
Submitted:
05 July 2024
Posted:
08 July 2024
You are already at the latest version
Abstract
Background: The antisense oligonucleotide against APOC3 mRNA volanesorsen was recently introduced to treat Familial Chylomicronemia Syndrome (FCS). Cases of decreased platelet count are reported among patients treated with volanesorsen. The aim of the study is to evaluate platelet function and thrombin generation (TG) assessment in FCS patients receiving volanesorsen. We performed a cross-sectional study on FCS patients treated with volanesorsen. Methods: changes in PLC were assessed from baseline to Tw12 and Tw36. To assess TG, samples were processed by CAT (with PPP-reagent LOW). The results were expressed by the trombogram graphic (thrombin variation over time); lagTime; Endogenous Thormbin Potential(ETP); peak; time to reach peak(ttpeak), startTail e velocity Index. Platelet aggregation was assessed by testing different agonists using turbidimetry method. Results: Four FCS patients and 4 matched healthy controls were included in the present study. Changes in PLC at Tw12 was 30% and 34% at T36w. Thrombin generation results show values in normal range (for patients and controls respectively lagTime:10,42±4,40 and 9,25±0,99; ttPeak:14,33± 4,01 and 13,10±0,67; startTail:32,13±3,54 and 29,46±1,69; velocityIndex:20,21±3,63 and 33,05±13,21; ETP 599,80±73,47 and 900,2±210,99; peak value 76,84±1,07 and 123,30±39,45) and no significant difference between cases and controls. Platelet aggregation test showed values in range with no significant difference with healthy controls Conclusions: Our study showed for the first time that no significant changes in the general haemostasis assessed by TG and in platelet function have been observed in FCS patients receiving volanesorsen.
Keywords:
Familial Chylomicronemia Syndrome
; severe hypertriglyceridemia
; Volanesorsen
; antisense oligonucleotide
; Platelets
; thrombin generation
1. Introduction
FCS is a rare monogenic disease (prevalence of 1-9:1000000) with autosomal recessive transmission. FCS is caused by loss of function mutations in lipoprotein-lipase (LPL) gene or in LPL co-factors genes such as apolipoprotein A5 (APOA5), lipase maturation factor 1 (LMF1), glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1) and glycerol- 3-phosphate dehydrogenase 1 (G3PDH1) [1].
Although, available lipid lowering therapies (fibrates, statins and omega-3 fatty acids) decrease serum triglycerides [2,3], a not negligible part of subjects with hypertriglyceridemia (HTG), particularly those with FCS, do not achieve therapeutic goals [4].
Targeting Apo-CIII is a promising approach to treat refractory HTG and to prevent acute pancreatitis in FCS subjects [5]. The second-generation antisense oligonucleotide (ASO) against APOC3 mRNA volanesorsen (previously called ISIS 304801, ISIS-ApoCIIIRx and IONIS-ApoCIIIRx) was recently introduced to treat sHTG [5,6,7,8].
A recent metanalysis including 4 RCTs showed a ≈ 92% mean reduction in TG in subjects receiving volanesorsen as compared to placebo-treated controls, accompanied by a significant reduction of 70% in Apo-B48 levels [5]. Cases of decreased platelet count (PLC) were reported during treatment with volanesorsen. The underlying mechanisms of this side effect is still unknown and further studies are needed to explore a potential change in platelet function and in overall hemostatic process.
Available data suggest that volanesorsen is quite well tolerated and cases of thrombocytopenia were found to be dose-related and reversible. Based on this, volanesorsen received approval by food drug administration (FDA) for FCS subjects treatment [9].
More in detail, the phase 3, double-blind, randomised, 52-week phase 3 study (APPROACH) conducted on a group of 66 individuals with FCS, with the primary outcome being the change in fasting TG percentage before and after 3 months of treatment, reported adverse effects mainly including injection site reactions and thrombocytopenia. A PLC lower than 100,000 per microlitre was reported in 16 out of 33 treated subjects, including 2 with a count lower than 25,000 per microlitre, with no bleeding occurring in any of the cases [10]. For this reason, the patients were treated with immunoglobulins and steroids with close platelet monitoring, accompanied by a dose reduction, with complete recovery [10].
However, it has been shown that thrombocytopenia is not just a ASO-related adverse event, but changes in platelets count are often reported in FCS patients [11]. Indeed, in a study conducted on homozygotes for variants in the LPL gene causing FCS (HoLPL), on heterozygotes for variants causing loss of function (HeLPL) and on a control group (WT), it was shown that in fasting condition, patients with FCS and HeLPL had a lower PLC than the normolipidemic control group (Larouche, Brisson et al. 2023). Following a meal consumption, PLC rises slightly in the control group, whereas in FCS patients, the levels remain low even 5 hours after consumption, and this drop is more pronounced in HoLPL than in HeLPL. These changes in PLC have been shown to correlate with plasma TG levels, neutrophil/lymphocyte ratio (NLR) and lymphocyte count, but not with platelet activity (Larouche, Brisson et al. 2023). Although, FCS patients show a lower PLC than healthy subjects, platelet activity seems not to be affected [11].
