.; Rahman, M.M.; Husna, A.A.; Arif, M.; Iwanaga, T.; Tsukiyama-Kohara, K.; Jasineviciute, I.; Kato, D.; Nakagawa, T.; Miura, N. Elevated Expression of miR-301a and Its Functional Roles in Canine Oral Melanoma. Vet. Comp. Oncol. 2024, 22, 78–88. [CrossRef]
- Inoue, K.; Ohashi, E.; Kadosawa, T.; Hong, S.H.; Matsunaga, S.; Mochizuki, M.; Nishimura, R.; Sasaki, N. Establishment and Characterization of Four Canine Melanoma Cell Lines. J. Vet. Med. Sci. 2004, 66, 1437–1440. [CrossRef]
- Hasan, M.N.; Rahman, M.M.; Husna, A.A.; Nozaki, N.; Yamato, O.; Miura, N. YRNA and tRNA Fragments Can Differentiate Benign from Malignant Canine Mammary Gland Tumors. Biochem. Biophys. Res. Commun. 2024, 691, 149336. [CrossRef]
- Rahman, M.M.; Lai, Y.C.; Husna, A.A.; Chen, H.W.; Tanaka, Y.; Kawaguchi, H.; Miyoshi, N.; Nakagawa, T.; Fukushima, R.; Miura, N. Micro RNA Transcriptome Profile in Canine Oral Melanoma. Int. J. Mol. Sci. 2019, 20, 4832. [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-Time PCR Data by the Comparative CT Method. Nat. Protoc. 2008, 3, 1101–1108. [CrossRef]
- Kim, W.S.; Vinayak, A.; Powers, B. Comparative Review of Malignant Melanoma and Histologically Well-Differentiated Melanocytic Neoplasm in the Oral Cavity of Dogs. Vet. Sci. 2021, 8, 261. [CrossRef]
- Gola, C.; Maniscalco, L.; Iussich, S.; Morello, E.; Olimpo, M.; Martignani, E.; Accornero, P.; Giacobino, D.; Mazzone, E.; Modesto, P.; Varello, K. Hypoxia-associated Markers in the Prognosis of Oral Canine Melanoma. Vet. Pathol. 2024, 13. [CrossRef]
- Obacz, J.; Pastorekova, S.; Vojtesek, B.; Hrstka, R. Cross-Talk between HIF and p53 as Mediators of Molecular Responses to Physiological and Genotoxic Stresses. Mol. Cancer 2013, 12, 93. [CrossRef]
- Ibrahim, A.A.; Schmithals, C.; Kowarz, E.; Köberle, V.; Kakoschky, B.; Pleli, T.; Kollmar, O.; Nitsch, S.; Waidmann, O.; Finkelmeier, F.; et al. Hypoxia Causes Downregulation of Dicer in Hepatocellular Carcinoma, Which Is Required for Upregulation of Hypoxia-Inducible Factor 1α and Epithelial–Mesenchymal Transition. Clin. Cancer Res. 2017, 23, 3896–3905. [CrossRef]
- Cheng, J.C.; Klausen, C.; Leung, P.C.K. Hypoxia-Inducible Factor 1 Alpha Mediates Epidermal Growth Factor-Induced Down-regulation of E-Cadherin Expression and Cell Invasion in Human Ovarian Cancer Cells. Cancer Lett. 2013, 329, 197–206. [CrossRef]
- Kalluri, R. The Biology and Function of Exosomes in Cancer. J. Clin. Invest. 2016, 126, 1208–1215. [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour Exosome Integrins Determine Organotropic Metastasis. Nature 2015, 527, 329–335. [CrossRef]
- Choudhry, H.; Harris, A.L.; McIntyre, A. The Tumour Hypoxia Induced Non-Coding Transcriptome. Mol. Aspects Med. 2016, 47–48, 35–53. [CrossRef]
- Leucci, E.; Coe, E.A.; Marine, J.C.; Vance, K.W. The Emerging Role of Long Non-Coding RNAs in Cutaneous Melanoma. Pigment Cell Melanoma Res. 2016, 29, 619–626. [CrossRef]
- Gutschner, T.; Diederichs, S. The Hallmarks of Cancer: A Long Non-Coding RNA Point of View. RNA Biol. 2012, 9, 703–719. [CrossRef]
Figure 1.
Differentially expressed ncRNAs (except miRNAs) in melanoma tissues and cell lines. (A-E) Volcano plot of differentially expressed upregulated and downregulated ncRNAs (except miRNAs) in Normal vs COM tissue (A), Normal tissue vs KMeC_hypoxia (B), Normal tissue vs LMeC_hypoxia (C), KMeC_normoxia vs KMeC_hypoxia (D), LMeC_normoxia vs LMeC_hypoxia (E). Red dots represent up and down-regulated ncRNAs, and blue dots represent ncRNAs that were not differentially expressed.
Figure 1.
Differentially expressed ncRNAs (except miRNAs) in melanoma tissues and cell lines. (A-E) Volcano plot of differentially expressed upregulated and downregulated ncRNAs (except miRNAs) in Normal vs COM tissue (A), Normal tissue vs KMeC_hypoxia (B), Normal tissue vs LMeC_hypoxia (C), KMeC_normoxia vs KMeC_hypoxia (D), LMeC_normoxia vs LMeC_hypoxia (E). Red dots represent up and down-regulated ncRNAs, and blue dots represent ncRNAs that were not differentially expressed.
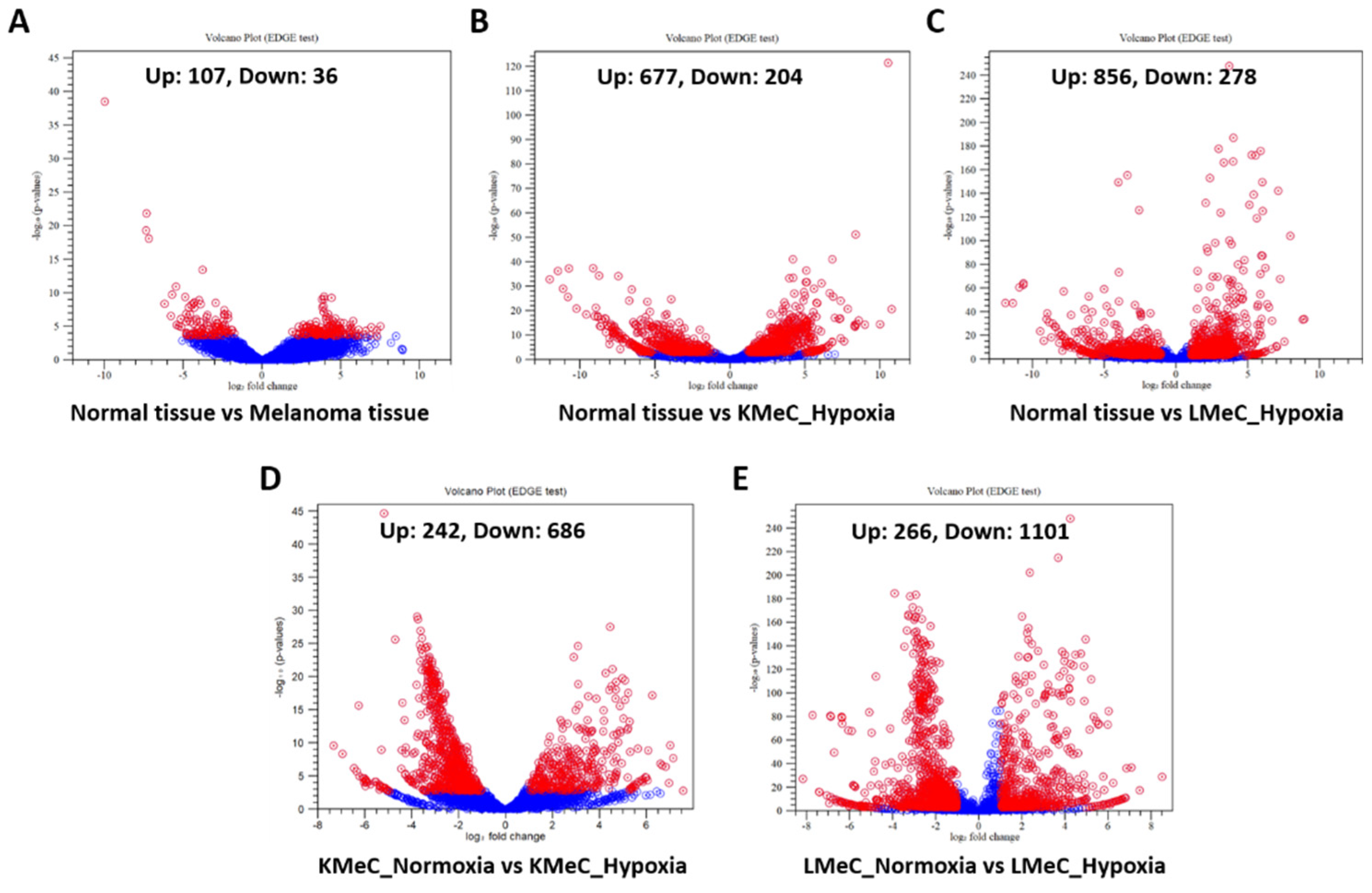
Figure 2.