To further evaluate key aspect of haemostasis and to observe and to estimate the impact of a potential adverse effect, the aim of our study was to assess platelet function by platelet aggregation test and thrombin generation assessed by the Calibrated Automated Thrombogram (CAT) test in patients with FCS, currently treated with Volanesorsen.
2. Materials and Methods
2.1. Study Population
Patients attending the Regional Reference Center for the Treatment of Dyslipidemia at the ‘Federico II’ University Hospital were recruited from January 2022 to September 2023, applying the following inclusion and exclusion criteria.
Inclusion criteria: diagnosis of FCS; eligibility for treatment with Volanesorsen with reference to the criteria identified by the Italian Medicines Agency (AIFA). The latter include genetic diagnosis of FCS, previous episode of acute pancreatitis in the last 5 years; TG > 885 mg/dl despite hypolipidic diet, fibrates and polyunsaturated fatty acids (PUFA); basal PLC >140,000 platelets per μl. Stable treatment with volanesorsen (> 12 weeks)
Exclusion criteria: Age < 18 years; Incompetence to understand or sign the consent form; high transaminase level (greater than 3 x upper limit of normal); Decompensated diabetes mellitus (HbA1C > 8%); End-stage renal failure (glomerular filtration rate <30 ml/min/m2); Ongoing neoplasm or diagnosis of malignant tumour within 2 years prior to first visit; Secondary HTG (induced by hypothyroidism, corticosteroids, hormone therapy).
2.2. Study Protocol
The patients gave their written informed consent to participate in the study, and all data of interest were entered anonymously into a special database. Following informed consent, a detailed medical history was taken for each patient: data were collected on age, sex, previous and/or current medical conditions, current and previous lipid lowering therapy with dosage and frequency of administration. Body weight was assessed using an electronic beam scale with digital readout and to the nearest 0.1 kg, following bladder emptying. Patients were measured barefoot and in light indoor clothing.
The study was approved by our ethics committee (protocol 2015/261) in accordance with the Declaration of Helsinki, and the remaining patients and guardians of the participants provided informed consent.
Before the start of the study, all enrolled patients received lipid-lowering therapy (fibrate and high-dose PUFA) and implemented treatment with Volanesorsen 285 mg (vials) administered subcutaneously every 7 days for the first 3 months (induction dosage) and then once every 14 days as maintenance.
2.3. Hematology Blood Parameteres
Periodic blood count monitoring (including PLC) was performed according to AIFA indication for patients monitoring during treatment with volanesorsen.
In the frame of the present cross-sectional study, we evaluated PLC on the same day of evaluation of other coagulative tests. We also evaluated changes in PLC from baseline values (before starting volanesorsen treatment), to week 12 (after concluding induction dose of volanesorsen) and to week 36 during treatment with volanesorsen.
2.4. Thrombin Generation
TG in plasma was measured with the calibrated automated thrombogram (CAT) assay as developed by Hemker and co-workers [12]. CAT was evaluated at week 36 during treatment with volanesorsen.
Briefly, 80 µl platelet poor plasma (PPP) was mixed with 20 µl of a mixture containing tissue factor (Dade-Behring) at a final concentration of 1 pM and phospholipid vesicles (f.c. 4 µM 20 mol% phosphatidylserine, 60 mol% phosphatidylcholine and 20 mol% phosphatidyl ethanolamine). To calibrator wells, 20 µl of calibrator (α2macroglobulin-thrombin complex, was added instead of TF and PL. After 10 minutes of incubation at 37°C, thrombin generation was initiated by the addition of 20 μl of the thrombin specific substrate, Z-Gly-Gly-Arg-7-amino-4-methylcoumarin (f.c. 416 µM, Bachem) and CaCl2 (f.c. 16.7 mM). Fluorescence was measured with a Fluoroscan Ascent reader (Thermo Labsystems) and data were analysed with dedicated software (Thrombinoscope, Stago). Thrombin generation was expressed based on endogenous thrombin potential (ETP); lagtime (LT); thrombin peak (TP), time-to-thrombin peak (TTP).