Hypoxia-regulated ncRNAs (except miRNAs) in canine oral melanoma (COM). (A-B) Venn diagram showing hypoxia-regulated up and down-regulated ncRNAs in KMeC cells. (C-D) Hypoxia-regulated up and down-regulated ncRNAs in LMeC cells. (F) Hypoxia-induced commonly expressed down-regulated ncRNAs in both cells. No up-regulated ncRNAs were commonly expressed in both cell lines (E). Arrows in the Venn diagram indicate the corresponding lists of ncRNAs, shared between or among the groups.
Figure 2.
Hypoxia-regulated ncRNAs (except miRNAs) in canine oral melanoma (COM). (A-B) Venn diagram showing hypoxia-regulated up and down-regulated ncRNAs in KMeC cells. (C-D) Hypoxia-regulated up and down-regulated ncRNAs in LMeC cells. (F) Hypoxia-induced commonly expressed down-regulated ncRNAs in both cells. No up-regulated ncRNAs were commonly expressed in both cell lines (E). Arrows in the Venn diagram indicate the corresponding lists of ncRNAs, shared between or among the groups.
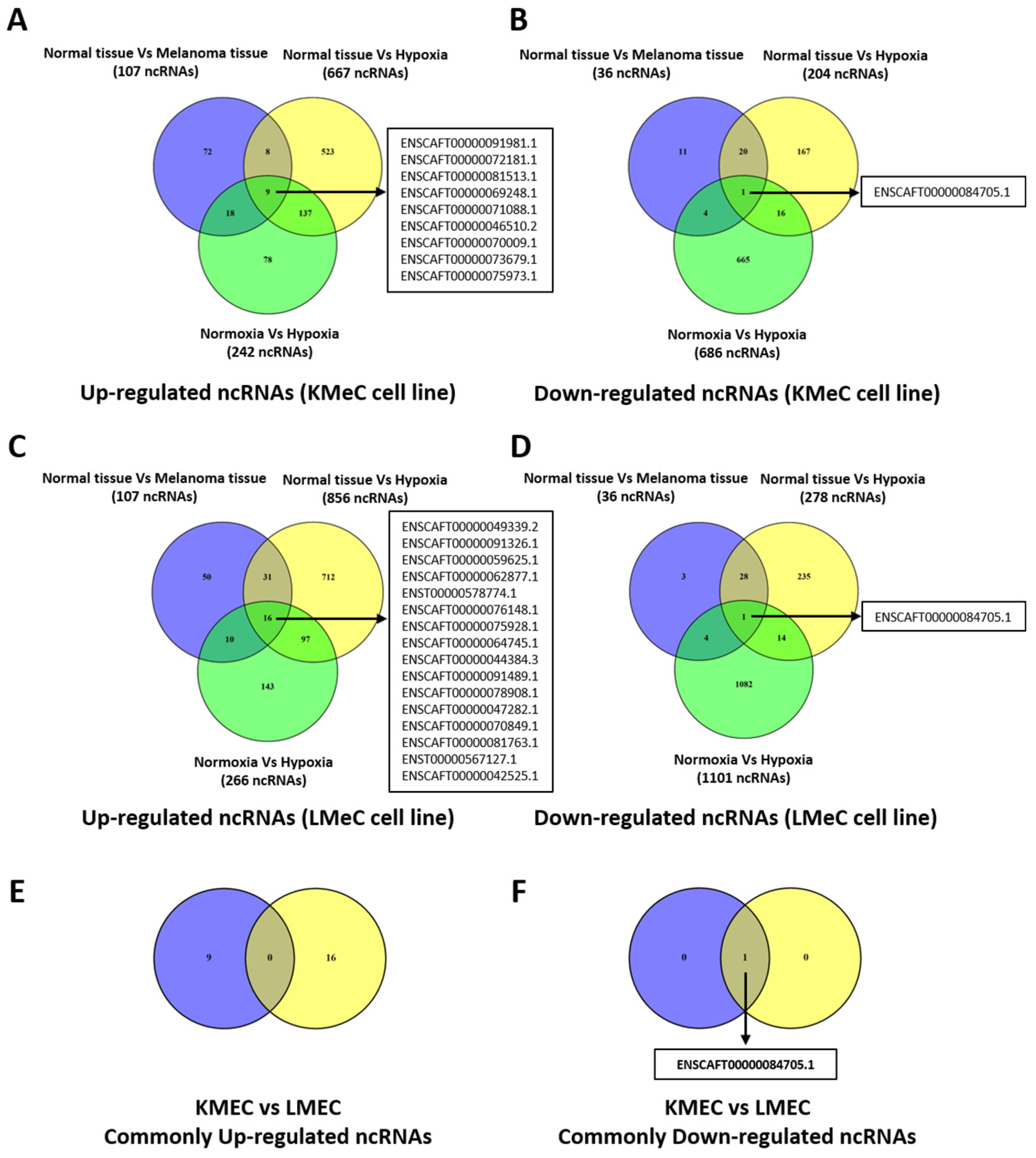
Figure 3.
Expression levels of the targeted ncRNA (ENSCAFT00000084705.1) in healthy oral tissue (n = 3), melanoma tissue (n = 8), normoxic KMeC cells (n = 3), hypoxic KMeC cells (n = 3), normoxic LMeC cells (n = 3), and hypoxic LMeC cells (n = 3) in NGS data.
Figure 3.
Expression levels of the targeted ncRNA (ENSCAFT00000084705.1) in healthy oral tissue (n = 3), melanoma tissue (n = 8), normoxic KMeC cells (n = 3), hypoxic KMeC cells (n = 3), normoxic LMeC cells (n = 3), and hypoxic LMeC cells (n = 3) in NGS data.
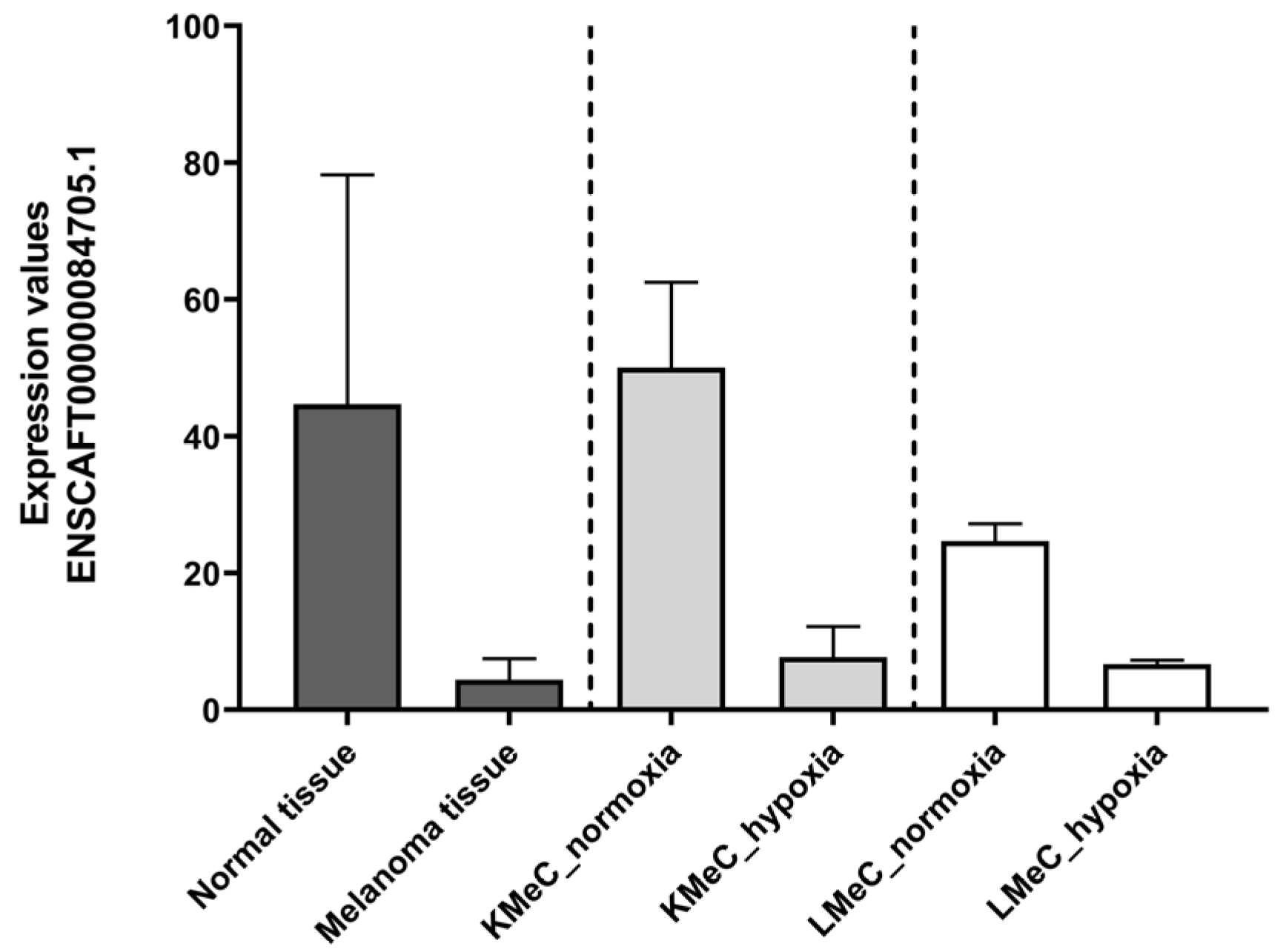
Figure 4.
Relative expression of the targeted ncRNA (ENSCAFT00000084705.1) at different time intervals in hypoxic KMeC and LMeC cells by qRT-PCR. Relative expression of ENSCAFT00000084705.1 at 12, 48, and 96h in hypoxic KMeC cells (A) and LMeC cells (B). The y-axis of the graph indicates the relative expression levels of ENSCAFT00000084705.1. Six replicates (n = 6) of KMeC and LMeC cells were analyzed at each time interval using the Kruskal–Wallis test. A p-value of less than 0.05 (p < 0.05) was considered statistically significant. * denotes a significant test.
Figure 4.
Relative expression of the targeted ncRNA (ENSCAFT00000084705.1) at different time intervals in hypoxic KMeC and LMeC cells by qRT-PCR. Relative expression of ENSCAFT00000084705.1 at 12, 48, and 96h in hypoxic KMeC cells (A) and LMeC cells (B). The y-axis of the graph indicates the relative expression levels of ENSCAFT00000084705.1. Six replicates (n = 6) of KMeC and LMeC cells were analyzed at each time interval using the Kruskal–Wallis test. A p-value of less than 0.05 (p < 0.05) was considered statistically significant. * denotes a significant test.
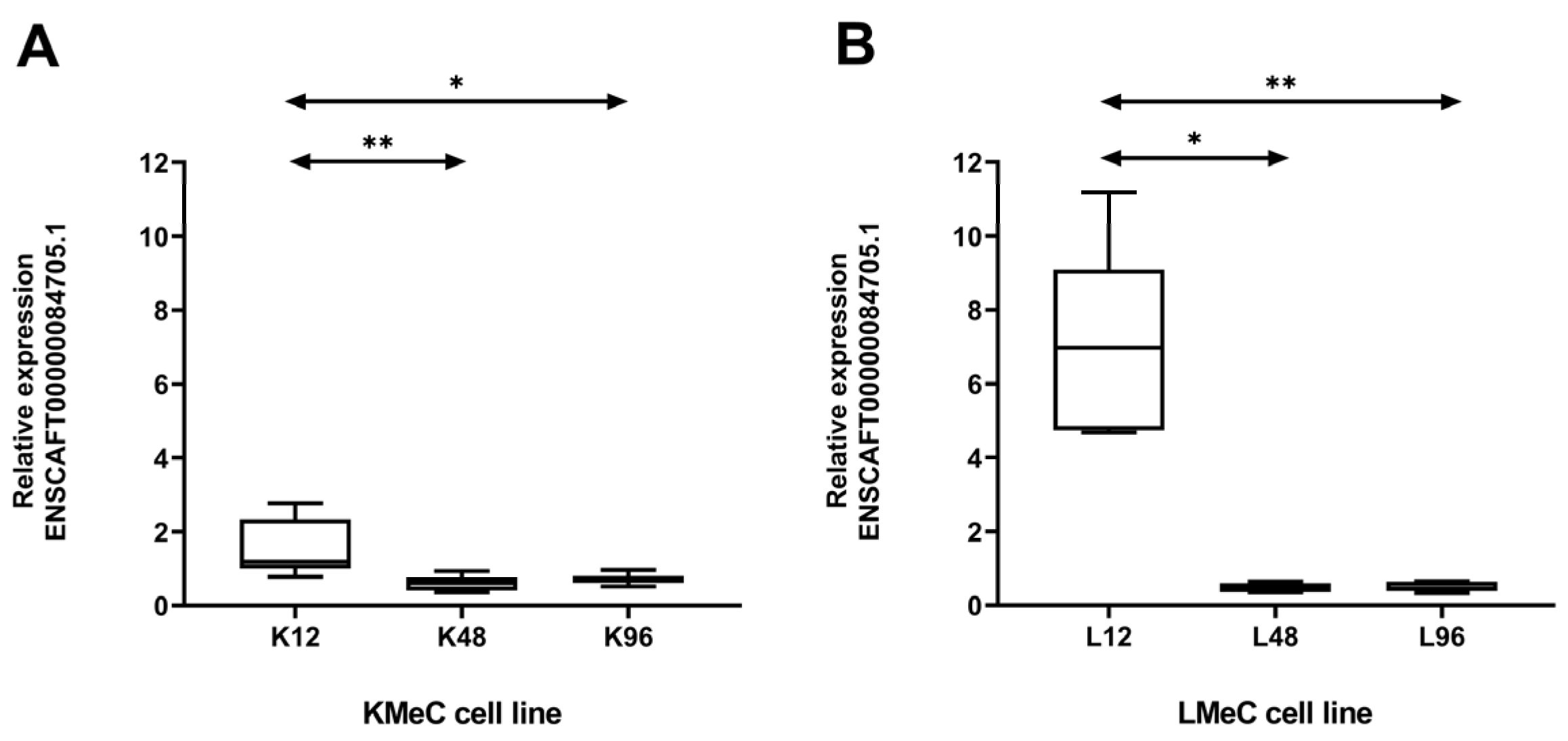
Figure 5.
Relative expression level of targeted ncRNA (ENSCAFT00000084705.1) between primary (KMeC, n = 6) and metastatic (LMeC, n = 6) COM cell lines under hypoxic condition. Dark grey color box indicates the relative expression level of ENSCAFT00000084705.1 in KMeC and LMeC cells after 48h and light grey color represents the expression levels after 96h. Differences between KMeC and LMeC cells at each time interval were analyzed using the Mann–Whitney U test. A p-value of less than 0.05 (p < 0.05) was considered statistically significant. * denotes a significant test; ns = non-significant test.
Figure 5.
Relative expression level of targeted ncRNA (ENSCAFT00000084705.1) between primary (KMeC, n = 6) and metastatic (LMeC, n = 6) COM cell lines under hypoxic condition. Dark grey color box indicates the relative expression level of ENSCAFT00000084705.1 in KMeC and LMeC cells after 48h and light grey color represents the expression levels after 96h. Differences between KMeC and LMeC cells at each time interval were analyzed using the Mann–Whitney U test. A p-value of less than 0.05 (p < 0.05) was considered statistically significant. * denotes a significant test; ns = non-significant test.