2.5. Platelet Aggregation
Platelet aggregation was evaluated by Born method of turbidimetric aggregation [13] using CHRONO-LOG® 700 Series Whole Blood/Optical Lumi-Aggregometer with AGGRO/LINK®8 Software at week 36 during treatment with volanesorsen. Platelet-rich plasma (PRP) was tested with four agonists (ADP, arachidonic acid, collagen and ristocetin) compared with a sample of platelet-poor plasma (PPP). The test was executed in two channels, adding two different concentrations for each agonist (1,8 μM and 4,0 μM ADP, 0,4 µg/ml and 0,8 µg/ml collagen, 0,8 mg/ml and 1,25 mg/ml ristocetin). Only arachidonic acid was tested once at 0,4 mM. Platelet aggregation induced by addiction of each agonist was evaluated through the percentage of light transmission: ≥50% within 3 minutes indicates no impairment of platelet aggregation. This test is effective to analyse platelet function abnormalities.
2.6. Statistical Analysis
Statistical analysis of the data was performed using the IBM SPSS 29 System (SPSS Inc., Chicago, IL, USA). Continuous data were expressed as mean value ± standard deviation (SD) or, in the case of variables with non-normal distribution, as median IQR range.
To compare continuous variables for paired and independent samples, the t-test was performed; in the case of values with a non-Gaussian skewed distribution, the Mann-Whitney U test was used. Categorical variables were expressed in % and analysed with the χ2 test or Fisher’s exact test, in the case where the minimum expected value was <5. Correlations were assessed using Pearson’s linear correlation coefficient (r) and in the case of non-parametric variables, Spearman’s correlation (rho) was calculated. All results were expressed as two-tailed values, with p-value < 0.05 to be statistically significant.
3. Results
We enrolled 4 FCS patients (reported by initials of first and last name, CG, CL, BA, PS), two men and two women, all diagnosed with FCS and treated with volanesorsen, and four healthy volunteers as control group, matched for gender and age.
3.1. Study Population
The clinical and demographic characteristics of the study population (as individual patients) are shown in Table 1. Overall, they have a mean age of 56.5 ± 0.5 years and a mean BMI of 22.1 ± 1.89. Three patients (BA, CG, CL) were carriers of HoLPL variant. CG and CL patients, who are related, present the same variant, secondary to inbreeding on the parents’ side. PS was carrier of pathogenic variants in the APOA5 cofactor in compound heterozygosity.
All patients were eligible to treatment with Volanesorsen according to the AIFA criteria. All patients were treated with hypolipidic diet plus MCT oil (20 grams/day). and lipid lowering drugs. In detail: micronised fenofibrate 145 mg CL, BA and PS and gemfibrozil 600 mg 2 cp per day CG, PUFA (6 grams/day).
All patients developed pancreatitis during their lifetime, the CG patient reported an episode during pregnancy. Patient BA presented more than 20 episodes of acute pancreatitis. Both CL and BA presented evolution towards chronic pancreatitis resulting in exocrine pancreas deficiency.
All patients complained of chronic abdominal pain and, with the exception of BA, developed diabetes secondary to pancreatitis.
3.2. Effects of Volanesorsen on Platelet Count
PLC was evaluated according to the AIFA recommendations for patient motoring every 2 weeks. PLC at baseline had a median value of 237.5 platelets/μl (IQR: 228 - 304) and after 12 weeks the median PLC value was 181 platelets/μl (IQR: 159 - 214.8), median PLC at week 36 was 178,5 platelets/μl (IQR: 162 - 211.75). All patients showed a PLC in the reference values (150,000-450,000 platelets/μl) at baseline. We observed a 30% reduction in PLC from baseline to week 12 and of 34% from baseline to week 36. However, none of patients reported PLC values lower than 100.000 platelets/μl and no clinically relevant bleeding episodes were reported. Individual and cumulative percent changes in PLC are reported in Figure 1a,b, respectively.
3.3. Thrombin Generation Analysis
The thrombin generation analyses shown in Table 2 resulted in mean lagTime values of 10.42 ± 4.40; ETP of 599.8 ± 73.47; peak of 76.84 ± 1.07; ttPeak of 14.33 ± 4.01; startTail of 32.13 ± 3.54; velocity Index of 20.21 ± 3.63. All values are within the estimated normal ranges and do not differ from the healthy controls whose values are shown in Table 2. However, comparing the cases and the controls, we can observe an interesting difference in the amount of thrombin obtained, i.e., the FTE (599.80 ± 73.47 for the cases and 900.2 ± 210.99 for the controls) and the maximum concentration of thrombin obtained, i.e., the peak (76.84 ± 1.07 for the cases and 123.30 ± 39.45 for the controls), as can be seen in Table 2. This can also be verified graphically in the thrombograms obtained (Figure 2). However, these differences are not statistically significant.
3.4. Platelet Aggregation Analysis
The platelet aggregation tests showed values in range: ADP (1,8 μM) 62%; Arachidonic Acid (0,4 mM) 66%; Collagen (0,8 μg/ml) 84%; Ristocetin (1,25 mg/ml) 90% as compared to heathy controls ADP (1,8 μM) 70%; Arachidonic Acid (0,4 mM) 72%; Collagen (0,8 μg/ml) 85%; Ristocetin (1,25 mg/ml) 92%. No significant difference was found between case and controls
4. Discussion
Results of our study show that volanesorsen treatment induced a mild reduction in PLC (never lower than 100,000 platelets/μl) in 4 FCS patients. These results are in line with those obtained from clinical trials that evaluated the efficacy and safety of volanesorsen [5]. Accordingly, in none of patients a dose delayed administration was needed. This effect on PLC reduction has also been documented for other second-generation ASOs [14]. However, the pathophysiological mechanism of this side effect is still unclear. One of the possible hypotheses is that the class of ASOs may promote the binding of neutrophils to platelets [15]. To the best of our knowledge, there are no studies aimed to evaluate the impact of ASOs-induced reduction of PLC on platelet aggregation and global hemostasis. Our study provides insight into hemostatic function beyond the clinical observation of PLC and hemorrhagic manifestations. The results of thrombin generation assessment reveal values within the estimated normal range and show no significant differences from healthy controls.
Platelet aggregation was performed in only one patient, due to the impossibility of performing the proposed method with lactescent samples. It showed normal results when compared to healthy controls, thus detecting an optimal status of global haemostasis.
To date, this represents the first study aimed to explore platelet function and thrombin generation capacity in FCS patients on volanesorsen therapy. Previous data from the literature show that PLC fluctuations are quite common in patients with FCS secondary to pathogenic variants in the LPL gene.
In one study, changes in PLC over 15 years in patients with such variants were reported, and it was observed that about 55% reported PLC <150,000 platelets/μl at least once. Paradoxically, it was also observed that approximately 12% presented thrombocytosis, with a PLC>450,000 platelets/μl and 3.6% presented both thrombocytosis and thrombocytopenia [11]. Furthermore, it was reported in a recent study that in HoLPL patients, PLC decreases after meals, but their function is not altered [15]. Consistently, patients carrying the pathogenic variant in the LPL gene were found to have lower PLC values than patients carrying the pathogenic variant in the APOA gene5.
The thrombin generation results would appear to be correlated with the type of APO A5 pathogenetic variant reported by each patient, with values being lower in relation to the type of causative genetic variant. Thus, a role of the underlying pathology could be hypothesized in contributing to the development of this potential side effect.
From a physiological point of view, the LPL enzyme is bound to endothelial cells via heparan sulphate proteoglycans (HSPGs), which makes it possible to identify the luminal surface of endothelial cells as the site of catalytic action [16]. Heparan sulphate (HS) is known to inhibit thrombin generation by inducing the inactivation of factor Xa and thrombin, via antithrombin III [17]. A more recent study also reveals how HS contained within the perlecan proteoglycan, synthesised and released by endothelial cells and the smooth muscle cells that make up the endothelium, induces the dimerisation of G6b-B, a transmembrane receptor containing ITIM motifs expressed by mature megakaryocytes (MK) and platelets. However, the involvement of other HSPGs besides perlecan is not excluded. This interaction has the effect of inhibiting platelet adhesion and activation [18]. Thus, the variants induce the formation of a protein, in this case LPL, which has no physiological interaction with HSPGs. As reported by the gnomAD and UniProt databases, in CL and CG patients the c.844G>T homozygosity variant (p.Glu282*) located in exon 6 of the LPL gene encodes for a stop codon, implying the production of a truncated protein, which lacks the interaction domain with HS present in the C-terminal portion of LPL [19]. For patient BA, the variant in homozygosis is c.621C>G (p.Asp207Glu) located in exon 5 of the LPL gene and encodes for a missense variant: as a result, we cannot establish with certainty whether this variant has an effect on the interaction with HSPGs as it does for CL and CG, it is only known that this variant impairs the lipolytic activity of the enzyme [19,20].
With regard to APOA5, it has been shown that the apolipoprotein not only binds heparin but also is able to facilitate the interaction between HSPGs and TRLs, thereby increasing the activity of LPL26. The PS patient is compound heterozygous for the c.427delC p.(Arg143Alafs*57) variant in exon 4 and c.49+5G>A in intron 2 of the APOA5 gene. The variant in exon 4 results in the production of a truncated protein that would not have part of a 42 amino acid positive stretch (residues 186-227), postulated in one study as a site of interaction with HSPGs. However, this interaction between lipoproteins, APOA5 and HSPGs appears to be more consistent with hepatic internalization of lipoproteins than hydrolysis [21].