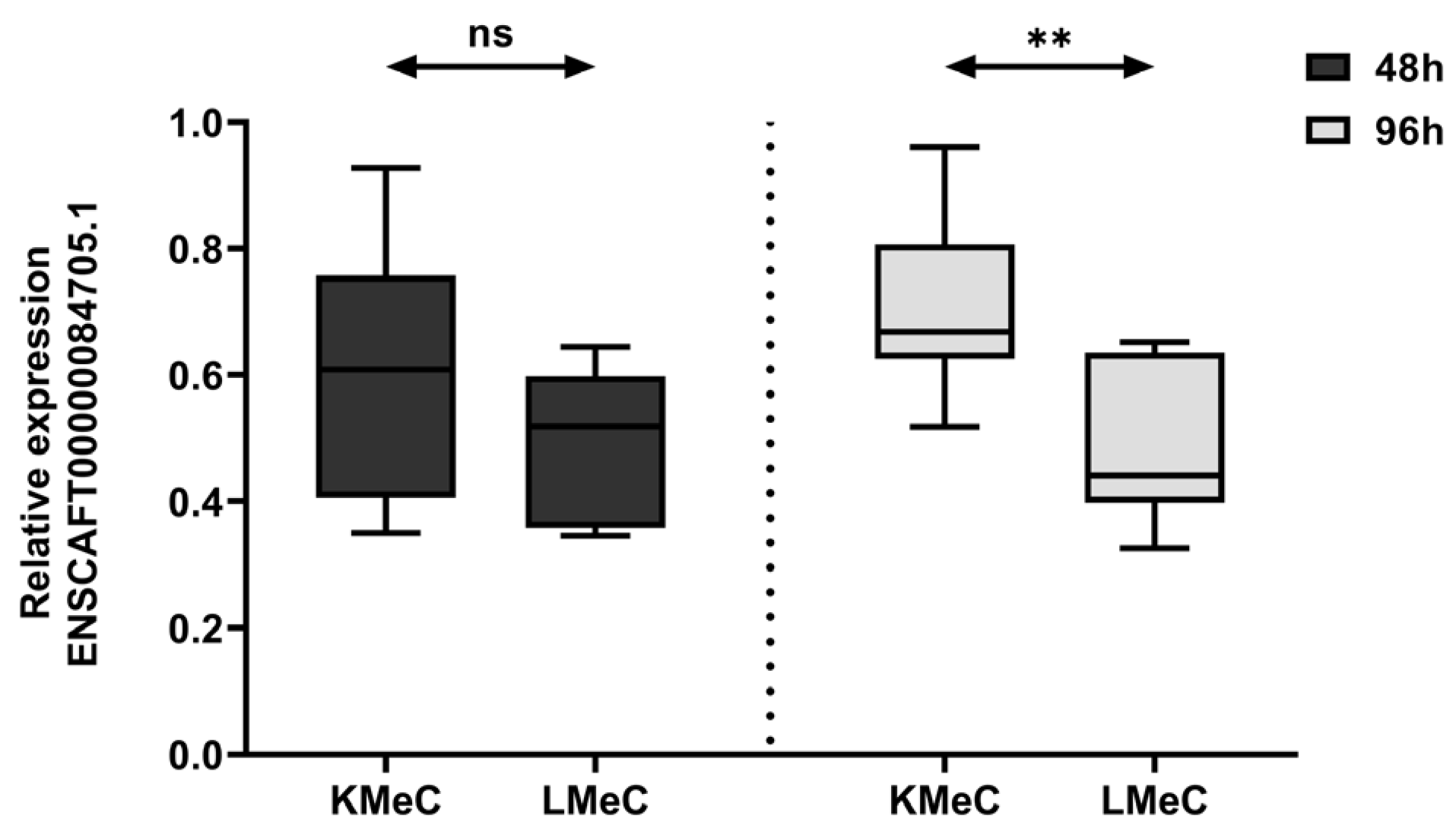
Figure 6.
Relative expression of the targeted ncRNA (ENSCAFT00000084705.1) in plasma and plasma EVs by qRT-PCR. The y-axis of the graph indicates the cycle threshold (Ct) values of miR-16 (internal control for plasma, n = 5) and ENSCAFT00000084705.1 (n = 20) in plasma samples, and miR-186 (internal control for plasma EVs, n = 5) and ENSCAFT00000084705.1 (n = 11) in plasma EVs. Average Ct value of ENSCAFT00000084705.1 was observed higher than 35 in both plasma and plasma EVs samples.
Figure 6.
Relative expression of the targeted ncRNA (ENSCAFT00000084705.1) in plasma and plasma EVs by qRT-PCR. The y-axis of the graph indicates the cycle threshold (Ct) values of miR-16 (internal control for plasma, n = 5) and ENSCAFT00000084705.1 (n = 20) in plasma samples, and miR-186 (internal control for plasma EVs, n = 5) and ENSCAFT00000084705.1 (n = 11) in plasma EVs. Average Ct value of ENSCAFT00000084705.1 was observed higher than 35 in both plasma and plasma EVs samples.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