In light of this, it could be assumed that reduced binding of LPL to HSPGs may favor the exposure of HS chains, thereby inhibiting thrombin generation. Similarly, a pathogenic variant in the APOA5 gene could have a similar effect to that seen for LPL variants, albeit to a lesser extent. This may explain how, although generation is within the estimated normal ranges, a slight variation in the amount of thrombin obtained and a slight shift in the peak was observed. In fact, the HTG condition would not affect thrombin generation, as demonstrated in a study carried out on patients with hypertriglyceridemia (TG = 330.5±24.5 mg/dl; p<0.01) [22]. From this point of view, our study shows an exploratory role in an area where there are grey areas in the literature; therefore, further studies are needed to further investigate this clinical question.
The main limitation of our study is represented by the small sample size. However, FCS is an extremely rare disease (1:1.000.000) and therefore our sample is representative of disease prevalence in our region according to Italian epidemiological data. Another limitation derives from the potential influence of chylomicrons on platelet aggregation test. However, results were entirely comparable to those obtained in healthy controls. It would be interesting to perform the same analyses in patients reporting more severe PLC reduction to explore the presence of functional changes in this setting. However, the extreme rarity of the disease and the low incidence of the side effect makes this difficult to be obtained.
5. Conclusions
Our study shows for the first time that in patients treated with Volanesorsen, despite a modest reduction in PLC, no significant alterations were observed in platelet function in terms of platelet aggregation and general haemostasis studied by means of the thrombin generation test. It is inferred that factors related to the underlying pathology, especially the genotype, may contribute to such manifestations that are not just a class effect of the drug. This study represents a starting point for a translational exploration of the global haemostatic pattern of patients with FCS. Therefore, further studies with larger study samples will be needed to better define this clinical issue.
Data Availability Statement:. Not applicable.
Author Contributions
Conceptualization, I.C.; methodology, I.C. and F.C.; validation, M.D.D.T., G.C. and G.F.; formal analysis, I.C.; investigation, N.V.; data curation, R.S..; writing—original draft preparation, I.C. and R.S.; writing—review and editing, GDE; and M.D.D.T.; visualization, G.I.; supervision, MDM; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of Federico II University Hospital (protocol code 2015/261).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
none.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moulin, P.; Dufour, R.; Averna, M.; Arca, M.; Cefalu, A.B.; Noto, D.; D’Erasmo, L.; Di Costanzo, A.; Marcais, C.; Alvarez-Sala Walther, L.A. , et al. Identification and diagnosis of patients with familial chylomicronaemia syndrome (FCS): Expert panel recommendations and proposal of an “FCS score”. Atherosclerosis 2018, 275, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Ooi, E.M.; Barrett, P.H.; Chan, D.C.; Watts, G.F. Apolipoprotein C-III: understanding an emerging cardiovascular risk factor. Clinical science 2008, 114, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Gouni-Berthold, I. The role of antisense oligonucleotide therapy against apolipoprotein-CIII in hypertriglyceridemia. Atherosclerosis. Supplements 2017, 30, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Warden, B.A.; Duell, P.B. Volanesorsen for treatment of patients with familial chylomicronemia syndrome. Drugs of today 2018, 54, 721–735. [Google Scholar] [CrossRef]
- Calcaterra, I.; Lupoli, R.; Di Minno, A.; Di Minno, M.N.D. Volanesorsen to treat severe hypertriglyceridaemia: A pooled analysis of randomized controlled trials. Eur J Clin Invest 2022, 52, e13841. [Google Scholar] [CrossRef]
- Gaudet, D.; Brisson, D.; Tremblay, K.; Alexander, V.J.; Singleton, W.; Hughes, S.G.; Geary, R.S.; Baker, B.F.; Graham, M.J.; Crooke, R.M. , et al. Targeting APOC3 in the familial chylomicronemia syndrome. The New England journal of medicine 2014, 371, 2200–2206. [Google Scholar] [CrossRef]
- L. D’Erasmo, S.D.F., A. Gelrud, A. Digenio, V.J. Alexander, A. Hsieh, I. Gouni-Berthold, E. Bruckert,E. Stroes, R.S. Gear y, S.G. Hughes, D. Gaudet. TREATMENT WITH VOLANESORSEN (VLN) REDUCED TRIGLYCERIDES AND PANCREATITIS IN PATIENTS WITH FAMILIAL CHYLOMICRONEMIA SYNDROME (FCS) AND SEVERE HYPERTRIGLYCERIDEMIA (SHTG) VS PLACEBO: RESULTS OF THE APPROACH AND COMPASS STUDIES. Giornale Italiano dell’arteriosclerosi 2019, 4, 100. [Google Scholar]
- Macchi, C.; Sirtori, C.R.; Corsini, A.; Santos, R.D.; Watts, G.F.; Ruscica, M. A new dawn for managing dyslipidemias: The era of rna-based therapies. Pharmacological research 2019, 150, 104413. [Google Scholar] [CrossRef]
- Paik, J.; Duggan, S. Volanesorsen: First Global Approval. Drugs 2019, 79, 1349–1354. [Google Scholar] [CrossRef]
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M. , et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. The New England journal of medicine 2019, 381, 531–542. [Google Scholar] [CrossRef]
- Gaudet, D.B. , Alexis & Tremblay, Karine & Brisson, Diane & Laflamme, Nathalie & Paquette, Martine & Dufour, Robert & Bergeron, Jean. Natural History (up to 15 years) of Platelet Count in 84 Patients with Familial Hyperchylomicronemia Due to Lipoprotein Lipase Deficiency. Journal of clinical lipidology 2017, 11, 797–798. [Google Scholar] [CrossRef]
- Tripodi, A. Thrombin Generation Assay and Its Application in the Clinical Laboratory. Clin Chem 2016, 62, 699–707. [Google Scholar] [CrossRef]
- Born, G.V. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962, 194, 927–929. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu Rev Med 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Larouche, M.; Brisson, D.; Morissette, M.C.; Gaudet, D. Post-prandial analysis of fluctuations in the platelet count and platelet function in patients with the familial chylomicronemia syndrome. Orphanet J Rare Dis 2023, 18, 167. [Google Scholar] [CrossRef]
- Mead, J.R.; Irvine, S.A.; Ramji, D.P. Lipoprotein lipase: structure, function, regulation, and role in disease. J Mol Med (Berl) 2002, 80, 753–769. [Google Scholar] [CrossRef]
- Ofosu, F.A.; Modi, G.J.; Smith, L.M.; Cerskus, A.L.; Hirsh, J.; Blajchman, M.A. Heparan sulfate and dermatan sulfate inhibit the generation of thrombin activity in plasma by complementary pathways. Blood 1984, 64, 742–747. [Google Scholar] [CrossRef]
- Vogtle, T.; Sharma, S.; Mori, J.; Nagy, Z.; Semeniak, D.; Scandola, C.; Geer, M.J.; Smith, C.W.; Lane, J.; Pollack, S. , et al. Heparan sulfates are critical regulators of the inhibitory megakaryocyte-platelet receptor G6b-B. Elife 2019, 8. [Google Scholar] [CrossRef]
- Lutz, E.P.; Merkel, M.; Kako, Y.; Melford, K.; Radner, H.; Breslow, J.L.; Bensadoun, A.; Goldberg, I.J. Heparin-binding defective lipoprotein lipase is unstable and causes abnormalities in lipid delivery to tissues. The Journal of clinical investigation 2001, 107, 1183–1192. [Google Scholar] [CrossRef]
- Haubenwallner, S.; Horl, G.; Shachter, N.S.; Presta, E.; Fried, S.K.; Hofler, G.; Kostner, G.M.; Breslow, J.L.; Zechner, R. A novel missense mutation in the gene for lipoprotein lipase resulting in a highly conservative amino acid substitution (Asp180-->Glu) causes familial chylomicronemia (type I hyperlipoproteinemia). Genomics 1993, 18, 392–396. [Google Scholar] [CrossRef]
- Lookene, A.; Beckstead, J.A.; Nilsson, S.; Olivecrona, G.; Ryan, R.O. Apolipoprotein A-V-heparin interactions: implications for plasma lipoprotein metabolism. J Biol Chem 2005, 280, 25383–25387. [Google Scholar] [CrossRef] [PubMed]
- Aoki, I.; Aoki, N.; Kawano, K.; Shimoyama, K.; Maki, A.; Homori, M.; Yanagisawa, A.; Yamamoto, M.; Kawai, Y.; Ishikawa, K. Platelet-dependent thrombin generation in patients with hyperlipidemia. J Am Coll Cardiol 1997, 30, 91–96. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Individual percent changes in PLC from baseline to T12w and T36w (a) Cumulative percent changes in PLC from baseline to T12w and T36w (b).
Figure 1.
Individual percent changes in PLC from baseline to T12w and T36w (a) Cumulative percent changes in PLC from baseline to T12w and T36w (b).
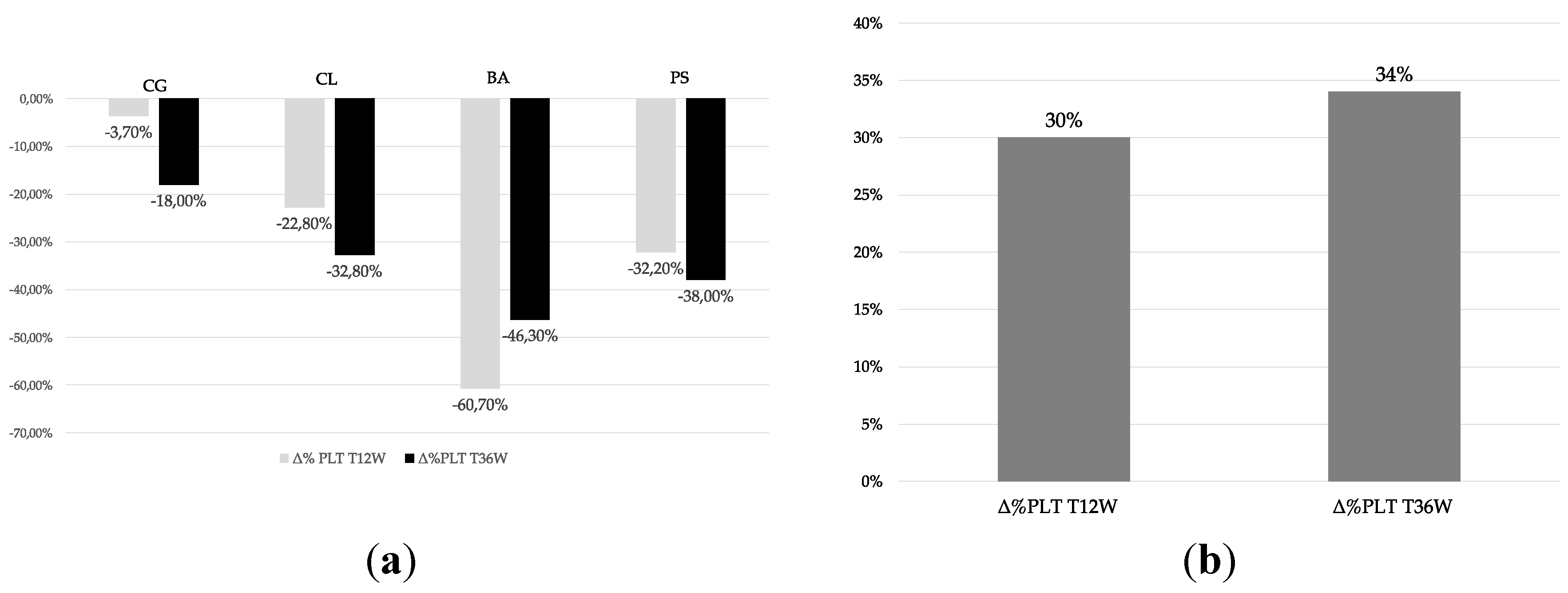
Figure 2.
Outcome of thrombin generation (CAT) showed for each patient.
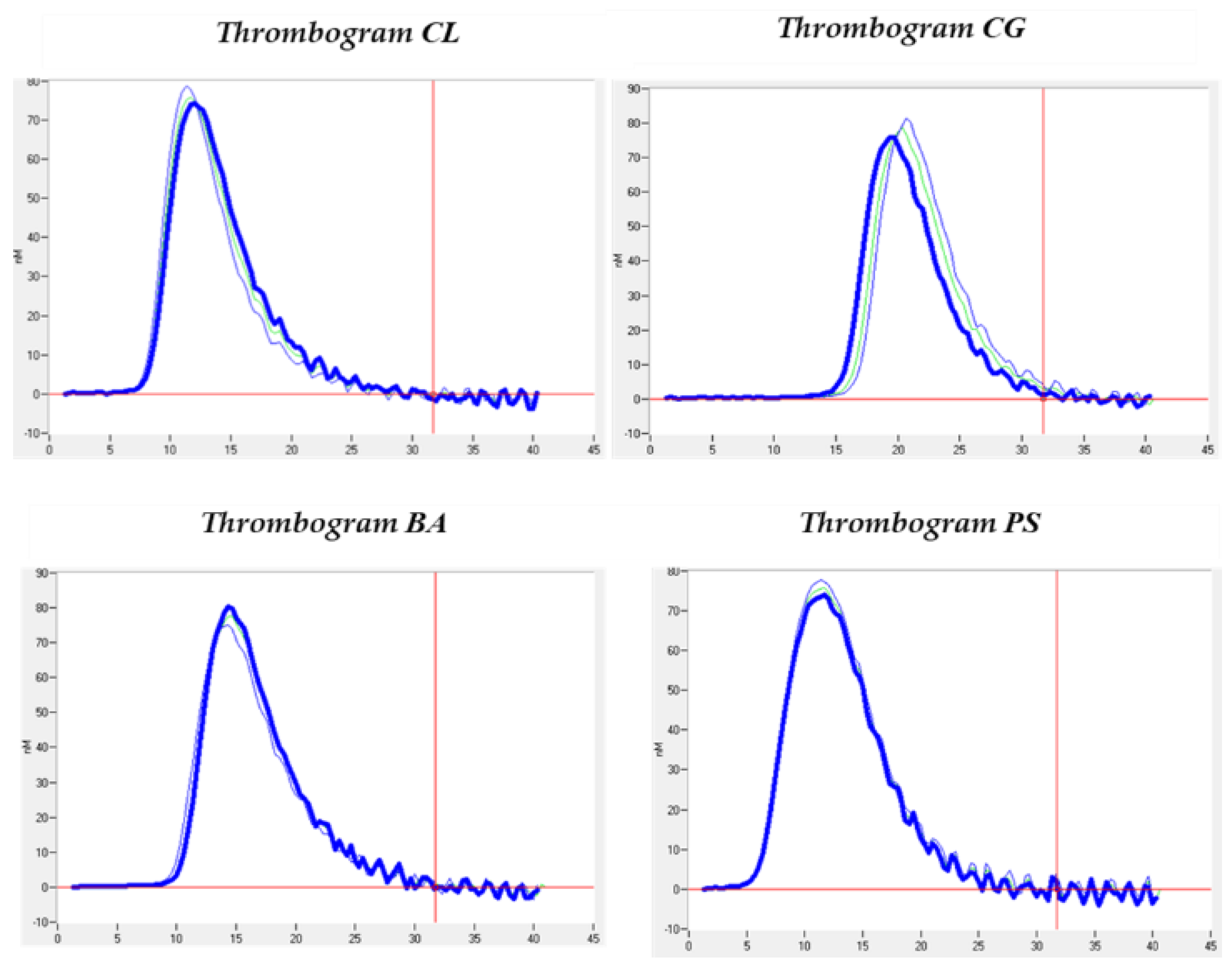

Table 1.
Clinical and demographic characteristics for each patient (CG, CL, BA and PS).
| Patients | CG | CL | BA | PS | Mean ± SD |
|---|---|---|---|---|---|
| Age | 62 | 71 | 53 | 40 | 56.5 ± 13.2 |
| Sex (M=1) | 0 | 1 | 1 | 0 | N.A. |
| BMI (kg/m2) | 20.8 | 22 | 24.8 | 20.8 | 22.1 ± 1.9 |
| Age at diagnosis | 20 | 22 | 6 | 4 | 13 ± 9.3 |
| Pathogenic variants | LPL c.844G>T (p.Glu282*) - stop gained |
LPL c.844G>T (p.Glu282*) - stop gained |
LPL c.621C>G (p.Asp207Glu) - missense |
APOA5 c.427delC p.(Arg143Alafs*57) c.49+5G>A |
|
| Previous pancreatitis | 4 | 10 | 20 | 3 | 9.25 |
| Pancreatitis in last 5 years | 1 | 2 | 10 | 2 | 3.75 |
| Chronic pancreatitis (Yes=1) | 0 | 1 | 1 | 0 | 50% |
| Diabetes | 1 | 1 | 0 | 1 | 75% |
| Treatment (fibrates, PUFA and MCT oil) | Gemfibrozil 600 mg bid, PUFA (6 gr), MCT oil | Fenofibrate 145 mg/daly, PUFA (6 gr), MCT oil | Fenofibrate 145 mg/daly, PUFA (6 gr) MCT oil | Fenofibrate 145 mg/daly, PUFA (6 gr) MCT oil |
-- |
Table 2.
Statistical analyses of thrombin generation (CAT) results in patients and healthy controls expressed as mean ± SD. Parameters analyzed are lagTime (indicates time interval between addition of trigger and thrombin generation), ETP (endogen thrombin potential indicates how much thrombin has been generated), peak (the maximum thrombin concentration obtained), ttPeak (time to peak indicates thrombin generation’s velocity), startTail (end of thrombin generation) and velocity Index (peak height/(time to peak – lag time). LagTime, ttPeak and startTail are expressed in minutes.
Table 2.
Statistical analyses of thrombin generation (CAT) results in patients and healthy controls expressed as mean ± SD. Parameters analyzed are lagTime (indicates time interval between addition of trigger and thrombin generation), ETP (endogen thrombin potential indicates how much thrombin has been generated), peak (the maximum thrombin concentration obtained), ttPeak (time to peak indicates thrombin generation’s velocity), startTail (end of thrombin generation) and velocity Index (peak height/(time to peak – lag time). LagTime, ttPeak and startTail are expressed in minutes.
| Parameter | FCS Patients | Controls | p-value |
| LagTime | 10.42 ± 4.40 | 9.25 ± 0.99 | 0.685 |
| ETP | 599.80 ± 73.47 | 900.2 ± 210.99 | 0.102 |
| Peak | 76.84 ± 1.07 | 123.30 ± 39.45 | 0.104 |
| ttPeak | 14.33 ± 4.01 | 13.10 ± 0.67 | 0.620 |
| StartTail | 32.13 ± 3.54 | 29.46 ± 1.69 | 0.143 |
| Velocity Index | 20.21 ± 3.63 | 33.05 ± 13.21 | 0.149 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



